Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and methods
Results
Discussion
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2024; 14(5):1841-1859. doi:10.7150/thno.89703 This issue Cite
Research Paper
Dual regulation of NEMO by Nrf2 and miR-125a inhibits ferroptosis and protects liver from endoplasmic reticulum stress-induced injury
Jihoon Tak1,4, Min Sung Joo1, Yun Seok Kim2, Hyun Woo Park1, Chang Hoon Lee4, Gil-Chun Park3, Shin Hwang3 ![]() , Sang Geon Kim4
, Sang Geon Kim4 ![]()
1. College of Pharmacy, Seoul National University, Seoul 08826, Republic of Korea.
2. Department of Clinical Pharmacology and Therapeutics, Seoul National University College of Medicine, Seoul 03080, Republic of Korea.
3. Department of Surgery, Asan Medical Center, University of Ulsan College of Medicine, Seoul, Republic of Korea.
4. College of Pharmacy and Integrated Research Institute for Drug Development, Dongguk University-Seoul, Goyang-si, Kyeonggi-do 10326, Republic of Korea.
Received 2023-8-31; Accepted 2024-2-13; Published 2024-2-24
Abstract

Rationale: The surge of severe liver damage underscores the necessity for identifying new targets and therapeutic agents. Endoplasmic reticulum (ER) stress induces ferroptosis with Gα12 overexpression. NF-κB essential modulator (NEMO) is a regulator of inflammation and necroptosis. Nonetheless, the regulatory basis of NEMO de novo synthesis and its impact on hepatocyte ferroptosis need to be established. This study investigated whether Nrf2 transcriptionally induces IKBKG (the NEMO gene) for ferroptosis inhibition and, if so, how NEMO induction protects hepatocytes against ER stress-induced ferroptosis.
Methods: Experiments were conducted using human liver tissues, hepatocytes, and injury models, incorporating NEMO overexpression and Gα12 gene modulations. RNA sequencing, immunoblotting, immunohistochemistry, reporter assays, and mutation analyses were done.
Results: NEMO downregulation connects closely to ER and oxidative stress, worsening liver damage via hepatocyte ferroptosis. NEMO overexpression protects hepatocytes from ferroptosis by promoting glutathione peroxidase 4 (GPX4) expression. This protective role extends to oxidative and ER stress. Similar shifts occur in nuclear factor erythroid-2-related factor-2 (Nrf2) expression alongside NEMO changes. Nrf2 is newly identified as an IKBKG (NEMO gene) transactivator. Gα12 changes, apart from Nrf2, impact NEMO expression, pointing to post-transcriptional control. Gα12 reduction lowers miR-125a, an inhibitor of NEMO, while overexpression has the opposite effect. NEMO also counters ER stress, which triggers Gα12 overexpression. Gα12's significance in NEMO-dependent hepatocyte survival is confirmed via ROCK1 inhibition, a Gα12 downstream kinase, and miR-125a. The verified alterations or associations within the targeted entities are validated in human liver specimens and datasets originating from livers subjected to exposure to other injurious agents.
Conclusions: Hepatic injury prompted by ER stress leads to the suppression of NEMO, thereby facilitating ferroptosis through the inhibition of GPX4. IKBKG is transactivated by Nrf2 against Gα12 overexpression responsible for the increase of miR-125a, an unprecedented NEMO inhibitor, resulting in GPX4 induction. Accordingly, the induction of NEMO mitigates ferroptotic liver injury.
Keywords: NEMO, Nrf2, miR-125a, Acute liver injury, Ferroptosis
Introduction
The emergence of acute liver injury (ALI) presents a clinical challenge and represents a key focal point within clinical practice and public health apprehensions [1, 2]. It encompasses hepatocellular impairment induced by pharmaceutical agents, herbal compounds, or other chemical entities. This spectrum extends from subtle, asymptomatic elevations in hepatic enzymes to profound hepatotoxicity, ultimately leading to acute hepatic failure or requiring transplantation [3]. Among many drugs and chemicals implicated in ALI, acetaminophen (APAP) intoxication has been a prominent cause [4, 5]. So, APAP-induced hepatotoxicity draws substantial public attention, prompting research to ascertain its pathology and underlying mechanisms. Nonetheless, the molecular basis of APAP-induced ALI remains incomplete.
Ferroptosis is a form of regulated cell death characterized by lipid peroxide accumulation and oxidative damage, which are the key features associated with APAP-induced liver injury. This liver injury also occurs when glutathione peroxidase 4 (GPX4) is suppressed in the cell, which leads to a decrease in reduced glutathione (GSH) content and impairs cellular defense against lipid peroxidation, exacerbating ferroptosis [6-9]. This type of cell death also relies on intracellular iron contents [10]. Moreover, ferroptosis is closely linked with endoplasmic reticulum (ER) stress, one of the mechanisms implicated in liver pathophysiology [11, 12]. Sustaining ER stress indeed enhances ferroptosis by promoting iron overload through autophagy [13]. Despite the link between ferroptosis and ER stress, the molecular association of ferroptosis and ER stress regulation should be characterized in the context of ALI progression.
The IκB kinase (IKK) complex, comprising subunits IKKα, IKKβ, and IKKγ (also called NF-κB essential modulator, NEMO), is a vital inflammation and cell death regulator [14]. Virus infection, ionizing radiation, TNFα-receptor activation, and other external stimuli form the IKKα/β/γ complex, inducing inflammatory cytokines and pathogen-associated molecular patterns. Of these isoforms, NEMO forms a high molecular complex with IKKα and IKKβ for IκBα degradation and NF-κB activation [15-17]. Decreased NEMO levels engender necroptosis through RIPK1/RIPK3. Thus, liver-specific NEMO knockout enhances liver injury [18, 19]. Similarly, NEMO knockout in the colon prompts epithelial cell death, facilitating inflammatory colon disease [20]. Thus, the pathogenesis of APAP-induced ferroptosis needs to be elucidated in conjunction with NEMO dysregulation.
Nuclear factor erythroid-2-related factor2 (Nrf2) protects cells from oxidative injury triggered by external stresses, including xenobiotic intoxication, through antioxidant enzyme induction and inflammatory stress inhibition [21-23]. Nrf2 also controls cell fate determination, induces cell survival molecules such as Bcl-2 and certain microRNAs (e.g., miR-125b), and inhibits pro-apoptotic molecules [24, 25]. In addition to apoptosis, Nrf2 activation attenuates necrosis or pyroptosis [26, 27]. Since the regulatory roles of Nrf2 in ER stress-induced ferroptosis and its associated targets remain elusive, the present study sought to explore antioxidant proteins that can be transcriptionally controlled by Nrf2 as part of an effort to find the regulator(s). This study investigated whether Nrf2 transcriptionally induces IKBKG (the NEMO gene) for ferroptosis inhibition and, if so, how NEMO induction protects hepatocytes against ER stress-induced ferroptosis. These findings reveal Nrf2's previously unrecognized role as a transcriptional IKBKG regulator, providing insights into the underlying mechanisms that govern ferroptosis via GPX4.
In our recent investigation, APAP-induced liver injury was characterized by ferroptosis via GPX4 from ER stress-mediated Gα12 overexpression [28]. These findings evidence a potential ferroptosis involvement in APAP-induced liver injury pathogenesis and highlight the need for further investigation into the APAP toxicity and ferroptosis relationship. Another objective of the present study was to ascertain whether Gα12 signaling regulates the Nrf2-NEMO axis, potentially influencing ferroptosis under APAP toxicity. Thus, apart from Nrf2's induction of IKBKG, we wondered if NEMO expression is under the control of the Gα12 signaling pathway. In this inquiry, this study intriguingly identified miR-125a as a newly determined NEMO inhibitor downstream of the Gα12-ROCK1 axis. Consequently, our research results support the dual regulatory pathways of NEMO expression by Nrf2 and miR-125a, which contributes to the inhibition of ferroptosis, protecting the liver from ER stress-induced ALI. The outcomes were validated through patient specimen examination, consolidating identified target relevance in human liver pathophysiology. Hence, this research may offer insights into discovering potential approaches for the treatment of ALI.
Materials and methods
Human sample analysis
Human liver specimens were obtained from donors and recipients undergoing liver transplantation from 2011 to 2020 after histologic examination and ultrasonography at Asan Medical Center (Seoul, South Korea) for ALI analysis. During procurement, an intraoperative liver assessment was systematically performed to rule out fibrosis, cirrhosis, steatosis, and other abnormalities before transplantation. All patients in this study provided written informed consent. This study was approved by Asan Medical Center's Institutional Review Board (IRB no. 2021-0839) and adhered to the 1975 Declaration of Helsinki ethical guidelines.
Animal models and experiments
The animal experiment protocol was approved by the Seoul National University's Institutional Review Board and conducted under the Institutional Animal Care and Use Committee (IACUC) at Seoul National University (No. SNU-171115-2, SNU-200723-2-2, SNU-201229-4, SNU-201229-5, SNU-201229-6) guidelines. The mice were housed in a 12 h light/dark cycle and relative humidity of 50% ± 5% under filtered, pathogen-free air, with food and water available ad libitum. Male mice at 8 to 12 weeks of age, unless otherwise indicated, were used. To mitigate environmental disparities, mice were acclimated for a minimum of one week preceding each experimental procedure. For the establishment of an acute liver injury model, male C57BL/6 mice underwent overnight fasting followed by a single intraperitoneal dose of APAP treatment (300 mg/kg BW, i.p.), and the liver and blood samples were collected 6 h afterward; In this experiment, we chose a 6 h time point because APAP treatment at the above dose increased serum ALT activities up to 24 h, which peaked 6 h post-treatment [29]. Where indicated, mice were given an injection of ferrostatin-1 (1 mg/kg BW, i.p.) 1 h prior to APAP treatment.
Separately, carbon tetrachloride (CCl4, 0.5 mL/kg BW, 1:20 in corn oil, i.p.) was administered twice weekly over the specified duration (6 weeks) as a liver injury model. For the induction of ER stress, male C57BL/6 mice were received a single intraperitoneal injection of 2 mg/kg tunicamycin (Tm, Sigma Aldrich) in 150 mM dextrose for 72 h, whereas only dextrose solution was injected into control mice. Male C57BL/6 mice underwent overnight fasting before a single dose of BSO treatment (1 g/kg BW, i.p.), and tissue and blood samples were obtained 6 h later. In the context of an in vivo rescue experiment, male C57BL/6 mice were fasted overnight and treated with a single dose of APAP (300 mg/kg BW, i.p.). After 1 h, the mice were subjected to ripasudil exposure (50 mg/kg BW, i.p.), and tissue samples were collected 5 h afterward.
Nrf2 knockout (KO) mice supplied by RIKEN BioResource Center (Tsukuba, Japan) were bred and maintained. Details of the generation of the Gna12 KO mice used in this study have been described previously [30].
Statistical analyses
Statistical significance was tested via two-tailed Student's t-tests, Mann-Whitney U test, one-way ANOVA coupled with Bonferroni's method, Tukey's honestly significant difference test, or the least significant difference multiple comparison procedure, when appropriate. Correlation coefficients (r) were determined via Pearson's or Spearman's correlation methods. Differences were considered significant at P < 0.05. Statistical analyses were performed using IBM SPSS Statistics 26 software or Prism version 8.0 (GraphPad Software).
Additional details regarding materials and experimental protocols are provided in the Supplementary Materials and Methods.
Results
Inhibition of IKBKG transcript levels in liver injury patients
As a first step toward identifying prominent liver injury regulators, pathways were analyzed using a public dataset (GSE99878); Tolvaptan, known for its capacity to cause ALI, induced a marked shift in the transcriptional heatmap profile (Figure 1A, left). Notably, NF-κB signaling process-associated gene sets were the most downregulated (first rank) in tolvaptan-treated human primary hepatocytes among hallmark pathways (Figure 1A, right). In addition, NF-κB-related gene sets were diminished among the top 15 Wikipathways in subsequent leading-edge analyses, which elucidated overlaps between NF-κB-related pathways and the associated genes (Figure 1B).
Inhibition of IKBKG transcript levels in ALI patients. (A) Heatmap and hierarchical correlation analysis of DEGs (absolute fold-change > 1.5 and P < 0.05) (left) and GSEA-enrichment plot of representative gene sets (NES = -2.584, FDR = 0.000) negatively correlated with tolvaptan treatment in primary human hepatocytes using hepatic transcriptome data (n = 3 each, GSE99878) (right). The top 20 genes comprising the enrichment score's leading edge are indicated in the corresponding heatmap (blue, low; red, high). (B) Leading-edge analysis (left) and bar graphs (right) of significantly enriched GSEA Wikipathways using the same data as in A. GSEA leading-edge analysis results are represented as a matrix where the green color intensity indicates the overlap degree between core genes in each gene set combination; the more intense the green color, the greater the overlap. NES and FDR are presented as a bar graph (NES < -1.82, FDR < 0.024). NF-κB signaling-related pathways were marked with red asterisks. (C) Volcano plot (upper) of RNA-seq data from a public dataset (GSE74000, n = 2 or 3 each). Horizontal and vertical lines indicate the filtering criteria (absolute fold-change > 1.5 and adjusted P < 0.05, respectively). Red and blue dots indicate upregulated or downregulated differentially expressed genes (DEGs) in acute liver failure patients. Reactome pathway analysis of DEGs identified NF-κB-related pathway enrichment in acute liver failure patients (lower). The Circos plots illustrate the enriched DEGs overlaps and specific responses in significant Reactome pathways. Hallmark genes and associated pathways are color-coded and are represented by a specific color in the inner ring. The ribbon/arc that originates from different genes and terminates at associated Reactome pathways demonstrates the connectivity between genes and Reactome pathways. (D) Fold change of IkappaB kinase (IKK) complex transcript levels in livers of healthy individuals or HBV-acute liver failure (ALF) patients. MHN, massive hepatic necrosis; SHN, submassive hepatic necrosis (n = 10 samples from 10 individual normal subjects; n = 8 samples from 2 HBV-ALF(SHN) patients; n = 9 samples from 2 HBV-ALF(MHN) patients, as described in the GSE38941 database). (E) IKBKG, CHUK, and IKBKB transcript levels in livers of healthy individuals (n = 10) or ALI patients (n = 22). For E, values were expressed as mean ± SEM (*P < 0.05, **P < 0.01). Statistical significance was tested via two-tailed Mann-Whitney U test.
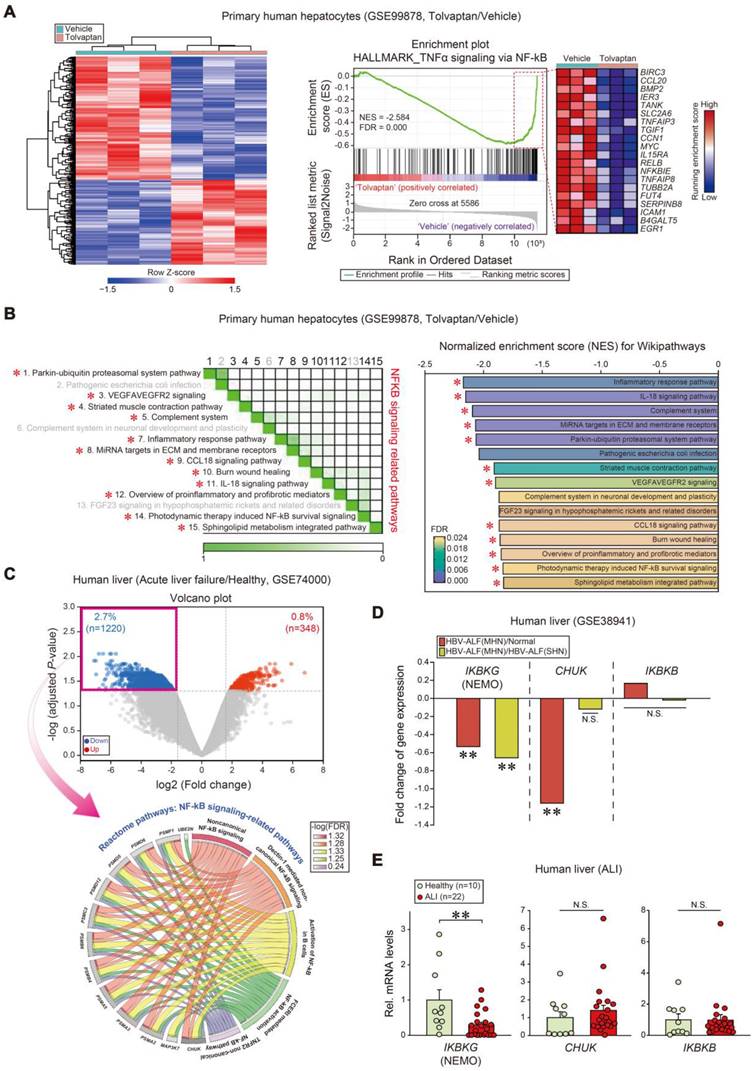
Next, we assessed gene ontology pathway changes in APAP-induced acute liver failure patients and healthy individuals (GSE74000); 1220 genes were significantly downregulated, whereas 348 genes were upregulated (Figure 1C, upper). Interestingly, the genes were tagged by several gene ontology (GO) terms analyzed in the Reactome pathway; 13 of each were clustered into five GO groups, belonging to the 'NF-κB signaling-related pathways' among downregulated genes in patients (Figure 1C, lower). GO and biological process analyses of the same dataset confirmed downregulated genes associated with the NF-κB pathway in the same patients (Figure S1A). Similarly, GO and biological process analyses indicated enhanced 'regulation of acute inflammatory response' gene expressions (Figure S1B).
To explore the new functional molecule(s), we focused on the IκB kinase (IKK) that participates in upstream NF-κB signaling and promotes NF-κB activation through pro-inflammatory stimuli [31].
In another public GEO database analysis regarding HBV-associated acute liver failure (HBV-ALF) patients (GSE38941), we assessed fold change values obtained from mRNA levels of enzyme complex IKK subunits (i.e., IKKα, IKKβ, and IKKγ). Relative levels in mRNA expression of IKBKG and CHUK were significantly downregulated in patients with hepatic necrosis, whereas IKBKB was unchanged (Figure 1D). To validate the IKBKG and CHUK association, we used our own samples of patients with ALI on drugs (i.e., liver intoxications from herbs, drug medications, or unknown origins), as described previously [28]. Among transcripts, IKBKG was the most significantly diminished in patients with ALI compared to healthy subjects (i.e., -70.7%) (Figure 1E). These results substantiate the inverse IKBKG transcript level and hepatic injury association.
Nrf2 and NEMO associations during liver injury
We first analyzed the RNA sequencing (RNA-seq) dataset from APAP-subjected mice (GSE104302) to explore NEMO regulation. Principal component analysis (PCA) exhibited a gene expression segregation between APAP and the vehicle (Figure 2A, left). Differentially expressed genes (DEGs) accounted for 11.5% of the entire transcriptomes; Among the 2652 DEGs, 816 were downregulated, whereas 1836 were upregulated (Figure 2A, middle). In the Biocarta analysis for the downregulated gene group, several biological processes expressed high-fold enrichments in 'Mechanism of APAP activity and toxicity' and 'Extrinsic prothrombin activation' (foremost affected), and 'Oxidative stress-induced gene expression via Nrf2' (second-most affected) (Figure 2A, right). Furthermore, we analyzed GO gene sets and GSEA hallmarks using the transcriptome dataset from mouse liver (GSE104302, GSE173595); APAP treatment affected protein refolding genes (Figure S2A) and those linked to the 'unfolded protein response' (Figure S2B).
The Nrf2 and NEMO relationship and association with ER stress responses. (A) RNA-seq dataset (GSE104302) obtained from the liver of APAP- or vehicle-treated mice. Principal component analysis (PCA) score (left) and volcano plots of RNA-seq data (middle) (mint color, downregulated; red color, upregulated; DEGs with P-value < 0.01 and absolute FC > 1.5). Enrichment bubble plot of the Biocarta pathway indicating that the 'Oxidative stress-induced gene expression via Nrf2' was significantly downregulated in response to APAP treatment (n = 3 each) (right). (B) NEMO and Nrf2 immunohistochemistry in the liver of the mice treated with a single APAP dose (300 mg/kg BW, i.p., 6 h) (n = 5 or 6 each). (C) NEMO, Nrf2, and Grp78 immunoblottings (upper left) in the livers of the same mice as in B. Band intensities represent values relative to each respective control (n = 5 or 6 each) (lower left). Nrf2 and NEMO correlations (upper right); Grp78 and NEMO correlations (n = 11) (lower right). (D, E) NEMO, Nrf2, and Grp78 immunoblottings (left) in the liver of CCl4-treated mice (0.5 mL/kg BW, i.p., 6 weeks) (D) or those treated with a single Tm dose (2 mg/kg BW, i.p., 72 h) (E). Band intensities represent values relative to each respective control (n = 7 each for D; and n = 5 or 6 each for E) (right). (F) ER stress markers (Hspa5, Ddit3) and Ikbkg transcript correlations using a public domain GSE75277 dataset concerning the liver of diclofenac-treated mice (30 mg/kg BW, i.p., 14 days) or the vehicle (n = 5 each). For C-E, values were expressed as mean ± SEM (*P < 0.05, **P < 0.01). Statistical significance was tested via two-tailed Student's t-tests and Pearson's correlation analyses.
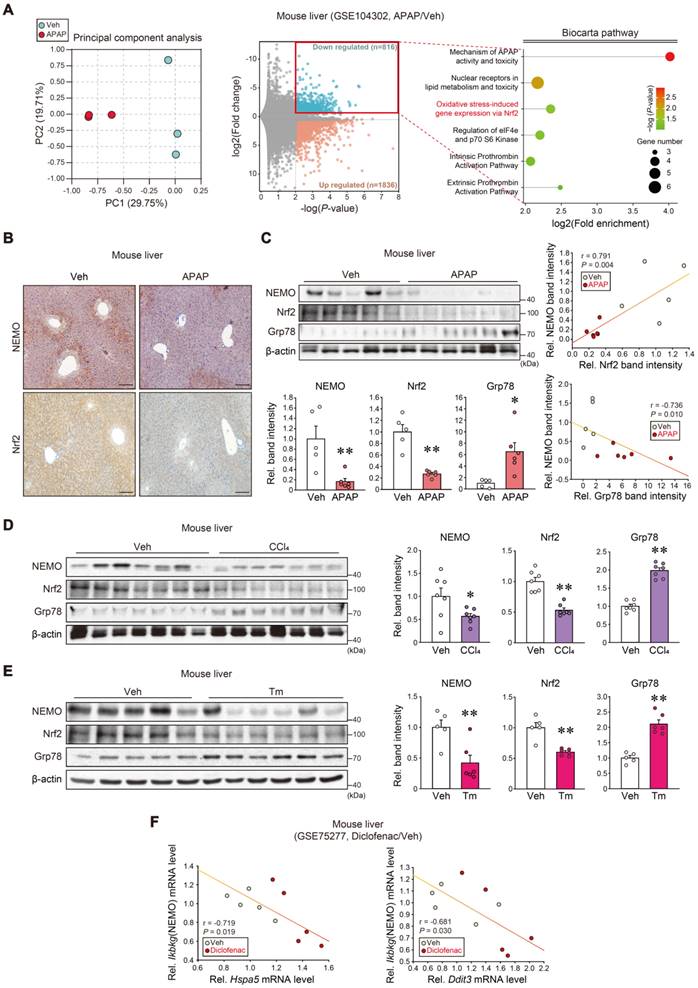
Considering the Nrf2 and hepatic injury association, we experimentally assessed the potential Nrf2 and NEMO correlation. APAP treatment in mice inhibited NEMO and Nrf2 in the liver with increased Grp78 levels (Figure 2B and C), as confirmed in primary hepatocytes (Figure S2C). Additionally, we examined other liver injury inducer effects (i.e., carbon tetrachloride [CCl4] and tunicamycin [Tm]), discovering that the toxicants treatment displayed similar effects (Figure 2D and E). In analyzing the diclofenac (i.e., an agent that causes liver injury) medication dataset, Ikbkg mRNA levels were inversely correlated with those of cellular stress markers (Hspa5 and Ddit3) (Figure 2F). These findings support Nrf2 and NEMO inhibition and their correlations with liver injury.
NEMO induction of GPX4 for the inhibition of ferroptosis
To understand the direct effect of chemical intoxication on NEMO's uncharacterized role in a different hepatocyte death pathway, we performed the KEGG analysis using our dataset (GSE104302), APAP intoxication promoted the ferroptosis pathway (second rank) among the different types of cell death (Figure 3A and Figure S3A). Moreover, NEMO ablation in hepatocytes upregulated ferroptosis-associated gene sets in other Biological processes, Reactome pathways, and Wikipathway analyses (Figure 3B). In subsequent experiments, NEMO's modulation of ferroptosis biomarkers in the liver and hepatocytes were examined. NEMO overexpression in hepatocytes, through hydrodynamic injection in mice [32], completely reversed the inhibitory effect of APAP on GPX4 along with decreases in 3-nitrotyrosine (3-NT) and 4-hydroxynonenal (4-HNE) levels (Figure 3C). Consistently, the reduced glutathione (GSH) content was recovered in the liver (Figure 3D). In AML12 cells, NEMO overexpression and siRNA knockdown affected GPX4 levels (Figure 3E), corroborating NEMO's direct antioxidant effect in hepatocytes. In addition, APAP treatment reduced Gpx4 transcript levels, which was not reversed by NEMO overexpression (Figure S3B), suggesting that NEMO is unlikely to transcriptionally activate GPX4. In the cycloheximide experiment, modulations of NEMO by overexpression and siRNA knockdown changed GPX4 levels, supportive of NEMO's effect on GPX4 stabilization (Figure S3C). Moreover, NEMO overexpression prevented APAP from increasing Fe2+ levels (Figure S3D). We further examined mitochondrial fusion and fission marker transcripts (i.e., Mfn1, Mfn2, and Opa1 for fusion; and Fis1, and Drp1 for fission); APAP treatment decreased all of the marker transcripts, but this effect was not changed by NEMO overexpression (Figure S3E).
Toxicant-induced ferroptosis inhibition through NEMO. (A) KEGG analysis using a cDNA microarray dataset obtained from WT mice livers treated with APAP or the vehicle. The ferroptosis pathway (red asterisk) exhibited high fold enrichment (second rank) (n = 3 each, DEGs of P-value < 0.01 and FC > 2, GSE104302). FDR is indicated in the bar graph. (B) Ferroptosis-related pathways based in Biological process, Reactome pathway, and Wikipathway obtained from NEMOf/f or NEMOΔhepa mice livers (n = 3 each, DEGs of P-value < 0.05 and FC > 2, GSE61100). (C) Immunoblottings for representative ferroptosis markers in WT mice livers treated with a single APAP dose (300 mg/kg BW, i.p., 6 h) 3 days post-hydrodynamic injection with NEMO or control plasmid DNA (25 μg each) via tail vein (left). Band intensities represent values relative to each respective control (n = 3 or 5 each) (right). (D) Reduced glutathione (GSH), oxidized glutathione disulfide (GSSG), and GSH/GSSG ratio measurements in WT mice livers treated with a single APAP dose (300 mg/kg BW, i.p., 6 h) 3 days post-hydrodynamic injection with NEMO or control plasmid DNA (25 μg each) via tail vein (n = 3 or 5 each). (E) GPX4 and NEMO immunoblottings for APAP-treated AML12 cells (10 mM, 12 h) after transfection with NEMO (Mock, 1 µg, 24 h) (upper) or siNEMO (siCon, 100 nM, 24 h) (lower) (n = 3; repeated three times with similar results). (F) Liver histopathology and immunohistochemistry for NEMO in the livers of mice treated with a single dose of BSO (1 g/kg BW, i.p., 6 h) (n = 3 each). Scale bar, 200 µm. (G) Immunoblottings for NEMO and GPX4 in AML12 cells treated with the indicated BSO (upper) and Erastin (lower) concentrations for 12 h (n = 3; repeated three times with similar results). (H) Liver histopathology and immunohistochemistry for NEMO in the livers of mice treated with APAP (300 mg/kg BW, i.p., 6 h) 1 h after vehicle or Fer-1 (1 mg/kg BW, i.p.) treatment. Scale bar, 200 µm. (I) Immunoblottings for NEMO and GPX4 in the same mice as in H (upper). Band intensities represent values relative to the respective control (n = 3 or 5 each) (lower). For C, D, and I values were expressed as mean ± SEM (*P < 0.05, **P < 0.01). Statistical significance was tested via one-way ANOVA coupled with Bonferroni's method or the LSD multiple comparison procedure when appropriate.
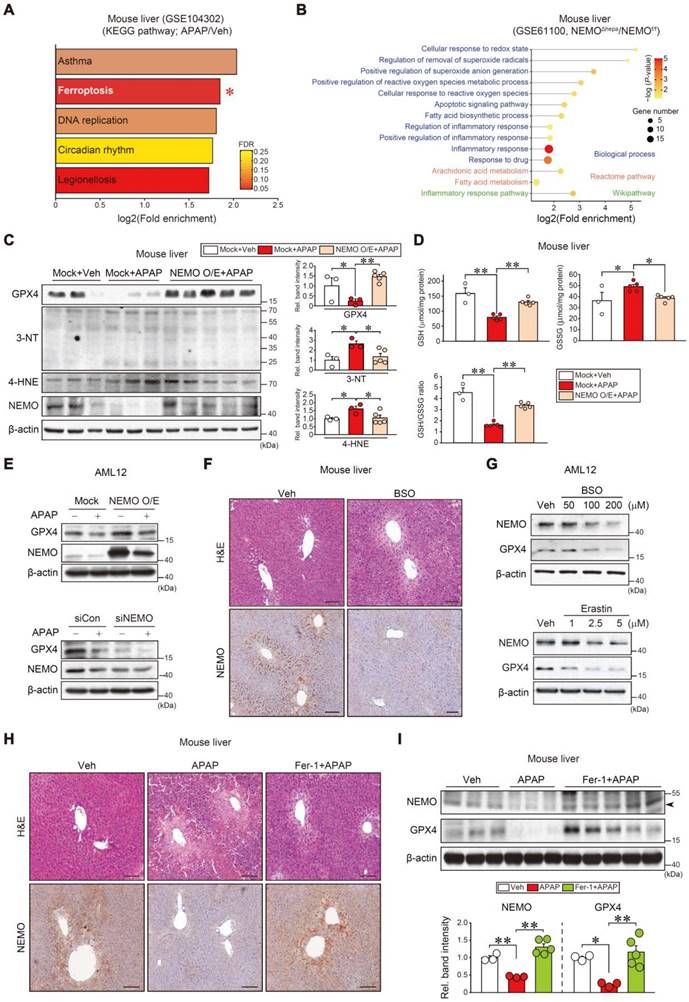
Subsequently, we confirmed that treatment with DL-buthionine-[S, R]-sulfoximine (BSO), an agent that induces ferroptosis and depletes cellular GSH through GPX4 inhibition [10], inhibited NEMO in the liver and AML12 cells. Erastin, another ferroptosis inducer, also inhibited NEMO and GPX4 (Figure 3F and G). Consistently, treatment with ferrostatin-1 (Fer-1), a specific antioxidative ferroptosis inhibitor [6, 33], ameliorated liver injury and enhanced NEMO and GPX4 levels against APAP (Figure 3H and I). Similar outcomes were obtained for Nrf2, NEMO, and GPX4 in mouse primary hepatocytes (Figure S3F), supporting the antioxidant spin-trapping effect of Fer-1. In this event, an increase of Nrf2 by Fer-1 may contribute to GPX4 expression against APAP. All of these results provide evidence that NEMO inhibits hepatic ferroptosis by upregulating GPX4 and is controlled by ferroptosis inducers and inhibitors in APAP-induced ALI.
Protective NEMO effects against oxidative- and ER stress-induced hepatic injury
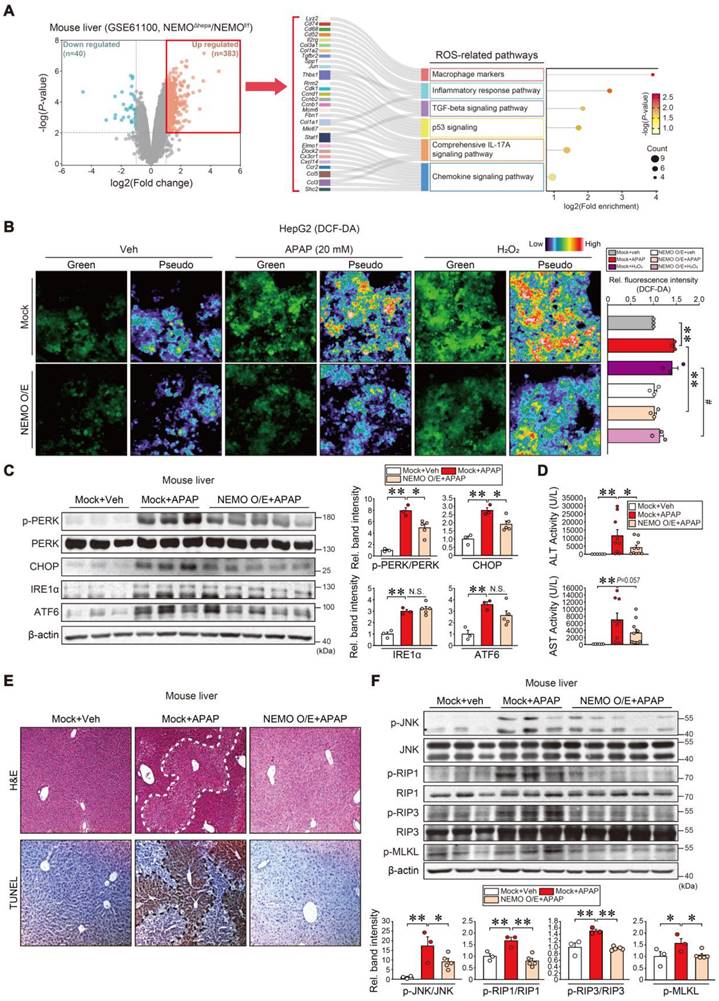
In subsequent studies, we examined NEMO effects on oxidative stress and ER stress in hepatocytes. In the public RNA-seq dataset analysis (GSE61100), NEMO gene knockout resulted in DEGs accounting for 1.7% of total transcriptomes; Among the 423 DEGs, 40 were downregulated, while 383 were upregulated (Figure 4A, left). NEMO abrogation upregulated 383 genes, of which 29 were associated with reactive oxygen species (ROS) pathways in the Sankey diagram visualization (i.e., macrophage markers and inflammatory response pathways) (Figure 4A, right). Conversely, downregulated genes were related to lipid metabolic processes in the dataset from hepatocyte-specific NEMO knockout mice (Figure S4A). Hallmark pathway analysis of our RNA-seq dataset (GSE173595) from APAP-treated mice conveyed upregulated gene sets associated with ROS and ER stress (Figure S4B and C) [34]. In our experiment, NEMO overexpression prevented APAP and H2O2 from increasing DCF fluorescent intensity in HepG2 cells (Figure 4B), confirming NEMO's ability to inhibit ROS. In addition, NEMO overexpression in hepatocytes through hydrodynamic gene deliveries attenuated p-PERK and CHOP intensities increased by APAP treatment (Figure 4C). IRE1α, ATF6, and other stress markers were largely unaffected. In addition, PERK overexpression enhanced APAP's inhibition on GPX4, whereas PERK siRNA exerted the opposite effect; However, PERK modulations did not change NEMO levels (Figure S5). These data support the notion that NEMO increases GPX4 by inhibiting PERK against ER stress. Furthermore, NEMO overexpression inhibited ALT and AST activities, ameliorating liver histopathology and TUNEL staining intensity (Figure 4D and E). Consistently, cell death markers including p-JNK, p-RIP1, p-RIP3, and p-MLKL were all inhibited (Figure 4F). Our results support the concept that NEMO inhibits oxidative stress and ER stress in association with liver protection.
Inhibition of toxicant-induced liver injury through NEMO. (A) Volcano plot (left) (mint, downregulation; red, upregulation) and ROS-related pathways based on the Sankey diagram (plot) of DEGs using hepatic transcriptome data from NEMOf/f or NEMOΔhepa mice (right). The Sankey diagram represents genes within each pathway; dot plots with sizes indicate gene numbers and dot colors display P-values (n = 3 each, DEGs of P-value < 0.01 and FC ≥ 2, FC ≤ -2, GSE61100). (B) Representative fluorescence images of DCF-DA (2′,7′-dichlorofluorescein diacetate) staining for ROS detection in HepG2 cells treated with APAP (20 mM, 12 h) or H2O2 (100 μM, 10 min) as a positive control after NEMO transfection (or Mock) (1 µg, 24h) (left). Green and pseudocolor images were captured, and the relative color scale indicates DCF fluorescence levels. Scale bar, 100 μm. DCF-DA-enhanced fluorescence-positive cells were analyzed with Image J software (right). (C) Immunoblottings for representative ER stress markers in the same samples as in Fig. 3C (left). Band intensities represent values relative to each respective control (n = 3 or 5 each) (right). (D) Serum alanine transaminase (ALT) and aspartate transaminase (AST) activities in WT mice livers treated with a single APAP dose (300 mg/kg BW, i.p., 6 h) 3 days post-hydrodynamic injection with NEMO or control plasmid DNA (25 μg each) via tail vein (n = 6-10 each). (E) Liver histopathology (H&E) and terminal deoxynucleotidyl transferase dUTP nick-end labeling (TUNEL) assays. H&E and TUNEL stainings were done on the same mice livers as in Fig. 3C. Scale bar, 200 µm. (F) Immunoblottings for representative necroptosis markers in the same samples as in Fig. 3C (upper). Band intensities represent values relative to each respective control (n = 3 or 5 each) (lower). For B-D and F, values were expressed as mean ± SEM (*P < 0.05, **P < 0.01). Statistical significance was tested via one-way ANOVA coupled with Bonferroni's method or the LSD multiple comparison procedure when appropriate.
Nrf2 as a transcriptional IKBKG gene regulator
We next extracted 84 genes from Nrf2 KO (GSE8969) and Keap1 KO mice (GSE11287) (Figure 5A, left), and analyzed the microarray datasets for functional GO enrichment to explore the Nrf2 and NEMO association (Figure 5A, middle). DAVID database analysis identified the 'lipid metabolic process' and 'positive regulation of gene expression' biological processes, which shared Ikbkg as a core gene in the gene-concept network (Figure 5A, right). In addition, the String program aided in determining Ikbkg and Map4k4 as affected TNFα pathway targets (Figure S6A). As predicted, Ikbkg mRNA levels were diminished in Nrf2 KO mice (GSE8969) but were increased in Keap1 KO mice (GSE11287) (Figure 5B). In APAP-induced acute liver failure patients (GSE74000), IKBKG levels decreased, and Nrf2 target genes were also repressed (i.e., HMOX1, PRDX1, TXNRD1, GPX4, GPX1, GCLC, GCLM, and NQO1) (Figure 5C).
Nrf2-mediated IKBKG (NEMO) transactivation. (A) Gene Venn diagrams of downregulated and upregulated genes in liver samples from Nrf2 and Keap1 knockout mouse models (n = 3 each, GSE8969 and GSE11287; DEGs of P-value < 0.05 and absolute FC > 1.5, respectively) (left). GO term enrichment of overlapped genes (84 genes, 4.5%) between two groups for biological processes, such as the lipid metabolic process (first rank) and positive gene expression regulation (second rank) (middle). The gene-concept network (cnetplot) of functional GO enrichment results (right) from the leading genes highlighted in the circles' red areas in the first and second biological process ranks. The cnetplot depicts gene and biological concept (GO terms) linkages as a network. Circle size indicates genes represented in a given biological process. (B) Ikbkg transcript levels obtained using a public dataset concerning livers of WT and Nrf2 KO (GSE8969) or Keap1 KO mice (GSE11287). (C) Heatmap of significantly down-regulated IKBKG transcript and Nrf2 target genes in APAP-intoxicated patients (n = 2 or 3, GSE74000). (D, E) Real-time RT-PCR assays for IKBKG, CHUK, and IKBKB in HepG2 cells treated with 10 μM sulforaphane (SFN) for the indicated times (n = 3) (D); or IKBKG in HepG2 cells treated with tBHQ and Olt for the indicated times (n = 3) (E). (F) ChIP-seq analysis for IKBKG antioxidant response element regions in sulforaphane-treated lymphoblastoid cells using the cDNA array database (GSE37589) (upper). IKBKG promoter-reporter assays. Luciferase activity was measured in HepG2 cells after Mock or hNrf2 transfection (n = 3 each) (lower left). WT or MT luciferase constructs were used in HepG2 cells after Mock or hNrf2 transfection (n = 3) (lower right). (G) NEMO and Nrf2 immunoblottings for SFN- and Olt-treated HepG2 cells for the indicated times (upper). Band intensities represent values relative to the respective control (n = 3 each) (lower). For B and D-G values were expressed as mean ± SEM (*P < 0.05, **P < 0.01). Statistical significance was tested via two-tailed Student's t-tests or one-way ANOVA coupled with Tukey HSD or the LSD multiple comparison procedure when appropriate.
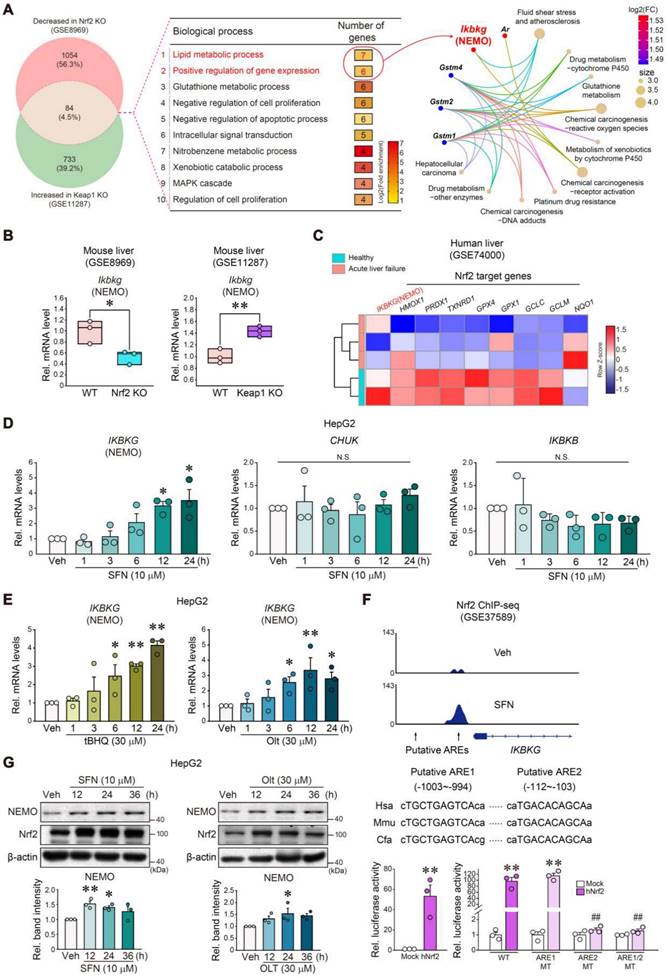
Then, we explored the potential transcriptional activation of Nrf2 for the IKBKG gene regulation. Sulforaphane (SFN, an Nrf2 inducer)-treated HepG2 cells exhibited increased IKBKG mRNA levels but not those of CHUK or IKBKB (Figure 5D). Other Nrf2 activators (i.e., tert-butylhydroquinone [tBHQ] and oltipraz [Olt]) exerted the same effects (Figure 5E). We also confirmed the nuclear accumulation of Nrf2 by the Nrf2 activators (SFN, Olt, and tBHQ) in HepG2 cells (Figure S6B). Likewise, these compounds increased Ikbkg mRNA levels in AML12 cells (Figure S6C).
Analyzing a publicly accessible Nrf2 ChIP-sequencing (ChIP-seq) dataset, two putative antioxidant response elements (AREs) were determined as well-conserved regions across the species. Additionally, SFN treatment induced a strong peak in ARE2 located in the proximal promoter (-112 bp ~ -103 bp) but not in ARE1 (-1003 bp ~ -994 bp) (Figure 5F, upper). Increased H3K27ac and Pol II ChIP intensities validated Nrf2 and ARE2 interactions (Figure S6D). Consistently, enforced Nrf2 expression sharply preponderated IKBKG promoter-driven luciferase activity (Figure 5F, lower left). We then crafted specific mutants with mutations at one or both ARE (i.e., ARE1 MT, ARE2 MT, or ARE1/2 MT) for reporter gene assays (Figure 5F, lower right); As expected, hNrf2-inducible luciferase expression was entirely attenuated in ARE2 MT (located between -112 and -103 bp) or ARE1/2 MT reporter assays but not ARE1 MT, verifying ARE2's functional role.
Furthermore, SFN, Olt, and tBHQ treatments begat NEMO protein induction in HepG2 cells (Figure 5G and S6E). Likewise, an Nrf2 deficiency inhibited hepatic NEMO expression (Figure S6F), corroborated by HepG2 cell immunoblottings (Figure S6G). Together, these results firmly evidence that Nrf2 transactivates the IKBKG gene.
The role of the Gα12-ROCK1 axis in NEMO regulation
Following the observation of significantly inhibited NEMO by APAP-induced ER stress, we further probed into the mechanistic underpinnings of NEMO downregulation induced by APAP, which aimed to elucidate factors beyond NEMO's transcriptional activity. Having previously identified Gα12 overexpression by ER stress influences ROCK1-dependent ferroptosis [28], we wondered whether Gα12 modulations affect NEMO expression. Notably, Gα12 KO enhanced gene sets associated with NF-κB-related pathways in the GSEA analysis regarding our RNA-seq data from WT and Gα12 KO mice (GSE173595) (Figure S7A). Also, Gα12 deficiency enhanced basal Ikbkg mRNA levels in the liver, preventing APAP from inhibiting Ikbkg (Figure 6A and Figure S7B). In addition, immunoblotting assays using primary hepatocytes demonstrated that Gα12 abrogation considerably reversed APAP's inhibition on NEMO (Figure 6B). Consistently, Gα12 overexpression using a liver-specific lentiviral albumin-Gα12 (Lv-Alb-Gα12) fortified APAP's inhibition on Ikbkg mRNA, reversing Gα12 KO plus APAP's promotion of the mRNA (Figure 6C). Immunohistochemistry and immunoblottings further confirmed that Gα12 overexpression inhibited NEMO in the liver of WT and Gα12 KO mice challenged by APAP (Figures 6D and E).
Gα12 modulation effects on NEMO expression. (A) qRT-PCR assays for Ikbkg in WT or Gα12 KO mice treated with APAP (300 mg/kg BW, i.p., 6 h) (n = 4-7 each). (B) Immunoblottings for NEMO and Gα12 in WT or Gα12 KO mouse primary hepatocytes treated with APAP (10 mM, 12 h) (n = 3; repeated three times with similar results). (C) qRT-PCR assays for Ikbkg in WT (n = 5 or 6 each) (left) or Gα12 KO mice (n = 4-6 each) (right) treated with APAP (300 mg/kg BW, 6 h) one-week post-injection with Lv-con or Lv-Gα12 via tail vein. (D, E) Immunohistochemistry (upper) and immunoblottings (lower) for NEMO and Gα12 for the same mice as in C. Band intensities represent values relative to the respective control (n = 3 or 4 each) (lower right). Scale bar, 200 µm. (F) Immunohistochemistry for NEMO in ripasudil-treated WT mice (50 mg/kg BW, 5 h) 1 h after APAP treatment (300 mg/kg BW, 6 h). Scale bar, 200 µm. (G) NEMO, Nrf2, and p-MLC immunoblottings (upper) for the same mice as in F. Band intensities represent values relative to the respective control (n = 3 or 4 each) (lower). For A, C-E, and G, values were expressed as mean ± SEM (*P < 0.05, **P < 0.01). Statistical significance was tested via one-way ANOVA coupled with Bonferroni's method or the LSD multiple comparison procedure when appropriate.
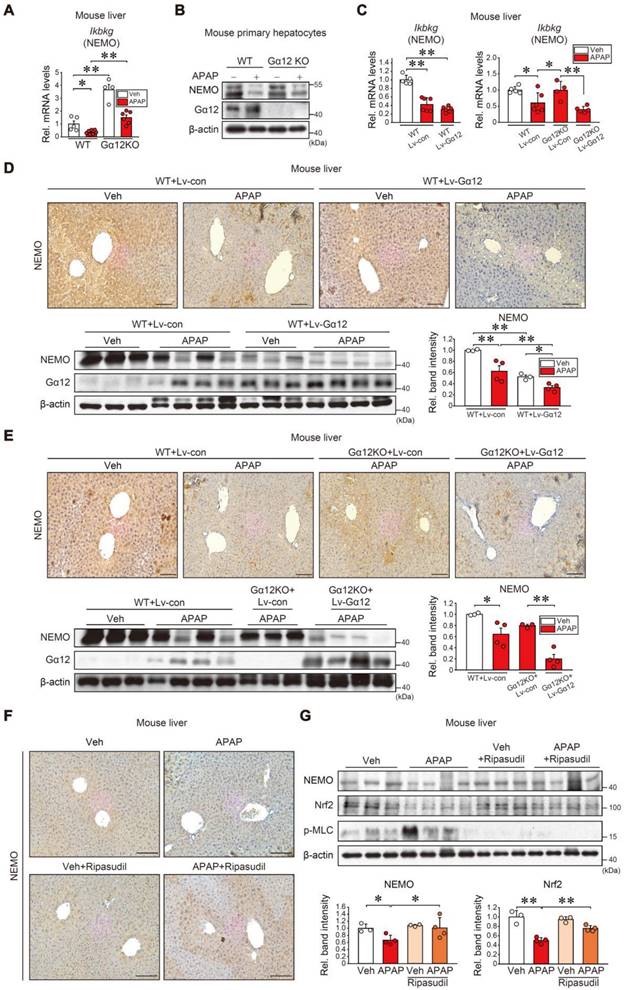
Considering that ROCK1 is a downstream Gα12 signaling effector, we further examined whether ROCK1 modulations impacted NEMO levels. ROCK1 knockdown and chemical inhibition (ripasudil) prevented APAP from inhibiting NEMO in primary hepatocytes (Figure S7C). Immunohistochemistry and immunoblottings corroborated ripasudil's influence on NEMO in the liver (Figure 6F and G) while also ameliorating liver injury (Figure S7D). Nrf2 and p-MLC levels were accordingly changed by a downstream ROCK1/2 effector; Ripasudil's ability to heighten NEMO levels was seemingly greater than Nrf2, indicating that the Gα12 pathway may additionally regulate NEMO (Figure 6G). Together, these results support that the Gα12-ROCK1 axis negatively controls NEMO under liver injury conditions.
miR-125a inhibition of NEMO downstream from Gα12
Next, we aimed to ascertain how microRNA (miRNAs) were involved in post-transcriptional NEMO regulation downstream from the Gα12 pathway. Four miRNAs with conserved sites and the potential to bind to IKBKG mRNA's 3'-UTR were selected using the 'Targetscan' database (Figure 7A). Out of the four, miR-125a was the most significantly elevated in ALI patient livers (Figure 7B). Then, miR-125a's effect on the IKBKG 3'-UTR was examined. IKBKG 3'-UTR and the miR-125a seed sequence exhibit a nearly complete pairing (Figure 7C, upper). When assessing miR-125a modulation effects on NEMO protein, miR-125a antisense oligonucleotides (ASOs) increased NEMO levels in HepG2 cells, whereas its mimic exerted the opposite (Figure 7C, lower). This effect was confirmed in primary hepatocytes (Figure 7D). Furthermore, GO analysis was accomplished to distinguish downstream pathways and the functional role of miR-125a-targeted genes. The target genes correlated with the MAPK and NF-κB pathways (Figure S8A, left), which were then implemented in a GO analysis (red color), establishing that they were correlated with the 'Apoptotic process' (the most significantly affected) (Figure S8A, right and S8B). Further, we verified miR-125a's functional role in ferroptosis elicited by APAP or Erastin treatment. As expected, APAP treatment induced morphological changes in HepG2 cells and primary hepatocytes, which were ameliorated by miR-125a ASO pretreatment (Figure 7E, left, S8C, and S8D). APAP's ferroptosis biomarkers (i.e., 4-HNE and 3-NT) were also impeded by miR-125a ASO transfection (Figure 7E, right), confirming miR-125a's effect on ferroptosis processes.
miR-125a as a downstream NEMO inhibitor regulated by Gα12. (A) Putative IKBKG-targeting miRNA candidates were obtained according to conserved branch length and context percentile scores using Targetscan. (B) qRT-PCR assays for miR-125a, miR-125b, miR-4319, and miR-365a in healthy individuals (n = 10) or ALI patients (n = 22). (C) Prediction of miR-125a binding to the 3'UTR of IKBKG mRNA (upper). Immunoblottings for NEMO in HepG2 cells transfected with miR-125a ASO (control ASO) (100 nM, 48 h) (lower left) or miR-125a mimic (control mimic) (100 nM, 48h) (lower right). Densitometric band intensities represent values relative to the respective control (n = 3 each). (D) Immunoblottings for NEMO in primary hepatocytes transfected with miR-125a ASO (control ASO) (100 nM, 48 h) (left) or miR-125a mimic (control mimic) (100 nM, 48 h) (right) (n = 3; repeated three times with similar results). (E) Representative cell morphology (left) and immunoblottings for 4-HNE and 3-NT (right) in HepG2 cells treated with APAP (20 mM, 12 or 24 h) after miR-125a ASO (or control ASO) transfection (100 nM, 48 h) (n = 3; repeated three times with similar results). Scale bar, 100 µm. (F) qRT-PCR assays for miR-125a, miR-125b, and miR-4319 in the same mice as in Fig. 6E (n = 4-6 each). (G) qRT-PCR assay for miR-125a in ripasudil-treated WT mice livers (50 mg/kg BW, 5 h) 1 h after APAP treatment (300 mg/kg BW, 6 h) (n = 3-7). (H) Immunoblottings for NEMO and Nrf2 in WT and Gα12 KO mouse primary hepatocyte treated with APAP (10 mM, 12 h) after transfection with miR-125a mimic (upper) or ASO (lower). Densitometric band intensities represent values relative to the respective control (n = 3 each) (right). For B, C, and F-H values were expressed as mean ± SEM (*P < 0.05, **P < 0.01). Statistical significance was tested via two-tailed Student's t-tests, Mann-Whitney U test, or one-way ANOVA with the LSD multiple comparison procedure when appropriate.
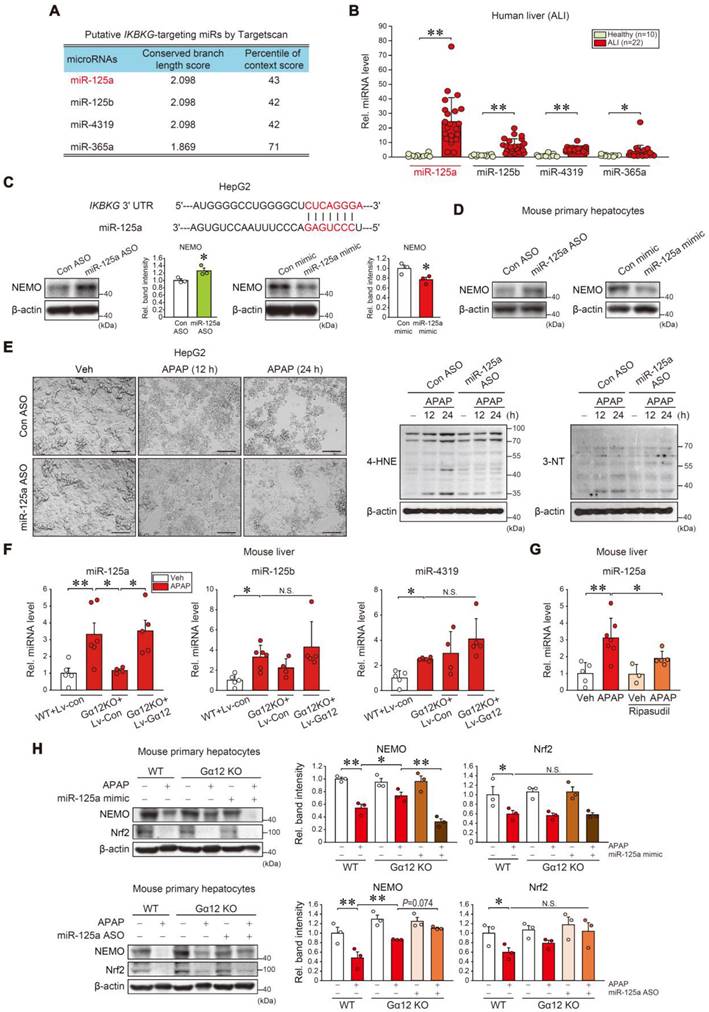
Then, we observed APAP treatment increased miR-125a, miR-125b, and miR-4319 levels in mice livers. Gα12 KO completely inhibited APAP's effect on miR-125a but not the others (Figure 7F); This finding is also evidenced by the absence of Gα12 regulatory effect on miR-125b in the Gα12QL microarray dataset analysis (Figure S9), indicating the specific Gα12 signaling effect on miR-125a. In addition, this inhibitory effect was entirely reversed through enforced Gα12 expression. Using ripasudil to inhibit ROCK1 resulted in similar outcomes (Figure 7G). In the experiment using WT and Gα12 KO primary hepatocytes, a deficiency of Gα12 prevented APAP from inhibiting NEMO, which was completely abrogated by miR-125a mimic transfection, but increased by miR-125a ASO (Figure 7H). Intriguingly, Nrf2 levels were not changed. Overall, our results fortify the concept of the dual NEMO regulatory pathways; (1) Nrf2 transcriptionally induces NEMO, and (2) ER stress-mediated Gα12 overexpression inhibits NEMO de novo synthesis by enhancing miR-125a level, which can be countered by ROCK1 inhibition.
Dysregulation of NEMO, Nrf2, GPX4 and miR-125a in patients with ALI
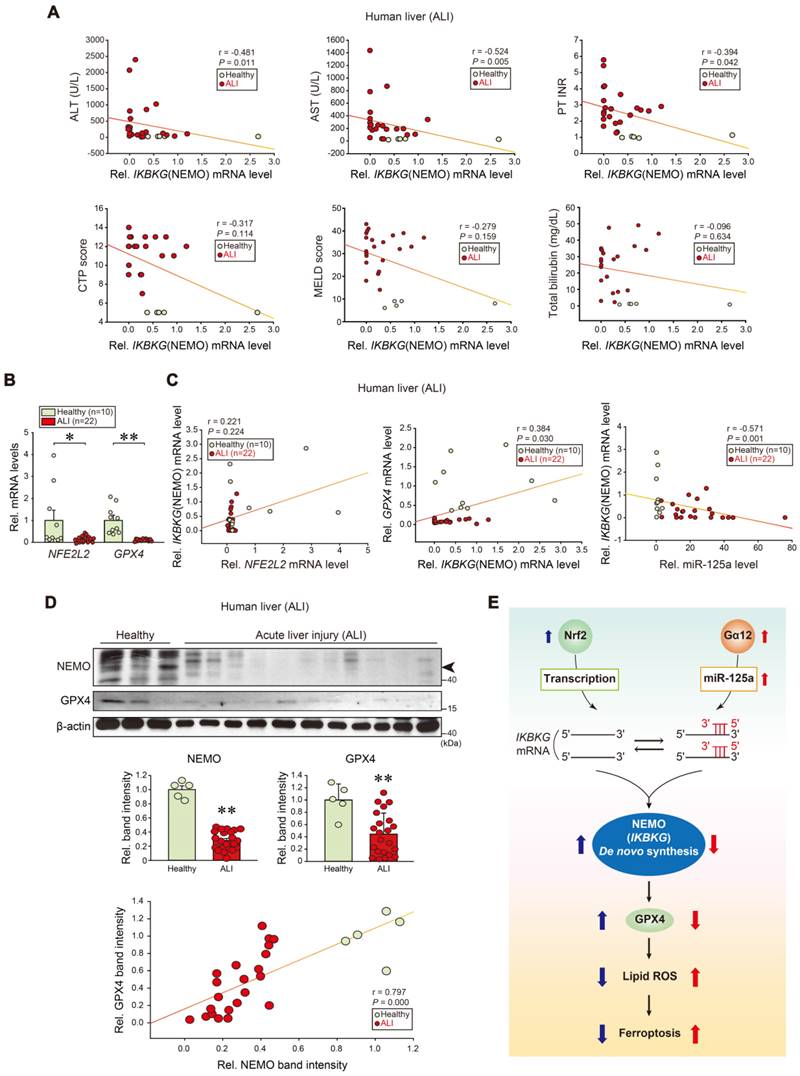
To further assess the association of IKBKG transcript levels and liver injury in clinical situations, we analyzed samples of patients with ALI on drugs. Significant negative correlations existed between IKBKG and each ALI marker: alanine aminotransferase (ALT), aspartate aminotransferase (AST), prothrombin time international normalized ratio (PT INR), Child-Turcotte-Pugh score (CTP score), MELD score, and total bilirubin contents (Figure 8A). As expected, NFE2L2 and GPX4 transcript levels were decreased in ALI patients (Figure 8B). In patients with ALI or fulminant liver failure, positive correlations existed between IKBKG and NFE2L2; or IKBKG and GPX4 transcripts. Hepatic miR-125a negatively correlated with IKBKG (Figure 8C). We also confirmed the inhibition of NEMO and GPX4 protein levels in patients with ALI, and correlations between NEMO and GPX4 (Figure 8D). In summary, NEMO and GPX4 levels were decreased in the patients through the dual regulatory pathways, exacerbating hepatocyte ferroptosis and ALI (Figure 8E).
Correlations between human NEMO and liver disease scores, and identified target levels in patients with ALI. (A) Correlations between IKBKG transcript and ALT, AST, PT INR, CTP, MELD scores, or total bilirubin contents in livers of healthy individuals or ALI patients (healthy: n = 5, ALI: n = 22). ALT, alanine aminotransferase; AST, aspartate aminotransferase; PT INR score, prothrombin time international normalized ratio. CTP, child-turcotte-pugh; MELD score, model for end-stage liver disease score. (B) NFE2L2 and GPX4 transcript levels in livers of healthy individuals (n = 10) or ALI patients (n = 22). (C) Correlations between NFE2L2 and IKBKG (left), or IKBKG and GPX4 (middle), or miR-125a and IKBKG transcripts (right) in livers of healthy individuals or ALI patients (healthy, n = 10; ALI, n = 22). (D) NEMO and GPX4 immunoblottings for healthy individuals or ALI patients (upper). Band intensities represent values relative to each respective control (healthy: n = 5, ALI: n = 22) (middle). Representative blots were shown. A correlation between NEMO and GPX4 protein levels (n = 27) (lower). (E) A schematic depicting how Nrf2-dependent NEMO induction inhibits liver injury through GPX4 against the Gα12-miR-125a axis. For B and D, values were expressed as mean ± SEM (*P < 0.05, **P < 0.01). Statistical significance was tested via two-tailed Mann-Whitney U test or Spearman's correlation analyses.
Discussion
The worldwide surge of severe liver injury highlights the necessity for identifying new drug targets and therapeutic compounds to treat this disease [11]. Recent reports have indicated that lipid peroxide and oxidative stress accumulations, as observed in ferroptosis, contribute to hepatocyte injury and trigger inflammatory responses, exacerbating liver disease progression [8]. Developing medications to regulate ferroptosis has garnered substantial attention [35]; however, studies on ferroptosis and its underlying mechanisms are currently insufficient. Hence, future investigations must identify novel regulators and elucidate the underlying mechanisms behind ferroptosis modulation relative to liver injury.
The NF-κB pathway is vital in regulating immune and inflammatory responses in the liver, thereby exerting considerable influence on liver homeostasis and disease processes. The canonical NF-κB activation pathway involves the IKK complex [36], whereas the non-canonical pathway incorporates NF-κB inducing kinase (NIK) upon receptor ligation [37]. However, emerging evidence has highlighted that NEMO, a prominent NF-κB activation protein, can protect hepatocytes independent of the canonical NF-κB pathway [38].
Our study uncovered a critical finding expounding upon NEMO's inhibitory role in ferroptosis during APAP intoxication, supporting NF-κB and its influence in further cell fate determination. Our findings showed that 3-NT and 4-HNE intensity changes and their reversal through NEMO overexpression support the ability of NEMO to protect hepatic cells from ferroptosis. Specifically, the outcomes of this study introduce a previously uncharacterized role of NEMO transcriptionally activated by Nrf2 and NEMO induction which inhibits hepatic ferroptosis via GPX4. Our findings indicate that NEMO levels are additionally affected by the Gα12 axis responsible for an increase of miR-125a as a previously unknown NEMO inhibitor. These findings establish significant links not only between Nrf2 and NEMO but also between NEMO and miR-125a controlled by the Gα12-ROCK1 axis.
We found that NEMO depletion causes upregulation of various gene clusters, including those involved in redox state and arachidonic acid metabolism, showing that NEMO may have a more substantial role in regulating cellular homeostasis beyond its well-known function in NF-κB [39]. The reactive intermediate NAPQI, produced from APAP biotransformation, depletes cellular GSH content and binds to cellular macromolecules in hepatocytes [8], effectuating ferroptosis, as confirmed in the present study. Our study also highlights the importance of GPX4 and its cofactor GSH in removing lipid peroxides and oxidation products, such as phospholipids and PUFAs [9], enabling hepatocytes to survive GSH depletion caused by BSO and Erastin treatment [40]. Furthermore, this study strongly evidenced that NEMO participates in cell fate determination mediated by GPX4, which alters the glutathione/oxidized glutathione disulfide (GSH/GSSH) ratio. This notion is corroborated by DCF-DA intensity alterations upon APAP exposure following NEMO overexpression.
The PERK/CHOP-dependent JNK pathway has been proven to induce cell death in response to ER stress [41, 42]. Our findings demonstrate that NEMO overexpression inhibits p-PERK and CHOP upregulation mediated by ER stress, suggesting NEMO's potential in attenuating ER stress-induced hepatocyte injury. However, ALT activity and TUNEL staining intensity changes after NEMO overexpression were more considerable than those in p-PERK and CHOP, indicating that other ER stress mediators may also be involved. IRE1α, a known Gα12 overexpression [28] and eIF2α regulator through PERK [43], potentially contributes to p-PERK/CHOP regulation under APAP intoxication conditions, even when NEMO is present. Therefore, NEMO likely regulates ferroptosis by inhibiting the PERK/CHOP-dependent JNK pathway and interacting with other mediators to regulate overall cellular responses to ER stress.
APAP-induced liver injury activates the apoptosis pathway and various other regulated necrosis types, namely necroptosis, and pyroptosis [44-46]. Among diverse and complex cell death pathways, JNK is an integral stress kinase in APAP-induced liver injury and hepatocyte death as it activates RIP1 and engenders necrosome complex formation [47, 48]. Furthermore, necroptosis is a programmed necrosis triggered by RIP1, RIP3, and MLKL. Previous studies have demonstrated that NEMO overexpression attenuates p-JNK and RIPK1 activation, preventing the RIPK1/FADD/caspase-8 complex from forming to inhibit necrotic cell death [18], consistent with our findings that NEMO overexpression prevents APAP from increasing p-JNK, p-RIP1, p-RIP3, and p-MLKL. In the present study, other gene clusters affected by NEMO depletion include inflammatory response pathways, being consistent with our previous observation confirming JNK's role in NF-κB-mediated inflammatory responses [33].
Our study identified Nrf2 as an IKBKG gene transcription factor, revealing a previously unknown NF-κB pathway regulation. Additionally, the observation that Nrf2 activation upregulates Bcl-2 and other Nrf2-target genes confirmed that Nrf2 could protect cells from chemical stresses while maintaining cellular homeostasis [24]. Nrf2 is not only imperative for cell survival as a nuclear receptor but also as a direct regulator of target genes, holding significant scientific importance concerning liver injuries. Moreover, Nrf2 can modulate key cytoprotective and antioxidant enzyme expressions pivotal in mitigating liver damage and promoting tissue repair [49]. Understanding the direct Nrf2 regulation of target genes provides insights into molecular mechanisms underlying liver injury and offers potential targets for developing interventions to combat liver pathologies.
Notably, the previous study demonstrated that Gα12 overexpression was associated with ER stress in acute liver injury through ROCK1, mediated by ALOX12 and miR-15a dysregulation [28]. This study also aimed to elucidate the newly acknowledged targets downstream from the Gα12 axis. Our results isolate prominent mechanisms underlying the Nrf2-dependent NEMO-GPX4 pathway's protective effect against ER stress, aligning with the previous report on Nrf2 activation through Gα12 deficiency [50]. Interestingly, ripasudil treatment prompted a substantial NEMO elevation against APAP intoxication but only weakly increased Nrf2 levels. This observation indicates that the Gα12-ROCK1 axis plays a distinct NEMO regulatory role, identified by ROCK1-mediated translational control separate from Nrf2-dependent NEMO induction.
Additional efforts enabled us to identify miR-125a as a functional NEMO inhibitor downstream from Gα12. Moreover, our data demonstrating miR-125a inhibition of IKBKG mRNA translation into proteins correlates with ROCK1-mediated inhibitory protein regulation. The different NEMO and Nrf2 levels after ripasudil treatment corroborate miR-125a involvement in NEMO expression downstream from Gα12. Also, apoptosis with JNK activation initiates from miR-125a targeted gene clusters associated with the NF-κB pathway, with IKBKG as the core gene. When analyzing miR-125a target genes using GO, we extracted the Biocarta pathway of the 'MAP kinase signaling pathway' and 'NF-κB signaling pathway' (red color) and determined IKBKG as a core gene clustering with the 'JNK phosphorylation and activation mediated by activated human TAK1' pathway (Figure S8B). Thus, the miR-125a-associated gene network may link to the JNK pathway for NEMO regulation.
Gaining a comprehensive understanding of non-canonical NEMO regulation pathways would be crucial to insights into therapeutic interventions. This study's RNA-seq analyses, human sample analyses, in vivo, and vitro-based outcomes established that ALI downregulated NF-κB and its related pathways. In addition, our findings confirmed that NEMO expression was diminished in ALI patients and was negatively correlated with representative liver injury markers. Consistently, we observed that miR-125a levels were significantly increased in the patient samples, matching miR-125a upregulation in the livers of mice with fibrosis [51]. Moreover, the close associations between NFE2L2 and IKBKG, or IKBKG and GPX4, or miR-125a and IKBKG in our ALI sample analyses further support these pathways' prominence in human liver health and diseases.
Collectively, our research reveals that NEMO mitigates hepatocytes ferroptosis by inducing GPX4. This effect is achieved through the inhibition of both chemical-induced ROS production and ER stress. Furthermore, the outcomes have illuminated the role of Nrf2 as a transcriptional regulator of NEMO. We have also uncovered miR-125a as an unprecedented suppressor of NEMO de novo synthesis, acting downstream from the Gα12-ROCK1 axis. Together, our data evidence the dual antagonistic NEMO regulation by Nrf2-dependent transcription and ROCK1-mediated microRNA regulation of protein synthesis (Figure 8E). Thus, the identified molecules will likely work together for ER stress-mediated ferroptosis. These newly identified pathways and targets present implications for developing therapeutic strategies aiming at ALI.
Abbreviations
3-NT: 3-nitrotyrosine; 4-HNE: 4-hydroxynonenal; ALF: acute liver failure; ALI: acute liver injury; ALT: alanine aminotransferase; APAP: acetaminophen; AREs: antioxidant response elements; ASK1: apoptosis signal-regulating kinase 1; ASO: antisense oligonucleotide; AST: aspartate aminotransferase; ATF6: activating transcription factor 6; BSO: buthionine sulfoximine; CCl4: carbon tetrachloride; ChIP-seq: chromatin immunoprecipitation; CHOP: C/EBP-homologous protein; CTP score: Child-Turcotte-Pugh score; CYP2E1: cytochrome P450 2E1; DCF-DA: 2′,7′-dichlorofluorescein diacetate; DEGs: differentially expressed genes; ER: endoplasmic reticulum; FADD: fas-associated death domain protein; FDR: false discovery rate; Fer-1: ferrostatin-1; Gα12: G protein subunit alpha 12; GEO: Gene Expression Omnibus; GO: Gene Ontology; GPX4: glutathione peroxidase 4; GRP78: glucose regulatory protein 78; GSH: glutathione; GSEA: gene set enrichment analysis; GSSG: oxidized glutathione disulfide; HBV-ALF: HBV-associated acute liver failure; IKK: IκB kinase; IRE1α: inositol-requiring enzyme 1α; JNK: c-Jun NH2-terminal kinase; KEGG: Kyoto Encyclopedia of Genes and Genomes; Lv-Alb- Gα12: lentiviral albumin-Gα12; MAPK: mitogen-activated protein kinase; MELD score: model for end-stage liver disease score; MHN: massive hepatic necrosis; miR-125a: microRNA-125a; miRNA: microRNA; MKK4: mitogen-activated protein kinase kinase 4; MLKL: mixed lineage kinase domain like pseudokinase; NAPQI: N-acetyl-p-benzoquinone imine; NES: normalized enrichment score; NEMO: NF-κB essential modulator; NF-κB: nuclear factor kappa-light-chain-enhancer of activated B cells; NIK: NF-κB inducing kinase; Nrf2: Nuclear factor erythroid-2-related factor2; Olt: oltipraz; PAMPs: pathogen-associated molecular patterns; PCA: principal component analysis; PERK: protein kinase R-like endoplasmic reticulum kinase; p-MLC: phosphor-myosin light chain; PT INR: prothrombin time international normalized ratio; PUFAs: polyunsaturated fatty acids; RIPK1: receptor-interacting protein kinase 1; RIPK3: receptor-interacting serine/threonine-protein kinase 3; RNA-seq: RNA sequencing; ROCK1: rho associated coiled-coil containing protein kinase 1; ROS: reactive oxygen species; SFN: sulforaphane; SHN: submassive hepatic necrosis; siRNA: small interfering RNA; TAK1: transforming growth factor-β-activated kinase 1; tBHQ: tert-butylhydroquinone; Tm: tunicamycin; TNFα: tumor necrosis factor-α; TUNEL: terminal deoxynucleotidyl transferase dUTP nick end labeling; UPR: unfolded protein response; UTR: untranslated region; WT: wild type.
Supplementary Material
Supplementary materials and methods, figures and table.
Acknowledgements
This research was supported by the National Research Foundation (NRF) grant funded by the Korean government (NRF-2021R1A2B5B03086265 to SGK, NRF-2018R1A5A2023127 to CHL, and NRF-2022R1A6A3A01086695 to JHT).
Author contributions
JHT: designed and performed experiments, analyzed and interpreted data, conducted the statistical analysis, obtained funding, and drafted the manuscript. MSJ, YSK, and HWP: performed the experiments and analyzed the data. CHL: analyzed the biological process dataset and critically reviewed the manuscript. GCP: collected and provided human samples. SH: collected, analyzed, and provided electronic medical records and human samples, and critically reviewed the manuscript. SGK: designed research, supervised experiments, drafted and critically reviewed the manuscript for important intellectual content, obtained funding, administrative, technical, or material support, and study supervision.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Watkins PB, Seeff LB. Drug-induced liver injury: Summary of a single topic clinical research conference. Hepatology. 2006;43:618-31
2. Bernal W, Auzinger G, Dhawan A, Wendon J. Acute liver failure. Lancet. 2010;376:190-201
3. David S, Hamilton JP. Drug-induced Liver Injury. US Gastroenterol Hepatol Rev. 2010;6:73-80
4. Bernal W, Wendon J. Acute liver failure. N Engl J Med. 2013;369:2525-34
5. Larson AM, Polson J, Fontana RJ, Davern TJ, Lalani E, Hynan LS. et al. Acetaminophen-induced acute liver failure: results of a United States multicenter, prospective study. Hepatology. 2005;42:1364-72
6. Stockwell BR, Friedmann Angeli JP, Bayir H, Bush AI, Conrad M, Dixon SJ. et al. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell. 2017;171:273-85
7. Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE. et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149:1060-72
8. Yamada N, Karasawa T, Kimura H, Watanabe S, Komada T, Kamata R. et al. Ferroptosis driven by radical oxidation of n-6 polyunsaturated fatty acids mediates acetaminophen-induced acute liver failure. Cell Death Dis. 2020;11:144
9. Yang WS, SriRamaratnam R, Welsch ME, Shimada K, Skouta R, Viswanathan VS. et al. Regulation of ferroptotic cancer cell death by GPX4. Cell. 2014;156:317-31
10. Xie Y, Hou W, Song X, Yu Y, Huang J, Sun X. et al. Ferroptosis: process and function. Cell Death Differ. 2016;23:369-79
11. Friedman SL, Neuschwander-Tetri BA, Rinella M, Sanyal AJ. Mechanisms of NAFLD development and therapeutic strategies. Nat Med. 2018;24:908-22
12. Zhang J, Guo J, Yang N, Huang Y, Hu T, Rao C. Endoplasmic reticulum stress-mediated cell death in liver injury. Cell Death Dis. 2022;13:1051
13. Zhao C, Yu D, He Z, Bao L, Feng L, Chen L. et al. Endoplasmic reticulum stress-mediated autophagy activation is involved in cadmium-induced ferroptosis of renal tubular epithelial cells. Free Radic Biol Med. 2021;175:236-48
14. Hsin IF, Montano E, Seki E. Finding a new role for NEMO: A key player in preventing hepatocyte apoptosis and liver tumorigenesis by inhibiting RIPK1. Hepatology. 2016;64:295-7
15. Criswell T, Leskov K, Miyamoto S, Luo G, Boothman DA. Transcription factors activated in mammalian cells after clinically relevant doses of ionizing radiation. Oncogene. 2003;22:5813-27
16. Huang TT, Wuerzberger-Davis SM, Wu ZH, Miyamoto S. Sequential modification of NEMO/IKKgamma by SUMO-1 and ubiquitin mediates NF-kappaB activation by genotoxic stress. Cell. 2003;115:565-76
17. Li N, Banin S, Ouyang H, Li GC, Courtois G, Shiloh Y. et al. ATM is required for IkappaB kinase (IKKk) activation in response to DNA double strand breaks. J Biol Chem. 2001;276:8898-903
18. Kondylis V, Polykratis A, Ehlken H, Ochoa-Callejero L, Straub BK, Krishna-Subramanian S. et al. NEMO Prevents Steatohepatitis and Hepatocellular Carcinoma by Inhibiting RIPK1 Kinase Activity-Mediated Hepatocyte Apoptosis. Cancer Cell. 2015;28:582-98
19. Pescatore A, Esposito E, Draber P, Walczak H, Ursini MV. NEMO regulates a cell death switch in TNF signaling by inhibiting recruitment of RIPK3 to the cell death-inducing complex II. Cell Death Dis. 2016;7:e2346
20. Vlantis K, Wullaert A, Polykratis A, Kondylis V, Dannappel M, Schwarzer R. et al. NEMO Prevents RIP Kinase 1-Mediated Epithelial Cell Death and Chronic Intestinal Inflammation by NF-κB-Dependent and -Independent Functions. Immunity. 2016;44:553-67
21. Zhang DD, Lo SC, Cross JV, Templeton DJ, Hannink M. Keap1 is a redox-regulated substrate adaptor protein for a Cul3-dependent ubiquitin ligase complex. Mol Cell Biol. 2004;24:10941-53
22. Villeneuve NF, Sun Z, Chen W, Zhang DD. Nrf2 and p21 regulate the fine balance between life and death by controlling ROS levels. Cell Cycle. 2009;8:3255-6
23. Hayes JD, McMahon M. Molecular basis for the contribution of the antioxidant responsive element to cancer chemoprevention. Cancer Lett. 2001;174:103-13
24. Niture SK, Jaiswal AK. Nrf2 protein up-regulates antiapoptotic protein Bcl-2 and prevents cellular apoptosis. J Biol Chem. 2012;287:9873-86
25. Joo MS, Lee CG, Koo JH, Kim SG. miR-125b transcriptionally increased by Nrf2 inhibits AhR repressor, which protects kidney from cisplatin-induced injury. Cell Death Dis. 2013;4:e899-e
26. Liu J, Wu KC, Lu Y-F, Ekuase E, Klaassen CD. Nrf2 protection against liver injury produced by various hepatotoxicants. Oxid Med Cell Longev. 2013;2013:305861 -
27. Cuevas S, Pelegrín P. Pyroptosis and redox balance in kidney diseases. Antioxid Redox Signal. 2021;35:40-60
28. Tak J, Kim YS, Kim TH, Park GC, Hwang S, Kim SG. Gα(12) overexpression in hepatocytes by ER stress exacerbates acute liver injury via ROCK1-mediated miR-15a and ALOX12 dysregulation. Theranostics. 2022;12:1570-88
29. Mohar I, Stamper BD, Rademacher PM, White CC, Nelson SD, Kavanagh TJ. Acetaminophen-induced liver damage in mice is associated with gender-specific adduction of peroxiredoxin-6. Redox Biol. 2014;2:377-87
30. Gu JL, Müller S, Mancino V, Offermanns S, Simon MI. Interaction of G alpha(12) with G alpha(13) and G alpha(q) signaling pathways. Proc Natl Acad Sci U S A. 2002;99:9352-7
31. Rothwarf DM, Zandi E, Natoli G, Karin M. IKK-gamma is an essential regulatory subunit of the IkappaB kinase complex. Nature. 1998;395:297-300
32. Chen X, Calvisi DF. Hydrodynamic transfection for generation of novel mouse models for liver cancer research. Am J Pathol. 2014;184:912-23
33. Miotto G, Rossetto M, Di Paolo ML, Orian L, Venerando R, Roveri A. et al. Insight into the mechanism of ferroptosis inhibition by ferrostatin-1. Redox Biol. 2020;28:101328
34. Dara L, Ji C, Kaplowitz N. The contribution of endoplasmic reticulum stress to liver diseases. Hepatology. 2011;53:1752-63
35. Li J, Cao F, Yin H-L, Huang Z-J, Lin Z-T, Mao N. et al. Ferroptosis: past, present and future. Cell Death Dis. 2020;11:88 -
36. Luo JL, Kamata H, Karin M. IKK/NF-kappaB signaling: balancing life and death-a new approach to cancer therapy. J Clin Invest. 2005;115:2625-32
37. Scheidereit C. IκB kinase complexes: gateways to NF-κB activation and transcription. Oncogene. 2006;25:6685-705
38. Luedde T, Beraza N, Kotsikoris V, van Loo G, Nenci A, De Vos R. et al. Deletion of NEMO/IKKgamma in liver parenchymal cells causes steatohepatitis and hepatocellular carcinoma. Cancer Cell. 2007;11:119-32
39. Cullinan SB, Diehl JA. PERK-dependent Activation of Nrf2 Contributes to Redox Homeostasis and Cell Survival following Endoplasmic Reticulum Stress*. J Biol Chem. 2004;279:20108-17
40. Seiler A, Schneider M, Förster H, Roth S, Wirth EK, Culmsee C. et al. Glutathione Peroxidase 4 Senses and Translates Oxidative Stress into 12/15-Lipoxygenase Dependent- and AIF-Mediated Cell Death. Cell Metab. 2008;8:237-48
41. Zhang W, Liu HT. MAPK signal pathways in the regulation of cell proliferation in mammalian cells. Cell Res. 2002;12:9-18
42. Oyadomari S, Mori M. Roles of CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ. 2004;11:381-9
43. Storniolo A, Alfano V, Carbotta S, Ferretti E, Di Renzo L. IRE1α deficiency promotes tumor cell death and eIF2α degradation through PERK dipendent autophagy. Cell Death Discov. 2018;4:3
44. Galluzzi L, Kepp O, Krautwald S, Kroemer G, Linkermann A. Molecular mechanisms of regulated necrosis. Semin Cell Dev Biol. 2014;35:24-32
45. Pasparakis M. Regulation of tissue homeostasis by NF-kappaB signalling: implications for inflammatory diseases. Nat Rev Immunol. 2009;9:778-88
46. Tak J, Kim SG. Effects of toxicants on endoplasmic reticulum stress and hepatic cell fate determination. Toxicol Res. 2023
47. Dara L, Johnson H, Suda J, Win S, Gaarde W, Han D. et al. Receptor interacting protein kinase 1 mediates murine acetaminophen toxicity independent of the necrosome and not through necroptosis. Hepatology. 2015;62:1847-57
48. Sun L, Wang X. A new kind of cell suicide: mechanisms and functions of programmed necrosis. Trends Biochem Sci. 2014;39:587-93
49. Ma Q. Role of nrf2 in oxidative stress and toxicity. Annu Rev Pharmacol Toxicol. 2013;53:401-26
50. Cho MK, Kim WD, Ki SH, Hwang JI, Choi S, Lee CH. et al. Role of Galpha12 and Galpha13 as novel switches for the activity of Nrf2, a key antioxidative transcription factor. Mol Cell Biol. 2007;27:6195-208
51. Li G, Li J, Li C, Qi H, Dong P, Zheng J. et al. MicroRNA-125a-5p Contributes to Hepatic Stellate Cell Activation through Targeting FIH1. Cell Physiol Biochem. 2016;38:1544-52
Author contact
![]() Corresponding authors: Prof. Sang Geon Kim, Ph.D., College of Pharmacy, Dongguk University-Seoul, Goyang-si, Kyeonggi-do 10326, Republic of Korea. E-mail: sgkimedu; Tel.: +8231-961-5218; Fax: +8231-961-5206. Prof. Shin Hwang, M.D., Ph.D., Department of Surgery, Asan Medical Center, University of Ulsan College of Medicine, Seoul, Republic of Korea; E-mail: shwangseoul.kr; Tel.: +822-3010-3930.
Corresponding authors: Prof. Sang Geon Kim, Ph.D., College of Pharmacy, Dongguk University-Seoul, Goyang-si, Kyeonggi-do 10326, Republic of Korea. E-mail: sgkimedu; Tel.: +8231-961-5218; Fax: +8231-961-5206. Prof. Shin Hwang, M.D., Ph.D., Department of Surgery, Asan Medical Center, University of Ulsan College of Medicine, Seoul, Republic of Korea; E-mail: shwangseoul.kr; Tel.: +822-3010-3930.