Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Methods
Results
Discussion
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2025; 15(14):6737-6752. doi:10.7150/thno.110162 This issue Cite
Research Paper
MTX2 facilitates PKM2 tetramerization to promote cardiac glucose metabolism and protects the heart against ischemia/reperfusion injury
Yueyang Li1,2*, Yu Li3*, Yang Li1,2, Yufan Jiang3, Miao Wang3, Mingyi Wang3, Jie Liu2, Mingrui Ma3, Xiaofeng Zhai4, Li Yi2, Tao Chen1 ![]() , Zhenyu Xiong1
, Zhenyu Xiong1 ![]() , Yundai Chen1,2
, Yundai Chen1,2 ![]()
1. Department of Cardiology, the Sixth Medical Centre, Chinese PLA General Hospital, Beijing, 100048, China.
2. Department of Cardiology, the First Medical Centre, Chinese PLA General Hospital, Beijing, 100853, China.
3. Medical School of Chinese PLA, Chinese PLA General Hospital, Beijing, 100853, China.
4. The Sixth Medical Centre, Chinese PLA General Hospital, Beijing, 100048, China.
*These authors have contributed equally to this work.
Received 2025-1-9; Accepted 2025-5-10; Published 2025-6-9
Abstract

Rationale: Myocardial ischemia reperfusion (I/R) injury is a major cause of adverse outcomes following revascularization therapy. Although alterations in metabolic activities during reperfusion have been implicated, the molecular mechanisms underlying the pathogenesis of I/R injury remain elusive. Metaxin 2 (MTX2), initially identified as a core component of protein import complexes, has recently been characterized in diverse cellular functions. Nevertheless, its involvement in myocardial I/R injury has yet to be fully elucidated. In this study, we aim to evaluate the role and the underlying mechanism of MTX2 in I/R injury.
Methods: The myocardial I/R model was established, and the protein levels of MTX2 were determined at different time points following coronary occlusion. Loss-of-function and gain-of-function strategies were applied via genetic ablation or intra-myocardial adenovirus injection to ascertain the role of MTX2 in myocardial I/R injury. RNA sequencing, seahorse metabolic analysis, and mass spectrometry were conducted to uncover the underlying molecular mechanisms.
Results: We observed that the expression of MTX2 was significantly decreased in I/R hearts. Tamoxifen-induced cardiomyocyte-specific deletion of Mtx2 led to aggravated myocardial I/R injury, resulting in impaired cardiac oxidative phosphorylation and glycolysis. Mechanistically, dimeric PKM2, a less active pyruvate kinase form compared with tetrameric PKM2, was found to be dramatically accumulated in Mtx2 deficiency mice after myocardial I/R surgery. The TOM37 domain of MTX2 interacted directly with PKM2 to promote PKM2 tetramerization, thereby modulating glucose metabolic flux. Pharmacological activation of PKM2 by a small-molecule PKM2 activator, TEPP-46, rescued the metabolic and functional outcomes of I/R in Mtx2 deficiency mice.
Conclusions: Our results identified, for the first time, a cardioprotective role of MTX2 in modulating cardiac glucose metabolism by facilitating PKM2 tetramerization. Targeting metabolic homeostasis by restoring MTX2 might be a promising therapeutic strategy to mitigate myocardial I/R injury.
Keywords: myocardial ischemia/reperfusion injury, metaxin 2, mitochondria, pyruvate kinase M2, metabolic homeostasis.
Introduction
Ischemic heart disease is among the leading causes of death in developed as well as developing countries [1]. Despite prompt and comprehensive revascularization by percutaneous coronary intervention, this reperfusion process paradoxically causes myocardial ischemia/ reperfusion (I/R) injury, pathological manifestations of which include enlarged myocardial necrotic areas, energy depletion, and ultimately contraction dysfunction [2]. To date, effective pharmacological interventions are yet to be developed to meet current clinical needs [3]. The heart is one of the most prominent energy-consuming organs and, thus, cardiac substrate metabolism is instrumental in maintaining the normal structure and function of the heart. Energy metabolic disturbance, in particular, glucose and fatty acid (FA) metabolism, has been recognized as a critical determinant of I/R damage [4]. However, the upstream event contributing to impaired substrate metabolism in the I/R hearts remains inadequately characterized.
As central hubs of cardiac ATP production, mitochondria are instrumental in maintaining metabolic balance during I/R. Thus, targeting molecules primarily located in mitochondria shows promise for intervening I/R-induced ATP defect and metabolic disturbance [5-8]. The Sorting and Assembly Machinery (SAM) complex, composed of metaxin 1 (MTX1), metaxin 2 (MTX2) and SAM50, have emerged as promising therapeutic targets through its coordinated governance of mitochondrial proteostasis, dynamic morphological remodeling, and cristae architecture preservation [9-12]. However, evidence for their functional implications in cardiovascular diseases remains strikingly limited. MTX2, also known as SAM37, was initially reported to collaborate with MTX1 and SAM50 for the accurate integration of β-barrel proteins into the mitochondrial outer membrane, thus ensuring proper assembly of essential proteins such as VDAC channels [13,14]. Unlike MTX1 and SAM50, MTX2 lacks a C-terminal mitochondrial outer membrane signal-anchor domain. Instead, MTX2 is oriented towards the cytosolic compartment of the mitochondrial outer membrane through its interaction with membrane-bound MTX1 [15,16], indicating a potential role as a mediator of mitochondrial-cytosolic crosstalk beyond its established transport function. Recent research has further expanded its role as a direct regulator of TNF-α induced apoptosis [16,17]. Furthermore, Mtx2 deficiency can lead to a spectrum of adverse effects, such as reduced oxidative phosphorylation, excessive mitochondrial fission, and altered nuclear morphology [12,18]. Despite these findings, it remains unclear whether MTX2 plays a role in cardiac energy metabolism and myocardial I/R injury. Furthermore, its subcellular and molecular mechanisms responsible for MTX2-related cardiac regulation have not been identified.
In this study, we generated mice with an inducible cardiomyocyte-specific deletion of Mtx2 using α-MHC-MerCreMer. Through the implementation of loss- and gain-of-function strategies, we sought to explore how MTX2 regulates cardiac metabolism in the pathogenesis of I/R injury and its therapeutic potential. Considering the important role of MTX2 in cardiac oxidative phosphorylation (OXPHOS) and glycolysis, these results provide proof of concept that targeting MTX2 might mitigate myocardial I/R injury and other cardiac diseases characterized by metabolic defects.
Methods
Extended methods sections are available in the Supplemental Material.
Animals
Animal studies were performed according to the National Institutes of Health Guidelines for the Care and Use of Laboratory Animals (NIH publication No. 85-23, revised 2011). Animal protocols and performances were approved by the Institutional Animal Care and Use Committee of PLA General Hospital. Euthanasia was performed in accordance with the American Veterinary Medical Association (AVMA) Guidelines for the Euthanasia of Animals (2020) and with the approval of local animal welfare committees.
Adult male C57BL/6J mice were purchased from Model Organisms Center, Inc. (Shanghai, China). Mice were anesthetized by inhalation of 2% isoflurane in oxygen throughout the cardiac I/R surgery, intra-myocardial injection of adenovirus, echocardiography and heart excision for adult cardiomyocyte isolation. Finally, mice were euthanized in their home cage via carbon dioxide inhalation. Carbon dioxide was introduced into the cage at a rate of 3 L/min, added to the existing air in the chamber, until the cessation of respiration in the mice was observed. Mice were then remained in cages for an additional 3 min. Cervical dislocation was performed thereafter to ensure death. The hearts were either stored at -80 °C for subsequent protein extraction or immersed in 4% paraformaldehyde for histological analyses.
Data availability
All supporting data in the study are available from the corresponding author on reasonable request. RNA sequencing data of this study have been uploaded to the National Center for Biotechnology Information (NCBI) BioProject database under the accession number PRJNA1136562.
Statistical analysis
All values were presented as mean ± SD. Data were analyzed using GraphPad Prism 8 statistic software. The normality of the data was assessed using the Shapiro-Wilk test. For normally distributed data, Student's t-test was carried out to compare differences between two groups, and one-way or two-way ANOVA followed by Tukey post-hoc test was used for multiple group comparisons. For non-normally distributed data, Mann-Whitney U test was used for two-group comparisons, and Kruskal-Wallis test with Dunn multiple comparison test was applied for comparisons involving multiple groups. P values < 0.05 were considered statistically significant.
Results
MTX2 is downregulated in myocardial I/R hearts
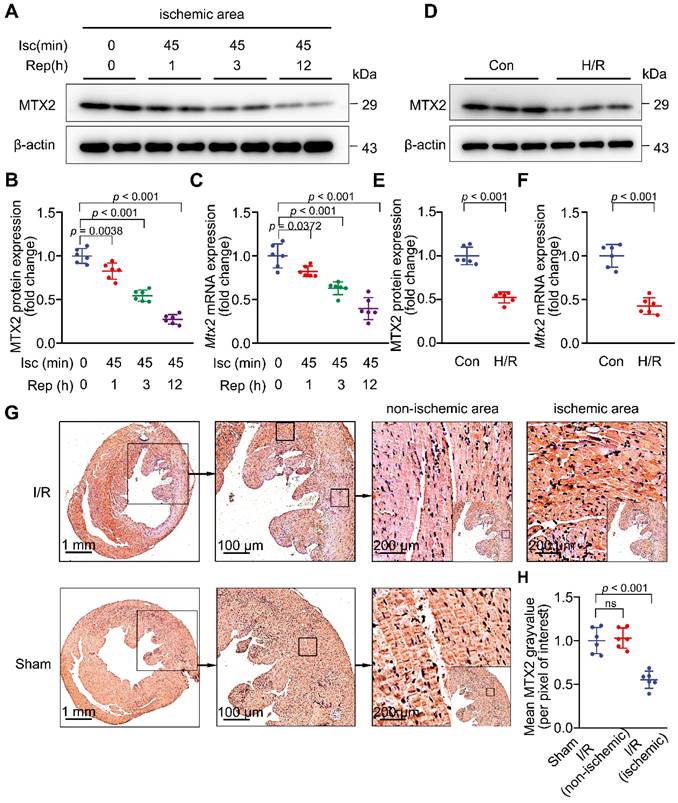
To investigate whether MTX2 plays a role in I/R injury, we first examined MTX2 protein and mRNA expression in adult mouse hearts at different time points of reperfusion. We found that MTX2 was abundantly expressed in the heart but significantly downregulated in response to 45-min ischemia followed by 1-h reperfusion. MTX2 protein was further decreased in the ischemic hearts as a consequence of 3-h and 12-h reperfusion (Figure 1A-1B). The protein expression pattern of MTX2 mirrored its mRNA pattern during myocardial reperfusion phase (Figure 1C). Moreover, we examined MTX2 protein and mRNA levels in non-ischemic myocardium after I/R surgery. By contrast, there was no statistically significant difference in MTX2 expression between non-ischemic myocardium and its sham control (Figure S1A-C). Consistently, declined cardiac MTX2 expression in ischemic myocardium was corroborated by immunohistochemical staining, in comparison to the sham-operated controls and non-ischemic myocardium (Figure 1G-H). Next, neonatal rat ventricular cardiomyocytes (NRVMs) were isolated and subjected to H/R attack. We found that MTX2 protein and mRNA expressions were significantly decreased in cardiomyocytes after H/R stimulation in vitro (Figure 1D-1F). However, MTX2 expression was not significantly changed at both protein and mRNA levels between the Con and H/R groups in both fibroblasts and endothelial cells (Figure S2A-F), suggesting that MTX2 expression was only decreased in myocardial cells in response to I/R attack. Taken together, given the decrease of MTX2 at both transcriptional and translational levels, a causative relationship between MTX2 and I/R insult is strongly indicated.
MTX2 is downregulated in myocardial I/R hearts. In vivo, 10-week WT mice were subjected to 45-min ischemia followed by reperfusion at different time points. In vitro, neonatal rat ventricular cardiomyocytes (NRVMs) were isolated and treated with 9-h of hypoxia and 3-h of reoxygenation (H/R) to simulate the ischemia/reperfusion (I/R) injury. (A-B) Western blot and associated quantification of MTX2 protein levels in ischemic myocardial tissue at the indicated time points after I/R induction (n = 6). (C) Quantitative real-time polymerase chain reaction (qPCR) analysis of heart Mtx2 levels in ischemic myocardial tissue at the indicated time points after I/R induction (n = 6). (D-E) Western blot and associated quantification of MTX2 protein levels in reoxygenated cardiomyocytes (n = 6). (F) qPCR analysis of Mtx2 mRNA levels in cardiomyocytes under H/R condition (n = 6). (G-H) Representative immunohistochemical staining of MTX2 in ischemic and non-ischemic myocardial tissue after 3-h reperfusion and its associated quantification (n = 6. Scale bars: 1 mm (left row), 100 μm (middle row), 200 μm (right row). (B, C, H) Data were analyzed by one-way ANOVA followed by Tukey post-hoc test. (E, F) Data were analyzed by Two-tailed unpaired Student t-test. All values are presented as mean ± SD. ns, not significant; MTX2, metaxin 2; Isc, ischemia; Rep, reperfusion; I/R, ischemia/reperfusion; H/R, hypoxia/reoxygenation.
Knockdown of Mtx2 exacerbates cardiac I/R injury
To mimic the effects of MTX2 reduction observed in I/R mice and further gain insight into the role of MTX2 in myocardial I/R injury, cardiac-specific Mtx2 knockout mice (cKO) were created using tamoxifen-inducible Cre recombinase system. Mtx2flox/flox mice were created by generating a floxed allele of Mtx2 with exon 3 flanked by loxp sites. These mice were then crossed with α-MHC-MerCreMer mice (Figure S3A). The genotypes of the offspring mice were verified and Mtx2flox/flox/MerCreMer mice were injected with tamoxifen (40 mg/kg/day) for 5 days to induce specific deletion of the Mtx2 gene in cardiomyocytes. Age-matched MerCreMer mice without the floxed Mtx2 allele were administered an equivalent dose of tamoxifen and served as controls (Ctrl) throughout the entire experiment. After a 2-week washing-out period, the knockout efficiency of Mtx2 in cKO mice was examined (Figure S3B). The genotype of cKO mice was confirmed by polymerase chain reaction (PCR) (Figure S3C). Moreover, the protein level of MTX2 was significantly decreased in the cKO group compared with Ctrl mice, validating the effective deletion of the Mtx2 gene in cardiomyocytes after tamoxifen induction (Figure S3D-E).
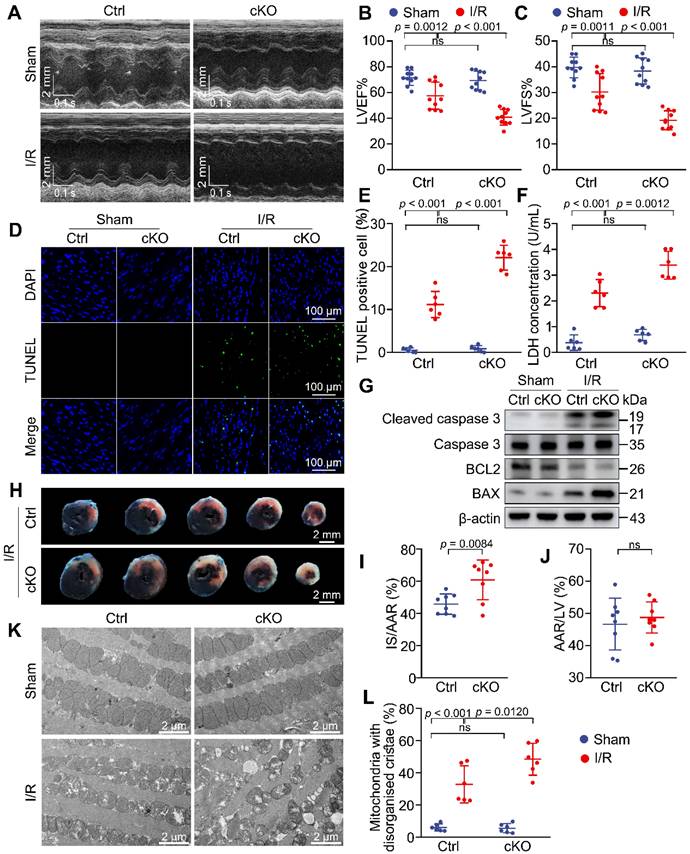
To assess the effects of Mtx2 knockout on cardiac function, echocardiography was performed on cKO and their Ctrl littermates with sham or I/R surgery. In sham-operated animals, cardiac function was comparable between the Ctrl and cKO groups. However, 24 h post-I/R procedure, cKO mice showed aggravated cardiac dysfunction compared with the Ctrl group, as indicated by a marked decline in LV ejection fractions (LVEF) and fractional shortenings (LVFS), as well as a significant increase in LV end-diastolic diameter (LVEDD) and LV end-systolic diameter (LVESD) (Figure 2A-C, Table S1). Moreover, cardiac apoptosis was significantly increased in cKO mice after I/R attack, as evidenced by cleaved caspase-3/caspase-3 ratio, BAX/BCL2 ratio and TUNEL/DAPI double staining (Figure 2D, 2E, 2G and Figure S4). In agreement with these findings, Mtx2-deficient hearts showed a remarkable elevation of cardiomyocyte injury measured by LDH release (Figure 2F). Compared with the Ctrl group, the cKO hearts exhibited a significantly enlarged I/R-induced infarct size (Figure 2H-J). Additionally, Mtx2 deletion aggravated I/R-induced mitochondria cristae disorder and the appearance of vacuoles (Figure 2K-L). Collectively, these in vivo molecular, structural, and functional experiments show that cardiac-specific deletion of Mtx2 detrimentally affects the outcome of I/R insult.
Knockdown of Mtx2 exacerbates cardiac I/R injury. 10-week control (Ctrl) mice or Mtx2 cKO (cKO) mice were subjected to sham or I/R surgery. (A-C) Representative M-Mode echocardiographic images and measurements of LV ejection fraction and LV fractional shortening at 24 h after reperfusion in the indicated groups (n = 10). (D-E) Representative cardiac apoptosis detected by TUNEL staining after 3-h reperfusion and TUNEL-positive cardiomyocyte quantification. Green: TUNEL-positive apoptotic nuclei; Blue: DAPI-stained total nuclei. (n = 6). Scale bar: 100 μm. (F) Cardiac damage by LDH release assay after 3-h reperfusion (n = 6). (G) Western blot of cleaved caspase 3, total caspase 3, BAX and BCL2 after 3-h reperfusion (n = 6). (H-J) Representative heart slices stained by Evans blue and tetrazolium chloride (TTC) after 24-h reperfusion. The infarcted size (IS, white), areas of the left ventricle (LV, red plus white plus blue), and areas at risk (AAR, red plus white) were calculated (n = 8). Scale bar: 2 mm. (K-L) Representative transmission electron microscopic (TEM) images of mitochondria in Ctrl or cKO mice subjected to sham or I/R surgery (magnification × 6000 [6 K]) (n=6). (B, C, E, F, L) Data were analyzed by two-way ANOVA, followed by a Tukey post-hoc test. (I, J) Data were analyzed by Two-tailed unpaired Student t-test. All values are presented as mean ± SD. ns, not significant; MTX2, metaxin 2; I/R, ischemia/reperfusion; cKO, cardiac knockout; Ctrl, control.
MTX2 is required for maintaining glucose metabolism in I/R hearts
To explore the molecular mechanisms responsible for the negative consequences of Mtx2 deletion, we performed RNA sequencing (RNA-Seq) of cKO hearts subjected to sham or I/R operation. RNA-seq analysis revealed 2032 differentially expressed genes between Ctrl + I/R and cKO + I/R samples (Figure 3A). Gene Ontology (GO) enrichment analysis of the downregulated genes in cKO hearts identified significant enrichment of genes related to substrate metabolism (Figure 3B). Particularly noteworthy was the gene set enrichment analysis (GSEA) which highlighted the TCA cycle and respiratory electron transport as among the most significantly downregulated terms in the cKO + I/R group (Figure 3C).
MTX2 is required for maintaining glucose metabolism in I/R hearts. (A) Cluster analysis of differentially expressed genes determined by RNA seq of I/R hearts in Ctrl and cKO mice (n = 4). (B) Top 10 Gene ontology (GO) pathways that were downregulated in heart tissue of cKO mice compared with Cre controls after I/R injury (n = 4). (C) Gene set enrichment analysis (GSEA) of RNA-seq data reveals Mtx2 deletion is negatively correlated with TCA cycle and respiratory electron transport compared to Ctrl mice (n = 4). (D) OCR curve measured using Seahorse flux analyzer in adult cardiomyocytes isolated from post-I/R injury Ctrl and cKO mice (n = 5). (E) Quantification of basal respiration, maximal respiration, ATP production, and spare capacity according to instruction (n = 5). (F) ECAR curve measured using Seahorse flux analyzer in adult cardiomyocytes isolated from post-I/R injury Ctrl and cKO mice (n = 5). (G) Quantification of glycolysis, glycolytic capacity, and glycolytic reserve according to instruction (n = 5). (H-K) Fatty acid oxidation (FAO) curve measured using Seahorse flux analyzer in adult cardiomyocytes isolated from post-I/R injury Ctrl and cKO mice (n = 5). (L) Quantification of basal respiration due to exogenous FAs according to instruction (n = 5). (M) Quantification of maximal respiration due to exogenous FAs according to instruction (n = 5). Data were analyzed by two-way ANOVA, followed by a Tukey post-hoc test. All values are presented as mean ± SD. ns, not significant; MTX2, metaxin 2; I/R, ischemia/reperfusion; cKO, cardiac knockout; Ctrl, control.
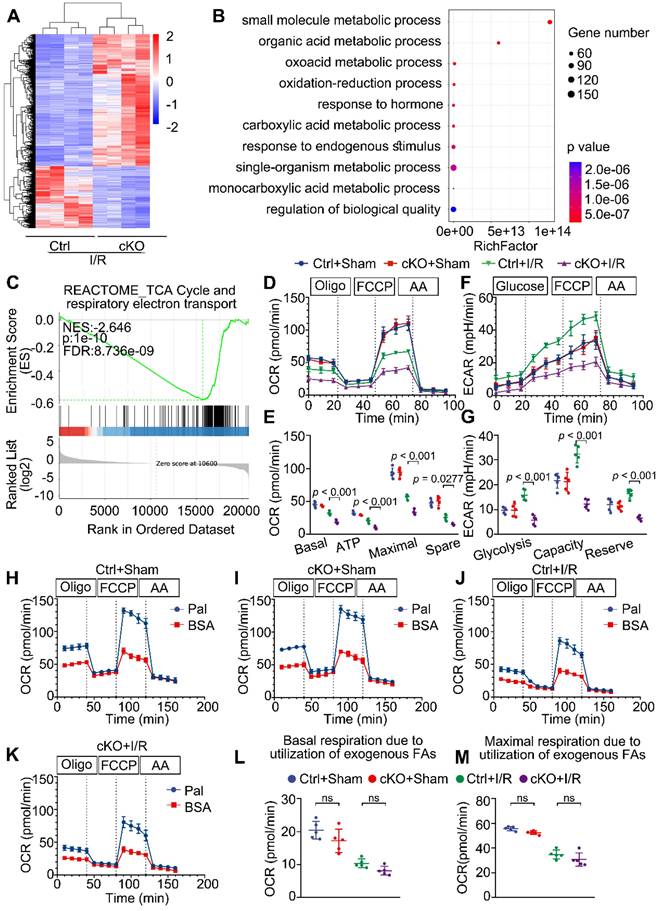
To confirm the effect of MTX2 on substrate metabolism, we tested the metabolic phenotypes of glucose oxidation, glycolysis, and fatty acid oxidation in adult mouse ventricular myocytes isolated from post-I/R injury Ctrl and cKO mice through utilizing the Seahorse flux analyzer. As shown in Figure 3D-E, there were no apparent differences in the energy metabolic phenotypes between Ctrl and cKO mice in the absence of I/R attack. However, upon I/R exposure, cKO hearts exhibited an approximately 50% reduction in glucose oxidation, as calculated by basal and maximal OCRs, ATP production, and spare respiratory capacity. Moreover, Mtx2 deficiency decreased glycolysis at both basal and maximal levels in cardiomyocytes under I/R condition (Figure 3F-G). Furthermore, when exogenous BSA or palmitate-BSA was added as an energy substrate, a slight decrease in both basal and maximal respiration due to utilization of exogenous palmitate-BSA was observed in cKO myocytes but with no significant difference (Figure 3H-M). These observations were consistent with the qPCR analysis of key glucose and fatty acid metabolic enzymes in Ctrl and cKO hearts under I/R attack (Figure S5A-C). Taken together, these results suggest that Mtx2 deletion leads to severe impairment in cardiac OXPHOS activity and glycolytic flux under I/R attack.
Identification of the kinase PKM2 (Pyruvate kinase M2) as an MTX2-interacting protein
To further search for the molecular mechanism of MTX2 regulating glucose metabolism, co-immunoprecipitation (Co-IP) was undertaken in heart tissues from mice with an intra-myocardial injection of flag-tagged MTX2. The IgG immunoprecipitation group served as the negative control. After electrophoresis, silver staining was performed. As shown in Figure 4A, differential spots were observed between samples under sham or I/R. The candidate MTX2-interacting proteins were detected by mass spectrometry (MS) in response to I/R treatment (Figure S6A). The top 10 MTX2-interacting functional proteins were listed in Figure 4B. We noticed PKM among the interacting proteins of MTX2, a key enzyme responsible for the irreversible conversion of phosphoenolpyruvate (PEP) to pyruvate to promote glycolytic flux or OXPHOS (Figure S6B). In light of the suppression of mitochondrial respiration and glycolytic flux, this interaction was quite intriguing and aligned with our disturbed metabolic phenotype. Next, the physical interaction of PKM with MTX2 in Ad-Flag-Mtx2 infected NRVMs was validated by immunoblotting after immunoprecipitation. PKM1 and PKM2 are two alternative splice isoforms encoded by the same Pkm gene and differ only by alternative mRNA splicing [19]. PKM2 was highly expressed in embryonic or stressed hearts, but displayed minimal expression in healthy adult hearts [20]. In contrast, PKM1 is abundantly expressed in normal adult cardiomyocytes [21]. As shown in Figure 4C, PKM2, rather than PKM1, was efficiently coimmunoprecipitated with MTX2, which was consistent with the aforementioned silver staining result of the band between 70 kDa and 55 kDa. We further substantiated the immunoprecipitation of MTX2 with PKM2 (Figure 4D). GST (glutathione S-transferase) pull-down assays were performed with purified recombinant GST-PKM2 and polyhistidine-MTX2, revealing a direct binding between the two proteins (Figure 4E). Consistently, the endogenous interaction of MTX2 with PKM2 was verified by immunofluorescence (Figure 4F).
MTX2 directly interacts with PKM2 to facilitate its tetramer formation hence promoting glucose metabolism. (A) Mice were received intra-myocardial injection with Flag-tagged Ad-Mtx2 7 days before sham or I/R operation. The heart lysates were immunoprecipitated with an anti-Flag antibody or IgG (negative control antibody). The eluates were resolved on SDS-PAGE and silver stained. (B) Top 10 MTX2-interacting functional proteins from mass spectrometry analysis of peptides pulled down using anti-FLAG antibody. (C) Co-immunoprecipitation of overexpressed Flag-Mtx2 and endogenous PKM1 and PKM2 in NRVMs. Lysates were immunoprecipitated with the anti-Flag antibody and immunoblotted with the indicated antibodies (n = 6). (D) Co-immunoprecipitation of overexpressed GST-PKM2 and endogenous MTX2 in NRVMs. Lysates were immunoprecipitated with the anti-GST antibody and immunoblotted with the indicated antibodies (n = 6). (E) Glutathione S-transferase (GST) pull-down assays with recombinant purified GST-PKM2 and His-MTX2. (F) Representative confocal microscopy images of immunofluorescence staining of MTX2, PKM2, Mitotracker and DAPI (4',6-diamidino-2-phenylindole) in NRVMs (n = 6). Scale bar, 20 μm. (G-H) Representative Western blot image and associated quantification of cardiac PKM2 oligomerization from Ctrl and cKO mice after 3-h reperfusion (n = 6). (I-J) Representative Western blot image and associated quantification of cardiac PKM2 oligomerization from cKO mice treated with TEPP-46 or vehicle 15 min before I/R surgery (n = 6). (K) PK activity in Ctrl and cKO mice under I/R attack (n = 6). (L-M) Ctrl or cKO mice were intraperitoneally injected with TEPP-46 15 min before I/R surgery. After 3-h reperfusion, adult cardiomyocytes were isolated for OCR and ECAR determination using a seahorse flux analyzer. (L) OCR curve of adult cardiomyocytes isolated from Ctrl and cKO mice treated with vehicle or TEPP-46 in the presence of I/R attack (n = 5). (M) ECAR curve of adult cardiomyocytes isolated from Ctrl and cKO mice treated with vehicle or TEPP-46 in the presence of I/R attack (n = 5). Data were analyzed by Two-tailed unpaired Student t-test. All values are presented as mean ± SD. ns, not significant; MTX2, metaxin 2; PKM2, pyruvate kinase M2; H/R, hypoxia/reoxygenation; I/R, ischemia/reperfusion; cKO, cardiac knockout; Ctrl, control.
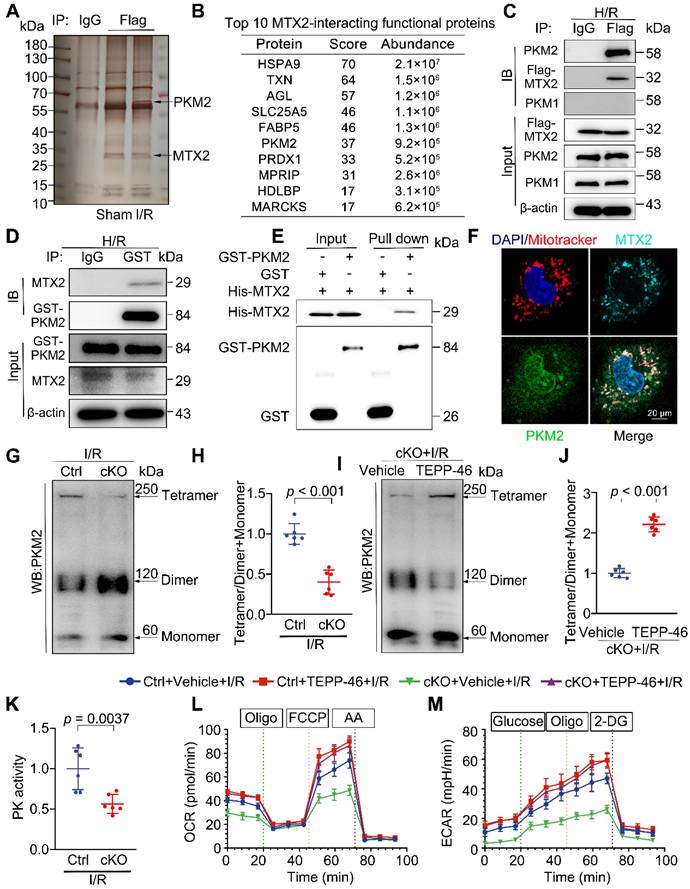
Given that PKM2 might be a potential target binding protein of MTX2 in NRVMs, we next sought to investigate whether the mRNA or protein level of PKM2 was changed in Mtx2-deficient hearts. The expression of PKM2 was scarcely detectable under sham surgery and was significantly induced in response to I/R attack (Figure S7A-B). As shown in Figure S8A-B, there was no significant difference in the protein and mRNA levels of PKM2 between the Ctrl and cKO groups following I/R exposure. However, compared with the control group, the cKO hearts showed a notable decrease in PK activity in response to I/R challenge, indicating that MTX2 modulates PKM2 activity without affecting Pkm2 transcription (Figure 4K).
MTX2 modulates PKM2 tetramer formation to promote glucose metabolism
PKM2 has been found to primarily exist in two multimeric forms in tumor cells: an enzymatically active tetrameric form and a nearly inactive dimeric form at physiological concentrations of PEP [22]. To identify the mechanism underlying MTX2's regulation of PKM2 activity, cross-linking studies were performed. As shown in Figure 4G-H, compared with the control hearts, the level of PKM2 tetramer was lower and the inactive dimer form was accumulated in the Mtx2 knockout hearts after I/R attack. This was consistent with the observed reduction in PK activity and the overall efficiency of glucose metabolism. We next sought to find a small molecule PKM2 activator capable of reversing the clinical phenotypes and metabolic abnormalities due to reduced PKM2 activity caused by Mtx2 deletion. TEPP-46 has been extensively characterized as a specific activator of PKM2, facilitating the conversion of PKM2 dimers to tetramers [23-25]. We observed that compared with the cKO group, TEPP-46 administration successfully upregulated the tetramer form of PKM2, as well as the ratio of the tetramer to dimer + monomer forms in the I/R hearts (Figure 4I-J). We next asked whether declined PKM2 tetramerization was responsible for the metabolic dysfunction in the hearts of cKO mice. TEPP-46 was administered via intraperitoneal injection 15 min before I/R surgery. OCR and ECAR in adult cardiomyocytes isolated from Ctrl or cKO mice with TEPP-46 treatment were determined via a seahorse metabolic flux analyzer after I/R surgery. As shown in Figure 4L-M and Figure S9A-G, the reduced level of glycolysis and oxidative phosphorylation caused by Mtx2 deletion was effectively rescued by TEPP-46. Importantly, the cKO and Ctrl mice pre-treated with TEPP-46 before I/R surgery exhibited comparable measurements of glucose oxidation and glycolysis, which suggests that activating PKM2 is sufficient to rescue the metabolic dysfunction caused by Mtx2 knockout. Together, these data further support that MTX2 regulates cardiac glucose metabolism by interfering with the dimer/tetramer equilibrium of PKM2.
MTX2 regulates PKM2 tetramerization via interacting with PKM2 through its TOM37 domain
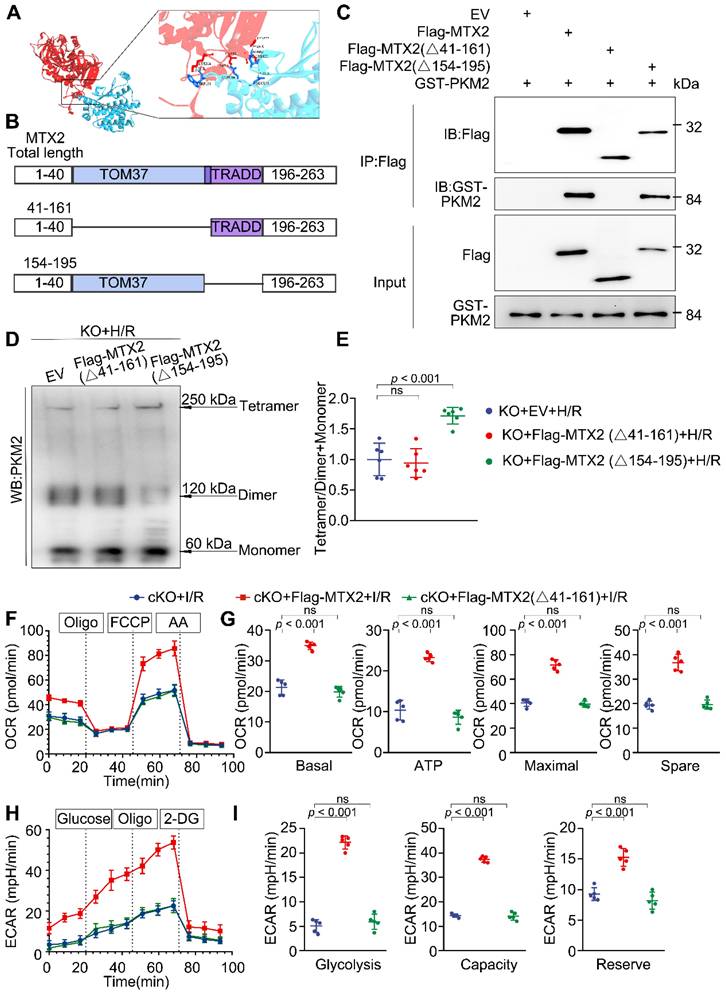
To delineate the molecular domains instrumental in MTX2 and PKM2 interaction, we utilized H-DOCK for molecular docking simulations of complexes MTX2 and PKM2. The interactive residues were predicted based on optimal free energy calculations and structure-based analysis of protein interaction interfaces. The docking results showed a favorable interaction between MTX2 and PKM2, with a calculated free energy of -14.14 kcal/mol (free energy < 0 was considered significant), indicating a positive interaction between MTX2 and PKM2. The predicted 3-dimensional crystal structures of the MTX2-PKM2 complex suggested the potential involvement of the MTX2 TOM37 domain or TRADD domain in its interaction with PKM2 (Figure 5A). Therefore, Flag-tagged MTX2 mutants based on its structural domain were constructed (Flag-MTX2 (Δ41-161 AA) plasmid, Flag-MTX2 (Δ154-195 AA) plasmid) (Figure 5B). Mtx2 knockout H9C2 cells were generated using the CRISPR-Cas9 technique. We performed immunoprecipitation experiments in Mtx2 KO H9C2 cells co-expressing plasmids of Flag-MTX2 mutants and GST-PKM. As shown in Figure 5C, PKM2 predominantly precipitated with Flag-MTX2 (full-length MTX2) and Flag-MTX2 (Δ154-195 AA) mutant. However, Flag-MTX2 (Δ41-161 AA) mutant transfection failed to capture the co-expressed GST-PKM2 in Mtx2 KO H9C2 cells, indicating that MTX2 might primarily interact with PKM2 via its TOM37 domain (41-161 AA). To elucidate whether MTX2 regulates PKM2 tetramerization via interaction with its TOM37 domain (41-161 AA), we overexpressed MTX2 mutants in Mtx2 KO H9C2 cells after H/R insult. Following glutaraldehyde cross-linking, the proportion of the tetramer in Flag-MTX2 (Δ154-195 AA) mutant overexpressing cells was notably increased, and the dimeric PKM2 reduced when compared with control cells (Figure 5D-E). However, deletion of the 41-161 AA domain in MTX2 abrogated its ability to promote PKM2 tetramerization (Figure 5D-E). To understand whether or not this sequence of MTX2 was associated with its regulation on cardiomyocyte glucose metabolism, we next determined the glycolytic and mitochondrial phenotypes in Mtx2-deficient cardiomyocytes overexpressing Flag-MTX2 (full-length MTX2) or Flag-MTX2 (Δ41-161 AA) mutant. Specifically, cKO mice were transfected with adenoviruses to overexpress a control vector, MTX2, or MTX2 (Δ41-161 AA). One week after adenovirus intra-myocardial injection, mice were subjected to I/R surgery. After 3-h reperfusion, cardiomyocytes were isolated for the OCR and ECAR determination. In line with the cross-linking studies, we found that overexpression of MTX2 resulted in a notable elevation in both oxidative phosphorylation and glycolysis when compared to the vector control, whereas MTX2 (Δ41-161 AA) overexpression failed to induce an obvious alteration in either ECAR or OCR (Figure 5F-I). Together, these results suggest that MTX2 orchestrates PKM2 tetramerization through its TOM37 domain (41-161 AA), thereby promoting glucose metabolism in cardiomyocytes.
MTX2 regulates PKM2 tetramerization via interacting with PKM2 through its TOM37 domain. (A) Structure-based representation of MTX2 (blue)-PKM2 (red) protein binding model. (B) Construction of wild-type (total length) and truncated mutants of MTX2. (C) Mtx2 KO H9C2 cells were transfected with Flag-tagged wild-type (total length) MTX2 and its truncated mutants. Whole-cell lysates were immunoprecipitated with anti-FLAG antibody and then immunoblotted with indicated proteins. (D-E) Representative Western blot image and associated quantification of cardiac PKM2 oligomerization from Mtx2 KO H9C2 cells overexpressing MTX2 mutants before H/R attack (n = 6). (F) OCR curve of adult cardiomyocytes isolated from cKO mice overexpressing Flag-tagged adenovirus carrying full-length MTX2 or MTX2 (△41-161 AA) in the presence of I/R attack (n = 5). (G) Quantification of basal respiration, maximal respiration, ATP production, and spare capacity according to instruction (n = 5). (H) ECAR curve of adult cardiomyocytes isolated from cKO mice overexpressing Flag-tagged adenovirus carrying full-length MTX2 or MTX2 (△41-161 AA) in the presence of I/R attack (n = 5). (I) Quantification of glycolysis, glycolytic capacity, and glycolytic reserve according to instruction (n = 5). Data were analyzed by one-way ANOVA followed by Tukey post-hoc test. All values are presented as mean ± SD. ns, not significant; MTX2, metaxin 2; PKM2, pyruvate kinase M2; H/R, hypoxia/reoxygenation; I/R, ischemia/reperfusion; cKO, cardiac knockout; KO, knockout.
Activating PKM2 rescues the exacerbated I/R injury in cKO hearts
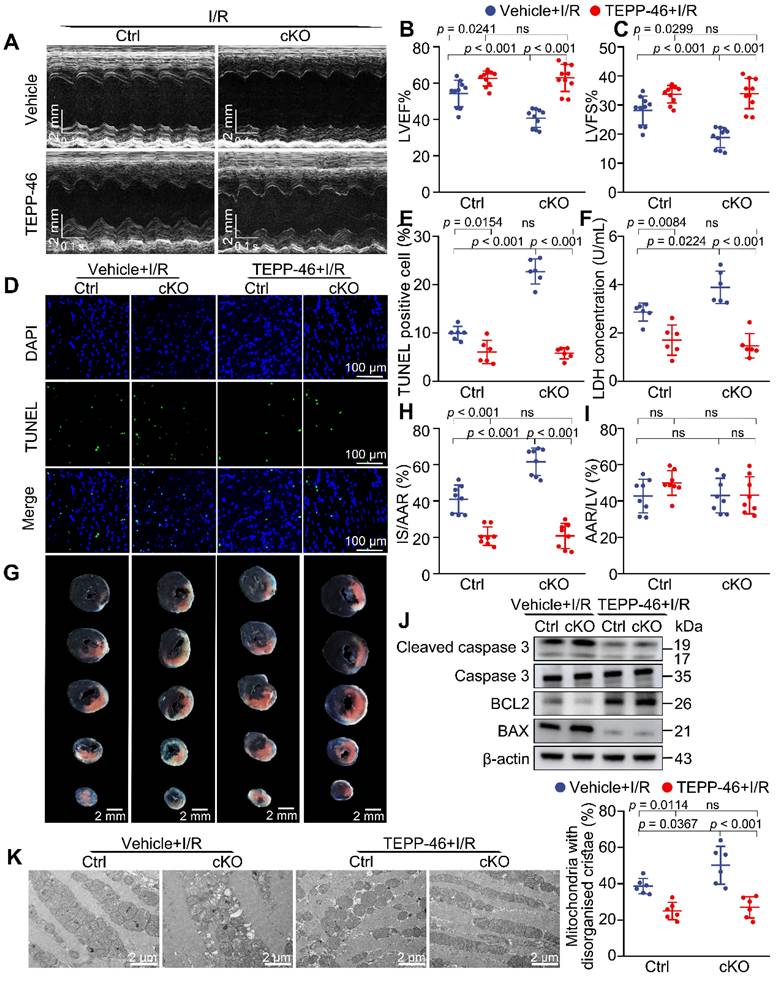
Given our findings that enhancing PKM2 activity could alleviate the metabolic defect caused by Mtx2 deficiency, we sought to examine whether reduced PKM2 activity is responsible for exacerbated I/R phenotype in cKO mice. TEPP-46 was intraperitoneally injected 15 min before I/R surgery. Both cKO and their Cre littermates were then subjected to 45-min ischemia and 24-h reperfusion for echocardiographic detection. The cardiac function of the TEPP-46-treated group was significantly improved in comparison with the untreated cKO group, as evidenced by a marked increase in LVEF and LVFS, along with a significant reduction in LVEDD and LVESD (Figure 6A-C, Table S2). Apoptosis induced by Mtx2 deficiency during I/R was decreased by TEPP-46 treatment, as indicated by decreased TUNEL-positive myocytes, cleaved caspase 3/ caspase 3 ratio and BAX/BCL2 ratio (Figure 6D, 6E, 6J and Figure S10). Furthermore, in response to I/R attack, MTX2 loss-of-function exacerbated LDH release, which was reversed by pharmacological PKM2 activation (Figure 6F). In agreement with these findings, TTC/ Evans blue staining showed that TEPP-46 treated mice exhibited a smaller infarct size compared with cKO mice (Figure 6G-I). Additionally, TEM analysis exhibited an increased proportion of mitochondria with disorganized cristae in cKO heart under I/R attack, which was significantly alleviated by TEPP-46 treatment (Figure 6K).
Activating PKM2 rescues the exacerbated I/R injury in cKO hearts. 10-week Ctrl mice or cKO mice were intraperitoneally injected with TEPP-46 15 min before sham or I/R surgery. (A-C) Representative M-Mode echocardiographic images from and measurements of LV ejection fraction and LV fractional shortening at 24 h after reperfusion in the Ctrl and cKO mice treated with vehicle or TEPP-46 (n = 10). (D-E) Representative cardiac apoptosis detected by TUNEL staining after 3-h reperfusion in the Ctrl and cKO mice treated with vehicle or TEPP-46 and TUNEL-positive cardiomyocyte quantification. Green: TUNEL-positive apoptotic nuclei; Blue: DAPI-stained total nuclei. (n = 6). Scale bar: 100 μm. (F) Cardiac damage by LDH release assay after 3 h reperfusion in the Ctrl and cKO mice treated with vehicle or TEPP-46 (n = 6). (G-I) Representative heart slices stained by Evans blue and tetrazolium chloride (TTC) after 24-h reperfusion in the Ctrl and cKO mice treated with vehicle or TEPP-46. The infarcted size (IS, white), areas of the left ventricle (LV, red plus white plus blue), and areas at risk (AAR, red plus white) were calculated (n = 8). Scale bar: 2 mm. (J) Western blot of cardiac cleaved caspase 3, total caspase 3, BAX and BCL2 after 3-h reperfusion in the Ctrl and cKO mice treated with vehicle or TEPP-46 (n = 6). (K) Representative transmission electron microscopy (TEM) images and quantitative analysis of the proportion of mitochondria with disorganized cristae in the Ctrl and cKO mice treated with vehicle or TEPP-46 (n = 6). Data were analyzed by two-way ANOVA, followed by a Tukey post-hoc test. All values are presented as mean ± SD. ns, not significant; MTX2, metaxin 2; I/R, ischemia/reperfusion; cKO, cardiac knockout; Ctrl, control.
Importantly, the cKO group recovered to the same level as the Ctrl mice after TEPP-46 administration in response to I/R attack, suggesting that reversing the disturbance of glucose metabolism is sufficient to rescue the pathological mechanisms caused by Mtx2 deficiency. Together, these findings demonstrate that activating PKM2, which is suppressed by MTX2 loss-of-function, can rescue myocardial apoptosis, infarction, and ventricular dysfunction during I/R.
Reconstitution of MTX2 in the heart alleviates I/R injury
Since MTX2 was downregulated in the I/R hearts, we proceeded to investigate whether MTX2 reconstitution could rescue the metabolic dysfunction under I/R stress. We utilized an adenovirus carrying Mtx2 gene to overexpress MTX2 specifically in cardiac tissue. This was achieved through intra-myocardial injection, as depicted in Figure S11A-B. A shift in the distribution of PKM2 from dimers to tetramers was observed in MTX2 overexpressed hearts (Figure S11C-D). Furthermore, by utilizing a seahorse flux analyzer, we found that MTX2 reconstitution improved I/R-induced decline of mitochondrial respiration and glycolytic flux in cardiomyocytes, and the baseline OCR and ECAR were comparable between the mice treated with Ad-EV and Ad-Mtx2 (Figure S11E-H).
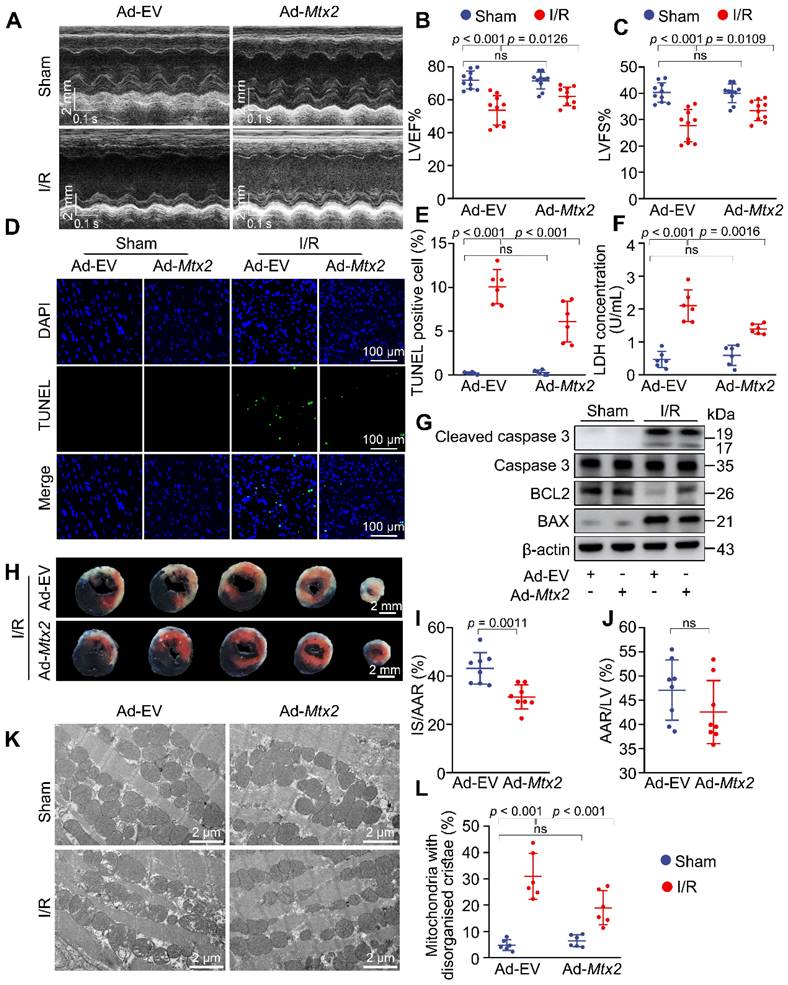
Given the pivotal role of MTX2 in cardiac PKM2 tetramerization and glucose metabolism, we next sought to determine whether overexpressing MTX2 could protect the heart against cardiac I/R injury. As shown in Figure 7A-C, MTX2 overexpression in the heart significantly alleviated cardiac dysfunction induced by I/R attack as indicated by elevated LVEF and LVFS. Both LVEDD and LVESD were significantly increased in response to I/R attack and Ad-Mtx2 administration before I/R attack restored the LVEDD to 3.93 ± 0.23 and LVESD to 2.61 ± 0.24 mm (Table S3). Moreover, MTX2 gain-of-function significantly reduced caspase 3 activation, BAX/BCL2 ratio and TUNEL-positive cells in I/R mice, suggesting the I/R-induced apoptosis was mitigated by overexpression of MTX2 (Figure 7D, 7E, 7G and Figure S12). Consistently, the MTX2-overexpressed hearts showed a reduced LDH release, which also confirmed a better recovery compared with the control group in response to I/R attack (Figure 7F). In addition, MTX2 upregulation led to a decrease in cardiac infarct size (Figure 7H-J). As shown in Figure 7K-L, MTX2 overexpression alleviated I/R-induced mitochondria cristae disorder and the appearance of vacuoles. Taken together, these data demonstrate that restoration of MTX2 is capable of ameliorating the detrimental effects of I/R injury.
Reconstitution of MTX2 in the heart alleviates I/R injury. 10-week mice received intra-myocardial injection with EV or Mtx2 adenovirus 7 d before sham or I/R operation. (A-C) Representative M-Mode echocardiographic images and measurements of LV ejection fraction and LV fractional shortening at 24 h after reperfusion in Ctrl and MTX2-overexpressed mice (n = 10). (D-E) Representative cardiac apoptosis detected by TUNEL staining after 3-h reperfusion in Ctrl and MTX2-overexpressed mice and TUNEL-positive cardiomyocyte quantification. Green: TUNEL-positive apoptotic nuclei; Blue: DAPI-stained total nuclei. (n = 6). Scale bar: 100 μm. (F) Cardiac damage by LDH release assay after 3-h reperfusion (n = 6). (G) Western blot of cardiac cleaved caspase 3, total caspase 3, BAX and BCL2 after 3-h reperfusion in Ctrl and MTX2-overexpressed mice (n = 6). (H-J) Representative heart slices stained by Evans blue and tetrazolium chloride (TTC) after 24-h reperfusion in Ctrl and MTX2-overexpressed mice. The infarcted size (IS, white), areas of the left ventricle (LV, red plus white plus blue), and areas at risk (AAR, red plus white) were calculated (n = 8). Scale bar: 2 mm. (K-L) Representative transmission electron microscopy (TEM) images and quantitative analysis of the proportion of mitochondria with disorganized cristae after 3-h reperfusion in Ctrl and MTX2-overexpressed mice (n = 6). (B, C, E, F, L) Data were analyzed by two-way ANOVA, followed by a Tukey post-hoc test. (I, J) Data were analyzed by Two-tailed unpaired Student t-test. All values are presented as mean ± SD. ns, not significant; MTX2, metaxin 2; I/R, ischemia/reperfusion; cKO, cardiac knockout; Ctrl, control.
Discussion
Myocardial reperfusion leads to compromised metabolism and dysfunctional cardiac performance [26,27]. Unfortunately, the underlying mechanism of this damage is far from clear. In this investigation, we report that loss of MTX2 contributes to the disruption of glucose metabolism in the heart in response to I/R stress. We have found that MTX2 interacts with PKM2 through its TOM37 domain to promote the tetramerization of PKM2, thus increasing PK activity. Both increasing MTX2 abundance and PKM2 agonist significantly rescued the reperfusion injury phenotypes. Our results, therefore, reveal for the first time a crucial role of MTX2 in regulating cardiac metabolism and I/R injury.
Metaxin proteins have been implicated in the integration of β-barrel proteins into the OMM [11,28]. Very recently, Cartron et al. showed a mechanistic role for MTX in TNF-induced cell death [16,17,29,30]. However, whether MTX2, a cytosolic peripherally-associated OMM protein of the MTX family with limited known functions, plays a role in I/R injury remained elusive. Here we have reported, for the first time, a significant decrease in MTX2 expression in the hearts of I/R mice, highlighting its pivotal role as an endogenous regulator of cardiac energy metabolism in response to I/R insult. Cardiac-specific knockdown of Mtx2 led to impaired mitochondrial OXPHOS and glycolysis in response to I/R. Conditional deletion of Mtx2 causes a deterioration in classic phenotypes of I/R injury, including impaired cardiac function, enlarged infarct size, increased cardiomyocyte apoptosis, and irregular mitochondrial cristae. To this end, we performed an MTX2 conditional overexpression by intra-myocardial injection adenovirus carrying Mtx2 and demonstrated a promising therapeutic effect on I/R injury.
Unlike MTX1, MTX2 lacks a mitochondrial C-terminal membrane anchor domain, and is peripherally associated with the outer mitochondrial membrane facing the cytoplasm [15,31], implicating other possible interactions with cytosolic proteins. Our results showed that MTX2 interacts with PKM2 through its TOM37 domain, and is required for the tetramerization of PKM2 in I/R mice. These observations collectively highlight a new role of MTX2 besides its conventional transport function.
The deprivation of oxygen and energy supply induces various myocardial damage during I/R [32,33]. Therefore, restoring ATP produced from the glucose metabolism may fulfill the energy demand of cardiomyocytes and is protective against I/R injury. Consistent with this notion, Li et al. found that interventions targeting the PDH flux at reperfusion could increase glucose oxidation and improve the recovery of myocardial function [26]. Furthermore, the ischemic pre-conditioning protective mechanism is strongly associated with activated aerobic glycolysis during early reperfusion [34,35]. In our study, results from the Seahorse flux analyzer support our hypothesis that MTX2 is instrumental in maintaining the mitochondrial OXPHOS and glycolytic flux during I/R, and PKM2 is identified for its regulation of glucose metabolism. These observations bolster the hypothesis that increased metabolic flux through glycolysis by activating PKM2 would mitigate the accumulation of toxic glucose-derived end products, augment flux through the TCA cycle, and improve mitochondrial function in cardiomyocytes.
As a rate-limiting enzyme for converting phosphoenolpyruvate to pyruvate, PKM2 occupies a crucial regulatory position in glycolytic flux [36,37]. Due to its low expression in the normal adult hearts, its function in cardiomyocytes has thus far remained largely unexplored. In this context, it is interesting that Mtx2 deficiency does not cause a significant change in PKM2 expression during I/R. Instead, the activity of PKM2 is decreased after Mtx2 knockdown in cardiomyocytes. The mechanism could be a shift in the distribution of PKM2 from tetramers to dimers, with the latter being less catalytically active. Interaction between MTX2 with PKM2 through its TOM37 domain prompts the formation of a tetrameric PKM2, thereby serving as a metabolic switch to augment glycolytic flux in cardiac tissue. Thus, our data revealed that PKM2 functions indispensably for the metabolic control of MTX2 in the I/R hearts.
However, the precise mechanism by which MTX2 regulates this conformational change in PKM2 remains elusive. Liang et.al propose that PKM2 phosphorylation may regulate this interesting transition [38], it is still unknown whether MTX2 induces PKM2 phosphorylation, thereby promoting increased tetramerization. To this end, we conducted additional experiments to investigate the underlying mechanisms. We isolated NRVMs, overexpressed MTX2, subjected them to hypoxia/reoxygenation (H/R) treatment, performed immunoprecipitation (IP) with PKM2 antibody, and immunoblotting (IB) with pan-phosphorylation antibody. The results demonstrated that MTX2 overexpression significantly increased PKM2 phosphorylation levels (Figure S13). Integrating this observation with our present study, we propose that MTX2 might facilitate the conversion of PKM2 dimers to tetramers by enhancing PKM2 phosphorylation. Nonetheless, the detailed mechanisms still await further investigation.
The concept of metabolic therapies has garnered great attention in the management of cardiovascular disease in recent years [39,40]. TEPP-46, a small molecule capable of regulating PKM2 polymerization, was reported to be widely implicated in various pathological conditions such as kidney diseases, immune system diseases, and cancer [24,41,42]. In our intervention studies, the activation of PKM2 by TEPP-46 improves the pathological glucose metabolism in cKO mice undergoing I/R surgery. Furthermore, our data demonstrated that TEPP-46 confers cardioprotective effects against cKO-exacerbated I/R injury, as evidenced by less cardiomyocyte apoptosis, reduced infarct size, and improved cardiac function. These findings provided a promising candidate for pharmacologically regulating PKM2 allostery in the treatment of cardiac diseases characterized by impaired glucose metabolism and decreased MTX2 expression.
In summary, the present study for the first time provides compelling evidence that MTX2 is an important regulator of maintaining metabolic homeostasis in the I/R hearts through modulating PKM2 tetramer formation. The direct interaction of MTX2 and PKM2 through its TOM37 domain provides new molecular insights into the regulation of PK activity. Considering that MTX2 is downregulated in response to I/R insult, our findings suggest that early intervention targeting MTX2 and PKM2 might be an effective strategy to protect against I/R injury by preserving metabolic homeostasis in the hearts.
Abbreviations
cKO: conditional knockout; ECAR: extracellular acidification rate; H/R: hypoxia/reoxygenation; I/R: ischemia-reperfusion; LVEF: left ventricular ejection fraction; LVFS: left ventricular fractional shortening; MTX2: metaxin 2; NRVMs: neonatal rat ventricular cardiomyocytes; OCR: oxygen consumption rate; PKM2: pyruvate kinase M2; RNA-seq: RNA sequencing; WT: wild type.
Supplementary Material
Supplementary methods, figures and tables.
Acknowledgements
We sincerely appreciate Professor Shunying Hu and Professor Qinglei Zhu from the Department of Cardiology, The Sixth Medical Center of Chinese PLA General Hospital, for their constructive suggestions.
Funding
This work was supported by the National Natural Science Foundation of China (Grant Nos. 81870178, 82200394, 82400421), and the Post-Doctoral Science Foundation of China (Grant No. 2022M713861).
Author contributions
Yundai Chen conceived the research and supervised the study. Yueyang Li, Zhenyu Xiong, and Tao Chen wrote the article. Yueyang Li, Zhenyu Xiong, and Yu Li conducted the experiments on mice. Yueyang Li, Zhenyu Xiong, and Yang Li performed the cell experiments. Yufan Jiang, Miao Wang and Mingyi Wang bred and maintained mouse lines. Jie Liu and Mingrui Ma collected echocardiographic images. Xiaofeng Zhai and Li Yi carried out RNA/protein extraction. All authors contributed to the experimental design, data analysis, and data interpretation.
Data availability
The data underlying this article will be shared on reasonable request to the corresponding author.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Roth GA, Johnson C, Abajobir A, Abd-Allah F, Abera SF, Abyu G. et al. Global, regional, and national burden of cardiovascular diseases for 10 causes, 1990 to 2015. J Am Coll Cardiol. 2017;70:1-25
2. Heusch G. Myocardial ischaemia-reperfusion injury and cardioprotection in perspective. Nat Rev Cardiol. 2020;17:773-89
3. Davidson SM, Ferdinandy P, Andreadou I, Bøtker HE, Heusch G, Ibáñez B. et al. Multitarget strategies to reduce myocardial ischemia/reperfusion injury: JACC review topic of the week. J Am Coll Cardiol. 2019;73:89-99
4. Zuurbier CJ, Bertrand L, Beauloye CR, Andreadou I, Ruiz-Meana M, Jespersen NR. et al. Cardiac metabolism as a driver and therapeutic target of myocardial infarction. J Cell Mol Med. 2020;24:5937-54
5. Ramachandra CJA, Hernandez-Resendiz S, Crespo-Avilan GE, Lin YH, Hausenloy DJ. Mitochondria in acute myocardial infarction and cardioprotection. EBioMedicine. 2020;57:102884
6. Prag HA, Aksentijevic D, Dannhorn A, Giles AV, Mulvey JF, Sauchanka O. et al. Ischemia-selective cardioprotection by malonate for ischemia/reperfusion injury. Circ Res. 2022;131:528-41
7. Chouchani ET, Pell VR, James AM, Work LM, Saeb-Parsy K, Frezza C. et al. A unifying mechanism for mitochondrial superoxide production during ischemia-reperfusion injury. Cell Metab. 2016;23:254-63
8. Russell RR, Li J, Coven DL, Pypaert M, Zechner C, Palmeri M. et al. AMP-activated protein kinase mediates ischemic glucose uptake and prevents postischemic cardiac dysfunction, apoptosis, and injury. J Clin Invest. 2004;114:495-503
9. Tang J, Zhang K, Dong J, Yan C, Hu C, Ji H. et al. Sam50-Mic19-Mic60 axis determines mitochondrial cristae architecture by mediating mitochondrial outer and inner membrane contact. Cell Death Differ. 2020;27:146-60
10. Jian F, Chen D, Chen L, Yan C, Lu B, Zhu Y. et al. Sam50 regulates PINK1-Parkin-mediated mitophagy by controlling PINK1 stability and mitochondrial morphology. Cell Rep. 2018;23:2989-3005
11. Zhao Y, Song E, Wang W, Hsieh CH, Wang X, Feng W. et al. Metaxins are core components of mitochondrial transport adaptor complexes. Nat Commun. 2021;12:83
12. Elouej S, Harhouri K, Le Mao M, Baujat G, Nampoothiri S, Kayserili H. et al. Loss of MTX2 causes mandibuloacral dysplasia and links mitochondrial dysfunction to altered nuclear morphology. Nat Commun. 2020;11:4589
13. Abudu YP, Shrestha BK, Zhang W, Palara A, Brenne HB, Larsen KB. et al. SAMM50 acts with p62 in piecemeal basal- and OXPHOS-induced mitophagy of SAM and MICOS components. J Cell Biol. 2021;220:e202009092
14. Takeda H, Tsutsumi A, Nishizawa T, Lindau C, Busto JV, Wenz LS. et al. Mitochondrial sorting and assembly machinery operates by β-barrel switching. Nature. 2021;590:163-69
15. Armstrong LC, Saenz AJ, Bornstein P. Metaxin 1 interacts with metaxin 2, a novel related protein associated with the mammalian mitochondrial outer membrane. J Cell Biochem. 1999;74:11-22
16. Ono K, Wang X, Kim SO, Armstrong LC, Bornstein P, Han J. Metaxin deficiency alters mitochondrial membrane permeability and leads to resistance to TNF-induced cell killing. Protein Cell. 2010;1:161-73
17. Cartron PF, Petit E, Bellot G, Oliver L, Vallette FM. Metaxins 1 and 2, two proteins of the mitochondrial protein sorting and assembly machinery, are essential for Bak activation during TNF alpha triggered apoptosis. Cell Signal. 2014;26:1928-34
18. He B, Yu H, Liu S, Wan H, Fu S, Liu S. et al. Mitochondrial cristae architecture protects against mtDNA release and inflammation. Cell Rep. 2022;41:111774
19. Ma WK, Voss DM, Scharner J, Costa ASH, Lin KT, Jeon HY. et al. ASO-based PKM splice-switching therapy inhibits hepatocellular carcinoma growth. Cancer Res. 2022;82:900-15
20. Li Q, Li C, Elnwasany A, Sharma G, An YA, Zhang G. et al. PKM1 exerts critical roles in cardiac remodeling under pressure overload in the heart. Circulation. 2021;144:712-27
21. Hauck L, Dadson K, Chauhan S, Grothe D, Billia F. Inhibiting the Pkm2/b-catenin axis drives in vivo replication of adult cardiomyocytes following experimental MI. Cell Death Differ. 2021;28:1398-417
22. Tang Y, Feng M, Su Y, Ma T, Zhang H, Wu H. et al. Jmjd4 facilitates Pkm2 degradation in cardiomyocytes and is protective against dilated cardiomyopathy. Circulation. 2023;147:1684-704
23. Qi W, Keenan HA, Li Q, Ishikado A, Kannt A, Sadowski T. et al. Pyruvate kinase M2 activation may protect against the progression of diabetic glomerular pathology and mitochondrial dysfunction. Nat Med. 2017;23:753-62
24. Seki SM, Posyniak K, McCloud R, Rosen DA, Fernández-Castañeda A, Beiter RM. et al. Modulation of PKM activity affects the differentiation of T(H)17 cells. Sci Signal. 2020;13:eaay9217
25. Veras FP, Publio GA, Melo BM, Prado DS, Norbiato T, Cecilio NT. et al. Pyruvate kinase M2 mediates IL-17 signaling in keratinocytes driving psoriatic skin inflammation. Cell Rep. 2022;41:111897
26. Li T, Zhang Z, Kolwicz SC, Abell L, Roe ND, Kim M. et al. Defective branched-chain amino acid catabolism disrupts glucose metabolism and sensitizes the heart to ischemia-reperfusion injury. Cell Metab. 2017;25:374-85
27. Yadid M, Lind JU, Ardoña HAM, Sheehy SP, Dickinson LE, Eweje F. et al. Endothelial extracellular vesicles contain protective proteins and rescue ischemia-reperfusion injury in a human heart-on-chip. Sci Transl Med. 2020;12:8005
28. Kozjak-Pavlovic V, Ross K, Benlasfer N, Kimmig S, Karlas A, Rudel T. Conserved roles of Sam50 and metaxins in VDAC biogenesis. EMBO Rep. 2007;8:576-82
29. Wang X, Ono K, Kim SO, Kravchenko V, Lin SC, Han J. Metaxin is required for tumor necrosis factor-induced cell death. EMBO Rep. 2001;2:628-33
30. Petit E, Cartron PF, Oliver L, Vallette FM. The phosphorylation of Metaxin 1 controls Bak activation during TNFα induced cell death. Cell Signal. 2017;30:171-8
31. Jo Y, Kim B, Shin DY. AREL1 E3 ubiquitin ligase inhibits TNF-induced necroptosis via the ubiquitination of MTX2. Exp Ther Med. 2021;22:1195
32. Kuznetsov AV, Javadov S, Margreiter R, Grimm M, Hagenbuchner J, Ausserlechner MJ. The role of mitochondria in the mechanisms of cardiac ischemia-reperfusion injury. Antioxidants (Basel). 2019;8:454
33. Wu R, Smeele KM, Wyatt E, Ichikawa Y, Eerbeek O, Sun L. et al. Reduction in hexokinase II levels results in decreased cardiac function and altered remodeling after ischemia/reperfusion injury. Circ Res. 2011;108:60-9
34. Abel ED. mTOR mediated metabolic rewiring and ischemic preconditioning, its complicated. Circ Res. 2021;128:652-4
35. Nadtochiy SM, Urciuoli W, Zhang J, Schafer X, Munger J, Brookes PS. Metabolomic profiling of the heart during acute ischemic preconditioning reveals a role for SIRT1 in rapid cardioprotective metabolic adaptation. J Mol Cell Cardiol. 2015;88:64-72
36. Zheng X, Boyer L, Jin M, Mertens J, Kim Y, Ma L. et al. Metabolic reprogramming during neuronal differentiation from aerobic glycolysis to neuronal oxidative phosphorylation. Elife. 2016;5:e13374
37. Palsson-McDermott EM, Curtis AM, Goel G, Lauterbach MA, Sheedy FJ, Gleeson LE. et al. Pyruvate kinase M2 regulates Hif-1α activity and IL-1β induction and is a critical determinant of the warburg effect in LPS-activated macrophages. Cell Metab. 2015;21:65-80
38. Liang LJ, Yang FY, Wang D, Zhang YF, Yu H, Wang Z. et al. CIP2A induces PKM2 tetramer formation and oxidative phosphorylation in non-small cell lung cancer. Cell Discov. 2024;10:13
39. Lopaschuk GD, Karwi QG, Tian R, Wende AR, Abel ED. Cardiac energy metabolism in heart failure. Circ Res. 2021;128:1487-513
40. Ussher JR, Elmariah S, Gerszten RE, Dyck JR. The emerging role of metabolomics in the diagnosis and prognosis of cardiovascular disease. J Am Coll Cardiol. 2016;68:2850-70
41. Chen Y, Bai X, Chen J, Huang M, Hong Q, Ouyang Q. et al. Pyruvate kinase M2 regulates kidney fibrosis through pericyte glycolysis during the progression from acute kidney injury to chronic kidney disease. Cell Prolif. 2024;57:e13548
42. Liu F, Ma M, Gao A, Ma F, Ma G, Liu P. et al. PKM2-TMEM33 axis regulates lipid homeostasis in cancer cells by controlling SCAP stability. Embo j. 2021;40:e108065
Author contact
![]() Corresponding author: Yundai Chen, M.D. E-mail: chenyundaicom.cn; Zhenyu Xiong, M.D. E-mail: drxiongzhenyucom. Tao Chen, M.D. E-mail: chentao1com.cn. Department of Cardiology, the Sixth Medical Centre, Chinese PLA General Hospital, 6 Fucheng Road, Beijing, 100048, China. Tel./Fax: +86-10-55499009.
Corresponding author: Yundai Chen, M.D. E-mail: chenyundaicom.cn; Zhenyu Xiong, M.D. E-mail: drxiongzhenyucom. Tao Chen, M.D. E-mail: chentao1com.cn. Department of Cardiology, the Sixth Medical Centre, Chinese PLA General Hospital, 6 Fucheng Road, Beijing, 100048, China. Tel./Fax: +86-10-55499009.