Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
1. Introduction
2. ROS and their effects in the...
3. Role of ROS in...
4. Do GLP-1RAs prevent oxidative...
5. Role of GLP-1RAs in...
6. Role of GLP-1RAs in dementia
7. Conclusion
Abbreviations
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2023; 13(14):4872-4884. doi:10.7150/thno.86831 This issue Cite
Review
Targeting redox imbalance in neurodegeneration: characterizing the role of GLP-1 receptor agonists
Puja Ghosh#1, Rosaria Anna Fontanella#1, Lucia Scisciola1, Ada Pesapane1, Fatemeh Taktaz1, Martina Franzese1, Armando Puocci1, Antonio Ceriello2, Francesco Prattichizzo2, Maria Rosaria Rizzo1, Giuseppe Paolisso1,3, Michelangela Barbieri1 ![]()
1. Department of Advanced Medical and Surgical Sciences, University of Campania Luigi Vanvitelli, Naples, Italy.
2. IRCCS MultiMedica, Milan, Italy.
3. UniCamillus, International Medical University, Rome Italy.
#Contributed equally.
Received 2023-6-5; Accepted 2023-7-6; Published 2023-9-4
Abstract

Reactive oxygen species (ROS) have emerged as essential signaling molecules regulating cell survival, death, inflammation, differentiation, growth, and immune response. Environmental factors, genetic factors, or many pathological condition such as diabetes increase the level of ROS generation by elevating the production of advanced glycation end products, reducing free radical scavengers, increasing mitochondrial oxidative stress, and by interfering with DAG-PKC-NADPH oxidase and xanthine oxidase pathways. Oxidative stress, and therefore the accumulation of intracellular ROS, determines the deregulation of several proteins and caspases, damages DNA and RNA, and interferes with normal neuronal function. Furthermore, ROS play an essential role in the polymerization, phosphorylation, and aggregation of tau and amyloid-beta, key mediators of cognitive function decline. At the neuronal level, ROS interfere with the DNA methylation pattern and various apoptotic factors related to cell death, promoting neurodegeneration. Only few drugs are able to quench ROS production in neurons. The cross-linking pathways between diabetes and dementia suggest that antidiabetic medications can potentially treat dementia. Among antidiabetic drugs, glucagon-like peptide-1 receptor agonists (GLP-1RAs) have been found to reduce ROS generation and ameliorate mitochondrial function, protein aggregation, neuroinflammation, synaptic plasticity, learning, and memory. The incretin hormone glucagon-like peptide-1 (GLP-1) is produced by the enteroendocrine L cells in the distal intestine after food ingestion. Upon interacting with its receptor (GLP-1R), it regulates blood glucose levels by inducing insulin secretion, inhibiting glucagon production, and slowing gastric emptying. No study has evidenced a specific GLP-1RA pathway that quenches ROS production. Here we summarize the effects of GLP-1RAs against ROS overproduction and discuss the putative efficacy of Exendin-4, Lixisenatide, and Liraglutide in treating dementia by decreasing ROS.
Keywords: ROS, Dementia, Diabetes, GLP-1RAs, Oxidative stress
1. Introduction
Environmental and genetic factors can promote neurodegeneration in the cerebral cortex, causing the development of dementia. Since the cerebral cortex controls personality, actions, memories, and thoughts, neurodegeneration in this area of the brain is associated with the development of cognitive impairment. Frontotemporal dementia, dementia with Lewy bodies, Parkinson's disease, and Alzheimer's disease (AD) are all conditions characterized by a progressive development and worsening of dementia [1]. Estimates suggest that 60-70% of dementia cases are AD, while only 25% are vascular dementia [2]. According to the World Health Organization (WHO), by 2030, the number of patients with dementia will increase up to 75 million, while the research output on dementia is expected to double between 2017 and 2025. Thus, the generic term dementia is a public health priority.
Although several factors cause the development of dementia, the redox balance plays a crucial role in the pathogenesis of neurodegeneration. The neurodegenerative process is accompanied by the inability of the cell to maintain the homeostasis of reactive oxygen species (ROS) and by an alteration of antioxidant molecules [3]. ROS are free radicals and highly reactive ions such as singlet oxygen, hydrogen peroxide, hypochlorite ion, hydroxyl radical, and superoxide radical. Depending on their concentration, localization, and persistence, they intervene in multiple cellular processes and can promote either harmful or beneficial effect. Indeed, low levels of ROS are essential for the correct functioning of multiple signaling pathways, while high concentrations of ROS can promote cellular damage and other noxious effects. Therefore, both low and high levels of ROS are held to play a role in the etiopathogenesis of a plethora of diseases.
Several studies have highlighted that the increase of ROS leads to the dysregulation of various proteins, such as tau and beta-amyloid, as well as to immune activation, events typical of dementia [3]. Numerous studies show that ROS also contributes to the oxidation of DNA, RNA, and lipid peroxidation, which causes neurodegeneration [4].
Diabetes mellitus is one of the main causes of increased oxidative stress in cells and tissues. In this regard, much scientific evidence demonstrates that hyperglycemia activates the electron transport chain, thus increasing ROS activity [5]. Many research groups have demonstrated an increased risk for diabetic patients to develop cognitive decline, possibly due to the common pathophysiological mechanisms shared by diabetes and the development of dementia [6]. In diabetic patients, the impaired insulin homeostasis in the brain, insulin resistance, and hyperinsulinemia contribute to amyloid proteolysis, the disruption of neurovascular coupling, and to impaired astrocyte trafficking, phenomena underlying the neurodegenerative process [7]. Therefore, antidiabetic drugs controlling plasma glucose levels and insulin resistance could theoretically have the ability to reduce the risk of dementia. Preliminary proofs of efficacy for some antidiabetic medications in the treatment of dementia has already been provided, e.g. sodium-glucose cotransporter-2 inhibitor (SGLT-2i), glucagon-like peptide-1 receptor agonists (GLP-1RA), dipeptidyl peptidase inhibitors- 4 (DPP-4i), metformin, thiazolidinediones. GLP-1 (glucagon-like peptide 1) is a hormone produced by the small and large intestines, pancreatic alpha cells, and the central nervous system [8]. Its release occurs after the meal when the L cells of the intestine perceive the presence of food in the gastrointestinal tract [9]. When GLP-1 binds to its receptor (GLP-1R), it exerts various effects on different organs through multiple signaling pathways, including cyclic adenosine monophosphate (cAMP) protein kinase A (PKA), mitogen-activated protein kinase (MAPK), epidermal growth factor receptor (EGFR), Phosphoinositide 3-kinases (PI3K) and Akt (protein kinase B). GLP-1R is expressed in various tissues, including pancreatic islets, pancreatic ducts, kidneys, lungs, heart, skin, immune cells, central and peripheral nervous systems, hypothalamus, hippocampus, and cortex [10]. The first oral formulation of the GLP-1RAs approved by the United States Food and Drug Administration was Rybelsus. Until then, only the injectable form was available. This is because GLP-1RAs are peptide or protein-based drugs, which can be easily degraded by the presence of proteolytic enzymes present in the gastrointestinal tract when consumed in tablet form [11].
Further, the GLP-1RAs can be characterized into i) long-acting agents, which include semaglutide, liraglutide, exenatide extended-release, and dulaglutide, and ii) Short-acting agents comprising lixisenatide and exenatide [12]. Liraglutide has 90% peptide sequence homology with GLP-1, and its half-life can also be increased up to 13 hours by adding a fatty chain to it [13]. That is why liraglutide is considered to be the full agonist of GLP-1. Researchers are trying different methods to develop an oral form of treatment. Bao and his team formulated a zein nano peptide hybrid loaded with exenatide and found promising results when administered to type 2 diabetic mice [14]. In phase 2 clinical trial, oral formulation of semaglutide on type 2 diabetic patients showed better glycemic control compared to placebo [11]. When loaded into chitosan-based nanoparticles and administered in mice, Liraglutide showed a significant increase in its bioavailability [15].
Diabetes mellitus and hyperglycemia promote broad damages in several organs. In particular, hyperglycemia induces the accumulation of intracellular ROS, causing oxidative stress in multiple tissues. This leads to a malfunction of the molecular mechanisms underlying cell survival, resulting in tissue damage. Multiple studies highlight promising results for GLP-1RAs in comparison to other antidiabetic drugs in the setting of attenuation of oxidative damage. However, the molecular mechanisms through which they exert their antioxidant action are only partially recognized.
2. ROS and their effects in the neurons
ROS are critical molecules in neuronal physiology and are involved in growth and development. In a balanced state, ROS play vital roles in several pathways, but when out of balance, they promote neurodegeneration. Even the cellular defense systems and the cellular metabolism determine an increase in ROS levels which, if not accurately balanced by the antioxidant systems, leads to oxidative stress. The latter is the cause of protein oxidation, DNA damage, and lipid peroxidation [16].
Ninenty percent of ROS are generated in the mitochondria, while only a minor part is generated via other cellular compartments, encompassing the endoplasmic reticulum, peroxisome, and the cytosol [17]. ROS mainly originate in the mitochondria from a) increased electron transport system (ETS) activity and b) decreased conversion of superoxide ions and hydrogen peroxide to water [18]. They can be produced by seven different enzymes (Complexes I and III, α-ketoglutarate dehydrogenase, aconitase, succinate dehydrogenase, α-glycerophosphate dehydrogenase, and dihydroorotate dehydrogenase), which reside in the inner membrane of the mitochondria and two others (monoamide oxidase and cytochrome b5 reductase) present in the outer membrane of the mitochondria [19]. Furthermore, free radicals are also produced by enzymes such as nicotinamide adenine dinucleotide hydrogen phosphate (NADPH) oxidase, mitochondrial (m) adenosine triphosphate (ATP) sensitive K+channels (mKATP channels) and nicotinamide adenine dinucleotide phosphate oxidase [20]. In physiological condition, ROS act as a critical regulator of neuronal plasticity and cognition [21]. Moreover, neuronal differentiation and proliferation are upregulated by ROS levels [22]. Further, the physiological concentration of ROS in neurons is involved in the development of hippocampal neuronal polarity [23].
Neurons are highly susceptible to slight variations in intracellular ROS levels as they have a high lipid content, a low amount of antioxidant enzymes, and a high oxygen intake [19]. While physiological ROS have also emerged as positive regulators of the processes driving brain growth, higher ROS levels result in neuronal pathophysiology [24]. Numerous scientific evidences highlight a strong correlation between oxidative stress, beta-amyloid aggregation, neuroinflammation, and neurodegeneration [25, 26]. Neurodegeneration is held to be promoted by the polymerization, phosphorylation, and aggregation of tau and beta-amyloid, all molecular mechanisms exacerbated by neuronal oxidative stress. During the neurodegenerative process, neuronal cells in some brain areas undergo a block of autophagy, a key mechanism to eliminate abnormal protein aggregates including the amyloid plaque. Persistent inhibition of autophagy results in the accumulation of protein aggregates causing loss of neuron function and cell death [27]. NADPH oxidase-dependent superoxide production leads to the loss of dopaminergic neurons in N27 rat and nigra neurons in adult mice and stimulates the formation of advanced oxidation protein products by triggering the production of excessive ROS [28, 29]. Increased ROS in BV2 microglia cell lines induced p38 MAPK and JNK phosphorylation, subsequently triggering NF-kB inflammatory pathway and mitophagy in neurons [30]. ROS upregulation has also been shown to activate NLRP3 inflammasome activation and cleavage of Gasdermin-d (GSDMD), causing pyroptosis. This was confirmed by cleavage of caspase-1, production of downstream mature interleukin (IL)-1β and IL-18, as well as rupture of the rapid cell membrane [29].
Oxidative stress can also alter the expression of the proteins of the Bcl2 family, which is involved in the anti-apoptotic process due to their ability to regulate mitochondrial permeability through modifications of the transition pore. In turn, neuronal apoptosis has been suggested as a relevant phenomenon in the development of cognitive impairment. ROS also plays an essential role in regulating epigenetic modifications by altering the expression of DNA methyltransferases, which catalyze DNA methylation. Similarly, ROS also led to the alteration of chromatin structure through changing histone acetylation, which led to the repression of different genes [31]. As a result, these alterations promote the expression of several genes responsible for dementia-associated pathogenesis [20].
3. Role of ROS in diabetes-induced dementia
According to the International Diabetes Federation, 537 million people of age between 20 to 79 were diagnosed with diabetes in 2021, and the number might rise to 783 million by 2045 [Diabetes Facts and Figures, International Diabetes Federation 2021]. Many studies highlight the strong correlation between diabetes mellitus and cognitive decline, with a substantial increase in the risk of developing dementia in people with diabetes [6]. A meta-analysis involving 1.4 million subjects evidenced a 40% increased risk of developing dementia in people with diabetes who have had hypoglycemic episodes [32]. The risk of vascular dementia and AD is also elevated in people with pre-diabetes and further augmented by a long duration of diabetes [33]. Indeed, a study on people aged between 70 and 89 suggests that both a long duration and an early onset of diabetes increase the predisposition to mild cognitive impairment [34]. Such a relationship is strengthened by comorbidities such as obesity, depression, dyslipidemia, and hypertension, which could synergistically lead to cognitive dysfunction [35].
Recently, to understand the genetic link between type 2 diabetes and AD, a study was performed to recognize the overlapping gene signatures. SLC2A2 was identified as the crosstalk gene possibly linking these two diseases [36]. Further, Hao and his colleagues carried out a genome-wide association study. They identified 395 shared single nucleotide polymorphisms between type 2 diabetes and AD, suggesting that the same pathogenic processes might underlie the onset of both [37]. Interestingly, based on pathway analysis utilizing a non-negative matrix factorization approach, 241 candidate genes connected to both AD and type 2 diabetes were identified, and it was predicted that these genes contribute to the shared pathogenic characteristics of AD and type 2 diabetes [38]. Due to these overlaps and correlations between type 2 diabetes and AD, the term "type 3 diabetes” was coined to refer to diabetes-induced AD [39].
Selected molecular markers are shared between diabetes and neurodegeneration, e.g. miRNAs. miRNAs are small non-coding RNAs that help regulate gene expression by silencing or activating mRNA transcripts [40]. Many microRNAs are dysregulated in diabetic patients and might play a role in cerebrovascular complications. These neuropathological conditions stimulate the cognitive decline associated with dementia. Several studies have highlighted high plasma levels of microRNAs involved in the redox balance. Salama et al. demonstrated that higher expression of miR-132 was detected among patients with mild cognitive impairment [41]. Furthermore, several studies have focused showed that cognitive decline and cerebrovascular disease are characterized by increased receptors for advanced glycation end product (RAGE) and by the reduction of brain-derived neurotrophic factor (BDNF) in conditions of hyperglycemia [42]. Hyperglycemia per se is one of the most common causes of cellular damage from oxidative stress, with the consequent dysregulation of several molecular pathways involved in cognitive decline [43].
Diabetic patients are characterized by impaired glycolytic capacity, impaired acetyl-CoA activity, and impaired glucose metabolism. These changes lead to the accumulation of ROS in the mitochondria, causing their dysfunction [44]. Mitochondrial ROS influence several physiological activities in the brain cells. Excessive mitochondrial ROS production can result in neuronal dysfunction and death, as discussed in the previous section [45, 46]. As a result, ROS have been identified to be a crucial factor for type 2 diabetes-induced dementia due to their involvement in neuroinflammation, neurodegeneration, and neuronal death. Additionally, diabetes promotes a marked increase in protein or lipid glycation due to elevated circulating glucose levels, leading to the formation of advanced glycation end products (AGEs). AGEs suppress the cellular antioxidant system and promote ROS production [47]. They were found to be involved in pathophysiological mechanisms leading to dementia and thus considered as a potential link between diabetes and neurodegeneration [48].
Hyperglcyemia fosters the activation of DAG-PKC-NADPH-oxidase (diacylglycerol-protein kinase C) pathways,promoting ROS generation. Hyperglycemia also activates phospholipases C and D, which aid in elevating DAG expression. This promotes the activation of PKC, which translocates the cytosolic elements of NADPH oxidase (Rac 1 and 2, low molecular weight G protein, p40phox, p67phox, and p47phox) to the plasma membrane. Here, these components combine with NOX2, which results in the production of ROS [49]. Interestingly, NADPH-oxidase has been found to be upregulated in the frontal and temporal cortex, which suggests that elevated NOX-associated redox pathways may play an integral part in the progression of dementia [50].
Glucose, in the presence of hexokinase, a key glycolysis enzyme, is converted to glucose-6-P, which enters the pentose phosphate pathway and helps in purine synthesis. In the presence of purine nucleotide phosphorylase, purines are converted to hypoxanthine. In turn, xanthine oxidase converts hypoxanthine to xanthine, which eventually progresses to uric acid but also produces superoxide ions as byproducts. Hyperglycemia prompts the production of xanthine oxidase, which contributes to the generation of ROS through several pathways, including calcium signaling and nitric oxide [51] (Figure 1). Of note, Miric and colleagues found in their study that xanthine oxidase-induced ROS in diabetes patients contribute to the development of peripheral neuropathy, which is consistently associated with cognitive impairment [52, 53].
Pathways leading to the production of hyperglycemia-induced ROS: The hyperglycemia determines oxidative stress in nueronal cells, resulting in pathogenesis of neurodegeneration, through i) glycation of proteins ii) mitochondrial oxidative stress iii) DAG-PKC-NADPH pathway activation iv) xanthine oxidase upregulation.
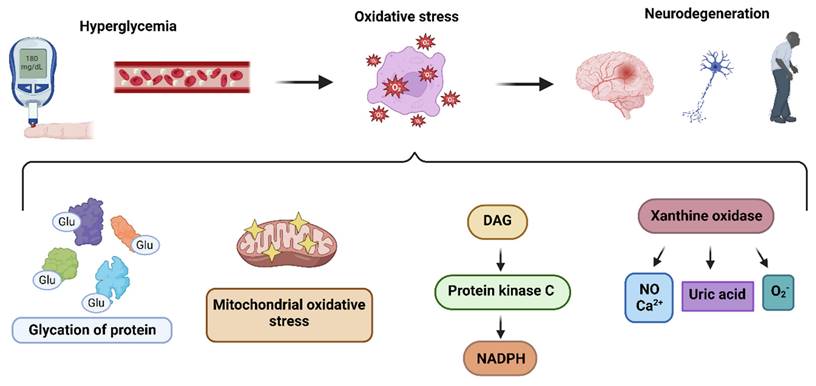
Hyperglycemiaalso interfers with the radical scavenging system. Different free radical scavenging mechanisms do exist such as zinc, copper, carotenoids, vitamin E, vitamin C, transferrin, albumin, lipoic acid, bilirubin, uric acid, and glutathione (GSH) [54]. These molecules react with free radicals to form stable molecules, preventing chain reactions from destabilizing neighboring molecules. Interestingly, the postmortem frontal cortex of patients with different stages of AD demonstrated a considerable drop in antioxidants level [55]. Selected evidences might also suggest that supplementing the diet with antioxidants might decrease the incidence of dementia [56].
4. Do GLP-1RAs prevent oxidative stress?
Recent studies highlighted many beneficial effects of GLP-1 in different tissues. It has been demonstrated that GLP-1 activates exchange proteins (Epac2), which stimulates insulin secretion and inhibits glucagon secretion by pancreatic cells [57]. Indeed, GLP-1 is able to reduce the hepatic production of glucose, shows a protective effect at the cardiac and neuronal levels, reduces the oxidative stress of the vascular system, and induces the proliferation of pancreatic beta cells [58]. Besides the well-known antihyperglycemic and protective effects in different cell types, GLP-1RAs regulate other cellular functions.
It is well known that diabetes mellitus, and therefore hyperglycemia, causes the accumulation of intracellular ROS in several organs and tissues. The ROS can affect physiological cellular functions, and all this causes diabetic complications. The accumulation of ROS appears to trigger cellular death and inflammatory molecules related to the pathogenesis of diabetes complications [49]. Furthermore, ROS-induced inactivation of anti-atherosclerosis enzymes, AGE overproduction, upregulation of stress-sensitive signaling cascades, and impairment of insulin signaling pathways lead to the development of macrovascular (cardiomyopathy) and microvascular (neuropathy, nephropathy, retinopathy, atherosclerosis) diabetic complications [59, 60].
GLP-1-RAs were found to reduce ROS generation in several experimental models (Table 1). It has been shown that GLP-1 treatment could reduce ROS in diabetic rats as it can restore the expression of manganese superoxide dismutase (SOD) and catalase [61, 62]. Because of this antioxidant property, GLP-1 is known to prevent other diabetic complications, including heart disease, neuropathies, peripheral vascular disease, and renal failure [63]. In vitro analysis of HUVECs (human umbilical vein endothelial cells) revealed that GLP-1 could prevent endothelial dysfunction caused by type 2 diabetes mellitus by decreasing the level of ROS via the GLP-1R-ERK1/2 pathway [64]. Furthermore, GLP-1RA treatment significantly reduced methylglyoxal-triggered ROS in cardiomyoblasts [65]. In addition, increases in antioxidant proteins such as SOD, glutathione peroxidase, and catalase were observed in murine cardiac cells (H9c2) after treatment with Exenatide [66]. Xiong et al. demonstrated that after GLP-1RA administration, a decrease in adiponectin expression was observed along with inhibition of ROS, resulting in improved vascular tone [67]. Exendin-4 may mediate cardioprotection in neonatal rats by inhibiting oxidative stress via the Epac-dependent pathway [68]. GLP-1RAs also reduce ROS levels in the diabetic rat aorta by inhibiting NOX4 and its subunits Ras-related C3 botulinum toxin substrate 1 (RAC-1) and p47phox. NOX4 is a major source of ROS in endothelial cells [61]. Furthermore, GLP-1RAs can counteract diabetic nephropathy by suppressing ROS production and inhibiting AGE accumulation [69]. Liljedahl and colleagues demonstrated the beneficial influence of GLP-1RA on oxidative stress in the kidney. They evaluated the renal tissue proteome of healthy mice and mice with streptozotocin-induced diabetes (STZ) receiving either vehicle or Liraglutide. After injection with STZ, there was a reduction of antioxidant enzymes such as catalase and glutathione peroxidase-3, critical enzymes for the response to oxidative stress. Liraglutide (GLP-1RA) restored antioxidant enzyme levels and improved renal histological lesions induced by diabetes [70].
Effect of GLP-1RAs on generating oxidative stress molecules in several tissues.
| Author | Experimental model | Targeted tissue in vivo | Antidiabetic drug | Pathway | Oxidative molecule |
|---|---|---|---|---|---|
| Ji et al., 2022 [120] | C57BL/6 J mice AML12 cells (Hepatocytes) | Hepatic stellate cells | Liraglutide | Reduction of RAGE/NOX2 | Hydrogen Peroxide |
| Cao et al., 2021 [121] | Sprague Dawley rats | Thoracic Aortas and Endothelial cells | Sitagliptin | Decrease in serum Malondialdehyde levels and increasing serum SOD | Reactive oxygen species |
| Chen et al., 2020 [122] | C57BL/6 mice | Cardiac fibroblasts | Liraglutide | Inhibition of Ang II-AT1R-ROS | Reactive oxygen species |
| Yang & Zhao, 2021 [123] | hRVECs cells (Human renital vascular endothelial cells) | - | Exenatide | Reduction of Sphingosine-1-phosphate receptor 2 | Reactive oxygen species |
| Li et al., 2020 [62] | H9c2 cells (Cardiomyocytes) | Semaglutide | Activation of AMPK pathway, improves autophagy. | Reactive oxygen species | |
| Wang et al., 2019 [124] | Diabetic Sprague-Dawley rat | Myocardial tissue | Exenatide | Inhibition of mammalian target of rapamycin complex 1/p70 ribosomal protein S6 kinase | Superoxide Radical |
| Ding et al., 2019 [125] | H9c2 cells (Cardiomyocytes) | - | Exenatide | Increase in the antioxidant enzymes manganese-dependent superoxide dismutase (MnSOD) and catalase. | Reactive oxygen species |
| Liljedahl et al., 2019 [70] | Male 129SV mice | Kidney | Liraglutide | Increase in protein glutathione peroxidase‐3 and catalase | Reactive oxygen species |
| Ke et al., 2017 [126] | HUVECs (Human umbilical vein endothelial cells) | - | Liraglutide + Metformin | Inhibition of PKC-NAD(P)H oxidase pathway | Reactive oxygen species |
| Chang et al., 2013 [66] | H9c2 cells (Cardiomyocytes) | - | Exenatide | Decrease in lactate dehydrogenase, creatine kinase, Malondialdehyde levels and increase in SOD, glutathione peroxidase & catalase | Hydrogen Peroxide |
5. Role of GLP-1RAs in controlling ROS in the neurons
5.1. Pathways by which GLP-1RAs regulate ROS
GLP-1 and its agonists can cross the blood-brain barrier (BBB) and affect central nervous system (CNS) functions [71]. Larsson H. and colleagues highlighted that GLP-1RA signaling induces neurogenesis, reduces apoptosis, and protects neuronal function [72, 73]. Besides the systemic level, GLP-1RAs are also able to reduce the production of ROS in neurons. ROS is essential in neurodegenerative diseases, especially AD pathogenesis and pathophysiology [74]. In neurons, GLP-1RAs act on the reduction of oxidative stress through different metabolic pathways. Indeed, GLP-1RAs trigger the activation of i) AC-cAMP-PKA-MEK-ERK (adenosine cyclase-cyclic adenosine monophosphate-mitogenic activated protein kinase-extracellular signal-regulated kinase), and ii) PI3K-Akt (phosphatidylinositol 3 kinase-protein kinase B) when it binds to its receptor in neurons [75]. These pathways activate the cAMP response element binding protein (CREB). CREB regulates the transcription of downstream genes BDNF, Apurinic endonuclease 1 (APE-1), and peroxisome proliferator-activated receptor 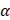 (PGC1α), which reduce ROS activity [76] (Figure 2). Furthermore, the multifunctional enzyme APE-1 has a nuclear localization signal and redox activity at its N-terminal end, thus decreasing nuclear ROS activity. Such later event interferes with the expression of several transcription factors such as signal transducer and activator of transcription 3 (STAT3), hypoxia-inducible factor 1-alpha (HIF-1α), AP-1, and nuclear factor-kappa B (NF-κβ), which in turn regulates inflammationand other cell signaling pathways [77].
(PGC1α), which reduce ROS activity [76] (Figure 2). Furthermore, the multifunctional enzyme APE-1 has a nuclear localization signal and redox activity at its N-terminal end, thus decreasing nuclear ROS activity. Such later event interferes with the expression of several transcription factors such as signal transducer and activator of transcription 3 (STAT3), hypoxia-inducible factor 1-alpha (HIF-1α), AP-1, and nuclear factor-kappa B (NF-κβ), which in turn regulates inflammationand other cell signaling pathways [77].
GLP-1RAs regulate different pathways preventing ROS production: GLP-1RA crosses blood brain barrier (BBB) and upon interacting with its receptor (GLP-1R) inhibits ROS upregulation mainly through two different metabolic pathways, i) AC-cAMP-PKA-MEK-ERK and ii) PI3K-Akt. Both these pathways activate CREB which in turn enhances the production of BDNF, APE1 and PGC1α. These molecules inhibit the upregulation of ROS by downregulating NF-kβ.
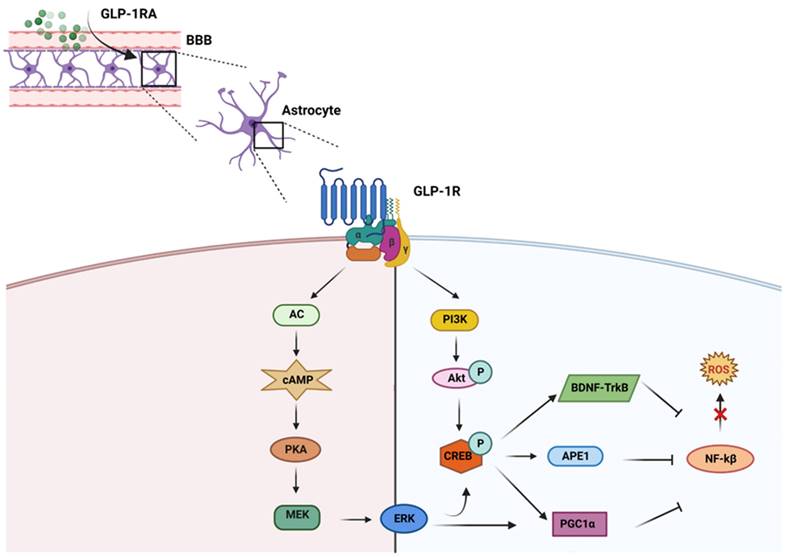
GLP-1RAs help in increasing the production of antioxidant molecules, including ϒ-glutamate cysteine ligase catalytic subunit, peroxiredoxin sulfotransferase, UDP-glucuronyl transferase, glutathione S-transferase, glutamate cysteine ligase, glutaredoxin, glutathione reductase, heme oxygenase-1, NADP quinone oxidoreductase-1, sulfiredoxin, thioredoxin reductase, thioredoxin, glutathione peroxidase, catalase and SOD, through CREB-BDNF-TrkB signaling pathway [8]. The activation of such antioxidant molecules blunts ROS [78]. Since neurons have a high energy demand to support physiological neuronal activities, mitochondrial alteration occur during the neurodegenerative process with consequent impairment of neuronal functions [79]. It is reported that GLP-1RAs can activate the PGC-1α signaling pathway, promote mitochondrial biogenesis, and reduce mitochondrial damage [80]. Moreover, CREB also enhances the production of PGC1α, which reduces NF-kB and helps to decrease mitochondrial ROS production [81]. Other researchers have also found that Exendin-4 significantly increases mitochondrial function, which is impaired by beta-amyloid accumulation [82].
GLP-1RA also interfere with activating a master regulator of cellular oxidative stress erythroid nuclear factor 2-related nuclear factor 2 (Nrf2) [8]. Under normal conditions, Nrf2 is bound to Kelch-like ECH-associated protein 1 (Keap1) in the cytoplasm. Keap1 regulates the degradation of Nrf2 through the ubiquitination system. In oxidative stress conditions, the Nrf2-Keap1 complex breaks down, and Nrf2 translocates to the nucleus and induces the transcription of antioxidant genes [83]. Interestingly, GLP-1RAs were discovered to enhance cellular antioxidant capacity through Nrf2 nuclear translocation via suppression of Keap1 and activation of the MAPKs, PKC and PI3K [84, 85].
5.2. Experimental evidence: GLP-1RAs controlling ROS
Recently, it has been demonstrated that GLP-1 shows positive neuro-regulation and protection effects in animal models [86] (Table 2). Liraglutide significantly decreased ROS overproduction in six months old 5x FAD mice and prevented other astrocyte mitochondrial dysfunctions [87]. These results suggest that GLP-1R agonists can improve cognitive function by attenuating oxidative stress and mitochondrial dysfunction in the CNS. In vitro studies in SHSY5Y cells (human neuroblastoma cell line) suggest a marked reduction in oxidative stress and increased SOD levels when treated with Liraglutide [88]. In the same cell line with silenced peptidyl-prolyl cis/trans isomerase (Pin1), another crucial ROS regulator, and treated with Liraglutide, there was an improvement in the insulin pathway and cell viability [89]. Zheng and colleagues also demonstrated the neuroprotective effect of Liraglutide in SHSY5Y cells exposed to hydrogen peroxide (H2O2) [90]. Further, Liraglutide in the AD mice model prevents beta-amyloid accumulation by rescuing oxidative stress [91]. Liraglutide has also been shown to reduce p62 levels, an adaptor of lysosomal-mediated autophagy, in female mice with early AD-like pathology. P62 is important in oxidative stress and lysosomal-mediated autophagy [92]. Other research showed that Liraglutide also downregulates 8-Hydroxy-2′-deoxyguanosine (8-OH-dG), a marker of oxidative DNA damage [93] and thiobarbituric acid reactive substances (TBARS), a significant marker for oxidative and nitrosative stress [94]. In HT22 cells (immortalized mouse hippocampal cell line), GLP-1 reduces hydrogen peroxide generation and prevents neuronal death by reducing beta-amyloid aggregation, thapsigargin, tunicamycin, and L-glutamate [95]. Incretins can also inhibit microglial death through the PKA pathway and upregulate several antioxidant enzymes' expression [96].
Effect of GLP-1RAs on the generation of oxidative stress molecules in neurons.
| Author | Experimental model | Antidiabetic drug | Pathway | Significance to human health | Oxidative molecule |
|---|---|---|---|---|---|
| Qi et al., 2022 [88] | SH-SY5Y cells (Neuroblastoma cells) | Liraglutide | Increase in SOD | Activation of energy metabolism and prevention of neuronal damage | Reactive oxygen species |
| Kornelius et al., 2022 [102] | RSC96 cells (Schwann cells) | Liraglutide | Inhibition of glucolipotoxicity | Attenuation of Schwann Cell's inflammation and dysfunction | Reactive oxygen species |
| Lambadiari et al., 2021 [127] | Type 2 diabetes clinical patients | Liraglutide + Empagliflozin | Induction of PKA & CREB, rise of glutathione | Increase of expression of neuroprotective proteins. | Reactive oxygen species |
| Xie et al., 2021 [87] | 5x FAD mice | Liraglutide | Activation of the cAMP/PKA pathway | Enhancement astrocyte's neural support abilities | Reactive oxygen species |
| Duarte et al., 2020 [91] | 3xTG-AD female mice | Liraglutide | Reduction of p62, 8-OH-dG, TBARS | Attenuation of beta amyloid accumulation and prevention of neuronal cells from oxidative and nitrosative stress | Reactive oxygen species |
| Bianchi et al., 2019 [89] | SH-SY5Y cells (Neuroblastoma cells) | Liraglutide | Induction of GSK3b/Akt | Reduction of neurotoxicity | Reactive oxygen species |
| Zheng et al., 2019 [90] | SH-SY5Y cells | Liraglutide | Influence Akt/GSK-3β | Prevention of AD induced neurodegeneration | Hydrogen peroxide |
| Spielman et al., 2017 [96] | THP-1 cells (human monocytes- microglia model) | Exendin | Activation of incretin receptors and PKA pathway | Upregulation of survival, and neurotrophins expression in microglia | Reactive oxygen species |
| Zhu et al., 2016 [97] | Neonatal Sprague-Dawley rats | Liraglutide | Activation of the PI3K/AKT and MAPK pathways | Prevention of neuronal apoptosis | Reactive oxygen species |
| Yoshino et al., 2015 [95] | HT22 cells (mouse hippocampal cell line) | GLP-1 (7-36) | Activation of Akt and ERK1/2 pathway | Protection of neurons from stressors | Hydrogen peroxide |
| An et al., 2015 [80] | PC12 cells (catecholamine cells) | Exendin-4 | Reduction of andvanced glycation end products and tau hyperphosphorylation | Enhancement of mitochondrial biogenesis and prevent tau hyperphosphorylation | Reactive oxygen species |
| Chen et al., 2012 [98] | PC12 cells (catecholamine cells) and male Wistar rats | Exendin-4 | Inhibiting high glucose-induced apoptosis | Protection of neurons against diabetes-related glucose metabolic dysfunction | Hydrogen peroxide |
Several experiments in animal models highlighted the GLP-1RAs beneficial effect on ROS homeostasis. In neonatal Sprague-Dawley rats, the neuroprotective nature of Liraglutide was exerted by suppressing ROS production [97]. In an AD-prone rat model, Exendin-4 administration was associated with remarkable cognitive performance and improved memory function [98]. Excessive ROS production, induced by various stimuli, promotes the activation of NF-κB and increases proinflammatory cytokine expression [99]. GLP-1 can reduce excessive ROS production through its anti-inflammatory properties. In neurons, microglia, and astrocytes, GLP-1 provides an anti-inflammatory effect by controlling receptor activation for advanced glycation end-products (RAGE), decreasing IL-1β (Interleukin-1 beta) and TNF-α (Tumor necrosis factor alpha) expression in the hippocampus, and inhibiting TLR4 (Toll-like receptor 4)/NF-kB signaling pathway [100]. Moreover, treatement with Liraglutide is associated with a reduction of the proinflammatory cytokines IL-6 and IL-12p70 in the brain [101]. In the Schwann cells isolated from a diabetic neuropathy rat model, treatment with Liraglutide decreased oxidative stress and attenuated inflammation [102]. Taken together, these findings suggest a potential role for GLP-1RA in the treatment of neurological disorders such as dementia.
6. Role of GLP-1RAs in dementia
Dementia, especially AD, is a devastating neurodegenerative disease and a major cause of disability worldwide [103, 104]. The available therapeutics for dementia can only manage some symptoms; therefore, researchers are focusing on developing a neuronal protective or regenerative drug that intercepts its pathogenesis. It has been demonstrated that, in several brain regions, like the hippocampus, nucleus accumbens, and striatum, the receptors of GLP-1 are expressed [105]. GLP-1RAs improves memory, learning, and synaptic plasticity and inhibits neuroinflammation, protein aggregation, mitochondrial functions, and neuronal apoptosis [106]. To deeply understand the role of GLP-1 in the brain, researchers generated knock-out mice for GLP-1 R, and they observed learning disabilities which got restored when the GLP-1 R gene was transferred to the hippocampal region [107].
Moreover, when a GLP-1RA is introduced into an AD rat model, a decrease in phosphorylated tauS396 and a reversal in memory impairment can be observed [108].
6.1. Evidence from human studies
Several studies were conducted to investigate whether GLP-1RAs prevent cognition impairment and functional deterioration in patients with dementia. Compared to the control group, the Exenatide group showed clinically significant improvements in cognitive decline [59]. Life quality, daily activities, mobility, and non-motor symptoms were improved in patients treated with Liraglutide [109]. In a clinical trial with 38 AD patients, treatment with Liraglutide was associated with a reduction in synaptic dysfunction and cognitive impairment [110]. Additionally, data collected from double-blind randomized control trial patients receiving Dulaglutide showed a 14% reduction in cognitive impairment compared to placebo [111]. Another study with 15000 participants also demonstrated the therapeutic benefit of GLP-1RAs in the decrease in the incidence of dementia [105]. Gejl and colleagues highlighted that the patients treated with Liraglutide for six months showed remarkable improvement in cognition and a significant reduction in beta-amyloid load in the brain when examined through "Brief cognitive examination" and positron emission tomography (PET) imaging [110]. In another clinical study, patients were enrolled after MINI International Neuropsychiatric Interview to understand their current mental problems and psychiatric history. Among them, patients with AD were treated with Liraglutide for 12 weeks. In order to investigate the neuroprotective effects of the drug, functional MRI (fMRI) of the brain was done both before and after the drug treatment. Results evidenced an increase in neuronal connectivity with the bilateral hippocampus in the Liraglutide-treated group compared to the placebo-receiving group [112].
Many clinical evidence highlighted the correlation between diabetes and neuropathology. Cheng et al. analyzed the fMRI of type 2 diabetes patients with cognitive decline. They showed remarkable restoration in the impaired cognitive domain after 16 weeks of Liraglutide treatment [113]. It was also reported that when Liraglutide was given to 50 subjects with diabetes who were susceptible to dementia, very significant activation of the orbitofrontal cortex and dorsolateral prefrontal cortex brain regions was observed through functional near-infrared spectroscopy. The result of cognitive tests carried out on these patients after 12 weeks of treatment also showed better scores in attention and memory [114].
Moreover, another study conducted in people with diabetes and obesity evidenced that a treatment with with GLP-1RA for three monthsimproved the Montreal Cognitive Assessment (MoCA) score, which is a highly sensitive tool for the diagnosis of mild cognitive decline [115]. Several studies demonstrated that insulin receptor desensitization (insulin resistance) is common in diabetes and dementia [116]. Therefore, antidiabetic drugs can be employed to restore the activity of insulin receptors. In order to activate the desensitized insulin receptor in the brain, researchers focused on incretin mimetics which stimulates insulin through parallel signaling pathways. A clinical study with 38 AD patients receiving Liraglutide for six months showed increased brain glucose transfer capacity from 0.72 to 1.1 umol/g/min [117]. Exenatide treatment on 15 male patients with type 2 diabetes resulted in an increase in glucose utilization in the brain which is required for glucose homeostasis regulation [118]. Moreover, in a clinical study on 10 Caucasian males after GLP-1RAs treatment, the researchers used a PET scan and observed a decrease in brain glucose fluctuation dependent on plasma glucose modification [119].
Overall, these data suggest that GLP-1RAs have the potential to prevent neurodegeneration, restorate brain glucose signaling, and improve memory, learning, synaptic plasticity, and other cognitive functions in patients with dementia.
7. Conclusion
Diabetes activates several ROS-producing pathways in different tissues. In particular, in neurons redox imbalance results in neuroinflammation and neurodegeneration. Hyperglycemia-induced oxidative stress plays a significant role in the onset and progression of diabetic neuropathology. As a result, conditions characterized by cognitive impairment, such as Alzheimer's disease and vascular dementia, are more common in diabetic patients. Consequently, antidiabetic medications, particularly GLP-1RAs, can be repurposed to improve cognitive impairments in Alzheimer's patients. Available data support a beneficial effect of GLP-1RAs in inhibiting neuronal oxidative stress and other detrimental pathways for neurodegeneration, sustaining their potential role as candidates for treating diabetes-related dementia, possibly through their ability to counteract ROS imbalances in the brain.
Abbreviations
8-OH-dG: 8-Hydroxy-2′-deoxyguanosine; AC: adenosine cyclase; AD: Alzheimer's disease; ADAS: exec -Alzheimer's disease assessment scale-cognitive subscale and exclusive domain scores of the Neuropsychological test battery; ADP: adenosine diphosphate; AGEs: advanced glycation end products; Akt: (PKB) protein kinase B; APE-1: apurinic endonuclease 1; ATP: adenosine triphosphate; BBB: blood brain barrier; Bcl2: B-cell lymphoma 2; BDNF: brain-derived neurotrophic factor; cAMP: cyclic adenosine monophosphate; CNS: central nervous system; CREB: cAMP response element binding protein; DAG: diacylglycerol; DPP-4i: dipeptidyl peptidase inhibitors- 4; EGFR: epidermal growth factor receptor; Epac: exchange protein activated by cyclic-AMP; ERK: extracellular signal-regulated kinase; ETS: electron transport system; FAD: flavin adenine nucleotide; fMRI: functional Magnetic resonance imaging; GLP-1: glucagon-like peptide 1; GLP-1RAs: Glucagon-like peptide 1 receptor agonists; GSH: glutathione; H2O2: hydrogen peroxide; HIF-1α: hypoxia-inducible factor 1-alpha; HUVECs: human umbilical vein endothelial cells; IL: interleukin; Keap1: Kelch-like ECH-associated protein 1; MEK: mitogenic activated protein kinase; mKATP: mitochondrial adenosine triphosphate sensitive K+ channels; MnSOD: manganese-dependent superoxide dismutase; MoCA: Montreal Cognitive Assessment; MRI: magnetic resonance imaging; NADPH: nicotinamide adenine dinucleotide hydrogen phosphate; NF-κβ: nuclear factor kappa B; NOX: NADPH oxidase; Nrf2: erythroid nuclear factor 2-related nuclear factor 2; PET: positron emission tomography; PGC-1α: peroxisome proliferator-activated receptor 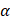 ; PI3K: phosphatidylinositol 3 kinase; Pin1: peptidyl-prolyl cis/trans isomerise; PKA: protein kinase A; PKC: protein kinase C; RAC-1: Ras-related C3 botulinum toxin substrate 1; RAGE: receptor for advanced glycation end-products; ROS: reactive oxygen species; SGLT-2i: sodium-glucose cotransporter-2 inhibitor; SOD: superoxide dismutase; STAT3: signal transducer and activator of transcription 3; STZ: streptozotocin; TBARS: thiobarbituric acid reactive substances; TLR4: toll-like receptor 4; TNF-α: tumor necrosis factor alpha; WHO: World Health Organization.
; PI3K: phosphatidylinositol 3 kinase; Pin1: peptidyl-prolyl cis/trans isomerise; PKA: protein kinase A; PKC: protein kinase C; RAC-1: Ras-related C3 botulinum toxin substrate 1; RAGE: receptor for advanced glycation end-products; ROS: reactive oxygen species; SGLT-2i: sodium-glucose cotransporter-2 inhibitor; SOD: superoxide dismutase; STAT3: signal transducer and activator of transcription 3; STZ: streptozotocin; TBARS: thiobarbituric acid reactive substances; TLR4: toll-like receptor 4; TNF-α: tumor necrosis factor alpha; WHO: World Health Organization.
Acknowledgements
Funding
This work has been supported by the Ministero dell′Istruzione, dell′Università e della Ricerca Scientifica (grants PRIN 2017), No. Prot. 2017CPLH32. This work was also supported by the Italian Ministry of Health- Ricerca Corrente to IRCCS MultiMedica.
Author Contributions
Puja Ghosh, Rosaria Anna Fontanella, Giuseppe Paolisso, Michelangela Barbieri: concept, design and writing; Lucia Scisciola, Ada Pesapane, Fatemeh Taktaz, Martina Franzese, Armando Puocci, Antonio Ceriello, Francesco Prattichizzo, Maria Rosaria Rizzo: drafting of the manuscript. All authors have read and agreed to the published version of the manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Delgado-Morales R, Esteller M. Opening up the DNA methylome of dementia. Mol Psychiatry. 2017;22:485-96
2. Hu HY, Wu BS, Ou YN, Ma YH, Huang YY, Cheng W. et al. Tea consumption and risk of incident dementia: A prospective cohort study of 377 592 UK Biobank participants. Transl Psychiatry. 2022;12:171
3. Simpson DSA, Oliver PL. ROS Generation in Microglia: Understanding Oxidative Stress and Inflammation in Neurodegenerative Disease. Antioxidants (Basel). 2020 9
4. Singh A, Kukreti R, Saso L, Kukreti S. Oxidative Stress: A Key Modulator in Neurodegenerative Diseases. Molecules. 2019 24
5. Yan LJ. Pathogenesis of chronic hyperglycemia: from reductive stress to oxidative stress. J Diabetes Res. 2014;2014:137919
6. Xue M, Xu W, Ou YN, Cao XP, Tan MS, Tan L. et al. Diabetes mellitus and risks of cognitive impairment and dementia: A systematic review and meta-analysis of 144 prospective studies. Ageing Res Rev. 2019;55:100944
7. Albai O, Frandes M, Timar R, Roman D, Timar B. Risk factors for developing dementia in type 2 diabetes mellitus patients with mild cognitive impairment. Neuropsychiatr Dis Treat. 2019;15:167-75
8. Oh YS, Jun HS. Effects of Glucagon-Like Peptide-1 on Oxidative Stress and Nrf2 Signaling. Int J Mol Sci. 2017 19
9. Drucker DJ, Habener JF, Holst JJ. Discovery, characterization, and clinical development of the glucagon-like peptides. J Clin Invest. 2017;127:4217-27
10. Kieffer TJ, Habener JF. The glucagon-like peptides. Endocr Rev. 1999;20:876-913
11. Davies M, Pieber TR, Hartoft-Nielsen ML, Hansen OKH, Jabbour S, Rosenstock J. Effect of Oral Semaglutide Compared With Placebo and Subcutaneous Semaglutide on Glycemic Control in Patients With Type 2 Diabetes: A Randomized Clinical Trial. JAMA. 2017;318:1460-70
12. Brunton SA, Wysham CH. GLP-1 receptor agonists in the treatment of type 2 diabetes: role and clinical experience to date. Postgrad Med. 2020;132:3-14
13. Agerso H, Jensen LB, Elbrond B, Rolan P, Zdravkovic M. The pharmacokinetics, pharmacodynamics, safety and tolerability of NN2211, a new long-acting GLP-1 derivative, in healthy men. Diabetologia. 2002;45:195-202
14. Bao X, Qian K, Yao P. Oral delivery of exenatide-loaded hybrid zein nanoparticles for stable blood glucose control and beta-cell repair of type 2 diabetes mice. J Nanobiotechnology. 2020;18:67
15. Shi Y, Yin M, Song Y, Wang T, Guo S, Zhang X. et al. Oral delivery of liraglutide-loaded Poly-N-(2-hydroxypropyl) methacrylamide/chitosan nanoparticles: Preparation, characterization, and pharmacokinetics. J Biomater Appl. 2021;35:754-61
16. Qu J, Chen W, Hu R, Feng H. The Injury and Therapy of Reactive Oxygen Species in Intracerebral Hemorrhage Looking at Mitochondria. Oxid Med Cell Longev. 2016;2016:2592935
17. Tarafdar A, Pula G. The Role of NADPH Oxidases and Oxidative Stress in Neurodegenerative Disorders. Int J Mol Sci. 2018 19
18. Koju N, Taleb A, Zhou J, Lv G, Yang J, Cao X. et al. Pharmacological strategies to lower crosstalk between nicotinamide adenine dinucleotide phosphate (NADPH) oxidase and mitochondria. Biomed Pharmacother. 2019;111:1478-98
19. Ahmad W, Ijaz B, Shabbiri K, Ahmed F, Rehman S. Oxidative toxicity in diabetes and Alzheimer's disease: mechanisms behind ROS/ RNS generation. J Biomed Sci. 2017;24:76
20. Di Meo S, Reed TT, Venditti P, Victor VM. Role of ROS and RNS Sources in Physiological and Pathological Conditions. Oxid Med Cell Longev. 2016;2016:1245049
21. Filosevic Vujnovic A, Rubinic M, Starcevic I, Andretic Waldowski R. Influence of Redox and Dopamine Regulation in Cocaine-Induced Phenotypes Using Drosophila. Antioxidants (Basel). 2023 12
22. Goldsmit Y, Erlich S, Pinkas-Kramarski R. Neuregulin induces sustained reactive oxygen species generation to mediate neuronal differentiation. Cell Mol Neurobiol. 2001;21:753-69
23. Wilson C, Nunez MT, Gonzalez-Billault C. Contribution of NADPH oxidase to the establishment of hippocampal neuronal polarity in culture. J Cell Sci. 2015;128:2989-95
24. Biswas K, Alexander K, Francis MM. Reactive Oxygen Species: Angels and Demons in the Life of a Neuron. NeuroSci. 2022;3(1):130-145
25. Pawlos A, Broncel M, Wozniak E, Gorzelak-Pabis P. Neuroprotective Effect of SGLT2 Inhibitors. Molecules. 2021 26
26. Du H, Meng X, Yao Y, Xu J. The mechanism and efficacy of GLP-1 receptor agonists in the treatment of Alzheimer's disease. Front Endocrinol (Lausanne). 2022;13:1033479
27. Zhang Z, Yang X, Song YQ, Tu J. Autophagy in Alzheimer's disease pathogenesis: Therapeutic potential and future perspectives. Ageing Res Rev. 2021;72:101464
28. Zawada WM, Banninger GP, Thornton J, Marriott B, Cantu D, Rachubinski AL. et al. Generation of reactive oxygen species in 1-methyl-4-phenylpyridinium (MPP+) treated dopaminergic neurons occurs as an NADPH oxidase-dependent two-wave cascade. J Neuroinflammation. 2011;8:129
29. Liu Z, Yao X, Jiang W, Li W, Zhu S, Liao C. et al. Advanced oxidation protein products induce microglia-mediated neuroinflammation via MAPKs-NF-kappaB signaling pathway and pyroptosis after secondary spinal cord injury. J Neuroinflammation. 2020;17:90
30. Zhou Q, Zhang Y, Lu L, Zhang H, Zhao C, Pu Y. et al. Copper induces microglia-mediated neuroinflammation through ROS/NF-kappaB pathway and mitophagy disorder. Food Chem Toxicol. 2022;168:113369
31. Kreuz S, Fischle W. Oxidative stress signaling to chromatin in health and disease. Epigenomics. 2016;8:843-62
32. Huang L, Zhu M, Ji J. Association between hypoglycemia and dementia in patients with diabetes: a systematic review and meta-analysis of 1.4 million patients. Diabetol Metab Syndr. 2022;14:31
33. Reinke C, Buchmann N, Fink A, Tegeler C, Demuth I, Doblhammer G. Diabetes duration and the risk of dementia: a cohort study based on German health claims data. Age Ageing. 2022 51
34. Roberts RO, Geda YE, Knopman DS, Christianson TJ, Pankratz VS, Boeve BF. et al. Association of duration and severity of diabetes mellitus with mild cognitive impairment. Arch Neurol. 2008;65:1066-73
35. Teixeira MM, Passos VMA, Barreto SM, Schmidt MI, Duncan BB, Beleigoli AMR. et al. Association between diabetes and cognitive function at baseline in the Brazilian Longitudinal Study of Adult Health (ELSA- Brasil). Sci Rep. 2020;10:1596
36. Kang P, Wang Z, Qiao D, Zhang B, Mu C, Cui H. et al. Dissecting genetic links between Alzheimer's disease and type 2 diabetes mellitus in a systems biology way. Front Genet. 2022;13:1019860
37. Hao K, Di Narzo AF, Ho L, Luo W, Li S, Chen R. et al. Shared genetic etiology underlying Alzheimer's disease and type 2 diabetes. Mol Aspects Med. 2015;43-44:66-76
38. Chung Y, Lee H, the Alzheimer's Disease Neuroimaging I. Correlation between Alzheimer's disease and type 2 diabetes using non-negative matrix factorization. Sci Rep. 2021;11:15265
39. de la Monte SM, Wands JR. Alzheimer's disease is type 3 diabetes-evidence reviewed. J Diabetes Sci Technol. 2008;2:1101-13
40. O'Brien J, Hayder H, Zayed Y, Peng C. Overview of MicroRNA Biogenesis, Mechanisms of Actions, and Circulation. Front Endocrinol (Lausanne). 2018;9:402
41. Salama II, Sami SM Abdellatif GA, Mohsen A Rasmy H, Kamel SA et al. Plasma microRNAs biomarkers in mild cognitive impairment among patients with type 2 diabetes mellitus. PLoS One. 2020;15:e0236453
42. Kim OY, Song J. The importance of BDNF and RAGE in diabetes-induced dementia. Pharmacol Res. 2020;160:105083
43. Nishikawa T, Edelstein D, Du XL, Yamagishi S, Matsumura T, Kaneda Y. et al. Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature. 2000;404:787-90
44. Sun Y, Ma C, Sun H, Wang H, Peng W, Zhou Z. et al. Metabolism: A Novel Shared Link between Diabetes Mellitus and Alzheimer's Disease. J Diabetes Res. 2020;2020:4981814
45. Angelova PR, Kasymov V, Christie I, Sheikhbahaei S, Turovsky E, Marina N. et al. Functional Oxygen Sensitivity of Astrocytes. J Neurosci. 2015;35:10460-73
46. Angelova PR, Abramov AY. Role of mitochondrial ROS in the brain: from physiology to neurodegeneration. FEBS Lett. 2018;592:692-702
47. Nowotny K, Jung T, Hohn A, Weber D, Grune T. Advanced glycation end products and oxidative stress in type 2 diabetes mellitus. Biomolecules. 2015;5:194-222
48. Chen J, Mooldijk SS, Licher S, Waqas K, Ikram MK, Uitterlinden AG. et al. Assessment of Advanced Glycation End Products and Receptors and the Risk of Dementia. JAMA Netw Open. 2021;4:e2033012
49. Volpe CMO, Villar-Delfino PH, Dos Anjos PMF, Nogueira-Machado JA. Cellular death, reactive oxygen species (ROS) and diabetic complications. Cell Death Dis. 2018;9:119
50. Ansari MA, Scheff SW. NADPH-oxidase activation and cognition in Alzheimer disease progression. Free Radic Biol Med. 2011;51:171-8
51. Yang Y, Zhao J, Qiu J, Li J, Liang X, Zhang Z. et al. Xanthine Oxidase Inhibitor Allopurinol Prevents Oxidative Stress-Mediated Atrial Remodeling in Alloxan-Induced Diabetes Mellitus Rabbits. J Am Heart Assoc. 2018 7
52. Miric DJ, Kisic BM, Filipovic-Danic S, Grbic R, Dragojevic I, Miric MB. et al. Xanthine Oxidase Activity in Type 2 Diabetes Mellitus Patients with and without Diabetic Peripheral Neuropathy. J Diabetes Res. 2016;2016:4370490
53. Lin YJ, Kao TW, Chen WL. Relationship between peripheral neuropathy and cognitive performance in the elderly population. Medicine (Baltimore). 2021;100:e26071
54. Ganjifrockwala FA, Joseph JT, George G. Decreased total antioxidant levels and increased oxidative stress in South African type 2 diabetes mellitus patients. Jemdsa. 2017;22:21-5
55. Ansari MA, Scheff SW. Oxidative stress in the progression of Alzheimer disease in the frontal cortex. J Neuropathol Exp Neurol. 2010;69:155-67
56. Beydoun MA, Beydoun HA, Fanelli-Kuczmarski MT, Weiss J, Hossain S, Canas JA. et al. Association of Serum Antioxidant Vitamins and Carotenoids With Incident Alzheimer Disease and All-Cause Dementia Among US Adults. Neurology. 2022;98:e2150-e62
57. Schmidt M, Dekker FJ, Maarsingh H. Exchange protein directly activated by cAMP (epac): a multidomain cAMP mediator in the regulation of diverse biological functions. Pharmacol Rev. 2013;65:670-709
58. Guglielmi V, Sbraccia P. GLP-1 receptor independent pathways: emerging beneficial effects of GLP-1 breakdown products. Eat Weight Disord. 2017;22:231-40
59. Giacco F, Brownlee M. Oxidative stress and diabetic complications. Circ Res. 2010;107:1058-70
60. Darenskaya MA, Kolesnikova LI, Kolesnikov SI. Oxidative Stress: Pathogenetic Role in Diabetes Mellitus and Its Complications and Therapeutic Approaches to Correction. Bull Exp Biol Med. 2021;171:179-89
61. Li Q, Lin Y, Wang S, Zhang L, Guo L. GLP-1 Inhibits High-Glucose-Induced Oxidative Injury of Vascular Endothelial Cells. Sci Rep. 2017;7:8008
62. Li Q, Tuo X, Li B, Deng Z, Qiu Y, Xie H. Semaglutide attenuates excessive exercise-induced myocardial injury through inhibiting oxidative stress and inflammation in rats. Life Sci. 2020;250:117531
63. Sivakumar PM, Premkumar B, Prabhawathi V, Prabhakar PK. Role of GLP-1 Analogs in the Management of Diabetes and its Secondary Complication. Mini Rev Med Chem. 2021;21:3166-82
64. Cai X, She M, Xu M, Chen H, Li J, Chen X. et al. GLP-1 treatment protects endothelial cells from oxidative stress-induced autophagy and endothelial dysfunction. Int J Biol Sci. 2018;14:1696-708
65. Nuamnaichati N, Mangmool S, Chattipakorn N, Parichatikanond W. Stimulation of GLP-1 Receptor Inhibits Methylglyoxal-Induced Mitochondrial Dysfunctions in H9c2 Cardiomyoblasts: Potential Role of Epac/PI3K/Akt Pathway. Front Pharmacol. 2020;11:805
66. Chang G, Zhang D, Yu H, Zhang P, Wang Y, Zheng A. et al. Cardioprotective effects of exenatide against oxidative stress-induced injury. Int J Mol Med. 2013;32:1011-20
67. Xiong X, Lu W, Qin X, Luo Q, Zhou W. Downregulation of the GLP-1/CREB/adiponectin pathway is partially responsible for diabetes-induced dysregulated vascular tone and VSMC dysfunction. Biomed Pharmacother. 2020;127:110218
68. Mangmool S, Hemplueksa P, Parichatikanond W, Chattipakorn N. Epac is required for GLP-1R-mediated inhibition of oxidative stress and apoptosis in cardiomyocytes. Mol Endocrinol. 2015;29:583-96
69. Ojima A, Ishibashi Y, Matsui T, Maeda S, Nishino Y, Takeuchi M. et al. Glucagon-like peptide-1 receptor agonist inhibits asymmetric dimethylarginine generation in the kidney of streptozotocin-induced diabetic rats by blocking advanced glycation end product-induced protein arginine methyltranferase-1 expression. Am J Pathol. 2013;182:132-41
70. Liljedahl L, Pedersen MH, McGuire JN, James P. The impact of the glucagon-like peptide 1 receptor agonist liraglutide on the streptozotocin-induced diabetic mouse kidney proteome. Physiol Rep. 2019;7:e13994
71. Yaribeygi H, Rashidy-Pour A, Atkin SL, Jamialahmadi T, Sahebkar A. GLP-1 mimetics and cognition. Life Sci. 2021;264:118645
72. Perry T, Lahiri DK, Sambamurti K, Chen D, Mattson MP, Egan JM. et al. Glucagon-like peptide-1 decreases endogenous amyloid-beta peptide (Abeta) levels and protects hippocampal neurons from death induced by Abeta and iron. J Neurosci Res. 2003;72:603-12
73. Perry T, Lahiri DK, Chen D, Zhou J, Shaw KT, Egan JM. et al. A novel neurotrophic property of glucagon-like peptide 1: a promoter of nerve growth factor-mediated differentiation in PC12 cells. J Pharmacol Exp Ther. 2002;300:958-66
74. Tobore TO. On the central role of mitochondria dysfunction and oxidative stress in Alzheimer's disease. Neurol Sci. 2019;40:1527-40
75. Yang JL, Lin YT, Chen WY, Yang YR, Sun SF, Chen SD. The Neurotrophic Function of Glucagon-Like Peptide-1 Promotes Human Neuroblastoma Differentiation via the PI3K-AKT Axis. Biology (Basel). 2020 9
76. Yang JL, Chen WY, Chen YP, Kuo CY, Chen SD. Activation of GLP-1 Receptor Enhances Neuronal Base Excision Repair via PI3K-AKT-Induced Expression of Apurinic/Apyrimidinic Endonuclease 1. Theranostics. 2016;6:2015-27
77. Oliveira TT, Coutinho LG, de Oliveira LOA, Timoteo ARS, Farias GC, Agnez-Lima LF. APE1/Ref-1 Role in Inflammation and Immune Response. Front Immunol. 2022;13:793096
78. Jin W. Regulation of BDNF-TrkB Signaling and Potential Therapeutic Strategies for Parkinson's Disease. J Clin Med. 2020 9
79. Ke J, Tian Q, Xu Q, Fu Z, Fu Q. Mitochondrial dysfunction: A potential target for Alzheimer's disease intervention and treatment. Drug Discov Today. 2021;26:1991-2002
80. An FM, Chen S, Xu Z, Yin L, Wang Y, Liu AR. et al. Glucagon-like peptide-1 regulates mitochondrial biogenesis and tau phosphorylation against advanced glycation end product-induced neuronal insult: Studies in vivo and in vitro. Neuroscience. 2015;300:75-84
81. Tetsi L, Charles AL, Paradis S, Lejay A, Talha S, Geny B. et al. Effects of cyclic nucleotide phosphodiesterases (PDEs) on mitochondrial skeletal muscle functions. Cell Mol Life Sci. 2017;74:1883-93
82. Garabadu D, Verma J. Exendin-4 attenuates brain mitochondrial toxicity through PI3K/Akt-dependent pathway in amyloid beta (1-42)-induced cognitive deficit rats. Neurochem Int. 2019;128:39-49
83. Huang HC, Nguyen T, Pickett CB. Phosphorylation of Nrf2 at Ser-40 by protein kinase C regulates antioxidant response element-mediated transcription. J Biol Chem. 2002;277:42769-74
84. Kanasaki K, Kawakita E, Koya D. Relevance of Autophagy Induction by Gastrointestinal Hormones: Focus on the Incretin-Based Drug Target and Glucagon. Front Pharmacol. 2019;10:476
85. Fernandez-Millan E, Martin MA, Goya L, Lizarraga-Mollinedo E, Escriva F, Ramos S. et al. Glucagon-like peptide-1 improves beta-cell antioxidant capacity via extracellular regulated kinases pathway and Nrf2 translocation. Free Radic Biol Med. 2016;95:16-26
86. Abd El-Rady NM, Ahmed A, Abdel-Rady MM, Ismail OI. Glucagon-like peptide-1 analog improves neuronal and behavioral impairment and promotes neuroprotection in a rat model of aluminum-induced dementia. Physiol Rep. 2021;8:e14651
87. Xie Y, Zheng J, Li S, Li H, Zhou Y, Zheng W. et al. GLP-1 improves the neuronal supportive ability of astrocytes in Alzheimer's disease by regulating mitochondrial dysfunction via the cAMP/PKA pathway. Biochem Pharmacol. 2021;188:114578
88. Qi L, Gao R, Chen Z, Lin D, Liu Z, Wang L. et al. Liraglutide reduces oxidative stress and improves energy metabolism in methylglyoxal-induced SH-SY5Y cells. Neurotoxicology. 2022;92:166-79
89. Bianchi M, D'Oria V, Braghini MR, Petrini S, Manco M. Liraglutide Treatment Ameliorates Neurotoxicity Induced by Stable Silencing of Pin1. Int J Mol Sci. 2019 20
90. Zheng C, Zhou M, Sun J, Xiong H, Peng P, Gu Z. et al. The protective effects of liraglutide on AD-like neurodegeneration induced by oxidative stress in human neuroblastoma SH-SY5Y cells. Chem Biol Interact. 2019;310:108688
91. Duarte AI, Candeias E, Alves IN, Mena D, Silva DF, Machado NJ. et al. Liraglutide Protects Against Brain Amyloid-beta(1-42) Accumulation in Female Mice with Early Alzheimer's Disease-Like Pathology by Partially Rescuing Oxidative/Nitrosative Stress and Inflammation. Int J Mol Sci. 2020 21
92. Zhang M, Yan W, Yu Y, Cheng J, Yi X, Guo T. et al. Liraglutide ameliorates diabetes-associated cognitive dysfunction via rescuing autophagic flux. J Pharmacol Sci. 2021;147:234-44
93. Zhang WQ, Tian Y, Chen XM, Wang LF, Chen CC, Qiu CM. Liraglutide ameliorates beta-cell function, alleviates oxidative stress and inhibits low grade inflammation in young patients with new-onset type 2 diabetes. Diabetol Metab Syndr. 2018;10:91
94. Shawki SM, Saad MA, Rahmo RM, Wadie W, El-Abhar HS. Liraglutide Improves Cognitive and Neuronal Function in 3-NP Rat Model of Huntington's Disease. Front Pharmacol. 2021;12:731483
95. Yoshino Y, Ishisaka M, Tsujii S, Shimazawa M, Hara H. Glucagon-like peptide-1 protects the murine hippocampus against stressors via Akt and ERK1/2 signaling. Biochem Biophys Res Commun. 2015;458:274-9
96. Spielman LJ, Gibson DL, Klegeris A. Incretin hormones regulate microglia oxidative stress, survival and expression of trophic factors. Eur J Cell Biol. 2017;96:240-53
97. Zhu H, Zhang Y, Shi Z, Lu D, Li T, Ding Y. et al. The Neuroprotection of Liraglutide Against Ischaemia-induced Apoptosis through the Activation of the PI3K/AKT and MAPK Pathways. Sci Rep. 2016;6:26859
98. Chen S, Liu AR, An FM, Yao WB, Gao XD. Amelioration of neurodegenerative changes in cellular and rat models of diabetes-related Alzheimer's disease by exendin-4. Age (Dordr). 2012;34:1211-24
99. Cominacini L, Pasini AF, Garbin U, Davoli A, Tosetti ML, Campagnola M. et al. Oxidized low density lipoprotein (ox-LDL) binding to ox-LDL receptor-1 in endothelial cells induces the activation of NF-kappaB through an increased production of intracellular reactive oxygen species. J Biol Chem. 2000;275:12633-8
100. Mohammed El Tabaa M, Mohammed El Tabaa M, Anis A, Mohamed Elgharabawy R, Borai El-Borai N. GLP-1 mediates the neuroprotective action of crocin against cigarette smoking-induced cognitive disorders via suppressing HMGB1-RAGE/TLR4-NF-kappaB pathway. Int Immunopharmacol. 2022;110:108995
101. Parthsarathy V, Holscher C. The type 2 diabetes drug liraglutide reduces chronic inflammation induced by irradiation in the mouse brain. Eur J Pharmacol. 2013;700:42-50
102. Kornelius E, Tsou SH, Chang CC, Ho YJ, Lin SC, Chen WL. et al. Liraglutide Attenuates Glucolipotoxicity-Induced RSC96 Schwann Cells' Inflammation and Dysfunction. Biomolecules. 2022 12
103. Kumar A, Sidhu J, Goyal A, Tsao JW. Alzheimer Disease. StatPearls. Treasure Island (FL). 2023
104. Akhondzadeh S. Medical Biotechnology and Alzheimer's Disease: New Hopes. Avicenna J Med Biotechnol. 2016;8:1
105. Norgaard CH, Friedrich S, Hansen CT, Gerds T, Ballard C, Moller DV. et al. Treatment with glucagon-like peptide-1 receptor agonists and incidence of dementia: Data from pooled double-blind randomized controlled trials and nationwide disease and prescription registers. Alzheimers Dement (N Y). 2022;8:e12268
106. Fu Z, Gong L, Liu J, Wu J, Barrett EJ, Aylor KW. et al. Brain Endothelial Cells Regulate Glucagon-Like Peptide 1 Entry Into the Brain via a Receptor-Mediated Process. Front Physiol. 2020;11:555
107. During MJ, Cao L, Zuzga DS, Francis JS, Fitzsimons HL, Jiao X. et al. Glucagon-like peptide-1 receptor is involved in learning and neuroprotection. Nat Med. 2003;9:1173-9
108. Li C, Liu W, Li X, Zhang Z, Qi H, Liu S. et al. The novel GLP-1/GIP analogue DA5-CH reduces tau phosphorylation and normalizes theta rhythm in the icv. STZ rat model of AD. Brain Behav. 2020;10:e01505
109. Hogg E, Wu T, Bresee C, Wertheimer J, Malatt C, Tan E. et al. A Phase II, Randomized, Double-Blinded, Placebo-Controlled Trial of Liraglutide in Parkinson's Disease. SSRN Electronic Journal. 2022
110. Gejl M, Gjedde A, Egefjord L, Moller A, Hansen SB, Vang K. et al. In Alzheimer's Disease, 6-Month Treatment with GLP-1 Analog Prevents Decline of Brain Glucose Metabolism: Randomized, Placebo-Controlled, Double-Blind Clinical Trial. Front Aging Neurosci. 2016;8:108
111. Cukierman-Yaffe T, Gerstein HC, Colhoun HM, Diaz R, Garcia-Perez LE, Lakshmanan M. et al. Effect of dulaglutide on cognitive impairment in type 2 diabetes: an exploratory analysis of the REWIND trial. Lancet Neurol. 2020;19:582-90
112. Watson KT, Wroolie TE, Tong G, Foland-Ross LC, Frangou S, Singh M. et al. Neural correlates of liraglutide effects in persons at risk for Alzheimer's disease. Behav Brain Res. 2019;356:271-8
113. Cheng H, Zhang Z, Zhang B, Zhang W, Wang J, Ni W. et al. Enhancement of Impaired Olfactory Neural Activation and Cognitive Capacity by Liraglutide, but Not Dapagliflozin or Acarbose, in Patients With Type 2 Diabetes: A 16-Week Randomized Parallel Comparative Study. Diabetes Care. 2022;45:1201-10
114. Li Q, Jia M, Yan Z, Li Q, Sun F, He C. et al. Activation of Glucagon-Like Peptide-1 Receptor Ameliorates Cognitive Decline in Type 2 Diabetes Mellitus Through a Metabolism-Independent Pathway. J Am Heart Assoc. 2021;10:e020734
115. Zhang Z, Zhang B, Wang X, Zhang X, Yang QX, Qing Z. et al. Olfactory Dysfunction Mediates Adiposity in Cognitive Impairment of Type 2 Diabetes: Insights From Clinical and Functional Neuroimaging Studies. Diabetes Care. 2019;42:1274-83
116. Duffy AM, Holscher C. The incretin analogue D-Ala2GIP reduces plaque load, astrogliosis and oxidative stress in an APP/PS1 mouse model of Alzheimer's disease. Neuroscience. 2013;228:294-300
117. Gejl M, Brock B, Egefjord L, Vang K, Rungby J, Gjedde A. Blood-Brain Glucose Transfer in Alzheimer's disease: Effect of GLP-1 Analog Treatment. Sci Rep. 2017;7:17490
118. Daniele G, Iozzo P, Molina-Carrion M, Lancaster J, Ciociaro D, Cersosimo E. et al. Exenatide Regulates Cerebral Glucose Metabolism in Brain Areas Associated With Glucose Homeostasis and Reward System. Diabetes. 2015;64:3406-12
119. Gejl M, Egefjord L, Lerche S, Vang K, Bibby BM, Holst JJ. et al. Glucagon-like peptide-1 decreases intracerebral glucose content by activating hexokinase and changing glucose clearance during hyperglycemia. J Cereb Blood Flow Metab. 2012;32:2146-52
120. Ji J, Feng M, Huang Y, Niu X. Liraglutide inhibits receptor for advanced glycation end products (RAGE)/reduced form of nicotinamide-adenine dinucleotide phosphate (NAPDH) signaling to ameliorate non-alcoholic fatty liver disease (NAFLD) in vivo and vitro. Bioengineered. 2022;13:5091-102
121. Cao Q, Xu D, Chen Y, Long Y, Dai F, Gui L. et al. Sitagliptin Reduces Endothelial Dysfunction and Apoptosis Induced by High-Fat Diet and Palmitate in Thoracic Aortas and Endothelial Cells via ROS-ER Stress-CHOP Pathway. Front Pharmacol. 2021;12:670389
122. Chen P, Yang F, Wang W, Li X, Liu D, Zhang Y. et al. Liraglutide Attenuates Myocardial Fibrosis via Inhibition of AT1R-Mediated ROS Production in Hypertensive Mice. J Cardiovasc Pharmacol Ther. 2021;26:179-88
123. Yang Y, Zhao Q. Exenatide regulates inflammation and the production of reactive oxygen species via inhibition of S1PR2 synthesis. Adv Clin Exp Med. 2021;30:555-61
124. Wang D, Jiang L, Feng B, He N, Zhang Y, Ye H. Protective effects of glucagon-like peptide-1 on cardiac remodeling by inhibiting oxidative stress through mammalian target of rapamy cin complex 1/p70 ribosomal protein S6 kinase pathway in diabetes mellitus. J Diabetes Investig. 2020;11:39-51
125. Ding W, Chang WG, Guo XC, Liu Y, Xiao DD, Ding D. et al. Exenatide Protects Against Cardiac Dysfunction by Attenuating Oxidative Stress in the Diabetic Mouse Heart. Front Endocrinol (Lausanne). 2019;10:202
126. Ke J, Liu Y, Yang J, Lu R, Tian Q, Hou W. et al. Synergistic effects of metformin with liraglutide against endothelial dysfunction through GLP-1 receptor and PKA signalling pathway. Sci Rep. 2017;7:41085
127. Lambadiari V, Thymis J, Kouretas D, Skaperda Z, Tekos F, Kousathana F. et al. Effects of a 12-Month Treatment with Glucagon-like Peptide-1 Receptor Agonists, Sodium-Glucose Cotransporter-2 Inhibitors, and Their Combination on Oxidant and Antioxidant Biomarkers in Patients with Type 2 Diabetes. Antioxidants (Basel). 2021 10
Author contact
![]() Corresponding author: Prof. Michelangela Barbieri. Department of Advanced Medical and Surgical Science, University of Campania "Luigi Vanvitelli" Napoli, (Italy). E-mail: michelangela.barbieriit.
Corresponding author: Prof. Michelangela Barbieri. Department of Advanced Medical and Surgical Science, University of Campania "Luigi Vanvitelli" Napoli, (Italy). E-mail: michelangela.barbieriit.