Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Background
Specific functions of lipids in...
Landscape of abnormal lipid...
Abnormal regulation of lipid...
Lipid metabolic reprogramming in...
Effect of lipid metabolic...
Abnormal cell death related to...
Clinical applications of lipid...
Conclusions
Abbreviations
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2023; 13(6):1774-1808. doi:10.7150/thno.82920 This issue Cite
Review
The role of lipid metabolic reprogramming in tumor microenvironment
Kai Yang1,2,*, Xiaokun Wang1,*, Chenghu Song1, Zhao He1, Ruixin Wang1, Yongrui Xu1, Guanyu Jiang1, Yuan Wan3, ![]() , Jie Mei2,
, Jie Mei2, ![]() , Wenjun Mao1,
, Wenjun Mao1, ![]()
1. Department of Thoracic Surgery, The Affiliated Wuxi People's Hospital of Nanjing Medical University, Wuxi, 214023, China.
2. Department of Oncology, The Affiliated Wuxi People's Hospital of Nanjing Medical University, Nanjing, 210029, China.
3. The Pq Laboratory of BiomeDx/Rx, Department of Biomedical Engineering, Binghamton University, Binghamton 13850, USA.
* equal contribution
Received 2023-1-25; Accepted 2023-3-7; Published 2023-3-13
Abstract

Metabolic reprogramming is one of the most important hallmarks of malignant tumors. Specifically, lipid metabolic reprogramming has marked impacts on cancer progression and therapeutic response by remodeling the tumor microenvironment (TME). In the past few decades, immunotherapy has revolutionized the treatment landscape for advanced cancers. Lipid metabolic reprogramming plays pivotal role in regulating the immune microenvironment and response to cancer immunotherapy. Here, we systematically reviewed the characteristics, mechanism, and role of lipid metabolic reprogramming in tumor and immune cells in the TME, appraised the effects of various cell death modes (specifically ferroptosis) on lipid metabolism, and summarized the antitumor therapies targeting lipid metabolism. Overall, lipid metabolic reprogramming has profound effects on cancer immunotherapy by regulating the immune microenvironment; therefore, targeting lipid metabolic reprogramming may lead to the development of innovative clinical applications including sensitizing immunotherapy.
Keywords: lipid metabolic reprogramming, tumor microenvironment, immunotherapy, therapeutic target
Background
The history of drug therapy for malignancies, such as cancer, has developed from chemotherapy to targeted therapy and then to immunotherapy. Currently, immunotherapy is the most promising antitumor treatment [1]. Unlike patients on conventional therapies, those who respond to immunotherapy are more likely to achieve high-quality long-term survival with fewer side effects [2]. The "immunosurveillance theory" states that the immune system has a complete surveillance function to distinguish and remove "non-self" tumors [3]. The core of tumor immunotherapy is to enhance the antitumor immunity by targeting the cancer-immune cycle. This mainly includes the release and presentation of tumor antigens to activate tumor-associated immune cells, and these antigens are delivered and infiltrated into the tumor tissues to enhance their recognition and subsequent elimination [4]. However, immune escape and immunosuppression reduce the effectiveness of immunotherapy because of abnormal regulations of the cancer-immune cycle [5]. The "tumor immunoediting theory" states that the immune system can not only eliminate tumors but also promote their growth, and this interaction occurs in three stages: clearance, equilibrium, and escape [6]. Tumors evade the clearance of the immune system and suppress the antitumor immune response by several mechanisms. Metabolic reprogramming is one of these mechanisms in which the metabolic patterns of tumor and immune cells are altered to meet their developmental requirements and adapt to the complex tumor microenvironment (TME) [7]. Therefore, correcting the metabolic reprogramming of cells can help to overcome immune escape and immunosuppression, thereby sensitizing immunotherapy.
Metabolic reprogramming is one of the 14 important hallmarks of tumorigenesis in which the energy metabolism network of tumor cells is reset to support their growth, proliferation, and metastasis under the metabolic stress of the TME and immune surveillance [8]. The "starved" tumor cells undergo adaptive metabolic reprogramming including glucose, lipid, and amino acid metabolism [9, 10]. Normal cells rely on mitochondrial respiration for energy supply but tumor cells rely mainly on glycolysis even under aerobic conditions; this phenomenon is known as the 'Warburg effect' [11]. This effect also modulates cellular lipid metabolism by providing raw materials for lipid synthesis, regulating lipid metabolic signaling pathways, and reducing lipid peroxidation. In addition, key enzymes of the gluconeogenesis pathway are also involved in the regulation of lipid metabolism [12]. However, compared with glucose metabolic reprogramming, lipid metabolic reprogramming needs to be further studied.
Lipids are classified as simple and complex lipids; the simple lipids include triglycerides (TAGs), commonly known as fats. The complex lipids include sterols and their esters, phospholipids (PL), and glycolipids. In addition, ketone bodies, although not lipids are important intermediates of fatty acid oxidation (FAO). Lipids play an important function in cellular activities. In addition to serving as a key energy source [13], lipids are the material basis for the synthesis of important biological components [14], involved in intra- and extracellular signal transduction [15], comprise the plasma membrane structure [16], and modify several biomolecules [17]. Cells obtain lipids through exogenous uptake and endogenous synthesis for metabolic activities and store excess lipids in lipid droplets (LDs), which finally undergo oxidative decomposition; this whole process is tightly regulated. Lipid metabolism in tumor cells is different from that in normal cells because of genetic mutations, mitochondrial damage, and TME [18]. This happens in three stages to meet the high energy demand during growth and to mediate immunosuppression. First, the uptake and de novo synthesis of lipids are enhanced through altered nutritional patterns [19, 20]. Then, the synthesized lipids are preferentially allocated to metabolic pathways that contribute to oncogenic modification. Finally, alterations in metabolism have long-term effects on the tumor and immune cells in the TME [21]. The key enzymes in lipid metabolism can have abnormal activity and even have new non-metabolic enzyme functions during this process [22].
Lipid metabolic reprogramming not only plays an important role in the oncogenesis and development of tumors but also modifies the TME by affecting the recruitment, survival, and function of immune cells [23]. Unlike monoclonal cell populations, tumor cells and the cells of TME interact to form a continuously evolving and heterogenous entity [24]. This dynamic change and heterogeneity results in varied lipid metabolism in different types of tumor and immune cells at different stages. Tumor cells in an active proliferative or metastatic state are more dependent on lipid metabolism. Moreover, protumor immune cells also depend on lipid metabolism, whereas antitumor immune cells depend on glucose metabolism [25]. In addition, autophagy induced by metabolic stress of the TME [26] and other cell death modes related to lipid peroxidation (including ferroptosis [27], cuproptosis [28], and calcium overload [29]) are also associated with cellular lipid metabolism and affect tumor immune responses. Therefore, these modes of cell death are the potential targets for tumor therapy.
Lipid metabolic reprogramming plays a critical role in sensitizing immunotherapy. Immunotherapy combined with therapies targeting lipid metabolism will provide a new direction for tumor treatment [23]. Here, we reviewed the characteristics, mechanism, and role of lipid metabolic reprogramming from the perspective of tumor and immune cells in the TME, elaborated on the effects of various cell death modes (especially ferroptosis) on lipid metabolism, and finally summarized the antitumor therapies targeting lipid metabolism to guide the development of new strategies for sensitizing immunotherapy.
Specific functions of lipids in tumor cells
Providing energy and material basis
The most prominent function of lipids is to provide energy for cellular activities. TAGs are an important energy supply and storage material for cells. TAGs break down to yield fatty acids (FA), which undergo FAO and mitochondrial oxidative phosphorylation (OXPHOS) to produce large amounts of ATP. Lipids are not preferentially used by cells. Tumor and other immune cells rely on glucose metabolism when the supply is sufficient. However, when cells are activated or starving, especially under the metabolic stress of the TME, they switch to lipids for energy and material basis [30].
In addition, the intermediates of FAO, ketone bodies, are also available for cellular utilization. Immune cells can use ketone bodies as an alternative energy source in the absence of glucose; however, some tumor cells, such as glioblastoma (GBM), are classified as "glycolysis subtype" for the lack of ketone body oxidases, such as 3-hydroxybutyrate dehydrogenase (BDH1) or 3-succinyl-CoA transferase (OXCT1), because of genetic mutations or mitochondrial damage [31]. In contrast, some cancer cells, such as breast or lung cancer cells, are classified as "ketone metabolic subtype" for their ability to use ketone bodies because of the high expression of ketone body oxidase [32]. A study also revealed that "ketone metabolic subtype" tumor cells were significantly associated with catenin 1 (CTNNB1) gene mutation [33]. Therefore, the use of ketone bodies instead of glucose as the main energy source can cut off the energy source of "glycolysis subtype" tumor cells with a minimal impact on the energy metabolism of immune cells, which provides a theoretical basis for the ketogenic diet (KD) therapy in such cases [34].
Involved in signal transduction
Lipids are the precursors of signaling molecules (lipid mediators), which participate in intra- and extra-cellular signal transduction. Arachidonic acid (AA) derivatives, including prostaglandins (PGs), thromboxanes (TXs), and leukotrienes (LTs), mediate the evolution of chronic inflammation into the tumor [35], such as Barrett's esophagus to esophageal adenocarcinoma, gastric ulcer to gastric cancer, and pancreatitis to pancreatic cancer. PGs reduce oxidative stress and prevent lipid peroxidation in tumor cells [36]. Notably, a study has established a paradigm for immune cell recruitment, described as lipid (PG and LT)-cytokine-chemokine cascade, as a driving force in the effector phase of immune responses [37]. Therefore, targeting the PG synthesis pathway is a major approach for sensitizing immunotherapy.
Metabolites of PL, including inositol triphosphate (IP3), diacylglycerol (DAG), and lysophosphatidic acid (LPA), are associated with multiple oncogenic signaling pathways and tumor immune responses [15]. IP3 promotes the release of intracellular Ca2+ and induce calcium overload, and DAG activates protein kinase C (PKC) leading to oxidative stress, thereby disrupting intracellular homeostasis. In addition, LPA binds to the G protein-coupled receptors (GPCRs) on the membranes of tumor and immune cells and activates RAS, RAC, and RHO signaling pathways [38].
Exosomes are one of the important forms of intercellular communication. Tumor cells can secrete numerous tumor-derived exosomes (TDEs) to transport signaling molecules to immune cells, thereby changing their metabolic state and inducing tumor immunosuppression. Lipids are the signaling molecules and also participate in the formation of the TDE membrane [39]. TDEs have a bidirectional effect on tumor immunity. TDEs increase the accumulation of LDs in immune cells and promote their FAO, which contributes to the formation of an immunosuppressive microenvironment [40]. Interestingly, TDEs carrying tumor biomolecules can mimic tumor antigens and shield the tumor cells from the attacks of the immune system or targeted drugs, which further promotes immune evasion and leads to treatment resistance [39]. In contrast, TDEs can present tumor antigens to T cells directly or through antigen-presenting cells (APCs) and activate macrophages, neutrophils, and natural killer (NK) cells to release antitumor factors, thereby promoting antitumor immunity [41]. Notably, immune cell-derived exosomes also induce an antitumor immune response. For example, exosomes secreted by APCs can stimulate the proliferation of T cells [42]. Exosomes derived from M2 macrophages can transfer key enzymes of the PG pathway to tumor cells, such as cyclo-oxygenase 1 (COX1) and TXA synthase 1 [43]. Therefore, exosomes have great potential in tumor therapy including immunotherapy.
Components of membrane structure
Lipids are important components of biological membranes and are involved in the transport of energy, materials, and information across membranes [44]. In addition, special domains in the membrane, such as lipid rafts, play an important role in the communication between tumor and immune cells [45].
The type and saturation of FAs in the membrane affect its structural stability and function [46]. On the one hand, saturated fatty acids (SFAs) are important for maintaining membrane fluidity and selective transmembrane transport. On the other hand, unsaturated fatty acids (UFAs), especially polyunsaturated fatty acids (PUFAs), are susceptible to reactive oxygen species (ROS), which results in lipid peroxidation and increases the sensitivity of cells to ferroptosis. In contrast, the peroxidation of monounsaturated fatty acids (MUFAs) plays a protective role [47]. Therefore, the tumor cell membrane is characterized by an increased ratio of MUFA/SFA and MUFA/UFA [48]. However, all PUFAs are not harmful to tumors. A study has reported that ω-3PUFA rather than ω-6PUFA has an antitumor effect, and the plasma levels of ω-3PUFA markedly decrease, whereas those of ω-6PUFA increase in patients with advanced tumors [49].
The key enzyme that regulates the type and saturation of FA in the membrane is stearoyl-CoA desaturase (SCD). Its Inhibition reduces the MUFA/SFA ratio and contributes to the induction of ferroptosis in tumor cells [47]. However, fatty acid desaturase (FADS) can replace SCD in partially SCD1-dependent or SCD1-independent tumor cells such as liver and lung cancer cells. Inhibition of FADS1/2 reduced the ratio of MUFA/SFA and ω-3/ω-6PUFA. Notably, FADS2 also produced the uncommon MUFA sapienate, whose level was abnormally increased upon SCD1 inhibition [50]. In addition, the PUFA/SFA ratio in the membrane is regulated by the remodeling of glycerol phosphatides (GPLs) remodeling through the Lands cycle [51]. Tumor cells undergo significant GPL remodeling and phospholipase A2 and lysophosphatidylcholine acyltransferase (LPCAT) are the key enzymes of this process, which can be used as tumor markers and potential targets for therapy [52].
Lipid rafts are microdomains in cell membranes that are rich in cholesterol and sphingomyelin with long SFA chains; these chains contribute to the anchoring of LPs and the formation of relatively stable liquid regions. High levels of lipid rafts are present in the tumor cell membranes, where many tumorigenic proteins, such as CD44, epidermal growth factor receptor (EGFR), and insulin-like growth factor receptor are localized [53]. Polyphenols can exert their antitumor effect by destroying lipid rafts [54]. Interestingly, lipid rafts also localize immune-recognition receptors, such as T-cell receptor (TCR), major histocompatibility complex II (MHC II) [55], and transmembrane tumor necrosis factor-α (TNF-α), and provide a specialized environment for the activation of signaling complexes in response to antigen recognition [56].
Involved in the modification of biomolecules
The great variability among lipids, in terms of their structure and properties, contributes to the remarkable diversity of lipid modifications. Among them, the most important is the acylation modification of proteins, in which FA is covalently linked to the peptide chain in the form of an acyl group [57]. In addition to classical lysine acetylation, many novel acylation modifications of histone and non-histone proteins, such as malonylation (Kma, mediated by malonyl-CoA), β-hydroxybutyrylation (Kbhb, mediated by β-hydroxybutyric acid; BHB), isoprene (mediated by intermediates of cholesterol synthesis), and long-chain fatty acid (LCFA) acylation, have been identified [58]. Histone acylation affects the epigenome through transcriptional regulation of gene expression, thereby linking epigenetics to lipid metabolism. Non-histone acylation mainly regulates protein stability, catalytic activity, membrane localization, and protein-protein interactions, thereby affecting protein function and linking lipid metabolism to the regulation of key proteins [57].
Metabolic reprogramming of tumor cells can lead to abnormal lipid modification. In turn, the changes in lipid modification affect tumor metabolism and the occurrence and development of tumors [59]. For example, crotonylation (Kcr) -mediated DNA damage repair and spindle localization help maintain genome integrity and prevent tumor progression. In addition, p53 can be modified by lipid modifications such as Kbhb, succinylation, propionylation, butyrylation, and Kcr, leading to decreased expression of p53 and its downstream genes and affecting p53-mediated tumor suppression. Notably, palmitoylation of programmed cell death ligand 1 (PD-L1) helps to maintain its stability, thereby inhibiting the cytotoxicity of CD8+T cells. In addition, palmitoylation also regulates tumor innate immunity and inflammatory responses [60].
Landscape of abnormal lipid metabolism in tumors
Lipid metabolism, especially anabolism, is highly active in tumor cells to promote their survival and development in the TME, which is characterized by the high expression of receptors and key enzymes involved in lipid uptake, synthesis, storage, and decomposition. However, the expression of key enzymes is different because of the different types, stages, and microenvironments of tumors (Table 1, Figure 1) [61].
Key enzymes of lipid metabolism and their expression in tumors.
| Category | Stage | Key enzyme | Function | Effect | Expression |
|---|---|---|---|---|---|
| FA | Uptake | CD36 | Absorb LCFA, TC and oxLDL | FAO, angiogenesis, EMT, lipid peroxidation, and chemoradiotherapy resistance | Highly expressed, positively correlated Tumor and immunity marker |
| FATP | Absorb and transport LCFA | ||||
| FABPpm/FABP | Absorb and transport FA | ||||
| Synthesis | ACLY | Catalyze AcCoA to citric acid | Glucose, glutamine, lipid metabolism, Wnt/β-catenin signaling, histone acetylation, and anti-tumor immunity | ||
| ACSS2 | Catalyze acetate to AcCoA | Compensate for ACLY, lysosomal synthesis and autophagy | Highly expressed except CRC and gastric cancer, positively correlated Tumor and immunity marker | ||
| ACC1/2 | Catalyze AcCoA to malonyl-CoA | Balance between FA synthesis and decomposition | Highly expressed except lung adenocarcinoma Tumor marker | ||
| FASN | Catalyze malonyl-CoA to palmitic acid | Glycolysis, amino acid metabolism, and ferroptosis | Highly expressed, positively correlated Tumor marker | ||
| Desaturation | SCD | Catalyze palmitic acid to palmitic acid, stearic acid to oleic acid | Ratio of MUFA/SFA in membrane, tumor proliferation, apoptosis, inflammation, invasion, and metastasis | High expressed, positively correlated with ccRCC but negatively with breast cancer and liver cancer Tumor marke | |
| FADS2 | Catalyze LA and ALA to PUFA, palmitic acid to sapienate | Compensate for SCD, ratio of SFA/UFA and ω-3/ω-6PUFA in membrane | Highly expressed in SCD independent or suppressed tumors | ||
| Elongation | ELOVL2/5/6 | Catalyze palmitic acid to stearic acid, palmitic acid to oleic acid, sapienate to coctadecenoate, LA and ALA to PUFA | Highly expressed in low malignancy but lowly expressed in high malignancy Tumor marke | ||
| Storage | ACSL | Catalyze FA and AcCoA to FA-CoA | TAG synthesis, FAO, autophagy, Lands cycle, and ferroptosis | ACSL3 highly expressed in AR+ tumors but lowly expressed in ER-ones ACSL4 highly expressed in ER- tumors but lowly expressed in ER+ and AR+ ones | |
| ACBP | Transport acyl-CoA | High expressed | |||
| GPAT | Catalyze glycerol 3-phosphate and acyl-CoA to LPA | Highly expressed in active TAG synthesis tumors, negatively correlated except LIPIN | |||
| AGPAT | Catalyze LPA and acyl-CoA to PA | ||||
| PAP (LIPIN) | Catalyze PA to DAG | Regulation of lipid metabolism | |||
| DAG | Catalyze DAG and acyl-CoA to TAG | ||||
| Lipolysis | ATGL | Catalyze TAG to DAG and FA | Cancer-associated cachexia | Highly expressed except lung cancer and head and neck squamous cell carcinoma | |
| HSL | Catalyze DAG to MG and FA | Highly expressed in high malignancy | |||
| MAGL | Catalyze MG to Gly and FA | Cancer stem cells marker | |||
| Decomposition | ACSL/ACBP | Ditto | Highly expressed Tumor and immunity marker | ||
| CAT1 | Transport LCFA-CoA into mitochondria | ||||
| β-oxidase system | Catalyze FA-CoA to AcCoA, NADH and FADH2 | ||||
| TC | Uptake | LDLR | Absorb TC | EGFR, PI3K/Akt and SREBP1 signaling | Highly expressed, positively correlated Immunity marker |
| Synthesis | HMGCR | Catalyze HMG-CoA to MVA | Lowly expressed, negatively correlated in lipid-rich tumors but highly expressed in other solid and hematological tumors Immunity marker | ||
| SQS | Catalyze FPP to squalene | Highly expressed especially tumors with low HMGCR, negatively correlated Immunity marker | |||
| SQLE | Catalyze squalene to 2,3-epoxy squalene | ||||
| Storage | ACAT1 | Catalyze TG to CE | TC uptake, synthesis, storage, flow to membrane, and tumor immunity | Highly expressed, positively correlated Tumor and immunity marker | |
| Efflux | ABCA1/G1 | Transport TC to ApoA1 and HDL-C | Tumor suppressor gene, tumor survival and EMT | Lowly expressed in p53-mutated tumors, highly expressed in lipid-rich tumors often with low HMGCR Tumor marker (including serum HDL-C and HDLR) | |
| Decomposition | CYP27A1 | Catalyze TC to 27-HC | Highly expressed in endothelial cells and macrophages | ||
| Ketone body | Synthesis | HMGCL | Catalyze HMG-CoA to AcAc | Highly expressed in melanoma and GBM | |
| β-hydroxybutyrate dehydrogenase | Catalyze AcAc to BHB | ||||
| Decomposition | BDH1 | Catalyze BHB to AcAc | Highly expressed in "ketone metabolic subtype" tumors | ||
| OXCT1 | Catalyze AcAc to AcAcCoA and succinic acid | ||||
| ACAT1 | Catalyze AcAcCoA to AcCoA | ||||
| PGs | Synthesis | COX-1/2 | Catalyze AA to PGG2 and PGH2 | Chronic inflammation into tumors, apoptosis, proliferation, invasion, metastasis, angiogenesis, immunosuppression and drug resistance | Highly expressed in solid and hematological tumors, especially Inflammation-associated ones except SCL |
| LOX | Produce LTs | ||||
| TXA2 synthetase | Catalyze PGG2 and PGH2 to TXA2 | ||||
| PGES | Catalyze PGG2 and PGH2 to PGE2 | ||||
| Decomposition | 15-PGDH | Decompose PGE2 Antagonize COX-2 | Lowly expressed | ||
| GPLs remodeling | Lands cycle | PLA2 | Participate in the deacylation of GPL | Vary with tumor types and stages, and subtypes of enzymes Tumor marker | |
| LPCAT | Participate in the reacylation of GPL | ||||
| Ferroptosis | Peroxidation | ACSL4 and LPCAT3 | Ditto | ||
| LOX | Participate in the generation of PLOOH | ||||
| Antioxidation | SXC | Transport cysteine intracellularly and pump glutamate out | Highly expressed | ||
| GPX4 | Scavenge PLOOH by catalyzing GSH to GSSG | ||||
| FSP1 | Catalyze CoQ10 to ubiquinol | ||||
| Transport | TFRC | Combine with TF and absorb Fe3+ | Highly expressed | ||
| FPN1 | Pump excessive Fe2+ extracellularly | ||||
Landscape of lipid metabolism. In the membrane, AA released from PL participates in the PG synthesis pathway, which affects inflammation and redox balance. The transmembrane transport of exogenous lipids, especially essential FAs, can be achieved through various receptors to form MUFAs. In the mitochondria, AcCoA can be generated from glucose, glutamine, acetate, or ketone bodies and subsequently enter the cytoplasm to continue the FA synthesis. In the endoplasmic reticulum, AcCoA is involved in the synthesis of TC, and its intermediates are associated with lipid modification and redox balance. In the cytoplasm, the products of the de novo synthesis of FA are SFAs, which are further elongated and desaturated to form MUFAs. The excess lipids are stored in LDs as TAG and CE, whereas FAs are used as substrates for lipid remodeling via the Lands cycle and then released when needed.
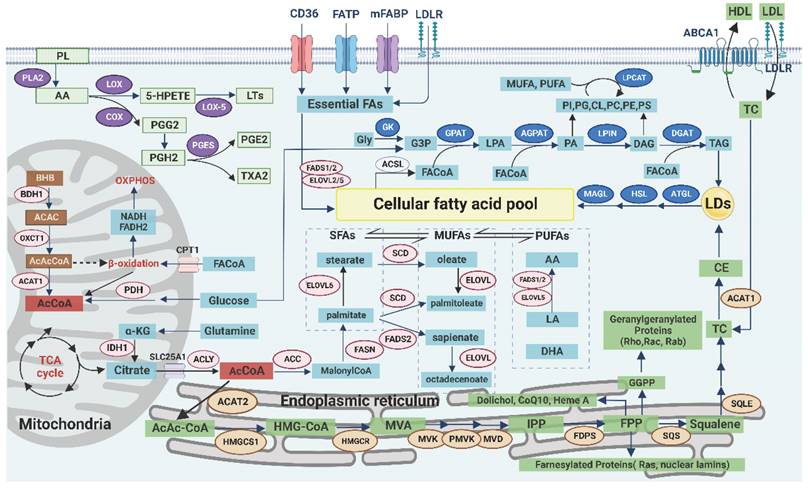
Exogenous uptake of lipids
The supply of exogenous lipids is easily affected by dietary intake. The occurrence and development of tumors are closely related to the amount and type of lipids consumed. High-fat diets (HFD, mainly containing SFA and cholesterol) can increase the risk of cancer [62], and a clinical study has found that lipid-lowering drugs can improve the prognosis of patients with cancer [63]. However, not all SFA are harmful. For example, palmitic acid can induce apoptosis of tumor cells under certain conditions [64]. In contrast, people who increase the intake of PUFA (specifically ω-3PUFA) have a reduced risk for cancer [65]. ω-3PUFA promotes lipid peroxidation and induces ferroptosis in tumor cells in the acidic TME [66]. However, not all PUFAs have antitumor effects. For example, high levels of ω-6PUFA intake combined with low levels of ω-3PUFA intake increase the risk of cancer [67]. This partially happens because the short-chain adducts derived from ω-3PUFA oxidation are less likely to cause mispairing of DNA, whereas the long-chain ones from ω-6PUFA preferentially bind to p53, thereby affecting p53-mediated tumor suppression and inducing carcinogenesis. ω-3PUFA competes with ω-6PUFA to bind COX and lipoxygenase (LOX) and reduces the generation of PGs; ω-3PUFA can also regulate autophagy and inhibit the NF-kB pathway [68]. Therefore, changes in dietary lipid intake can be a selective antitumor therapy and provide a new strategy for sensitizing immunotherapy.
The transmembrane transport of exogenous lipids can be achieved through a variety of pathways. Low-density lipoprotein (LDL) enters the cell through endocytosis mediated by LDL receptor (LDLR), and it is subsequently digested by lysosomal acid lipase (LAL) to release free fatty acids (FFA) and cholesterol. LDLR is highly expressed in tumors and enhances the EGFR, PI3K/Akt, and sterol regulatory element-binding protein (SREBP) signaling pathways. Tumor metastasis is also associated with persistently high expression of LDLR [69]. LCFA can be transported by fatty acid translocase (CD36, belonging to scavenger receptor) or fatty acid transporter (FATP, belonging to solute carrier family SLC27A) [70]. In addition, CD36 mediates the uptake of oxidized low-density lipoprotein (ox-LDL) and cholesterol. The high expression or structural abnormalities of CD36 were closely related to tumor growth, invasion, and metastasis. CD36 promotes FAO, angiogenesis, and tumor chemoradiotherapy resistance by regulating the Src/PI3K/Akt signaling pathway. CD36 can also activate Wnt/TGF-β signaling pathway through epithelial-to-mesenchymal transition (EMT) to promote metastasis [71]. Plasma membrane fatty acid binding proteins (FABPpm) contribute to FA uptake, whereas intracellular FABPs mediate the transport of FA within the cell. The potential protumor role of FABP4 has been demonstrated, and inhibition of FABP affects tumor growth and invasion. Therefore, FABP has been used as a marker for colorectal cancer (CRC) and renal cell carcinoma (RCC) [61, 72]. The ability of tumor cells to absorb lipids is affected by oncogene types, and not all lipids are taken up equally. For example, tumor cells selectively take up specific lysophospholipids (lysoPLs, PL containing MUFA, and PUFA acyl chains) from the TME [73]. Similarly, tumor-associated immune cells can selectively take up lipids according to the state of differentiation and activation but can also passively ingest some harmful lipids in the TME, such as ox-LDL [74].
Synthesis of lipids
De novo synthesis and desaturation of fatty acids
De novo synthesis of FA begins with acetyl coenzyme A (AcCoA), which is mainly produced by glucose, glutamine [75], and acetate [76] in the cytoplasm. The citric acid in mitochondria is transported into cells through SLC25A1 (carrier protein), and then AcCoA is generated by ATP-citrate lyase (ACLY) [20]. SLC25A1 plays a key role in the maintenance of the mitochondrial citric acid pool and redox balance, and its inhibition leads to the accumulation of ROS [77]. ACLY promotes tumor invasion and metastasis by regulating the Wnt/β-catenin signaling pathway [78]. Additionally, ACLY promotes the early activation of CD8+T cells and their lipid metabolic reprogramming [79]. In addition to being involved in the synthesis of FA and cholesterol, AcCoA participates in acylation modifications of proteins, and its expression level is dynamically associated with histone acetylation [80]. The expression of AcCoA is also related to programmed cell death and autophagy. Therefore, targeting AcCoA metabolism has potential in tumor treatment and may also have implications in immunotherapy [81].
Acetyl-CoA carboxylase (ACC/ACAC) catalyzes the conversion of AcCoA to malonyl-CoA, which can inhibit carnitine palmitoyltransferase 1 (CPT1)-mediated FAO in mitochondria, thereby regulating the balance between synthesis and decomposition of FA. In addition, activated AMP-activated protein kinase (AMPK) inhibits the activity of ACC1/2 [82]. Then, fatty acid synthase (FASN) catalyzes the conversion of malonyl-CoA to palmitic acid. In addition to being involved in synthesis of FA, FASN can impact other metabolic pathways such as glycolysis, amino acid metabolism, and ferroptosis [83]. Therefore, FASN and other key enzymes can be used as potential diagnostic markers for molecular subsets of tumors with high levels of lipid metabolism [84].
Palmitic acid is subsequently elongated by the activity of elongation of the very long chain fatty acid (ELOVL) family of enzymes and converted to MUFA by SCD1. SCD1 can relieve the inhibitory effect of SFAs (including palmitic acid) on the activity of upstream ACC by reducing the SFA levels and subsequently increasing the production of malonyl-CoA, which affects FAO [26]. In addition to affecting the membrane composition, SCD1 can damage the mitochondrial electron transport chain (ETC) and reduce the binding of MUFA to cardiolipin, which increases the production of intracellular ROS and induces mitochondria-dependent apoptosis. Inhibition of SCD1 also leads to endoplasmic reticulum stress, which, in turn, induces antitumor immunity [85].
Furthermore, SCD1 affects the sensitivity of the cells to ferroptosis. On the one hand, inhibition of SCD decreases the MUFA/UFA in the membrane, and MUFA has an antiperoxidative effect, which, in turn, promotes ferroptosis [47]. On the other hand, inhibition of SCD1/FADS2 directly downregulates key enzymes and proteins, such as glutathione peroxidase 4 (GPX4) and reduced glutathione (GSH), respectively, in the ferroptosis-related antioxidant defense system, and disrupts cellular/mitochondrial redox balance, which leads to iron-mediated lipid peroxidation and results in mitochondrial dysfunction [86].
De novo synthesis of cholesterol
A high level of cholesterol is essential for the proliferation and metastasis of tumor cells to fulfill the requirement for membrane structure. cholesterol also stabilizes PD-L1 on the tumor cell surface, thus inhibiting tumor immunity [87]. In addition, high levels of cholesterol in the TME also affect the phenotype and activity of tumor-associated immune cells [88].
Cholesterol is primarily synthesized through the mevalonate (MVA) pathway. The key enzyme is HMG-CoA reductase (HMGCR), which is highly expressed in many cancers, and targeted inhibition of this enzyme has emerged as a potential therapeutic approach [89]. In addition to the end product cholesterol, the intermediates of the MVA pathway are equally important for the survival and development of tumor cells and antitumor immune response including ferroptosis [90]. For example, phosphate polyterpene alcohols are essential for the synthesis of lipid-linked oligosaccharides and are used for the N-glycosylation of proteins [91]. Farnicyl pyrophosphate and geranyl pyrophosphate, as isoprene derivatives, participate in the isoprenylation modification of protein fa such as Ras and Rho. They can also upregulate the expression of PD-1 on the surface of immune cells such as regulatory T cells (Treg cells) [92]. Coenzyme Q10 (CoQ10/ubiquinone) is a coenzyme of cytochrome C oxidase in the mitochondrial respiratory chain complex IV. Under the metabolic stress of the TME, impairment of the respiratory chain by inhibiting the expression of CoQ10 can cause tumor cell death. In addition, CoQ10 inhibits iron action suppressor protein 1 (FSP1), consequently suppressing ferroptosis. This process is positively correlated with the activity of SCD1 [93]. Squalene acts as a free radical trapping antioxidant to protect membrane lipids from peroxidation [90]. Isopentenylpyrophosphate is required for the maturation of selenocysteinyl-tRNA and GPX4 synthesis, which, in turn, inhibits ferroptosis [94]. Taken together, these findings suggest that de novo cholesterol synthesis may play a protective role against ferroptosis in tumor cells.
Storage of lipids
Excess lipids are stored in LDs in the form of TAG and cholesterol ester (CE). The number of LDs and the amount of stored TAG and CE inside them abnormally increases in tumor cells and tumor-associated immune cells (especially macrophages) [95]. The low pH of the TME enhances the formation of LDs in tumor cells [96]. Therefore, LD accumulation is considered a prominent feature of tumors. Phosphoenolpyruvate carboxykinase (PCK1), the key enzyme in the gluconeogenesis pathway, promotes the continuous formation of LDs through the activation of the SREBP signaling pathway in human hepatocellular carcinoma (HCC) cells [12, 97].
The core of an LD consists of neutral fats (mainly including TAG and CE) and is surrounded by a single layer of PLs and several proteins. Various enzymes involved in the synthesis of TAG and lipolysis are localized on the surface of LDs [98]. In addition, LDs have a selective form of autophagy (lipophagy). Therefore, LDs not only simply store lipids but also actively participate in the lipid metabolism pathway. Furthermore, the formation or alteration of LDs is tightly regulated by the cell to meet the requirements of lipid metabolism.
LD is a dynamically changing and multifunctional organelle [96]. As a reservoir of lipids, LD not only protects its contents from enzymatic decomposition but also protects the cell from the toxic effects of excess FFA, cholesterol, and ROS [99]. Especially for immune cells in the acidic and lipid-rich TME, LD mediates the flow of cholesterol to membrane, thereby regulating the intracellular distribution of cholesterol, and helps cholesterol participate in the formation of immune synapse [100]. In addition, LDs move along the cytoskeleton and interact with other organelles, thereby playing an important role in transmembrane transport, protein degradation, and signal transduction [99]. LDs also act as a hub of host innate immunity and affect the tumor immune response [101].
Decomposition of lipids
Decomposition of fatty acids
The main function of FAO, known as β-oxidation, is to provide energy. In addition, it serves as a source of NADPH especially when glucose is deficient or the pentose phosphate pathway (PPP, mainly producing NADPH) is compromised [102]. FAO is highly active in many tumor cells, such as lung cancer cells with K-Ras mutation [103] and tumor-associated immune cells, indicating their adaptation to the metabolic stress of TME. CPT1 is critical for the transport of LCFAs into the mitochondria for β-oxidation. However, FAs need to be activated by the attachment of CoA through the activity of long-chain acyl-CoA synthetase 3 (ACSL3), leading to the formation of FA-CoA [104]. In addition, ACSL participates in TAG synthesis, autophagy, the Lands cycle, and ferroptosis and promotes the proliferation and progression of tumors. Therefore, ACSL can be a potential marker and therapeutic target for tumors [105].
Decomposition of cholesterol
The basic structure of cholesterol is cyclopentane polyhydrophenanthrene, which cannot be degraded completely. However, its oxidative metabolites, such as 27-hydroxycholesterol (27-HC; an oxysterol), affect tumor development. In the TME, 27-HC is mainly derived from macrophages and endothelial cells. 27-HC inhibits the uptake and synthesis of FA and cholesterol by inhibiting the SREBP pathway and activating the liver X receptor (LXR) pathway [106]. In contrast, 27-HC can competitively inhibit the effect of estrogen because of its selective estrogen receptor (ER) regulatory function and promotes the proliferation of high-ER-expressing tumor cells, such as breast and ovarian cancer cells [107]. It can also induce an increase in GPX4 expression, thereby protecting tumor cells from ferroptosis. This phenomenon partially explains why metastatic breast tumor cells are not damaged by ferroptosis induced by lipid accumulation while taking up lipids in large quantities [108].
Decomposition of ketone bodies
The catabolism of ketone bodies in tumor cells of the "ketone metabolic subtype" serves as an important energy source [32]. The ketone bodies, especially BHB, can serve as a negative feedback signal to promote lipid metabolism and regulate the tricarboxylic acid (TCA) cycle. For example, BHB decreases the LDL level and increases the high-density lipoprotein level. As a key endogenous GPCR signaling modulator, BHB can also inhibit the process of fat mobilization by activating GPCR109A and reducing the supply of extracellular lipids [109]. In addition, BHB can regulate the respiratory chain and upregulate genes involved in protecting cells from oxidative stress as an endogenous inhibitor of histone deacetylase. Notably, BHB also promotes tumor immune response by downregulating the PD-L1 levels and promoting the proliferation of T cells [109].
Abnormal regulation of lipid metabolism
The expression of various key enzymes in lipid metabolism can be regulated by several signaling pathways, including AMPK and mammalian target of rapamycin (mTOR), peroxisome proliferator-activated receptors (PPAR), SREBP, and LXR signaling pathways. These pathways are further regulated by upstream signaling pathways such as p53, BRAF, PTEN, Ras, and Myc [110]. However, the lipid metabolic pathway is not a passive recipient of signals. Some metabolic intermediates can feedback information about the metabolic state of cells to the genes and proteins responsible for regulation through histone acylation modification [111]. The close relationship between the lipid metabolism and signal regulation networks plays an important role in the survival and growth of tumor cells and tumor-associated immune cells (Figure 2).
Regulation of lipid metabolism. The expression of various key enzymes of lipid metabolism can be regulated by signaling pathways, including AMPK, mTOR, PPAR, SREBP, and LXR signaling pathways. In the endoplasmic reticulum, SREBPs, as inactive precursors, reside as transmembrane proteins where they are associated with a chaperone, the SREBP cleavage activating protein (SCAP). When intracellular cholesterol levels are reduced, SREBPs are transported to the Golgi, where a two-step proteolytic process releases the N-terminal half of the protein, and then mature SREBPs translocate to the nucleus. In the nucleus, transcriptional regulators that control the synthesis and transport of FA and TC are SREBPs, LXRs, and PPARs, which bind as homodimers or heterodimers to SREs, LXRE, and PPREs, respectively, in the promoter regions of their target genes. In the TME, energy stress, increased Ca2+ level, and oxidative stress increase AMP/ATP or ADP/ATP ratio to activate AMPK but inhibit mTOR, which promotes autophagy.
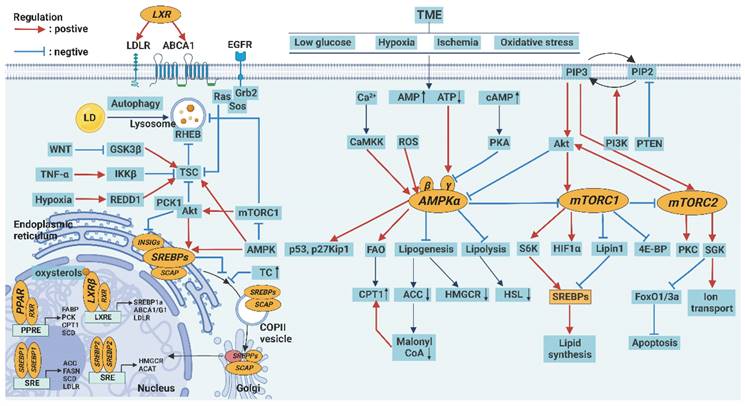
AMPK and mTOR signaling pathways
AMPK and mTOR are mutually antagonistic key molecules in the regulation of energy metabolism. The factors, such as energy stress, increased Ca2+ level, and oxidative stress increase AMP/ATP or ADP/ATP ratio and activate AMPK in cells, which promote catabolism and inhibit anabolism and confer resistance to metabolic stress [112].
Specifically, AMPK enhances FA uptake through the upregulation of CD36 and other lipid transporters and inhibits SREBP signaling. In addition, AMPK activates autophagy directly or indirectly by inhibiting mTORC1 [113]. AMPK activation also inhibits cell proliferation by promoting the p53 pathway and inhibiting the cyclin-dependent kinase (CDK) pathway [112]. Notably, loss of AMPK in Treg cells promotes tumor immunosuppression by increasing the expression of PD-1 [114]. In contrast, the activation of AMPK in NK cells mediates antitumor immunity and enhances the effect of anti-PD-L1 therapy [115]. Therefore, AMPK is considered an important tumor suppressor.
However, AMPK can also promote tumor growth and metastasis in some cases. For example, AMPK can prevent lipid peroxidation and inhibit antitumor immune responses by activating the production of antioxidants and inhibiting inflammatory responses through the NF-κB pathway [116].
When nutrients are sufficient, mTOR is activated instead of AMPK to promote anabolism and inhibit catabolism. mTORC1 and mTORC2 are two different complexes in the cell. mTORC1 promotes anabolism by activating ribosomal protein S6K (S6K) and inhibiting EIF4E-binding protein (4EBP) and is sensitive to rapamycin, whereas the mTORC2 promotes cell survival and proliferation by activating protein kinases such as AKT, SGK, and PKC and is not sensitive to rapamycin [117]. In addition to regulating metabolism, S6K and AKT can activate hypoxia inducible factor 1 α (HIF-1α) and SREBP1, thus promoting angiogenesis and FA synthesis [118]. mTOR also inhibits apoptosis, autophagy, and ferroptosis [119].
However, over-activation of mTOR can accelerate cell senescence and even induce cell death. Contrary to AMPK, mTORC1 induces mitochondrial oxidative stress by inhibiting the transcription factor PGC-1α, which promotes lipid peroxidation and calcium overload [120]. The continuous activation of mTOR can also disrupt the balance of pro- and anti-inflammatory immune responses, leading to the generation of chronic inflammation [121].
PPAR signaling pathway
PPARs are ligand-induced nuclear receptors and are involved in several peroxide proliferation reactions, including metabolic and apoptotic pathways related to lipid metabolism. PPARs are generally highly expressed on tumor cells, and their expression is dependent on several factors such as PPAR subtype and type and stage of the tumor [122]. When activated by ligands such as AA derivatives, PUFA, and non-steroidal anti-inflammatory drugs (NSAIDs), PPARs receptors enter the nucleus to regulate the transcription of genes encoding enzymes of lipid metabolism, such as FABP, CPT1, and SCD1, and peroxisome genes related to lipid peroxidation [123]. A zinc-finger protein interacts with PPARs to regulate their activity. Zinc deficiency upregulates the expression of PPARc, FABP, and SREBP1, which results in lipid accumulation [124]. In addition, PPARγ can induce apoptosis of tumor cells through the exogenous Fas/FasL death receptor pathway or endogenous mitochondrial apoptosis pathway [125]. Notably, PPARγ regulates the innate immune system by promoting the apoptosis of N1 neutrophils and activating M2 macrophages. PPARα also plays an important role in inhibiting inflammatory pathways such as NF-κB and JAK/STAT pathways [126]. Therefore, the PPAR signaling pathway can be a potential target for tumor immunotherapy.
SREBP and LXR signaling pathways
SREBPs are members of the nuclear transcription factor family. SREBP1 regulates FA metabolism [127], whereas SREBP2 regulates cholesterol metabolism [128]. When the level of intracellular lipid decreases, the precursor proteins of SREBPs residing on the endoplasmic reticulum membrane are transported to the Golgi apparatus, where they are cleaved, released into the nucleus, and promote the expression of genes encoding key enzymes of lipid metabolism. SREBP1 promotes ACC, FANS, and SCD, whereas SREBP2 promotes HMGCR and ACAT [90].
The SREBP signaling pathway is abnormally activated in tumor and immune cells. Abnormally activated intracellular PI3K/Akt/mTOR signaling pathway promotes the expression of SREBP1 [129]. TP53 gene mutations can also promote the activation of the SREBP signaling pathway [130]. In addition, SREBP signaling interacts with glucose metabolism pathways, thereby linking glucose and lipid metabolism. Pyruvate kinase M2 (PKM2), a key enzyme in glycolysis, modifies SREBP1a to enhance its stability [131]. In turn, SREBP1a also promoted the expression of isocitrate dehydrogenase 1 (IDH1), a key enzyme of the TCA cycle, by binding to its promoter region [132]. Notably, SREBP2 is involved in the inflammatory response associated with Nod-like receptor protein 3 (NLRP3), which affects the tumor immune response [133].
Similarly, LXRs synergize with SREBPs as key regulators of cholesterol homeostasis in cells. LXRs are activated by intracellular cholesterol or 27-HC and subsequently promote transcription of target genes encoding ATP-binding cassette transporter A1/G1 (ABCA1/G1), SREBP1a, and LDLR [134]. When the intracellular cholesterol level is too high, LXRs inhibit the SREBP2 signaling pathway. They promote the efflux of cholesterol by increasing the expression of ABCA1/G1 and apolipoprotein A1/E (ApoA1/E) and inhibit the uptake of cholesterol by enhancing the degradation of LDLR [135]. Activation of LXRs can also induce tumor cell apoptosis by blocking key growth pathways such as the EGFR pathway and downregulating survival signals such as Akt [136]. Notably, the activation of LXR receptors induced the differentiation of macrophages into an M2-like phenotype [137].
Lipid metabolic reprogramming in tumor-associated immune cells
Immune cells can recognize and destroy tumor cells during immune surveillance. However, in the process of tumor immune editing, the products of tumor lipid metabolism not only modify the TME but also affect the recruitment and function of tumor-associated immune cells. Therefore, immune cells can act as guardians (antitumor immunity) as well as bystanders or even supporters (protumor immunity) [138].
Immune cells can be divided into two types based on their effect on tumors. Antitumor immune cells include effector CD8+T (Teff) cells, antitumor CD4+ (CD4+conv) cells, memory CD8+T (Tmem) cells, B cells, natural killer T (NKT) cells, NK cells, M1 macrophages, dendritic cells (DCs), and N1 neutrophils. Protumor immune cells include Treg cells, regulatory B (Breg) cells, M2 macrophages, myeloid-derived suppressor cells (MDSCs), and N2 neutrophils. Lipid metabolic reprogramming plays a crucial role in regulating the phenotype and function of immune cells. Although certain types of antitumor/protumor cells rely on both types of metabolism, in general, antitumor immune cells tend to rely on glucose metabolism to exert anti-tumor function, whereas protumor immune cells tend to rely on lipid metabolism to exert tumor immunosuppressive function (Table 2) [25, 139-141].
Metabolic reprogramming in tumor-associated immune cells.
| Cell type | Functions | Reprogramming | ||
|---|---|---|---|---|
| Anti-tumor immune cells | Adaptive immunity | Teff cells | Antigen-specific cytotoxic effects, perforin, granzyme, γ-IFN, TNF-α, and FasL | Enhance glycolysis, TCA cycle, OXPHOS, and PPP, use lactic acids Enhance FA synthesis, FAO, and TC uptake, use ketone bodies |
| CD4+ conv cells | γ-IFN and TNF-α Activate Teff cells, NK cells, and B cells | |||
| CD8+T mem cells | Long-term tumor immunity | Enhance OXPHOS Enhance FAO, CD36-mediated FA uptake, acetate-mediated FA synthesis | ||
| B cells | Tumor-specific antibodies, ADCC effect, CDC effect, granzyme B, and TRAIL Activate T cells | Increase oxygen consumption Enhance glycolysis and PPP, upregulate GLUT | ||
| Innate immunity | NKT cells | γ-IFN, perforin, FasL and TRAIL Activate NK cells, T cells, and DCs | Depend on FA and TC | |
| NK cells | ADCC effect, perforin, granzyme, γ-IFN, TNF-α, FasL, and TRAIL | Enhance glycolysis and OXPHOS, inhibit gluconeogenesis Upregulate LDLR | ||
| M1 macrophages | ADCC effect, TNF-α, IL-1β, COX-2, iNOS, ROS, NO, and antigen presentation | Enhance glycolysis and PPP Tend to PUFAs | ||
| DCs | Antigen presentation, recognize DAMP, and PAMP Activate NK cells, NKT cells, and Teff cells | Enhance glycolysis and inhibit OXPHOS Tend to PL and TAG (especially PUFA) but TC and CE | ||
| N1 neutrophils | Phagocytosis, ROS, RNS, and α/β-IFN Activate NK cells and inhibit IL-17 T cells | Enhance glycolysis, PPP, and glycogen metabolism | ||
| Pro-tumor immune cells | Adaptive immunity | Treg cells | IL-4, IL-10, TGF-β, adenosine, CTLA-4, PD-1, and LAG-3 Inhibit APCs and T cells | Enhance OXPHOS, FAO, FA and TC synthesis, upregulate CD36 and FANS Downregulate GLUT1 and upregulate MCT1, use lactic acids |
| Breg cells | IL-10, TGF-β, ROS, PD-L1 and CTLA-4 Inhibit T cells and NK cell, and activate Treg cells, MDSCs and M2 macrophages | Abnormal TC metabolism Use lactic acid, enhance glycolysis | ||
| Innate immunity | M2 macrophages | IL-10, TGF-β, VEGF, EGFR, and MMPs | Enhance FA synthesis, FAO, and OXPHOS, upregulate CD36 and ABCG1 | |
| MDSCs | IL-10, TGF-β, Arg1, NO, and ROS Inhibit T cells and activate Treg cells | Enhance FAO, OXPHOS, upregulate CD36, LDLR and FATP2 Enhanced glycolysis | ||
| N2 neutrophils | ROS, RNS, Arg1, MPO, NE, and NETs Inhibit T cells and NK cells | Enhance lipid uptake, LD formation, and inhibit FAO and LD degradation Upregulate GLUT1 | ||
Abnormal metabolism of antitumor immune cells
Adaptive immunity: Teff, CD4+conv, Tmem, and B cells
Teff cells directly kill tumor cells by inducing apoptosis and secreting cytokines, such as interferon-γ (IFN-γ) and TNF-α, and CD4+conv cells mainly exert an indirect antitumor effect by assisting Teff cells. Teff cells require an adequate supply of glucose for their antitumor functions. Although enhanced glycolysis is critical for T-cell activation, the upregulation of the TCA cycle and OXPHOS is equally important, and specifically, FAO also maintains the antitumor function of Teff cells [142]. In addition, lipid metabolism is enhanced in activated T cells leading to an increase in FA synthesis and cholesterol uptake, which contributes to the maintenance of membrane structure and immune receptor signaling.
Similarly, the metabolic bypass of glycolysis, that is PPP, is also upregulated with the activation of T cells to provide NADPH for lipid synthesis. However, the dependence of CD4+T cells on FAO promotes their differentiation into Treg cells [138]. The activation of T cells depends on the mitochondrial activity (similar to tumor cells), mitochondria in tumor-associated T cells are defective, which predisposes to intracellular oxidative stress and impairs their antitumor effect [143].
Notably, CD8+T cells can generate a memory cell population (Tmem cells), which can persist after the tumor response fades; therefore, these cells are the key to long-term tumor control. Unlike Teff cells, Tmem cells, as quiescent cells, preferentially rely on FAO instead of glycolysis. These cells have significant mitochondrial reserves of FAs and further upregulate OXPHOS after activation [139]. Under the metabolic stress of the TME, Tmem cells tend to increase their uptake of FAs and enhance acetate metabolism to promote lipid synthesis, thereby reversing the apoptosis induced by lipid deprivation and retaining their antitumor function [144].
B cells differentiate into plasma cells and produce tumor-specific antibodies to recognize and mediate tumor lysis through antibody-dependent cytotoxicity (ADCC) or complement-dependent cytotoxicity. B cells also promote the activation of T cells. In addition, B cells can directly kill tumor cells by secreting granzyme B and tumor necrosis factor-related apoptosis-inducing ligand [145]. Toll-like receptor (TLR) and B-cell receptor (BCR) signaling can lead to the activation of B cells and increase oxygen consumption and glucose transport [146]. However, unlike T cells, B cells mainly utilize glucose through PPP to produce NADPH and ribose 5-phosphate, which are required for the synthesis of antibodies. In addition, activation of TLR and BCR signaling induces differentiation of Breg cells and promotes their glycolytic metabolism [147].
Innate immunity: NKT cells, NK cells, M1 macrophages, DCs, and N1 neutrophils
NKT cells have the characteristics of both adaptive and innate immune cells and mainly recognize lipid antigens to play an immunomodulatory role. Therefore, lipid metabolism can promote the activation of NKT cells and regulate their cytotoxic effect. High levels of lactic acid in the TME reduced the expression of PPARγ in NKT cells, thereby decreasing lipid synthesis and production of IFN-γ. In addition, high levels of cholesterol in the TME can promote the secretion of IFN -γ by NKT cells and enhance the TCR signaling at immunological synapses of NKT cells [139, 148].
NK cells can directly kill tumor cells by releasing cytotoxic granules and inflammatory cytokines or through ADCC. Glycolysis and OXPHOS are upregulated during the activation of NK cells, which is mediated by Myc, SREBP, and mTORC1 signaling pathways. Myc is regulated by mTORC1 and promotes glycolysis and OXPHOS in NK cells by promoting the expression of glucose transporter protein (GLUT) and key enzymes of glycolysis [140]. However, an increase in endogenous SREBP inhibitors such as 27-HC in the TME inhibits glycolysis and OXPHOS in NK cells by reducing the expression of SLC25A1 and ACLY; moreover, the secretion of IFN-γ and granzyme-B is also inhibited. In addition, glucose-1, 6-bisphosphatase (FBP), the key enzyme of gluconeogenesis, significantly inhibits glycolysis in NK cells and leads to their dysfunction [149].
M1 macrophages exert antitumor immune responses by producing pro-inflammatory cytokines and inducible nitric oxide synthase and participating in ADCC. When TLR is activated, M1 macrophages show increased glucose uptake and lactic acid production. However, the lack of glucose and the accumulation of lactic acids in the TME promotes the transformation of macrophages from antitumor M1 to protumor M2 phenotype. In addition, M1 macrophages depend on NADPH produced through PPP. In the TME, PUFAs promote the aggregation of M1 macrophages accompanied by an increase in ROS, while SFAs inhibit the antitumor function of M1 macrophages. Notably, exogenously modified lipids, such as phosphotyrosine cholesterol, can polarize or even re-induce the antitumor M1 phenotype [150, 151].
DCs, an important APC in antitumor response, switch from OXPHOS to glycolysis after activation. The transition process is mainly related to LPS/HIF-1α and TLR/PI3K/AKT pathways. In the lipid-rich TME, DCs tend to accumulate PL and TAG (especially PUFA) rather than cholesterol and CE, which enhances the secretion of pro-inflammatory cytokines and chemokines, thereby playing a stronger antigen cross-presentation function and effectively activating NK, NKT, and Teff cells. In addition, 27-HC was found to induce the maturation of DCs. However, high levels of oxidized lipids and high accumulation of LDs limit the formation of the MHC I complex and impair the ability of DCs to present antigens and activate T cells. In addition, liver kinase B1 is an important metabolic regulator of DCs and the loss of its expression can activate the mTOR signaling pathway, resulting in metabolic disorders in DCs [151, 152].
N1 neutrophils can exert antitumor effects by producing ROS, reactive nitrogen species, and IFN-α/β and phagocytosis. They can activate NK cells and inhibit IL-17-producing T cells [153]. N1 neutrophils mainly rely on glycolysis, and they also upregulate PPP to meet the NADPH requirement for respiratory burst. In addition, N1 neutrophils have active glycogen metabolism to fulfill their antitumor function [154].
Abnormal metabolism of protumor immune cells
Adaptive immunity: Treg and Breg cells
Treg cells are the key cell population involved in adaptive tumor immunosuppression. Treg cells secrete inhibitory factors such as IL-4, IL-10, and transforming growth factor-β (TGF-β) and express inhibitory molecules such as CTLA-4, PD-1, and LAG-3 to inhibit the activation and function of other immune cells. Treg cells preferentially utilize the TCA cycle and OXPHOS for their metabolism. However, Treg cells are more dependent on FAO under the metabolic stress of the TME. In contrast to Teff cells, Treg cells show lower levels of GLUT1 and higher levels of monocarboxylate transporter 1 (MCT1), CD36, and FANS, which increase lactic acid and lipid metabolism to replace glucose metabolism. Similarly, the expression of genes and proteins related to glucose metabolism is downregulated in Treg cells, which are activated by TLR8 [141]. In addition, the activation of the AMPK and SREBP pathways enhances the synthesis of FA and cholesterol, which, in turn, increases the expression of PD-1 in Treg cells [114]. However, the activity of ACLY decreases significantly during the differentiation of Treg cells stimulated by TGF-β, which leads to the transformation from lipid anabolism to catabolism [155]. Of note, some highly active Treg cells still rely on glycolysis. Therefore, Treg cells have good metabolic adaptability, which is conducive to their survival under the metabolic stress of the TME [156].
Breg cells also have an immunosuppressive function and inhibit the activation of T and NK cells. They also promote the protumor effect of Treg cells, MDSCs, and M2 macrophages by secreting IL-10, TGF-β, and ROS and upregulating PD-L1 and CTLA-4 [157]. High levels of lactic acid in the TME can induce the differentiation of antitumor B cells into protumor Breg cells through the GPR81/PPARs/NADPH oxidase signaling pathway [158]. Like other B cell populations, glycolysis is enhanced in activated Breg cells, and the hypoxic TME and HIF-1α can maintain the function of Breg cells by promoting glycolysis. In addition, changes in cholesterol metabolism in B cells regulate IL-10 production [159].
Innate immunity: M2 macrophages, MDSCs, and N2 neutrophils
The phenotype of tumor-associated macrophages (TAMs) is mainly the immunosuppressive M2 phenotype. M2 macrophages secrete various protumor factors such as arginase 1 (Arg1). M2 macrophages, like Treg cells, are mainly dependent on FAO. M2 macrophages tend to take up more lipids from the TME and enhance the synthesis of FAs to maintain their protumor function. For example, oleic acid can induce M2 polarization through the mTOR signaling pathway [138]. Cholesterol is an inflammatory mediator in M2 macrophages and ABCG1 (a transporter protein involved in cholesterol homeostasis and efflux) is highly expressed in M2 macrophages, thereby promoting cholesterol efflux and IL-4 secretion. In addition, 27-HC induces polarization of macrophages to the M2 phenotype, which may be related to the activation of LXRs. Besides, hypoxia increases the infiltration of M2 macrophages and inhibits M1 macrophages by producing angiogenic factors, mitogenic factors, and adenosine. Similarly, lactic acid in the TME enhanced the activity of HIF-2α by inhibiting the expression of the M2 macrophage-specific vacuolar ATP subunit (ATP6V0d2). Lactic acid also upregulates HIF-1α stable lncRNA in M2 macrophages [150, 160].
MDSCs are also important immunosuppressive cells that establish a complex tumor immunosuppressive network with other immune cells. Although glycolysis and OXPHOS are upregulated in MDSCs, these cells rely on FAO to maintain their protumor function [161]. Serine/threonine kinase (PIM1) regulates FAO through PPARγ. The lipid-rich TME (having 27-HC) can promote the survival and infiltration of MDSCs, and these cells also upregulate CD36 and LDLR to further increase lipid uptake. Similarly, tumor-derived granulocyte-macrophage colony-stimulating factor (GM-CSF) promotes lipid accumulation in MDSCs by inducing the expression of FATP2 through STAT3 signaling. However, excessive lipid accumulation inhibits their immunosuppressive function. UFAs promote the formation of LDs, and LDs inhibit the activity of MDSCs [162, 163].
Tumor-associated neutrophils (TANs) have protumor N2 phenotype. These neutrophils secrete Arg1, myeloperoxidase, neutrophil elastase, matrix metalloproteases, and neutrophil extracellular traps to promote tumor development and inhibit the activation and proliferation of T and NK cells [153]. Tumors can enhance the FAO and maintain intracellular redox balance in N2 neutrophils through the stem cell factor/c-Kit signaling pathway, which allows them to exert their immunosuppressive effects for a longer period. Cholesterol and oxysterols such as 27-HC promote the aggregation of N2 neutrophils and the formation of an immunosuppressive microenvironment in the TME [139]. In addition, N2 neutrophils show a high expression of genes associated with lipid uptake and LD formation, and a low expression of genes related to the degradation of LDs and β-oxidation, which results in lipid accumulation in them. N2 neutrophils also demonstrate high expression of GLUT1. Notably, the accumulation of nutrients in N2 neutrophils may be related to the prolongation of their life span, thereby allowing them to exert immunosuppressive effects in the TME for a longer duration [164].
Effect of lipid metabolic reprogramming on the TME
The TME plays an important role in tumor immunity and immunotherapy. Tumor cells can alter the metabolism of the cells present in the TME, especially tumor-associated immune cells, which, in turn, interfere with the metabolism of tumor cells. Tumor cells have a high nutrient requirement; however, limited nutrient supply leads to metabolic stress in the TME, which mainly manifests as a hypoxic environment caused by a lack of oxygen and as energy competition caused by a lack of glucose. In addition, metabolites from tumor cells or other cells accumulate to create a low pH environment caused by lactic acid accumulation and a lipid peroxidative environment caused by lipid and ROS accumulation. Therefore, tumor and immune cells undergo metabolic reprogramming to circumvent these limitations, thereby affecting tumor immune response (Figure 3) [165].
Lipid metabolic reprogramming in the TME. Metabolic stresses, such as glucose and oxygen deprivation, lactic acid accumulation, and lipid peroxidation, induce lipid metabolic reprogramming of tumor and immune cells in the TME to circumvent these limitations.
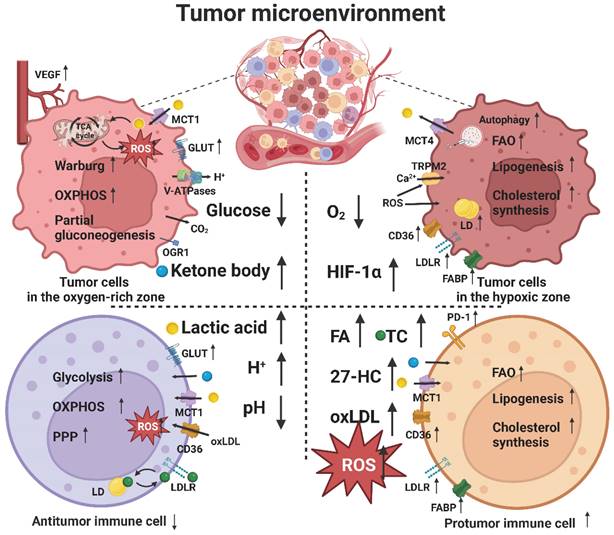
The number and composition of immune cells in the TME are heterogeneous. Some tumors are classified as “cold tumors” because of a lack of immune cell infiltration and present an “immune desert.” However, some tumors are classified as "hot tumors" because they are rich in immune cells. Apparently, a “hot tumor” is more responsive to immunotherapy than a “cold tumor” [5]. The difference lies in the mechanism by which immune cells find tumor cells. Here, chemokines play an important guiding role, and different subsets of lymphocytes and APCs are recruited to the TME through different chemokine-chemokine receptor signaling pathways [37]. In addition, chemokines can directly target non-immune cells including tumor cells and vascular endothelial cells, resulting in differential effects on tumor progression and immunotherapy outcomes [166]. Notably, some authors have reported that high-density lipoprotein (HDL) [167] and oxidized lipids [168] can affect the expression of chemokines and their receptors, indicating that lipid metabolism in the TME can affect the expression of chemokines. In turn, some other researchers have shown that chemokines can affect the lipid metabolism of cells; however, the specific mechanism is unknown [169].
Lipid metabolic reprogramming in cancer-associated fibroblasts
In addition to immune cells, cancer-associated fibroblasts (CAFs) are the most important component of stromal cells in the TME and are the center of cross-communication between various cells. CAFs can secrete several cytokines, chemokines, and extracellular matrix proteins, thereby promoting the occurrence and development of tumors [170]. Tumor cells are anabolic, whereas CAFs are catabolic. Some studies have pointed out that their metabolism is coupled, implying that the catabolism of CAFs can provide important metabolic substances for the growth of tumor cells, thus establishing a symbiotic relationship. CAFs demonstrate the reverse "Warburg effect," which is characterized by aerobic glycolysis and leads to lactic acid secretion; the lactic acid is then absorbed and utilized by cancer cells. In addition, CAFs increase the endogenous level of ROS in the TME. In terms of lipid metabolism, CAFs undergo reprogramming, which is manifested as the secretion of lysophosphatidylcholines into the TME, where they exert diverse effects on tumor cells and other cells in the TME [171]. On the contrary, CAFs also regulate immune cell-mediated antitumor immunity, including promoting the activation of immunosuppressive cells, enhancing the expression of immune checkpoint molecules, and limiting the function of effector immune cells [172].
Hypoxic environment
Tumor cells have high oxygen requirements to support their rapid proliferation but the relatively slow and abnormal angiogenesis causes insufficient oxygen supply, which often leads to local hypoxia in tumor tissues. Necrosis, occurring in the hypoxic area, releases tumor antigens to activate the tumor immune response [173]. The differences in blood supply to the tumor tissue make hypoxic and oxygen-rich areas coexist and change dynamically, which results in metabolic heterogeneity and the occurrence of a unique phenomenon of "metabolic symbiosis". For example, tumor cells in the hypoxic area away from blood vessels enhance aerobic glycolysis by upregulating GLUT and glycolysis-related enzymes, whereas those in the normal oxygen supply area near blood vessels tend to undergo TCA cycle and OXPHOS to efficiently use glucose and transport saved glucose to the hypoxic area [174].
The functions of some key enzymes of lipid metabolism are affected in a hypoxic environment. For example, SCD1 needs oxygen to catalyze the formation of carbon double bonds to desaturate FAs. Therefore, SFAs are accumulated in the absence of oxygen by preventing the formation of UFAs. Tumor cells enhance the uptake of UFA and its release from LD to restore the SFA/UFA balance. However, hypoxia can also increase the expression of SCD1 through the regulation of SREBP1 [175].
HIF-1α and hypoxia-related genes, such as EPAS1, are highly expressed in various hypoxic tumor tissues, which play an important role in tumor angiogenesis, metabolism, and immunity [176]. In addition to promoting glycolysis, hypoxia can enhance lipid metabolism through the activation of the PI3K/Akt/mTOR pathway. HIF-1α can inhibit lipid peroxidation for inducing ferroptosis by increasing the expression of FABP3/7, LDLR, and LD to promote lipid uptake and storage [177]. Notably, the activation of T cells increases the expression of HIF-1 and Myc, thereby promoting the activity of key enzymes in glycolysis such as GLUT. Hypoxia also induces the persistent activation of dynamin-related protein 1 in NK cells [178], which is involved in the mTOR signaling pathway and leads to mitochondrial fission, thereby decreasing the survival and cytotoxicity of NK cells. In addition, hypoxia can increase the expression of Foxp3, a key transcription factor in Treg cells, which promotes lipid uptake by CD36, OXPHOS, and FAO [179]. Meanwhile, hypoxia also promotes the infiltration of Treg cells into tumor tissues. However, HIF-1α can also destroy the stability of Treg cells [180]. This contradiction can be attributed to different degrees of hypoxia, implying that mild hypoxia stimulates cell growth, whereas severe hypoxia induces cell death.
Glucose-deficient environment
Tumor cells are more sensitive to glucose deficiency than other cells in the TME because of their high energy requirements. Tumor-associated immune cells can partially use ketone bodies as alternative energy sources but "glycolysis subtype" tumor cells cannot use ketone bodies instead of glucose [31]. Therefore, such tumor cells rely on lipids as the main energy source, which makes them more aggressive and induces immunosuppression.
However, glucose metabolism plays an irreplaceable role in the occurrence and development of tumors. As an important carbon source, intermediates of glycolysis and aerobic oxidation, such as glucose-6-phosphate and pyruvate, can be used as the essential material basis. Specifically, the TCA cycle and PPP provide AcCoA and NADPH for lipid synthesis, respectively. Glycolysis also provides glycerol 3-phosphate for the synthesis of TAG. Compared with FAO and OXPHOS, the rapid energy supply provided by glycolysis can meet the emergency energy needs of tumor cells during proliferation or metastasis. Glycolysis reduces the dependence on oxygen and mitochondrial function, which reduces the production of ETC-derived ROS and inhibits lipid peroxidation. The lactic acid produced by glycolysis can acidify the TME and induce tumor immunosuppression. Hexokinase (HK), a key glycolytic enzyme, inhibits the apoptosis of tumor cells [181, 182].
Therefore, tumor cells compete for glucose and outdo immune cells by upregulating GLUT and other pathways [11]. For example, the partial gluconeogenesis pathway is activated in tumor cells, which allows more intermediate flow into the glycolysis pathway to compensate for the lack of glucose. In non-gluconeogenesis tissues, such as lung cancer and colorectal cancer, tumor cells with the expression of PCK1 can use non-sugar energy sources to carry out partial gluconeogenesis because of the lack of downstream key enzymes of gluconeogenesis, such as FBP and glucose-6-phosphatase (G6P). On the contrary, in some cancers, such as liver and kidney cancer, tumor cells expressing key enzymes of gluconeogenesis, including PCK1, FBP, and G6P, can promote the whole gluconeogenesis pathway to produce glucose, which, in turn, inhibits the glycolytic pathway. Notably, FBP can also inhibit PPARα-mediated FAO, thereby inhibiting tumor development [12].
The supply of glucose is equally important for the activation of immune cells, specifically antitumor immune cells. The lack of glucose hinders the activation of T cells, and T cells undergo metabolic reprogramming and shift toward lipid metabolism for survival by activating the AMPK pathway, PPARα signaling pathway, and FAO. In addition, T cells differentiate into Treg cells in response to low glucose levels, which is dependent on the activity of AMPK and PPARα signaling pathways [140]. Low glucose levels also inhibit glycolysis induced by mTORC1 signaling, which, in turn, inhibits the production of IFN-γ and granzyme by NK cells [183] and the ability of DCs [152] to activate tumor immune responses.
Low pH environment
Accumulation of lactic acid produced by glycolysis lowers the pH of the TME. The acidic environment can theoretically lead to apoptosis, thus playing an antitumor role. However, lactic acid transported to the stroma is quickly taken up by tumor cells through MCT1 and utilized in the normal oxygen supply area, thereby maintaining optimal lactic acid levels in the TME for protumor effect. Moreover, metabolic stress of the TME can promote the expression of MCT1, which can also activate the NF-κB signaling pathway [184].
Lactic acid has traditionally been considered a metabolic waste; however, a study has indicated that it can upregulate the expression of vascular endothelial growth factor (VEGF) and HIF-1 [185]. In addition, tumor cells can use lactic acid as an energy source to generate ATP through a partial TCA cycle, which further strengthens the concept of “metabolic symbiosis”. For example, lactic acid produced by tumor cells in the hypoxic area is excreted into the stroma through MCT4 and then transported to the oxygen-rich area, where tumor cells uptake lactic acid through MCT1 and activate the mTOR signaling pathway. In this process, cells in the oxygen-rich area reduce the consumption of oxygen and glucose, while those in the hypoxic area avoid excessive accumulation of lactic acid. In addition, lactic acid increases the expression of PD-L1 through the lactic acid receptor (GPR81) pathway, eventually leading to tumor immunosuppression [186].
Lactic acid and H+ have long been regarded as negative regulators of immunity, but recent studies have indicated that lactic acid can have immune-promoting effects. Lactic acid and H+ can inhibit the survival and function of T cells by inhibiting the MPAK signaling pathway [187]. In addition, the increased lactic acid uptake by NK cells l decreases intracellular pH, which impairs their energy metabolism and function [188]. Lactic acid can promote tumor development by inducing immunosuppression-related cells, such as Treg cells and M2 macrophages [160], which may be related to histone lactonization (Kla). It also enhances the expression of PD-1 in Treg cells [187]. In contrast, lactic acid can partially replace glucose as an energy source and increase the stem cell properties of CD8+T cells to enhance antitumor immunity [189]. Overall, these observations suggest that the different effects of lactic acid and H+ should be distinguished while evaluating the effect of low pH on antitumor responses.
In addition to lactic acid, H+ produced by redox reactions occurring in tumor cells can be mainly transported out of the cell via V-ATPases to prevent acidosis and maintain a low pH environment [190]. In addition, the CO2 produced by intracellular decarboxylation reaction can diffuse to the TME, which results in a low pH environment and affects the survival and function of immune cells [191]. A low pH environment can activate lysosomes and proteolytic enzymes to promote the degradation of the extracellular matrix, thereby promoting the invasion and metastasis of the tumor. Therefore, protein carriers such as V-ATPases can be potential targets for cancer therapy [192].
For lipid metabolism, acidic conditions mediate the transport of FA by CD36 and promote the storage of PUFA in LDs, thereby increasing lipid accumulation in tumor cells [184]. Acid sensory receptors on the membrane of tumor cells, such as G protein-coupled receptor 1 (OGR1), can sense the low pH and promote the formation of LDs [193]. The low pH environment also promotes the lipid peroxidation of ω-3 and ω-6 PUFAs, which increases the toxicity of PUFAs and the sensitivity of tumor cells to ferroptosis. In addition, low pH regulates the expression of SREBP2 target genes, such as HMGCR and ACAT, and increases the biosynthesis of cholesterol [185].
Lipid peroxidation environment
Most solid tumors are rich in lipids, which are obtained from the surrounding cells or bloodstream. Tumor cells can induce metabolic changes in adjacent adipocytes to release FFA and cholesterol into the TME and then absorb them through FATP [194]. The TME is often in a state of oxidative stress. The "respiratory burst" of infiltrating neutrophils and catalytic reactions of LOX, COX, and xanthine oxidase produces a large amount of ROS. Together, they create a lipid peroxidation environment in the TME and produce many "harmful lipids" such as ox-LDL, malondialdehyde (MDA), 4-hydroxynonenoic acid (HNE), and 27-HC [195].
Tumor cells often upregulate lipid uptake and storage to adapt to the low-glucose and high-fat TME. The uptake of ox-LDL by tumor cells promotes the occurrence and progression of tumors because it affects mitosis, regulates the expression of cyclin, and induces the expression of endoplasmic reticulum localization protein (Nogo-B) [196]. However, excessive accumulation of intracellular lipids, particularly PUFA, can also lead to lipid peroxidation and ferroptosis. But interestingly, mammary adipocytes can protect triple-negative breast cancer (TNBC) cells from ferroptosis [197].
The high-fat environment has a bidirectional effect on tumor immune response. The high level of FA can activate PPARα signaling in T cells, which, in turn, promotes FAO and OXPHOS; however, the depletion of FA in T cells hinders their proliferation and antitumor effect [198]. Likewise, the activation of PD-1 signaling inhibited the TCR and CD28-mediated PI3K/AKT/mTOR pathway to promote FAO [199]. In addition, high levels of cholesterol can enhance the number and antitumor effect of NK cells by upregulating LDLR [149]. In contrast, excessive FAs, including oxidized lipids, inhibit the survival and antitumor effect of T cells. Specifically, T cells take up ox-LDL and AA via CD36, leading to intracellular lipid peroxidation [74]. Similarly, high levels of cholesterol can induce endoplasmic reticulum stress in T cells and upregulate PD-1 expression [200]. However, an increase in the intracellular cholesterol level by inhibiting ACAT1 can promote the activation of T cells [201]. This contradictory result may be attributed to the distribution of intracellular cholesterol, implying that inhibition of cholesterol storage may increase the flow of free cholesterol to the cell membrane, which promotes the formation of lipid rafts and the aggregation of TCR. Overall, these observations suggest that lowering the level of cholesterol in the TME is not necessarily beneficial.
Tumor microenvironment induces autophagy
Autophagy is considered a self-protective mechanism of cells. In the TME, metabolic stress, oxidative stress, and immune signals can induce autophagy by increasing the activity of autophagy-related genes and the ability of intracellular lysosome transport, which allows for efficient degradation of cellular components. Autophagy is regulated by two antagonistic regulatory kinases, AMPK (activator) and mTORC1 (inhibitor) [113].
Tumor cells can achieve energy recycling and substance renewal through autophagy, which increases the lipid pool and helps tumor cells adapt to the nutrient competition [202]. In addition, autophagy can reduce intracellular oxidative stress and inflammation and prevent DNA damage, thereby reducing lipid peroxidation. Notably, the resistance of tumor cells to chemotherapeutic and targeted drugs is partly caused by abnormal activation of autophagy [203]. However, the use of autophagy inhibitors alone has not achieved ideal antitumor effects in clinical trials. Therefore, autophagy inhibitors are used in combination with chemotherapy or immunotherapy [204]. In contrast, excessive autophagy induces apoptosis and can promote ferroptosis in tumor cells. Although autophagy can scavenge peroxides, the phagocytosis of ferritin releases large amounts of Fe2+ and increases the intracellular labile iron pool. In addition, mtDNA stress triggered autophagy-dependent ferroptosis [205].
Autophagy also affects the tumor immune response. Autophagy can selectively degrade MHC I molecules, thereby reducing the immunogenicity of tumor cells. In contrast, the inhibition of autophagy in tumor cells can increase the expression of tumor surface antigens. Autophagy in MDSCs degrades MHC II molecules and inhibits the activation of T cells [206]. However, autophagy of immune cells can support their adaptive survival in the TME [207].
Overall, autophagy is a double-edged sword for tumor and immune cells. Inhibition of autophagy promotes antitumor immune responses to a certain extent and represents a novel direction for cancer immunotherapy [208].
Abnormal cell death related to lipid peroxidation and tumor immunity
Lipid peroxidation is the process by which ROS reacts with PL, enzymes, membrane receptors, and other PUFA side chains to form lipid peroxidation products (LPO) such as MDA and HNE. It eventually destroys membranes and important cellular components such as proteins and nucleic acids [209]. Cell death related to lipid peroxidation involves ferroptosis, cuproptosis, and calcium overload. Tumor cell death can promote the infiltration of immune cells and activate antitumor immune responses through the expression of damage-related molecular patterns such as calreticulin and the release of immune effectors, such as tumor antigens, high mobility group box 1, ATP, and heat shock proteins (HSP70 and HSP90). This process is also called tumor immunogenic cell death (Figure 4) [210].
Lipid peroxidation-related cell death. The cell death modes related to lipid peroxidation mainly include ferroptosis, cuproptosis, and calcium overload. Notably, intermediates in the synthesis of TC can play a protective role against ferroptosis. In ferroptosis, ACSL4 is involved in the attachment of PUFAs to coenzyme A, and LPCAT3 promotes the esterification and incorporation of PUFA-CoA into the membrane phospholipids. The resulting PUFA-PL, specifically AA-PL and AdA-PL, are major peroxidation substrates for ferroptosis. PUFA-PL are readily oxidized by ROS (specifically HO•) to generate PLOOH, which damages the structure of membrane lipids if not effectively neutralized. The peroxidation could be initiated in cells both enzymatically and nonenzymatically. Nonenzymatic peroxidation of lipids is mostly catalyzed by LIP. Enzymatic peroxidation is driven by multiple LOX and POR. PL with AA-PL is also susceptible to LOX-mediated radical oxidation to PLOOH and 5-HPETE. SXC/GSH/GPX4 axis and FSP1/CoQ10/NADPH axis are two major monitoring systems to inhibit PL peroxidation and prevent ferroptosis. In cuproptosis, excessive Cu2+ directly binds to the lipoacylated component of the TCA cycle, such as dihydrolipoic acid transacetylase (DLAT), and results in the abnormal aggregation of insoluble DLAT and subsequent downregulation of Fe-S cluster proteins. In calcium overload, lipid peroxidation increases the permeability of the membrane. ROS can activate TRPM2 and cause massive Ca2+ influx. Then, the excessive intracellular Ca2+ further increases the Ca2+ uptake by mitochondria, which leads to the formation of calcium phosphate deposits and inhibits the synthesis of ATP, thereby decreasing the activity of the Ca2+ pump in the endoplasmic reticulum and cell membrane.
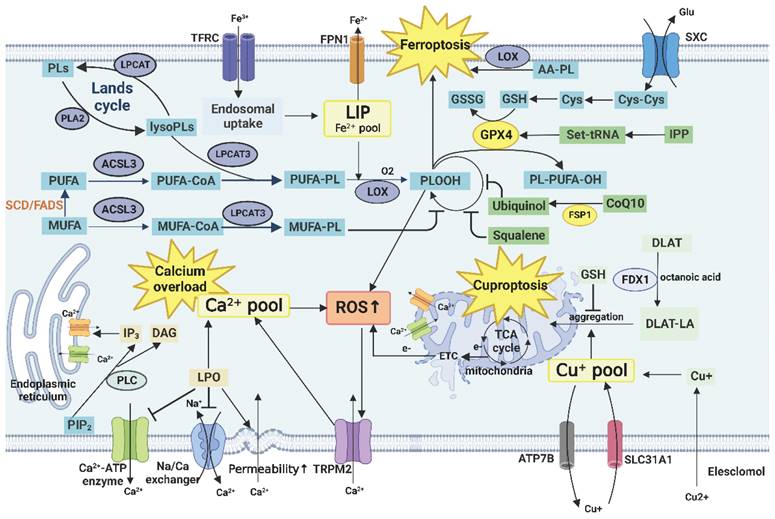
Lipid peroxidation in tumor cells
Tumor cells are more susceptible to lipid peroxidation than normal cells. The production rate of ROS is higher in tumor cells. ETC is prone to leak out ROS during OXPHOS because of the defects in intracellular mitochondria and the dependence of energy metabolism on them. In addition, intracellular NADPH oxidase can produce superoxide, which is rapidly converted to H2O2 by superoxide dismutase (SOD). Moreover, the clearance rate of ROS is lower in tumor cells. Unlike normal cells, tumor cells have low levels of antioxidant enzymes and their antioxidant barrier is defective. Therefore, tumor cells inhibit lipid peroxidation by reducing mitochondrial activity (Warburg effect), increasing the production of PGs, and reducing the level of PUFA in the membrane. Low levels of ROS are often maintained in tumor stem cells and tumor cells in the proliferative state or with drug resistance [209, 211].
However, long-term maintenance of moderate levels of ROS is beneficial for tumor progression. ROS-induced DNA damage leads to genomic instability, which drives the accumulation of oncogenic mutations. A certain level of ROS and LPO act as secondary messengers to activate related transcription factors and participate in the regulation of signaling pathways, such as PI3k/Akt, RTK/Ras, VEGF, NF-κB, and COX-2, to promote tumor inflammation and immune response [212]. In addition, lipid peroxidation induces non-enzymatic oxidative modification of proteins and makes them immunogenic [209].
Taken together, the bidirectional effect of lipid peroxidation on tumors suggests that different types and stages of tumors and immune responses have different requirements for the expression of ROS and LPO; therefore, tumor immunotherapy targeting lipid peroxidation should be carefully designed [213].
Ferroptosis
Ferroptosis, a newly emerged form of non-apoptotic regulatory cell death caused by LPO overload in the membrane, is considered a by-product of abnormal lipid metabolic reprogramming. The balance between ferroptosis-related peroxidation and antioxidation systems determines whether the cell undergoes apoptosis or not [214]. Activated CD8+T cells accelerate the ferroptosis of tumor cells by hypersecreting IFN-γ during anti-PD-L1 immunotherapy [215]. Ferroptosis inhibition in tumor cells is also one of the resistance mechanisms of PD-1/PD-L1 inhibitors. In addition, both radiotherapy and targeted therapy with TYRO3 receptor tyrosine kinase inhibitors promote ferroptosis in tumor cells, and exert excellent tumor-control effects in combination with immunotherapy [216].
Oxygen and iron are essential factors in the ferroptosis-related peroxidation system that drive lipid peroxidation to produce phospholipid peroxides (PLOOH), which can damage the structure of membrane lipids if not effectively neutralized. ACSL4 converts PUFAs into their respective acyl-CoA, while LPCAT3 promotes the esterification and incorporation of PUFA-CoA into membrane PLs [217]. The resulting PUFA-PLs, specifically AA-PL and adrenocorticotropic acid (AdA)-PL, are the major peroxidation substrates for ferroptosis, which are easily oxidized by ROS to produce PLOOH [218]. The peroxidation could be initiated enzymatically by multiple LOXs and P450 oxidoreductases and nonenzymatically by LIP [219]. CD8+T cell-derived IFN-γ stimulates ACSL4 and increases the incorporation of AA into C16 and C18 acyl chain-containing phospholipids. Therefore, IFN-γ combined with AA has become one of the means to control immunogenic tumors [220].
The System X-(SXC)/GSH /GPX4 and FSP1/CoQ10/NADPH axes are two major monitoring systems in the ferroptosis-related antioxidant defense system to inhibit PL peroxidation and prevent ferroptosis. SXC, GPX4, and FSP1 are highly expressed in tumors and are related to the poor prognosis of patients, suggesting that tumor cells tend to inhibit their ferroptosis [221]. IFN-γ derived from immunotherapy-activated CD8+T cells and the radiotherapy-activated ataxia telangiectasia mutated gene synergistically repress SLC7A11, and this combination strongly promotes lipid oxidation and ferroptosis of tumor cells. A decreased expression of SLC3A2 and an increase in IFN-γ and CD8+T cells are associated with clinical benefits [222]. The androgen receptor promotes GPX4 overexpression in the luminal androgen receptor subtype of TNBC, which assists tumor cells in escaping ferroptosis [223].
Notably, ferroptosis may also occur in immune cells, and this may be a specific mechanism of tumor immunosuppression. For example, CD36-mediated uptake of FA by CD8+T cells in the TME can induce lipid peroxidation and ferroptosis, which reduce the production of cytotoxic cytokines and decreases their antitumor ability [224]. GPX4-deficient T cells can rapidly accumulate membrane LPO and lead to ferroptosis, and therapeutic induction of ferroptosis in tumor cells by GPX4 inhibitors may have unnecessary effects on T cells. ACSL4 is also essential for the ferroptosis and the maintenance of immune function of CD8+T cells [225]. Knockout of GPX4 promotes the occurrence of ferroptosis in Treg cells, inhibits the immune escape function, and enhances the response of TH17 cells by secreting IL-1β to promote the antitumor immune function [226].
Cuproptosis
Cu2+ is a necessary cofactor for enzymes that mediate basic cellular functions, including mitochondrial respiration and redox reactions. Although Cu2+ does not participate directly in the mitochondrial respiratory chain, it promotes the activity of key enzymes in the TCA cycle and inhibits the leakage of ROS from the respiratory chain. Therefore, compared with normal cells, tumor cells need higher levels of Cu2+ to maintain mitochondrial metabolic activity and intracellular redox balance. The level of Cu2+ in serum or tumor cells of tumor patients is higher than that in normal individuals, which can induce cuproptosis in cells [227].
Excessive Cu2+ can directly bind to the lipoacylated components of the TCA cycle, such as dihydrolipoic acid transacetylase (DLAT), and result in the abnormal aggregation of insoluble DLAT and subsequent downregulation of Fe-S cluster proteins, eventually leading to proteotoxic stress and ultimately cell death. Therefore, cells dependent on mitochondrial respiration are more sensitive to Cu2+ ions than cells that rely on glycolysis [228]. Therefore, tumor cells reduce the activity of mitochondria through the Warburg effect to avoid cuproptosis [229]. In addition, Cu2+ promotes the accumulation of ROS and induces lipid peroxidation. Cu2+ can also directly activate various angiogenic factors, including VEGF, fibroblast growth factor 2, TNF-α, and IL-1, thereby promoting tumor angiogenesis. Cu2+ regulates autophagy through ULK1 and ULK2 (serine/threonine kinases). High levels of Cu2+ are associated with an increased expression of PD-L1, which may help tumors resist immunotherapy [230]. Therefore, several researchers have analyzed the relationship between Cu2+ and tumor-associated immune cells and established models to predict the effect of Cu2+ on tumor immunotherapy.
Calcium overload
Mitochondria, as an intracellular calcium pool, are involved in the regulation of intracellular calcium homeostasis. Lipid peroxidation increases the permeability of the membrane under oxidative stress. ROS activate TRPM2 (H2O2 -dependent Ca2+ channel) on the surface of tumor cells in the TME leading to massive Ca2+ influx. The excessive intracellular Ca2+ further increases the uptake of Ca2+ by mitochondria, which leads to the formation of calcium phosphate deposits and inhibits ATP synthesis. Consequently, the activity of the Ca2+ pump in the endoplasmic reticulum and cell membrane further decreases, which, in turn, increases intracellular Ca2+ levels. Then, excessive Ca2+ can abnormally activate various Ca2+-dependent degradation enzymes. For example, the phospholipase activation promotes the hydrolysis of PLs, resulting in membrane destruction [231].
However, cell migration and invasion in some tumors, such as glioblastoma and esophageal squamous cell carcinoma (ESCC), are critically dependent on Ca2+ signaling [232]. The hyperactive intracellular calcium oscillations caused by the Ca2+ influx in the endoplasmic reticulum from the cytoplasm or the excessive opening of some store-operated Ca2+ entry (SOCE) channels are associated with the proliferation of tumor cells. Notably, extracellular zinc exerted rapid inhibitory effects on SOCE and intracellular Ca2+ oscillations in the ESCC cells, thereby suggesting a possible molecular basis for zinc-induced cancer prevention [233].
Calcium overload can activate the GSDME protein, which induces pyroptosis in tumor cells and sensitizes immunotherapy. Currently, several methods, such as Ca2+ nanoparticles and calcium hydride (CaH2) administration, are being evaluated to induce calcium overload in tumor cells to assist immunotherapy and develop a new tumor treatment strategy, namely ion interference therapy [234, 235].
Clinical applications of lipid metabolic reprogramming
Targeting lipid metabolism is more sensitive and beneficial than targeting glucose metabolism for tumor immunotherapy. Tumors with metabolic heterogeneity are more adaptable to interventions in glucose metabolism. Further, drugs that target glucose metabolism can also inhibit antitumor immune cells because of their dependence on glucose. In contrast, protumor immune cells are mainly dependent on lipid metabolism and tumor cells are more sensitive to lipids than glucose. In addition, existing drugs for disorders of lipid metabolism and NSAIDs can also be reused to treat abnormal lipid metabolism in the TME [236]. Ferroptosis and autophagy are also linked to lipid metabolism, which can further improve the effect of immunotherapy (Table 3).
Therapies targeting lipid metabolism to sensitize immunotherapy.
| Target | Drug | Phase | Tumor type | Trial number | Effect | |
|---|---|---|---|---|---|---|
| FA | Reduce SFA and TC in blood | PCSK9 inhibitor, statins, fibrates, and ezetimibe | Preclinical, phase I and II | Melanoma, Colon cancer, and breast cancer | NA | Promote Teff cells and inhibit Treg cells |
| Increase ω-3PUFA in blood | Diet therapy | Phase II and III | Head and Neck Cancer | NCT05101889 | Inhibit PGs and NF-kB pathway, regulate autophagy | |
| Inhibit CD36 | CD36 antibody, siRNA, siRNA, and CD36 inhibitor | Preclinical | Melanoma, colon cancer, and lymphoma | NA | Promote DCs and Teff cells, inhibit Treg cells, and induce ferroptosis | |
| Inhibit FATP | FATP inhibitor: Lipofermata and (Z)-Ligustilide | Preclinical | NSCLC, colon cancer, and lymphoma | NA | Inhibit MDSCs | |
| Inhibit ACLY | ACLY inhibitor: LY294002, SB-204990, and NDI-091143 | Preclinical | GBM | NA | Affect Treg cells, T cells | |
| Inhibit ACSS2 | ACSS2 inhibitor: VY-3-135 | Preclinical | CESC and TNBC | NA | Affect PD-L1, B cells, T cells, and CAFs | |
| Inhibit ACSL4 | ACSL4 inhibitor: troglitazone, and PRGL493 | Preclinical | Liposarcoma | NA | Inhibit ferroptosis of Teff cells | |
| Inhibit ACC | ACC inhibitor: PF-05175157, Firsocostat, and CP-640186 | Phase II | Esophageal cancer | ChiCTR- ICR-15005940 | Promote T cells and inhibit M2 macrophages | |
| Inhibit FASN | FASN inhibitor: omeprazole, C75, orlistat, and UCM05 | Phase II | TNBC, thyroid cancer, neuroblastoma | NA | Induce ferroptosis, inhibit M2 macrophages, and Treg cells | |
| Inhibit SCD1 | SCD1 inhibitor: BZ36, A939572, MK-8245, and XEN723 | Phase II | Breast cancer | NA | Induce ferroptosis, endoplasmic reticulum stress, mitochondrial apoptosis, and autophagy | |
| Inhibit FADS | FADS inhibitor: SC-26196 | Preclinical | Colon cancer | NA | Reduce ω-3/ω-6PUFA and induce ferroptosis | |
| Inhibit MAGL | MAGL inhibitor: JZL184, JJKK048, and JW 642 | Preclinical | Multiple tumors | NA | Increase LPA, LPC, and PGE2 | |
| Inhibit CPT1 | CPT1 inhibitor: etomoxir, McN3716, teglicar, and perhexiline | Preclinical | Breast cancer, colon cancer, lung cancer, and prostate cancer | NA | Inhibit TAMs and MDSCs, and enhance T cells | |
| PGs | Inhibit COX /PGES | COX1/2 inhibitor: NSAIDs mPGES1 inhibitor | Preclinical and phase II | Melanoma, CRC, prostate cancer, and GBM | NA | Affect IDO1 |
| TC | Inhibit HMGCR | HMGCR inhibitor: statins and HMG499 | Phase II | Breast cancer, leukemia, liver cancer, and myeloma | NA | Reduce ROS and PD-L1, enhance T cells, and inhibit ferroptosis |
| Inhibit SQS | SQS inhibitor: zalagozic acid A | Preclinical | Liver cancer | NA | Increase TNF-α, NF-κB, and MMP1 | |
| Inhibit SQLE | SQLE inhibitor | Preclinical | PAAD | NA | Affect immune cell and immune checkpoint | |
| Inhibit ACAT1 | ACAT1 inhibitor: avamaib | Preclinical | Melanoma and cholangiocarcinoma | NA | Enhance Teff cells | |
| Regulation | Activate AMPK | AMPK agonist: metformin, acardixin (AICAR) | Phase III | Breast cancer | NCT01101438 | Reduce PD-1 |
| Inhibit mTOR | mTOR inhibitor: rapamycin analogue and everolimus | Phase III | AML and breast cancer | ChiCTR-OPC-14005488 | Inhibit Treg cells, Teff cells, and NK cells | |
| Activate PPARγ | PPARγagonist: bezafibrate and troglitazone | Preclinical, phase I, and II | Thyroid tumor and breast cancer | NA | Enhance Teff cells | |
| Inhibit PPARα | PPARαinhibitor: adriamycin and chophylline | Preclinical | Breast cancer, liver cancer, and colorectal cancer | NA | Enhance DCs | |
| Inhibit SREBP1 | SREBP1 inhibitor: siRNA, pseudoprotodioscin, and fatostatin | Preclinical | CRC and TNBC | NA | Inhibit Treg cells and M2 macrophages | |
| Activate LXR | LXR agonist: RGX-104 | Preclinical and phase I | Breast cancer | NCT02922764 | Inhibit MDSCs and enhance M1 macrophages | |
| Ketone body | Increase BHB | KD therapy | Phase II and I | Breast cancer, ovarian cancer, endometrial cancer, and breast cancer | NCT03795493 | Enhance anti-oxidation of immune cells, regulate p53 and mTOR pathways, and control inflammation |
| Supplement BHB | ||||||
| Inhibit autophagy | Autophagy inhibitor: hydroxychloroquine (HCQ) and autophinib | Phase II | CRC, pancreatic cancer, and melanoma | NCT02316340 | Increase tumor immunogenicity and enhance T cells | |
| Ferroptosis | Inhibit SLC7A11 | Sorafenib | Clinal | AML, HCC, neuroblastoma, NSCLC, and RCC | NCT03247088, NCT02559778, and NCT00064350 | Inhibit ferroptosis-related antioxidant system |
| Sulfasalazine | Phase II | Breast cancer and GBM | NCT04205357, NCT01577966, and NCT03847311 | |||
| Inhibit GPX4 | Altretamine | Clinical | Lymphoma and sarcoma | NCT00002936 | ||
| Withaferin A | Phase II | Breast cancer, osteosarcoma, and HCC | NCT04092647 and NCT00689195 | |||
| Inhibit GCL | Buthionine sulfoxide | Phase I | Melanoma and neuroblastoma | NCT00002730, NCT00005835, and NCT00661336 | ||
| Activate iron metabolism | Neratinib | Clinical | Breast cancer and CRC | NCT04366713, NCT03377387, and NCT03457896 | Enhance ferroptosis-related peroxidation system | |
| Salinomycin | Clinical | Multiple cancer | NA | |||
| Lapatinib | Clinical | Breast cancer | NCT03085368, NCT00356811, and NCT00667251 | |||
| Drug carriers | Exosome | TAE-DC | Preclinical | Pancreatic cancer | NA | Enhance T cells |
| Exosome-derived miRNA | Preclinical | Breast cancer | NA | Inhibit M2 macrophages and induce M1 macrophages | ||
| Liposome | Antigen-capture spike liposomes | Preclinical | Multiple cancer | NA | Capture and transport tumor antigens to DCs | |
However, although lipid metabolic reprogramming leads to an immunosuppressive TME, lipid remains the important energy and material basis of the cells for antitumor immune response. Therefore, therapies targeting lipid metabolism should be specific and targeted to certain components and pathways of lipid metabolism in different types and stages of tumor and immune cells to maximize their sensitizing effects on immunotherapy [237]. Presently, most lipid-related antitumor drugs are still in preclinical studies and have yet not achieved satisfactory therapeutic effects or even have contradictory results (especially when used alone). Nevertheless, the research on such drugs mainly provides new possibilities for sensitizing tumor immunotherapy and their limitations prompt us to further elucidate the associated mechanisms [60].
Targeting fatty acid metabolism
Interventions in the uptake of fatty acids
CD36 inhibitors hinder tumor development, specifically metastasis and angiogenesis in the TME [238] and have shown synergistic effects in combination with the FASN inhibitors and anti-PD-1 therapy [239]. CD36 inhibitors can not only enhance the antigen presentation ability of DC but also reduce the number of Treg cells and upregulate PD-1 by inhibiting the PPAR pathway, thereby promoting the survival and function of CD8+T cells. CD36 inhibitors are related to the inhibition of lipid peroxidation and ferroptosis in immune cells [224].
In addition, the potential protumor role of the FABP has been demonstrated. The inhibition of FATP reduces lipid uptake, invasion, and growth in tumors such as melanoma [240, 241]. Notably, FATP2 can promote the utilization of AA and the synthesis of PGE2 in MDSCs, thereby mediating immunosuppression [242]. Therefore, FATP inhibitors can also improve the effect of immunotherapy.
Interventions in the synthesis of fatty acids
The development of tumor cells and immune cells is affected by the expression of ACLY and its product AcCoA. Inhibition of ACLY can reduce the levels of intracellular FA and cholesterol and inhibit upstream glucose metabolism [243]. Notably, ACLY is involved in the IL-2-mediated activation of Treg, CD4+T, and CD8+T cells [244, 245]. However, the in vivo efficacy of existing ACLY inhibitors in tumor models (specifically on tumor immune response) has not been definitive; however, the recently identified 3D structure of ACLY holoenzyme tetramer is expected to provide new insights into the mechanism of ACLY inhibition leading to the development of effective ACLY inhibitors [246].
The pathway of AcCoA synthesis by acetyl-CoA synthetase (ACSS) can bypass the inhibition of ACLY, which makes tumor cells resistant to ACLY inhibitors. Therefore, ACSS2 inhibitors not only hinder the growth of acetate-dependent tumors but also improve the antitumor effect of ACLY inhibitors. In addition, ACSS2 inhibitors promote antitumor immune responses. The expression of ACSS2 in cervical squamous cell carcinoma is proportional to that of PD-L1 and is associated with the infiltration of B cells, CD4+T cells, CD8+T cells, and CAFs [247].
ACC inhibitors can impede the growth of the tumor and enhance the efficacy of chemotherapy and targeted drugs [248]. However, inhibition of ACC increases the expression of SREBP1 and leads to the accumulation of TAGs in blood. Notably, diacylglycerol acyltransferase 2 (DGAT2) inhibitors can downregulate the activity of SREBP1 and inhibit the expression of genes related to lipid synthesis. Therefore, the combination of ACC and DGAT2 inhibitors can improve the antitumor effect and reduce the side effects, such as hyperlipidemia, of ACC treatment [249]. Given that AMPK is a key inhibitor of ACC, a classical agonist of AMPK metformin can block lipid metabolism in tumor cells. Metformin has an important role in tumor immunomodulation. For example, metformin can promote the transformation within the TME to an antitumor response by increasing the number of CD8+T cells and decreasing that of M2 macrophages [250].
FASN inhibitors inhibit the energy supply of tumor cells and disrupt their membrane composition. Specifically, FASN inhibitors can induce ferroptosis in tumor cells by reducing the levels of intracellular TAG and PC and increasing the levels of PUFA in the membrane through the Lands cycle. In addition, FASN inhibitors promote the formation of lipid rafts and the activation of the TLR4 signaling pathway [251]. Notably, FASN inhibitors markedly reduce the release of ROS, IL-10, and TNF-α by M2 macrophages and inhibit the activation of Treg cells. FASN inhibitors also prevent tumor metastasis after the withdrawal of antiangiogenic therapy [252]. Several FASN inhibitors have shown fewer side effects and better antitumor effects in preclinical studies; however, none of them has yet been approved for clinical use.
Interventions in the desaturation of fatty acids
Currently, SCD1 inhibitors are in the preclinical stage, and the existence of SCD1 compensatory or alternative pathways can easily make tumor cells resistant to SCD1 inhibitors. The expression of the transcription factor FOSB, which regulates the activity of SCD1, rapidly restores the SCD1 level and induces drug resistance [253]. Tumor cells with high expression of FADS1/2 can bypass the effect of SCD1 inhibition [254]. Therefore, a combination of SCD1 and FADS inhibitors effectively blocks the proliferation of tumor cells. Notably, SCD1 inhibitors have also demonstrated synergistic antitumor effects with targeted therapies. These inhibitors can increase the infiltration and activation of DC and CD8+T cells and enhance the efficacy of anti-PD-1 therapy [60].
Interventions in the lipolysis of triglycerides
The expression of monoacylglycerol lipase (MAGL) is markedly elevated in many tumor cells, which leads to an increase in intracellular FFA levels that are closely related to cancer-associated cachexia [255]. Through the MAGL-FFA pathway, tumor cells can promote the production of lipid signaling molecules such as LPA, LPC, and PGE2, which promote tumor development and affect tumor immune response [256]. Therefore, in preclinical studies, MAGL inhibitors have reduced the invasiveness of tumors and improved the efficacy of tumor immunotherapy.
Interventions in the decomposition of fatty acids
The drug-resistant tumor cells, such as TNBC, exhibit dependence on FAO [257]; therefore, CPT1 inhibitors can synergistically enhance the antitumor effects of antiangiogenic, chemotherapy, and immune drugs. CPT1 inhibitors can eliminate the protumor effect of TAMs, inhibit the infiltration of MDSCs, and enhance the antitumor function of T cells [258]. Further, CPT1 expression can be used as a potential marker of tumor immunosuppression.
Targeting the prostaglandin pathway
COX-2 inhibitors are effective in the prevention and treatment of tumors (specifically gastrointestinal tumors) and prostate cancer [259]. However, they are beneficial only when combined with chemotherapy drugs or immune checkpoint inhibitors (ICIs). Notably, the EGFR signaling pathway reduces the expression of intracellular 15-PGDH; therefore, the combined blocking effect of EGFR and COX-2 may also enhance the antitumor effect of COX-2 inhibitors [260]. In addition, the expression of indoleamine 2, 3-dioxygenase 1, an important immunomodulatory enzyme in tumor cells, is related to the activation of the COX2/PGE2 axis; therefore, COX-2 inhibitors can enhance the effect of the anti-PD-1 therapy to some extent [261].
The mechanism of COX-2 inhibitors is not entirely dependent on the role of COX-2 but is related to COX-1 or even independent of the PGE2 pathway. COX-1 is highly expressed (even more than COX-2) in ovarian and cervical cancers. Therefore, COX-1 inhibitors are more effective in treating these COX-1-dominated tumors [262]. However, both COX-1 and COX-2 inhibitors may increase the risk of cardiovascular diseases and gastrointestinal mucosal injury in patients. Therefore, it is necessary to further determine the indications for COX inhibitors or develop specific inhibitors against downstream targets of COX, such as PGES and prostaglandin receptors to reduce their side effects and achieve better efficacy. For example, mPGES-1 inhibitors reduce PGE2 while avoiding adverse reactions such as cardiovascular diseases, and they even demonstrate antithrombotic and antiatherosclerotic effects [259].
Targeting cholesterol metabolism
Interventions in the uptake of cholesterol
LDLR is upregulated through EGFR/PI3K/AKT/SREBP-1 signaling, which enhances the uptake of LDL-C and plays an important role in the growth of cancers such as lung cancer and breast cancer [136]. In addition, LDLR is involved in the regulation of tumor immunity. It can interact with the TCR complex and regulate its recycling and signaling, thus enhancing the cytotoxic effect of CD8+T cells. Therefore, LDLR inhibitors can enhance the efficacy of tumor immunotherapy. For example, PCSK9, as a regulator of cholesterol metabolism, can not only reduce the level of cholesterol in blood but also negatively regulate the expression of LDLR, thereby synergistically enhancing its antitumor effect with PD-1 inhibitors [63].
Interventions in the synthesis of cholesterol
Statins, in addition to lowering blood lipids, can inhibit the activity of HMGCR. They can remodel the cytoskeleton and lipid rafts by reducing cholesterol in the membrane, thereby inhibiting the proliferation and metastasis of the tumor [263]. In addition, statins can downregulate the TNF-α receptor and inhibit the NF-κB signaling pathway to induce apoptosis of tumor cells. Although the protective effect of the MVA pathway on ferroptosis is weakened, statins can exert obvious antioxidant effects through other pathways, including PI3K/Akt, AMPK, Rho/ROCK, and NO signaling pathways, and reduce ROS-induced tumorigenesis [264]. Further, statins block the stabilizing effects of cholesterol on PD-L1 and PD-1, which are expressed on T, NKT, and Treg cells. Statins can also reduce the level of PD-L1 by inhibiting AKT and β-catenin-mediated signaling pathways and the expression of long non-coding RNA (SNHG29) [265]. Similarly, inhibition of SREBP signaling also downregulates the expression of PD-1 on Treg cells [156].
Of note, fat-soluble statins have better antitumor effects than water-soluble statins but the mechanism is unknown [266]. However, some clinical trials of statins have not achieved ideal effects, possibly because the doses used are similar to those used for lipid-lowering therapy and fail to achieve sufficient concentrations. Similarly, some studies have reported that after removing the effect of cholesterol, statins may not affect the prevention and prognosis of some cancers, especially liver cancer, and even promote their development. This may be attributed to statin-mediated activation of the SREBP signaling pathway by negative feedback, thus resisting or even reversing the effect of statins on lowering cholesterol [267]. Therefore, the combined therapy with SREBPs inhibitors and statins may have improved efficacy. In addition, p53 mutation can be used as a marker to reflect the sensitivity of statins; however, the use of statins worsens the prognosis of patients with wild-type p53 [130].
Squalene synthase (SQS) and squalene epoxidase (SQLE) also affect the synthesis of cholesterol and tumor immune response. SQS inhibitors not only interfere with the squalene synthesis downstream but also increase the CoQ10 synthesis upstream, which may diminish their antitumor effect [268]. Similarly, SQLE was upregulated in pancreatic adenocarcinoma, and its levels were significantly correlated with tumor-associated immune cells, immune checkpoints, and biomarkers of the TME. Specifically, the expression of SQLE was negatively correlated with CD4+T cells but positively correlated with CD8+T cells and neutrophils [269]. Therefore, SQLE can be used as a potential marker of tumor immune response.
Interventions in the storage of cholesterol
The high expression of ACAT1 in tumor-associated immune cells can reduce the level of intracellular free cholesterol and promote the composition and function of the membrane. For example, ACAT1 inhibitors can enhance the cholesterol level in the membranes of CD8+T cells and promote the formation of immune synapses, which enhances immune recognition and cytotoxic effects of CD8+T cells [270]. Therefore, ACAT1 can be used as a marker to predict tumor immune response to a certain extent.
Targeting pathways regulating lipid metabolism
In contrast to the key enzymes and receptors specific for lipid metabolism, targeting the upstream regulatory pathways is more complicated because such pathways are also involved in regulating other cellular processes, such as glucose metabolism, amino acid metabolism, and autophagy. The drugs targeting the regulation of lipid metabolism fail to achieve ideal therapeutic effects and show various side effects or even the paradoxical phenomenon that both forward and reverse regulation of the same signaling pathway will produce antitumor effects. Therefore, underlying mechanisms need to be further studied [248].
Interventions in the AMPK and mTOR signaling pathways
Presently, some AMPK agonists, such as metformin, have been reported to have good antitumor effects and are being tested in clinical trials [250]. However, AMPK has a bidirectional effect on tumors and appears to promote the survival of tumor cells under the metabolic stress of the TME. Therefore, attempts are being made to develop AMPK agonists that target specific types and stages of tumors [112]. Furthermore, the AMPK targeting therapeutic strategies should shift from simple agonists to partial inhibitors in certain tumors to achieve the best efficacy.
AMPK pathways act through mTORC1 inhibition; therefore, inhibition of mTOR signaling has been explored as an antitumor strategy but the results were not optimal [271]. Rapamycin analogs cannot completely inhibit the phosphorylation of mTORC1 effector 4EBP and instead lead to compensatory upregulation of mTORC2/AKT activity. Since PI3K/Akt mediates the main pathway of mTOR, a new generation of dual PI3K/mTOR inhibitors are currently being evaluated preclinically, which have achieved better antitumor effects [272]. The survival and function of tumor-associated immune cells require mTOR. Therefore, mTOR inhibitors not only hinder the immunosuppressive function of Treg cells but also hinder the antitumor effect of CD8+T and NKT cells. This implies that mTOR inhibitors inhibit antitumor immunity even while positively promoting the formation of immunosuppressive TME. Furthermore, mTOR inhibitors may increase the possibility of developing autoimmunity in the long term because of the suppression of Treg cells [273]. Therefore, attempts can be made to develop mTOR inhibitors that target specific immune cells.
Interventions in PPAR signaling pathway
The function and expression of PPARs are heterogeneous in different types and stages of tumors. PPARα/γ agonists exert antitumor effects by lowering blood lipids and promoting FAO. In addition, PPARγ agonists have been found to inhibit tumor proliferation, invasion, and angiogenesis. PPARγ agonists can also enhance the function of CD8+T cells and promote the effectiveness of anti-PD-1 therapy [274]. Interestingly, PPARγ inhibitors are effective in the treatment of some cancers, such as liver, colorectal, and gastric cancers. PPARα inhibitor can inhibit the proliferation of tumor cells by regulating CPT1 (essential for fatty acid metabolism and energy production). In addition, PPARα inhibitors were demonstrated to eliminate the TDE-induced dysfunction of DC immune function [275]. Overall, agonists are effective in treating tumors with low expression of PPARs, whereas inhibitors are effective in treating tumors with high expression of PPARs.
Interventions in SREBP and LXR signaling pathways
SREBP1 inhibitors can significantly inhibit the growth of tumor cells. Currently, most of the SREBP1 inhibitors are in the preclinical evaluation stage. SREBP1 inhibitors can enhance the effect of radiotherapy, anti-PD-1 therapy, and polyadenosine diphosphate ribose polymerase inhibitors. SREBP1 inhibitors have also been found to reverse the immunosuppressive effect of Treg cells and reduce the protumor effect of M2 macrophages [276].
LXR agonists can cause the depletion of cholesterol in tumor cells and thus reduce lipid raft signaling. In addition, LXR agonists can induce caspase-dependent apoptosis and block survival signals such as EGFR and Akt [136]. LXR agonists can inhibit the survival and immunosuppressive effect of MDSCs while promoting the differentiation of M1 macrophages, resulting in an effective antitumor response [277]. Therefore, LXR agonists combined with ICIs may demonstrate an enhanced antitumor effect. In addition, the activation of LXRs inhibited PPARα and then inhibited the expression of FAO-related genes. Therefore, a combination of LXR agonist and PPARα inhibitor can synergistically enhance tumor growth inhibition [278].
Targeting ketone body metabolism
KD therapy activates ketogenesis in the body through a high-fat low-carbohydrate diet, thereby increasing the concentration of ketone bodies in the blood to replace glucose as the main energy source. This therapy inhibits "glycolysis subtype" tumor cells but is less effective against immune cells. Furthermore, KD therapy can improve the ability of immune cells to resist oxidative stress and thus promote antitumor immunity [279]. In addition, KD therapy regulates p53 and mTOR signaling pathways, controls inflammatory responses, and inhibits tumor angiogenesis [280]. Notably, KD therapy involves high-fat intake but it does not cause disorders of lipid metabolism such as hyperlipidemia (which is related to the high intake of ω-3PUFA). KD includes "good lipids" such as PUFA, whereas HFD includes "bad lipids" such as SFA and cholesterol [62]. However, KD therapy has a low compliance rate. A study has pointed out that BHB is the key to the antitumor effect of KD therapy. BHB supplementation to the level of KD therapy can achieve a similar antitumor effect and enhance the efficacy of ICIs and can solve the problem of poor compliance and enhance the efficacy of tumor immunotherapy. However, the role of BHB in tumors is still unclear, and a study even indicates that BHB can promote the development of some tumors [281].
Targeting ferroptosis
Interventions in phospholipid unsaturation
In addition to directly targeting tumor cells, CD8+T cells can also promote the ferroptosis of tumor cells. T-cell-derived IFN-γ combines with arachidonic acid to activate ACSL4, and the activated enzyme increases the levels of PUFA in the cell membrane, thereby changing the lipid distribution pattern of tumor cell membranes. These changes increase the susceptibility of membranes to lipid peroxidation and naturally induce ferroptosis of tumor cells. However, ACSL4 is the key enzyme of lipid metabolism, which mediates the remodeling of GPLs and the activation of FAs [282]. ACSL4 is upregulated in some tumor cells and is related to the invasion of tumors, such as breast and colon tumors [283]. Therefore, cancers can be of two types according to the role of ACSL4 in promoting lipid metabolism and ferroptosis. The first type of cancers has ferroptosis dominance patterns such as lung [284], ER+ breast [285], and cervical cancers [286]. A study has suggested that targeting the ACSL4 pathway is a potential anticancer strategy; however, this has not yet been confirmed. The second type of cancers have lipid metabolism dominance patterns including ER- breast cancer [287], HCC [288], CRC [289], and prostate cancer [287]. ACSL4 inhibitors, such as triacsin C, thiazolidinediones, and troglitazone, have a potent antitumor effect in these malignant tumors [290].
LPCAT3 further increases the ferroptosis sensitivity of tumor cells. LPCAT3 knockout reduced the composition of PUFA-PL in tumor cells, thereby inhibiting ferroptosis and promoting tumor progression [291]. In addition, LPCAT3 limited ER stress induced by lipid overload [292]. In macrophages, LXR-mediated activation of LPCAT3 promotes antitumor immunity. Although the activation of LPCAT3 can inhibit tumor progression, there are currently no antitumor drugs that target LPCAT3, and further research is needed to determine the mechanism of action by which LPCAT3 promotes antitumor immunity and develop LPCAT3-targeted therapies [293].
Balance of regulation between peroxidation and antioxidation
Iron-catalyzed oxidation and enzymatic oxidation ultimately lead to ferroptosis and are the two targets in the context of current research on ferroptosis-targeting therapies. The growth of tumor cells (especially tumor stem cells) depends on the role of iron, and high iron intake increases the risk of tumors (such as HCC and breast cancer) [294]. Increasing iron absorption [295], reducing iron storage [296], or restricting iron outflow will lead to increased iron accumulation and metabolism, thus directly or indirectly enhancing LPO. Salinomycin, an antibacterial drug, actively triggers the degradation of ferritin in lysosomes, leading to further iron loading in organelles, thus activating ferroptosis [297]. Neratinib and lapatinib increase the expression of transferrin, which is responsible for iron transfer into cells, and decrease the expression of ferroportin, which is responsible for clearing iron from cells. Therefore, these drugs may induce ferroptosis by modulating iron metabolism in the cells [298, 299].
GPX4 and SXC are key enzymes that inhibit ferroptosis. Knockout of GXP4 or inhibition of SXC leads to the accumulation of intracellular LPO, resulting in ferroptosis of tumor cells. Many antineoplastic drugs targeting GPX4 and SXC are also being studied or used clinically; these drugs include SLC7A11 inhibitors (sorafenib and sulfasalazine) [300, 301], GPX4 inhibitors (altretamine and withaferin A) [302, 303], and GCL inhibitors (sulfoximine) [304]. Combining GPX4 inhibitors with immunotherapy can further activate T cells in the TME, subsequently enhancing their killing effect on tumor cells. Cyst(e)inase, an engineered enzyme that degrades cystine and cysteine, could be combined with a PD-L1 blocker to significantly improve LPO in tumor cells [305].
Targeting ferroptosis of immune cells
Targeting ferroptosis in tumor cells may exert unintended effects on CD8+T cells in the TME. CD8+T cells are more sensitive to ferroptosis induced by GPX4 inhibitors, and overexpression of FSP1 induces ferroptosis resistance and maintains CD8+T cell function (antitumor activity) in the TME. Therefore, FSP1 may have potential therapeutic applications in cancer immunotherapy. Promoting the ferroptosis of immunosuppressive cells, such as Treg cells, M2 macrophages, and MDSC, is a potential antitumor strategy and ferroptosis-targeting drugs may emerge in the future [306].
Lipids as drug carriers
The therapies targeting lipid metabolism exploit the biological characteristics of lipids, whereas the physical and chemical properties of lipids, such as liposolubility and polarity, determine their use as effective drug carriers, including exosomes and liposomes.
Currently, the immunotherapy strategies that target exosomes mainly inhibit the production of TDEs or interfere with the exosomes released from immune cells. GW4869 is an inhibitor of neutral sphingomyelinase, which inhibits the production of TDEs and exerts an antitumor effect. DC-derived exosomes contain tumor antigens; therefore, TAE-DC vaccines have been developed to activate T cells in the TME [307]. Macrophage-derived exosomes can also be used to reverse the immunosuppressive effect of M2 macrophages and induce polarization of macrophages to the M1 phenotype [308]. In addition, artificially developed antigen-capturing liposomes can capture and transport tumor antigens to DC, subsequently enhancing antigen cross-presentation [309].
Tumor biomarkers related to lipid metabolism
Several key enzymes and products of lipid metabolic pathways can be used as markers for the development of tumors and tumor immunity [310]. The function, characteristics, and expression of lipid metabolism-related markers in different types and stages of tumors are summarized in Table 1. Notably, an enhanced expression of cholesterol-related genes is associated with low immunotherapy response, which indicates their potential value as immunotherapy biomarkers [87].
Conclusions
Lipid metabolic reprogramming plays an essential role in TME and cancer immunotherapy. Here, we described the characteristics, mechanism, and role of lipid metabolic reprogramming in tumor and immune cells interacting in the TME. Importantly, lipid metabolic reprogramming even could determine cell fate, which is manifested in the regulation of different cell death modes, especially ferroptosis. Further, we summarized the potential clinical applications of targeting lipid metabolic reprogramming for antitumor therapies and as a tumor biomarker. Given the complexity of lipid metabolic reprogramming and the continuous research efforts to identify the critical genes and pathways involved, researchers will discover several candidate genes/proteins for diagnostic and therapeutic purposes. In addition, lipid metabolic biomarkers have shown potential for monitoring immunotherapy; therefore, they should be the primary focus of future research. Overall, an improved understanding of lipid metabolic reprogramming will provide useful insights into mechanisms of tumorigenicity and may lead to the development of novel therapeutic approaches for patients with cancer.
Abbreviations
27-HC: 27-hydroxycholesterol
4EBP: EIF4E binding protein
5-HPETE: 5-hydroxyperoxyeicosatetraenoic acid
AAD: Anti-angiogenic drug
ABCA1: ATP-binding cassette transporter A1
ABCG1: ATP-binding cassette transporter G1
ACAT: Cholesterol acyltransferase/acetoacetyl-CoA sulfhydrylase
ACC/ACACA: Acetyl-CoA carboxylase
ACLY: ATP-citrate lyase
ACSL: Acyl-CoA synthase long-chain family
ACSS: Acetyl-CoA synthase
AdA: Adrenocorticotropic acid
ADCC: Antibody dependent cell mediated cytotoxicity
AGPAT: acylglycerol-3-phosphate acyltransferase
ALOX: Arachidonic acid lipoxygenase
AML: Angiomyolipoma
AMPK: AMP-activated protein kinase
APCs: Antigen-presenting cells
AR: Androgen receptor
ATGL: Adipose triglyceride lipase
AcAc: Acetoacetic acid
AcAcCoA: Acetoacetyl-CoA
AcCoA: Acetyl coenzyme A
Apo: Apolipoprotein
Arg1: Arginase 1
BCR: B-cell receptor
BDH1: 3-hydroxybutyrate dehydrogenase
BHB: β-hydroxybutyrate
Breg: Regulatory B
CAFs: Cancer-associated fibroblasts
CALR: Calreticulin
CD4+conv: Antitumor CD4+
CDC: Complement dependent cytotoxicity
CDK: Cell cycle protein-dependent kinase
CE: Cholesterol ester
CESC: Cervical squamous cell carcinoma
CL: Cardiolipin
COX: Cyclo-oxyganese
CPT1: Carnitine lipid acyltransferase 1
CRC: Colorectal cancer
CoQ10: Coenzyme Q10
Cys: Cysteine
DAG: Diacylglycerol
DAMP: Damage-associated molecular pattern
DC: Dendritic cells
DGAT: Diacylglycerol acyltransferase
DHA: Docosahexaenoic acid
DLAT: Dihydrolipoyl Transacetylase
Drp1: Hairpin-associated protein 1
ECM: Extracellular matrix
EGFR: Epidermal growth factor receptor
ELOVL: Elongation of very long chain fatty acid
EMT: Epithelial-mesenchymal transition
ER: Estrogen receptor
ETC: Electron transport chain
Eps: PGE receptor
FA: Fatty acid
FABPpm: Plasma membrane fatty acid binding protein
FACoA: Acyl-CoA
FADS: Fatty acid desaturase
FAO: Fatty acid oxidation
FASN: Fatty acid synthase
FATP: Fatty acid transport protein
FBP: Fructose-1,6-bisphosphatase
FDPS: Farnesyl diphosphate synthase
FDX1: Iron oxygen reducing protein 1
FFA: Free fatty acid
FGF2: Fibroblast growth factor 2
Foxp3: Forkhead box protein 3
FPN: Iron transport protein
FPP: 15-carbon pyrophosphate farnesylate
FSP1: Iron action suppressor protein 1
G3P: 3-phosphoglycerol
G6P: Glucose-6-phosphatase
GBM: Glioblastoma
GCL: Glutamate cysteine ligase
GGPP: Geranyl geranyl pyrophosphate
GK: Glycerol kinase
GLUT: Glucose transporter protein
Gly: Glycerin
GM-CSF: Granulocyte-macrophage colony-stimulating factor
GPAT: Glycerol-3-phosphate acyltransferase
GPCR: G protein-coupled receptor
GPLs: Glycerol phospholipids
GPX4: Glutathione peroxidase 4
GSH: Reduced glutathione
GSSG: Oxidized glutathione
Glu: Glutamate
HCC: Hepatocellular carcinoma
HDAC: Histone deacetylase
HDL: High density lipoproteins
HFD: High-fat diet
HIF-1α: Hypoxia-inducible factor-1α
HISLA: HIF-1α stable lncRNA
HK: Hexokinase
HMGB1: High mobility group protein 1
HMGCR: HMGCoA reductase
HMGCS1: HMGCoA synthase 1
HMGCoA: 3-hydroxy-3-methylglutaryl-CoA
HNE: 4-hydroxynonenoic acid
HSL: Hormone-sensitive lipase
HSP: Heat shock protein
ICD: Immunogenic cell death
ICIs: Immune checkpoint inhibitors
IDH1: Isocitrate dehydrogenase 1
IDO1: Indoleamine 2,3-dioxygenase 1
IGFR: Insulin-like growth factor receptor
iNOS: Inducible nitric oxide synthase
INSIG: Insulin-inducible gene protein
IP3: Inositol 1,4,5-triphosphate
IPP: Isopentenyl pyrophosphate
lysoPLs: Lysophospholipids
KD: Ketogenic diet
Kbhb: β-hydroxybutyrylation
Kbu: Butyrylation
Kcr: Crotonylation
Kla: Lactonization
Kma: Malonylation
Kpr: Malonylation
Ksucc: Succinylation
LA: Linoleic acid
LAL: Lysosomal acid lipase
LAR: Luminal androgen receptor
LCFA: Long chain fatty acids
LD: Lipid droplet
LDLR: Low density lipoprotein receptor
LIP: Unstable iron pool
LKB1: Liver kinase B1
LLO: Lipid-linked oligosaccharide
LOX: Lipoxygenase
LPIN1/PAP: Phosphatidate phosphatase
LPA: Lysophosphatidic acid
LPC: Lysophosphatidylcholine
LPCAT: Lysophosphatidylcholine acyltransferase
LPO: Lipid peroxidation
LTs: Leukotrienes
LXRE: LXR response element
LXRs: Liver X receptors
MAGL: Monoacylglycerol lipase
MCT1: Monocarboxylate transporter 1
MDA: Malondialdehyde
MDSCs: Myeloid suppressor cells
MG: Monoglyceride
MHC I: Major histocompatibility complex 1
MPO: Myeloperoxidase
mTOR: Mammalian target of rapamycin protein
MUFA: Monounsaturated fatty acids
MVA: Mevalonate
MVD: Mevalonate diphosphate decarboxylase
MVK: Mevalonate kinase
NE: Neutrophil elastase
NETs: Neutrophil extracellular trap network
NK: Natural killer
NLRP3: Nod-like receptor protein 3
NSAIDs: Non-steroidal anti-inflammatory drugs
NSCLC: Non-small cell lung cancer
OGR1: G protein-coupled receptor 1
OXCT1: 3-succinyl coenzyme A transferase
oxLDL: Oxidized low-density lipoprotein
OXPHOS: Oxidative phosphorylation
PA: Phosphatidic acid
PAAD: Pancreatic adenocarcinoma
PARP: Polyadenosine diphosphate ribose polymerase
PC: Phosphatidylcholine
PCK1: Phosphoenolpyruvate carboxykinase
PDH: Pyruvate dehydrogenase
PE: Phosphatidylethanolamine
PGES: Prostaglandin E2 synthase
PGs: Prostaglandins
PI: Phosphatidylinositol
PI3K: Phosphoinositide 3-kinase
PIM1: Serine/threonine kinase
PIP2: Phosphati-dylinositol-4,5-bisphosphate
PIP3: Phosphatidylinositol-3,4,5-trisphosphate
PKC: Protein kinase C
PKM2: Pyruvate kinase M2
PL: Phospholipids
PLA2: Phospholipase A2
PLC: Phospholipase C
PLOOH: Phospholipid peroxide
PMVK: Mevalonate phosphate kinase
POR: P450 oxidoreductase
PPARs: Peroxisome proliferator-activated receptors
PPP: Pentose phosphate pathway
PPRE: PPAR response element
PS: Phosphatidylserine
PUFA: Polyunsaturated fatty acid
RCC: Renal cell carcinoma
RCD: Regulatory cell death
RNS: Reactive nitrogen
ROS: Reactive oxygen species
RTA: Free radical trapping antioxidant
RTK: Receptor tyrosine kinase
RXR: Retinol-like X receptor
S6K: Ribosomal protein S6 kinase
SCAP: SREBP cleavage-activating protein
SCD: Stearoyl-CoA desaturase
SCF: Stem cell factor
sec-tRNA: Selenocysteine-tRNA
SGK: Serum and glucocorticoid induced kinase
SOD: Superoxide dismutase
SQLE: Squalene epoxidase
SQS: Squalene synthase
SRE: Sterol regulatory element
SREBPs: Sterol regulatory element binding proteins
SXC: System x-
TAG: Triglycerides
TAMs: Tumor-associated macrophages
TANs: Tumor-associated neutrophils
TC: Total cholesterol
TCA: Tricarboxylic acid
TCR: T-cell antigen receptor
TDE: Tumor-derived exosomes
TFRC: Transferrin receptor
TGF-β: Transforming growth factor-β
TLR: Toll-like receptor
TME: Tumor microenvironment
tmTNF-α: Transmembrane tumor necrosis factor alpha
TNBC: Triple-negative breast cancer
TRAIL: Tumor necrosis factor-associated apoptosis-inducing ligand
TXs: Thromboxane
TZDs: Thiazolidinedione
Teff: Effector CD8+ T
Tmem: Memory CD8+ T
Treg: Regulatory T
UFA: Saturated fatty acids
VEGF: Vascular endothelial growth factor
XO: Xanthine oxidase
α-KG: α-ketoglutarate
γ-IFN: γ-interferon
Acknowledgements
This work was supported by the Natural Science Foundation of Jiangsu Province (BK20210068), the Top Talent Support Program for Young and Middle-aged People of Wuxi Municipal Health Commission (HB2020003), Mega-project of Wuxi Commission of Health (Z202216), and the High-end Medical Expert Team of the 2019 Taihu Talent Plan(2019-THRCTD-1).
Author contributions
Wenjun Mao, Jie Mei, and Yuan Wan conceptualized the review. Kai Yang, Xiaokun Wang, Chenghu Song, Zhao He, Ruixin Wang, Yongrui Xu, and Guanyu Jiang wrote the manuscript. Kai Yang and Xiaokun Wang prepared the figures and tables. Wenjun Mao, Jie Mei, and Yuan Wan critically reviewed and edited the manuscript. Wenjun Mao got funding support. All authors read and approved the final manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Helmy KY, Patel SA, Nahas GR, Rameshwar P. Cancer immunotherapy: accomplishments to date and future promise. Ther Deliv. 2013;4:1307-20
2. Yang Y. Cancer immunotherapy: harnessing the immune system to battle cancer. J Clin Invest. 2015;125:3335-7
3. Ribatti D. The concept of immune surveillance against tumors. The first theories. Oncotarget. 2017;8:7175-80
4. Chen DS, Mellman I. Oncology meets immunology: the cancer-immunity cycle. Immunity. 2013;39:1-10
5. Hiam-Galvez KJ, Allen BM, Spitzer MH. Systemic immunity in cancer. Nat Rev Cancer. 2021;21:345-59
6. O'Donnell JS, Teng MWL, Smyth MJ. Cancer immunoediting and resistance to T cell-based immunotherapy. Nat Rev Clin Oncol. 2019;16:151-67
7. Liu X, Hartman CL, Li L, Albert CJ, Si F, Gao A. et al. Reprogramming lipid metabolism prevents effector T cell senescence and enhances tumor immunotherapy. Sci Transl Med. 2021 13
8. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646-74
9. Yoo HC, Yu YC, Sung Y, Han JM. Glutamine reliance in cell metabolism. Exp Mol Med. 2020;52:1496-516
10. Faubert B, Solmonson A, DeBerardinis RJ. Metabolic reprogramming and cancer progression. Science. 2020 368
11. Vaupel P, Schmidberger H, Mayer A. The Warburg effect: essential part of metabolic reprogramming and central contributor to cancer progression. Int J Radiat Biol. 2019;95:912-9
12. Wang Z, Dong C. Gluconeogenesis in Cancer: Function and Regulation of PEPCK, FBPase, and G6Pase. Trends Cancer. 2019;5:30-45
13. Wen YA, Xing X, Harris JW, Zaytseva YY, Mitov MI, Napier DL. et al. Adipocytes activate mitochondrial fatty acid oxidation and autophagy to promote tumor growth in colon cancer. Cell Death Dis. 2017;8:e2593
14. Currie E, Schulze A, Zechner R, Walther TC, Farese RV Jr. Cellular fatty acid metabolism and cancer. Cell Metab. 2013;18:153-61
15. Park JB, Lee CS, Jang JH, Ghim J, Kim YJ, You S. et al. Phospholipase signalling networks in cancer. Nat Rev Cancer. 2012;12:782-92
16. Yao CH, Fowle-Grider R, Mahieu NG, Liu GY, Chen YJ, Wang R. et al. Exogenous Fatty Acids Are the Preferred Source of Membrane Lipids in Proliferating Fibroblasts. Cell Chem Biol. 2016;23:483-93
17. Ko PJ, Dixon SJ. Protein palmitoylation and cancer. EMBO Rep. 2018 19
18. Maan M, Peters JM, Dutta M, Patterson AD. Lipid metabolism and lipophagy in cancer. Biochem Biophys Res Commun. 2018;504:582-9
19. Zhao J, Zhi Z, Wang C, Xing H, Song G, Yu X. et al. Exogenous lipids promote the growth of breast cancer cells via CD36. Oncol Rep. 2017;38:2105-15
20. Röhrig F, Schulze A. The multifaceted roles of fatty acid synthesis in cancer. Nat Rev Cancer. 2016;16:732-49
21. Pavlova NN, Thompson CB. The Emerging Hallmarks of Cancer Metabolism. Cell Metab. 2016;23:27-47
22. Matsushita Y, Nakagawa H, Koike K. Lipid Metabolism in Oncology: Why It Matters, How to Research, and How to Treat. Cancers (Basel). 2021 13
23. Corn KC, Windham MA, Rafat M. Lipids in the tumor microenvironment: From cancer progression to treatment. Prog Lipid Res. 2020;80:101055
24. da Silva JL, Dos Santos ALS, Nunes NCC, de Moraes Lino da Silva F, Ferreira CGM, de Melo AC. Cancer immunotherapy: the art of targeting the tumor immune microenvironment. Cancer Chemother Pharmacol. 2019;84:227-40
25. Yu W, Lei Q, Yang L, Qin G, Liu S, Wang D. et al. Contradictory roles of lipid metabolism in immune response within the tumor microenvironment. J Hematol Oncol. 2021;14:187
26. Ascenzi F, De Vitis C, Maugeri-Saccà M, Napoli C, Ciliberto G, Mancini R. SCD1, autophagy and cancer: implications for therapy. J Exp Clin Cancer Res. 2021;40:265
27. Doll S, Proneth B, Tyurina YY, Panzilius E, Kobayashi S, Ingold I. et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat Chem Biol. 2017;13:91-8
28. Li Z, Zhang H, Wang X, Wang Q, Xue J, Shi Y. et al. Identification of cuproptosis-related subtypes, characterization of tumor microenvironment infiltration, and development of a prognosis model in breast cancer. Front Immunol. 2022;13:996836
29. Kong H, Chu Q, Fang C, Cao G, Han G, Li X. Cu-Ferrocene-Functionalized CaO(2) Nanoparticles to Enable Tumor-Specific Synergistic Therapy with GSH Depletion and Calcium Overload. Adv Sci (Weinh). 2021;8:e2100241
30. Xia L, Oyang L, Lin J, Tan S, Han Y, Wu N. et al. The cancer metabolic reprogramming and immune response. Mol Cancer. 2021;20:28
31. Saraon P, Cretu D, Musrap N, Karagiannis GS, Batruch I, Drabovich AP. et al. Quantitative proteomics reveals that enzymes of the ketogenic pathway are associated with prostate cancer progression. Mol Cell Proteomics. 2013;12:1589-601
32. Guzmán M, Blázquez C. Is there an astrocyte-neuron ketone body shuttle? Trends Endocrinol Metab. 2001;12:169-73
33. Yang F, Fang E, Mei H, Chen Y, Li H, Li D. et al. Cis-Acting circ-CTNNB1 Promotes β-Catenin Signaling and Cancer Progression via DDX3-Mediated Transactivation of YY1. Cancer Res. 2019;79:557-71
34. Okechukwu CE. Cross Talk between the Ketogenic Diet and Metastatic Prostate Cancer Cells. World J Mens Health. 2022;40:162-3
35. Yang L, Lin PC. Mechanisms that drive inflammatory tumor microenvironment, tumor heterogeneity, and metastatic progression. Semin Cancer Biol. 2017;47:185-95
36. Wang Q, Morris RJ, Bode AM, Zhang T. Prostaglandin Pathways: Opportunities for Cancer Prevention and Therapy. Cancer Res. 2022;82:949-65
37. Sadik CD, Luster AD. Lipid-cytokine-chemokine cascades orchestrate leukocyte recruitment in inflammation. J Leukoc Biol. 2012;91:207-15
38. Liu S, Paknejad N, Zhu L, Kihara Y, Ray M, Chun J. et al. Differential activation mechanisms of lipid GPCRs by lysophosphatidic acid and sphingosine 1-phosphate. Nat Commun. 2022;13:731
39. Mashouri L, Yousefi H, Aref AR, Ahadi AM, Molaei F, Alahari SK. Exosomes: composition, biogenesis, and mechanisms in cancer metastasis and drug resistance. Mol Cancer. 2019;18:75
40. Morrissey SM, Zhang F, Ding C, Montoya-Durango DE, Hu X, Yang C. et al. Tumor-derived exosomes drive immunosuppressive macrophages in a pre-metastatic niche through glycolytic dominant metabolic reprogramming. Cell Metab. 2021;33:2040-58.e10
41. Paskeh MDA, Entezari M, Mirzaei S, Zabolian A, Saleki H, Naghdi MJ. et al. Emerging role of exosomes in cancer progression and tumor microenvironment remodeling. J Hematol Oncol. 2022;15:83
42. Maybruck BT, Pfannenstiel LW, Diaz-Montero M, Gastman BR. Tumor-derived exosomes induce CD8(+) T cell suppressors. J Immunother Cancer. 2017;5:65
43. Li Y, Chen ZK, Duan X, Zhang HJ, Xiao BL, Wang KM. et al. Targeted inhibition of tumor-derived exosomes as a novel therapeutic option for cancer. Exp Mol Med. 2022;54:1379-89
44. Chang J, Chaudhuri O. Beyond proteases: Basement membrane mechanics and cancer invasion. J Cell Biol. 2019;218:2456-69
45. Li B, Qin Y, Yu X, Xu X, Yu W. Lipid raft involvement in signal transduction in cancer cell survival, cell death and metastasis. Cell Prolif. 2022;55:e13167
46. Casares D, Escribá PV, Rosselló CA. Membrane Lipid Composition: Effect on Membrane and Organelle Structure, Function and Compartmentalization and Therapeutic Avenues. Int J Mol Sci. 2019 20
47. Tesfay L, Paul BT, Konstorum A, Deng Z, Cox AO, Lee J. et al. Stearoyl-CoA Desaturase 1 Protects Ovarian Cancer Cells from Ferroptotic Cell Death. Cancer Res. 2019;79:5355-66
48. Hoy AJ, Nagarajan SR, Butler LM. Tumour fatty acid metabolism in the context of therapy resistance and obesity. Nat Rev Cancer. 2021;21:753-66
49. Fuentes NR, Kim E, Fan YY, Chapkin RS. Omega-3 fatty acids, membrane remodeling and cancer prevention. Mol Aspects Med. 2018;64:79-91
50. Vriens K, Christen S, Parik S, Broekaert D, Yoshinaga K, Talebi A. et al. Evidence for an alternative fatty acid desaturation pathway increasing cancer plasticity. Nature. 2019;566:403-6
51. Wang B, Tontonoz P. Phospholipid Remodeling in Physiology and Disease. Annu Rev Physiol. 2019;81:165-88
52. Peng Z, Chang Y, Fan J, Ji W, Su C. Phospholipase A2 superfamily in cancer. Cancer Lett. 2021;497:165-77
53. Murai T. Lipid Raft-Mediated Regulation of Hyaluronan-CD44 Interactions in Inflammation and Cancer. Front Immunol. 2015;6:420
54. Colin D, Limagne E, Jeanningros S, Jacquel A, Lizard G, Athias A. et al. Endocytosis of resveratrol via lipid rafts and activation of downstream signaling pathways in cancer cells. Cancer Prev Res (Phila). 2011;4:1095-106
55. Oh J, Perry JSA, Pua H, Irgens-Möller N, Ishido S, Hsieh CS. et al. MARCH1 protects the lipid raft and tetraspanin web from MHCII proteotoxicity in dendritic cells. J Cell Biol. 2018;217:1395-410
56. Sun L, Su Y, Wang JG, Xia F, Xu Y, Li D. DNA nanotweezers for stabilizing and dynamically lighting up a lipid raft on living cell membranes and the activation of T cells. Chem Sci. 2020;11:1581-6
57. Fu Y, Yu J, Li F, Ge S. Oncometabolites drive tumorigenesis by enhancing protein acylation: from chromosomal remodelling to nonhistone modification. J Exp Clin Cancer Res. 2022;41:144
58. Shvedunova M, Akhtar A. Modulation of cellular processes by histone and non-histone protein acetylation. Nat Rev Mol Cell Biol. 2022;23:329-49
59. Zhao S, Zhang X, Li H. Beyond histone acetylation-writing and erasing histone acylations. Curr Opin Struct Biol. 2018;53:169-77
60. Zheng S, Song Q, Zhang P. Metabolic Modifications, Inflammation, and Cancer Immunotherapy. Front Oncol. 2021;11:703681
61. Koundouros N, Poulogiannis G. Reprogramming of fatty acid metabolism in cancer. Br J Cancer. 2020;122:4-22
62. Chlebowski RT, Aragaki AK, Anderson GL, Pan K, Neuhouser ML, Manson JE. et al. Dietary Modification and Breast Cancer Mortality: Long-Term Follow-Up of the Women's Health Initiative Randomized Trial. J Clin Oncol. 2020;38:1419-28
63. Liu X, Bao X, Hu M, Chang H, Jiao M, Cheng J. et al. Inhibition of PCSK9 potentiates immune checkpoint therapy for cancer. Nature. 2020;588:693-8
64. Bojková B, Winklewski PJ, Wszedybyl-Winklewska M. Dietary Fat and Cancer-Which Is Good, Which Is Bad, and the Body of Evidence. Int J Mol Sci. 2020 21
65. Dechaphunkul T, Arundon T, Raungkhajon P, Jiratrachu R, Geater SL, Dechaphunkul A. Benefits of immunonutrition in patients with head and neck cancer receiving chemoradiation: A phase II randomized, double-blind study. Clin Nutr. 2022;41:433-40
66. Yee LD, Lester JL, Cole RM, Richardson JR, Hsu JC, Li Y. et al. Omega-3 fatty acid supplements in women at high risk of breast cancer have dose-dependent effects on breast adipose tissue fatty acid composition. Am J Clin Nutr. 2010;91:1185-94
67. Dierge E, Debock E, Guilbaud C, Corbet C, Mignolet E, Mignard L. et al. Peroxidation of n-3 and n-6 polyunsaturated fatty acids in the acidic tumor environment leads to ferroptosis-mediated anticancer effects. Cell Metab. 2021;33:1701-15.e5
68. Kanarek N, Petrova B, Sabatini DM. Dietary modifications for enhanced cancer therapy. Nature. 2020;579:507-17
69. Mahboobnia K, Pirro M, Marini E, Grignani F, Bezsonov EE, Jamialahmadi T. et al. PCSK9 and cancer: Rethinking the link. Biomed Pharmacother. 2021;140:111758
70. Acharya R, Shetty SS, Kumari NS. Fatty acid transport proteins (FATPs) in cancer. Chem Phys Lipids. 2023;250:105269
71. Wang J, Li Y. CD36 tango in cancer: signaling pathways and functions. Theranostics. 2019;9:4893-908
72. Mallick R, Basak S, Duttaroy AK. Fatty acids and evolving roles of their proteins in neurological, cardiovascular disorders and cancers. Prog Lipid Res. 2021;83:101116
73. Ma K, Zhang L. Overview: Lipid Metabolism in the Tumor Microenvironment. Adv Exp Med Biol. 2021;1316:41-7
74. Xu S, Chaudhary O, Rodríguez-Morales P, Sun X, Chen D, Zappasodi R. et al. Uptake of oxidized lipids by the scavenger receptor CD36 promotes lipid peroxidation and dysfunction in CD8(+) T cells in tumors. Immunity. 2021;54:1561-77.e7
75. Metallo CM, Gameiro PA, Bell EL, Mattaini KR, Yang J, Hiller K. et al. Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature. 2011;481:380-4
76. Comerford SA, Huang Z, Du X, Wang Y, Cai L, Witkiewicz AK. et al. Acetate dependence of tumors. Cell. 2014;159:1591-602
77. Fernandez HR, Gadre SM, Tan M, Graham GT, Mosaoa R, Ongkeko MS. et al. The mitochondrial citrate carrier, SLC25A1, drives stemness and therapy resistance in non-small cell lung cancer. Cell Death Differ. 2018;25:1239-58
78. Han Q, Chen CA, Yang W, Liang D, Lv HW, Lv GS. et al. ATP-citrate lyase regulates stemness and metastasis in hepatocellular carcinoma via the Wnt/β-catenin signaling pathway. Hepatobiliary Pancreat Dis Int. 2021;20:251-61
79. Vaughn N, Haviland DL. Acly promotes metabolic reprogramming and induction of IRF4 during early CD8(+) T cell activation. Cytometry A. 2021;99:825-31
80. Xu Y, Liao W, Luo Q, Yang D, Pan M. Histone Acetylation Regulator-Mediated Acetylation Patterns Define Tumor Malignant Pathways and Tumor Microenvironment in Hepatocellular Carcinoma. Front Immunol. 2022;13:761046
81. Chowdhury S, Kar A, Bhowmik D, Gautam A, Basak D, Sarkar I. et al. Intracellular Acetyl CoA Potentiates the Therapeutic Efficacy of Antitumor CD8+ T Cells. Cancer Res. 2022;82:2640-55
82. Chen L, Duan Y, Wei H, Ning H, Bi C, Zhao Y. et al. Acetyl-CoA carboxylase (ACC) as a therapeutic target for metabolic syndrome and recent developments in ACC1/2 inhibitors. Expert Opin Investig Drugs. 2019;28:917-30
83. Menendez JA, Lupu R. Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nat Rev Cancer. 2007;7:763-77
84. Fhu CW, Ali A. Fatty Acid Synthase: An Emerging Target in Cancer. Molecules. 2020 25
85. Zhang Q, Yu S, Lam MMT, Poon TCW, Sun L, Jiao Y. et al. Angiotensin II promotes ovarian cancer spheroid formation and metastasis by upregulation of lipid desaturation and suppression of endoplasmic reticulum stress. J Exp Clin Cancer Res. 2019;38:116
86. Xuan Y, Wang H, Yung MM, Chen F, Chan WS, Chan YS. et al. SCD1/FADS2 fatty acid desaturases equipoise lipid metabolic activity and redox-driven ferroptosis in ascites-derived ovarian cancer cells. Theranostics. 2022;12:3534-52
87. Mao W, Cai Y, Chen D, Jiang G, Xu Y, Chen R. et al. Statin shapes inflamed tumor microenvironment and enhances immune checkpoint blockade in non-small cell lung cancer. JCI Insight. 2022 7
88. Huang B, Song BL, Xu C. Cholesterol metabolism in cancer: mechanisms and therapeutic opportunities. Nat Metab. 2020;2:132-41
89. Kong Y, Cheng L, Mao F, Zhang Z, Zhang Y, Farah E. et al. Inhibition of cholesterol biosynthesis overcomes enzalutamide resistance in castration-resistant prostate cancer (CRPC). J Biol Chem. 2018;293:14328-41
90. Snaebjornsson MT, Janaki-Raman S, Schulze A. Greasing the Wheels of the Cancer Machine: The Role of Lipid Metabolism in Cancer. Cell Metab. 2020;31:62-76
91. Kuk ACY, Hao A, Lee SY. Structure and Mechanism of the Lipid Flippase MurJ. Annu Rev Biochem. 2022;91:705-29
92. Waller DD, Park J, Tsantrizos YS. Inhibition of farnesyl pyrophosphate (FPP) and/or geranylgeranyl pyrophosphate (GGPP) biosynthesis and its implication in the treatment of cancers. Crit Rev Biochem Mol Biol. 2019;54:41-60
93. Bersuker K, Hendricks JM, Li Z, Magtanong L, Ford B, Tang PH. et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature. 2019;575:688-92
94. Fradejas-Villar N, Zhao W, Reuter U, Doengi M, Ingold I, Bohleber S. et al. Missense mutation in selenocysteine synthase causes cardio-respiratory failure and perinatal death in mice which can be compensated by selenium-independent GPX4. Redox Biol. 2021;48:102188
95. Cheng C, Geng F, Cheng X, Guo D. Lipid metabolism reprogramming and its potential targets in cancer. Cancer Commun (Lond). 2018;38:27
96. Jin C, Yuan P. Implications of lipid droplets in lung cancer: Associations with drug resistance. Oncol Lett. 2020;20:2091-104
97. Xu D, Wang Z, Xia Y, Shao F, Xia W, Wei Y. et al. The gluconeogenic enzyme PCK1 phosphorylates INSIG1/2 for lipogenesis. Nature. 2020;580:530-5
98. Martin S. Caveolae, lipid droplets, and adipose tissue biology: pathophysiological aspects. Horm Mol Biol Clin Investig. 2013;15:11-8
99. Walther TC, Chung J, Farese RV Jr. Lipid Droplet Biogenesis. Annu Rev Cell Dev Biol. 2017;33:491-510
100. Trempolec N, Degavre C, Doix B, Brusa D, Corbet C, Feron O. Acidosis-Induced TGF-β2 Production Promotes Lipid Droplet Formation in Dendritic Cells and Alters Their Potential to Support Anti-Mesothelioma T Cell Response. Cancers (Basel). 2020 12
101. Antunes P, Cruz A, Barbosa J, Bonifácio VDB, Pinto SN. Lipid Droplets in Cancer: From Composition and Role to Imaging and Therapeutics. Molecules. 2022 27
102. Carracedo A, Cantley LC, Pandolfi PP. Cancer metabolism: fatty acid oxidation in the limelight. Nat Rev Cancer. 2013;13:227-32
103. Padanad MS, Konstantinidou G, Venkateswaran N, Melegari M, Rindhe S, Mitsche M. et al. Fatty Acid Oxidation Mediated by Acyl-CoA Synthetase Long Chain 3 Is Required for Mutant KRAS Lung Tumorigenesis. Cell Rep. 2016;16:1614-28
104. Zaugg K, Yao Y, Reilly PT, Kannan K, Kiarash R, Mason J. et al. Carnitine palmitoyltransferase 1C promotes cell survival and tumor growth under conditions of metabolic stress. Genes Dev. 2011;25:1041-51
105. Liao P, Wang W, Wang W, Kryczek I, Li X, Bian Y. et al. CD8(+) T cells and fatty acids orchestrate tumor ferroptosis and immunity via ACSL4. Cancer Cell. 2022;40:365-78.e6
106. Abdalkareem Jasim S, Kzar HH, Haider Hamad M, Ahmad I, Al-Gazally ME, Ziyadullaev S. et al. The emerging role of 27-hydroxycholesterol in cancer development and progression: An update. Int Immunopharmacol. 2022;110:109074
107. Vini R, Rajavelu A, Sreeharshan S. 27-Hydroxycholesterol, The Estrogen Receptor Modulator, Alters DNA Methylation in Breast Cancer. Front Endocrinol (Lausanne). 2022;13:783823
108. Liu W, Chakraborty B, Safi R, Kazmin D, Chang CY, McDonnell DP. Dysregulated cholesterol homeostasis results in resistance to ferroptosis increasing tumorigenicity and metastasis in cancer. Nat Commun. 2021;12:5103
109. Dmitrieva-Posocco O, Wong AC, Lundgren P, Golos AM, Descamps HC, Dohnalová L. et al. β-Hydroxybutyrate suppresses colorectal cancer. Nature. 2022;605:160-5
110. Bian X, Liu R, Meng Y, Xing D, Xu D, Lu Z. Lipid metabolism and cancer. J Exp Med. 2021 218
111. Sun L, Zhang H, Gao P. Metabolic reprogramming and epigenetic modifications on the path to cancer. Protein Cell. 2022;13:877-919
112. Hsu CC, Peng D, Cai Z, Lin HK. AMPK signaling and its targeting in cancer progression and treatment. Semin Cancer Biol. 2022;85:52-68
113. Saikia R, Joseph J. AMPK: a key regulator of energy stress and calcium-induced autophagy. J Mol Med (Berl). 2021;99:1539-51
114. Pokhrel RH, Acharya S, Ahn JH, Gu Y, Pandit M, Kim JO. et al. AMPK promotes antitumor immunity by downregulating PD-1 in regulatory T cells via the HMGCR/p38 signaling pathway. Mol Cancer. 2021;20:133
115. Wu L, Xie W, Li Y, Ni Q, Timashev P, Lyu M. et al. Biomimetic Nanocarriers Guide Extracellular ATP Homeostasis to Remodel Energy Metabolism for Activating Innate and Adaptive Immunity System. Adv Sci (Weinh). 2022;9:e2105376
116. Trefts E, Shaw RJ. AMPK: restoring metabolic homeostasis over space and time. Mol Cell. 2021;81:3677-90
117. Revathidevi S, Munirajan AK. Akt in cancer: Mediator and more. Semin Cancer Biol. 2019;59:80-91
118. Peterson TR, Sengupta SS, Harris TE, Carmack AE, Kang SA, Balderas E. et al. mTOR complex 1 regulates lipin 1 localization to control the SREBP pathway. Cell. 2011;146:408-20
119. Liu Y, Wang Y, Liu J, Kang R, Tang D. Interplay between MTOR and GPX4 signaling modulates autophagy-dependent ferroptotic cancer cell death. Cancer Gene Ther. 2021;28:55-63
120. Summer R, Shaghaghi H, Schriner D, Roque W, Sales D, Cuevas-Mora K. et al. Activation of the mTORC1/PGC-1 axis promotes mitochondrial biogenesis and induces cellular senescence in the lung epithelium. Am J Physiol Lung Cell Mol Physiol. 2019;316:L1049-l60
121. Mossmann D, Park S, Hall MN. mTOR signalling and cellular metabolism are mutual determinants in cancer. Nat Rev Cancer. 2018;18:744-57
122. Porcuna J, Mínguez-Martínez J, Ricote M. The PPARα and PPARγ Epigenetic Landscape in Cancer and Immune and Metabolic Disorders. Int J Mol Sci. 2021 22
123. Mirza AZ, Althagafi, II, Shamshad H. Role of PPAR receptor in different diseases and their ligands: Physiological importance and clinical implications. Eur J Med Chem. 2019;166:502-13
124. Zhou T, Yan X, Wang G, Liu H, Gan X, Zhang T. et al. Evolutionary Pattern and Regulation Analysis to Support Why Diversity Functions Existed within PPAR Gene Family Members. Biomed Res Int. 2015;2015:613910
125. Yoon JS, Lee HJ, Sim DY, Im E, Park JE, Park WY. et al. Moracin D induces apoptosis in prostate cancer cells via activation of PPAR gamma/PKC delta and inhibition of PKC alpha. Phytother Res. 2021;35:6944-53
126. Song S, Attia RR, Connaughton S, Niesen MI, Ness GC, Elam MB. et al. Peroxisome proliferator activated receptor alpha (PPARalpha) and PPAR gamma coactivator (PGC-1alpha) induce carnitine palmitoyltransferase IA (CPT-1A) via independent gene elements. Mol Cell Endocrinol. 2010;325:54-63
127. Shimano H, Sato R. SREBP-regulated lipid metabolism: convergent physiology - divergent pathophysiology. Nat Rev Endocrinol. 2017;13:710-30
128. Wei M, Nurjanah U, Herkilini A, Huang C, Li Y, Miyagishi M. et al. Unspliced XBP1 contributes to cholesterol biosynthesis and tumorigenesis by stabilizing SREBP2 in hepatocellular carcinoma. Cell Mol Life Sci. 2022;79:472
129. Ricoult SJ, Yecies JL, Ben-Sahra I, Manning BD. Oncogenic PI3K and K-Ras stimulate de novo lipid synthesis through mTORC1 and SREBP. Oncogene. 2016;35:1250-60
130. Moon SH, Huang CH, Houlihan SL, Regunath K, Freed-Pastor WA, Morris JPt. et al. p53 Represses the Mevalonate Pathway to Mediate Tumor Suppression. Cell. 2019;176:564-80.e19
131. Cheng C, Ru P, Geng F, Liu J, Yoo JY, Wu X. et al. Glucose-Mediated N-glycosylation of SCAP Is Essential for SREBP-1 Activation and Tumor Growth. Cancer Cell. 2015;28:569-81
132. Shechter I, Dai P, Huo L, Guan G. IDH1 gene transcription is sterol regulated and activated by SREBP-1a and SREBP-2 in human hepatoma HepG2 cells: evidence that IDH1 may regulate lipogenesis in hepatic cells. J Lipid Res. 2003;44:2169-80
133. Guo C, Chi Z, Jiang D, Xu T, Yu W, Wang Z. et al. Cholesterol Homeostatic Regulator SCAP-SREBP2 Integrates NLRP3 Inflammasome Activation and Cholesterol Biosynthetic Signaling in Macrophages. Immunity. 2018;49:842-56.e7
134. Hampton RY. A cholesterol toggle switch. Cell Metab. 2008;8:451-3
135. Zelcer N, Hong C, Boyadjian R, Tontonoz P. LXR regulates cholesterol uptake through Idol-dependent ubiquitination of the LDL receptor. Science. 2009;325:100-4
136. Guo D, Reinitz F, Youssef M, Hong C, Nathanson D, Akhavan D. et al. An LXR agonist promotes glioblastoma cell death through inhibition of an EGFR/AKT/SREBP-1/LDLR-dependent pathway. Cancer Discov. 2011;1:442-56
137. Feng K, Ma C, Liu Y, Yang X, Yang Z, Chen Y. et al. Encapsulation of LXR ligand by D-Nap-GFFY hydrogel enhances anti-tumorigenic actions of LXR and removes LXR-induced lipogenesis. Theranostics. 2021;11:2634-54
138. Peña-Romero AC, Orenes-Piñero E. Dual Effect of Immune Cells within Tumour Microenvironment: Pro- and Anti-Tumour Effects and Their Triggers. Cancers (Basel). 2022 14
139. Lian X, Yang K, Li R, Li M, Zuo J, Zheng B. et al. Immunometabolic rewiring in tumorigenesis and anti-tumor immunotherapy. Mol Cancer. 2022;21:27
140. Biswas SK. Metabolic Reprogramming of Immune Cells in Cancer Progression. Immunity. 2015;43:435-49
141. Fan C, Zhang S, Gong Z, Li X, Xiang B, Deng H. et al. Emerging role of metabolic reprogramming in tumor immune evasion and immunotherapy. Sci China Life Sci. 2021;64:534-47
142. Lin R, Zhang H, Yuan Y, He Q, Zhou J, Li S. et al. Fatty Acid Oxidation Controls CD8(+) Tissue-Resident Memory T-cell Survival in Gastric Adenocarcinoma. Cancer Immunol Res. 2020;8:479-92
143. Kishton RJ, Sukumar M, Restifo NP. Metabolic Regulation of T Cell Longevity and Function in Tumor Immunotherapy. Cell Metab. 2017;26:94-109
144. Kalia V, Yuzefpolskiy Y, Vegaraju A, Xiao H, Baumann F, Jatav S. et al. Metabolic regulation by PD-1 signaling promotes long-lived quiescent CD8 T cell memory in mice. Sci Transl Med. 2021;13:eaba6006
145. Engelhard V, Conejo-Garcia JR, Ahmed R, Nelson BH, Willard-Gallo K, Bruno TC. et al. B cells and cancer. Cancer Cell. 2021;39:1293-6
146. Waters LR, Ahsan FM, Wolf DM, Shirihai O, Teitell MA. Initial B Cell Activation Induces Metabolic Reprogramming and Mitochondrial Remodeling. iScience. 2018;5:99-109
147. Yang F, Wan F. Lipid Metabolism in Tumor-Associated B Cells. Adv Exp Med Biol. 2021;1316:133-47
148. Terabe M, Berzofsky JA. Tissue-Specific Roles of NKT Cells in Tumor Immunity. Front Immunol. 2018;9:1838
149. Chen Y, Sui M. Lipid Metabolism in Tumor-Associated Natural Killer Cells. Adv Exp Med Biol. 2021;1316:71-85
150. Chen D, Zhang X, Li Z, Zhu B. Metabolic regulatory crosstalk between tumor microenvironment and tumor-associated macrophages. Theranostics. 2021;11:1016-30
151. Kelly B, O'Neill LA. Metabolic reprogramming in macrophages and dendritic cells in innate immunity. Cell Res. 2015;25:771-84
152. Peng X, He Y, Huang J, Tao Y, Liu S. Metabolism of Dendritic Cells in Tumor Microenvironment: For Immunotherapy. Front Immunol. 2021;12:613492
153. Giese MA, Hind LE, Huttenlocher A. Neutrophil plasticity in the tumor microenvironment. Blood. 2019;133:2159-67
154. Bodac A, Meylan E. Neutrophil metabolism in the cancer context. Semin Immunol. 2021;57:101583
155. Tian M, Hao F, Jin X, Sun X, Jiang Y, Wang Y. et al. ACLY ubiquitination by CUL3-KLHL25 induces the reprogramming of fatty acid metabolism to facilitate iTreg differentiation. Elife. 2021 10
156. Lim SA, Wei J, Nguyen TM, Shi H, Su W, Palacios G. et al. Lipid signalling enforces functional specialization of T(reg) cells in tumours. Nature. 2021;591:306-11
157. Rosser EC, Mauri C. Regulatory B cells: origin, phenotype, and function. Immunity. 2015;42:607-12
158. Rosser EC, Mauri C. The emerging field of regulatory B cell immunometabolism. Cell Metab. 2021;33:1088-97
159. Michaud D, Steward CR, Mirlekar B, Pylayeva-Gupta Y. Regulatory B cells in cancer. Immunol Rev. 2021;299:74-92
160. Noe JT, Rendon BE, Geller AE, Conroy LR, Morrissey SM, Young LEA. et al. Lactate supports a metabolic-epigenetic link in macrophage polarization. Sci Adv. 2021;7:eabi8602
161. Bleve A, Durante B, Sica A, Consonni FM. Lipid Metabolism and Cancer Immunotherapy: Immunosuppressive Myeloid Cells at the Crossroad. Int J Mol Sci. 2020 21
162. Zhu L, Zhu X, Wu Y. Effects of Glucose Metabolism, Lipid Metabolism, and Glutamine Metabolism on Tumor Microenvironment and Clinical Implications. Biomolecules. 2022 12
163. Liu W, Song H, Li X, Ren D, Ding S, Li Y. Lipid Metabolism in Tumor-Associated Myeloid-Derived Suppressor Cells. Adv Exp Med Biol. 2021;1316:103-15
164. Xue R, Zhang Q, Cao Q, Kong R, Xiang X, Liu H. et al. Liver tumour immune microenvironment subtypes and neutrophil heterogeneity. Nature. 2022;612:141-7
165. Bader JE, Voss K, Rathmell JC. Targeting Metabolism to Improve the Tumor Microenvironment for Cancer Immunotherapy. Mol Cell. 2020;78:1019-33
166. Nagarsheth N, Wicha MS, Zou W. Chemokines in the cancer microenvironment and their relevance in cancer immunotherapy. Nat Rev Immunol. 2017;17:559-72
167. Bursill CA, Castro ML, Beattie DT, Nakhla S, van der Vorst E, Heather AK. et al. High-density lipoproteins suppress chemokines and chemokine receptors in vitro and in vivo. Arterioscler Thromb Vasc Biol. 2010;30:1773-8
168. Barlic J, Murphy PM. Chemokine regulation of atherosclerosis. J Leukoc Biol. 2007;82:226-36
169. Dey P, Kimmelman AC, DePinho RA. Metabolic Codependencies in the Tumor Microenvironment. Cancer Discov. 2021;11:1067-81
170. Park D, Sahai E, Rullan A. SnapShot: Cancer-Associated Fibroblasts. Cell. 2020;181:486-e1
171. Li Z, Sun C, Qin Z. Metabolic reprogramming of cancer-associated fibroblasts and its effect on cancer cell reprogramming. Theranostics. 2021;11:8322-36
172. Saw PE, Chen J, Song E. Targeting CAFs to overcome anticancer therapeutic resistance. Trends Cancer. 2022;8:527-55
173. Riera-Domingo C, Audigé A, Granja S, Cheng WC, Ho PC, Baltazar F. et al. Immunity, Hypoxia, and Metabolism-the Ménage à Trois of Cancer: Implications for Immunotherapy. Physiol Rev. 2020;100:1-102
174. Yuen VW, Wong CC. Hypoxia-inducible factors and innate immunity in liver cancer. J Clin Invest. 2020;130:5052-62
175. Lee HJ, Ryu JM, Jung YH, Oh SY, Lee SJ, Han HJ. Novel Pathway for Hypoxia-Induced Proliferation and Migration in Human Mesenchymal Stem Cells: Involvement of HIF-1α, FASN, and mTORC1. Stem Cells. 2015;33:2182-95
176. Wicks EE, Semenza GL. Hypoxia-inducible factors: cancer progression and clinical translation. J Clin Invest. 2022 132
177. Menard JA, Christianson HC, Kucharzewska P, Bourseau-Guilmain E, Svensson KJ, Lindqvist E. et al. Metastasis Stimulation by Hypoxia and Acidosis-Induced Extracellular Lipid Uptake Is Mediated by Proteoglycan-Dependent Endocytosis. Cancer Res. 2016;76:4828-40
178. Duan C, Kuang L, Hong C, Xiang X, Liu J, Li Q. et al. Mitochondrial Drp1 recognizes and induces excessive mPTP opening after hypoxia through BAX-PiC and LRRK2-HK2. Cell Death Dis. 2021;12:1050
179. Angelin A, Gil-de-Gómez L, Dahiya S, Jiao J, Guo L, Levine MH. et al. Foxp3 Reprograms T Cell Metabolism to Function in Low-Glucose, High-Lactate Environments. Cell Metab. 2017;25:1282-93.e7
180. Hsu TS, Lin YL, Wang YA, Mo ST, Chi PY, Lai AC. et al. HIF-2α is indispensable for regulatory T cell function. Nat Commun. 2020;11:5005
181. Hay N. Reprogramming glucose metabolism in cancer: can it be exploited for cancer therapy? Nat Rev Cancer. 2016;16:635-49
182. Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324:1029-33
183. Terrén I, Orrantia A, Vitallé J, Zenarruzabeitia O, Borrego F. NK Cell Metabolism and Tumor Microenvironment. Front Immunol. 2019;10:2278
184. Wang JX, Choi SYC, Niu X, Kang N, Xue H, Killam J. et al. Lactic Acid and an Acidic Tumor Microenvironment suppress Anticancer Immunity. Int J Mol Sci. 2020 21
185. Hirschhaeuser F, Sattler UG, Mueller-Klieser W. Lactate: a metabolic key player in cancer. Cancer Res. 2011;71:6921-5
186. Feng J, Yang H, Zhang Y, Wei H, Zhu Z, Zhu B. et al. Tumor cell-derived lactate induces TAZ-dependent upregulation of PD-L1 through GPR81 in human lung cancer cells. Oncogene. 2017;36:5829-39
187. Kumagai S, Koyama S, Itahashi K, Tanegashima T, Lin YT, Togashi Y. et al. Lactic acid promotes PD-1 expression in regulatory T cells in highly glycolytic tumor microenvironments. Cancer Cell. 2022;40:201-18.e9
188. Brand A, Singer K, Koehl GE, Kolitzus M, Schoenhammer G, Thiel A. et al. LDHA-Associated Lactic Acid Production Blunts Tumor Immunosurveillance by T and NK Cells. Cell Metab. 2016;24:657-71
189. Feng Q, Liu Z, Yu X, Huang T, Chen J, Wang J. et al. Lactate increases stemness of CD8 + T cells to augment anti-tumor immunity. Nat Commun. 2022;13:4981
190. Gillies RJ, Pilot C, Marunaka Y, Fais S. Targeting acidity in cancer and diabetes. Biochim Biophys Acta, Rev Cancer. 2019;1871:273-80
191. Queen A, Bhutto HN, Yousuf M, Syed MA, Hassan MI. Carbonic anhydrase IX: A tumor acidification switch in heterogeneity and chemokine regulation. Semin Cancer Biol. 2022;86:899-913
192. Webb BA, Chimenti M, Jacobson MP, Barber DL. Dysregulated pH: a perfect storm for cancer progression. Nat Rev Cancer. 2011;11:671-7
193. Pillai S, Mahmud I, Mahar R, Griffith C, Langsen M, Nguyen J. et al. Lipogenesis mediated by OGR1 regulates metabolic adaptation to acid stress in cancer cells via autophagy. Cell Rep. 2022;39:110796
194. Panaroni C, Fulzele K, Mori T, Siu KT, Onyewadume C, Maebius A. et al. Multiple myeloma cells induce lipolysis in adipocytes and uptake fatty acids through fatty acid transporter proteins. Blood. 2022;139:876-88
195. Raman P, Dewitt DL, Nair MG. Lipid peroxidation and cyclooxygenase enzyme inhibitory activities of acidic aqueous extracts of some dietary supplements. Phytother Res. 2008;22:204-12
196. Yang L, Sun J, Li M, Long Y, Zhang D, Guo H. et al. Oxidized Low-Density Lipoprotein Links Hypercholesterolemia and Bladder Cancer Aggressiveness by Promoting Cancer Stemness. Cancer Res. 2021;81:5720-32
197. Xie Y, Wang B, Zhao Y, Tao Z, Wang Y, Chen G. et al. Mammary adipocytes protect triple-negative breast cancer cells from ferroptosis. J Hematol Oncol. 2022;15:72
198. Aspichueta P. Lipid-rich environment: a key role promoting carcinogenesis in obesity-related non-alcoholic fatty liver disease. Gut. 2018;67:1376-7
199. Wu D, Hu L, Han M, Deng Y, Zhang Y, Ren G. et al. PD-1 signaling facilitates activation of lymphoid tissue inducer cells by restraining fatty acid oxidation. Nat Metab. 2022;4:867-82
200. Ma X, Bi E, Lu Y, Su P, Huang C, Liu L. et al. Cholesterol Induces CD8(+) T Cell Exhaustion in the Tumor Microenvironment. Cell Metab. 2019;30:143-56.e5
201. Zhao Y, Wang H, Yang Y, Jia W, Su T, Che Y. et al. Mannose-Modified Liposome Co-Delivery of Human Papillomavirus Type 16 E7 Peptide and CpG Oligodeoxynucleotide Adjuvant Enhances Antitumor Activity Against Established Large TC-1 Grafted Tumors in Mice. Int J Nanomedicine. 2020;15:9571-86
202. White E, Mehnert JM, Chan CS. Autophagy, Metabolism, and Cancer. Clin Cancer Res. 2015;21:5037-46
203. Li X, He S, Ma B. Autophagy and autophagy-related proteins in cancer. Mol Cancer. 2020;19:12
204. Devis-Jauregui L, Eritja N, Davis ML, Matias-Guiu X, Llobet-Navàs D. Autophagy in the physiological endometrium and cancer. Autophagy. 2021;17:1077-95
205. Liu J, Kuang F, Kroemer G, Klionsky DJ, Kang R, Tang D. Autophagy-Dependent Ferroptosis: Machinery and Regulation. Cell Chem Biol. 2020;27:420-35
206. Valečka J, Almeida CR, Su B, Pierre P, Gatti E. Autophagy and MHC-restricted antigen presentation. Mol Immunol. 2018;99:163-70
207. Xia H, Green DR, Zou W. Autophagy in tumour immunity and therapy. Nat Rev Cancer. 2021;21:281-97
208. Arora SP, Tenner L, Sarantopoulos J, Morris J, Liu Q, Mendez JA. et al. Modulation of autophagy: a Phase II study of vorinostat plus hydroxychloroquine versus regorafenib in chemotherapy-refractory metastatic colorectal cancer (mCRC). Br J Cancer. 2022;127:1153-61
209. Martín-Sierra C, Laranjeira P, Domingues MR, Paiva A. Lipoxidation and cancer immunity. Redox Biol. 2019;23:101103
210. Kroemer G, Galassi C, Zitvogel L, Galluzzi L. Immunogenic cell stress and death. Nat Immunol. 2022;23:487-500
211. Gago-Dominguez M, Jiang X, Castelao JE. Lipid peroxidation, oxidative stress genes and dietary factors in breast cancer protection: a hypothesis. Breast Cancer Res. 2007;9:201
212. Zhao Y, Hu X, Liu Y, Dong S, Wen Z, He W. et al. ROS signaling under metabolic stress: cross-talk between AMPK and AKT pathway. Mol Cancer. 2017;16:79
213. Clemente SM, Martínez-Costa OH, Monsalve M, Samhan-Arias AK. Targeting Lipid Peroxidation for Cancer Treatment. Molecules. 2020 25
214. Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE. et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149:1060-72
215. Sun LL, Linghu DL, Hung MC. Ferroptosis: a promising target for cancer immunotherapy. Am J Cancer Res. 2021;11:5856-63
216. Jiang Z, Lim SO, Yan M, Hsu JL, Yao J, Wei Y. et al. TYRO3 induces anti-PD-1/PD-L1 therapy resistance by limiting innate immunity and tumoral ferroptosis. J Clin Invest. 2021 131
217. Ishibashi M, Varin A, Filomenko R, Lopez T, Athias A, Gambert P. et al. Liver x receptor regulates arachidonic acid distribution and eicosanoid release in human macrophages: a key role for lysophosphatidylcholine acyltransferase 3. Arterioscler Thromb Vasc Biol. 2013;33:1171-9
218. Kagan VE, Mao G, Qu F, Angeli JP, Doll S, Croix CS. et al. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat Chem Biol. 2017;13:81-90
219. Shureiqi I, Wojno KJ, Poore JA, Reddy RG, Moussalli MJ, Spindler SA. et al. Decreased 13-S-hydroxyoctadecadienoic acid levels and 15-lipoxygenase-1 expression in human colon cancers. Carcinogenesis. 1999;20:1985-95
220. Zou Y, Henry WS, Ricq EL, Graham ET, Phadnis VV, Maretich P. et al. Plasticity of ether lipids promotes ferroptosis susceptibility and evasion. Nature. 2020;585:603-8
221. Ursini F, Maiorino M, Valente M, Ferri L, Gregolin C. Purification from pig liver of a protein which protects liposomes and biomembranes from peroxidative degradation and exhibits glutathione peroxidase activity on phosphatidylcholine hydroperoxides. Biochim Biophys Acta. 1982;710:197-211
222. Lang X, Green MD, Wang W, Yu J, Choi JE, Jiang L. et al. Radiotherapy and Immunotherapy Promote Tumoral Lipid Oxidation and Ferroptosis via Synergistic Repression of SLC7A11. Cancer Discov. 2019;9:1673-85
223. Yang F, Xiao Y, Ding JH, Jin X, Ma D, Li DQ. et al. Ferroptosis heterogeneity in triple-negative breast cancer reveals an innovative immunotherapy combination strategy. Cell Metab. 2023;35:84-100.e8
224. Ma X, Xiao L, Liu L, Ye L, Su P, Bi E. et al. CD36-mediated ferroptosis dampens intratumoral CD8(+) T cell effector function and impairs their antitumor ability. Cell Metab. 2021;33:1001-12.e5
225. Xu C, Sun S, Johnson T, Qi R, Zhang S, Zhang J. et al. The glutathione peroxidase Gpx4 prevents lipid peroxidation and ferroptosis to sustain Treg cell activation and suppression of antitumor immunity. Cell Rep. 2021;35:109235
226. Kapralov AA, Yang Q, Dar HH, Tyurina YY, Anthonymuthu TS, Kim R. et al. Redox lipid reprogramming commands susceptibility of macrophages and microglia to ferroptotic death. Nat Chem Biol. 2020;16:278-90
227. Chen L, Min J, Wang F. Copper homeostasis and cuproptosis in health and disease. Signal Transduct Target Ther. 2022;7:378
228. Tsvetkov P, Coy S, Petrova B, Dreishpoon M, Verma A, Abdusamad M. et al. Copper induces cell death by targeting lipoylated TCA cycle proteins. Science. 2022;375:1254-61
229. Cobine PA, Brady DC. Cuproptosis: Cellular and molecular mechanisms underlying copper-induced cell death. Mol Cell. 2022;82:1786-7
230. Song Q, Zhou R, Shu F, Fu W. Cuproptosis scoring system to predict the clinical outcome and immune response in bladder cancer. Front Immunol. 2022;13:958368
231. Lin Y, Jiang M, Chen W, Zhao T, Wei Y. Cancer and ER stress: Mutual crosstalk between autophagy, oxidative stress and inflammatory response. Biomed Pharmacother. 2019;118:109249
232. Catacuzzeno L, Franciolini F. Role of KCa3.1 Channels in Modulating Ca(2+) Oscillations during Glioblastoma Cell Migration and Invasion. Int J Mol Sci. 2018; 19
233. Choi S, Cui C, Luo Y, Kim SH, Ko JK, Huo X. et al. Selective inhibitory effects of zinc on cell proliferation in esophageal squamous cell carcinoma through Orai1. FASEB J. 2018;32:404-16
234. Yao J, Peng H, Qiu Y, Li S, Xu X, Wu A. et al. Nanoplatform-mediated calcium overload for cancer therapy. J Mater Chem B. 2022;10:1508-19
235. Bai S, Lan Y, Fu S, Cheng H, Lu Z, Liu G. Connecting Calcium-Based Nanomaterials and Cancer: From Diagnosis to Therapy. Nanomicro Lett. 2022;14:145
236. Ogino S, Nowak JA, Hamada T, Phipps AI, Peters U, Milner DA Jr. et al. Integrative analysis of exogenous, endogenous, tumour and immune factors for precision medicine. Gut. 2018;67:1168-80
237. Stine ZE, Schug ZT, Salvino JM, Dang CV. Targeting cancer metabolism in the era of precision oncology. Nat Rev Drug Discov. 2022;21:141-62
238. Pascual G, Avgustinova A, Mejetta S, Martín M, Castellanos A, Attolini CS. et al. Targeting metastasis-initiating cells through the fatty acid receptor CD36. Nature. 2017;541:41-5
239. Watt MJ, Clark AK, Selth LA, Haynes VR, Lister N, Rebello R. et al. Suppressing fatty acid uptake has therapeutic effects in preclinical models of prostate cancer. Sci Transl Med. 2019 11
240. Zhang M, Di Martino JS, Bowman RL, Campbell NR, Baksh SC, Simon-Vermot T. et al. Adipocyte-Derived Lipids Mediate Melanoma Progression via FATP Proteins. Cancer Discov. 2018;8:1006-25
241. Thompson KJ, Austin RG, Nazari SS, Gersin KS, Iannitti DA, McKillop IH. Altered fatty acid-binding protein 4 (FABP4) expression and function in human and animal models of hepatocellular carcinoma. Liver Int. 2018;38:1074-83
242. Veglia F, Tyurin VA, Blasi M, De Leo A, Kossenkov AV, Donthireddy L. et al. Fatty acid transport protein 2 reprograms neutrophils in cancer. Nature. 2019;569:73-8
243. Granchi C. ATP citrate lyase (ACLY) inhibitors: An anti-cancer strategy at the crossroads of glucose and lipid metabolism. Eur J Med Chem. 2018;157:1276-91
244. Osinalde N, Mitxelena J, Sánchez-Quiles V, Akimov V, Aloria K, Arizmendi JM. et al. Nuclear Phosphoproteomic Screen Uncovers ACLY as Mediator of IL-2-induced Proliferation of CD4+ T lymphocytes. Mol Cell Proteomics. 2016;15:2076-92
245. Dominguez M, Brüne B, Namgaladze D. Exploring the Role of ATP-Citrate Lyase in the Immune System. Front Immunol. 2021;12:632526
246. Verschueren KHG, Blanchet C, Felix J, Dansercoer A, De Vos D, Bloch Y. et al. Structure of ATP citrate lyase and the origin of citrate synthase in the Krebs cycle. Nature. 2019;568:571-5
247. Li CJ, Chiu YH, Chang C, Chang YI, Sheu JJ, Chiang AJ. Acetyl Coenzyme A Synthase 2 Acts as a Prognostic Biomarker Associated with Immune Infiltration in Cervical Squamous Cell Carcinoma. Cancers (Basel). 2021 13
248. Hardie DG, Pan DA. Regulation of fatty acid synthesis and oxidation by the AMP-activated protein kinase. Biochem Soc Trans. 2002;30:1064-70
249. Calle RA, Amin NB, Carvajal-Gonzalez S, Ross TT, Bergman A, Aggarwal S. et al. ACC inhibitor alone or co-administered with a DGAT2 inhibitor in patients with non-alcoholic fatty liver disease: two parallel, placebo-controlled, randomized phase 2a trials. Nat Med. 2021;27:1836-48
250. Goodwin PJ, Chen BE, Gelmon KA, Whelan TJ, Ennis M, Lemieux J. et al. Effect of Metformin vs Placebo on Invasive Disease-Free Survival in Patients With Breast Cancer: The MA.32 Randomized Clinical Trial. JAMA. 2022;327:1963-73
251. Cao D, Yang J, Deng Y, Su M, Wang Y, Feng X. et al. Discovery of a mammalian FASN inhibitor against xenografts of non-small cell lung cancer and melanoma. Signal Transduct Target Ther. 2022;7:273
252. Sardesai SD, Thomas A, Gallagher C, Lynce F, Ottaviano YL, Ballinger TJ. et al. Inhibiting Fatty Acid Synthase with Omeprazole to Improve Efficacy of Neoadjuvant Chemotherapy in Patients with Operable TNBC. Clin Cancer Res. 2021;27:5810-7
253. Manni A, Richie JP, Schetter SE, Calcagnotto A, Trushin N, Aliaga C. et al. Stearoyl-CoA desaturase-1, a novel target of omega-3 fatty acids for reducing breast cancer risk in obese postmenopausal women. Eur J Clin Nutr. 2017;71:762-5
254. Rifkin SB, Shrubsole MJ, Cai Q, Smalley WE, Ness RM, Swift LL. et al. Differences in erythrocyte phospholipid membrane long-chain polyunsaturated fatty acids and the prevalence of fatty acid desaturase genotype among African Americans and European Americans. Prostaglandins Leukot Essent Fatty Acids. 2021;164:102216
255. Das SK, Eder S, Schauer S, Diwoky C, Temmel H, Guertl B. et al. Adipose triglyceride lipase contributes to cancer-associated cachexia. Science. 2011;333:233-8
256. Gil-Ordóñez A, Martín-Fontecha M, Ortega-Gutiérrez S, López-Rodríguez ML. Monoacylglycerol lipase (MAGL) as a promising therapeutic target. Biochem Pharmacol. 2018;157:18-32
257. Camarda R, Zhou AY, Kohnz RA, Balakrishnan S, Mahieu C, Anderton B. et al. Inhibition of fatty acid oxidation as a therapy for MYC-overexpressing triple-negative breast cancer. Nat Med. 2016;22:427-32
258. Hossain F, Al-Khami AA, Wyczechowska D, Hernandez C, Zheng L, Reiss K. et al. Inhibition of Fatty Acid Oxidation Modulates Immunosuppressive Functions of Myeloid-Derived Suppressor Cells and Enhances Cancer Therapies. Cancer Immunol Res. 2015;3:1236-47
259. Ko CJ, Lan SW, Lu YC, Cheng TS, Lai PF, Tsai CH. et al. Inhibition of cyclooxygenase-2-mediated matriptase activation contributes to the suppression of prostate cancer cell motility and metastasis. Oncogene. 2017;36:4597-609
260. Musa A, Mostafa EM, Bukhari SNA, Alotaibi NH, El-Ghorab AH, Farouk A. et al. EGFR and COX-2 Dual Inhibitor: The Design, Synthesis, and Biological Evaluation of Novel Chalcones. Molecules. 2022 27
261. Hennequart M, Pilotte L, Cane S, Hoffmann D, Stroobant V, Plaen E. et al. Constitutive IDO1 Expression in Human Tumors Is Driven by Cyclooxygenase-2 and Mediates Intrinsic Immune Resistance. Cancer Immunol Res. 2017;5:695-709
262. Barnard ME, Hecht JL, Rice MS, Gupta M, Harris HR, Eliassen AH. et al. Anti-Inflammatory Drug Use and Ovarian Cancer Risk by COX1/COX2 Expression and Infiltration of Tumor-Associated Macrophages. Cancer Epidemiol Biomarkers Prev. 2018;27:1509-17
263. Bjarnadottir O, Feldt M, Inasu M, Bendahl PO, Elebro K, Kimbung S. et al. Statin use, HMGCR expression, and breast cancer survival - The Malmö Diet and Cancer Study. Sci Rep. 2020;10:558
264. Gorabi AM, Kiaie N, Hajighasemi S, Banach M, Penson PE, Jamialahmadi T. et al. Statin-Induced Nitric Oxide Signaling: Mechanisms and Therapeutic Implications. J Clin Med. 2019 8
265. Bjarnadottir O, Romero Q, Bendahl PO, Jirström K, Rydén L, Loman N. et al. Targeting HMG-CoA reductase with statins in a window-of-opportunity breast cancer trial. Breast Cancer Res Treat. 2013;138:499-508
266. Kubatka P, Kruzliak P, Rotrekl V, Jelinkova S, Mladosievicova B. Statins in oncological research: from experimental studies to clinical practice. Crit Rev Oncol Hematol. 2014;92:296-311
267. Kimbung S, Lettiero B, Feldt M, Bosch A, Borgquist S. High expression of cholesterol biosynthesis genes is associated with resistance to statin treatment and inferior survival in breast cancer. Oncotarget. 2016;7:59640-51
268. Yang YF, Jan YH, Liu YP, Yang CJ, Su CY, Chang YC. et al. Squalene synthase induces tumor necrosis factor receptor 1 enrichment in lipid rafts to promote lung cancer metastasis. Am J Respir Crit Care Med. 2014;190:675-87
269. You W, Ke J, Chen Y, Cai Z, Huang ZP, Hu P. et al. SQLE, A Key Enzyme in Cholesterol Metabolism, Correlates With Tumor Immune Infiltration and Immunotherapy Outcome of Pancreatic Adenocarcinoma. Front Immunol. 2022;13:864244
270. Yang W, Bai Y, Xiong Y, Zhang J, Chen S, Zheng X. et al. Potentiating the antitumour response of CD8(+) T cells by modulating cholesterol metabolism. Nature. 2016;531:651-5
271. Cai Y, Guo H, Wang W, Li H, Sun H, Shi B. et al. Assessing the outcomes of everolimus on renal angiomyolipoma associated with tuberous sclerosis complex in China: a two years trial. Orphanet J Rare Dis. 2018;13:43
272. Guerrero-Zotano A, Mayer IA, Arteaga CL. PI3K/AKT/mTOR: role in breast cancer progression, drug resistance, and treatment. Cancer Metastasis Rev. 2016;35:515-24
273. Ediriweera MK, Tennekoon KH, Samarakoon SR. Role of the PI3K/AKT/mTOR signaling pathway in ovarian cancer: Biological and therapeutic significance. Semin Cancer Biol. 2019;59:147-60
274. Chowdhury PS, Chamoto K, Kumar A, Honjo T. PPAR-Induced Fatty Acid Oxidation in T Cells Increases the Number of Tumor-Reactive CD8(+) T Cells and Facilitates Anti-PD-1 Therapy. Cancer Immunol Res. 2018;6:1375-87
275. Yin X, Zeng W, Wu B, Wang L, Wang Z, Tian H. et al. PPARα Inhibition Overcomes Tumor-Derived Exosomal Lipid-Induced Dendritic Cell Dysfunction. Cell Rep. 2020;33:108278
276. Liu C, Chikina M, Deshpande R, Menk AV, Wang T, Tabib T. et al. Treg Cells Promote the SREBP1-Dependent Metabolic Fitness of Tumor-Promoting Macrophages via Repression of CD8(+) T Cell-Derived Interferon-γ. Immunity. 2019;51:381-97.e6
277. Wan D, Yang Y, Liu Y, Cun X, Li M, Xu S. et al. Sequential depletion of myeloid-derived suppressor cells and tumor cells with a dual-pH-sensitive conjugated micelle system for cancer chemoimmunotherapy. J Control Release. 2020;317:43-56
278. Chinetti G, Zawadski C, Fruchart JC, Staels B. Expression of adiponectin receptors in human macrophages and regulation by agonists of the nuclear receptors PPARalpha, PPARgamma, and LXR. Biochem Biophys Res Commun. 2004;314:151-8
279. Mann SD, Sidhu MD, Gowin KD. Understanding the Mechanisms of Diet and Outcomes in Colon, Prostate, and Breast Cancer; Malignant Gliomas; and Cancer Patients on Immunotherapy. Nutrients. 2020 12
280. Kirkham AA, King K, Joy AA, Pelletier AB, Mackey JR, Young K. et al. Rationale and design of the Diet Restriction and Exercise-induced Adaptations in Metastatic breast cancer (DREAM) study: a 2-arm, parallel-group, phase II, randomized control trial of a short-term, calorie-restricted, and ketogenic diet plus exercise during intravenous chemotherapy versus usual care. BMC Cancer. 2021;21:1093
281. Dowis K, Banga S. The Potential Health Benefits of the Ketogenic Diet: A Narrative Review. Nutrients. 2021 13
282. Cao Y, Dave KB, Doan TP, Prescott SM. Fatty acid CoA ligase 4 is up-regulated in colon adenocarcinoma. Cancer Res. 2001;61:8429-34
283. Liang YC, Wu CH, Chu JS, Wang CK, Hung LF, Wang YJ. et al. Involvement of fatty acid-CoA ligase 4 in hepatocellular carcinoma growth: roles of cyclic AMP and p38 mitogen-activated protein kinase. World J Gastroenterol. 2005;11:2557-63
284. Zhang Y, Li S, Li F, Lv C, Yang QK. High-fat diet impairs ferroptosis and promotes cancer invasiveness via downregulating tumor suppressor ACSL4 in lung adenocarcinoma. Biol Direct. 2021;16:10
285. Yen MC, Kan JY, Hsieh CJ, Kuo PL, Hou MF, Hsu YL. Association of long-chain acyl-coenzyme A synthetase 5 expression in human breast cancer by estrogen receptor status and its clinical significance. Oncol Rep. 2017;37:3253-60
286. Xiaofei J, Mingqing S, Miao S, Yizhen Y, Shuang Z, Qinhua X. et al. Oleanolic acid inhibits cervical cancer Hela cell proliferation through modulation of the ACSL4 ferroptosis signaling pathway. Biochem Biophys Res Commun. 2021;545:81-8
287. Castillo AF, Orlando UD, Maloberti PM, Prada JG, Dattilo MA, Solano AR. et al. New inhibitor targeting Acyl-CoA synthetase 4 reduces breast and prostate tumor growth, therapeutic resistance and steroidogenesis. Cell Mol Life Sci. 2021;78:2893-910
288. Calvisi DF, Wang C, Ho C, Ladu S, Lee SA, Mattu S. et al. Increased lipogenesis, induced by AKT-mTORC1-RPS6 signaling, promotes development of human hepatocellular carcinoma. Gastroenterology. 2011;140:1071-83
289. Park S, Oh J, Kim M, Jin EJ. Bromelain effectively suppresses Kras-mutant colorectal cancer by stimulating ferroptosis. Anim Cells Syst (Seoul). 2018;22:334-40
290. Demetri GD, Fletcher CD, Mueller E, Sarraf P, Naujoks R, Campbell N. et al. Induction of solid tumor differentiation by the peroxisome proliferator-activated receptor-gamma ligand troglitazone in patients with liposarcoma. Proc Natl Acad Sci U S A. 1999;96:3951-6
291. Wang B, Rong X, Palladino END, Wang J, Fogelman AM, Martín MG. et al. Phospholipid Remodeling and Cholesterol Availability Regulate Intestinal Stemness and Tumorigenesis. Cell Stem Cell. 2018;22:206-20.e4
292. Rong X, Albert CJ, Hong C, Duerr MA, Chamberlain BT, Tarling EJ. et al. LXRs regulate ER stress and inflammation through dynamic modulation of membrane phospholipid composition. Cell Metab. 2013;18:685-97
293. Di Conza G, Tsai CH, Gallart-Ayala H, Yu YR, Franco F, Zaffalon L. et al. Tumor-induced reshuffling of lipid composition on the endoplasmic reticulum membrane sustains macrophage survival and pro-tumorigenic activity. Nat Immunol. 2021;22:1403-15
294. Fonseca-Nunes A, Jakszyn P, Agudo A. Iron and cancer risk-a systematic review and meta-analysis of the epidemiological evidence. Cancer Epidemiol Biomarkers Prev. 2014;23:12-31
295. Gao M, Monian P, Quadri N, Ramasamy R, Jiang X. Glutaminolysis and Transferrin Regulate Ferroptosis. Mol Cell. 2015;59:298-308
296. Hou W, Xie Y, Song X, Sun X, Lotze MT, Zeh HJ 3rd. et al. Autophagy promotes ferroptosis by degradation of ferritin. Autophagy. 2016;12:1425-8
297. Mai TT, Hamaï A, Hienzsch A, Cañeque T, Müller S, Wicinski J. et al. Salinomycin kills cancer stem cells by sequestering iron in lysosomes. Nat Chem. 2017;9:1025-33
298. Nagpal A, Redvers RP, Ling X, Ayton S, Fuentes M, Tavancheh E. et al. Neoadjuvant neratinib promotes ferroptosis and inhibits brain metastasis in a novel syngeneic model of spontaneous HER2(+ve) breast cancer metastasis. Breast Cancer Res. 2019;21:94
299. Ma S, Henson ES, Chen Y, Gibson SB. Ferroptosis is induced following siramesine and lapatinib treatment of breast cancer cells. Cell Death Dis. 2016;7:e2307
300. Louandre C, Ezzoukhry Z, Godin C, Barbare JC, Mazière JC, Chauffert B. et al. Iron-dependent cell death of hepatocellular carcinoma cells exposed to sorafenib. Int J Cancer. 2013;133:1732-42
301. Gout PW, Buckley AR, Simms CR, Bruchovsky N. Sulfasalazine, a potent suppressor of lymphoma growth by inhibition of the x(c)- cystine transporter: a new action for an old drug. Leukemia. 2001;15:1633-40
302. Hassannia B, Wiernicki B, Ingold I, Qu F, Van Herck S, Tyurina YY. et al. Nano-targeted induction of dual ferroptotic mechanisms eradicates high-risk neuroblastoma. J Clin Invest. 2018;128:3341-55
303. Zhang Y, Tan Y, Liu S, Yin H, Duan J, Fan L. et al. Implications of Withaferin A for the metastatic potential and drug resistance in hepatocellular carcinoma cells via Nrf2-mediated EMT and ferroptosis. Toxicol Mech Methods. 2023;33:47-55
304. O'Dwyer PJ, Hamilton TC, LaCreta FP, Gallo JM, Kilpatrick D, Halbherr T. et al. Phase I trial of buthionine sulfoximine in combination with melphalan in patients with cancer. J Clin Oncol. 1996;14:249-56
305. Wang W, Green M, Choi JE, Gijón M, Kennedy PD, Johnson JK. et al. CD8(+) T cells regulate tumour ferroptosis during cancer immunotherapy. Nature. 2019;569:270-4
306. Sacco A, Battaglia AM, Botta C, Aversa I, Mancuso S, Costanzo F. et al. Iron Metabolism in the Tumor Microenvironment-Implications for Anti-Cancer Immune Response. Cells. 2021 10
307. Xiao L, Erb U, Zhao K, Hackert T, Zöller M. Efficacy of vaccination with tumor-exosome loaded dendritic cells combined with cytotoxic drug treatment in pancreatic cancer. Oncoimmunology. 2017;6:e1319044
308. Kanlikilicer P, Bayraktar R, Denizli M, Rashed MH, Ivan C, Aslan B. et al. Exosomal miRNA confers chemo resistance via targeting Cav1/p-gp/M2-type macrophage axis in ovarian cancer. EBioMedicine. 2018;38:100-12
309. Zhao Y, Hou X, Chai J, Zhang Z, Xue X, Huang F. et al. Stapled Liposomes Enhance Cross-Priming of Radio-Immunotherapy. Adv Mater. 2022;34:e2107161
310. Cummins G, Yung DE, Cox BF, Koulaouzidis A, Desmulliez MPY, Cochran S. Luminally expressed gastrointestinal biomarkers. Expert Rev Gastroenterol Hepatol. 2017;11:1119-34
Author contact
![]() Corresponding authors: Wenjun Mao, M.D., Department of Cardiothoracic Surgery, The Affiliated Wuxi People's Hospital of Nanjing Medical University, No. 299 Qingyang Rd., Wuxi, 214023, China. E-mail: maowenjun1edu.cn. Jie Mei, M.D., Department of Oncology, The Affiliated Wuxi People's Hospital of Nanjing Medical University, No. 299 Qingyang Rd., Wuxi, 214023, China. E-mail: meijie1996edu.cn. Yuan Wan, Ph.D., The Pq Laboratory of BiomeDx/Rx, Department of Biomedical Engineering, Binghamton University, No. 65 Murray Hill Rd., Binghamton, 13850, USA. E-mail: ywanedu
Corresponding authors: Wenjun Mao, M.D., Department of Cardiothoracic Surgery, The Affiliated Wuxi People's Hospital of Nanjing Medical University, No. 299 Qingyang Rd., Wuxi, 214023, China. E-mail: maowenjun1edu.cn. Jie Mei, M.D., Department of Oncology, The Affiliated Wuxi People's Hospital of Nanjing Medical University, No. 299 Qingyang Rd., Wuxi, 214023, China. E-mail: meijie1996edu.cn. Yuan Wan, Ph.D., The Pq Laboratory of BiomeDx/Rx, Department of Biomedical Engineering, Binghamton University, No. 65 Murray Hill Rd., Binghamton, 13850, USA. E-mail: ywanedu