Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Material and Methods
Results
Discussion
Conclusion
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2025; 15(15):7489-7500. doi:10.7150/thno.116062 This issue Cite
Research Paper
Comparison of targeting two antigens (GPA33 versus HER2) for 225Ac-pretargeted alpha-radioimmunotherapy of colorectal cancer
Sara S. Rinne1,2, Daniela Burnes Vargas1, Brett A. Vaughn1,3, Sumudu Katugampola4, Brian W. Miller5, Darren R. Veach1,6, Blesida Punzalan6, Elisa de Stanchina3,7, Rona Yaeger8, Ileana C. Miranda9, Hong Xu2, Hongfen Guo2, Jazmin Schwartz10, Edward K. Fung1, Roger W. Howell4, Steven M. Larson1,3,6, Nai-Kong V. Cheung2, Sarah M. Cheal1,3 ![]()
1. Department of Radiology, Weill Cornell Medicine, New York, NY, USA.
2. Department of Pediatrics, Memorial Sloan Kettering Cancer Center (MSKCC), New York, NY, USA
3. Molecular Pharmacology Program, MSKCC, New York, NY, USA.
4. Division of Radiation Research, Department of Radiology and Center for Cell Signaling, New Jersey Medical School, Rutgers University, Newark, NJ, USA.
5. Department of Radiation Oncology, Department of Medical Imaging, College of Medicine, University of Arizona, Tucson, AZ, USA.
6. Department of Radiology, MSKCC, New York, NY, USA.
7. Antitumor Assessment Core Facility, MSKCC, New York, NY, USA.
8. Department of Medicine, MSKCC, New York, NY, USA.
9. Laboratory of Comparative Pathology, Weill Cornell Medicine, MSKCC, The Rockefeller University, New York, NY, USA.
10. Department of Medical Physics, MSKCC, New York, NY, USA.
Received 2025-4-19; Accepted 2025-6-4; Published 2025-6-20
Abstract

Purpose: Curative therapies remain a significant unmet need for advanced human colorectal cancer (CRC). The aim of this study was to establish a 3-step 225Ac-DOTA pretargeted radioimmunotherapy (PRIT) system for human CRC, targeting two individual antigens: glycoprotein A33 (GPA33) and human epidermal growth factor receptor 2 (HER2).
Methods: In vitro cellular uptake and internalization assays, as well as survival assays (colony forming) were performed in GPA33- and HER2-positive SW1222 human CRC cells. In vivo biodistribution and therapy studies were performed with two human CRC xenograft mouse models.
Results: For both antigens, treatment with up to 74 kBq 225Ac-DOTA-PRIT in SW1222-tumored mice significantly enhanced overall survival in comparison to controls, including histologic cures. The uptake of 225Ac at the tumor correlated with antigen expression (antigen expression for GPA33:HER2 is 5:1). Cellular assays showed no significant differences in internalized fraction or nucleus-associated radioactivity between the two targets. GPA33 had a higher absorbed dose to the nucleus (1.3 Gy vs. 0.65 Gy for HER2), resulting in reduced clonogenic survival. A single cycle of either GPA33 or HER2 DOTA-PRIT (37 kBq; 193.31 Gy and 113.91 Gy (relative biological effectiveness [RBE] = 5), respectively) was equally effective. No chronic nephrotoxicity was seen at ≤ 20 Gy (RBE = 5). The efficacy of GPA33-directed 225Ac-DOTA-PRIT was also confirmed in a patient-derived xenograft model.
Conclusion: In summary, 225Ac-DOTA-PRIT to GPA33 or HER2 was highly effective and safe in preclinical models of human CRC. Tumor response to treatment could not be predicted by nuclear absorbed dose alone, highlighting the importance of comprehensive micro- and macro-dosimetry.
Keywords: targeted alpha therapy, Actinium-225, glycoprotein A33, human epidermal growth factor receptor 2, pretargeted radioimmunotherapy
Introduction
Colorectal cancer (CRC) is a significant global health concern, accounting for about 10% of cancer cases worldwide, according to the WHO. Treatment options depend on factors such as cancer stage and patient health and can include surgery, chemotherapy, radiation therapy, and targeted therapy, often in combination. Although cures are possible with localized disease, durable control of metastatic CRC remains an urgent currently unmet challenge [1]. Radioimmunotherapy (RIT) offers an alternative approach, using antibodies to deliver a radioactive, cytotoxic payload specifically to tumor cells. This method is advantageous as it can target both macroscopic tumors and micrometastases while minimizing radiation exposure to healthy tissue.
The radiation doses necessary for tumor ablation are limited by the absorbed doses to healthy tissue. One of the major hurdles in RIT is the long circulatory half-life of antibodies leading to unintended myelotoxicity. Radiolabeled peptides with much faster pharmacokinetics for peptide receptor radionuclide therapy (PRRT) can reduce the radiotoxicity to blood. However, renal trapping of peptides in the proximal tubules of the kidney after glomerular filtration leads to an increased risk for nephrotoxicity with PRRT [2]. Pretargeted radioimmunotherapy (PRIT) can circumvent the limitations of traditional RIT and PRRT by separating the antigen binding from the radioactive payload delivery in a multistep approach. In the first step, a bispecific antibody (BsAb) with specificities for the tumor and for the payload is allowed to bind to the tumor. With time, unbound BsAb is cleared from circulation or actively removed by a clearing agent (CA). In the last step, the radioactive payload is delivered in the form of a small molecule with rapid tumor penetration and renal clearance, achieving exceptional therapeutic indices (TIs; tumor-to-normal tissue-absorbed dose ratios) for RIT. Several recently published reviews provide excellent overviews of different PRIT approaches and illustrate the modularity of PRIT in targeting a wide range of tumor antigens with diverse payloads [3-5].
From a radiobiological perspective, ⍺-emitters such as 225Ac, 211At, 212Bi, or 212Pb are extremely potent payloads for targeted radiotherapy, including PRIT. Due to their high linear energy transfer, they create dense and complex ionizations along their path, resulting in DNA double-strand breaks and damage estimated to be several folds more effective than the damage caused by β-emitters and external beam radiation. Recently, we have adapted a BsAb/DOTA-radiometal-based PRIT system (DOTA-PRIT) for targeted ⍺-therapy (TAT) with 225Ac [6]. This PRIT approach uses a BsAb in the IgG-scFv format (210 kDa) [7], which contains sequences for an anti-tumor antigen IgG and an anti-DOTA(metal) scFv. The anti-DOTA(metal) scFv is an affinity-matured version of the anti-DOTA(metal) antibody 2D12.5, with affinities of approximately 10-15 picomolar for lutetium or yttrium complexes [8]. To achieve high TIs, we use a CA (either DOTA-dendron [9] or DOTA-dextran [10]) to reduce circulating IgG-scFv prior to injection of the DOTA-radioligand. A typical regimen consists of three steps: administration of BsAb at t = -28 h, followed with administration of CA (t = -4 h) and radioligand (t = 0 h). Initial therapy experiments using a specialized carrier for 225Ac ([225Ac]Ac-Proteus-DOTA or [225Ac]Ac-Pr) demonstrated complete responses and histological cures in subcutaneous breast cancer, CRC, and neuroblastoma xenografts, as well as an ovarian peritoneal carcinomatosis model, with only mild toxicity [6, 11].
Most RIT approaches against CRC have been based on targeting the human carcinoembryonic antigen using IgG antibodies, for example, 90Y-M5A and 131I-labetuzumab [12, 13]. Two other molecular markers holding promise for RIT of CRC are glycoprotein A33 (GPA33) and human epidermal growth factor receptor 2 (HER2). GPA33 is expressed in 95% of CRC patients [14]. Because of its limited expression in healthy tissue (colon and bowel epithelium), it has been extensively studied for RIT of CRC, which our team and others have previously demonstrated [15, 16]. Clinical studies of GPA33-immunoPET with nanobody-based probes are ongoing (NCT06203587). HER2 is an emerging target in metastatic CRC [17]. Although high levels of HER2 overexpression is found in only 6% of tumors [18], low HER2 density CRC appears to be relatively frequent [19]. Despite the low expression of HER2, clinical trials have demonstrated the activity of dual HER2-targeted therapy with trastuzumab plus tucatinib, which is approved for HER2-positive metastatic CRC, and of the antibody-drug conjugate trastuzumab deruxtecan, which is approved for metastatic CRC with 3+ HER2 expression by immunohistochemistry (IHC).
In the present study we report high-TI 225Ac-DOTA-PRIT in a preclinical model of CRC targeting two different tumor-associated antigens: GPA33 and HER2. GPA33 is known to have 5- times as many binding sites on SW1222 CRC as HER2. Our hypothesis was that this difference would directly correlate with the relative effectiveness of the in vivo response and the curative potential. We hypothesized that targeting GPA33, the antigen with the greatest tumor expression and thus the most favorable tumor-absorbed dose, would lead to greater tumor control in comparison to HER2 during head-to-head studies. In a comprehensive approach including micro- and macro-dosimetry, in vitro and in vivo efficacy studies, and evaluation of in xenograft and PDX models study, we aimed to refine and better understand the underlying mechanisms contributing to the exceptional properties of 225Ac-DOTA-PRIT.
Material and Methods
General
The bispecific anti-GPA33/anti-DOTA (anti-huA33-C825 BsAb, 210 kDa) and anti-HER2/anti-DOTA (anti-HER2-C825 BsAb, 210 kDa) antibodies were produced as previously described [16, 20]. Please see Table S1 for BsAb binding affinities to target antigens measured by surface plasmon resonance (Biacore T100). The DOTA-dendron CA CCA α-16-DOTA-Y3+ (MW: 9059 Da), consisting of a nonradioactive yttrium-DOTA-Bn molecule attached via a linker to a glycodendron displaying 16 terminal α-thio-N-acetylgalactosamine (α-SGalNAc) units, was prepared according to reported methods [9]. Proteus-DOTA (Pr), the precursor to the 111In and 225Ac-labeled radioligand, consists of a three-arm DOTA radiometal-chelating agent (1,4,7,10-tetraazacyclododecane-1,4,7-triacetic acid; DO3A) separated by a tetraethylene glycol (PEG4) linker to a 175Lu (natural) lutetium complex of benzyl-DOTA (MW: 1350 Da) [6]. All PRIT reagents were formulated in sterile isotonic saline solution and administered intravenously via the tail vein. The GPA33- and HER2- expressing human CRC cell lines SW1222 and LS174T were obtained from the Ludwig Institute for Cancer Immunotherapy (New York, NY) or purchased from ATCC (Manassas, VA, USA), respectively. SW1222 cells were maintained in IMDM media supplemented with 10% FBS, 2 mM glutamine, 100 units/mL penicillin, and 100 units/mL streptomycin. LS174T cells were maintained in RPMI1640 with 10% FBS, 2 mM glutamine, 100 units/mL penicillin, and 100 units/mL streptomycin. GPA33 and HER2 antigen density of SW1222 and LS174T cells was measured by fluorescent-activated cell sorting with Quantum™ Simply Cellular® beads (Bangs Laboratories, Inc.) according to the manufacturer's instructions.
Radiochemistry
Chemical synthesis of Pr and subsequent radiolabeling with [225Ac]Ac(NO3)3 yielding the [225Ac]Ac-Pr radioligand was performed according to previous methods [6].
For 111In-labeling of Pr, 4 nmol of Pr was added to 100 µL of 0.2 M ammonium acetate buffer (pH 5.3) in a polypropylene vial. [111In]InCl3 solution (7 µL, 92.5 MBq [2.50 mCi]), obtained from Nuclear Diagnostic Products, Rockaway, NJ, USA) was added to the vial and mixed gently by pipetting. The mixture was heated for 30 min at 90 °C and cooled. A small amount (2 µL) was removed to determine radiochemical yield (RCY) and radiochemical purity (RCP) by HPLC (>99% RCY and RCP). The product was used without further purification.
GPA33 and HER2 antigen density
The GPA33 and HER2 antigen levels determined by fluorescent-activated cell sorting for SW1222 were an average of 151904 and 56765 sites/cell, respectively. For LS174T, the GPA33 antigen levels and HER2 antigen levels were an average of 216461 and 57582 sites/cell, respectively.
In vitro uptake and internalization assays
The in vitro uptake and internalization kinetics of the anti-GPA33- and anti-HER2- pretargeted tracer radioligand were investigated. [111In]In-Pr was used as a surrogate for [225Ac]Ac-Pr [6]. In brief, CRC cells (SW1222 or LS174T) were pre-incubated with 64 nM of anti-GPA33 or anti-HER2 BsAb for 1 h at 37 °C. Following removal of the antibody-containing media, cells were incubated with 22 nM [111In]In-Pr for up to 24 h. At selected timepoints, the membrane-bound activity, activity in the cytoplasm, and the activity in the nuclei were collected. Cells were first incubated with 0.2 M glycine buffer with 0.15 M NaCl and 4 M urea [21] to collect the membrane-bound activity. A nuclei isolation kit (Nuclei EZ Prep, Sigma Aldrich) was used according to the manufacturer's instructions to separate the nuclei from the cytoplasm. A more detailed description is provided in the supplementary material.
In vitro cell survival experiments
SW1222 cells (500,000 cells/mL in 0.6 mL, in suspension in vented culture tubes) were incubated with 64 nM of BsAb for 1 h at 37 °C on a rocker-roller. Next, cells were washed twice with serum-free media. The cells were resuspended in 1 mL of serum-free medium with [225Ac]Ac-Pr (0-20 kBq/mL) and suspensions were placed back on the rocker roller for 90 min at 37 °C. As a final step, cells were washed twice with serum-free media, counted, and plated for colony formation at different densities. After 21 d, cells were fixed and stained using 6% glutaraldehyde with 0.5% crystal violet [22]. Colonies (>50 cells) were counted, and surviving fractions were calculated. Additional details are in Figure S1 and the supplementary data.
Multicellular dosimetry
Cellular-level dosimetry of the SW1222 cells was performed using the multicellular dosimetry software tool, MIRDcell V4.14 [23-25]. For dosimetry purposes, within MIRDcell, SW1222 cells were modeled with a concentric spherical geometry with 6 μm as a cellular radius and 4 μm as nuclear radius. The uptake and the percentage of internalized activity of [111In]In-Pr were used to calculate the 225Ac disintegrations in each cell compartment (surface (CS), cytoplasm (Cy), nucleus (N)) at measured time points (1 h, 2 h, 6 h, and 24 h). It was assumed that the uptake and subcellular activity distribution of 111In were the same as those of 225Ac. The total absorbed dose to the nucleus of the SW1222 cells from 225Ac was calculated as a sum of the self-dose from activity in the cells, the cross-dose from the medium during the labeling period, and the cross-dose from the other cells during the colony-forming period. A detailed description can be found in Figure S2 and the supplementary data.
Animal experiments
All animal experiments were done in accordance with protocols approved by the Institutional Animal Care and Use Committee of Memorial Sloan Kettering Cancer Center following National Institutes of Health guidelines for animal welfare.
To prepare the SW1222 CRC animal model used in biodistribution and therapy experiments, female athymic nude mice (strain: Hsd:Athymic Nude-Foxn1nu, Envigo, aged 6-8 weeks) were inoculated in the flank with SW1222 cells (5 x 106 cells/mouse in 50% Matrigel (Corning)) 7-11 d prior to the experiments. The CLR37 PDX model originated from the biopsy of a liver metastasis from a patient with BRAF V600E CRC who had progressed through all standard treatment and was on trials of RAF + EGFR inhibitors. Notable findings from MSK IMPACT targeted exon sequencing included the BRAF V600E mutation + acquired NRAS G13R mutation [26]. GPA33 expression in the original tumor and early-passage PDX was confirmed via GPA33-IHC. For CLR37 experiments, Balb/c Rag2-/-IL2Rγ-/- mice (derived from the colony of Dr. Mamoru Ito, CIEA, Kawasaki, Japan, and now commercially available from Taconic as CIEA BRG mice) were used. Mice were randomly assigned to treatment groups.
225Ac-DOTA-PRIT regimen
225Ac-DOTA-PRIT was administered according to previously published protocol [6]. In brief, 250 µg (1.19 nmol) of BsAb was administered 28 h before administration of [225Ac]Ac-Pr. Twenty-five μg (2.76 nmol) of CA were injected 24 h after BsAb injection and 4 h prior to [225Ac]Ac-Pr.
Biodistribution and tissue dosimetry
To investigate the influence of administered [225Ac]Ac-Pr mass on in vivo PRIT performance, SW1222-xenografted mice were treated with anti-GPA33 BsAb, CA, and increasing amounts of [225Ac]Ac-Pr (7.8 kBq [0.21 µCi], 100-10000 pmol). A biodistribution study was performed 24 h post-injection (pi) of [225Ac]Ac-Pr (n = 4-5/mass dose). Mice were euthanized via CO2 asphyxiation, relevant tissues were collected, and activity content was measured at secular equilibrium.
For serial biodistribution studies, SW1222 xenograft-bearing mice were injected with anti-GPA33 or anti-HER2 BsAb and 37 kBq [1 µCi] of [225Ac]Ac-Pr (700 pmol) according to the protocol described above. Biodistribution was performed at 2 h, 24 h, 72 h, and 168 h pi of [225Ac]Ac-Pr (n = 5 mice/timepoint).
Time-integrated activity (TIA) was calculated by trapezoidal integration over the period from 0 to 168 h. Tissue activity concentration was assumed to be 0 Bq/mL at time 0 h. Time-activity curves were fit to exponential decay models, and tissue TIAs from 168 h to infinity were calculated using the exponential fits. The exception was the liver which had an increasing time-activity curve at 168 h. In this case, only physical decay was assumed to occur after 168 h. For all tissues, absorbed dose was calculated based on total local absorption of alpha and beta emissions only. Cross-dose from gamma emissions was excluded.
In vivo therapy in a flank model of CRC
On 11 d post-inoculation of SW1222 xenografts, mice were randomized into groups of n = 10 and treated with anti-HER2 and anti-GPA33 225Ac-DOTA-PRIT (37 kBq [1 µCi], 740 pmol) with appropriate controls. For GPA33, additional dose escalation therapy studies were conducted (all groups n = 10). These studies included a two-cycle treatment (total administered [225Ac]Ac-Pr: 74 kBq [2 μCi], cycle 1: 740 pmol, cycle 2: 790 pmol) separated by 1 week. Additionally, the efficacy of anti-GPA33 225Ac-DOTA-PRIT (74 kBq [2 µCi], 700 pmol) was evaluated. Median survival (MS; in d after treatment initiation) was defined as time to tumor progression to a diameter of 15 mm or death for any reason. Tumor sizes were manually measured 1-2 times per week using a caliper, and tumor volumes were computed assuming ellipsoidal geometry.
Body weight and overall well-being of the mice were monitored to assess treatment-related toxicity. Mice were subjected to regular blood sampling, and samples were analyzed for white blood count (WBC), red blood cell count (RBC), and platelet count (PLT) using the Heska Element HT5 (Heska Corporation, Loveland, CO, USA).
Animals alive 124 d post treatment were evaluated for radiation-induced histological organ damage by board-certified veterinary pathologists (Laboratory for Comparative Pathology at Memorial Sloan Kettering Cancer Center and Weill Cornell Medical College, New York, NY, USA). If available, residual tumors of surviving mice were stained for expression of GPA33 and HER2. For details see supplemental material.
Biodistribution and experimental therapy in a PDX model
In vivo biodistribution and therapy experiments were also performed with the CLR37 PDX model. For biodistribution, mice (n = 4) were injected with the anti-GPA33 225Ac-DOTA-PRIT (74 kBq [2 μCi] of [225Ac]Ac-Pr) and sacrificed at 24 h pi. For the experimental therapy, mice (n = 5 mice/group) were treated with a single cycle of anti-GPA33 225Ac-DOTA-PRIT (700 pmol, 37 kBq [1 µCi] or 74 kBq [2 μCi]). A [225Ac]Ac-Pr only (74 kBq [2 μCi], 700 pmol) and a non-treated control group were included. Survival criteria were the same as for the SW1222 flank model.
Tumor and kidney autoradiography using iQID camera
Activity distribution in SW1222 xenografts and kidneys was visualized using an ionizing-radiation Quantum Imaging Detector (iQID) imaging system (QScint Imaging Solutions). For sample preparations, SW1222-tumor bearing mice were injected with anti-GPA33 or anti-HER2 225Ac-DOTA-PRIT. Xenografts and kidneys were collected 2 and 24 h after [225Ac]Ac-Pr administration and immediately frozen in OCT. Frozen tissue sections (12 µm) were prepared using a cryostat microtome and placed on a ZnS:Ag phosphor Screen (EJ-440-100-3.2, Eljen Technology, Sweetwater, TX, USA). iQID digital autoradiographs (effective pixel size 19.5 microns) were generated by imaging tissue sections for 24 h in an alpha-particle counting mode. Counts images and ROI selections and statistics were processed using ImageJ.
Statistical analysis
For biodistribution data, statistically significant differences (p < 0.05) were determined by one-way ANOVA with post-hoc t-test corrected for multiple comparisons with the Tukey method using GraphPad Prism 9 (GraphPad, Inc.), unless stated otherwise. Survival analysis was performed using GraphPad Prism 9. Kaplan-Meier survival curves were analyzed with the log-rank (Mantel-Cox) test. A p-value of < 0.05 was considered statistically significant.
Results
Multicellular dosimetry using MIRDcell and in vitro cell survival
Table 1 shows the summary of cellular dosimetry results for the clonogenic cell survival assay for each antibody type. For both GPA33 and HER2, the self-dose and cross-dose from the medium are about 72% and 28%, respectively. Overall, the estimated absorbed dose to cells treated with the anti-GPA33 225Ac-DOTA-PRIT regimen was double that with the anti-HER2 regimen.
Summary of absorbed doses to cell nucleus of SW1222 cells based on uptake and internalization data and calculated using MIRDcell V4.
| Source of Dose and Stage of Colony Forming Assay | D (Gy per kBq/mL) | |
|---|---|---|
| GPA33 | HER2 | |
| Self-dose during labeling | 0.04 | 0.02 |
| Cross-dose from medium during labeling | 0.024 | 0.024 |
| Self-dose during colony forming | 0.90 | 0.45 |
| Cross-dose during colony forming | 0.371 | 0.185 |
| Total absorbed dose (self + cross) | 1.3 | 0.65 |
Exposure of SW1222 to anti-GPA33 and anti-HER2 225Ac-DOTA-PRIT resulted in increased cell death (Figure 1). Greater potency was observed with the GPA33-targeting regimen. After incubation with 20 kBq/mL [225Ac]Ac-Pr the surviving fraction was 4-fold higher for cells that were pre-targeted with the HER2 BsAb. See Table S2 and Figure S3 for mean absorbed doses received by SW1222 cell nucleus during clonogenic survival assay.
In vitro characterization of the anti-GPA33 and anti-HER2 DOTA-PRIT systems in SW1222 CRC cells. (A, C, D) Cells were incubated with 64 nM BsAb for 1 h at 37 ºC, followed by continuous incubation with 22 nM of [111In]In-Pr at 37 ºC (as a surrogate for 225Ac). (A) Uptake and internalization in adherent cells as % added activity (average ± SD). (C, D) activity internalized in adherent cells and normalized to total bound activity at each timepoint and separated into nucleus and cytoplasm. (B) Surviving fraction (SF) of SW1222 cells in suspension after incubation with [225Ac]Ac-Pr in the presence or absence of BsAb (average ± SD). Cells were incubated with 64 nM BsAb for 1 h on a rocker roller, followed by 90 min incubation with [225Ac]Ac-Pr before they were plated for colony formation. n = 3-6 per data point. Data from two independent experiments.
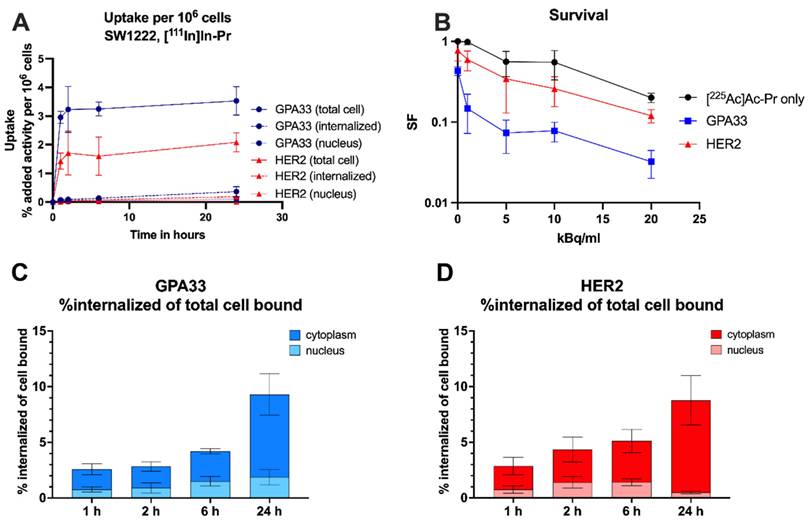
Slow internalization of [111In]In-Pr independent of the targeted antigen
Similar uptake and internalization patterns were observed for [111In]In-Pr in the presence of anti-GPA33 and anti-HER2 BsAb in SW1222 cells with stable total uptake of [111In]In-Pr after 2 h but increasing internalization with time (Figure 1, Table S3). There were no significant differences in the internalized fraction or nucleus-associated radioactivity between the two targets at any of the investigated time points. At 24 h, the internalized fractions were 9 ± 1% and 9 ± 2% of the total cell-bound activity for GPA33 and HER2, respectively. Absolute uptake was 1.86-fold higher (p < 0.05) when targeting GPA33, attributed to its higher expression level in SW1222 cells.
In LS174T cells (Figure S4, Table S3), the total cell uptake continued to increase until the 24 h timepoints for both GPA33 and HER2. While there was no significant difference in internalization between cell lines for HER2, the internalization of [111In]In-Pr and its localization in the nucleus were significantly higher for GPA33 BsAb at all time points, except at 6 h.
Refer to Table S3 for a detailed analysis of uptake and internalization results, including statistical comparisons for the SW1222 and LS174T cell lines.
In vivo targeting and dosimetry
The [225Ac]Ac-Pr dosing experiment targeting GPA33 showed that the highest tumor-to-kidney ratios could be achieved at [225Ac]Ac-Pr mass doses between 350 and 700 pmol (Figure S5, Tables S4 and S5). A radiohapten dose of 700 pmol was used for serial biodistribution in mice bearing SW1222 xenografts using either the anti-GPA33 or anti-HER2 225Ac-DOTA-PRIT regimen (Figure 2, Figure S6, Tables S6 and S7). Peak tumor uptake was achieved rapidly, as a maximum average uptake of 31 ± 12 %ID/g or 11 ± 1 %ID/g was observed at 2 h pi for targeting GPA33 and HER2, respectively. Tumor retention of [225Ac]Ac-Pr was higher when targeting HER2 with no significant release of activity over the investigation period. Uptake of [225Ac]Ac-Pr in mice pre-targeted with anti-HER2 BsAb was significantly higher in most tissue at 2 h, 24 h, and 72 h. Most notably, the concentration of 225Ac in blood was 2- to 4-fold higher in HER2-targeted mice than in GPA33 between 2 h and 72 h pi. Kidney uptake of [225Ac]Ac-Pr for the anti-HER2 225Ac-DOTA-PRIT was twice the uptake of mice that received the anti-GPA33 225Ac-DOTA-PRIT regimen. See Tables S6 and S7 for all statistical comparisons. Autoradiography of SW1222 xenografts revealed an apparent uniform distribution of 225Ac for both target antigens (Figure 2). Ratios of tumor to kidney activity (Table 2) were obtained via analysis of iQID alpha counts digital autoradiography images (Table S8). Based on the iQID kidney images collected at 2 and 24 h pi, the 225Ac uptake is concentrated in the cortex. In general, tumor-to-kidney ratios obtained via ex vivo biodistribution tended to be higher.
Quantification of iQID autoradiography images. Ratios are based on ROI intensities. Data in column 1-3 represents one animal per row. Absolute ROI intensities can be found in Table S8.
| Target and timepoint | Mean IntensityTumor/Cortex Ratio | Mean Intensity Tumor/Medulla Ratio | Mean Intensity Tumor/Kidney Ratio |
|---|---|---|---|
| GPA33 2 h pi | 19.1 | 68.4 | 14.9 |
| 7.6 | 43.7 | 6.5 | |
| GPA33 24 h pi | 7.5 | 68.0 | 6.7 |
| 7.1 | 32.7 | 5.8 | |
| HER2 2 h pi | 1.6 | 2.4 | 1.0 |
| HER2 24 h pi | 1.8 | 3.8 | 1.2 |
| 1.5 | 4.6 | 1.1 |
(A) Serial biodistribution of [225Ac]Ac-Pr (700 pmol) in SW1222 xenografted mice (n = 4-5 mice/time point, average ± SD). Uptake over time in tumors, blood, and kidney after pre-targeting with anti-GPA33 or anti-HER2 BsAb. Full biodistribution data can be found in Figure S6 and Tables S6 and S7. (B) Tumor and kidney autoradiography using iQID camera 2 h and 24 h after injection of [225Ac]Ac-Pr. Tissues from two mice per timepoint, and target, except for anti-HER2 2 h (only 1 mouse). Scalebar: 5 mm. Colorbar is in units of alpha counts acquired during 24 h imaging acquisition.
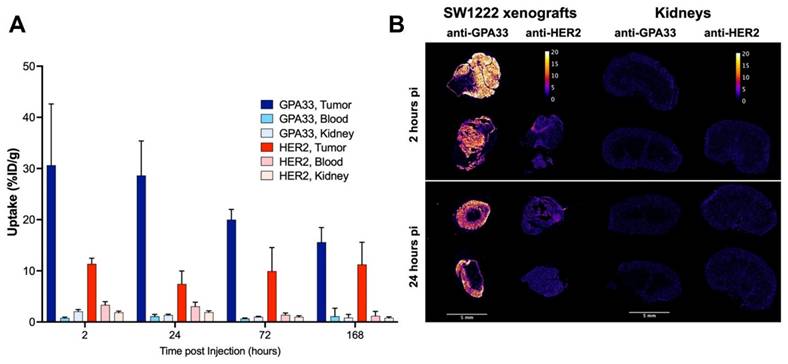
The ex vivo serial biodistribution data was used to estimate the absorbed doses to tumors and healthy tissues (Table 3, Table S9). The estimated dose to the SW1222 xenograft was about 1.7-fold higher when targeting GPA33 compared with HER2. Not considering the tumors, the absorbed doses were highest for blood, kidney, and liver, with doses being generally lower with the GPA33 targeting regimen than the HER2 regimen.
Mean absorbed doses delivered to tissues and therapeutic indices based on serial biodistribution of 225Ac-anti-GPA33 and 225Ac-anti-HER2-DOTA-PRIT in SW1222 xenografted mice. Dose shown as Gy/37 kBq. Estimation does not include cross-dose between organs, total local absorption assumed. RBE: relative biological effectiveness
| Tissue | Tissue doses (Gy/37 kBq) | |||||
|---|---|---|---|---|---|---|
| GPA33 | HER2 | TI | ||||
| 225Ac + all daughters | 225Ac + all daughters (RBE = 5) | 225Ac + all daughters | 225Ac + all daughters (RBE = 5) | GPA33 | HER2 | |
| Blood | 2.18 | 10.89 | 3.16 | 15.78 | 17.7 | 7.2 |
| Tumor | 38.66 | 193.31 | 22.78 | 113.91 | ||
| Heart | 0.64 | 3.19 | 1.14 | 5.70 | 60.5 | 20.0 |
| Lungs | 1.08 | 5.38 | 1.70 | 8.50 | 35.9 | 13.4 |
| Liver | 1.70 | 8.50 | 2.77 | 13.85 | 22.8 | 8.2 |
| Spleen | 0.88 | 4.40 | 1.65 | 8.24 | 44.0 | 13.8 |
| Stomach | 0.17 | 0.85 | 0.29 | 1.45 | 228.1 | 78.5 |
| Sm. Intestine | 0.26 | 1.31 | 0.46 | 2.28 | 148.1 | 49.9 |
| Lg. Intestine | 0.35 | 1.73 | 0.41 | 2.05 | 111.6 | 55.6 |
| Kidneys | 2.02 | 10.10 | 2.10 | 10.50 | 19.1 | 10.9 |
| Muscle | 0.34 | 1.69 | 0.51 | 2.54 | 114.7 | 44.9 |
| Bone | 0.73 | 3.63 | 0.76 | 3.81 | 53.3 | 29.9 |
225Ac-DOTA-PRIT results in significantly prolonged survival in a flank model of human CRC with histological cures
Survival curves, tumor growth curves, median survival, and residual tumor volumes of animals alive 124 d post-treatment are shown in Figure 3 and Table 4. All groups treated with the 225Ac-DOTA-PRIT regimen, including the off-target control, had significantly prolonged survival compared with the no treatment group. All treatments were well tolerated, with only a transient average body weight loss of less than 10% (Figure S7). WBC, RBC, and PLT counts remained within the reference range for nude mice during the first month after treatment, indicating the absence of acute toxicity (Figure S8).
Experimental therapy results from mice bearing SW1222 xenografts after different 225Ac-DOTA-PRIT treatment regimens (n = 10/group). (A) Percent survival (B) Tumor volumes (mm3). Graphs were discontinued after exclusion of the first animal in each group. Please see Table 4 for detailed information on long-term survivors and tumor status.
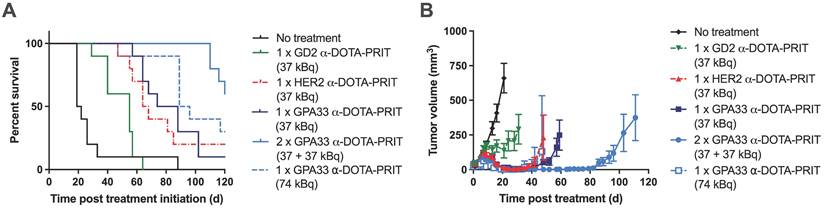
Summary of experimental therapy results from SW1222-xenografted mice: MS, residual tumor volume, and tumor status from mice surviving until 124 d post-treatment. Histological cures were determined at study endpoint based on hematoxylin and eosin staining and assessment by a board-certified veterinary pathologist.
| Treatment Group | MS (d) | Alive at 124 d (residual tumor volume) | Tumor Status |
|---|---|---|---|
| No treatment | 20.5a | 0/10 | n/a |
| 1 x 225Ac-anti-GD2-DOTA-PRIT (37 kBq) | 55a,b | 0/10 | n/a |
| 1 x 225Ac-anti-HER2-DOTA-PRIT (37 kBq) | 66b | 2/10 (0 mm3, 141.7 mm3) | 1: Histologic “cure” 2: Carcinoma, with necrosis |
| 1 x 225Ac-anti-GPA33-DOTA-PRIT (37 kBq) | 81b | 1/10 (96.2 mm3) | 1: Carcinoma, with necrosis |
| 2 x 225Ac-anti-GPA33-DOTA-PRIT (37 + 37 kBq) | 123a,b | 5/10 (0 mm3, 0 mm3, 11.5 mm3, 62.8 mm3, 172.5 mm3) | 1 and 2: Histologic “cure” 3: Carcinoma, with lymphocytic infiltrates 4: Carcinoma, with necrosis 5: Carcinoma, with necrosis |
| 1 x 225Ac-anti-GPA33-DOTA-PRIT (74 kBq) | 92.5b | 3/10 (0 mm3, 0 mm3, 19.2 mm3) | 1,2,3: Histologic “cure” 3: Mass of with intratumoral necrosis, mucinous material, and mineralized concretions, surrounded by chronic inflammation and fibrosis (no neoplastic cells identified) |
a significant difference p < 0.05 compared with 1 x 225Ac-anti-GPA33-DOTA-PRIT (37 kBq) by Log-rank (Mantel-Cox) test; b significantly prolonged survival compared to no treatment group by Log-rank (Mantel-Cox) test.
For single-cycle treatments (37 kBq) of either anti-GPA33 225Ac-DOTA-PRIT or anti-HER2 225Ac-DOTA-PRIT, all treated mice showed complete response (CR; tumor volume ≤10 mm3) at approximately 20 d post-treatment; however, tumor escape was observed for most of the animals. For anti-GPA33 therapy, a single mouse was still alive at 124 d with a tumor volume of 96.2 mm3. For anti-HER2 therapy, 2/10 were still alive with tumor volumes of 0 mm3 (i.e., no palpable tumor), and 141.7 mm3. There was no significant difference in MS between the single-cycle anti-HER2 and anti-GPA33 treatments (all individual p-values in Table S10). Two cycles of anti-GPA33 225Ac-DOTA-PRIT significantly prolonged MS compared with all other groups, except the high single dose (74 kBq) of anti-GPA33 225Ac-DOTA-PRIT (all individual p-values in Table S10). Similar to single-cycle treatment (37 kBq), all treated mice (10/10) showed CR, and 5/10 mice of this group were still alive after 124 d, two of which had histological cures. Histological cures at 124 d post treatment were also observed in the 1 x 74 kBq anti-GPA33 (3/5 mice alive at endpoint) and 1 x 37 kBq anti-HER2 (1/2 mice alive at endpoint) 225Ac-DOTA-PRIT groups. Examples of hematoxylin and eosin-stained, GPA33-stained, and HER2-stained residual tumors are shown in Figure S9.
Kidney damage was scored according to criteria in Jaggi et al. [27]. Results can be found in Table S11. No major pathological changes were observed in the surviving mice, and there was no difference between groups. The minimal tubulointerstitial changes were interpreted as incidental changes that cannot be differentiated from spontaneous background lesions commonly seen in murine kidneys. These findings were not considered adverse as they did not appear to significantly affect renal function or general health of the animals, based on the low percentage of the renal parenchyma affected (<1%), normal serum chemistry parameters, and absence of clinical signs or alterations in body weights. A representative kidney image can be found in Figure S10.
Anti-GPA33 225Ac-DOTA-PRIT shows efficacy in a GPA33 positive patient derived xenograft model
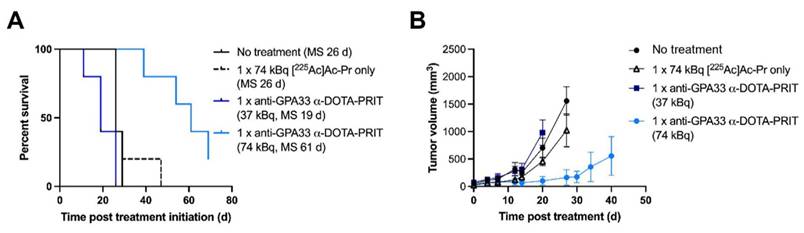
Expression of GPA33 in the CLR37 PDX was confirmed by IHC (Figure S11). Biodistribution of [225Ac]Ac-Pr 24 h pi (Figure S12) showed efficient targeting of GPA33 in CLR37 PDX, with the tumor uptake being 16 ± 7 %IA/g 24 h pi.
Treatment of CLR37 PDX with 74 kBq anti-GPA33 225Ac-DOTA-PRIT significantly delayed growth of CLR37 PDX and significantly prolonged survival compared with all other treatment groups (Figure 4). There was no significant difference in MS between no treatment, [225Ac]Ac-Pr only, and 37 kBq anti-GPA33 225Ac-DOTA-PRIT groups. No changes in body weight were observed (Figure S12).
Therapy of mice bearing GPA33 expressing PDX. (A) Survival curves after experimental anti-GPA33 225Ac-DOTA-PRIT of CLR37 PDX in mice (n = 5 mice/group). (B) Tumor volumes (mm3). Graphs were discontinued upon exclusion of the first animal.
Discussion
Herein we evaluated 225Ac-DOTA-PRIT against GPA33, an antigen highly expressed in CRC, and HER2, an antigen with only 20% of the comparable antigen concentration, in terms of treatment efficacy. Despite significant differences in tumor uptake, we found that a single cycle of pretargeting with 37 kBq of [225Ac]Ac-Pr achieved similar tumor control efficacy. Treatment of subcutaneous CRC SW1222 xenografts in mice with either anti-GPA33 or anti-HER2 225Ac-DOTA-PRIT resulted in CRs and histological cures in a subset of the long-term survivors without apparent toxicity to healthy organs. Due to the excellent TIs, more aggressive treatment regimens (either 1 x 74 kBq or 2 x 37 kBq) further enhanced therapeutic efficacy and prolonged survival without detectable toxicity. The MTD has not yet been reached. We also verified the efficacy of anti-GPA33 225Ac-DOTA-PRIT in a CRC PDX. This model represents heavily pre-treated CRC and was generated from a patient with resistant disease to both chemotherapy and targeted therapy. In summary, these studies establish anti-GPA33 and anti-HER2 225Ac-DOTA-PRIT as safe and effective treatments for CRC co-expressing GPA33 and HER2.
The potency of the anti-HER2 regimen in comparison with the anti-GPA33 regimen was somewhat surprising. This phenomenon has been documented previously by Boudousq et al. during RIT treatment of small volume carcinomatosis using two different 212Pb-IgG; the 212Pb-35A7 mAbs (anti-carcinoembryonic antigen) were less efficient than 212Pb-trastuzumab (anti-HER2), although the dose absorbed by the tumor was higher for 212Pb-35A7 [28]. Several potential mechanisms may be at play in this setting. One possibility is that the relative biological effectiveness (RBE) differs due to the intrinsic biology of the two antibody-antigen systems, perhaps leading to increased bystander effects [29-32]. Another possibility is that this reflects the intrinsic heterogeneity of each tumor-associated antigen on individual tumor cells [33]. Nonetheless, for a given cGy delivered to the nucleus, HER2 appears to be more effective than GPA33 as a carrier for delivering cytocidal α-radiation.
The pathophysiology of the SW1222 solid tumor model provides a unique opportunity to evaluate the radiation dose-response relationships of these two antigen targets. This model is highly vascularized and well-differentiated [34]. We investigated the intratumoral distribution of 225Ac using autoradiography with an iQID alpha camera [35, 36]. Images indicated relatively homogenous uptake independent of the targeted antigen. This targeting pattern allows for a fair comparison of the two antigens, especially in the context of TAT. Therefore, differences in the effectiveness of HER2 and GPA33 could not be attributed to obvious heterogeneity of uptake within the tumor mass.
To understand the radiation biology underlying the enhanced effectiveness of anti-HER2 225Ac-DOTA-PRIT compared to anti-GPA33 225Ac-DOTA-PRIT, we conducted in vitro studies and estimated the nuclear doses using MIRDcell techniques. We observed a two-fold difference in doses: 1.3 Gy for GPA33 and 0.65 Gy for HER2 for the same activity concentration in the medium. Notably, both regimens showed similar in vivo efficacy, despite this apparent difference in nuclear dose and the saturating in vitro cell survival curves. This observation supports a testable hypothesis: a threshold radiation dose to the nucleus may be required to achieve a CR. To further investigate this phenomenon, we intend to repeat these studies with larger cohorts and a broader range of administered activities, with the specific objective of determining whether this effect is maintained at lower 225Ac doses or when using a low linear energy transfer radioisotope with a broader dynamic dosing range. We hypothesize that, despite likely differences in antibody-antigen complex trafficking within the cell due to GPA33 or HER2 antigen, tumor cell death can be expected if the radiation dose delivered to the nucleus exceeds a critical threshold. A corollary of this idea is that the delivery of the increased amount of radiation absorbed dose from α-particles beyond this threshold is in fact redundant and probably toxic.
While α-dosimetry and the prediction of radiobiological effects are complex and continuously discussed within the TAT community [37, 38], we attempted to establish dose-response relationships for the two regimens. In terms of the average number of sites per cell, there is a difference on the order of 5-fold between the two antigens. Our macrodosimetry estimation predicted a 1.7-higher dose to be delivered to GPA33-targeted tumors (193.31 Gy/37 kBq; RBE = 5) compared to HER2-targeted tumors (113.91 Gy/37 kBq; RBE = 5). However, at equal administered [225Ac]Ac-Pr, the therapeutic outcomes were similar. This suggests that relative efficacy could not be predicted by absolute radiation absorbed dose (Gy).
A key factor that could explain the enhanced potency of HER2 compared to GPA33 likely relates to differences at the cellular level and the contrasting intrinsic biology of the two tumor-associated antigens. GPA33 is associated with tight junctions between cells and is minimally endocytosing [14, 15]. In contrast, HER2 is a part of the cell's growth and proliferation signal transduction cascade and is known to be an endocytosing antigen [39]. Additionally, since the anti-HER2 BsAb contains the sequence for trastuzumab, it may have a similar signal-blocking function, providing an additional anti-tumor effect [40]. This was previously observed in high HER2-expressing BT474 human breast cancer xenografts, although the standalone effect of the anti-HER2 BsAb was small (MS: 62 d for no treatment control, 72 d for BsAb only, and >130 d for anti-HER2 225Ac-DOTA-PRIT) [6]. These biological differences likely have a significant impact on the RBE.
Efficacy and therapeutic index (dose-toxicity relationships) are equally important. The data presented here is another step towards establishing dose-toxicity benchmarks for TAT and continues to push preclinical boundaries. Despite careful histologic examination of the kidneys, nephrotoxicity was not observed, and thus further dose escalation is feasible. Our dosimetry predictions, based on serial biodistribution data, estimated the dose to kidneys to be 10.5 Gy/37 kBq (RBE = 5). We acknowledge that microdosimetry of key organs at risk, including the kidneys, is very important, and we will continue to characterize nephrotoxicity with great care in future studies.
The findings and paradigms established in this study open the door to several possible future directions. While GPA33 and HER2 remain compelling CRC targets, our murine xenograft models do not fully capture potential human toxicity profiles. Further preclinical and early clinical trials in man are needed to evaluate on-target, off-tumor effects—such as those that may arise from GPA33 expression in normal gut tissue. In humans, radiation absorbed dose delivered by the GPA33-directed radioimmunoconjugate 131I-huA33 was readily tolerated in the range of 150 cGy or less to the red marrow in most patients [41]. We will keep these threshold numbers in mind as we plan for early clinical trials in man with GPA33-DOTA-PRIT. Furthermore, co-targeting these or additional co-expressed tumor antigens may help overcome the issue of antigen and uptake heterogeneity. The versatility of our DOTA-PRIT makes it an excellent candidate for co-targeting multiple antigens using an antibody cocktail, with the same radioligand binding to each BsAb. Optimized antibody cocktails can substantially enhance cell-absorbed doses compared to conventional single-target TAT [33]. Additionally, our recently developed self-assembling and disassembling (SADA) BsAb platform [42] offers further potential to improve TIs and eliminate the need for a CA.
Conclusion
In conclusion, we have demonstrated comparable safety and effectiveness for 225Ac-DOTA-PRIT against two different antigen systems, GPA33 and HER2. These treatment regimens resulted in significant prolonged survival and histologic cures in a subset of human CRC models growing as xenografts in immunodeficient mice. These outcomes were achieved with well- tolerated doses of 225Ac-DOTA-PRIT. These findings point to the potential of effective TAT regimens in advanced CRC using one or more tumor-associated antigen targets, either sequentially or in combination.
Supplementary Material
Supplementary materials and methods, figures and tables.
Acknowledgements
The 225Ac used in this research was supplied by the United States Department of Energy Isotope Program, managed by the Office of Isotope R&D and Production.
Funding
This work was supported by the Hedvig Hricak Chair in Radiology (to Steven Larson), Enid A. Haupt Chair (to Nai Kong Cheung), The Center for Targeted Radioimmunotherapy and Theranostics, Ludwig Center for Cancer Immunotherapy of Memorial Sloan Kettering Cancer Center (to Steven Larson), and Mr. William H. Goodwin and Mrs. Alice Goodwin and the Commonwealth Foundation for Cancer Research and The Experimental Therapeutics Center of Memorial Sloan Kettering Cancer Center (to Steven Larson). Steven Larson was also supported in part by P50-CA86438. This study also received support from R01-CA233896 (to Sarah Cheal). We also acknowledge P30-CA008748 for: use of the Tri-Institutional Laboratory of Comparative Pathology, Memorial Sloan Kettering Cancer Center, Weill Cornell Medicine, and The Rockefeller University, New York, NY; technical services provided by the Memorial Sloan Kettering Cancer Center Small-Animal Imaging Core Facility and Laboratory of Comparative Pathology; as well as the Molecular Cytology Core Facility.
Data availability
The datasets generated and/or analyzed during the current study are available from the corresponding author upon reasonable request.
Ethics approval
All animal experiments were done in accordance with protocols approved by the Institutional Animal Care and Use Committee of Memorial Sloan Kettering Cancer Center following National Institutes of Health guidelines for animal welfare.
Author contributions
SML, NKVC, and SMC: Study design, project supervision, and data interpretation. SSR: study design, in vitro and in vivo studies, data interpretation, and writing of the initial manuscript. SML and SMC contributed to the writing of the initial manuscript. DBV and BP: Coordinated and conduction of in vivo studies. BAV and DRV: 225Ac and 111In radiochemistry. SK and RWH: Study design, multicellular dosimetry, and data interpretation. BWM and JS: Data analysis. ES and RY: In vivo studies. ICM: Histopathology and data interpretation. HX and HG: Bispecific antibody production and characterization. EKF: Tissue dosimetry and data interpretation. All authors contributed to manuscript editing and approved the final version.
Competing Interests
Both Memorial Sloan Kettering Cancer Center (MSKCC) and Nai Kong Cheung have financial interests in Y-mAbs Therapeutics, Inc., Abpro-Labs, and Lallemand-Biotec Pharmacon. Nai Kong Cheung reports receiving commercial research grants from Y-mAbs Therapeutics, Inc. and Abpro-Labs. Nai Kong Cheung was named as inventor on multiple patents filed by MSKCC, including those licensed to Y-mAbs Therapeutics, Inc., Lallemand-Biotec Pharmacon, and Abpro-Labs. Nai Kong Cheung is a scientific advisory board member for Eureka Therapeutics. Nai Kong Cheung, Steven Larson, and Sarah Cheal were named as inventors in the following patent applications relating to GPA33: SK2014-074, SK2015-091, SK2017-079, SK2018-045, SK2014-116, SK2016-052, and SK2018-068 filed by MSKCC. Steven Larson reports receiving commercial research grants from Genentech, Inc., WILEX AG, Telix Pharmaceuticals Limited, and Regeneron Pharmaceuticals, Inc.; holding ownership interest/equity in Elucida Oncology, Inc. and Y-mAbs Therapeutics, Inc., and holding stock in ImaginAb, Inc. Steven Larson is the inventor and owner of issued patents, both currently unlicensed and licensed by MSKCC to Samus Therapeutics, Inc., Elucida Oncology, Inc., and Y-mAbs Therapeutics, Inc. Steven Larson serves or has served as a consultant to Y-mAbs Therapeutics, Inc., Cynvec LLC, Eli Lilly & Co., Prescient Therapeutics Limited, Advanced Innovative Partners, LLC, Gerson Lehrman Group, Progenics Pharmaceuticals, Inc., and Janssen Pharmaceuticals, Inc. Steven Larson, Nai Kong Cheung, Darren Veach, and Sarah Cheal were named as inventors in PCT/US2021/039418 (THOR cell (tumor homing radio-emitting cell)). Sarah Cheal serves or has served as a consultant to Affibody AB and Primary Insight. All other authors have no competing interests.
References
1. Abedizadeh R, Majidi F, Khorasani HR, Abedi H, Sabour D. Colorectal cancer: a comprehensive review of carcinogenesis, diagnosis, and novel strategies for classified treatments. Cancer Metastasis Rev. 2023;43(2):729-753
2. Melis M, Krenning EP, Bernard BF, Barone R, Visser TJ, de Jong M. Localisation and mechanism of renal retention of radiolabelled somatostatin analogues. Eur J Nucl Med Mol Imaging. 2005;32(10):1136-43
3. Cheal SM, Chung SK, Vaughn BA, Cheung NV, Larson SM. Pretargeting: a path forward for radioimmunotherapy. J Nucl Med. 2022;63(9):1302-15
4. Altai M, Membreno R, Cook B, Tolmachev V, Zeglis BM. Pretargeted imaging and therapy. J Nucl Med. 2017;58(10):1553-9
5. Verhoeven M, Seimbille Y, Dalm SU. Therapeutic applications of pretargeting. Pharmaceutics. 2019;11(9):434
6. Cheal SM, McDevitt MR, Santich BH, Patel M, Yang G, Fung EK. et al. Alpha radioimmunotherapy using (225)Ac-proteus-DOTA for solid tumors - safety at curative doses. Theranostics. 2020;10(25):11359-75
7. Orcutt KD, Ackerman ME, Cieslewicz M, Quiroz E, Slusarczyk AL, Frangioni JV. et al. A modular IgG-scFv bispecific antibody topology. Protein Eng Des Sel. 2010;23(4):221-8
8. Orcutt KD, Slusarczyk AL, Cieslewicz M, Ruiz-Yi B, Bhushan KR, Frangioni JV. et al. Engineering an antibody with picomolar affinity to DOTA chelates of multiple radionuclides for pretargeted radioimmunotherapy and imaging. Nucl Med Biol. 2011;38(2):223-33
9. Cheal SM, Patel M, Yang G, Veach D, Xu H, Guo HF. et al. An N-acetylgalactosamino dendron-clearing agent for high-therapeutic-index DOTA-hapten pretargeted radioimmunotherapy. Bioconjug Chem. 2020;31(3):501-6
10. Orcutt KD, Rhoden JJ, Ruiz-Yi B, Frangioni JV, Wittrup KD. Effect of small-molecule-binding affinity on tumor uptake in vivo: a systematic study using a pretargeted bispecific antibody. Mol Cancer Ther. 2012;11(6):1365-72
11. Chung SK, Vargas DB, Chandler CS, Katugampola S, Veach DR, McDevitt MR. et al. Efficacy of HER2-targeted intraperitoneal (225)Ac α-pretargeted radioimmunotherapy for small-volume ovarian peritoneal carcinomatosis. J Nucl Med. 2023;64(9):1439-1445
12. Akhavan D, Yazaki P, Yamauchi D, Simpson J, Frankel PH, Bading J. et al. Phase I study of yttrium-90 radiolabeled M5A anti-carcinoembryonic antigen humanized antibody in patients with advanced carcinoembryonic antigen producing malignancies. Cancer Biother Radiopharm. 2020;35(1):10-5
13. Sahlmann CO, Homayounfar K, Niessner M, Dyczkowski J, Conradi LC, Braulke F. et al. Repeated adjuvant anti-CEA radioimmunotherapy after resection of colorectal liver metastases: Safety, feasibility, and long-term efficacy results of a prospective phase 2 study. Cancer. 2017;123(4):638-49
14. Garinchesa P, Sakamoto J, Welt S, Real F, Rettig W, Old L. Organ-specific expression of the colon cancer antigen A33, a cell surface target for antibody-based therapy. Int J Oncol. 1996;9(3):465-71
15. Murer P, Plüss L, Neri D. A novel human monoclonal antibody specific to the A33 glycoprotein recognizes colorectal cancer and inhibits metastasis. MAbs. 2020;12(1):1714371
16. Cheal SM, Xu H, Guo HF, Lee SG, Punzalan B, Chalasani S. et al. Theranostic pretargeted radioimmunotherapy of colorectal cancer xenografts in mice using picomolar affinity ⁸⁶Y- or ¹⁷⁷Lu-DOTA-Bn binding scFv C825/GPA33 IgG bispecific immunoconjugates. Eur J Nucl Med Mol Imaging. 2016;43(5):925-37
17. Ahcene Djaballah S, Daniel F, Milani A, Ricagno G, Lonardi S. HER2 in colorectal cancer: The long and winding road from negative predictive factor to positive actionable target. Am Soc Clin Oncol Educ Book. 2022;42:1-14
18. Sur D, Lungulescu C, Dumitrescu EA, Afrăsânie V, Spînu Ș, Lungulescu CV. et al. A systematic review of clinical trials of treatment regimens in HER2-amplified metastatic colorectal cancer. Expert Rev Anticancer Ther. 2023;23(6):633-641
19. Lang-Schwarz C, Vieth M, Dregelies T, Sterlacci W. Frequency of Her2-low in colorectal cancer and its relations with the tumor microenvironment. Pathol Res Pract. 2023;244:154417
20. Cheal SM, Xu H, Guo HF, Patel M, Punzalan B, Fung EK. et al. Theranostic pretargeted radioimmunotherapy of internalizing solid tumor antigens in human tumor xenografts in mice: Curative treatment of HER2-positive breast carcinoma. Theranostics. 2018;8(18):5106-25
21. Wållberg H, Orlova A. Slow internalization of anti-HER2 synthetic affibody monomer 111In-DOTA-ZHER2:342-pep2: implications for development of labeled tracers. Cancer Biother Radiopharm. 2008;23(4):435-42
22. Franken NAP, Rodermond HM, Stap J, Haveman J, van Bree C. Clonogenic assay of cells in vitro. Nature Protocols. 2006;1(5):2315-9
23. Vaziri B, Wu H, Dhawan AP, Du P, Howell RW. MIRD pamphlet No. 25: MIRDcell V2.0 software tool for dosimetric analysis of biologic response of multicellular populations. J Nucl Med. 2014;55(9):1557-64
24. Katugampola S, Wang J, Prasad A, Sofou S, Howell RW. Predicting response of micrometastases with MIRDcell V3: proof of principle with (225)Ac-DOTA encapsulating liposomes that produce different activity distributions in tumor spheroids. Eur J Nucl Med Mol Imaging. 2022;49(12):3989-3999
25. Katugampola S, Wang J, Howell RW. MIRD pamphlet no. 31: MIRDcell V4—Artificial intelligence tools to formulate optimized radiopharmaceutical cocktails for therapy. J Nucl Med. 2024;65(12):1965-1973
26. Yaeger R, Yao Z, Hyman DM, Hechtman JF, Vakiani E, Zhao H. et al. Mechanisms of acquired resistance to BRAF V600E inhibition in colon cancers converge on RAF dimerization and are sensitive to its inhibition. Cancer Res. 2017;77(23):6513-23
27. Jaggi JS, Seshan SV, McDevitt MR, LaPerle K, Sgouros G, Scheinberg DA. Renal tubulointerstitial changes after internal irradiation with alpha-particle-emitting actinium daughters. J Am Soc Nephrol. 2005;16(9):2677-89
28. Boudousq V, Bobyk L, Busson M, Garambois V, Jarlier M, Charalambatou P. et al. Comparison between internalizing anti-HER2 mAbs and non-internalizing anti-CEA mAbs in alpha-radioimmunotherapy of small volume peritoneal carcinomatosis using 212Pb. PLoS One. 2013;8(7):e69613
29. Pouget JP, Constanzo J. Revisiting the radiobiology of targeted alpha therapy. Front Med (Lausanne). 2021;8:692436
30. Canter BS, Leung CN, Fritton JC, Back T, Rajon D, Azzam EI. et al. Radium-223-induced bystander effects cause DNA damage and apoptosis in disseminated tumor cells in bone marrow. Mol Cancer Res. 2021;19(10):1739-50
31. Rajon DA, Canter BS, Leung CN, Back TA, Fritton JC, Azzam EI. et al. Modeling bystander effects that cause growth delay of breast cancer xenografts in bone marrow of mice treated with radium-223. Int J Radiat Biol. 2021;97(9):1217-28
32. Leung CN, Canter BS, Rajon D, Back TA, Fritton JC, Azzam EI. et al. Dose-dependent growth delay of breast cancer xenografts in the bone marrow of mice treated with (223)Ra: The role of bystander effects and their potential for therapy. J Nucl Med. 2020;61(1):89-95
33. Pasternack JB, Domogauer JD, Khullar A, Akudugu JM, Howell RW. The advantage of antibody cocktails for targeted alpha therapy depends on specific activity. J Nucl Med. 2014;55(12):2012-9
34. El Emir E, Qureshi U, Dearling JL, Boxer GM, Clatworthy I, Folarin AA. et al. Predicting response to radioimmunotherapy from the tumor microenvironment of colorectal carcinomas. Cancer Res. 2007;67(24):11896-905
35. Miller BW, Frost SH, Frayo SL, Kenoyer AL, Santos E, Jones JC. et al. Quantitative single-particle digital autoradiography with α-particle emitters for targeted radionuclide therapy using the iQID camera. Med Phys. 2015;42(7):4094-105
36. Miller BW, Gregory SJ, Fuller ES, Barrett HH, Barber HB, Furenlid LR. The iQID camera: An ionizing-radiation quantum imaging detector. Nucl Instrum Methods Phys Res A. 2014;767:146-52
37. Sgouros G. Dosimetry, Radiobiology and synthetic lethality: Radiopharmaceutical therapy (RPT) with alpha-particle-emitters. Semin Nucl Med. 2020;50(2):124-32
38. Tronchin S, Forster JC, Hickson K, Bezak E. Dosimetry in targeted alpha therapy. A systematic review: current findings and what is needed. Phys Med Biol. 2022 67(9)
39. Ram S, Kim D, Ober RJ, Ward ES. The level of HER2 expression is a predictor of antibody-HER2 trafficking behavior in cancer cells. MAbs. 2014;6(5):1211-9
40. Baselga J, Albanell J, Molina MA, Arribas J. Mechanism of action of trastuzumab and scientific update. Semin Oncol. 2001;28(5 Suppl 16):4-11
41. Chong G, Lee FT, Hopkins W, Tebbutt N, Cebon JS, Mountain AJ. et al. Phase I trial of 131I-huA33 in patients with advanced colorectal carcinoma. Clin Cancer Res. 2005;11(13):4818-26
42. Santich BH, Cheal SM, Ahmed M, McDevitt MR, Ouerfelli O, Yang G. et al. A self-assembling and disassembling (SADA) bispecific antibody (BsAb) platform for curative two-step pretargeted radioimmunotherapy. Clin Cancer Res. 2021;27(2):532-41
Author contact
![]() Corresponding author: Sarah M. Cheal, Weill Cornell Medicine, Belfer Research Building, 413 East 69th Street, 16th Floor, New York, NY 10065; Phone: 646-962-6148; Fax: 646-962-0367; smc4002cornell.edu.
Corresponding author: Sarah M. Cheal, Weill Cornell Medicine, Belfer Research Building, 413 East 69th Street, 16th Floor, New York, NY 10065; Phone: 646-962-6148; Fax: 646-962-0367; smc4002cornell.edu.