Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Results
Discussion
Materials and Methods
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2023; 13(9):2843-2862. doi:10.7150/thno.83942 This issue Cite
Research Paper
Mesenchymal stromal cells and alpha-1 antitrypsin have a strong synergy in modulating inflammation and its resolution
Li Han1,2, Xinran Wu1, Ou Wang3, Xiao Luan4, William H. Velander3, Michael Aynardi5, E. Scott Halstead6, Anthony S. Bonavia7, Rong Jin8, Guohong Li8, Yulong Li9, Yong Wang1, Cheng Dong1, Yuguo Lei1,2 ![]()
1. Department of Biomedical Engineering, Pennsylvania State University; University Park, PA, 16802, USA.
2. Huck Institutes of the Life Sciences, Pennsylvania State University; University Park, PA, 16802, USA.
3. Department of Chemical and Biomolecular Engineering, University of Nebraska-Lincoln; Lincoln, NE, 68588, USA.
4. Biomedical Center of Qingdao University; Qingdao, Shandong, 266000, China.
5. Department of Orthopedics Surgery, Pennsylvania State University College of Medicine; Hershey, PA, 17033, USA.
6. Division of Pediatric Critical Care Medicine, Department of Pediatrics, Pennsylvania State Milton S Hershey Medical Center; Hershey, PA, 17033, USA.
7. Division of Critical Care Medicine, Department of Anesthesiology and Perioperative Medicine, Pennsylvania State Milton S Hershey Medical Center; Hershey, PA, 17033, USA.
8. Department of Neurosurgery, Pennsylvania State Milton S Hershey Medical Center; Hershey, PA, 17033, USA.
9. Department of Emergency Medicine, University of Nebraska Medical Center; Omaha, NE, 68105, USA.
Received 2023-3-1; Accepted 2023-4-25; Published 2023-5-8
Abstract

Rationale: Trauma, surgery, and infection can cause severe inflammation. Both dysregulated inflammation intensity and duration can lead to significant tissue injuries, organ dysfunction, mortality, and morbidity. Anti-inflammatory drugs such as steroids and immunosuppressants can dampen inflammation intensity, but they derail inflammation resolution, compromise normal immunity, and have significant adverse effects. The natural inflammation regulator mesenchymal stromal cells (MSCs) have high therapeutic potential because of their unique capabilities to mitigate inflammation intensity, enhance normal immunity, and accelerate inflammation resolution and tissue healing. Furthermore, clinical studies have shown that MSCs are safe and effective. However, they are not potent enough, alone, to completely resolve severe inflammation and injuries. One approach to boost the potency of MSCs is to combine them with synergistic agents. We hypothesized that alpha-1 antitrypsin (A1AT), a plasma protein used clinically and has an excellent safety profile, was a promising candidate for synergism.
Methods: This investigation examined the efficacy and synergy of MSCs and A1AT to mitigate inflammation and promote resolution, using in vitro inflammatory assay and in vivo mouse acute lung injury model. The in vitro assay measured cytokine releases, inflammatory pathways, reactive oxygen species (ROS), and neutrophil extracellular traps (NETs) production by neutrophils and phagocytosis in different immune cell lines. The in vivo model monitored inflammation resolution, tissue healing, and animal survival.
Results: We found that the combination of MSCs and A1AT was much more effective than each component alone in i) modulating cytokine releases and inflammatory pathways, ii) inhibiting ROS and NETs production by neutrophils, iii) enhancing phagocytosis and, iv) promoting inflammation resolution, tissue healing, and animal survival.
Conclusion: These results support the combined use of MSCs, and A1AT is a promising approach for managing severe, acute inflammation.
Keywords: inflammation, mesenchymal stromal cells, alpha-1 antitrypsin, combination therapy
Introduction
Many conditions, including infection, trauma, and surgery, can cause severe inflammation. Immune cells are expected to recognize pathogens (or triggers), respond proportionally to the pathogen burden, and effectively eliminate them [1,2]. Subsequently, they initiate a process leading to the resolution of inflammation and restoration of homeostasis [3,4]. Cytokines play critical roles in coordinating immune cell function, ensuring that the initiation, amplification, and resolution of inflammation occurs in an organized manner. Cytokines have a short life span and often remain at the injury site to avoid systemic immune activation. However, under certain conditions, such as an overwhelming pathogen burden, immune cell activation, and cytokine production become dysregulated, excessive, persistent, and systemic (i.e., cytokine storm) [5]. Hyperinflammation can rapidly progress to disseminated intravascular coagulation, vascular leakage, acute respiratory distress syndrome (ARDS), multi-organ dysfunction (MODS), and death [6,7].
Clinical strategies used to treat patients with severe inflammation include supportive care to maintain critical organ functions and elimination of inflammatory stimuli, such as antibiotics. Additionally, steroids and immunosuppressants can be used to suppress immune cells and targeted biologics (e.g., monoclonal antibodies) can be used to neutralize specific cytokines [5]. However, steroids derail inflammation resolution pathways, compromise antibacterial host defenses, and have significant adverse effects [8-10]. Therefore, there is a clinical need for safe therapies that can mitigate hyper-inflammation while boosting normal immunity and accelerating inflammation resolution.
Our body has multiple types of negative regulators of inflammation, including cells (e.g., Treg) [11], proteins (e.g., IL10) [12,13], and special lipid mediators (e.g., lipoxin A4) [3,8,14-17]. These mechanisms, designed to work together to prevent severe inflammation, often fail in patients with severe medical comorbidities and/or compromised immunity [3,4]. It follows that augmenting these inflammatory regulators may offer a promising therapeutic approach. Among various inflammatory regulators, mesenchymal stromal cells (MSCs) are of particular interest since they possess unique and multi-faceted capabilities to mitigate severe inflammation. They can balance the inflammatory environment by downregulating pro-inflammatory cytokines, such as IL6 and TNFα, while upregulating anti-inflammatory or/and pro-resolving cytokines, such as IL10 and IL4 [18-33]. Using secreted mediators and direct interactions, MSCs can program monocytes and macrophages into the anti-inflammatory and pro-resolving M2 phenotype [19,33-36]. They reduce the adherence of leukocytes to endothelium [37]. MSCs can inhibit tissue infiltration as well as ROS and NETs production by neutrophils [19,20,27,30,37-39]. MSCs can also enhance 'normal' immunity by boosting the phagocytosis, bacterial killing, and efferocytosis of monocytes and macrophages [34,36,37,40-44]. MSCs also secrete antibacterial peptides such as LL-37, lipocalin-2, and hepcidin [18,23,29,45]. Finally, MSCs can protect organs from inflammation-associated damage while promoting organ healing [20,21,23,24,31,45-49]. MSCs can reduce cell death and improve barrier functions of endothelium and epiththium [19,22,24,25,37,46,49-51].
In addition to these multiple beneficial functions, MSCs have low immunogenicity. Therefore, allogeneic MSCs can be administered without significant side effects [52]. MSCs can be isolated from various tissues, such as the placenta, umbilical cord, and adipose tissue, and they can be efficiently expanded in vitro. It is therefore hardly surprising that MSCs have been studied in varying disease contexts, including ARDS, sepsis, GvHD, stroke, spinal cord injury, myocardial infarction, organ transplantation, and COVID-19[53-65]. MSCs have also recently been used to treat severe COVID-19 patients [66], reducing disease mortality significantly [67-71]. However, one shortcoming of MSCs is that monotherapy is not potent enough to fully resolve severe inflammation [72]. Therefore, approaches to boost MSCs' potency are necessary. One proposed strategy is to combine MSCs with FDA-approved drugs that have excellent safety profiles and can synergize with MSCs.
We propose that protein alpha-1 antitrypsin (A1AT) possesses properties well suited to synergize with MSCs and increase their therapeutic efficacy. A1AT is an acute-phase protein whose concentration increases five-fold when the body is injured or infected. A1AT has anti-inflammatory, anti-protease, pro-resolution, cytoprotective, and pro-angiogenic properties [73-83]. It selectively inhibits neutrophil recruitment and cytokine production and neutralizes many pro-inflammatory cytokines [82,84-91]. It suppresses M1 macrophages while promoting M2 macrophages and Treg cells [73,92-99]. It also reduces bacterial and viral burden [100-108]. In addition, it protects cells from various stress [75,109-112] and promotes angiogenesis [113,114]. A1AT purified from plasma has been used to treat alpha-1 antitrypsin deficiency for decades, with an excellent safety profile [115,116]. Most recently, A1AT has been studied to treat severe COVID-19 patients with positive outcomes [117-121]. However, like MSCs, A1AT alone is insufficient to completely resolve severe inflammation [117-121]. In this investigation, we examined MSCs-A1AT synergism using both in vitro cell cultures and a murine acute lung injury and inflammation model.
Results
Isolating MSCs from placenta
The full-term placenta was cut into small pieces, treated with TrypLE for 30 mins, and placed in a cell culture flask (Figure S1A). Cells migrated from the tissues, adhered to the flask surface, and expanded (Figure S1B). When cells reached about 70% confluence, tissues were removed, and cells were allowed to grow until full confluence. These cells were cryopreserved or sub-cultured (Figure S1C). Cells had the classical spindle-like morphology. Above 95% of passage 4 (P4) cells expressed MSC surface markers including CD73, CD90, CD105, CD44, and CD166. The expression of negative markers, including CD45, CD34, CD11b, CD79A, and HLA-DR, was negligible (Figure S1D). In addition, MSCs could be differentiated into FABP4+ adipocytes and osteocalcin+ osteocytes (Figure S1E). In summary, we successfully isolated MSCs from the placenta.
MSCs modulate cytokine release
To test if our cultured cells could similarly suppress inflammation, we stimulated mouse Raw 264.7 macrophages (MΦs) with LPS and IFNγ to induce intense inflammation. We optimized the concentrations of stimulants such that 100 ng/mL LPS + 10 ng/mL IFNγ induced maximal cytokine release while not causing rapid and significant cell death. Inflamed cells were treated with MSCs at three different ratios: one MSC for 1, 5, or 10 macrophages (1/1, 1/5, 1/10). 1 µg/mL dexamethasone, a clinically relevant dose used to treat severe inflammation, was used to benchmark MSC's capability. In addition, one sample was treated with MSCs conditioned medium (CCM) to assess if factors secreted by MSCs were effective. After 24 hs, the pro-inflammatory (IL6 and TNFα) and anti-inflammatory (IL10) cytokines in the medium were measured with ELISA. The antibodies are specific to mouse proteins to avoid interference from human cytokines secreted by human placenta-derived MSCs.
All treatments reduced the IL6 concentration (Figure S2A). MSCs also decreased TNFα secretion, similar to IL6 (Figure S2B). All treatments except dexamethasone increased IL10 levels. MSCs were better than their conditioned medium (Figure S2C). The IL6/IL10 or TNFα/IL10 ratio can be used to assess inflammation/anti-inflammation balance. Dexamethasone decreased IL6/IL10 from 8 to 3.5, and MSCs decreased IL6/IL10 to 1.5 for 1/10 dosage and to < 0.5 for 1/5 and 1/1 dosages. The conditioned medium reduced the ratio to 1.5 (Figure S2D). Dexamethasone decreased TNFα/IL10 from 38 to 18. MSCs decreased TNFα/IL10 to ~ 5, while the conditioned medium reduced the ratio to ~ 10 (Figure S2E). In summary, the data showed that i) MSCs could dampen pro-inflammatory cytokine secretion while promoting anti-inflammatory or pro-resolving cytokine secretion; ii) cells were better than their conditioned medium alone and better than dexamethasone; iii) there was no huge difference between the 1/10, 1/5 and 1/1 dose for MSCs in terms of IL6/IL10 or TNFα/IL10 ratios. Thus, we decided to perform subsequent experiments using MSCs at a 1/10 ratio.
A1AT modulates cytokine release
We evaluated A1AT's ability to suppress inflammation in Raw 264.7 macrophages. Inflamed cells were treated with A1AT (isolated from human plasma) with concentrations ranging from 0.1 to 2.0 mg/mL. A1AT reduced the IL6 and TNFα levels in a dose-dependent manner (Figure S3A-B). A1AT at a concentration ≥ 0.5 mg/mL significantly increased IL10 expression, while dexamethasone did not (Figure S3C). These findings were concordant with previously published data [122]. Dexamethasone decreased IL6/IL10 from 7.5 to 2.2, while A1AT decreased IL6/IL10 to < 0.5 when ≥ 0.5 mg/mL protein was used [122]. Dexamethasone decreased IL6/IL10 from 7.5 to 2.2, which A1AT decreased IL6/IL10 to < 0.5 when ≥ 0.5 mg/mL protein was used (Figure S3D). Dexamethasone decreased TNFα/IL10 from 30 to 15, while A1AT decreased the ratio to ~ 2 when the protein was ≥ 0.5 mg/mL (Figure S3E). In summary, we found that i) A1AT could inhibit pro-inflammatory cytokine secretion while promoting anti-inflammatory/pro-resolving cytokine secretion; ii) there was no significant difference between 0.5, 1.0, and 2.0 mg/mL A1AT in terms of IL6/IL10 or TNFα/IL10 ratios. Therefore, 0.5 mg/mL A1AT was used to perform subsequent experiments.
MSCs and A1AT have synergy to modulate cytokine release
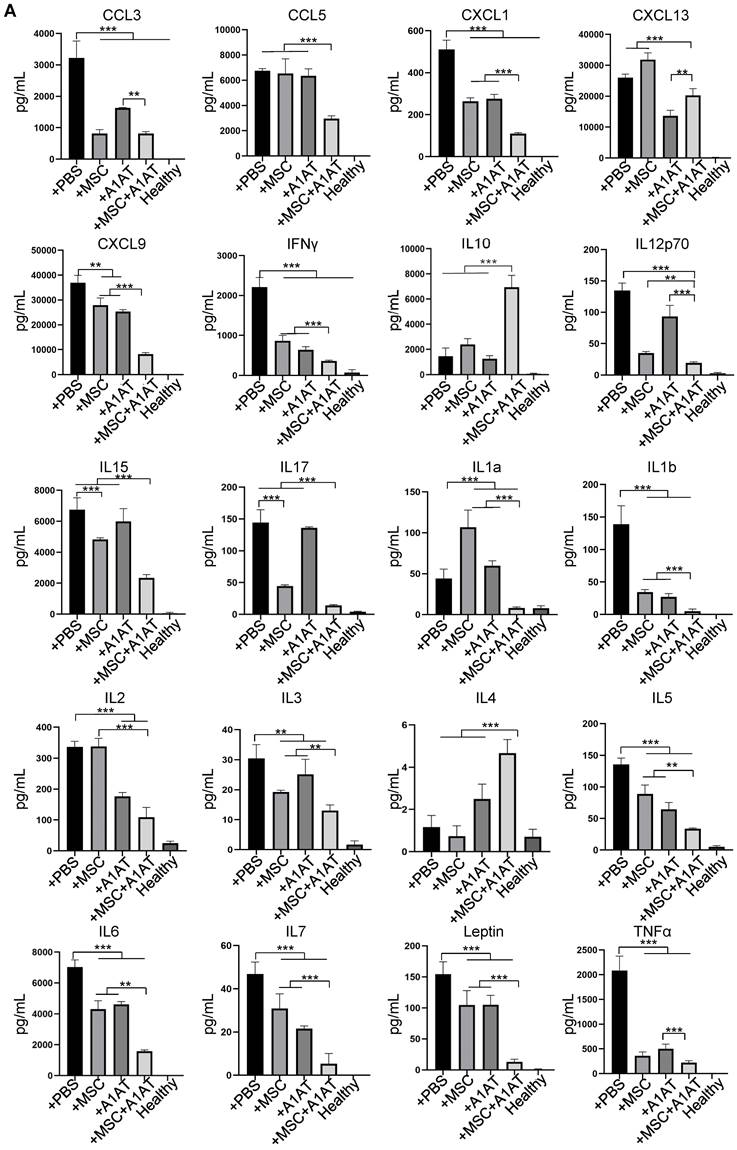
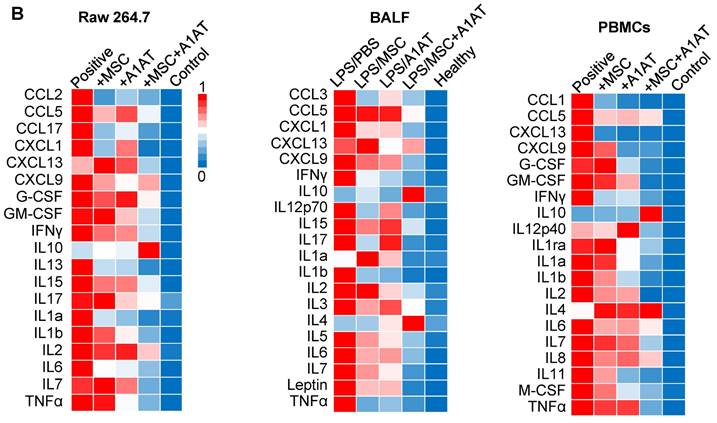
Next, we studied if MSCs and A1AT exhibited synergistic properties. We treated inflamed Raw 264.7 macrophages with 0.5 mg/mL A1AT alone, 1/10 MSCs alone, or their combination. All treatments reduced IL6 and TNFα levels while increasing IL10 levels, with the MSCs + A1AT combination demonstrating the most significant effect (Figure 1A). Furthermore, we measured 40 inflammation-related cytokines using an antibody array. The treatments affected the expression of 19 cytokines (Figure S4). A1AT reduced the expression of CCL2 (MCP-1), CCL5 (RANTES), CCL17, CXCL1, CXCL9, IFNγ, IL13, IL15, IL1a and IL6 (Figure S4). MSCs reduced the expression of CCL2, CCL17, CXCL9, GM-CSF, IFNγ, IL13, IL15, IL17, IL1a, IL1b, IL6 and TNFα. A1AT and MSCs showed strong synergism in regulating the expression of CCL5, CCL17, CXCL1, CXCL13, CXCL9, G-CSF, GM-CSF, IFNγ, IL10, IL13, IL15, IL1a, IL1b, IL2, IL6, IL7, and TNFα (Figure S4). In summary, the results showed that i) MSCs and A1AT had synergistic effects on regulating many cytokines, and ii) the cytokines affected by A1AT and MSCs were not identical, indicating their mechanisms of action were not identical.
MSCs synergized with A1AT to modulate inflammation in Raw 264.7 macrophages. Cells were stimulated with 100 ng/mL LPS plus 10 ng/mL IFNγ and treated with 0.5 mg/mL A1AT or MSCs (MSC/MΦ = 1/10) or their combination. Dexamethasone (Dex, 1 µg/mL) was used as a benchmark. Pro-inflammatory mouse cytokine IL6 (A), TNFα (B), and anti-inflammatory mouse cytokine IL10 (C) were measured via ELISA. The IL6/IL10 (D) and TNFα/IL10 ratio (E) was also shown. *:p < 0.05, **:p < 0.01, ***:p < 0.001.
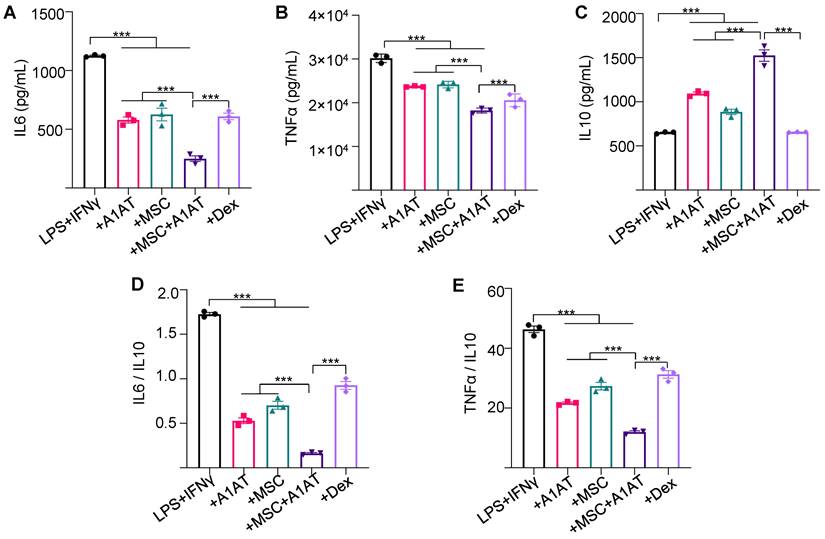
We then tested whether the findings could be replicated using human macrophages. THP-1 monocytes were first differentiated into macrophages. Inflammation was then induced using LPS and IFNγ. The effects of MSCs, A1AT and their combination on dampening cytokine release (Figure S5) were similar to Raw 264.7 macrophages (Figure 1). All treatments reduced IL6 and TNFα levels, but only the MSCs + A1AT increased IL10 release. The MSCs and A1AT combination was much more effective than the individual components. The results again showed that MSCs and A1AT could concomitantly downregulate the pro-inflammatory program and upregulate the anti-inflammatory or pro-resolving program.
We also used primary PBMCs to confirm the findings. To avoid donor-to-donor variations, we used PBMCs pooled from multiple donors. We added LPS and IFNγ to activate innate immune cells and anti-CD3 and anti-CD28 antibodies to activate T cells. All treatments reduced IFNγ and TNFα secretion while increasing IL10 production. Again, MSC and A1AT combination was much more effective than the individual components (Figure 2). dexamethasone increased IL10 levels in PBMCs, which is different from the findings using macrophages (Figure 1 and Figure S5). Therefore, we used flow cytometry to assess the cytokine production of monocytes and T cells in PBMCs (Figure S6). Monocytes and T cells were identified with CD14 and CD3 surface markers, respectively. All treatments reduced the %TNFα+ and %IFNγ+ monocytes and their mean fluorescence intensity (Figure S6A). Only MSCs and MSCs + A1AT increased the %IL10+ monocytes and their mean fluorescence intensity. Similar results were found for T cells, except that only MSCs + A1AT increased the %IL10+ monocytes and their mean fluorescence intensity. The results indicated that dexamethasone boosted IL10 production from cell types other than monocytes and T cells in PBMCs.
MSCs synergized with A1AT to modulate inflammation in primary human PBMCs. Cells were stimulated with 100 ng/mL LPS + anti-CD3/CD28 antibodies (positive) and treated with 0.5 mg/mL A1AT or MSCs (MSC/PBMC = 1/10) or their combination for 24 hs. Dexamethasone (Dex, 1 µg/mL) was used as a benchmark. PBMCs without activation and treatment were used as a negative control. Pro-inflammatory human cytokine IL6 (A), TNFα (B), and anti-inflammatory human cytokine IL10 (C) were measured via ELISA. The IL6/IL10 (D) and TNFα/IL10 ratio (E) was also shown. *:p < 0.05, **:p < 0.01, ***:p < 0.001.
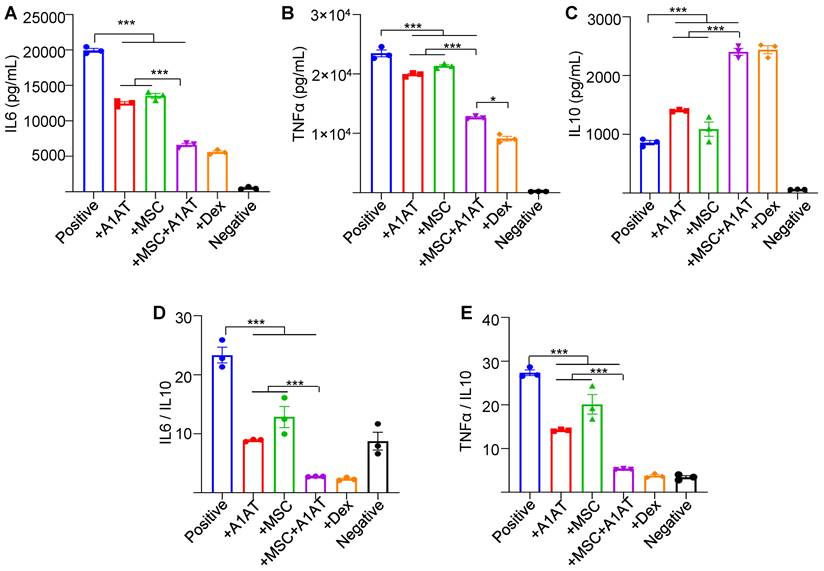
Furthermore, we measured 40 human inflammation-related cytokines in the PBMCs medium using an antibody array (Figure S7). The treatments affected the expression of 20 cytokines. MSCs reduced the expression of CCL1, CCL5 (RANTES), CXCL13, IFNγ, IL1b, IL2, IL6, IL7 and IL11, while increased IL4 production. A1AT reduced the expression of CCL1, CCL5, CXCL13, CXCL9, G-CSF, CM-CSF, IFNγ, IL12p40, IL1ra, IL1a, IL1b, IL2, IL6, IL7, IL11 and M-CSF, while increased IL10 and IL4 production. A1AT and MSCs showed a strong synergy in regulating the expression of CCL1, CCL5, G-CSF, CM-CSF, IFNγ, IL10, IL12p40, IL1ra, IL1a, IL1b, IL2, IL6, IL7, IL8, IL11, M-CSF and TNFα (Figure S7). The results confirmed the findings using macrophages that i) MSCs synergized with A1AT in regulating many cytokines, and ii) the cytokines affected by A1AT and MSCs were not identical.
MSCs synergize with A1AT to modulate neutrophil ROS and NETs production
MSCs and A1AT each can inhibit ROS and NETs production [20,123]. We hypothesized that combination therapy would provide synergistic anti-ROS and anti-NET properties when co-incubated with neutrophils. Indeed, MSCs + A1AT demonstrated significant synergism in reducing ROS production (Figure 3A-B) and NET production (Figure 3C-D). All treatments also reduced IL6 and TNFα concentrations in the culture medium while increasing the concentration of IL10. In addition, the MSC and A1AT combination worked much better than each treatment alone (Figure S8). In summary, MSCs and A1AT showed a substantial synergy to modulate inflammation and ROS and NETs production in neutrophils.
MSCs and A1AT combination treatment reduced neutrophil ROS and NETs production. HL-60 cells derived neutrophils were stimulated with 100 nM PMA and treated with 0.5 mg/mL A1AT or MSCs (MSC/neutrophil = 1/10) or their combination for 4 hs. Reactive oxygen species (ROS) (A-B) and neutrophil extracellular traps (NETs) production (C-D) were analyzed. *:p < 0.05, **:p < 0.01, ***:p < 0.001.
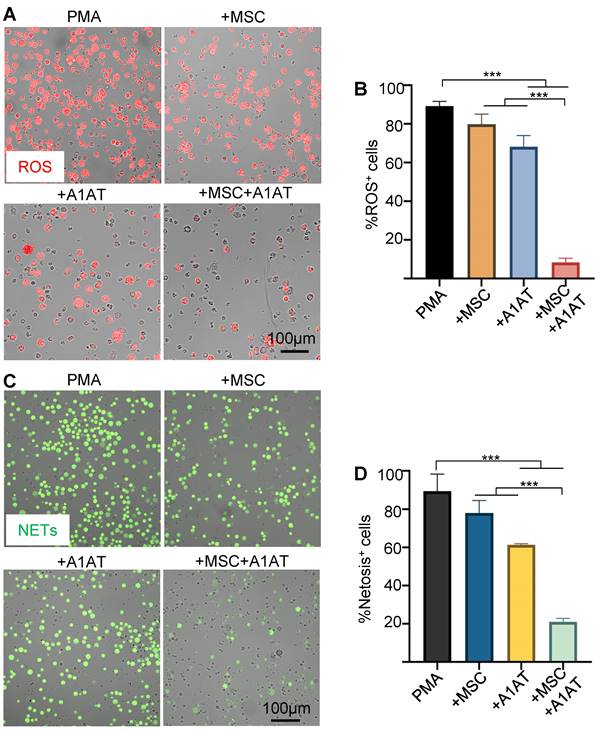
MSCs synergize with A1AT to modulate macrophage phagocytosis and inflammation pathways
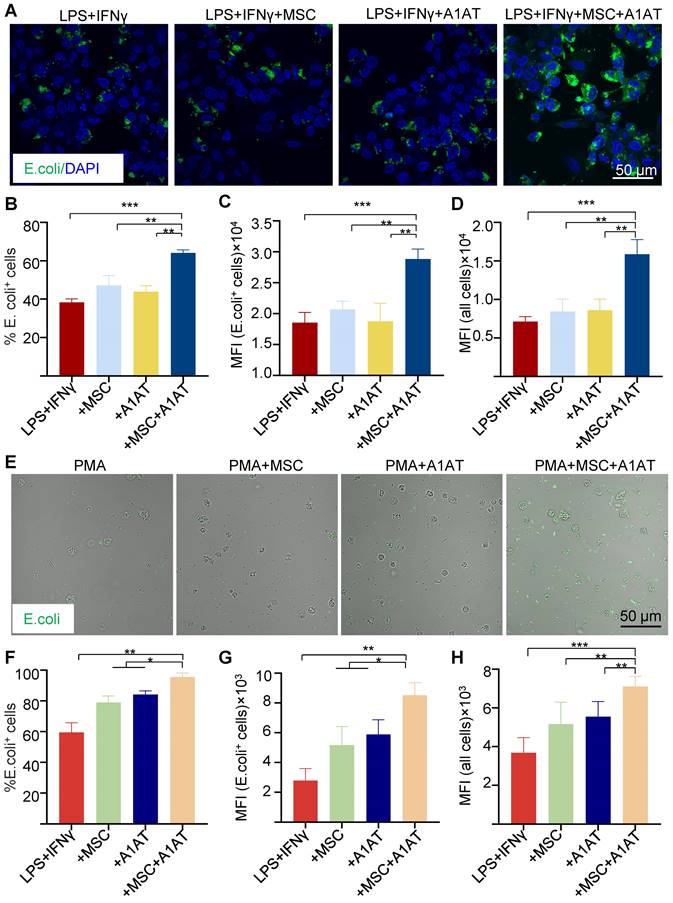
Severe inflammation compromises phagocytosis by innate immune cells, preventing pathogen clearance and inflammation resolution [124-126]. MSCs and A1AT can boost macrophage phagocytosis [33,34,36,37,40-44,95,127]. We thus tested if MSCs and A1AT synergize to enhance phagocytosis in macrophages and neutrophils. We measured the % of cells phagocytosing E. Coli particles, mean fluorescence intensity (MFI) per cell for all cells, and MFI per cell for cells phagocytosing particles. MSCs or A1AT alone did not significantly increase any of these measurements. However, MSCs plus A1AT led to a substantial increase in all these parameters in macrophages (Figure 4A-D) and neutrophils (Figure 4E-H).
MSC and A1AT combination treatment enhanced phagocytosis in THP-1 derived macrophages (A-D) and HL-60 cells derived neutrophils (E-H). Macrophages were stimulated with 100 ng/mL LPS plus 10 ng/mL IFNγ for 24 hs. Neutrophils were stimulated with 100 nM PMA for 4 hs. Cells were treated with 0.5 mg/mL A1AT or MSCs (MSC/MΦ = 1/10) or their combination during the stimulation. E. coli particles were added for 3 hs after treatment. (A, E) E. coli particles emitted green fluorescence after being phagocyted. (B, F) The % E. coli+ cells. (C, G) MFI per cell for all cells. (D, H) MFI per cell for cells with E. coli particles. *:p < 0.05, **:p < 0.01, ***:p < 0.001.
Nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) and the interferon regulatory factors (IRF) signaling are critical components of pro-inflammatory pathways. Raw 264.7 and THP-1 cells engineered to express a secreted embryonic alkaline phosphatase (SEAP) reporter for the NF-κB pathway and a secreted luciferase reporter for the IRF pathway were used to evaluate if MSCs and A1AT could regulate these pathways. THP-1 monocytes were differentiated into macrophages before testing. MSCs and A1AT inhibited both pathways in both macrophage types, again demonstrating strong synergistic effects (Figure S9).
MSCs synergize with A1AT to suppress inflammation and promote inflammation resolution in vivo
We then used the LPS-induced acute lung injury and inflammation mouse model to test if the in vitro results could be replicated in vivo. Treatments were administered 30 mins after the injury (Figure 5A). A lethal dosage (20 mg LPS/kg body weight) was administrated to the first cohort of mice for survival tests. All mice died in 3 days without treatment. MSCs or A1AT alone increased the survival rate, but only their combination wholly protected mice from death (Figure 5B). Furthermore, mice with the combination treatment had significantly less body weight reduction (Figure 5C). A non-lethal dosage (10 mg LPS/kg body weight) was administrated to the second cohort of mice to test inflammation and tissue healing. Tissues were harvested on day 3 for analysis. First, we analyzed lung injury via H&E staining. The lung injury was scored based on five criteria, including i) the number of neutrophils in alveolar space; ii) the number of neutrophils in interstitial space; iii) the amount of hyaline membranes; iv) the amount of proteinaceous debris in airspaces, and v) the alveolar septal thickening. The treatment groups had much less lung injury. The combination therapy group showed the least tissue injury (Figure 5D-E).
MSCs synergized with A1AT to improve survival rate and reduce lung injury in mice. (A) Illustration of the model. (B) The survival rate and (C) body weight development. N = 6. (D) H&E staining and (E) lung injury scores. The lung injury scores were calculated based on the five criteria shown in (D). *:p < 0.05, **:p < 0.01, ***:p < 0.001.
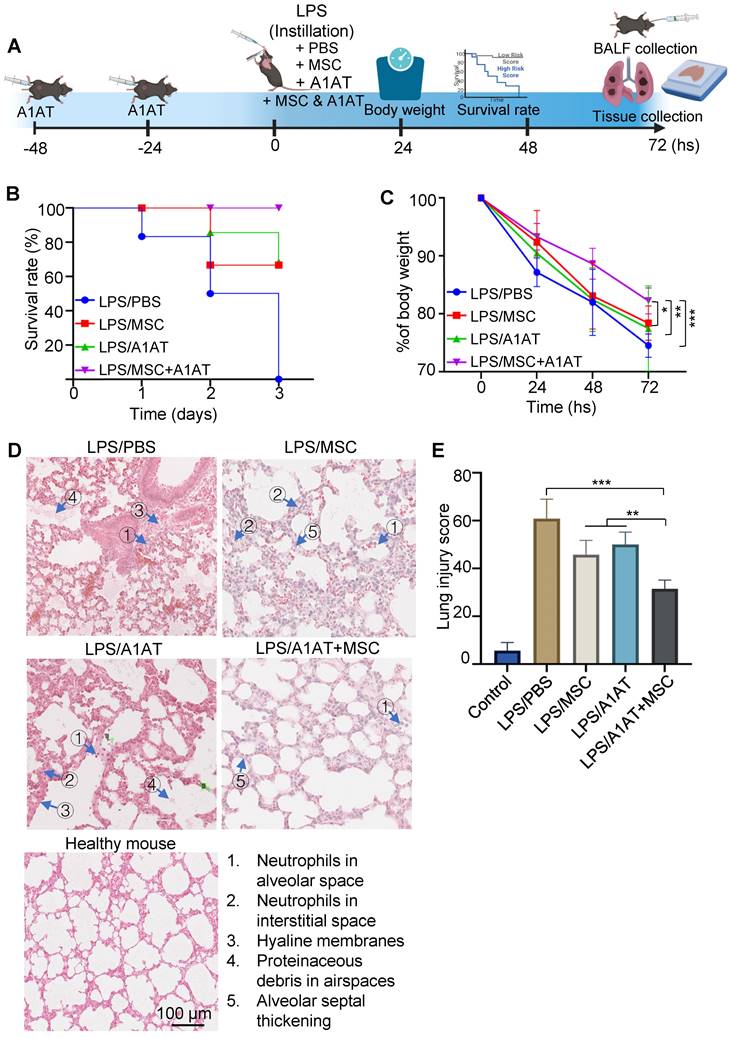
We harvested the bronchoalveolar lavage fluid (BALF) for protein and immune cell analyses. A high total protein concentration indicates the disruption of the endothelium and epithelium. MSCs and A1AT reduced the total protein level, and their combination worked significantly better (Figure 6A). Similar to the in vitro results, MSCs and A1AT reduced IL6 and sTNFαR levels while increasing IL10 levels significantly. Their combination was much more effective than the individual components (Figure 6B-F). We measured 40 inflammation-related cytokines with an antibody array. The treatments affected the expression of 21 cytokines. MSCs and A1AT showed a strong synergy on regulating the expression of CCL5, CXCL1, CXCL9, IFNγ, IL10, IL12p70, IL15, IL17, IL1a, IL1b, IL2, IL3, IL4, IL5, IL6, IL7, Leptin and TNFα (Figure 7A). The cytokine array results from BALF (Figure 7A), in vitro mouse macrophage study (Figure S4), and in vitro human PBMCs study (Figure S7) were similar (Figure 7B).
MSCs and A1AT synergized in reducing total protein (A) and pro-inflammatory cytokines while increasing anti-inflammatory cytokine IL10 (B-F) in BALF. *:p < 0.05, **:p < 0.01, ***:p < 0.001.
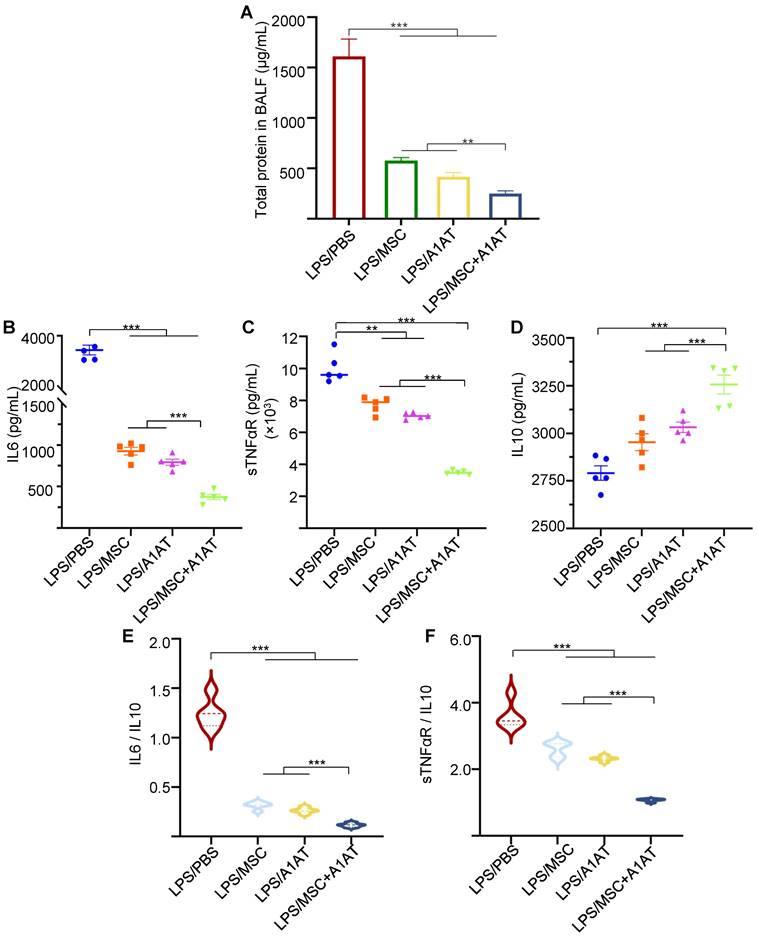
(A) MSC and A1AT combination treatment reduced pro-inflammatory cytokines while increasing anti-inflammatory cytokines in BALF as measured using an inflammation antibody array. Healthy: healthy mouse sample. (B) Heatmaps of cytokine levels in Raw 264.7 medium (from Figure S4), PBMCs medium (from Figure S7), and BALF (from Figure 7A). For each cytokine, the highest expression is set as 1 (red). Other groups are normalized to the highest expression. *:p < 0.05, **:p < 0.01, ***:p < 0.001.
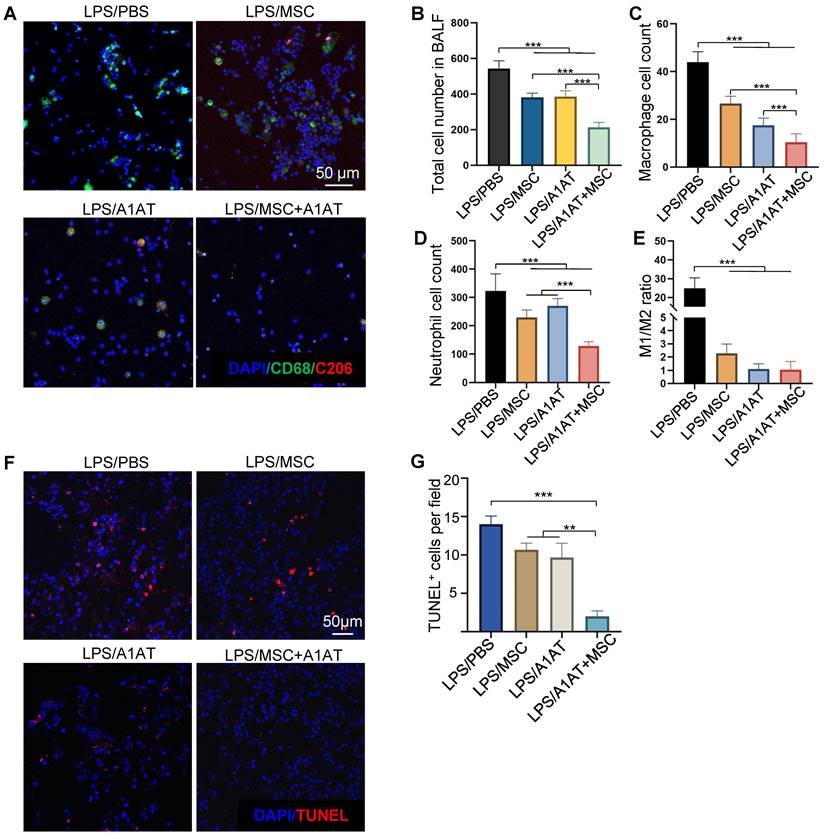
We also analyzed immune cells in BALF. MSCs, A1AT, and especially their combination reduced the number of total cells, macrophages, and neutrophils in BALF. The MSC + A1AT treatment functioned better than the individual components (Figure 8A-D). The M1/M2 ratio of macrophages was reduced by all treatments (Figure 8E). We used TUNEL staining to identify dead cells in lung tissue. Both MSCs and A1AT reduced the number of dead cells. Dead cells were scarce in the combination treatment group (Figure 8F-G).
MSCs synergized with A1AT to reduce total cell (A-B), macrophage (C), and neutrophil number (D) in BALF. The M1/M2 macrophage ratio was reduced by all treatments (E). *:p < 0.05, **:p < 0.01, ***:p < 0.001. (F-G) MSCs synergized with A1AT to reduce cell death as identified via TUNEL staining.
Discussion
Due to their unique ability to mitigate inflammation, boost normal immunity, and promote inflammation resolution and tissue healing, MSCs have been extensively studied in clinical trials for treating severe inflammatory diseases, such as ARDS, sepsis, GvHD, stroke, spinal cord injury, myocardial infarction, multiple sclerosis, organ transplantation, rheumatoid arthritis, Crohn's, systemic lupus erythematosus, ulcerative colitis and COVID-19[53-65]. A meta-analysis including 55 randomized clinical studies with 2696 patients reported that MSCs induce minor adverse effects while significantly reducing the risk of death [52]. Additionally, no signs of increased tumorgenicity and pro-thrombotic effect were reported [52]. There are about 10 clinical studies on using MSCs to treat ARDS and sepsis [72]. Published results show MSCs are safe and effective in reducing inflammation, epithelial and endothelial damage, and risk of death [55-57,59,62-65,128]. Since the pandemic, > 106 registered clinical trials using MSCs to treat severe COVID-19 patients have been initiated [66-69,71,128-135]. Published data show that MSCs can reduce the levels of inflammation biomarkers, pro-inflammatory cytokines, and NETs while increasing the levels of anti-inflammatory cytokines and reducing mortality and morbidity significantly [66,136]. Further, critically ill patients benefitted more from MSC treatment than non-critically ill patients. This finding indicates an additional, unique characteristic of MSCs: they may be able to appropriately respond to the level of inflammation [130] and are suitable for treating severely ill patients [69].
A1AT is used to treat alpha-1 antitrypsin deficiency [115,116]. A1AT has also been studied for treating COVID-19[117-121]. Clinical data shows that A1AT concentration is elevated in all COVID-19 patients as a mechanism to counteract inflammation. However, the A1AT response alone is insufficient to resolve the cytokine storm [118]. The IL6/A1AT ratio is significantly higher in severe patients compared to middle patients [118]. A higher IL6/A1AT predicts a prolonged ICU stay and higher mortality [118]. An improvement in A1AT/IL6 is associated with better clinical outcomes [118]. A published clinical study finds that A1AT injection can significantly reduce blood IL6 and sTNFR1 levels [120,121]. However, clinical data show that MSCs or A1AT alone are not potent enough to completely resolve hyperinflammation and prevent organ damage [66,120,121,136]. Our data show that MSCs and A1AT demonstrate strong synergy in suppressing pro-inflammatory cytokines, pathways, and NETosis while boosting anti-inflammatory/pro-resolving factors, normal immunity, and tissue healing. Our study provides strong evidence to support the combined use of MSCs and A1AT for treating severe inflammation in diverse disease states.
Complex networks of cells, cytokines, and signaling pathways are involved in hyperinflammation and cytokine storm [5]. Macrophages are major cytokine producers [137-140]. Our data demonstrate that MSCs and A1AT can individually suppress cytokine release from inflamed macrophages and monocytes (Figure 1-2 and Figure S1-7), confirming previously reported results [19,33-36]. We further demonstrate that combination therapy exceeds the performance of each component (Figure 1-2 and Figure S1-7). Neutrophils also play a critical role in hyperinflammation [141-150]. Activated neutrophils release NETs and ROS to eradicate bacteria [151]. However, excessive NETs can cause collateral damage to the endothelium, epithelium, and surrounding tissues [152-154], amplify the cytokine storm [152-154], and induce disseminated intravascular coagulation [143,155-158]. Our data show that MSCs and A1AT reduce the production of cytokines, ROS, and NETs from neutrophils (Figure 3 and Figure S8), with combination therapy, again exceeding the performance of each individual component. IFNγ release from T cells is crucial to activating macrophages [137-140]. We show that the combination of MSCs and A1AT can significantly suppress TNFα and IFNγ production by T cells (Figure S6). In short, MSCs can synergize with A1AT to effectively modulate the major immune cell types involved in hyperinflammation.
Cytokines IFNγ, IL1, IL6, TNFα, and IL18 play a central role in hyperinflammation [5]. IFNγ is mainly produced by T cells and NK cells and is critical for activating macrophages [137-140]. A recent study finds that IFNγ and TNFα synergistically induce cytokine shock, MODS, and mortality in mice [159]. IL1a/1b bind to IL1 receptors and activate NF-kB to express multiple pro-inflammatory cytokines [160,161]. IL6 acts on both immune and non-immune cells [162-165]. IL6 causes inflammation in endothelial cells, leading to barrier function loss, vascular permeability, hypotension, ARDS, and MODS. TNFα, a potent, multifunctional, pro-inflammatory cytokine, plays a crucial role in a cytokine storm, as shown by the effectiveness of anti-TNF therapies in certain cytokine storm conditions [166-168]. IL10 inhibits the production of TNFα, IL1, IL6, and IL12 and promotes inflammation resolution [169,170]. Our data show that MSCs synergize with A1AT to simultaneously modulate the major immune cells, cytokines, and pathways involved in severe inflammation (Figure 7 and Figure S4-7), implying an advantage of this therapy over targeted biologic agents [5]. Neutralizing a particular cytokine with targeted biologics may not always be effective since there is redundancy in pro- and anti-inflammatory pathways [5].
It should be noted that cytokines modulated by MSCs and A1AT are not identical (Figure 7 and Figure S4-7), indicating that the cell types and signaling pathways affected by MSCs and A1AT may have differences. This may partly explain their synergism. Our data from mouse macrophages, human macrophages, and PBMCs are congruent in demonstrating the robust efficacy and synergism between MSCs and A1AT (Figure 1-8 and Figure S2-9). Furthermore, the in vivo data agree well with the in vitro results, indicating that the mechanisms of action in vivo can be modeled by the in vitro assays.
The NF-kB pathway plays a pivotal role in inflammation and cytokine storm [171,172]. It can be activated by various ligand-receptor binding such as the binding of LPS to Toll-like receptor 4 (TLR4), the binding of single-stranded viral RNA to TLR7/8 and double-stranded viral RNA to TLR3, and the binding of IL1 and TNFα to their corresponding receptors [171,172]. These lead to the p50/p65 protein translocation to the nucleus to initiate the expression of many pro-inflammatory cytokines, chemokines, adhesion molecules, and growth factors [171,172]. Inhibiting the NF-kB pathway can significantly reduce the cytokine storm, ARDS, MODS, and mortality in animal models with different triggers [171,172]. Glucocorticoids such as dexamethasone and immunosuppressive agents such as Cyclosporin A and tacrolimus are potent NF-kB blockers; however, they have significant adverse effects [173-175]. The IRF pathways also contribute to a cytokine storm. Knocking down the IRF3 and ISGF3 complex in myeloid cells significantly reduces inflammation and mortality in LPS-induced severe inflammation in mice [176,177]. MSCs can inhibit NF-kB signaling [178-181], which is confirmed by our study. Additionally, we show that the MSCs synergize with A1AT to block both pathways effectively (Figure S9).
An overwhelming pathogen burden often triggers hyperinflammation. Phagocytosis, a major way to clear pathogens, thus represents a valuable therapeutic target to dampen and resolve severe inflammation [125]. Increasing monocytes and macrophage phagocytosis can reduce bacterial burden, cytokine levels, MODS, and mortality [126,182,183]. Clinically, immunoglobulins infused to opsonize and neutralize bacteria and bacterial products have met modest success [184-187]. G-CSF and GM-CSF have also been studied to increase the neutrophil and macrophage numbers to enhance bacterial clearance with similarly modest success [188-191]. MSCs can boost phagocytosis and bacterial killing of macrophages, thus reducing bacterial burden [34,36,37,40-44]. Our data show that combined MSCs and A1AT can maximally enhance phagocytosis (Figure 4).
Severe inflammation causes ARDS and MODS [5,192-196]. Circulating cytokines upregulate adhesion molecules such as VCAM-1 and ICAM-1 on the endothelium surface while downregulating the tight junction proteins. The adhesion of leukocytes to the endothelium and their trans-endothelium migration is enhanced during severe inflammation. Consequently, large amounts of plasma proteins, cytokines, and immune cells are leaked into parenchymal tissues. They activate the resident immune cells, causing inflammation in distal tissues/organs. The released cytokines and chemokines recruit more immune cells to the tissues. Cytokines, ROS, and proteases cause significant tissue damage. Our data show that MSCs and A1AT reduce BALF's total protein and immune cells (Figure 8), indicating they can protect the endothelial and epithelial barrier functions. In addition, the total TUNEL+ cells were significantly reduced. Thus, MSCs and A1AT synergize to protect the endothelium, epithelium, and parenchymal tissues. However, since the tissues were harvested 3 days after injury and treatment, the improvement in tissue structure may be because MSCs and A1AT accelerated the inflammation resolution and tissue healing. The higher M2/M1 macrophage ratio and low dead cell number in treatment groups may support this mechanism (Figure 8). Future work should clarify the treatment's action model and time.
There are a few limitations to the study. First, MSCs and A1AT were only tested in a sterile acute lung injury and inflammation mouse model. Whether the treatment can effectively mitigate severe inflammation caused by infection is unclear, although the features of severe inflammation caused by different triggers are similar. Infection models such as cecal ligation and puncture mice can be used to test the treatment in the future. Testing with large animal models will also be necessary before clinical studies. Second, the molecular mechanisms leading to the MSCs and A1AT synergy are not fully understood. Our data show that MSCs synergize with A1AT to modulate the NF-kB and IFR pathways. We expect there are other pathways contributing to the synergy. Future studies can apply RNA-Seq technology to fully characterize the changes in global gene expressions and signaling pathways caused by the treatments.
In summary, we showed that the MSCs and A1AT combination was much more effective than individual components in i) downregulating pro-inflammatory cytokines while upregulating pro-resolving cytokines, ii) turning off the NF-kB and IRF inflammation pathways, iii) inhibiting neutrophil ROS and NETs production, iv) enhancing macrophage phagocytosis in vitro, and v) reducing the levels of pro-inflammatory cytokines, neutrophils, M1 macrophages, M1/M2 ratio, and tissue injury and mortality significantly in a mouse lung injury model. Our results provide evidence supporting the combined use of MSCs and A1AT as anti-inflammatory therapy. Further investigations are warranted to investigate their combined utility in treating human disease.
Materials and Methods
Study design
The study was designed to investigate the combinational use of MSCs and A1AT for modulating severe acute inflammation response in vitro and in vivo. All experiments performed in this study had at least three replicates to demonstrate biological reproducibility and to ensure adequate statistical power for comparisons. All animals were randomly allocated to the control and treatment groups. Details for the number of mice, number of cells used, duration, and statistical tests are described below and in the figure legends.
MSC isolation
Full-term human placentas were purchased from ZenBio Inc. The procedure for isolating and expanding MSCs is similar to a published protocol with minor modifications [197,198]. Briefly, the placenta was washed and cut into 0.5 cm3 pieces that were treated with TrypLE select solution (Gibco) at 37 ˚C for 30 mins for partial digestion. 15-20 partially digested pieces were then plated in a 75 cm2 tissue flask with 9 mL of EBM-2 complete cell culture medium (EBM-2 + 10% FBS + 1% antibiotic). The flasks were placed in an incubator without disturbance for three days to allow tissues to adhere to the flask surface. After that, the medium was changed every three days until cells reached 70% confluence. These cells were considered passage 0 (P0). They were cryopreserved or subcultured at a seeding density of 5,000 cells/cm2 with EBM-2 complete medium.
MSC surface marker characterization
P4 MSCs were characterized with the Human Mesenchymal Stem Cell Verification Flow Kit (R&D Systems), including antibodies for positive markers CD90, CD73, CD105, and negative markers CD45, CD34, CD11b, CD79A, HLA-DR, as well as the Human Mesenchymal Stem Cells Multi-Color Flow Kit (R&D Systems) including antibodies for positive markers CD44, CD106, CD146, and CD166. Cells were analyzed with the BD FACSCanto™ II System.
MSC differentiation
P4 MSCs were assessed using the Human Mesenchymal Stem Cell Functional Identification Kit (R&D System) following the product instruction. After 21 days, cells were fixed and stained with FABP-4 antibody to identify adipocytes and osteocalcin antibody to identify osteocytes.
Immune cell culture
Raw 264.7 cells (RAW-dual cells from InvivoGen) were cultured in DMEM (with 4.5 g/L glucose, 2 mM L-glutamine, 10% heat-inactivated FBS, 100 µg/mL Normocin and 1% Pen-Strep) at a seeding density of 1.5 × 104 cells/cm2. The medium was renewed twice a week. THP-1 cells (THP1-dual cells from InvivoGen) were maintained in RPMI 1640 (with 2 mM L-glutamine, 25 mM HEPES,10% heat-inactivated FBS, 100 μg/mL Normocin, and 1% Pen-Strep). HL-60 cells were cultured in IMEM with 20% FBS.
Macrophage inflammation assay
Raw 264.7 cells were stimulated with 100 ng/mL LPS (O111:B4, Sigma) plus 10 ng/mL murine IFNγ (Peprotech). Human M0 macrophages were differentiated from THP-1 monocytes by incubating cells with 100 ng/mL PMA (Sigma) for 24 hs. Macrophages were then stimulated with 100 ng/mL LPS plus 10 ng/mL human IFNγ. For treatment, A1AT was added to the medium, and P4 MSCs were co-cultured with macrophages. Condition medium was harvested after 18 hs, and cytokines were measured by ELISA. The quantitative levels of 40 mouse (for Raw 264.7 and BALF) or human (for PBMCs) cytokines were evaluated with the Mouse or Human Inflammation Arrays (RayBiotech) following the product instructions. Array scanning and data extraction were done by RayBiotech using InnoScan 700/710 Microarray Scanner (Innopsys).
Neutrophil ROS production
HL-60 cells were differentiated into neutrophil-like cells with 0.1 μM ATRA and 1.25% DMSO in RPMI1640 (with 10% FBS and 2 mM L-Glutamine) for 5 days. Cells were preloaded with 5 μM CellROX deep red reagent (Invitrogen) for 15 mins at 37 °C. After washing, cells were resuspended in fresh medium and seeded into 96-well plates (100 µL of 200,000 cells/mL/well). Next, cells were activated with 100 nM PMA and treated with 0.5 mg/mL A1AT or 1/10 MSCs or their combination. The fluorescent and phase contrast images were taken with an FV3000 confocal laser scanning microscope (Olympus).
Neutrophil NETs production
The Incucyte Cytotox Red Dye was used to measure NETs production. HL-60 cells were differentiated into neutrophil-like cells with 0.1 μM ATRA and 1.25% DMSO in RPMI1640 (with 10% FBS and 2 mM L-Glutamine) for 5 days. Cells were preloaded with Cytotox Red Dye and seeded into 96-well plates (100 µL of 200,000 cells/mL/well). Cells were immediately stimulated with PMA and treated with 0.5 mg/mL A1AT or 1/10 MSCs or their combination. The fluorescent and phase contrast images were taken by the FV3000 confocal laser scanning microscope (Olympus).
PBMC flow cytometry assay
Pooled human PBMCs were purchased from Zenbio and recovered overnight before stimulation. LPS (100 ng/mL) and 25 uL human CD3/CD28 activator solution / million cells and the treatments were added for 72 hours. Then PBMCs were cultured with 1 x Cell Stimulation Cocktail plus protein transport inhibitors (Invitrogen) for 4 hs. Single cells were harvested and stained with anti-human CD3-APCcy7 and CD14-FITC for 15 mins at room temperature. After that, the cells were fixed and permeabilized with the BD Cytofix/Cytoperm™ Fixation/Permeabilization Solution Kit (BD Bioscience) and labeled intracellularly with anti-human IFNγ-APC, TNFα-BV605 (Biolegend) and IL10-PE (ebioscience). Data were collected on Attune NxT Flow Cytometer (Thermofisher) and analyzed using FlowJo software.
Phagocytosis analysis
FITC-labeled pHrodo E. coli Bioparticles® Conjugate (Thermo Fisher) were used to assess phagocytosis of THP-1 derived macrophage and HL-60 derived neutrophils. The stimulation and treatment methods were described in their inflammation assay paragraph. E. coli particles were resuspended in PBS and coated with rabbit polyclonal IgG antibodies (Escherichia coli BioParticles™ Opsonizing Reagent, Thermo Fisher) at 37 °C for 1 h. Next, cells were incubated with 0.1 mg/mL coated E. coli particles at 37 °C for 3 hs. Non-phagocytosed E. coli bioparticles were removed by washing with PBS (PH = 7.4). Next, cells were fixed with 4% PFA, permeabilized with 0.05% TritonX-100, and stained in DAPI solution. Cells were imaged with Olympus FV3000 confocal microscope and analyzed using ImageJ software.
Acute lung injury and inflammation mice
All animal experiments were approved by the Animal Care and Use Committee of the University of Nebraska-Lincoln. 10-week old male C57BL/6 mice (25 g) were purchased from Jackson Lab. For A1AT treatment, 2 mg A1AT (in 200 µL PBS) was injected intraperitoneally (i.p.) at 48 hs, 24 hs, and 0 h before the LPS challenge (three doses). Mice were anesthetized with ketamine (120 mg/kg body weight or BW, i.p.) and xylazine (16 mg/kg BW, i.p.). Mice were placed in the prone position. A 22 gauge (G) venous catheter was gently inserted into the trachea along the tongue's root in the vertical direction. Approximately 10 mm of the catheter was inserted. 50 µL of LPS was instilled. For survival rate assay, 20 mg LPS/kg BW was used. For lung tissue injury and cytokine production studies, 10 mg LPS/kg BW was used. Using a pipette, 1 × 106 MSCs were instilled via the catheter 30 mins after the LPS challenge. Next, 1 mL air was instilled to ensure LPS and cells were distributed well in the lung. The mouse's upper body was kept upright for 30 seconds to avoid fluid leakage. The body temperature was maintained at 37 °C until full awareness. The mouse was transferred to ventilated cage individually with free access to food and water. The survival rate and body weight were monitored and recorded twice a day.
Bronchoalveolar lavage fluid (BALF) and tissue harvest
Anesthesia was induced. The trachea was carefully exposed, and a 22 G venous catheter was inserted after a 5 mm cut to the trachea. 0.5 mL PBS was instilled, followed by 0.1 mL of air. After 60 s, the fluid was aspirated. This process was repeated three times to collect all BALF. Cells in BALF were harvested by centrifuging at 300 g for 10 mins. BALF cells were resuspended using 90% FBS plus 10% DMSO and frozen in a Mr. Frost at - 80 °C before long-term storage in liquid nitrogen. The supernatant was frozen at - 80 °C for cytokine analysis. After collecting BALF, lungs and other organs were harvested and fixed in 4% PFA for histology analyses.
Histology and immune staining
The fixed tissues were embedded in paraffin and sectioned (5 μm thickness). Sections were dewaxed with the Leica Auto Stainer XL and soaked in EDTA pH 8.0 (Abcam) or 10 mM Sodium Citrate solution pH 6.0 (Invitrogen) for antigen retrieval. The TBS superblock blocking buffer (Thermo Fisher) was applied to the slide for 1 h, followed by primary antibody incubation overnight at 4 °C. Slides were washed with PBS and incubated with secondary antibody and DAPI at room temperature in the dark.
BALF cells staining
Cells collected from BALF were thawed, resuspended in PBS, and fixed in 4% PFA for 20 mins. Next, cells were washed in dd H2O, placed on a Poly-Prep Slide (Sigma), and heated until dry. Slides were blocked and stained as the tissue immune staining.
TUNEL staining
The One-step TUNEL In situ Apoptosis AF 594 Kit (Elabscience) was used. Paraffin sections were dewaxed and treated with 1 x proteinase K solution at 37 °C for 20 mins. Next, sections were labeled by TDT reaction mixture for 2 hs at 37 °C. The reaction was stopped with PBS and stained with DAPI before mounting and imaging.
Statistical analysis
All the data were analyzed using GraphPad Prism 8 statistical software and shown as mean ± standard error of the mean. P value was determined by one-way analysis of variance (ANOVA) for comparison between the means of three or more groups, log-rank test for survival, or unpaired two-tailed t-tests for two groups analysis. The significance levels are indicated by p-value, *: p < 0.05, **: p < 0.01, ***: p < 0.001.
Supplementary Material
Supplementary figures.
Acknowledgements
Financial Disclosure
YL received funding from the National Heart, Lung, And Blood Institute of the National Institutes of Health under Award Number R33HL163711 and the National Cancer Institute under Award Number R33CA235326. ASB received funding from the National Institutes of General Medical Sciences under Award Number K08 GM138825.
Author Contributions
Conceptualization: YL, WV, CD, YW, MA, EH, AS, YL, RJ, GL; Investigation: LH, XW, OW; Data analysis: LH, XW, XL. Writing-original draft: YL, LH. Writing-review and editing: LH, EH, AS, YL.
Competing Interests
Dr. Lei owns equity in CellGro Technologies, LLC. This financial interest has been reviewed by the University's Individual Conflict of Interest Committee and is currently being managed by the University.
References
1. A current view on inflammation. Nat Immunol. 2017; 18: 825.
2. Netea MG, Balkwill F, Chonchol M. et al. A guiding map for inflammation. Nat Immunol [Internet]. 2017;18:826-31 Available at: http://dx.doi.org/10.1038/ni.3790
3. Fullerton JN, Gilroy DW. Resolution of inflammation: A new therapeutic frontier. Nat Rev Drug Discov. 2016;15:551-67
4. Feehan KT, Gilroy DW. Is Resolution the End of Inflammation? Trends Mol Med. 2019;25:198-214
5. Fajgenbaum DC, June CH. Cytokine Storm. N Engl J Med. 2020;383:2255-73
6. Mahmudpour M, Roozbeh J, Keshavarz M, Farrokhi S, Nabipour I. COVID-19 cytokine storm: The anger of inflammation. Cytokine. 2020;133:155151
7. Ghanbarpour R, Saghafinia M, Binabaj MR, Madani SJ, Tadressi D, Forozanmehr MJ. Pulmonary infections in ICU patients without underlying disease on ventilators. Trauma Mon. 2014;19:41-4
8. Motwani MP, Bennett F, Norris PC. et al. Potent Anti-Inflammatory and Pro-Resolving Effects of Anabasum in a Human Model of Self-Resolving Acute Inflammation. Clin Pharmacol Ther. 2018;104:675-86
9. Zhang Z, Tian H, Yang C. et al. Mesenchymal stem cells promote the resolution of cardiac inflammation after ischemia reperfusion via enhancing efferocytosis of neutrophils. J Am Heart Assoc. 2020 9
10. Giugliano GR, Giugliano RP, Gibson CM, Kuntz RE. Meta-analysis of corticosteroid treatment in acute myocardial infarction. Am J Cardiol. 2003;91:1055-9
11. Proto JD, Doran AC, Gusarova G. et al. Regulatory T Cells Promote Macrophage Efferocytosis during Inflammation Resolution. Immunity [Internet]. 2018;49:666-677.e6 Available at: https://doi.org/10.1016/j.immuni.2018.07.015
12. Mietto BS, Kroner A, Girolami EI, Santos-Nogueira E, Zhang J, David S. Role of IL-10 in resolution of inflammation and functional recovery after peripheral nerve injury. J Neurosci. 2015;35:16431-42
13. Hutchins AP, Diez D, Miranda-Saavedra D. The IL-10/STAT3-mediated anti-inflammatory response: Recent developments and future challenges. Brief Funct Genomics. 2013;12:489-98
14. Gu Z, Lamont GJ, Lamont RJ, Uriarte SM, Wang H, Scott DA. Resolvin D1, resolvin D2 and maresin 1 activate the GSK3β anti-inflammatory axis in TLR4-engaged human monocytes. Innate Immun. 2016;22:186-95
15. Cioccari L, Luethi N, Masoodi M. Lipid Mediators in Critically Ill Patients: A Step Towards Precision Medicine. Front Immunol. 2020;11:1-10
16. Serhan CN, Chiang N, Dalli J, Levy BD. Lipid mediators in the resolution of inflammation. Cold Spring Harb Perspect Biol. 2015;7:1-20
17. Fonseca MT, Moretti EH, Marques LMM. et al. A leukotriene-dependent spleen-liver axis drives TNF production in systemic inflammation. Sci Signal. 2021 14
18. Gupta N, Krasnodembskaya A, Kapetanaki M. et al. Mesenchymal stem cells enhance survival and bacterial clearance in murine Escherichia coli pneumonia. Thorax. 2012;67:533-9
19. Németh K, Leelahavanichkul A, Yuen PST. et al. Bone marrow stromal cells attenuate sepsis via prostaglandin E 2-dependent reprogramming of host macrophages to increase their interleukin-10 production. Nat Med. 2009;15:42-9
20. Pedrazza L, Cunha AA, Luft C. et al. Mesenchymal stem cells improves survival in LPS-induced acute lung injury acting through inhibition of NETs formation. J Cell Physiol. 2017;232:3552-64
21. Perlee D, de Vos AF, Scicluna BP. et al. Human Adipose-Derived Mesenchymal Stem Cells Modify Lung Immunity and Improve Antibacterial Defense in Pneumosepsis Caused by Klebsiella pneumoniae. Stem Cells Transl Med. 2019;8:785-96
22. Shin S, Kim Y, Jeong S. et al. The therapeutic effect of human adult stem cells derived from adipose tissue in endotoxemic rat model. Int J Med Sci. 2012;10:8-18
23. Devaney J, Horie S, Masterson C. et al. Human mesenchymal stromal cells decrease the severity of acute lung injury induced by E. Coli in the rat. Thorax. 2015;70:625-35
24. Yang Y, Hu S, Xu X. et al. The Vascular Endothelial Growth Factors-Expressing Character of Mesenchymal Stem Cells Plays a Positive Role in Treatment of Acute Lung Injury In vivo. Mediators Inflamm. 2016. 2016
25. José M Cóndor, Camila E Rodrigues, Roberto de Sousa Moreira, Daniele Canale, Rildo A Volpini, Maria H M Shimizu, Niels O S Camara, Irene de L Noronha LA. Treatment With Human Wharton's Jelly-Derived Mesenchymal Stem Cells Attenuates Sepsis-Induced Kidney Injury, Liver Injury, and Endothelial Dysfunction. Stem Cells Transl Med. 2016;5:1048-57
26. Danchuk S, Ylostalo JH, Hossain F. et al. Human multipotent stromal cells attenuate lipopolysaccharide-induced acute lung injury in mice via secretion of tumor necrosis factor-α-induced protein 6. Stem Cell Res Ther. 2011;2:1-15
27. Curley GF, Hayes M, Ansari B. et al. Mesenchymal stem cells enhance recovery and repair following ventilator-induced lung injury in the rat. Thorax. 2012;67:496-501
28. Hackstein H, Lippitsch A, Krug P. et al. Prospectively defined murine mesenchymal stem cells inhibit Klebsiella pneumoniae-induced acute lung injury and improve pneumonia survival. Respir Res [Internet]. 2015;16:1-12 Available at: http://dx.doi.org/10.1186/s12931-015-0288-1
29. Alcayaga-Miranda F, Cuenca J, Martin A, Contreras L, Figueroa FE, Khoury M. Combination therapy of menstrual derived mesenchymal stem cells and antibiotics ameliorates survival in sepsis. Stem Cell Res Ther [Internet]. 2015;6:1-13 Available at: http://dx.doi.org/10.1186/s13287-015-0192-0
30. Curley GF, Ansari B, Hayes M. et al. Effects of intratracheal mesenchymal stromal cell therapy during recovery and resolution after ventilator-induced lung injury. Anesthesiology. 2013;118:924-33
31. Hayes M, Masterson C, Devaney J. et al. Therapeutic efficacy of human mesenchymal stromal cells in the repair of established ventilator-induced lung injury in the rat. Anesthesiology. 2015;122:363-73
32. Rocheteau P, Chatre L, Briand D. et al. Sepsis induces long-term metabolic and mitochondrial muscle stem cell dysfunction amenable by mesenchymal stem cell therapy. Nat Commun. 2015;6:1-12
33. Jackson M V, Morrison TJ, Doherty DF. et al. Mitochondrial Transfer via Tunneling Nanotubes is an Important Mechanism by Which Mesenchymal Stem Cells Enhance Macrophage Phagocytosis in the In vitro and In vivo Models of ARDS. Stem Cells. 2016;34:2210-23
34. Krasnodembskaya A, Samarani G, Song Y. et al. Human mesenchymal stem cells reduce mortality and bacteremia in gram-negative sepsis in mice in part by enhancing the phagocytic activity of blood monocytes. Am J Physiol - Lung Cell Mol Physiol. 2012;302:1003-13
35. Morrison TJ, Jackson M V, Cunningham EK. et al. Mesenchymal stromal cells modulate macrophages in clinically relevant lung injury models by extracellular vesicle mitochondrial transfer. Am J Respir Crit Care Med. 2017;196:1275-86
36. Li B, Zhang H, Zeng M. et al. Bone marrow mesenchymal stem cells protect alveolar macrophages from lipopolysaccharide-induced apoptosis partially by inhibiting the Wnt/β-catenin pathway. Cell Biol Int. 2015;39:192-200
37. Lee JW, Krasnodembskaya A, McKenna DH, Song Y, Abbott J, Matthay MA. Therapeutic effects of human mesenchymal stem cells in ex vivo human lungs injured with live bacteria. Am J Respir Crit Care Med. 2013;187:751-60
38. Hall SRR, Tsoyi K, Ith B. et al. Mesenchymal stromal cells improve survival during sepsis in the absence of heme oxygenase-1: The importance of neutrophils. Stem Cells. 2013;31:397-407
39. Laroye C, Boufenzer A, Jolly L. et al. Bone marrow vs Wharton's jelly mesenchymal stem cells in experimental sepsis: A comparative study. Stem Cell Res Ther. 2019;10:1-11
40. Jerkic M, Gagnon S, Rabani R. et al. Human Umbilical Cord Mesenchymal Stromal Cells Attenuate Systemic Sepsis in Part by Enhancing Peritoneal Macrophage Bacterial Killing via Heme Oxygenase-1 Induction in Rats. Anesthesiology. 2020;132:140-54
41. Rabani R, Volchuk A, Jerkic M. et al. Mesenchymal stem cells enhance NOX2-dependent reactive oxygen species production and bacterial killing in macrophages during sepsis. Eur Respir J [Internet]. 2018;51:1-14 Available at: http://dx.doi.org/10.1183/13993003.02021-2017
42. Kim J, Hematti P. Mesenchymal stem cell-educated macrophages: A novel type of alternatively activated macrophages. Exp Hematol [Internet]. 2009;37:1445-53 Available at: http://dx.doi.org/10.1016/j.exphem.2009.09.004
43. Mao YX, Xu JF, Seeley EJ. et al. Adipose tissue-derived mesenchymal stem cells attenuate pulmonary infection caused by Pseudomonas aeruginosa via inhibiting overproduction of prostaglandin E2. Stem Cells. 2015;33:2331-42
44. Mei SHJ, Haitsma JJ, Dos Santos CC. et al. Mesenchymal stem cells reduce inflammation while enhancing bacterial clearance and improving survival in sepsis. Am J Respir Crit Care Med. 2010;182:1047-57
45. Sung DK, Chang YS, Sung SI, Yoo HS, Ahn SY, Park WS. Antibacterial effect of mesenchymal stem cells against Escherichia coli is mediated by secretion of beta- defensin- 2 via toll- like receptor 4 signalling. Cell Microbiol. 2016;18:424-36
46. Lu Z, Chang W, Meng S. et al. Mesenchymal stem cells induce dendritic cell immune tolerance via paracrine hepatocyte growth factor to alleviate acute lung injury. Stem Cell Res Ther. 2019;10:1-16
47. Silva JD, Lopes-Pacheco M, De Castro LL. et al. Eicosapentaenoic acid potentiates the therapeutic effects of adipose tissue-derived mesenchymal stromal cells on lung and distal organ injury in experimental sepsis. Stem Cell Res Ther. 2019;10:1-16
48. Marrow B, Masterson C, Ph D. et al. induced Acute Lung Injury and Enhance Resolution of Ventilator-induced Lung Injury in Rats. 2018; 502-16.
49. Zhang Z, Li W, Heng Z. et al. Combination therapy of human umbilical cord mesenchymal stem cells and FTY720 attenuates acute lung injury induced by lipopolysaccharide in a murine model. Oncotarget. 2017;8:77407-14
50. Pati S, Gerber MH, Menge TD. et al. Bone marrow derived mesenchymal stem cells inhibit inflammation and preserve vascular endothelial integrity in the lungs after hemorrhagic shock. PLoS One. 2011 6
51. Asmussen S, Ito H, Traber DL. et al. Human mesenchymal stem cells reduce the severity of acute lung injury in a sheep model of bacterial pneumonia. Thorax. 2014;69:819-25
52. Thompson M, Mei SHJ, Wolfe D. et al. Cell therapy with intravascular administration of mesenchymal stromal cells continues to appear safe: An updated systematic review and meta-analysis. EClinicalMedicine. 2020 19
53. Le Blanc K, Frassoni F, Ball L. et al. Mesenchymal stem cells for treatment of steroid-resistant, severe, acute graft-versus-host disease: a phase II study. Lancet. 2008;371:1579-86
54. Karussis D, Karageorgiou C, Vaknin-Dembinsky A. et al. Safety and immunological effects of mesenchymal stem cell transplantation in patients with multiple sclerosis and amyotrophic lateral sclerosis. Arch Neurol. 2010;67:1187-94
55. He X, Ai S, Guo W. et al. Umbilical cord-derived mesenchymal stem (stromal) cells for treatment of severe sepsis: aphase 1 clinical trial. Transl Res [Internet]. 2018;199:52-61 Available at: https://doi.org/10.1016/j.trsl.2018.04.006
56. McIntyre LA, Stewart DJ, Mei SHJ. et al. Cellular immunotherapy for septic shock: A phase I clinical trial. Am J Respir Crit Care Med. 2018;197:337-47
57. Chen J, Hu C, Chen L. et al. Clinical Study of Mesenchymal Stem Cell Treatment for Acute Respiratory Distress Syndrome Induced by Epidemic Influenza A (H7N9) Infection: A Hint for COVID-19 Treatment. Engineering [Internet]. 2020;6:1153-61 Available at: https://doi.org/10.1016/j.eng.2020.02.006
58. Connick P, Kolappan M, Crawley C. et al. Autologous mesenchymal stem cells for the treatment of secondary progressive multiple sclerosis: An open-label phase 2a proof-of-concept study. Lancet Neurol. 2012;11:150-6
59. Wilson JG, Liu KD, Zhuo H. et al. Mesenchymal stem (stromal) cells for treatment of ARDS: A phase 1 clinical trial. Lancet Respir Med. 2015;3:24-32
60. Panés J, García-Olmo D, Van Assche G. et al. Expanded allogeneic adipose-derived mesenchymal stem cells (Cx601) for complex perianal fistulas in Crohn's disease: a phase 3 randomised, double-blind controlled trial. Lancet. 2016;388:1281-90
61. Kebriaei P, Hayes J, Daly A. et al. A Phase 3 Randomized Study of Remestemcel-L versus Placebo Added to Second-Line Therapy in Patients with Steroid-Refractory Acute Graft-versus-Host Disease. Biol Blood Marrow Transplant [Internet]. 2020;26:835-44 Available at: https://doi.org/10.1016/j.bbmt.2019.08.029
62. Zheng G, Huang L, Tong H. et al. Treatment of acute respiratory distress syndrome with allogeneic adipose-derived mesenchymal stem cells: A randomized, placebo-controlled pilot study. Respir Res. 2014;15:1-10
63. Lv H, Chen W, Xiang AP. et al. Mesenchymal stromal cells as a salvage treatment for confirmed acute respiratory distress syndrome: preliminary data from a single-arm study. Intensive Care Med. 2020;46:1944-7
64. Matthay MA, Calfee CS, Zhuo H. et al. Treatment with allogeneic mesenchymal stromal cells for moderate to severe acute respiratory distress syndrome (START study): a randomised phase 2a safety trial. Lancet Respir Med [Internet]. 2019;7:154-62 Available at: http://dx.doi.org/10.1016/S2213-2600(18)30418-1
65. Gennadiy G, Polina M, Elena P. et al. The Results of the Single Center Pilot Randomized Russian Clinical Trial of Mesenchymal Stromal Cells in Severe Neutropenic Patients with Septic Shock (RUMCESS). Int J Blood Res Disord. 2018 5
66. Rossello-Gelabert M, Gonzalez-Pujana A, Igartua M, Santos-Vizcaino E, Hernandez RM. Clinical progress in MSC-based therapies for the management of severe COVID-19. Cytokine Growth Factor Rev [Internet]. 2022;68:25-36 Available at: https://doi.org/10.1016/j.cytogfr.2022.07.002
67. Liang B, Chen J, Li T. et al. Clinical remission of a critically ill COVID-19 patient treated by human umbilical cord mesenchymal stem cells. Medicine (Baltimore). 2020;99:e21429
68. Leng Z, Zhu R, Hou W. et al. Transplantation of ACE2- Mesenchymal stem cells improves the outcome of patients with covid-19 pneumonia. Aging Dis. 2020;11:216-28
69. Lanzoni G, Linetsky E, Correa D. et al. Umbilical cord mesenchymal stem cells for COVID-19 acute respiratory distress syndrome: A double-blind, phase 1/2a, randomized controlled trial. Stem Cells Transl Med. 2021;10:660-73
70. Sánchez-Guijo F, García-Arranz M, López-Parra M. et al. Adipose-derived mesenchymal stromal cells for the treatment of patients with severe SARS-CoV-2 pneumonia requiring mechanical ventilation. A proof of concept study. EClinicalMedicine. 2020 25
71. Chen X, Shan Y, Wen Y, Sun J, Du H. Mesenchymal stem cell therapy in severe COVID-19: A retrospective study of short-term treatment efficacy and side effects. Vol. 81, Journal of Infection. 2020
72. Gorman E, Millar J, McAuley D, O'Kane C. Mesenchymal stromal cells for acute respiratory distress syndrome (ARDS), sepsis, and COVID-19 infection: optimizing the therapeutic potential. Expert Rev Respir Med [Internet]. 2021;15:301-24 Available at: https://doi.org/10.1080/17476348.2021.1848555
73. Guttman O, S Freixo-Lima G, C Lewis E. Alpha1-antitrypsin, an endogenous immunoregulatory molecule: distinction between local and systemic effects on tumor immunology. Integr Cancer Sci Ther. 2016;2:272-80
74. Bergin DA, Hurley K, McElvaney NG, Reeves EP. Alpha-1 antitrypsin: A potent anti-inflammatory and potential novel therapeutic agent. Arch Immunol Ther Exp (Warsz). 2012;60:81-97
75. Toldo S, Seropian IM, Mezzaroma E. et al. Alpha-1 antitrypsin inhibits caspase-1 and protects from acute myocardial ischemia-reperfusion injury. J Mol Cell Cardiol. 2011;51:244-51
76. Janciauskiene S, Wrenger S, Immenschuh S. et al. The Multifaceted Effects of Alpha1-Antitrypsin on Neutrophil Functions. Front Pharmacol. 2018;17:341
77. Marcondes AM, Li X, Tabellini L. et al. Inhibition of IL-32 activation by α-1 antitrypsin suppresses alloreactivity and increases survival in an allogeneic murine marrow transplantation model. Blood. 2011;118:5031-9
78. Shapiro SD, Goldstein NM, Houghton AM, Kobayashi DK, Kelley D, Belaaouaj A. Neutrophil Elastase Contributes to Cigarette Smoke-Induced Emphysema in Mice. Am J Pathol. 2003;163:2329-35
79. Kidokoro Y, Kravis TC, Moser KM, Taylor JC CI. Relationship of Leukocyte Elastase Concentration to Severity of Emphysema in Homozygous α1-Antitrypsin-Deficient Persons. Am Rev Respir Dis. 1977;115:793-803
80. Yuan-Ping Han, Chunli Yan, WLG. Proteolytic Activation of Matrix Metalloproteinase-9 in Skin Wound Healing Is Inhibited by α-1-Antichymotrypsin. J Invest Dermatol. 2008;128:2334-42
81. He S, Chen H, Zheng J. Inhibition of tryptase and chymase induced nucleated cell infiltration by proteinase inhibitors 1. Acta Pharmacol Sin. 2004;25:1677-84
82. Bergin DA, Reeves EP, Meleady P. et al. α-1 antitrypsin regulates human neutrophil chemotaxis induced by soluble immune complexes and IL-8. J Clin Invest. 2010;120:4236-50
83. Jedicke N, Struever N, Aggrawal N. et al. α-1-antitrypsin inhibits acute liver failure in mice. Hepatology. 2014;59:2299-308
84. Libert C, Molle W Van, Brouckaert P, Fiers W. Alpha-1-Antitrypsin Inhibits the Lethal Response to TNF in Mice. J Immunol. 1996;157:5126-9
85. Subramaniyam D, Steele C, Köhnlein T. et al. Effects of alpha 1-antitrypsin on endotoxin-induced lung inflammation in vivo. Inflamm Res. 2010;59:571-8
86. Griese M, Latzin P, Kappler M. et al. α1-Antitrypsin inhalation reduces airway inflammation in cystic fibrosis patients. Eur Respir J. 2007;29:240-50
87. Pott GB, Chan ED, Dinarello CA, Shapiro L. α-1-Antitrypsin is an endogenous inhibitor of proinflammatory cytokine production in whole blood. J Leukoc Biol. 2009;85:886-95
88. Ochayon DE, Mizrahi M, Shahaf G, Baranovski BM, Lewis EC. Human α1-Antitrypsin Binds to Heat-Shock Protein gp96 and Protects from Endogenous gp96-Mediated Injury In vivo. Front Immunol. 2013;4:320
89. Tilg BH, Vannier E, Vachino G, Dinardlo CA, Mier JW. Antiinflammatory properties of hepatic acute phase proteins: preferential induction of interleukin 1 (IL-1) receptor antagonist over IL-1 beta synthesis by human peripheral blood mononuclear cells. J Exp Med. 1993;178:1629-36
90. Finotti P, Pagetta A. A heat shock protein70 fusion protein with alpha1-antitrypsin in plasma of type 1 diabetic subjects. Biochem Biophys Res Commun. 2004;315:297-305
91. Lockett AD, Kimani S, Ddungu G. et al. α₁-Antitrypsin modulates lung endothelial cell inflammatory responses to TNF-α. Am J Respir Cell Mol Biol. 2013;49:143-50
92. Chan ED, Pott GB, Silkoff PE, Ralston AH, Bryan CL, Shapiro L. Alpha-1-antitrypsin inhibits nitric oxide production. J Leukoc Biol. 2012;92:1251-60
93. Zhou T, Huang Z, Zhu X. et al. Alpha-1 antitrypsin attenuates M1 microglia-mediated neuroinflammation in retinal degeneration. Front Immunol. 2018;9:1202
94. Jonigk D, Al-Omari M, Maegel L. et al. Anti-inflammatory and immunomodulatory properties of 1-antitrypsin without inhibition of elastase. Proc Natl Acad Sci. 2013;110:15007-12
95. Serban KA, Petrusca DN, Mikosz A. et al. Alpha-1 antitrypsin supplementation improves alveolar macrophages efferocytosis and phagocytosis following cigarette smoke exposure. PLoS One. 2017;12:1-17
96. Nita IM, Serapinas D, Janciauskiene SM. α1-Antitrypsin regulates CD14 expression and soluble CD14 levels in human monocytes in vitro. Int J Biochem Cell Biol. 2007;39:1165-76
97. Janciauskiene SM, Nita IM, Stevens T. α1-antitrypsin, old dog, new tricks: α1- antitrypsin exerts in vitro anti-inflammatory activity in human monocytes by elevating cAMP. J Biol Chem. 2007;282:8573-82
98. Ozeri E, Mizrahi M, Shahaf G, Lewis EC. -1 Antitrypsin Promotes Semimature, IL-10-Producing and Readily Migrating Tolerogenic Dendritic Cells. J Immunol. 2012;189:146-53
99. Churg A, Dai J, Zay K. et al. Alpha-1-Antitrypsin and a Broad Spectrum Acute Anti-Inflammatory Effects. Lab Invest. 2001;81:1119-31
100. Kaner Z, Ochayon DE, Shahaf G. et al. Acute Phase Protein α1-Antitrypsin Reduces the Bacterial Burden in Mice by Selective Modulation of Innate Cell Responses. J Infect Dis. 2015;211:1489-98
101. Pott GB, Beard KS, Bryan CL, Merrick DT, Shapiro L. Alpha-1 Antitrypsin Reduces Severity of Pseudomonas Pneumonia in Mice and Inhibits Epithelial Barrier Disruption and Pseudomonas Invasion of Respiratory Epithelial Cells. Front Public Heal. 2013;1:1-13
102. Wanner A, Arce A De, Pardee E. Novel therapeutic uses of alpha-1 antitrypsin: A window to the future. COPD J Chronic Obstr Pulm Dis. 2012;9:583-8
103. Jia Q, Jiang X, Yu F. et al. Short cyclic peptides derived from the C-terminal sequence of α1-antitrypsin exhibit significant anti-HIV-1 activity. Bioorganic Med Chem Lett. 2012;22:2393-5
104. Bristow CL, Modarresi R, Babayeva MA, LaBrunda M, Mukhtarzad R, Trucy M, Franklin A, Reeves RE, Long A, Mullen MP, Cortes J WR. A feedback regulatory pathway between LDL and alpha-1 proteinase inhibitor in chronic inflammation and infection. Discov Med. 2013;16:201-18
105. Bristow CL, Babayeva MA, LaBrunda M, Mullen MP, Winston R. α 1proteinase inhibitor regulates CD4 + lymphocyte levels and is rate limiting in HIV-1 disease. PLoS One. 2012;7:1-10
106. Abdulsalam SI, Abdulatif A, Joyal J, Wisam G, Ajayeb A. Increased Prevalence of the Alpha-1-Antitrypsin (A1AT) Deficiency-Related S Gene in Patients Infected With Human Immunodeficiency Virus Type 1. J Med Virol. 2009;81:1047-51
107. Münch J, Ständker L, Adermann K. et al. Discovery and Optimization of a Natural HIV-1 Entry Inhibitor Targeting the gp41 Fusion Peptide. Cell. 2007;129:263-75
108. SHAPIRO L, POTT GB, RALSTON AH. Alpha-1-antitrypsin inhibits human immunodeficiency virus type 1. FASEB J. 2002;15:115-22
109. Moldthan HL, Hirko AC, Thinschmidt JS. et al. Alpha 1-antitrypsin therapy mitigated ischemic stroke damage in rats. J Stroke Cerebrovasc Dis. 2014;23:e355-63
110. Koulmanda M, Bhasin M, Fan Z. et al. Alpha 1-antitrypsin reduces inflammation and enhances mouse pancreatic islet transplant survival. Proc Natl Acad Sci. 2012;109:15443-8
111. Petrache I, Fijalkowska I, Medler TR. et al. Alpha-1 Antitrypsin Inhibits Antitrypsin Inhibits Caspase-3 Activity, Preventing Lung Endothelial Cell Apoptosis. Am J Pathol. 2006;169:1155-66
112. Kalis M, Kumar R, Janciauskiene S, Salehi A, Cilio CM. Α 1-Antitrypsin Enhances Insulin Secretion and Prevents Cytokine-Mediated Apoptosis in Pancreatic Β-Cells. Islets. 2010;2:185-9
113. Bellacen K, Kalay N, Ozeri E, Shahaf G, Lewis EC. Revascularization of pancreatic islet allografts is enhanced by α-1-Antitrypsin under anti-inflammatory conditions. Cell Transplant. 2013;22:2119-33
114. Aldonyte R, Hutchinson ET, Jin B. et al. Endothelial alpha-1-antitrypsin attenuates cigarette smoke induced apoptosis in vitro. COPD J Chronic Obstr Pulm Dis. 2008;5:153-62
115. Janciauskiene S, Welte T. Well-known and less well-known functions of Alpha-1 antitrypsin: Its role in chronic obstructive pulmonary disease and other disease developments. Ann Am Thorac Soc. 2016;13:S280-S288
116. Kim M, Cai Q, Oh Y. Therapeutic potential of alpha-1 antitrypsin in human disease. Ann Pediatr Endocrinol Metab. 2018;23:131-5
117. Ritzmann F, Chitirala P, Krüger N. et al. Therapeutic application of alpha-1 antitrypsin in COVID-19. Am J Respir Crit Care Med. 2021;204:224-7
118. Philippe A, Puel M, Narjoz C. et al. Imbalance between alpha-1-antitrypsin and interleukin 6 is associated with in-hospital mortality and thrombosis during COVID-19. Biochimie. 2022 202
119. Bai X, Hippensteel J, Leavitt A. et al. Hypothesis: Alpha-1-antitrypsin is a promising treatment option for COVID-19. Med Hypotheses [Internet]. 2021;146:110394. Available at: https://doi.org/10.1016/j.mehy.2020.110394
120. McEvoy NL, Clarke JL, Mc Elvaney OJ. et al. A randomised, double-blind, placebo-controlled, pilot trial of intravenous plasma purified alpha-1 antitrypsin for SARS-CoV-2-induced Acute Respiratory Distress Syndrome: a structured summary of a study protocol for a randomised, controlled trial. Trials. 2021;22:22-4
121. McElvaney OJ, McEvoy NL, Boland F. et al. A randomized, double-blind, placebo-controlled trial of intravenous alpha-1 antitrypsin for ARDS secondary to COVID-19. Med. 2022;3:233-248.e6
122. Schuster R, Motola-Kalay N, Baranovski BM. et al. Distinct anti-inflammatory properties of alpha1-antitrypsin and corticosteroids reveal unique underlying mechanisms of action. Cell Immunol [Internet]. 2020;356:104177. Available at: https://doi.org/10.1016/j.cellimm.2020.104177
123. Jiang D, Muschhammer J, Qi Y. et al. Suppression of Neutrophil-Mediated Tissue Damage—A Novel Skill of Mesenchymal Stem Cells. Stem Cells. 2016;34:2393-406
124. Kim EY, Ner-Gaon H, Varon J. et al. Post-sepsis immunosuppression depends on NKT cell regulation of mTOR/IFN-γ in NK cells. J Clin Invest. 2020;130:3238-52
125. Hortová-Kohoutková M, Tidu F, De Zuani M, Šrámek V, Helán M, Frič J. Phagocytosis-Inflammation Crosstalk in Sepsis: New Avenues for Therapeutic Intervention. Shock. 2020;54:606-14
126. Jin Z, Zhu Z, Liu S. et al. TRIM59 Protects Mice From Sepsis by Regulating Inflammation and Phagocytosis in Macrophages. Front Immunol. 2020;11:1-12
127. de Witte SFH, Luk F, Sierra Parraga JM. et al. Immunomodulation By Therapeutic Mesenchymal Stromal Cells (MSC) Is Triggered Through Phagocytosis of MSC By Monocytic Cells. Stem Cells. 2018;36:602-15
128. Yip HK, Fang WF, Li YC. et al. Human Umbilical Cord-Derived Mesenchymal Stem Cells for Acute Respiratory Distress Syndrome. Crit Care Med. 2020 E391-9
129. Barkama R, Mayo A, Paz A. et al. Placenta-Derived Cell Therapy to Treat Patients With Respiratory Failure Due to Coronavirus Disease 2019. Crit Care Explor. 2020;2:e0207
130. Zhu R, Yan T, Feng Y. et al. Mesenchymal stem cell treatment improves outcome of COVID-19 patients via multiple immunomodulatory mechanisms. Cell Res. 2021;31:1244-62
131. Tang L, Jiang Y, Zhu M. et al. Clinical study using mesenchymal stem cells for the treatment of patients with severe COVID-19. Front Med. 2020;14:664-73
132. Shu L, Niu C, Li R. et al. Treatment of severe COVID-19 with human umbilical cord mesenchymal stem cells. Stem Cell Res Ther. 2020;11:1-11
133. Adas G, Cukurova Z, Yasar KK. et al. The Systematic Effect of Mesenchymal Stem Cell Therapy in Critical COVID-19 Patients: A Prospective Double Controlled Trial. Cell Transplant. 2021;30:1-14
134. Xu X, Jiang W, Chen L. et al. Evaluation of the safety and efficacy of using human menstrual blood-derived mesenchymal stromal cells in treating severe and critically ill COVID-19 patients: An exploratory clinical trial. Clin Transl Med. 2021 11
135. Meng F, Xu R, Wang S. et al. Human umbilical cord-derived mesenchymal stem cell therapy in patients with COVID-19: a phase 1 clinical trial. Signal Transduct Target Ther [Internet]. 2020 5. Available at: http://dx.doi.org/10.1038/s41392-020-00286-5
136. Kavianpour M, Saleh M, Verdi J. The role of mesenchymal stromal cells in immune modulation of COVID-19: Focus on cytokine storm. Stem Cell Res Ther. 2020 11
137. Grom AA, Horne A, De Benedetti F. Macrophage activation syndrome in the era of biologic therapy. Nat Rev Rheumatol. 2016;12:259-68
138. Grigorieva KN, Bitsadze VO, Khizroeva JK. et al. Macrophage activation syndrome in COVID-19. Obstet Gynecol Reprod. 2021;15:313-20
139. Crayne CB, Albeituni S, Nichols KE, Cron RQ. The immunology of macrophage activation syndrome. Front Immunol. 2019;10:1-11
140. McGonagle D, Ramanan A V, Bridgewood C. Immune cartography of macrophage activation syndrome in the COVID-19 era. Nat Rev Rheumatol [Internet]. 2021;17:145-57 Available at: http://dx.doi.org/10.1038/s41584-020-00571-1
141. Ackermann M, Anders HJ, Bilyy R. et al. Patients with COVID-19: in the dark-NETs of neutrophils. Cell Death Differ. 2021;28:3125-39
142. Chiang CC, Korinek M, Cheng WJ, Hwang TL. Targeting Neutrophils to Treat Acute Respiratory Distress Syndrome in Coronavirus Disease. Front Pharmacol. 2020 11
143. Skendros P, Mitsios A, Chrysanthopoulou A. et al. Complement and tissue factor-enriched neutrophil extracellular traps are key drivers in COVID-19 immunothrombosis. J Clin Invest. 2020;130:6151-7
144. McKenna E, Wubben R, Isaza-Correa JM. et al. Neutrophils in COVID-19: Not Innocent Bystanders. Front Immunol. 2022;13:1-12
145. Reusch N, De Domenico E, Bonaguro L. et al. Neutrophils in COVID-19. Front Immunol. 2021;12:1-9
146. Meizlish ML, Pine AB, Bishai JD. et al. A neutrophil activation signature predicts critical illness and mortality in COVID-19. Blood Adv. 2021;5:1164-77
147. Masso-Silva JA, Moshensky A, Lam MTY. et al. Increased Peripheral Blood Neutrophil Activation Phenotypes and Neutrophil Extracellular Trap Formation in Critically Ill Coronavirus Disease 2019 (COVID-19) Patients: A Case Series and Review of the Literature. Clin Infect Dis. 2022;74:479-89
148. Dowey R, Cole J, Roger Thompson AA. et al. Enhanced neutrophil extracellular trap formation in COVID-19 is inhibited by the protein kinase C inhibitor ruboxistaurin. ERJ Open Res [Internet]. 2022 8. Available at: http://dx.doi.org/10.1183/23120541.00596-2021
149. Narasaraju T, Tang BM, Herrmann M, Muller S, Chow VTK, Radic M. Neutrophilia and NETopathy as Key Pathologic Drivers of Progressive Lung Impairment in Patients With COVID-19. Front Pharmacol. 2020;11:1-8
150. Laforge M, Elbim C, Frère C. et al. Tissue damage from neutrophil-induced oxidative stress in COVID-19. Nat Rev Immunol [Internet]. 2020;20:515-6 Available at: http://dx.doi.org/10.1038/s41577-020-0407-1
151. Al-Kuraishy HM, Al-Gareeb AI, Al-hussaniy HA, Al-Harcan NAH, Alexiou A, Batiha GES. Neutrophil Extracellular Traps (NETs) and Covid-19: A new frontiers for therapeutic modality. Int Immunopharmacol. 2022 104
152. Yaqinuddin A, Kvietys P, Kashir J. COVID-19: Role of neutrophil extracellular traps in acute lung injury. Respir Investig. 2020 419
153. Lefrançais E, Mallavia B, Zhuo H, Calfee CS, Looney MR. Maladaptive role of neutrophil extracellular traps in pathogen-induced lung injury. JCI insight. 2018;3:1-15
154. Yaqinuddin A, Kashir J. Novel therapeutic targets for SARS-CoV-2-induced acute lung injury: Targeting a potential IL-1β/neutrophil extracellular traps feedback loop. Med Hypotheses [Internet]. 2020;143:109906. Available at: https://doi.org/10.1016/j.mehy.2020.109906
155. Gould TJ, Vu TT, Swystun LL. et al. Neutrophil extracellular traps promote thrombin generation through platelet-dependent and platelet-independent mechanisms. Arterioscler Thromb Vasc Biol. 2014;34:1977-84
156. Wang Y, Luo L, Braun O. et al. Neutrophil extracellular trap-microparticle complexes enhance thrombin generation via the intrinsic pathway of coagulation in mice. Sci Rep. 2018;8:1-14
157. Zuo Y, Zuo M, Yalavarthi S. et al. Neutrophil extracellular traps and thrombosis in COVID-19. J Thromb Thrombolysis [Internet]. 2021;51:446-53 Available at: https://doi.org/10.1007/s11239-020-02324-z
158. Hisada Y, Grover SP, Maqsood A. et al. Neutrophils and neutrophil extracellular traps enhance venous thrombosis in mice bearing human pancreatic tumors. Haematologica. 2020;105:218-25
159. Karki R, Sharma BR, Tuladhar S. et al. Synergism of TNF-α and IFN-γ Triggers Inflammatory Cell Death, Tissue Damage, and Mortality in SARS-CoV-2 Infection and Cytokine Shock Syndromes. Cell [Internet]. 2021;184:149-168.e17 Available at: https://doi.org/10.1016/j.cell.2020.11.025
160. Eloseily EM, Weiser P, Crayne CB. et al. Benefit of Anakinra in Treating Pediatric Secondary Hemophagocytic Lymphohistiocytosis. Arthritis Rheumatol. 2020;72:326-34
161. Durand M, Troyanov Y, Laflamme P, Gregoire G. Macrophage activation syndrome treated with anakinra. J Rheumatol. 2010;37:879-80
162. Kang S, Tanaka T, Narazaki M, Kishimoto T. Targeting Interleukin-6 Signaling in Clinic. Immunity [Internet]. 2019;50:1007-23 Available at: https://doi.org/10.1016/j.immuni.2019.03.026
163. van der Stegen SJC, Davies DM, Wilkie S. et al. Preclinical In vivo Modeling of Cytokine Release Syndrome Induced by ErbB-Retargeted Human T Cells: Identifying a Window of Therapeutic Opportunity? J Immunol. 2013;191:4589-98
164. Teachey DT, Rheingold SR, Maude SL. et al. Cytokine release syndrome after blinatumomab treatment related to abnormal macrophage activation and ameliorated with cytokine-directed therapy. Blood. 2013;121:5154-7
165. Winkler U, Jensen M, Manzke O, Schulz H, Diehl V, Engert A. Cytokine-release syndrome in patients with B-cell chronic lymphocytic leukemia and high lymphocyte counts after treatment with an anti-CD20 monoclonal antibody (rituximab, IDEC-C2B8). Blood. 1999;94:2217-24
166. Faulkner L, Cooper A, Fantino C, Altmann DM, Sriskandan S. The Mechanism of Superantigen-Mediated Toxic Shock: Not a Simple Th1 Cytokine Storm. J Immunol. 2005;175:6870-7
167. Ablamunits V, Lepsy C. Blocking TNF signaling may save lives in COVID-19 infection. Mol Biol Rep [Internet]. 2022;49:2303-9 Available at: https://doi.org/10.1007/s11033-022-07166-x
168. Guo Y, Hu K, Li Y. et al. Targeting TNF-α for COVID-19: Recent Advanced and Controversies. Front Public Heal. 2022;10:1-9
169. Saraiva M, Saraiva M, Vieira P. et al. Biology and therapeutic potential of interleukin-10. J Exp Med. 2020;217:1-19
170. Fioranelli M, Roccia MG. Twenty-five years of studies and trials for the therapeutic application of IL-10 immunomodulating properties. From high doses administration to low dose medicine new paradigm. J Integr Cardiol. 2014;1:2-6
171. Kircheis R, Haasbach E, Lueftenegger D, Heyken WT, Ocker M, Planz O. NF-κB Pathway as a Potential Target for Treatment of Critical Stage COVID-19 Patients. Front Immunol. 2020;11:1-11
172. Coldewey SM, Rogazzo M, Collino M, Patel NSA, Thiemermann C. Inhibition of IκB kinase reduces the multiple organ dysfunction caused by sepsis in the mouse. DMM Dis Model Mech. 2013;6:1031-42
173. Yamamoto Y, Gaynor RB. Therapeutic potential of inhibition of the NF-κB pathway in the treatment of inflammation and cancer. J Clin Invest. 2001;107:135-42
174. Ponta H, Kanno T, Franzoso G, Helmberg A, Karin M. GR could physically associate with NF-KB Immunosuppression by Glucocorticoids : Inhibition of NF-KB Activity Through IKB Synthesis. Science (80- ). 1993;270:286-90
175. Fan P, Siwak DR, Abderrahman B, Agboke FA, Yerrum S, Jordan VC. Suppression of nuclear factor-kB by glucocorticoid receptor blocks estrogen-induced apoptosis in estrogen-deprived breast cancer cells. Mol Cancer Ther. 2019;18:1684-95
176. McComb S, Cessford E, Alturki NA. et al. Type-I interferon signaling through ISGF3 complex is required for sustained Rip3 activation and necroptosis in macrophages. Proc Natl Acad Sci U S A. 2014;111:3206-13
177. Yanai H, Chiba S, Hangai S. et al. Revisiting the role of IRF3 in inflammation and immunity by conditional and specifically targeted gene ablation in mice. Proc Natl Acad Sci U S A. 2018;115:5253-8
178. Xu X, Wang W, Lin L, Chen P. Liraglutide in combination with human umbilical cord mesenchymal stem cell could improve liver lesions by modulating TLR4/NF-kB inflammatory pathway and oxidative stress in T2DM/NAFLD rats. Tissue Cell [Internet]. 2020;66:101382. Available at: https://doi.org/10.1016/j.tice.2020.101382
179. Jiang Z, Zhang J. Mesenchymal stem cell-derived exosomes containing miR-145-5p reduce inflammation in spinal cord injury by regulating the TLR4/NF-κB signaling pathway. Cell Cycle [Internet]. 2021;20:993-1009 Available at: https://doi.org/10.1080/15384101.2021.1919825
180. Su VYF, Lin CS, Hung SC, Yang KY. Mesenchymal stem cell-conditioned medium induces neutrophil apoptosis associated with inhibition of the NF-κb pathway in endotoxin- induced acute lung injury. Int J Mol Sci. 2019 20
181. Liu Y, Zhang X, Hu Y. et al. Human placental mesenchymal stem cells regulate inflammation via the NF-κB signaling pathway. Exp Ther Med. 2022;24:1-11
182. Yang X, Yin Y, Yan X, Yu Z, Liu Y, Cao J. Flagellin attenuates experimental sepsis in a macrophage-dependent manner. Crit Care. 2019;23:1-14
183. Belikoff BG, Hatfield S, Georgiev P. et al. A2B Adenosine Receptor Blockade Enhances Macrophage-Mediated Bacterial Phagocytosis and Improves Polymicrobial Sepsis Survival in Mice. J Immunol. 2011;186:2444-53
184. Cui J, Wei X, Lv H. et al. The clinical efficacy of intravenous IgM-enriched immunoglobulin (pentaglobin) in sepsis or septic shock: a meta-analysis with trial sequential analysis. Ann Intensive Care [Internet]. 2019 9. Available at: https://doi.org/10.1186/s13613-019-0501-3
185. Busani S, Damiani E, Cavazzuti I, Donati A, Girardis M. Intravenous immunoglobulin in septic shock: Review of the mechanisms of action and meta-analysis of the clinical effectiveness. Minerva Anestesiol. 2016;82:559-72
186. Akdag A, Dilmen U, Haque K, Dilli D, Erdeve O, Goekmen T. Role of Pentoxifylline and/or IgM-Enriched Intravenous Immunoglobulin in the Management of Neonatal Sepsis. Am J Perinatol. 2014;31:905-12
187. Greenfield KG, Badovinac VP, Griffith TS, Knoop KA. Sepsis, Cytokine Storms, and Immunopathology: The Divide between Neonates and Adults. ImmunoHorizons. 2021;5:512-22
188. Kuhn P, Messer J, Paupe A. et al. A Multicenter, Randomized, Placebo-Controlled Trial of Prophylactic Recombinant Granulocyte-Colony Stimulating Factor in Preterm Neonates with Neutropenia. J Pediatr. 2009 155
189. Miura E, Procianoy RS, Bittar C, Miura CS. With the Clinical Diagnosis of Early-Onset Sepsis. 2015; 107.
190. Bo L, Wang F, Zhu J, Li J, Deng X. Granulocyte-colony stimulating factor (G-CSF) and granulocyte-macrophage colony stimulating factor (GM-CSF) for sepsis: A meta-analysis. Crit Care. 2011;15:1-12
191. Mathias B, Szpila BE, Moore FA, Efron PA, Moldawer LL. A review of GM-CSF therapy in sepsis. Med (United States). 2015;94:1-10
192. Pool R, Gomez H, Kellum JA. Mechanisms of Organ Dysfunction in Sepsis. Crit Care Clin [Internet]. 2018;34:63-80 Available at: https://doi.org/10.1016/j.ccc.2017.08.003
193. Spapen HD, Jacobs R, Honoré PM. Sepsis-induced multi-organ dysfunction syndrome—a mechanistic approach. J Emerg Crit Care Med. 2017;1:27-27
194. Tamburro RF, Jenkins TL. Multiple organ dysfunction syndrome: A challenge for the pediatric critical care community. Pediatr Crit Care Med. 2017;18:S1-3
195. Wang H, Ma S. The cytokine storm and factors determining the sequence and severity of organ dysfunction in multiple organ dysfunction syndrome. Am J Emerg Med. 2008;26:711-5
196. Lelubre C, Vincent JL. Mechanisms and treatment of organ failure in sepsis. Nat Rev Nephrol [Internet]. 2018;14:417-27 Available at: http://dx.doi.org/10.1038/s41581-018-0005-7
197. Pelekanos RA, Sardesai VS, Futrega K, Lott WB, Kuhn M, Doran MR. Isolation and expansion of mesenchymal stem/stromal cells derived from human placenta tissue. J Vis Exp. 2016;2016:1-13
198. Grove K. Avoidance of Maternal Cell Contamination and Overgrowth in Isolating Fetal Chorionic Villi Mesenchymal Stem Cells from Human Term Placenta. Stem Cells Transl Med. 2017;6:1070-84
Author contact
![]() Corresponding author: Yuguo Lei, The Pennsylvania State University, PA, USA. Email: yxl6034edu
Corresponding author: Yuguo Lei, The Pennsylvania State University, PA, USA. Email: yxl6034edu