Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Background
Tumor specific expression of...
Cellular functions regulated by...
Signaling pathways regulated by...
PRL3-targeted therapy
Perspectives
Conclusions
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2023; 13(6):1876-1891. doi:10.7150/thno.79265 This issue Cite
Review
PRL3 as a therapeutic target for novel cancer immunotherapy in multiple cancer types
Pei Ling Chia†, Koon Hwee Ang†, Min Thura, Qi Zeng ![]()
Institute of Molecular and Cell Biology, Agency for Science, Technology and Research (A*STAR), Singapore 138673; chiapl@imcb.a-star.edu.sg; angkh@imcb.a-star.edu.sg; mthura@imcb.a-star.edu.sg
† These authors contributed equally to this work.
Received 2022-9-26; Accepted 2022-12-20; Published 2023-3-21
Abstract

Phosphatase of Regenerating Liver-3 (PRL3) was discovered in 1998 and was subsequently found to be correlated with cancer progression and metastasis in 2001. Extensive research in the past two decades has produced significant findings on PRL3-mediated cancer signaling and functions, as well as its clinical relevance in diverse types of cancer. PRL3 has been established to play a role in many cancer-related functions, including but not limited to metastasis, proliferation, and angiogenesis. Importantly, the tumor-specific expression of PRL3 protein in multiple cancer types has made it an attractive therapeutic target. Much effort has been made in developing PRL3-targeted therapy with small chemical inhibitors against intracellular PRL3, and notably, the development of PRL3-zumab as a novel cancer immunotherapy against PRL3. In this review, we summarize the current understanding of the role of PRL3 in cancer-related cellular functions, its prognostic value, as well as perspectives on PRL3 as a target for unconventional immunotherapy in the clinic with PRL3-zumab.
Keywords: Phosphatase of Regenerating Liver-3 (PRL3), cancer metastasis, monoclonal antibody (mAb), immunotherapy, clinical trials
Background
PRL3, encoded by Protein Tyrosine Phosphatase 4A3 (PTP4A3), is a member of the class of dual-specificity protein tyrosine phosphatases known as the phosphatases of the regenerating liver (PRLs). These phosphatases facilitate reversible dephosphorylation of proteins, a key post-translational modification that regulates multiple cellular processes, including but not limited to transcription, cell cycle progression, and important signaling pathways [1]. PRL3 was identified by Zeng et al in 1998 [2]. It is composed of 173 amino acids with a molecular mass of 19,535 Da. PRL3 is prenylated in vivo and in vitro, a post-translational modification that enables the association of PRL3 with the cell membrane [2, 3]. The active site of PRL3 contains a catalytic cysteine residue at position 104 that is important for phosphatase activity, and a single amino acid substitution of this cysteine for serine could render PRL3 inactive, thereby perturbing its cellular functions [4, 5]. Importantly, PRL3 was first linked to cancer progression and metastasis by Vogelstein in 2001 [6]. Since then, numerous studies have firmly established PRL3 as an important oncoprotein. In the subsequent sections, we will discuss the tumor-specific expression of PRL3 in various cancer types, its central role in regulating cellular functions and signaling pathways in cancer, as well as therapeutic targeting of PRL3 via unconventional antibody-based immunotherapy.
Tumor specific expression of PRL3 protein in multiple cancer types
To date, there has been extensive research done on PRL3 regarding its expression in primary and metastatic tumors, prognostic value, and function in cancer cells. High PRL3 expression has been associated with both primary and secondary tumors of approximately twenty different cancer types, a higher risk of metastatic outcome, and poor prognosis.
The Zeng group analyzed PRL3 protein expression in multiple human tumor tissues (n = 1008) by immunohistochemistry (IHC) of formalin fixed paraffin embedded samples and detected an average of 22.3% PRL3 overexpression [7]. Many other labs have also assessed PRL3 protein expression in tumor tissue sections mainly using IHC and found a highly variable positivity rate of 16-90% (Table 1). By western blotting (WB) of fresh-frozen tumor samples, it was found that PRL3 protein was consistently highly expressed (80.6%) across 11 common cancer types but not in patient-matched normal tissues [8]. This suggests PRL3 is a cancer specific therapeutic oncotarget. The same highly specific antibody against PRL3 [9] yielded a differential positivity rate when used in WB (80.6%) and IHC (22.3%) analyses. This indicates a potential high chance of false negatives (~58%) with IHC detection of PRL3 (Table 2). Such a discrepancy was attributed in part to non-representative tumor sections and failed antigen retrieval on formalin-fixed paraffin-embedded (FFPE) sections. Because formalin-fixed tissue undergoes tissue processing before embedment in paraffin (wax), antigen cross-linking, and epitope masking could occur. Variable protocols between laboratories and/or individuals as well as protocols non-optimized for different tissue types are additional confounding factors [10]. Furthermore, the use of different commercially available antibodies can impact results as some antibodies have low sensitivity and/or cross-react with other PRLs. Therefore, potential false negatives in IHC assessment suggests the percentage of tumors expressing PRL3 protein is at the higher end of the range derived from numerous studies. It also implies that a full WB to detect the 20 kDa PRL3 protein band could provide a more accurate assessment of PRL3 protein expression in clinical diagnosis. Importantly, studies have consistently found minimal or no PRL3 protein expression in patient-matched normal tissues, which affirms PRL3 as a highly tumor-specific target and suggest targeting PRL3 is a promising strategy for effective treatment of many cancer types with potentially few side effects.
PRL3 expression in primary and metastatic tumors
| Year of first report | Type of cancer | Expression | |||
|---|---|---|---|---|---|
| Primary | Metastatic | ||||
| % of tumors | Method | % of tumors | Method | ||
| 2001 [Saha S. et al] | Colorectal | 44.6 [11] | ISH | Lymph nodes: 100; Liver: 91 Lung: 86; Brain: 100; Ovary: 100 [12] | ISH |
| 2004 [Wu X. et al] | Liver | Hepatocellular carcinoma (HCC) p = <0.001 in T vs N [13, 14] p = <0.01 in T vs N [15] | RT [13, 15] IHC [14] | ||
| Intrahepatic cholangiocarcinoma (ICC) 47.1 [16] | IHC | Lymph nodes: 80.6 [16] | IHC | ||
| 2004 [Miskad UA. et al] | Gastric | 70.4 [17, 18] | IHC | Lymph nodes: 74.1 [18] | ISH |
| 2004 [Parker BS. et al] | Breast | 70.7 [19] | IHC | Positively correlated with lymph node metastases [19] | |
| 2005 [Polato F. et al] | Ovarian | Stage III>Stage I [20] | RT | ||
| 2007 [Kong L. et al] | Brain | Grade IV: 50 [21] | IHC | ||
| 2008 [Liu YQ. et al] | Esophagus | 78 [22] | IHC | 96 [22] | IHC |
| 2008 [Fagerli UM. et al] | Myeloma | 90 [23] | IHC | ||
| 2009 [Ming J. et al] | Lung | Stage III: 79.2 [24] | IHC | Lymph nodes: 81.5 [24] | IHC |
| 2011 [Ma Y. et al] | Cervix | 100 [25] | IHC | Lymph nodes: 100 [25] | IHC |
| 2011 [Laurent C. et al] | Eye | High in metastases [26] | FC | ||
| 2011 [Hassan NM. et al] | Oral | Stage III/IV> Stage I/II [27] | IHC | ||
| 2012 [Zhou J. et al] | Myeloid Leukemia | 47 [28] | IHC | ||
| 2013 [Guzińska-Ustymowicz K. et al] | Endometroid | 100 [29] | IHC | Lymph nodes: 100 [29] | IHC |
| 2015 [Fang XY. et al] | Skin | 48.6% of patients - high expression correlates with melanoma-specific death [30] | GE | ||
| 2016 [Vandsemb EN. et al] | Prostate | 100 [31] | IHC | Lymph nodes: 100 [31] | IHC |
| 2017 [Hjort MA. et al] | Lymphoblastic Leukemia | Higher in ALL cells than normal cells [32] | RT, FC | ||
| 2017 [Sun F. et al] | Kidney [Wilms'] | 19 [33] | IHC | ||
| 2018 [Hjort MA. et al] | Hodgkin Lymphoma | 16 [34] | IHC | ||
RT: RT-PCR; ISH: In situ hybridization; GE: Gene expression (Bioinformatics); IHC: Immunohistochemistry; FC: Flow cytometry
Comparison between IHC and WB in the detection of PRL3 protein expression in tumor samples
| Type of cancer | IHC | WB | ||
|---|---|---|---|---|
| n | PRL3+ [%] | n | PRL3+ [%] | |
| Liver | 14 | 14.3 | 20 | 80 |
| Lung | 155 | 29 | 10 | 90 |
| Colorectal | 301 | 20.6 | 10 | 70 |
| Breast | 173 | 12.7 | 10 | 90 |
| Stomach | 18 | 16.7 | 14 | 86 |
| Kidney | 12 | 8.3 | 18 | 72 |
| Prostate | 53 | 9.4 | 4 | 100 |
| Pancreas | 126 | 33.3 | 7 | 86 |
| Bladder | 14 | 21.4 | 34 | 71 |
| Brain | 20 | 10 | ||
| Esophagus | 18 | 27.8 | ||
| Cervix | 15 | 20 | ||
| Squamous | 66 | 37.7 | ||
| Ovary | 12 | 8.3 | ||
| Skin | 8 | 37.5 | ||
| Total/Average [%] | 1008 | 22.3 | 151 | 80.6 |
IHC: Immunohistochemistry; WB: Western blotting
Cellular functions regulated by PRL3
In addition to the assessment of clinical samples, studies utilizing genetic and chemical perturbation methods in both in vitro and in vivo models have established a causative role for PRL3 in promoting cancer progression. PRL3 overexpression perturbs many cellular functions that are deregulated in cancers, leading to phenotypes such as increased cell proliferation, migration, invasion, metastasis, cell cycle deregulation, evasion of apoptosis, genomic instability, angiogenesis, deregulation of cellular metabolism, phenotypic plasticity, as well as alterations in the tumor microenvironment that are permissive for tumor growth and metastasis (Figure 1). These features are widely considered to be key hallmarks of cancer [35-37], thereby attesting to the role of PRL3 as an oncogenic hub regulating a web of downstream processes that facilitate cancer progression and metastasis. In the sections below, we provide an update from earlier publications and reviews that have discussed the cellular functions and signaling pathways disrupted by PRL3 overexpression [38, 39].
PRL3-mediated cancer signaling and functions. Overview of cellular functions perturbed by PRL3 in cancer progression. Created with BioRender.com
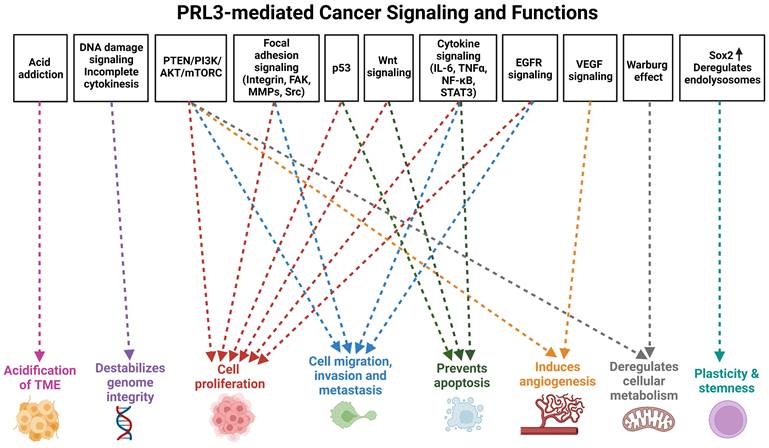
Cell proliferation and tumorigenesis
In 2001, Matter et al reported a role for PRL3 in promoting cell proliferation; HEK293 cells transfected to overexpress PRL3 exhibited increased growth rate, and this was dependent on PRL3 phosphatase activity [40]. Since then, several papers have further demonstrated that PRL3 promotes proliferation and tumorigenicity in vitro and in vivo in various cancer cell models, including mouse melanoma cells [41, 42], Chinese Hamster Ovary (CHO) cells, and human cancer cell lines including liver [14], ovarian [20, 43-45], gastric [46, 47], lung [24, 48], colon [49-53], breast [54], prostate [31, 55], esophageal [22], multiple myeloma (MM) [56, 57], glioma [58], classical Hodgkin lymphoma [34], Acute Myeloid Leukemia (AML) [59-61], Chronic Myeloid Leukemia (CML) [62], and T cell acute lymphoblastic leukemia (T-ALL) [63].
Cell migration, invasion and metastasis
The causative role of PRL3 in mediating cell migration, invasion, and cancer metastasis is well established. The Zeng group reported this phenomenon in 2003; stable expression of PRL3 in CHO cells increased cell migration and invasion in vitro, and promoted tumor formation and cancer metastasis in vivo [4]. Subsequently, these observations were confirmed in other cancer cell models both in vitro and in vivo, including liver [14], gastric [17, 46, 47, 64-66], prostate [31, 55], colorectal [11, 51, 53, 67-72], esophageal [22], lung [24], diffuse glioma [58], breast [54, 73], melanoma [41, 74-76], ovarian [77], nasopharyngeal [78], B-cell Acute lymphoblastic leukemia (B-ALL) [32], AML [79], MM [23], and classical Hodgkin lymphoma [34].
In parallel, a clinical association between PRL3 and cancer metastasis has also been established. This association was first made by the Vogelstein lab, who found that PRL3 mRNA was expressed in metastatic colorectal cancer (CRC) but not in benign tumors or normal colorectal epithelium in 2001 [6]. Their findings were corroborated by various studies involving human samples, including hepatocellular carcinoma (HCC) [13-15], intrahepatic cholangiocarcinoma [16], CRC [11, 51], gastric cancer [64, 80], prostate cancer [31], esophageal squamous cell carcinoma [22], non-small cell lung cancer [24], and cervical cancer [25].
PRL3 induces changes in cellular morphology characteristic of epithelial-mesenchymal transition (EMT), a process important for metastasis [81-85]. Cells display an epithelial to fibroblast/mesenchymal-like morphology that is more elongated and spread out, with increased formation of filopodia [11, 41, 69, 86]. Furthermore, PRL3 enhances cell adhesion to extracellular matrix (ECM) substrates such as fibronectin and laminin [14, 32, 41, 54, 74, 81]. Interestingly, some studies found that co-culture of PRL3 expressing CRC cells with tumor associated macrophages (TAMs) further promoted cell invasion [70, 72], thus emphasizing the importance of crosstalk between PRL3 expressing cells and the surrounding tumor microenvironment.
Cell cycle deregulation and evasion of apoptosis
Studies have reported arrest at various cell cycle checkpoints - G1/S, S, and G2/M, upon knockdown or overexpression of PRL3 [53, 57, 59, 61, 79, 87, 88]. However, others have also found no significant changes in the cell cycle upon PRL3 perturbation [23, 74]. These highly varied and conflicting findings are likely due to an endogenous function of PRL3 regulating multiple cell cycle checkpoints. Hence, PRL3 perturbs the cell cycle in a context-dependent manner, given the myriad of genetic mutations in different cell lines that can independently influence the cell cycle.
Though the precise details on how PRL3 influences the cell cycle remains to be fully uncovered, there is consistent evidence that PRL3 has an anti-apoptotic function [22, 34, 47, 53, 57, 59, 61, 67, 79, 87-90]. PRL3 protects cells from apoptosis induced by growth factor deprivation stress [67], cytokine deprivation [59], as well as DNA-damaging chemotherapeutic drugs such as doxorubicin, cytarabine [61], and 5-fluorouracil (5-FU) [22, 89]. Notably, PRL3 positively regulates the expression of the anti-apoptotic protein, Mcl-1, and loss of PRL3 leads to activation of the intrinsic apoptotic pathway [34, 57, 90].
Genome instability and mutation
Genome instability has been widely considered a hallmark of cancer that is advantageous for tumor progression, and is characterized by features such as chromosomal instability (CIN), DNA damage and repair defects, as well as loss of telomeric DNA [36]. In that regard, PRL3 has been associated with indicators of genome instability in human tumor samples. In PRL3-positive colorectal cancer tissues, markers of CIN were observed [53]. A study by Lian et al found that overexpression of PRL3 in primary fibroblasts as well as CRC cells leads to abnormalities in telomeric structure and telomere deprotection, which induces a persistent DNA damage response (DDR) that leads to CIN and senescence [91]. The study also made a similar observation in transgenic mice, where induction of PRL3 in colon and liver tissues led to telomere deprotection, and this was associated with colon tumorigenesis. In addition, correlation of PRL3 expression with indicators of telomere deprotection and senescence was also seen in tumor samples. The acquisition of further mutations that promote cell cycle checkpoint deregulation can enable senescent cells to achieve replicative immortality, which facilitates cancer progression [36].
In addition, PRL3 also promotes the formation of grossly hyperdiploid and multinucleated cancer cells known as polyploid giant cells (PGCCs) [92]. This is facilitated by an increased incidence of incomplete cytokinesis, suppression of DNA damage signaling, which leads to an accumulation of double stranded breaks and genomic instability [92].
Inducing angiogenesis
Clinically, PRL3 is more significantly expressed in the vasculature of tumor tissue vs normal tissue, such as in the colon [93] and the mammary gland [94]. A study of endometrial adenocarcinoma also found a correlation between PRL3 overexpression and Vascular Endothelial Growth Factor (VEGF)-A and VEGF-C expression, as well as greater microvessel density (MVD) and lymphatic vascular density (LVD) [95]. In addition, PRL3 expression is associated with vascular invasion in HCC tumors [14], which often display hypervascularity and marked vascular abnormalities [96, 97].
Mechanistically, the earliest evidence that PRL3 was involved in tumor angiogenesis was the observation that PRL3 expressing CHO cells formed micro- and macro-metastatic solid tumors in blood vessels [5]. A later study showed that PRL3 expressing cells were able to recruit and enhance vascular formation by human umbilical vein endothelial cells (HUVECs) in vitro, as well as recruit host endothelial cells and promote tumor angiogenesis in vivo [93]. Furthermore, genetic and chemical inhibition of PRL3 abrogates tube formation by HUVEC cultured on Matrigel [98]. In addition, inhibition of PRL3 also reduces vascular endothelial permeability of primary human lung microvascular endothelial cells (MVEC) [45]. Conversely, overexpression of PRL3 increased migration of cancer cells for metastasis [94]. Collectively, these studies point to a pro-angiogenic function of PRL3.
Deregulating cellular metabolism
Many cancers exhibit the Warburg effect, which is characterized by increased glucose consumption and lactate production [99]. PRL3 has been shown to mediate the Warburg effect in CRC cells and multiple myeloma cells [56, 100]. In CRC cells, PRL3 promotes increased expression of glycolytic enzymes glucose transporter 1 (GLUT1), hexokinase 2 (HK2), pyruvate kinase M2 (PKM2), and lactate dehydrogenase A (LDHA). Cancer cell proliferation and invasion were abrogated by the lactate dehydrogenase (LDH) inhibitor oxamate, suggesting PRL3-induced metabolic reprogramming contributes to its oncogenic function [100]. In MM, cells overexpressing PRL3 were more metabolically active, upregulating both oxidative phosphorylation (OXPHOS) in addition to glycolysis. Enzymes in the serine/glycine pathway were also upregulated, which suggest increased macromolecule biosynthesis [56]. Interestingly, the study in MM found these alterations in cancer metabolism were independent of PRL3 phosphatase activity [56].
In addition, a study found that PRL3 knockdown promotes production of mitochondrial superoxide anion with a concomitant increase in mitochondrial membrane potential and induction of cell cycle arrest [101]. PRL3 acts as a transcription factor, binding to the promoter of repressor activator protein 1 (RAP1), a protein that regulates the master regulator of mitochondrial biogenesis, peroxisome proliferator-activated receptor gamma coactivator 1 alpha (PGC-1α) [101]. Thus, knockdown of PRL3 decreases PGC-1α as well as its downstream genes superoxide dismutase 2 (SOD2) and uncoupling protein 2 (UCP2), both reactive oxygen species (ROS)-detoxifying proteins that prevent the accumulation of ROS. The regulation of a key mediator of mitochondrial function (PGC-1α) by PRL3 suggests that PRL3 could play a critical role in the cellular metabolism. Furthermore, the function of PRL3 as a transcription factor could potentially be a novel mechanism by which PRL3 regulates cellular processes, though this warrants further investigation.
Emerging research has also found PRL3 affects energy metabolism by regulating Mg2+ efflux [102-104]. The PRL-family (PRL1, PRL2, and PRL3) in general, was shown by both the Tremblay group and Hiroaki group to directly bind and inhibit Mg2+ efflux by cyclin M (CNNM) magnesium regulators [105]. The interaction between PRL3 and CNNM4 results in decreased Mg2+ efflux and increased intracellular Mg2+ [104, 105]. As intracellular ATP exists in complex with Mg2+ and Mg2+ is also needed for ATP synthesis, PRL3 overexpressing cells have increased intracellular ATP. Consequently, PRL3 overexpressing cells have a proliferative advantage under glucose-limiting conditions that decrease intracellular ATP levels [104]. Interestingly, Hardy et al found that Mg2+ levels regulate PRL1 and PRL2 mRNA translation through a Mg2+-sensitive upstream ORF in the 5-UTR [106]. Mg2+ depletion and corresponding decrease in intracellular ATP activates the AMPK/mTOR2C pathway, which ultimately leads to increased translation of PRL1 and -2. As this study was done in cell lines that did not express PRL3, it is possible that PRL3 could be regulated by a similar mechanism, but this would need to be confirmed in suitable models expressing PRL3. Nevertheless, the abovementioned findings on PRLs and CNNM/Mg2+ regulation open a new area for future studies in defining an exciting mechanism of action of PRL3.
Conversely, others have found that PRL3-overexpressing MM cells were more sensitive to the anti-proliferative effects resulting from glucose uptake blockade by the GLUT1 inhibitor STF-31 [56, 107], indicating possible glucose dependency. This finding has potential therapeutic relevance; as GLUT inhibitors have variable efficacy due to metabolic heterogeneity, identifying mutations that predispose tumors to the anti-proliferative and/or cytotoxic effects of glucose uptake blockade are of interest [108]. Preliminary evidence demonstrating PRL3 deregulates glucose metabolism and increases sensitivity to GLUT inhibitors suggests it is worth exploring PRL3 expression as a possible biomarker. The interesting insights on the role of PRL3 in deregulating cellular metabolism warrants further study, especially given the seemingly contradictory findings, which are likely because existing studies have been limited to one cancer type (MM). In-depth study into how PRL3 regulates the diverse aspects of cellular metabolism in various cancers would help to explore PRL3 as a biomarker for response to metabolic inhibitors.
Plasticity and stemness
Recent studies have highlighted a role of PRL3 in cellular plasticity and acquisition of a cancer stem cell (CSC) like state [36, 109, 110]. Using a zebrafish model, Johansson et al uncovered an endogenous function of PRL3 in preventing differentiation of melanocyte stem cells (MSCs) during regeneration [110]. By extension, PRL3 overexpression in melanoma deregulates cellular differentiation to maintain cells in a stem-like state, which is a key feature of cancer progression in general [35]. Mechanistically, PRL3 restricts transcription of master transcription factor (MITF)-regulated endolysosomal genes by dephosphorylating and impairing binding of the RNA helicase DDX21. Clinically, dysregulation of endolysosomal pathways has been linked to melanoma progression. Furthermore, the endolysosomal pathway has been implicated in stem cell fate determination as well as cancer stem cells [111, 112].
PRL3 has also been found to promote the transition of ovarian cancer cells to a CSC-like state by increasing the transcription of SRY-Box transcription factor 2 (SOX2), a well-characterized pluripotent factor that maintains cancer stemness [109, 113]. PRL3 does so by sequestering histone deacetylase 4 (HDAC4) in the cytoplasm, thereby preventing histone deacetylation and increasing accessibility of the SOX2 promoter to its transcription factor myocyte enhancer factor 2A (MEF2A). Interestingly, this process is independent of PRL3 phosphatase activity, but dependent on the cytoplasmic translocation of PRL3 [109].
Tumor microenvironment
Cancer cells exist in a tumor microenvironment (TME) that has undergone changes to create a niche that is permissive for cancer progression. Of note, increased acidity of the TME drives malignant progression of cancer by promoting ECM degradation and remodeling, cell invasion, as well as creating an immune evasive environment [114, 115]. A study by Funato et al found that PRL3 stimulates lysosomal exocytosis, which extrudes H+ into the extracellular space. Consequently, PRL3 overexpressing cells become acid addicted, a phenomenon important for PRL3-mediated metastasis [103]. A previous study by Machado et al had also found a role for increased lysosomal exocytosis in aggressive sarcomas, where the release of soluble hydrolases promotes degradation of the ECM and tumor cell invasion [116].
As previously described, PRL3 is also involved in inhibiting the transcription of endolysosomal genes to promote stemness; this further suggests PRL3 plays a significant role in regulating the endolysosomal system - a hub for multiple biological processes that are perturbed in cancer, such as autophagy, apoptosis, cell adhesion and migration [117].
Signaling pathways regulated by PRL3
In this section, we highlight how PRL3 is a central node of many oncogenic pathways [118] that are often deregulated in many cancer types, including but not limited to transmembrane growth factor signaling (eg. ErbB), focal adhesion/integrin signaling, cytokine signaling, as well as pathways downstream of p53 and Wnt [71, 86] (Figure 1). This emphasizes PRL3 as a critical player in mediating the hallmarks of cancer and by extension, its relevance as a therapeutic target.
Receptor Tyrosine Kinase (RTK) signaling
PRL3 has been implicated in RTK signaling, perturbing key downstream pathways commonly deregulated in cancer, such as MAPK/ERK and PI3K/AKT signaling [36]. A few examples are highlighted below.
Epidermal growth factor receptor (EGFR)
Hyperactivation of EGFR by PRL3 initiates multiple downstream signaling cascades, including MAPK/ERK, JAK/STAT, PI3K/AKT, and p38/JNK. PRL3 does so by transcriptionally downregulating PTP1B, a phosphatase that inhibits EGFR. Consequently, PRL3-overexpressing cells are addicted to EGFR signaling and hypersensitive to EGFR inhibitors, thereby highlighting the potential of PRL3 as a biomarker for response to anti-EGFR therapy such as Cetuximab [48].
Platelet-derived growth factor (PDGF)
PDGF signaling is involved in facilitating PRL3 mediated metastasis. PDGF can stimulate PRL3 phosphorylation in a Src-dependent manner [69], a modification that is required for PRL3 mediated cell migration and invasion. In addition, expression of PRL3 induces Src-dependent phosphorylation and activation of PDGFR-β [72], a key player in vascular remodeling and angiogenesis [98].
Vascular endothelial growth factor (VEGF)
VEGF, a master regulator of tumor angiogenesis [119, 120], has been implicated in PRL3-induced angiogenesis [24, 95, 98]. Clinically, PRL3 overexpression is associated with VEGF expression, tumor angiogenesis and lymph node metastasis [24, 95]. VEGF induces transcription of PRL3, which is needed for tube formation by HUVEC cultured on Matrigel [98]. Conversely, inhibiting PRL3 downregulated VEGF by downregulating ERK1/2 signaling through reduction of phospho-ERK1/2 [24, 95].
Focal adhesion signaling
There is significant evidence that PRL3-mediated migration, invasion, and metastasis is regulated by integrin/Rho signaling, with the involvement of kinases like Src and focal adhesion kinase (FAK), as well as various matrix metalloproteinases (MMPs).
PRL3 was shown to be co-amplified and co-expressed with FAK in HCC. The involvement of PRL3 results in the phosphorylation of FAK which then activates the p38 and PI3K/AKT pathway, mediating the oncogenic effect of PRL3 in HCC cells [14].
In addition, PRL3-induced cell migration is Src-dependent and can be abolished using a Src inhibitor [81, 121]. Mechanistically, PRL3 downregulates C-terminal Src kinase (Csk), a negative regulator of Src, to induce Src activation [81]. This leads to increased activity of p130Cas - a scaffold protein which localizes to focal adhesion complexes to coordinate downstream pathways mediating cell migration, as well as ERK1/2, signal transducer and activator of transcription 3 (STAT3), which promote cell proliferation and invasion [81, 122, 123]. Conversely, PRL3 can be phosphorylated and activated by Src, triggering downstream signaling mediated by Rho GTPases to promote cell migration and invasion [69, 124]. Interestingly, a study found PRL3 dephosphorylates and inactivates vav guanine nucleotide exchange factor 1 (VAV1), a regulator of Rho GTPases that is critical for T cell receptor (TCR) signaling. PRL3 is coamplified with the known oncogenic driver MYC, and they synergize to sustain T-ALL tumor growth. The re-activation of TCR signaling upon PRL3 inhibition kills T-cell acute lymphoblastic leukemia (T-ALL) cells [63].
Furthermore, there is a significant correlation of PRL3 with the expression of various MMPs, such as MMP2 and MMP9 in HCC [13, 125]. These zinc-dependent endoproteinases break down ECM of surrounding tissues to facilitate the invasion of tumor cells and metastasis [126, 127]. Mechanistically, studies in CRC cells found that PRL3-mediated invasion and migration was dependent on MMP7 [128], and PRL3 also enhanced the gelatinolytic function of MMP2 [122].
Cytokine signaling
PRL3 has also been implicated in cytokine signaling, such as pathways downstream of pro-inflammatory cytokines interleukin-6 (IL-6) and tumor necrosis factor alpha (TNFα), with the involvement of key transcription factors regulating the inflammatory response, nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) and STAT3 [50, 66, 70-72, 83, 85, 129].
STAT3 is a direct transcriptional regulator of PRL3 and mediates IL-6-dependent increase in PRL3. Conversely, inhibition of PRL3 reduces nuclear accumulation of STAT3, thereby downregulating downstream targets c-myc, cyclin D1, and the anti-apoptotic proteins Mcl-1, Bcl-xL and Bcl-2. This leads to decreased cell migration and survival in both in vitro and in vivo systems [57, 130].
Ectopic expression of PRL3 in CRC cells increases secretion of the cytokine TNFα, which activates NF-κB in an autocrine manner, leading to cell growth via increased G2/M transition [50], induction of EMT [83], as well as increased secretion of angiogenesis associated proteins such as VEGF-A [85]. This is achieved in part by NF-κB-dependent upregulation of potassium calcium-activated channel subfamily N member 4 (KCNN4), a Ca2+-activated K+ channel which has been shown to promote cell proliferation, invasion, and metastasis by activating MAPK/ERK and PI3K/AKT signaling [131]. Furthermore, TNFα secreted by PRL3 overexpressing cells can stimulate surrounding TAMs to produce proinflammatory cytokines IL-6 and IL-8, creating a paracrine loop that further induces EMT [85] and promotes metastasis [70]. PRL3 overexpressing cells also produce more chemokine ligand 26 (CCL26), which engages the chemokine receptor type 3 (CCR3) on TAMs, inducing TAM infiltration in vivo to promote tumor growth [72, 85]. In addition, NF-κB transcriptionally regulates PRL3 expression downstream of TNFα. Knockdown of PRL3 induces G1 cell cycle arrest and senescence in triple-negative breast cancer (TNBC) cells, and continued suppression leads to increased secretion of TNFα, which activates the extrinsic apoptotic pathway in an NF-κB dependent manner [54].
Evidently, the reciprocal regulation between PRL3 and TNFα/IL-6 illustrates the important role of cytokine signaling in PRL3 expressing tumors as well as their surrounding microenvironment.
p53 signaling
Mutual regulation exists between PRL3 and the tumor suppressor p53. PRL3 is known to be a direct transcriptional target of p53, whereas PRL3 negatively regulates p53 protein levels [89, 132, 133]. In non-transformed primary cells, both knockdown and overexpression of PRL3 levels triggers G1 arrest, alluding to a requirement for basal levels of PRL3 for proper cell cycle progression [132]. In cancer cells - which often deregulate p53, PRL3 downregulates p53, which can lead to increased cell proliferation and clonogenicity [133], increased G1-S cell cycle progression [61], as well as inhibition of apoptosis [89]. In general, PRL3 promotes proteasomal degradation of p53 protein through increased activity of mouse double minute 2 homolog (Mdm2), the negative regulator of p53 [89]. Several mechanisms have been described; in breast cancer cells, the Mdm2 inhibitor p14ARF is downregulated [133], and activated Akt phosphorylates and stabilizes Mdm2 in colorectal HCT116 p53 (+/+) cells [89]. Upregulation of PIRH2, another p53-induced ubiquitin-protein ligase which promotes its degradation, was also observed [89]. Thus, we can see that deregulation of PRL3 and p53 in cancer cells interact to disrupt homeostatic control of the cell cycle and cell proliferation.
PTEN/PI3K/AKT/mTOR signaling
PRL3 activates the PI3K/AKT pathway by downregulating its key negative regulator PTEN, leading to increased cell proliferation, EMT, migration, and invasion [15, 82]. In the context of melanoma progression, PRL3 dephosphorylates Na+/H+ exchanger regulating factor 1 (NHERF1), an interactor of PTEN, promoting the nuclear to cytoplasmic translocation of both NHERF1 and PTEN. The differential localization of PTEN leads to phosphorylation and activation of AKT [134]. In addition, AKT activation was also observed upon knockdown of polyC-RNA-binding protein 1 (PCBP1), which inhibits translation of PRL3 mRNA by preventing its incorporation into polyribosomes. In HCC tumors, a negative regulator of PCBP1, the long non-coding RNA (lncRNA) PCBP1-AS1, is highly expressed compared to adjacent normal tissue, and correlates with increased metastasis and poorer patient survival. The increase in cell proliferation and metastasis induced by PCBP1-AS1 overexpression was shown to be dependent on the PCBP1-PRL3-AKT axis, which consequently promotes tumor growth and metastasis in vivo [7, 135].
Activation of mechanistic target of rapamycin complex 1 (mTORC1) signaling by PRL3 drives motility, invasion, as well as tumorigenesis [104, 136]. PRL3 increases binding of mTOR to RagB/C GTPases, which facilitates mTORC1 recruitment to lysosomal membranes [136]. In turn, RagB/C inhibit both autophagic and proteasomal degradation of PRL3 [137]. Furthermore, PRL3 promotes canonical autophagic flux in addition to serving as an autophagic substrate, thereby creating a negative feedback loop [44]. Consequently, PRL3-driven autophagy is associated with AKT activation and increased cell growth [44].
Wnt signaling
Research has shown PRL3 mediates its oncogenic function in AML by activating canonical Wnt signaling [60, 138], Consequently, AML cells overexpressing PRL3 have enhanced sensitivity towards β-catenin inhibition both in vitro and in vivo [138]. In addition, combinatorial inhibition of PI3K/AKT/mTOR and Wnt/β-catenin signaling is synthetic lethal specifically in cells with high PRL3 levels [139]. Thus, Wnt/β-catenin signaling, in conjunction with other signaling pathways, plays a significant role in mediating downstream effects of PRL3.
PRL3-targeted therapy
The tumor-specific expression of PRL3 across a broad range of cancer types makes it an attractive therapeutic target. Significant work has been done in developing PRL3-targeted therapy, which includes the identification and optimization of small molecule inhibitors against PRL3 [22, 47, 49, 68, 140-149], as well as the development of antibody-based therapy [9, 150-153]. We will briefly discuss the usage of PRL3 inhibitors, which has been reviewed in detail by Wei et al [154], and highlight recent advances in anti-PRL3 immunotherapy, where a first-in-class humanized monoclonal antibody (mAb) known as PRL3-zumab is currently in Phase II clinical trials (ClinicalTrials.gov identifier: NCT04118114; NCT04452955).
Small molecule inhibitors against intracellular PRL3
Much work on PRL3 inhibitors has focused on computationally screening small molecule libraries to identify potential lead compounds for further in vitro validation and optimization [45, 49, 77, 141, 144-149, 155]. Rhodanine-based compounds have been of key interest after this chemotype was shown to have inhibitory activity against PRL3 through a high throughput screening of a chemical library in 2006 [141]. Rhodanine derivatives have been demonstrated to inhibit PRL3-mediated cell proliferation, migration, invasion, as well as induce cell cycle arrest and apoptosis in cancer cells [22, 47, 68, 143]. Most notably, the compound BR-1 was shown to reduce tumor growth in vivo and synergize in vitro with the multikinase inhibitor Sorafenib [143]. Despite these promising results, an overwhelming majority of studies on small molecule inhibitors against PRL3 have only been done in cell line models, and further work in vivo needs to be done before PRL3 inhibitors can move into clinical trials. This involves overcoming the challenge of limited selectivity in the design of PRL3 inhibitors; the high degree of similarity between PRL3 and its other family members PRL1 and PRL2 makes it challenging to develop an inhibitor specific to PRL3 [147, 156]. Furthermore, PRL3 inhibitors could also target other structurally similar phosphatases like the tumor suppressor PTEN [156]. Hence, there is a need to tap on existing knowledge around PRL3 structural biology to address the issue of off-target effects.
Antibody-based therapy against intracellular PRL3
A major challenge in cancer therapy is a lack of target specificity for cancer cells, resulting in damage to normal tissues. Antibody-based therapy have greater specificity over standard chemotherapy regimens and thus improved efficacy. Hence, the research efforts of the Zeng group have focused on using PRL3 antibodies to target PRL3 expressing tumors, as opposed to small inhibitors. Extensive work has led to the development of a first-in-class humanized monoclonal antibody (PRL3-zumab) against PRL3, a novel cancer immunotherapy against an intracellular antigen [153]. Using orthotopic cancer mouse models, it was shown that PRL3-zumab reduces tumor growth, inhibits metastatic tumors, reduces tumor recurrence, and increases survival [8, 150, 151, 153]. In addition, PRL3-zumab can serve as a form of adjuvant immunotherapy to eliminate stem-cell like PGCCs that remain after tumor removal, thereby preventing relapse [92]. The abovementioned effects are seen in tumors that express PRL3 but not in PRL3-negative tumors, and PRL3-zumab also showed no cross-reactivity with the closely related homologs PRL1 and PRL2. Hence, this confirms the specific binding of PRL3-zumab to its target antigen, PRL3 [8, 9, 150, 151, 153]. The substantial pre-clinical data paved the way for PRL3-zumab to enter clinical trials, and the immunotherapeutic is currently in Phase II clinical trials in Singapore (ClinicalTrials.gov identifier: NCT04118114), USA (ClinicalTrials.gov identifier: NCT04452955) and China (China Drugtrials.org.cn identifier: CTR20211180), with the Phase II clinical trial in the US almost completed. In both the Phase I and Phase II trials, PRL3-zumab has shown a strong safety profile with no serious adverse events (SAEs), infusion reactions, dose-limiting toxicities (DLTs), or drug-related deaths.
PRL3-zumab targets PRL3 intracellular oncoprotein, which begets the question of the antibody's mechanism of action (MOA), given the general assumption that antibodies can only target antigens located on the cell surface as they are too large to enter the cell [157].
However, several lines of evidence have challenged this assumption [158, 159]. In a major proof-of-concept study, Guo et al generated three mAbs against their respective intracellular proteins - PRL3, EGFP, and the polyomavirus middle T oncoprotein (mT) [160, 161]. The study found the mAbs induced tumor regression in immunocompetent wild type C57BL/6 mice specifically expressing the target protein (PRL3 or EGFP), as well as in MMTV-PyMT transgenic spontaneous breast cancer models specifically expressing the target intracellular mT. For example, anti-PRL3 mAb induced tumor regression of PRL3-positive tumors but not PRL3-negative tumors, thereby highlighting the specificity of mAbs for their respective targets. Furthermore, proteins that are located intracellularly in normal cells have been shown to be externalized on the surface of tumor cells. These include viable therapeutic targets such as heat-shock protein 70 (HSP70), heat-shock protein 90 (HSP90), glucose-regulated protein 78 and 94 (GRP78 and GRP94), vimentin, estrogen receptor-alpha variant 36 (ER-α36), and feto-acinar pancreatic protein (FAPP) [162]. In the pre-clinical setting, mAbs designed specifically against known externalized epitopes have been developed by GRP94 [163], HSP70 [164]. In addition, early studies have shown that tumor cell membranes are generally more permeable than normal cells [165], which is favorable for secretion and externalization of intracellular antigens.
In the case of PRL3, Min et al found evidence that the intracellular PRL3 oncoprotein is externalized on the cell surface in tumor cells in vivo and serum-starved cells in vitro; this phenomenon is not observed in normal cultured cells in vitro, suggesting that stresses in the tumor microenvironment promote externalization of PRL3, and this can be recapitulated in vitro by subjecting cells to serum starvation [8]. PRL3 mini-body (lacking Fc domain) showed no therapeutic effect, suggesting PRL3-zumab recruits immune cells such as B cells, natural killer (NK) cells, and macrophages in an Fc receptor (FcR)-dependent manner, which implies the involvement of antibody-dependent cellular cytotoxicity (ADCC) and antibody-dependent cellular phagocytosis (ADCP) in the elimination of cancer cells [8]. Notably, studies utilizing more than 4000 mice from 8 different strains demonstrated the therapeutic efficacy of anti-PRL3 mAb against PRL3+ tumors could be T-cell independent in nude mice models [166]. In addition, PRL3 is secreted extracellularly on the surface of exosomes, a possible mechanism behind the 'inside-out' phenomenon of PRL3, which also serves as bait for PRL3-zumab. It has been hypothesized by Ferrone et al [161] that resultant antigen-antibody complexes can be processed by dendritic cells for antigen presentation and subsequent T cell and NK cell activation, though there has thus far been found no evidence of T cell involvement. [8, 153, 159]. As PRL3 can be found on the cell membrane and endosome (where exosomes can originate) when prenylated [3], it is plausible that PRL3-zumab mediates its effects by targeting PRL3 antigen externalized on the cell membrane and exosomes.
Perspectives
PRL3 - an attractive therapeutic target for a broad range of cancer types and beyond
There is compelling evidence that PRL3 is an attractive therapeutic target for cancer. PRL3 is a tumor-specific antigen overexpressed across a broad range of tumor types and largely absent in matched normal tissue. PRL3 protein perturbs a myriad of cellular functions and signaling pathways commonly deregulated in cancer, establishing it as an oncogenic driver with a key role in cancer progression.
Recent findings have uncovered previously undescribed functions of PRL3, such as its endogenous function in development, and its ability to deregulate cellular metabolism and alter the TME. Aside from advancing basic knowledge on PRL3, understanding the signaling pathways perturbed by PRL3 can help expand its potential as a biomarker. For example, the association of PRL3 expression with addiction to EGFR signaling highlights the potential of PRL3 to serve as a biomarker for response to anti-EGFR therapy. Work demonstrating PRL3-induced activation of mTORC1 signaling as well as incipient evidence that PRL3 mediates the Warburg effect and glucose dependency suggests PRL3 could contribute to the growing need to identify genetic mutations associated with metabolic vulnerabilities that predict sensitivity to inhibitors targeting cancer metabolism [167].
In addition, several recent studies have uncovered phosphatase-independent functions of PRL3, which has a greater diversity of binding partners apart from enzymatic substrates. Some have even proposed that PRL3 phosphatase activity is dispensable for its oncogenic function [76, 109, 168]. For example, ablating CNNM binding function of PRL3 alone prevented metastasis of B16 mouse melanoma cells, whereas a phosphatase-dead PRL3 mutant that could still bind CNNM showed no significant change in tumorigenesis [104]. The suggestion that PRL3 phosphatase activity is dispensable for its oncogenic function is provocative, given the overwhelming amount of contradictory evidence. However, Kozlov et al pointed out many studies utilized PRL3 mutants deficient in both CNNM binding and phosphatase activity, making it difficult to dissect the contribution of each individual function. By leveraging the wealth of information on PRL3 structure [157-159], future studies utilizing PRL-3 mutants that perturb specific domains could help better address this question [169-171]. Importantly, the discovery of these novel phosphatase-independent functions of PRL3 suggests that PRL3 could regulate a broader range of biological processes that have yet to be uncovered [76, 103, 109]. In particular, the role of PRL3 in regulating magnesium transport is an emerging area of interest, with several studies demonstrating it is a mechanism by which PRL3 alters cellular metabolism to promote cell proliferation and tumorigenesis. Overall, further investigation would help identify contexts in which PRL3 could influence response to therapy targeting a particular deregulated biological process.
By identifying diseases in which PRL3-regulated pathways are perturbed, it is tempting to speculate we could apply PRL3-targeted therapy to other relevant diseases. For example, PRL3 directly binds to CNNM magnesium regulators, whose suppression has been implicated not only in cancer, but also in other diseases such as hypertension and schizophrenia. In fact, preliminary efforts have been made to indirectly enhance CNNM expression via treatment with PRL3 inhibitors as a therapeutic strategy [102-104]. Thus, it would be worth exploring the therapeutic potential of targeting PRL3 in the context of other diseases, thereby broadening the scope of its application in the clinic.
PRL3-zumab - a different take on immunotherapy
PRL3-zumab, the first-in-class humanized antibody, is currently in Phase II clinical trials in Singapore, USA, China, and Malaysia. PRL3-zumab is a novel cancer immunotherapy at the forefront of PRL3-targeted therapy (Figure 2). As a therapeutic mAb, PRL3-zumab has demonstrated a strong safety profile in Phase I and Phase II clinical trials. In general, therapeutic mAbs avoid the toxic side effects associated with conventional chemotherapy. Furthermore, targeted therapy is more specific than small molecule inhibitors, thereby avoiding toxic side effects [172]. However, the safety profile and effectiveness of therapeutic mAbs are dependent on the suitability of the targeted antigen, i.e. its specificity and prevalence in tumor cells. A tumor-specific target antigen is critical to prevent the drug from attacking normal tissues, thereby reducing adverse side effects in patients.
Schematic illustrations of the MOAs of immune checkpoint inhibitors, conventional immunotherapy, and PRL3-zumab. A) Immune checkpoints to prevent T cell activation (left). Immune checkpoint inhibitors block CTLA-4, PD-1 and PD-L1 and this blockade allows the binding of MHC with TCR in Treg and T cells, leading to the clonal expansion and activation of T cells. Activated T cells release granzymes and perforin which consequently kill the tumor cells (right). B) Tumor cells with extracellular targets (left). Binding of antibody to extracellular target triggers the antibody-dependent cellular cytotoxicity and phagocytosis (ADCC/ADCP) cascade, resulting in the killing of tumor cells by immune cells. C) Tumor cells with externalized intracellular PRL3 (left). Binding of PRL3-zumab to the externalized intracellular PRL3 in TME leads to ADCC/ADCP cascade and killing of the tumor cells akin to conventional immunotherapy. PRL3-zumab can also bind to PRL3 on the surface of secreted exosomes for clearance of exosomes to prevent metastasis. D) Chemo-resistant PRL3+ polyploid giant cancer cells (PGCCs) have stem-cell like properties, which remain after tumor removal and adjuvant chemotherapy, can initiate tumor relapse. PRL3-zumab could serve as an adjuvant therapy to eliminate PGCCs and prevent tumor relapse. Created with BioRender.com.
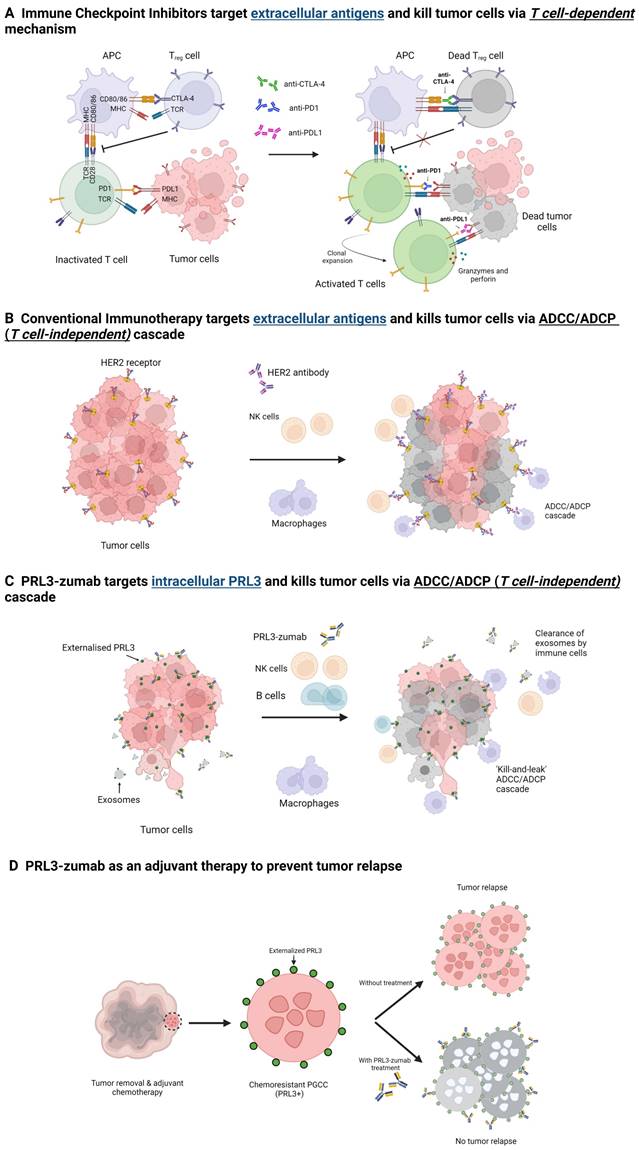
The ubiquitous and tumor specific expression implies that targeting PRL3 would be appropriate in multiple cancer types with few side effects. PRL3 plays an essential role not just in the biology of tumor cells, but also the tumor microenvironment. Hypothetically, targeting PRL3 would yield a two-pronged approach of attacking tumor cells as well as the TME, thereby shrinking the tumor as well as creating an unfavorable niche for cells to proliferate. Furthermore, PRL3-zumab can act as adjuvant therapy to eliminate residual tumor cells refractory to other forms of treatment. Targeting PRL3 would thus be a unique therapeutic strategy for eradicating a tumor in its entirety.
Notably, PRL3 is an intracellular oncoprotein, which challenges the widespread assumption that antibodies can only target extracellular proteins [159, 166]. Because of this assumption, the search for tumor associated antigens have focused on surface proteins which include cell surface differentiation antigens, growth factors, as well as vascular targets involved in angiogenesis [173]. PRL3-zumab is a proof of concept that intracellular oncoproteins are viable targets for therapeutic mAbs. Hence, by expanding the repertoire of potential targets to include intracellular tumor antigens, we can further the use of therapeutic mAbs in cancer immunotherapy.
In recent years, much focus has been on developing immune checkpoint inhibitors (ICIs), monoclonal antibodies that target negative regulators of T cell activation, namely CTLA-4 and PD-1 on Treg cells or T cells and PD-L1 on tumor cells [173-176]. Antibodies against CTLA-4, PD-1 and PD-L1 can block the binding of CTLA-4 & CD80/86 on antigen presenting cells (APC) or the binding of PD-1 and PD-L1 on tumor cells, which can lead to binding of TCR and MHC causing T cell activation. Activated T cells release granzymes and perforin which ultimately results in tumor cell killing and decreased metastasis [173-177]. The use of ICIs such as anti-CTLA-4 and notably anti-PD-1/PD-L1 - which has been shown to have a better safety profile - has contributed significantly to the improvement of patient outcome [175-178]. However, there are two key issues/limitations that need to be addressed.
Firstly, ICIs can result in immune-related adverse events (irAEs) due to activated T cell populations that can infiltrate a broad spectrum of organs. The most fatal irAEs of major concern include encephalitis, hepatitis, myocarditis, and pneumonitis. As these side effects can affect many different organs, and also differ from those associated with conventional chemotherapy, clinicians face the challenge of learning how to diagnose and manage irAEs with the growing use of ICIs [177, 179].
Secondly, there is a heterogeneous benefit rate for ICIs. The benefit rate is 10-20% in solid tumors, 40-50% in melanoma and non-small cell lung cancers (NSCLC), and 60-75% in Hodgkin lymphoma [180].
Multiple factors influence response to ICIs, include degree of tumor infiltrating T cells, target expression, as well as tumor mutational burden (TMB). In addition, different patients also have variable risk for irAEs. Hence, much research has focused on identifying biomarkers to predict patients that will benefit from ICIs [178, 181-183].
To this end, we propose that PRL3-zumab, which targets a tumor specific antigen widely expressed in various tumor types, can address these issues through its excellent safety profile, providing an alternative for patients that do not respond well to ICIs. PRL3-zumab could also benefit a broad set of patients given its expression in a wide range of tumor types. The MOA of PRL3-zumab involves NK cells, macrophages, and B cells [159], in contrast to ICIs, which rely on T-cell activity. Diversifying strategies by targeting both humoral and cell immunity provides a more comprehensive approach to immunotherapy and would ultimately translate to more treatment options for patients, thereby increasing the chances of finding a suitable therapy to improve clinical outcome. With further clinical studies, PRL3-zumab could potentially serve as an alternative immunotherapy option for patients, including those who are resistant and/or experience adverse side effects to ICIs.
Conclusions
PRL3 has been established as a key oncogenic driver that promotes cancer progression and metastasis. PRL3 overexpression in multiple cancers has pleiotropic effects, causing cells to acquire hallmark traits such as sustained proliferative signaling, replicative immortality, genome instability and mutation, resistance to cell death, angiogenesis etc. Recent findings have also expanded our knowledge of PRL3-mediated functions, including its endogenous function in regulating stem cell differentiation during development, and its influence on the TME. Importantly, its tumor specific expression in a broad range of cancers and absence in normal tissues make it an attractive therapeutic target. PRL3-zumab, a first-in-class humanized monoclonal antibody targeting PRL3, has shown promising results, including an excellent safety profile, in Phase I and II clinical trials. This novel immunotherapeutic against an intracellular target challenges a fundamental assumption that only cell surface proteins are candidates for monoclonal antibody therapy, alluding to the untapped potential of other tumor-specific antigens that have yet to be discovered. Importantly, PRL3-zumab could potentially serve as an alternative immunotherapy option for patients who are unsuitable for treatment with ICIs, thereby addressing an urgent unmet medical need.
Acknowledgements
We would like to acknowledge the support from our lab mates Jie Li and Kam Yew Kuan.
Funding
This research was funded by Agency for Science, Technology and Research, Institute of Molecular and Cell Biology core fund and Career Development Fund [C210812005].
Author Contributions
Conceptualization, P.L.C. and K.H.A.; writing—original draft preparation, P.L.C. and K.H.A.; writing—review and editing, P.L.C., K.H.A. and M.T.; supervision, Q.Z.; project administration, Q.Z.; All authors have read and agreed to the published version of the manuscript.
Competing Interests
Q.Z. is the founder of Intra-Immu SG Pte Ltd., an Agency for Science, Technology and Research (A*STAR) spin-off company granted licensing rights for the PRL3-zumab IP portfolio. The other authors declare no conflict of interest.
References
1. Gao PP, Qi XW, Sun N, Sun YY, Zhang Y, Tan XN. et al. The emerging roles of dual-specificity phosphatases and their specific characteristics in human cancer. Biochim Biophys Acta Rev Cancer. 2021;1876:188562
2. Zeng Q, Hong W, Tan YH. Mouse PRL-2 and PRL-3, two potentially prenylated protein tyrosine phosphatases homologous to PRL-1. Biochem Biophys Res Commun. 1998;244:421-7
3. Zeng Q, Si X, Horstmann H, Xu Y, Hong W, Pallen CJ. Prenylation-dependent association of protein-tyrosine phosphatases PRL-1, -2, and -3 with the plasma membrane and the early endosome. J Biol Chem. 2000;275:21444-52
4. Zeng Q, Dong JM, Guo K, Li J, Tan HX, Koh V. et al. PRL-3 and PRL-1 promote cell migration, invasion, and metastasis. Cancer Res. 2003;63:2716-22
5. Guo K, Li J, Tang JP, Koh V, Gan BQ, Zeng Q. Catalytic domain of PRL-3 plays an essential role in tumor metastasis: formation of PRL-3 tumors inside the blood vessels. Cancer Biol Ther. 2004;3:945-51
6. Saha S, Bardelli A, Buckhaults P, Velculescu VE, Rago C, St Croix B. et al. A phosphatase associated with metastasis of colorectal cancer. Science. 2001;294:1343-6
7. Wang H, Vardy LA, Tan CP, Loo JM, Guo K, Li J. et al. PCBP1 suppresses the translation of metastasis-associated PRL-3 phosphatase. Cancer Cell. 2010;18:52-62
8. Thura M, Al-Aidaroos AQ, Gupta A, Chee CE, Lee SC, Hui KM. et al. PRL3-zumab as an immunotherapy to inhibit tumors expressing PRL3 oncoprotein. Nature Communications. 2019;10:2484
9. Li J, Guo K, Koh VW, Tang JP, Gan BQ, Shi H. et al. Generation of PRL-3- and PRL-1-specific monoclonal antibodies as potential diagnostic markers for cancer metastases. Clin Cancer Res. 2005;11:2195-204
10. Gown AM. Diagnostic Immunohistochemistry: What Can Go Wrong and How to Prevent It. Archives of Pathology & Laboratory Medicine. 2016;140:893-8
11. Kato H, Semba S, Miskad UA, Seo Y, Kasuga M, Yokozaki H. High expression of PRL-3 promotes cancer cell motility and liver metastasis in human colorectal cancer: a predictive molecular marker of metachronous liver and lung metastases. Clin Cancer Res. 2004;10:7318-28
12. Bardelli A, Saha S, Sager JA, Romans KE, Xin B, Markowitz SD. et al. PRL-3 expression in metastatic cancers. Clin Cancer Res. 2003;9:5607-15
13. Zhao WB, Li Y, Liu X, Zhang LY, Wang X. Evaluation of PRL-3 expression, and its correlation with angiogenesis and invasion in hepatocellular carcinoma. Int J Mol Med. 2008;22:187-92
14. Zhou Q, Zhou Q, Liu Q, He Z, Yan Y, Lin J. et al. PRL-3 facilitates Hepatocellular Carcinoma progression by co-amplifying with and activating FAK. Theranostics. 2020;10:10345-59
15. Li BH, Wang Y, Wang CY, Zhao MJ, Deng T, Ren XQ. Up-Regulation of Phosphatase in Regenerating Liver-3 (PRL-3) Contributes to Malignant Progression of Hepatocellular Carcinoma by Activating Phosphatase and Tensin Homolog Deleted on Chromosome Ten (PTEN)/Phosphoinositide 3-Kinase (PI3K)/AKT Signaling Pathway. Med Sci Monit. 2018;24:8105-14
16. Xu Y, Zhu M, Zhang S, Liu H, Li T, Qin C. Expression and prognostic value of PRL-3 in human intrahepatic cholangiocarcinoma. Pathol Oncol Res. 2010;16:169-75
17. Li Z-R, Wang Z, Zhu B-H, He Y-L, Peng J-S, Cai S-R. et al. Association of Tyrosine PRL-3 Phosphatase Protein Expression with Peritoneal Metastasis of Gastric Carcinoma and Prognosis. Surgery Today. 2007;37:646-51
18. Miskad UA, Semba S, Kato H, Matsukawa Y, Kodama Y, Mizuuchi E. et al. High PRL-3 expression in human gastric cancer is a marker of metastasis and grades of malignancies: an in situ hybridization study. Virchows Archiv. 2007;450:303-10
19. Hao R-T, Zhang X-H, Pan Y-F, Liu H-G, Xiang Y-Q, Wan L. et al. Prognostic and metastatic value of phosphatase of regenerating liver-3 in invasive breast cancer. Journal of Cancer Research and Clinical Oncology. 2010;136:1349-57
20. Polato F, Codegoni A, Fruscio R, Perego P, Mangioni C, Saha S. et al. PRL-3 phosphatase is implicated in ovarian cancer growth. Clin Cancer Res. 2005;11:6835-9
21. Kong L, Li Q, Wang L, Liu Z, Sun T. The value and correlation between PRL-3 expression and matrix metalloproteinase activity and expression in human gliomas. Neuropathology. 2007;27:516-21
22. Ooki A, Yamashita K, Kikuchi S, Sakuramoto S, Katada N, Watanabe M. Phosphatase of regenerating liver-3 as a convergent therapeutic target for lymph node metastasis in esophageal squamous cell carcinoma. Int J Cancer. 2010;127:543-54
23. Fagerli UM, Holt RU, Holien T, Vaatsveen TK, Zhan F, Egeberg KW. et al. Overexpression and involvement in migration by the metastasis-associated phosphatase PRL-3 in human myeloma cells. Blood. 2008;111:806-15
24. Ming J, Liu N, Gu Y, Qiu X, Wang EH. PRL-3 facilitates angiogenesis and metastasis by increasing ERK phosphorylation and up-regulating the levels and activities of Rho-A/C in lung cancer. Pathology. 2009;41:118-26
25. Ma Y, Li B. Expression of phosphatase of regenerating liver-3 in squamous cell carcinoma of the cervix. Med Oncol. 2011;28:775-80
26. Laurent C, Valet F, Planque N, Silveri L, Maacha S, Anezo O. et al. High PTP4A3 phosphatase expression correlates with metastatic risk in uveal melanoma patients. Cancer Res. 2011;71:666-74
27. Dong Q, Ding X, Chang B, Wang H, Wang A. PRL-3 promotes migration and invasion and is associated with poor prognosis in salivary adenoid cystic carcinoma. J Oral Pathol Med. 2016;45:111-8
28. Zhou J, Bi C, Chng WJ, Cheong LL, Liu SC, Mahara S. et al. PRL-3, a metastasis associated tyrosine phosphatase, is involved in FLT3-ITD signaling and implicated in anti-AML therapy. PLoS One. 2011;6:e19798
29. Guzińska-Ustymowicz K, Kiśluk J, Terlikowski SJ, Pryczynicz A, Niewiarowska K, Ustymowicz M. et al. Expression of phosphatase of regenerating liver-3 (PRL-3) in endometrioid cancer and lymph nodes metastases. Adv Med Sci. 2013;58:221-6
30. Johansson JA, Marie KL, Lu Y, Brombin A, Santoriello C, Zeng Z. et al. PRL3-DDX21 Transcriptional Control of Endolysosomal Genes Restricts Melanocyte Stem Cell Differentiation. Dev Cell. 2020;54:317-32.e9
31. Vandsemb EN, Bertilsson H, Abdollahi P, Storkersen O, Vatsveen TK, Rye MB. et al. Phosphatase of regenerating liver 3 (PRL-3) is overexpressed in human prostate cancer tissue and promotes growth and migration. J Transl Med. 2016;14:71
32. Hjort MA, Abdollahi P, Vandsemb EN, Fenstad MH, Lund B, Slørdahl TS. et al. Phosphatase of regenerating liver-3 is expressed in acute lymphoblastic leukemia and mediates leukemic cell adhesion, migration and drug resistance. Oncotarget. 2018;9:3549-61
33. Sun F, Li W, Wang L, Jiao C. Expression of phosphatase of regenerating liver-3 is associated with prognosis of Wilms' tumor. Onco Targets Ther. 2017;10:311-7
34. Hjort MA, Hov H, Abdollahi P, Vandsemb EN, Fagerli UM, Lund B. et al. Phosphatase of regenerating liver-3 (PRL-3) is overexpressed in classical Hodgkin lymphoma and promotes survival and migration. Exp Hematol Oncol. 2018;7:8
35. Hanahan D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022;12:31-46
36. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646-74
37. Hanahan D, Weinberg RA. The Hallmarks of Cancer. Cell. 2000;100:57-70
38. Duciel L, Monraz Gomez LC, Kondratova M, Kuperstein I, Saule S. The Phosphatase PRL-3 Is Involved in Key Steps of Cancer Metastasis. J Mol Biol. 2019;431:3056-67
39. Al-Aidaroos AQO, Zeng Q. PRL-3 phosphatase and cancer metastasis. Journal of Cellular Biochemistry. 2010;111:1087-98
40. Matter WF, Estridge T, Zhang C, Belagaje R, Stancato L, Dixon J. et al. Role of PRL-3, a human muscle-specific tyrosine phosphatase, in angiotensin-II signaling. Biochem Biophys Res Commun. 2001;283:1061-8
41. Wu X, Zeng H, Zhang X, Zhao Y, Sha H, Ge X. et al. Phosphatase of Regenerating Liver-3 Promotes Motility and Metastasis of Mouse Melanoma Cells. The American Journal of Pathology. 2004;164:2039-54
42. Csoboz B, Gombos I, Tatrai E, Tovari J, Kiss AL, Horvath I. et al. Chemotherapy induced PRL3 expression promotes cancer growth via plasma membrane remodeling and specific alterations of caveolae-associated signaling. Cell Commun Signal. 2018;16:51
43. Liu H, Al-aidaroos AQ, Wang H, Guo K, Li J, Zhang HF. et al. PRL-3 suppresses c-Fos and integrin α2 expression in ovarian cancer cells. BMC Cancer. 2013;13:80
44. Huang YH, Al-Aidaroos AQ, Yuen HF, Zhang SD, Shen HM, Rozycka E. et al. A role of autophagy in PTP4A3-driven cancer progression. Autophagy. 2014;10:1787-800
45. McQueeney KE, Salamoun JM, Burnett JC, Barabutis N, Pekic P, Lewandowski SL. et al. Targeting ovarian cancer and endothelium with an allosteric PTP4A3 phosphatase inhibitor. Oncotarget. 2018;9:8223-40
46. Matsukawa Y, Semba S, Kato H, Koma Y, Yanagihara K, Yokozaki H. Constitutive suppression of PRL-3 inhibits invasion and proliferation of gastric cancer cell in vitro and in vivo. Pathobiology. 2010;77:155-62
47. Ooki A, Yamashita K, Kikuchi S, Sakuramoto S, Katada N, Waraya M. et al. Therapeutic potential of PRL-3 targeting and clinical significance of PRL-3 genomic amplification in gastric cancer. BMC Cancer. 2011;11:122
48. Al-Aidaroos AQ, Yuen HF, Guo K, Zhang SD, Chung TH, Chng WJ. et al. Metastasis-associated PRL-3 induces EGFR activation and addiction in cancer cells. J Clin Invest. 2013;123:3459-71
49. Daouti S, Li WH, Qian H, Huang KS, Holmgren J, Levin W. et al. A selective phosphatase of regenerating liver phosphatase inhibitor suppresses tumor cell anchorage-independent growth by a novel mechanism involving p130Cas cleavage. Cancer Res. 2008;68:1162-9
50. Lai W, Chen S, Wu H, Guan Y, Liu L, Zeng Y. et al. PRL-3 promotes the proliferation of LoVo cells via the upregulation of KCNN4 channels. Oncol Rep. 2011;26:909-17
51. Zhang J, Xiao Z, Lai D, Sun J, He C, Chu Z. et al. miR-21, miR-17 and miR-19a induced by phosphatase of regenerating liver-3 promote the proliferation and metastasis of colon cancer. Br J Cancer. 2012;107:352-9
52. Zimmerman MW, Homanics GE, Lazo JS. Targeted deletion of the metastasis-associated phosphatase Ptp4a3 (PRL-3) suppresses murine colon cancer. PLoS One. 2013;8:e58300
53. Zhang C, Qu L, Lian S, Meng L, Min L, Liu J. et al. PRL-3 Promotes Ubiquitination and Degradation of AURKA and Colorectal Cancer Progression via Dephosphorylation of FZR1. Cancer Res. 2019;79:928-40
54. Gari HH, DeGala GD, Ray R, Lucia MS, Lambert JR. PRL-3 engages the focal adhesion pathway in triple-negative breast cancer cells to alter actin structure and substrate adhesion properties critical for cell migration and invasion. Cancer Lett. 2016;380:505-12
55. Edwards DR, Moroz K, Zhang H, Mulholland D, Abdel-Mageed AB, Mondal D. PRL3 increases the aggressive phenotype of prostate cancer cells in vitro and its expression correlates with high-grade prostate tumors in patients. Int J Oncol. 2018;52:402-12
56. Abdollahi P, Vandsemb EN, Elsaadi S, Rost LM, Yang R, Hjort MA. et al. Phosphatase of regenerating liver-3 regulates cancer cell metabolism in multiple myeloma. FASEB J. 2021;35:e21344
57. Slørdahl TS, Abdollahi P, Vandsemb EN, Rampa C, Misund K, Baranowska KA. et al. The phosphatase of regenerating liver-3 (PRL-3) is important for IL-6-mediated survival of myeloma cells. Oncotarget. 2016;7:27295-306
58. Mu N, Gu J, Liu N, Xue X, Shu Z, Zhang K. et al. PRL-3 is a potential glioblastoma prognostic marker and promotes glioblastoma progression by enhancing MMP7 through the ERK and JNK pathways. Theranostics. 2018;8:1527-39
59. Park JE, Yuen HF, Zhou JB, Al-Aidaroos AQ, Guo K, Valk PJ. et al. Oncogenic roles of PRL-3 in FLT3-ITD induced acute myeloid leukaemia. EMBO Mol Med. 2013;5:1351-66
60. Chong PS, Zhou J, Cheong LL, Liu SC, Qian J, Guo T. et al. LEO1 is regulated by PRL-3 and mediates its oncogenic properties in acute myelogenous leukemia. Cancer Res. 2014;74:3043-53
61. Qu S, Liu B, Guo X, Shi H, Zhou M, Li L. et al. Independent oncogenic and therapeutic significance of phosphatase PRL-3 in FLT3-ITD-negative acute myeloid leukemia. Cancer. 2014;120:2130-41
62. Zhou J, Cheong LL, Liu SC, Chong PS, Mahara S, Bi C. et al. The pro-metastasis tyrosine phosphatase, PRL-3 (PTP4A3), is a novel mediator of oncogenic function of BCR-ABL in human chronic myeloid leukemia. Mol Cancer. 2012;11:72
63. Garcia EG, Veloso A, Oliveira ML, Allen JR, Loontiens S, Brunson D. et al. PRL3 enhances T-cell acute lymphoblastic leukemia growth through suppressing T-cell signaling pathways and apoptosis. Leukemia. 2021;35:679-90
64. Xing X, Lian S, Hu Y, Li Z, Zhang L, Wen X. et al. Phosphatase of regenerating liver-3 (PRL-3) is associated with metastasis and poor prognosis in gastric carcinoma. J Transl Med. 2013;11:309
65. Xiong J, Li Z, Zhang Y, Li D, Zhang G, Luo X. et al. PRL-3 promotes the peritoneal metastasis of gastric cancer through the PI3K/Akt signaling pathway by regulating PTEN. Oncol Rep. 2016;36:1819-28
66. Zhang C, Tian W, Meng L, Qu L, Shou C. PRL-3 promotes gastric cancer migration and invasion through a NF-kappaB-HIF-1alpha-miR-210 axis. J Mol Med (Berl). 2016;94:401-15
67. Jiang Y, Liu XQ, Rajput A, Geng L, Ongchin M, Zeng Q. et al. Phosphatase PRL-3 is a direct regulatory target of TGFbeta in colon cancer metastasis. Cancer Res. 2011;71:234-44
68. Min G, Lee SK, Kim HN, Han YM, Lee RH, Jeong DG. et al. Rhodanine-based PRL-3 inhibitors blocked the migration and invasion of metastatic cancer cells. Bioorg Med Chem Lett. 2013;23:3769-74
69. Fiordalisi JJ, Dewar BJ, Graves LM, Madigan JP, Cox AD. Src-mediated phosphorylation of the tyrosine phosphatase PRL-3 is required for PRL-3 promotion of Rho activation, motility and invasion. PLoS One. 2013;8:e64309
70. Xu H, Lai W, Zhang Y, Liu L, Luo X, Zeng Y. et al. Tumor-associated macrophage-derived IL-6 and IL-8 enhance invasive activity of LoVo cells induced by PRL-3 in a KCNN4 channel-dependent manner. BMC Cancer. 2014;14:330
71. Yang Y, Lian S, Meng L, Qu L, Shou C. Antibody Array Revealed PRL-3 Affects Protein Phosphorylation and Cytokine Secretion. PLoS One. 2017;12:e0169665
72. Lan Q, Lai W, Zeng Y, Liu L, Li S, Jin S. et al. CCL26 Participates in the PRL-3-Induced Promotion of Colorectal Cancer Invasion by Stimulating Tumor-Associated Macrophage Infiltration. Mol Cancer Ther. 2018;17:276-89
73. Gari HH, Gearheart CM, Fosmire S, DeGala GD, Fan Z, Torkko KC. et al. Genome-wide functional genetic screen with the anticancer agent AMPI-109 identifies PRL-3 as an oncogenic driver in triple-negative breast cancers. Oncotarget. 2016;7:15757-71
74. Qian F, Li YP, Sheng X, Zhang ZC, Song R, Dong W. et al. PRL-3 siRNA inhibits the metastasis of B16-BL6 mouse melanoma cells in vitro and in vivo. Mol Med. 2007;13:151-9
75. Song R, Qian F, Li YP, Sheng X, Cao SX, Xu Q. Phosphatase of regenerating liver-3 localizes to cyto-membrane and is required for B16F1 melanoma cell metastasis in vitro and in vivo. PLoS One. 2009;4:e4450
76. Kozlov G, Funato Y, Chen YS, Zhang Z, Illes K, Miki H. et al. PRL3 pseudophosphatase activity is necessary and sufficient to promote metastatic growth. J Biol Chem. 2020;295:11682-92
77. Lazo JS, Sharlow ER, Cornelison R, Hart DJ, Llaneza DC, Mendelson AJ. et al. Credentialing and Pharmacologically Targeting PTP4A3 Phosphatase as a Molecular Target for Ovarian Cancer. Biomolecules. 2021 11
78. Zhou J, Wang S, Lu J, Li J, Ding Y. Over-expression of phosphatase of regenerating liver-3 correlates with tumor progression and poor prognosis in nasopharyngeal carcinoma. Int J Cancer. 2009;124:1879-86
79. Xu J, Wu W, Tang Y, Lin Y, Xue Y, Hu J. et al. PRL-3 exerts oncogenic functions in myeloid leukemia cells via aberrant dephosphorylation of stathmin and activation of STAT3 signaling. Aging (Albany NY). 2019;11:7817-29
80. Li Z, Zhan W, Wang Z, Zhu B, He Y, Peng J. et al. Inhibition of PRL-3 gene expression in gastric cancer cell line SGC7901 via microRNA suppressed reduces peritoneal metastasis. Biochem Biophys Res Commun. 2006;348:229-37
81. Liang F, Liang J, Wang WQ, Sun JP, Udho E, Zhang ZY. PRL3 promotes cell invasion and proliferation by down-regulation of Csk leading to Src activation. J Biol Chem. 2007;282:5413-9
82. Wang H, Quah SY, Dong JM, Manser E, Tang JP, Zeng Q. PRL-3 down-regulates PTEN expression and signals through PI3K to promote epithelial-mesenchymal transition. Cancer Res. 2007;67:2922-6
83. Lai W, Liu L, Zeng Y, Wu H, Xu H, Chen S. et al. KCNN4 channels participate in the EMT induced by PRL-3 in colorectal cancer. Med Oncol. 2013;30:566
84. Liu Y, Zhou J, Chen J, Gao W, Le Y, Ding Y. et al. PRL-3 promotes epithelial mesenchymal transition by regulating cadherin directly. Cancer Biol Ther. 2009;8:1352-9
85. Zhang T, Liu L, Lai W, Zeng Y, Xu H, Lan Q. et al. Interaction with tumorassociated macrophages promotes PRL3induced invasion of colorectal cancer cells via MAPK pathwayinduced EMT and NFkappaB signalinginduced angiogenesis. Oncol Rep. 2019;41:2790-802
86. Walls CD, Iliuk A, Bai Y, Wang M, Tao WA, Zhang ZY. Phosphatase of regenerating liver 3 (PRL3) provokes a tyrosine phosphoproteome to drive prometastatic signal transduction. Mol Cell Proteomics. 2013;12:3759-77
87. Gari HH, DeGala GD, Lucia MS, Lambert JR. Loss of the oncogenic phosphatase PRL-3 promotes a TNF-R1 feedback loop that mediates triple-negative breast cancer growth. Oncogenesis. 2016;5:e255
88. den Hollander P, Rawls K, Tsimelzon A, Shepherd J, Mazumdar A, Hill J. et al. Phosphatase PTP4A3 Promotes Triple-Negative Breast Cancer Growth and Predicts Poor Patient Survival. Cancer Res. 2016;76:1942-53
89. Min SH, Kim DM, Heo YS, Kim HM, Kim IC, Yoo OJ. Downregulation of p53 by phosphatase of regenerating liver 3 is mediated by MDM2 and PIRH2. Life Sci. 2010;86:66-72
90. Abdollahi P, Vandsemb EN, Hjort MA, Misund K, Holien T, Sponaas AM. et al. Src Family Kinases Are Regulated in Multiple Myeloma Cells by Phosphatase of Regenerating Liver-3. Mol Cancer Res. 2017;15:69-77
91. Lian S, Meng L, Yang Y, Ma T, Xing X, Feng Q. et al. PRL-3 promotes telomere deprotection and chromosomal instability. Nucleic Acids Res. 2017;45:6546-71
92. Thura M, Ye Z, Al-Aidaroos AQ, Xiong Q, Ong JY, Gupta A. et al. PRL3 induces polypoid giant cancer cells eliminated by PRL3-zumab to reduce tumor relapse. Commun Biol. 2021;4:923
93. Guo K, Li J, Wang H, Osato M, Tang JP, Quah SY. et al. PRL-3 initiates tumor angiogenesis by recruiting endothelial cells in vitro and in vivo. Cancer Res. 2006;66:9625-35
94. Parker BS, Argani P, Cook BP, Liangfeng H, Chartrand SD, Zhang M. et al. Alterations in vascular gene expression in invasive breast carcinoma. Cancer Res. 2004;64:7857-66
95. Ming J, Jiang Y, Jiang G, Zheng H. Phosphatase of regenerating liver-3 induces angiogenesis by increasing extracellular signal-regulated kinase phosphorylation in endometrial adenocarcinoma. Pathobiology. 2014;81:1-7
96. Morse MA, Sun W, Kim R, He AR, Abada PB, Mynderse M. et al. The Role of Angiogenesis in Hepatocellular Carcinoma. Clinical Cancer Research. 2019;25:912-20
97. Zhu AX, Duda DG, Sahani DV, Jain RK. HCC and angiogenesis: possible targets and future directions. Nature Reviews Clinical Oncology. 2011;8:292-301
98. Xu J, Cao S, Wang L, Xu R, Chen G, Xu Q. VEGF promotes the transcription of the human PRL-3 gene in HUVEC through transcription factor MEF2C. PLoS One. 2011;6:e27165
99. Liberti MV, Locasale JW. The Warburg Effect: How Does it Benefit Cancer Cells? Trends Biochem Sci. 2016;41:211-8
100. Xu H, Zeng Y, Liu L, Gao Q, Jin S, Lan Q. et al. PRL-3 improves colorectal cancer cell proliferation and invasion through IL-8 mediated glycolysis metabolism. Int J Oncol. 2017;51:1271-9
101. Yang Y, Lian S, Meng L, Tian Z, Feng Q, Wang Y. et al. Knockdown of PRL-3 increases mitochondrial superoxide anion production through transcriptional regulation of RAP1. Cancer Manag Res. 2018;10:5071-81
102. Funato Y, Miki H. The emerging roles and therapeutic potential of cyclin M/CorC family of Mg(2+) transporters. J Pharmacol Sci. 2022;148:14-8
103. Funato Y, Yoshida A, Hirata Y, Hashizume O, Yamazaki D, Miki H. The Oncogenic PRL Protein Causes Acid Addiction of Cells by Stimulating Lysosomal Exocytosis. Dev Cell. 2020;55:387-97 e8
104. Funato Y, Yamazaki D, Mizukami S, Du L, Kikuchi K, Miki H. Membrane protein CNNM4-dependent Mg2+ efflux suppresses tumor progression. J Clin Invest. 2014;124:5398-410
105. Hardy S, Uetani N, Wong N, Kostantin E, Labbé DP, Bégin LR. et al. The protein tyrosine phosphatase PRL-2 interacts with the magnesium transporter CNNM3 to promote oncogenesis. Oncogene. 2015;34:986-95
106. Hardy S, Kostantin E, Wang SJ, Hristova T, Galicia-Vazquez G, Baranov PV. et al. Magnesium-sensitive upstream ORF controls PRL phosphatase expression to mediate energy metabolism. Proc Natl Acad Sci U S A. 2019;116:2925-34
107. Chan DA, Sutphin PD, Nguyen P, Turcotte S, Lai EW, Banh A. et al. Targeting GLUT1 and the Warburg effect in renal cell carcinoma by chemical synthetic lethality. Sci Transl Med. 2011;3:94ra70
108. Reckzeh ES, Waldmann H. Small-Molecule Inhibition of Glucose Transporters GLUT-1-4. Chembiochem. 2020;21:45-52
109. Zhang M, Wei Y, Liu Y, Guan W, Zhang X, Kong J. et al. Metastatic Phosphatase PRL-3 Induces Ovarian Cancer Stem Cell Sub-population through Phosphatase-Independent Deacetylation Modulations. iScience. 2020;23:100766
110. Johansson JA, Marie KL, Lu Y, Brombin A, Santoriello C, Zeng Z. et al. PRL3-DDX21 Transcriptional Control of Endolysosomal Genes Restricts Melanocyte Stem Cell Differentiation. Dev Cell. 2020;54:317-32 e9
111. Takeda M, Koseki J, Takahashi H, Miyoshi N, Nishida N, Nishimura J. et al. Disruption of Endolysosomal RAB5/7 Efficiently Eliminates Colorectal Cancer Stem Cells. Cancer Res. 2019;79:1426-37
112. Garcia-Prat L, Kaufmann KB, Schneiter F, Voisin V, Murison A, Chen J. et al. TFEB-mediated endolysosomal activity controls human hematopoietic stem cell fate. Cell Stem Cell. 2021;28:1838-50 e10
113. Zhang S, Xiong X, Sun Y. Functional characterization of SOX2 as an anticancer target. Signal Transduct Target Ther. 2020;5:135
114. Boedtkjer E, Pedersen SF. The Acidic Tumor Microenvironment as a Driver of Cancer. Annu Rev Physiol. 2020;82:103-26
115. Baghban R, Roshangar L, Jahanban-Esfahlan R, Seidi K, Ebrahimi-Kalan A, Jaymand M. et al. Tumor microenvironment complexity and therapeutic implications at a glance. Cell Commun Signal. 2020;18:59
116. Machado E, White-Gilbertson S, Vlekkert Dvd, Janke L, Moshiach S, Campos Y. et al. Regulated lysosomal exocytosis mediates cancer progression. Science Advances. 2015;1:e1500603
117. Tang T, Yang Z-y, Wang D, Yang X-y, Wang J, Li L. et al. The role of lysosomes in cancer development and progression. Cell & Bioscience. 2020;10:131
118. Sanchez-Vega F, Mina M, Armenia J, Chatila WK, Luna A, La KC. et al. Oncogenic Signaling Pathways in The Cancer Genome Atlas. Cell. 2018;173:321-37 e10
119. Ferrara N. VEGF as a therapeutic target in cancer. Oncology. 2005;69(Suppl 3):11-6
120. Ferrara N, Gerber HP, LeCouter J. The biology of VEGF and its receptors. Nat Med. 2003;9:669-76
121. Wei M, Haney MG, Rivas DR, Blackburn JS. Protein tyrosine phosphatase 4A3 (PTP4A3/PRL-3) drives migration and progression of T-cell acute lymphoblastic leukemia in vitro and in vivo. Oncogenesis. 2020;9:6
122. Peng L, Xing X, Li W, Qu L, Meng L, Lian S. et al. PRL-3 promotes the motility, invasion, and metastasis of LoVo colon cancer cells through PRL-3-integrin beta1-ERK1/2 and-MMP2 signaling. Mol Cancer. 2009;8:110
123. Peng L, Jin G, Wang L, Guo J, Meng L, Shou C. Identification of integrin alpha1 as an interacting protein of protein tyrosine phosphatase PRL-3. Biochem Biophys Res Commun. 2006;342:179-83
124. Fiordalisi JJ, Keller PJ, Cox AD. PRL tyrosine phosphatases regulate rho family GTPases to promote invasion and motility. Cancer Res. 2006;66:3153-61
125. Zhan H, Ma J, Ruan F, Bedaiwy MA, Peng B, Wu R. et al. Elevated phosphatase of regenerating liver 3 (PRL-3) promotes cytoskeleton reorganization, cell migration and invasion in endometrial stromal cells from endometrioma. Hum Reprod. 2016;31:723-33
126. Kleiner DE, Stetler-Stevenson WG. Matrix metalloproteinases and metastasis. Cancer Chemother Pharmacol. 1999;43(Suppl):S42-51
127. Stamenkovic I. Matrix metalloproteinases in tumor invasion and metastasis. Semin Cancer Biol. 2000;10:415-33
128. Lee SK, Han YM, Yun J, Lee CW, Shin DS, Ha YR. et al. Phosphatase of regenerating liver-3 promotes migration and invasion by upregulating matrix metalloproteinases-7 in human colorectal cancer cells. Int J Cancer. 2012;131:E190-203
129. Yu L-X, Ling Y, Wang H-Y. Role of nonresolving inflammation in hepatocellular carcinoma development and progression. npj Precision Oncology. 2018;2:6
130. Chong PSY, Zhou J, Lim JSL, Hee YT, Chooi JY, Chung TH. et al. IL6 Promotes a STAT3-PRL3 Feedforward Loop via SHP2 Repression in Multiple Myeloma. Cancer Res. 2019;79:4679-88
131. Li QT, Feng YM, Ke ZH, Qiu MJ, He XX, Wang MM. et al. KCNN4 promotes invasion and metastasis through the MAPK/ERK pathway in hepatocellular carcinoma. J Investig Med. 2020;68:68-74
132. Basak S, Jacobs SB, Krieg AJ, Pathak N, Zeng Q, Kaldis P. et al. The metastasis-associated gene Prl-3 is a p53 target involved in cell-cycle regulation. Mol Cell. 2008;30:303-14
133. Xie H, Wang H. PRL-3 promotes breast cancer progression by downregulating p14(ARF)-mediated p53 expression. Oncol Lett. 2018;15:2795-800
134. Fang XY, Song R, Chen W, Yang YY, Gu YH, Shu YQ. et al. PRL-3 Promotes the Malignant Progression of Melanoma via Triggering Dephosphorylation and Cytoplasmic Localization of NHERF1. J Invest Dermatol. 2015;135:2273-82
135. Luo T, Gao Y, Zhangyuan G, Xu X, Xue C, Jin L. et al. lncRNA PCBP1-AS1 Aggravates the Progression of Hepatocellular Carcinoma via Regulating PCBP1/PRL-3/AKT Pathway. Cancer Manag Res. 2020;12:5395-408
136. Ye Z, Al-Aidaroos AQ, Park JE, Yuen HF, Zhang SD, Gupta A. et al. PRL-3 activates mTORC1 in Cancer Progression. Sci Rep. 2015;5:17046
137. Shi Y, Xu S, Ngoi NYL, Hui Y, Ye Z. Rag GTPases suppress PRL-3 degradation and predict poor clinical diagnosis of cancer patients with low PRL-3 mRNA expression. Biochem Biophys Res Commun. 2021;576:108-16
138. Chong PSY, Zhou J, Chooi JY, Chan ZL, Toh SHM, Tan TZ. et al. Non-canonical activation of beta-catenin by PRL-3 phosphatase in acute myeloid leukemia. Oncogene. 2019;38:1508-19
139. Zhou J, Toh SH, Chan ZL, Quah JY, Chooi JY, Tan TZ. et al. A loss-of-function genetic screening reveals synergistic targeting of AKT/mTOR and WTN/beta-catenin pathways for treatment of AML with high PRL-3 phosphatase. J Hematol Oncol. 2018;11:36
140. Pathak MK, Dhawan D, Lindner DJ, Borden EC, Farver C, Yi T. Pentamidine is an inhibitor of PRL phosphatases with anticancer activity. Mol Cancer Ther. 2002;1:1255-64
141. Ahn JH, Kim SJ, Park WS, Cho SY, Ha JD, Kim SS. et al. Synthesis and biological evaluation of rhodanine derivatives as PRL-3 inhibitors. Bioorg Med Chem Lett. 2006;16:2996-9
142. Zimmerman MW, McQueeney KE, Isenberg JS, Pitt BR, Wasserloos KA, Homanics GE. et al. Protein-tyrosine phosphatase 4A3 (PTP4A3) promotes vascular endothelial growth factor signaling and enables endothelial cell motility. J Biol Chem. 2014;289:5904-13
143. Chen X, Rong D, Cai W, Tong X, Wang H. The combination of PRL-3 inhibitor with sorafenib synergistically promotes AML apoptosis. Neoplasma. 2021;68:832-41
144. Lin L, Lu L, Yuan C, Wang A, Zhu M, Fu X. et al. The dual inhibition against the activity and expression of tyrosine phosphatase PRL-3 from a rhodanine derivative. Bioorg Med Chem Lett. 2021;41:127981
145. Qi L, Zhang Y, Zhang W, Wang Y, Han Y, Ding Y. The inhibition of colorectal cancer growth by the natural product macrocarpal I. Free Radic Biol Med. 2021;162:383-91
146. Park H, Jung SK, Jeong DG, Ryu SE, Kim SJ. Discovery of novel PRL-3 inhibitors based on the structure-based virtual screening. Bioorg Med Chem Lett. 2008;18:2250-5
147. Hoeger B, Diether M, Ballester PJ, Kohn M. Biochemical evaluation of virtual screening methods reveals a cell-active inhibitor of the cancer-promoting phosphatases of regenerating liver. Eur J Med Chem. 2014;88:89-100
148. Leung WH, Vong QP, Lin W, Bouck D, Wendt S, Sullivan E. et al. PRL-3 mediates the protein maturation of ULBP2 by regulating the tyrosine phosphorylation of HSP60. J Immunol. 2015;194:2930-41
149. Salamoun JM, McQueeney KE, Patil K, Geib SJ, Sharlow ER, Lazo JS. et al. Photooxygenation of an amino-thienopyridone yields a more potent PTP4A3 inhibitor. Org Biomol Chem. 2016;14:6398-402
150. Guo K, Tang JP, Tan CP, Wang H, Zeng Q. Monoclonal antibodies target intracellular PRL phosphatases to inhibit cancer metastases in mice. Cancer Biol Ther. 2008;7:750-7
151. Guo K, Tang JP, Jie L, Al-Aidaroos AQ, Hong CW, Tan CP. et al. Engineering the first chimeric antibody in targeting intracellular PRL-3 oncoprotein for cancer therapy in mice. Oncotarget. 2012;3:158-71
152. Thura M, Al-Aidaroos AQ, Gupta A, Chee CE, Lee SC, Hui KM. et al. PRL3-zumab as an immunotherapy to inhibit tumors expressing PRL3 oncoprotein. Nat Commun. 2019;10:2484
153. Thura M, Al-Aidaroos AQO, Yong WP, Kono K, Gupta A, Lin YB. et al. PRL3-zumab, a first-in-class humanized antibody for cancer therapy. JCI Insight. 2016;1:e87607
154. Wei M, Korotkov KV, Blackburn JS. Targeting phosphatases of regenerating liver (PRLs) in cancer. Pharmacol Ther. 2018;190:128-38
155. Rivas DR, Dela Cerna MVC, Smith CN, Sampathi S, Patty BG, Lee D. et al. A screen of FDA-approved drugs identifies inhibitors of protein tyrosine phosphatase 4A3 (PTP4A3 or PRL-3). Sci Rep. 2021;11:10302
156. Stephens BJ, Han H, Gokhale V, Von Hoff DD. PRL phosphatases as potential molecular targets in cancer. Mol Cancer Ther. 2005;4:1653-61
157. Chames P, Van Regenmortel M, Weiss E, Baty D. Therapeutic antibodies: successes, limitations and hopes for the future. Br J Pharmacol. 2009;157:220-33
158. Trenevska I, Li D, Banham AH. Therapeutic Antibodies against Intracellular Tumor Antigens. Frontiers in Immunology. 2017 8
159. Wang Y, Wang X, Ferrone CR, Schwab JH, Ferrone S. Intracellular antigens as targets for antibody based immunotherapy of malignant diseases. Mol Oncol. 2015;9:1982-93
160. Guo K, Li J, Tang JP, Tan CPB, Hong CW, Al-Aidaroos AQO. et al. Targeting Intracellular Oncoproteins with Antibody Therapy or Vaccination. Science Translational Medicine. 2011;3:99ra85-99ra85
161. Ferrone S. Hidden Immunotherapy Targets Challenge Dogma. Science Translational Medicine. 2011;3:99ps38-99ps38
162. Weidle UH, Maisel D, Klostermann S, Schiller C, Weiss EH. Intracellular proteins displayed on the surface of tumor cells as targets for therapeutic intervention with antibody-related agents. Cancer Genomics Proteomics. 2011;8:49-63
163. Li X, Sun L, Hou J, Gui M, Ying J, Zhao H. et al. Cell membrane gp96 facilitates HER2 dimerization and serves as a novel target in breast cancer. International Journal of Cancer. 2015;137:512-24
164. Stangl S, Gehrmann M, Riegger J, Kuhs K, Riederer I, Sievert W. et al. Targeting membrane heat-shock protein 70 (Hsp70) on tumors by cmHsp70.1 antibody. Proceedings of the National Academy of Sciences. 2011;108:733-8
165. Holmberg B. On the in vitro Release of Cytoplasmic Enzymes from Ascites Tumor Cells as Compared with Strain L Cells*. Cancer Research. 1961;21:1386-93
166. Hong CW, Zeng Q. Tapping the treasure of intracellular oncotargets with immunotherapy. FEBS Lett. 2014;588:350-5
167. Stine ZE, Schug ZT, Salvino JM, Dang CV. Targeting cancer metabolism in the era of precision oncology. Nat Rev Drug Discov. 2022;21:141-62
168. Funato Y, Yoshida A, Hirata Y, Hashizume O, Yamazaki D, Miki H. The Oncogenic PRL Protein Causes Acid Addiction of Cells by Stimulating Lysosomal Exocytosis. Dev Cell. 2020;55:387-97.e8
169. Hoeger B, Rios P, Berteotti A, Hoermann B, Duan G, Kohn M. Mutational Analysis of a Conserved Glutamate Reveals Unique Mechanistic and Structural Features of the Phosphatase PRL-3. ACS Omega. 2017;2:9171-80
170. Jeong KW, Kang DI, Lee E, Shin A, Jin B, Park YG. et al. Structure and backbone dynamics of vanadate-bound PRL-3: comparison of 15N nuclear magnetic resonance relaxation profiles of free and vanadate-bound PRL-3. Biochemistry. 2014;53:4814-25
171. Kozlov G, Cheng J, Ziomek E, Banville D, Gehring K, Ekiel I. Structural insights into molecular function of the metastasis-associated phosphatase PRL-3. J Biol Chem. 2004;279:11882-9
172. Imai K, Takaoka A. Comparing antibody and small-molecule therapies for cancer. Nat Rev Cancer. 2006;6:714-27
173. Scott AM, Allison Jp Fau - Wolchok JD, Wolchok JD. Monoclonal antibodies in cancer therapy
174. Zahavi D, Weiner L. Monoclonal Antibodies in Cancer Therapy. Antibodies. 2020 9
175. Waldman AD, Fritz JM, Lenardo MJ. A guide to cancer immunotherapy: from T cell basic science to clinical practice. Nat Rev Immunol. 2020;20:651-68
176. Kraehenbuehl L, Weng CH, Eghbali S, Wolchok JD, Merghoub T. Enhancing immunotherapy in cancer by targeting emerging immunomodulatory pathways. Nat Rev Clin Oncol. 2022;19:37-50
177. Martins F, Sofiya L, Sykiotis GP, Lamine F, Maillard M, Fraga M. et al. Adverse effects of immune-checkpoint inhibitors: epidemiology, management and surveillance. Nat Rev Clin Oncol. 2019;16:563-80
178. Robert CA-O. A decade of immune-checkpoint inhibitors in cancer therapy
179. Conroy M, Naidoo J. Immune-related adverse events and the balancing act of immunotherapy. Nat Commun. 2022;13:392
180. Robert C, Ribas A, Hamid O, Daud A, Wolchok JD, Joshua AM, et al. Durable Complete Response After Discontinuation of Pembrolizumab in Patients With Metastatic Melanoma
181. Alsaab HO, Sau S, Alzhrani R, Tatiparti K, Bhise K, Kashaw SK. et al. PD-1 and PD-L1 Checkpoint Signaling Inhibition for Cancer Immunotherapy: Mechanism, Combinations, and Clinical Outcome. Front Pharmacol. 2017;8:561
182. Darvin P, Toor SM, Sasidharan Nair V, Elkord E. Immune checkpoint inhibitors: recent progress and potential biomarkers. Exp Mol Med. 2018;50:1-11
183. Johnson DB, Sullivan RJ, Menzies AM. Immune checkpoint inhibitors in challenging populations. Cancer. 2017;123:1904-11
Author contact
![]() Corresponding author: Qi Zeng, Institute of Molecular and Cell Biology, Agency for Science, Technology and Research (A*STAR), Singapore 138673. E-mail: mcbzengqa-star.edu.sg
Corresponding author: Qi Zeng, Institute of Molecular and Cell Biology, Agency for Science, Technology and Research (A*STAR), Singapore 138673. E-mail: mcbzengqa-star.edu.sg