Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2025; 15(16):8176-8201. doi:10.7150/thno.113599 This issue Cite
Research Paper
An unrecognized mechanism of self-protection in degenerating neurons mediated by astrocytic YAP through Wnts/β-catenin/EAAT2 signaling in C9orf72-poly-GA mice
Dongmei Li1#, Yan Wei1#, Rui Yang1#, Xuan Luo1, Yanzhu Liu1,2, Weiqiao Zhao3, Hui Yang1,2, Yumin Wu4, Ying Wang2 ![]() , Zhihui Huang1
, Zhihui Huang1 ![]()
1. School of Pharmacy, Hangzhou Normal University, Hangzhou 311121, Zhejiang, China.
2. Department of Clinical Research Center, Affiliated Hangzhou First People's Hospital, Westlake University School of Medicine, Hangzhou 310006, Zhejiang, China.
3. School of Basic Medicine, Hangzhou Normal University, Hangzhou 311121, Zhejiang, China.
4. Department of Orthopedics (Spine Surgery), the First Affiliated Hospital of Wenzhou Medical University, Wenzhou 325000, Zhejiang, China.
#These authors contributed equally this work.
Received 2025-3-11; Accepted 2025-7-8; Published 2025-7-24
Abstract

Rationale: Amyotrophic lateral sclerosis (ALS) is a fatal neurodegenerative disorder characterized by the progressive loss of motor neurons in the central nervous system (CNS). Non-neuronal cells, particularly astrocytes, have been recognized as pivotal contributors to ALS onset and progression. However, the underlying mechanisms of interactions between astrocytes and motor neurons during ALS remain unclear. Recent studies have identified the neuronal Hippo kinase mammalian sterile 20-like kinase 1 (MST1) as a key regulator of neurodegeneration in ALS. Yes-associated protein (YAP), a major downstream effector of the Hippo pathway, is predominantly expressed in astrocytes. However, the role of astrocytic YAP in ALS and its underlying mechanisms remain unexplored.
Methods: To evaluate the function of YAP in ALS, we established a C9orf72-poly-GA mouse model (ALS mice) via intracerebroventricular injection of AAV viruses. Furthermore, mice with conditional knockout (CKO) of YAP in astrocytes (YAPGFAP-CKO mice) were generated and then YAPGFAP-CKO ALS mice and their littermate controls (YAPf/f ALS mice) were used as experimental subjects. Behavioral tests, immunostaining, Nissl staining, quantitative real-time PCR (qPCR), and Western blotting were used to assess the effects of astrocytic YAP deletion in ALS progression. In addition, we investigated the role and mechanism of astrocytic YAP in the pathogenesis of ALS by integrating RNA sequencing (RNA-seq) from primary cultured astrocytes with single-nucleus transcriptomic (snRNA-seq) from C9orf72-ALS/FTD patients. Then, in vitro experiments including primary cultured astrocytes and neurons were used to further elucidate the potential molecular mechanism of astrocytic YAP in ALS. Finally, we evaluated the therapeutic effects of the excitatory amino acid transporter-2 (EAAT2) activator LDN-212320 and the Hippo kinase MST1/2 inhibitor XMU-MP-1 as candidate treatments for ALS.
Results: We found that YAP was upregulated and activated specifically in astrocytes, but not in neurons or microglia, within the motor cortex of ALS mice. Conditional knockout of YAP in astrocytes exacerbated motor deficits, neuronal loss, pathological translocation of TDP-43, inflammatory infiltration, and reduced astrocytic proliferation in ALS mice. Mechanistically, Wnts secreted by degenerating neurons and astrocytes activated YAP/β-catenin signaling and further promoted the expression of EAAT2 in astrocytes, which prevented neuronal glutamate excitotoxicity, neuronal loss, and motor dysfunction in ALS mice. Interestingly, treatment with LDN-212320 promoted EAAT2 expression and partially restored motor deficits and neuronal loss in YAPGFAP-CKO ALS mice. Finally, activation of YAP by XMU-MP-1 upregulated β-catenin and EAAT2 expression, and partially alleviated motor deficits and neurodegeneration in ALS mice.
Conclusions: These results identify an unrecognized mechanism of self-protection in degenerating neurons mediated by astrocytic YAP through Wnts/β-catenin/EAAT2 signaling to prevent glutamate excitotoxicity of neurons in ALS mice, and provide a novel drug target for ALS.
Keywords: YAP, ALS, EAAT2, neurodegeneration, motor dysfunction
Introduction
Amyotrophic lateral sclerosis (ALS) is a fatal neurodegenerative disorder characterized by the progressive degeneration of motor neurons in the brain and spinal cord leading to muscle weakness, eventual paralysis, and death from respiratory paralysis within 3 to 5 years [1-3]. Despite decades of intensive research, there is no cure for ALS. Currently, three pharmaceutical compounds with an effect on disease progression are approved: Riluzole (a glutamate antagonist), Edaravone (a free radical scavenger) and Sodium phenylbutyrate/Taurursodiol [4]. In addition, a combination of dextromethorphan hydrobromide and quinidine sulfate (Nuedexta) is used for the treatment of pseudobulbar affect (PBA) in ALS pathogenesis [5]. Globally, efforts are underway to prevent or alleviate the symptoms of this neurodegenerative disease, including the implementation of antisense oligonucleotides (ASOs), induced pluripotent stem cells (iPSCs), CRISPR-Cas9 technology, non-invasive brain stimulation or ALS-on-a-chip technology [6]. However, the treatment of ALS is largely limited to palliative care, so it is urgent to develop new and effective disease improvement therapies.
ALS is affected by many pathological mechanisms such as glutamate-induced excitotoxicity, apoptosis, mitochondrial dysfunction, oxidative stress, protein misfolding, neuroinflammation, altered RNA metabolism and neurofilament abnormalities [7-9]. Approximately 90~95% of ALS cases are sporadic (sALS), with no obvious genetic link, while the remaining 5~10% are familial (fALS) [10]. Many important gene mutations have been identified in fALS, including mutations in Cu/Zn superoxide dismutase 1 gene (SOD1), Chromosome 9 Open Reading Frame 72 (C9ORF72), TAR DNA Binding Protein (TARDBP), Fused in Sarcoma (FUS) gene and TANK-binding kinase 1 (TBK1) [11]. The above studies mainly focus on the pathogenic mechanisms of motor neuron degeneration in ALS, however, non-neuronal cells such as astrocytes have been recognized to play a pivotal role in disease onset and progression of ALS [12-14]. Astrocytes have critical roles in adult central nervous system (CNS) homeostasis, including synaptic glutamate uptake and synapse plasticity, maintenance of extracellular potassium and nutrient support of neurons, and regulation of blood brain barrier, water flux, ion and pH homeostasis [15]. Initial evidence for astrocytic contribution to motor neuron damage and ALS progression came from studies of decrease and dysfunction of astrocyte glutamate transporter GLT1 (EAAT2 in human) in ALS patients or ALS animal models [16, 17]. The decrease and dysfunction of EAAT2 in astrocytes is the main cause of glutamate-mediated excitotoxicity in ALS [18]. Furthermore, transplantation of wild-type lineage-restricted astrocyte precursors around the cervical spinal cord of SOD1G93A rats slowed the progression of ALS and prolonged survival probability [19]. Oppositely, transplantation of astrocyte precursors carrying SOD1G93A mutation induced the local degeneration of motor neurons in wild-type spinal cord [20]. Nowadays, although several mechanisms of astrocytic contribution to ALS pathology and progression have been addressed, including loss of support mechanisms and toxic gain of function mechanisms [14], the detailed molecular mechanisms of interactions between astrocytes and motor neurons during ALS remain unclear.
The Hippo pathway is critical in organ size and tissue homeostasis, and its dysregulation has been implicated in several diseases including tumorigenesis, cardiac diseases, pulmonary diseases, renal diseases and immune dysfunction [21]. The core of the Hippo pathway, including mammalian sterile 20-like kinases 1/2 (MST1/2), salvador homolog 1 (SAV1), Mps one binder 1A and B (MOB1A/B), large tumor suppressor 1/2 (LATS1/2), Yes-associated protein (YAP) and transcriptional co-activator with PDZ binding motif (TAZ), constitutes a kinase cascade reaction, with YAP acting as a major downstream effector of the Hippo pathway [21, 22]. After the Hippo signal is activated, the MST1/2 and SAV1 phosphorylate and activate the downstream kinase LATS1/2, and then LATS1/2 and MOB1A/B phosphorylate the transcriptional co-activators YAP and TAZ, leading to degradation or retention of YAP in the cytoplasm [23, 24]. When the Hippo signaling is inhibited, MST1/2 and LATS1/2 are deactivated, which promotes YAP to accumulate in the nucleus and combine with the transcriptional enhanced associate domain (TEAD) family, and finally induces the expression of genes [25]. Interestingly, motor neurons in sALS patients and SOD1G93A mice exhibit the elevated MST1 activity, and MST1 knockout delays the onset of disease and prolongs the survival of SOD1G93A mice [26]. In addition, loss of function of Hippo (Drosophila MST) rescues the deficits in the FUS-ALS Drosophila model [27]. Another study has also clarified that inactivation of Hippo signaling and JNK signaling alleviates FUS-mediated neurodegeneration in Drosophila eyes [28]. Collectively, these studies demonstrate that neuronal MST1/Hippo in a YAP-independent manner contributes to neurodegeneration in ALS. YAP is abundant in astrocytes and plays a critical role in regulating astrocytic proliferation and differentiation [29-32]. Growing evidence has revealed the function of astrocytic YAP under pathological conditions, such as promoting the formation of glial scars and neural regeneration of mice after spinal cord injury [33], promoting cholesterol synthesis in experimental autoimmune encephalomyelitis (EAE) to prevent demyelination [34], and upregulating TGF-β signaling to prevent neuroinflammatory infiltration and demyelination in multiple sclerosis-related optic neuritis (MS-ON) [35]. However, the roles and mechanisms of astrocytic YAP signaling in ALS remain poorly understood.
Wnt signaling is mediated through β-catenin dependent pathway, also known as the canonical pathway (Wnt/β-catenin pathway) and β-catenin independent pathway, referred to as the noncanonical pathway (Wnt/Ca2+ and Wnt/planar cell polarity) [36]. Canonical pathway is very important for neurogenesis, cell proliferation and neurogenesis, while noncanonical pathway is responsible for cell polarity, calcium maintenance and cell migration [37]. Recent studies have shown that Wnts, their receptors, and core components of the Wnt signaling pathway are expressed in ALS transgenic mice and ALS patients [38]. The activation of Wnt signaling pathway was identified in hiPSC and mouse ALS astrocytes [39]. Abnormal activation of the Wnt/β-catenin signaling pathway is related to neuronal degeneration and glial cell proliferation in ALS [40]. In addition, there are hypotheses proposing that the activation of the Wnt/β-catenin pathway in astrocytes could be a neuroprotective attempt to counteract motor neuron pathology in ALS [41]. Interestingly, the crosstalk between Hippo-YAP/TAZ and Wnt/β-catenin signaling pathways has been variously inferred in different physiological and pathophysiological conditions [42-44]. For example, YAP/TAZ as critical mediators of alternative Wnt signaling regulates its biological responses [45], and Wnt/β-catenin signaling regulates YAP/TAZ activity during embryonic development in zebrafish [46]. Also, Wnt/β-catenin pathway has been defined as an essential signaling system not only for neurodevelopment, but especially for the survival, neuroprotection and self-repair against neurodegeneration, inflammation and oxidative stress, via astrocyte-microglial-neuron crosstalks mediated by Wnts [47-50]. However, the potential crosstalk between Hippo-YAP/TAZ and Wnt/β-catenin signaling pathways in ALS, and its contribution to ALS remain unclear.
In this study, we established a C9orf72-poly-GA mouse model (ALS mice) to investigate the function of astrocytic YAP in ALS. We found that YAP was upregulated and activated in astrocytes, but not in neurons or microglia, within the motor cortex of ALS mice, and the knockout of astrocytic YAP exacerbated the disease progression of ALS. Mechanistically, we found that degenerating neurons and astrocytes secreted Wnts, which activated YAP/β-catenin signaling and further promoted EAAT2 expression in astrocytes, thereby preventing neuronal glutamate excitotoxicity, neuronal loss, and motor dysfunction in ALS mice. Our study reveals an unrecognized function of astrocytic YAP in preventing glutamate excitotoxicity of neurons through Wnts/β-catenin/EAAT2 signaling in ALS mice, and provide a new drug target for ALS.
Materials and Methods
Animals
YAPGFAP-CKO mice were generated by crossing floxed YAP allele (YAPf/f) with GFAP-Cre mice (from the Jackson Laboratory), as described previously [35]. Both YAPf/f and YAPGFAP-CKO mice were maintained under the background of C57BL/6 strain and these mice were genotyped. The use of animals and all relevant experimental protocols have acquired the approval of the Animal Care and Use Committee of Hangzhou Normal University (HSD20220911).
Intracerebroventricular injections
C9orf72-poly-GA mice were described previously [51]. Briefly, pups from YAPf/f and YAPGFAP-CKO mice were cryoanesthetized on ice at postnatal day 0 (P0). Then, a 32-gauge Hamilton needle was attached to a 10 µl Hamilton syringe and inserted vertically at two-fifths of the distance between the pup's eyes and lambda. The vertical depth of that needle was kept at 2 mm. 4 µl (2 µl per side) of AAV8 viruses (AAV-GFP-GA5, 6.60 × 10¹³ vg/ml; AAV-GFP-GA100, 4.11 × 10¹³ vg/ml; WZ Biosciences Inc, viral sequences gifted from Prof. Chengyong Shen, Zhejiang University) were slowly injected into the cerebral ventricle. After the injection, the pups were placed on a heating pad until they recovered from anesthesia and then were returned to their home cages. Behavioral analyses were conducted when the mice reached 3 months of age.
Behavioral analysis
A series of behavioral tests were conducted on 3-month-old YAPf/f ALS mice, YAPGFAP-CKO ALS mice, and their littermate YAPf/f and YAPGFAP-CKO control mice to evaluate motor function [52]. All mice were acclimated to the laboratory for at least 1 h before the formal experiment and were returned to their home cages after each experiment. No more than two tests were performed each day. The testing room was kept quietly and forbidden to disturb the spontaneous activity of mice.
Tail-suspension test
Briefly, 3-month-old YAPf/f and YAPGFAP-CKO control mice, YAPf/f ALS and YAPGFAP-CKO ALS mice were placed in a tail-suspension tester. The duration of hind limb clasping was recorded for 3 min.
Cage behavior test
Cage behavior test was an experiment to test the movement balance and coordination ability of mice. Briefly, the mice were put on the edge of the cage to walk on the premise that should never be subjected to this test before. The motor performance of mice was recorded with in 3 min.
Rotarod test
The exercise ability of mice was evaluated using the rotarod test. Briefly, one day before the test, the mice were placed on the rod for 2 min at a constant speed of 4 rpm to adapt and train. On the test day, the rod was set to accelerate from 4 rpm to 40 rpm over 5 min, and the latency to fall was recorded for each mouse. The test was repeated three times for each mouse with an interval of 15 min.
Balance beam test
The balance beam test was used to test the balance ability, muscles and coordination of mice. On the first day, each mouse was trained to run across the beam without interruption. On the second day, the camera was used to record the process of each mouse crossing the beam and the number of times each mouse's hind limbs slipped.
Footprint test
The footprint test was used to assess the motor function of the forelimbs and hind limbs. Briefly, the mice ran successfully along the paper track during training. In the formal experiment, the fore paws and hind paws of mice were coated with red and blue dyes, respectively. The gait of the mice was recorded. The center of each paw was marked, and the distance between the fore paw and hind paw at the same side was measured as back-front distance, and the distance between hind paws at the same side was measured as back stride distance.
Immunostaining
The cultured cells were washed with phosphate buffered saline (PBS) three times, fixed with 4% paraformaldehyde (PFA) for 20 min, and then permeabilized and blocked in PBS containing 0.1% Triton X-100 (#T8200, Solarbio) and 5% bovine serum albumin (BSA, #V900933, Sigma-Aldrich) for 1 h. The cells were subsequently incubated overnight at 4℃ with primary antibody, including rat anti-EAAT2 (1:200, OB-PRT026, Oasis Biofarm), mouse anti-GFAP (1:500, MAB360, Millipore), rabbit anti-YAP (1:100, #14074, CST), and rabbit anti-MAP2 (1:400, #4542, CST). The cells were washed three times with PBS (5 min each time), and then incubated with appropriate secondary antibodies [(1: 1, 000, Invitrogen Alexa Fluor™ :Plus 488 goat anti-rabbit IgG (H+L), #A32731; 546 donkey anti-rabbit IgG (H+L), #A11040; 546 goat anti-mouse IgG (H+L), #A11030; 1:1, 000, Oasis Biofarm: 647 goat anti-Rat IgG (H+L), #G-RT647; 1: 1, 000, Beyotime Alexa Fluor: 488 goat anti-mouse IgG (H+L), #A0428)] and DAPI for labeling of nuclei (1:1, 000, #4083S, CST) in 5% BSA at room temperature incubation for 1 h. After PBS washing, the cells were mounted. Image acquisition was done by Nikon single-photon and two-photon integrated confocal microscope (Nikon AX R MP), and analyzed by the ImageJ software.
For tissue section staining, mice were perfused with PBS followed by 4% PFA through the heart. The brains were then extracted and immersed in 4% PFA for 24-48 h. After fixation, the brains were transferred to 15% and 30% sucrose solution for continuous dehydration for 24-48 h. Subsequently, 20 μm sections were cut using a cryosectioner (CryoStar NX50, Thermo Scientific). The brain tissue sections were stained, fixed for 30 min, and then repaired with sodium citrate antigen repair solution at 65℃ for 45 min, permeabilized and blocked with 5% BSA (#V900933, Sigma-Aldrich) containing 0.3% Triton X-100 (#T8200, Solarbio) at room temperature for 1.5 h, followed by overnight incubation with primary antibodies at 4℃. The next day, the sections were washed three times and incubated with the appropriate secondary antibody [(1: 1, 000, Invitrogen Alexa Fluor™ :Plus 488 goat anti-rabbit IgG (H+L), #A32731; 546 donkey anti-rabbit IgG (H+L), #A11040; 546 goat anti-mouse IgG (H+L), #A11030; 1: 1, 000, Beyotime Alexa Fluor: 647 goat anti-rabbit IgG (H+L), #A0468; 647 goat anti-mouse IgG (H+L), #A0473; 488 goat anti-mouse IgG (H+L), #A0428; 1:1, 000, Oasis Biofarm: 647 goat anti-Rat IgG (H+L), #G-RT647; 647 goat anti-Guine a pig IgG, #G-GP647)] and DAPI for labeling of nuclei (1:1, 000, #4083S, CST) in 5% BSA at room temperature incubation for 1 h. The primary antibodies included rabbit anti-YAP (1:500, GTX129151, GeneTex), rabbit anti-YAP (1:100, #14074, CST), mouse anti-GFAP (1:500, MAB360, Millipore), mouse anti-NeuN (1:400, #94403S, CST), pig anti-Iba1 (1:500, OB-PGP049-01, Oasis Biofarm), rabbit anti-TDP-43 (1:100, #3448S, CST), rabbit anti-NF (1:500, ab8135, Abcam), rabbit anti-Iba1 (1:500, ab178846, Abcam), rabbit anti-CD45 (1:500, ab10558, Abcam), rabbit anti-CD206 (1:400, #24595, CST), rabbit anti-Vimentin (1:200, ET1610-39, HUABIO), rabbit anti-S100β (1:200, OB-PRB050-01, Oasis Biofarm), rabbit anti-ALDH1L1 (1:500, OB-PRB001-01, Oasis Biofarm), rabbit anti-Ki67 (1:400, #9129S, CST), rat anti-EAAT2 (1:200, OB-PRT026, Oasis Biofarm), and rabbit anti-Cleaved-caspase-3 (1:400, #9661S, CST). Images were acquired with fluorescence microscope (VS200, Olympus) or Nikon confocal microscope (Nikon A1) or Nikon single-photon and two-photon integrated confocal microscope (Nikon AX R MP), and analyzed by the ImageJ software.
Immunohistochemistry
Brain tissue sections were fixed for 30 min and incubated in 3% hydrogen peroxide to block endogenous peroxidase activity, and then repaired with sodium citrate antigen repair solution at 90℃ for 30 min. Next, the brain tissue sections were permeabilized and blocked with 5% BSA (#V900933, Sigma-Aldrich) and 0.3% Triton X-100 (#T8200, Solarbio) at room temperature for 1.5 h. The sections were subsequently incubated overnight at 4℃ with rabbit anti-TDP-43 (1:2, 000, 10782-2-AP, Proteintech) primary antibody. The following day, after washing with PBS three times, the sections were incubated with universal secondary antibodies labeled with HRP (PV-9000, 2-step plus® Poly-HRP Anti Mouse/Rabbit IgG Detection System, ZSGB-Bio) for 1 h at room temperature. Immunoreactivity was visualized using DAB reagents (ZSGB-Bio). Finally, the nuclei were stained with hematoxylin staining solution (C0107, Beyotime), and images were captured using an Olympus VS200 microscope.
Nissl staining
Brain tissue sections were incubated with 0.1% cresyl violet solution (#C5042-10G, Sigma) at room temperature for 10 min, followed by washing with distilled water. The sections were then dehydrated in 75%, 95% and anhydrous ethanol, cleared with xylene twice (the first time for 5 min and the second time for 10 min), and finally covered with neutral resin. Images were captured using a microscope (Olympus, VS200), and the density of Nissl bodies in the cerebral motor cortex was quantified using ImageJ software.
Western blotting
Brain tissue or cultured cells were lysed with ice-cold RIPA solution (P0013B, Beyotime) for 30 min, and tissue lysates or cell lysates were centrifuged 12, 000 rpm for 30 min at 4℃. Then the extracted protein was mixed with the loading buffer and boiled at 100℃ for 15 min. Subsequently, the samples were separated by 8-10% SDS-PAGE gel electrophoresis and transferred to western blotting membrane (Millipore, ISEQ00010). The blotted membranes were blocked in 5% skim milk at room temperature for 1.5 h and incubated with primary antibodies overnight at 4℃. The primary antibodies included rabbit anti-YAP (1:1, 000, #14074, CST), rabbit anti-p-YAP (1:1, 000, #130084, CST), mouse anti-GFAP (1:1, 000, MAB360, Millipore), rat anti-EAAT2 (1:1, 000, OB-PRT026, Oasis Biofarm), rabbit anti-EAAT2 (1:1, 000, 22515-1-AP, Proteintech, for Figure 6I and Figure 7M), rabbit anti-β-catenin (1:1, 000, #8480S, CST), rabbit anti-β-catenin (1:100, ET1601-5, HUABIO), rabbit anti-TNF-α (1:400, GB11188-100, Servicebio), rabbit anti-Bax (1:5, 000, ET1603-34, HUABIO), rabbit anti-Bcl-2 (1:2, 000, ab182858, Abcam), rabbit anti-Bcl-2 (1:5, 000, ET1702-53, HUABIO). Rabbit anti-GAPDH (1:5, 000, ET1601-4, HUABIO) or mouse anti-β-actin (1:10, 000, EM21002, HUABIO) or rabbit anti-Lamin B1 (1: 1, 000, ab16048, Abcam) were served as loading controls. After washing in 1 x tris buffered saline with Tween 20 (TBST) for three times and incubating with appropriate secondary antibody (Goat anti-rabbit HRP, 1: 10, 000, #FDR007, Fdbio; Anti-mouse IgG HRP, 1: 10, 000, #A9044, Sigma; Goat anti-rat IgG, HRP, 1:5, 000, #RT-HRP, Oasis Biofarm) at room temperature for 1 h. The blots were then washed three times with 1 x TBST, and the protein blots were detected by Fdbio-Dura ECL kit (#FD8020, Fdbio) on the electrochemiluminescence imaging analysis system (GelViev 6000Plus, BLT). The gray values were subsequently analyzed using ImageJ software.
Cytosol-nuclei fractionation
We used the Minte™ Cytosolic and Nuclear Extraction kit for Frozen/Fresh Tissues (#NT-032, Invent Biotechnologies) to dissociate cytoplasmic and nuclear proteins from the cortex according to manufacturer's instructions. The fractions were analyzed by SDS-PAGE and Western blotting with specific antibodies.
RNA extraction and quantitative real-time PCR (qPCR)
Total RNA was extracted from the cerebral cortex, Neuro-2a (N2a) cells, primary cultured astrocytes and neurons using RNA-easy Isolation Reagent (R701-01, Vazyme) according to the manufacturer's instructions. The expression levels of EAAT2, β-catenin, TNF-α, IL-1β, IL-6, CXCL10, CD206, Wnt1, Wnt2, Wnt3a, Wnt4, Wnt5a and Wnt7a mRNA were quantified using ChamQ Universal SYBR qPCR Master Mix (Q711-02, Vazyme) on a CFX96 Touch Real-Time PCR Detection System (Bio-Rad). β-actin was used as endogenous control. Δ CT = CT gene - CT reference was used to refer to the relative mRNA expression, and 2-ΔΔ CT method was used to calculate the change of gene expression multiple. The primers used in this study were synthesized by Tsingke Biotechnology Company and expressed as followed:
EAAT2 primer: forward, 5'-CTGATGTGGTCATGTTGATAGCC-3', reverse, 5'-AACTGGAGATGATAAGAGGGAGG-3'.
β-catenin primer: forward, 5'-ATGGAGCCGGACAGAAAAGC-3', reverse, 5'-TGGGAGGTGTCAACATCTTCTT-3'.
TNF-α primer: forward, 5'-TCTCATGCACCACCATCAAGGACT-3', reverse, 5'- ACCACTCTCCCTTTGCAGAACTCA -3'.
IL-1β primer: forward, 5'-AAGGGCTGCTTCCAAACCTTTGAC-3', reverse, 5'-ATACTGCCTGCCTGAAGCTCTTGT-3'.
IL-6 primer: forward, 5'-ATCCAGTTGCCTTCTTGGGACTGA-3', reverse, 5'-TAAGCCTCCGACTTGTGAAGTGGT-3'.
CXCL10 primer: forward, 5'-GGAAATCGTGCGTGACATTA-3', reverse, 5'-AGGAAGGAAGGCTGGAAGAG-3'.
CD206 primer: forward, 5'-TCAGCTATTGGACGCGAGGCA-3', reverse, 5'-TCCGGGTTGCAAGTTGCCGT-3'.
Wnt1 primer: forward, 5'-GGTTTCTACTACGTTGCTACTGG-3', reverse, 5'- GGAATCCGTCAACAGGTTCGT-3'.
Wnt2 primer: forward, 5'-CTCGGTGGAATCTGGCTCTG-3', reverse, 5'- CACATTGTCACACATCACCCT-3'.
Wnt3a primer: forward, 5'-CAGGAACTACGTGGAGATCATGC-3', reverse, 5'- CGTGTCACTGCGAAAGCTACT-3'.
Wnt4 primer: forward, 5'-GTCAGGATGCTCGGACAACAT-3', reverse, 5'- CACGTCTTTACCTCGCAGGA-3'.
Wnt5a primer: forward, 5'-CTGCGGAGACAACATCGACTA-3', reverse, 5'- CGTGGATTCGTTCCCTTTCTCTA-3'.
Wnt7a primer: forward, 5'-TGAACTTACACAATAACGAGGCG-3', reverse, 5'- GTGGTCCAGCACGTCTTAGT-3'.
β-actin primer: forward, 5'-GGCACCACACCTTCTACAATG-3', reverse, 5'-GGGGTGTTGAAGGTCTCAAAC-3'.
Cell lines and transfection
Mouse Neuro-2a (N2a) cells were obtained from Procell (CL-0168) and cultured in DMEM (C11995500BT, Gibco), containing 10% fetal bovine serum (FBS, S-FBS-MX-015, SERANA) and 1% penicillin/streptomycin (PS, C0222, Beyotime). GFP-GA5 or GFP-GA100 plasmids (kindly provided by Prof. Chengyong Shen, Zhejiang University) were separately transfected into N2a cells using Lipofectamine® 3000 Transfection Kit (L3000-015, Invitrogen) according to the manufacturer's protocol. Cells and conditioned medium (CM) were collected after transfection for subsequent experiments.
Primary neuron culture
For primary neuron culture, we isolated cells from the cortex and hippocampus of wild- type (WT) mouse embryos at days 14-16 of gestation. Cells were digested with 0.25% trypsin (C0201-500 ml, Beyotime) for 12 min at 37°C, resuspended with DMEM containing 10% FBS and 1% PS, and plated on poly-L-lysine coated six-well plates or coverslips. After 2-4 h, the medium was replaced with neuronal basal medium [Neurobasal medium (21103049, Gibco)/2% B27 (17504044, Gibco)/1% GlutaMAX (35050-061, Gibco)/1% PS]. Neurons were treated with 2 µM cytosine arabinoside at 2 days in vitro (DIV2) for 24 h to suppress glial proliferation. The medium was changed once every 2 days. To mimic the pathology of ALS in vitro, neurons at DIV3 were transduced with AAV-GFP-GA5 virus or AAV-GFP-GA100 virus at multiplicities of infection (MOIs) of 2×10⁵ or 4×10⁵ vg/cell, and the GFP expression was confirmed 48 h post-transduction [53]. CM from the transduced neurons was collected every 2 days.
Cell viability assay
Cell viability was assayed by Cell Counting Kit-8 (CCK-8, FD3788, Fdbio). Briefly, primary cultured neurons were inoculated into 96-well plates coated with poly-L-lysine. The cell density in each well was approximately 10, 000-50, 000 cells. Primary cultured neurons at DIV3 were transduced with AAV-GFP-GA100 virus. 48 h post-transduction, GFP expression was confirmed, and neurons were treated with CM from YAP+/+ or YAP-/- astrocytes for 24 h. Subsequently, 10 µl of CCK-8 solution was added to each well and incubated at 37°C for 2 h. The absorbance of each well was assessed at 450 nm using a microplate reader (Multiskan FC, Thermo Scientific).
Astrocyte culture
YAP+/+ and YAP-/- astrocyte cultures were derived from the cortex and hippocampus of YAPf/f and YAPGFAP-CKO mice, respectively, as described previously [54]. Briefly, the cortex and hippocampus of P1-P3 mice were minced, digested with 0.25% trypsin (C0201-500 ml, Beyotime) for 12 min at 37℃, and mechanically disrupted to separate into single-cell suspensions. Cells were then cultured in DMEM containing 10% FBS and 1% PS in a humidified atmosphere with 5% CO2 at 37℃. After 6-10 days of culture, microglia and oligodendrocytes were removed by shaking at 250 rpm for 4-6 h. Then, the astrocytes were plated on poly-L-lysine coated dishes or coverslips for subsequent experiments.
Collecting astrocyte conditioned medium (ACM)
Astrocyte cultures were prepared from YAPf/f and YAPGFAP-CKO mice. Cultures were maintained at 37°C and 5% CO2 in DMEM containing 10% FBS and 1% PS. Cultures were passaged to reach confluence and contained >95% glial fibrillary acidic protein (GFAP)-positive astrocytes. ACM was collected every 2 days, filtered and stored at -80℃ for treatment of N2a cells. For ACM used in primary cultures for neuronal survival experiments, neuronal basal medium was substituted for the previous medium and ACM was collected every 2 days, filtered, and stored at -80°C [55].
Flow cytometry analysis
The apoptosis rate of transfected N2a cells was analyzed by the Annexin V-APC/PI apoptosis kit (AP107-30-kit, Multi Sciences). GFP-GA5 or GFP-GA100 plasmids were transfected into N2a cells using the Lipofectamine® 3000 Transfection Kit (L3000-015, Invitrogen), and then conditioned medium from YAP+/+ or YAP-/- astrocytes was used to culture the transfected N2a cells. Cells were collected and washed, resuspended with 500 µl 1x binding buffer, and then incubated with Annexin V-APC and PI staining solution. After incubation in the dark for 5 min at room temperature, the apoptosis rate of cells was detected by flow cytometer (Beckman, CytoFLEX S) and analyzed using FlowJo software.
Glutamate uptake assay
The glutamate clearance capacity was evaluated, and the glutamate uptake assay was determined according to the manufacturer's instructions (#BC1580, Solarbio) and the previous description [54]. In brief, the medium for YAP+/+ and YAP-/- astrocytes was replaced with L-glutamine-free DMEM (Gibco, 10313-021) containing 2 mM glutamate (H-Glu-OH, GA10750, GlpBio). After 30 min of culture, 100 µl of astrocyte culture supernatant was collected and mixed with the reagents provided by the kit. Then, 100 µl of mixed solution was transferred to each well of a 96-well culture plate. The absorbance of the solution was measured at 340 nm using an enzyme-labeled instrument (Infinite M Plex, Tecan). A standard curve of glutamate concentration was constructed using culture medium with known glutamic acid concentration and the glutamate content was calculated according to the manufacturer's instructions.
Injection of drugs and reagents
After the model of C9orf72-poly-GA was established, LDN-212320 (HY-12741, MedChemExpress) was dissolved in 0.9% NaCl containing 1% DMSO, 1% polyethylene glycol 400, and 0.2% Tween 80 and administered continuously for two weeks starting at P75 (40 mg/kg intraperitoneally). XMU-MP-1 (GC11245, GlpBio or GY-100526, MedChemExpress) was dissolved in DMSO and continuously administered for two weeks starting at P75 (1 mg/kg intraperitoneally). Behavioral analysis was subsequently performed.
MSAB (HY-120697, MedChemExpress) was dissolved in DMSO and applied at a concentration of 10 µM for 20 h, unless otherwise specified.
Data analysis and statistics
All data were presented as mean ± SEM from at least three independent experiments. Statistical analysis was performed using GraphPad Prism 8.0.1 and ImageJ. Comparisons between two groups were performed using an unpaired two-tailed Student's t-test; for comparisons between multiple groups, one-way ANOVA or two-way ANOVA with Tukey's multiple comparisons test was used. P<0.05 was considered statistically significant and details of each statistical test were described in the Figure legends.
Results
YAP is upregulated and activated in astrocytes, but not in neurons or microglia, and YAP knockout in astrocytes exacerbates motor deficits in C9orf72-poly-GA mice
To explore the potential role of YAP in ALS, we established the C9orf72-poly-GA pathological ALS mouse model as described previously (ALS mice) [51]. AAV-GFP-GA5 and AAV-GFP-GA100 viruses were injected into the cerebral ventricles of P0 neonatal pups, respectively (Supplementary Figure S1A-B). To determine whether AAV-GFP-GA100-infected mice exhibited ALS-associated phenotypes, behavioral analyses were performed at 3 months of age, followed by histological and biochemical analyses at 104 days (Supplementary Figure S1B). As expected, in the tail-suspension test, ALS mice injected with AAV-GFP-GA100 virus exhibited hind limb clasping and persistent trembling, while control mice injected with AAV-GFP-GA5 virus did not display these phenotypes (Supplementary Figure S1C-D). Subsequently, in the balance beam test, ALS mice exhibited significantly increased hind limb slips compared to control mice, indicating serious deficits in motor balance in mice expressing poly-GA100 (Supplementary Figure S1E). On day 104, it was found that the cerebral cortex of mice injected with AAV-GFP-GA5 and AAV-GFP-GA100 showed abundant GFP fluorescence signals (Supplementary Figure S1F). Compared to control mice, the brains of ALS mice showed abundant GFP-GA100 puncta (arrow) (Supplementary Figure S1F), similar to the neuronal cytoplasmic inclusion bodies observed in the brains of C9orf72-ALS/FTD patients [51]. In contrast, these puncta were scarcely observed in the brains of control mice. In summary, these results suggest that an ALS mouse model of C9orf72-poly-GA has been successfully established.
Next, we examined the expression pattern of YAP in ALS mice. Cortex tissues were collected from control and ALS mice. Western blot analysis showed that YAP expression was significantly increased in the cortex of ALS mice (Figure 1A-B), while the relative ratio of phosphorylated YAP (p-YAP)/YAP was significantly decreased (Figure 1A, C). Additionally, the expression of GFAP (astrocyte marker) was also significantly upregulated in the cortex of ALS mice (Figure 1A, D). These results indicate that YAP is upregulated and activated in the cortex of C9orf72-poly-GA mice. Furthermore, double immunostaining of YAP with several cellular markers, such as GFAP (astrocyte marker), Iba1 (microglia marker) and NeuN (neuronal marker) in the motor cortex of control mice and ALS mice showed that YAP was predominantly expressed in GFAP+ astrocytes (Figure 1E), but not in Iba1+ microglia (Figure 1F) or NeuN+ neurons (Figure 1G), and YAP was primarily located in the nucleus of GFAP+ astrocytes in ALS mice (Figure 1E), indicating activation of YAP signaling in astrocytes. Taken together, these results suggest that YAP is upregulated and activated in motor cortex astrocytes of C9orf72-poly-GA mice.
YAP is upregulated and activated in astrocytes, but not in neurons or microglia, and YAP knockout in astrocytes exacerbates motor deficits in C9orf72-poly-GA mice. (A) Western blot analysis of YAP, p-YAP/YAP and GFAP expression in the cortex of control mice and C9orf72-poly-GA mice (ALS mice). (B-D) Quantitative analysis of the relative YAP (B), p-YAP/YAP (C) and GFAP (D) expression levels as shown in (A) (n = 4, normalized to control mice, Student's t-test). (E-G) Representative staining images of YAP (green) and GFAP (red) (E), YAP (green) and Iba1 (red) (F), and YAP (green) and NeuN (red) (G) in the motor cortex of control mice and ALS mice. (H) Representative images of YAPf/f and YAPGFAP-CKO control mice, YAPf/f ALS and YAPGFAP-CKO ALS mice in tail-suspension test. The red arrows indicate the hind limbs of mice. (I) Quantitative analysis of hind limb clasping time of YAPf/f and YAPGFAP-CKO control mice, YAPf/f ALS and YAPGFAP-CKO ALS mice within 3 min in tail-suspension test. (J) Quantitative analysis of time keeping on the edges of cages of YAPf/f and YAPGFAP-CKO control mice, YAPf/f ALS and YAPGFAP-CKO ALS mice. (K) Quantitative analysis of the numbers of hind limb foot slips in the balance beam test in YAPf/f and YAPGFAP-CKO control mice, YAPf/f ALS and YAPGFAP-CKO ALS mice. (L) Quantitative analysis of the latency to fall from the accelerated rotating rod of YAPf/f and YAPGFAP-CKO control mice, YAPf/f ALS and YAPGFAP-CKO ALS mice. (M) Representative images of YAPf/f and YAPGFAP-CKO control mice, YAPf/f ALS and YAPGFAP-CKO ALS mice in footprint test. Fore paws (red), hind paws (blue). Black squares indicate localization of hind paws. The red line indicates the distance between the hind paws. (N-P) Quantitative analysis of back stride length (right) (N), back stride length (left) (O), and back front distance (P) of YAPf/f and YAPGFAP-CKO control mice, YAPf/f ALS and YAPGFAP-CKO ALS mice in footprint test. For behavioral tests, YAPf/f control mice, n = 6 each group; YAPGFAP-CKO control mice, n = 8 each group; YAPf/f ALS and YAPGFAP-CKO ALS mice, n = 13 each group. Scale bars, 50 µm. Data were presented as mean ± SEM. Two-way ANOVA with Tukey's multiple comparisons test unless otherwise indicated, n.s., not significant (p > 0.05), *p < 0.05, **p < 0.01.
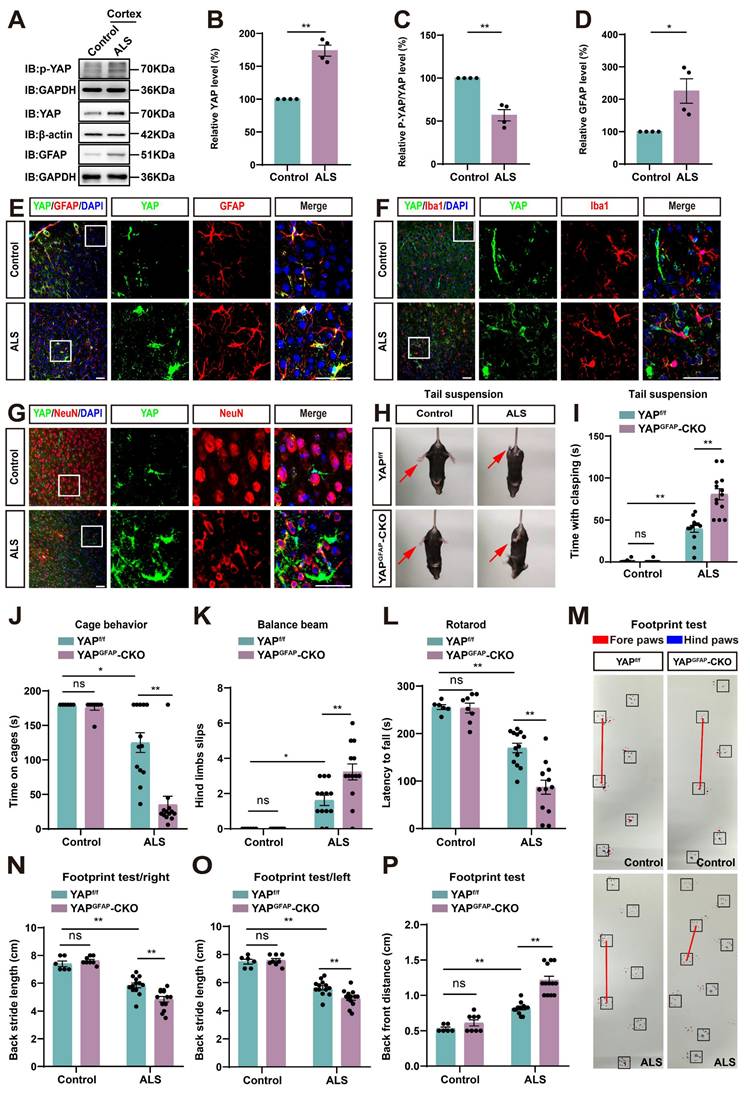
To further study the role of astrocytic YAP in ALS, YAPGFAP-CKO mice with conditional knockout of YAP in astrocytes were generated (Supplementary Figure S2A). In YAPGFAP-CKO mice, YAP expression was significantly decreased in brain regions, including the cortex, hippocampus, cerebellum, and spinal cord (Supplementary Figure S2B-C). Immunostaining also showed that YAP signal was not detected in GFAP+ astrocytes in the cerebral cortex of YAPGFAP-CKO mice and in primary cultured astrocytes (Supplementary Figure S2D-E). However, no significant difference in body size was observed between YAPf/f and YAPGFAP-CKO mice (Supplementary Figure S2F). Behavioral tests, including the tail-suspension test (Supplementary Figure S2G-H), cage behavior test (Supplementary Figure S2I), balance beam test (Supplementary Figure S2J), and rotarod test (Supplementary Figure S2K), showed that YAP deficiency in astrocytes had no significant effect on motor functions of mice. These results suggest that the knockout of astrocytic YAP does not affect the motor function of the mice.
Next, we examined the potential phenotypic differences between YAPf/f C9orf72-poly-GA mice (YAPf/f ALS mice) and YAPGFAP-CKO C9orf72-poly-GA mice (YAPGFAP-CKO ALS mice). The tail-suspension test showed that compared to YAPf/f ALS mice, YAPGFAP-CKO ALS mice exhibited more pronounced hind limb clasping and continuous trembling (Figure 1H-I). Furthermore, the cage behavior test showed that YAPGFAP-CKO ALS mice were more likely to fall within 3 min while walking along the edge of the cage (Figure 1J), indicating more severe deficits in motor balance in YAPGFAP-CKO ALS mice. To further confirm these results, we used three widely utilized assays to test motor function: the balance beam test, rotarod test, and footprint test. The results showed that the hind limb slips on the balance beam were significantly increased in YAPGFAP-CKO ALS mice (Figure 1K), and the latency to fall off the rotating rod was significantly decreased in YAPGFAP-CKO ALS mice (Figure 1L), and the footprint test showed more pronounced gait abnormalities in YAPGFAP-CKO ALS mice (Figure 1M-P). Taken together, these results suggest that YAP knockout in astrocytes exacerbates motor deficits in C9orf72-poly-GA mice.
YAP knockout in astrocytes exacerbates neurodegeneration and pathological TDP-43 cytoplasmic translocation in C9orf72-poly-GA mice
Since motor behavioral deficits in ALS result from the neurodegeneration of motor neurons, we next examined the loss of neurons in the motor cortex of YAPGFAP-CKO ALS mice. Notably, both Nissl staining and immunostaining of NeuN showed a significant reduction in the density of Nissl bodies (Figure 2A-B) and NeuN+ neurons (Figure 2C-D) in the motor cortex of YAPGFAP-CKO ALS mice, compared to YAPf/f ALS mice. In addition, double immunostaining for NeuN and Cleaved-caspase-3 (c-caspase-3) further demonstrated a significant increase in neuronal apoptosis in the motor cortex of YAPGFAP-CKO ALS mice (Figure 2E-F).
YAP knockout in astrocytes exacerbates neurodegeneration and pathological TDP-43 cytoplasmic translocation in C9orf72-poly-GA mice. (A, C) Representative staining images of Nissl staining (A) and NeuN (red) (C) in the motor cortex of YAPf/f ALS mice and YAPGFAP-CKO ALS mice. (B, D) Quantitative analysis of the density of Nissl bodies (B) and NeuN+ cells (D) as shown in (A) and (C), respectively (n = 6 each group). (E) Representative staining images of NeuN (red) and c-caspase-3 (far-red) in the motor cortex of YAPf/f ALS mice and YAPGFAP-CKO ALS mice. (F) Quantitative analysis of the density of c-caspase-3+ in NeuN+ neurons as shown in (E) (n = 6 each group). (G-H) Representative staining images of NF (red) (G) and TDP-43 (far-red) (H) in the motor cortex of YAPf/f ALS mice and YAPGFAP-CKO ALS mice. White arrows indicate cytoplasmic TDP-43 expression. (I) Quantitative analysis of the relative intensity of NF as shown in (G) (n = 6 each group). (J) Quantitative analysis of the density of cytoplasmic TDP-43 in NeuN+ neurons as shown in (H) (n = 6 each group). (K) Immunohistochemistry of TDP-43 aggregation in the motor cortex of YAPf/f ALS mice and YAPGFAP-CKO ALS mice. Red arrows indicate the cytoplasmic inclusion bodies of TDP-43. Scale bars, 50 µm. Data were presented as mean ± SEM. Student's t-test, **p < 0.01.
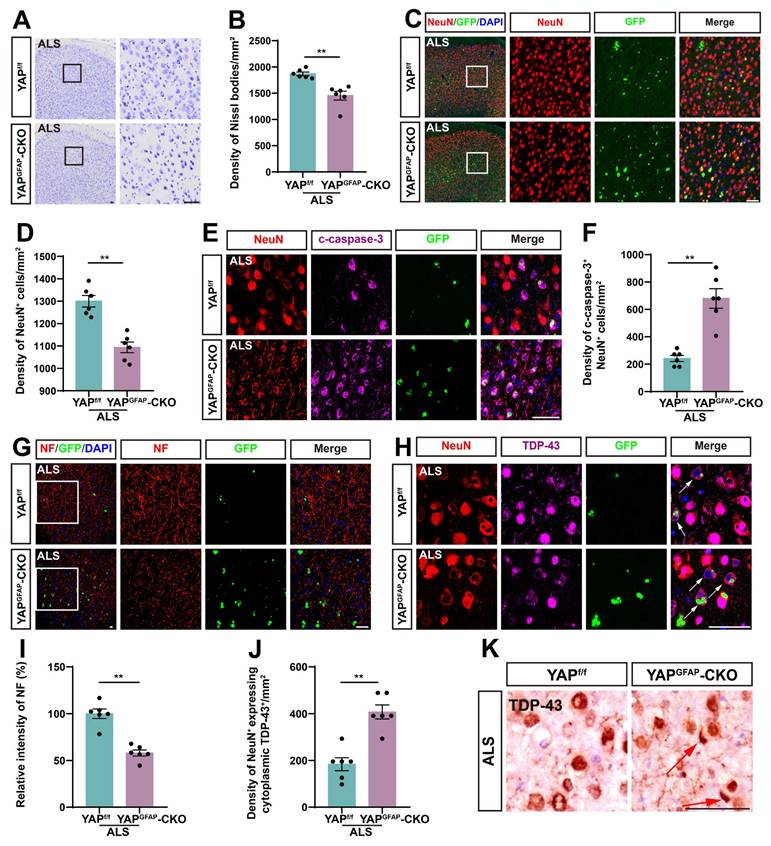
Previous studies have shown that neurofilaments, as the main intermediate filament system in mature neurons, are essential for neuronal function, and abnormalities in neurofilament can directly lead to motor neuron dysfunction [56-58]. Immunostaining for NF (neurofilament heavy polypeptide) showed that the intensity of NF was significantly lower in YAPGFAP-CKO ALS mice (Figure 2G, I). Collectively, these results suggest that knockout of astrocytic YAP exacerbates neurodegeneration in C9orf72-poly-GA mice. TDP-43 cytoplasmic inclusions are major neuropathological feature of C9orf72-ALS/FTD [51, 59-61]. Therefore, we examined TDP-43 translocation in the motor cortex of C9orf72-poly-GA mice. As expected, double immunostaining showed a significant increase in cytoplasmic TDP-43 aggregation in the neurons of YAPGFAP-CKO ALS mice (Figure 2H, J-K). Taken together, these results suggest that YAP knockout in astrocytes exacerbates neurodegeneration and pathological TDP-43 cytoplasmic translocation in C9orf72-poly-GA mice.
YAP knockout in astrocytes exacerbates the inflammatory infiltration in the motor cortex of C9orf72-poly-GA mice
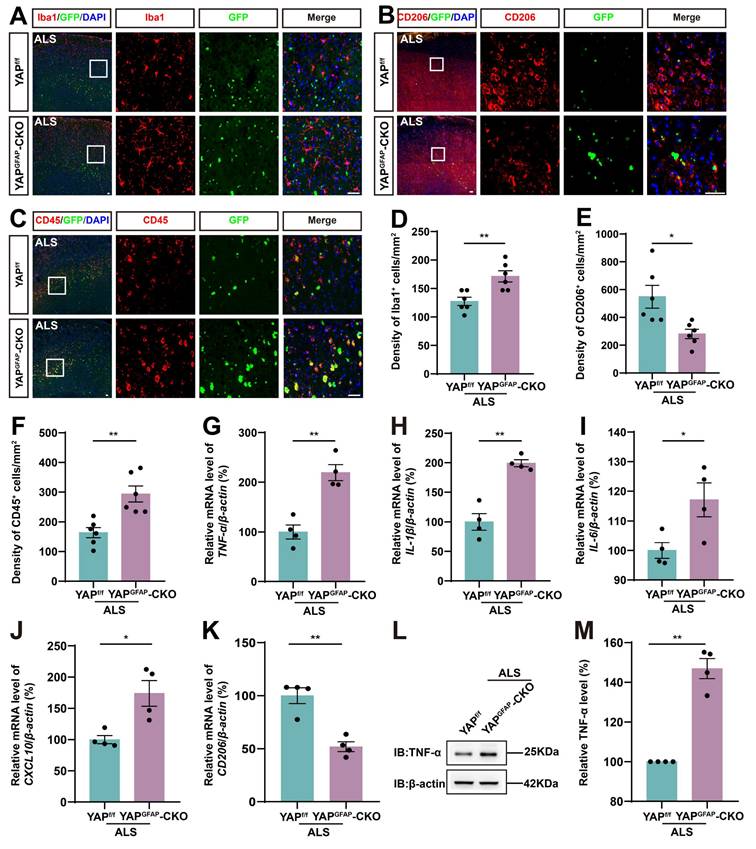
Neuroinflammation is a prominent pathological manifestation of ALS, characterized by microglia activation, astrocyte proliferation, and monocyte and T-cell infiltration [62]. Therefore, we next examined the inflammatory infiltration in YAPGFAP-CKO ALS mice. The results showed that Iba1+ microglia were activated, and the density of Iba1+ microglia was significantly increased in the motor cortex of YAPGFAP-CKO ALS mice (Figure 3A, D). M2 macrophages produce high levels of anti-inflammatory cytokines and neurotrophic factors [63]. We found the density of CD206+ (an M2 macrophage marker) cells was significantly decreased in YAPGFAP-CKO ALS mice (Figure 3B, E). Similarly, immunostaining of CD45+ (a marker of common leukocyte antigen) showed that a significant increase in the density of CD45+ cells in the motor cortex of YAPGFAP-CKO ALS mice (Figure 3C, F). Additionally, we examined the expression of inflammatory factors in the cortex of these mice. The qPCR results showed that pro-inflammatory markers, including TNF-α (Figure 3G), IL-1β (Figure 3H), IL-6 (Figure 3I), and CXCL10 (Figure 3J) mRNA, were significantly upregulated in the cortex of YAPGFAP-CKO ALS mice, whereas the mRNA of anti-inflammatory marker CD206 (Figure 3K) was significantly downregulated. Western blot analysis further confirmed that the expression of TNF-α was upregulated in the cortex of YAPGFAP-CKO ALS mice (Figure 3L-M). Taken together, these results suggest that the inflammatory infiltration is exacerbated in the motor cortex of YAPGFAP-CKO ALS mice.
YAP knockout in astrocytes exacerbates the inflammatory infiltration in the motor cortex of C9orf72-poly-GA mice. (A-C) Representative staining images of Iba1(red) (A), CD206 (red) (B)and CD45 (red) (C) in the motor cortex of YAPf/f ALS mice and YAPGFAP-CKO ALS mice. (D-F) Quantitative analysis of the density of Iba1+ cells (D), CD206+ cells (E) and CD45+ cells (F) as shown in (A), (B) and (C), respectively (n = 6 each group). (G-K) qPCR analysis of the relative mRNA levels of TNF-α (G), IL-1β (H), IL-6 (I), CXCL10 (J) and CD206 (K) in the cortex of YAPf/f ALS mice and YAPGFAP-CKO ALS mice (n = 4 each group). (L) Western blot analysis of TNF-α expression in the cortex of YAPf/f ALS mice and YAPGFAP-CKO ALS mice. (M) Quantitative analysis of the relative TNF-α expression level as shown in (L) (n = 4 each group, normalized to YAPf/f ALS mice). Scale bars, 50 µm. Data were presented as mean ± SEM. Student's t-test, *p < 0.05, **p < 0.01.
The density and proliferation of astrocytes are reduced in the motor cortex of YAPGFAP-CKO ALS mice
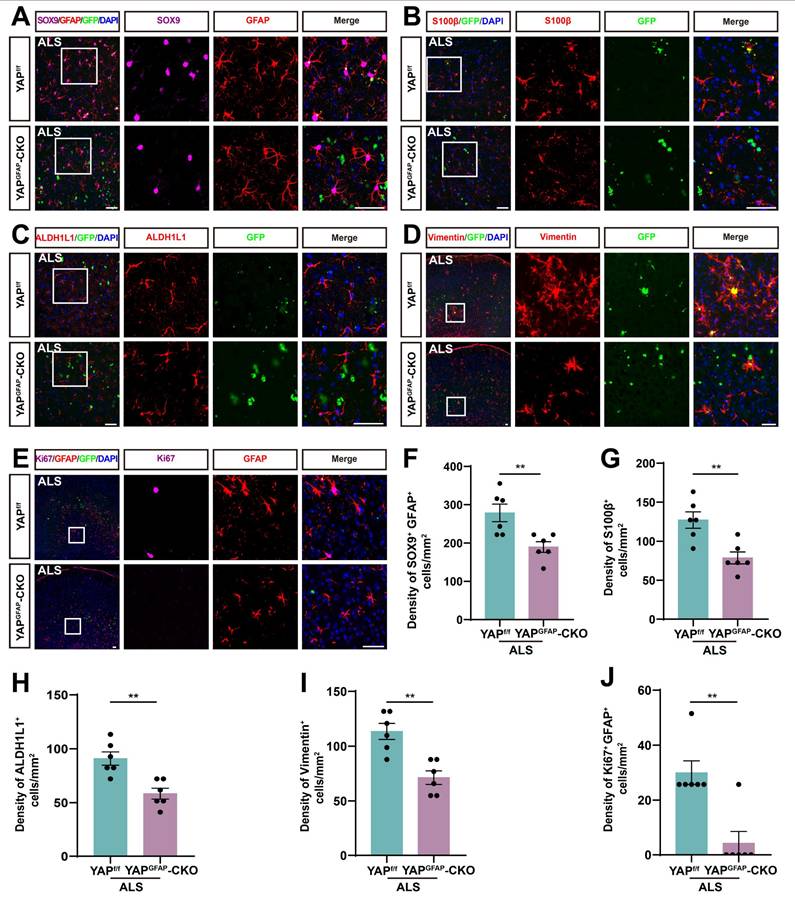
Studies have shown that reactive astrocytes are a key feature of ALS [64, 65]. To investigate astrocyte changes in the motor cortex of YAPGFAP-CKO ALS mice, we performed immunostaining for astrocyte markers. SOX9, a transcription factor known to support the development of glial cells in the peripheral nervous system, has been reported to promote differentiation into astrocytes [66, 67]. Therefore, we performed double immunostaining for SOX9 and GFAP in the motor cortex of YAPf/f ALS mice and YAPGFAP-CKO ALS mice. The results showed that the density of SOX9+ astrocytes was significantly reduced in the motor cortex of YAPGFAP-CKO ALS mice (Figure 4A, F). To further confirm these results, we stained for other astrocyte markers, including S100β, ALDH1L1 and Vimentin, and found that the density of S100β+ (Figure 4B, G), ALDH1L1+ (Figure 4C, H) and Vimentin+ (Figure 4D, I) astrocytes was significantly decreased in the motor cortex of YAPGFAP-CKO ALS mice, suggesting that the density of astrocytes was reduced in the motor cortex of YAPGFAP-CKO ALS mice. In addition, double immunostaining for Ki67 (a cell proliferation marker) and GFAP further showed that astrocytic proliferation was significantly reduced in the motor cortex of YAPGFAP-CKO ALS mice (Figure 4E, J). Taken together, these results suggest that YAP knockout in astrocytes reduces the density and proliferation of astrocytes in the motor cortex of YAPGFAP-CKO ALS mice.
The density and proliferation of astrocytes are reduced in the motor cortex of YAPGFAP-CKO ALS mice. (A) Representative staining images of SOX9 (far-red) and GFAP (red) in the motor cortex of YAPf/f ALS mice and YAPGFAP-CKO ALS mice. (B-D) Representative staining images of S100β (red) (B), ALDH1L1 (red) (C)and Vimentin (red) (D) in the motor cortex of YAPf/f ALS mice and YAPGFAP-CKO ALS mice. (E) Representative staining images of Ki67 (far-red) and GFAP (red) in the motor cortex of YAPf/f ALS mice and YAPGFAP-CKO ALS mice. (F) Quantitative analysis of the density of SOX9 + and GFAP+ double positive cells as shown in (A) (n = 6 each group). (G-I) Quantitative analysis of the density of S100β+ cells (G), ALDH1L1+ cells (H) and Vimentin+ cells (I) as shown in (B), (C) and (D), respectively (n = 6 each group). (J) Quantitative analysis of Ki67+ and GFAP+ cells as shown in (E) (n = 6 each group). Scale bars, 50 µm. Data were presented as mean ± SEM. Student's t-test, **p < 0.01.
Astrocytic EAAT2 expression is downregulated in the motor cortex of YAPGFAP-CKO ALS mice through downregulating β-catenin signaling
To investigate the downstream effector mechanisms through which astrocytic YAP regulates neuronal loss and motor deficits in C9orf72-poly-GA mice, we analyzed single-nucleus transcriptomic (snRNA-seq) data from motor cortex samples of C9orf72-ALS/FTD patients with low and high YAP expression [68], along with RNA sequencing (RNA-seq) results from cultured YAP+/+ and YAP-/- (YAP knockout) astrocytes [54]. We then identified their overlapping differentially expressed genes, including both upregulated and downregulated genes (Figure 5A). The results showed that there were 2 upregulated genes and 102 downregulated genes. Since YAP is believed to be a transcriptional activator, it is unlikely to directly suppress gene expression. Therefore, we next performed Gene Ontology (GO) enrichment analysis on these 102 downregulated genes and found that the most significantly altered pathway was related to neurotransmitter uptake (Figure 5B). The M-versus-A plot (MA-plot) for YAP+/+ and YAP-/- astrocytes showed that genes related to neurotransmitter uptake, such as Slc6a11, Slc1a2 (EAAT2) and Slc1a3 (EAAT1), were downregulated (Figure 5C). Given that the loss of EAAT2 protein and function significantly contributes to excitotoxicity in ALS [8], and that EAAT2 is responsible for clearing approximately 90% of glutamate from the synaptic cleft and plays a key role in regulating excitotoxicity and neuronal death [69, 70], we further examined EAAT2 expression in the motor cortex of C9orf72-ALS/FTD patients. The results showed that EAAT2 expression was significantly downregulated in the cortex of patients with low YAP expression compared to those with high YAP expression (Figure 5D). Recent studies have shown that YAP signaling in astrocytes promotes EAAT2 expression by regulating the transcriptional activity of β-catenin [54]. To further explore whether the deletion of astrocytic YAP in the motor cortex of C9orf72-poly-GA mice also reduced the expression of EAAT2 through decreased transcriptional activity of β-catenin, we conducted qPCR analysis. The results confirmed a reduction in the levels of EAAT2 and β-catenin mRNA in YAPGFAP-CKO ALS mice (Figure 5E-F). Western blot analysis further showed that EAAT2 and β-catenin protein levels were significantly downregulated in YAPGFAP-CKO ALS mice (Figure 5G-I). In addition, cytosol-nuclei fractionation experiments showed that the nucleus/cytoplasm ratio of β-catenin was significantly increased in the cortex of YAPf/f ALS mice, suggesting the Wnt/β-catenin pathway was activated in YAPf/f ALS mice. However, activation of the Wnt/β-catenin pathway was significantly decreased in the cortex of YAPGFAP-CKO ALS mice (Figure 5J-K). Double immunostaining further confirmed a significant reduction of EAAT2 expression in astrocytes of the motor cortex of YAPGFAP-CKO ALS mice (Figure 5L-M). Taken together, these results suggest that astrocytic EAAT2 expression is downregulated in the motor cortex of YAPGFAP-CKO ALS mice through downregulating the β-catenin signaling.
Astrocytic EAAT2 expression is downregulated in the motor cortex of YAPGFAP-CKO ALS mice through downregulating β-catenin signaling. (A) Overlapping differentially expressed genes between the C9orf72-ALS/FTD patient (snRNA-seq) database and RNA-seq from YAP-/- and YAP+/+ astrocytes, including upregulated and downregulated genes. (B) Gene ontology (GO) enrichment of overlapping downregulated differentially expressed genes between C9orf72-ALS/FTD patient (snRNA-seq) database and RNA-seq results from YAP+/+ and YAP-/-astrocytes. (C) Differential gene MA-plot of YAP+/+ and YAP-/- astrocytes. (D) Expression of EAAT2 in the motor cortex of C9orf72-ALS/FTD patients with high YAP expression and low YAP expression (high YAP expression group, n = 809; low YAP expression group, n = 776; Student's t-test). (E-F) qPCR analysis of the relative mRNA levels of EAAT2 and β-catenin in the cortex of YAPf/f ALS mice and YAPGFAP-CKO ALS mice (n = 4 each group, Student's t-test). (G) Western blot analysis of EAAT2 and β-catenin expression in the cortex of YAPf/f and YAPGFAP-CKO control mice, YAPf/f ALS and YAPGFAP-CKO ALS mice. (H-I) Quantitative analysis of the relative EAAT2 (H) and β-catenin (I) expression levels as shown in (G) (n = 4 each group, normalized to YAPf/f control mice). (J) Western blot analysis of the cytoplasmic and nuclear expression of β-catenin in the cortex of YAPf/f and YAPGFAP-CKO control mice, YAPf/f ALS and YAPGFAP-CKO ALS mice. (K) Quantitative analysis of the relative nucleus/cytoplasm ratio of β-catenin expression level as shown in (J) (n = 3 each group, normalized to YAPf/f control mice). (L) Representative staining images of EAAT2 (green) and GFAP (red) in the motor cortex of YAPf/f and YAPGFAP-CKO control mice, YAPf/f ALS and YAPGFAP-CKO ALS mice. (M) Quantitative analysis of the density of EAAT2+ and GFAP+ double positive cells as shown in (L) (n = 6 each group). Scale bars, 50 µm. Data were presented as mean ± SEM. Two-way ANOVA with Tukey's multiple comparisons test unless otherwise indicated, *p < 0.05, **p < 0.01.
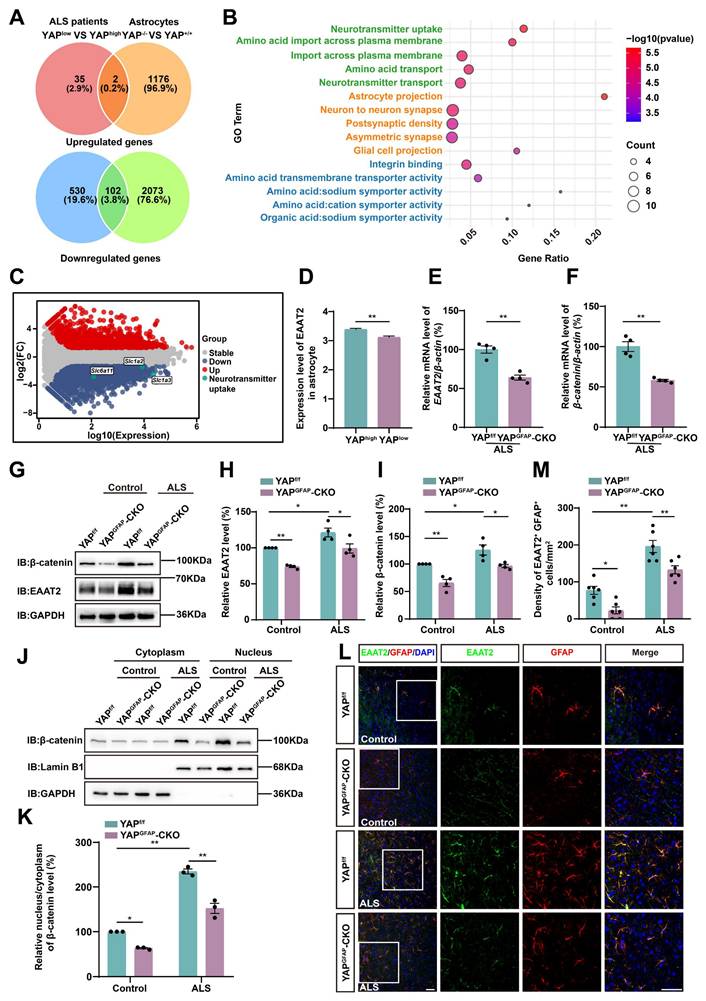
Impaired glutamate uptake in YAP-/- astrocytes exacerbates glutamate excitotoxicity in neuronal cells in ALS in vitro model
We next examined whether impaired glutamate uptake in YAP-deficient astrocytes resulted in neuronal death under the C9orf72-poly-GA model in vitro. GFP-GA5 (Control) and GFP-GA100 plasmids were transfected into N2a cells (mouse neuroblastoma cell line) to mimic the C9orf72-poly-GA model in vitro. We first assessed whether YAP knockout affected glutamate uptake in astrocytes by measuring extracellular glutamate concentration using a glutamate uptake assay (Figure 6A). As expected, the results showed that extracellular glutamate concentration was significantly higher in YAP-/- astrocytes (Figure 6B), suggesting impaired glutamate uptake in YAP-/- astrocytes. We speculated that this was due to reduced EAAT2 expression in YAP-/- astrocytes. To examine whether increased glutamate concentration caused toxic effects on neurons, we collected conditioned medium (CM) from YAP+/+ and YAP-/- astrocytes, described as YAP+/+ ACM and YAP-/- ACM, respectively, and applied it to N2a cells transfected with GFP-GA5 plasmid or GFP-GA100 plasmid (Supplementary Figure S3A). Flow cytometry analysis using Annexin V-APC/PI showed that the percentage of early and late apoptotic cells was significantly increased in N2a-GFP-GA100 cells treated with YAP-/- ACM (Supplementary Figure S3B-C). Furthermore, western blot analysis showed that the ratio of anti-apoptotic protein Bcl-2/pro-apoptotic protein Bax was significantly downregulated in N2a-GFP-GA100 cells treated with YAP-/- ACM (Supplementary Figure S3D-E).
Impaired glutamate uptake in YAP-/- astrocytes exacerbates glutamate excitotoxicity in primary cultured neurons and the YAP/β-catenin/EAAT2 signaling pathway is activated in astrocytes in ALS in vitro model. (A) Flowchart of the glutamate uptake assay. (B) Quantitative analysis of the relative extracellular glutamate levels in YAP+/+ and YAP-/- astrocytes (n = 4 each group, Student's t-test). (C) Representative images of immunostaining of primary cultured neurons transduced with AAV-GFP-GA5 virus or AAV-GFP-GA100 virus. The arrows indicated neurons that expressed GFP-GA5 or GFP-GA100. GFP-GA5 and GFP-GA100, green; MAP2, red; DAPI, blue. (D) Flowchart of conditioned medium collected from YAP+/+ (YAP+/+ ACM) and YAP-/- (YAP-/- ACM) astrocytes and applied to primary cultured neurons transduced with AAV-GFP-GA100 virus for relevant experiments. (E) Neuronal viability was assessed by CCK-8 assay in primary cultured neurons transduced with AAV-GFP-GA100 virus and treated with YAP+/+ ACM or YAP-/- ACM (n = 5 each group, Student's t-test). (F) Western blot analysis of Bcl-2 and Bax expression in primary cultured neurons transduced with AAV-GFP-GA100 virus and treated with YAP+/+ ACM or YAP-/- ACM. (G) Quantitative analysis of the relative Bax/Bcl-2 expression level in primary cultured neurons as shown in (F) (n = 4 each group, normalized to GA100-YAP+/+ACM, Student's t-test). (H) Flowchart of conditioned medium collected from primary cultured neurons transduced with AAV-GFP-GA5 virus (GA5 CM) or AAV-GFP-GA100 virus (GA100 CM) and applied to YAP+/+ and YAP-/- astrocytes for relevant experiments. (I) Western blot analysis of YAP, β-catenin and EAAT2 expression in YAP+/+ and YAP-/- astrocytes treated with GA5 CM or GA100 CM. (J-L) Quantitative analysis of the relative YAP (J, n = 4 each group, normalized to YAP+/+-GA5 CM), β-catenin (K, n = 4 each group, normalized to YAP+/+-GA5 CM) and EAAT2 (L, n = 3 each group, normalized to YAP+/+-GA5 CM) expression levels in astrocytes as shown in (I). (M) Double immunostaining of EAAT2 (green) and GFAP (red) in YAP+/+ and YAP-/- astrocytes treated with GA5 CM or GA100 CM. (N) Quantitative analysis of the relative intensity of EAAT2 as shown in (M) (n = 14 each group). Scale bars, 50 µm. Data were presented as mean ± SEM. Two-way ANOVA with Tukey's multiple comparisons test unless otherwise indicated, n.s., not significant (p > 0.05), *p < 0.05, **p < 0.01.
In addition, we cultured primary neurons from the cortex and hippocampus of WT mice. AAV-GFP-GA5 (control) virus and AAV-GFP-GA100 virus transduced these neurons to mimic the ALS model in vitro (Figure 6C). Similarly, we collected conditioned medium (CM) from YAP+/+ (YAP+/+ ACM) and YAP-/- (YAP-/- ACM) astrocytes to treat primary cultured neurons transduced with AAV-GFP-GA100 virus (Figure 6D). As shown in Figure 6E, we found that the viability of neurons transduced with AAV-GFP-GA100 was significantly decreased after treatment with YAP-/- ACM. Western blot results further confirmed the ratio of pro-apoptotic protein Bax/anti-apoptotic protein Bcl-2 was significantly increased in these neurons (Figure 6F-G). Collectively, these results suggest that impaired glutamate uptake in YAP-/- astrocytes contributes to increased neuronal apoptosis following glutamate excitotoxicity under the pathology of C9orf72-ALS.
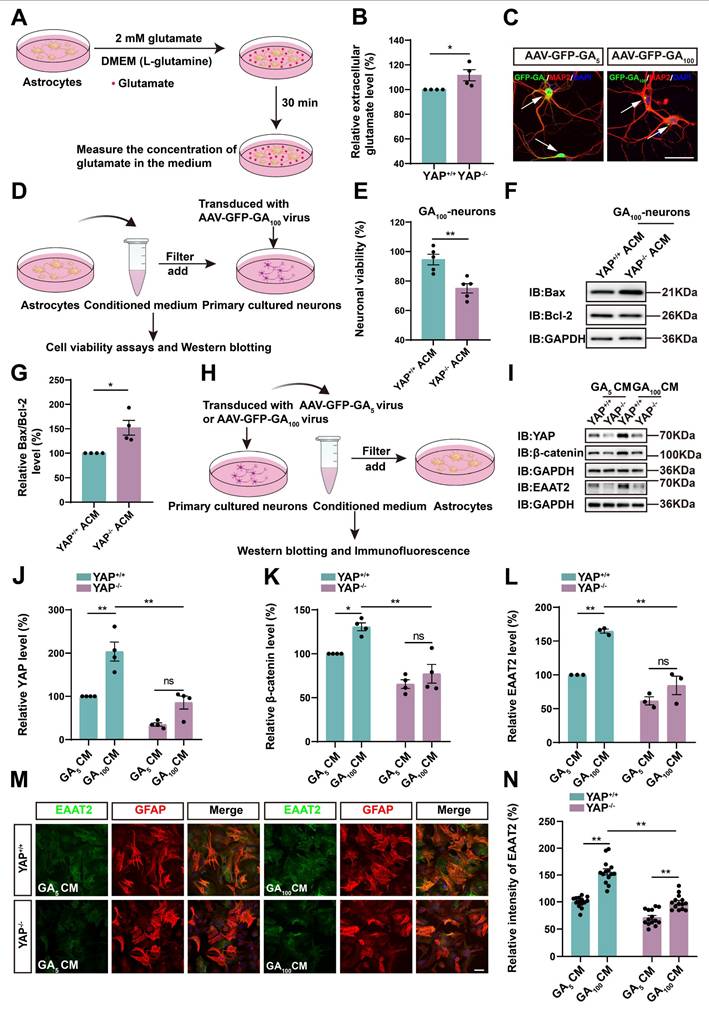
Wnts secreted by degenerating neurons and/or astrocytes activate the YAP/β-catenin/EAAT2 signaling pathway in astrocytes
To further investigate the molecular mechanism of astrocytic YAP in C9orf72-poly-GA ALS, we collected CM from N2a cells transfected with GFP-GA5 plasmid and GFP-GA100 plasmid, described as GA5 CM and GA100 CM, respectively, and treated YAP+/+ and YAP-/- astrocytes with them (Supplementary Figure S3F). Immunostaining showed that YAP was upregulated and translocated to the nucleus in YAP+/+ astrocytes treated with GA100 CM (Supplementary Figure S3G). We speculated that GFP-GA100-transfected N2a cells (degenerating neuronal cells), might secrete specific factors to activate YAP in astrocytes. Furthermore, western blot analysis demonstrated that the expression of β-catenin was upregulated in YAP+/+ astrocytes treated with GA100 CM. In contrast, the expression of β-catenin was significantly downregulated in YAP-/- astrocytes treated with GA100 CM (Supplementary Figure S3H-I). Immunostaining further showed that the intensity of EAAT2 was significantly downregulated in YAP-/- astrocytes treated with GA100 CM (Supplementary Figure S3J-K).
In addition, we collected CM from primary cultured neurons transduced with AAV-GFP-GA5 virus (GA5 CM) and AAV-GFP-GA100 virus (GA100 CM) to treat YAP+/+ and YAP-/- astrocytes (Figure 6H). As shown in Figure 6I-L, the expression of YAP, EAAT2 and β-catenin were upregulated in YAP+/+ astrocytes treated with GA100 CM, however, GA100 CM failed to induce the upregulation of YAP, EAAT2 and β-catenin in YAP-/- astrocytes. Immunostaining further confirmed that the upregulation of EAAT2 induced by GA100 CM was significantly reduced in YAP-/- astrocytes (Figure 6M-N). These results suggest that degenerating neurons may secrete factors that activate YAP/β-catenin/EAAT2 signaling in WT astrocytes.
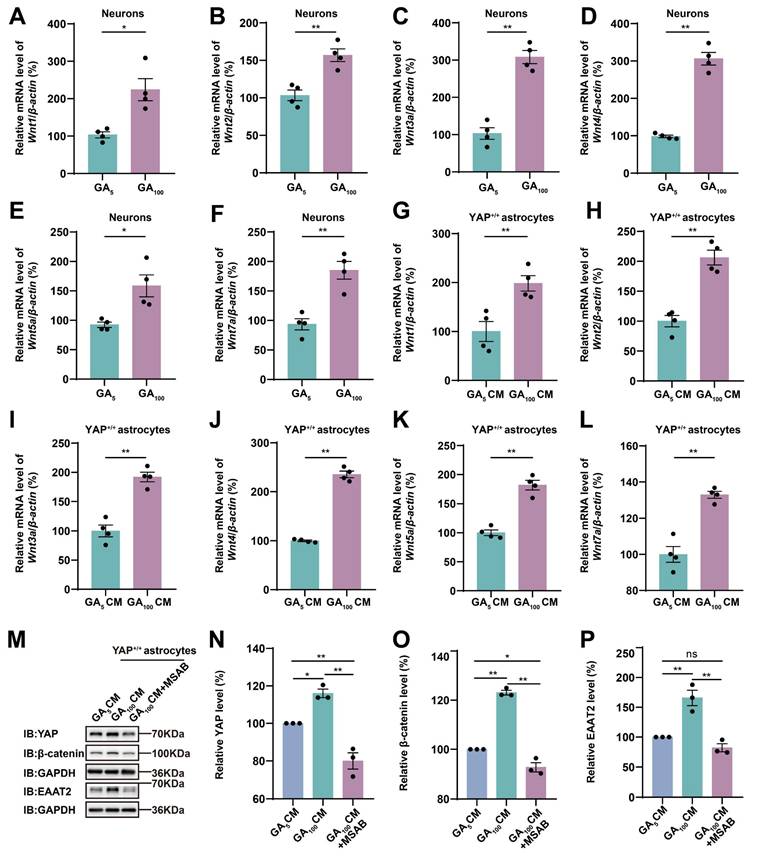
The Wnt signaling pathway plays an important role in the physiological and pathophysiological processes of ALS [38], and interacts with Hippo/YAP signaling [43]. Therefore, we next examined whether N2a cells transfected with GFP-GA100 could secrete Wnt-related ligands. qPCR results showed that Wnt1, Wnt3a, Wnt4, and Wnt7a mRNA were significantly upregulated in N2a cells transfected with GFP-GA100 (Supplementary Figure S3L, N-O, Q), but no significant differences were observed in Wnt2 and Wnt5a mRNA (Supplementary Figure S3M, P). Furthermore, in primary cultured neurons transduced with AAV-GFP-GA5 virus or AAV-GFP-GA100 virus, we found that Wnt1, Wnt2, Wnt3a, Wnt4, Wnt5a, and Wnt7a mRNA were significantly upregulated in the latter (Figure 7A-F). Multiple studies have shown activation of the Wnt/β-catenin pathway in astrocytes under ALS conditions [39-41]. Therefore, we treated YAP+/+ astrocytes with CM collected from primary cultured neurons transduced with AAV-GFP-GA5 virus or AAV-GFP-GA100 virus. qPCR results showed that Wnt1, Wnt2, Wnt3a, Wnt4, Wnt5a, and Wnt7a mRNA were significantly upregulated in YAP+/+ astrocytes treated with GA100 CM (Figure 7G-L). In addition, to determine whether GA100 CM induced upregulation of EAAT2 was through the Wnt/β-catenin pathway, we used MSAB, a potent and selective inhibitor of the Wnt/β-catenin pathway. Interestingly, the upregulation of YAP, β-catenin and EAAT2 expression induced by GA100 CM was significantly blocked by MSAB (Figure 7M-P). These results suggest that degenerating neurons and/or astrocytes secrete Wnts, such as Wnt1 and/or Wnt3a to activate the YAP/β-catenin/EAAT2 signaling pathway in astrocytes. Taken together, these results indicate that impaired glutamate uptake in YAP-/- astrocytes exacerbates glutamate excitotoxicity in neuronal cells in ALS by downregulating the astrocytic Wnt/YAP/β-catenin/EAAT2 signaling pathway in vitro.
Wnts secreted by degenerating neurons transfected with GFP-GA100 and/or astrocytes activate the YAP/β-catenin/EAAT2 signaling pathway in astrocytes. (A-F) qPCR analysis of the relative mRNA levels of Wnt1 (A), Wnt2 (B), Wnt3a (C), Wnt4 (D), Wnt5a (E) and Wnt7a (F) in primary cultured neurons transduced with AAV-GFP-GA5 virus or AAV-GFP-GA100 virus (n = 4 each group, Student's t-test). (G-L) qPCR analysis of the relative mRNA levels of Wnt1 (G), Wnt2 (H), Wnt3a (I), Wnt4 (J), Wnt5a (K) and Wnt7a (L) in YAP+/+ astrocytes treated with GA5 CM or GA100 CM (n = 4 each group, Student's t-test). (M) Western blot analysis detected YAP, β-catenin and EAAT2 expression in YAP+/+ astrocytes 20 h after treatment with GA5 CM, GA100 CM or GA100 CM supplemented with 10 µM MSAB. (N-P) Quantitative analysis of the relative YAP (N), β-catenin (O) and EAAT2 (P) expression levels in YAP+/+ astrocytes as shown in (M) (n = 3 each group, normalized to YAP+/+-GA5 CM). Data were presented as mean ± SEM. One-way ANOVA with Tukey's multiple comparisons test unless otherwise indicated, n.s., not significant (p > 0.05), *p < 0.05, **p < 0.01.
Activation of EAAT2 partially restores motor deficits and neuronal loss in YAPGFAP-CKO ALS mice
EAAT2 can be upregulated through transcriptional or translational activation, and previous studies have also shown that treatment with LDN/OSU-212320 (an agonist of EAAT2) restores EAAT2 protein levels, enhances motor function, and prolongs lifespan in SOD1G93A mice [71]. To investigate whether EAAT2 activation restored motor deficits and neuronal loss in YAPGFAP-CKO ALS mice, we performed several experiments. As expected, the tail-suspension test showed that the hind limb clasping time of YAPGFAP-CKO ALS mice was significantly reduced after LDN-212320 treatment (Figure 8A-B). Meanwhile, the cage behavior test further showed that the walking time along the edge of the cage was significantly prolonged in LDN-212320-treated YAPGFAP-CKO ALS mice (Figure 8C). The balance beam test showed that LDN-212320-treated YAPGFAP-CKO ALS mice had fewer hind limb slips on the balance beam (Figure 8D), and the rotarod test showed that the latency to fall from the rotating rod was prolonged in LDN-212320-treated YAPGFAP-CKO ALS mice (Figure 8E). Collectively, these results suggest that activation of EAAT2 by LDN-212320 treatment partially restores behavioral deficits in the motor coordination and balance in YAPGFAP-CKO ALS mice.
Activation of EAAT2 partially restores motor deficits and neuronal loss in YAPGFAP-CKO ALS mice. (A) Representative images of YAPf/f ALS mice and YAPGFAP-CKO ALS mice treated with or without LDN-212320 treatment in tail-suspension test. The red arrows indicate the hind limbs of mice. (B) Quantitative analysis of hind limb clasping time of YAPf/f ALS mice and YAPGFAP-CKO ALS mice treated with or without LDN-212320 treatment within 3 min in tail-suspension test (n = 5 each group). (C) Quantitative analysis of time keeping on the edges of cages of YAPf/f ALS mice and YAPGFAP-CKO ALS mice treated with or without LDN-212320 treatment (n = 5 each group). (D) Quantitative analysis of the numbers of hind limb foot slips in the balance beam test of YAPf/f ALS mice and YAPGFAP-CKO ALS mice treated with or without LDN-212320 (n = 5 each group). (E) Quantitative analysis of the latency to fall from the accelerated rotating rod of YAPf/f ALS mice and YAPGFAP-CKO ALS mice treated with or without LDN-212320 (n = 5 each group). (F) Western blot analysis of EAAT2, Bax and Bcl-2 expression in the cortex of YAPf/f ALS mice and YAPGFAP-CKO ALS mice treated with or without LDN-212320. (G) Quantitative analysis of the relative EAAT2 expression level as shown in (F) (n = 5 each group, normalized to control YAPf/f ALS mice). (H) Quantitative analysis of the relative Bax/Bcl-2 expression level as shown in (F) (n = 4 each group, normalized to control YAPf/f ALS mice). (I) Representative staining images of Nissl staining in the motor cortex of YAPf/f ALS mice and YAPGFAP-CKO ALS mice treated with or without LDN-212320. (J) Quantitative analysis of the density of Nissl bodies as shown in (I) (n = 6 each group). (K-L) Double immunostaining of NeuN (red) and c-caspase-3 (far-red) (K), and NeuN (red) and TDP-43 (far-red) (L) in the motor cortex of YAPf/f ALS mice and YAPGFAP-CKO ALS mice treated with or without LDN-212320. White arrows indicate cytoplasmic TDP-43 expression. (M-N) Quantitative analysis of the density of c-caspase-3+ cells in NeuN+ neurons (M) and the density of cytoplasmic TDP-43 in NeuN+ neurons (N) as shown in (K) and (L), respectively (n = 6 each group). Scale bars, 50 µm. Data were presented as mean ± SEM. Two-way ANOVA with Tukey's multiple comparisons test, n.s., not significant (p > 0.05), *p < 0.05, **p < 0.01.
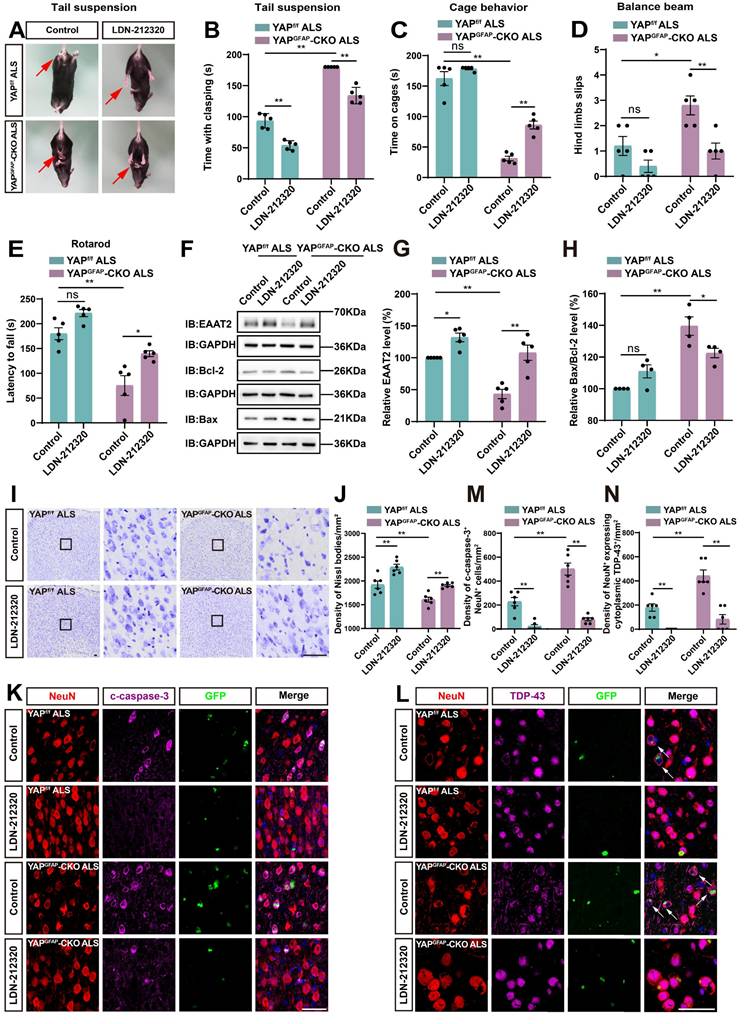
Next, western blot analysis showed that after treatment with LDN-212320, the level of EAAT2 protein was significantly upregulated (Figure 8F-G), and the ratio of Bax/Bcl-2 was significantly decreased in YAPGFAP-CKO ALS mice (Figure 8F, H). In addition, Nissl staining showed that neuronal loss was significantly inhibited in the motor cortex of LDN-212320-treated YAPGFAP-CKO ALS mice (Figure 8I-J). Double immunostaining for NeuN and c-caspase-3 showed that LDN-212320 treatment significantly reduced the density of c-caspase-3 positive neurons in the motor cortex of YAPGFAP-CKO ALS mice (Figure 8K, M). We then examined the translocation of TDP-43 in the motor cortex of control and LDN-212320-treated mice. Interestingly, double immunostaining showed that pathological translocation of TDP-43 was significantly reduced in LDN-212320-treated YAPGFAP-CKO ALS mice, and TDP-43 was primarily expressed in the nucleus of neurons (Figure 8L, N). Taken together, these results suggest that activation of EAAT2 partially restores motor deficits and neuronal loss in YAPGFAP-CKO ALS mice.
Activation of astrocytic YAP/β-catenin/EAAT2 signaling alleviates motor deficits and neurodegeneration in C9orf72-poly-GA mice
To investigate whether activation of astrocytic YAP could alleviate the symptoms of C9orf72-poly-GA mice by upregulating EAAT2, we treated ALS mice with XMU-MP-1, a compound that activates the downstream effector YAP by blocking MST1/2 kinase activity [72]. Interestingly, the tail-suspension test showed that the hind limb clasping time was significantly reduced in C9orf72-poly-GA mice after XMU-MP-1 treatment (Figure 9A-B). The balance beam test further showed that the number of hind limb slips were significantly decreased in XMU-MP-1-treated C9orf72-poly-GA mice (Figure 9C), and the rotarod test showed that the latency to fall from the rotating rod was prolonged in XMU-MP-1-treated C9orf72-poly-GA mice (Figure 9D). These results indicate that XMU-MP-1 improves the recovery of motor function in C9orf72-poly-GA mice.
Activation of astrocytic YAP/β-catenin/EAAT2 signaling alleviates motor deficits and neurodegeneration in C9orf72-poly-GA mice. (A) Representative images of ALS mice and XMU-MP-1-treated ALS mice in tail-suspension test. The red arrows indicate the hind limbs of mice. (B) Quantitative analysis of hind limb clasping time of ALS mice and XMU-MP-1-treated ALS mice within 3 min in tail-suspension test (n = 10 each group, Student's t-test). (C) Quantitative analysis of the numbers of hind limb foot slips in the balance beam test of ALS mice and XMU-MP-1-treated ALS mice (n = 10 each group, Student's t-test). (D) Quantitative analysis of latencies to fall from the accelerated rotating beams of ALS mice and XMU-MP-1-treated ALS mice (n = 10 each group, Student's t-test). (E) Western blot analysis of YAP, β-catenin and EAAT2 expression in the cortex of control mice, ALS mice and XMU-MP-1-treated ALS mice. (F-H) Quantitative analysis of the relative YAP (F), β-catenin (G) and EAAT2 (H) expression levels as shown in (E) (n = 4 each group, normalized to control mice). (I) Western blot analysis of cytoplasmic and nuclear expression of β-catenin in the cortex of control mice, ALS mice, and XMU-MP-1-treated ALS mice. (J) Quantitative analysis of the relative nucleus/cytoplasm ratio of β-catenin expression level as shown in (I) (n = 5 each group, normalized to control mice). (K) Representative staining images of Nissl staining in the motor cortex of control mice, ALS mice and XMU-MP-1-treated ALS mice. (L) Quantitative analysis of the density of Nissl bodies as shown in (I) (n = 6 each group). (M, O) Double immunostaining of NeuN (red) and c-caspase-3 (far-red) (M), and NeuN (red) and TDP-43 (far-red) (O) in the motor cortex of control mice, ALS mice and XMU-MP-1-treated ALS mice. White arrows indicate cytoplasmic TDP-43 expression. (N, P) Quantitative analysis of the density of c-caspase-3+ cells in NeuN+ neurons (N) and the density of cytoplasmic TDP-43 in NeuN+ neurons (P) as shown in (M) and (O), respectively (n = 6 each group). Scale bars, 50 µm. Data were presented as mean ± SEM. One-way ANOVA with Tukey's multiple comparisons test unless otherwise indicated, *p < 0.05, **p < 0.01.
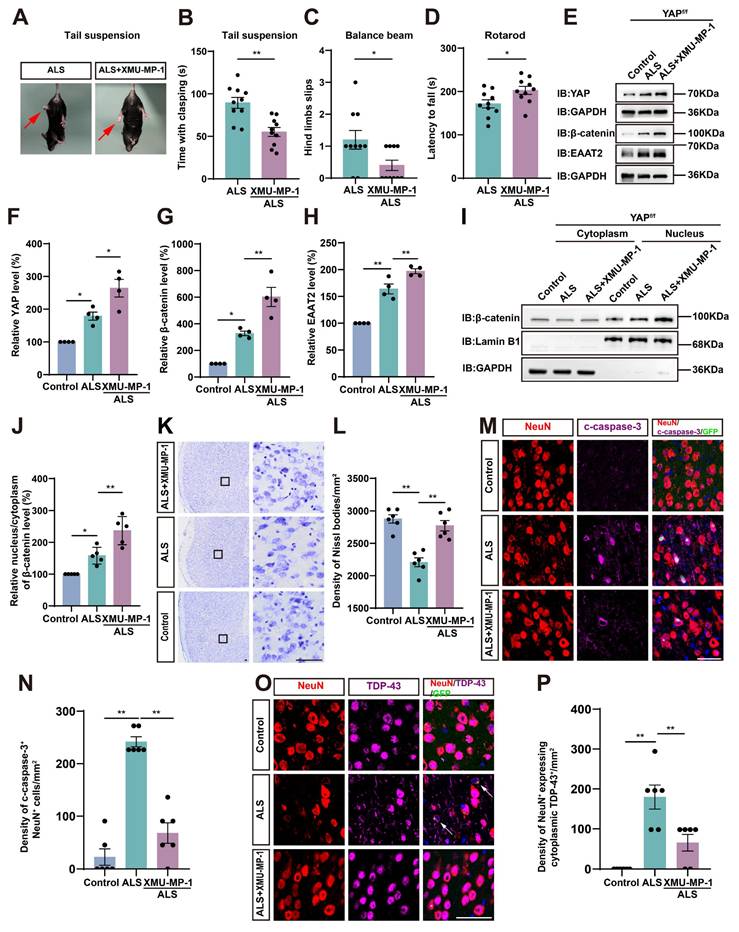
As expected, western blot analysis further showed that YAP signaling was activated in the cortex of C9orf72-poly-GA mice treated with XMU-MP-1(Figure 9E-F), with significant increases in β-catenin and EAAT2 protein levels (Figure 9E, G-H). In addition, cytosol-nuclei fractionation experiments showed that XMU-MP-1 treatment increased the nucleus/cytoplasm ratio of β-catenin in the cortex of C9orf72-poly-GA mice (Figure 9I-J), indicating that the Wnt/β-catenin pathway was activated in these mice. Nissl staining showed that the loss of Nissl bodies in the motor cortex was significantly reduced in XMU-MP-1-treated C9orf72-poly-GA mice (Figure 9K-L). Double immunostaining for NeuN and c-caspase-3 further showed that neuronal death in the motor cortex of C9orf72-poly-GA mice was significantly reduced after XMU-MP-1 treatment (Figure 9M-N). Moreover, double immunostaining for TDP-43 and NeuN showed that TDP-43 was primarily localized in the nucleus of neurons in the motor cortex of C9orf72-poly-GA mice treated with XMU-MP-1 (Figure 9O-P). Taken together, these results suggest that activation of astrocytic YAP/β-catenin /EAAT2 signaling alleviates motor deficits and neurodegeneration in C9orf72-poly-GA mice.
Discussion
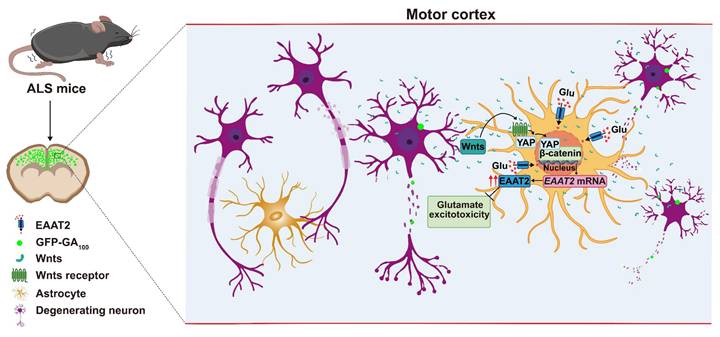
In this study, we provide evidence supporting the role and mechanism of astrocytic YAP-EAAT2 signaling in C9orf72-poly-GA mice and propose a working model (Figure 10). In this model, degenerating neurons and/or astrocytes in ALS mice may release the help signals by secreting factors such as Wnts, which activate the YAP/β-catenin signaling in astrocytes to upregulate EAAT2. This mechanism mitigates neuronal glutamate excitotoxicity, neuronal loss, and motor dysfunction in C9orf72-poly-GA mice. In addition, activation of YAP signaling upregulates β-catenin transcriptional activity and increases EAAT2 expression, thereby preventing neuronal loss and motor dysfunction in C9orf72-poly-GA mice. These findings reveal a previously unrecognized mechanism of self-protection in degenerating neurons mediated by astrocytic YAP through Wnts/β-catenin/EAAT2 signaling to prevent glutamate excitotoxicity of neurons in ALS mice, and provide a potential new drug target for ALS.
A working model of astrocytic YAP's function in C9orf72-poly-GA mice. In the motor cortex of C9orf72-poly-GA mice, degenerating neurons and/or astrocytes release the help signals by secreting factors such as Wnts to activate YAP/β-catenin signaling, and further upregulate EAAT2 expression in astrocytes. This cascade prevents glutamate excitotoxicity, neuronal loss, and motor dysfunction in C9orf72-poly-GA mice.
Recent studies have shown that YAP is not expressed in spinal cord neurons, but is upregulated and activated in astrocytes in a Hippo kinase-dependent manner, promoting the formation of glial scars and neural regeneration of mice after spinal cord injury [33]. In EAE mice, astrocytic YAP also prevents the demyelination by inhibiting the upregulation and activation of Hippo pathway in astrocytes and promotes the expression of cholesterol synthesis gene in EAE [34]. Consistent with these previous studies, this study showed that YAP was upregulated and activated in astrocytes in C9orf72-poly-GA mice, preventing motor deficits and neuronal loss by promoting the expression of EAAT2, suggesting that YAP in astrocytes may be activated and play conservatively protective roles in various disease models. Notably, a previous study has shown that YAP deltaCs (pro-survival isoforms of YAP) is progressively decreased in spinal cord neurons of SOD1G93A transgenic ALS mice [73]. These differing observations may be attributed to the use of different mouse models (C9orf72-poly-GA versus SOD1G93A) or the distinct brain regions examined (motor cortex versus spinal cord). In the future, it is interesting to test whether astrocytic YAP signaling is activated in a manner dependent on the inactivation of Hippo kinases in ALS.
By integrating the snRNA-seq of C9orf72-ALS/FTD patients with low and high YAP expression, and RNA-seq from cultured YAP+/+ and YAP-/- astrocytes, we found that the most differential pathway was enriched in neurotransmitter uptake (Figure 5A-B). The MA-plot of YAP+/+ and YAP-/- astrocytes showed that neurotransmitter uptake-related genes such as Slc6a11, Slc1a2 (EAAT2), and Slc1a3 (EAAT1) were significantly downregulated (Figure 5C). Indeed, glutamate excitotoxicity caused by dysfunction of EAAT2 is considered to play a crucial role in the pathogenesis of ALS [8, 70]. Riluzole is a drug approved by the FDA for the treatment of ALS, which has been shown to inhibit excitotoxicity [74, 75]. Previous studies have shown that glutamate transport is impaired in the brain and spinal cord of ALS patients [76]. In addition, it has been reported that EAAT2 protein levels are decreased in the motor cortex and spinal cord of ALS [17, 77]. However, some studies have also shown that EAAT2 expression was increased in the middle laminae of the motor cortex in patients [78], and EAAT2 was significantly upregulated in astrocytes induced by ALS patient-derived iPSC lines after co-culturing with neurons in vitro [79]. In SOD1G93A mice, no changes in EAAT2 expression were observed at 10 weeks of age; however, a progressive decrease in EAAT2 expression was observed in the ventral, but not in the dorsal, horn of the lumbar spinal cord at 14 and 18 weeks of age [80]. In our study, we found that EAAT2 expression was increased in 3-month-old C9orf72-poly-GA mice and decreased in YAPGFAP-CKO ALS mice. This discrepancy may potentially be explained by the different roles of EAAT2 in the disease progression of ALS.
Several lines of evidence suggest that glutamate excitotoxicity in ALS is regulated by astrocytic YAP-EAAT2 signaling. Firstly, the extracellular glutamate concentration was significantly higher in YAP-/- astrocytes (Figure 6B). Secondly, the apoptosis of primary cultured neurons transduced with AAV-GFP-GA100 (Figure 6E-G) or N2a cells transfected with GFP-GA100 (Supplementary Figure 3B-E) was aggravated after treatment with conditioned medium from YAP-/- astrocytes. Thirdly, treatment with LDN-212320, an agonist of EAAT2, promoted EAAT2 expression and partially restored motor deficits and neuronal loss in YAPGFAP-CKO ALS mice (Figure 8). Finally, activation of YAP by XMU-MP-1 upregulated β-catenin and EAAT2 expression, which partially alleviated neurodegeneration and motor deficits in ALS mice (Figure 9). How is YAP/EAAT2 signaling activated in ALS mice? Our recent studies have shown that YAP signaling in astrocytes promotes EAAT2 expression by regulating the transcriptional activity of β-catenin [54]. Consistent with these findings, in our studies, we found that β-catenin was downregulated in YAPGFAP-CKO ALS mice and YAP-/- astrocytes, and upregulated and translocated into the nucleus by the activation of YAP signaling. Interestingly, YAP/EAAT2 signaling was activated by conditioned medium from N2a cells transfected with GFP-GA100 or primary cultured degenerating neurons, suggesting that degenerating neurons with GA aggregates might secrete factors that activate YAP/β-catenin/EAAT2 signaling in astrocytes.
It has been reported that the expression of Wnt1, Wnt2, Wnt3a and Wnt7a is upregulated in SOD1G93A mice, and in ALS, motor neurons, astrocytes, microglia and oligodendrocytes exhibit high levels of Wnt/β-catenin signaling and β-catenin activation [37, 38]. The Wnt/β-catenin signaling pathway activated by neurodegeneration is related to the proliferation of spinal glial cells in SOD1G93A transgenic mice [40]. In fact, previous studies have shown that Wnts such as Wnt1 and Wnt3a are upregulated in astrocytes in ALS [81]. In this study, primary cultured neurons were transduced with AAV-GFP-GA5 and AAV-GFP-GA100 viruses to establish an in vitro ALS model (Figure 6C). qPCR results showed that Wnt1, Wnt2, Wnt3a, Wnt4, Wnt5a, and Wnt7a mRNA were significantly upregulated in primary cultured neurons transduced with AAV-GFP-GA100 virus (Figure 7A-F), suggesting that degenerating neurons upregulated Wnts mRNA, consistent with the results in N2a cell line. Meanwhile, as shown in Figure 7G-L, we found that Wnt1, Wnt2, Wnt3a, Wnt4, Wnt5a, and Wnt7a mRNA were significantly upregulated in YAP+/+ astrocytes treated with conditioned medium from primary cultured neurons transduced with AAV-GFP-GA100 virus, suggesting that degenerating neurons somehow promoted the upregulation expression of Wnts in astrocytes. Thus, astrocytes both receive Wnt signals and express Wnts in ALS. Interestingly, in the MPTP mouse model of Parkinson's disease, previous studies documented the early up-regulation of astrocytic Wnt1 in the ventral midbrain of degenerating substantia nigra neurons as a self-repair intersystem neuroprotective crosstalk [47-50]. Based on these previous studies and our results (Figure 7 and Supplementary Figure S3), it is possible that early in ALS pathogenesis, Wnts are secreted from neurons and/or astrocytes during neurodegeneration, then in turn activate the YAP/β-catenin/EAAT2 signaling pathway in astrocytes. However, YAP may regulate EAAT2 expression through its interaction with β-catenin, and the precise mechanism underlying this process requires further investigation in ALS. Moreover, we also observed an intriguing phenomenon in which GFP-GA100 aggregates increased significantly in the cortex of YAPGFAP-CKO ALS mice. We hypothesize that an additional mechanism may be influencing the C9orf72-poly-GA mice, and we plan to further explore the mechanisms involved in future studies.
Riluzole has been shown to reduce the release of glutamate from nerve endings [74, 75]. Interestingly, Riluzole treatment upregulates the Wnt3a expression in melanoma cells in vitro, and enhances the Wnt/β-catenin signaling in both HT22 neuronal cells and adult hippocampal progenitor cells [82, 83], suggesting that Riluzole is an enhancer of the Wnt/β-catenin signaling. Thus, it is interesting to test whether Riluzole also activates Wnt/β-catenin/YAP/EAAT2 signaling in astrocytes to perform neuroprotective effects in ALS in future. LDN/OSU-212320 can increase the expression of EAAT2 through translational activation, prevent excitotoxicity, delay the decline of motor function, and prolong the lifespan of SOD1G93A mice [71]. Consequently, increasing the expression of EAAT2 could serve as a potential treatment to prevent excitotoxicity in neurodegenerative diseases. Consistent with the previous results, treatment with LDN-212320 upregulated the expression of EAAT2 in the motor cortex of YAPGFAP-CKO ALS mice, preventing neuronal loss and motor deficits (Figure 8). XMU-MP-1 (an inhibitor of Hippo kinase MST1/2) is used to activate YAP signaling [72]. Our results showed that XMU-MP-1 activated YAP signaling and partially improved motor function recovery in C9orf72-poly-GA mice (Figure 9). These results suggest that the Hippo/YAP pathway plays a critical role in regulating the glutamate transporter EAAT2 and maintaining glutamate homeostasis in astrocytes. Therefore, it will be interesting to test the combination of these two drugs (LDN-212320 or Riluzole with XMU-MP-1) for potential synergistic therapeutic effects in ALS.
In summary, our studies identify an unrecognized mechanism of self-protection in degenerating neurons, mediated by astrocytic YAP through Wnt/β-catenin/EAAT2 signaling, which prevents glutamate excitotoxicity in neurons of ALS mice. This highlights the therapeutic potential of the YAP/Wnt/β-catenin/EAAT2 signaling pathway in delaying and treating ALS diseases, which may contribute to the development of new therapeutic methods for ALS.
Abbreviations
ALS: Amyotrophic lateral sclerosis; CNS: central nervous system; YAP: Yes-associated protein; CKO: conditional knockout; qPCR: quantitative real-time PCR; EAAT2: excitatory amino acid transporter-2; PBA: pseudobulbar affect; ASOs: antisense oligonucleotides; iPSCs: induced pluripotent stem cells; SOD1: Cu/Zn superoxide dismutase 1 gene; C9ORF72: Chromosome 9 Open Reading Frame 72; TARDBP: TAR DNA Binding Protein; FUS: Fused in Sarcoma; TBK1: TANK-binding kinase 1; MST1/2: mammalian Sterile 20-like kinases 1/2; SAV1: salvador homolog 1; MOB1A/B: Mps one binder 1A and B; LATS1/2: large tumor suppressor 1/2; TAZ: transcriptional co-activator with PDZ binding motif; TEAD: transcriptional enhanced associate domain; EAE: experimental autoimmune encephalomyelitis; MS-ON: multiple sclerosis-related optic neuritis; P0: postnatal day 0; PBS: phosphate buffered saline; PFA: paraformaldehyde; BSA: bovine serum albumin; TBST: tris buffered saline with Tween 20; N2a: Neuro-2a; FBS: fetal bovine serum; PS: penicillin/streptomycin; CM: conditioned medium; WT: wild-type; DIV: days in vitro; MOIs: multiplicities of infection; CCK-8: Cell Counting Kit-8; ACM: astrocyte conditioned medium; GFAP: glial fibrillary acidic protein; p-YAP: phosphorylated YAP; c-caspase-3: Cleaved-caspase-3; NF: neurofilament heavy polypeptide; snRNA-seq: single-nucleus transcriptomic; RNA-seq: RNA sequencing; GO: Gene Ontology; MA-plot: M-versus-A plot.
Supplementary Material
Supplementary figures.
Acknowledgements
We thank Prof. Chengyong Shen (Zhejiang University)'s lab for kind help in ALS mice model in this study. We also thank the School of Pharmacy of Hangzhou Normal University for providing technical support for this work.
Funding
This work was supported by the Natural Science Foundation of Zhejiang Province (LZ23C090001), Major Projects of Hangzhou Health Science and Technology (No. Z20220024) and the Zhejiang Province Leading Geese Plan (2024C03099), Key R&D Program of Hangzhou Science and Technology Bureau (20241203A17).
Author contributions
Conceptualization: Zhihui Huang, Ying Wang; methodology: Dongmei Li, Yan Wei, Rui Yang; validation: Dongmei Li, Yan Wei, Rui Yang, Xuan Luo and Yanzhu Liu; formal analysis: Dongmei Li, Hui Yang; resources: Zhihui Huang, Ying Wang and Weiqiao Zhao; data curation and investigation: Dongmei Li, Yan Wei, Rui Yang; writing-original draft preparation: Dongmei Li; writing-review & editing: Zhihui Huang, Ying Wang, Dongmei Li; visualization: Dongmei Li, Yan Wei, Rui Yang, Xuan Luo, Yanzhu Liu, Hui Yang and Yumin Wu; supervision: Ying Wang, Zhihui Huang; funding acquisition: Ying Wang, Zhihui Huang. All authors have read and agreed to the version of the manuscript.
Ethics approval statement
The use of experimental animals and all relevant experimental protocols were approved by the Animal Care and Use Committee of Hangzhou Normal University (HSD20220911).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Brown RH, Al-Chalabi A. Amyotrophic Lateral Sclerosis. New England Journal of Medicine. 2017;377:162-72
2. Hardiman O, Al-Chalabi A, Chio A. et al. Amyotrophic lateral sclerosis. Nat Rev Dis Primers. 2017;3:1-19
3. Taylor JP, Brown RH, Cleveland DW. Decoding ALS: from genes to mechanism. Nature. 2016;539:197-206
4. Tzeplaeff L, Wilfling S, Requardt MV, Herdick M. Current State and Future Directions in the Therapy of ALS. Cells. 2023;12:1523
5. Sun Y, Benatar M, Mascías Cadavid J. et al. ALSUntangled #71: Nuedexta. Amyotrophic Lateral Sclerosis and Frontotemporal Degeneration. 2024;25:218-22
6. Sever B, Ciftci H, DeMirci H. et al. Comprehensive Research on Past and Future Therapeutic Strategies Devoted to Treatment of Amyotrophic Lateral Sclerosis. Int J Mol Sci. 2022;23:2400
7. Mejzini R, Flynn LL, Pitout IL, Fletcher S, Wilton SD, Akkari PA. ALS Genetics, Mechanisms, and Therapeutics: Where Are We Now? Front Neurosci. 2019;13:1310
8. van den Bos MAJ, Geevasinga N, Higashihara M, Menon P, Vucic S. Pathophysiology and Diagnosis of ALS: Insights from Advances in Neurophysiological Techniques. Int J Mol Sci. 2019;20:2818
9. Masrori P, Van Damme P. Amyotrophic lateral sclerosis: a clinical review. Eur J Neurol. 2020;27:1918-29
10. Turner MR, Hardiman O, Benatar M. et al. Controversies and priorities in amyotrophic lateral sclerosis. The Lancet Neurology. 2013;12:310-22
11. Nijs M, Van Damme P. The genetics of amyotrophic lateral sclerosis. Curr Opin Neurol. 2024;37:560-9
12. Provenzano F, Torazza C, Bonifacino T, Bonanno G, Milanese M. The Key Role of Astrocytes in Amyotrophic Lateral Sclerosis and Their Commitment to Glutamate Excitotoxicity. Int J Mol Sci. 2023;24:15430
13. Yamanaka K, Chun SJ, Boillee S. et al. Astrocytes as determinants of disease progression in inherited ALS. Nat Neurosci. 2008;11:251-3
14. Stoklund Dittlau K, Van Den Bosch L. Why should we care about astrocytes in a motor neuron disease? Front Mol Med. 2023;3:1047540
15. Verkhratsky A, Nedergaard M. Physiology of Astroglia. Physiol Rev. 2018;98:239-389
16. Howland DS, Liu J, She Y. et al. Focal loss of the glutamate transporter EAAT2 in a transgenic rat model of SOD1 mutant-mediated amyotrophic lateral sclerosis (ALS). Proc Natl Acad Sci U S A. 2002;99:1604-9
17. Rothstein JD, Van Kammen M, Levey AI, Martin LJ, Kuncl RW. Selective loss of glial glutamate transporter GLT-1 in amyotrophic lateral sclerosis. Annals of Neurology. 1995;38:73-84
18. Van Den Bosch L, Van Damme P, Bogaert E, Robberecht W. The role of excitotoxicity in the pathogenesis of amyotrophic lateral sclerosis. Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease. 2006;1762:1068-82
19. Lepore AC, Rauck B, Dejea C. et al. Focal Transplantation-based Astrocyte Replacement is Neuroprotective in a Model of Motor Neuron Disease. Nat Neurosci. 2008;11:1294-301
20. Papadeas ST, Kraig SE, O'Banion C, Lepore AC, Maragakis NJ. Astrocytes carrying the superoxide dismutase 1 (SOD1G93A) mutation induce wild-type motor neuron degeneration in vivo. Proc Natl Acad Sci U S A. 2011;108:17803-8
21. Fu M, Hu Y, Lan T, Guan K-L, Luo T, Luo M. The Hippo signalling pathway and its implications in human health and diseases. Signal Transduct Target Ther. 2022;7:376
22. Cheng J, Wang S, Dong Y, Yuan Z. The Role and Regulatory Mechanism of Hippo Signaling Components in the Neuronal System. Front Immunol. 2020;11:281
23. Zhao B, Wei X, Li W. et al. Inactivation of YAP oncoprotein by the Hippo pathway is involved in cell contact inhibition and tissue growth control. Genes Dev. 2007;21:2747-61
24. Meng Z, Moroishi T, Guan K-L. Mechanisms of Hippo pathway regulation. Genes & Development. 2016;30:1
25. Wang S, Zhou L, Ling L. et al. The Crosstalk Between Hippo-YAP Pathway and Innate Immunity. Front Immunol. 2020;11:323
26. Lee JK, Shin JH, Hwang SG. et al. MST1 functions as a key modulator of neurodegeneration in a mouse model of ALS. Proceedings of the National Academy of Sciences. 2013;110:12066-71
27. Azuma Y, Tokuda T, Kushimura Y. et al. Hippo, Drosophila MST, is a novel modifier of motor neuron degeneration induced by knockdown of Caz, Drosophila FUS. Experimental Cell Research. 2018;371:311-21
28. Gogia N, Sarkar A, Mehta AS. et al. Inactivation of Hippo and cJun-N-terminal Kinase (JNK) signaling mitigate FUS mediated neurodegeneration in vivo. Neurobiol Dis. 2020;140:104837
29. Huang Z, Wang Y, Hu G, Zhou J, Mei L, Xiong W-C. YAP Is a Critical Inducer of SOCS3, Preventing Reactive Astrogliosis. Cereb Cortex. 2016;26:2299-310
30. Huang Z, Xiong W-C. Neogenin-YAP signaling in neocortical astrocytic differentiation. Neurogenesis (Austin). 2016;3:e1248735
31. Huang Z, Sun D, Hu J-X. et al. Neogenin Promotes BMP2 Activation of YAP and Smad1 and Enhances Astrocytic Differentiation in Developing Mouse Neocortex. J Neurosci. 2016;36:5833-49
32. Huang Z, Hu J, Pan J. et al. YAP stabilizes SMAD1 and promotes BMP2-induced neocortical astrocytic differentiation. Development. 2016;143:2398-409
33. Xie C, Shen X, Xu X. et al. Astrocytic YAP Promotes the Formation of Glia Scars and Neural Regeneration after Spinal Cord Injury. J Neurosci. 2020;40:2644-62
34. Zhang J, Xu X, Liu H. et al. Astrocytic YAP prevents the demyelination through promoting expression of cholesterol synthesis genes in experimental autoimmune encephalomyelitis. Cell Death Dis. 2021;12:907
35. Wu Q, Miao X, Zhang J. et al. Astrocytic YAP protects the optic nerve and retina in an experimental autoimmune encephalomyelitis model through TGF-β signaling. Theranostics. 2021;11:8480-99
36. Soumya BS, Shreenidhi VP, Agarwal A, Gandhirajan RK, Dharmarajan A, Warrier S. Unwinding the role of Wnt signaling cascade and molecular triggers of motor neuron degeneration in amyotrophic lateral sclerosis (ALS). Cellular Signalling. 2023;110:110807
37. Ramakrishna K, Nalla LV, Naresh D. et al. WNT-β Catenin Signaling as a Potential Therapeutic Target for Neurodegenerative Diseases: Current Status and Future Perspective. Diseases. 2023;11:89
38. Jiang X, Guan Y, Zhao Z. et al. Potential Roles of the WNT Signaling Pathway in Amyotrophic Lateral Sclerosis. Cells. 2021;10:839
39. Ziff OJ, Clarke BE, Taha DM, Crerar H, Luscombe NM, Patani R. Meta-analysis of human and mouse ALS astrocytes reveals multi-omic signatures of inflammatory reactive states. Genome Res. 2022;32:71-84
40. Chen Y, Guan Y, Liu H. et al. Activation of the Wnt/β-catenin signaling pathway is associated with glial proliferation in the adult spinal cord of ALS transgenic mice. Biochemical and Biophysical Research Communications. 2012;420:397-403
41. Stoklund Dittlau K, Terrie L, Baatsen P. et al. FUS-ALS hiPSC-derived astrocytes impair human motor units through both gain-of-toxicity and loss-of-support mechanisms. Mol Neurodegener. 2023;18:5
42. Sileo P, Simonin C, Melnyk P, Chartier-Harlin M-C, Cotelle P. Crosstalk between the Hippo Pathway and the Wnt Pathway in Huntington's Disease and Other Neurodegenerative Disorders. Cells. 2022;11:3631
43. Konsavage WM, Yochum GS. Intersection of Hippo/YAP and Wnt/β-catenin signaling pathways. ABBS. 2013;45:71-9
44. Azzolin L, Panciera T, Soligo S. et al. YAP/TAZ Incorporation in the β-Catenin Destruction Complex Orchestrates the Wnt Response. Cell. 2014;158:157-70
45. Park HW, Kim YC, Yu B. et al. Alternative Wnt Signaling Activates YAP/TAZ. Cell. 2015;162:780-94
46. Astone M, Tesoriero C, Schiavone M. et al. Wnt/β-Catenin Signaling Regulates Yap/Taz Activity during Embryonic Development in Zebrafish. Int J Mol Sci. 2024;25:10005
47. L'Episcopo F, Tirolo C, Testa N. et al. Reactive astrocytes and Wnt/β-catenin signaling link nigrostriatal injury to repair in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine model of Parkinson's disease. Neurobiol Dis. 2011;41:508-27
48. Marchetti B, Pluchino S. Wnt your brain be inflamed? Yes, it Wnt!. Trends Mol Med. 2013;19:144-56
49. Marchetti B, L'Episcopo F, Morale MC. et al. Uncovering novel actors in astrocyte-neuron crosstalk in Parkinson's disease: the Wnt/β-catenin signaling cascade as the common final pathway for neuroprotection and self-repair. Eur J Neurosci. 2013;37:1550-63
50. Marchetti B. Nrf2/Wnt resilience orchestrates rejuvenation of glia-neuron dialogue in Parkinson's disease. Redox Biol. 2020;36:101664
51. Zhang K, Wang A, Zhong K. et al. UBQLN2-HSP70 axis reduces poly-Gly-Ala aggregates and alleviates behavioral defects in the C9ORF72 animal model. Neuron. 2021;109:1949-1962.e6
52. Hao Z, Liu L, Tao Z. et al. Motor dysfunction and neurodegeneration in a C9orf72 mouse line expressing poly-PR. Nat Commun. 2019;10:2906
53. Howard DB, Powers K, Wang Y, Harvey BK. Tropism and toxicity of adeno-associated viral vector serotypes 1,2,5,6,7,8,9 in rat neurons and glia in vitro. Virology. 2008;372:24-34
54. Xu X, Wang J, Du S. et al. Yes-associated protein regulates glutamate homeostasis through promoting the expression of excitatory amino acid transporter-2 in astrocytes via β-catenin signaling. Glia. 2023;71:1197-216
55. Birger A, Ben-Dor I, Ottolenghi M. et al. Human iPSC-derived astrocytes from ALS patients with mutated C9ORF72 show increased oxidative stress and neurotoxicity. EBioMedicine. 2019;50:274-89
56. Julien J, Couillard-Després S, Meier J. Transgenic Mice in the Study of ALS: The Role of Neurofilaments. Brain Pathol. 2006;8:759-69
57. Poesen K, Van Damme P. Diagnostic and Prognostic Performance of Neurofilaments in ALS. Front Neurol. 2019;9:1167
58. Lee MK, Cleveland DW. Neurofilament function and dysfunction: involvement in axonal growth and neuronal disease. Current Opinion in Cell Biology. 1994;6:34-40
59. Neumann M, Sampathu DM, Kwong LK. et al. Ubiquitinated TDP-43 in Frontotemporal Lobar Degeneration and Amyotrophic Lateral Sclerosis. Science. 2006;314:130-3
60. Murray ME, DeJesus-Hernandez M, Rutherford NJ. et al. Clinical and neuropathologic heterogeneity of c9FTD/ALS associated with hexanucleotide repeat expansion in C9ORF72. Acta Neuropathol. 2011;122:673-90
61. Wong JK, Roselle AK, Shue TM. et al. Apolipoprotein B-100-mediated motor neuron degeneration in sporadic amyotrophic lateral sclerosis. Brain Commun. 2022;4:fcac207
62. Beers DR, Appel SH. Immune dysregulation in amyotrophic lateral sclerosis: mechanisms and emerging therapies. The Lancet Neurology. 2019;18:211-20
63. Guo S, Wang H, Yin Y. Microglia Polarization From M1 to M2 in Neurodegenerative Diseases. Front Aging Neurosci. 2022;14:815347
64. Schiffer D, Cordera S, Cavalla P, Migheli A. Reactive astrogliosis of the spinal cord in amyotrophic lateral sclerosis. Journal of the Neurological Sciences. 1996;139:27-33
65. Vargas MR, Johnson JA. Astrogliosis in amyotrophic lateral sclerosis: Role and therapeutic potential of astrocytes. Neurotherapeutics. 2010;7:471-81
66. Balint V, Peric M, Dacic S. et al. The Role of SOX2 and SOX9 Transcription Factors in the Reactivation-Related Functional Properties of NT2/D1-Derived Astrocytes. Biomedicines. 2024;12:796
67. Neyrinck K, Van Den Daele J, Vervliet T. et al. SOX9-induced Generation of Functional Astrocytes Supporting Neuronal Maturation in an All-human System. Stem Cell Rev Rep. 2021;17:1855-73
68. Li J, Jaiswal MK, Chien J-F. et al. Divergent single cell transcriptome and epigenome alterations in ALS and FTD patients with C9orf72 mutation. Nat Commun. 2023;14:5714
69. Haugeto O, Ullensvang K, Levy LM. et al. Brain Glutamate Transporter Proteins Form Homomultimers*. Journal of Biological Chemistry. 1996;271:27715-22
70. Pajarillo E, Rizor A, Lee J, Aschner M, Leea E. The role of astrocytic glutamate transporters GLT-1 and GLAST in neurological disorders: potential targets for neurotherapeutics. Neuropharmacology. 2019;161:107559
71. Kong Q, Chang L-C, Takahashi K. et al. Small-molecule activator of glutamate transporter EAAT2 translation provides neuroprotection. J Clin Invest. 2014;124:1255-67
72. Fan F, He Z, Kong L-L. et al. Pharmacological targeting of kinases MST1 and MST2 augments tissue repair and regeneration. Science Translational Medicine. 2016;8:352ra108-352ra108
73. Morimoto N, Nagai M, Miyazaki K. et al. Progressive decrease in the level of YAPdeltaCs, prosurvival isoforms of YAP, in the spinal cord of transgenic mouse carrying a mutant SOD1 gene. Journal of Neuroscience Research. 2009;87:928-36
74. Jaiswal MK. Riluzole and edaravone: A tale of two amyotrophic lateral sclerosis drugs. Medicinal Research Reviews. 2019;39:733-48
75. Bellingham MC. A Review of the Neural Mechanisms of Action and Clinical Efficiency of Riluzole in Treating Amyotrophic Lateral Sclerosis: What have we Learned in the Last Decade? CNS Neuroscience & Therapeutics. 2011;17:4-31
76. Rothstein JD, Martin LJ, Kuncl RW. Decreased Glutamate Transport by the Brain and Spinal Cord in Amyotrophic Lateral Sclerosis. New England Journal of Medicine. 1992;326:1464-8
77. Bristol LA, Rothstein JD. Glutamate transporter gene expression in amyotrophic lateral sclerosis motor cortex. Annals of Neurology. 1996;39:676-9
78. Fray AE, Ince PG, Banner SJ. et al. The expression of the glial glutamate transporter protein EAAT2 in motor neuron disease: an immunohistochemical study. European Journal of Neuroscience. 1998;10:2481-9
79. Zhang P-W, Haidet-Phillips AM, Pham JT. et al. Generation of GFAP::GFP astrocyte reporter lines from human adult fibroblast-derived iPS cells using zinc-finger nuclease technology. Glia. 2016;64:63-75
80. Bendotti C, Tortarolo M, Suchak SK. et al. Transgenic SOD1 G93A mice develop reduced GLT-1 in spinal cord without alterations in cerebrospinal fluid glutamate levels. Journal of Neurochemistry. 2001;79:737-46
81. González-Fernández C, González P, Rodríguez FJ. New insights into Wnt signaling alterations in amyotrophic lateral sclerosis: a potential therapeutic target? Neural Regen Res. 2020;15:1580-9
82. Biechele TL, Camp ND, Fass DM. et al. Chemical-Genetic Screen Identifies Riluzole as an Enhancer of Wnt/β-catenin Signaling in Melanoma. Chemistry & Biology. 2010;17:1177-82
83. Lecarpentier Y, Vallée A. Opposite Interplay between PPAR Gamma and Canonical Wnt/Beta-Catenin Pathway in Amyotrophic Lateral Sclerosis. Front Neurol. 2016;7:100
Author contact
![]() Corresponding authors: Ying Wang, Email: nancywangyingcom. Zhihui Huang, Email: huang0069edu.cn.
Corresponding authors: Ying Wang, Email: nancywangyingcom. Zhihui Huang, Email: huang0069edu.cn.