Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Methods
Results
Discussion
Conclusions
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2024; 14(9):3760-3776. doi:10.7150/thno.95197 This issue Cite
Research Paper
Sleep deprivation causes gut dysbiosis impacting on systemic metabolomics leading to premature ovarian insufficiency in adolescent mice
Jiamao Yan1,2, Xiaoyuan Zhang1,2, Kexin Zhu1, Mubin Yu1, Qingchun Liu1, Massimo De Felici3, Teng Zhang2, ![]() , Junjie Wang1,
, Junjie Wang1, ![]() , Wei Shen1,
, Wei Shen1, ![]()
1. College of Life Sciences, Qingdao Agricultural University, Qingdao 266109, China.
2. State Key Laboratory of Reproductive Regulation and Breeding of Grassland Livestock, College of Life Sciences, Inner Mongolia University, Hohhot 010021, China.
3. Department of Biomedicine and Prevention, University of Rome Tor Vergata, Rome 00133, Italy.
Received 2024-2-8; Accepted 2024-6-6; Published 2024-6-17
Abstract

Rationale: Currently, there are occasional reports of health problems caused by sleep deprivation (SD). However, to date, there remains a lack of in-depth research regarding the effects of SD on the growth and development of oocytes in females. The present work aimed to investigate whether SD influences ovarian folliculogenesis in adolescent female mice.
Methods: Using a dedicated device, SD conditions were established in 3-week old female mice (a critical stage of follicular development) for 6 weeks and gut microbiota and systemic metabolomics were analyzed. Analyses were related to parameters of folliculogenesis and reproductive performance of SD females.
Results: We found that the gut microbiota and systemic metabolomics were severely altered in SD females and that these were associated with parameters of premature ovarian insufficiency (POI). These included increased granulosa cell apoptosis, reduced numbers of primordial follicles (PmFs), correlation with decreased AMH, E2, and increased LH in blood serum, and a parallel increased number of growing follicles and changes in protein expression compatible with PmF activation. SD also reduced oocyte maturation and reproductive performance. Notably, fecal microbial transplantation from SD females into normal females induced POI parameters in the latter while niacinamide (NAM) supplementation alleviated such symptoms in SD females.
Conclusion: Gut microbiota and alterations in systemic metabolomics caused by SD induced POI features in juvenile females that could be counteracted with NAM supplementation.
Keywords: Sleep deprivation, Premature ovarian insufficiency, Gut microbiota, Niacinamide metabolism, Mouse
Introduction
Sleep plays a vital role in human physical and mental health. A lack of high-quality sleep weakens bodily defenses against disease and medical conditions. Numerous studies have highlighted the negative health effects of acute and chronic sleep deprivation (SD), including cognitive developmental impairment, decline of creative ability, and disorders of the gut microbiota, endocrine system, and immune function, thereby inducing various physical and mental diseases and even death [1-9]. Meanwhile, reproductive disfunctions are also connected with SD. In animal models, the impaired secretion of sex hormones caused by SD is reported to decrease testosterone levels, reduced sperm motility, and cause apoptosis of Leydig cells in the testis [10]. In female mice, SD reduces the expression of uterine circadian genes and affects early pregnancy outcomes through inhibition of IL-6-dependent endometrium decidualization [7]. Furthermore, pregnancy and perinatal disorders are reported to be associated with insomnia in humans [11]. Moreover, sleep disturbances are increasingly recognized as important factors in women's reproductive health, particularly the menstrual cycle, pregnancy, and menopause [12, 13]. However, the effects of sleep deprivation on fertility in female animals is largely unknown.
Premature ovarian insufficiency (POI) is an infertility disorder characterized by cessation of ovarian function that affects 3.7% of women before the age of 40 [14, 15]. POI is a highly heterogeneous disease that may be a result of the following: a small pool of primordial follicles, impaired follicular development, follicle dysfunction, or premature follicle depletion due to accelerated atresia [16, 17]. The etiology of POI is diverse, and may be of genetic, iatrogenic, autoimmune, infectious environmental, or metabolic origin; these can be grouped into two categories: genetic and non-genetic causes [18]. For genetic aspects of POI, approximately 110 causative genes have been identified based on either a physiological role in ovaries or the phenotype of mutant mouse models. These include genes involved in DNA damage repair (Mcm9, Spidr), follicle development and maturation (Gdf9, Bmp15, Wt1, Cpeb1, and Foxl2), ovulation (Alox12), and hormones and receptors (Amh, Esr2, Fshr, and Lhcgr) [14, 17]. Among non-genetic causes for POI, the effects of metabolic disorders cannot be ignored. Accordingly, mutation of the GALT gene would impair the function of galactose-1-phosphate uridylotransferase, and trigger a metabolic change in galactose, leading to POI [19]. In addition, 17-hydroxylase deficiency can block synthesis of all 19-carbon steroids, causing POI [20]. In addition, gut microbiota dysbiosis, such as disturbance of core microbiota (Bacteroides and Lachnoclostridium) or an increase of opportunistic pathogenic bacteria, has been suggested to be connected with POI [21, 22]. Therefore, the pathogenesis of POI is relatively complex, and understanding its origins will be conducive to the clinical treatment of POI patients.
Recent studies indicate that sleep deprivation and disruption are associated with impaired reproductive function and poor reproductive outcomes in women [23]. Furthermore, hypothalamic-pituitary-adrenal axis (HPA) activation and suppression or augmentation of reproductive hormones and alteration of gut microbiota, are important ways in which, SD can affect female reproduction and fertility [12, 24]. Importantly, the gut microbiota creates a bridge between external environmental stimuli and host metabolism since it participates in the transformation and absorption of nutrients, and directly or indirectly impacts on a variety of tissue and organ functions. Studies have shown that the intestinal microbiota plays an important role in the occurrence and development of reproductive endocrine diseases, and is closely related to host health [25-27]. For example, studies have found that gut microbiome imbalance in a metabolic syndrome model significantly lowers bile acid levels and causes abnormal metabolism of vitamin A, which in turn affects testicular cells through circulation and impaired spermatogenesis [28]. Recently, it has been suggested that the gut microbiota is associated with metabolic disorders in women with PCOS, and that its modification alters bile acid metabolism and/or increases IL-22 levels which may be of value for the treatment of PCOS [29]. In addition, dysregulation of the gut flora leading to increased beta-glucuronidase activity, has been reported to elevate estrogen levels, leading to reproductive diseases and cancer [30].
In the present study, we established a mouse model of SD in adolescent female mice and analyzed the effects of SD on ovarian follicle dynamics and reproductive performance from the perspective of gut microbiota and systemic metabolism changes induced under this condition. Results indicated that SD was capable of changing gut microbiota and systemic metabolism, and these reduced female reproductive performance; this can be alleviated by dietary supplementation of NAM metabolite.
Methods
Animal treatment and sample collection
Three-week-old C57BL/6J female mice were purchased from Jinan Pengyue Experimental Animal Breeding Center (Jinan, China). They were housed under a standard 12-h light/12-h dark cycle at 23±1°C, 50%-60% humidity, and ad libitum food and water consumption for 3 days before experimental treatment. All procedures were approved by the Animal Ethics Committee of Qingdao Agricultural University (approval No. 2022-0021).
The mouse model of sleep deprivation (SD) was established for 20 h per day for 6 weeks by using the dedicated device KW-BD, purchased from NJKEW Biotechnology Co., Ltd (Nanjing, China). Briefly, a disturbance bar at the bottom of the SD device, rotates and slides at a constant frequency with a working speed of 1.0 r/min, moving under the mouse's body, thereby preventing sleep (for a detailed description of the instrument, see http://www.kw689.com/product/yd/info128.html). SD time was set from 18:00 to 14:00 h of the next day. Animals were weighed every 2 days and at the end of SD treatment blood samples were collected from the eye and the mice were sacrificed by neck dislocation. Ovaries, small intestine, and small intestinal contents (gut microbiota) were rapidly collected in a sterile environment, snap frozen in liquid nitrogen, and stored at -80°C for further analysis.
Fecal microbiota transplantation
Fecal microbiota transplantation (FMT) was performed as previously described [31, 32]. Briefly, small intestinal content was collected and thoroughly mixed with 20% glycerol at a ratio of 1:1, quick frozen in liquid nitrogen, and stored at -80°C. Before use, the sample was diluted with saline solution to a working concentration of 50 mg/ml and filtered through a 70 μm cell sieve. Each recipient mouse received 100 μL/d filtrate by oral gavage for 2 weeks followed by regular feeding for 3 weeks.
Niacinamide supplementation
Niacinamide (NAM) powder was stored and sealed at -20°C, to ensure the consistency of experimental results. NAM was diluted with water to the required concentration (100, 200, 400 mg/ml) before use, so that similar volumes were administered to mice daily at a working dose of 100, 200, or 400 mg/kg body weight. Intragastric NAM was administered for 6 weeks under SD conditions.
Histology and immunohistochemistry
The procedures for ovarian histology and immunohistochemistry (IHC) have been previously described in detail [33]. Briefly, ovaries were fixed overnight in paraformaldehyde (Solarbio, P1110), flushed with water, dehydrated with graded alcohols, and transferred into xylene before paraffin embedding. Ovaries were serially sectioned to a thickness of 5 µm and sections were placed onto slides for drying on a heated plate at 42°C. Samples were then left in an incubator at 60°C for 2 h and treated with xylene twice for 15 min each at room temperature (RT). After rehydration in graded alcohols, the sections were washed in PBS for 10 min, placed in citric acid antigen retrieval solution at 96°C for 10 min, and after cooling, sequentially treated with tris-buffered saline (TBS) and TBST (TBS with Tween-20) for 5 min. Samples were then treated with 3% H2O2 to remove endogenous peroxidase activity, reacted with BDT (TBS containing 3% BSA and 10% goat serum) at RT for 30 min, and incubated in primary antibodies overnight at 4°C. The next day, slides were incubated with a peroxidase-conjugated secondary antibody (Beyotime, A0208, Shanghai, China) at 37°C for 45 min. Finally, the samples were sequentially stained using a DAB kit (ZSGB-BIO, ZLI-9017, Beijing, China) and hematoxylin (Sangon Biotech, E607317), sealed with neutral resin, and observed under an Olympus BX51 microscope.
Starting from the middle section of the ovary, additional sections were selected every 15-20, both forward and backward, to give a total of 9 sections/ovary that were scored. Follicles were classified as i) primordial if they contained an oocyte surrounded by a single layer of flattened granulosa cells; ii) primary if they contained an oocyte surrounded by a single layer of flattened and cuboidal granulosa cells; iii) secondary if they contained an oocyte surrounded by 2 to 3 layers of cuboidal granulosa cells and no antral space; and iv) antral if they contained an oocyte surrounded by >3 layers of granulosa cells and an antral space. To avoid double-counting, only follicles containing oocytes with visible nuclei were considered [34].
Small intestine histology and immunofluorescence staining
Similar to ovarian tissues, small intestine tissues were fixed, embedded, and then continuously sectioned at 7 μm thickness. After treatment with xylene and alcohol, the samples were stained with hematoxylin and eosin (Solarbio, G1100) for histological observation.
For small intestine tissue immunofluorescence (IF) staining, slides carrying sections were sequentially immersed in xylene, alcohol, PBS, and sodium citrate buffers before staining. After cooling, the slides were reacted with BDT at RT for 30 min; subsequently, they were incubated with the primary antibody at 4°C overnight. The next day, the slides were incubated with secondary antibodies in a dark room at 37°C for 45 min, and then the nuclei were stained with Hoechst 33342 at RT for 5 min. Finally, each slide was sealed with an anti-fluorescence attenuation quencher. The slides were then viewed under a fluorescence microscope.
ELISA
The levels of NAM, FSH, LH, AMH, and E2 in blood serum samples were measured using dedicated enzyme-linked immunosorbent assay (ELISA) kits: JM-11773M2, JM-02838M2, JM-11607M2 (Jingmei Biological Technology, Jiangsu, China), and JB1334-Mu, JB973-Mu (Jinma Biotechnology, Shanghai, China), respectively, according to the manufacturer's instructions.
Western blotting
Ovarian tissues were lysed in appropriate amounts of RIPA (Beyotime, P0013C) and sodium dodecyl sulfate (SDS) buffers. The extracted proteins were denatured by boiling the samples for 5 min and then stored at -20°C. Electrophoresis was conducted using a BioRad-PowerPacBasic electrophoresis apparatus (BIO-RAD, Taiwan, China). Proteins were transferred from the gel to a poly (vinylidene fluoride) membrane (PVDF, Millipore, ISEQ00010) at 200 mA using the wet-transfer method. After blocking for 4 h in TBST containing 5% BSA, the membranes were incubated with primary antibodies overnight at 4°C. The next morning, after washing with TBST, they were incubated with horseradish peroxidase-conjugated secondary antibodies for 90 min at RT and target protein bands were detected using a BeyoECL Plus kit (Beyotime, P0018). Proteins were quantified using ImageJ software. The intensities of the protein bands were normalized to GAPDH, and the relative amounts of the target proteins compared to that of the control. The antibody information used is listed in Table S1.
TUNEL staining
Paraffin ovary sections were dewaxed with xylene and rehydrated with graded alcohols. TUNEL staining was conducted using a TUNEL BrightGreen Apoptosis Detection Kit (Vazyme, A113-02, Nanjing, China), following the manufacturer's instructions and counterstained with Hoechst 33342 (Beyotime, C1022) for 5 min. Finally, after the addition of anti-fluorescence quencher, the slides were sealed and stored in the dark at 4°C or directly observed under a microscope.
In vitro maturation and meiotic spindle staining of the oocytes
Fully-grown germinal vesicle (GV) oocytes were collected and cultured following standard protocols as described in previous papers [33]. The number of oocytes undergoing germinal vesicle break-down (GVBD) and first polar body extrusion (PBE) were counted after 4-6 h and 12-16 h, respectively. In order to visualize the meiotic spindle, oocytes were fixed in 4% paraformaldehyde for 30 min at RT, and permeabilizated in PBS supplemented with 0.5% Triton X-100 for 20 min. After blocking in PBS supplemented with 1% BSA for 1 h, the oocytes were incubated with α-tubulin antibody (Invitrogen, 322588) for 2 h and finally rinsed in washing buffer (PBS plus 0.1% Tween 20 and 0.01% Triton X-100) and stained with Hoechst 33342 for 15 min at RT. A laser-scanning confocal microscope (Leica TCS SP5 II, Mannheim, Germany) was used to observe and capture representative images.
Real-Time Quantitative PCR
Total RNA was extracted from ovarian tissue according to the instructions of a SPARKeasy Improved Tissue/Cell RNA Extraction Kit (Sparkjade, AC0202, Shandong, China), followed by reverse transcription using SPARKscript II RT Plus (Sparkjade, AG0304, Shandong, China). Real Time quantitative PCRs (RT-qPCRs) were conducted using a SYBR Premix Ex TaqTM fluorescent dye and CFX96 Real-Time PCR instruments (BioRad-CFX96, USA). The primers used are listed in the Table S2.
Single cell RNA sequencing (scRNA-seq)
Oocytes were individually lysed, and cDNA synthesis and amplification performed following the SMART-seq2 protocol. The cDNA libraries were then checked on a Bioanalyzer instrument (Agilent), and the passed samples were finally chosen for sequencing. Sequence libraries were constructed using Annoroad Gene Technology Corporation (Beijing, China), and were sequenced by Illumina NovaSeq 6000 using the 150 bp paired-end strategy. Oocyte samples from 3 different ovaries were mixed and treated as one replicate, with 2 replicates per group for scRNA-seq. The scRNA-seq data were treated with FastQC (v0.11.8) for quality control and then aligned with the reference genome sequence [35]. FPKM (fragment per kilobase of transcript per million mapped reads) values were calculated to quantify gene expression abundance. Differential expression RNA analysis was performed using DESeq2 software comparing 2 different groups [36]. Differentially expressed genes (DEGs) were identified with a parameter of false discovery rate (FDR) <0.05 and absolute fold change (FC) ≥2. Pathway enrichment analysis was performed using Metascape [37].
RNA sequencing (RNA-seq)
Total RNA was extracted from ovaries according to the manufacturer's instructions. High quality RNA was used for cDNA library construction and Illumina sequencing by Annoroad Gene Technology Corporation (Beijing, China). Firstly, FastQC (v0.11.8) was used to check the quality of sequencing raw data. FASTP (v0.23.1) software was used to remove low-quality data, including joint contaminated reads and low-quality reads. High-quality data were aligned to a reference genome using STAR (v2.7.0f) software, and gene counts were calculated using FeatureCounts (v1.6.3) software.
16S rRNA sequencing and analysis
16S rRNA sequencing analysis was performed with 15 samples in each group (30 in total). First, genomic DNA was extracted from the small intestine contents. The amplified V3-V4 region sequences were pooled and purified using a Qiagen gel extraction kit. Sequencing libraries were generated using TruSeq® DNA PCR-Free Sample Preparation Kit (Illumina, USA) following manufacturer's instructions. The library was sequenced using the Illumina NovaSeq platform (Novogene, Beijing, China) and 250 bp paired-end reads were generated. Subsequently, we used QIIME2 software (v2020.6) to merge paired ends and perform quality control; the DADA2 amplicon algorithm was used to remove sequencing noise, delete chimera sequences; and amplicon sequence variants (ASVs) were selected to generate signatures of each sample (feature table). Sequences with a similarity ≥97% were assigned to the same operational taxonomic unit (OTU). Based on bioinformatics, the alpha diversity indices of samples in groups were analyzed (including ACE, Chao1, Shannon, and Simpson). ANOSIM similarity analysis using the Bray-Curtis algorithm was used to determine differences between groups. Representative species were determined using an online tool (https://huttenhower.sph.harvard.edu/galaxy/) for linear discriminant analysis effect size (LEfSe) analysis.
Non-targeted metabolomics
Non-targeted metabolomics were performed in collaboration with Shanghai Applied Protein Technology Co., Ltd (Shanghai, China). First, an appropriate amount of serum sample was added to pre-cooled methanol/acetonitrile/water solution (2:2:1, v/v), and vortexed, and mixed; it was then exposed to ultrasound at low temperature for 30 min, subsequently the centrifugal supernatant was taken and dried in a vacuum. During mass spectrometry analysis, 100 μL of acetonitrile aqueous solution (acetonitrile: water =1:1, v/v) was added for resolution. After vortex centrifugation, the supernatant was taken for sample analysis. Then the samples were separated by Agilent 1290 Infinity LC ultra-high performance liquid chromatography (UHPLC) HILIC column: column temperature was 25°C; flow rate was 0.5 mL/min; injection volume was 2 μL. After separation, mass spectrometry was performed with a Triple TOF 6600 mass spectrometer (AB SCIEX) using positive and negative ion modes of electrospray ionization (ESI), respectively. The raw data were converted into files in mzXML format by ProteoWizard, and then XCMS software was used for peak alignment, retention time correction, and peak area extraction. Structures of metabolites in biological samples were identified by matching with information of metabolite retention time, molecular mass (molecular mass error is less than 10 ppm), second-order fragmentation spectrum, and collision energy [38]. According to the results of metabolite identification, the quality of experimental data was evaluated, and then the data were analyzed. Metabolites measured by positive and negative ion channels were combined, and the dimension reduction of the data was analyzed based on partial least squares discriminant analysis (PLS-DA). According to the results of metabolite comparison, all the differentiated metabolites were displayed in a volcano map. MetaboAnalyst (v5.0) was used for pathway enrichment analysis to screen metabolic pathways with significant changes [39].
Statistical analysis
All data contained at least 3 independent replicates and the results are presented as mean ± standard error of the mean (SEM). Significant differences were determined by t-test or one-way analysis of variance (ANOVA) using GraphPad Prism 8 software (GraphPad Software Inc., San Diego, CA); values with *P < 0.05 and **P < 0.01 were considered to have significant and highly significant differences, respectively.
Results
Sleep deprivation caused a reduction of body weight and gut dysbiosis
SD was induced in female mice from 3 to 9 weeks of age, the period of adolescence critical for establishment of the ovarian reserve and its dynamic control [40, 41]. During the 6-week experimental period, we registered a constant body weight increase and decrease in control (CTRL) and SD mice, respectively (Figure 1A-B). Accordingly, at the end of the treatment, the body weight of SD females was significantly lower than CTRL (Figure 1C).
Body weight and gut microbiota changes caused by sleep deprivation. (A) Schematic diagram of the sleep deprivation (SD) experiment. (B) Changes of body weight in control (CTRL) and SD females during the experimental time (n=12). (C) Body weight of CTRL and SD females at the end of the experimental time (n=12). (D) ANOSIM similarity analysis of gut microbiota obtained from CTRL and SD females at the end of the experimental time. (E) PCA of the data shown in D. (F) α-Diversity indexes, including Shannon, Simpson, Chao1, and ACE of the data shown in D. (G) Relative abundance of the bacterial phyla in the gut microbiota of CTRL and SD females. (H) Relative abundance of bacterial genera in the gut microbiota of CTRL and SD females. (I) LDA algorithm of CTRL vs SD gut microbiota bacterial genera with an LDA score threshold of 2.0. (J) Cladogram of the main taxa of microbiota in CTRL and SD females based on LEfSe analysis.
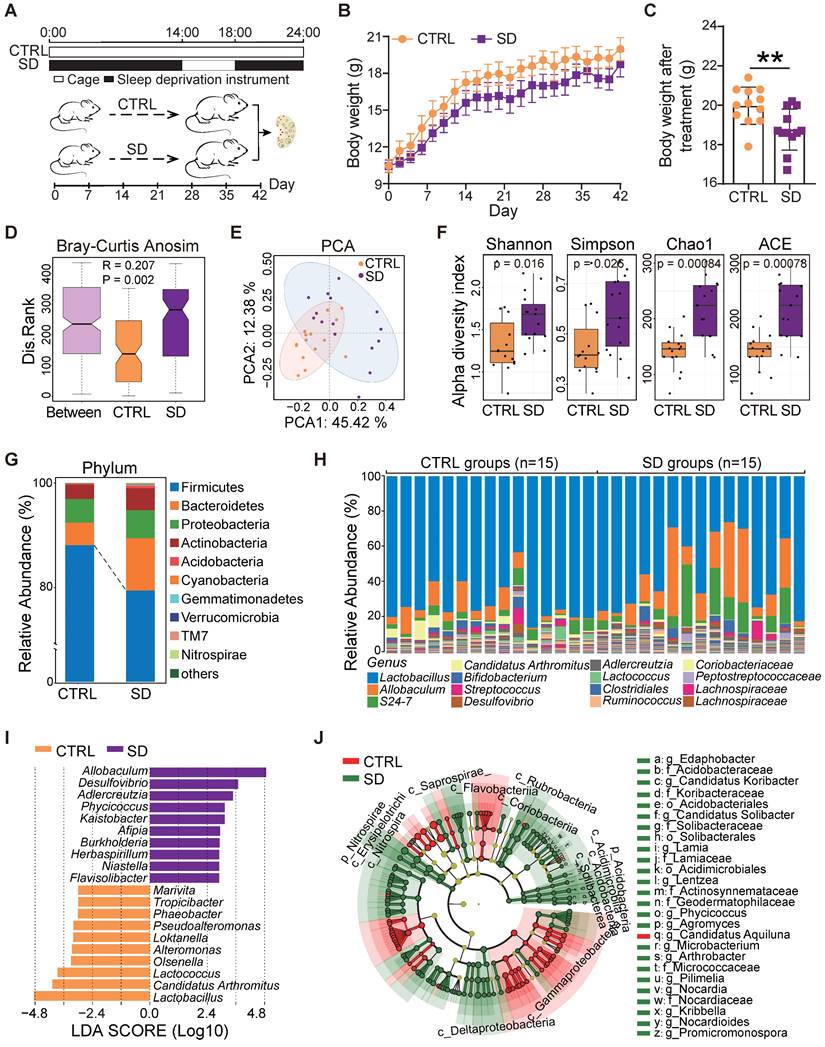
The impact of SD on the gut microbiota was investigated by 16S rRNA sequencing of the small intestine contents obtained from CTRL and SD mice at the end of treatment. Analysis of similarities (ANOSIM) indicated that differences between CTRL and SD groups were significantly greater than differences within groups (Figure 1D). The PCA scatter plot confirmed significant changes within the gut microbiota caused by SD (Figure 1E). Moreover, the α-diversity analysis, including Shannon, Simpson, Chao1, and ACE indices, confirmed distinct differences in the abundance of microbial species between CTRL and SD groups (Figure 1F). Namely, bacterial species annotation data showed that SD altered the abundance of gut microbes both at the phylum and genus level (Figure 1G-H). Among phyla, Firmicutes and Verrucomicrbia decreased while Bacteroides, Proteobacteria, and Actinobacteria increased. To identify the specific microbiota related to SD, and using the linear discriminant analysis (LDA) effect size (LEfSe) method, we screened out 120 biomarkers. The top genera in SD were Allobaculum, Desulfovibrio, and Adlercreuzia; whereas in CTRL, Lactobacillus, Candidatus Arthromitus, and Lactococcus prevailed (Figure 1I-J). On this basis, we also employed STAMP software to conduct the difference analysis based on the abundance of gut microbes. In total, 102 types of differential bacterial genera were found, of which 64 genera were up-regulated after SD, including Kaistobacter, Streptomyces, Flavisolibacter, Arthrobacter, and Promicromonospora; meanwhile, 38 genera were down-regulated, including Pseudoalteromonas, vibrio, Alteromonas, Octadecabacter, and Phaeobacter (Figure S1A). Moreover, based on analysis of LEfSe and STAMP results, 96 bacteria genera were found in both groups (Figure S1B and Table S3). Among them, 12 types of the genera were selected and displayed, 6 of which were up-regulated and 6 down-regulated by SD (Figure S1C). These results indicated that there were substantial differences in the gut microbiome between SD and CTRL groups.
Sleep deprivation impacted on intestinal immunity and the integrity of the intestinal epithelial barrier
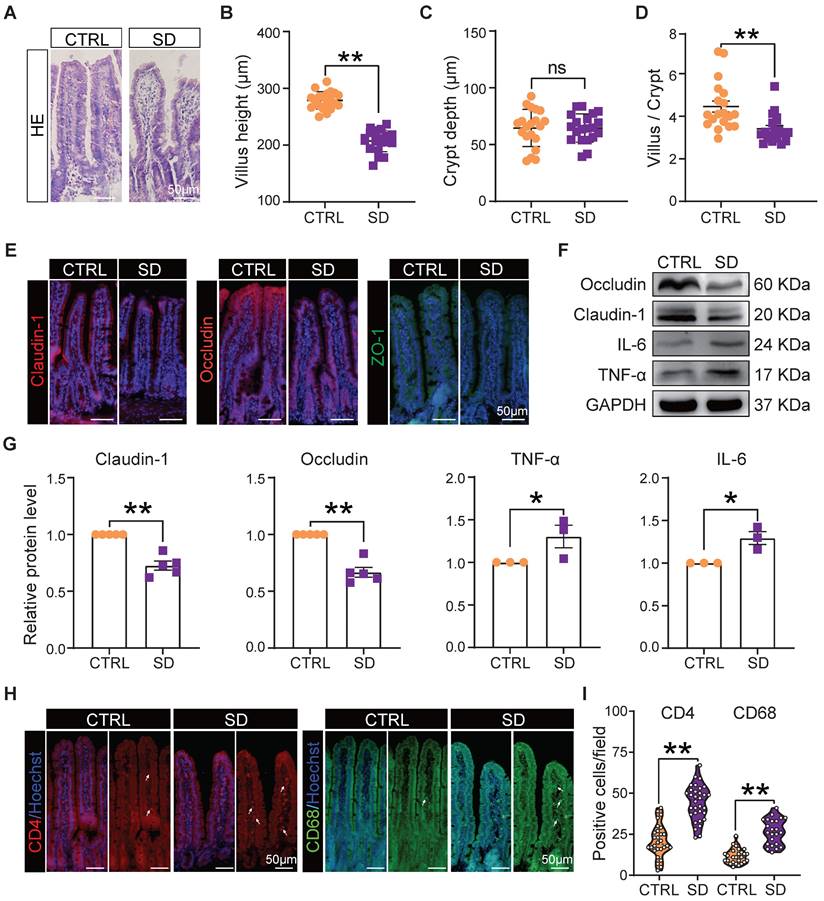
It is well known that the integrity of the intestinal epithelial barrier is essential for maintaining a healthy symbiosis between the epithelial cells and the gut bacteria, and that tight junction proteins play a key role in maintaining barrier integrity. Thus, we collected histological samples of the small intestine and evaluated the expression of tight junction proteins in this organ of CTRL and SD mice. Histological sections showed that, after SD, the height of intestinal villi decreased significantly and, while the crypt depth did not change significantly, the ratio of villi height to crypt depth decreased significantly. Moreover, the epithelial layer appeared less compact, and cells showed hill-defined borders (Figure 2A-D). Most importantly, after SD, the fluorescence intensity of tight junction proteins such as Claudin-1, Occludin, and ZO-1 in tissue sections decreased, while their presence, as detected by WB, was reduced (Figure 2E-G). Interestingly, these changes were associated with increased levels of the inflammatory proteins interleukin-6 (IL-6) and tumor necrosis factor-alpha (TNF-α) (Figure 2F-G), a further indication of altered integrity of the epithelial barrier. In addition, an accumulation of CD4+ T cells and CD68+ macrophages was found in the lamina propria of the small intestine in sleep-deprived mice (Figure 2H-I), suggesting that SD increased immune cell infiltration in the lamina propria of the small intestine.
Intestinal barrier disturbance caused by sleep deprivation. (A) Representative H&E histological sections of small intestine samples collected from CTRL and SD females. (B-D) Comparison of villi height, crypt depth, and villi height/crypt depth of small intestine samples of CTRL and SD females. (E) Fluorescent immunostaining (IF) for the indicated tight junction proteins on small intestinal sections of CTRL and SD females. (F-G) Representative WB and relative densitometric evaluation of the amount of the indicated tight junction proteins and inflammatory cytokines in the small intestine of CTRL and SD females. (H-I) IF quantitative statistics of CD4 and CD68 proteins associated with intestinal immune cells.
Sleep deprivation caused systemic metabolic changes
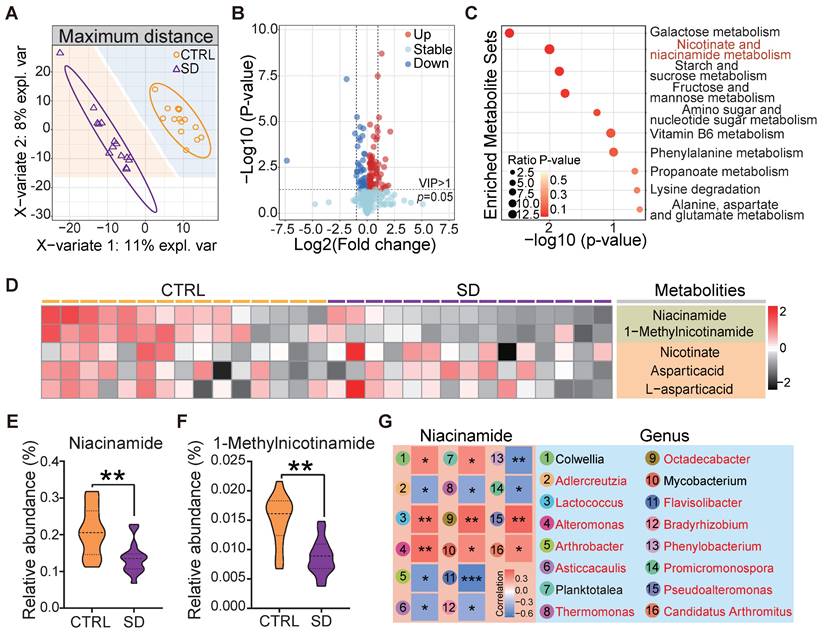
The gut microbiota plays a vital role in its host's metabolism [28]. Several studies support the notion that metabolic processes are correlated with sleep and that SD can cause maladaptive metabolic changes [42]. In order to verify how our SD model impacted on systemic metabolism, we examined the untargeted metabolome profile of blood serum samples obtained from CTRL and SD mice. Partial least squares discriminant analysis (PLS-DA) of the 1022 identified metabolites resulted in distinct metabolite clustering in CTRL and SD groups (Figure 3A). Using an orthogonal PLS-DA (OPLS-DA) model to obtain the variable importance for the projection (VIP) with OPLS-DA VIP >1 and P-value < 0.05, 122 differential metabolites were identified, 82 of which were up-regulated and 40 down-regulated after SD (Figure 3B). According to pathway annotation results of the Kyoto Encyclopedia of Genes and Genomes (KEGG) database, a total of 18 metabolic pathways were enriched in the differential metabolites between the 2 groups, including the metabolism of galactose, nicotinate and nicotinamiade/niacinamide (NAM), starch and sucrose, and fructose and mannose. Among them, nicotinate and NAM metabolic pathways had the highest enrichment rates (Figure 3C). The nicotinate and NAM pathways in the CTRL and SD groups are shown in Figure 3D. Namely, the relative abundance of serum NAM and 1-Methylnicotinamide was higher in CTRL than in the SD group (Figure 3E-F). To investigate the functional relationship between gut microbial dysbiosis and NAM metabolism, Spearman's correlation analysis was performed. As shown in Figure 3G, Spearman's correlation analysis between NAM and genus level bacteria showed that a total of 16 bacteria genera were significantly correlated with NAM, among which 13 bacteria genera were significantly different between CTRL and SD groups (Figure S1B).
Impact of sleep deprivation on the metabolome. (A) PLS-DA discriminant analysis of the metabolome in blood serum from CTRL and SD females. (B) Volcano map of metabolite changes caused by SD. (C) KEGG metabolic pathway enrichment analysis. (D) Heatmap of metabolites in niacinamide (NAM) metabolic pathways. (E-F) Comparison of the relative abundance of NAM and 1-Methylnicotinamide in the metabolome of CTRL and SD females. (G) Spearman correlation analysis between NAM and different bacterial genera. The red font represents the bacteria genera with significant differences shown between CTRL and SD groups.
Sleep deprivation affected follicle number and dynamics and increased granulosa cell apoptosis
At the end of treatment of SD females, the blood concentration of AMH and E2 decreased, while that of LH increased compared to CTRL (Figure 4A). Moreover, in parallel to the decrease in body weight, we found a significant reduction in ovary size and index (ovary and body weight ratio) in comparison to CTRL (Figure 4B-C). The SD effects on the ovary were further investigated by comparing the number of follicles and the percent of each follicle class in CTRL and SD females (Figure 4D and Figure S2A).
Effects of sleep deprivation on ovary index and follicle dynamics. (A) Blood serum concentrations of AMH, E2, FSH, and LH hormones at the end of the experimental time. (B) Representative picture of the CTRL and SD ovary at the end of the experimental time, bar =1 mm. (C) Ovary and body weight ratio (ovary index) in CTRL and SD females at the end of the experimental time (n=12). (D) Representative IHC for the oocyte-specific MVH protein on CTRL and SD ovary sections, bar = 200 μm. (E) Number of different classes of follicles in the 9 sections of CTRL and SD ovaries at the end of the experimental time. (F) Percent of primordial follicles and growing follicles (all other follicle classes, see below) for sections of CTRL and SD ovaries at the end of the experimental time. (G-I) Representative WB and relative densitometric evaluation of the amount of the indicated proteins from CTRL and SD ovaries at the end of the experimental time. PmF: Primordial follicle, PF: Primary follicle, SF: Secondary follicle, AF: Antral follicle.
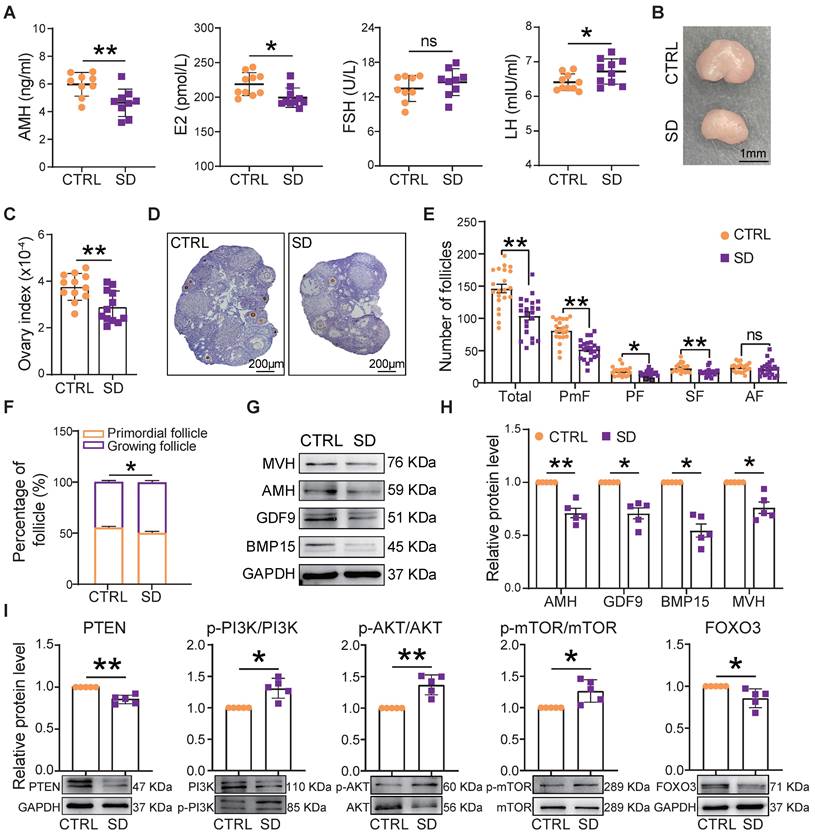
Compared with CTRL, after only 4 weeks of SD treatment, the number of follicles in the 9 scored sections of SD ovaries had decreased significantly, mostly through a marked reduction in the number of PmF, representing the ovarian reserve (Figure S2B). At 6 weeks, this trend was even more pronounced (Figure 4E), and the percent of growing follicles appeared slightly higher in SD (49.66%±1.50%) than CTRL (44.92%±1.19%) ovaries (Figure 4F), suggesting an increment in PmF activation. In line with such a possibility, serum levels of AMH (a marker of ovarian reserve) in SD females were significantly lower than in CTRL (Figure 4A). Moreover, both for mRNA and proteins, the expression of AMH as well that of oocyte specific proteins [growth differentiation factor 9 (GDF9), bone morphogenetic protein 15 (BMP15), and mouse vasa homolog (MVH)] were reduced in SD compared with CTRL ovaries (Figure 4G-H and Figure S2C). At the same time, markers of PmF activation including phosphorylated PI3K, AKT, and mTOR proteins were increased whereas the levels of critical inhibitors of PmF activation including PTEN and FOXO3 proteins, decreased (Figure 4I).
In SD ovaries, besides indication of PmF activation, we found that the number of TUNEL-positive cells, mainly among granulosa cells, were significantly increase in comparison to CTRL (Figure S3A-B). Raised amounts of γ-H2AX protein, a marker of DNA damage, and proapoptotic markers such as BAX/BCL2, P53 and cleaved Caspase 9 paralleled this increase (Figure S3C-D).
Sleep deprivation impaired oocyte mitochondrial function and meiotic maturation
To measure the influence of SD on oocyte quality, we compared the ability of fully grown germinal vesicle (GV) oocytes cultured in vitro from CTRL and SD ovaries to resume meiosis [as germinal vesicle break-down (GVBD)], reach metaphase II (extrusion of PB1), and to correctly assemble the meiotic spindle. The results reported in Figures 5A and 5B show that a smaller number of SD oocytes underwent GVBD and extruded PB1 in comparison to CTRL. Moreover, the percent of spindle (Metaphase I) abnormalities and chromosome misalignments increased significantly in SD oocytes after 8 h of culture (Figure 5C-D).
Sleep deprivation affected oocyte maturation, spindle assembly, and mitochondrial function. (A) Representative pictures of GV, GVBD, and PB1. Scale bar = 100 μm. (B) Percent of GVBD and PB1 in CTRL and SD oocytes after in vitro culture for 4-6 h and 12-16 h (n>90). (C) Representative pictures of normal and abnormal spindle morphologies and chromosome alignment. (D) Percent of aberrant spindles and misaligned chromosomes in CTRL and SD oocytes after in vitro culture for 8 h (n>90). (E) Volcano map of gene transcript differences in CTRL and SD oocytes. (F) Enrichment terms of down-regulated genes. (G) Gene expression levels of the indicated genes related to mitochondrial function. (H) RT-qPCR of the same genes showed in G. (I) Enrichment terms of up-regulated genes.
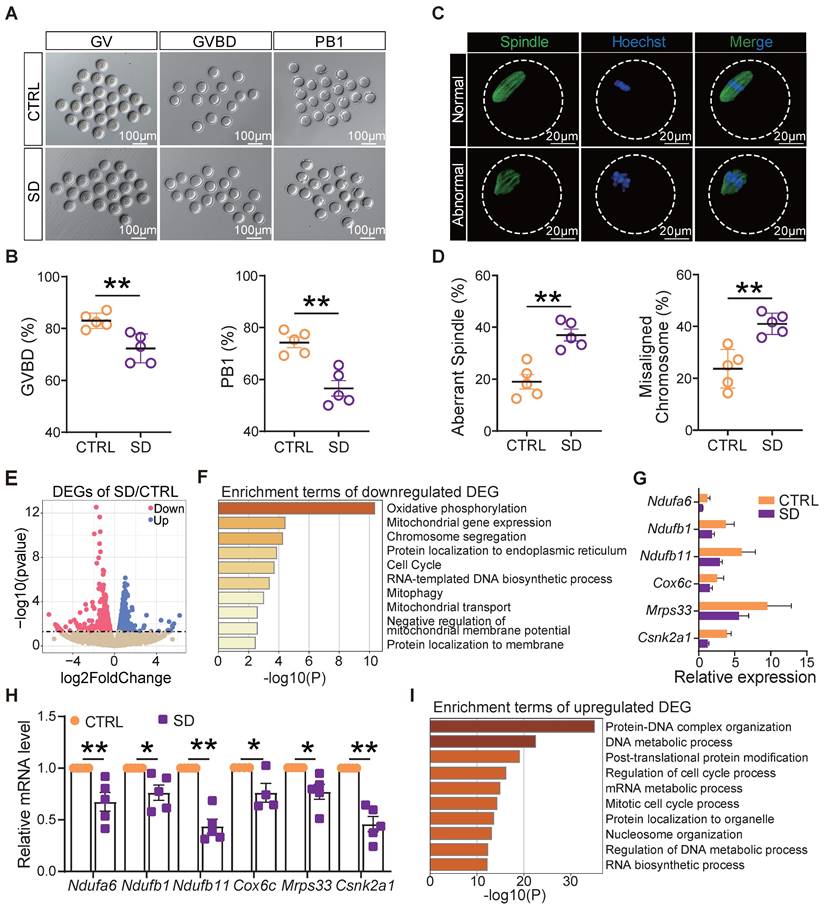
In order to identify molecular pathways or altered functions, we next performed single-cell transcriptome sequencing (scRNA-seq) analyses of CTRL and SD oocytes. Among 1640 differentially expressed genes, we found 1006 genes up-regulated and 634 genes down-regulated in SD oocytes (Figure 5E). Interestingly, down-regulated genes were mainly enriched in terms of oxidative phosphorylation, mitochondrial gene expression, chromosome segregation, and mitophagy signaling pathways (Figure 5F). Deleterious effects of SD on the mitochondrial function of oocytes were supported by reductions in mRNA levels of Ndufa6, Ndufb1, Ndufb11, Cox6c, Mrps33, and Csnk2a1 involved in oxidative phosphorylation and respiratory chain complex assembly, mitochondrial translation, and mitophagy (Figure 5G-H). Up-regulated genes were mainly enriched in terms of protein-DNA complex organization, DNA metabolic process, and regulation of cell cycle process pathways (Figure 5I).
In addition, using RNA-seq analysis of the whole ovaries and c-means clustering algorithm, 8 gene modules were generated (Figure S4A). Notably, most of the genes in Cluster 1, mainly enriched in mitochondrial function, appeared downregulated in SD ovaries (Figure S4B), which is consistent with scRNA-seq results of oocytes, while several genes in cluster 7 were mainly enriched in histone modification, and mRNA processing was up-regulated in SD ovaries (Figure S4C). We also identified 133 common differential genes both in RNA-seq and scRNA-seq (Figure S4D), which are involved in the regulation of growth and development, chromosome organization, and ovarian infertility (Figure S4E). In addition, the expression pattern of POI-related genes in the ovaries also changed significantly after SD (Figure S4F).
Fecal microbiota transplantation in host females recapitulated ovarian damage induced by sleep deprivation
Fecal microbiota transplantation (FMT) is used to directly change the recipient's gut microbiota [31, 43]. In order to understand whether changes of gut microbiota caused by SD could be responsible for the ovary disorders described in the previous sections, we transplanted the fecal microbiota of SD females to normal mice by oral gavage (Figure 6A). After 2 weeks of feeding and 3 weeks of colonization, we found that in FMT females blood serum concentrations of NAM were significantly lower than in CTRL (Figure 6D). Furthermore, the body and ovary weights of recipient females were lower than those of CTRL and comparable to those of SD mice reported in Figure 1C and Figure 4C (Figure 6B-C). Notably, the ovary indices of CTRL and FMT females were not significantly different (not shown).
Fecal microbiota transplantation from SD females reproduces the effects of SD in normal host females. (A) Study design of fecal microbiota transplantation (FMT). (B-C) Body and ovary weights of females under the indicated experimental conditions. (D) Concentration of niacinamide (NAM) in the blood serum obtained from females under the indicated experimental conditions. (E) Total number and different follicle class number for sections of ovaries from females under the indicated experimental conditions. (F) Percent of PmF and growing follicles in the ovaries of females under the indicated experimental conditions. (G) Representative WB and relative densitometric evaluation of the amount of the indicated proteins in the ovaries of females under the indicated experimental conditions.
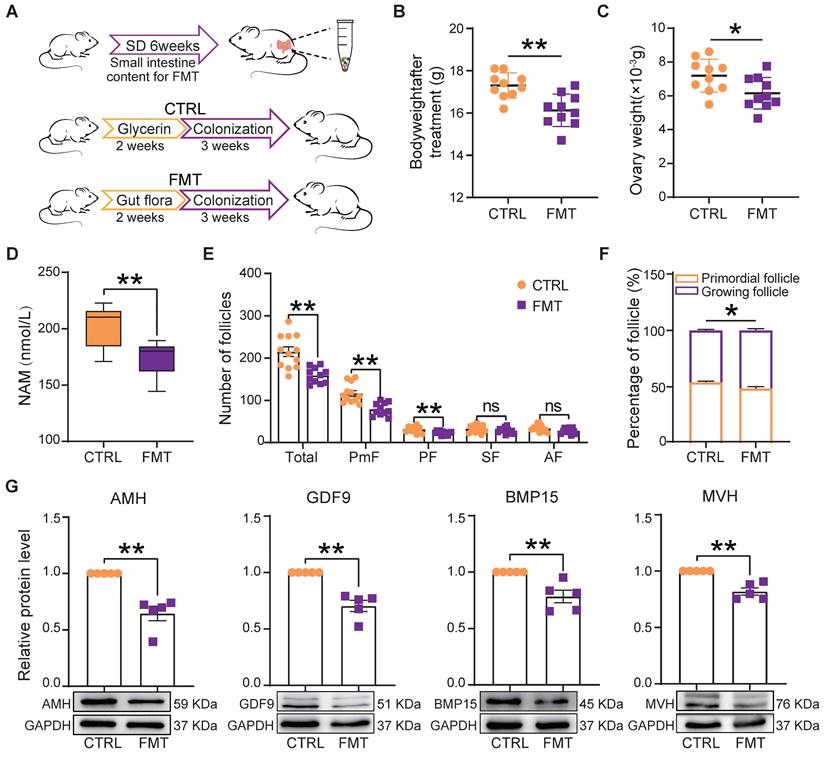
Moreover, FMT females showed follicle alterations similar to those induced by SD including reduced number (mainly PmF), and altered dynamics (Figure 6E-F), associated with decreased expression of AMH, GDF9, BMP15, and MVH proteins (Figure 6G).
Supplementation of niacinamide alleviated the effects of sleep deprivation on follicle dynamics and female reproductive performance
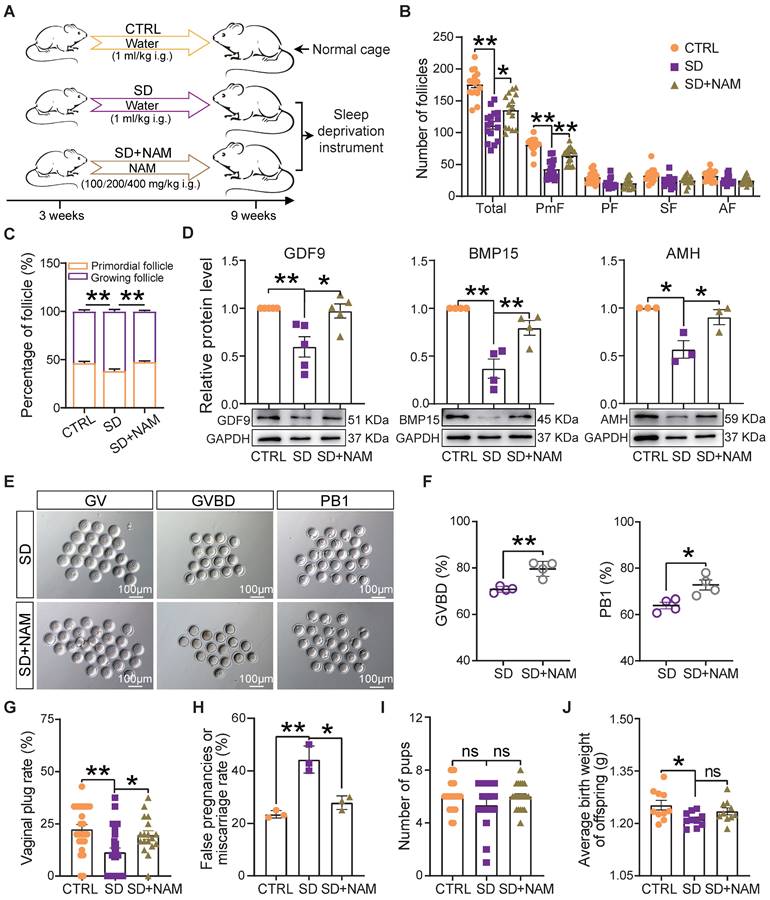
It is well known that NAM, a nicotinamide adenine dinucleotide (NAD+) precursor, is an essential redox cofactor for cellular reactions in mammals and microbes. This is mostly due to the fact that circulating NAM enters the gut lumen and is able to support microbial NAD+ synthesis [44]. For this reason and since we found NAM deficiency to be attributable to a specific gut microbiota imbalance, as one of the most evident effects of our SD treatment in the female mice, we performed experiments to investigate whether NAM supplementation could counteract the effects of SD on follicle dynamics described in the previous sections (Figure 7A). The results showed that although the feeding of 100-400 mg/kg NAM for 6 weeks had no significant effect on body and ovary weight of mice (Figure S5A), the ovaries of SD females fed with 200 mg/kg NAM had follicle numbers and percent of PmFs and of the other follicle classes comparable to those of CTRL (Figure 7B-C and Figure S5B-D). In these ovaries, the levels of AMH and the oocyte secreted factors GDF9 and BMP15 were at concentrations comparable to those of CTRL (Figure 7D). In addition, a higher proportion of oocytes from female mice supplemented with NAM reached GVBD and expelled PB1 compared to SD (Figure 7E-F). In terms of reproductive performance, we found a significant reduction in the rate of pregnant (marked by the presence of vaginal plug) and a significant increase in the rate of false pregnancy or miscarriage in SD female mice, which were effectively improved after NAM supplementation, although the indexes of litter size and average birth weight showed no significant difference (Figure 7G-J).
Dietary niacinamide supplementation alleviated the effects of sleep deprivation on the ovarian follicle reserve. (A) Study design of niacinamide (NAM) supplementation. (B) Total number and different follicle class number for sections of ovaries of females under the indicated experimental conditions. (C) Percent of PmF and growing follicles in the ovaries of females under the indicated experimental conditions. (D) Representative WB and relative densitometric evaluation of the amounts of the indicated proteins in the ovaries of females under the indicated experimental conditions. (E) Representative pictures of oocytes at GV, GVBD, and PB1 stages in SD and SD+NAM groups. Scale bar = 100 μm. (F) Percentage of oocytes at GVBD and PB1 stages in SD, and SD+NAM groups after in vitro culture (n>90). (G) The statistical effect of vaginal plug rate in CTRL, SD, and SD+NAM groups, also including false pregnancies or miscarriage rate (H, number of female mice with vaginal plugs in each group >29), pup numbers (I), and average birth weight of offspring (J).
Discussion
SD is a stressor that affects multiple tissues throughout the body. In recent years, research has shown that abnormal sleep patterns and duration affect systemic metabolism and gut microbiota composition. This latter in turn provides substrates for supplying and regulating multiple compounds reaching the circulation and influencing the function of distal organs. In the present study, using a validated device and female mice, we investigated the effects of SD on murine ovaries during the adolescence period when follicles, the ovarian reserve, and the basic dynamics and control of folliculogenesis are established.
We first confirmed that SD caused gut dysbiosis and systemic metabolic changes in the treated females. In line with other studies reporting mass death of intestinal epithelial cells, reduction of goblet cells, decreased expression of tight junction proteins, and impaired intestinal barrier function after SD [45-47], we obtained histological evidence suggesting the presence of an altered intestinal immunity and integrity of the intestinal epithelial barrier. Moreover, after performing 16S rRNA sequencing, relevant changes in the microbiota were detected. Our data showed that SD caused relevant changes in the abundance of gut microbes at the phylum and genus level. For example, following SD, bacteria genera including Lactobacillus, Lactococcus, Candidatus arthromitus, and Pseudoalteromonas decreased whereas Allobaculum, Kaistobacter, Desulfovibrio, and Adlercreutzia increased. As an important component of the digestive and female reproductive microbiota, Lactobacillus maintains microbiota balance, improves digestive function, assists the synthesis of B vitamins and vitamin K, inhibits pro-inflammatory mediators, and produces antibacterial activity against pathogens [48]. Candidatus arthromitus is a commensal bacterium necessary for inducing the postnatal maturation of homeostatic innate and adaptive immune responses in the mouse gut [49]. Interestingly, an increase in the relative abundance of Allobaculum after SD has also been observed in the intestines of mice fed a high-fat diet while an increase in Desulfovibrio is indicative of intestinal inflammation and altered sensitivity [50]. Furthermore, Desulfovibrio is increased in a rotenone-induced Parkinson's mouse model and in a high-cholesterol-induced non-alcoholic fatty liver mouse model [51, 52].
SD females showed a significant body weight reduction and 18 metabolic pathways were enriched in differential metabolites in the blood of CTRL and SD animals. Among them, nicotinate and NAM pathways had the highest enrichment rates. The higher amounts of NAM present in CTRL in comparison to SD, were correlated to decreased numbers of Lactococcus, Alteromonas, Pseudoalteromonas, Candidatus arthromitus, and Octadecabacter, and an increase in Arthrobacter, Adlercreutzia, Flavisolibacter, Phenylobacterium, and Promicromonospora (Figure 3G). The decreased NAM content after FMT also confirmed the association between NAM and gut microbiome changes (Figure 6D). In this regard, the Firmicutes/Bacteroidetes (F/B) ratio is widely believed to play an important role in maintaining normal intestinal homeostasis [53-55]. At the phylum level, higher Bacteroides and lower Firmicutes abundances were observed in POI patients [22], which coincided with the sequencing data after SD. In addition, studies on mice with chronic colitis found that NAM could increase the proportion of beneficial bacteria and restore the function of gut microbiota [56]. Recent results indicate that intestinal bacteria can promote host NAD+ pools through deamidation and de novo synthesis pathways [57, 58]. Our investigation, however, did not allow us to distinguish between these possibilities.
During the female reproductive cycle, only a limited number of primordial follicles are recruited, therefore disruption of primordial follicle activation inevitably leads to abnormal follicle development. When primordial follicles are activated too quickly, the size of the ovarian reserve is impacted [18]. In general, POI is often induced by abnormal follicular oocyte development and ovarian dysfunction caused by premature depletion of follicle reserve and increased follicle atresia. Histological observations showed that SD reduced the follicle reserve, likely by increasing both primordial follicle activation and follicle atresia. Therefore, we suggest that SD induced the POI phenotype in adolescent female mice; these findings are also supported by the abnormal genetic changes associated with POI. At the same time, decreased ovarian size and changes in the levels of circulating sex hormones were observed. Similar to our findings, some studies suggest that a lack of sleep contributes to obesity and insulin resistance, which in turn promotes PCOS [59, 60]. Furthermore, SD may cause harmful alterations to endocrine systems (including the ovarian axis) [61]. In menstruating women, short sleep duration is associated with lower estradiol and luteal phase progesterone levels and an increased incidence of anovulation, which can also decrease reproductive capacity by impairing the arrival of morulas in the uterus [62].
In our study, the ability of fully grown oocytes to resume and complete meiosis I was also observed to be impaired. scRNA-seq analyses performed on SD oocytes indicated that mitochondrial disfunction was one cause of the reduced quality of such oocytes. This likely impacted on the reproductive performance of the SD females and was significantly reduced, as shown by the lower number of vaginal plugs and higher levels of false pregnancies or miscarriage.
The central role of microbiota impacting on systemic NAM levels and inducing disordered ovarian function, following the SD reported here, was clearly supported by FMT. In female hosts, FMT resulted in a marked reduction in NAM concentration in the blood, and ovarian disorders-including weight reduction, reduced follicle number (mainly PmF), and altered folliculogenesis dynamics-associated with the decrease in AMH, GDF9, BMP15, and MVH proteins.
Finally, the ability of NAM supplementation to counteract the effects of SD on follicle numbers and dynamics and the decrease of AMH, GDF9, and BMP15, points to the reduction of this vitamin as one of the main causes of ovarian disfunction resulting from SD. As reported above, NAM, as a precursor of NAD+, is a key component of the NAD+ salvage pathway and an essential redox cofactor for cellular reactions in mammals and microbes. Challapa et al., using isotope labeling to track NAD metabolism in host tissues and gut microbiota, found that circulating host NAM enters the gut lumen and supports microbial NAD synthesis; then the microbiota subsequently converts host-derived NAM into nicotinic acid, which is used for NAD synthesis in host tissue [63]. Relevant to the present results, continuous administration of NAM has been shown to maintain DNA integrity and prevent apoptosis [64]. NAM treatment can increase the expression of proteins associated with mitochondrial function, including oxidative phosphorylation, fatty acid oxidation, and TCA cycling [65]. Studies in patients with poor ovarian response (POR) have shown that this disfunction is related to nicotinate and NAM metabolic pathways and appears to impact the ovarian reserve [66]. Moreover, NAM is very important for oocyte maturation, it is involved in the salvage pathway of NAD+ synthesis in cumulus cells, and is increased substantially throughout the course of oocyte maturation [67]. We speculate that the mitochondrial disfunction observed in oocytes can be due, at least in part, to a decrease in NAM availability. Increasing NAD+ levels by supplementing NAD+ preforms, such as NAM, niacinamide ribonucleoside (NR), or niacinamide mononucleotide (NMN), may enhance mitochondrial function and promote nuclear and cytoplasmic maturation to mitigate fertility damage associated with ovarian aging [68]. Interestingly, some studies show that the level of NAM in preovulatory follicles can be significantly higher than that in small follicles and that NAM concentration in the follicular fluid of antral follicles is positively correlated with oocyte maturation and fertilization rate. These findings support the notion that NAM may favor follicular development, oocyte quality, and reproductive performance by increasing endogenous NAD+ levels to improve mitochondrial function [69]. In line with the present results, NAM supplementation improved oocyte quality and offspring development by modulating mitochondrial function in an aged Caenorhabditis elegans model [70].
Conclusions
In conclusion, our results support the notion that, in adolescent females, SD can cause changes in the gut microbiota that impact on systemic metabolism centered on NAM pathways. In the ovary, this can impair oocyte mitochondrial energy pathways resulting in POI and reduced oocyte meiotic maturation. Finally, NAM supplementation can be used to counteract the deleterious effects of SD on the ovary.
Abbreviations
POI: premature ovarian insufficiency; PmF: primordial follicles; SD: sleep deprivation; FMT: fecal microbiota transplantation; NAM: nicotinamiade/niacinamide; NAD+: nicotinamide adenine dinucleotide; GV: germinal vesicle; GVBD: germinal vesicle break-down; PBE: polar body extrusion; ANOSIM: analysis of similarities; IHC: immunochemistry; RT-qPCR: real-time quantitative PCR; OTUs: operation altaxonomic units; PCoA: principal coordinate analysis; LEfSe: linear discriminate analysis effect size; VIP: variable importance for the projection; PmF: primordial follicle; PF: primary follicle; SF: secondary follicle; AF: antral follicle; LH: luteinizing hormone.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
This work was supported by the National Nature Science Foundation of China (32270903 and 32200699), the Start-up Fund for High-level Talents of Qingdao Agricultural University (6651122011), and the Taishan Scholar Construction Foundation of Shandong Province of China (ts20190946).
Ethics approval and consent to participate
All procedures involved in this experiment conformed to the standards of the Animal Ethics Committee of Qingdao Agricultural University (approval No. 2022-0021).
Availability of data and materials
Data are available in a public, open access repository. Single-cell RNA sequencing data, RNA sequencing data, 16S rRNA gene sequencing data, and Metabolome sequencing data generated in this study are deposited in the Genome Sequence Archive and OMIX at the National Genomics Data Center with the accession number: CRA012931, CRA012907, CRA012983, and OMIX005078.
Author contributions
JY, JW, TZ, and WS contributed to the study design; WS obtained funding; JY, KZ, and XZ performed the experiments; JW, MDF, and MY analyzed the data; JY, JW, TZ, MDF, and WS wrote the manuscript. All authors read and approved the final manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Kecklund G, Axelsson J. Health consequences of shift work and insufficient sleep. BMJ. 2016;355:i5210
2. Lv YN, Cui Y, Zhang B, Huang SM. Sleep deficiency promotes Alzheimer's disease development and progression. Front Neurol. 2022;13:1053942
3. Krause AJ, Vallat R, Simon EB, Walker MP. Sleep Loss Influences the Interconnected Brain-Body Regulation of Cardiovascular Function in Humans. Psychosom Med. 2023;85:34-41
4. McHill AW, Wright KP Jr. Role of sleep and circadian disruption on energy expenditure and in metabolic predisposition to human obesity and metabolic disease. Obes Rev. 2017;18(Suppl 1):15-24
5. Qian Y, Xia J, Liu KQ, Xu L, Xie SY, Chen GB. et al. Observational and genetic evidence highlight the association of human sleep behaviors with the incidence of fracture. Commun Biol. 2021;4:1339
6. Dominguez-Salazar E, Hurtado-Alvarado G, Medina-Flores F, Dorantes J, Gonzalez-Flores O, Contis-Montes de Oca A. et al. Chronic sleep loss disrupts blood-testis and blood-epididymis barriers, and reduces male fertility. J Sleep Res. 2020;29:e12907
7. Cui L, Xu F, Xu C, Ding Y, Wang S, Du M. Circadian gene Rev-erbalpha influenced by sleep conduces to pregnancy by promoting endometrial decidualization via IL-6-PR-C/EBPbeta axis. J Biomed Sci. 2022;29:101
8. Wang X, Wang Z, Cao J, Dong Y, Chen Y. Gut microbiota-derived metabolites mediate the neuroprotective effect of melatonin in cognitive impairment induced by sleep deprivation. Microbiome. 2023;11:17
9. Vaccaro A, Kaplan Dor Y, Nambara K, Pollina EA, Lin C, Greenberg ME. et al. Sleep Loss Can Cause Death through Accumulation of Reactive Oxygen Species in the Gut. Cell. 2020;181:1307-28 e15
10. Lateef OM, Akintubosun MO. Sleep and Reproductive Health. J Circadian Rhythms. 2020;18:1
11. Yang Q, Borges MC, Sanderson E, Magnus MC, Kilpi F, Collings PJ. et al. Associations between insomnia and pregnancy and perinatal outcomes: Evidence from mendelian randomization and multivariable regression analyses. PLoS Med. 2022;19:e1004090
12. Kloss JD, Perlis ML, Zamzow JA, Culnan EJ, Gracia CR. Sleep, sleep disturbance, and fertility in women. Sleep Med Rev. 2015;22:78-87
13. Pengo MF, Won CH, Bourjeily G. Sleep in Women Across the Life Span. Chest. 2018;154:196-206
14. Ke H, Tang S, Guo T, Hou D, Jiao X, Li S. et al. Landscape of pathogenic mutations in premature ovarian insufficiency. Nat Med. 2023;29:483-92
15. Golezar S, Ramezani Tehrani F, Khazaei S, Ebadi A, Keshavarz Z. The global prevalence of primary ovarian insufficiency and early menopause: a meta-analysis. Climacteric. 2019;22:403-11
16. Visser JA, Schipper I, Laven JS, Themmen AP. Anti-Mullerian hormone: an ovarian reserve marker in primary ovarian insufficiency. Nat Rev Endocrinol. 2012;8:331-41
17. Jiao X, Ke H, Qin Y, Chen ZJ. Molecular Genetics of Premature Ovarian Insufficiency. Trends Endocrinol Metab. 2018;29:795-807
18. Chon SJ, Umair Z, Yoon MS. Premature Ovarian Insufficiency: Past, Present, and Future. Front Cell Dev Biol. 2021;9:672890
19. Auchus RJ. Steroid 17-hydroxylase and 17,20-lyase deficiencies, genetic and pharmacologic. J Steroid Biochem Mol Biol. 2017;165:71-8
20. Fridovich-Keil JL, Gubbels CS, Spencer JB, Sanders RD, Land JA, Rubio-Gozalbo E. Ovarian function in girls and women with GALT-deficiency galactosemia. J Inherit Metab Dis. 2011;34:357-66
21. Jiang L, Fei H, Tong J, Zhou J, Zhu J, Jin X. et al. Hormone Replacement Therapy Reverses Gut Microbiome and Serum Metabolome Alterations in Premature Ovarian Insufficiency. Front Endocrinol (Lausanne). 2021;12:794496
22. Huang F, Cao Y, Liang J, Tang R, Wu S, Zhang P. et al. The influence of the gut microbiome on ovarian aging. Gut Microbes. 2024;16:2295394
23. Beroukhim G, Esencan E, Seifer DB. Impact of sleep patterns upon female neuroendocrinology and reproductive outcomes: a comprehensive review. Reprod Biol Endocrinol. 2022;20:16
24. Al-Nasiry S, Ambrosino E, Schlaepfer M, Morre SA, Wieten L, Voncken JW. et al. The Interplay Between Reproductive Tract Microbiota and Immunological System in Human Reproduction. Front Immunol. 2020;11:378
25. Valdes AM, Walter J, Segal E, Spector TD. Role of the gut microbiota in nutrition and health. BMJ. 2018;361:k2179
26. Bosch JA, Nieuwdorp M, Zwinderman AH, Deschasaux M, Radjabzadeh D, Kraaij R. et al. The gut microbiota and depressive symptoms across ethnic groups. Nat Commun. 2022;13:7129
27. Pellegrini C, Fornai M, D'Antongiovanni V, Antonioli L, Bernardini N, Derkinderen P. The intestinal barrier in disorders of the central nervous system. Lancet Gastroenterol Hepatol. 2023;8:66-80
28. Zhang T, Sun P, Geng Q, Fan H, Gong Y, Hu Y. et al. Disrupted spermatogenesis in a metabolic syndrome model: the role of vitamin A metabolism in the gut-testis axis. Gut. 2022;71:78-87
29. Qi X, Yun C, Sun L, Xia J, Wu Q, Wang Y. et al. Gut microbiota-bile acid-interleukin-22 axis orchestrates polycystic ovary syndrome. Nat Med. 2019;25:1225-33
30. Chadchan SB, Singh V, Kommagani R. Female reproductive dysfunctions and the gut microbiota. J Mol Endocrinol. 2022;69:R81-R94
31. Fang H, Feng X, Xu T, Zhong R, Lu D, Zhang H. et al. Gut-Spleen Axis: Microbiota via Vascular and Immune Pathways Improve Busulfan-Induced Spleen Disruption. mSphere. 2023;8:e0058122
32. Zhang P, Feng Y, Li L, Ge W, Yu S, Hao Y. et al. Improvement in sperm quality and spermatogenesis following faecal microbiota transplantation from alginate oligosaccharide dosed mice. Gut. 2021;70:222-5
33. Yan J, Kong L, Zhang X, Yu M, Zhu K, Zhao A. et al. Maternal Zearalenone Exposure Affects Gut Microbiota and Follicular Development in Suckled Offspring. J Agric Food Chem. 2022;70:15570-82
34. Hernandez-Ochoa I, Barnett-Ringgold KR, Dehlinger SL, Gupta RK, Leslie TC, Roby KF. et al. The ability of the aryl hydrocarbon receptor to regulate ovarian follicle growth and estradiol biosynthesis in mice depends on stage of sexual maturity. Biol Reprod. 2010;83:698-706
35. Brown J, Pirrung M, McCue LA. FQC Dashboard: integrates FastQC results into a web-based, interactive, and extensible FASTQ quality control tool. Bioinformatics. 2017;33:3137-9
36. Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550
37. Zhou Y, Zhou B, Pache L, Chang M, Khodabakhshi AH, Tanaseichuk O. et al. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat Commun. 2019;10:1523
38. Shen X, Wang R, Xiong X, Yin Y, Cai Y, Ma Z. et al. Metabolic reaction network-based recursive metabolite annotation for untargeted metabolomics. Nat Commun. 2019;10:1516
39. Pang Z, Zhou G, Ewald J, Chang L, Hacariz O, Basu N. et al. Using MetaboAnalyst 5.0 for LC-HRMS spectra processing, multi-omics integration and covariate adjustment of global metabolomics data. Nat Protoc. 2022;17:1735-61
40. Laviola G, Macri S, Morley-Fletcher S, Adriani W. Risk-taking behavior in adolescent mice: psychobiological determinants and early epigenetic influence. Neurosci Biobehav Rev. 2003;27:19-31
41. Barnett KR, Schilling C, Greenfeld CR, Tomic D, Flaws JA. Ovarian follicle development and transgenic mouse models. Hum Reprod Update. 2006;12:537-55
42. Humer E, Pieh C, Brandmayr G. Metabolomics in Sleep, Insomnia and Sleep Apnea. Int J Mol Sci. 2020;21:7244
43. Hamamah S, Gheorghita R, Lobiuc A, Sirbu IO, Covasa M. Fecal microbiota transplantation in non-communicable diseases: Recent advances and protocols. Front Med (Lausanne). 2022;9:1060581
44. Ren Z, Xu Y, Li T, Sun W, Tang Z, Wang Y. et al. NAD(+) and its possible role in gut microbiota: Insights on the mechanisms by which gut microbes influence host metabolism. Anim Nutr. 2022;10:360-71
45. Gao T, Wang Z, Cao J, Dong Y, Chen Y. Melatonin alleviates oxidative stress in sleep deprived mice: Involvement of small intestinal mucosa injury. Int Immunopharmacol. 2020;78:106041
46. Gao T, Wang Z, Dong Y, Cao J, Lin R, Wang X. et al. Role of melatonin in sleep deprivation-induced intestinal barrier dysfunction in mice. J Pineal Res. 2019;67:e12574
47. Yang DF, Huang WC, Wu CW, Huang CY, Yang YSH, Tung YT. Acute sleep deprivation exacerbates systemic inflammation and psychiatry disorders through gut microbiota dysbiosis and disruption of circadian rhythms. Microbiol Res. 2023;268:127292
48. Duar RM, Lin XB, Zheng J, Martino ME, Grenier T, Perez-Munoz ME. et al. Lifestyles in transition: evolution and natural history of the genus Lactobacillus. FEMS Microbiol Rev. 2017;41:S27-S48
49. Bolotin A, de Wouters T, Schnupf P, Bouchier C, Loux V, Rhimi M. et al. Genome Sequence of "Candidatus Arthromitus" sp. Strain SFB-Mouse-NL, a Commensal Bacterium with a Key Role in Postnatal Maturation of Gut Immune Functions. Genome Announc. 2014;2:e00705-14
50. Kain V, Van Der Pol W, Mariappan N, Ahmad A, Eipers P, Gibson DL. et al. Obesogenic diet in aging mice disrupts gut microbe composition and alters neutrophil:lymphocyte ratio, leading to inflamed milieu in acute heart failure. FASEB J. 2019;33:6456-69
51. Zhao Z, Ning J, Bao XQ, Shang M, Ma J, Li G. et al. Fecal microbiota transplantation protects rotenone-induced Parkinson's disease mice via suppressing inflammation mediated by the lipopolysaccharide-TLR4 signaling pathway through the microbiota-gut-brain axis. Microbiome. 2021;9:226
52. Zhang X, Coker OO, Chu ES, Fu K, Lau HCH, Wang YX. et al. Dietary cholesterol drives fatty liver-associated liver cancer by modulating gut microbiota and metabolites. Gut. 2021;70:761-74
53. Li J, Zhao F, Wang Y, Chen J, Tao J, Tian G. et al. Gut microbiota dysbiosis contributes to the development of hypertension. Microbiome. 2017;5:14
54. Crovesy L, Masterson D, Rosado EL. Profile of the gut microbiota of adults with obesity: a systematic review. Eur J Clin Nutr. 2020;74:1251-62
55. Quaglio AEV, Grillo TG, De Oliveira ECS, Di Stasi LC, Sassaki LY. Gut microbiota, inflammatory bowel disease and colorectal cancer. World J Gastroenterol. 2022;28:4053-60
56. Kang K, Sun Y, Pan D, Chang B, Sang LX. Nicotinamide Ameliorates Dextran Sulfate Sodium-Induced Chronic Colitis in Mice through Its Anti-Inflammatory Properties and Modulates the Gut Microbiota. J Immunol Res. 2021;2021:5084713
57. Magnusdottir S, Ravcheev D, de Crecy-Lagard V, Thiele I. Systematic genome assessment of B-vitamin biosynthesis suggests co-operation among gut microbes. Front Genet. 2015;6:148
58. Shats I, Williams JG, Liu J, Makarov MV, Wu X, Lih FB. et al. Bacteria Boost Mammalian Host NAD Metabolism by Engaging the Deamidated Biosynthesis Pathway. Cell Metab. 2020;31:564-79 e7
59. Reutrakul S, Van Cauter E. Sleep influences on obesity, insulin resistance, and risk of type 2 diabetes. Metabolism. 2018;84:56-66
60. Brower MA, Hai Y, Jones MR, Guo X, Chen YI, Rotter JI. et al. Bidirectional Mendelian randomization to explore the causal relationships between body mass index and polycystic ovary syndrome. Hum Reprod. 2019;34:127-36
61. Michels KA, Mendola P, Schliep KC, Yeung EH, Ye A, Dunietz GL. et al. The influences of sleep duration, chronotype, and nightwork on the ovarian cycle. Chronobiol Int. 2020;37:260-71
62. Calegare BFA, Azzolini A, da Silva Vallim JR, Turco EGL, Tempaku PF, da Silva VC. et al. Sleep deprivation decreases the reproductive capacity by affecting the arrival of morulas in the uterus. Genesis. 2020;58:e23350
63. Chellappa K, McReynolds MR, Lu W, Zeng X, Makarov M, Hayat F. et al. NAD precursors cycle between host tissues and the gut microbiome. Cell Metab. 2022;34:1947-59 e5
64. Lin SH, Vincent A, Shaw T, Maynard KI, Maiese K. Prevention of nitric oxide-induced neuronal injury through the modulation of independent pathways of programmed cell death. J Cereb Blood Flow Metab. 2000;20:1380-91
65. Luo C, Yang C, Wang X, Chen Y, Liu X, Deng H. Nicotinamide reprograms adipose cellular metabolism and increases mitochondrial biogenesis to ameliorate obesity. J Nutr Biochem. 2022;107:109056
66. Song H, Qin Q, Yuan C, Li H, Zhang F, Fan L. Metabolomic Profiling of Poor Ovarian Response Identifies Potential Predictive Biomarkers. Front Endocrinol (Lausanne). 2021;12:774667
67. Moyses-Oliveira M, Scaff AMC, Adami LNG, Hachul H, Andersen ML, Tufik S. Genetic factors underlying insomnia and ovarian insufficiency. Climacteric. 2023;26:510-2
68. Yang Q, Li H, Wang H, Chen W, Zeng X, Luo X. et al. Deletion of enzymes for de novo NAD(+) biosynthesis accelerated ovarian aging. Aging Cell. 2023;22:e13904
69. Guo Z, Yang J, Yang G, Feng T, Zhang X, Chen Y. et al. Effects of nicotinamide on follicular development and the quality of oocytes. Reprod Biol Endocrinol. 2022;20:70
70. Min H, Lee M, Cho KS, Lim HJ, Shim YH. Nicotinamide Supplementation Improves Oocyte Quality and Offspring Development by Modulating Mitochondrial Function in an Aged Caenorhabditis elegans Model. Antioxidants (Basel). 2021;10:519
Author contact
![]() Corresponding authors: Prof. Wei Shen; E-mail: wshenedu.cn; Prof. Jun-Jie Wang; E-mail: junjieseven0717com; Prof. Teng Zhang; E-mail: zhangteng428com.
Corresponding authors: Prof. Wei Shen; E-mail: wshenedu.cn; Prof. Jun-Jie Wang; E-mail: junjieseven0717com; Prof. Teng Zhang; E-mail: zhangteng428com.