Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
1. Origins of TAMs
2. Phenotypic and functional...
3. Metabolism of TAMs
4. Interactions between TAMs and...
5. Advances in Targeting TAMs in...
6. Concluding Remarks and...
Abbreviations
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2025; 15(15):7378-7408. doi:10.7150/thno.113727 This issue Cite
Review
Focusing on the interplay between tumor-associated macrophages and tumor microenvironment: from mechanism to intervention
Hancheng Wu1,2,3,4,#, Jing Li5,#, Ruilin Yao1,2,3,4, Jing Liu1,2,3,4, Lili Su1,2,3,4, ![]() , Wenjie You1,2,3,4,6,
, Wenjie You1,2,3,4,6, ![]()
1. Department of Respiratory and Critical Care Medicine, Shandong Provincial Hospital affiliated to Shandong First Medical University, Jinan, Shandong 250021, China.
2. Department of Respiratory and Critical Care Medicine, Shandong Provincial Hospital, Shandong University, Jinan, Shandong 250021, China.
3. Shandong Key Laboratory of Infectious Respiratory Disease, Jinan, Shandong 250000, China.
4. Medical Science and Technology Innovation Center, Shandong First Medical University & Shandong Academy of Medical Sciences, Jinan, Shandong 250000, China.
5. Department of Respiratory and Critical Care Medicine, Shandong Provincial Public Health Clinical Center, Jinan, Shandong 250013, China.
6. Departments of Thoracic Surgery and State Key Laboratory of Genetic Engineering, Fudan University Shanghai Cancer Center, Shanghai 200032, China.
# These authors contributed equally to this work and share first authorship.
Received 2025-3-13; Accepted 2025-5-26; Published 2025-6-20
Abstract

Immunotherapy has generated promising outcomes in cancer treatment; however, therapeutic responses are hampered by immunosuppression in the tumor microenvironment (TME). This has resulted in increased study of key immune cells in the TME as therapeutic interventions. Tumor-associated macrophages (TAMs), a major component of infiltrating immune cells in the TME, display high plasticity, largely dependent on cues received from their surroundings. Although significant progress in metabolomics and single-cell omics has unraveled the metabolic and functional heterogeneity of TAMs across several types of cancer, the development of TAM-targeted therapy remains challenging. In the present review, the crosstalk between TAMs and other components in TME, such as tumor cells, immune cells, cancer-associated fibroblasts, and extracellular matrix is highlighted. Additionally, updated insights into the origin, heterogeneity, and metabolic reprogramming of TAMs are discussed, and relevant approaches of targeting TAMs in clinical investigations are summarized. The present review provides a deeper understanding of TAMs within the microenvironment network, aimed at identifying candidate targets to improve cancer immunotherapy.
Keywords: cancer immunotherapy, tumor-associated macrophages, tumor microenvironment, heterogeneity, interaction
Introduction
Cancer immunotherapy, which is designed to counter immune tolerance and induce antitumor immunity, has contributed substantially to cancer treatment in the past decade. Immunotherapeutic approaches involving immune checkpoint blockade (ICB), chimeric antigen receptor (CAR) T-cell engineering therapy, and tumor vaccines have made significant progress in preclinical and clinical studies [1-3]. However, the responsiveness of patients to immunotherapy varies significantly across different tumor types and among individuals. Only a small fraction of patients fully respond to immunotherapy, and the factors underlying responses remain largely unknown [4]. Therefore, a deeper understanding of the cancer-immunity cycle is of great importance to increase the efficacy of cancer immunotherapy.
The tumor microenvironment (TME) is composed of tumor cells and nonmalignant immune cells, stromal cells, vascular structures, and extracellular components. Certain immune populations serve as antitumor effectors, such as cytotoxic T lymphocytes (CTLs) and natural killer (NK) cells, while other cell types, including regulatory T (Treg) cells, myeloid-derived suppressor cells (MDSCs), and alternatively activated type 2 (M2) tumor-associated macrophages (TAMs), suppress immune-mediated tumor cytotoxicity and facilitate tumor progression [5]. Emerging evidence has indicated that the poor efficiency of cancer immunotherapy is tightly associated with the immunosuppressive status driven by immune cell subsets of a myeloid lineage in the TME [6]. Among myeloid cells, TAMs, recruited from circulating monocytes or derived from embryonic precursors and bone marrow as tissue-resident macrophages (TRMs), are abundantly present in the TME of most tumors [7]. TAMs are a highly plastic and heterogeneous immune cell type. High-throughput techniques, such as single-cell sequencing, have shown a previously unrecognized image of TAM heterogeneity and complexity in multiple tumor types. Novel insights into the metabolic profiles of TAM subpopulations pave the way for attractive methods for TAM metabolic reprogramming [8]. The phenotypic and metabolic versatility underlies their interactions with other components in the TME, including tumor cells, infiltrating immune cells, cancer-associated fibroblasts (CAFs), endothelial cells (ECs), adipocytes, extracellular matrix (ECM), etc. In general, TAMs fuel cancer malignancy to promote survival, proliferation, angiogenesis, distant spreading, stemness, immunosuppression, and therapeutic response [9]. All the above features of TAMs highlight them as attractive therapeutic targets to aid cancer immunotherapy, which have been extensively investigated in preclinical and clinical studies [9]. A broad range of strategies have been developed, from targeting the recruitment, heterogeneity, and metabolism of TAMs, depleting and enhancing the phagocytosis and reprogramming of TAMs, to genetically engineered macrophages. There is intense interest in understanding the biology of TAMs, with the ultimate goal of overcoming the limitations of cancer immunotherapy.
In this review, we first describe the origins of TAMs in TME, based on recruitment, differentiation, and polarization. The heterogeneity, diversity, functional profiles, and metabolic characteristics of TAMs are also reviewed in the setting of various tumors. We summarize the interaction and crosstalk between TAMs and surrounding cellular and noncellular components in the TME to comprehensively show how TAMs influence tumorigenesis and therapeutic efficiency. Finally, the relationship between TAMs and current antitumor treatments, as well as the emerging strategies of targeting TAMs as a therapeutic tool, are discussed.
1. Origins of TAMs
1.1 Recruitment
As shown in Figure 1, there are various chemoattractants, including chemokines and growth factors, that influence the recruitment of circulating monocytes into neoplastic tissues. It is widely accepted that the CCL2-CCR2 signal serves as a key determinant for monocyte and TAM recruitment. Bottazzi et al. showed that TAMs are recruited by tumor cell-derived chemotactic factors, which were later identified to be CCL2 [10]. Accordingly, targeting CCL2 with neutralizing antibody in vivo or shRNA in tumor cells significantly reduced the infiltration of TAMs in mouse renal cell carcinoma xenografts [11]. Notably, CCL2 can originate from other stromal cells within the TME, in addition to tumor cells. Immunohistochemical analysis showed a positive correlation between stromal CCL2 expression and the number of TAMs in human breast cancer samples [12]. Consistently, deletion of CCL2 in the host, rather than in tumor cells, significantly decreased TAM infiltration, angiogenesis, and lung metastasis in mouse orthotopical 4T1 tumor models [13]. Despite the dominant role of the CCL2-CCR2 axis in TAM recruitment, CCL2 deletion resulted in an approximately 60% TAM reduction in mouse models of endometrial cancer [14], suggesting the involvement of additional chemoattractant signals to attract TAMs. Another study showed that the interaction of CCL5 with CCR1 and CCR5 promotes monocyte adhesion and immobilization to activated endothelium [15], in line with the chemotactic activity of CCL5 in the recruitment of pro-metastatic TAMs in triple-negative breast cancer (TNBC) [16]. It was demonstrated that the blockade of CCL20-CCR6 interaction in mouse breast cancer models significantly inhibited TAM recruitment in tumors [17]. In mouse models of glioma, TAMs were recruited to tumors when treated with radiation in part through the interaction between CXCL12 (stromal cell-derived factor-1, SDF-1) and its receptor CXCR4 [18]. Hypoxia induced the production of CCL11 in breast cancer cells, which recruited TAMs to hypoxic regions possibly via interacting with CCR3 [19].
Chemoattractant signals that influence the recruitment of TAMs. TAMs are mainly recruited by the interaction of CCL2, derived from tumor cells and stromal cells within the TME, and CCR2. Other chemoattractant-receptor axis, including CCL20-CCR6, CXCL12-CXCR4 and CCL11-CCR3, also attract macrophages to TME. The interaction of CCL5 with CCR1 and CCR5 promotes monocyte adhesion and immobilization to activated endothelium. As for distinct TAM subsets, M1 TAMs can be recruited by CCL19, CCL21, CCL24, CCL25, CXCL8, CXCL10, XCL2 and CCL7, while M2 TAMs are recruited by CCL7. CCL12 induces chemotaxis of MRC1+TIE2highCXCR4high TAM subset to the perivascular area. CCL12 and CCL7 recruit inflammatory Ly6Chigh monocytes via interaction with their co-receptor CCR2. As for different processes in TAM recruitment, while CCL2-CCR2 axis induces the recruitment of MAMs, CCL3-CCR1 axis promotes MAM-cancer cell interaction and MAM retention at the site of metastasis. In advanced stage of metastasis, MAMs migrate to the metastatic site via the CCL3-CCR5 axis.
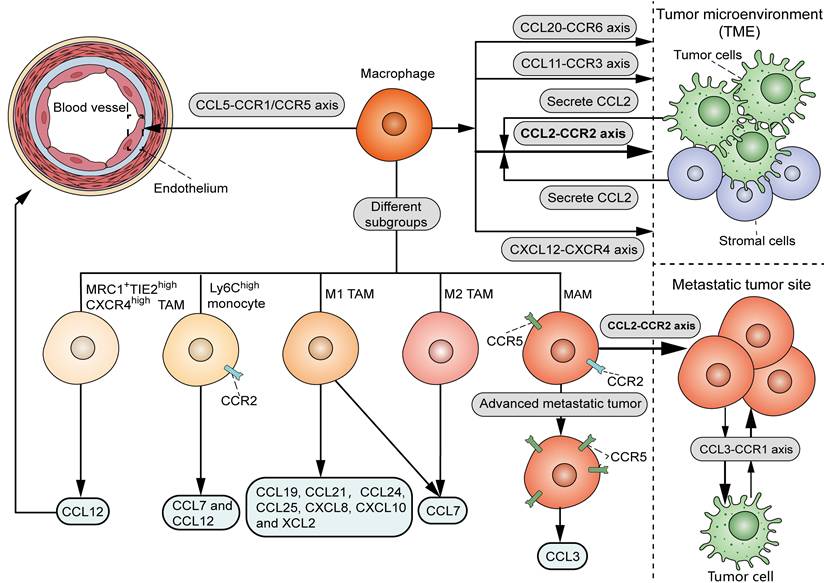
Interestingly, it was found that different chemokines attract distinct TAM subsets, which provides a theoretical foundation for isolating specific subsets of TAMs from the TME (Figure 1). Xuan et al. screened chemokines that differentially recruit classically activated type 1 (M1) or M2 TAMs [20]. Their data showed that CCL19, CCL21, CCL24, CCL25, CXCL8, CXCL10 and XCL2 selectively recruited M1 macrophages, while CCL7 induced chemotaxis of both M1 and M2 macrophages. In mouse LLC tumor models, chemotherapy induced CCL12 production, which was specifically chemotactic for the MRC1+TIE2highCXCR4high TAM subset to the perivascular area [21]. Another study showed that CCL12 and CCL7 could recruit inflammatory Ly6Chigh monocytes via interaction with their co-receptor CCR2 [22]. It should be noted that different chemokine signals may regulate a specific process involved in TAM recruitment. In murine breast cancer models, the CCL2-CCR2 axis played a vital role in the recruitment of metastasis-associated macrophage (MAM) in metastatic tumors, whereas the CCL3-CCR1 signals specifically promoted MAM-cancer cell interaction and subsequent MAM retention at the site of metastasis [23]. Compared to the early MAM accumulation in pulmonary metastasis in a mouse renal tumor model, MAMs increased CCR5 expression in the late stage of metastasis, and migrated to the metastatic site via involvement of the CCL3-CCR5 axis [24]. Therefore, targeting TAMs by interfering with certain chemoattractant-receptor interactions may serve as a precise therapeutic approach for cancer management.
1.2 Differentiation
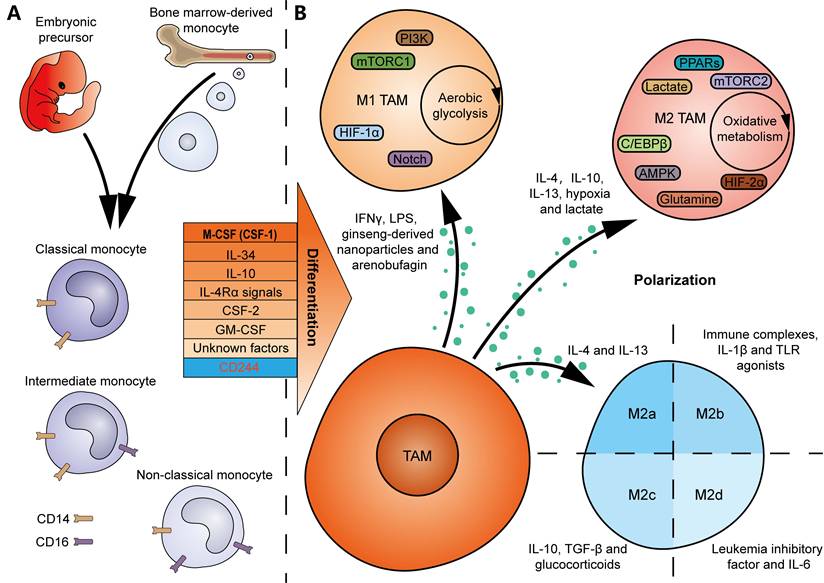
Bone marrow-derived monocytes are considered the origins of multiple TRMs. And TRM-derived TAMs are abundantly present in the TME of several tumor types [25]. Many colony-stimulating factors contribute to monocyte-to-macrophage differentiation in vitro and in vivo (Figure 2A). A null mutation in colony-stimulating factor 1 (CSF1) resulted in defective TRMs in mice, suggesting CSF1 as a major regulator of macrophage differentiation [26]. IL-34, secreted by keratinocytes in skin epidermis and neurons in central nervous system, regulates the development of local Langerhans cells and microglia [27]. Notably, deficiency in CSF1R, the receptor gene for CSF1 and IL-34, led to a greater decline both in the number and diversity of macrophages in mice [28], indicating that macrophages are primarily regulated by the CSF1R signaling. In addition, IL-10 is an inducer of decidual macrophages by promoting oxidative phosphorylation (OXPHOS) in bone marrow-derived monocytes [29]. IL-4 receptor α (IL-4Rα) signals in T cells, which is activated following pleural-dwelling nematode infection in C57BL/6 mice, enables the differentiation of resident large-cavity macrophages [30]. Dong et al. characterized the necessity of CSF-2 in alveolar CD44+ macrophage development and maintenance [31]. GM-CSF, secreted by intestinal PDGFRA+CD142-/low fibroblasts in response to inflammation in inflammatory bowel disease, promotes the transition of monocytes to local CCR2+CD206+ macrophages [32]. Nevertheless, the differentiation of macrophages may depend on unknown growth factors or cytokines, since mice deficient in G-CSF, GM-CSF, and M-CSF still generate macrophages when challenged with sterile peritonitis [33]. The above findings suggest that tissue-specific factors or microenvironmental cues should enable local differentiation of macrophages. In contrast, deletion of CD244 in monocytes using Cre-Lox recombination in mice resulted in a higher infiltration of anti-tumor Ly6Clow macrophages, demonstrating an inhibitory role for CD244 in anti-tumor macrophage generation [34].
Factors that drive TAM differentiation and polarization. (A) TAMs can originate either from embryonic precursors in yolk sac and fetal liver, or from bone marrow-derived monocytes. Many factors, including CSF-1, IL-34, IL-10, IL-4Rα signals, CSF-2, GM-CSF and unknown factors, promote monocyte-to-macrophage differentiation. In contrast, CD244 inhibits the differentiation of anti-tumor macrophages. Human monocytes can be divided into the classical CD14++CD16-, intermediate CD14++CD16+, and non-classical CD14+CD16++ monocytes. (B) IFN-γ, LPS, ginseng-derived nanoparticles and arenobufagin drive M1 TAM polarization, metabolically characterized by aerobic glycolysis, whereas IL-4, IL-10, IL-13, hypoxia and lactate drive M2 TAM polarization, characterized by oxidative metabolism. Intrinsic signaling pathways, including PI3K, mTORC1, HIF-1α and Notch signals, are involved in M1 TAM polarization, whereas mTORC2, HIF-2α, AMPK, PPARs, glutamine, lactate and C/EBPβ signals are involved in M2 TAM polarization. Moreover, M2 TAMs can be divided into 4 subgroups: M2a macrophages induced by IL-4 and IL-13, M2b macrophages induced by immune complexes, IL-1β and TLR agonists, M2c macrophages induced by IL-10, TGF-β and glucocorticoids, and M2d macrophages induced by leukemia inhibitory factor and IL-6.
Human monocytes can be classified as follows: (1) classical CD14++CD16- monocytes, with proinflammatory and antimicrobial roles; (2) intermediate CD14++CD16+ monocytes, with proinflammatory roles; and (3) non-classical CD14+CD16++ monocytes, with patrolling and antiviral roles (Figure 2A). An investigation of ovarian cancer revealed a positive correlation between the proportion of intermediate monocytes in peripheral blood and TAMs exhibiting a CCR2highCD163highCD206highCD86low profile in TME [35]. Meanwhile, another study on B-cell acute lymphoblastic leukemia suggested that non-classical monocytes in peripheral blood are likely to be the primary source of TAMs in TME [36].
With the development of modern lineage tracing techniques, our understanding of the origins of macrophages has been determined. Several TAMs are derived from TRMs that originate from embryonic precursors in the yolk sac or fetal liver, rather than bone marrow-derived monocytes (Figure 2A) [37]. Zhu et al. characterized the sources of TAMs in mouse pancreatic ductal adenocarcinoma (PDAC) models, showing that pancreas-resident macrophages can originate from embryonic development and increase by in situ proliferation during tumor progression [38]. The dual origins are believed to determine the heterogeneity of TAM functions: while monocyte-derived TAMs contribute to antigen presentation in immune reactions, embryo-derived TAMs may play a more potent role in the production of the ECM in tumors [38].
1.3 Polarization
The phenotype of macrophages is plastic and dynamic, which depends on environmental cues (Figure 2B) [39]. For example, TAMs can polarize towards an M1 phenotype in response to IFN-γ and lipopolysaccharide (LPS) stimulation, while IL-4, IL-10, and IL-13 are classical cytokines that drive TAM polarization towards an M2 state [39, 40]. It should be noted that non-cytokine factors also contribute to TAM polarization, for example, hypoxia and lactate, which drive M2 polarization [39]. Additionally, traditional Chinese medicines, such as ginseng-derived nanoparticles and arenobufagin, have been found to promote the polarization of M1 TAMs, thereby achieving anti-tumor effects [41, 42]. The intrinsic signaling pathways that influence M1 TAM polarization include PI3K, mTORC1, hypoxia inducible factor-1α (HIF-1α), and the Notch signaling pathway [40]. In contrast, the pathways involved in M2 TAM polarization are composed of mTORC2, HIF-2α, AMPK, PPARs, glutamine, lactate, and the C/EBPβ signaling pathway [40]. Moreover, metabolic events may play a crucial role in determining their polarization. Typically, M1 macrophages predominantly rely on aerobic glycolysis, whereas M2 macrophages are more dependent on oxidative metabolism, providing potential opportunities to induce reprogramming of TAMs in tumors [43]. Furthermore, the M2 macrophages are divided into 4 subgroups, characterized by different inducers (M2a macrophages induced by IL-4 and IL-13, M2b macrophages induced by immune complexes, IL-1β and Toll-like receptor (TLR) agonists, M2c macrophages induced by IL-10, TGF-β and glucocorticoids, and M2d macrophages induced by IL-6 and leukemia inhibitory factor) (Figure 2B) [44, 45]. These adjustments in classifications better reflect the heterogeneity and diversity of TAMs in vivo.
2. Phenotypic and functional heterogeneity of TAMs
TAMs in the TME are not a homogenous cell population. In contrast, subgroups of these TAMs may even have antagonistic functions. The current mainstream classification of TAMs comes from the suggestion put forward by Mills' team in 2000, that TAMs can be divided into M1 and M2 macrophages (Figure 3A) [46]. In comparison, the undifferentiated macrophages are called M0 macrophages (Figure 3A). M1 macrophages upregulate genes involved in antigen processing, presentation, and co-stimulatory signals [39, 47]. On one hand, M1 macrophages can kill tumor cells through direct phagocytosis and antibody-dependent cell-mediated cytotoxicity (ADCC). Briefly, when phagocytized by macrophages, tumor cells can be processed into antigen peptides, which induce adaptive antitumor immunity with the assistance of MHC and co-stimulatory molecules [47]. On the other hand, M1 macrophages can stimulate Th1-type cytotoxic T cells and recruit Th1, Th17 and cytotoxic T cells [47, 48]. M2 macrophages express high levels of CD163, stabilin-1, CD206, CD301, detin-1 and CD209 [39, 47]. While M2 macrophages play an important role in tissue remodeling, wound healing and homeostasis, certain subsets of M2 macrophages possess tumor-supportive characteristics, such as promoting tumor cell survival, growth, motility, invasion, angiogenesis, immune evasion, stemness, metabolic reprogramming, and therapeutic resistance [49]. In addition, M2 macrophages attract Treg and tumor cells [47, 48]. However, it is acknowledged that M1 and M2 states do not represent two completely separate subpopulations, but rather two extreme phenotypes that macrophages specifically adopt for the needs of the body. Under certain circumstances, the coexistence of cells with different gene signatures and the presence of a mixture of M1 and M2 macrophages is observed [50]. The plasticity of TAMs enables their phenotype and functionality to further lean towards M1 or M2 phenotypes in response to various stimuli from the TME [39]. Thus, the M1 and M2 paradigm is inadequate for further analysis of TAM subpopulations.
Phenotypic and functional heterogeneity of TAMs. (A) Tradditionally, TAMs can be divided into M1 and M2 macrophages, which have different phenotypes and functions. MHC-II, CD80 and CD86 are upregulated in M1 macrophages, while CD163, stabilin-1, CD206, CD301, detin-1 and CD209 are upregulated in M2 macrophages. M1 macrophages can kill tumor cells via direct phagocytosis and ADCC. M1 macrophages also stimulate Th1-type cytotoxic T cells, and recruit Th1, Th17 and cytotoxic T cells. M2 macrophages play an important role in tissue remodeling, wound healing and homeostasis. M2 macrophages promote tumor cell survival, growth, motility, invasion, angiogenesis, immune evasion, stemness, metabolic reprogramming and therapeutic resistance, and attract Treg and tumor cells. Furthermore, M2 macrophages can be divided into four subgroups: M2a, characterized by IL-1R, mannose receptor, CCL17, fibronectin and TGF-β; M2b, characterized by TNFSF14 and CCL1; M2c, characterized by IL-10, TGF-β, CCL16, CCL18 and MerTK; M2d, characterized by CD86 and iNOS. M0, undifferentiated macrophages. (B) Current single-cell sequencing have identified seven major subsets of TAMs in tumors: IFN-TAMs, Reg-TAMs, Inflam-TAMs, LA-TAMs, Angio-TAMs, RTM-TAMs and Prolif-TAMs. Moreover, TAMs can be classified into eight subtypes based on the indicated gene signatures and functions: SPP1+ TAMs, FOLR2+ TAMs, TIE2+ TAMs, TREM2+ TAMs, MARCO+ TAMs, FCN1+ TAMs, C1QC+ TAMs, and ISG15+ TAMs.
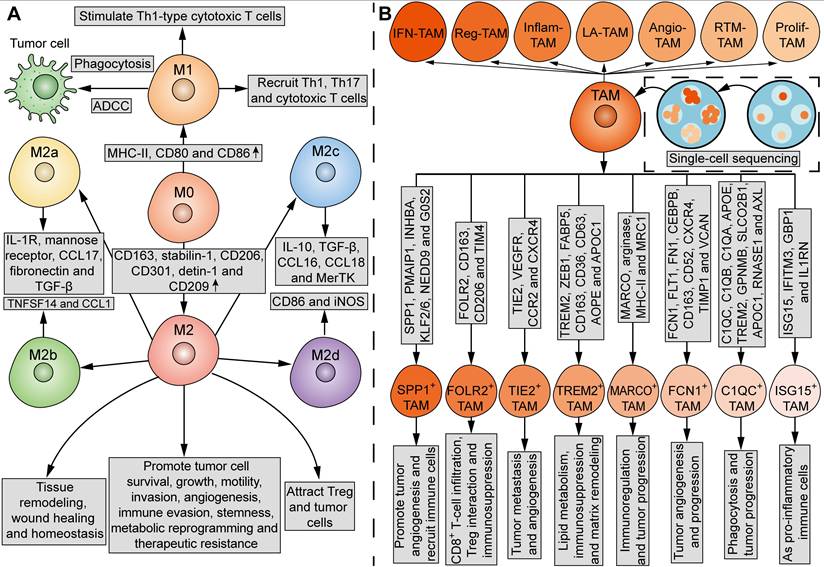
Mantovani and his colleagues in 2004 proposed to further divide M2 macrophages into M2a (characterized by IL-1R, mannose receptor, CCL17, fibronectin, and TGF-β), M2b (characterized by TNFSF14 and CCL1) and M2c (characterized by IL-10, TGF-β, CCL16, CCL18 and MerTK) macrophages, which exhibit different functions and behaviors (Figure 3A) [44]. In 2007, Duluc et al. discovered a new M2 subpopulation and named it M2d macrophages [45]. Compared with M2a-c, M2d macrophages express CD86 and inducible nitric oxide synthetase (iNOS), and exhibit most ovarian TAM phenotypic and functional characteristics, which inhibit T-cell proliferation more effectively [45]. Functionally, while M2a and M2b macrophages play immunomodulatory roles, M2c and M2d subpopulations are involved in immune suppression and tissue remodeling [40, 44]. In 2015, Igor Malyshev and colleagues proposed the M3 switching phenotype, which is characterized by reprogramming towards the M2 phenotype in response to pro-inflammatory factors or reprogramming towards the M1 phenotype in response to anti-inflammatory factors, inconsistent with traditional views [51]. Another study identified a new subgroup of macrophages that can be induced by CXCL4 in atherosclerosis, which was named M4 macrophages [52]. However, until now, there has been no literature report on M4 macrophages in the field of tumor biology.
Current single-cell multi-omics approaches have highlighted the heterogeneity of TAMs. A review of single-cell sequencing studies noticed that certain subsets of TAMs are prevalent in almost all cancer types, which fall into seven major classes: interferon-stimulated (IFN-TAMs), immune-modulated (Reg-TAMs), inflammatory cytokine-enriched (Inflam-TAMs), lipid-associated (LA-TAMs), pro-angiogenic (Angio-TAMs), RTM-like (RTM-TAMs), and proliferative TAMs (Prolif-TAMs) (Figure 3B) [53]. In other studies, TAMs are classified into eight subgroups based on their specific gene signatures and functions: SPP1+ TAMs, which are characterized by SPP1, PMAIP1, INHBA, KLF2/6, NEDD9, and G0S2, and act to promote tumor angiogenesis and recruit immune cells, FOLR2+ TAMs, which are characterized by FOLR2, CD163, CD206, and TIM4, and are involved in CD8+ T-cell infiltration, Treg interaction and immunosuppression, TIE2+ TAMs, which are characterized by TIE2, VEGFR, CCR2, and CXCR4, and are involved in tumor metastasis and angiogenesis, TREM2+ TAMs, which are characterized by TREM2, ZEB1, FABP5, CD163, CD36, CD63, AOPE, and APOC1, and are involved in lipid metabolism, immunosuppression and matrix remodeling, MARCO+ TAMs, which are characterized by MARCO, arginase, MHC-II, and MRC1, and are involved in immunoregulation and tumor progression, FCN1+ TAMs, which are characterized by FCN1, FLT1, FN1, CEBPB, CD163, CD52, CXCR4, TIMP1, and VCAN, and are involved in tumor angiogenesis and progression, C1QC+ TAMs, which are characterized by C1QC, C1QB, C1QA, APOE, TREM2, GPNMB, SLCO2B1, APOC1, RNASE1, and AXL, and are involved in phagocytosis and tumor progression, and ISG15+ TAMs, which are characterized by ISG15, IFITM3, GBP1, and IL1RN, and function as pro-inflammatory immune cells (Figure 3B) [54, 55]. However, the spectrum of TAM subpopulations may expand beyond current classifications, as the transcriptome signatures of macrophages in tumors are more like continuous and dynamic variables, rather than discrete and fixed phenotypes. Spatial localization and tissue-specific programming may play a major role in determining the phenotypic and functional heterogeneity of TAMs, since the macrophages adopt different functional states in response to local stimulations. By defining the precise TAM subgroups, it may be possible to correlate different TAM subgroups with tumor progression for therapeutic purposes.
3. Metabolism of TAMs
Similar to the Warburg effect in tumor cells, cellular metabolism is reprogrammed in TAMs, not only to meet the increased energy demands and biosynthesis, but also to support effector functions, differentiation, and gene expression [40]. Increasing evidence suggests a potential correlation between metabolic profiles and the phenotypic and functional characteristics of TAMs [8, 43]. Understanding the specific metabolic events in distinct TAM subpopulations is indispensable for metabolic modulation of TAM-associated activity in tumors.
3.1 Glucose metabolism
Traditionally, the M1 and M2 macrophages display distinct glycometabolic signatures (Figure 4). The M1 TAMs rely on glycolysis to fight pathogens and tumor cells, accompanied by a truncated TCA cycle [43]. Specifically, the metabolic intermediates of glycolysis, through which glucose is converted to pyruvate and lactate, are rerouted to the oxidative pentose phosphate pathway (PPP) to produce nicotinamide adenine dinucleotide phosphate (NADPH). Then, ROS is generated by NADPH oxidases (NOXs), which are critical for the phagocytic and tumoricidal effects of M1 macrophages [56]. The truncated TCA cycle is characterized by the downregulated levels of isocitrate dehydrogenase (IDH) and upregulated levels of aconitate decarboxylase 1 (ACOD1, also known as IRG1) in M1 macrophage polarization, which increase the production of itaconate (ITA) [57]. ITA inhibits succinate dehydrogenase (SDH), resulting in the accumulation of succinate to stabilize HIF-1α, which supports the inflammatory response and further strengthens glycolysis [58]. Meanwhile, the production of L-arginine is increased in M1 TAMs due to interruption of the TCA cycle, which induces the synthesis of nitric oxide (NO). M2 macrophages exhibit a complete TCA cycle and OXPHOS, with lower glycolytic activity compared to M1 macrophages [43]. M2 macrophages tend to utilize β-oxidation of fatty acids and glutaminolysis, rather than glycolysis, to fuel the TCA cycle turnover [59]. Furthermore, unlike L-arginine-dependent NO generation in M1 macrophages, the M2 macrophages produce polyamines and L-proline from L-arginine to facilitate tissue homeostasis and tumorigenesis [60].
Distinct metabolic profiles of M1 TAMs and M2 TAMs. M1 TAMs are featured by glycolysis, in which G6P is rerouted to PPP to produce NADPH. Then, ROS is generated by NOXs to eliminate pathogens and tumor cells. In M1 macrophage polarization, IDH is downregulated, while IRG1 is upregulated, leading to the production of ITA. ITA inhibits SDH, resulting in the accumulation of succinate to stabilize HIF-1α, which supports the inflammatory response. Meanwhile, L-arginine is increased in M1 TAMs due to interruption of TCA cycle, inducing the synthesis of NO. M2 TAMs are featured by a complete TCA cycle and OXPHOS, fueled by β-oxidation of fatty acids and glutaminolysis. In M2 macrophage polarization, fatty acids can be taken up by CD36 and FATP1, and then lysed by LAL, providing carbon source for FAO to drive TCA cycle and support OXPHOS. Meanwhile, IL-4 increases the uptake of glutamine in macrophages via glutamine transporters. GS and GLUL are upregulated to supply intracellular glutamine for M2 macrophage polarization. Then, glutamine is hydrolysed by glutaminase 1 to produce glutamate, which is transformed into α-KG by GLUD1, in order to fuel TCA cycle and increase FAO and OXPHOS. M2 macrophages can also promote α-KG accumulation via suppressing α-KG dehydrogenase. L-arginine can be converted by Arg-1 into L-ornithine in M2 macrophages. Then, polyamines and L-proline are generated from L-arginine to facilitate tumorigenesis. Tryptophan can be catalyzed by IDO into kynurenine, contributing to immune suppression in M2 macrophages. In response to inflammatory stimuli, glycolytic product ITA is upregulated to potentiate tumor growth by increasing OXPHOS and OXPHOS-driven ROS production. However, glycolysis inhibitor 2-DG can decrease M2 TAM polarization via an AMPK-HIF-1ɑ-dependent pathway. G6P, Glucose-6-phosphate.
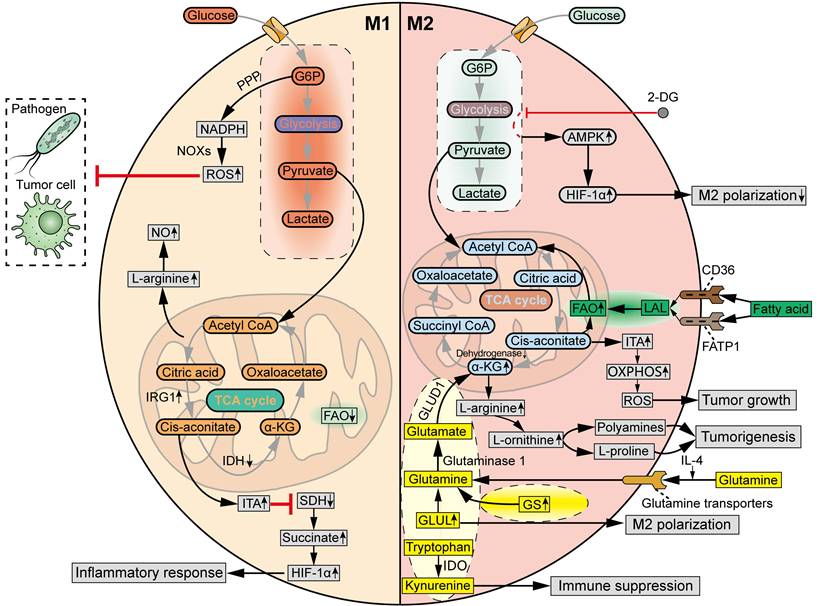
Even so, the glycometabolism of TAMs exhibits increasingly complex and diverse properties (Figure 4). Emerging data indicate that glycolysis is also required for M2 polarization of TAMs. For example, inhibition of glycolysis with 2-Deoxy-d-glucose (2-DG) decreased M2 TAM polarization via an AMPK-HIF-1ɑ-dependent pathway in mouse models [61]. Meanwhile, the hypoxic TME induces metabolic shift of TAMs from oxidative metabolism to the glycolytic pathway, which promotes the pro-tumoral M2 phenotype in TAMs and contributes to immune evasion and tumor progression [62]. In addition, another glycolytic product, ITA, in response to stimuli such as LPS, TLR, and IFN-γ, was reported to potentiate tumor growth by increasing OXPHOS and OXPHOS-driven ROS production [63].
The heterogeneity of TAMs can be attributed to their ability to modulate key regulators of energy metabolism. MHC-IIhigh TAM subgroup exhibits a hampered TCA cycle. In comparison, low MHC-II expression levels result in higher oxidative and glycolytic metabolism and increased L-arginine metabolism [64]. Tim-4+ TAMs display higher levels of OXPHOS and arginase-1 (Arg-1), and respond to mitosis to reduce oxidative stress as compared to Tim-4- TAMs [65]. Tim-4+ TAMs, but not Tim-4- TAMs, promote the peritoneal metastasis of ovarian cancer [65]. At present, there remain large gaps in our understanding of the signatures of glucose metabolism in different subgroups of TAMs.
3.2 Fatty acid and lipid metabolism
Elevated lipid synthesis in TAMs is tightly associated with tumorigenesis (Figure 4). The enhanced fatty acid oxidation (FAO) is one of the most important metabolic features of M2 TAMs. In detail, during M2 macrophage polarization, fatty acids are taken up by the scavenger receptor CD36 and FATP1, and then lysed by lysosomal acid lipase (LAL), to provide a source of carbons for FAO to drive the TCA cycle and support OXPHOS [66]. However, other research has suggested that simultaneous induction of fatty acid biosynthesis and FAO may instead direct the polarization of TAMs towards an antitumor phenotype. For example, TLR9 agonism evokes the antitumor potential of TAMs against CD47+ cancer cells through activating the FAO and shunting the TCA cycle intermediates to de novo lipogenesis [67]. Thereafter, it remains to be determined whether and how the coordination of lipid anabolism and catabolism regulates the activities of TAMs within the TME.
High-throughput techniques have depicted the lipid metabolic features in specific subgroups of TAMs. Single-cell and spatially resolved transcriptomics analysis of breast cancer identified two subsets of lipid-associated macrophages (LAM1 and LAM2) [68]. LAM1 showed a high expression of FABP5 and abundantly present in invasive cancer areas, while LAM2 showed a high expression of APOE and was primarily located in areas with high stroma, adipocyte, lymphocyte, and high PD-1/PD-L1 staining, indicating that those LAMs are associated with immunosuppressive functions. Another single-cell analysis of early-stage smoking-associated non-small cell lung cancer (NSCLC) patients identified two different immunosuppressive TAM subsets within the TME of NSCLC [54]. Specifically, the CCL18+ macrophages were characterized by a higher level of fatty acid OXPHOS and exerted immunosuppressive effects by inhibiting the expression of inflammatory factors. A deeper and more comprehensive exploration is required to understand the lipid metabolic reprogramming in those TAM subsets.
3.3 Amino acid metabolism
Glutamine, the most abundant circulating amino acid in the blood, is tightly associated with metabolic needs both in tumor cells and M2 macrophages (Figure 4). In general, glutamine metabolism is higher in M2 macrophages than in M1 macrophages. IL-4 results in an increased uptake of glutamine in macrophages via glutamine transporters [69]. Additionally, glutamine synthetase (GS) is upregulated in glutamine-deprived conditions to replenish the cellular levels of glutamine [70]. Glutamate-ammonia ligase (GLUL) was found to be upregulated to maintain the supply of glutamine in M2 TAMs, and inhibition of GLUL decreased glutamine metabolism and resulted in the repolarization of macrophages towards the M1 phenotype [69]. Glutamine is hydrolyzed by glutaminase 1 to generate glutamate, which is transformed into α-ketoglutarate (α-KG) by glutamate dehydrogenase 1 (GLUD1), to fuel the TCA cycle and increase FAO and OXPHOS in M2 macrophages [71]. Moreover, M2 macrophages can also promote α-KG accumulation by suppressing the enzymatic activity of α-KG dehydrogenase.
The metabolic pathways of other amino acids also exert a significant influence on the phenotypic and functional characteristics of TAMs (Figure 4). Tryptophan is catalyzed by the enzyme indoleamine 2,3-dioxygenase (IDO) into kynurenine, also contributing to the immunosuppressive phenotype of TAMs [72]. However, further studies are warranted to elucidate the diverse functions of other amino acids in metabolic reprogramming of TAM subpopulations at the cellular level.
4. Interactions between TAMs and TME components
The TME is composed of tumor cells, infiltrating immune cells, including TAMs, MDSCs, dendritic cells (DCs), neutrophils, and lymphocytes, stromal cells, such as CAFs, ECs, and the ECM [5]. Research on the interactions between certain members of the TME, such as DCs and mast cells, with TAMs is still limited. Therefore, this review only provides an overview of members that have been extensively studied thus far.
4.1 Tumor cells
Under steady-state conditions, macrophages are capable of recognizing the “eat me” signal to engulf pathogens, apoptotic cells, or fragments. Conversely, the “do not eat me” signals, such as CD47, PD-L1, and CD24, can inhibit the phagocytic ability of macrophages upon contact. Tumor cells can evade the phagocytic activity of macrophages by increasing the expression of “do not eat me” signals and decreasing the expression of “eat me” signals.
As mentioned above, TAMs, specifically M2 macrophages, participate in tumorigenesis and cancer progression [49]. A variety of factors, including protein, metabolites, and non-coding RNAs from TAM-derived extracellular vesicles (EVs), function in the interactions between TAMs and tumor cells (Figure 5). (1) Proliferation: Exosomal circ-0020256 from TAMs was found to promote the proliferation of cholangiocarcinoma cells by regulating the miR-432-5p/E2F3 axis pathway [73]. However, exosomal miR-628-5p from M1 TAMs was reported to inhibit hepatocellular carcinoma (HCC) proliferation by suppressing the m6A modification of circFUT8 [74]. In addition, exosomal a disintegrin and metalloproteinase 15 (ADAM15) from TAMs can slow tumor growth and improve survival when co-injected with tumor cells into nude mice [75]. (2) Metastasis: By using mass spectrometry, Zheng et al. demonstrated that M2 TAM-derived exosomes transfer functional apolipoprotein E (ApoE) to gastric cancer cells, activate PI3K-Akt signaling pathway, and promote tumor migration [76]. Exosomal miR-21-5p and miR-155-5p from M2 TAMs boosted the metastasis of colorectal cancer [77]. However, PROS1, a protein derived from macrophages, has been identified to exhibit anti-metastatic activities [78]. Mechanistically, PROS1 modulated the peripheral inflammation and immune responses, rather than the TAM-related signals within tumor cells, which ultimately decreased tumor metastasis. (3) Immune evasion: Typically, M2 TAMs can facilitate tumor immune evasion by expressing immunosuppressive cytokines and enzymes, and immune checkpoints. Moreover, GATA3 from TAM-derived EVs supported immune evasion of ovarian cancer cells through the CD24/Siglec-10 axis [79]. Chiara et al. characterized the proteomic and lipidomic profiles of EVs released from mouse TAMs and showed that while TAMs are immunosuppressive, EVs from TAMs show molecular profiles of a Th1/M1 polarization signature and have the potential to stimulate anti-tumor immunity [80]. (4) Chemotherapy resistance: TGF-β1 secreted by TAMs can drive cisplatin resistance in TNBC through hepatocyte leukemia factor (HLF) [81]. Exosomal lncRNA CRNDE from M2 TAMs promotes the resistance of gastric cancer cells to cisplatin by facilitating neural precursor cell expressed developmentally downregulated protein 4-1 (NEDD4-1)-mediated PTEN ubiquitination [82]. (5) Tumor stemness: Cancer stem cells (CSCs) have significant potential for self-renewal and reversible differentiation. TAMs can secrete different products, such as soluble glycoprotein myosone (GPNMB) and CXCL7, to restore the stemness of differentiated tumor cells [83, 84]. (6) Tumor metabolism: It was reported that TAM-derived IL-6 promotes 3-phosphoinositide-dependent protein kinase 1 (PDPK1)-mediated phosphoglycerate kinase 1 (PGK1) phosphorylation, promoting glycolysis and malignant behaviors in tumor cells [85]. lncMMPA from TAM-derived exosomes interacted with miR-548 to target ALDH1A3, promoting aerobic glycolysis and tumor progression in HCC [86]. TAM-derived exosomal HIF-1α-stabilizing long noncoding RNA (HISLA) facilitated the process of aerobic glycolysis and apoptotic resistance in breast cancer cells by activating HIF-1α [87]. All the above findings collectively emphasize the important and complex role of TAMs in tumor progression.
Interactions between TAMs and tumor cells. TAM-derived exosomal circ-0020256 promotes tumor proliferation, while exosomal miR-628-5p and ADAM15 inhibit tumor proliferation. TAM-derived exosomal miR-21-5p and miR-155-5p promote tumor invasion and metastasis, compared to PROS1, which inhibits tumor metastasis. Exosomal ApoE from TAMs promotes tumor migration via activating PI3K-Akt pathway. GATA3 from TAM-derived EVs induces tumor immune evasion via upregulating CD24 and Siglec-10. TAM-derived TGF-β1 and lncRNA CRNDE drive tumor resistance to cisplatin through HLF and NEDD4-1-mediated PTEN ubiquitination, respectively. TAM-secreted GPNMB and CXCL7 restore the stemness of differentiated tumor cells. TAM-derived IL-6, exosomal lncMMPA, and HISLA promote glycolysis in tumor cells via PDPK1, ALDH1A3, and HIF-1α, respectively. Reciprocally, tumor cells recruit circulating monocytes via secreting IL-6, IL-34, CSF-1 and CSF-2. Tumor cell-derived IL-4, IL-10, CSF-1, lactic acid, succinate and TDEs induce M2 TAM polarization. TDEs also induce PD-L1 expression in monocyte-derived TAMs. At last, metastatic TDEs can reduce the phagocytosis, efferocytosis and cytotoxicity of macrophages on tumor cells through TGFB2 signals.
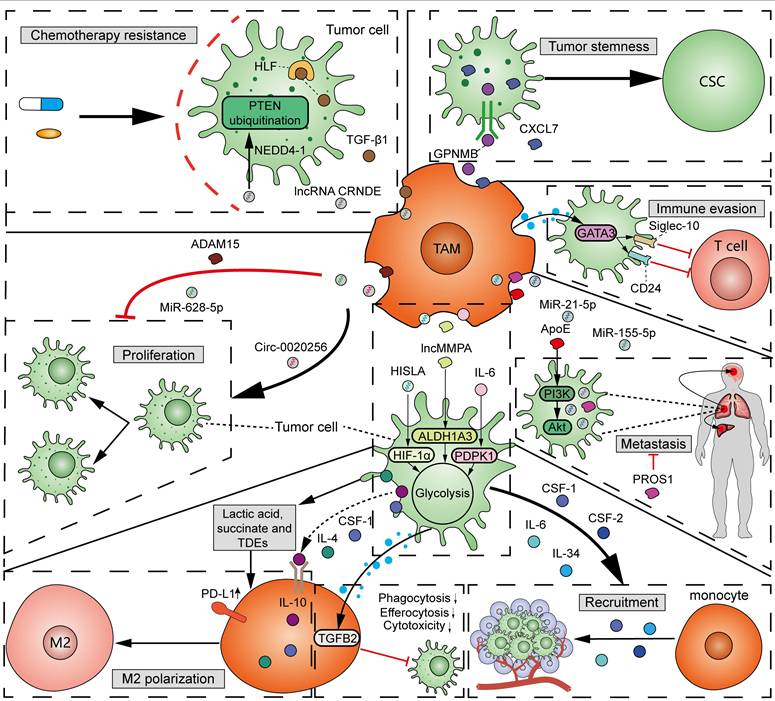
It should be noted that the fitness of tumor cells and TAMs sense with each other (Figure 5). Tumor cells can induce the recruitment of circulating monocytes into tumor tissues through secreting a variety of cytokines and chemokines, such as IL-6, IL-34, CSF1, and CSF2 [88]. In addition, tumor-cell-derived cytokines, metabolites, and exosomes affect the polarization and phagocytosis of TAMs, which, in-turn, determine tumor progression and immune evasion. It was reported that tumor cell-derived IL-4, IL-10, CSF-1, lactic acid, and succinate can induce M2 TAM polarization [89, 90]. Additionally, several studies have shown that tumor-derived exosomes (TDEs) promote the polarization towards an immunosuppressive M2 phenotype and PD-L1 expression in monocyte-derived TAMs [91]. Wolf et al. demonstrated that the adoption of exosomes derived from metastatic osteosarcoma cells into mouse alveolar macrophages can reduce the phagocytosis, efferocytosis, and cytotoxicity of macrophages on tumor cells through induction of the TGFB2 signaling pathway (Figure 5) [92]. The high infiltration of M2 macrophages induced by members of the TME, in addition to tumor cells, is often associated with unfavorable outcomes.
4.2 Infiltrating immune cells
4.2.1 T cells
The importance of lymphocytes, which consist of T cells, B cells, and innate lymphoid cells (ILCs) within the TME, is comparable to that of myeloid cells such as TAMs. Among them, T cells are the focus of attention, both in mechanistic and translational studies of immunotherapies, such as ICB and CAR-T cell therapies [1, 2]. T cells can be divided by T cell receptor (TCR) subunits into TCRαβ+ T cells, which recognize major histocompatibility complex (MHC) class I or class II, and TCRγδ+ T cells, which are almost independent of MHC class I and II. In addition, there is another mainstream classification of T cells into CD8+ T cells and CD4+ T cells according to cell surface CD expression. After recognizing tumor antigens, CD8+ T cells can be activated and exert cytotoxic effects by producing perforin, granzyme B, IFN-γ, and FasL/Fas pathways [93]. CD4+ T cells, which produce cytokines that generate a durable immune response to eliminate pathogens or tumor cells, are primarily referred to as “helper T cells”. Interestingly, another study has shown that CD4+ T cells can also exert antitumor activities by direct cytotoxicity [94].
The attitude of TAMs towards T cells can range from “friendly to unfriendly”, which is contingent on the phenotype of both TAMs and T cells. In the process of tumorigenesis, TAMs exert notable influence on the recruitment, activation, proliferation, and effector functions of T cells through the production of chemokines, cytokines, exosomes, or surface immune ligands/receptors (Figure 6A). It has been reported that TAMs can suppress CD8+ CTLs via the inhibitory B7x (B7-H4/B7S1) molecule in a cell-cell contact manner [95]. Moreover, infiltrating CD4+ T and CD8+ T cells in mouse PDAC models displayed activated IL-10 promoter and repressed T-bet activity (IL-10high/IFN-γlow, PD-1high phenotype), whereas the infiltrating T cells in TAM-inhibited mouse tumor models exhibited reduced IL-10 and PD-1 levels and activated T-bet promoter activity (IL-10low/IFN-γhigh, PD-1low phenotype), suggesting the epigenetic modulation of infiltrating T cells by TAMs [96]. Besides, when CD169+ macrophages are adjacent to the tumor, they play an immunosuppressive role by recruiting Treg cells [97]. However, M1hot TAMs were found to be positively correlated with CD8+ tissue-resident memory T cells, which predicted improved survival in lung cancer [98]. M1hot TAMs may induce sustained CD8+ tissue-resident memory T-cell recruitment via CXCL9 and essential fatty acids. Using co-culture assays, Zhuang et al. showed that the polarized M1 TAMs promoted the proliferation and cytotoxic function of CD8+ T by increasing granzyme-B, TNF-α, and perforin expression, and downregulating PD-1, Tim-3, and Lag-3 [99]. It should be noted that TAMs can influence the differentiation of T-cell subsets. Specifically, NLRP3 signaling in TAMs drives the differentiation of CD4+ T cells into tumor-promoting Th2, Th17, and Treg cells, while inhibiting the polarization of Th1 cells [100]. The heterogeneity of TAMs in the regulation of T cells has become more apparent. A study on breast cancer showed that IL-15Rα+ TAMs reduced tumor infiltration of CD8+ T cells by expressing the IL-15/IL-15Rα complex (IL-15Rc) [101]. In comparison, a subset of TRMs, defined as FOLR2+ TAMs, was found to interact with CD8+ T cells and prime effector CD8+ T cells in breast cancer [102].
Interactions between TAMs and lymphoid cells in TME. (A) Crosstalk of TAMs and T cells: TAMs inhibit the infiltration of CD8+ T cells, and suppress CD8+ CTLs via the inhibitory B7-H4/B7S1 and PD-L1/PD-1 pathways. CD169+ TAMs can recruit Treg cells to suppress CTLs. On the other hand, M1 TAMs induce the recruitment of tissue-resident memory CD8+ T cells via CXCL9 and fatty acids. M1 TAMs promote the proliferation and cytotoxicity of CD8+ CTLs by upregulating granzyme-B, TNF-α and perforin, and downregulating PD-1, Tim-3 and Lag-3. In addition, TAMs epigenetically modulate tumor-infiltrating T cells into an IL-10high/IFN-γlow, PD-1high phenotype. TAMs drive the differentiation of Th2, Th17 and Treg cells, and inhibit the polarization of Th1 cells via NLRP3 signals. IL-15Rα+ TAMs reduce the infiltration of CD8+ T cells by expressing IL-15Rc, while FOLR2+ TAMs interact with CTLs and prime effector CD8+ T cells. Reciprocally, CD4+ Th1 and NK-T cells induce the polarization of M1 TAMs by secreting IFN-γ. CAR-T cells recruit peripheral F4/80lowLy-6C+ myeloid cells to the TME via secreting GM-CSF, and activate NO production and phagocytosis in F4/80high macrophages via secreting IFN-γ. In contrast, Treg cells support M2 TAMs. (B) Crosstalk of TAMs and B cells: CXCL12+ TAMs recruit PD-L1+ Breg cells via the CXCL12/CXCR4 axis. The M2 polarization of TAMs is promoted by B cell-derived GABA, Breg cell-secreted TGF-β, and B1 cell-derived IL-10 and TRIF/STAT1 pathway. Meanwhile, B cell-derived anti-tumor antibodies bind TAMs' Fc receptors to induce ADCC towards tumor cells. (C) Crosstalk of TAMs and ILCs: TAMs induce ILC3 to produce IL-22 via secreting IL-7, thereby promoting tumor development. M1 TAMs promote the proliferation of ILC3s via secreting IL-23. TAMs inhibit the function of NK cells by TGF-β-dependent mechanisms. MACRO+ TAMs suppress tumoricidal NK cells. Reciprocally, ILC1s, ILC3s and NK cells drive M1 TAM polarization, while ILC2s drive M2 TAM polarization.
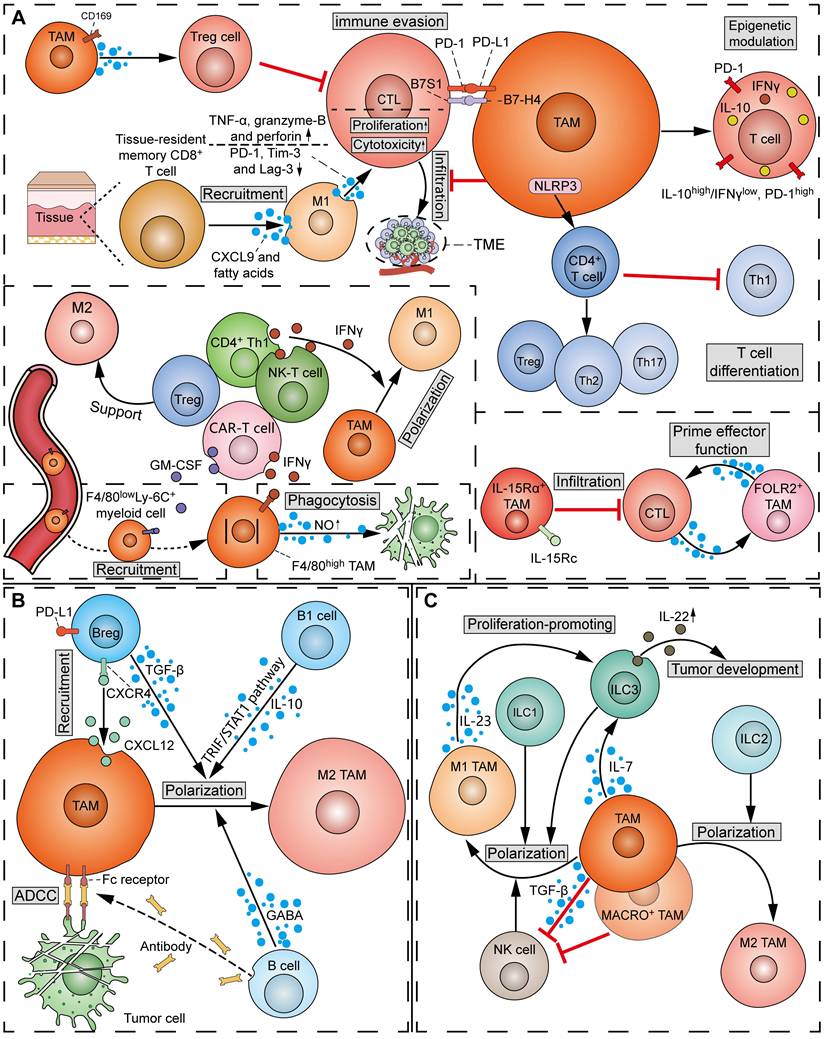
Conversely, T cells also play a crucial role in TAM regulation (Figure 6A). Emerging studies report that T-cell subsets capable of releasing IFN-γ, such as CD4+ Th1 cells and NK-T cells, can induce M1 polarization of TAMs [103]. In a mouse tumor model, the adoptive transfer of T cells expressing a CAR recruited peripheral F4/80lowLy-6C+ myeloid cells to the TME by secreting GM-CSF, and activated NO production and phagocytosis against tumor cells in F4/80high macrophages by secreting IFN-γ [104]. Conversely, Treg cells can facilitate the SREBP1-dependent metabolic fitness, mitochondrial integrity, and survival of M2 TAMs by suppressing CD8+ T cell-derived IFN-γ [105].
The relationship between TAMs and T cells in the TME is currently a hot topic of research. However, their interactions are complex, as the phenotypes of both parties and the influence of the surrounding environment should be considered. On one hand, TAMs can directly modulate the activities of anti-tumor T cells. On the other hand, there exists an indirect mechanism through which TAMs determine the immune functions of tumor-infiltrating T cells via their impact on other immune cells. The development of advanced technologies, including single-cell RNA sequencing, spatial transcriptomics and multi-omics, and SpaTial Enhanced REsolution Omics-sequencing (Stereo-seq) [106-108], may assist in understanding the heterogeneity of TAM subtypes, examining cellular variations across different tumor regions, and characterizing intercellular interactions between TAMs and T cells within the TME.
4.2.2 B cells
B cells are predominantly associated with tertiary lymphoid structures (TLSs) in the TME, which can differentiate into plasma cells to produce IgG/IgA in response to tumor-associated antigens [109]. Otherwise, B cells can differentiate into regulatory B (Breg) cells to produce immunosuppressive cytokines, including IL-10, IL-35, and TGF-β. The density of TLS and B cell content varies considerably across different tumor types. During tumorigenesis, the effect of B cells has generally been considered as tumor-promoting, either via Breg cell-mediated immunosuppression or via IgG-mediated macrophage activation. On the other hand, though B cells may not have direct effector roles in anti-tumor immunity, a few studies suggested that B cells are involved in anti-tumor immunity by regulating the activation and effector functions of T cells, NK cells, DCs, neutrophils, and TAMs [110].
The crosstalk between myeloid-derived TAMs and B cells has attracted notable attention recently (Figure 6B). Lian et al. observed co-localization between CXCL12+ TAMs and PD-L1+ Breg cells in adjacent HCC tissues. Mechanistically, CXCL12+ TAMs recruited PD-L1+ Breg cells via the CXCL12/CXCR4 axis [111]. It is hypothesized that certain glucose-regulated metabolites, for example, cyclic adenosine monophosphate (cAMP), also play an important role in mediating the interaction between TAMs and B cells.
Reciprocally, Zhang et al. showed that B cell-derived GABA drives anti-inflammatory polarization of TAMs to weaken the cytotoxic functions of CD8+ T cells (Figure 6B) [112]. As a canonical immunomodulatory factor, TGF-β produced by Breg cells has been shown to skew TAMs towards an immunosuppressive M2 phenotype [113]. Another study demonstrated that B1 cells drive TAM polarization towards an M2 phenotype via B1 cell-derived IL-10 and TRIF/STAT1 pathway activation [114]. However, the action of B cells on TAMs can be anti-tumorigenic, since the binding of TAMs' Fc receptors with constant regions of anti-tumor antibodies can induce ADCC towards tumor cells [109]. Despite the above findings, a landmark discovery is desperately required regarding the functional role of B cells in tumorigenesis, especially with the identification of diverse B-cell clones and subsets through high-throughput technologies. This may be an exciting area of research to uncover the underlying mechanisms governing interactions between tumor-infiltrating B-cell subsets and TAMs in tumor progression.
4.2.3 ILCs
ILCs are derived from the same lymphoid progenitor as T cells, but they differ in structure and function. Currently, the ILC family is classified into five subgroups based on a comprehensive categorization method incorporating cytokines and transcription factors. These subgroups include the helper-like ILC1s, ILC2s, and ILC3s, as well as NK cells and lymphoid tissue-inducer (LTi) cells [115]. Notably, both ILC1s and NK cells express high levels of IFN-γ and cytotoxic molecules, which are closely associated with tumor elimination.
In patients with rectal cancer, TAMs can express more IL-7 under the stimulation of Candida albicans, and then further induce ILC3 to produce IL-22, ultimately promoting tumor development (Figure 6C) [116]. Additionally, targeting MARCO, a receptor on the surface of TAM, can alter MACRO+ TAM metabolism and enhance the tumor-killing function of NK cells [117]. Similar results were obtained in metastatic carcinoma, where TAMs inhibited NK cell function by TGF-β-dependent mechanisms; by contrast, the absence of MAMs promotes the activation, maturation, and number of NK cells, thereby enhancing tumor rejection [118]. Instead, IL-23 secreted by M1 macrophages has been shown to promote the proliferation of ILC3s [119].
In turn, ILCs can induce M1/M2 polarization in TAMs, depending on the ILC subpopulation. Specifically, ILC1s, ILC3s, and NK cells can induce the expression of M1 macrophage-related genes, while M2 genes in macrophages can be induced by ILC2s (Figure 6C) [120].
4.2.4 MDSCs
MDSCs are a unique type of activated myeloid cells that accumulate in pathological conditions of neutrophil and monocyte accumulation, such as persistent inflammation or tumors. In mice, there are two major subpopulations of MDSC: monocytic (M)-MDSCs (CD11b+Ly6G-Ly6Chigh) and polymorphonuclear (PMN)-MDSCs (CD11b+Ly6GhighLy6C-). In humans, three subpopulations have been found: major M-MDSCs (CD11b+HLA-DR-/lowCD14+CD15-), PMN-MDSCs (CD11b+HLA-DR-/lowCD14-CD15+), and minor early MDSCs (Lin-HLA-DR-CD33+, eMDSCs). Research has indicated that the sole inhibition of TAMs did not lead to a reduction in tumor progression, potentially due to the compensatory emergence of immunosuppressive MDSCs [121].
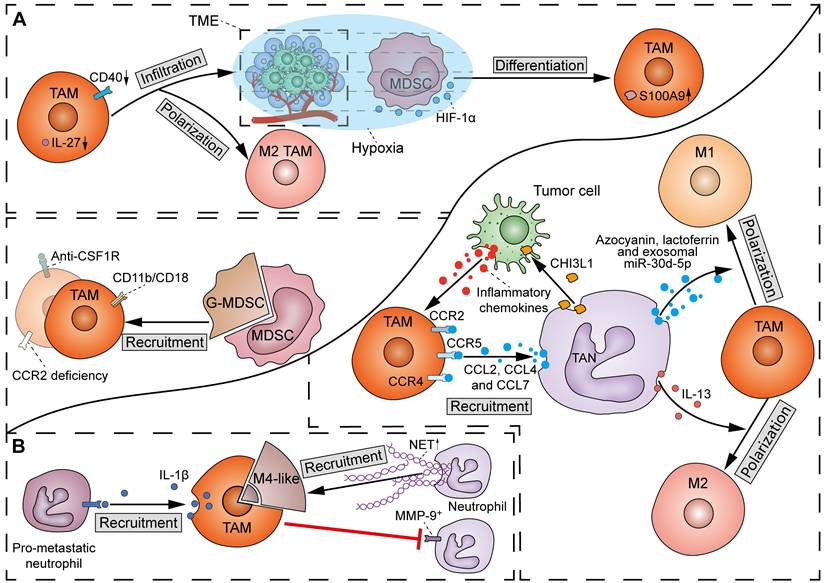
There has been a scarcity of molecular studies concerning the crosstalk between TAMs and MDSCs (Figure 7A). TAMs play a crucial role in facilitating the migration and recruitment of MDSCs to the TME. The M2 metabolic status of TAMs positively correlates with MDSC infiltration. Mechanistically, TAMs express CD11b/CD18 integrin heterodimer (Mac-1; αMβ2), which is one of the essential tools required by MDSCs for their migration and recruitment [122]. However, others observed that inhibition of TAM recruitment by CCR2 deficiency or anti-CSF1R agent resulted in a compensatory increase of immunosuppressive G-MDSCs to impair T-cell responses in cholangiocarcinoma [121]. Therefore, in most cases, dual inhibition of TAMs and G-MDSCs elicits a more potent effect in potentiating immunotherapy in cancer. Conversely, MDSCs can induce macrophage polarization and infiltration by suppressing CD40/IL-27 signaling to drive melanoma progression and ICB resistance [123]. Of note, due to the homogeneity between MDSCs and monocytes, M-MDSCs can differentiate into TAMs. It was found that hypoxia via HIF-1α dramatically induced the differentiation of MDSCs into TAMs within the TME [124]. Specifically, TAMs derived from M-MDSCs exhibit potent immunosuppressive properties and M2 polarization tendency due to sustained expression of S100A9 protein.
Interactions between TAMs and myeloid cells in the TME. (A) Crosstalk of TAMs and MDSCs: TAMs induce the recruitment of MDSCs via expressing CD11b/CD18 integrin heterodimer. However, TAM inhibition by CCR2 deficiency or anti-CSF1R agent also increases G-MDSCs. Reciprocally, MDSCs can induce M2 TAM polarization and infiltration by suppressing CD40/IL-27 signals. Additionally, hypoxia via HIF-1α induces the differentiation of MDSCs into TAMs, with a sustained expression of S100A9. (B) Crosstalk of TAMs and TANs: TAMs inhibit the recruitment of MMP-9+ neutrophils, except for M4 macrophages, which promote the recruitment of neutrophils and induce the secretion of NETs. On the other hand, TAMs drive the recruitment of pro-metastatic neutrophils to metastatic lesions via producing IL-1β. Reciprocally, TANs promote the recruitment of TAMs through CCL2-CCR2, CCL4-CCR5 and CCL7-CCR4 pathways. Neutrophil-derived CHI3L1 indirectly enhance TAM recruitment by stimulating tumor cells to release inflammatory chemokines. At last, TAN-derived azocyanin, lactoferrin and exosomal miR-30d-5p drive M1 polarization of TAMs, whereas IL-13 initiates M2 TAM polarization.
4.2.5 Neutrophils
Neutrophils are traditionally considered the backbone of primary immune defense. Despite the most abundant leukocytes in human peripheral blood, neutrophils have a short half-life and lifespan, except in the context of inflammatory stimulation, wherein the half-life of neutrophils can be increased by 3.3-fold by 200 U/ml IL-1β [125]. The presence of neutrophils in tumors often predicts poor clinical outcomes. For instance, neutrophil extracellular traps (NETs) from dying neutrophils are correlated with tumor progression and metastasis in some studies [126]. Nevertheless, neutrophils are highly plastic and heterogeneous cells, some of which exert antitumor functions [127]. Neutrophils in the TME, referred to as PMN-MDSCs or tumor-associated neutrophils (TANs), have a simplified bipolar classification: IFN-β signaling pathway induced anti-tumor (N1) TANs and TGF-β signaling pathway induced pro-tumor (N2) TANs. The following studies support the presence of interactions between TAMs and TANs and are also relevant to tumor progression.
There has been emerging evidence regarding the influence of TAMs on TANs, even though the detailed molecular mechanisms are still lacking (Figure 7B). A previous study showed that defects in TAM infiltration led to a compensatory recruitment of MMP-9+ neutrophils in mouse tumors [128]. However, another study found an exception that M4 macrophages derived from M2 macrophages and Kupffer cells promoted the recruitment of neutrophils and induced the secretion of NETs [129]. Moreover, Wellenstein et al. observed that loss of p53 in breast cancer cells induces the secretion of WNT ligands to increase IL-1β production in TAMs, thereby recruiting pro-metastatic neutrophils to metastatic lesions [130]. The above discrepancy may be due to the considerable heterogeneity in intrinsic properties between TAM and TAN subtypes.
TANs can directly promote the recruitment of TAMs by releasing cytokines such as CCL2, CCL4, and CCL7, which bind to surface receptors CCR2, CCR5, and CCR4, respectively (Figure 7B) [131]. Furthermore, non-enzymatic chitinase-3-like-protein-1 (CHI3L1), a glycoprotein synthesized and released by neutrophils, can also indirectly enhance TAM recruitment by stimulating tumor cells to release inflammatory chemokines [132]. TANs can also produce different proteins to influence the polarization of macrophages. Specifically, macrophages can phagocytose TAN-derived azocyanin, lactoferrin, and exosomal miR-30d-5p to promote M1 polarization, or initiate M2 polarization after phagocytosis of IL-13 from the same source [133].
As mentioned previously, TANs can be considered as functional substitutes for TAMs from a certain perspective. Therefore, solely targeting TAMs may not yield effective antitumor effects. It has been demonstrated that simultaneously targeting TAMs and TANs holds significant advantages, such as enhancing chemotherapy response and reducing allergic reactions caused by the use of anti-PD-L1 antibodies. However, targeting TANs is more challenging than targeting TAMs or M-MDSCs due to a lack of tractable targets. The impact of TAMs on the heterogeneity of TANs cannot be overlooked.
4.3 CAFs
CAFs, a kind of activated stromal cell capable of secreting collagen, are recognized as a predominant component, as well as the center of communications between different cell types in the TME. CAFs have a variety of precursor cell types, and its molecular markers include but are not limited to α-smooth muscle actin (α-SMA), fibroblast-specific protein 1 (FSP1), fibroblast activation protein (FAP), platelet-derived growth factor receptor-α (PDGFR-α), PDGFR-β, and vimentin. Numerous studies have shown that the interplay between CAFs and TAMs is essential for the formation of immunosuppressive TME.
Experiments on NSCLC models have confirmed that TAMs can transform CAFs, a process known as macrophage-myofibroblast transition (MMT) [134]. Additionally, TAMs have the potential to induce myoCAF transformation of CAFs through the CXCL3/CXCR2 axis [135]. CCL18 secreted by TAMs induced the conversion of normal breast-resident fibroblasts into CD10+GPR77+ CAFs, resulting in the enrichment of CSCs and chemoresistance in breast cancer [136].
To date, research has focused on the regulation of CAFs on TAMs. There is ample experimental data showing that CAFs can recruit circulating monocytes into the TME, as well as induce the polarization of TAMs towards M2 phenotype. It was reported that CAFs recruit monocytes and induce STAB1+TREM2high LAM via the CXCL12-CXCR4 axis, which supports an immunosuppressive TME in breast cancer [137]. In orthotopic and syngeneic colon carcinoma mouse models, IL-6 and GM-CSF from CAFs synergically induce the polarization of monocytes into pre-invasive M2 macrophages [138].
At present, there are far fewer studies on the regulation of CAFs by TAMs than expected. Although CAFs seem to have more regulatory mechanisms for TAMs, crosstalk may exist between pathways in the same direction, although this needs to be experimentally confirmed. From a therapeutic perspective, among these regulatory mechanisms, it may be preferable to select targets that are also essential in other mechanisms. For example, CXCL12 not only promotes M2 polarization of TAMs mediated by CAFs, but also recruits circulating monocytes to TME.
4.4 ECs
The process by which new blood vessels sprout from pre-existing blood vessels is called angiogenesis, which not only provides oxygen and nutrients for tumor growth, but is also an important method for tumor metastasis. In particular, angiogenesis plays a vital role in addressing the high metabolic demands of growing tumors. ECs are present in the 80-nm-thick basal lamina located in the innermost layer of blood vessels. Functionally, ECs are not only regulators of vascular tension, but also serve as an important physical barrier and endocrine organ.
TAMs possess the ability to regulate ECs at various levels, including phenotypic and functional aspects. Research by Yang et al. indicates that exosomal miR-155-5p and miR-221-5p released by M2 TAMs could be transferred into ECs to further promote angiogenesis [139]. Interestingly, in a study on breast cancer, M2 macrophages, but not M1 macrophages, were found to inhibit vascular cellular adhesion molecule-1 (VCAM-1) expression in ECs, which improved vascular integrity [140]. Finally, TAMs were reported to induce EC inflammation and promote EC adhesion under hypoxic conditions, which is closely related to inflammation and tumor metastasis [141].
Conversely, ECs in the TME can recruit circulating monocytes, promote monocyte-macrophage transformation, and regulate the polarization of TAMs through different methods. The co-culture of CCR2+ monocytes with TNFR2+ ECs results in the acquisition of macrophage phenotype [142].
4.5 ECM
ECM represents the complex 3D network of structures that surround and support cells within organs and tissues. Furthermore, ECMs also play an important role in regulating cell signaling, function, properties, and morphology [143]. In tumors, the four major mechanisms of ECM remodeling, including ECM deposition, post-translational chemical modification, proteolytic degradation, and forced physical remodeling, are disrupted, leading to a tumorigenic ECM [143]. CAFs, cancer cells, and certain immune cells (such as TAMs) are the main sources of ECM molecules in the TME. Therefore, exploring the interaction between ECMs and other components may help us to achieve more effective and specialized immunotherapeutic strategies.
Proteoglycan HSPG2 is one of the classic members of several types of tumor ECM and promotes tumor growth by various means. Intriguingly, TAMs have been found to increase the stiffness of the ECM through HSPG2 deposition, which induces immune escape in breast cancer [144]. Specifically, TAMs derived from CCR2+ monocytes can degrade collagen through receptor-mediated endocytosis via Mrc1 [145]. TAMs can also indirectly regulate the physical properties and external arrangement of fibers in the ECM through CAFs, depending on the ratio of M1/M2 macrophages. For example, ECM in M2 macrophage-conditioned media had more aligned and thinner fibers than those in mixed M1/M2 macrophage-conditioned media [146].
An earlier study demonstrated that the ECM of different tissues can induce contrasting phenotypes in macrophages, indicating the therapeutic value of targeting ECM molecules [147]. The regulatory role of major ECM molecules on TAMs has been gradually elucidated with the advancement of research. For example, collagen internalization can upregulate the expression of Arg-1 and iNOS in TAMs [148]. Another fact that demonstrates the potential value of ECM molecules is that, compared with ECM in lean individuals, ECM associated with obesity is more effective in promoting M2 macrophage functions [149].
The process of interaction between TAMs and ECM is characterized by complexity and dynamism. However, it is important to note that ECM is a heterogeneous structure and should not be regarded as a unified entity. It is necessary to divide ECM into different components and explore their respective relationships with TAMs. Targeting TAMs to reshape the tumor-promoting ECM should be a potential antitumor strategy.
5. Advances in Targeting TAMs in Cancer Treatment
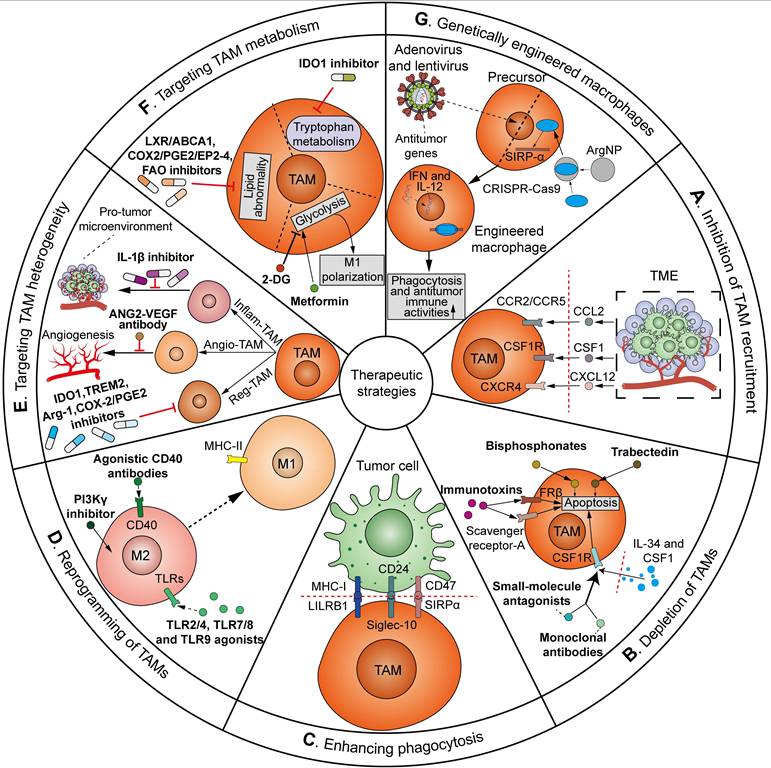
As mentioned above, TAMs play a critical role throughout all stages of cancer, from tumor initiation, metastatic cascade, immune evasion, to cancer therapy resistance, across various cancer types. Importantly, given the prevalence of tumor immunotherapy, there is growing interest in targeting TAMs in clinical trials as adjuvants to current immunotherapies. The means by which TAMs have been manipulated for therapeutic applications can be sorted into the following approaches: (1) inhibition of TAM recruitment; (2) depletion of TAMs in the TME; (3) enhancing phagocytosis; (4) reprogramming of TAMs; (5) targeting TAM heterogeneity; (6) targeting TAM metabolism; and (7) genetically engineered macrophages (Figure 8). Many of these strategies have been translated from preclinical models to clinical trials. Herein, we summarize the clinical investigations of agents targeting TAMs in tumors (Table 1).
Clinical trials of anti-tumor agents targeting TAMs.
| Agent | Target | NCT/UMIN | Phase | Tumor type | Combine with other medicine/therapy | Result | Ref. |
|---|---|---|---|---|---|---|---|
| Carlumab (CNTO 888) | CCL2 | NCT00537368 | Phase 1 | Solid tumors | Well tolerated, transient free CCL2 suppression, preliminary antitumor activity | [150] | |
| NCT01204996 | Phase 1b | Solid tumors | Chemotherapy | Well tolerated, neither long-term serum CCL2 suppression nor significant tumor responses | [151] | ||
| Propagermanium | CCL2 | UMIN000022494 | Phase 1 | Breast cancer | Low-grade adverse events, decreased serum IL-6 | [152] | |
| PF-04136309 | CCR2 | NCT02732938 | Phase 1b | PDAC | Gemcitabine and nab-paclitaxel | Increased pulmonary toxicity, no combination therapy advantage | [153] |
| CCX872-B | CCR2 | NCT02345408 | Phase 1b | Pancreatic cancer | FOLFIRINOX | 18-month OS rate: 29% for all subjects. no safety issues observed | [154] |
| Maraviroc | CCR5 | NCT03274804 | Phase 1 | Colorectal cancer | Pembrolizumab | ORR 5.3%, median PFS 2.1 months, median OS 9.83 months | [155] |
| Emactuzumab (RG7155) | CSF1R | NCT01494688 | Phase 1a/b | Solid tumors | Paclitaxel | Reduced immunosuppressive TAMs, no antitumor efficacy | [156] |
| NCT02323191 | Phase 1b | Solid tumors | Atezolizumab | Elevated ORR, enhanced CD8+ T-cell infiltration | [157] | ||
| Lacnotuzumab (MCS110) | CSF1 | NCT02435680 | Phase 2 | Breast cancer | Gemcitabine and Carboplatin | No combination therapy advantage | [158] |
| Plerixafor (AMD3100) | CXCR4 | NCT00943943 | Phase 1 | FLT3-ITD-mutated AML | Sorafenib and G-CSF | Improved disease response rate | [159] |
| BL-8040 | CXCR4 | NCT01838395 | Phase 2a | AML | Cytarabine | Well tolerated, clinical responses observed | [160] |
| Olaptesed pegol (NOX-A12) | CXCL12 | NCT01486797 | Phase 2a | CLL | Bendamustine and rituximab | Well tolerated, ORR 86%, the median PFS 15.4 months | [161] |
| Zoledronate | NCT02181101 | Phase 3 | Breast cancer | Chemotherapy | No prognosis benefit from prolonged adjuvant zoledronate (> 2 years) | [162] | |
| Magrolimab (Hu5F9-G4) | CD47 | NCT04892446 | Phase 2 | Multiple myeloma | Daratumumab, pomalidomide/dexamethasone, carfilzomib/dexamethasone, or bortezomib/dexamethasone | NA | [163] |
| NCT02953509 | Phase 1b | Non-Hodgkin's lymphoma | Rituximab | Promising therapeutic effect with no significant safety concerns | [164] | ||
| NCT03869190 | Phase 1b/2 | Urothelial carcinoma | Atezolizumab | Tolerable, no additive benefit in ORR, PFS, or OS with the addition of magrolimab to atezolizumab | [165] | ||
| CC-90002 | CD47 | NCT02641002 | Phase 1 | AML and MDS | No objective responses with CC-90002 monotherapy | [166] | |
| TTI-621 | CD47 | NCT02663518 | Phase 1 | Hematologic malignancies | Rituximab or nivolumab | Well tolerated, therapeutic activity in TTI-621 monotherapy or in combination with rituximab | [167] |
| CP-870893 | CD40 | NA | Phase 1 | Solid tumors | Well tolerated with induced objective responses and antitumor activity | [168] | |
| NCT00607048 | Phase 1 | Solid tumors | Paclitaxel and carboplatin | Safe profile, biological/clinical responses observed | [169] | ||
| BCG | TLR2/4 | CUETO 93009 | Bladder cancer | Mitomycin C | Reduced disease relapse, higher toxicity profile | [170] | |
| 852A | TLR7 | NCT0018933 | Phase 2 | Melanoma | Well tolerated, prolonged stable disease in stage IV metastatic patients | [171] | |
| NA | Phase 2 | Hematologic malignancies | Safe dosing up to 1.2 mg/m2 twice weekly, sustained tolerability/clinical activity | [172] | |||
| Imiquimod | TLR7/8 | NCT00899574 | Phase 2 | Breast cancer skin metastases | Well tolerated with 20% partial response in patients | [173] | |
| Eganelisib (IPI-549) | PI3Kγ | NCT02637531 | Phase 1/1b | Solid tumors | Nivolumab | Antitumor activity in combination group | [174] |
| Navoximod (GDC-0919) | IDO1 | NCT02048709 | Phase 1a | Solid tumors | Favorable tolerability with significant therapeutic efficacy | [175] | |
| PY314 | TREM2 | NCT04691375 | Phase 1b | Renal cell carcinoma | Pembrolizumab | Limited antitumor efficacy | [176] |
| Celecoxib | COX-2 | NCT03926338 | Phase 2 | Colorectal cancer | Toripalimab | Therapeutic efficacy, a high pathological complete response rate and an acceptable safety profile | [177] |
| BI 836880 | ANG2-VEGF | NCT03972150 | Phase 1 | Solid tumors | Ezabenlimab | Manageable safety profile with preliminary clinical activity | [178] |
| Canakinumab | IL-1β | NCT03631199 | Phase 3 | NSCLC | Pembrolizumab and platinum-based chemotherapy | No PFS/OS benefit with combination therapy | [179] |
| 2-DG | Glycolysis | NCT00096707 | Phase 1 | Solid tumors | Docetaxel | Well tolerated for 63 mg/kg/day 2-DG and docetaxel | [180] |
| Metformin | Mitochondrial respiratory chain complex I | NCT01243385 | Phase 2 | Prostate cancer | Tolerable in non-diabetics, objective prostate-specific antigen responses | [181] | |
| UMIN 000002210 | Phase 2 | Endometrial cancer | MPA | Disease relapse suppression | [182] | ||
| Epacadostat | IDO1 | NCT03414229 | Phase 2 | Sarcoma | Pembrolizumab | Favorable tolerability, limited anti-tumor activity | [183] |
| NCT02752074 | Phase 3 | Melanoma | Pembrolizumab | Unimproved PFS and OS | [184] | ||
| Tefinostat (CHR-2845) | HDAC | NCT00820508 | Phase 1 | Hematologic malignancies | Monocyte-selective histone deacetylase inhibition, well tolerated, no dose-limiting toxicities observed | [185] |
PDAC: pancreatic ductal adenocarcinoma; FOLFIRINOX: 5-FU, leucovorin, irinotecan and oxaliplatin; OS: overall survival; ORR: objective response rate; PFS: progression free survival; AML: acute myeloid leukemia; CLL: chromic lymphocytic leukemia; NA: not available; MDS: myelodysplastic syndrome; MPA: medroxyprogesterone acetate; HDAC: histone deacetylase.
An overview of strategies for TAM-targeting antitumor therapeutics. The ways TAMs have been manipulated can be sorted as follows: (A) Inhibition of TAM recruitment. Targeting CCL2-CCR2/CCR5, CSF1-CSF1R, or CXCL12-CXCR4 axis via inhibitors or antibodies can inhibit the recruitment of TAMs to the TME; (B) Depletion of TAMs in the TME. CSF1/IL-34-CSF1R blockade via monoclonal antibodies or small-molecule antagonists, bisphosphonates and trabectedin, and targeting scavenger receptor-A or FRβ via immunotoxins can reduce TAM count in TME by inducing apoptosis; (C) Enhancing phagocytosis. Blockade of the “do not eat me” signals, e.g. SIRPα/CD47 pathway, CD24/Siglec-10 pathway and MHC-I/LILRB1 pathway, can evoke the phagocytic activities of TAMs against tumor cells; (D) Reprogramming of TAMs. Agonistic CD40 antibodies, TLR2/4, TLR7/8 and TLR9 agonists, and PI3Kγ inhibitors induce the repolarization of TAMs from M2 towards pro-inflammatory and tumoricidal M1 phenotype; (E) Targeting TAM heterogeneity. The heterogeneous TAM subgroups can be targeted by distinct therapeutics. Reg-TAMs are inhibited via IDO1, TREM2, Arg-1 and COX-2/PGE2 inhibitors to blunt immunosuppression. Angio-TAMs are inhibited by ANG2-VEGF antibody to reduce tumor angiogenesis and therapeutic resistance. Inflam-TAMs are inhibited by IL-1β inhibitor to suppress pro-tumor inflammatory and immunosuppressive microenvironment; (F) Targeting TAM metabolism. Manipulation of TAMs' metabolism can be achieved via glycolytic modulators 2-DG and metformin to induce M1 TAM polarization, LXR/ABCA1, COX2/PGE2/EP2-4 and FAO inhibitors to treat lipid abnormality, and IDO1 inhibitor to block the tryptophan metabolism; (G) Genetically engineered macrophages. Macrophage precursors are genetically engineered through adenoviral or lentiviral transduction of antitumor IFN and IL-12, or ArgNP-mediated CRISPR/Cas9 delivery to knockout SIRP-α gene. Adoptive transfer of genetically engineered macrophages represents a new approach to enhance the phagocytosis and antitumor immune activities of TAMs.
5.1 Inhibition of TAM recruitment
To date, the mediators involved in TAM recruitment are diverse and remain incompletely understood. Despite this, a consensus has been reached that the recruitment of circulating monocytes/macrophages is highly dependent on several chemokine signals, some of which may be targeted in antitumor therapy.
The CCL2-CCR2/CCR5 axis. The universality of the CCL2-CCR2/CCR5 axis makes it the most attractive target for inhibition of TAM recruitment. Preclinically, targeting CCL2-CCR2 via neutralizing antibodies has yielded encouraging results in delaying tumor progression [186]. CCL2-CCR2/CCR5 inhibitors that have been assessed in clinical trials include carlumab (CNTO 888), propagermanium, PF-04136309, CCX872-B, and maraviroc. A Phase 1 trial demonstrated that CNTO 888, a human anti-CCL2 mono-antibody, was well-tolerated with transient free CCL2 suppression and preliminary antitumor activity in solid tumors [150]. However, another Phase 1b clinical study of solid tumors found that although no severe adverse events were reported, CNTO 888 did not show long-term CCL2-CCR2 blockade or antitumor effect when combined with chemotherapeutic regimens [151]. The reasons may lie in the inadequate clearance of circulating CCL2, since free CCL2 in serum decreased immediately after CNTO 888 treatment but increased with chemotherapy administration. A Phase 1 dose-escalation trial evaluating the effects of propagermanium, an oral organogermanium CCL2 antagonist, in breast cancer patients found that serum IL-6 was downregulated in a dose-dependent manner in the propagermanium-treated patients, implying the promising therapeutic efficacy of propagermanium in cancer angiogenesis and metastasis [152]. PF-04136309 is a CCR2 inhibitor, which was assessed in a Phase 1 study of metastatic pancreatic cancer as a combined regimen with gemcitabine and nab-paclitaxel [153]. There was a 23.8% ORR in all 21 patients who received PF-04136309; however, the incidence of pulmonary toxicity was relatively high (24%). CCX872-B is a specific CCR2 antagonist. An ongoing Phase 1b trial of pancreatic cancer showed that the overall surviva (OS) rate at 18 months for all patients, including those receiving FOLFIRINOX plus CCX872-B, was 29%, whereas the 18-month OS rate was only 18.6% for FOLFIRINOX treatment [154]. Maraviroc, an antagonist of CCR5, has been tested in a Phase 1 clinical trial as an adjuvant to pembrolizumab, which resulted in 5.3% objective response rate (ORR), 2.1-month median progression free survival (PFS), and 9.83-month median OS in refractory mismatch repair proficient colorectal cancer [155]. Significantly, another advantage of targeting CCL2-CCR2/CCR5 is the potential for inhibiting the recruitment and function of other immunosuppressive cells, including MDSCs and Treg cells [187], and this may result in a better response.
The CSF1-CSF1R axis. The CSF1-CSF1R axis is also critical in the recruitment of TAMs. CSF1R is exclusively expressed by cells of the monocytic lineage, making it a potential target for inhibition of immunosuppressive myeloid-derived cells, especially TAMs. The therapeutic implications of CSF1-CSF1R inhibition in reducing TAM recruitment have been reported in mouse tumor models [188]. Antibodies or small-molecule antagonists selectively targeting CSF1-CSF1R have entered Phase 1 and Phase 2 clinical testing, including emactuzumab (RG7155) and lacnotuzumab (MCS110). The humanized anti-CSF1R antibody RG7155 was assessed in a Phase 1 clinical study, which showed that RG7155 effectively blocked the recruitment of immunosuppressive TAMs, but did not result in significant antitumor activities in solid tumors [156]. However, another study of diffuse-type tenosynovial giant cell tumor revealed that although facial oedema was found as the most common adverse events, 24 (86%) of all 28 patients treated with RG7155 showed an objective response and two patients achieved a complete response [189], which was predicted to be attributed to the intrinsic feature of diffuse-type tenosynovial giant cell tumors, driven by aberrant CSF1 expression. Roca et al. assessed the combinatorial blockade of TAM recruitment with checkpoint immunotherapy in solid tumors [157]. They found that RG7155 resulted in a considerable ORR and increased CD8+ T-cell infiltration in anti-PD-L1-treated NSCLC patients, suggesting a synergistic antitumor immune response. A Phase 2 clinical study investigated the combinatorial effect of MCS110 with chemotherapy in advanced TNBC [158]. However, results showed that while a decrease in CD163+ TAMs was observed in lacnotuzumab-treated patients, no additional benefit in terms of increased ORR or improved PFS was observed in the combination treatment group [158]. These unexpected results may be attributed to the inadequate selection with regard to high TAM content, since CD163 is a pan-macrophage and monocyte biomarker rather than a specific M2 macrophage biomarker. Therefore, strategies for targeting specific tumorigenic TAM subsets are warranted.
The CXCL12-CXCR4 axis. CXCL12-CXCR4 is another signaling axis involved in TAM recruitment and tumor progression. More importantly, since CXCL12 can be induced by conventional cancer treatments, targeting CXCL12-CXCR4 may be a promising adjuvant alongside chemotherapy, radiotherapy, anti-angiogenesis, or immunotherapy by blocking CXCR4+ macrophage trafficking. Currently, the majority of drugs targeting CXCL12-CXCR4 in clinical trials are CXCR4 inhibitors, including plerixafor (AMD3100) and BL-8040. The only drug targeting CXCL12 in cancer treatment is olaptesed pegol (NOX-A12). Apart from the mobilization of hematopoietic cells, AMD3100, BL-8040, and NOX-A12 have been evaluated as sensitizing treatment strategies in combination with chemotherapeutic agents or kinase-specific inhibitors in Phase 1 and 2 clinical trials of hematologic tumors [159-161]. As for solid tumors, a Phase 1 clinical study found that AMD3100 may exhibit synergistic effects with anti-angiogenic bevacizumab in recurrent high-grade glioma patients [190]. Another Phase 2a study showed that BL-8040 increased the clinical benefits of immunotherapy in chemotherapy-resistant PDACs [191].
Although significant achievements have been made in inhibiting the recruitment of TAM-precursors to tumors, the degree and duration of TAM reduction need to be determined and improved. Inhibition of TAM recruitment may lead to compensatory infiltration of TANs and expansion of TRMs, either as monotherapy or in combination therapies, which may compromise the long-term efficacy of this therapeutic approach [128]. Notably, the incidence of adverse events likely increases with dose, and this should be considered in clinical use on a per-patient basis.
5.2 Depletion of TAMs in TME
Since CSF1R signaling promotes the proliferation, survival, activation, and differentiation of macrophages, CSF1R blockade via monoclonal antibodies or small-molecule antagonists, as listed in Table 1, also results in TAM depletion in clinical studies.
Other approaches to reduce TAM counts in the TME employed cytotoxic compounds, including bisphosphonates and trabectedin. Bisphosphonate, a traditional drug for treating cancer bone metastasis and resorption, induces apoptosis after being phagocytosed by TAMs [192]. At present, three generations of bisphosphonates have been developed to induce apoptosis by different mechanisms: The first generation etidronate, clodronate, and tiludronate are non-nitrogen-containing bisphosphonates, which can be converted into non-hydrolyzable ATP analogues intracellularly and result in apoptosis. The second-generation tiludronate and third-generation zoledronate are bisphosphonates that induce apoptosis by inhibiting the farnesyl diphosphate (FPP) synthase. Clinically, clodronate and zoledronate have been evaluated as adjuvant agents for treating breast cancer and multiple myeloma [162, 193]. Trabectedin is an anticancer agent originally isolated from the Caribbean tunicate Ecteinascidia turbinata. Apart from triggering tumor-cell apoptosis, trabectedin was found to selectively induce caspase-8-dependent apoptosis in monocytic cell lineage, including TAMs, through TNF-related apoptosis-inducing ligand (TRAIL) receptors [194]. A preclinical study demonstrated that trabectedin administration before anti-PD-1 immunotherapy could alleviate TAM-mediated immunosuppression and thus improve anti-tumor efficacy, which provides a theoretical basis for the combination of trabectedin and immunotherapy [87].
Other approaches to deplete TAMs in tumors have employed immunotoxins that target scavenger receptor-A or folate receptor β (FRβ) expressed on TAMs. Altogether, the major concern about TAM depletion is non-specificity. General depletion of TAMs via the above approaches may lead to the loss of TRMs, which are vital in maintaining homeostasis and bacterial clearance. It is more reasonable and ideal to target specific TAM subsets with high immunosuppressive properties. In support of this notion, depletion of CD163+ macrophages resulted in a massive infiltration of activated T cells and tumor reduction in an experimental melanoma model that is insensitive to anti-PD-1 therapy, while the pan-targeting of TAMs did not have therapeutic effects [195].
5.3 Enhancing phagocytosis
Macrophages play an essential role via phagocytosis in host defense against pathogens and damaged or aged cells. Normal cells avoid being engulfed by macrophages through the expression of certain molecules, so-called “do not eat me” signals, which can be utilized by tumor cells to evade immune surveillance. There are three regulatory pathways in macrophage phagocytic activities, including the signal-regulated protein α (SIRPα)/CD47 pathway, the CD24/Siglec-10 pathway, and the MHC class I/leukocyte immunoglobulin-like receptor subfamily B member 1 (MHC-I/LILRB1) pathway [196-198]. Blockade of the “do not eat me” signals has long been regarded an important therapeutic strategy in evoking the phagocytic function of TAMs to eliminate tumor cells.
CD47 functions as a ligand for SIRPα, which induces a downstream anti-phagocytic cascade in myeloid cells, including TAMs [197]. A preclinical study showed the therapeutic efficacy of an anti-CD47 antibody via macrophage phagocytosis in mouse tumor models [199]. CD47-SIRPα-targeted agents that have been evaluated in clinical trials include magrolimab (Hu5F9-G4), CC-90002, TTI-621, and BMS-986351. Phase 1 and 2 clinical trials have demonstrated the promising therapeutic efficacy and good tolerability of Hu5F9-G4 in combination with chemotherapeutic agents and anti-CD20 antibody for treating multiple myeloma and non-Hodgkin lymphoma [163, 164]. In theory, the bridging between innate and adaptive immunity paves the way for combining phagocytosis-related drugs with ICI-based immunotherapy to elicit more potent antitumor immunity. Drakaki et al. evaluated the combination of Hu5F9-G4 with atezolizumab in a Phase 1b/2 open-label, multicenter study of platinum-refractory locally advanced or metastatic urothelial carcinomas [165]. Although no improvement in ORR, PFS, or OS in patients treated with atezolizumab plus magrolimab was observed, a trend was observed for increased therapeutic efficacy of atezolizumab plus Hu5F9-G4 in immune-excluded tumors. Maybe the pretreatment selection of CD47 and/or TAM-enriched tumors may improve the efficacy of PD-L1 and CD47 inhibition in this tumor type. Another anti-CD47 antibody, CC-90002, has been evaluated in a Phase 1 study for relapsed/refractory acute myeloid leukemia (AML) and high-risk myelodysplastic syndrome (MDS) [166]. However, no objective responses were observed for CC-90002 as a monotherapy. TTI-621 is a SIRPα-IgG1 Fc fusion protein designed to block CD47. A Phase 1 trial showed that TTI-621 was well-tolerated and demonstrated favorable effects both as monotherapy in relapsed/refractory B-cell and T-cell non-Hodgkin lymphomas, and when combined with rituximab in relapsed/refractory B-cell non-Hodgkin lymphomas [167]. It is worth noting that CD47 is expressed not only on tumor cells but also on erythrocytes, platelets, and neutrophils. Accordingly, anti-CD47 antibodies inevitably led to the depletion of these normal cells in patients [164]. In comparison, approaches to block its counterreceptor SIRPα, expressed on myeloid cells, neutrophils, and microglial cells, are less toxic, but can result in neutropenia and neurotoxicity [200]. BMS-986351, a novel anti-SIRPα mAb, is being evaluated in clinical trials for the treatment of advanced solid and hematologic malignancies.
Additional phagocytic checkpoints also regulate the phagocytosis of macrophages. CD24 generates an inhibitory effect on phagocytosis by binding to Siglec-10 [198]. LILRB1, an inhibitory receptor expressed on macrophages, can interact with MHC-I to inhibit phagocytosis of tumor cells [196]. Novel regulators of phagocytosis may be identified via genome-wide overexpression and knockout CRISPR screens in both cancer cells and macrophages, which may serve as promising targets for the development of therapeutic agents to enhance macrophage phagocytosis in the future.
5.4 Reprogramming of TAMs
The high plasticity of macrophages provides a rationale for TAM reprogramming in cancer treatment. Emerging studies suggest that re-educating TAMs from tumor-supportive M2 phenotype into antitumor phagocytic and cytotoxic M1 macrophages can be more effective than TAM depletion or recruitment inhibition with regard to killing tumor cells. Additionally, TAM reprogramming is associated with the rebalance of immune infiltrates within the TME. At present, therapeutic approaches primarily focus on the activation of CD40 receptors and TLRs, and the inhibition of phosphatidylinositol-3-kinase-γ (PI3Kγ) pathway.
CD40, a member of the TNF receptor superfamily, is primarily expressed by monocytes, macrophages, DCs, B cells, and epithelial cells [201]. Upon binding CD40L, CD40 triggers the upregulation of MHC molecules and the secretion of pro-inflammatory cytokines. Agonistic CD40 antibodies increase TAM infiltration and induce the repolarization of TAMs to favor pro-inflammatory or M1 phenotype in preclinical tumor models [202]. Thus, clinical studies of several anti-CD40 agonists, either as monotherapy or in combination with chemotherapy, targeted therapy, and ICB agents in advanced solid tumors have been performed. A Phase 1 dose-escalation study showed that a single intravenous dose of CP-870893, a selective CD40 agonist mAb, was well tolerated and induced objective response and antitumor activity in solid tumors [168]. A Phase 1 trial tested whether there were synergetic treatment effects between CP-870893 and chemotherapeutics in advanced solid tumors [169]. Both biological responses, such as the depletion of peripheral B cells and the upregulation of immune co-stimulatory molecules, and clinical responses were observed when combining treatment groups. The most common toxicity associated with CP-870893 treatment was cytokine release syndrome (CRS).
TLRs represent one of the major receptor families that polarize macrophages towards a pro-inflammatory and tumoricidal phenotype [203]. Preclinical models of cancer have investigated the antitumor immune responses of TLR2/4, TLR7/8, and TLR9 agonists (BCG, 852A, and imiquimod) owing to their properties in TAM modulation. BCG is one of the first TLR2/4 agonists approved for treating bladder cancer patients based on the results of clinical trials [170]. A Phase 2 study reported that systemically administered 852A induced systemic immune activation, leading to prolonged disease stabilization in chemotherapy-refractory metastatic melanomas [171]. Unfortunately, systemic toxicity, such as fatigue and constitutional symptoms, prevented the use of injections with high levels of TLR ligands in cancer patients. Instead, locally or intratumorally administrated TLR agonists are under evaluation in different tumor models, which have shown the need for achieving a fine balance between effectiveness and toxicity. Weigel et al. evaluated subcutaneously delivered 852A in patients with recurrent hematologic malignancies [172]. The local 852A treatment resulted in objective responses in 15.4% of hematologic cancer patients. A prospective clinical trial showed that topical imiquimod, a TLR7/8 agonist, was well tolerated and achieved a partial response in 20% of breast cancer skin metastases patients [173]. The responders displayed histologic tumor regression and increased tumor lymphocytic infiltration and local cytokine production. It is worth noting that TLR-stimulated TAM is often accompanied by the upregulation of PD-L1 [204], which theoretically enables the future combined use of TLR agonists and PD-1/PD-L1 inhibitors in clinical trials.
PI3Kγ is involved in the pro-tumoral activities of TAMs. PI3Kγ inhibition in TAMs induced the expression of MHC-II and pro-inflammatory cytokines and reduced the immunosuppressive molecules, including IL-10 and Arg-1, which contributed to TAM reprogramming. A preclinical study showed that the PI3Kγ inhibitor eganelisib (IPI-549) reprogrammed TAMs and increased CD8+ T-cell recruitment, achieving tumor growth inhibition when combined with checkpoint inhibitors [205]. Subsequently, a Phase 1/1b MARIO-1 trial demonstrated that IPI-549 achieved antitumor activity when combined with nivolumab in solid tumors, including those who progressed when receiving PD-1/PD-L1 inhibitors [174].
Certain chemotherapeutics, irradiation, or oncolytic virus therapy can induce immunogenic cell death (ICD) of tumor cells, which stimulates antitumor immune responses, and in particular, TAM re-education towards an M1 phenotype, and thus results in additional therapeutic efficiency [206, 207]. In this respect, a transcription factor, RORC1/RORγ, orchestrates cancer-driven myelopoiesis, predominantly of TAMs and MDSCs, by promoting C/EBPβ [208]. Additionally, several specific inhibitors targeting RORC1/RORγ are under evaluation in preclinical models.
5.5 Targeting TAM heterogeneity
New insights into the heterogeneity of TAMs enable the development of novel therapeutics to inhibit TAM-mediated immunosuppression, angiogenesis, and inflammation by targeting immune-related enzymes, ligands, receptors, or signaling transducers. At present, three subgroups of TAMs, including reg-TAMs, angio-TAMs, and inflam-TAMs, are being targeted in clinical trials.
Reg-TAMs express IDO1, TREM2, Arg-1, and the COX-2/PGE2 pathway to induce T-cell exhaustion and Treg infiltration. In a Phase 1a trial, targeting IDO1 enzyme with a small-molecule inhibitor, navoximod (GDC-0919), displayed promising effects in recurrent/advanced solid tumors [175]. TREM is an essential immunosuppressive gene in Reg-TAMs. Molgora et al. reported that Trem2-/- mice are more resistant to the growth of various cancers and more sensitive to PD-1 blockade therapy than wild-type mice [209]. However, targeting TREM2+ Reg-TAMs with PY314, a humanized anti-TREM2 mAb, in combination with PD-1 blockade yielded limited anti-tumor effect in patients with checkpoint inhibition-refractory renal cell carcinoma in the setting of a Phase 1b clinical trial [176]. Arg-1 represents an immunosuppressive enzyme in myeloid cells and induces depletion of L-arginine, an essential nutrient for T cell and NK cell proliferation. A preclinical study found that CB-1158, a small-molecule Arg-1 inhibitor, shifted the immune landscape towards a pro-inflammatory environment, blunted myeloid cell-mediated immunosuppression, and reduced tumor growth in multiple mouse models of cancer [210]. The inflammatory COX-2/PGE2 pathway has been implicated in eliciting immune escape and tumor progression by recruiting and activating Reg-TAMs. In a Phase 2 study, COX-2 inhibitor celecoxib led to a higher pathological complete response rate and an acceptable safety profile when combined with toripalimab as neoadjuvant drugs for mismatch repair-deficient or microsatellite instability-high metastatic colorectal cancer patients [177].
Angio-TAMs are prevalent in the hypoxic core of solid tumors, which facilitate angiogenesis and mediate therapeutic resistance to anti-VEGF agents. ANG-2 confers resistance to anti-VEGF treatment by recruiting angio-TAMs [211]. In a preclinical setting, dual blockade of VEGF and ANG2 enhanced the normalization of tumor vasculature and suppressed tumor progression compared with each therapy alone in mouse tumor models [212]. A Phase 1 study even showed that when combined with ezabenlimab, BI 836880, a humanized bispecific ANG2-VEGF antibody, had a manageable safety profile with preliminary clinical activity in advanced solid tumors [178]. Further clinical trials are warranted to explore the efficiency of targeting angio-TAMs to improve resistance to anti-VEGF or ICI-based treatments.
Inflam-TAMs play an essential role in tumorigenic processes by maintaining a pro-tumor inflammatory and immunosuppressive microenvironment. Numerous inflammatory genes, such as IL-1β, from inflam-TAMs have been evaluated as anti-tumor targets in preclinical investigations [213]. A Phase 3 clinical trial evaluated the effect of first-line canakinumab, an IL-1β-blocking antibody, in conjunction with chemotherapy and immunotherapy in advanced NSCLC [179]. The addition of canakinumab did not prolong PFS or OS in NSCLC patients. Several other approaches targeting inflam-TAMs are still in preclinical investigations for cancer treatment [214].
5.6 Targeting TAM metabolism
There is a growing consensus on the importance of metabolic regulation of immune cells, including TAMs, in reactivating anti-tumor immunity. A list of metabolic intermediates, by-products, and enzymes, generated or activated in the TME in terms of nutrient deprivation, hypoxia, and an acidic environment, underlie the recruitment, activation, expansion, and function of TAMs, which serve as potential targets to reprogram TAMs.
Given that glycolysis and intermediate metabolite lactate are required for the polarization of M2 TAMs, glycolytic inhibitors, such as 2-DG, have been investigated to revert macrophage polarization in a preclinical study [61]. A Phase 1 trial showed that 2-DG at 63 mg/kg combined with docetaxel was well-tolerated and resulted in a partial response in one metastatic breast cancer patient and stable disease in eleven solid tumor patients [180]. However, since glycolysis is fundamental for the phagocytic and tumoricidal function of M1 TAMs, glycolytic pathway blockade may result in undesirable M1 TAM suppression, which may explain the above limited clinical benefits of 2-DG treatment. Metformin, a mitochondrial respiratory chain complex I inhibitor to promote glucose uptake and glycolysis, has also emerged as a therapeutic candidate in TAM repolarization [215]. Preclinically, metformin was found to reeducate M2 TAMs towards an M1 phenotype, which reversed a tumor immunosuppressive microenvironment and synergized with anti-PD-1 immunotherapy in mouse tumor models [216]. Mounting evidence from clinical trials has dissected the encouraging antitumor effects of metformin in prostate and endometrial cancer [181, 182]. The mechanism of antitumor activities of metformin is partially attributed to tumor immune microenvironment reprogramming, based on the repolarization of macrophages to an antitumoral M1 phenotype and increased infiltration of CD8+ T cells and CD20+ B cells in metformin-treated mouse esophageal squamous cell carcinoma models [217]. However, conflicting data and inconclusive results have also been reported. The addition of anti-diabetic doses of metformin did not improve outcomes in early breast cancer and advanced-stage NSCLC treated with standard therapies [218, 219]. Differences in the patient selection, including the diabetic conditions and diets and sensitivity of tumors to energetic stress, constitute major determinants of their responses to metformin and antitumor efficiency. Further trials are required to validate the beneficial effects of metformin in different cancer types.
TAMs display impaired lipid handling, which correlates with the activation of immunosuppressive pathways and the emergence of therapeutic resistance. A preclinical study showed that the cholesterol-lowering simvastatin can induce TAM repolarization from an M2 to M1 phenotype via cholesterol-associated LXR/ABCA1 regulation, resulting in the reversion of EMT-associated resistance to chemotherapy [220]. Future clinical studies are required to evaluate cholesterol metabolism modulators in cancer treatment. Additionally, tumor-derived prostaglandin E2 (PGE2) induces the transformation of myeloid cells toward an immunosuppressive phenotype. Targeting of COX2/PGE2/EP2-4 pathway with nonsteroidal and steroidal anti-inflammatory drugs enhanced the efficacy of immune checkpoint inhibitors in mouse tumor models [221]. FAO represents another metabolic hallmark in immunosuppressive TAMs. A previous study showed that inhibition of free fatty acid production can repolarize TAMs to a pro-inflammatory phenotype, promoting secretion of tumor-killing cytokines in pancreatic adenocarcinoma tumor models [222]. However, another study showed that TLR9 agonist, CpG oligodeoxynucleotides, increased the membrane fluidity of macrophages and enhanced the phagocytosis of tumor cells through promoting intracellular FAO by activating carnitine palmitoyltransferase 1A (CPT1A) and citrate lyase [67].
Abnormal glutamine, arginine, and tryptophan metabolism is intricately associated with the immunosuppressive activities of M2 TAMs, highlighting the possibility of targeting amino acid metabolism as an anti-tumor strategy. Several small-molecule inhibitors targeting IDO1 have been assessed in clinical studies, including epacadostat and indoximod. However, the addition of IDO1 inhibitors to immunotherapy yielded limited antitumor activity in sarcoma and melanoma [183, 184]. It is hypothesized that the compensatory expression of other immunosuppressive enzymes, including tryptophan 2,3-dioxygenase (TDO) and IDO2, may limit the effects of IDO1 inhibitors.
Other approaches include the epigenetic modulation and DNA damage repair of TAMs, among other mechanisms. A Phase 1 study showed early signs of efficacy of a histone deacetylase (HDAC) inhibitor, tefinostat (CHR-2845), in patients with advanced hematologic malignancies [185]. Another preclinical study showed that PARP inhibitors enhanced both anti- and pro-tumor properties of TAMs through glucose and lipid metabolic reprogramming in BRCA-deficient TNBC models [223]. Of note, non-specific drugs affecting shared metabolic pathways can impact numerous cellular components within the TME, potentially resulting in unpredictable side-effects and poor effectiveness in cancer treatment. Identification of more specific metabolic transcription factors, pathways, or byproducts involved in TAM reprogramming is required to better strategically manage the various types of cancer.
5.7 Genetically engineered macrophages
Advances in cellular engineering methods hold notable potential, including reprogramming macrophages, as a promising anti-tumor strategy. In comparison with adoptive T cells, genetically engineered macrophages infiltrate the TME more efficiently, functioning not only in tumor cell phagocytosis, but also in neo-antigen presentation to tumoricidal immune cells. Macrophage precursors, including circulating monocytes and isolated hematopoietic stem cells (HSC), can be genetically modified by using adenoviral or lentiviral transduction, or gene editing using CRISPR-Cas9 technology [224].
Genetically engineered macrophages were obtained through lentiviral transduction of antitumor genes, such as IL-12, into macrophage precursors to activate antitumor immune responses [225]. By using viral protein X (VPX)-containing lentivirus, Klichinsky et al. engineered macrophages with an anti-human EGF receptor 2 (HER2) CAR with a CD3ζ cytosolic domain that recognized tumoral HER2 antigen, and this transformed macrophages into a pro-inflammatory phenotype, enhanced antigen-specific phagocytosis, and reduced tumor growth and metastasis in xenograft mouse models [226]. Nowadays, various combinations of CAR constructs based on antigen-binding receptors and cytosolic domains have been evaluated preclinically for their phagocytic and immuno-stimulating capacities in macrophages [227]. As for the CRISPR-Cas9 approach, arginine nanoparticles (ArgNPs) have been used to deliver CRISPR-Cas9 into macrophages to knockout SIRP-α, and this increased their capacity to phagocytose the U2OS osteosarcoma cells 4-fold [228].
One of the biggest challenges for genetically engineered macrophages is the lack of expansion in vitro and self-renewal in vivo following adoptive transfer. Moreover, macrophages cannot react to HLA and are less effective than CAR-T cells at direct target cell killing. Genetic changes in HSC may lead to off-target effects, such as leukemia or lymphoma. New technologies to expand macrophages, or to identify specific tumor antigens or immuno-stimulatory targets, will have to be implemented in the future.
6. Concluding Remarks and Perspectives
In recent years, tumor immunotherapy has seen significant progress. However, patient responsiveness varies significantly across different tumor types and among individuals, and the TME plays a pivotal role in the response to immunotherapy. TAMs represent one of the predominant immune cell types within the TME and interact with their surroundings to influence the immune outcomes. It is essential to elucidate the precise regulatory mechanisms and identify specific targets of TAMs to enhance the efficacy of current immunotherapies. Current research on TAMs continues to face numerous challenges: 1) there is a need for a unified and more scientific approach to identify TAM subtypes; 2) investigations into the mechanisms underlying the interactions between TAMs and the TME remain insufficient; 3) the clinical responses and adverse events associated with TAM-targeted therapies require further evaluation. It is important to note that there may be intrinsic connections among the aforementioned challenges. Despite these limitations, it is encouraging to note that research on TAMs has made significant strides in recent years. For example, single-cell sequencing technologies have provided an opportunity to identify subtypes of TAMs. The metabolic pathways of TAMs have emerged as a novel perspective for subtype identification and as new targets for reprogramming phenotypes. Targeting TAMs, either as a monotherapy or as an adjuvant to chemotherapy and targeted therapies, has received positive feedback from clinical studies. In summary, immunotherapy centered on TAMs is experiencing robust development. The ultimate goal is to reverse the immunosuppressive TME by targeting TAMs, thereby enhancing the efficacy of immunotherapy and ultimately benefiting cancer patients.
Abbreviations
ICB: immune checkpoint blockade; CAR: chimeric antigen receptor; TME: tumor microenvironment; CTL: cytotoxic T lymphocyte; NK: natural killer; Treg: regulatory T; MDSC: myeloid-derived suppressor cell; TAM: tumor-associated macrophage; M2 TAM: alternatively activated type 2 TAM; TRM: tissue-resident macrophage; CAF: cancer-associated fibroblast; ECM: extracellular matrix; TNBC: triple-negative breast cancer; SDF-1: stromal cell-derived factor-1; M1 TAM: classically activated type 1 TAM; MAM: metastasis-associated macrophage; CSF1: colony-stimulating factor 1; OXPHOS: oxidative phosphorylation; IL-4Rα: IL-4 receptor α; LPS: lipopolysaccharide; HIF-1α: hypoxia inducible factor-1α; TLR: Toll-like receptor; ADCC: antibody-dependent cell-mediated cytotoxicity; iNOS: inducible nitric oxide synthetase; G6P: glucose-6-phosphate; PPP: pentose phosphate pathway; NADPH: nicotinamide adenine dinucleotide phosphate; NOX: NADPH oxidase; DH: isocitrate dehydrogenase; ACOD1: aconitate decarboxylase 1; ITA: itaconate; SDH: succinate dehydrogenase; NO: nitric oxide; 2-DG: 2-Deoxy-d-glucose; Arg-1: arginase-1; FAO: fatty acid oxidation; LAL: lysosomal acid lipase; LAM: lipid-associated macrophage; NSCLC: non-small cell lung cancer; GS: glutamine synthetase; GLUL: glutamate-ammonia ligase; α-KG: α-ketoglutarate; GLUD1: glutamate dehydrogenase 1; IDO: indoleamine 2,3-dioxygenase; DC: dendritic cell; EV: extracellular vesicle; HCC: hepatocellular carcinoma; ADAM15: a disintegrin and metalloproteinase 15; ApoE: apolipoprotein E; HLF: hepatocyte leukemia factor; NEDD4-1: neural precursor cell expressed developmentally downregulated protein 4-1; CSC: cancer stem cell; GPNMB: glycoprotein myosone; PDPK1: phosphoinositide-dependent protein kinase 1; PGK1: phosphoglycerate kinase 1; HISLA: HIF-1α-stabilizing long noncoding RNA; TDE: tumor-derived exosome; TCR: T cell receptor; MHC: major histocompatibility complex; IL-15Rc: IL-15Rα complex; Stereo-seq: SpaTial Enhanced REsolution Omics-sequencing; TLS: tertiary lymphoid structure; Breg: regulatory B; cAMP: cyclic adenosine monophosphate; ILC: innate lymphoid cell; LTi: lymphoid tissue-inducer; M-MDSC: monocytic MDSC; PMN-MDSC: polymorphonuclear MDSC; NET: neutrophil extracellular trap; TAN: tumor-associated neutrophil; N1 TAN: anti-tumor TAN; N2 TAN: pro-tumor TAN; CHI3L1: chitinase-3-like-protein-1; α-SMA: α-smooth muscle actin; FSP1: fibroblast-specific protein 1; FAP: fibroblast activation protein; PDGFR: platelet-derived growth factor receptor; MMT: macrophage-myofibroblast transition; EC: endothelial cell; VCAM-1: vascular cellular adhesion molecule-1; PDAC: pancreatic ductal adenocarcinoma; FOLFIRINOX: 5-FU, leucovorin, irinotecan and oxaliplatin; OS: overall survival; ORR: objective response rate; PFS: progression free survival; AML: acute myeloid leukemia; CLL: chromic lymphocytic leukemia; MDS: myelodysplastic syndrome; MPA: medroxyprogesterone acetate; HDAC: histone deacetylase; FPP: farnesyl diphosphate; TRAIL: TNF-related apoptosis-inducing ligand; FRβ: folate receptor β; SIRPα: signal-regulated protein α; LILRB1: leukocyte immunoglobulin-like receptor subfamily B member 1; PI3Kγ: phosphatidylinositol-3-kinase-γ; CRS: cytokine release syndrome; ICD: immunogenic cell death; PGE2: prostaglandin E2; CPT1A: carnitine palmitoyltransferase 1A; TDO: tryptophan 2,3-dioxygenase; HSC: hematopoietic stem cell; VPX: viral protein X; HER2: human EGF receptor 2; ArgNP: arginine nanoparticle.
Acknowledgements
We would like to thank Meiyu Geng from Chinese Academy of Sciences, Handong Jiang from Shanghai Jiao Tong University, and Haiquan Chen from Fudan University Shanghai Cancer Center for their invaluable guidance and support in the conception of this manuscript.
Funding
This work was supported by the Shandong Provincial Natural Science Foundation (grants ZR2023QH021 and ZR2023MH250) in China, the Promotion Foundation of Shandong Provincial Hospital (grant 2024TS04) in China, the National Natural Science Foundation of China (grant 81902325), the China Postdoctoral Science Foundation (grant 2023M740684), the Jinan Science and Technology Plan Project (grants 202019201 and 202225051) in China, and the Key Research and Development Plan of Shandong Province (grant 2019GSF107051) in China.
Author contributions
Hancheng Wu: Methodology, Formal analysis, and Writing-Original Draft; Jing Li: Software, Investigation, Data curation, and Funding acquisition; Ruilin Yao: Methodology, Software, Investigation, and Visualization; Jing Liu: Methodology, Software, Validation, and Visualization; Lili Su: Validation, Resources, Writing-Editing, Supervision, Project administration, and Funding acquisition; Wenjie You: Conceptualization, Validation, Resources, Data Curation, Writing-Review & Editing, Supervision, Project administration, and Funding acquisition. All authors read and approved the final manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Topalian SL, Drake CG, Pardoll DM. Immune checkpoint blockade: a common denominator approach to cancer therapy. Cancer Cell. 2015;27:450-61
2. Xu X, Li T, Shen S, Wang J, Abdou P, Gu Z. et al. Advances in Engineering Cells for Cancer Immunotherapy. Theranostics. 2019;9:7889-7905
3. Lin MJ, Svensson-Arvelund J, Lubitz GS, Marabelle A, Melero I, Brown BD. et al. Cancer vaccines: the next immunotherapy frontier. Nat Cancer. 2022;3:911-926
4. Haslam A, Prasad V. Estimation of the Percentage of US Patients With Cancer Who Are Eligible for and Respond to Checkpoint Inhibitor Immunotherapy Drugs. JAMA Netw Open. 2019;2:e192535
5. Binnewies M, Roberts EW, Kersten K, Chan V, Fearon DF, Merad M. et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat Med. 2018;24:541-550
6. Engblom C, Pfirschke C, Pittet MJ. The role of myeloid cells in cancer therapies. Nat Rev Cancer. 2016;16:447-62
7. Xiang X, Wang J, Lu D, Xu X. Targeting tumor-associated macrophages to synergize tumor immunotherapy. Signal Transduct Target Ther. 2021;6:75
8. Vitale I, Manic G, Coussens LM, Kroemer G, Galluzzi L. Macrophages and Metabolism in the Tumor Microenvironment. Cell Metab. 2019;30:36-50
9. Mantovani A, Marchesi F, Malesci A, Laghi L, Allavena P. Tumour-associated macrophages as treatment targets in oncology. Nat Rev Clin Oncol. 2017;14:399-416
10. Bottazzi B, Polentarutti N, Acero R, Balsari A, Boraschi D, Ghezzi P. et al. Regulation of the macrophage content of neoplasms by chemoattractants. Science. 1983;220:210-2
11. Arakaki R, Yamasaki T, Kanno T, Shibasaki N, Sakamoto H, Utsunomiya N. et al. CCL2 as a potential therapeutic target for clear cell renal cell carcinoma. Cancer Med. 2016;5:2920-2933
12. Fujimoto H, Sangai T, Ishii G, Ikehara A, Nagashima T, Miyazaki M. et al. Stromal MCP-1 in mammary tumors induces tumor-associated macrophage infiltration and contributes to tumor progression. Int J Cancer. 2009;125:1276-84
13. Yoshimura T, Howard OM, Ito T, Kuwabara M, Matsukawa A, Chen K. et al. Monocyte chemoattractant protein-1/CCL2 produced by stromal cells promotes lung metastasis of 4T1 murine breast cancer cells. PLoS One. 2013;8:e58791
14. Pena CG, Nakada Y, Saatcioglu HD, Aloisio GM, Cuevas I, Zhang S. et al. LKB1 loss promotes endometrial cancer progression via CCL2-dependent macrophage recruitment. J Clin Invest. 2015;125:4063-76
15. Weber C, Weber KS, Klier C, Gu S, Wank R, Horuk R. et al. Specialized roles of the chemokine receptors CCR1 and CCR5 in the recruitment of monocytes and T(H)1-like/CD45RO(+) T cells. Blood. 2001;97:1144-6
16. Frankenberger C, Rabe D, Bainer R, Sankarasharma D, Chada K, Krausz T. et al. Metastasis Suppressors Regulate the Tumor Microenvironment by Blocking Recruitment of Prometastatic Tumor-Associated Macrophages. Cancer Res. 2015;75:4063-73
17. Boyle ST, Faulkner JW, McColl SR, Kochetkova M. The chemokine receptor CCR6 facilitates the onset of mammary neoplasia in the MMTV-PyMT mouse model via recruitment of tumor-promoting macrophages. Mol Cancer. 2015;14:115
18. Wang SC, Yu CF, Hong JH, Tsai CS, Chiang CS. Radiation therapy-induced tumor invasiveness is associated with SDF-1-regulated macrophage mobilization and vasculogenesis. PLoS One. 2013;8:e69182
19. Tripathi C, Tewari BN, Kanchan RK, Baghel KS, Nautiyal N, Shrivastava R. et al. Macrophages are recruited to hypoxic tumor areas and acquire a pro-angiogenic M2-polarized phenotype via hypoxic cancer cell derived cytokines Oncostatin M and Eotaxin. Oncotarget. 2014;5:5350-68
20. Xuan W, Qu Q, Zheng B, Xiong S, Fan GH. The chemotaxis of M1 and M2 macrophages is regulated by different chemokines. J Leukoc Biol. 2015;97:61-9
21. Hughes R, Qian BZ, Rowan C, Muthana M, Keklikoglou I, Olson OC. et al. Perivascular M2 Macrophages Stimulate Tumor Relapse after Chemotherapy. Cancer Res. 2015;75:3479-91
22. Lee PY, Li Y, Kumagai Y, Xu Y, Weinstein JS, Kellner ES. et al. Type I interferon modulates monocyte recruitment and maturation in chronic inflammation. Am J Pathol. 2009;175:2023-33
23. Kitamura T, Qian BZ, Soong D, Cassetta L, Noy R, Sugano G. et al. CCL2-induced chemokine cascade promotes breast cancer metastasis by enhancing retention of metastasis-associated macrophages. J Exp Med. 2015;212:1043-59
24. Wu Y, Li YY, Matsushima K, Baba T, Mukaida N. CCL3-CCR5 axis regulates intratumoral accumulation of leukocytes and fibroblasts and promotes angiogenesis in murine lung metastasis process. J Immunol. 2008;181:6384-93
25. Loyher PL, Hamon P, Laviron M, Meghraoui-Kheddar A, Goncalves E, Deng Z. et al. Macrophages of distinct origins contribute to tumor development in the lung. J Exp Med. 2018;215:2536-2553
26. Wiktor-Jedrzejczak W, Bartocci A, Ferrante AW Jr, Ahmed-Ansari A, Sell KW, Pollard JW. et al. Total absence of colony-stimulating factor 1 in the macrophage-deficient osteopetrotic (op/op) mouse. Proc Natl Acad Sci U S A. 1990;87:4828-32
27. Wang Y, Szretter KJ, Vermi W, Gilfillan S, Rossini C, Cella M. et al. IL-34 is a tissue-restricted ligand of CSF1R required for the development of Langerhans cells and microglia. Nat Immunol. 2012;13:753-60
28. Dai XM, Ryan GR, Hapel AJ, Dominguez MG, Russell RG, Kapp S. et al. Targeted disruption of the mouse colony-stimulating factor 1 receptor gene results in osteopetrosis, mononuclear phagocyte deficiency, increased primitive progenitor cell frequencies, and reproductive defects. Blood. 2002;99:111-20
29. Wang H, Wang L, Gong G, Lin X, Luo J, Liu C. et al. Interleukin-10: a novel metabolic inducer of macrophage differentiation and subsequently contributing to improved pregnancy outcomes of mice by orchestrating oxidative phosphorylation metabolismdagger. Biol Reprod. 2024;111:76-91
30. Finlay CM, Parkinson JE, Zhang L, Chan BHK, Ajendra J, Chenery A. et al. T helper 2 cells control monocyte to tissue-resident macrophage differentiation during nematode infection of the pleural cavity. Immunity. 2023;56:1064-1081.e10
31. Dong Y, Arif AA, Guo J, Ha Z, Lee-Sayer SSM, Poon GFT. et al. CD44 Loss Disrupts Lung Lipid Surfactant Homeostasis and Exacerbates Oxidized Lipid-Induced Lung Inflammation. Front Immunol. 2020;11:29
32. Kvedaraite E, Lourda M, Mouratidou N, Duking T, Padhi A, Moll K. et al. Intestinal stroma guides monocyte differentiation to macrophages through GM-CSF. Nat Commun. 2024;15:1752
33. Hibbs ML, Quilici C, Kountouri N, Seymour JF, Armes JE, Burgess AW. et al. Mice lacking three myeloid colony-stimulating factors (G-CSF, GM-CSF, and M-CSF) still produce macrophages and granulocytes and mount an inflammatory response in a sterile model of peritonitis. J Immunol. 2007;178:6435-43
34. Kim J, Kim TJ, Chae S, Ha H, Park Y, Park S. et al. Targeted deletion of CD244 on monocytes promotes differentiation into anti-tumorigenic macrophages and potentiates PD-L1 blockade in melanoma. Mol Cancer. 2024;23:45
35. Prat M, Le Naour A, Coulson K, Lemee F, Leray H, Jacquemin G. et al. Circulating CD14(high) CD16(low) intermediate blood monocytes as a biomarker of ascites immune status and ovarian cancer progression. J Immunother Cancer. 2020;8:e000472
36. Dander E, Fallati A, Gulic T, Pagni F, Gaspari S, Silvestri D. et al. Monocyte-macrophage polarization and recruitment pathways in the tumour microenvironment of B-cell acute lymphoblastic leukaemia. Br J Haematol. 2021;193:1157-1171
37. Schulz C, Gomez Perdiguero E, Chorro L, Szabo-Rogers H, Cagnard N, Kierdorf K. et al. A lineage of myeloid cells independent of Myb and hematopoietic stem cells. Science. 2012;336:86-90
38. Zhu Y, Herndon JM, Sojka DK, Kim KW, Knolhoff BL, Zuo C. et al. Tissue-Resident Macrophages in Pancreatic Ductal Adenocarcinoma Originate from Embryonic Hematopoiesis and Promote Tumor Progression. Immunity. 2017;47:597
39. Boutilier AJ, Elsawa SF. Macrophage Polarization States in the Tumor Microenvironment. Int J Mol Sci. 2021;22:6995
40. Wang S, Liu G, Li Y, Pan Y. Metabolic Reprogramming Induces Macrophage Polarization in the Tumor Microenvironment. Front Immunol. 2022;13:840029
41. Cao M, Yan H, Han X, Weng L, Wei Q, Sun X. et al. Ginseng-derived nanoparticles alter macrophage polarization to inhibit melanoma growth. J Immunother Cancer. 2019;7:326
42. Li Y, Chen Y, Zhao C, Yang Y, Zhang M, Cheng H. et al. Arenobufagin modulation of PCSK9-mediated cholesterol metabolism induces tumor-associated macrophages polarisation to inhibit hepatocellular carcinoma progression. Phytomedicine. 2024;128:155532
43. Li M, Yang Y, Xiong L, Jiang P, Wang J, Li C. Metabolism, metabolites, and macrophages in cancer. J Hematol Oncol. 2023;16:80
44. Mantovani A, Sica A, Sozzani S, Allavena P, Vecchi A, Locati M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 2004;25:677-86
45. Duluc D, Delneste Y, Tan F, Moles MP, Grimaud L, Lenoir J. et al. Tumor-associated leukemia inhibitory factor and IL-6 skew monocyte differentiation into tumor-associated macrophage-like cells. Blood. 2007;110:4319-30
46. Mills CD, Kincaid K, Alt JM, Heilman MJ, Hill AM. M-1/M-2 macrophages and the Th1/Th2 paradigm. J Immunol. 2000;164:6166-73
47. Shapouri-Moghaddam A, Mohammadian S, Vazini H, Taghadosi M, Esmaeili SA, Mardani F. et al. Macrophage plasticity, polarization, and function in health and disease. J Cell Physiol. 2018;233:6425-6440
48. Mills CD, Lenz LL, Harris RA. A Breakthrough: Macrophage-Directed Cancer Immunotherapy. Cancer Res. 2016;76:513-6
49. Nielsen SR, Schmid MC. Macrophages as Key Drivers of Cancer Progression and Metastasis. Mediators Inflamm. 2017;2017:9624760
50. Sica A, Mantovani A. Macrophage plasticity and polarization: in vivo veritas. J Clin Invest. 2012;122:787-95
51. Malyshev I, Malyshev Y. Current Concept and Update of the Macrophage Plasticity Concept: Intracellular Mechanisms of Reprogramming and M3 Macrophage "Switch" Phenotype. Biomed Res Int. 2015;2015:341308
52. Erbel C, Tyka M, Helmes CM, Akhavanpoor M, Rupp G, Domschke G. et al. CXCL4-induced plaque macrophages can be specifically identified by co-expression of MMP7+S100A8+ in vitro and in vivo. Innate Immun. 2015;21:255-65
53. Yi C, Li Z, Zhao Q, Gong D, Zhao S, Chen Z. et al. Single-Cell RNA Sequencing Pro-angiogenic Macrophage Profiles Reveal Novel Prognostic Biomarkers and Therapeutic Targets for Osteosarcoma. Biochem Genet. 2024;62:1325-1346
54. Yang Q, Zhang H, Wei T, Lin A, Sun Y, Luo P. et al. Single-Cell RNA Sequencing Reveals the Heterogeneity of Tumor-Associated Macrophage in Non-Small Cell Lung Cancer and Differences Between Sexes. Front Immunol. 2021;12:756722
55. Cheng S, Li Z, Gao R, Xing B, Gao Y, Yang Y. et al. A pan-cancer single-cell transcriptional atlas of tumor infiltrating myeloid cells. Cell. 2021;184:792-809.e23
56. Tsai TL, Zhou TA, Hsieh YT, Wang JC, Cheng HK, Huang CH. et al. Multiomics reveal the central role of pentose phosphate pathway in resident thymic macrophages to cope with efferocytosis-associated stress. Cell Rep. 2022;40:111065
57. Michelucci A, Cordes T, Ghelfi J, Pailot A, Reiling N, Goldmann O. et al. Immune-responsive gene 1 protein links metabolism to immunity by catalyzing itaconic acid production. Proc Natl Acad Sci U S A. 2013;110:7820-5
58. Lampropoulou V, Sergushichev A, Bambouskova M, Nair S, Vincent EE, Loginicheva E. et al. Itaconate Links Inhibition of Succinate Dehydrogenase with Macrophage Metabolic Remodeling and Regulation of Inflammation. Cell Metab. 2016;24:158-66
59. Wang F, Zhang S, Vuckovic I, Jeon R, Lerman A, Folmes CD. et al. Glycolytic Stimulation Is Not a Requirement for M2 Macrophage Differentiation. Cell Metab. 2018;28:463-475.e4
60. Latour YL, Gobert AP, Wilson KT. The role of polyamines in the regulation of macrophage polarization and function. Amino Acids. 2020;52:151-160
61. Zhao Q, Chu Z, Zhu L, Yang T, Wang P, Liu F. et al. 2-Deoxy-d-Glucose Treatment Decreases Anti-inflammatory M2 Macrophage Polarization in Mice with Tumor and Allergic Airway Inflammation. Front Immunol. 2017;8:637
62. He Z, Zhang S. Tumor-Associated Macrophages and Their Functional Transformation in the Hypoxic Tumor Microenvironment. Front Immunol. 2021;12:741305
63. Weiss JM, Davies LC, Karwan M, Ileva L, Ozaki MK, Cheng RY. et al. Itaconic acid mediates crosstalk between macrophage metabolism and peritoneal tumors. J Clin Invest. 2018;128:3794-3805
64. Geeraerts X, Fernandez-Garcia J, Hartmann FJ, de Goede KE, Martens L, Elkrim Y. et al. Macrophages are metabolically heterogeneous within the tumor microenvironment. Cell Rep. 2021;37:110171
65. Xia H, Li S, Li X, Wang W, Bian Y, Wei S. et al. Autophagic adaptation to oxidative stress alters peritoneal residential macrophage survival and ovarian cancer metastasis. JCI Insight. 2020;5:e141115
66. Huang SC, Everts B, Ivanova Y, O'Sullivan D, Nascimento M, Smith AM. et al. Cell-intrinsic lysosomal lipolysis is essential for alternative activation of macrophages. Nat Immunol. 2014;15:846-55
67. Liu M, O'Connor RS, Trefely S, Graham K, Snyder NW, Beatty GL. Metabolic rewiring of macrophages by CpG potentiates clearance of cancer cells and overcomes tumor-expressed CD47-mediated 'don't-eat-me' signal. Nat Immunol. 2019;20:265-275
68. Wu SZ, Al-Eryani G, Roden DL, Junankar S, Harvey K, Andersson A. et al. A single-cell and spatially resolved atlas of human breast cancers. Nat Genet. 2021;53:1334-1347
69. Yang L, Venneti S, Nagrath D. Glutaminolysis: A Hallmark of Cancer Metabolism. Annu Rev Biomed Eng. 2017;19:163-194
70. Shang M, Cappellesso F, Amorim R, Serneels J, Virga F, Eelen G. et al. Macrophage-derived glutamine boosts satellite cells and muscle regeneration. Nature. 2020;587:626-631
71. Zhou W, Hu G, He J, Wang T, Zuo Y, Cao Y. et al. SENP1-Sirt3 signaling promotes alpha-ketoglutarate production during M2 macrophage polarization. Cell Rep. 2022;39:110660
72. Labadie BW, Bao R, Luke JJ. Reimagining IDO Pathway Inhibition in Cancer Immunotherapy via Downstream Focus on the Tryptophan-Kynurenine-Aryl Hydrocarbon Axis. Clin Cancer Res. 2019;25:1462-1471
73. Chen S, Chen Z, Li Z, Li S, Wen Z, Cao L. et al. Tumor-associated macrophages promote cholangiocarcinoma progression via exosomal Circ_0020256. Cell Death Dis. 2022;13:94
74. Wang L, Yi X, Xiao X, Zheng Q, Ma L, Li B. Exosomal miR-628-5p from M1 polarized macrophages hinders m6A modification of circFUT8 to suppress hepatocellular carcinoma progression. Cell Mol Biol Lett. 2022;27:106
75. Lee HD, Koo BH, Kim YH, Jeon OH, Kim DS. Exosome release of ADAM15 and the functional implications of human macrophage-derived ADAM15 exosomes. FASEB J. 2012;26:3084-95
76. Zheng P, Luo Q, Wang W, Li J, Wang T, Wang P. et al. Tumor-associated macrophages-derived exosomes promote the migration of gastric cancer cells by transfer of functional Apolipoprotein E. Cell Death Dis. 2018;9:434
77. Lan J, Sun L, Xu F, Liu L, Hu F, Song D. et al. M2 Macrophage-Derived Exosomes Promote Cell Migration and Invasion in Colon Cancer. Cancer Res. 2019;79:146-158
78. Maimon A, Levi-Yahid V, Ben-Meir K, Halpern A, Talmi Z, Priya S. et al. Myeloid cell-derived PROS1 inhibits tumor metastasis by regulating inflammatory and immune responses via IL-10. J Clin Invest. 2021;131:e126089
79. Chen C, Zhang L, Ruan Z. GATA3 Encapsulated by Tumor-Associated Macrophage-Derived Extracellular Vesicles Promotes Immune Escape and Chemotherapy Resistance of Ovarian Cancer Cells by Upregulating the CD24/Siglec-10 Axis. Mol Pharm. 2023;20:971-986
80. Cianciaruso C, Beltraminelli T, Duval F, Nassiri S, Hamelin R, Mozes A. et al. Molecular Profiling and Functional Analysis of Macrophage-Derived Tumor Extracellular Vesicles. Cell Rep. 2019;27:3062-3080.e11
81. Li H, Yang P, Wang J, Zhang J, Ma Q, Jiang Y. et al. HLF regulates ferroptosis, development and chemoresistance of triple-negative breast cancer by activating tumor cell-macrophage crosstalk. J Hematol Oncol. 2022;15:2
82. Xin L, Zhou LQ, Liu C, Zeng F, Yuan YW, Zhou Q. et al. Transfer of LncRNA CRNDE in TAM-derived exosomes is linked with cisplatin resistance in gastric cancer. EMBO Rep. 2021;22:e52124
83. Liguori M, Digifico E, Vacchini A, Avigni R, Colombo FS, Borroni EM. et al. The soluble glycoprotein NMB (GPNMB) produced by macrophages induces cancer stemness and metastasis via CD44 and IL-33. Cell Mol Immunol. 2021;18:711-722
84. Yan J, Zhao Q, Wang J, Tian X, Wang J, Xia X. et al. FGL2-wired macrophages secrete CXCL7 to regulate the stem-like functionality of glioma cells. Cancer Lett. 2021;506:83-94
85. Zhang Y, Yu G, Chu H, Wang X, Xiong L, Cai G. et al. Macrophage-Associated PGK1 Phosphorylation Promotes Aerobic Glycolysis and Tumorigenesis. Mol Cell. 2018;71:201-215.e7
86. Xu M, Zhou C, Weng J, Chen Z, Zhou Q, Gao J. et al. Tumor associated macrophages-derived exosomes facilitate hepatocellular carcinoma malignance by transferring lncMMPA to tumor cells and activating glycolysis pathway. J Exp Clin Cancer Res. 2022;41:253
87. Chen F, Chen J, Yang L, Liu J, Zhang X, Zhang Y. et al. Extracellular vesicle-packaged HIF-1alpha-stabilizing lncRNA from tumour-associated macrophages regulates aerobic glycolysis of breast cancer cells. Nat Cell Biol. 2019;21:498-510
88. Wang H, Shao Q, Sun J, Ma C, Gao W, Wang Q. et al. Interactions between colon cancer cells and tumor-infiltrated macrophages depending on cancer cell-derived colony stimulating factor 1. Oncoimmunology. 2016;5:e1122157
89. Colegio OR, Chu NQ, Szabo AL, Chu T, Rhebergen AM, Jairam V. et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature. 2014;513:559-63
90. Wu JY, Huang TW, Hsieh YT, Wang YF, Yen CC, Lee GL. et al. Cancer-Derived Succinate Promotes Macrophage Polarization and Cancer Metastasis via Succinate Receptor. Mol Cell. 2020;77:213-227.e5
91. Gabrusiewicz K, Li X, Wei J, Hashimoto Y, Marisetty AL, Ott M. et al. Glioblastoma stem cell-derived exosomes induce M2 macrophages and PD-L1 expression on human monocytes. Oncoimmunology. 2018;7:e1412909
92. Wolf-Dennen K, Gordon N, Kleinerman ES. Exosomal communication by metastatic osteosarcoma cells modulates alveolar macrophages to an M2 tumor-promoting phenotype and inhibits tumoricidal functions. Oncoimmunology. 2020;9:1747677
93. Hossain MA, Liu G, Dai B, Si Y, Yang Q, Wazir J. et al. Reinvigorating exhausted CD8(+) cytotoxic T lymphocytes in the tumor microenvironment and current strategies in cancer immunotherapy. Med Res Rev. 2021;41:156-201
94. Oh DY, Kwek SS, Raju SS, Li T, McCarthy E, Chow E. et al. Intratumoral CD4(+) T Cells Mediate Anti-tumor Cytotoxicity in Human Bladder Cancer. Cell. 2020;181:1612-1625.e13
95. Li L, Han L, Sun F, Zhou J, Ohaegbulam KC, Tang X. et al. NF-kappaB RelA renders tumor-associated macrophages resistant to and capable of directly suppressing CD8(+) T cells for tumor promotion. Oncoimmunology. 2018;7:e1435250
96. Borgoni S, Iannello A, Cutrupi S, Allavena P, D'Incalci M, Novelli F. et al. Depletion of tumor-associated macrophages switches the epigenetic profile of pancreatic cancer infiltrating T cells and restores their anti-tumor phenotype. Oncoimmunology. 2018;7:e1393596
97. Briem O, Kallberg E, Kimbung S, Veerla S, Stenstrom J, Hatschek T. et al. CD169(+) Macrophages in Primary Breast Tumors Associate with Tertiary Lymphoid Structures, T(regs) and a Worse Prognosis for Patients with Advanced Breast Cancer. Cancers (Basel). 2023;15:1262
98. Garrido-Martin EM, Mellows TWP, Clarke J, Ganesan AP, Wood O, Cazaly A. et al. M1(hot) tumor-associated macrophages boost tissue-resident memory T cells infiltration and survival in human lung cancer. J Immunother Cancer. 2020;8:e000778
99. Zhuang H, Dai X, Zhang X, Mao Z, Huang H. Sophoridine suppresses macrophage-mediated immunosuppression through TLR4/IRF3 pathway and subsequently upregulates CD8(+) T cytotoxic function against gastric cancer. Biomed Pharmacother. 2020;121:109636
100. Daley D, Mani VR, Mohan N, Akkad N, Pandian G, Savadkar S. et al. NLRP3 signaling drives macrophage-induced adaptive immune suppression in pancreatic carcinoma. J Exp Med. 2017;214:1711-1724
101. Zhang W, Zhang Q, Yang N, Shi Q, Su H, Lin T. et al. Crosstalk between IL-15Ralpha(+) tumor-associated macrophages and breast cancer cells reduces CD8(+) T cell recruitment. Cancer Commun (Lond). 2022;42:536-557
102. Nalio Ramos R, Missolo-Koussou Y, Gerber-Ferder Y, Bromley CP, Bugatti M, Nunez NG. et al. Tissue-resident FOLR2(+) macrophages associate with CD8(+) T cell infiltration in human breast cancer. Cell. 2022;185:1189-1207.e25
103. Eisel D, Das K, Dickes E, Konig R, Osen W, Eichmuller SB. Cognate Interaction With CD4(+) T Cells Instructs Tumor-Associated Macrophages to Acquire M1-Like Phenotype. Front Immunol. 2019;10:219
104. Spear P, Barber A, Rynda-Apple A, Sentman CL. Chimeric antigen receptor T cells shape myeloid cell function within the tumor microenvironment through IFN-gamma and GM-CSF. J Immunol. 2012;188:6389-98
105. Liu C, Chikina M, Deshpande R, Menk AV, Wang T, Tabib T. et al. Treg Cells Promote the SREBP1-Dependent Metabolic Fitness of Tumor-Promoting Macrophages via Repression of CD8(+) T Cell-Derived Interferon-gamma. Immunity. 2019;51:381-397.e6
106. Kurten CHL, Kulkarni A, Cillo AR, Santos PM, Roble AK, Onkar S. et al. Investigating immune and non-immune cell interactions in head and neck tumors by single-cell RNA sequencing. Nat Commun. 2021;12:7338
107. Guilliams M, Bonnardel J, Haest B, Vanderborght B, Wagner C, Remmerie A. et al. Spatial proteogenomics reveals distinct and evolutionarily conserved hepatic macrophage niches. Cell. 2022;185:379-396.e38
108. Wu L, Yan J, Bai Y, Chen F, Zou X, Xu J. et al. An invasive zone in human liver cancer identified by Stereo-seq promotes hepatocyte-tumor cell crosstalk, local immunosuppression and tumor progression. Cell Res. 2023;33:585-603
109. Fridman WH, Meylan M, Petitprez F, Sun CM, Italiano A, Sautes-Fridman C. B cells and tertiary lymphoid structures as determinants of tumour immune contexture and clinical outcome. Nat Rev Clin Oncol. 2022;19:441-457
110. DiLillo DJ, Yanaba K, Tedder TF. B cells are required for optimal CD4+ and CD8+ T cell tumor immunity: therapeutic B cell depletion enhances B16 melanoma growth in mice. J Immunol. 2010;184:4006-16
111. Lian SL, Lu YT, Lu YJ, Yao YL, Wang XL, Jiang RQ. Tumor-associated macrophages promoting PD-L1 expression in infiltrating B cells through the CXCL12/CXCR4 axis in human hepatocellular carcinoma. Am J Cancer Res. 2024;14:832-853
112. Zhang B, Vogelzang A, Miyajima M, Sugiura Y, Wu Y, Chamoto K. et al. B cell-derived GABA elicits IL-10(+) macrophages to limit anti-tumour immunity. Nature. 2021;599:471-476
113. Mantovani A, Sozzani S, Locati M, Allavena P, Sica A. Macrophage polarization: tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. 2002;23:549-55
114. Wong SC, Puaux AL, Chittezhath M, Shalova I, Kajiji TS, Wang X. et al. Macrophage polarization to a unique phenotype driven by B cells. Eur J Immunol. 2010;40:2296-307
115. Qi J, Crinier A, Escaliere B, Ye Y, Wang Z, Zhang T. et al. Single-cell transcriptomic landscape reveals tumor specific innate lymphoid cells associated with colorectal cancer progression. Cell Rep Med. 2021;2:100353
116. Zhu Y, Shi T, Lu X, Xu Z, Qu J, Zhang Z. et al. Fungal-induced glycolysis in macrophages promotes colon cancer by enhancing innate lymphoid cell secretion of IL-22. EMBO J. 2021;40:e105320
117. Eisinger S, Sarhan D, Boura VF, Ibarlucea-Benitez I, Tyystjarvi S, Oliynyk G. et al. Targeting a scavenger receptor on tumor-associated macrophages activates tumor cell killing by natural killer cells. Proc Natl Acad Sci U S A. 2020;117:32005-32016
118. Brownlie D, Doughty-Shenton D, Yh Soong D, Nixon C, N OC, L MC. et al. Metastasis-associated macrophages constrain antitumor capability of natural killer cells in the metastatic site at least partially by membrane bound transforming growth factor beta. J Immunother Cancer. 2021;9:e001740
119. Hamaguchi M, Okamura T, Fukuda T, Nishida K, Yoshimura Y, Hashimoto Y. et al. Group 3 Innate Lymphoid Cells Protect Steatohepatitis From High-Fat Diet Induced Toxicity. Front Immunol. 2021;12:648754
120. Kim J, Chang Y, Bae B, Sohn KH, Cho SH, Chung DH. et al. Innate immune crosstalk in asthmatic airways: Innate lymphoid cells coordinate polarization of lung macrophages. J Allergy Clin Immunol. 2019;143:1769-1782.e11
121. Loeuillard E, Yang J, Buckarma E, Wang J, Liu Y, Conboy C. et al. Targeting tumor-associated macrophages and granulocytic myeloid-derived suppressor cells augments PD-1 blockade in cholangiocarcinoma. J Clin Invest. 2020;130:5380-5396
122. DeNardo DG, Galkin A, Dupont J, Zhou L, Bendell J. GB1275, a first-in-class CD11b modulator: rationale for immunotherapeutic combinations in solid tumors. J Immunother Cancer. 2021;9:e003005
123. Valencia JC, Erwin-Cohen RA, Clavijo PE, Allen C, Sanford ME, Day CP. et al. Myeloid-Derived Suppressive Cell Expansion Promotes Melanoma Growth and Autoimmunity by Inhibiting CD40/IL27 Regulation in Macrophages. Cancer Res. 2021;81:5977-5990
124. Corzo CA, Condamine T, Lu L, Cotter MJ, Youn JI, Cheng P. et al. HIF-1alpha regulates function and differentiation of myeloid-derived suppressor cells in the tumor microenvironment. J Exp Med. 2010;207:2439-53
125. Colotta F, Re F, Polentarutti N, Sozzani S, Mantovani A. Modulation of granulocyte survival and programmed cell death by cytoki nes and bacterial products. Blood. 1992;80:2012-20
126. Masucci MT, Minopoli M, Del Vecchio S, Carriero MV. The Emerging Role of Neutrophil Extracellular Traps (NETs) in Tumor Progression and Metastasis. Front Immunol. 2020;11:1749
127. Blaisdell A, Crequer A, Columbus D, Daikoku T, Mittal K, Dey SK. et al. Neutrophils Oppose Uterine Epithelial Carcinogenesis via Debridement of Hypoxic Tumor Cells. Cancer Cell. 2015;28:785-799
128. Pahler JC, Tazzyman S, Erez N, Chen YY, Murdoch C, Nozawa H. et al. Plasticity in tumor-promoting inflammation: impairment of macrophage recruitment evokes a compensatory neutrophil response. Neoplasia. 2008;10:329-40
129. Maretti-Mira AC, Golden-Mason L, Salomon MP, Kaplan MJ, Rosen HR. Cholesterol-Induced M4-Like Macrophages Recruit Neutrophils and Induce NETosis. Front Immunol. 2021;12:671073
130. Wellenstein MD, Coffelt SB, Duits DEM, van Miltenburg MH, Slagter M, de Rink I. et al. Loss of p53 triggers WNT-dependent systemic inflammation to drive breast cancer metastasis. Nature. 2019;572:538-542
131. Zhou SL, Zhou ZJ, Hu ZQ, Huang XW, Wang Z, Chen EB. et al. Tumor-Associated Neutrophils Recruit Macrophages and T-Regulatory Cells to Promote Progression of Hepatocellular Carcinoma and Resistance to Sorafenib. Gastroenterology. 2016;150:1646-1658.e17
132. Zhao T, Su Z, Li Y, Zhang X, You Q. Chitinase-3 like-protein-1 function and its role in diseases. Signal Transduct Target Ther. 2020;5:201
133. Jiao Y, Zhang T, Zhang C, Ji H, Tong X, Xia R. et al. Exosomal miR-30d-5p of neutrophils induces M1 macrophage polarization and primes macrophage pyroptosis in sepsis-related acute lung injury. Crit Care. 2021;25:356
134. Tang PC, Chung JY, Xue VW, Xiao J, Meng XM, Huang XR. et al. Smad3 Promotes Cancer-Associated Fibroblasts Generation via Macrophage-Myofibroblast Transition. Adv Sci (Weinh). 2022;9:e2101235
135. Sun X, He X, Zhang Y, Hosaka K, Andersson P, Wu J. et al. Inflammatory cell-derived CXCL3 promotes pancreatic cancer metastasis through a novel myofibroblast-hijacked cancer escape mechanism. Gut. 2022;71:129-147
136. Zeng W, Xiong L, Wu W, Li S, Liu J, Yang L. et al. CCL18 signaling from tumor-associated macrophages activates fibroblasts to adopt a chemoresistance-inducing phenotype. Oncogene. 2023;42:224-237
137. Timperi E, Gueguen P, Molgora M, Magagna I, Kieffer Y, Lopez-Lastra S. et al. Lipid-Associated Macrophages Are Induced by Cancer-Associated Fibroblasts and Mediate Immune Suppression in Breast Cancer. Cancer Res. 2022;82:3291-3306
138. Cho H, Seo Y, Loke KM, Kim SW, Oh SM, Kim JH. et al. Cancer-Stimulated CAFs Enhance Monocyte Differentiation and Protumoral TAM Activation via IL6 and GM-CSF Secretion. Clin Cancer Res. 2018;24:5407-5421
139. Yang Y, Guo Z, Chen W, Wang X, Cao M, Han X. et al. M2 Macrophage-Derived Exosomes Promote Angiogenesis and Growth of Pancreatic Ductal Adenocarcinoma by Targeting E2F2. Mol Ther. 2021;29:1226-1238
140. Zhang S, Xie B, Wang L, Yang H, Zhang H, Chen Y. et al. Macrophage-mediated vascular permeability via VLA4/VCAM1 pathway dictates ascites development in ovarian cancer. J Clin Invest. 2021;131:e140315
141. Delprat V, Huart C, Feron O, Soncin F, Michiels C. The impact of macrophages on endothelial cells is potentiated by cycling hypoxia: Enhanced tumor inflammation and metastasis. Front Oncol. 2022;12:961753
142. Mysore V, Tahir S, Furuhashi K, Arora J, Rosetti F, Cullere X. et al. Monocytes transition to macrophages within the inflamed vasculature via monocyte CCR2 and endothelial TNFR2. J Exp Med. 2022;219:e20210562
143. Marozzi M, Parnigoni A, Negri A, Viola M, Vigetti D, Passi A. et al. Inflammation, Extracellular Matrix Remodeling, and Proteostasis in Tumor Microenvironment. Int J Mol Sci. 2021;22:8102
144. De Paolis V, Maiullari F, Chirivi M, Milan M, Cordiglieri C, Pagano F. et al. Unusual Association of NF-kappaB Components in Tumor-Associated Macrophages (TAMs) Promotes HSPG2-Mediated Immune-Escaping Mechanism in Breast Cancer. Int J Mol Sci. 2022;23:7902
145. Madsen DH, Jurgensen HJ, Siersbaek MS, Kuczek DE, Grey Cloud L, Liu S. et al. Tumor-Associated Macrophages Derived from Circulating Inflammatory Monocytes Degrade Collagen through Cellular Uptake. Cell Rep. 2017;21:3662-3671
146. Witherel CE, Sao K, Brisson BK, Han B, Volk SW, Petrie RJ. et al. Regulation of extracellular matrix assembly and structure by hybrid M1/M2 macrophages. Biomaterials. 2021;269:120667
147. Meng FW, Slivka PF, Dearth CL, Badylak SF. Solubilized extracellular matrix from brain and urinary bladder elicits distinct functional and phenotypic responses in macrophages. Biomaterials. 2015;46:131-40
148. LaRue MM, Parker S, Puccini J, Cammer M, Kimmelman AC, Bar-Sagi D. Metabolic reprogramming of tumor-associated macrophages by collagen turnover promotes fibrosis in pancreatic cancer. Proc Natl Acad Sci U S A. 2022;119:e2119168119
149. Springer NL, Iyengar NM, Bareja R, Verma A, Jochelson MS, Giri DD. et al. Obesity-Associated Extracellular Matrix Remodeling Promotes a Macrophage Phenotype Similar to Tumor-Associated Macrophages. Am J Pathol. 2019;189:2019-2035
150. Sandhu SK, Papadopoulos K, Fong PC, Patnaik A, Messiou C, Olmos D. et al. A first-in-human, first-in-class, phase I study of carlumab (CNTO 888), a human monoclonal antibody against CC-chemokine ligand 2 in patients with solid tumors. Cancer Chemother Pharmacol. 2013;71:1041-50
151. Brana I, Calles A, LoRusso PM, Yee LK, Puchalski TA, Seetharam S. et al. Carlumab, an anti-C-C chemokine ligand 2 monoclonal antibody, in combination with four chemotherapy regimens for the treatment of patients with solid tumors: an open-label, multicenter phase 1b study. Target Oncol. 2015;10:111-23
152. Masuda T, Noda M, Kogawa T, Kitagawa D, Hayashi N, Jomori T. et al. Phase I dose-escalation trial to repurpose propagermanium, an oral CCL2 inhibitor, in patients with breast cancer. Cancer Sci. 2020;111:924-931
153. Noel M, O'Reilly EM, Wolpin BM, Ryan DP, Bullock AJ, Britten CD. et al. Phase 1b study of a small molecule antagonist of human chemokine (C-C motif) receptor 2 (PF-04136309) in combination with nab-paclitaxel/gemcitabine in first-line treatment of metastatic pancreatic ductal adenocarcinoma. Invest New Drugs. 2020;38:800-811
154. Linehan D, Noel MS, Hezel AF, Wang-Gillam A, Eskens F, Sleijfer S. et al. Overall survival in a trial of orally administered CCR2 inhibitor CCX872 in locally advanced/metastatic pancreatic cancer: Correlation with blood monocyte counts. Journal of Clinical Oncology. 2018;36:92-92
155. Haag GM, Springfeld C, Grun B, Apostolidis L, Zschabitz S, Dietrich M. et al. Pembrolizumab and maraviroc in refractory mismatch repair proficient/microsatellite-stable metastatic colorectal cancer - The PICCASSO phase I trial. Eur J Cancer. 2022;167:112-122
156. Gomez-Roca CA, Italiano A, Le Tourneau C, Cassier PA, Toulmonde M, D'Angelo SP. et al. Phase I study of emactuzumab single agent or in combination with paclitaxel in patients with advanced/metastatic solid tumors reveals depletion of immunosuppressive M2-like macrophages. Ann Oncol. 2019;30:1381-1392
157. Gomez-Roca C, Cassier P, Zamarin D, Machiels JP, Perez Gracia JL, Stephen Hodi F. et al. Anti-CSF-1R emactuzumab in combination with anti-PD-L1 atezolizumab in advanced solid tumor patients naive or experienced for immune checkpoint blockade. J Immunother Cancer. 2022;10:e004076
158. Kuemmel S, Campone M, Loirat D, Lopez RL, Beck JT, De Laurentiis M. et al. A Randomized Phase II Study of Anti-CSF1 Monoclonal Antibody Lacnotuzumab (MCS110) Combined with Gemcitabine and Carboplatin in Advanced Triple-Negative Breast Cancer. Clin Cancer Res. 2022;28:106-115
159. Borthakur G, Zeng Z, Cortes JE, Chen HC, Huang X, Konopleva M. et al. Phase 1 study of combinatorial sorafenib, G-CSF, and plerixafor treatment in relapsed/refractory, FLT3-ITD-mutated acute myelogenous leukemia patients. Am J Hematol. 2020;95:1296-1303
160. Borthakur G, Ofran Y, Tallman MS, Foran J, Uy GL, DiPersio JF. et al. BL-8040 CXCR4 antagonist is safe and demonstrates antileukemic activity in combination with cytarabine for the treatment of relapsed/refractory acute myelogenous leukemia: An open-label safety and efficacy phase 2a study. Cancer. 2021;127:1246-1259
161. Steurer M, Montillo M, Scarfo L, Mauro FR, Andel J, Wildner S. et al. Olaptesed pegol (NOX-A12) with bendamustine and rituximab: a phase IIa study in patients with relapsed/refractory chronic lymphocytic leukemia. Haematologica. 2019;104:2053-2060
162. Friedl TWP, Fehm T, Muller V, Lichtenegger W, Blohmer J, Lorenz R. et al. Prognosis of Patients With Early Breast Cancer Receiving 5 Years vs 2 Years of Adjuvant Bisphosphonate Treatment: A Phase 3 Randomized Clinical Trial. JAMA Oncol. 2021;7:1149-1157
163. Paul B, Liedtke M, Khouri J, Rifkin R, Gandhi MD, Kin A. et al. A phase II multi-arm study of magrolimab combinations in patients with relapsed/refractory multiple myeloma. Future Oncol. 2023;19:7-17
164. Advani R, Flinn I, Popplewell L, Forero A, Bartlett NL, Ghosh N. et al. CD47 Blockade by Hu5F9-G4 and Rituximab in Non-Hodgkin's Lymphoma. N Engl J Med. 2018;379:1711-1721
165. Drakaki A, Powles T, Bamias A, Martin-Liberal J, Shin SJ, Friedlander T. et al. Atezolizumab plus Magrolimab, Niraparib, or Tocilizumab versus Atezolizumab Monotherapy in Platinum-Refractory Metastatic Urothelial Carcinoma: A Phase Ib/II Open-Label, Multicenter, Randomized Umbrella Study (MORPHEUS Urothelial Carcinoma). Clin Cancer Res. 2023;29:4373-4384
166. Zeidan AM, DeAngelo DJ, Palmer J, Seet CS, Tallman MS, Wei X. et al. Phase 1 study of anti-CD47 monoclonal antibody CC-90002 in patients with relapsed/refractory acute myeloid leukemia and high-risk myelodysplastic syndromes. Ann Hematol. 2022;101:557-569
167. Ansell SM, Maris MB, Lesokhin AM, Chen RW, Flinn IW, Sawas A. et al. Phase I Study of the CD47 Blocker TTI-621 in Patients with Relapsed or Refractory Hematologic Malignancies. Clin Cancer Res. 2021;27:2190-2199
168. Vonderheide RH, Flaherty KT, Khalil M, Stumacher MS, Bajor DL, Hutnick NA. et al. Clinical activity and immune modulation in cancer patients treated with CP-870,893, a novel CD40 agonist monoclonal antibody. J Clin Oncol. 2007;25:876-83
169. Vonderheide RH, Burg JM, Mick R, Trosko JA, Li D, Shaik MN. et al. Phase I study of the CD40 agonist antibody CP-870,893 combined with carboplatin and paclitaxel in patients with advanced solid tumors. Oncoimmunology. 2013;2:e23033
170. Solsona E, Madero R, Chantada V, Fernandez JM, Zabala JA, Portillo JA. et al. Sequential combination of mitomycin C plus bacillus Calmette-Guerin (BCG) is more effective but more toxic than BCG alone in patients with non-muscle-invasive bladder cancer in intermediate- and high-risk patients: final outcome of CUETO 93009, a randomized prospective trial. Eur Urol. 2015;67:508-16
171. Dummer R, Hauschild A, Becker JC, Grob JJ, Schadendorf D, Tebbs V. et al. An exploratory study of systemic administration of the toll-like receptor-7 agonist 852A in patients with refractory metastatic melanoma. Clin Cancer Res. 2008;14:856-64
172. Weigel BJ, Cooley S, DeFor T, Weisdorf DJ, Panoskaltsis-Mortari A, Chen W. et al. Prolonged subcutaneous administration of 852A, a novel systemic toll-like receptor 7 agonist, to activate innate immune responses in patients with advanced hematologic malignancies. Am J Hematol. 2012;87:953-6
173. Adams S, Kozhaya L, Martiniuk F, Meng TC, Chiriboga L, Liebes L. et al. Topical TLR7 agonist imiquimod can induce immune-mediated rejection of skin metastases in patients with breast cancer. Clin Cancer Res. 2012;18:6748-57
174. Hong DS, Postow M, Chmielowski B, Sullivan R, Patnaik A, Cohen EEW. et al. Eganelisib, a First-in-Class PI3Kgamma Inhibitor, in Patients with Advanced Solid Tumors: Results of the Phase 1/1b MARIO-1 Trial. Clin Cancer Res. 2023;29:2210-2219
175. Nayak-Kapoor A, Hao Z, Sadek R, Dobbins R, Marshall L, Vahanian NN. et al. Phase Ia study of the indoleamine 2,3-dioxygenase 1 (IDO1) inhibitor navoximod (GDC-0919) in patients with recurrent advanced solid tumors. J Immunother Cancer. 2018;6:61
176. Beckermann KE, Patnaik A, Winer I, Tan W, Bashir B, Kyriakopoulos CE. et al. A phase 1b open-label study to evaluate the safety, tolerability, pharmacokinetics, and pharmacodynamics of py314 in combination with pembrolizumab in patients with advanced renal cell carcinoma. Invest New Drugs. 2024;42:179-184
177. Hu H, Kang L, Zhang J, Wu Z, Wang H, Huang M. et al. Neoadjuvant PD-1 blockade with toripalimab, with or without celecoxib, in mismatch repair-deficient or microsatellite instability-high, locally advanced, colorectal cancer (PICC): a single-centre, parallel-group, non-comparative, randomised, phase 2 trial. Lancet Gastroenterol Hepatol. 2022;7:38-48
178. Yamamoto N, Koyama T, Shimizu T, Todaka A, Kawakami T, Erzen D. et al. Phase I study of the VEGF/Ang-2 inhibitor BI 836880 alone or combined with the anti-programmed cell death protein-1 antibody ezabenlimab in Japanese patients with advanced solid tumors. Cancer Chemother Pharmacol. 2023;91:469-480
179. Tan DSW, Felip E, de Castro G, Solomon BJ, Greystoke A, Cho BC. et al. Canakinumab Versus Placebo in Combination With First-Line Pembrolizumab Plus Chemotherapy for Advanced Non-Small-Cell Lung Cancer: Results From the CANOPY-1 Trial. J Clin Oncol. 2024;42:192-204
180. Raez LE, Papadopoulos K, Ricart AD, Chiorean EG, Dipaola RS, Stein MN. et al. A phase I dose-escalation trial of 2-deoxy-D-glucose alone or combined with docetaxel in patients with advanced solid tumors. Cancer Chemother Pharmacol. 2013;71:523-30
181. Rothermundt C, Hayoz S, Templeton AJ, Winterhalder R, Strebel RT, Bartschi D. et al. Metformin in chemotherapy-naive castration-resistant prostate cancer: a multicenter phase 2 trial (SAKK 08/09). Eur Urol. 2014;66:468-74
182. Mitsuhashi A, Sato Y, Kiyokawa T, Koshizaka M, Hanaoka H, Shozu M. Phase II study of medroxyprogesterone acetate plus metformin as a fertility-sparing treatment for atypical endometrial hyperplasia and endometrial cancer. Ann Oncol. 2016;27:262-6
183. Kelly CM, Qin LX, Whiting KA, Richards AL, Avutu V, Chan JE. et al. A Phase II Study of Epacadostat and Pembrolizumab in Patients with Advanced Sarcoma. Clin Cancer Res. 2023;29:2043-2051
184. Long GV, Dummer R, Hamid O, Gajewski TF, Caglevic C, Dalle S. et al. Epacadostat plus pembrolizumab versus placebo plus pembrolizumab in patients with unresectable or metastatic melanoma (ECHO-301/KEYNOTE-252): a phase 3, randomised, double-blind study. Lancet Oncol. 2019;20:1083-1097
185. Ossenkoppele GJ, Lowenberg B, Zachee P, Vey N, Breems D, Van de Loosdrecht AA. et al. A phase I first-in-human study with tefinostat - a monocyte/macrophage targeted histone deacetylase inhibitor - in patients with advanced haematological malignancies. Br J Haematol. 2013;162:191-201
186. Teng KY, Han J, Zhang X, Hsu SH, He S, Wani NA. et al. Blocking the CCL2-CCR2 Axis Using CCL2-Neutralizing Antibody Is an Effective Therapy for Hepatocellular Cancer in a Mouse Model. Mol Cancer Ther. 2017;16:312-322
187. Kadomoto S, Izumi K, Mizokami A. Roles of CCL2-CCR2 Axis in the Tumor Microenvironment. Int J Mol Sci. 2021;22:8530
188. Ries CH, Cannarile MA, Hoves S, Benz J, Wartha K, Runza V. et al. Targeting tumor-associated macrophages with anti-CSF-1R antibody reveals a strategy for cancer therapy. Cancer Cell. 2014;25:846-59
189. Cassier PA, Italiano A, Gomez-Roca CA, Le Tourneau C, Toulmonde M, Cannarile MA. et al. CSF1R inhibition with emactuzumab in locally advanced diffuse-type tenosynovial giant cell tumours of the soft tissue: a dose-escalation and dose-expansion phase 1 study. Lancet Oncol. 2015;16:949-56
190. Lee EQ, Duda DG, Muzikansky A, Gerstner ER, Kuhn JG, Reardon DA. et al. Phase I and Biomarker Study of Plerixafor and Bevacizumab in Recurrent High-Grade Glioma. Clin Cancer Res. 2018;24:4643-4649
191. Bockorny B, Semenisty V, Macarulla T, Borazanci E, Wolpin BM, Stemmer SM. et al. BL-8040, a CXCR4 antagonist, in combination with pembrolizumab and chemotherapy for pancreatic cancer: the COMBAT trial. Nat Med. 2020;26:878-885
192. Junankar S, Shay G, Jurczyluk J, Ali N, Down J, Pocock N. et al. Real-time intravital imaging establishes tumor-associated macrophages as the extraskeletal target of bisphosphonate action in cancer. Cancer Discov. 2015;5:35-42
193. Morgan GJ, Davies FE, Gregory WM, Szubert AJ, Bell SE, Drayson MT. et al. Effects of induction and maintenance plus long-term bisphosphonates on bone disease in patients with multiple myeloma: the Medical Research Council Myeloma IX Trial. Blood. 2012;119:5374-83
194. Germano G, Frapolli R, Belgiovine C, Anselmo A, Pesce S, Liguori M. et al. Role of macrophage targeting in the antitumor activity of trabectedin. Cancer Cell. 2013;23:249-62
195. Etzerodt A, Tsalkitzi K, Maniecki M, Damsky W, Delfini M, Baudoin E. et al. Specific targeting of CD163(+) TAMs mobilizes inflammatory monocytes and promotes T cell-mediated tumor regression. J Exp Med. 2019;216:2394-2411
196. Barkal AA, Weiskopf K, Kao KS, Gordon SR, Rosental B, Yiu YY. et al. Engagement of MHC class I by the inhibitory receptor LILRB1 suppresses macrophages and is a target of cancer immunotherapy. Nat Immunol. 2018;19:76-84
197. Jaiswal S, Jamieson CH, Pang WW, Park CY, Chao MP, Majeti R. et al. CD47 is upregulated on circulating hematopoietic stem cells and leukemia cells to avoid phagocytosis. Cell. 2009;138:271-85
198. Barkal AA, Brewer RE, Markovic M, Kowarsky M, Barkal SA, Zaro BW. et al. CD24 signalling through macrophage Siglec-10 is a target for cancer immunotherapy. Nature. 2019;572:392-396
199. Gholamin S, Mitra SS, Feroze AH, Liu J, Kahn SA, Zhang M. et al. Disrupting the CD47-SIRPalpha anti-phagocytic axis by a humanized anti-CD47 antibody is an efficacious treatment for malignant pediatric brain tumors. Sci Transl Med. 2017;9:eaaf2968
200. Stenberg A, Sehlin J, Oldenborg PA. Neutrophil apoptosis is associated with loss of signal regulatory protein alpha (SIRPalpha) from the cell surface. J Leukoc Biol. 2013;93:403-12
201. Tang T, Cheng X, Truong B, Sun L, Yang X, Wang H. Molecular basis and therapeutic implications of CD40/CD40L immune checkpoint. Pharmacol Ther. 2021;219:107709
202. Lim CY, Chang JH, Lee WS, Kim J, Park IY. CD40 Agonists Alter the Pancreatic Cancer Microenvironment by Shifting the Macrophage Phenotype toward M1 and Suppress Human Pancreatic Cancer in Organotypic Slice Cultures. Gut Liver. 2022;16:645-659
203. Fitzgerald KA, Kagan JC. Toll-like Receptors and the Control of Immunity. Cell. 2020;180:1044-1066
204. Altves S, Guclu E, Yetisgin E, Bilecen K, Vural H. Upregulation of Immune checkpoint PD-L1 in Colon cancer cell lines and activation of T cells by Leuconostoc mesenteroides. World J Microbiol Biotechnol. 2024;40:204
205. De Henau O, Rausch M, Winkler D, Campesato LF, Liu C, Cymerman DH. et al. Overcoming resistance to checkpoint blockade therapy by targeting PI3Kgamma in myeloid cells. Nature. 2016;539:443-447
206. Wanderley CW, Colon DF, Luiz JPM, Oliveira FF, Viacava PR, Leite CA. et al. Paclitaxel Reduces Tumor Growth by Reprogramming Tumor-Associated Macrophages to an M1 Profile in a TLR4-Dependent Manner. Cancer Res. 2018;78:5891-5900
207. Klug F, Prakash H, Huber PE, Seibel T, Bender N, Halama N. et al. Low-dose irradiation programs macrophage differentiation to an iNOS(+)/M1 phenotype that orchestrates effective T cell immunotherapy. Cancer Cell. 2013;24:589-602
208. Strauss L, Sangaletti S, Consonni FM, Szebeni G, Morlacchi S, Totaro MG. et al. RORC1 Regulates Tumor-Promoting "Emergency" Granulo-Monocytopoiesis. Cancer Cell. 2015;28:253-69
209. Molgora M, Esaulova E, Vermi W, Hou J, Chen Y, Luo J. et al. TREM2 Modulation Remodels the Tumor Myeloid Landscape Enhancing Anti-PD-1 Immunotherapy. Cell. 2020;182:886-900.e17
210. Steggerda SM, Bennett MK, Chen J, Emberley E, Huang T, Janes JR. et al. Inhibition of arginase by CB-1158 blocks myeloid cell-mediated immune suppression in the tumor microenvironment. J Immunother Cancer. 2017;5:101
211. Rigamonti N, Kadioglu E, Keklikoglou I, Wyser Rmili C, Leow CC, De Palma M. Role of angiopoietin-2 in adaptive tumor resistance to VEGF signaling blockade. Cell Rep. 2014;8:696-706
212. Brown JL, Cao ZA, Pinzon-Ortiz M, Kendrew J, Reimer C, Wen S. et al. A human monoclonal anti-ANG2 antibody leads to broad antitumor activity in combination with VEGF inhibitors and chemotherapy agents in preclinical models. Mol Cancer Ther. 2010;9:145-56
213. Kaplanov I, Carmi Y, Kornetsky R, Shemesh A, Shurin GV, Shurin MR. et al. Blocking IL-1beta reverses the immunosuppression in mouse breast cancer and synergizes with anti-PD-1 for tumor abrogation. Proc Natl Acad Sci U S A. 2019;116:1361-1369
214. Wang YC, Wang X, Yu J, Ma F, Li Z, Zhou Y. et al. Targeting monoamine oxidase A-regulated tumor-associated macrophage polarization for cancer immunotherapy. Nat Commun. 2021;12:3530
215. Chen GG, Woo PYM, Ng SCP, Wong GKC, Chan DTM, van Hasselt CA. et al. Impact of metformin on immunological markers: Implication in its anti-tumor mechanism. Pharmacol Ther. 2020;213:107585
216. Wei Z, Zhang X, Yong T, Bie N, Zhan G, Li X. et al. Boosting anti-PD-1 therapy with metformin-loaded macrophage-derived microparticles. Nat Commun. 2021;12:440
217. Wang S, Lin Y, Xiong X, Wang L, Guo Y, Chen Y. et al. Low-Dose Metformin Reprograms the Tumor Immune Microenvironment in Human Esophageal Cancer: Results of a Phase II Clinical Trial. Clin Cancer Res. 2020;26:4921-4932
218. Goodwin PJ, Chen BE, Gelmon KA, Whelan TJ, Ennis M, Lemieux J. et al. Effect of Metformin vs Placebo on Invasive Disease-Free Survival in Patients With Breast Cancer: The MA.32 Randomized Clinical Trial. JAMA. 2022;327:1963-1973
219. Skinner H, Hu C, Tsakiridis T, Santana-Davila R, Lu B, Erasmus JJ. et al. Addition of Metformin to Concurrent Chemoradiation in Patients With Locally Advanced Non-Small Cell Lung Cancer: The NRG-LU001 Phase 2 Randomized Clinical Trial. JAMA Oncol. 2021;7:1324-1332
220. Jin H, He Y, Zhao P, Hu Y, Tao J, Chen J. et al. Targeting lipid metabolism to overcome EMT-associated drug resistance via integrin beta3/FAK pathway and tumor-associated macrophage repolarization using legumain-activatable delivery. Theranostics. 2019;9:265-278
221. Pelly VS, Moeini A, Roelofsen LM, Bonavita E, Bell CR, Hutton C. et al. Anti-Inflammatory Drugs Remodel the Tumor Immune Environment to Enhance Immune Checkpoint Blockade Efficacy. Cancer Discov. 2021;11:2602-2619
222. Cao S, Saw PE, Shen Q, Li R, Liu Y, Xu X. Reduction-responsive RNAi nanoplatform to reprogram tumor lipid metabolism and repolarize macrophage for combination pancreatic cancer therapy. Biomaterials. 2022;280:121264
223. Mehta AK, Cheney EM, Hartl CA, Pantelidou C, Oliwa M, Castrillon JA. et al. Targeting immunosuppressive macrophages overcomes PARP inhibitor resistance in BRCA1-associated triple-negative breast cancer. Nat Cancer. 2021;2:66-82
224. Cannac M, Nikolic J, Benaroch P. Cancer Immunotherapies Based on Genetically Engineered Macrophages. Cancer Immunol Res. 2022;10:1156-1166
225. Brempelis KJ, Cowan CM, Kreuser SA, Labadie KP, Prieskorn BM, Lieberman NAP. et al. Genetically engineered macrophages persist in solid tumors and locally deliver therapeutic proteins to activate immune responses. J Immunother Cancer. 2020;8:e001356
226. Klichinsky M, Ruella M, Shestova O, Lu XM, Best A, Zeeman M. et al. Human chimeric antigen receptor macrophages for cancer immunotherapy. Nat Biotechnol. 2020;38:947-953
227. Morrissey MA, Williamson AP, Steinbach AM, Roberts EW, Kern N, Headley MB. et al. Chimeric antigen receptors that trigger phagocytosis. Elife. 2018;7:e36688
228. Ray M, Lee YW, Hardie J, Mout R, Yesilbag Tonga G, Farkas ME. et al. CRISPRed Macrophages for Cell-Based Cancer Immunotherapy. Bioconjug Chem. 2018;29:445-450
Author contact
![]() Corresponding authors: Prof. Wenjie You, Department of Respiratory and Critical Care Medicine, Shandong Provincial Hospital affiliated to Shandong First Medical University, Jingwu Road 324#, Jinan 250021, China 86; Tel.: +86-(0)531-68776360; E-mail: youwenjieedu.cn; Fax: +86-(0)531-68776072; ORCID: 0000-0002-8044-3914. Prof. Lili Su, Department of Respiratory and Critical Care Medicine, Shandong Provincial Hospital affiliated to Shandong First Medical University, Jingwu Road 324#, Jinan 250021, China 86; Tel.: +86-(0)531-68776360; E-mail: suliliedu.cn.
Corresponding authors: Prof. Wenjie You, Department of Respiratory and Critical Care Medicine, Shandong Provincial Hospital affiliated to Shandong First Medical University, Jingwu Road 324#, Jinan 250021, China 86; Tel.: +86-(0)531-68776360; E-mail: youwenjieedu.cn; Fax: +86-(0)531-68776072; ORCID: 0000-0002-8044-3914. Prof. Lili Su, Department of Respiratory and Critical Care Medicine, Shandong Provincial Hospital affiliated to Shandong First Medical University, Jingwu Road 324#, Jinan 250021, China 86; Tel.: +86-(0)531-68776360; E-mail: suliliedu.cn.