Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
1. Introduction
2. Regulation and functions of...
3. TGase2 as a therapeutic...
4. Inhibition of TGase2 as a...
5. Conclusion and future...
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2024; 14(6):2329-2344. doi:10.7150/thno.95742 This issue Cite
Review
Transglutaminase 2 in diabetes mellitus: Unraveling its multifaceted role and therapeutic implications for vascular complications
Kwon-Soo Ha ![]()
Department of Molecular and Cellular Biochemistry, Kangwon National University School of Medicine, Chuncheon, Kangwon-do 24341, Korea.
Received 2024-2-27; Accepted 2024-3-17; Published 2024-3-25
Abstract

Diabetes, a severe metabolic disease characterized by chronic hypoglycemia, poses debilitating and life-threatening risks of microvascular and macrovascular complications, including blindness, kidney failure, heart attacks, and limb amputation. Addressing these complications is paramount, urging the development of interventions targeting diabetes-associated vascular dysfunctions. To effectively combat diabetes, a comprehensive understanding of the pathological mechanisms underlying complications and identification of precise therapeutic targets are imperative. Transglutaminase 2 (TGase2) is a multifunctional enzyme implicated in the pathogenesis of diverse diseases such as neurodegenerative disorders, fibrosis, and inflammatory conditions. TGase2 has recently emerged as a key player in both the pathogenesis and therapeutic intervention of diabetic complications. This review highlights TGase2 as a therapeutic target for diabetic complications and explores TGase2 inhibition as a promising therapeutic approach in their treatment.
Keywords: diabetic complications, hyperglycemic memory, inhibitors, transglutaminase 2, vascular leakage
1. Introduction
Diabetes, a serious metabolic disorder arising from chronic hypoglycemia, induces progressive vascular damage and dysfunction, leading to both microvascular and macrovascular complications [1, 2]. Microvascular complications include diabetic retinopathy (DR), diabetic nephropathy (DN), diabetic peripheral neuropathy, and diabetic pulmonopathy (DP), arising from chronic hyperglycemia affecting the retina, renal glomerulus, peripheral nerves, and lungs, respectively [3, 4]. Macrovascular complications stem from accelerated cardiovascular diseases (CVDs), culminating in myocardial infarction and strokes [5]. Given that diabetes significantly contributes to blindness, kidney failure, heart attacks, and limb amputation, the imperative to address diabetes and its associated vasculopathy is substantial [2, 3].
While the primary treatment for diabetes focuses on normalizing blood glucose levels [6], both experimental and clinical studies have demonstrated that glucose normalization alone does not suffice in preventing diabetic complications. This phenomenon, termed hyperglycemic memory (HGM) or the legacy effect, underscores the impact of persistent hyperglycemic stress [7-10]. Effectively treating diabetes necessitates not only the normalization of hyperglycemia but also the prevention of hyperglycemic memory-induced diabetic complications. Therefore, understanding the pathological mechanism(s) underlying microvascular and macrovascular diabetic complications and identifying therapeutic targets for these complications are pivotal for developing comprehensive treatment strategies.
Transglutaminase 2 (tissue transglutaminase; TGase2) belongs to the transglutaminase family and catalyzes Ca2+-dependent protein crosslinking through the transamidation of glutamine residues to lysine residues [11]. Ubiquitously expressed, TGase2 is a multifunctional enzyme, acting as a transamidase, serine/threonine kinase, protein disulfide isomerase, and GTPase [11-14]. Additionally, TGase2 demonstrates non-enzymatic functions through interactions with extracellular proteins, such as fibronectin, integrins, and syndecans, promoting matrix stabilization, which contributes to fibrosis in the kidney, liver, heart, and lung [11, 15].
Participating in various physiological processes, TGase2 influences apoptosis, inflammation, epithelial-mesenchymal transition, fibrogenic reactions, and mitochondrial dysfunction through post-translational modifications of several substrate proteins, including collagen, gluten, tau, α-synuclein, and β-crystallin [11, 13, 15-17]. Given its multifaceted roles, TGase2 is implicated in the pathogenesis of diverse diseases, including celiac disease [18], neurodegenerative diseases [13, 19], cancers [16, 20], fibrosis [17, 21], inflammatory diseases [22], and heart failure [23].
Recently, TGase2 has emerged as a key enzyme in the pathogenesis and therapeutic investigation of diabetic vascular complications, including DR, DN, DP, and CVD [4, 24-27]. This review focuses on TGase2's multifaceted role in diabetic complications, exploring its potential as a therapeutic target and discussing the emerging landscape of TGase2 inhibitors for addressing these intricate vascular complications.
2. Regulation and functions of TGase2 transamidase and kinase activities
Among the four distinct enzymatic activities of TGase2, the transamidase and kinase activities are most likely to be involved in the pathogenesis of diabetic complications [24, 28-31]. Consequently, this review focuses on the regulation and functions of these two enzymatic activities. The intricate interplay between these activities is influenced by various regulators, including thiol compounds, divalent metal cations, and phosphorylation [11, 14, 32] (Table 1). Additionally, they share several common proteins as substrates, such as nuclear factor-κB (NF-κB), retinoblastoma protein (pRb), E-cadherin, and p53 [11, 30, 33] (Table 2). Thus, to grasp the role of TGase2 in the pathogenesis of diabetic complications, a thorough understanding of the regulatory mechanisms governing TGase2 transamidase and kinase activities, along with their respective target proteins, becomes imperative.
Regulation of TGase2 transamidase and kinase activities.
| Regulators | Transamidase | Kinase |
|---|---|---|
| Calcium | Activation [11] | Inhibition [52, 54] |
| GDP/GTP | Inhibition [176, 177] | No effect [54] |
| ATP | Inhibition [14, 52] | Activation [14, 32, 52] |
| Cystamine | Inhibition [14] | Activation [14] |
| Amine compoundsa | Inhibition [17, 37, 38] | N.D. |
| Thiol compoundsb | Activation [14] | Inhibition [14, 32] |
| Nitric oxide | Inhibition [39] | N.D. |
| Mg2+ | No effect [14] | Activation [14] |
| Divalent metal cationsc | N.D. | Inhibition [32] |
| Phosphorylation | Inhibition [14] | Activation [14] |
| Acetylation | No effect [14], inhibition [42] | Activation [14] |
| C277S mutation | Complete inhibition [41, 178, 179] | No effect [41] |
| K444A mutation | Complete inhibition [41] | Partial inhibition [41] |
| K663A mutation | No effect [41] | Partial inhibition [41] |
aputrescine, monodansylcadaverine (MDC), 5-(biotinamido)pentylamine (BAPA), spermine, spermidine, and histamine; bglutathione, dithiothreitol, mercaptoethanol, and S-nitroso-N-acetylpenicillamine; cCu2+, Mn2+, Ni2+, and Zn2+; N.D.; not determined.
Substrate proteins of TGase2 transamidase and kinase: functions and regulatory actions.
| Substrate proteins | Transamidase activity | Kinase activity |
|---|---|---|
| RhoA | Activation: cell differentiation [43], stress fiber formation [48] | N.D. |
| NF-κB | Activation by IκB polymerization: inflammation [44] | Activation: PTEN downregulation and anti-apoptosis in cancer [56] |
| GAPDH | Inhibition: interfering in energy metabolism [3, 45, 50]; deamination: actin cytoskeleton remodeling [180] | N.D. |
| pRB | Protection of pRB degradation: anti-apoptosis [33, 46, 48] | Phosphorylation: disturbing E2F1 interactions and pro-apoptosis [52] |
| E-cadherin | Transamidation [33] | Phosphorylation: ECM breakdown and metastasis [30, 64] |
| p53 oncoprotein | Inhibition: tumorigenicity [47] | Phosphorylation: pro-apoptosis [58] |
| IGFBP-3 | N.D. | Phosphorylation: regulation of IGF functions [54] |
| Histone proteins | N.D. | Phosphorylation: regulation of chromatin structure and function [53] |
| G6PD | N.D. | Phosphorylation: oxidative stress [30] |
| Cytochrome C | N.D. | Phosphorylation: pro-apoptosis [30, 61] |
| Calmodulin | N.D. | Phosphorylation: Ca2+ signaling [30, 62] |
| S100A7 | Small effect [33] | Phosphorylation: Ca2+ signaling [30] |
| cMMP-3 | N.D. | Phosphorylation: ECM breakdown and metastasis [30, 63] |
cMMP-3, catalytic domain of human matrix metalloproteinase-3; ECM, extracellular matrix; GAPDH, glyceraldehyde 3-phosphate dehydrogenase; G6PD, glucose 6-phosphate dehydrogenase; IGF, insulin-like growth factor; IGFBP-3, insulin-like growth factor-binding protein-3; NFκ-B, nuclear factor-κB; pRb, retinoblastoma protein; PTEN, phosphatase and tensin homolog; S100A7, S100 calcium-binding protein A7; N.D., not determined.
2.1. TGase2 transamidase activity
2.1.1. Regulation of TGase2 transamidase activity
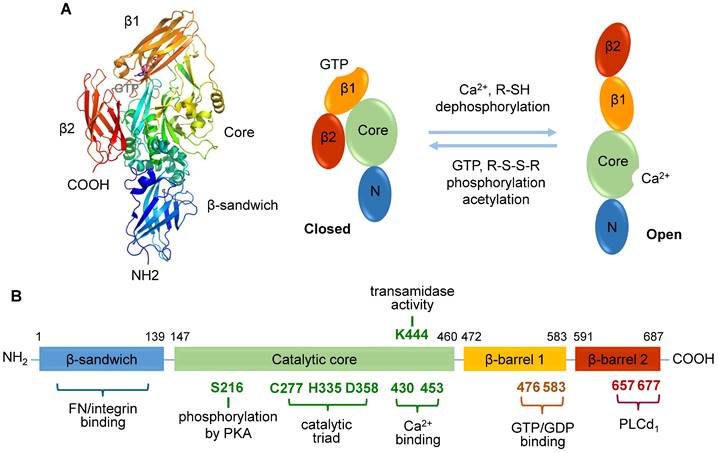
TGase2 comprises four distinct domains: an N-terminal β-sandwich domain, a catalytic core domain, and two C-terminal β-barrel domains [34, 35] (Figure 1A). While the N-terminal β-sandwich domain lacks catalytic activity, it is recognized for its binding affinity to fibronectin and integrin [35]. The catalytic core domain, housing a catalytic triad (Cys277, His335, and Asp358), possesses five calcium-binding sites [36], and β-barrel 1 features a GTP/GDP-binding site (Figure 1B). Three-dimensional structural analysis has revealed the reciprocal regulation of transamidase activity by Ca2+, nucleotides, and various regulators [11]. TGase2 bound with GDP adopts a closed conformation, negatively regulating its transamidase activity. In contrast, the transamidase activity is induced through Ca2+ binding, resulting in an open conformation in which the transamidase active site is exposed to substrates.
Human transglutaminase 2 (TGase2) open and closed structures and amino acid regulation map. A. TGase2 exhibits open and closed structural formations. The closed TGase2 structure was prepared with Pymol 3.0 using the PBD file 4PYG. B. A comprehensive amino acid map illustrating the regulatory mechanisms of TGase2 activity. β1; β-barrel 1 domain; β2, β-barrel 2 domain; Core, core catalytic domain; N, N-terminal β-sandwich domain; FN, fibronectin; PKA, protein kinase A; PLCδ1, phospholipase C δ1.
In addition to Ca2+ and nucleotides, the transamidase activity of TGase2 is subject to regulation by amine compounds, nitric oxide, and thiol compounds (Table 1) [11, 14]. Amine compounds, such as putrescine, monodansylcadaverine (MDC), 5-(biotinamido)pentylamine (BAPA), spermidine, and histamine, act as amine donors to inhibit transamidase activity by competing with natural substrates [17, 37, 38]. Nitric oxide also inhibits the transamidase activity through S-nitrosylation of the cysteine residue in the active site [39]. Thiol compounds, including glutathione (GSH), cystamine, and cysteamine, potentially regulate transamidase activity by affecting the redox state of TGase2 cysteine residues, particularly the catalytic core residue (Cys277) and three cysteine residues (Cys230, Cys370, and Cys371) [14, 25, 40].
Several amino acids in TGase2 play a role in regulating transamidase activity [41] (Figure 1B). Catalytic Cys277 is essential for transamidase activity, and this function is further influenced by disulfide bond formation among three cysteine residues (Cys230, Cys370, and Cys371) [40, 41]. A site-directed mutagenesis study identified Lys444 in the catalytic domain as significant for transamidase activity [41]. Interestingly, the phosphorylation levels of TGase2 also regulate its transamidase activity [14]. Dephosphorylation of TGase2 with alkaline phosphatase enhances transamidase activity, whereas phosphorylation using the catalytic subunit of protein kinase A (PKA) inhibits this enzyme [14]. However, the role of acetylation in the regulation of transamidase activity remains controversial [14, 42].
2.1.2. Functions of TGase2 transamidase activity
TGase2 transamidase plays a pivotal role in the regulation of various proteins, including RhoA [43], NF-κB [44], glyceraldehyde 3-phosphate dehydrogenase (GAPDH) [45], pRb [46], and the oncoprotein p53 [47] (Table 2). Retinoic acid-induced activation of the transamidase leads to increased transamidation of RhoA at Gln63, a process inhibited by MDC [43, 48]. Transamidated RhoA, acting as a constitutively active G-protein, enhances its binding to RhoA-associated kinase-2, fostering the formation of stress fibers and focal adhesion complexes. This suggests the involvement of transamidase activity in cell differentiation, including that of human leukemia cells and neurons [43, 48, 49]. The transamidase also activates NF-κB by inducing the polymerization of I-κB, resulting in subsequent NF-κB dissociation and translocation into the nucleus. This activation is capable of upregulating inflammatory genes such as inducible NO synthase and tumor necrosis factor α [44].
Furthermore, the transamidase inhibits GAPDH, a glycolytic enzyme, through crosslinking lysine residues in the C-terminal region of GAPDH with polyglutamine repeats. This interference leads to disruptions in energy metabolism [45, 50]. Inhibition of GAPDH activity elevates the levels of glycolysis intermediates or their metabolites, resulting in the activation of pathological vascular complications, including diabetic complications [3]. Modification of pRb by the transamidase protects from apoptosis by inhibiting caspase-induced degradation of polymerized pRb [46, 51]. Integrative proteomic profiling of the transamidase activity using protein arrays has identified several potential substrates, including osteopontin and globular actin [33], suggesting potential roles of the transamidase in cytoskeleton and bone remodeling. The TGase2 transamidase activity is also implicated in tumorigenicity [47] and pulmonary fibrosis [21].
2.2. TGase2 kinase activity
2.2.1. Regulation of TGase2 kinase activity
The regulatory mechanism of TGase2 kinase activity is less understood compared to the transamidase activity, primarily due to limitations in suitable assays for determining in situ and in vivo kinase activity [30]. Determination of kinase activity has traditionally involved detecting ATP incorporation into substrate proteins using radioactive isotopes [52, 53] or antibodies against phosphoamino acids [52]. However, these methods have drawbacks, such as the use of hazardous radioactive probes or relatively low affinities for phosphorylated substrates [30]. To overcome these limits, a well-type array-based kinase activity assay has been introduced using Pro-Q Diamond phosphoprotein stain [30].
Investigations into the regulation mechanism of TGase2 kinase have utilized on-chip kinase activity assays [14, 32]. The kinase is regulated by thiol compounds, divalent metal cations, phosphorylation, and acetylation [14, 32] (Table 1). The kinase activity assay, employing a cysteine-modified insulin-like growth factor-binding protein-3 (IGFBP-3)-derived peptide, revealed that thiol compounds, such as 5,5′-dithio-bis-(2-nitrobenzoic acid), S-nitroso-N-acetylpenicillamine, dithiothreitol, GSH, and mercaptoethanol, inhibit the kinase (Table 1). Thus, the modification of TGase2 cysteine residues may play a critical role in regulating the TGase2 kinase [32]. Notably, cystamine, a disulfide compound with amine moieties at both ends, enhances the kinase activity [14], even though this disulfide compound inhibits TGase2-induced phosphorylation of IGFBP-3 [54]. Divalent metal cations also play a role in the regulation of TGase2 kinase, as on-chip activity assays demonstrate kinase inhibition by Cu2+, Mn2+, Ni2+, and Zn2+, but not by Ca2+ [32]. Furthermore, phosphorylation and acetylation of TGase2 enhance the kinase activity, with acetylation of TGase2 not affecting transamidase activity [14].
2.2.2. Functions of TGase2 kinase activity
TGase2 can phosphorylate serine and threonine, but not tyrosine, residues of substrate proteins [55]. The intrinsic kinase activity of TGase2 was revealed by Mishra and Murphy [54], who demonstrated the phosphorylation of IGFBP-3 by TGase2 on breast cancer cell membranes. TGase2 undergoes phosphorylation at Ser216 by PKA, resulting in activation of NF-κB and protein kinase B, downregulation of phosphatase and tensin homolog (PTEN) [56], phosphorylation of pRb [52], and enhanced TGase2 binding to 14-3-3 [57] (Table 2). TGase2 kinase further phosphorylates p53 [58] and histone proteins [53], indicating its role in apoptosis regulation and chromatin structure and function, respectively. p53 is a key tumor suppressor protein that has multiple biological functions, including DNA damage repair, cell cycle arrest, apoptosis, and senescence [59]. Additionally, the kinase phosphorylates β-catenin at Tyr654 through a c-Src-dependent mechanism, leading to the proliferation of ovarian cancer cells. However, the precise mechanism by which TGase2 activates c-Src remains unclear [31].
An on-chip kinase activity assay using Pro-Q Diamond stain, suitable for screening the kinase substrates, was employed to investigate potential substrates of TGase2 kinase [30]. This high-throughput activity assay revealed several kinase substrate proteins based on substrate affinity (Km), including glucose 6-phosphate dehydrogenase, cytochrome C, calmodulin, and S100 calcium-binding protein A7 in the cytosol. This suggests potential roles of TGase2 kinase in regulating oxidative stress [60], apoptosis [61], and intracellular Ca2+ signaling events [62]. TGase2 kinase also phosphorylates E-cadherin in the plasma membrane and matrix metalloproteinase-3 (MMP-3) in the extracellular matrix, indicating its potential involvement in the breakdown of extracellular matrix and cancer metastasis [63, 64].
3. TGase2 as a therapeutic target for diabetic complications
Oxidative stress plays a pivotal role in the pathogenesis of diabetic complications, leading to progressive vascular damage and dysfunction [1, 65, 66]. These vascular complications are associated with various pathological pathways, including increased polyol pathway flux, elevated hexosamine biosynthesis, activation of protein kinase C, and augmented formation of advanced glycation end products [3, 8]. These pathways are triggered by hyperglycemia-induced inhibition of GAPDH through poly ADP-ribosylation [67]. Recent reports emphasize that hyperglycemia-induced generation of intracellular reactive oxygen species (ROS) plays a crucial role in activating TGase2, contributing to diabetic microvascular and macrovascular complications [3, 24, 27]. Importantly, a vicious cycle exists between hyperglycemia-induced ROS generation and TGase2 activation, which significantly contributes to HGM-induced endothelial dysfunction [28, 65].
This section delves into the pivotal role of TGase2 in diabetic complications, specifically DR, DN, DP, and CVD. Furthermore, we explore the role of TGase2 in HGM, a significant phenomenon in the development and progression of diabetic complications in both type 1 and type 2 diabetes [28, 65]. It is important to note that this review excludes discussions on diabetic neuropathy, impaired wound healing, and stroke due to the unclear role of TGase2 in the pathogenesis of these complications.
3.1. Diabetic retinopathy
DR is the most common diabetic microvascular complication and remains the leading cause of blindness in working-age populations [68, 69]. This metabolic disease progresses from early non-proliferative DR to late proliferative DR [70, 71]. DR is influenced by various risk factors such as poor glucose control, diabetes duration, hypertension, and plasma glucose fluctuations [72]. Non-proliferative DR represents the early stages, characterized by thickening of the basement membrane, pericyte loss, formation of acellular capillaries, microaneurysms, and microvascular leakage [5, 70]. Proliferative DR, on the other hand, features pathological neovascularization and eventual diabetic macular edema, contributing to vision loss [73, 74].
A predominant cause of microvascular leakage in the diabetic retina is hyperglycemia-induced overexpression of vascular endothelial growth factor (VEGF) [24, 75, 76] (Figure 2). Elevated VEGF levels in the retina activate TGase2 through sequential elevation of intracellular Ca2+ and ROS, leading to microvascular leakage through stress fiber formation and disassembly of adherens junctions [24, 76]. Thus, ROS-mediated activation of TGase2 plays a key role in VEGF-induced retinal vascular leakage, positioning TGase2 as a potential therapeutic target for DR treatment in several reports [3, 25, 28, 71, 77].
The role of transglutaminase 2 (TGase2) in the pathogenesis of diabetic retinopathy, pulmonopathy, and nephropathy. Reactive oxygen species (ROS)-mediated activation of TGase2 contributes to hyperglycemia-induced microvascular leakage and neovascularization in the retina, cancer metastasis and fibrosis in the lung, and glomerular dysfunction in the kidney. GABAA, γ-aminobutyric acid type A; K9-C-peptide, human C-peptide conjugated with nine repeats of lysine-containing elastin-like polypeptide; VEGF, vascular endothelial growth factor.
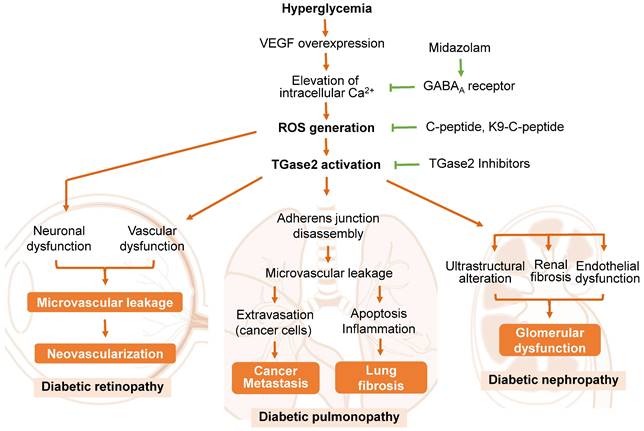
Proinsulin C-peptide, administered systemically or intravitreally, alleviates VEGF-induced retinal microvascular leakage by inhibiting ROS-mediated activation of TGase2, subsequently preventing stress fiber formation and vascular endothelial (VE)-cadherin disassembly in diabetic mice [24, 71, 76]. Cysteamine, an aminothiol derived from coenzyme A degradation, attenuates vascular leakage by inhibiting VEGF-induced activation of TGase2 and VE-cadherin disruption in diabetic retinas [25]. The benzodiazepine anesthetic midazolam reduces hyperglycemia-induced vascular leakage by inhibiting VEGF-induced elevation of intracellular Ca2+ and subsequent ROS generation and TGase2 activation through the GABAA receptor in the retinas of diabetic mice [77]. While TGase2 plays a crucial role in VEGF-induced retinal vascular dysfunction, it is not associated with diabetes-induced retinal neurodegeneration [71]. Thus, TGase2 has emerged as a significant enzyme in the pathogenesis of retinal vascular dysfunction in diabetic retinas and represents a promising therapeutic target for DR.
3.2. Diabetic pulmonopathy
DP is a newly recognized microvascular complication of diabetes [3, 78]. While diabetes is well-established as a systemic disease with chronic oxidative stress and inflammation affecting various organs, such as the eyes, kidneys, nerves, skin, and the vascular system [79, 80], the pulmonary complications have been comparatively overlooked. This disregard is attributed to the lung's significant physiological reserve and the presence of subclinical pulmonary abnormalities in diabetic patients [78, 80, 81]. Nevertheless, recent clinical and experimental investigations highlight the lung as a potential target organ affected by diabetes [78, 82, 83]. The intricate alveolar-capillary network and the abundance of collagen and elastin in the lung render it susceptible to diabetic microvascular damage [78, 80, 84]. Type 1 and type 2 diabetes patients exhibit respiratory abnormalities, including reduced diffusing capacity, lung volume, control of ventilation, and elastic recoil [81, 85]. Clinical studies also suggest associations between diabetes and conditions such as asthma, idiopathic fibrosis, chronic obstructive pulmonary disease, and hypertension in the lungs of diabetic patients [80, 82, 83, 86].
Idiopathic pulmonary fibrosis, characterized by chronic, progressive fibrotic lung disease with high mortality and limited therapeutic options [84, 85], shows abnormal extracellular matrix accumulation in the lung destroying alveolar architecture, which results in pulmonary dysfunction and respiratory failure [87]. Emerging research indicates a potential link between idiopathic pulmonary fibrosis and diabetes [80, 82-85], with epidemiological research identifying diabetes as an independent risk factor for idiopathic pulmonary fibrosis [88].
The underlying mechanisms of DP have been investigated in rodent models of type 1 diabetes [28, 78, 79]. In diabetic rats, hyperglycemia induces fibrotic changes in the lung by activating TGF-β-mediated epithelial-mesenchymal transition [79]. Notably, recent findings by Ha and colleagues [4] highlight that chronic hyperglycemia promotes vascular leakage in the lungs through VEGF-induced ROS generation and subsequent TGase2 activation, leading to melanoma cell metastasis in diabetic mice (Figure 2). The hyperglycemia-induced vascular leakage and melanoma cell metastasis were mitigated by inhibiting TGase2 through systemic supplementation of proinsulin C-peptide [4], insulin [26], or midazolam [89].
TGase2 is also implicated in hyperglycemia-induced inflammation and apoptosis, contributing to pulmonary fibrosis in the diabetic lung. This pathological process was ameliorated through long-term systemic supplementation of human C-peptide, achieved by subcutaneous implantation of a thermosensitive biopolymer-conjugated C-peptide (K9-C-peptide) depot [28, 78]. The involvement of TGase2 in pulmonary fibrosis was further confirmed in a mouse model of bleomycin-induced pulmonary fibrosis [21]. Consequently, TGase2 is a promising therapeutic target for addressing multiple facets of DP, including microvascular leakage, inflammation, metastasis, and idiopathic fibrosis.
3.3. Diabetic nephropathy
DN, a significant microvascular complication affecting approximately 40% of diabetic patients, is the primary cause of chronic kidney disease leading to end-stage renal disease in developed countries [90, 91]. Clinically, DN manifests as albuminuria, a progressive decrease in glomerular filtration rate, and elevated blood pressure [92]. Pathologically, this chronic kidney disease is characterized by glomerulosclerosis, tubulointerstitial fibrosis, thickening of glomerular and tubular membranes, and vascular dysfunction [93, 94]. While the prevalence of diabetic kidney disease has surged over the past decades, currently available treatments are limited to those preventing or delaying disease progression [90].
Microalbuminuria, widely acknowledged as the initial clinical sign of DN, is induced by the disruption of the glomerular filtration barrier due to glomerular basement membrane thickening, foot process effacement of podocytes, and microvascular dysfunction [93]. Notably, microvascular dysfunction, arising from VEGF overexpression and subsequent vascular integrity disruption, plays a pivotal role in the pathogenesis of diabetic microvascular complications in the retina and lungs [4, 76, 95]. In the kidney, glomerular endothelial dysfunction has been implicated in the pathogenesis of diabetic kidney disease [95, 96]. However, the underlying mechanism by which hyperglycemia induces alterations in glomerular endothelial permeability and subsequent microalbuminuria remains unclear. Recent studies have highlighted the role of TGase2 in glomerular endothelial dysfunction and renal fibrosis in diabetic animal models [27, 93, 97] (Figure 2).
In the renal cortex of type 1 diabetic mice, hyperglycemia activates the transamidase activity of TGase2 and induces apoptosis, both of which are suppressed by systemic supplementation of human C-peptide [27]. TGase2's involvement has been reported in mouse models of renal fibrosis induced by unilateral ureteral obstruction or streptozotocin [93, 98]. TGase2 contributes to interstitial renal fibrosis through TGF-β activation and cell infiltration in unilateral ureteral obstruction mouse models [98]. Additionally, TGase2 plays a role in hyperglycemia-induced pathological alterations of glomerular ultrastructure and renal fibrosis, with midazolam attenuating glomerular endothelial dysfunction in the kidneys of diabetic mice [93]. Despite the formidable challenge of elucidating the pathological mechanisms of DN, TGase2 is a potential therapeutic target for its treatment.
3.4. Cardiovascular disease
CVD, the most prevalent diabetic complication, is the leading cause of death in patients with either type 1 or type 2 diabetes [3, 99, 100]. CVD encompasses a group of disorders affecting the heart and blood vessels, including premature atherosclerosis leading to myocardial infarction, stroke, and compromised cardiac function [5, 101]. Its high prevalence in type 1 diabetes patients significantly impairs life expectancy [100, 102], with chronic kidney disease and cardiac autonomic neuropathy being associated with its progression in these patients [100, 103]. In type 2 diabetes, kidney disease remains a major risk factor for CVD, accompanied by other CVD risk factors, including dyslipidemia, poor glycemic control, and persistent high blood pressure [5, 101].
Several reports propose the involvement of TGase2 in the pathogenesis of CVDs, including atherosclerosis, myocardial fibrosis, and associated heart diseases [23, 104-106]. However, studies specifically addressing the role of TGase2 in diabetes-associated CVDs are limited. Investigations into hyperglycemia-induced progression to CVD in the aorta of type 1 diabetic mice [27, 65] indicate that hyperglycemia stimulates transamidating activity and endothelial cell apoptosis in the aortic endothelium, processes inhibited by systemic supplementation of human C-peptide [27]. Administering human C-peptide through osmotic pumps protects endothelial cells from hyperglycemia-induced apoptosis by blocking ROS-mediated activation of TGase2. This ROS generation and TGase2 activation form a vicious cycle implicated in hyperglycemia-induced vascular dysfunction, including the expression of inflammatory adhesion molecules and apoptosis [65]. Nevertheless, further research is essential to comprehensively understand the role of TGase2 in the pathogenesis of diabetes-associated CVD.
3.5. Hyperglycemic memory
HGM, signifying the persistence of hyperglycemic stress post-glucose normalization, is a crucial factor in the pathogenesis and progression of both diabetic microvascular and macrovascular diseases in type 1 and type 2 diabetes [3, 10, 28]. Engerman and Kern's seminal work [7] reveals that intensive glycemic control fails to arrest the progression of DR in diabetic dogs initially subjected to poor glycemic control, characterized by capillary aneurysm, acellular capillaries, and pericyte loss. Large-scale clinical studies, such as the Diabetes Control and Complications Trial (DCCT)-Epidemiology of Diabetes Interventions and Complications (EDIC) study in type 1 diabetes patients [10, 107] and the UK Prospective Diabetes Study (UKPDS) in type 2 diabetes patients [108, 109], demonstrate that initial tight glycemic control diminishes the incidence of diabetic complications. Conversely, initial poor glycemic control can lead to the long-term development of diabetic complications [110].
To unravel the underlying mechanism of persistent hyperglycemic stress and understand the pathophysiology of diabetic complications, diabetic animal models have been employed [9, 65, 111, 112]. Persistent oxidative stress plays a central role in the long-lasting vascular complications induced by HGM [8, 65, 110, 113]. Hyperglycemia induces sustained upregulation of pro-oxidant enzymes, namely protein kinase C β and p47phox, a NADPH oxidase subunit, post-glucose normalization in the retina of diabetic rats [112]. The prolonged activation of p66Shc by protein kinase C βII maintains ROS generation after normoglycemia, resulting in vascular apoptosis in the mouse aorta [110]. p66Shc, a 66 kDa Src homology/collagen adaptor protein, is a key regulator of mitochondrial function, oxidative stress, and apoptosis [114].
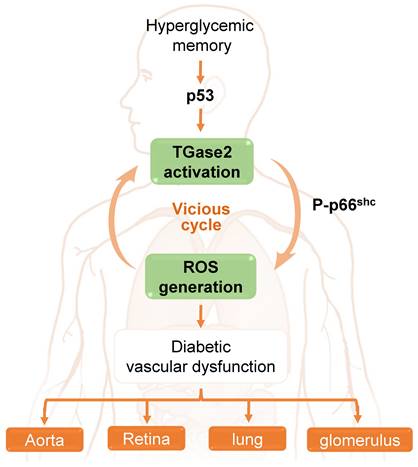
In the aorta of diabetic mice, Ha and colleagues [65] elucidated that hyperglycemia induces sustained ROS generation and TGase2 activation post-return to normoglycemia, initiating a detrimental cycle. This ROS-TGase2 cycle, regulated by the mitochondrial adaptor p66Shc and p53, is implicated in the sustained expression of inflammatory adhesion molecules and apoptosis in the aortic endothelium [65, 111]. Furthermore, this vicious cycle is associated with HGM-induced complications in the retina, including vascular leakage and neurodegeneration, as well as in the lungs, where it leads to vascular leakage and fibrosis. Additionally, it contributes to other complications such as glomerular adherens junction disruption and vascular leakage in diabetic mice [28]. Thus, TGase2, activated by persistent oxidative stress and associated with p66Shc and p53, assumes a pivotal role in HGM-induced vascular complications in the aorta, retina, lung, and kidney (Figure 3).
The role of a transglutaminase 2 (TGase2)-reactive oxygen species (ROS) vicious cycle in hyperglycemic memory-induced vascular dysfunction in the aorta, retina, lung, and glomerulus.
4. Inhibition of TGase2 as a potential therapeutic strategy for diabetic complications
TGase2 plays a pivotal role in the pathogenesis of diabetic complications, such as DR, DN, DP, CVD, and HGM. These microvascular and macrovascular complications, stemming from chronic hyperglycemia and hyperglycemia memory, can be addressed through the direct or indirect inhibition of TGase2. Direct inhibition encompasses the use of competitive, reversible, and irreversible inhibitors that affect substrate binding to the active site of TGase2. Indirect inhibition by G-protein-coupled receptor agonists, such as C-peptide, C-peptide conjugates, and midazolam, regulate TGase2 by binding to its receptors.
4.1. Direct inhibition
4.1.1. Competitive inhibitors: monoamines
Competitive inhibitors, which consist of biogenic monoamines and polyamines, regulate TGase2 activity by competing with its natural substrates rather than preventing the enzyme activity through covalent modification or allosteric regulation during the transamidation reaction, leading to the formation of isopeptide bonds between the natural glutamine substrates and the inhibitors [37]. Amine inhibitors have been intensively used in cellular and organismal studies due to their commercial availability, stability, and relative non-toxicity [37, 97]. Uteroglobin, a protein substrate of TGase2, has also been employed as a competitive TGase2 inhibitor in murine experimental crescentic glomerulonephritis [115].
Monoamine inhibitors, including catecholamines and amino acid derivatives, are characterized by a primary amine linked to alkyl chains [37, 97, 116]. Among the biogenic monoamines, cysteamine, MDC, and dopamine have demonstrated efficacy against diabetic complications (Table 3). Cysteamine, also known as β-mercaptoethylamine, an aminothiol with a primary amine group and a sulfhydryl group, is endogenously derived from coenzyme A degradation and metabolized into taurine [25, 117]. Cysteamine exhibits therapeutic potential for DR and vascular leakage-associated diseases [25]. Additionally, the aminothiol is beneficial in conditions such as cystinosis [117], nonalcoholic fatty liver disease [118], neurodegenerative diseases like Alzheimer's, Huntington's, and Parkinson's diseases [119, 120], and postinflammatory hyperpigmentation [121]. There is a recent report from three clinical trial programs [122], suggesting long-term clinical benefits of delayed-release cysteamine bitartrate capsules in patients with nephropathic cystinosis.
TGase2 inhibitors for the treatment of diabetic complications.
| Group | Inhibitor | Potential target diseases or functions |
|---|---|---|
| Monoamines | Cysteamine | Diabetic retinopathy [25], cystinosis [117], neurodegenerative diseases [119, 120] |
| Monodansylcadaverine | Diabetic retinopathy [24, 77], pulmonary disease [4], nephropathy [93] | |
| Dopamine | Diabetic retinopathy [113, 124, 125] | |
| 5-(biotinamido)pentylamine | TGase pseudosubstrate [24, 27, 28, 33, 126, 127, 129] | |
| Polyamines | Cystamine | Diabetic retinopathy [24, 71], pulmonopathy [4, 78], nephropathy [89, 93], aortic dysfunction [65] |
| Putrescine | β-cell function [135], TGase substrate [133] | |
| Spermidine | β-cell function [135], TGase substrate [133] | |
| Spermine | β-cell function [135], TGase substrate [133] | |
| Irreversible inhibitors | Dihydroisoxazole derivatives | Anti-inflammation [141], antioxidant [144], glioblastoma [142, 143] |
| Michael acceptors | β-cell function [181], Celiac disease [137], neurodegenerative diseases [116] | |
| Gluten peptide analogs | Celiac disease [150] | |
| Acetylsalicylic acid | Acetylation of lysine residues [14, 42] | |
| Reversible inhibitors | LDN27219 | Vascular dysfunction [154] |
| TGase2 siRNA | Diabetic retinopathy [24], pulmonary disease [4], nephropathy [93] |
LDN27219, thieno[2,3-d]pyrimidin-4-one acylhydrazides.
MDC has been used to investigate the role of TGase2 in the pathogenesis of diabetic complications [4, 24, 93]. MDC inhibits VEGF-induced stress fiber formation, adherens junction disassembly, and subsequent endothelial permeability in human retinal endothelial cells, attenuating hyperglycemia-induced microvascular leakage in the retinas of diabetic mice [24, 77]. Moreover, MDC suppresses VEGF or high glucose-induced VE-cadherin disassembly and endothelial permeability in human pulmonary microvascular endothelial cells [4] or human glomerular microvascular endothelial cells [93], indicating its benefit against DR, DP, and DN.
Dopamine, a neurotransmitter in the nervous system, has beneficial effects against DR [113]. In the retina, dopamine, the most abundant catecholamine, is released from dopaminergic neurons identified as dopaminergic amacrine cells and interplexiform cells [123]. In type 1 diabetic mice, dopamine deficiency is associated with early visual dysfunction, which is improved by intraperitoneal treatment with dopamine receptor agonists or the dopamine precursor levodopa [124, 125]. Furthermore, in the retinas of type 1 diabetic mice, HGM-induced persistent stresses, including oxidative stress, mitochondrial membrane potential collapse and fission, and adherens junction disassembly leading to vascular leakage, are attenuated by intravitreal injection of levodopa [113].
Moreover, BAPA holds therapeutic promise for diabetic complications. This is attributed to its extensive utilization as a pseudosubstrate in in vitro [4, 33, 126, 127], in situ [27, 28, 128, 129], and in vivo [4, 24, 28] TGase activity assays.
4.1.2. Competitive inhibitors: polyamines
There exist various biogenic polyamine inhibitors of TGase2, including cystamine, putrescine, spermine, and spermidine, that compete with substrate proteins in the transamidation reaction (Table 3). Cystamine is a frequently utilized polyamine for investigating TGase2's function in the pathogenesis of diabetic vascular dysfunctions in multiple tissues, including the aorta [65], eye [24, 71], lung [4, 78], and kidney [89, 93]. Distinguished by its intricate regulatory mechanisms compared to other competitive inhibitors [37, 97], cystamine is reduced into cysteamine due to the high molar ratio of GSH to GSSG (100:1) in resting cells [130]. Cysteamine, the reduced form of cystamine, functions as a competitive amine inhibitor. It can irreversibly bind to the catalytic cysteine residues of TGase2, forming disulfide bonds [37]. Additionally, cystamine elevates intracellular GSH levels [131], influencing oxidative stress-associated pathophysiological responses [97]. Furthermore, cystamine increases brain and serum levels of the neuronal survival factor brain-derived neurotrophic factor and has therapeutic potential for neurodegenerative diseases and schizophrenia [132].
Polyamines derived from ornithine, such as putrescine, spermidine, and spermine, serve as TGase2 substrates [133]. Ornithine is converted into putrescine by ornithine decarboxylase, followed by sequential conversion into spermidine and spermine by spermidine synthase and spermine synthase, respectively [134]. These ornithine-derived polyamines play a crucial role in normal β-cell functions, influencing intracellular Ca2+ levels and inflammation. Consequently, alterations in polyamine levels are implicated in the pathogenesis of diabetes [135]. Due to their positive charge at physiological pH, these polyamines strongly interact with negatively charged molecules like DNA and RNA, participating in DNA replication, gene expression, and mRNA translation [135]. Elevated serum levels of putrescine and spermine in type 2 diabetic patients suggest the potential involvement of polyamine metabolism in the pathogenesis of type 2 diabetes [136]. However, the precise association between the roles of putrescine, spermidine, and spermine in normal β-cell functions and the pathogenesis of type 2 diabetes remains unclear, particularly regarding their impact on TGase2 inhibition.
4.1.3. Irreversible TGase2 inhibitors
Several irreversible TGase2 inhibitors have been reported by various research groups, including chloromethyl ketones, dihydroisoxazole derivatives, Michael acceptors, and gluten peptide analogs [37, 116, 137] (Table 3). Irreversible TGase2 inhibitors, commonly termed suicide inhibitors, exert their effect by covalently modifying the enzyme, primarily at the active site, thus hindering substrate binding to the active site [37].
Iodoacetamide, an early irreversible inhibitor, was employed to inhibit guinea pig TGase2 by forming a thioester bond with the cysteine residue of the enzyme's active site [138]. A series of chloromethyl ketones, synthesized based on a peptidic (carbobenzyloxy-phenylalanine) scaffold, underwent testing for reactivity toward glutathione [139]. Dihydroisoxazole derivatives, derived from the natural product acivicin, represent a well-studied class of TGase2 irreversible inhibitors. Exhibiting various biological activities [140], including anti-inflammatory [141], anticancer [142-144], and antioxidant effects [145], dihydroisoxazoles hold potential for treating diabetic complications. NTU compounds, such as NTU281 (N-benzyloxycarbonyl-L-phenylalanyl-6-dimethylsulfonium-5-oxo-L-norleucine) and NTU283 (1,dimethyl-2[(oxopropyl)thio]imidazolium), bind irreversibly to the TGase2 catalytic cysteine residues [97, 146]. NTU281 showed beneficial effects against hyperglycemia-induced glomerular dysfunction in diabetic rats [147, 148].
Michael acceptors, as reported by Keillor and colleagues [116], belong to a family of irreversible TGase2 inhibitors with the carbobenzyloxy-phenylalanine scaffold and acrylamide warhead. Zedira also documented several peptidomimetic TGase2 inhibitors featuring an α,β-unsaturated ester, designed for treating celiac disease and TGase2-associated ailments [137]. Gluten is a complex protein mixture with immunogenic peptide sequences triggering autoimmune responses in patients with celiac disease [149]. Gluten peptides have been utilized as peptidomimetic irreversible inhibitors due to its high affinity toward TGase2. The glutamine in these peptides has been substituted with acivicin or 6-diazo-5-oxo-norleucine (DON) to enhance the inhibitory potency [150, 151], while the DON peptide was shown to be more potent in inhibiting TGase2 than the acivicin analogue [150]. Acetylsalicylic acid, commonly known as aspirin, impacts the transamidation or kinase activity of TGase2 through lysine residue acetylation [14, 42]. However, its effect on diabetic complications remains poorly understood.
4-1-4. Reversible inhibitors
Reversible TGase2 inhibitors curtail enzyme activity by impeding substrate access to the active site without instigating covalent modifications [37]. Allosteric regulation of TGase2 is achieved through GTP analogs and thieno[2,3-d]pyrimidin-4-one acylhydrazides (LDN27219) [37, 116]. Nonhydrolyzable GTP analogs, such as GTPγS and GMP-PCP, reversibly inhibit TGase2 [129, 152]. LDN27219, categorized as a reversible inhibitor, exhibits slow binding to the GTP site of TGase2, inducing a closed conformation and inhibiting transamidase activity [153]. In arteries, LDN 27219 shows promise in lowering blood pressure and enhancing endothelium-dependent vasorelaxation, suggesting its potential in addressing vascular dysfunction [154] (Table 3).
TGase2-specific siRNA has emerged as a potential therapeutic avenue for diabetic complications. Intravitreal injection of TGase2 siRNA attenuates hyperglycemia-induced retinal vascular leakage in type 1 diabetic mice [24]. Moreover, TGase2 siRNA inhibits VEGF and high glucose-induced oxidative stress, VE-cadherin disassembly, and endothelial permeability in human retinal endothelial cells, human pulmonary microvascular endothelial cells, and human glomerular microvascular endothelial cells [4, 24, 93], underscoring its potential role in treating DR, DP, and DN (Table 3).
Various other reversible TGase2 inhibitors have been reported, including trans-cinnamoyl derivatives, cinnamoyl benzotriazolyl amides and azachalcones [155], ZM39923 and ZM449829 [156], acylideneoxoindoles [157], and quinoxaline derivatives [158]. However, the precise roles of these inhibitors in diabetic complications remain unclear. Further investigations are warranted to elucidate their impact on mitigating the effects of diabetes-related pathologies.
4.2. Indirect inhibition
4.2.1. Proinsulin C-peptide
Human proinsulin C-peptide, a 31-amino acid peptide, is released from pancreatic β-cells into the portal circulation in equimolar concentrations with insulin [159]. Discovered in 1967, C-peptide serves as an indicator of β-cell function. Since the early 1990s, C-peptide has been recognized for its potential benefits in treating diabetic microvascular and macrovascular complications [3, 160] (Table 4).
Therapeutic agents for diabetic complications.
| Inhibitor | Diabetic complication | Study models | Delivery system |
|---|---|---|---|
| Human C-peptide | Retinopathy | HRECs [24], type 1 diabetic mice [24, 71] | Intravitreal injection [24], Osmotic pumps [71] |
| Pulmonopathy | HPMECs [4], type 1 diabetic mice [4, 78] | Osmotic pumps [4, 78] | |
| Nephropathy | Type 1 diabetic mice [27], type 1 diabetic patients [182] | Osmotic pumps [27], subcutaneous injection [182] | |
| Cardiovascular disease | HAECs [65], type 1 diabetic mice [27, 65] | Osmotic pumps [27, 65], | |
| Peripheral neuropathy | Type 1 diabetic patients [182, 183] | Subcutaneous injection [182, 183] | |
| PEG-C-peptide | Peripheral neuropathy | Type 1 diabetic mice [184], type 1 diabetic patients [165, 185] | Subcutaneous injection [165, 184, 185] |
| K9-C-peptide | Retinopathy | HRECs [28], type 1 diabetic mice [28, 74] | Intravitreal injection [74], subcutaneous injection [28] |
| Pulmonopathy | Type 1 diabetic mice [28] | Subcutaneous injection [28] | |
| Nephropathy | Type 1 diabetic mice [28] | Subcutaneous injection [28] | |
| Cardiovascular disease | Type 1 diabetic mice [167] | Subcutaneous injection [167] | |
| Midazolam | Retinopathy | HRECs [77], type 1 diabetic mice [77] | Intravitreal injection [77] |
| Pulmonopathy | HPMECs [89], type 1 diabetic mice [89] | Subcutaneous injection [89] | |
| Nephropathy | HGMECs [93], type 1 diabetic mice [93] | Subcutaneous injection [93] |
HAECs, human aortic endothelial cells; HGMECs, human glomerular microvascular endothelial cells; HPMECs, human pulmonary microvascular endothelial cells; HRECs, human retinal endothelial cells; K9-C-peptide, human C-peptide conjugated with nine repeats of lysine-containing elastin-like polypeptide; PEG-C-peptide, PEGylated human C-peptide.
C-peptide exhibits a reduction in ROS generation by attenuating NADPH oxidase activation in human aortic endothelial cells [161], potentially mediated through binding to G protein-coupled receptor 146 [162]. In various human endothelial cells (retina, lung, aorta, and umbilical vein), C-peptide inhibits ROS-mediated TGase2 activation, preventing high glucose- or VEGF-induced VE-cadherin disassembly and endothelial permeability [4, 24, 27, 65]. C-peptide replacement ameliorates hyperglycemia-induced vascular dysfunction in the aorta [27, 65], heart [27], renal cortex [27], retina [24], and lung [4, 78], highlighting its beneficial impact on diabetic microvascular and macrovascular complications. Additionally, systemic C-peptide supplementation is effective in preventing impaired wound healing by stimulating angiogenesis and inhibiting inflammation [163]. Furthermore, C-peptide improves renal dysfunction, such as hyperfiltration and albumin excretion, and alleviates autonomic and sensory nerve dysfunction in patients and animal models with type 1 diabetes [159, 164].
A drawback of C-peptide in clinical applications is its short circulating half-time (approximately 30 min). However, two approaches-PEGylation and conjugation with a thermosensitive biopolymer-have addressed this limitation. Wahren and colleagues [165] reported a PEGylated C-peptide, extending the half-life of C-peptide into 6-7 days, and applied this C-peptide to a clinical trial involving 250 patients with type 1 diabetes and peripheral neuropathy. Results indicate that subcutaneous administration of this PEGylated C-peptide for 52 weeks improves the vibration perception threshold but does not improve sural nerve conduction velocity, suggesting that C-peptide may not fully restore hyperglycemia-induced neuronal damage. Ha and colleagues [166] designed a controlled releasable human C-peptide, K9-C-peptide, by recombinantly conjugating human C-peptide with a lysine-containing elastin-like polypeptide. Subcutaneous injection of K9-C-peptide gradually releases human C-peptide from a hydrogel depot into circulation for 19 days, ameliorating aortic dysfunction in diabetic mice [167]. K9-C-peptide simultaneously attenuates hyperglycemia-induced retinal, pulmonary, and glomerular dysfunctions in type 1 diabetic mice [28]. Moreover, intravitreal injection of K9-C-peptide maintains physiological C-peptide levels in the intraocular space for at least 56 days, normalizing diabetic retinal neovascularization [74].
4.2.2. Midazolam
Midazolam, a short-acting benzodiazepine medication widely employed for anesthesia, procedural sedation, and anxiolysis [168], demonstrates notable benefits against diabetic complications, including DR [77], DP [89], and DN [93]. This is achieved by inhibiting VEGF-induced elevation of intracellular Ca2+, subsequent ROS generation, and TGase2 activation in a GAVAA receptor-dependent manner (Table 4).
In the retina of type 1 diabetic mice, intravitreal injection of midazolam mitigates hyperglycemia-induced vascular leakage by inhibiting ROS-mediated TGase2 activation. These inhibitory effects are reversed with flumazenil, a GAVAA receptor antagonist [77]. Subcutaneous injection of midazolam attenuates hyperglycemia-induced microvascular leakage and cancer metastasis by inhibiting TGase2-mediated VE-cadherin disassembly in the lungs of type 1 diabetic mice [89]. Moreover, subcutaneous injection of midazolam in type 1 diabetic mice improves hyperglycemia-induced glomerular dysfunction, such as pathological alterations in glomerular ultrastructure and renal fibrosis.
While midazolam holds promise as a potential therapy for diabetic complications, the optimization of dosage and delivery routes is imperative to minimize potential side effects, including allergenic effects and dose-related respiratory depression. Careful consideration of these factors is essential for harnessing the full therapeutic potential of midazolam in mitigating the diverse complications associated with diabetes.
4.2.3. Other ligands
A couple of ligands, including pigment epithelium-derived factor (PEDF), norrin, and somatostatin, have potential for therapeutic intervention of diabetic vascular complications, although their action mechanisms involving TGase2 remains unclear. PEDF is a member of the serine protease inhibitor superfamily with multiple biological functions and is known as an anti-angiogenic and neurotropic factor [169]. PEDF binds to its multiple receptors, including laminin receptor, F1-ATP synthase, and low-density lipoprotein receptor-related protein 6, and exerts protective effects against diabetic microvascular complications by inhibiting oxidative stress and inflammation in the retina and kidney. [74, 170].
Norrin, a secreted 131-amino acid protein, utilizes the Wnt signaling pathway by binding to the frizzled class receptor 4 and the low-density lipoprotein receptor-related protein 5/6 co-receptor [171]. Norrin attenuated hyperglycemia-induced vascular leakage by restoring disrupted blood-retinal barrier properties in diabetic retinas [171, 172]. Somatostatin, which is a neuroprotective peptide, also ameliorated hyperglycemia-induced inflammation and neurodegeneration in the retina of diabetic rodent models [173, 174]. However, further investigations to elucidate the underlying mechanisms of their beneficial effects can warrant clinical application for diabetic complications.
5. Conclusion and future perspectives
TGase2 stands as a multifunctional enzyme with transamidase, serine/threonine kinase, disulfide isomerase, and GTPase activities. Its non-enzymatic functions involve interacting with extracellular proteins, enhancing its role across various diseases, encompassing neurodegenerative disorders, pulmonary and kidney fibrosis, tumor initiation and progression, inflammatory conditions, and diabetic complications. This comprehensive review underscores TGase2's pivotal involvement in the pathogenesis of diabetic complications, including DR, DP, DN, CVD, and HGM. The proposed therapeutic strategies involve TGase2 inhibition, offering a promising avenue for managing diabetic complications.
Two primary approaches exist for TGase2 inhibition: direct and indirect. The direct inhibition strategy encompasses three groups-competitive, reversible, and irreversible inhibitors-each strategically impeding substrate binding to the TGase2 active site. Additionally, indirect TGase2 inhibition involves ligands binding to specific receptors, orchestrating TGase2 modulation through intricate intracellular signaling pathways. While both approaches hold considerable promise for treating diabetic complications, their clinical viability hinges on meticulous assessments of safety, efficacy, and possible adverse effects.
Direct TGase2 inhibitors exhibit distinct potential, with cystamine and its reduced form, cysteamine, emerging as particularly promising candidates. These amine inhibitors showcase compelling protective effects in preclinical studies, specifically ameliorating diabetic vascular dysfunctions in various organs, including the aorta, eye, lung, and kidney. Notably, these inhibitors have undergone clinical trials, demonstrating safety in conditions such as cystinosis and neurodegenerative disorders [120, 175].
Indirect TGase2 inhibition via ligands presents an attractive, comparatively safe avenue for treating diabetic complications. The proinsulin C-peptide stands out as an encouraging candidate, demonstrating beneficial effects against diabetic microvascular and macrovascular dysfunctions in both type 1 diabetic patients and animal models. The inherent endogeneity of human C-peptide secreted from pancreatic β-cells suggests minimal side effects. Further potential enhancements through PEGylation or conjugation with a thermosensitive polymer open avenues for extending its therapeutic half-life. Although human C-peptide may not fully address diabetic peripheral neuropathy, rigorous clinical trials are imperative to evaluate its efficacy in mitigating diabetic vascular complications. Similarly, midazolam, a short-acting benzodiazepine, shows promise, especially in treating DR through localized delivery. However, optimizing dosage and delivery routes and ensuring safety warrant exploration through clinical studies.
Clinical utilization of TGase2 inhibitors for diabetic complications necessitates an intricate understanding of TGase2's four enzymatic activities in the pathogenesis of hyperglycemia-induced vascular and neuronal dysfunctions. TGase2 transamidase activity orchestrates complex interactions with key proteins, including RhoA, NFκB, GAPDH, and E-cadherin, resulting in actin cytoskeleton rearrangement, inflammation, and disrupted energy metabolism, which are crucial factors in hyperglycemia-induced vascular dysfunctions. The TGase2 kinase activity, with its extensive substrate phosphorylation repertoire including NFκB, pRB, E-cadherin, and p53 oncoprotein, demands further study to elucidate its specific functions in diabetic complications. A nuanced understanding of the reciprocal regulation of vascular dysfunction-associated proteins by TGase2 transamidase and kinase activities introduces a compelling layer to the investigation. Furthermore, untangling the functions of TGase2 disulfide isomerase and GTPase activities introduces complexity, urging further investigations to unravel their roles in the pathogenesis of diabetic complications. The challenge and excitement lie in deciphering these intricate facets, expanding our understanding, and paving the way for potential therapeutic interventions that could transform the landscape of diabetic care.
Acknowledgements
The author thanks Chan-Hee Moon and Sung-Hoon Jung for the support in preparing figures.
Funding
This work was supported by a grant from the National Research Foundation of Korea (2021R1A2C2091794).
Author Contributions
K.-S.H. is the guarantor of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Bhatt MP, Lim YC, Ha KS. C-peptide replacement therapy as an emerging strategy for preventing diabetic vasculopathy. Cardiovascular Research. 2014;104:234-44
2. Harding JL, Pavkov ME, Magliano DJ, Shaw JE, Gregg EW. Global trends in diabetes complications: a review of current evidence. Diabetologia. 2019;62:3-16
3. Jeon HY, Lee AJ, Ha KS. Polymer-Based Delivery of Peptide Drugs to Treat Diabetes: Normalizing Hyperglycemia and Preventing Diabetic Complications. Biochip Journal. 2022;16:111-27
4. Jeon HY, Lee YJ, Kim YS, Kim SY, Han ET, Park WS. et al. Proinsulin C-peptide prevents hyperglycemia-induced vascular leakage and metastasis of melanoma cells in the lungs of diabetic mice. FASEB J. 2019;33:750-62
5. Forbes JM, Cooper ME. Mechanisms of diabetic complications. Physiol Rev. 2013;93:137-88
6. Zaykov AN, Mayer JP, DiMarchi RD. Pursuit of a perfect insulin. Nat Rev Drug Discov. 2016;15:425-39
7. Engerman RL, Kern TS. Progression of incipient diabetic retinopathy during good glycemic control. Diabetes. 1987;36:808-12
8. Giacco F, Brownlee M. Oxidative Stress and Diabetic Complications. Circulation Research. 2010;107:1058-70
9. Paneni F, Mocharla P, Akhmedov A, Costantino S, Osto E, Volpe M. et al. Gene Silencing of the Mitochondrial Adaptor p66(Shc) Suppresses Vascular Hyperglycemic Memory in Diabetes. Circulation Research. 2012;111:278-89
10. Pirola L, Balcerczyk A, Okabe J, El-Osta A. Epigenetic phenomena linked to diabetic complications. Nature Reviews Endocrinology. 2010;6:665-75
11. Park D, Choi SS, Ha KS. Transglutaminase 2: a multi-functional protein in multiple subcellular compartments. Amino Acids. 2010;39:619-31
12. Gundemir S, Colak G, Tucholski J, Johnson GV. Transglutaminase 2: a molecular Swiss army knife. Biochim Biophys Acta. 2012;1823:406-19
13. Lai TS, Lin CJ, Wu YT, Wu CJ. Tissue transglutaminase (TG2) and mitochondrial function and dysfunction. Front Biosci (Landmark Ed). 2017;22:1114-37
14. Jung SH, Jeon HY, Lee SH, Han ET, Park WS, Hong SH. et al. On-chip dual enzyme activity assay to investigate regulation of the transamidase and kinase activities of transglutaminase 2. Anal Chim Acta. 2018;1027:92-100
15. Furini G, Verderio EAM. Spotlight on the Transglutaminase 2-Heparan Sulfate Interaction. Med Sci (Basel). 2019 7
16. Tabolacci C, De Martino A, Mischiati C, Feriotto G, Beninati S. The Role of Tissue Transglutaminase in Cancer Cell Initiation, Survival and Progression. Med Sci (Basel). 2019 7
17. Caccamo D, Curro M, Ientile R. Potential of transglutaminase 2 as a therapeutic target. Expert Opinion on Therapeutic Targets. 2010;14:989-1003
18. Reif S, Lerner A. Tissue transglutaminase-the key player in celiac disease: a review. Autoimmun Rev. 2004;3:40-5
19. Min B, Chung KC. New insight into transglutaminase 2 and link to neurodegenerative diseases. Bmb Reports. 2018;51:5-13
20. Huang L, Xu AM, Liu W. Transglutaminase 2 in cancer. Am J Cancer Res. 2015;5:2756-76
21. Olsen KC, Sapinoro RE, Kottmann RM, Kulkarni AA, Iismaa SE, Johnson GV. et al. Transglutaminase 2 and its role in pulmonary fibrosis. Am J Respir Crit Care Med. 2011;184:699-707
22. Kim SY. Transglutaminase 2 in inflammation. Front Biosci. 2006;11:3026-35
23. Shinde AV, Frangogiannis NG. Tissue transglutaminase in the pathogenesis of heart failure. Cell Death Differ. 2017;25:453-6
24. Lee YJ, Jung SH, Kim SH, Kim MS, Lee S, Hwang J. et al. Essential Role of Transglutaminase 2 in Vascular Endothelial Growth Factor-Induced Vascular Leakage in the Retina of Diabetic Mice. Diabetes. 2016;65:2414-28
25. Lee YJ, Jung SH, Hwang J, Jeon S, Han ET, Park WS. et al. Cysteamine prevents vascular leakage through inhibiting transglutaminase in diabetic retina. J Endocrinol. 2017;235:39-48
26. Jeon HY, Seo JA, Jung SH, Lee YJ, Han ET, Park WS. et al. Insulin prevents pulmonary vascular leakage by inhibiting transglutaminase 2 in diabetic mice. Life Sciences. 2019;233:116711
27. Bhatt MP, Lim YC, Hwang J, Na S, Kim YM, Ha KS. C-Peptide Prevents Hyperglycemia-Induced Endothelial Apoptosis Through Inhibition of Reactive Oxygen Species-Mediated Transglutaminase 2 Activation. Diabetes. 2013;62:243-53
28. Jeon HY, Moon CH, Kim EB, Sayyed ND, Lee AJ, Ha KS. Simultaneous attenuation of hyperglycemic memory-induced retinal, pulmonary, and glomerular dysfunctions by proinsulin C-peptide in diabetes. BMC Med. 2023;21:49
29. Bhatt MP, Lim YC, Kim YM, Ha KS. C-peptide activates AMPKalpha and prevents ROS-mediated mitochondrial fission and endothelial apoptosis in diabetes. Diabetes. 2013;62:3851-62
30. Jung SH, Kong DH, Jeon HY, Ji SH, Han ET, Park WS. et al. Identification of transglutaminase 2 kinase substrates using a novel on-chip activity assay. Biosens Bioelectron. 2016;82:40-8
31. Condello S, Cao L, Matei D. Tissue transglutaminase regulates beta-catenin signaling through a c-Src-dependent mechanism. FASEB J. 2013;27:3100-12
32. Jung SH, Kwon MH, Lee SH, Han ET, Park WS, Hong SH. et al. High-throughput investigation of transglutaminase 2 kinase regulation using a novel cysteine-modified peptide array. Anal Biochem. 2018;559:62-70
33. Jung SH, Lee K, Kong DH, Kim WJ, Kim YM, Ha KS. Integrative proteomic profiling of protein activity and interactions using protein arrays. Mol Cell Proteomics. 2012;11:1167-76
34. Pinkas DM, Strop P, Brunger AT, Khosla C. Transglutaminase 2 undergoes a large conformational change upon activation. Plos Biology. 2007;5:2788-96
35. Pietsch M, Wodtke R, Pietzsch J, Loser R. Tissue transglutaminase: An emerging target for therapy and imaging. Bioorganic & Medicinal Chemistry Letters. 2013;23:6528-43
36. Kiraly R, Csosz E, Kurtan T, Antus S, Szigeti K, Simon-Vecsei Z. et al. Functional significance of five noncanonical Ca2+-binding sites of human transglutaminase 2 characterized by site-directed mutagenesis. FEBS J. 2009;276:7083-96
37. Siegel M, Khosla C. Transglutaminase 2 inhibitors and their therapeutic role in disease states. Pharmacology & Therapeutics. 2007;115:232-45
38. Kim GE, Park HH. Structures of Human Transglutaminase 2: Finding Clues for Interference in Cross-linking Mediated Activity. Int J Mol Sci. 2020 21
39. Bernassola F, Rossi A, Melino G. Regulation of transglutaminases by nitric oxide. Ann N Y Acad Sci. 1999;887:83-91
40. Zhuang R, Khosla C. Substrates, inhibitors, and probes of mammalian transglutaminase 2. Anal Biochem. 2020;591:113560
41. Jung SH, Kwon MH, Han ET, Park WS, Hong SH, Kim YM. et al. Array-based Investigation of Amino Acids Responsible for Regulation of Transamidase and Kinase Activities of Transglutaminase 2. Biochip Journal. 2019 13
42. Lai TS, Davies C, Greenberg CS. Human tissue transglutaminase is inhibited by pharmacologic and chemical acetylation. Protein Science. 2010;19:229-35
43. Singh US, Kunar MT, Kao YL, Baker KM. Role of transglutaminase II in retinoic acid-induced activation of RhoA-associated kinase-2. EMBO J. 2001;20:2413-23
44. Lee J, Kim YS, Choi DH, Bang MS, Han TR, Joh TH. et al. Transglutaminase 2 induces nuclear factor-kappaB activation via a novel pathway in BV-2 microglia. J Biol Chem. 2004;279:53725-35
45. Cooper AJ, Sheu KR, Burke JR, Onodera O, Strittmatter WJ, Roses AD. et al. Transglutaminase-catalyzed inactivation of glyceraldehyde 3-phosphate dehydrogenase and alpha-ketoglutarate dehydrogenase complex by polyglutamine domains of pathological length. Proc Natl Acad Sci U S A. 1997;94:12604-9
46. Boehm JE, Singh U, Combs C, Antonyak MA, Cerione RA. Tissue transglutaminase protects against apoptosis by modifying the tumor suppressor protein p110 Rb. J Biol Chem. 2002;277:20127-30
47. Malkomes P, Lunger I, Oppermann E, Abou-El-Ardat K, Oellerich T, Gunther S. et al. Transglutaminase 2 promotes tumorigenicity of colon cancer cells by inactivation of the tumor suppressor p53. Oncogene. 2021;40:4352-67
48. Singh US, Pan J, Kao YL, Joshi S, Young KL, Baker KM. Tissue transglutaminase mediates activation of RhoA and MAP kinase pathways during retinoic acid-induced neuronal differentiation of SH-SY5Y cells. J Biol Chem. 2003;278:391-9
49. Antonyak MA, Singh US, Lee DA, Boehm JE, Combs C, Zgola MM. et al. Effects of tissue transglutaminase on retinoic acid-induced cellular differentiation and protection against apoptosis. J Biol Chem. 2001;276:33582-7
50. Ruoppolo M, Orru S, Francese S, Caputo I, Esposito C. Structural characterization of transglutaminase-catalyzed cross-linking between glyceraldehyde 3-phosphate dehydrogenase and polyglutamine repeats. Protein Sci. 2003;12:170-9
51. Oliverio S, Amendola A, Di Sano F, Farrace MG, Fesus L, Nemes Z. et al. Tissue transglutaminase-dependent posttranslational modification of the retinoblastoma gene product in promonocytic cells undergoing apoptosis. Mol Cell Biol. 1997;17:6040-8
52. Mishra S, Melino G, Murphy LJ. Transglutaminase 2 kinase activity facilitates protein kinase A-induced phosphorylation of retinoblastoma protein. Journal of Biological Chemistry. 2007;282:18108-15
53. Mishra S, Saleh A, Espino PS, Davie JR, Murphy LJ. Phosphorylation of histones by tissue transglutaminase. Journal of Biological Chemistry. 2006;281:5532-8
54. Mishra S, Murphy LJ. Tissue transglutaminase has intrinsic kinase activity - Identification of transglutaminase 2 as an insulin-like growth factor-binding protein-3 kinase. Journal of Biological Chemistry. 2004;279:23863-8
55. Nurminskaya MV, Belkin AM. Cellular Functions of Tissue Transglutaminase. International Review of Cell and Molecular Biology, Vol 294. 2012;294:1-97
56. Wang Y, Ande SR, Mishra S. Phosphorylation of transglutaminase 2 (TG2) at serine-216 has a role in TG2 mediated activation of nuclear factor-kappa B and in the downregulation of PTEN. Bmc Cancer. 2012 12
57. Mishra S, Murphy LJ. Phosphorylation of transglutaminase 2 by PKA at Ser216 creates 14-3-3 binding sites. Biochemical and Biophysical Research Communications. 2006;347:1166-70
58. Mishra S, Murphy LJ. The p53 oncoprotein is a substrate for tissue transglutaminase kinase activity. Biochemical and Biophysical Research Communications. 2006;339:726-30
59. Hafner A, Bulyk ML, Jambhekar A, Lahav G. The multiple mechanisms that regulate p53 activity and cell fate. Nat Rev Mol Cell Biol. 2019;20:199-210
60. Jung SH, Ji SH, Han ET, Park WS, Hong SH, Kim YM. et al. Real-time monitoring of glucose-6-phosphate dehydrogenase activity using liquid droplet arrays and its application to human plasma samples. Biosens Bioelectron. 2016;79:930-7
61. Garrido C, Galluzzi L, Brunet M, Puig PE, Didelot C, Kroemer G. Mechanisms of cytochrome c release from mitochondria. Cell Death Differ. 2006;13:1423-33
62. Johnson CK. Calmodulin, conformational states, and calcium signaling. A single-molecule perspective. Biochemistry. 2006;45:14233-46
63. Jung SH, Kong DH, Park JH, Lee ST, Hyun J, Kim YM. et al. Rapid analysis of matrix metalloproteinase-3 activity by gelatin arrays using a spectral surface plasmon resonance biosensor. Analyst. 2010;135:1050-7
64. Kaszak I, Witkowska-Pilaszewicz O, Niewiadomska Z, Dworecka-Kaszak B, Ngosa Toka F, Jurka P. Role of Cadherins in Cancer-A Review. Int J Mol Sci. 2020 21
65. Lee JY, Lee YJ, Jeon HY, Han ET, Park WS, Hong SH. et al. The vicious cycle between transglutaminase 2 and reactive oxygen species in hyperglycemic memory-induced endothelial dysfunction. FASEB Journal. 2019;33:12655-67
66. Nishikawa T, Edelstein D, Du XL, Yamagishi S, Matsumura T, Kaneda Y. et al. Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature. 2000;404:787-90
67. Brownlee M. The pathobiology of diabetic complications: a unifying mechanism. Diabetes. 2005;54:1615-25
68. Antonetti DA, Silva PS, Stitt AW. Current understanding of the molecular and cellular pathology of diabetic retinopathy. Nature Reviews Endocrinology. 2021;17:195-206
69. Duh EJ, Sun JK, Stitt AW. Diabetic retinopathy: current understanding, mechanisms, and treatment strategies. Jci Insight. 2017;2:e93751
70. Wang W, Lo ACY. Diabetic Retinopathy: Pathophysiology and Treatments. International Journal of Molecular Sciences. 2018;19:1816
71. Lee AJ, Moon CH, Lee YJ, Jeon HY, Park WS, Ha KS. Systemic C-peptide supplementation ameliorates retinal neurodegeneration by inhibiting VEGF-induced pathological events in diabetes. FASEB J. 2023;37:e22763
72. Simo-Servat O, Hernandez C, Simo R. Diabetic Retinopathy in the Context of Patients with Diabetes. Ophthalmic Research. 2019;62:211-7
73. Crawford TN, Alfaro DV 3rd, Kerrison JB, Jablon EP. Diabetic retinopathy and angiogenesis. Curr Diabetes Rev. 2009;5:8-13
74. Moon CH, Lee AJ, Jeon HY, Kim EB, Ha KS. Therapeutic effect of ultra-long-lasting human C-peptide delivery against hyperglycemia-induced neovascularization in diabetic retinopathy. Theranostics. 2023;13:2424-38
75. Caldwell RB, Bartoli M, Behzadian MA, El-Remessy AE, Al-Shabrawey M, Platt DH. et al. Vascular endothelial growth factor and diabetic retinopathy: role of oxidative stress. Curr Drug Targets. 2005;6:511-24
76. Lim YC, Bhatt MP, Kwon MH, Park D, Lee S, Choe J. et al. Prevention of VEGF-mediated microvascular permeability by C-peptide in diabetic mice. Cardiovascular Research. 2014;101:155-64
77. Lee YJ, Kim M, Lee JY, Jung SH, Jeon HY, Lee SA. et al. The benzodiazepine anesthetic midazolam prevents hyperglycemia-induced microvascular leakage in the retinas of diabetic mice. FASEB J. 2018;32:6089-99
78. Jeon H-Y, Lee A-J, Kim E-B, Kim M, Park WS, Ha K-S. C-peptide attenuates hyperglycemia-induced pulmonary fibrosis by inhibiting transglutaminase 2. Journal of Molecular Endocrinology. 2022;68:209-23
79. Talakatta G, Sarikhani M, Muhamed J, Dhanya K, Somashekar BS, Mahesh PA. et al. Diabetes induces fibrotic changes in the lung through the activation of TGF-beta signaling pathways. Sci Rep. 2018;8:11920
80. Khateeb J, Fuchs E, Khamaisi M. Diabetes and Lung Disease: A Neglected Relationship. Rev Diabet Stud. 2019;15:1-15
81. Pitocco D, Fuso L, Conte EG, Zaccardi F, Condoluci C, Scavone G. et al. The diabetic lung-a new target organ? Rev Diabet Stud. 2012;9:23-35
82. Bai L, Li Z, Pan TY, Wang W, Wang DA, Turner C. et al. Idiopathic pulmonary fibrosis and diabetes mellitus: a meta-analysis and systematic review. Respiratory Research. 2021;22:175
83. Jagadapillai R, Rane MJ, Lin X, Roberts AM, Hoyle GW, Cai L. et al. Diabetic Microvascular Disease and Pulmonary Fibrosis: The Contribution of Platelets and Systemic Inflammation. Int J Mol Sci. 2016 17
84. Wang DG, Ma Y, Tong X, Zhang YG, Fan H. Diabetes Mellitus Contributes to Idiopathic Pulmonary Fibrosis: A Review From Clinical Appearance to Possible Pathogenesis. Frontiers in Public Health. 2020;8:196
85. Kolahian S, Leiss V, Nurnberg B. Diabetic lung disease: fact or fiction? Rev Endocr Metab Disord. 2019;20:303-19
86. Ehrlich SF, Quesenberry CP Jr, Van Den Eeden SK, Shan J, Ferrara A. Patients diagnosed with diabetes are at increased risk for asthma, chronic obstructive pulmonary disease, pulmonary fibrosis, and pneumonia but not lung cancer. Diabetes Care. 2010;33:55-60
87. Richeldi L, Collard HR, Jones MG. Idiopathic pulmonary fibrosis. Lancet. 2017;389:1941-52
88. Hyldgaard C, Hilberg O, Bendstrup E. How does comorbidity influence survival in idiopathic pulmonary fibrosis? Respir Med. 2014;108:647-53
89. Seo JA, Jeon HY, Kim M, Lee YJ, Han ET, Park WS. et al. Anti-metastatic effect of midazolam on melanoma B16F10 cells in the lungs of diabetic mice. Biochem Pharmacol. 2020;178:114052
90. Wang G, Ouyang J, Lie S, Wang H, Lian BF, Liu ZH. et al. The analysis of risk factors for diabetic nephropathy progression and the construction of a prognostic database for chronic kidney diseases. Journal of Translational Medicine. 2019;17:264
91. Hussain S, Jamali MC, Habib A, Hussain MS, Akhtar M, Najmi AK. Diabetic kidney disease: An overview of prevalence, risk factors, and biomarkers. Clinical Epidemiology and Global Health. 2021;9:2-6
92. Said SM, Nasr SH. Silent diabetic nephropathy. Kidney International. 2016;90:24-6
93. Seo J-A, Sayyed ND, Lee Y-J, Jeon H-Y, Kim E-B, Hong S-H. et al. Midazolam ameliorates hyperglycemia-induced glomerular endothelial dysfunction by inhibiting transglutaminase 2 in diabetes. International Journal of Molecular Sciences. 2022;23:753
94. Bulow RD, Boor P. Extracellular Matrix in Kidney Fibrosis: More Than Just a Scaffold. J Histochem Cytochem. 2019;67:643-61
95. Stehouwer CDA. Microvascular Dysfunction and Hyperglycemia: A Vicious Cycle With Widespread Consequences. Diabetes. 2018;67:1729-41
96. Cai A, Chatziantoniou C, Calmont A. Vascular Permeability: Regulation Pathways and Role in Kidney Diseases. Nephron. 2021;145:297-310
97. Prat-Duran J, Pinilla E, Norregaard R, Simonsen U, Buus NH. Transglutaminase 2 as a novel target in chronic kidney disease-Methods, mechanisms and pharmacological inhibition. Pharmacology & Therapeutics. 2021;222:107787
98. Shweke N, Boulos N, Jouanneau C, Vandermeersch S, Melino G, Dussaule JC. et al. Tissue transglutaminase contributes to interstitial renal fibrosis by favoring accumulation of fibrillar collagen through TGF-beta activation and cell infiltration. Am J Pathol. 2008;173:631-42
99. Regassa LD, Tola A, Ayele Y. Prevalence of Cardiovascular Disease and Associated Factors Among Type 2 Diabetes Patients in Selected Hospitals of Harari Region, Eastern Ethiopia. Frontiers in Public Health. 2021;8:532719
100. Verges B. Cardiovascular disease in type 1 diabetes: A review of epidemiological data and underlying mechanisms. Diabetes Metab. 2020;46:442-9
101. Einarson TR, Acs A, Ludwig C, Panton UH. Prevalence of cardiovascular disease in type 2 diabetes: a systematic literature review of scientific evidence from across the world in 2007-2017. Cardiovascular Diabetology. 2018;17:83
102. Schnell O, Cappuccio F, Genovese S, Standl E, Valensi P, Ceriello A. Type 1 diabetes and cardiovascular disease. Cardiovasc Diabetol. 2013;12:156
103. Groop PH, Thomas MC, Moran JL, Waden J, Thorn LM, Makinen VP. et al. The presence and severity of chronic kidney disease predicts all-cause mortality in type 1 diabetes. Diabetes. 2009;58:1651-8
104. Al-U'datt D GF, Tranchant CC, Al-Dwairi A, Alqudah M, Al-Shboul O, Hiram R. et al. Implications of enigmatic transglutaminase 2 (TG2) in cardiac diseases and therapeutic developments. Biochem Pharmacol. 2022;201:115104
105. Iismaa SE, Mearns BM, Lorand L, Graham RM. Transglutaminases and Disease: Lessons From Genetically Engineered Mouse Models and Inherited Disorders. Physiological Reviews. 2009;89:991-1023
106. Szondy Z, Korponay-Szabo I, Kiraly R, Sarang Z, Tsay GJ. Transglutaminase 2 in human diseases. Biomedicine (Taipei). 2017;7:15
107. Nathan DM. Realising the long-term promise of insulin therapy: the DCCT/EDIC study. Diabetologia. 2021;64:1049-58
108. Holman RR, Paul SK, Bethel MA, Matthews DR, Neil HA. 10-year follow-up of intensive glucose control in type 2 diabetes. N Engl J Med. 2008;359:1577-89
109. King P, Peacock I, Donnelly R. The UK prospective diabetes study (UKPDS): clinical and therapeutic implications for type 2 diabetes. Br J Clin Pharmacol. 1999;48:643-8
110. Paneni F, Volpe M, Luscher TF, Cosentino F. SIRT1, p66(Shc), and Set7/9 in Vascular Hyperglycemic Memory. Diabetes. 2013;62:1800-7
111. Bhatt MP, Lee YJ, Jung SH, Kim YH, Hwang JY, Han ET. et al. C-peptide protects against hyperglycemic memory and vascular endothelial cell apoptosis. J Endocrinol. 2016;231:97-108
112. Ihnat MA, Thorpe JE, Kamat CD, Szabo C, Green DE, Warnke LA. et al. Reactive oxygen species mediate a cellular 'memory' of high glucose stress signalling. Diabetologia. 2007;50:1523-31
113. Lee YJ, Jeon HY, Lee AJ, Kim M, Ha KS. Dopamine ameliorates hyperglycemic memory-induced microvascular dysfunction in diabetic retinopathy. FASEB J. 2022;36:e22643
114. Lone A, Harris RA, Singh O, Betts DH, Cumming RC. p66Shc activation promotes increased oxidative phosphorylation and renders CNS cells more vulnerable to amyloid beta toxicity. Sci Rep. 2018;8:17081
115. Yang SH, Shin SJ, Oh JE, Jin JZ, Chung NH, Lim CS. et al. The protective role of uteroglobin through the modulation of tissue transglutaminase in the experimental crescentic glomerulonephritis. Nephrol Dial Transplant. 2008;23:3437-45
116. Keillor JW, Apperley KY, Akbar A. Inhibitors of tissue transglutaminase. Trends Pharmacol Sci. 2015;36:32-40
117. Besouw M, Masereeuw R, van den Heuvel L, Levtchenko E. Cysteamine: an old drug with new potential. Drug Discov Today. 2013;18:785-92
118. Schwimmer JB, Lavine JE, Wilson LA, Neuschwander-Tetri BA, Xanthakos SA, Kohli R. et al. In Children With Nonalcoholic Fatty Liver Disease, Cysteamine Bitartrate Delayed Release Improves Liver Enzymes but Does Not Reduce Disease Activity Scores. Gastroenterology. 2016;151:1141-54 e9
119. Arbez N, Roby E, Akimov S, Eddings C, Ren M, Wang X. et al. Cysteamine Protects Neurons from Mutant Huntingtin Toxicity. J Huntingtons Dis. 2019;8:129-43
120. Paul BD, Snyder SH. Therapeutic Applications of Cysteamine and Cystamine in Neurodegenerative and Neuropsychiatric Diseases. Front Neurol. 2019;10:1315
121. Liu RTL, Tsai TF, Lai YJ, Ng CY. Efficacy and safety of cysteamine-isobionicamide complex in postinflammatory hyperpigmentation: A 16-week, randomized, double-blinded, vehicle-controlled trial. Dermatol Sin. 2023 41
122. Langman CB. Long-term clinical benefits of delayed-release cysteamine bitartrate capsules in patients with nephropathic cystinosis (response to "A comparison of immediate release and delayed release cysteamine in 17 patients with nephropathic cystinosis"). Orphanet J Rare Dis. 2023;18:162
123. Popova E. Role of dopamine in distal retina. J Comp Physiol A Neuroethol Sens Neural Behav Physiol. 2014;200:333-58
124. Aung MH, Park HN, Han MK, Obertone TS, Abey J, Aseem F. et al. Dopamine Deficiency Contributes to Early Visual Dysfunction in a Rodent Model of Type 1 Diabetes. J Neurosci. 2014;34:726-36
125. Kim MK, Aung MH, Mees L, Olson DE, Pozdeyev N, Iuvone PM. et al. Dopamine Deficiency Mediates Early Rod-Driven Inner Retinal Dysfunction in Diabetic Mice. Invest Ophthalmol Vis Sci. 2018;59:572-81
126. Kwon MH, Jung JW, Jung SH, Park JY, Kim YM, Ha KS. Quantitative and rapid analysis of transglutaminase activity using protein arrays in mammalian cells. Mol Cells. 2009;27:337-43
127. Kwon MH, Jung SH, Kim YM, Ha KS. Simultaneous activity assay of two transglutaminase isozymes, blood coagulation factor XIII and transglutaminase 2, by use of fibrinogen arrays. Anal Chem. 2011;83:8718-24
128. Kim SW, Lee ZW, Lee C, Im KS, Ha KS. The role of tissue transglutaminase in the germinal vesicle breakdown of mouse oocytes. Biochem Biophys Res Commun. 2001;286:229-34
129. Lee ZW, Kwon SM, Kim SW, Yi SJ, Kim YM, Ha KS. Activation of in situ tissue transglutaminase by intracellular reactive oxygen species. Biochem Biophys Res Commun. 2003;305:633-40
130. Zitka O, Skalickova S, Gumulec J, Masarik M, Adam V, Hubalek J. et al. Redox status expressed as GSH:GSSG ratio as a marker for oxidative stress in paediatric tumour patients. Oncol Lett. 2012;4:1247-53
131. Jokay I, Kelemenics K, Gyuris A, Minarovits J. S-methylthio-cysteine and cystamine are potent stimulators of thiol production and glutathione synthesis. Life Sci. 1998;62:PL27-33
132. Paul BD, Snyder SH. Therapeutic Applications of Cysteamine and Cystamine in Neurodegenerative and Neuropsychiatric Diseases. Frontiers in Neurology. 2019 10
133. Agostinelli E. Polyamines and transglutaminases: future perspectives. Amino Acids. 2016;48:2273-81
134. Kambis TN, Tofilau HMN, Gawargi FI, Chandra S, Mishra PK. Regulating Polyamine Metabolism by miRNAs in Diabetic Cardiomyopathy. Curr Diab Rep. 2021;21:52
135. Kulkarni A, Anderson CM, Mirmira RG, Tersey SA. Role of Polyamines and Hypusine in β Cells and Diabetes Pathogenesis. Metabolites. 2022 12
136. Fernandez-Garcia JC, Delpino-Rius A, Samarra I, Castellano-Castillo D, Muñoz-Garach A, Bernal-Lopez MR. et al. Type 2 Diabetes Is Associated with a Different Pattern of Serum Polyamines: A Case-Control Study from the PREDIMED-Plus Trial. J Clin Med. 2019 8
137. Song M, Hwang H, Im CY, Kim SY. Recent Progress in the Development of Transglutaminase 2 (TGase2) Inhibitors. J Med Chem. 2017;60:554-67
138. Folk JE, Cole PW. Mechanism of action of guinea pig liver transglutaminase. I. Purification and properties of the enzyme: identification of a functional cysteine essential for activity. J Biol Chem. 1966;241:5518-25
139. Pardin C, Gillet SM, Keillor JW. Synthesis and evaluation of peptidic irreversible inhibitors of tissue transglutaminase. Bioorg Med Chem. 2006;14:8379-85
140. Yang C, Hu S, Pan X, Yang K, Zhang K, Liu Q. et al. Novel Synthesis of Dihydroisoxazoles by p-TsOH-Participated 1,3-Dipolar Cycloaddition of Dipolarophiles withalpha-Nitroketones. Molecules. 2023 28
141. Lopes EF, Penteado F, Thurow S, Pinz M, Reis AS, Wilhelm EA. et al. Synthesis of Isoxazolines by the Electrophilic Chalcogenation of beta,gamma-Unsaturated Oximes: Fishing Novel Anti-Inflammatory Agents. J Org Chem. 2019;84:12452-62
142. Yuan L, Siegel M, Choi K, Khosla C, Miller CR, Jackson EN. et al. Transglutaminase 2 inhibitor, KCC009, disrupts fibronectin assembly in the extracellular matrix and sensitizes orthotopic glioblastomas to chemotherapy. Oncogene. 2007;26:2563-73
143. Choi K, Siegel M, Piper JL, Yuan L, Cho E, Strnad P. et al. Chemistry and biology of dihydroisoxazole derivatives: selective inhibitors of human transglutaminase 2. Chem Biol. 2005;12:469-75
144. Kaur K, Kumar V, Sharma AK, Gupta GK. Isoxazoline containing natural products as anticancer agents: A review. Eur J Med Chem. 2014;77:121-33
145. Alshamari A, Al-Qudah M, Hamadeh F, Al-Momani L, Abu-Orabi S. Synthesis, Antimicrobial and Antioxidant Activities of 2-Isoxazoline Derivatives. Molecules. 2020 25
146. Griffin M, Mongeot A, Collighan R, Saint RE, Jones RA, Coutts IG. et al. Synthesis of potent water-soluble tissue transglutaminase inhibitors. Bioorg Med Chem Lett. 2008;18:5559-62
147. Hornigold N, Johnson TS, Huang LH, Haylor JL, Griffin M, Mooney A. Inhibition of collagen I accumulation reduces glomerulosclerosis by a Hic-5-dependent mechanism in experimental diabetic nephropathy. Lab Invest. 2013;93:553-65
148. Huang L, Haylor JL, Hau Z, Jones RA, Vickers ME, Wagner B. et al. Transglutaminase inhibition ameliorates experimental diabetic nephropathy. Kidney Int. 2009;76:383-94
149. Coto L, Mendia I, Sousa C, Bai JC, Cebolla A. Determination of gluten immunogenic peptides for the management of the treatment adherence of celiac disease: A systematic review. World J Gastroenterol. 2021;27:6306-21
150. Hausch F, Halttunen T, Maki M, Khosla C. Design, synthesis, and evaluation of gluten peptide analogs as selective inhibitors of human tissue transglutaminase. Chem Biol. 2003;10:225-31
151. Piper JL, Gray GM, Khosla C. High selectivity of human tissue transglutaminase for immunoactive gliadin peptides: implications for celiac sprue. Biochemistry. 2002;41:386-93
152. Lai TS, Slaughter TF, Peoples KA, Hettasch JM, Greenberg CS. Regulation of human tissue transglutaminase function by magnesium-nucleotide complexes. Identification of distinct binding sites for Mg-GTP and Mg-ATP. J Biol Chem. 1998;273:1776-81
153. Case A, Stein RL. Kinetic analysis of the interaction of tissue transglutaminase with a nonpeptidic slow-binding inhibitor. Biochemistry. 2007;46:1106-15
154. Pinilla E, Comerma-Steffensen S, Prat-Duran J, Rivera L, Matchkov VV, Buus NH. et al. Transglutaminase 2 Inhibitor LDN 27219 Age-Dependently Lowers Blood Pressure and Improves Endothelium-Dependent Vasodilation in Resistance Arteries. Hypertension. 2021;77:216-27
155. Pardin C, Pelletier JN, Lubell WD, Keillor JW. Cinnamoyl inhibitors of tissue transglutaminase. J Org Chem. 2008;73:5766-75
156. Lai TS, Liu Y, Tucker T, Daniel KR, Sane DC, Toone E. et al. Identification of chemical inhibitors to human tissue transglutaminase by screening existing drug libraries. Chem Biol. 2008;15:969-78
157. Klock C, Jin X, Choi K, Khosla C, Madrid PB, Spencer A. et al. Acylideneoxoindoles: a new class of reversible inhibitors of human transglutaminase 2. Bioorg Med Chem Lett. 2011;21:2692-6
158. Lee SH, Kim N, Kim SJ, Song J, Gong YD, Kim SY. Anti-cancer effect of a quinoxaline derivative GK13 as a transglutaminase 2 inhibitor. J Cancer Res Clin. 2013;139:1279-94
159. Wahren J, Kallas A, Sima AAF. The Clinical Potential of C-Peptide Replacement in Type 1 Diabetes. Diabetes. 2012;61:761-72
160. Chen J, Huang Y, Liu C, Chi J, Wang Y, Xu L. The role of C-peptide in diabetes and its complications: an updated review. Front Endocrinol (Lausanne). 2023;14:1256093
161. Cifarelli V, Geng X, Styche A, Lakomy R, Trucco M, Luppi P. C-peptide reduces high-glucose-induced apoptosis of endothelial cells and decreases NAD(P)H-oxidase reactive oxygen species generation in human aortic endothelial cells. Diabetologia. 2011;54:2702-12
162. Yosten GL, Kolar GR, Redlinger LJ, Samson WK. Evidence for an interaction between proinsulin C-peptide and GPR146. J Endocrinol. 2013;218:B1-8
163. Lim YC, Bhatt MP, Kwon MH, Park D, Na S, Kim YM. et al. Proinsulin C-peptide prevents impaired wound healing by activating angiogenesis in diabetes. J Invest Dermatol. 2015;135:269-78
164. Luppi P, Cifarelli V, Wahren J. C-peptide and long-term complications of diabetes. Pediatr Diabetes. 2011;12:276-92
165. Wahren J, Foyt H, Daniels M, Arezzo JC. Long-Acting C-Peptide and Neuropathy in Type 1 Diabetes: A 12-Month Clinical Trial. Diabetes Care. 2016;39:596-602
166. Jung SH, Lee JY, Lee SH, Kwon MH, Han ET, Park WS. et al. Preventive Effects of Thermosensitive Biopolymer-Conjugated C-Peptide against High Glucose-Induced Endothelial Cell Dysfunction. Macromol Biosci. 2019;19:e1900129
167. Lee AJ, Lee YJ, Jeon HY, Kim M, Han ET, Park WS. et al. Application of elastin-like biopolymer-conjugated C-peptide hydrogel for systemic long-term delivery against diabetic aortic dysfunction. Acta Biomater. 2020;118:32-43
168. Olkkola KT, Ahonen J. Midazolam and other benzodiazepines. Handb Exp Pharmacol. 2008:335-60
169. He X, Cheng R, Benyajati S, Ma JX. PEDF and its roles in physiological and pathological conditions: implication in diabetic and hypoxia-induced angiogenic diseases. Clin Sci (Lond). 2015;128:805-23
170. Wang Y, Liu X, Quan X, Qin X, Zhou Y, Liu Z. et al. Pigment epithelium-derived factor and its role in microvascular-related diseases. Biochimie. 2022;200:153-71
171. Diaz-Coranguez M, Lin CM, Liebner S, Antonetti DA. Norrin restores blood-retinal barrier properties after vascular endothelial growth factor-induced permeability. J Biol Chem. 2020;295:4647-60
172. Díaz-Coránguez M, González-González L, Wang A, Liu XW, Antonetti DA. Disheveled-1 Interacts with Claudin-5 and Contributes to Norrin-Induced Endothelial Barrier Restoration. Cells-Basel. 2023 12
173. Hernández C, Arroba AI, Bogdanov P, Ramos H, Simó-Servat O, Simó R. et al. Effect of Topical Administration of Somatostatin on Retinal Inflammation and Neurodegeneration in an Experimental Model of Diabetes. J Clin Med. 2020 9
174. Hernández C, García-Ramírez M, Corraliza L, Fernández-Carneado J, Farrera-Sinfreu J, Ponsati B. et al. Topical Administration of Somatostatin Prevents Retinal Neurodegeneration in Experimental Diabetes. Diabetes. 2013;62:2569-78
175. Cicchetti F, David LS, Siddu A, Denis HL. Cysteamine as a novel disease-modifying compound for Parkinson's disease: Over a decade of research supporting a clinical trial. Neurobiol Dis. 2019 130
176. Nakaoka H, Perez DM, Baek KJ, Das T, Husain A, Misono K. et al. Gh: a GTP-binding protein with transglutaminase activity and receptor signaling function. Science. 1994;264:1593-6
177. Achyuthan KE, Greenberg CS. Identification of a guanosine triphosphate-binding site on guinea pig liver transglutaminase. Role of GTP and calcium ions in modulating activity. J Biol Chem. 1987;262:1901-6
178. Lee KN, Arnold SA, Birckbichler PJ, Patterson MK Jr, Fraij BM, Takeuchi Y. et al. Site-directed mutagenesis of human tissue transglutaminase: Cys-277 is essential for transglutaminase activity but not for GTPase activity. Biochim Biophys Acta. 1993;1202:1-6
179. Chen S, Lin F, Iismaa S, Lee KN, Birckbichler PJ, Graham RM. Alpha1-adrenergic receptor signaling via Gh is subtype specific and independent of its transglutaminase activity. J Biol Chem. 1996;271:32385-91
180. Iwai K, Shibukawa Y, Yamazaki N, Wada Y. Transglutaminase 2-dependent deamidation of glyceraldehyde-3-phosphate dehydrogenase promotes trophoblastic cell fusion. J Biol Chem. 2014;289:4989-99
181. Liang ST, Chen C, Chen RX, Li R, Chen WL, Jiang GH. et al. Michael acceptor molecules in natural products and their mechanism of action. Front Pharmacol. 2022;13:1033003
182. Johansson BL, Borg K, Fernqvist-Forbes E, Kernell A, Odergren T, Wahren J. Beneficial effects of C-peptide on incipient nephropathy and neuropathy in patients with Type 1 diabetes mellitus. Diabet Med. 2000;17:181-9
183. Ekberg K, Brismar T, Johansson BL, Lindstrom P, Juntti-Berggren L, Norrby A. et al. C-Peptide replacement therapy and sensory nerve function in type 1 diabetic neuropathy. Diabetes Care. 2007;30:71-6
184. Jolivalt CG, Rodriguez M, Wahren J, Calcutt NA. Efficacy of a long-acting C-peptide analogue against peripheral neuropathy in streptozotocin-diabetic mice. Diabetes Obes Metab. 2015;17:781-8
185. Ekberg K, Brismar T, Johansson BL, Jonsson B, Lindstrom P, Wahren J. Amelioration of sensory nerve dysfunction by C-Peptide in patients with type 1 diabetes. Diabetes. 2003;52:536-41
Author contact
![]() Corresponding author: Department of Molecular and Cellular Biochemistry, Kangwon National University School of Medicine, Chuncheon, Kangwon-do 24341, Korea. Tel.: +82-33-250-8833; E-mail address: kshaac.kr.
Corresponding author: Department of Molecular and Cellular Biochemistry, Kangwon National University School of Medicine, Chuncheon, Kangwon-do 24341, Korea. Tel.: +82-33-250-8833; E-mail address: kshaac.kr.