Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
1. Introduction
2. Structure, Ligands, and...
3. Upstream Modulators of LILRBs
4. LILRBs and Immune System
5. Roles of LILRBs in various...
6. Clinical Trials of Targeting...
7. Conclusions and Perspectives
Abbreviations
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2025; 15(16):8222-8258. doi:10.7150/thno.116951 This issue Cite
Review
Inhibitory leukocyte immunoglobulin-like receptors, subfamily B (LILRBs) in human diseases: structure, roles, mechanisms, and clinical applications
Yuxiu Zhang1,2,#, Yuanyuan Xu1,#, Qihui Wu3,4,#, Xiaodan Fu4,5, ![]() , Yimin Li1,
, Yimin Li1, ![]() , Anqi Li1,
, Anqi Li1, ![]()
1. Department of Pathology, Ruijin Hospital, Shanghai Jiaotong University School of Medicine, Shanghai, China, 200025.
2. Department of Pathology, Zhongshan Hospital, Fudan University, Shanghai, China, 200032.
3. Department of Gynecology, Xiangya Hospital, Central South University, Changsha, China, 410008.
4. National Clinical Research Center for Geriatric Disorders, Xiangya Hospital, Changsha, China, 410008.
5. Department of Pathology, Xiangya Hospital, Central South University, Changsha, China, 410008.
# These authors have contributed equally to this work.
Received 2025-5-5; Accepted 2025-7-18; Published 2025-7-25
Abstract

Leukocyte immunoglobulin-like receptors, subfamily B (LILRBs), are a class of critical immunosuppressive receptors that contribute to immune homeostasis by transmitting suppressive signals upon binding to ligands such as major histocompatibility complex class I molecules. They play key roles in modulating both innate and adaptive immune responses. This review summarizes the structural features, ligand interactions, signaling pathways, and expression regulation of LILRBs, and discusses their roles in immune cell function and disease progression, particularly in the tumor microenvironment. We also review current progress in the development of LILRB-targeted therapies for hematological malignancies and solid tumors and outline the challenges and future directions in translating these findings into clinical applications. By integrating recent advances, this review provides a framework for understanding the potential of LILRBs as therapeutic targets in cancer and immune-related disorders.
Keywords: immunosuppressive receptors, hematological malignancies, solid tumors, immune regulation, immunotherapy
1. Introduction
The immune system maintains the ability to distinguish self from non-self through precise recognition and signaling mechanisms, enabling the clearance of pathogens while preserving host tissue integrity and immune homeostasis [1]. However, dysregulation of this recognition process can trigger aberrant immune responses, leading to autoimmunity and chronic inflammation [1]. From an immunological perspective, tumors can be viewed as non-self or altered self-entities. While the immune system is capable of recognizing and eliminating early-stage malignant cells, tumor cells often evade immune surveillance by expressing antigens shared with normal tissues or by downregulating antigen presentation machinery [1, 2]. Additionally, they actively suppress immune cell functions and remodel the immune microenvironment to support immune evasion and tumor progression [1, 2].
In recent years, immunotherapy has revolutionized the treatment of infectious diseases, autoimmune conditions, and cancer by modulating immune responses with high precision [3]. Core strategies, including monoclonal antibodies (mAbs), chimeric antigen receptor (CAR) T-cell therapy, therapeutic vaccines, and immune checkpoint blockade, have achieved notable clinical success [4-7]. The remarkable efficacy of checkpoint blockade, in particular, has driven extensive research into its potential to mitigate autoimmune responses and inhibit tumor progression [8, 9]. Nonetheless, challenges remain, including poor tissue penetration, off-target effects, and treatment-related toxicities, which limit the broader application of these therapies [10-13]. Additionally, T-cell expansion and the abnormal elevation of inflammatory factors can lead to immune resistance or hypersensitivity reactions, further complicating therapeutic outcomes [14-16].
Inhibitory immune receptors serve as essential modulators that fine-tune immune activation and maintain immune homeostasis [17]. Among these, the leukocyte immunoglobulin (Ig)-like receptors, subfamily B (LILRBs), represent a prominent class of inhibitory receptors with broad immunoregulatory roles. Accumulating evidence implicates LILRBs in the development and progression of infectious diseases, autoimmune disorders, and various malignancies [18-20]. The human LILRB family consists of five members: LILRB1 (also known as LIR-1, ILT2, CD85j), LILRB2 (LIR-2, ILT4, CD85d), LILRB3 (LIR-3, ILT5, CD85a), LILRB4 (LIR-5, ILT3, CD85k), and LILRB5 (LIR-8, CD85c) [21]. These receptors contain two to four cytoplasmic immunoreceptor tyrosine-based inhibitory motifs (ITIMs), which recruit Src homology 2 domain-containing phosphatases 1/2 (SHP1/2) to downregulate activating signaling pathways [18]. LILRBs are predominantly expressed on myeloid lineage cells, including monocytes, macrophages, neutrophils, and dendritic cells (DCs), as well as certain tumor cells [18, 20, 21]. LILRB1 represents the exception, with additional expression documented on lymphocytes such as B cells and specific subsets of natural killer (NK) cells and T cells [18, 20, 21]. In mice, paired Ig-like receptor B (PirB) is considered the functional ortholog of human LILRB2/3, while Lilrb4a (also known as gp49b) shares homology with LILRB4 [20]. These murine receptors have been widely used in preclinical studies to investigate LILRB functions in immune regulation and disease pathology. Emerging evidence has revealed aberrant expression of LILRBs across multiple tumor types, where they regulate diverse oncogenic processes, including tumor proliferation, invasion, metastasis, and immune evasion [22-25]. These findings highlight LILRBs as potential diagnostic biomarkers and therapeutic targets in oncology, warranting further translational investigation.
This review systematically summarizes the structure features, ligand interactions, signaling mechanisms, and regulatory pathways governing LILRB expression. We further explore their expression and functional implications in the immune system and diseases, with particular emphasis on their impact on tumor cells and the tumor microenvironment (TME). Additionally, the review compiles information on clinical trial drugs targeting LILRBs, covering drug types, mechanisms of action, and trial stages. By integrating recent advances, this review aims to provide a theoretical foundation for the development of novel LILRB-targeted therapies and to foster innovation in disease treatment strategies.
2. Structure, Ligands, and Signaling Pathways of LILRBs
2.1 Structure of LILRBs
LILRB family consist of inhibitory transmembrane glycoproteins characterized by extracellular Ig-like domains, a single transmembrane segment, and multiple intracellular ITIMs. These receptors are encoded within the leukocyte receptor complex gene cluster on chromosome 19q13.4 and were first cloned in 1997 [26-29]. LILRB1-3 and LILRB5 share a conserved domain architecture consisting of four extracellular Ig-like domains (D1-D4), whereas LILRB4 contains only two such domains (Figure 1A) [21]. Structural analyses have revealed distinct interdomain geometries in LILRB1 and LILRB2, especially at the D2-D3 hinge (LILRB1: ~60°; LILRB2: ~50°), a difference attributed to a single amino acid substitution (W284 in LILRB1 vs. C283 in LILRB2) that affects hydrophobic packing and steric compatibility [30]. While the D3-D4 domains acted as a rigid scaffold without direct human leukocyte antigen (HLA)-G1 contact, the staggered assembly of both receptors restricted interdomain flexibility, confining ligand recognition exclusively to D1 and D2 [30, 31]. Specifically, the D1 domain engaged the α3 domain of HLA-G1, and the D2 domain interacted primarily with β₂-microglobulin (β2m), forming a conserved binding interface essential for HLA class I (HLA-I) recognition. Notably, the D1 and D2 domains exhibited significant conformational plasticity, enabling adaptation to diverse HLA ligands (e.g., HLA-A2, HLA-F, UL18) [32, 33], which facilitates robust immune regulation across varied HLA polymorphisms and pathogenic contexts. LILRB4 adopted a similar overall fold but displayed unique structural features. Its D2 domain most closely resembled the D4 domains of other LILRB members and uniquely contains 3₁₀-helices [34]. The reduced interdomain contacted between D1 and D2 result in a wider interdomain angle (~107°), producing a more open conformation [34]. Due to both conformational and electrostatic incompatibility, LILRB4 is poorly suited for MHC class I (MHC I) binding. Instead, distinct surface patches on its D1 domain and hinge region suggested interaction with non-MHC-I ligands [34].
Structure, ligands, signal transduction pathways, and upstream regulatory mechanisms of LILRBs. (A) Top: Bubble chart displaying ligands of LILRB1-5, ordered by their introduction sequence in text, with columns representing corresponding LILRB receptors. Bottom: Domain architecture of individual LILRB receptors: extracellular Ig-like domains (red), transmembrane regions, and intracellular ITIMs (yellow). The dashed circle indicates that the functional role of this ligand-receptor interaction remains controversial. (B) Canonical LILRB-mediated downstream signaling pathway: ① Ligand binding to LILRBs triggers ② Lyn kinase autophosphorylation; ③ phosphorylates ITIM motifs in intracellular domains, leads to ④ recruitment and activation of SHP1/2, which ⑤ dephosphorylate downstream signaling proteins, ultimately ⑥ modulating downstream signaling cascades and cellular functions. (C) A polymorphism at c.59G in the 5'-UTR region modulates LILRB2 expression. (D) Differential promoter usage of LILRB1 in ① lymphocyte versus ② monocyte results in an additional exon in lymphocyte that suppresses protein translation. (E) Transcriptional regulation of LILRB1 and LILRB2 by epigenetic modifications, including ① histone acetylation and ② DNA methylation in promoter regions. (F) Post-translational regulatory mechanisms: ① FTO enhances LILRB4 expression via m6A demethylation, ② miR-103a-2-5p suppresses LILRB3 expression through mRNA degradation. (G) LILRB expression modulation by intercellular crosstalk, cytokines, pharmacological agents, and microbial infections.
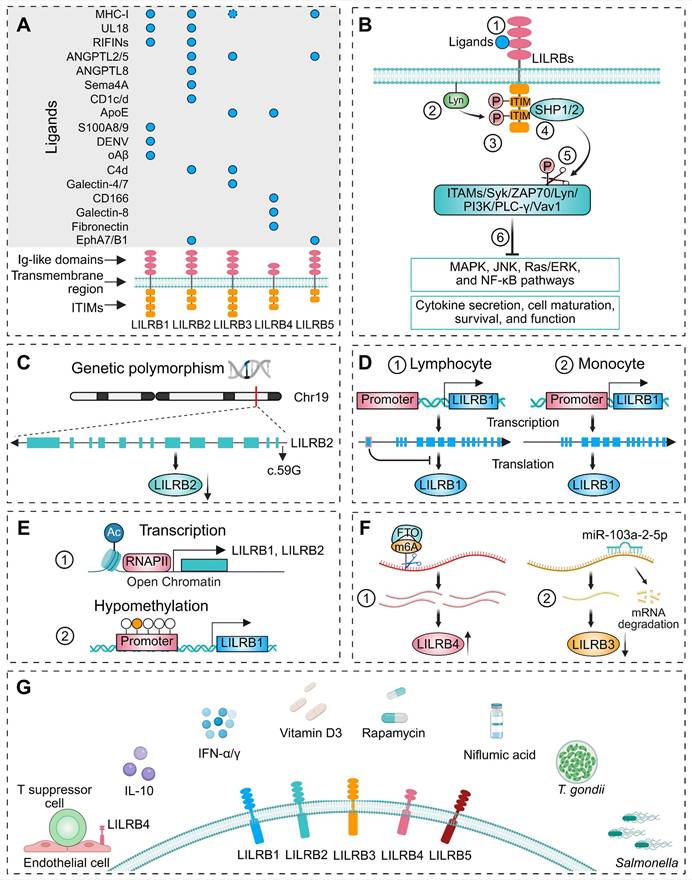
Anchored by a specific transmembrane region, LILRBs connect their extracellular ligand-binding domains to cytoplasmic ITIM-containing tails (Figure 1A). ITIMs are a class of conserved amino acid sequences located in the cytoplasmic tails of certain transmembrane receptors (Figure 1A), characterized by unique structural features. The canonical ITIM sequence is S/I/V/LxYxxI/V/L, where "x" denotes any amino acid [35]. Although the intracellular tails of LILRBs lack typical kinase domains, they exert inhibitory signaling by recruiting phosphatases such as SHP1 and SHP2 upon phosphorylation, thereby modulating immune responses.
2.2 Ligands of LILRBs
MHC-I molecules
MHC-I molecules play a pivotal role in immune surveillance by presenting intracellular peptides to T cells and NK cells [36]. These molecules are typically expressed on the surface of nucleated cells and consist of a trimeric complex formed by heavy chains (α1-α3) and an invariant light chain, β2m. The heavy chains span the cell membrane, while the domains distant from the membrane, including α1 and α2, form the peptide-binding groove [37]. The heavy chains of MHC-I molecules in human are part of the HLA system. Classical MHC-I molecules are encoded by classical MHC-I genes: HLA-A, HLA-B, and HLA-C, while non-classical MHC class I genes include HLA-E, HLA-F, and HLA-G [37]. β2m binds non-covalently to the extracellular portion of the heavy chain but does not directly interact with the cell membrane [38]. LILRB1 and LILRB2 transmit inhibitory immune signals by interacting with the α3 domain and the β2m subunit of classical MHC-I molecules (e.g., HLA-A and HLA-B) on the surface of target cells [38-40]. MHC-I molecules exist in two forms on the cell surface: the β2m-associated form and a free heavy chain with an open conformation [41]. Notably, LILRB1 specifically recognizes the β2m-associated form and fails to bind to MHC-I molecules lacking β2m [42]. In contrast, LILRB2 has broader ligand recognition and can interact with β2m-free heavy chains, such as those of HLA-B27 [43], HLA-C [44] and HLA-G [42, 45]. In contrast, the interaction between LILRB3 and MHC-I remains controversial. Several studies have reported that MHC-I molecules do not bind to LILRB3 [43, 44, 46], or that HLA-G fails to induce the conformational changes necessary for LILRB3 activation in myeloid cells, as shown in a recent study [47]. However, Ayukawa et al. demonstrated that LILRB3 on healthy epithelial cells binds to the α3 domain of MHC-I on transformed cells, triggering mechanical force via the SHP2/Rho-associated protein kinase 2 (ROCK2) pathway to promote extrusion and apoptosis of precancerous cells [48]. These findings suggest that the binding affinity and functional consequences of the LILRB3-MHC-I interaction are likely to be highly context-dependent, varying according to cell type and microenvironmental cues. LILRB5 has also been shown to bind to certain classical MHC-I molecules. Zhang et al. reported that the heavy chains of HLA-B7 and HLA-B27 can bind to LILRB5 in transduced B and rat basophil RBL cell lines [49].
Similar to classical HLA-I molecules, HLA-G comprises a heavy chain, β2m, and an 8-10 amino acid peptide derived from intracellular proteolytic degradation products [42]. However, it binds to LILRB1 and LILRB2 with greater affinity than classical MHC-I molecules [42, 45]. Through these interactions, HLA-G can inhibit cytotoxic T cells, NK cells, and B cells, induce T-cell anergy, regulate myeloid cell functions, and promote the generation and activation of regulatory T cells (Tregs) [50]. HLA-G exists in multiple membrane-bound and soluble isoforms, generated through alternative splicing or proteolytic cleavage [41]. Notably, HLA-G can form disulfide-linked homodimers, which are more effective than monomers at engaging LILRBs and transmitting inhibitory signals [51-54]. HLA-F, one of the least characterized non-classical MHC-I molecules, is primarily retained intracellularly in resting leukocytes. However, its surface expression has been observed in B lymphoblastoid and monocyte-derived cell lines [55]. Surface plasmon resonance studies have shown that tetrameric complexes of HLA-F can directly interact with LILRB1 and LILRB2 [56].
Membrane glycoprotein UL18 (UL18)
The human cytomegalovirus (CMV) UL18 gene encodes a highly glycosylated transmembrane protein that shares approximately 25% sequence homology with host MHC-I molecules [57]. UL18 is proposed to act as a viral decoy, mimicking MHC-I structure to engage killer-cell immunoglobulin-like receptors (KIRs) on NK cells and thereby inhibit NK cell-mediated cytotoxicity [58]. Similar to host MHC-I molecules, UL18 can bind β2m and endogenous peptides [28]. Structurally, UL18 contains α1, α2, and α3 domains in its extracellular region, with the α3 domain primarily interacting with the D1 domain of LILRB1 [59]. Although the binding mode of LILRB1 to UL18 resembles its interaction with classical MHC-I molecules, LILRB1 exhibits an affinity for UL18 that is over 1,000-fold higher than for either classical or non-classical MHC-I ligands [59, 60]. In addition to the binding sites for LILRB1 and the host-derived light chain, crystal structure analyses revealed that UL18 is extensively shielded by carbohydrate moieties, which prevent interactions with many potential binding partners [60]. Such structural adaptations enabled UL18 to outcompete host MHC molecules for LILRB1 binding, thereby disrupting immune surveillance mechanisms [60]. Despite the high sequence homology among LILRB family members, only LILRB1, and to a lesser extent LILRB2, can bind UL18 [26, 33, 61].
Repetitive interspersed families of polypeptides (RIFINs)
RIFINs are variant antigens encoded by the rif gene families in Plasmodium falciparum (P. falciparum) and expressed on the surface of infected red blood cells [62]. These proteins contribute to immune evasion by binding to LILRB1, thereby inhibiting the activation of B cells and NK cells [63]. The extracellular domain of RIFINs comprises a relatively conserved N-terminal region, which includes the FHEYDER sequence, and a highly variable C-terminal region [63]. Although RIFINs share no sequence or structural homology with HLA-I molecules, Harrison et al. demonstrated that their variable regions can interact with the D1 and D2 domains of LILRB1, mimicking the binding pattern of classical MHC-I molecules [64]. Further structural analyses by Chen et al. revealed that RIFINs also directly engage the D3 domain of LILRB1 via specific amino acid residues or conformational features within their variable regions [65]. These findings suggest that the polygenicity and polymorphism of the RIFIN family reflect strong selective pressure to evade the antibody response while evolving multiple mechanisms to engage inhibitory receptors [65]. Although LILRB2 shares significant structural similarity with LILRB1, exhibiting 81% amino acid homology in the extracellular region [33, 42], functional assays indicate a distinct binding pattern. Blocking assays with anti-LILRB2 antibodies and binding assays with recombinant LILRB2 proteins have shown that RIFINs primarily interact with the D3 domain of LILRB2, rather than D1 or D2 domain [66]. Elucidating the interactions between RIFINs and LILRB1/LILRB2 not only sheds light on the immune evasion strategies of P. falciparum but also provide novel insights for the development of malaria treatments and vaccines.
Angiopoietin-like proteins (ANGPTLs)
ANGPTLs are a class of proteins structurally similar to angiopoietins and play a significant role in regulating angiogenesis [67]. Unlike angiopoietins, ANGPTLs do not bind to the angiopoietin receptors Tie1 or Tie2, which led to their initial classification as "orphan ligands" [68, 69]. In 2012, Zheng et al. identified LILRB2 as the receptor for ANGPTL2 and ANGPTL5, demonstrating that signaling through this receptor maintains hematopoietic stem cell (HSC) stemness and promotes leukemia development [70]. A subsequent study identified that the HGY*C motifs within the D1 and D4 domains of LILRB2 are essential for binding ANGPTL2 and activating downstream signaling pathways [71]. Notably, the interaction between LILRB2 and ANGPTL2 exhibited a higher binding affinity than that with HLA-G, with partially overlapping yet distinct binding sites on LILRB2 [71]. Although ANGPTL2 and ANGPTL5 also interacted with LILRB3 and LILRB5, the binding affinity was lower than that of ANGPTL2 and LILRB2 [72]. ANGPTL8 is predominantly expressed in human liver [73] and is closely associated with non-alcoholic fatty liver disease in both mice and humans [74]. ANGPTL8 bound its receptor PirB/LILRB2 to regulate macrophage migration in nonalcoholic steatohepatitis (NASH), and its hepatocyte-specific deletion reduced macrophage infiltration, lipid accumulation, and fibrosis progression in NASH mice [75].
Semaphorin-4A (Sema4A)
Sema4A was initially identified in developing embryos, with its transcriptional levels progressively increasing throughout embryonic development [76]. This protein plays critical roles in neural development and immune regulation [77]. Lu et al. demonstrated that human SEMA4A co-stimulates CD4+ T cells and regulates T helper 2 (Th2) cell differentiation by binding to LILRB2 on activated CD4+ T cells [77].
CD1 family
The human CD1 family consists of four functional subtypes: CD1a, CD1b, CD1c, and CD1d. These glycoproteins are expressed on the cell surface and function as the third class of antigen-presenting molecules [78]. Structurally, CD1 molecules resemble MHC-I molecules, notably through the non-covalent association of their heavy chains with β2m [79, 80]. LILRB2 functions as an inhibitory receptor for CD1c and CD1d, suppressing immune responses triggered by these molecules [80, 81].
Apolipoprotein E (ApoE)
ApoE is a plasma protein that acts as a ligand for the low-density lipoprotein (LDL) receptor, facilitating the transport of cholesterol and other lipids between diverse cell types [82]. In terms of immune regulation, studies have presented conflicting conclusions regarding the relationship between ApoE and immune receptors. One study demonstrated that among ApoE isoforms, ApoE4 can bind a variant of LILRB3 and activate human microglia HMC3 cells in a LILRB3-dependent manner, promoting a proinflammatory state [83]. However, Huang et al. reported that no ApoE isoform, including ApoE4, activate LILRB3 reporter cells [47]. In contrast, the same study found that all ApoE isoforms could activate LILRB4 reporter cells, consistent with prior research [22, 47, 84]. Given these discrepancies, Huang et al. proposed that while ApoE may bind LILRB3 with measurable affinity, this interaction—under their experimental conditions—failed to induce the functional conformational changes necessary for LILRB3 activation [47].
Other ligands
Several studies have demonstrated that LILRB1 can bind not only to the calcium-binding proteins S100 calcium binding protein A8 (S100A8) and S100A9 [85], but also to products of the Dengue virus (DENV) [86]. In a mouse model of Alzheimer's disease (AD), oligomeric β-amyloid (Aβ) interacted with LILRB2, leading to synaptic loss [87]. Additionally, LILRB2 and LILRB3 can interact with soluble monomeric complement component C4d, mediating its endocytosis [88]. Using a LILRB3 reporter cell system, Huang et al. demonstrated that galectin-4 and galectin-7 can activate LILRB3 and induce the downstream SHP1/2 signaling [47]. LILRB4 binds ligands including activated leukocyte cell adhesion molecule (ALCAM)/CD166 [89], galectin-8 [90], and fibronectin [91, 92], regulating downstream cellular activities. A recent study demonstrated that LILRB5 (and partially LILRB2) forms specific interactions with Eph receptors (EphA7 and EphB1), triggering bidirectional signaling through phosphorylation of both ITIM domains in LILRB5 and Eph receptors [93]. This crosstalk induced an immunosuppressive phenotype in myeloid cells, and suppresses antitumor T cell responses, therefore supporting tumor progression.
2.3 LILRBs and signal transduction pathways
LILRB receptors interact with various ligands, including MHC-I molecules and other immune-related ligands, initiating inhibitory signal transduction through ITIMs within their cytoplasmic domains [18, 23, 94, 95]. The Src family kinases, particularly Lck/Yes-related novel protein tyrosine kinase (Lyn), play a central role in LILRB signaling [96, 97]. Upon receptor engagement, Lyn undergoes autophosphorylation and subsequently phosphorylates the tyrosine residues within the ITIMs (Figure 1B) [20]. These phosphorylated residues serve as docking sites for critical negative regulators SHP1/2 [98], which, upon recruitment, dephosphorylate key signaling molecules [99, 100]. These molecules include immunoreceptor tyrosine-based activation motifs (ITAMs), Src family kinases, spleen tyrosine kinase (Syk), zeta-chain-associated protein kinase 70 kDa (ZAP70), Lyn, phosphatidylinositol-4-phosphate 3-kinase (PI3K), phospholipase C gamma (PLC-γ), and Vav guanine nucleotide exchange factor 1 (Vav1) (Figure 1B) [97, 101-104]. This dephosphorylation inhibits downstream signaling pathways, such as mitogen-activated protein kinase (MAPK), c-Jun N-terminal kinase (JNK), Ras/extracellular signal-regulated kinase (ERK), and NF-κB pathways, ultimately suppressing cytokine secretion, effector cell maturation, survival, and function (Figure 1B) [97, 98]. The following sections will delve into the interactions between LILRBs, their ligands, and downstream signaling molecules in specific diseases and cell types.
3. Upstream Modulators of LILRBs
Recent studies have demonstrated that polymorphisms in LILRBs significantly contribute to the pathogenesis and progression of multiple diseases [32, 94, 105-111]. Polymorphisms can alter gene sequences, affecting transcription factor binding, mRNA processing, and translation, thereby modulating gene expression levels. Additionally, polymorphisms in coding regions may lead to amino acid substitutions that disrupt protein folding and structural stability. Notably, these disease-associated polymorphisms exhibit distinct ethnic distributions [110, 112-114]. For instance, Hirayasu et al. identified LILRB2 polymorphisms within 5'-UTR and signal sequence regions in northeast Asian populations that directly influence its expression (Figure 1C) [113]. Flow cytometry confirmed the association between the common LILRB2 c.59G allele and its lower expression [113]. In Brazilian populations, next-generation sequencing identified six amino acid substitutions in LILRB1 and five in LILRB2 [114]. These allelic variations may affect the structure and/or stability of these molecules, with LILRB2 demonstrating higher average stability. Collectively, these findings provide critical insights into population-specific functional diversification of LILRBs. LILRBs are most highly expressed in myeloid hematopoietic cells, with relatively lower expression in non-hematopoietic tissues, though the precise mechanisms underlying these patterns remain incompletely understood [18, 21]. LILRB expression is regulated at multiple levels, including transcriptional, epigenetic, and post-transcriptional mechanisms, each contributing to the cell type-specific expression profiles of these receptors. For instance, LILRB1 expression was controlled by cell-specific promoters: a lymphocyte-specific promoter located approximately 13 kb upstream of the monocyte-specific promoter introduced a unique exon into the 5'-UTR, which inhibited protein translation, resulting in lower protein levels in lymphocytes compared to monocytes (Figure 1D) [115]. In multiple myeloma (MM), the transcription factor Ikaros family zinc finger protein 1 (IKZF1) directly activated LILRB4 transcription [116]. Beyond transcriptional regulation, epigenetic modifications also play a crucial role in controlling LILRB expression by modulating chromatin accessibility at gene promoters. In addition to transcriptional regulation, epigenetic modifications further fine-tune LILRB expression. These modifications, such as histone acetylation and DNA methylation, contribute to gene expression by altering chromatin structure and accessibility. The promoters of LILRB1 and LILRB2 share several cis-elements and trans-factors, but their activation mechanisms differ. LILRB1 transcription relies on Sp1-family binding to its GC-box, while LILRB2 is tightly regulated by histone acetylation at its core promoter, which restricts its expression to myeloid lineage cells (Figure 1E) [117]. Another critical epigenetic modification is DNA methylation, with TCGA analysis revealing a significant association between LILRB1 promoter hypomethylation and its elevated expression in low-grade glioma and glioblastoma (Figure 1E) [118]. Post-transcriptional mechanisms, including RNA modifications and microRNA-mediated regulation, also contribute to the fine-tuning of LILRB expression. For instance, the N6-methyladenosine (m6A) demethylase FTO stabilized LILRB4 mRNA by removing m6A modifications, thereby promoting LILRB4 expression and contributing to leukemia stem cell self-renewal and immune evasion (Figure 1F) [119]. Similarly, in acute myeloid leukemia (AML), overexpression of miR-103a-2-5p suppressed LILRB3 expression, inhibiting AML cell growth and reducing apoptosis in CD8+ T cells (Figure 1F) [120].
LILRB expression is dynamically regulated through cell-cell interactions, immune signaling, and exogenous factors (Figure 1G). Alloantigen-specific CD8+CD28- T suppressor cells upregulated LILRB4 in endothelial cells to promote immune tolerance (Figure 1G) [121]. Mechanistic studies revealed that in quiescent endothelial cells, LILRB4 pre-mRNA is retained in the nucleus. Upon interaction with T suppressor cells or exposure to interleukin-10 (IL-10) and interferon-alpha (IFN-α), LILRB4 pre-mRNA processing was activated, leading to LILRB4 protein production [122]. Beyond cell-cell interactions, immune signals further modulate LILRB expression via inflammatory stimuli, cytokines, and growth factors (Figure 1G) [123]. In DCs, IL-10 [124] and IFN-γ [125, 126] have been shown to enhance LILRB expression, as well as pharmacological agents such as vitamin D3 [126, 127], rapamycin [128], and certain non-steroidal anti-inflammatory drugs (e.g., niflumic acid) [129] can also upregulate LILRB expression (Figure 1G). Microbial infections, including Toxoplasma gondii (T. gondii) [130] and Salmonella [131], also regulate LILRB expression (Figure 1G). During pregnancy, T. gondii infection downregulated macrophage LILRB4, driving proinflammatory M1 polarization while suppressing M2-type immunoregulatory tolerance [130, 132, 133]. This shift altered M1/M2 surface markers, dysregulated arginine metabolic enzymes, and modified cytokine secretion profiles, ultimately disrupting fetal-maternal tolerance and contributing to adverse outcomes including preterm birth, miscarriage, and anencephaly [130].
These findings collectively delineate the multifaceted regulatory landscape of LILRB expression, spanning genetic polymorphisms, post-transcriptional modifications, immune cell interactions, and environmental stimuli. Such intricate regulatory networks underscore the pivotal role of LILRBs in modulating immune homeostasis, providing mechanistic insights into immune evasion, immune tolerance, and related biological processes. This refined understanding advances fundamental knowledge of immune regulation and elucidates the mechanisms underlying immune-related disorders, highlighting LILRBs as potential therapeutic targets.
4. LILRBs and Immune System
LILRBs constitute a class of inhibitory receptors broadly expressed on immune cells—including monocytes, macrophages, NK cells, T cells, and B cells [134]—that critically modulate both innate and adaptive immunity [18, 27, 135]. Emerging evidence reveals their dual role in regulating antigen-presenting cell (APC) functions while simultaneously suppressing cytotoxic anti-tumor responses and actively remodeling the TME [21]. Notably, certain LILRBs are also expressed on tumor cells, where they directly influence cancer initiation, progression, therapeutic resistance, recurrence, and cancer stem cell behavior [97]. This section explores LILRB-mediated immunomodulation, with a particular focus on their mechanisms of action within the TME.
4.1 Innate immunity
HSCs in the bone marrow (BM) serve as the progenitors of immature myeloid cells (IMCs), which subsequently differentiate into monocytes—capable of further maturing into macrophages and DCs—as well as granulocytes, including neutrophils, basophils, and eosinophils [136]. Innate immune cells, encompassing myeloid-derived cells (e.g., monocytes, DCs, and macrophages) and innate lymphoid cells such as NK cells, critically mediate innate immunity and orchestrate inflammatory responses [137-139].
DCs serve as pivotal APCs bridging innate and adaptive immunity, initiating immune responses while maintaining immune tolerance [140, 141]. DCs exist in various subsets and functional states, with immature forms primarily involved in establishing immune tolerance and mature forms critical for inducing protective T-cell-mediated immunity [142]. LILRB expression on DCs is dynamically regulated by inflammatory stimuli, cytokines, and growth factors, and is typically downregulated upon DC activation [123]. For instance, HLA-G binding to LILRB2 suppressed DC activation and downregulated co-stimulatory molecules (CD80, CD86, CD83, and MHC class II) (Figure 2A) [143], recruiting SHP1/2 to inhibit monocyte-to-DC differentiation via the IL-6/STAT3 signaling pathway (Figure 2A) [144]. Similarly, LILRB1 and LILRB4 were downregulated upon DC activation, suggesting their involvement in regulating DC maturation [123]. Persistent LILRB1 ligation during monocyte culture generated tolerogenic DCs (tDCs), which interacts with Tregs and suppresses T-cell responses [145]. Complementary to this, LILRB2+ DCs inhibited the differentiation of T helper 1 (Th1) cells and cytotoxic T lymphocytes (CTLs), while promoting the generation of Th2 and type 2 cytokine-secreting CD8+ T cells (Figure 2A) [146-148]. Additionally, LILRB2 contributed to the induction of various Treg subsets, including CD4+CD25+ Tregs, CD8+CD28- inhibitory T cells, and type 1 Tregs, which are critical for maintaining immune tolerance to both self- and non-self-antigens (Figure 2A) [149-151]. In the dermis, CD14+ DCs expressing LILRB1 and LILRB2 exhibited a reduced capacity to induce cytotoxic CD8+ T cells, compared to LILRB-negative Langerhans cells, and blockade of LILRB1 and LILRB2 significantly enhanced CTL generation [148].
Roles of LILRBs in innate immunity, adaptive immunity, and tumor-immune crosstalk. (A) Distribution of LILRBs across innate immune cells (left; including dendritic cells (DCs), macrophages, myeloid-derived suppressor cells (MDSCs), neutrophils, and natural killer (NK) cells) and adaptive immune cells (right; including T and B lymphocytes). The diagram highlights associated ligands, signaling pathways, and immunomodulatory outcomes. (B) LILRB-mediated tumor-immune crosstalk. The left panel depicts tumor cell-derived ligands engaging LILRB-expressing immune cells, modulating anti-tumor responses. The right panel illustrates how tumor cells, upon stimulation by exogenous ligands, activate intracellular signaling pathways (e.g., SHP2/CaMK1, PI3K/AKT, MAPK, NF-κB, and JAK/STAT) to regulate tumor progression and immune evasion.
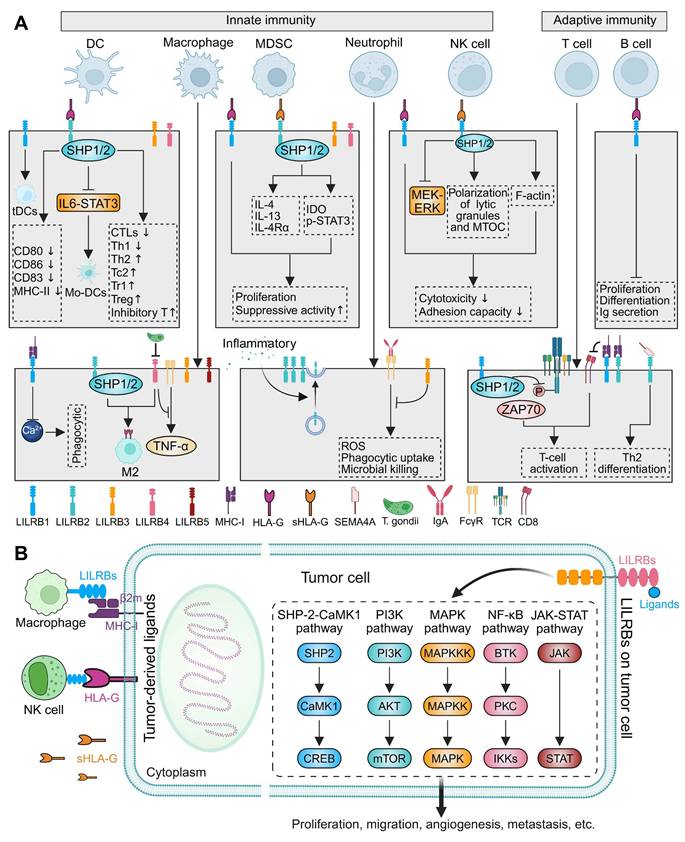
Macrophages, another pivotal mediator of innate and adaptive immunity, primarily derive from circulating monocytes and polarize into M1 or M2 subtypes in response to various stimuli [152, 153]. M1 macrophages are closely associated with inflammatory responses, while M2 macrophages are primarily involved in anti-inflammatory processes, tissue repair and immunoregulation [153]. The M2 spectrum includes tumor-associated macrophages (TAMs/M2d), which drive tumor progression, angiogenesis, and metastasis [153]. LILRBs critically regulate macrophage function by modulating polarization, phagocytic capacity, and intercellular signaling. In TAMs, LILRB1 and LILRB2 are significantly upregulated, while blocking these receptors enhances macrophage phagocytosis (Figure 2A) [154, 155]. LILRB1 inhibition may disrupt calcium mobilization following MHC-I engagement, impairing macrophage activation [27]. LILRB2 promotes the polarization of TAMs toward M2-like phenotypes, which are associated with immune suppression and tumor progression (Figure 2A). In contrast, LILRB2 blockade repolarized TAMs to M1-like phenotypes by suppressing SHP1/2 phosphorylation and activating the NF-κB pathway, thereby augmenting anti-tumor immunity [155, 156]. LILRB3, predominantly expressed on myeloid cells, especially monocytes, when agonistically bound by monoclonal antibodies to Ig-like domains 2 or 4, suppresses M1-inflammatory pathways and induces an M2-like immunosuppressive phenotype, leading to reduced T cell proliferation and immune suppression [157]. LILRB4 and LILRB5 are also expressed in macrophages [21, 99, 158]. In a midgestation murine model, macrophage-expressed gp49B (LILRB4 homolog) colligated with FcγRI to suppress tumor necrosis factor (TNF)-α production, maintaining maternal-fetal immune tolerance (Figure 2A) [159]. During T. gondii infection, LILRB4 downregulation on decidual macrophages promoted M1 polarization and impaired M2-mediated tolerance functions (Figure 2A) [130]. Conversely, NLRP12-induced LILRB4 upregulation in TAMs promoted M2 polarization within tumors [160].
Myeloid-derived suppressor cells (MDSCs) are a population of cells derived from IMCs under conditions of chronic inflammation, cancer, and autoimmune diseases. In these pathological states, persistent inflammatory signals disrupt their normal differentiation pathways and induce pathological activation. MDSCs exhibit an immature phenotype, reduced phagocytic activity, and potent immunosuppressive functions, enabling them to effectively suppress immune responses [136]. HLA-G has been proven to promote MDSC expansion and suppressive function through its interaction with LILRB1, which increases the secretion of IL-4 and IL-13 and upregulates IL-4Rα, leading to T cell proliferation inhibition (Figure 2A) [161]. Additionally, soluble HLA-G (sHLA-G) binding to LILRB2 on granulocytic MDSCs, enhancing indoleamine-2,3-dioxygenase (IDO) expression and promoting STAT3 phosphorylation, thus facilitating MDSC accumulation and their immunosuppressive activity (Figure 2A) [162]. LILRB3 is functionally expressed on immunosuppressive myeloid cells from cancer patients, and targeted inhibition of this receptor has been shown to reverse MDSC-mediated immunosuppression [47, 163]. Furthermore, LILRB4 expression on monocytic MDSCs plays a crucial role in mediating cancer-related immunosuppression, and its blockade can reverse this suppression, thereby enhancing the efficacy of immune checkpoint inhibitors (ICIs) [164].
Neutrophils play activating, regulatory, and effector roles in both innate and adaptive immunity [165, 166]. As the most abundant granulocytes, their activation and function are finely regulated by LILRBs [20]. Neutrophil granules contain various membrane receptors, which can be translocated to cell surface through exocytosis [167]. In response to inflammatory stimulation, LILRB2 expression on the neutrophil surface was rapidly upregulated via granule exocytosis, thereby enhancing HLA-G-mediated suppression of neutrophil phagocytic function (Figure 2A) [167]. Additionally, LILRB3 is highly expressed on resting neutrophils and is shed following activation [168, 169]. Sustained LILRB3 ligation inhibited key IgA-mediated effector functions, including reactive oxygen species (ROS) production, phagocytosis, and microbial killing (Figure 2A) [168, 169].
NK cells defend against infections and tumors through direct lysis of target cells and the secretion of immune mediators [170]. The effector functions of NK cells are regulated by MHC-I molecules, whereby target cells expressing MHC-I exhibit heightened resistance to NK-mediated killing relative to MHC-I-deficient cells [171]. This regulatory mechanism is mediated by the interaction between HLA-I molecules and inhibitory NK receptors [172]. Upon ligand binding, these inhibitory receptors undergo tyrosine phosphorylation at their intracellular ITIM domains, recruiting the tyrosine phosphatase SHP1, which suppresses the effector functions driven by activating receptors on NK cells [171]. Conversely, in the absence of inhibitory signals, the engagement of activating receptors triggers target cell lysis and cytokine secretion [171]. LILRB1 inhibited NK cell cytotoxicity by binding HLA-G1, and its blockade conferred partial protection against NK cells (Figure 2A) [173-175]. In vitro, soluble HLA-G1α binding to LILRB1 inhibited downstream signaling molecules, including MEK and ERK, thereby suppressed NK cell cytotoxicity (Figure 2A) [176]. Moreover, the interaction between HLA-G and LILRB1 disrupted polarization of NK cell lytic granules and the microtubule organizing center (MTOC), along with filamentous actin (F-actin) accumulation at the contact site, severely impaired NK cell cytotoxicity (Figure 2A) [177]. Additionally, LILRB1 binding HLA-G expressed on endothelial cells attenuated the rolling adhesion capacity of NK cells (Figure 2A) [178].
4.2 Adaptive immunity
Innate immunity serves as the first line of defense, rapidly detecting and eliminating pathogens. In contrast, adaptive immunity employs sophisticated mechanisms to distinguish self from non-self, ensuring precise and specific immune responses [179, 180]. This system relies on the orchestrated interplay of APCs, T lymphocytes, and B lymphocytes [180]. Crucially, LILRBs serve as key immunoregulators, modulating both T- and B-cell functions through inhibitory signaling.
LILRB1 is broadly expressed on the surface of most T cells and within the cytoplasmic vesicles of all T lymphocytes [181, 182]. When LILRB1 was blocked with specific mAbs, the proliferation of antigen-specific polyclonal CD4+ T cells significantly increased, along with enhanced secretion of IL-2, IFN-γ, and IL-13 [183]. Mechanistically, LILRB1 suppresses T-cell activation by recruiting SHP1/2, which dephosphorylate the TCR-ζ chain, thereby inhibiting the ZAP70-mediated downstream signaling cascade (Figure 2A) [184, 185]. In addition, human SEMA4A, expressed on APCs, bound to LILRB2 expressed on CD4+ T cells, enhancing their activation and facilitating Th2 cell differentiation (Figure 2A) [77]. Both LILRB1 and LILRB2 can compete with CD8 to bind MHC-I, thereby restricting T-cell activation signals (Figure 2A) [186]. While immunogenic DCs drove naïve T-cell differentiation into effector subsets, high LILRB2 expression promoted a tDC phenotype [147]. As previously discussed, these tDCs potently suppress T-cell differentiation and function, reinforcing immune tolerance. In summary, LILRBs regulate T-cell differentiation, activation, and function via multiple pathways, maintaining a critical balance between immune tolerance and inflammatory responses.
B lymphocytes play a central role in immunity through antibody production and secretion of immunomodulatory cytokines that regulate both innate and adaptive immune cells [187]. Emerging evidence implicates LILRBs in B-cell biology, with elevated expression closely linked to B-cell differentiation [188]. While LILRB1 is ubiquitously expressed in peripheral B cells, it indirectly regulates B-cell activity via modulating APC and T-cell responses [20, 188]. To date, LILRB1 remains the only family with well-defined functions in B cells [20]. Both in vitro and in vivo studies demonstrated that HLA-G binds to LILRB1 to inhibit B-cell proliferation, differentiation, and Ig secretion (Figure 2A) [189]. In addition, elevated sHLA-G in acquired aplastic anemia suppressed BM B-cell proliferation via LILRB1 engagement. Importantly, blocking HLA-G/LILRB1 interactions restores normal proliferative capacity of B cells in BM [190].
4.3 Tumor immunity
Cancer represents a complex ecosystem comprising tumor cells and diverse non-tumor elements. These non-tumor cells, embedded within the tumor extracellular matrix, collectively form a dynamic environment that includes immune cells, cancer-associated fibroblasts, endothelial cells, pericytes, and other tissue-specific cell types [191]. Dynamic interactions between tumor cells and the TME critically drive tumorigenesis and malignant progression [192]. While effective immune responses can eliminate or suppress malignant cells, tumor frequently evade immune surveillance through diverse mechanisms that compromise immune effector functions and subvert anti-tumor immunity [9, 193, 194]. LILRBs are predominantly expressed on myeloid-derived cells within the TME, and their signaling activation can promote tumor immune escape [20, 156, 195, 196]. Aberrant LILRB expression has also been observed in various cancers, particularly hematological malignancies [22, 47, 116, 156, 195-200]. In such contexts, LILRBs may directly regulate cancer initiation, drug resistance, recurrence, and the activity of cancer stem cell [97]. Below, we concisely overview LILRB and ligand expression patterns, multifaceted roles, and potential mechanisms in both tumor immune cells and tumor cells.
Crosstalk between tumor cell-derived ligands and LILRB-expressing immune cells
Cancer arises from genomic instability, which generates tumor antigens through mutations and structural alterations, thereby eliciting cellular immune responses [9]. Effective tumor-specific CD8+ T-cell responses depend on recognition of tumor antigens presented by MHC-I or HLA molecules [201]. The interaction between MHC-I and immune cells within the TME exhibits a dual role. While tumors commonly evade CD8+ T-cell surveillance by downregulating classical MHC-I expression, overexpression of β2m-complexed MHC-I heavy chains can engage LILRBs on immune cells—including NK cells and TAMs, releasing a "Don't Eat Me" signal that suppresses innate immune functions (Figure 2B) [37, 38]. Although classical HLA I molecule downregulation is a common feature in many cancers, elevated surface HLA-G and increased plasma sHLA-G are observed in both hematological and solid malignancies (Figure 2B) [202, 203]. For instance, a proportion of gastric cancers (GCs) exhibited HLA-G/sHLA-G expression [204-206]. HLA-G overexpression corelated with decreased number of NK cells through inhibiting the cell proliferation and cytotoxic activity, as well as reducing IFN-γ and TNF-α secretion through its interaction with LILRB1 [205]. Similarly, elevated sHLA-G in pancreatic cancer inversely correlated with peripheral activated T cells (Figure 2B) [207]. Collectively, tumor cells exploit interactions with LILRB-expressing immune cells via β2m, sHLA-G, and membrane-bound HLA-G to induce immune tolerance at various stages of the immune response [50, 202].
The dual role of LILRBs in tumor immunity and progression
LILRBs are primarily expressed on myeloid and certain hematopoietic cells, where they critically regulate immune responses [20, 21]. However, recent studies revealed their abnormal expression in tumor cells themselves, directly contributing to tumor maintenance and progression [208]. Aberrant LILRB expression is well-documented in hematological malignancies, including AML [209-212], chronic monocytic leukemia [213], and MM [214, 215], where genetic knockdown of LILRBs significantly inhibits proliferation and migration. Conversely, lymphocyte-derived tumors, such as chronic lymphocytic leukemia (CLL) [216], acute lymphoblastic leukemia (ALL) [217], and acute T-cell leukemia (ATL) [89] exhibit reduced LILRB expression. In solid tumors, LILRBs are specifically expressed or upregulated across diverse lineages: digestive [218-221], respiratory [222-224], reproductive [219, 225], urinary [226, 227], and nervous system [118], with expression levels correlating closely with disease progression and prognosis. Mechanistically, tumor-expressed LILRBs drive key oncogenic functions, including proliferation, migration, angiogenesis, and metastasis, through activation of multiple pathways such as SHP2/calcium/calmodulin-dependent protein kinase 1 (CaMK1) [72, 223, 228], PI3K/AKT [224, 229], MAPK/ERK [230, 231], NF-kB [230, 232, 233], and JAK/STAT [118, 197] (Figure 2B).
In summary, LILRBs exhibit a complex dual immunoregulatory role in tumor immunity. As inhibitory receptors on immune cells, they engage tumor-expressed ligands, to trigger immunosuppressive signaling that establishes an immunosuppressive milieu. Conversely, aberrant LILRB expression in tumor cells directly drives tumor proliferation, migration, and metastatic dissemination through activation of diverse oncogenic signaling cascades. These findings collectively establish LILRBs as critical determinants of TME dynamics and highlight their significant therapeutic promise in oncology.
5. Roles of LILRBs in various diseases
5.1 LILRBs and infectious diseases
LILRBs critically regulate immune responses by suppressing effector functions of DCs, macrophages, MDSCs, and NK cells. Pathogens have evolved mechanisms to exploit this immunosuppressive axis to evade the host immunity. Disruption of pathogen-associated ligand-LILRBs interactions can shorten infection cycles and reduces pathogen survival [18]. Understanding how LILRB-mediated immune evasion provides valuable insights into the pathogenesis of infectious disease and informs the development of potential immune-based therapeutic strategies. The following sections detail the major mechanisms by which LILRBs modulate infectious disease outcomes (Table 1).
The role of LILRB family in infectious, autoimmune and neurodegenerative diseases.
| Diseases | LILRBs | Ligands | Expressing cells | Mechanisms | References |
|---|---|---|---|---|---|
| Viral infections | |||||
| CMV | LILRB1 | UL18 | Cytotoxic T cell | Lysis of CMV-infected cells | [238] |
| UL18 | NK cell | Inhibit NK cytotoxic activity | [236] | ||
| UL18 | DC | Inhibit DC migration, maturation, and allogeneic T cell proliferation | [145, 237] | ||
| Lymphocyte | CMV reactivation after lung transplantation | [239] | |||
| HIV-1 | LILRB2 | Monocyte | Impair monocyte antigen presentation | [124] | |
| HLA-B | Myelomonocytic cell | Induce myelomonocytic tolerogenesis | [241] | ||
| HLA-I | DC | Dysregulation of cDCs | [243] | ||
| HLA-G | Myeloid DC | Modulate DC antigen presentation and proinflammatory cytokine secretion | [244] | ||
| LILRB1, LILRB3 | Myeloid DC | NA | [247] | ||
| LILRB1 | S100A9 | NK cell | Enhance NK cell anti-HIV-1 activity | [85] | |
| DENV | LILRB1 | Enhance DENV replication | [86] | ||
| Overcome cell-autonomous immunity, boosting DENV intracellular survival | [251] | ||||
| Resistant to TLR/anti-IgM stimulation in CD19+CD27+ naïve B cells | [252] | ||||
| SARS-CoV-2 | LILRB4 | MDSC | Elevated inflammation and regulatory B/T cell accumulation | [255] | |
| EBV | LILRB1 | CD8+ T cell | Inhibiting IFN-γ production to impair immunity | [182, 257] | |
| CHB virus | LILRB1 | NK cell | Reduce NK cell degranulation and IFN-γ production | [258] | |
| Zika virus | LILRB1, LILRB2 | Mutations impact in utero transmission chances | [108] | ||
| Reovirus | LILRB2/PirB | Essential for reovirus replication and neuropathogenicity | [259] | ||
| Bacterial infections | |||||
| Mycobacterium tuberculosis | LILRB1 | NK cell | Decrease CD107a and IFN-γ expression | [260] | |
| LILRB2 | MDSC | Transition between immunosuppressive and proinflammatory states | [261] | ||
| LILRB5 | Cytotoxic T cell | Enhance cytotoxic T cell proliferation | [263] | ||
| Staphylococcus aureus | LILRB1, LILRB3/PirB | Macrophage | Influence TLR-mediated cytokine production | [264] | |
| LILRB3 | Neutrophil | Regulate neutrophil activation and antimicrobial functions | [169] | ||
| Salmonella | LILRB2, LILRB4 | Antigen presenting cell | Induce tolerogenic antigen-presenting cells | [131] | |
| Escherichia coli | LILRB1, LILRB4, PirB | Macrophage | Enhanced proinflammatory cytokine production and MAPK/NF-kB activation | [265] | |
| Parasite infections | |||||
| Plasmodium falciparum | LILRB1 | RIFIN | NK and B cells | Regulate IgM production and NK-mediated killing | [63, 64] |
| B cell | Modulates inflammatory cytokine release | [274] | |||
| LILRB2 | RIFIN | Modulate immune response for immune evasion | [66] | ||
| Toxoplasma gondii | LILRB1 | HLA-G | NK cell | NA | [277] |
| LILRB4 | MDSC | Alter ARG1 and IL-10 expression via SHP2/STAT6, leading to MDSC dysfunction | [278] | ||
| Trypanosoma cruzi | LILRB1 | CD4+ T cell | Modulate parasite-specific T cell response | [279] | |
| Autoimmune diseases | |||||
| Autoimmune thyroid diseases | LILRB1 | CD4+/CD8+ T cell | Disrupt IL-10 synthesis and cell proliferation | [281] | |
| Rheumatoid arthritis | LILRB1 | HLA-G | CD8+ T cell | Regulate CD8+ T cell activation | [283] |
| Soluble HLA-G | Binding capacity dictates anti-inflammatory protection | [284] | |||
| Systemic lupus erythematosus | LILRB1 | Peripheral blood B cell | Impaired LILRB1 expression and function | [285] | |
| LILRB4 | Monocytoid DC | Loss-of-function polymorphisms affect inflammatory cytokine levels | [106] | ||
| LILRB4 | Plasmablast and plasma cell | Ectopically expressed | [286] | ||
| Multiple sclerosis | LILRB1 | HLA-G | Modulate T-cell infiltration and immune-inflammatory response | [287] | |
| LILRB4 | Influence Th1/Th17 proliferation and proinflammatory factor release | [289] | |||
| Neurodegenerative disease | |||||
| Alzheimer's disease | LILRB2/PirB | Aβ oligomers | Causes synaptic toxicity and impaired developmental plasticity | [291] | |
| LILRB2 | Aβ oligomers, L-α-phosphatidylserine | Microglia | Inhibits TREM2 signaling | [292] | |
| LILRB4 | ApoE | Microglia | Dampening microglia phagocytosis of amyloid plaques | [84] |
Aβ, β-amyloid; ApoE, apolipoprotein E; ARG, Arginase-1; CHB, chronic hepatitis B; CMV, cytomegalovirus; DC, dendritic cell; DENV, dengue virus; EBV, Epstein-Barr virus; HIV-1, human immunodeficiency virus type 1; HLA, human leukocyte antigen; IFN-α, interferon-alpha; IL-10, interleukin-10; MDSC, myeloid-derived suppressor cell; NK, natural killer; RIFINs, repetitive interspersed families of polypeptides; S100A9, S100 calcium binding protein A9; SARS-CoV-2, severe acute respiratory syndrome coronavirus 2; TLR, toll-like receptor.
Viral infections
Cytomegalovirus (CMV)
CMV, a β-herpesvirus, encodes immunomodulatory genes that subvert both innate and adaptive immunity [234]. While establishing lifelong latency in immunocompetent hosts, CMV caused severe complications in immunocompromised individuals [235]. The viral UL18 protein shares structural homology with MHC-I and exhibits high-affinity binding to LILRB1 (but weak binding to LILRB2) on immune cells [59, 61]. During early infection, UL18 inhibited cytotoxicity of LILRB1+ NK cells, thereby protecting infected cells from destruction [109]. Notably, UL18 also activated a subset of LILRB1-NK cells through a LILRB1-independent mechanism, which can counterbalance the inhibitory effect of UL18 on LILRB1+ NK cells in the global NK cell response [236]. Furthermore, UL18 engaged LILRB1 on DCs, impairing DC maturation and migration, and inhibiting subsequent allogeneic T-cell proliferation [237]. Beyond immune evasion, CMV manipulates cell death pathways to facilitate viral spread. Crucially, LILRB1+ cytotoxic T cells lysed UL18+ CMV-infected cells, while cells lacking UL18 were resistant to this effect. This process was independent of antigen specificity and MHC restriction, and it can be blocked by mAbs targeting LILRB1 and UL18 [238]. Clinically, CMV infection is a common complication following organ transplantation, and LILRB1 upregulation on lymphocytes was associated with CMV reactivation post-lung transplantation [239], suggesting its utility as an early biomarker in transplant recipients.
Human immunodeficiency virus type 1 (HIV-1)
HIV-1 infection triggered profound immune dysregulation in its early stages [240]. Alterations in LILRB expression critically impaired DC and NK cell function in HIV-1+ individuals [241, 242]. During early infection, specific HLA-B variants engaged LILRB2 on monocytes, inducing tolerance that exacerbates DC dysfunction in chronic infection, impairing DC maturation and expression of co-stimulatory molecules [241]. The co-expression of LILRB2/MHC-I on classical DCs during HIV/simian immunodeficiency virus (SIV) infection drove cellular dysregulation, blunting adaptive immunity and hindering viral control [243]. As the infection progresses, elevated secretion of HLA-G from monocytes/DCs bound to LILRB2 on DCs, suppressing antigen presentation while promoting proinflammatory cytokine release [244]. In untreated HIV-1 patients, strong LILRB2-HLA-I binding correlated with increased viral replication and further DC dysfunction [40]. Strikingly, elite controllers, a subset of HIV-1-infected individuals who maintained undetectable viral loads without antiretroviral therapy, exhibited upregulated LILRB1 and LILRB3 on circulating myeloid DCs (mDCs) compared to progressive-stage patients [245-247]. Concurrently, expanded LILRB1+ NK cells emerged during viremia and treatment interruptions [19, 85, 248]—consistent with NK cells' critical role in killing infected cells and cytokine-mediated viral control. These findings elucidate key HIV-1 pathogenesis mechanisms and reveal potential LILRB-targeted strategies for immune intervention.
Dengue virus (DENV)
DENV exploited Fc-gamma receptors (FcγR) for cellular entry via antibody-dependent enhancement, amplifying viral load [249, 250]. Concurrently, DENV engaged LILRB1 to activate SHP1, which suppressed FcγR-mediated Syk pathway. This inhibition downregulated interferon-stimulated genes, enabling DENV early immune evasion and enhanced viral replication [86]. Furthermore, LILRB1 impaired lysosomal acidification necessary for enzyme activation following DENV uptake via SHP1-dependent mechanisms, facilitating evasion of cell-autonomous immunity and promoting intracellular survival [251]. Clinically, DENV-infected patients exhibited hyporesponsive CD19+CD27- naïve B cells to Toll-like receptor (TLR)/anti-IgM stimulation. This functional impairment correlated with upregulated expression of inhibitory receptors CD32 (FcγRIIb) and LILRB1 on B cells, likely contributing to disrupted humoral immunity [252].
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2)
SARS-CoV-2, the causative agent of COVID-19, typically manifests with clinical symptoms such as fever, cough, and myalgia [253]. Critically, disease severity in patients correlated with upregulated LILRB4 expression [254]. MDSCs exacerbated pathological inflammation through the co-expression of programmed cell death ligand 1 (PD-L1), LILRB4, and IDO-1, functioning as significant producers of proinflammatory cytokines IL-6 and IL-10 [255]. In murine SARS-CoV-2 infection models, rapid disease progression characterized by neurotropic infection and encephalitis coincided with marked LILRB4 upregulation [256].
Other viruses
Beyond the established viral models, LILRBs critically regulate immune evasion and viral dissemination in various other viral infections. In Epstein-Barr virus (EBV)-infected individuals, elevated LILRB1 expression on virus-specific CD8+ T cells impaired anti-viral immunity by suppressing IFN-γ production [182, 257]. During chronic hepatitis B progression—particularly in immune-tolerant, active hepatitis, and HBeAg-negative phases—CD56dimCD16+ NK cells exhibited significant LILRB1 upregulation. This correlated with reduced cytolytic capacity, decreased IFN-γ production, and increased apoptosis [258]. Genetic analyses further indicated that LILRB1/HLA-G variants increase the risk for mother-to-fetus Zika virus transmission, while protective LILRB2 polymorphisms reduce in utero transmission likelihood [108]. Notably, the murine LILRB2 homolog, PirB facilitated binding and internalization of neurotropic reovirus serotype 3, promoting central nervous system (CNS) replication and pathogenicity [259].
Bacterial infections
Mycobacterium tuberculosis (M. tuberculosis)
In active pulmonary tuberculosis, CD56dimCD16+ NK cells exhibited significantly elevated LILRB1 expression versus latent tuberculosis infection controls. This upregulation mediated functional suppression—evidenced by reduced CD107a degranulation, impaired IFN-γ production, and increased spontaneous apoptosis—directly impairing anti-mycobacterial immunity [260]. Therapeutically, monoclonal antibody blockade of LILRB2 reprogramed human MDSCs toward a proinflammatory phenotype, enhancing M. tuberculosis killing capacity and nominating LILRB2 as a therapeutic target [261]. Notably, LILRB4 upregulation in latent tuberculosis served as both a diagnostic biomarker and reactivation predictor [262]. Furthermore, exposure to M. tuberculosis induced monocyte LILRB5 expression, which facilitates direct bacterial binding and activates cytotoxic T-cell proliferation through downstream immunomodulatory signaling [263].
Staphylococcus aureus
LILRBs play a crucial role in Staphylococcus aureus infections. Murine PirB, along with human LILRB1 and LILRB3, function as direct pathogen recognition receptors for Staphylococcus aureus. PirB bound to Staphylococcus aureus and interacted with TLRs, promoting inhibitory cytokine release, actively suppressing anti-inflammatory responses [264]. Furthermore, LILRB3 impaired neutrophil-mediated defense by suppressing effector functions and microbicidal activity, attenuating IgA-induced ROS production, and inhibiting antimicrobial peptide release [169].
Other bacterial infections
LILRBs play pivotal roles in modulating immune responses and inflammation during infections with Salmonella [131], Escherichia coli [265], and leprosy [266]. Following TLR recognition of Salmonella or its components, upregulated LILRB2 and LILRB4 expression on APCs promoted immune tolerance [131], with LILRB4 additionally suppressing T-cell activation and reducing IL-8 production through cell-contact-dependent mechanisms [131]. During Escherichia coli infections, lipopolysaccharide (LPS) induced macrophage expression of PirB, subsequently downregulating proinflammatory cytokine production [265].
Sepsis
Sepsis induces profound immune paralysis, impairing bacterial clearance while triggering dysregulated inflammation. Critically, LILRB2 upregulation on monocytes correlated with immunosuppression and organ failure in patients, characterized by diminished CD86 expression and elevated IL-10/IL-12 ratios [267]. Paradoxically, neutrophils failed to upregulate LILRB2, impairing phagocytosis and suggesting LILRB2 modulation could prevent neutrophil dysfunction [167]. In septic shock, LPS-induced dysregulation of LILRB2 predicted mortality risk, establishing its central immunomodulatory role [268]. Concurrently, heightened LILRB3 expression in peripheral blood mononuclear cells (PBMCs) and macrophages suppressed bacterial killing, ROS production, and antigen presentation, dampening Th1 immune responses [269]. Therapeutic blockade of LILRB3 enhanced bacterial clearance, ROS generation, and Th1 differentiation, improving survival in preclinical models [269]. Neonatal sepsis further demonstrated significant LILRB2, LILRB3, and LILRB4 upregulation, with LILRB4 driving immune suppression through conversion of effector T cells into suppressive phenotypes [270].
Parasite infections
Plasmodium falciparum (P. falciparum)
P. falciparum malaria remains a leading global infectious threat, with severe disease manifestations posing significant clinical challenges [271]. The parasite evades the host immune system by expressing variant RIFIN proteins on erythrocytes, which interact with immune inhibitory receptors to facilitate escape [272]. Specifically, RIFIN mimicked HLA-I structures to bind LILRB1, suppressing NK cell cytotoxicity and inhibiting B-cell IgM production, enabling immune detection avoidance in severe malaria [63, 64]. RIFINs additionally bound LILRB2, amplifying evasion through redundant HLA-I mimicry [66]. Chronic exposure expanded atypical CD56- NK cells that exhibited elevated LILRB1 expression and enhanced P. falciparum-specific antibody-dependent cellular cytotoxicity compared to CD56dim NK cells [273]. In malaria patients, increased LILRB1 on CD19+ B cells correlated with early apoptosis markers and excessive cytokine release, potentially impairing immunological memory [274]. Additionally, infants born to mothers with placental malaria showed LILRB2 overexpression on non-classical monocytes, highlighting its potential role in mediating malaria-induced immune tolerance [275].
Toxoplasma gondii (T. gondii)
T. gondii infection is typically asymptomatic in immunocompetent individuals but can cause adverse pregnancy outcomes in immunocompromised hosts, particularly pregnant women [276]. T. gondii infection dysregulated KIR2DL4/LILRB1 on decidual NK cells and HLA-G on trophoblasts, inducing excessive immunotolerance that suppressed NK function and facilitated fetal infection, ultimately contributing to adverse pregnancy outcomes [277]. Furthermore, T. gondii infection downregulated LILRB4 on decidual MDSCs by inhibiting STAT3 phosphorylation and altered arginase-1 and IL-10 expression via the SHP2/STAT6 pathway. These alterations disrupted decidual MDSC immunosuppressive function, suggesting a pivotal role in adverse pregnancy outcomes [278].
Trypanosoma cruzi
In chronic Trypanosoma cruzi infection, LILRB1 expression was increased during specific CD4+ T cell differentiation. This upregulation impaired T cell function, compromising anti-parasite immune responses and potentially fostering persistent chronic infection [279].
5.2 LILRBs and autoimmune diseases
Autoimmune diseases arise from immune dysregulation, triggering activation of autoreactive immune cells and subsequent tissue damage [280]. LILRBs critically modulate immune tolerance and effector responses, contributing to autoimmune pathogenesis. Genetic polymorphisms and deletions in LILRBs are correlated with disease susceptibility and altered Treg development [18, 94]. Below, we detail the pathogenic roles of LILRBs in major autoimmune disorders (Table 1).
Thyroid-related immune diseases
Pathogenic dysregulation of LILRBs is especially pronounced in thyroid diseases. In autoimmune thyroid diseases (AITD), LILRB1 expression was elevated on lymphocytes, but its inhibitory capacity on CD4+ and CD8+ T cell proliferation was impaired. This functional impairment extended to IL-10 regulation, a critical anti-inflammatory cytokine that suppresses cellular immunity. AITD patients demonstrated reduced LILRB1-mediated IL-10 synthesis, potentially disrupting immune homeostasis [281]. Notably, the LILRB1 missense variant c.479G>A (p.G160E) was associated with familial autoimmune disorders including Graves' disease, Hashimoto's thyroiditis, and systemic lupus erythematosus (SLE). This variant reduced LILRB1 expression on Treg cells, disrupting SHP1 signaling and promoting pro-inflammatory M1-like macrophage expansion, thus disturbing the M1/M2 balance [282].
Rheumatoid arthritis (RA)
In RA patients with CMV co-infection, the interactions between UL18, HLA-G, classical MHC-I molecules, and LILRB1 were disrupted. Declining sHLA-G levels impaired LILRB1-mediated suppression of CD8+ T cells, potentially triggering pathogenic hyperactivation and accelerating RA progression [283]. Paradoxically, some research has demonstrated that even when plasma sHLA-G concentrations increase in RA, LILRB1 often fails to recognize it due to HLA-G's preferential formation of monomeric or non-canonical conformations, rather than the dimeric structure required for productive receptor engagement. This structural mismatch prevented LILRB1 from exerting its immunosuppressive functions, perpetuating chronic inflammation [284].
Systemic lupus erythematosus (SLE)
In SLE, LILRB1 function was compromised in PBMCs, with significantly reduced surface expression on B lymphocytes—a perturbation implicated in SLE pathogenesis [285]. Furthermore, the LILRB4 rs11540761 single nucleotide polymorphism (SNP) associated with downregulated receptor expression on circulating monocytoid DCs, establishing LILRB4 dysregulation as a key mechanism in SLE [106]. Elevated LILRB4 expression on plasmablasts and plasma cells in untreated SLE patients suggested that LILRB4 inhibition could suppress pathological autoantibody production, presenting a novel therapeutic strategy [286].
Multiple sclerosis (MS)
In healthy CNS tissue, LILRB1 expression was minimal or absent, but in MS lesions, both LILRB1 and its ligand HLA-G were upregulated, modulating neuroinflammatory responses [287]. Glatiramer acetate (GA; Copaxone), a copolymer therapeutic agent approved by the US Food and Drug Administration (FDA) for relapsing-remitting MS, engaged murine PirB on MDSCs to regulate myeloid function. Crucially, human LILRB2 and LILRB3 serve as functional orthologs of PirB mediating GA binding. GA competitively occupied LILRB2/LILRB3 in dose-dependent fashion, restoring immune homeostasis [288]. Additionally, recombinant human LILRB4 protein has shown promise in MS mouse models by suppressing proinflammatory cytokine release and inhibiting pathogenic Th1 and Th17 cell proliferation, preventing autoimmune neuroinflammation and suggesting a potential treatment modality for MS [289].
5.3 LILRBs and neurodegenerative diseases
In AD, soluble Aβ oligomers mediated cognitive dysfunction and synaptic loss [290]. Both murine PirB and its human homolog LILRB2 functioned as receptors for Aβ1-42 oligomers in CNS tissue (Table 1). This interaction mediated synaptic toxicity and induced early deficits in developmental visual cortex plasticity in AD models, suggesting that blocking LILRB2 could be a promising therapeutic strategy for AD [291]. Additionally, microglia contributed to AD progression through LILRB2-TREM2 interactions, which inhibited TREM2 signaling and compromised microglial function [292]. Notably, LILRB4 mAbs have been shown to enhance microglial activation and Aβ phagocytosis while suppressing interferon pathways [84], positioning LILRB4 blockade as a complementary immunomodulatory approach for AD.
5.4 LILRBs and tumors
LILRBs exert central immunomodulatory functions across diverse malignancies, critically influencing tumor development and progression. This section comprehensively analyzes LILRB expression patterns in human cancers, evaluates their associations with clinicopathological features and survival outcomes, and elucidates their mechanistic contributions to tumorigenesis and metastatic dissemination (Table 2). Collectively, these findings establish LILRBs as master regulators of cancer immunoediting and highlight their potential as therapeutic targets and prognostic biomarkers.
LILRB expression and function in human malignancies.
| Cancer types | LILRBs | Expression* | Clinical features | Survival (Prognosis) | Cellular roles | References |
|---|---|---|---|---|---|---|
| Hematological malignancies | ||||||
| AML | LILRB1 | Upregulated | OS (Poor) | [210] | ||
| LILRB2 | Upregulated | OS (Poor) | Support HSC expansion | [72, 210, 211, 212, 293] | ||
| LILRB3 | Upregulated | OS (Poor) | Enhance tumor cell survival, inhibit T cell activity | [198, 210, 212, 233] | ||
| LILRB4 | Upregulated | OS (Poor) | Support tumor cell infiltration and suppress T cell activity | [22, 210, 212] | ||
| LILRB5 | NS | OS (Favorable) | [97, 212] | |||
| CMML | LILRB4 | Upregulated | [213] | |||
| MM | LILRB1 | (a) Downregulated; (b) Upregulated | Cytogenetic abnormality t(4;14) translocation | OS (Poor) | Increase susceptibility to T/NK-mediated killing; Regulate cholesterol metabolism and protect tumor cells from ferroptosis | [214, 297] |
| LILRB2 | Downregulated | [214] | ||||
| LILRB3 | Downregulated | [214] | ||||
| LILRB4 | (a) NS; (b) Upregulated | Bone damage | OS (Poor) | Support tumor cell proliferation; Promote osteolytic lesions | [116, 214, 215, 232, 299] | |
| LILRB5 | NS | [214] | ||||
| CLL | LILRB1 | Downregulated | [216] | |||
| LILRB2 | 54.5% expression | [300] | ||||
| LILRB4 | 48.9% expression | Lymphoid tissue involvement | Regulate tumor progression | [300, 301] | ||
| ALL | LILRB2 | Downregulated | [302] | |||
| KMT2A-rearranged ALL | LILRB4 | Upregulated | [217] | |||
| ATL | LILRB4 | Inhibit tumor cell growth | [89] | |||
| CTCL | LILRB1 | 100% expression | Inhibit tumor cell proliferation | [303, 304, 305] | ||
| cHL | LILRB2 | Upregulated | OS (Poor) | [309] | ||
| Digestive system | ||||||
| EC | LILRB1 | Upregulated | [221] | |||
| LILRB2 | Upregulated | Tumor stage | [312] | |||
| HCC | LILRB1 | Downregulated | TILs | RFS/PFS (Favorable) | [313] | |
| LILRB2 | (a) Upregulated;(b) Downregulated | Gender, tumor size, cell differentiation, TILs | (a) OS (Poor); (b) OS/RFS/PFS/DFS (Favorable) | Promote macrophage polarization to M2 phenotype and recruit immunosuppressive T cells | [196, 313, 314] | |
| LILRB3 | Downregulated | TILs | RFS/PFS (Favorable) | [313] | ||
| LILRB4 | Upregulated | TILs | RFS/PFS (Favorable) | [313] | ||
| LILRB5 | Downregulated | TILs | OS/RFS/PFS/DFS (Favorable) | [313] | ||
| GC | LILRB1 | Upregulated | Pathological stage, cell differentiation, tumor size | OS (Poor) | Inhibit the anti-tumor effect of NK cells | [205, 219, 220] |
| LILRB2 | Upregulated | [317] | ||||
| LILRB4 | Upregulated | [220] | ||||
| PDAC | LILRB1 | Upregulated; Downregulated | Pathological stage | OS (Poor) | [219, 321] | |
| LILRB2 | Upregulated; Downregulated | Sustain EMT and the early metastatic behavior of tumor cells | [319, 321] | |||
| LILRB3 | Downregulated | [321] | ||||
| LILRB4 | Downregulated | OS/RFS (Favorable) | [321] | |||
| CRC | LILRB1 | Upregulated | OS (Poor) | [219] | ||
| LILRB2 | Upregulated | Gender, cell differentiation, vascular involvement, LN metastasis, tumor stage | OS (Poor) | Regulate tumor cell proliferation, invasion, migration, and angiogenesis | [218, 231, 322, 323] | |
| LILRB3 | Upregulated | LN metastasis, tumor stage | OS/PFS (Poor) | Inhibit T-cell infiltration and promote M2-like TAM accumulation | [324] | |
| LILRB4 | Upregulated | LN metastasis, tumor stage, CD45RO+ T cell count | OS (Poor) | [325] | ||
| Respiratory system | ||||||
| NSCLC | LILRB1 | 51.5% expression | Tumor stage | [107] | ||
| LILRB2 | Upregulated | Cell differentiation, LN metastasis, tumor stage, age, TILs | OS/PFS (Poor) | Promote tumor growth, invasion, migration, and angiogenesis; Recruit M2-like TAMs and impair T cell response; Enhance resistance to radiation | [197, 199, 222, 223, 224, 327, 328, 329] | |
| LILRB4 | Upregulated | Cell differentiation, tumor size, vascular involvement, tumor stage | OS (Poor) | Enhance tumor cell migration, invasion, and angiogenesis | [230] | |
| Reproductive system | ||||||
| BC | LILRB1 | Upregulated | Pathological stage | OS (Poor) | [219] | |
| LILRB2 | Upregulated | TILs, LN metastasis | OS/PFS (Poor) | Promote immune evasion; Promote tumor growth, and induce effector T cell senescence; Reprogram toward aerobic glycolysis | [195, 229, 336, 337] | |
| OC | LILRB1 | Upregulated | Age, tumor stage | [225] | ||
| Endometrial cancer | LILRB2 | Upregulated | OS (Poor) | Support tumor cell expansion and migration | [228] | |
| Urinary system | ||||||
| ccRCC | LILRB1 | 60% expression | [227] | |||
| LILRB2 | 60% expression | [227] | ||||
| LILRB3 | Upregulated | OS (Poor) | [344] | |||
| Nervous system | ||||||
| Glioma | LILRB1 | Upregulated | Tumor size | OS (Poor) | Enhance tumor cell proliferation, migration and invasion | [118] |
| Glioblastoma | LILRB1 | Upregulated | [349] | |||
| LILRB2 | Upregulated | Pathological stage | OS/DFS (Poor) | Induce MDSC formation and expansion | [349, 350] | |
| LILRB3 | Upregulated | [349] | ||||
| LILRB4 | Upregulated | [349] | ||||
| Cutaneous tumors | ||||||
| Melanoma | LILRB2 | Upregulated | Promote tumor growth; Induce effector T cell senescence | [229] | ||
| Tumors of other system | ||||||
| OSCC | LILRB1 | Downregulated | [352] | |||
| TC | LILRB1 | Upregulated | Pathological stage | OS (Poor) | [219] |
ALL, acute lymphoblastic leukemia; AML, acute myeloid leukemia; ATL, acute T cell leukemia; BC, breast cancer; ccRCC, clear cell renal cell carcinoma; cHL, classical Hodgkin lymphoma; CLL, chronic lymphocytic leukemia; CMML, chronic myelomonocytic leukemia; CRC, colorectal cancer; CTCL, cutaneous T-cell lymphoma; DFS, disease-free survival; EC, esophageal carcinoma; EMT, epithelial-mesenchymal transition; GC, gastric cancer; HCC, hepatocellular carcinoma; HSC, hematopoietic stem cell; LN, lymph node; MDS, myelodysplastic syndrome; MDSC, myeloid-derived suppressor cell; MM, multiple myeloma; NS, no significance; NSCLC, non-small cell lung cancer; OC, ovarian cancer; OS, overall survival; OSCC, oral squamous cell carcinoma; PDAC, pancreatic ductal adenocarcinoma; PFS, progression-free survival; RFS, recurrence-free survival; TAMs, tumor-associated macrophages; TC, thyroid cancer; TILs, tumor-infiltrating lymphocytes; VEGF-C, vascular endothelial growth factor C.
*Compare the expression levels of LILRBs between tumor cells and normal cells.
Hematological malignancies
Acute myeloid leukemia (AML)
LILRBs serve as critical pathogenic regulators in AML, orchestrating tumor progression and immune remodeling within the TME. Multiple studies have demonstrated that LILRB1-4 are highly expressed in AML cells, particularly in monocytic differentiation-associated AML (M-AML) [22, 72, 198, 210-212, 233, 293-295]. Among these, LILRB1 and LILRB4 co-expression served as a specific diagnostic marker to differentiate M-AML from other subtypes [209]. Elevated LILRB1-4 correlated with poor prognosis and promoted leukemogenesis, while conversely, higher LILRB5 expression was associated with improved outcomes [97, 212]. Within the AML TME, NK cells exhibited marked upregulation of LILRB2 and LILRB3 [296]. Mechanistically, IL-10-driven LILRB2 expression in HSCs facilitated ANGPTL-mediated activation of SHP2 and CaMK pathways, promoting leukemic progenitor expansion and disease progression (Figure 3A) [72, 211, 293]. LILRB3 recruited TNF receptor-associated factor 2 (TRAF2) and cellular FLICE-inhibitory protein (cFLIP) to activate NF-κB, supporting AML survival while suppressing T-cell function (Figure 3A) [233]. LILRB3 agonism enhanced cholesterol metabolism, supporting AML cell survival [198]. Conversely, LILRB3-targeted approaches—including monoclonal antibodies and CAR T cells—have demonstrated potent anti-AML cytotoxicity in vitro. Moreover, LILRB3-specific CAR T cells induced durable remission in vivo, with transgenic models showing only mild monocytopenia and no significant hematologic or metabolic toxicity [198]. Additionally, miR-103a-2-5p has been shown to downregulate LILRB3 and inhibit AML cell growth (Figure 3A) [120]. LILRB4 expression was regulated post-transcriptionally by m6A methylation, with FTO promoting its upregulation (Figure 3A) [119]. Another study suggested that APOE activates LILRB4 and modulates the SHP2/NF-κB/urokinase plasminogen activator receptor (UPAR)/Arginase-1 (ARG1) axis (Figure 3A), facilitating AML cell migration, infiltration, and T-cell suppression [22]. Clinically, the rs1048801 variant in LILRB4 was associated with refractory disease, poorer treatment response, reduced overall survival (OS), and may serve as an independent prognostic risk factor in AML [111].
Roles of LILRBs in hematological malignancies. (A-E) The schematic diagrams illustrate the functional roles of LILRBs in hematologic malignancies, depicting their interactions with specific ligands, activation of downstream signaling pathways, and subsequent modulation of tumor cell proliferation, migration, and T-cell function across distinct disease contexts: (A) acute myeloid leukemia (AML), (B) multiple myeloma (MM), (C) chronic lymphocytic leukemia (CLL), (D) acute T-cell leukemia (ATL), and (E) cutaneous T-cell lymphoma (CTCL).
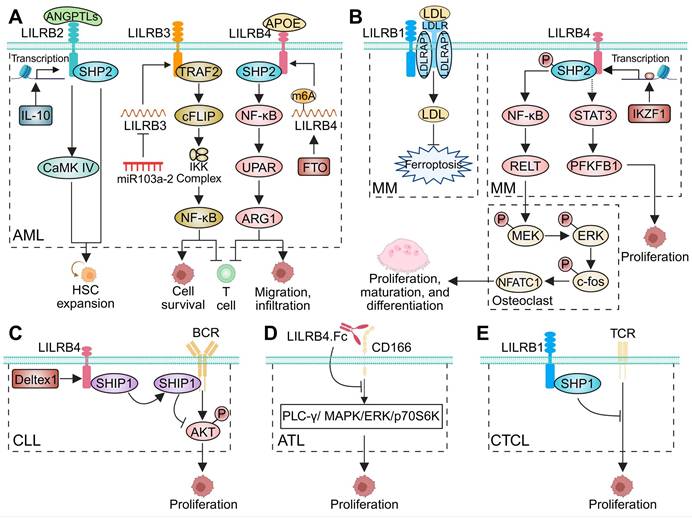
Chronic myelomonocytic leukemia (CMML) and myelodysplastic syndromes (MDS)
LILRB4 mRNA expression was significantly upregulated in CMML compared to MDS and healthy controls. This upregulation was closely associated with activation of pivotal immune checkpoint pathways, including PD-1, cytotoxic T-lymphocyte antigen 4 (CTLA-4), IFN-γ response, and inflammatory signaling, indicating LILRB4-mediated immune dysregulation in CMML pathogenesis [213]. While similar associations were observed in MDS, they appeared less pronounced. Additionally, LILRB4 expression positively correlated with CTLA-4 levels, suggesting its involvement in immune evasion via CTLA-4-dependent mechanisms [213].
Multiple myeloma (MM)
Patients with active MM and monoclonal gammopathy of undetermined significance (MGUS) exhibited reduced expression of LILRB1 and its ligand S100A9. This impaired LILRB1/S100A9 interaction compromised NK and T-cell cytotoxic activity, contributing to immune evasion and disease progression [214]. Abnormal plasma cells from MM patients also exhibited decreased LILRB2 and LILRB3 expression, while LILRB4 and LILRB5 levels remained largely unchanged [214]. Contrastingly, a recent study indicated that myeloma cells from MGUS and MM patients exhibit significantly higher LILRB1 levels compared to normal plasma cells from healthy donors [297]. Elevated LILRB1 expression was associated with advanced disease stages, high-risk cytogenetic abnormalities (such as t(4;14) translocation), and unfavorable survival outcomes [297]. Mechanistically, LILRB1 promoted LDL uptake through interactions with the LDL receptor (LDLR) and LDL receptor adapter protein 1 (LDLRAP1), maintaining metabolic homeostasis and conferring ferroptosis resistance (Figure 3B) [297]. Furthermore, LILRB1 was highly expressed on NK cells in MM patients, and its blockade significantly enhanced NK cell cytotoxicity [298].
Concurrently, LILRB4 expression was notably high in MM and linked to worse prognosis [116, 215, 232, 299]. Functionally, LILRB4 promoted myeloma cell proliferation and migration, whereas its silencing inhibited disease progression both in vitro and in vivo [116, 215]. Transcription factor IKZF1 activated LILRB4 expression, driving MM cell proliferation via the STAT3/PFKFB1 pathway (Figure 3B) [116]. Additionally, high LILRB4 expression correlated with bone destruction severity. Mechanistically, LILRB4 recruited phosphorylated SHP2 to activate NF-κB signaling, promoting multiple myeloma cells to secret RELT. This, in turn, stimulated osteoclast proliferation, maturation, and differentiation via the phosphorylation of MEK/ERK/c-fos and NFATC1 pathways (Figure 3B) [232].
Chronic lymphocytic leukemia (CLL)
LILRB1 expression was significantly reduced on leukemia cells in CLL patients, but notably increased on NK cells, particularly in those with advanced disease or adverse prognostic features, such as del(11q) and trisomy 12 [216]. Additionally, LILRB4, absent in normal B cells, was detected in approximately 50% of CLL patients and was typically associated with advanced lymphoid involvement. In LILRB4-expressing CLL cells, co-expression of LILRB2 was frequently observed, displaying similar expression patterns [300]. Mechanistically, LILRB4 expression in CLL was regulated by Deltex1, an inhibitor of antigen receptor signaling. LILRB4 recruited Src homology 2 domain-containing inositol phosphatase 1 (SHIP1), inhibiting B-cell receptor (BCR)-dependent AKT signaling and thereby driving tumor progression (Figure 3C) [301].
Acute lymphoblastic leukemia (ALL)
Gene expression profile and experimental validation of CD10+CD19+ precursor B lymphoblasts in pediatric ALL revealed significant LILRB2 downregulation in leukemia cells [302]. In contrast, KMT2A-rearranged ALL cells that developed resistance to the DOT1L inhibitor pinometostat exhibited upregulated LILRB4, suggesting myeloid-like phenotypic shifts in resistant populations [217].
Acute T-cell leukemia (ATL)
In ATL, LILRB4.Fc inhibited tumor growth by suppressing the PLC-γ/MAPK/ERK/p70S6K signaling pathway (Figure 3D) and disrupting the homophilic CD166-CD166 intercellular adhesion, thereby interfering with the production and acquisition of growth factors [89].
Cutaneous T-cell lymphoma (CTCL)
LILRB1 was highly expressed in CTCL subtypes, including CD56+, CD8+, CD56+CD4+, and Sézary syndrome [303, 304]. Functional study demonstrated that LILRB1 activation recruits SHP1, which specifically inhibited CTCL cell proliferation induced by CD3/TCR stimulation (Figure 3E) [305].
Other lymphomas
LILRB expression has been confirmed in multiple lymphoma subtypes [306-308]. Bioinformatics analyses revealed significantly elevated LILRB2 expression in classical Hodgkin lymphoma, correlating with poor prognosis [309]. Furthermore, M2-like TAMs within the TME of follicular lymphoma and diffuse large B-cell lymphoma harboring BCL6 translocations exhibited high LILRB3 expression [310]. In elderly EBV-positive diffuse large B-cell lymphoma patients, a higher frequency of LILRB1 mutations was observed [311].
Digestive system
Esophageal cancer
LILRB1 expression was significantly upregulated in esophageal adenocarcinoma (Figure 4A) [221]. Additionally, LILRB2 was notably elevated in esophageal squamous cell carcinoma and early-stage esophageal adenocarcinoma (pT1-2) (Figure 4A) [312].
Roles of LILRBs in digestive, respiratory and reproductive cancers. (A) Expression patterns and functional roles of LILRBs in esophageal carcinoma (EC), hepatocellular carcinoma (HCC), gastric cancer (GC), pancreatic ductal adenocarcinoma (PDAC), and colorectal cancer (CRC). The diagrams illustrate their distribution and functions in tumor cells (left) and immune cells (right). In HCC and CRC, LILRB2 regulates proliferation, migration, invasion, epithelial-mesenchymal transition (EMT), and angiogenesis through the ROS/ERK, AKT/ERK, and MAPK/ERK signaling pathways. In CRC, LILRB3 promotes immune escape by modulating the spatial distribution of CD8+ T cells and M2-type tumor-associated macrophages (TAMs) within the tumor microenvironment. (B) In NSCLC, LILRB2 regulates tumor proliferation, migration, invasion, and influences the immune microenvironment through the ERK, SHP2-CaMK1, JAK-STAT3, and PI3K-AKT signaling pathways, thereby promoting tumor progression. Additionally, LILRB2 expression is regulated by EGFR and cGAS-STING pathways. LILRB4 promotes EMT and angiogenesis in NSCLC by activating ERK pathway. In MDSCs, LILRB4 enhances MDSC-mediated tumor metastasis by regulating M2-type macrophage polarization and suppressing the secretion of miR-1 family miRNAs. (C-E) LILRB expression patterns in (C) breast cancer, (D) ovarian cancer, and (E) endometrial cancer, along with their key downstream signaling pathways.
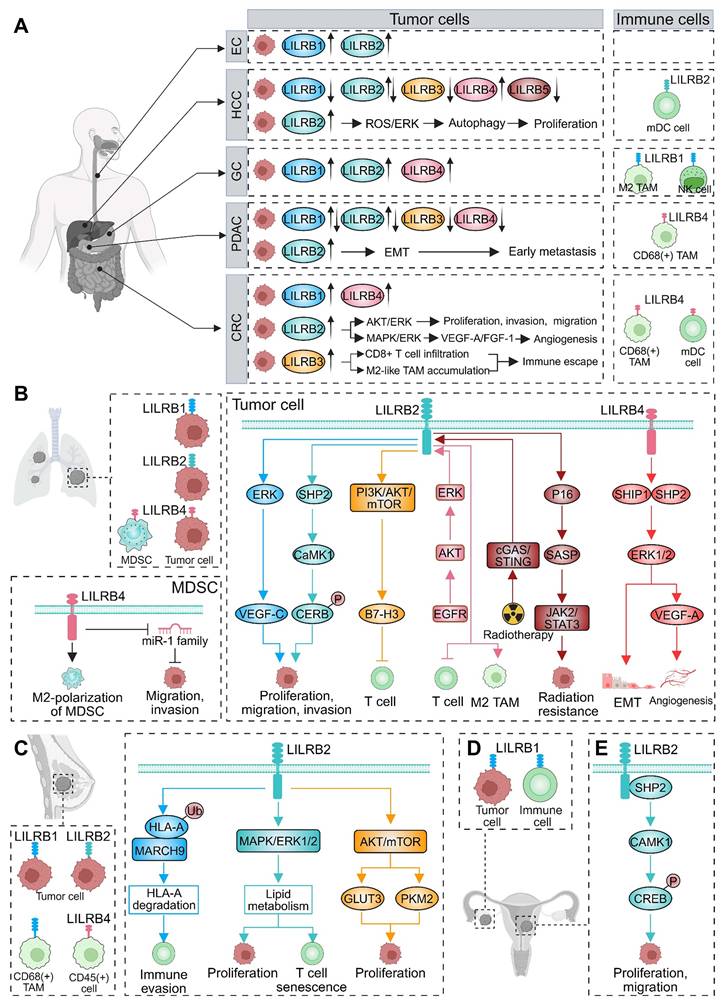
Hepatocellular carcinoma (HCC)
Bioinformatics analyses revealed significant downregulation of LILRB1, LILRB2, LILRB3, and LILRB5 mRNA in HCC tissues, whereas LILRB4 was notably upregulated (Figure 4A) [313]. Additionally, the expression of LILRB family members correlated with patient prognosis and immune cell infiltration [313]. Contradicting this, another study reported LILRB2 overexpression in HCC tissues, which was associated with poor differentiation, larger tumor size, and reduced OS [314]. Subsequent investigations demonstrated that LILRB2/PirB interacts with ANGPTL8 to activate the ROS/ERK pathway, thereby enhancing autophagy and promoting HCC proliferation (Figure 4A) [196]. Moreover, LILRB2 expression was significantly elevated in the CD1c+ mDC subpopulation within tumor tissues, particularly in poorly differentiated tumors, suggesting its potential role in suppressing immune responses [315, 316].
Gastric cancer (GC)
LILRB1 and LILRB4 were highly expressed in poorly differentiated and undifferentiated GC cells, with LILRB1 levels showing a clear correlation with tumor size and degree of differentiation (Figure 4A) [220]. In gastric mixed adenocarcinoma, LILRB1 expression associated with advanced pathological stage and reduced OS [219]. LILRB2 was significantly upregulated in GC cells and linked to neutrophil extracellular trap-related transcriptional reprogramming (Figure 4A) [317]. Within the TME, LILRB1 was predominantly localized to M2-type TAMs, with high expression predicting advanced tumor stage, increased recurrence risk, and unfavorable prognosis [318]. Additionally, HLA-G interacted with LILRB1 to inhibit the proliferation and cytotoxic activity of NK cells, thereby facilitating immune escape in GC [205].
Pancreatic ductal adenocarcinoma (PDAC)
In PDAC, LILRB1 expression was significantly elevated in tumor tissue versus adjacent normal tissue, correlating with advanced pathological stage and poor OS [219]. During multistep tumorigenesis in PDAC models, LILRB2 and its ligand ANGPTL2 were progressively upregulated. Subsequent studies showed that the ANGPTL2/LILRB2 autocrine axis promotes epithelial-to-mesenchymal transition (EMT) and early metastasis (Figure 4A) [319]. Soluble LILRB4, mainly derived from CD68+ TAMs, induced inhibitory differentiation of CD8+ T cells, thereby suppressing antitumor immunity [320]. Interestingly, another study found reduced expression of LILRB1-4 in early-stage PDAC tumors compared to adjacent normal tissues, with high LILRB4 levels correlating with improved relapse-free survival (RFS) [321].
Colorectal cancer (CRC)
LILRB1+ cells were significantly more abundant in colorectal mucinous adenocarcinoma tumors and inversely correlated with OS [219]. Elevated LILRB2 expression was associated with male, poor differentiation, vascular invasion, lymph node (LN) metastasis, and advanced clinical stage [218, 231, 322, 323], and served as an independent prognostic factor for poorer outcomes [231, 323]. Mechanistically, LILRB2 promoted CRC progression by interacting with HLA-G, activating AKT and ERK signaling (Figure 4A) [322], enhanced tumor angiogenesis via MAPK/ERK-mediated upregulation of vascular endothelial growth factor (VEGF)-A and fibroblast growth factor 1 (FGF-1, Figure 4A) [231]. LILRB3 expression was significantly higher in CRC versus normal colorectal epithelium and associated with LN metastasis, advanced tumor stages and reduced survival [324]. Tumor-derived LILRB3 inhibited CD8+ T cell infiltration and polarized TAMs toward an M2-like phenotype, mediating immune escape (Figure 4A) [324]. Similarly, high LILRB4 expression correlated with LN metastasis, advanced disease, reduced CD45RO+ T cell infiltration, poor OS, and was an independent prognostic factor [325]. In the TME, LILRB4 was prominently expressed on mDCs and CD68+ TAMs, implicating its role in immune suppression (Figure 4A) [320, 326].
Respiratory system
Non-small cell lung cancer (NSCLC)
LILRB1 expression was detected in over half of NSCLC samples, with levels escalating alongside advancing tumor stage [107]. The LILRB1 gene SNP c.5724G>A (p.E625K) was more commonly found in NSCLC patients and significantly associated with regional LN metastasis, although it does not appear to influence LILRB1 expression [107]. Compared to adjacent normal tissue, LILRB2 expression was significantly elevated in NSCLC tumor tissue [222-224, 327], and associated with adverse clinicopathological features including poor differentiation, regional LN metastasis, advanced tumor stage, older age, reduced tumor-infiltrating lymphocytes (TILs), and poorer prognosis [222-224, 327-329]. Functionally, LILRB2 activated ERK signaling pathway, increasing VEGF-C expression and promoting tumor cell malignancy, migration, and invasion (Figure 4B) [222, 223]. Additionally, LILRB2 interacted with ANGPTL2 to activate the SHP2/CaMK1/CREB signaling pathway, further supporting tumor cell proliferation (Figure 4B) [223]. In terms of immune evasion, LILRB2 promoted B7-H3 expression via PI3K/AKT/mTOR signaling to suppress T cell-mediated anti-tumor immunity, aiding NSCLC progression (Figure 4B) [224]. LILRB2 expression was further regulated by EGFR/AKT/ERK pathways, facilitating recruitment of M2-type TAMs and inhibiting T cell activity (Figure 4B) [199]. Notably, radiotherapy increased LILRB2 expression through cGAS/STING pathway, promoting cellular senescence and activating the JAK2/STAT3 axis, which drives tumor progression and radiation resistance (Figure 4B) [197]. LILRB4 was also highly expressed in NSCLC and strongly associated with adverse clinical outcomes [230]. LILRB4 recruited SHP2 and SHIP1 to activate ERK1/2 pathway, thereby promoting EMT and upregulating VEGF-A expression, which in turn enhances tumor cell migration, invasion, and angiogenesis (Figure 4B) [230]. Within the TME, high LILRB4 expression on tumor-infiltrating immune cells predicted increased risk of postoperative recurrence, shorter OS, and reduced RFS, establishing it as an independent prognostic factor [330]. LILRB4 was predominantly expressed on MDSCs and its blockade significantly inhibited NSCLC cell migration in co-culture systems with CD33+ MDSCs [330, 331]. Furthermore, LILRB4/gp49b regulated MDSCs M2 polarization and suppressed miR-1 family miRNAs secretion, thereby promoting MDSC-mediated metastasis and enhancing tumor migration and invasion (Figure 4B) [332].
Reproductive system
Breast cancer (BC)
In invasive ductal carcinoma of the breast, LILRB1 expression was significantly higher in tumor cells than in adjacent normal tissue, positively correlating with tumor stage and inversely correlating with OS [219]. In the TME of BC, LILRB1 was predominantly expressed by infiltrating CD68+ macrophages [333], and was also notably upregulated in NK cells, particularly in patients with invasive BC relative to healthy donors and individuals with noninvasive (in situ) lesions [334, 335]. LILRB2 expression was detected in BC cell lines, as well as in primary ductal and lobular carcinomas, but was absent in normal breast tissue. Its upregulation was associated with reduced TILs, increased LN metastasis [336], and poor prognosis [195, 229]. Mechanistically, LILRB2 promoted immune evasion by facilitating the interaction between HLA-A and the E3 ubiquitin ligase membrane-associated RING-CH finger protein 9 (MARCH9), leading to HLA-A degradation (Figure 4C) [195]. Furthermore, LILRB2 activated MAPK/ERK1/2 signaling, which increased fatty acid synthesis and lipid accumulation to support tumor progression while inducing T cell senescence (Figure 4C) [229]. In triple-negative BC, elevated LILRB2 activated AKT/mTOR signaling to upregulate glucose transporter 3 (GLUT3) and pyruvate kinase muscle 2 (PKM2), thereby reprogramming aerobic glycolysis and enhancing tumor aggressiveness (Figure 4C) [337]. LILRB4 expression was notably enriched in infiltrating CD45+ cells in BC relative to other cancers [338]. By regulating cytokines like IL-6, IL-1β, and TNF, LILRB4 influenced BC incidence, tumor growth, and patient survival outcomes [339].
Ovarian cancer
In ovarian cancer, LILRB1 was highly expressed in both tumor cells and infiltrating immune cells (Figure 4D). A high density of LILRB1+ immune cells correlated significantly with younger age, advanced FIGO stage, shorter survival, and chemoresistance [225]. Additionally, LILRB1 expression was linked to increased infiltration of M2-type macrophages, reduced DC activation, and impaired CD8+ T cell function. Notably, the combined assessment of LILRB1+ immune cells and CD8+ T cell levels effectively stratified patient prognosis [225].
Endometrial cancer and cervical cancer
LILRB2 was significantly upregulated in endometrial cancer, where it predicted poor prognosis and functionally promoted tumor cell proliferation, colony formation, and migration. Mechanistically, LILRB2 promoted tumor progression by activating the SHP2/CaMK1/CREB axis (Figure 4E) [228]. In cervical cancer, transcriptomic profiling identified LILRB2 as a robust diagnostic biomarker capable of distinguishing patients from healthy individuals [340].
Urinary system
Clear cell renal cell carcinoma (ccRCC)
In a preliminary cohort of ten ccRCC patients, LILRB1 and LILRB2 were each expressed in tumor cells from six cases, with markedly elevated LILRB1 levels in three [227]. Both receptors were abundantly expressed within the ccRCC TME. Tumor-infiltrating CD8+ T cells expressed LILRB1, with their function inhibited by HLA-G [341]. Additionally, infiltrating LILRB1+CD4+ T cells exhibited high cytolytic properties, but were selectively inhibited by HLA-G+ targets [342]. Elevated LILRB2 in peritumoral macrophages enhanced infiltration and stimulated VEGF-C production, driving angiogenesis [343]. Bioinformatics analyses revealed LILRB3 overexpression in ccRCC, correlating with poor prognosis and increased immune infiltration [344].
Bladder cancer
Soluble LILRB1 and LILRB2 levels were low in healthy individuals but significantly elevated in patients with non-muscle invasive bladder cancer (NMIBC) [345]. Additionally, patients with recurrent NMIBC exhibited higher frequencies of circulating LILRB1+CD8+ T cells compared to non-recurrent cases, with this increase emerging as an independent prognostic marker for disease recurrence [226].
Prostate cancer (PC)
In PC, LILRB1 expression was elevated in NK cell-infiltrated regions of metastatic patients compared to localized disease and healthy controls. Critically, tumor cells induced LILRB1 expression in NK cells while downregulating activating receptors, including NKp46, NKG2D, and CD16, thereby impairing tumor recognition [346]. Additionally, elevated mRNA levels of LILRB2, LILRB3, and LILRB5 were associated with shorter RFS [347].
Nervous system
Glioma
In glioma, high LILRB1 expression correlated with increased tumor volume and independently predicted poor prognosis, confirming its role as a pathogenic driver. Functionally, LILRB1 promoted tumor cell proliferation, migration, and invasion [118]. In a bioinformatics study, increased LILRB1 expression was positively associated with hypomethylation, M2 macrophage infiltration, upregulation of immune checkpoints, and activation of the JAK/STAT signaling pathway [118].
Glioblastoma
Glioblastoma, a World Health Organization grade IV malignant glioma characterized by poorly differentiated and highly proliferative pleomorphic astrocytes [348], demonstrated significant upregulation of LILRB1-4 expression in tumor tissues while LILRB5 remains unaltered [349]. Among these, LILRB2 expression positively correlated with tumor grade, with high expression independently predicting shorter disease-free survival and OS [350]. Within the glioblastoma TME, LILRB1 was predominantly expressed on NK cells, where co-culture with tumor cells induced its expression and impaired NK cell cytotoxicity [349]. Concurrently, LILRB2/PirB promoted MDSC formation and expansion through small extracellular vesicle secretion, establishing an immunosuppressive microenvironment [350]. In addition, LILRB3 was highly expressed in a subset of monocyte-derived TAMs, with its expression closely associated with the mesenchymal-like cellular state and significantly correlated with poor prognosis [163]. Mechanistically, LILRB3 bound to APOE and activated the SHP1 signaling pathway, contributing to the formation of an immunosuppressive TME [163].
Cutaneous tumors
Melanoma
In melanoma, LILRBs are primarily expressed in immune cells within the TME, with individual members fulfilling distinct roles. LILRB2 showed marked overexpression in melanoma cells compared to normal epithelium [229]. Its murine ortholog, PirB, drove tumor proliferation, adhesion, and migration through MAPK/ERK1/2-mediated fatty acid synthesis and lipid accumulation, concurrently driving tumor progression while inducing T cell exhaustion [229]. LILRB3 was functionally expressed on MDSCs in patients with melanoma, with its activity ligand-dependently activated by galectin-4 and galectin-7 [47]. Notably, therapeutic blockade of LILRB3 successfully inhibited immunosuppressive activity of MDSC from melanoma patients [47]. LILRB4 predominantly localized to CD68+ TAMs in melanoma, facilitating immune escape by inducing CD8+ T suppressor cells and blunting T cell responses [320]. Additionally, LILRB4 demonstrated upregulation on CD45+ immune infiltrates, with expression levels increasing progressively during tumor advancement [338].
Merkel cell carcinoma (MCC)
MCC, a rare and aggressive skin cancer typically arising in sun-exposed areas, presents a complex TME enriched with diverse immune infiltrates. Strikingly, all TAM and DC subsets in MCC exhibited significantly elevated expression of LILRB1, LILRB2, and LILRB4 [351].
Tumors of other systems
Oral squamous cell carcinoma (OSCC)
In OSCC, LILRB1 mRNA expression was significantly downregulated compared to control tissues [352]. This reduced expression inversely correlated with oxidative stress markers, suggesting that oxidative stress may suppress LILRB1 to promote tumor immune evasion [352].
Head and neck squamous cell carcinoma
Cancer stem cells in head and neck squamous cell carcinoma are key drivers of multidrug resistance, metastasis, recurrence, and immunosuppression, with LILRB2 expression notably upregulated in these cells [353]. However, the functional role and underlying mechanisms of LILRB2 in cancer stem cells require further investigation.
Thyroid carcinoma
In papillary thyroid carcinoma, LILRB1 expression was significantly elevated in tumor tissues relative to matched non-tumor tissues. Its expression positively correlated with pathological stage and inversely with OS [219]. In anaplastic thyroid carcinoma, LILRB2 identified as a critical immune checkpoint, with high expression in TAMs which drives immune evasion [354].
6. Clinical Trials of Targeting LILRBs in Cancer Therapy
LILRBs serve dual roles as immune checkpoint regulators and tumor-promoting factors, highlighting them as compelling therapeutic targets in oncology. Although LILRB-targeted therapies remain in early stages of development, accumulating preclinical evidence provides a robust theoretical foundation for clinical translation. Currently, mAbs and engineered T cell therapies targeting LILRBs are under clinical investigation, with active clinical evaluation primarily focus on hematologic malignancies and advanced solid tumors. This section summarizes ongoing clinical trials, detailing therapeutic modalities, mechanisms of action, and stages of development (Table 3).
Clinical trials targeting LILRB family in hematological malignancies and solid tumors.
| Target | Agent | Drug type | Disease indication | Therapy | Intervention | Trial number | Phase and status | Trial duration |
|---|---|---|---|---|---|---|---|---|
| Hematological malignancies | ||||||||
| LILRB4 | IO-202 | IgG1 mAb | M-AML, CMML | Monotherapy and combination therapy | IO-202 alone; IO-202 and azacitidine (chemotherapy); IO-202 and azacitidine (chemotherapy) + venetoclax (BCL-2 inhibitor) | NCT04372433 | Phase I (Completed) | Sep 2020 - Jan 2025 |
| LILRB4 CAR T | CAR T (Specific effector T-cells) | AML (M4/M5) | Monotherapy | Anti-LILRB4 CAR T cells | NCT04803929 | Phase I (Recruiting) | Mar 2021 - Mar 2026 | |
| LILRB4 STAR‑T | STAR-T (Specific effector T-cells) | AML | Monotherapy | LILRB4 STAR-T cells | NCT05518357 | Phase I (Completed) | Jun 2022 - Nov 2022 | |
| LILRB4 STAR-T | STAR-T (Specific effector T-cells) | AML | Monotherapy | LILRB4 STAR-T cells | NCT05548088 | Phase I (Completed) | May 2023 - Aug 2024 | |
| LILRB4 STAR-T | STAR-T (Specific effector T-cells) | AML, CMML | Monotherapy | LILRB4 STAR-T cells | NCT05739409 | NA (Unknown status) | Feb 2023 - Aug 2024 | |
| Solid tumors | ||||||||
| LILRB1 | BND-22/SAR444881 | IgG4 mAb | Advanced solid tumors: BC, CC, CRC, EC, GC, HNSCC, HCC, gallbladder cancer, CCA, NSCLC, RCC, CSCC, PC, OC, and urothelial carcinoma | Monotherapy and combination therapy | BND-22/SAR444881 alone; BND-22/SAR444881 + pembrolizumab (anti-PD-1); BND-22/SAR444881 + cetuximab (anti-EGFR) | NCT04717375 | Phase I/II (Active, not recruiting) | Apr 2021 - Jun 2025 |
| AGEN1571 | IgG4 mAb | Advanced solid tumors | Monotherapy and combination therapy | AGEN1571 alone;AGEN1571 + balstilimab (anti-PD-1);AGEN1571 + botensilimab (anti-CTLA-4);AGEN1571 + balstilimab (anti-PD-1) + botensilimab (anti-CTLA-4) | NCT05377528 | Phase I (Completed) | Jul 2022 - Dec 2024 | |
| ADA-011 | mAb | Advanced solid tumors | Monotherapy and combination therapy | ADA-011 alone; ADA-011 + PD-L1 inhibitor | NCT05601219 | Phase I (Terminated) | Nov 2022 - Oct 2024 | |
| LILRB2 | JTX-8064 | IgG4 mAb | Advanced solid tumors: ccRCC, TNBC, OC, HNSCC, NSCLC, CSCC, UPS, liposarcoma, BTC | Monotherapy and combination therapy | JTX-8064 alone;JTX-8064 + pembrolizumab (anti-PD-1) | NCT04669899 | Phase I/II (Completed) | Jan 2021 - Nov 2023 |
| IO-108 | IgG4 mAb | Advanced solid tumors | Monotherapy and combination therapy | IO-108 alone; IO-108 + pembrolizumab (anti-PD-1);IO-108 + cemiplimab (anti-PD-1); | NCT05054348 | Phase Ib (Completed) | Sep 2021 - May 2024 | |
| MK-4830 | IgG4 mAb | Advanced solid tumors: PC, glioblastoma, NSCLC, HNSCC, CCRCC, GC, OC, FTC, PPC, BC, mesothelioma | Monotherapy and combination therapy | MK-4830 alone; MK-4830 + pembrolizumab (anti-PD-1); MK-4830 + pembrolizumab (anti-PD-1) + lenvatinib (tyrosine kinase inhibitor), carboplatin, pemetrexed, paclitaxel and cisplatin (chemotherapies) | NCT03564691 | Phase I (Active, not recruiting) | Jul 2018 - Sep 2025 | |
| MK-4830 | IgG4 mAb | High-grade serous OC | Combination therapy | MK-4830 + pembrolizumab (anti-PD-1) + chemotherapy | NCT05446870 | Phase II (Completed) | Jul 2022 - Oct 2024 | |
| ES009 | IgG4 mAb | Advanced solid tumors | Monotherapy | ES009 alone | NCT06007482 | Phase I (Completed) | Sep 2023 - Feb 2025 | |
| LILRB4 | IO-202 | IgG1 mAb | Advanced solid tumors | Monotherapy and combination therapy | IO-202 alone;IO-202 + pembrolizumab (anti-PD-1); RP2D of IO-202 + pembrolizumab (anti-PD-1) | NCT05309187 | Phase I (Completed) | Apr 2022 - May 2024 |
| NGM831 | IgG1 mAb | Advanced solid tumors: PC, BC, GC, NSCLC, CC, endocervical cancer, HNSCC, BUC, CRC, EC, OC, RCC, PCa, Melanoma, Mesothelioma, CCA | Monotherapy and combination therapy | NGM831; NGM831 + pembrolizumab (anti-PD-1); NGM831 + NGM438 (anti-LAIR1) + pembrolizumab (anti-PD-1) | NCT05215574 | Phase I (Active, not recruiting) | Mar 2022 - Mar 2026 | |
| MK-0482 | IgG4 mAb | Advanced solid tumors | Monotherapy and combination therapy | MK-0482 alone; MK-0482 + pembrolizumab (anti-PD-1); MK-0482 + pembrolizumab (anti-PD-1) + paclitaxel, nab-paclitaxel, gemcitabine, carboplatin and pemetrexed (chemotherapies) | NCT03918278 | Phase I (Completed) | Jun 2019 - Jun 2025 | |
| MK-0482 | IgG4 mAb | NSCLC | Combination therapy | MK-4830 + pembrolizumab (anti-PD-1) | NCT04165096 | Phase II (Completed) | Jan 2020 - May 2025 | |
| LILRB1 and LILRB2 | NGM707 | BsAb (mAb) | Advanced solid tumors: Mesothelioma, glioblastoma, RCC, NSCLC, Melanoma, PC, GC, HNSCC, CCA, endocervical cancer, BC, OC, CC, CRC, EC | Monotherapy and combination therapy | NGM707 alone; NGM707 + pembrolizumab (anti-PD-1) | NCT04913337 | Phase I/II (Active, not recruiting) | Jun 2021 - Jul 2025 |
| LILRB2 and PD-1 | CDX-585 | BsAb (IgG-scFv) | Advanced solid tumors: NSCLC, GC, HNSCC, OC, PPC, FTC, BUC, CRC, EC, HCC, RCC, CCA, PC | Monotherapy | CDX-585 alone | NCT05788484 | Phase I (Completed) | May 2023 - May 2025 |
| LILRB1, LILRB2 and KIR3DL1 | IOS-1002 | TsAb (HLA-B57- B2m-IgG4_Fc fusion protein) | Advanced solid tumors | Monotherapy and combination therapy | IOS-1002 alone; IOS-1002 + pembrolizumab (anti-PD-1) | NCT05763004 | Phase Ia/Ib (Recruiting) | Mar 2023 - May 2025 |
AML, acute myeloid leukemia; BC, breast cancer; BCL-2, B-cell lymphoma 2; BsAb, bispecific antibody; BTC, biliary tract cancer; BUC, bladder urothelial cancer; CAR T, chimeric antigen receptor T-cell; CC, cervical cancer; CCA, cholangiocarcinoma; ccRCC, clear cell renal cell carcinoma; CMML, chronic myelomonocytic leukemia; CRC, colorectal cancer; CSCC, cutaneous squamous cell carcinoma; CTLA-4, cytotoxic T-lymphocyte antigen 4; EC, esophageal carcinoma; EGFR, epidermal growth factor receptor; FTC, fallopian tube cancer; GC, gastric cancer; HCC, hepatocellular carcinoma; HNSCC, head and neck squamous cell carcinoma; IgG-scFv, immunoglobulin G single-chain variable fragment; LAIR1, leukocyte-associated immunoglobulin-like receptor 1; mAb, monoclonal antibody; M-AML, acute myeloid leukemia with monocytic differentiation; NA, not available; NSCLC, non-small cell lung cancer; OC, ovarian cancer; PC, pancreatic cancer; PCa, prostate cancer; PD-1, programmed cell death protein 1; PD-L1, programmed death ligand 1; PPC, primary peritoneal carcinoma; RCC, renal cell carcinoma; STAR‑T, synthetic T-cell receptor and antigen receptor; TNBC, triple-negative breast cancer; TsAb, trispecific antibody; UPS, undifferentiated pleomorphic sarcoma.
6.1 Hematological malignancies
LILRB4
LILRB4 has emerged as a promising therapeutic target in hematologic malignancies, particularly AML, driving active development of targeted agents. IO-202, a humanized IgG1 mAb, specifically binds LILRB4 and antagonizes its interaction with ApoE, effectively antagonizing downstream signaling and functional effects (Figure 5) [355, 356]. This agent has completed evaluation by a Phase I clinical trial (NCT04372433) as a monotherapy or combined with pembrolizumab in AML and CMML patients [355, 356]. CAR T-cell therapy—which utilizes genetically engineered T cells expressing CARs to recognize and eliminate tumor cells expressing surface antigens [357]. Although six CAR T products are FDA-approved for hematologic malignancies, their development for myeloid malignancies remains limited by the challenge of identifying safe and effective myeloid-restricted targets. LILRB4 represents a compelling CAR T target in AML, due to its selective expression on both leukemia cells and HSCs. A 2018 study demonstrated potent anti-leukemic activity of LILRB4-directed CAR T cells in vitro and in vivo, with no observed toxicity to normal HSCs (Figure 5) [295]. Based on these findings, a Phase I trial (NCT04803929) is currently evaluating this strategy in relapsed or refractory M4/M5 AML subtypes. Recent advances include LILRB4-specific nanoantibodies engineered for Synthetic T-cell Receptor and Antigen Receptor (STAR)-T cells (Figure 5). Preclinically, LILRB4 STAR-T cells effectively eliminated LILRB4+ AML cells [358], with dual-epitope targeting demonstrating enhanced anti-leukemia activity in vivo. Following completion of two Phase I STAR-T trials (NCT05518357 and NCT05548088), an additional trial (NCT05739409) is assessing this approach in AML and CMML.
Clinical trials of LILRB-targeted cancer therapies. In hematological malignancies, LILRB4-targeted therapeutic strategies include monoclonal antibodies, chimeric antigen receptor (CAR) T-cell therapy, and synthetic T-cell receptor and antigen receptor (STAR-T) therapy, all of which demonstrate potent tumor cell-killing activity. In solid tumors, current approaches employ monoclonal antibodies targeting LILRB1, LILRB2, and LILRB4 to inhibit tumor progression. Additionally, emerging bispecific and trispecific antibodies—designed to simultaneously engage multiple LILRBs or other immune-related proteins—are under development for solid tumors, aiming to enhance antitumor efficacy through synergistic mechanisms.
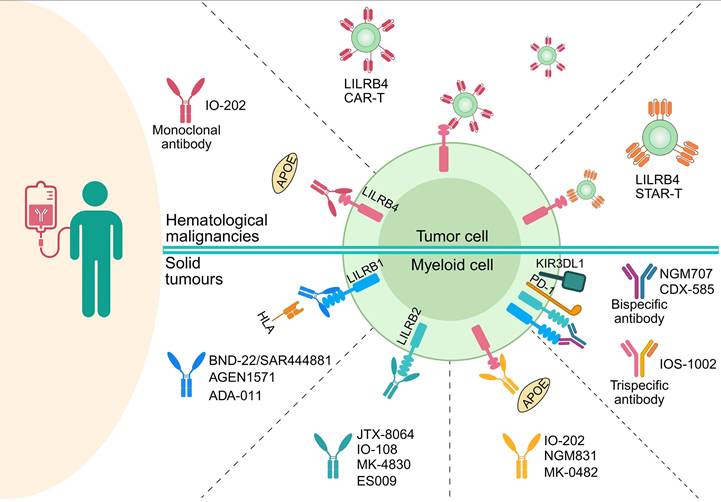
6.2 Solid tumors
Development of LILRB-targeted therapies for advanced solid tumors has predominantly focused on LILRB1, LILRB2, and LILRB4. Therapeutic agents against these receptors are progressing through clinical development, with several now in active clinical trials. Current research prioritizes evaluating these therapies as monotherapies, while parallel efforts explore their potential in combination with ICIs, such as anti-PD-1 antibodies.
LILRB1
LILRB1 inhibits immune cell activity and amplifies myeloid-driven immunosuppression via interactions with HLA-G. To disrupt this axis, the anti-LILRB1 antibody BND-22/SAR444881 has been developed to block LILRB1 binding to these ligands, thereby restoring immune cell activity (Figure 5) [359]. BND-22/SAR444881 is under evaluation in a Phase I/II trial (NCT04717375) to determine its safety, tolerability, and antitumor efficacy, both as a monotherapy and in combination with the anti-EGFR antibody cetuximab or the anti-PD-1 antibody pembrolizumab in patients with advanced solid tumors [359]. Several other novel immunotherapies, including combinations with anti-CTLA-4 or anti-PD-1 antibodies, are under clinical evaluation for advanced solid tumors [360]. AGEN1571 has completed clinical evaluation in trial NCT05377528, which investigated its efficacy both as monotherapy and in combination with PD-1 inhibitor balstilimab and/or anti-CTLA-4 antibody botensilimab for advanced solid tumors (Figure 5). ADA-011, the third candidate, was evaluated in a Phase I trial (NCT05601219) with/without PD-L1 inhibition before trial termination (Figure 5). ADA-011, the third candidate, was evaluated in a Phase I trial (NCT05601219) with/without PD-L1 inhibition before trial termination. These clinical programs underscore LILRB1 as a therapeutic target and highlight strategic combinations to overcome immune suppression in solid tumors.
LILRB2
JTX-8064, a mAb targeting LILRB2, disrupts its interaction with ligands and reprograms macrophages and DCs toward immunostimulatory phenotypes (Figure 5), enhancing anti-PD-1 efficacy in preclinical models [361, 362]. Its Phase I trial (NCT04669899) evaluating monotherapy and PD-1 combinations remains unpublished. Similarly, IO-108 induces myeloid proinflammatory reprogramming and synergizes with PD-1 blockade (Figure 5), with Phase I safety/efficacy data (NCT05054348) pending [363]. MK-4830, a third LILRB2-targeting mAb, blocks ligand-induced immunosuppression (Figure 5). Early trial data (NCT03564691 and NCT05446870) showed favorable tolerability and dose-dependent target engagement in advanced solid tumors [364]. Finally, the high-affinity antibody ES009 reprograms myeloid cells to enhance T-cell activation and potentiates PD-1 inhibitors (Figure 5), has completed Phase I evaluation (NCT06007482) [365]. Collectively, these clinical programs validate LILRB2 targeting to reverse myeloid-mediated immunosuppression through phenotypic reprogramming and rational ICI combinations.
LILRB4
IO-202 has completed clinical evaluation for AML and CMML, with a separate Phase I trial (NCT05309187) demonstrating its efficacy both as monotherapy and in combination with pembrolizumab for advanced solid tumors (Figure 5). NGM831, designed to disrupt LILRB4 interactions with fibronectin and APOE, attenuating immunosuppression and potentiating myeloid cell activation (Figure 5). It is currently being assessed in a Phase I trial (NCT05215574) for solid tumors. Additionally, MK-0482, an IgG4 mAb targeting LILRB4 has been evaluated across multiple completed trials (NCT03918278 and NCT04165096) to assess safety, pharmacokinetics, and therapeutic efficacy in advanced and metastatic solid tumors (Figure 5). These efforts highlight the therapeutic potential of LILRB4 targeting to reverse tumor immunosuppression and enhance antitumor immunity.
Antibodies targeting multiple LILRBs
Bispecific and multi-specific antibodies are advancing clinically for solid tumors, frequently combined with ICIs [366]. NGM707, a humanized IgG antibody with dual specificity for LILRB1 and LILRB2, reprograms MDSCs to enhance immune activity (Figure 5). When combined with anti-PD-1 therapy, it further amplifies macrophage-mediated CD4+ T cell activation. NGM707, administered as monotherapy or in combination with pembrolizumab (NCT04913337), is being actively investigated in advanced or metastatic solid tumors. Although the trial is no longer enrolling participants, its results are highly anticipated. CDX-585, a tetravalent bispecific antibody targeting both PD-1 and LILRB2, drives M1 polarization and induces proinflammatory cytokine secretion following treatment with LPS or CD40 agonist mAbs (Figure 5). The Phase I trial of CDX‑585 (NCT05788484) in advanced solid tumors was recently completed, and its forthcoming results are highly anticipated for insights into its clinical efficacy and safety [367]. IOS-1002, a trispecific mAb targeting LILRB1, LILRB2, and KIR3DL1, blocks HLA-G/ANGPTL engagement to inhibit ITIM signaling and polarize macrophages toward immunostimulatory phenotype, enhancing tumor phagocytosis (Figure 5) [368]. A Phase Ia/Ib clinical trial (NCT05763004) is currently evaluating IOS-1002 both as a monotherapy and in combination with anti-PD-1 antibodies.
Early-phase clinical trials demonstrate promising antitumor activity of LILRB-targeted therapies through remodeling TME, yet their development faces significant challenges. The broad expression of LILRBs across immune and non-immune tissues raises legitimate concerns regarding off-target toxicity and autoimmune sequelae. Compounding this risk, the dual immunomodulatory roles of LILRBs mean that therapeutic interference could disrupt immune homeostasis, leading to unintended immune dysregulation. To advance this therapeutic class, future research must prioritize four critical domains: 1) comprehensive mapping of ligand-receptor interaction networks, 2) single-cell resolution expression profiling across physiological and pathological contexts, 3) mechanism-guided biomarker development for patient stratification, and 4) engineering solutions including optimized Fc domains and conditional activation systems to enhance target specificity. Rigorous safety monitoring integrated with mechanistic pharmacodynamic studies will be essential to maximize therapeutic efficacy while mitigating risks, ultimately enabling transformative cancer immunotherapies.
7. Conclusions and Perspectives
This review delineates the pivotal immunoregulatory functions of LILRB family members and their multifaceted contributions to disease pathogenesis. We systematically dissect mechanistic insights into LILRB-mediated TME reprogramming and tumor cell functional modulation across malignancies, while synthesizing clinical advances in LILRB-targeted therapeutic strategies. The compelling capacity of LILRBs to architect immunosuppressive TME and subvert antitumor immunity underscores their emerging potential as therapeutic targets. Future interrogation of LILRB signaling networks promises to unveil novel diagnostic biomarkers, prognostic indicators, and precision immunotherapy paradigms.
Despite advances in understanding LILRB in infectious diseases and tumor, knowledge of their ligands remains limited, hindering a comprehensive analysis of their immune-regulatory mechanisms [18]. Current ligand identification methods, including expression cloning, chemical cross-linking with mass spectrometry, high-throughput protein arrays, candidate molecule validation, cell microarrays, and light-based receptor capture techniques, face significant challenges [97, 369, 370]. Ligand discovery is complicated by context-dependent expression patterns within specific tumor or disease microenvironments. Moreover, LILRB polymorphisms may alter ligand-binding specificities and contribute to diverse pathological roles. For example, a specific LILRB3 haplotype has been linked to kidney transplant failure and early onset of end-stage renal disease [371], highlighting the broader clinical significance of LILRB genetic variations. These gaps hinder a full mechanistic understanding of LILRB functions and immune regulation [18]. Although several downstream signaling pathways activated by LILRB-ligand interactions have been identified [18-20, 24, 372], the overall signaling network remains incompletely defined, particularly in cancer. Future studies elucidating LILRB ligands and their integrated signaling networks will be crucial for unraveling immune-regulatory mechanisms and optimizing cancer treatment strategies.
LILRB expression varies significantly across tumor types and correlates strongly with disease progression [197, 224, 229, 231, 301, 322, 350]. However, the dynamic spatiotemporal regulation of LILRBs throughout tumor evolution—from initiation to metastasis—and how different LILRBs cooperate in driving malignancy remain poorly understood [214]. Additionally, while LILRBs engage in multiple signaling pathways with extensive crosstalk, the integrated mechanisms through which these networks collectively regulate tumor cell behaviors—including proliferation, apoptosis, invasion, and metastasis—require comprehensive elucidation [72, 116, 215, 222, 223, 228, 232, 305, 321]. Furthermore, the mechanisms underlying aberrant LILRB expression in malignancies remain undefined, representing a fundamental barrier to therapeutic development.
LILRBs are widely expressed in immune cells within both hematological and solid tumors [20, 372], where they orchestrate highly complex immunomodulatory functions. Critical knowledge gaps persist regarding how LILRB-mediated intercellular signaling coordinates immune cell crosstalk to balance immune surveillance and tumor evasion [195, 205, 213, 214, 320, 324, 352, 354]. The crosstalk between LILRBs and other cytokines or signaling pathways in the TME further complicates understanding their role in immune escape and therapy resistance [296, 310, 320, 324, 326, 333, 342].
Methodological limitations remain a major barrier to advancing LILRB research. The lack of suitable animal models severely restricts the ability to investigate the dynamic regulation and functional roles of LILRBs during disease progression. This challenge is further exacerbated by the limited availability of high-quality research reagents—particularly for understudied members such as LILRB5 [49, 313, 373, 374]—which significantly hampers mechanistic and functional studies [20, 21]. Overcoming these technical deficiencies is essential for deepening our understanding of LILRB biology and enabling its therapeutic exploitation.
To overcome these challenges, future research could prioritize four interconnected areas: First, improving ligand discovery with enhanced detection platforms and integrated methodologies. Second, leveraging single-cell and spatial transcriptomics to map stage-specific LILRB expression trajectories throughout tumor evolution. Third, applying multiplexed proteomic and phosphoproteomic approaches to delineate downstream signaling pathways. Finally, developing 3D organotypic cultures and precision gene-edited in vivo models to dissect intercellular LILRB signaling networks. Collectively, these efforts will provide a comprehensive framework for understanding LILRB biology and guiding therapeutic innovation.
Therapeutically, despite the complexity and biomarker challenges, LILRBs remain promising immuno-oncology targets. Rational drug design—such as bispecific antibodies or allosteric inhibitors targeting key LILRB-ligand interactions—may disrupt tumor-immune crosstalk. Clinically, combining LILRB-targeted agents with immune checkpoint blockade or conventional therapies holds potential to overcome resistance and expand durable response rates. As research advances, LILRB-targeted strategies are poised to make transformative impacts in cancer treatment, offering new hope for patients.
Abbreviations
AD: Alzheimer's disease; AITD: autoimmune thyroid disease; ALCAM: activated leukocyte cell adhesion molecule; ALL: acute lymphoblastic leukemia; AML: acute myeloid leukemia; ANGPTL: angiopoietin-like protein; APC: antigen-presenting cell; ApoE: apolipoprotein E; ARG1: arginase-1; ATL: acute T-cell leukemia; Aβ: β-amyloid; BC: breast cancer; BCR: B-cell receptor; BM: bone marrow; CaMK: calcium/calmodulin-dependent protein kinase; CAR: chimeric antigen receptor; ccRCC: clear cell renal cell carcinoma; cFLIP: cellular FLICE-inhibitory protein; CLL: chronic lymphocytic leukemia; CMML: chronic myelomonocytic leukemia; CMV: cytomegalovirus; CNS: central nervous system; CRC: colorectal cancer; CREB: cAMP response element-binding protein; CTCL: cutaneous T-cell lymphoma; CTLA-4: cytotoxic T-lymphocyte antigen 4; CTL: cytotoxic T lymphocyte; DC: dendritic cell; DENV: Dengue virus; EBV: Epstein-Barr virus; ERK: extracellular signal-regulated kinase; F-actin: filamentous actin; FDA: food and drug administration; FcγR: Fc-gamma receptor; FGF-1: fibroblast growth factor 1; FTO: fat mass and obesity-associated protein; GA: glatiramer acetate; GC: gastric cancer; GLUT3: glucose transporter 3; HCC: hepatocellular carcinoma; HIV-1: Human Immunodeficiency Virus Type 1; HLA: human leukocyte antigen; HLA-I: HLA class I; HSC: hematopoietic stem cell; ICI: immune checkpoint inhibitor; IDO: indoleamine 2, 3-dioxygenase; IFN: interferon; Ig-like: immunoglobulin-like; IL-10: interleukin-10; IMC: immature myeloid cell; ITAMs: immunoreceptor tyrosine-based activation motifs; ITIMs: immunoreceptor tyrosine-based inhibitory motifs; JNK: c-Jun N-terminal kinase; KIRs: killer-cell immunoglobulin-like receptors; LDL: low-density lipoprotein; LDLR: LDL receptor; LDLRAP1: LDL receptor adapter protein 1; LILRBs: leukocyte immunoglobulin-like receptors, subfamily B; LN: lymph node; LPS: lipopolysaccharide; Lyn: Lck/Yes-related novel protein tyrosine kinase; MCC: merkel cell carcinoma; M.tuberculosis: Mycobacterium tuberculosis; m6A: N6-methyladenosine; mAbs: monoclonal antibodies; M-AML: monocytic differentiation-associated AML; MAPK: mitogen-activated protein kinase; MARCH9: membrane-associated RING-CH finger protein 9; mDCs: myeloid DCs; MDS: myelodysplastic syndrome; MDSC: myeloid-derived suppressor cell; MGUS: monoclonal gammopathy of undetermined significance; MHC-I: Major Histocompatibility Complex class I; MM: multiple myeloma; MS: multiple sclerosis; MTOC: microtubule organizing center; NASH: non-alcoholic steatohepatitis; NK: natural killer; NMIBC: non-muscle invasive bladder cancer; NSCLC: non-small cell lung cancer; OS: overall survival; OSCC: oral squamous cell carcinoma; P.falciparum: Plasmodium falciparum; PBMC: peripheral blood mononuclear cell; PC: prostate cancer; PDAC: pancreatic ductal adenocarcinoma; PD-L1: programmed cell death ligand 1; PI3K: phosphatidylinositol-4-phosphate 3-kinase; PirB: paired immunoglobulin-like receptor B; PKM2: pyruvate kinase muscle 2; PLC-γ: phospholipase C gamma; RA: rheumatoid arthritis; RFS: relapse-free survival; RIFINs: repetitive interspersed families of polypeptides; ROCK2: Rho-associated protein kinase 2; ROS: reactive oxygen species; S100A8: S100 calcium binding protein A8; S100A9: S100 calcium binding protein A9; SARS-CoV-2: severe acute respiratory syndrome coronavirus 2; Sema4A: semaphorin-4A; SHIP1: Src homology 2 domain-containing inositol phosphatase 1; sHLA-G: soluble HLA-G; SHP1/2: Src homology 2 domain-containing phosphatase 1/2; SIV: simian immunodeficiency virus; SLE: systemic lupus erythematosus; SNP: single nucleotide polymorphism; STAR: synthetic T-cell receptor and antigen receptor; Syk: Src family kinases: spleen tyrosine kinase; tDCs: tolerogenic DCs; T.gondii: Toxoplasma gondii; TAMs: tumor-associated macrophages; Th1: T helper 1; Th2: T helper 2; TILs: tumor-infiltrating lymphocytes; TLR: Toll-like receptor; TME: tumor microenvironment; TNF: tumor necrosis factor; Tr1: type 1 T regulatory; TRAF2: TNF receptor-associated factor 2; Treg: regulatory T cell; UPAR: urokinase plasminogen activator receptor; Vav1: Vav guanine nucleotide exchange factor 1; VEGF: vascular endothelial growth factor; ZAP70: zeta-chain-associated protein kinase 70 kDa; β2m: β2-microglobulin.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (82300212 and 82403198) and the Science and Technology Commission of Shanghai Municipality (24ZR1447300).
Author contributions
L.A, L.Y and F.X. supervised, coordinated and participated in the literature search, in the writing and figure generation; Z.Y, X.Y and W.Q participated in the literature search and in the writing. All authors have read and approved the article.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Houghton AN, Guevara-Patino JA. Immune recognition of self in immunity against cancer. J Clin Invest. 2004;114:468-71
2. Haynes NM, Chadwick TB, Parker BS. The complexity of immune evasion mechanisms throughout the metastatic cascade. Nat Immunol. 2024;25:1793-808
3. Cesano A, Augustin R, Barrea L, Bedognetti D, Bruno TC, Carturan A. et al. Advances in the understanding and therapeutic manipulation of cancer immune responsiveness: A society for immunotherapy of cancer (sitc) review. J Immunother Cancer. 2025;13:e008876
4. Zhu D, Kim WJ, Lee H, Bao X, Kim P. Engineering CAR-T therapeutics for enhanced solid tumor targeting. Adv Mater. 2025;37:e2414882
5. Poudel K, Vithiananthan T, Kim JO, Tsao H. Recent progress in cancer vaccines and nanovaccines. Biomaterials. 2025;314:122856
6. Yaremenko AV, Khan MM, Zhen X, Tang Y, Tao W. Clinical advances of mRNA vaccines for cancer immunotherapy. Med. 2025;6:100562
7. Larson RC, Maus MV. Recent advances and discoveries in the mechanisms and functions of CAR T cells. Nat Rev Cancer. 2021;21:145-61
8. Wei SC, Duffy CR, Allison JP. Fundamental mechanisms of immune checkpoint blockade therapy. Cancer Discov. 2018;8:1069-86
9. Zhang Y, Zhang Z. The history and advances in cancer immunotherapy: Understanding the characteristics of tumor-infiltrating immune cells and their therapeutic implications. Cell Mol Immunol. 2020;17:807-21
10. Grosser R, Cherkassky L, Chintala N, Adusumilli PS. Combination immunotherapy with CAR T cells and checkpoint blockade for the treatment of solid tumors. Cancer Cell. 2019;36:471-82
11. Sharma P, Goswami S, Raychaudhuri D, Siddiqui BA, Singh P, Nagarajan A. et al. Immune checkpoint therapy-current perspectives and future directions. Cell. 2023;186:1652-69
12. Sharma P, Siddiqui BA, Anandhan S, Yadav SS, Subudhi SK, Gao J. et al. The next decade of immune checkpoint therapy. Cancer Discov. 2021;11:838-57
13. Alsaafeen BH, Ali BR, Elkord E. Resistance mechanisms to immune checkpoint inhibitors: Updated insights. Mol Cancer. 2025;24:20
14. Sanmamed MF, Nie X, Desai SS, Villaroel-Espindola F, Badri T, Zhao D. et al. A burned-out CD8(+) T-cell subset expands in the tumor microenvironment and curbs cancer immunotherapy. Cancer Discov. 2021;11:1700-15
15. A cocktail of kinase inhibitors that enhance the antitumor effects of CAR-T cell therapy. Nat Immunol. 2025;26:170-1
16. Pauken KE, Sammons MA, Odorizzi PM, Manne S, Godec J, Khan O. et al. Epigenetic stability of exhausted T cells limits durability of reinvigoration by PD-1 blockade. Science. 2016;354:1160-5
17. Tay C, Tanaka A, Sakaguchi S. Tumor-infiltrating regulatory T cells as targets of cancer immunotherapy. Cancer Cell. 2023;41:450-65
18. De Louche CD, Roghanian A. Human inhibitory leukocyte Ig-like receptors: From immunotolerance to immunotherapy. JCI Insight. 2022;7:e2414882
19. Abdallah F, Coindre S, Gardet M, Meurisse F, Naji A, Suganuma N. et al. Leukocyte immunoglobulin-like receptors in regulating the immune response in infectious diseases: A window of opportunity to pathogen persistence and a sound target in therapeutics. Front Immunol. 2021;12:717998
20. Redondo-García S, Barritt C, Papagregoriou C, Yeboah M, Frendeus B, Cragg MS. et al. Human leukocyte immunoglobulin-like receptors in health and disease. Front Immunol. 2023;14:1282874
21. Hodges A, Dubuque R, Chen SH, Pan PY. The LILRB family in hematologic malignancies: Prognostic associations, mechanistic considerations, and therapeutic implications. Biomark Res. 2024;12:159
22. Deng M, Gui X, Kim J, Xie L, Chen W, Li Z. et al. LILRB4 signalling in leukaemia cells mediates T cell suppression and tumour infiltration. Nature. 2018;562:605-9
23. Li Z, Deng M, Huang F, Jin C, Sun S, Chen H. et al. LILRB4 itims mediate the T cell suppression and infiltration of acute myeloid leukemia cells. Cell Mol Immunol. 2020;17:272-82
24. Zeller T, Münnich IA, Windisch R, Hilger P, Schewe DM, Humpe A. et al. Perspectives of targeting LILRB1 in innate and adaptive immune checkpoint therapy of cancer. Front Immunol. 2023;14:1240275
25. Zhang CC. A perspective on LILRBs and LAIR as immune checkpoint targets for cancer treatment. Biochem Biophys Res Commun. 2022;633:64-7
26. Borges L, Hsu ML, Fanger N, Kubin M, Cosman D. A family of human lymphoid and myeloid Ig-like receptors, some of which bind to MHC class I molecules. J Immunol. 1997;159:5192-6
27. Colonna M, Navarro F, Bellon T, Llano M, Garcia P, Samaridis J. et al. A common inhibitory receptor for major histocompatibility complex class I molecules on human lymphoid and myelomonocytic cells. J Exp Med. 1997;186:1809-18
28. Cosman D, Fanger N, Borges L, Kubin M, Chin W, Peterson L. et al. A novel immunoglobulin superfamily receptor for cellular and viral MHC class I molecules. Immunity. 1997;7:273-82
29. Wende H, Colonna M, Ziegler A, Volz A. Organization of the leukocyte receptor cluster (LRC) on human chromosome 19q13.4. Mamm Genome. 1999;10:154-60
30. Wang Q, Song H, Cheng H, Qi J, Nam G, Tan S. et al. Structures of the four Ig-like domain LILRB2 and the four-domain LILRB1 and HLA-G1 complex. Cell Mol Immunol. 2020;17:966-75
31. Nam G, Shi Y, Ryu M, Wang Q, Song H, Liu J. et al. Crystal structures of the two membrane-proximal Ig-like domains (D3D4) of LILRB1/B2: Alternative models for their involvement in peptide-HLA binding. Protein Cell. 2013;4:761-70
32. Liu F, Cocker ATH, Pugh JL, Djaoud Z, Parham P, Guethlein LA. Natural LILRB1 D1-D2 variants show frequency differences in populations and bind to HLA class I with various avidities. Immunogenetics. 2022;74:513-25
33. Chapman TL, Heikema AP, West AP Jr, Bjorkman PJ. Crystal structure and ligand binding properties of the D1D2 region of the inhibitory receptor LIR-1 (ILT2). Immunity. 2000;13:727-36
34. Cheng H, Mohammed F, Nam G, Chen Y, Qi J, Garner LI. et al. Crystal structure of leukocyte Ig-like receptor LILRB4 (ILT3/LIR-5/CD85k): A myeloid inhibitory receptor involved in immune tolerance. J Biol Chem. 2011;286:18013-25
35. Daeron M, Latour S, Malbec O, Espinosa E, Pina P, Pasmans S. et al. The same tyrosine-based inhibition motif, in the intracytoplasmic domain of FC gamma RIIB, regulates negatively BCR-, TCR-, and FCR-dependent cell activation. Immunity. 1995;3:635-46
36. Pymm P, Illing PT, Ramarathinam SH, O'Connor GM, Hughes VA, Hitchen C. et al. MHC-I peptides get out of the groove and enable a novel mechanism of HIV-1 escape. Nat Struct Mol Biol. 2017;24:387-94
37. Wu X, Li T, Jiang R, Yang X, Guo H, Yang R. Targeting MHC-I molecules for cancer: Function, mechanism, and therapeutic prospects. Mol Cancer. 2023;22:194
38. Zhao J, Zhong S, Niu X, Jiang J, Zhang R, Li Q. The MHC class I-LILRB1 signalling axis as a promising target in cancer therapy. Scand J Immunol. 2019;90:e12804
39. Hu Z, Zhang Q, He Z, Jia X, Zhang W, Cao X. MHC1/LILRB1 axis as an innate immune checkpoint for cancer therapy. Front Immunol. 2024;15:1421092
40. Barsh GS, Bashirova AA, Martin-Gayo E, Jones DC, Qi Y, Apps R. et al. LILRB2 interaction with HLA class I correlates with control of HIV-1 infection. PLoS Genet. 2014;10:e1004196
41. Marchesi M, Andersson E, Villabona L, Seliger B, Lundqvist A, Kiessling R. et al. HLA-dependent tumour development: A role for tumour associate macrophages? J Transl Med. 2013;11:247
42. Shiroishi M, Kuroki K, Rasubala L, Tsumoto K, Kumagai I, Kurimoto E. et al. Structural basis for recognition of the nonclassical MHC molecule HLA-G by the leukocyte Ig-like receptor B2 (LILRB2/LIR2/ILT4/CD85d). Proc Natl Acad Sci U S A. 2006;103:16412-7
43. Allen RL, Raine T, Haude A, Trowsdale J, Wilson MJ. Leukocyte receptor complex-encoded immunomodulatory receptors show differing specificity for alternative HLA-B27 structures. J Immunol. 2001;167:5543-7
44. Jones DC, Kosmoliaptsis V, Apps R, Lapaque N, Smith I, Kono A. et al. HLA class I allelic sequence and conformation regulate leukocyte Ig-like receptor binding. J Immunol. 2011;186:2990-7
45. Kuroki K, Matsubara H, Kanda R, Miyashita N, Shiroishi M, Fukunaga Y. et al. Structural and functional basis for LILRB immune checkpoint receptor recognition of HLA-G isoforms. J Immunol. 2019;203:3386-94
46. Willcox BE, Thomas LM, Bjorkman PJ. Crystal structure of HLA-A2 bound to LIR-1, a host and viral major histocompatibility complex receptor. Nat Immunol. 2003;4:913-9
47. Huang R, Liu X, Kim J, Deng H, Deng M, Gui X. et al. LILRB3 supports immunosuppressive activity of myeloid cells and tumor development. Cancer Immunol Res. 2024;12:350-62
48. Ayukawa S, Kamoshita N, Nakayama J, Teramoto R, Pishesha N, Ohba K. et al. Epithelial cells remove precancerous cells by cell competition via MHC class I-LILRB3 interaction. Nat Immunol. 2021;22:1391-402
49. Zhang Z, Hatano H, Shaw J, Olde Nordkamp M, Jiang G, Li D. et al. The leukocyte immunoglobulin-like receptor family member LILRB5 binds to HLA-class I heavy chains. PLoS One. 2015;10:e0129063
50. Carosella ED, Gregori S, Tronik-Le Roux D. HLA-G/LILRBs: A cancer immunotherapy challenge. Trends Cancer. 2021;7:389-92
51. Boyson JE, Erskine R, Whitman MC, Chiu M, Lau JM, Koopman LA. et al. Disulfide bond-mediated dimerization of HLA-G on the cell surface. Proc Natl Acad Sci U S A. 2002;99:16180-5
52. Clements CS, Kjer-Nielsen L, Kostenko L, Hoare HL, Dunstone MA, Moses E. et al. Crystal structure of HLA-G: A nonclassical MHC class I molecule expressed at the fetal-maternal interface. Proc Natl Acad Sci U S A. 2005;102:3360-5
53. Gonen-Gross T, Achdout H, Gazit R, Hanna J, Mizrahi S, Markel G. et al. Complexes of HLA-G protein on the cell surface are important for leukocyte Ig-like receptor-1 function. J Immunol. 2003;171:1343-51
54. Shiroishi M, Kuroki K, Ose T, Rasubala L, Shiratori I, Arase H. et al. Efficient leukocyte Ig-like receptor signaling and crystal structure of disulfide-linked HLA-G dimer. J Biol Chem. 2006;281:10439-47
55. Lee N, Geraghty DE. HLA-F surface expression on B cell and monocyte cell lines is partially independent from tapasin and completely independent from TAP. J Immunol. 2003;171:5264-71
56. Lepin EJM, Bastin JM, Allan DSJ, Roncador G, Braud VM, Mason DY. et al. Functional characterization of HLA-F and binding of HLA-F tetramers to ILT2 and ILT4 receptors. Eur J Immunol. 2000;30:3552-61
57. Beck S, Barrell BG. Human cytomegalovirus encodes a glycoprotein homologous to MHC class-I antigens. Nature. 1988;331:269-72
58. Fahnestock ML, Johnson JL, Feldman RM, Neveu JM, Lane WS, Bjorkman PJ. The MHC class I homolog encoded by human cytomegalovirus binds endogenous peptides. Immunity. 1995;3:583-90
59. Chapman TL, Heikeman AP, Bjorkman PJ. The inhibitory receptor LIR-1 uses a common binding interaction to recognize class I MHC molecules and the viral homolog UL18. Immunity. 1999;11:603-13
60. Yang Z, Bjorkman PJ. Structure of UL18, a peptide-binding viral MHC mimic, bound to a host inhibitory receptor. Proc Natl Acad Sci U S A. 2008;105:10095-100
61. Willcox BE, Thomas LM, Chapman TL, Heikema AP, West AP Jr, Bjorkman PJ. Crystal structure of LIR-2 (ILT4) at 1.8 A: Differences from LIR-1 (ILT2) in regions implicated in the binding of the human cytomegalovirus class I MHC homolog UL18. BMC Struct Biol. 2002;2:6
62. Wahlgren M, Goel S, Akhouri RR. Variant surface antigens of Plasmodium falciparum and their roles in severe malaria. Nat Rev Microbiol. 2017;15:479-91
63. Saito F, Hirayasu K, Satoh T, Wang CW, Lusingu J, Arimori T. et al. Immune evasion of Plasmodium falciparum by RIFIN via inhibitory receptors. Nature. 2017;552:101-5
64. Harrison TE, Mørch AM, Felce JH, Sakoguchi A, Reid AJ, Arase H. et al. Structural basis for RIFIN-mediated activation of LILRB1 in malaria. Nature. 2020;587:309-12
65. Chen Y, Xu K, Piccoli L, Foglierini M, Tan J, Jin W. et al. Structural basis of malaria RIFIN binding by LILRB1-containing antibodies. Nature. 2021;592:639-43
66. Sakoguchi A, Saito F, Hirayasu K, Shida K, Matsuoka S, Itagaki S. et al. Plasmodium falciparum RIFIN is a novel ligand for inhibitory immune receptor LILRB2. Biochem Biophys Res Commun. 2021;548:167-73
67. Kersten S. Angiopoietin-like 3 in lipoprotein metabolism. Nat Rev Endocrinol. 2017;13:731-9
68. Kim I, Moon SO, Koh KN, Kim H, Uhm CS, Kwak HJ. et al. Molecular cloning, expression, and characterization of angiopoietin-related protein. Angiopoietin-related protein induces endothelial cell sprouting. J Biol Chem. 1999;274:26523-8
69. Horiguchi H, Kadomatsu T, Oike Y. The two faces of angiopoietin-like protein 2 in cancer. Cancer Sci. 2025;116:592-9
70. Zhang CC, Kaba M, Ge G, Xie K, Tong W, Hug C. et al. Angiopoietin-like proteins stimulate ex vivo expansion of hematopoietic stem cells. Nat Med. 2006;12:240-5
71. Deng M, Lu Z, Zheng J, Wan X, Chen X, Hirayasu K. et al. A motif in LILRB2 critical for ANGPTL2 binding and activation. Blood. 2014;124:924-35
72. Zheng J, Umikawa M, Cui C, Li J, Chen X, Zhang C. et al. Inhibitory receptors bind angptls and support blood stem cells and leukaemia development. Nature. 2012;485:656-60
73. Abu-Farha M, Ghosh A, Al-Khairi I, Madiraju SRM, Abubaker J, Prentki M. The multi-faces of ANGPTL8 in health and disease: Novel functions beyond lipoprotein lipase modulation. Prog Lipid Res. 2020;80:101067
74. Lee YH, Lee SG, Lee CJ, Kim SH, Song YM, Yoon MR. et al. Association between betatrophin/ANGPTL8 and non-alcoholic fatty liver disease: Animal and human studies. Sci Rep. 2016;6:24013
75. Li DP, Huang L, Kan RR, Meng XY, Wang SY, Zou HJ. et al. LILRB2/PirB mediates macrophage recruitment in fibrogenesis of nonalcoholic steatohepatitis. Nat Commun. 2023;14:4436
76. Puschel AW, Adams RH, Betz H. Murine semaphorin D/collapsin is a member of a diverse gene family and creates domains inhibitory for axonal extension. Neuron. 1995;14:941-8
77. Lu N, Li Y, Zhang Z, Xing J, Sun Y, Yao S. et al. Human Semaphorin-4A drives Th2 responses by binding to receptor ILT-4. Nat Commun. 2018;9:742
78. Dutronc Y, Porcelli SA. The CD1 family and T cell recognition of lipid antigens. Tissue Antigens. 2002;60:337-53
79. Porcelli SA. The CD1 family: A third lineage of antigen-presenting molecules. Adv Immunol. 1995;59:1-98
80. Li D, Hong A, Lu Q, Gao GF, Jin B, Screaton GR. et al. A novel role of CD1c in regulating CD1d-mediated NKT cell recognition by competitive binding to Ig-like transcript 4. Int Immunol. 2012;24:729-37
81. Li D, Wang L, Yu L, Freundt EC, Jin B, Screaton GR. et al. Ig-like transcript 4 inhibits lipid antigen presentation through direct CD1d interaction. J Immunol. 2009;182:1033-40
82. Mahley RW. Apolipoprotein e: Cholesterol transport protein with expanding role in cell biology. Science. 1988;240:622-30
83. Zhou J, Wang Y, Huang G, Yang M, Zhu Y, Jin C. et al. LILRB3 is a putative cell surface receptor of APOE4. Cell Res. 2023;33:116-30
84. Hou J, Chen Y, Cai Z, Heo GS, Yuede CM, Wang Z. et al. Antibody-mediated targeting of human microglial leukocyte Ig-like receptor B4 attenuates amyloid pathology in a mouse model. Sci Transl Med. 2024;16:eadj9052
85. Arnold V, Cummings JS, Moreno-Nieves UY, Didier C, Gilbert A, Barre-Sinoussi F. et al. S100A9 protein is a novel ligand for the CD85j receptor and its interaction is implicated in the control of HIV-1 replication by NK cells. Retrovirology. 2013;10:122
86. Chan KR, Ong EZ, Tan HC, Zhang SL-X, Zhang Q, Tang KF. et al. Leukocyte immunoglobulin-like receptor B1 is critical for antibody-dependent dengue. Proc Natl Acad Sci U S A. 2014;111:2722-7
87. Cao Q, Shin WS, Chan H, Vuong CK, Dubois B, Li B. et al. Inhibiting amyloid-β cytotoxicity through its interaction with the cell surface receptor LILRB2 by structure-based design. Nat Chem. 2018;10:1213-21
88. Hofer J, Forster F, Isenman DE, Wahrmann M, Leitner J, Holzl MA. et al. Ig-like transcript 4 as a cellular receptor for soluble complement fragment C4d. FASEB J. 2016;30:1492-503
89. Xu Z, Chang C-C, Li M, Zhang Q-Y, Vasilescu E-RM, D'Agati V. et al. ILT3.Fc-CD166 interaction induces inactivation of p70 s6 kinase and inhibits tumor cell growth. J Immunol. 2018;200:1207-19
90. Wang Y, Sun Y, Deng S, Liu J, Yu J, Chi H. et al. Discovery of galectin-8 as an LILRB4 ligand driving m-MDSCS defines a class of antibodies to fight solid tumors. Cell Rep Med. 2024;5:101374
91. Su MT, Inui M, Wong YL, Takahashi M, Sugahara-Tobinai A, Ono K. et al. Blockade of checkpoint ILT3/LILRB4/gp49b binding to fibronectin ameliorates autoimmune disease in BXSB/Yaa mice. Int Immunol. 2021;33:447-58
92. Paavola KJ, Roda JM, Lin VY, Chen P, O'Hollaren KP, Ventura R. et al. The fibronectin-ILT3 interaction functions as a stromal checkpoint that suppresses myeloid cells. Cancer Immunol Res. 2021;9:1283-97
93. He Y, Zhang C, Tan L, Deng M, Liu X, Huang R. et al. Eph receptors activate myeloid checkpoint receptor LILRB5 to support tumor development. Cancer Immunol Res. 2025;13:821-35
94. Thomas R, Matthias T, Witte T. Leukocyte immunoglobulin-like receptors as new players in autoimmunity. Clin Rev Allergy Immunol. 2009;38:159-62
95. Storm L, Bruijnesteijn J, de Groot NG, Bontrop RE. The genomic organization of the LILR region remained largely conserved throughout primate evolution: Implications for health and disease. Front Immunol. 2021;12:716289
96. Daeron M, Jaeger S, Du Pasquier L, Vivier E. Immunoreceptor tyrosine-based inhibition motifs: A quest in the past and future. Immunol Rev. 2008;224:11-43
97. Zhang F, Zheng J, Kang X, Deng M, Lu Z, Kim J. et al. Inhibitory leukocyte immunoglobulin-like receptors in cancer development. Sci China Life Sci. 2015;58:1216-25
98. van der Touw W, Chen HM, Pan PY, Chen SH. LILRB receptor-mediated regulation of myeloid cell maturation and function. Cancer Immunol Immunother. 2017;66:1079-87
99. Colonna M, Nakajima H, Navarro F, Lopez-Botet M. A novel family of Ig-like receptors for HLA class I molecules that modulate function of lymphoid and myeloid cells. J Leukoc Biol. 1999;66:375-81
100. Christofides A, Katopodi XL, Cao C, Karagkouni D, Aliazis K, Yenyuwadee S. et al. SHP-2 and PD-1-SHP-2 signaling regulate myeloid cell differentiation and antitumor responses. Nat Immunol. 2023;24:55-68
101. Binstadt BA, Brumbaugh KM, Dick CJ, Scharenberg AM, Williams BL, Colonna M. et al. Sequential involvement of lck and SHP-1 with MHC-recognizing receptors on NK cells inhibits FCR-initiated tyrosine kinase activation. Immunity. 1996;5:629-38
102. Daigle I, Yousefi S, Colonna M, Green DR, Simon HU. Death receptors bind SHP-1 and block cytokine-induced anti-apoptotic signaling in neutrophils. Nat Med. 2002;8:61-7
103. Huang ZY, Hunter S, Kim MK, Indik ZK, Schreiber AD. The effect of phosphatases SHP-1 and SHIP-1 on signaling by the ITIM- and ITAM-containing Fcgamma receptors FcgammaRIIB and FcgammaRIIA. J Leukoc Biol. 2003;73:823-9
104. Stebbins CC, Watzl C, Billadeau DD, Leibson PJ, Burshtyn DN, Long EO. Vav1 dephosphorylation by the tyrosine phosphatase SHP-1 as a mechanism for inhibition of cellular cytotoxicity. Mol Cell Biol. 2003;23:6291-9
105. Lichterfeld M, Yu XG. The emerging role of leukocyte immunoglobulin-like receptors (LILRs) in HIV-1 infection. J Leukoc Biol. 2012;91:27-33
106. Jensen MA, Patterson KC, Kumar AA, Kumabe M, Franek BS, Niewold TB. Functional genetic polymorphisms in ILT3 are associated with decreased surface expression on dendritic cells and increased serum cytokines in lupus patients. Ann Rheum Dis. 2013;72:596-601
107. Wiśniewski A, Kowal A, Wyrodek E, Nowak I, Majorczyk E, Wagner M. et al. Genetic polymorphisms and expression of HLA-G and its receptors, KIR2DL4 and LILRB1, in non-small cell lung cancer. Tissue Antigens. 2015;85:466-75
108. Moraes AGd, Ayo CM, Elpídio LNS, Souza VHd, Yamanaka AHU, Nogueira ML. et al. HLA-G, LILRB1 and LILRB2 variants in Zika virus transmission from mother to child in a population from south and southeast of Brazil. Curr Issues Mol Biol. 2022;44:2783-93
109. Yu K, Davidson CL, Wójtowicz A, Lisboa L, Wang T, Airo AM. et al. LILRB1 polymorphisms influence posttransplant hcmv susceptibility and ligand interactions. J Clin Invest. 2018;128:1523-37
110. Papanikolaou NA, Vasilescu ER, Suciu-Foca N. Novel single nucleotide polymorphisms in the human immune inhibitory immunoglobulin-like T cell receptor type 4. Hum Immunol. 2004;65:700-5
111. Li M, Ye J, Chang M, Feng L, Liu T, Zhang D. et al. Polymorphisms in immunosuppression-related genes are associated with AML. Front Immunol. 2025;16:1530510
112. Wilson MJ, Torkar M, Haude A, Milne S, Jones T, Sheer D. et al. Plasticity in the organization and sequences of human KIR/ILT gene families. Proc Natl Acad Sci U S A. 2000;97:4778-83
113. Hirayasu K, Ohashi J, Tanaka H, Kashiwase K, Ogawa A, Takanashi M. et al. Evidence for natural selection on leukocyte immunoglobulin-like receptors for HLA class I in Northeast Asians. Am J Hum Genet. 2008;82:1075-83
114. Oliveira MLG, Castelli EC, Veiga-Castelli LC, Pereira ALE, Marcorin L, Carratto TMT. et al. Genetic diversity of the LILRB1 and LILRB2 coding regions in an admixed Brazilian population sample. HLA. 2022;100:325-48
115. Lamar DL, Weyand CM, Goronzy JJ. Promoter choice and translational repression determine cell type-specific cell surface density of the inhibitory receptor CD85j expressed on different hematopoietic lineages. Blood. 2010;115:3278-86
116. Xie L, Chen C, Zhang T, Yang W, Zheng D, Cao L. et al. LILRB4 regulates multiple myeloma development through STAT3-PFKFB1 pathway. Cell Death Dis. 2024;15:515
117. Nakajima H, Asai A, Okada A, Ping L, Hamajima F, Sata T. et al. Transcriptional regulation of ILT family receptors. J Immunol. 2003;171:6611-20
118. Zou R, Zhong X, Liang K, Zhi C, Chen D, Xu Z. et al. Elevated LILRB1 expression predicts poor prognosis and is associated with tumor immune infiltration in patients with glioma. BMC Cancer. 2023;23:403
119. Su R, Dong L, Li Y, Gao M, Han L, Wunderlich M. et al. Targeting FTO suppresses cancer stem cell maintenance and immune evasion. Cancer Cell. 2020;38:79-96.e11
120. Cen Q, Chen J, Guo J, Chen M, Wang H, Wu S. et al. CLPs-miR-103a-2-5p inhibits proliferation and promotes cell apoptosis in AML cells by targeting LILRB3 and Nrf2/HO-1 axis, regulating CD8+ T cell response. J Transl Med. 2024;22:278
121. Manavalan JS, Kim-Schulze S, Scotto L, Naiyer AJ, Vlad G, Colombo PC. et al. Alloantigen specific CD8+CD28- Foxp3+ T suppressor cells induce ILT3+ ILT4+ tolerogenic endothelial cells, inhibiting alloreactivity. Int Immunol. 2004;16:1055-68
122. Kim-Schulze S, Seki T, Vlad G, Scotto L, Fan J, Colombo PC. et al. Regulation of ILT3 gene expression by processing of precursor transcripts in human endothelial cells. Am J Transplant. 2006;6:76-82
123. Ju XS, Hacker C, Scherer B, Redecke V, Berger T, Schuler G. et al. Immunoglobulin-like transcripts ILT2, ILT3 and ILT7 are expressed by human dendritic cells and down-regulated following activation. Gene. 2004;331:159-64
124. Vlad G, Piazza F, Colovai A, Cortesini R, Della Pietra F, Suciu-Foca N. et al. Interleukin-10 induces the upregulation of the inhibitory receptor ILT4 in monocytes from HIV positive individuals. Hum Immunol. 2003;64:483-9
125. Svajger U, Obermajer N, Jeras M. IFN-gamma-rich environment programs dendritic cells toward silencing of cytotoxic immune responses. J Leukoc Biol. 2014;95:33-46
126. Svajger U, Rozman PJ. Synergistic effects of interferon-gamma and vitamin D(3) signaling in induction of ILT-3(high)PDL-1(high) tolerogenic dendritic cells. Front Immunol. 2019;10:2627
127. Penna G, Roncari A, Amuchastegui S, Daniel KC, Berti E, Colonna M. et al. Expression of the inhibitory receptor ILT3 on dendritic cells is dispensable for induction of CD4+Foxp3+ regulatory T cells by 1,25-dihydroxyvitamin D3. Blood. 2005;106:3490-7
128. Fedoric B, Krishnan R. Rapamycin downregulates the inhibitory receptors ILT2, ILT3, ILT4 on human dendritic cells and yet induces T cell hyporesponsiveness independent of FoxP3 induction. Immunol Lett. 2008;120:49-56
129. Svajger U, Vidmar A, Jeras M. Niflumic acid renders dendritic cells tolerogenic and up-regulates inhibitory molecules ILT3 and ILT4. Int Immunopharmacol. 2008;8:997-1005
130. Li Z, Zhao M, Li T, Zheng J, Liu X, Jiang Y. et al. Decidual macrophage functional polarization during abnormal pregnancy due to toxoplasma gondii: Role for LILRB4. Front Immunol. 2017;8:1013
131. Brown DP, Jones DC, Anderson KJ, Lapaque N, Buerki RA, Trowsdale J. et al. The inhibitory receptor LILRB4 (ILT3) modulates antigen presenting cell phenotype and, along with LILRB2 (ILT4), is upregulated in response to salmonella infection. BMC Immunol. 2009;10:56
132. Montoya JG, Liesenfeld O. Toxoplasmosis. Lancet. 2004;363:1965-76
133. Garnaud C, Fricker-Hidalgo H, Evengard B, Alvarez-Martinez MJ, Petersen E, Kortbeek LM. et al. Toxoplasma gondii-specific IgG avidity testing in pregnant women. Clin Microbiol Infect. 2020;26:1155-60
134. Wang J, Zhao SJ, Wang LL, Lin XX, Mor G, Liao AH. Leukocyte immunoglobulin-like receptor subfamily B: A novel immune checkpoint molecule at the maternal-fetal interface. J Reprod Immunol. 2023;155:103764
135. Colonna M, Samaridis J, Cella M, Angman L, Allen RL, O'Callaghan CA. et al. Human myelomonocytic cells express an inhibitory receptor for classical and nonclassical MHC class I molecules. J Immunol. 1998;160:3096-100
136. Li K, Shi H, Zhang B, Ou X, Ma Q, Chen Y. et al. Myeloid-derived suppressor cells as immunosuppressive regulators and therapeutic targets in cancer. Signal Transduct Target Ther. 2021;6:362
137. Medzhitov R, Janeway CA Jr. Innate immune induction of the adaptive immune response. Cold Spring Harb Symp Quant Biol. 1999;64:429-35
138. Vivier E, Artis D, Colonna M, Diefenbach A, Di Santo JP, Eberl G. et al. Innate lymphoid cells: 10 years on. Cell. 2018;174:1054-66
139. Demaria O, Cornen S, Daeron M, Morel Y, Medzhitov R, Vivier E. Harnessing innate immunity in cancer therapy. Nature. 2019;574:45-56
140. Banchereau J, Steinman RM. Dendritic cells and the control of immunity. Nature. 1998;392:245-52
141. Wu L, Liu YJ. Development of dendritic-cell lineages. Immunity. 2007;26:741-50
142. Bosteels V, Janssens S. Striking a balance: New perspectives on homeostatic dendritic cell maturation. Nat Rev Immunol. 2025;25:125-40
143. Liang S, Horuzsko A. Mobilizing dendritic cells for tolerance by engagement of immune inhibitory receptors for HLA-G. Hum Immunol. 2003;64:1025-32
144. Liang S, Ristich V, Arase H, Dausset J, Carosella ED, Horuzsko A. Modulation of dendritic cell differentiation by HLA-G and ILT4 requires the IL-6-STAT3 signaling pathway. Proc Natl Acad Sci U S A. 2008;105:8357-62
145. Young NT, Waller EC, Patel R, Roghanian A, Austyn JM, Trowsdale J. The inhibitory receptor LILRB1 modulates the differentiation and regulatory potential of human dendritic cells. Blood. 2008;111:3090-6
146. Stallone G, Pontrelli P, Infante B, Gigante M, Netti GS, Ranieri E. et al. Rapamycin induces ILT3(high)ILT4(high) dendritic cells promoting a new immunoregulatory pathway. Kidney Int. 2014;85:888-97
147. Gao A, Sun Y, Peng G. ILT4 functions as a potential checkpoint molecule for tumor immunotherapy. Biochim Biophys Acta Rev Cancer. 2018;1869:278-85
148. Banchereau J, Zurawski S, Thompson-Snipes L, Blanck J-P, Clayton S, Munk A. et al. Immunoglobulin-like transcript receptors on human dermal CD14+dendritic cells act as a CD8-antagonist to control cytotoxic T cell priming. Proc Natl Acad Sci U S A. 2012;109:18885-90
149. Manavalan JS, Rossi PC, Vlad G, Piazza F, Yarilina A, Cortesini R. et al. High expression of ILT3 and ILT4 is a general feature of tolerogenic dendritic cells. Transpl Immunol. 2003;11:245-58
150. Ristich V, Liang S, Zhang W, Wu J, Horuzsko A. Tolerization of dendritic cells by HLA-G. Eur J Immunol. 2005;35:1133-42
151. Gregori S, Tomasoni D, Pacciani V, Scirpoli M, Battaglia M, Magnani CF. et al. Differentiation of type 1 T regulatory cells (Tr1) by tolerogenic DC-10 requires the IL-10-dependent ILT4/HLA-G pathway. Blood. 2010;116:935-44
152. Ngambenjawong C, Gustafson HH, Pun SH. Progress in tumor-associated macrophage (TAM)-targeted therapeutics. Adv Drug Deliv Rev. 2017;114:206-21
153. Li M, Yang Y, Xiong L, Jiang P, Wang J, Li C. Metabolism, metabolites, and macrophages in cancer. J Hematol Oncol. 2023;16:80
154. Barkal AA, Weiskopf K, Kao KS, Gordon SR, Rosental B, Yiu YY. et al. Engagement of MHC class I by the inhibitory receptor LILRB1 suppresses macrophages and is a target of cancer immunotherapy. Nat Immunol. 2017;19:76-84
155. Chen HM, van der Touw W, Wang YS, Kang K, Mai S, Zhang J. et al. Blocking immunoinhibitory receptor LILRB2 reprograms tumor-associated myeloid cells and promotes antitumor immunity. J Clin Invest. 2018;128:5647-62
156. Jones DC, Irving L, Dudley R, Blumli S, Wolny M, Chatzopoulou EI. et al. LILRB2 blockade facilitates macrophage repolarization and enhances T cell-mediated antitumor immunity. J Immunother Cancer. 2025;13:e010012
157. Yeboah M, Papagregoriou C, Jones DC, Chan HTC, Hu G, McPartlan JS. et al. LILRB3 (ILT5) is a myeloid cell checkpoint that elicits profound immunomodulation. JCI Insight. 2020;5:e141593
158. Yang T, Qian Y, Liang X, Wu J, Zou M, Deng M. LILRB4, an immune checkpoint on myeloid cells. Blood Sci. 2022;4:49-56
159. Matsumoto Y, Wang LL, Yokoyama WM, Aso T. Uterine macrophages express the gp49b inhibitory receptor in midgestation. J Immunol. 2001;166:781-6
160. Yin J, Song Y, Fu Y, Wang J, Zhang Z, Ruan S. et al. NLRP12/C1qA positive feedback in tumor-associated macrophages regulates immunosuppression through LILRB4/NF-κB pathway in lung adenocarcinoma. Cancer Immunol Immunother. 2024;74:16
161. Zhang W, Liang S, Wu J, Horuzsko A. Human inhibitory receptor immunoglobulin-like transcript 2 amplifies CD11b+Gr1+ myeloid-derived suppressor cells that promote long-term survival of allografts. Transplantation. 2008;86:1125-34
162. Kostlin N, Ostermeir AL, Spring B, Schwarz J, Marme A, Walter CB. et al. HLA-G promotes myeloid-derived suppressor cell accumulation and suppressive activity during human pregnancy through engagement of the receptor ILT4. Eur J Immunol. 2017;47:374-84
163. Zhuang Q, Liu Y, Wang H, Lin Z, Sun L, Liu Y. et al. LILRB3 suppresses immunity in glioma and is associated with poor prognosis. Clin Transl Med. 2023;13:e1396
164. Singh L, Muise ES, Bhattacharya A, Grein J, Javaid S, Stivers P. et al. ILT3 (LILRB4) promotes the immunosuppressive function of tumor-educated human monocytic myeloid-derived suppressor cells. Mol Cancer Res. 2021;19:702-16
165. Mantovani A, Cassatella MA, Costantini C, Jaillon S. Neutrophils in the activation and regulation of innate and adaptive immunity. Nat Rev Immunol. 2011;11:519-31
166. Zhang F, Xia Y, Su J, Quan F, Zhou H, Li Q. et al. Neutrophil diversity and function in health and disease. Signal Transduct Target Ther. 2024;9:343
167. Baudhuin J, Migraine J, Faivre V, Loumagne L, Lukaszewicz A-C, Payen D. et al. Exocytosis acts as a modulator of the ILT4-mediated inhibition of neutrophil functions. Proc Natl Acad Sci U S A. 2013;110:17957-62
168. Lewis Marffy AL, McCarthy AJ. Leukocyte immunoglobulin-like receptors (LILRs) on human neutrophils: Modulators of infection and immunity. Front Immunol. 2020;11:857
169. Zhao Y, van Woudenbergh E, Zhu J, Heck AJR, van Kessel KPM, de Haas CJC. et al. The orphan immune receptor LILRB3 modulates Fc receptor-mediated functions of neutrophils. J Immunol. 2020;204:954-66
170. Orr MT, Lanier LL. Natural killer cell education and tolerance. Cell. 2010;142:847-56
171. Yokoyama WM, Kim S. How do natural killer cells find self to achieve tolerance? Immunity. 2006;24:249-57
172. Parham P, Guethlein LA. Genetics of natural killer cells in human health, disease, and survival. Annu Rev Immunol. 2018;36:519-48
173. Ponte M, Cantoni C, Biassoni R, Tradori-Cappai A, Bentivoglio G, Vitale C. et al. Inhibitory receptors sensing HLA-G1 molecules in pregnancy: Decidua-associated natural killer cells express LIR-1 and CD94/NKG2A and acquire p49, an HLA-G1-specific receptor. Proc Natl Acad Sci U S A. 1999;96:5674-9
174. Wang J, Chai Q, Lei Z, Wang Y, He J, Ge P. et al. LILRB1-HLA-G axis defines a checkpoint driving natural killer cell exhaustion in tuberculosis. EMBO Mol Med. 2024;16:1755-90
175. Nunez SY, Ziblat A, Secchiari F, Torres NI, Sierra JM, Raffo Iraolagoitia XL. et al. Human M2 macrophages limit NK cell effector functions through secretion of TGF-beta and engagement of CD85j. J Immunol. 2018;200:1008-15
176. Yao AY, Tang HY, Wang Y, Feng MF, Zhou RL. Inhibition of the activating signals in NK92 cells by recombinant GST-sHLA-G1a chain. Cell Res. 2004;14:155-60
177. Favier B, Lemaoult J, Lesport E, Carosella ED. ILT2/HLA-G interaction impairs NK-cell functions through the inhibition of the late but not the early events of the nk-cell activating synapse. FASEB J. 2010;24:689-99
178. Forte P, Pazmany L, Matter-Reissmann UB, Stussi G, Schneider MK, Seebach JD. HLA-G inhibits rolling adhesion of activated human NK cells on porcine endothelial cells. J Immunol. 2001;167:6002-8
179. Bonilla FA, Oettgen HC. Adaptive immunity. J Allergy Clin Immunol. 2010;125:S33-40
180. Metur SP, Klionsky DJ. Adaptive immunity at the crossroads of autophagy and metabolism. Cell Mol Immunol. 2021;18:1096-105
181. Saverino D, Fabbi M, Ghiotto F, Merlo A, Bruno S, Zarcone D. et al. The CD85/LIR-1/ILT2 inhibitory receptor is expressed by all human T lymphocytes and down-regulates their functions. J Immunol. 2000;165:3742-55
182. Ince MN, Harnisch B, Xu Z, Lee SK, Lange C, Moretta L. et al. Increased expression of the natural killer cell inhibitory receptor CD85j/ILT2 on antigen-specific effector CD8 T cells and its impact on CD8 T-cell function. Immunology. 2004;112:531-42
183. Saverino D, Merlo A, Bruno S, Pistoia V, Grossi CE, Ciccone E. Dual effect of CD85/leukocyte Ig-like receptor-1/Ig-like transcript 2 and CD152 (CTLA-4) on cytokine production by antigen-stimulated human T cells. J Immunol. 2002;168:207-15
184. Purbhoo MA, Liu H, Oddos S, Owen DM, Neil MA, Pageon SV. et al. Dynamics of subsynaptic vesicles and surface microclusters at the immunological synapse. Sci Signal. 2010;3:ra36
185. Dietrich J, Cella M, Colonna M. Ig-like transcript 2 (ILT2)/leukocyte Ig-like receptor 1 (LIR1) inhibits tcr signaling and actin cytoskeleton reorganization. J Immunol. 2001;166:2514-21
186. Shiroishi M, Tsumoto K, Amano K, Shirakihara Y, Colonna M, Braud VM. et al. Human inhibitory receptors Ig-like transcript 2 (ILT2) and ILT4 compete with CD8 for MHC class I binding and bind preferentially to HLA-G. Proc Natl Acad Sci U S A. 2003;100:8856-61
187. Fillatreau S. Cytokine-producing B cells as regulators of pathogenic and protective immune responses. Ann Rheum Dis. 2013;72(Suppl 2):ii80-4
188. Banham AH, Colonna M, Cella M, Micklem KJ, Pulford K, Willis AC. et al. Identification of the CD85 antigen as ILT2, an inhibitory MHC class I receptor of the immunoglobulin superfamily. J Leukoc Biol. 1999;65:841-5
189. Naji A, Menier C, Morandi F, Agaugue S, Maki G, Ferretti E. et al. Binding of HLA-G to ITIM-bearing Ig-like transcript 2 receptor suppresses B cell responses. J Immunol. 2014;192:1536-46
190. Sun YX, Feng Q, Wang SW, Li X, Sheng Z, Peng J. HLA-G-ILT2 interaction contributes to suppression of bone marrow B cell proliferation in acquired aplastic anemia. Ann Hematol. 2022;101:739-48
191. de Visser KE, Joyce JA. The evolving tumor microenvironment: From cancer initiation to metastatic outgrowth. Cancer Cell. 2023;41:374-403
192. Hanahan D. Hallmarks of cancer: New dimensions. Cancer Discov. 2022;12:31-46
193. Chen L, Flies DB. Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat Rev Immunol. 2013;13:227-42
194. Liu Y, Wang Y, Yang Y, Weng L, Wu Q, Zhang J. et al. Emerging phagocytosis checkpoints in cancer immunotherapy. Signal Transduct Target Ther. 2023;8:104
195. Jiang Z, Huang Q, Chang Y, Qiu Y, Cheng H, Yang M. et al. LILRB2 promotes immune escape in breast cancer cells via enhanced HLA-A degradation. Cell Oncol. 2024;47:1679-96
196. Gao Y, Yuan Y, Wen S, Chen Y, Zhang Z, Feng Y. et al. Dual role of ANGPTL8 in promoting tumor cell proliferation and immune escape during hepatocarcinogenesis. Oncogenesis. 2023;12:26
197. Chen X, Yuan M, Zhong T, Wang M, Wu F, Lu J. et al. LILRB2 inhibition enhances radiation sensitivity in non-small cell lung cancer by attenuating radiation-induced senescence. Cancer Lett. 2024;593:216930
198. Mai S, Hodges A, Chen HM, Zhang J, Wang YL, Liu Y. et al. LILRB3 modulates acute myeloid leukemia progression and acts as an effective target for CAR T-cell therapy. Cancer Res. 2023;83:4047-62
199. Chen X, Gao A, Zhang F, Yang Z, Wang S, Fang Y. et al. ILT4 inhibition prevents TAM- and dysfunctional T cell-mediated immunosuppression and enhances the efficacy of anti-PD-L1 therapy in nsclc with EGFR activation. Theranostics. 2021;11:3392-416
200. Zhao WZ, Wang HG, Yang XZ. Leukocyte immunoglobulin-like receptor B2: A promising biomarker for colorectal cancer. World J Gastroenterol. 2024;30:421-3
201. Wang J, Lu Q, Chen X, Aifantis I. Targeting MHC-I inhibitory pathways for cancer immunotherapy. Trends Immunol. 2024;45:177-87
202. Curigliano G, Criscitiello C, Gelao L, Goldhirsch A. Molecular pathways: Human leukocyte antigen G (HLA-G). Clin Cancer Res. 2013;19:5564-71
203. da Silva IL, Montero-Montero L, Ferreira E, Quintanilla M. New insights into the role of Qa-2 and HLA-G non-classical MHC-I complexes in malignancy. Front Immunol. 2018;9:2894
204. Chen QY, Zhou WJ, Zhang JG, Zhang X, Han QY, Lin A. et al. Prognostic significance of the immune checkpoint HLA-G/ILT-4 in the survival of patients with gastric cancer. Int Immunopharmacol. 2022;109:108798
205. Wan R, Wang ZW, Li H, Peng XD, Liu GY, Ou JM. et al. Human leukocyte antigen-G inhibits the anti-tumor effect of natural killer cells via immunoglobulin-like transcript 2 in gastric cancer. Cell Physiol Biochem. 2017;44:1828-41
206. Mejia-Guarnizo LV, Monroy-Camacho PS, Rincon-Rodriguez DE, Rincon-Riveros A, Martinez-Vargas DA, Huertas-Caro CA. et al. Soluble HLA-G (sHLA-G) measurement might be useful as an early diagnostic biomarker and screening test for gastric cancer. Sci Rep. 2023;13:13119
207. Xu YF, Lu Y, Cheng H, Jiang J, Xu J, Long J. et al. High expression of human leukocyte antigen-G is associated with a poor prognosis in patients with PDAC. Curr Mol Med. 2015;15:360-7
208. Kang X, Kim J, Deng M, John S, Chen H, Wu G. et al. Inhibitory leukocyte immunoglobulin-like receptors: Immune checkpoint proteins and tumor sustaining factors. Cell Cycle. 2016;15:25-40
209. Churchill HRO, Fuda FS, Xu J, Deng M, Zhang CC, An Z. et al. Leukocyte immunoglobulin-like receptor B1 and B4 (LILRB1 and LILRB4): Highly sensitive and specific markers of acute myeloid leukemia with monocytic differentiation. Cytometry B Clin Cytom. 2020;100:476-87
210. Kang X, Lu Z, Cui C, Deng M, Fan Y, Dong B. et al. The ITIM-containing receptor LAIR1 is essential for acute myeloid leukaemia development. Nat Cell Biol. 2015;17:665-77
211. Perna F, Berman SH, Soni RK, Mansilla-Soto J, Eyquem J, Hamieh M. et al. Integrating proteomics and transcriptomics for systematic combinatorial chimeric antigen receptor therapy of AML. Cancer Cell. 2017;32:506-19.e5
212. Xu ZJ, Zhang XL, Jin Y, Wang SS, Gu Y, Ma JC. et al. Pan-cancer analysis reveals distinct clinical, genomic, and immunological features of the LILRB immune checkpoint family in acute myeloid leukemia. Mol Ther Oncolytics. 2022;26:88-104
213. Chien KS, Class CA, Montalban-Bravo G, Wei Y, Sasaki K, Naqvi K. et al. LILRB4 expression in chronic myelomonocytic leukemia and myelodysplastic syndrome based on response to hypomethylating agents. Leuk Lymphoma. 2020;61:1493-9
214. Lozano E, Díaz T, Mena MP, Suñe G, Calvo X, Calderón M. et al. Loss of the immune checkpoint CD85j/LILRB1 on malignant plasma cells contributes to immune escape in multiple myeloma. J Immunol. 2018;200:2581-91
215. Sun Z, Ji J, Li Y, Cui Y, Fan L, Li J. et al. Identification of evolutionary mechanisms of myelomatous effusion by single-cell RNA sequencing. Blood Adv. 2023;7:4148-59
216. Villa-Álvarez M, Sordo-Bahamonde C, Lorenzo-Herrero S, Gonzalez-Rodriguez AP, Payer AR, Gonzalez-Garcia E. et al. Ig-like transcript 2 (ILT2) blockade and lenalidomide restore NK cell function in chronic lymphocytic leukemia. Front Immunol. 2018;9:2917
217. Schneider P, Crump NT, Arentsen-Peters STCJM, Smith AL, Hagelaar R, Adriaanse FRS. et al. Modelling acquired resistance to DOT1L inhibition exhibits the adaptive potential of KMT2A-rearranged acute lymphoblastic leukemia. Exp Hematol Oncol. 2023;12:81
218. He J, Xu J, Yu X, Zhu H, Zeng Y, Fan D. et al. Overexpression of ANGPTL2 and LILRB2 as predictive and therapeutic biomarkers for metastasis and prognosis in colorectal cancer. Int J Clin Exp Pathol. 2018;11:2281-94
219. Cheng J, Gao X, Zhang X, Guo H, Chen S, Gou X. Leukocyte immunoglobulin-like receptor subfamily B member 1 potentially acts as a diagnostic and prognostic target in certain subtypes of adenocarcinoma. Med Hypotheses. 2020;144:109863
220. Zhang YI, Lu NAN, Xue Y, Zhang MIN, Li Y, Si Y. et al. Expression of immunoglobulin-like transcript (ILT)2 and ILT3 in human gastric cancer and its clinical significance. Mol Med Rep. 2012;5:910-6
221. Wagener-Ryczek S, Schoemmel M, Kraemer M, Bruns C, Schroeder W, Zander T. et al. Immune profile and immunosurveillance in treatment-naive and neoadjuvantly treated esophageal adenocarcinoma. Cancer Immunol Immunother. 2020;69:523-33
222. Zhang P, Guo X, Li J, Yu S, Wang L, Jiang G. et al. Immunoglobulin-like transcript 4 promotes tumor progression and metastasis and up-regulates VEGF-C expression via ERK signaling pathway in non-small cell lung cancer. Oncotarget. 2015;6:13550-63
223. Liu X, Yu X, Xie J, Zhan M, Yu Z, Xie L. et al. ANGPTL2/LILRB2 signaling promotes the propagation of lung cancer cells. Oncotarget. 2015;6:21004-15
224. Zhang P, Yu S, Li H, Liu C, Li J, Lin W. et al. ILT4 drives B7-H3 expression via PI3K/AKT/MTOR signalling and ILT4/B7-H3 co-expression correlates with poor prognosis in non-small cell lung cancer. FEBS Lett. 2015;589:2248-56
225. Xu X, Yin S, Wang Y, Zhu Q, Zheng G, Lu Y. et al. LILRB1+ immune cell infiltration identifies immunosuppressive microenvironment and dismal outcomes of patients with ovarian cancer. Int Immunopharmacol. 2023;119:110162
226. Desgrandchamps F, LeMaoult J, Goujon A, Riviere A, Rivero-Juarez A, Djouadou M. et al. Prediction of non-muscle-invasive bladder cancer recurrence by measurement of checkpoint HLAG's receptor ILT2 on peripheral CD8(+) T cells. Oncotarget. 2018;9:33160-9
227. Tronik-Le Roux D, Sautreuil M, Bentriou M, Vérine J, Palma MB, Daouya M. et al. Comprehensive landscape of immune-checkpoints uncovered in clear cell renal cell carcinoma reveals new and emerging therapeutic targets. Cancer Immunol Immunother. 2020;69:1237-52
228. Shao H, Ma L, Jin F, Zhou Y, Tao M, Teng Y. Immune inhibitory receptor LILRB2 is critical for the endometrial cancer progression. Biochem Biophys Res Commun. 2018;506:243-50
229. Gao A, Liu X, Lin W, Wang J, Wang S, Si F. et al. Tumor-derived ILT4 induces T cell senescence and suppresses tumor immunity. J Immunother Cancer. 2021;9:e001536
230. Li J, Gao A, Zhang F, Wang S, Wang J, Wang J. et al. ILT3 promotes tumor cell motility and angiogenesis in non-small cell lung cancer. Cancer Lett. 2021;501:263-76
231. Liu J, Zhang F, He J, Wang S, Wang L, Li J. et al. Tumor-derived immunoglobulin-like transcript 4 facilitates angiogenesis of colorectal cancer. Am J Cancer Res. 2023;13:419-35
232. Wang H, Wang L, Luan H, Xiao J, Zhao Z, Yu P. et al. LILRB4 on multiple myeloma cells promotes bone lesion by p-SHP2/NF-kB/RELT signal pathway. J Exp Clin Cancer Res. 2024;43:183
233. Wu G, Xu Y, Schultz RD, Chen H, Xie J, Deng M. et al. LILRB3 supports acute myeloid leukemia development and regulates T-cell antitumor immune responses through the TRAF2-cFLIP-NF-κB signaling axis. Nat Cancer. 2021;2:1170-84
234. Mendez-Lagares G, Chin N, Chang WLW, Lee J, Rosas-Umbert M, Kieu HT. et al. Cytomegalovirus mediates expansion of IL-15-responsive innate-memory cells with SIV killing function. J Clin Invest. 2021 131
235. Klenerman P, Oxenius A. T cell responses to cytomegalovirus. Nat Rev Immunol. 2016;16:367-77
236. Prod'homme V, Griffin C, Aicheler RJ, Wang EC, McSharry BP, Rickards CR. et al. The human cytomegalovirus MHC class I homolog UL18 inhibits LIR-1+ but activates LIR-1- NK cells. J Immunol. 2007;178:4473-81
237. Wagner CS, Walther-Jallow L, Buentke E, Ljunggren H-G, Achour A, Chambers BJ. Human cytomegalovirus-derived protein UL18 alters the phenotype and function of monocyte-derived dendritic cells. J Leukoc Biol. 2008;83:56-63
238. Saverino D, Ghiotto F, Merlo A, Bruno S, Battini L, Occhino M. et al. Specific recognition of the viral protein UL18 by CD85j/LIR-1/ILT2 on CD8+ T cells mediates the non-MHC-restricted lysis of human cytomegalovirus-infected cells. J Immunol. 2004;172:5629-37
239. Berg L, Riise GC, Cosman D, Bergström T, Olofsson S, Kärre K. et al. LIR-1 expression on lymphocytes, and cytomegalovirus disease in lung-transplant recipients. Lancet. 2003;361:1099-101
240. Altfeld M, Fadda L, Frleta D, Bhardwaj N. DCs and NK cells: Critical effectors in the immune response to HIV-1. Nat Rev Immunol. 2011;11:176-86
241. Lichterfeld M, Kavanagh DG, Williams KL, Moza B, Mui SK, Miura T. et al. A viral CTL escape mutation leading to immunoglobulin-like transcript 4-mediated functional inhibition of myelomonocytic cells. J Exp Med. 2007;204:2813-24
242. O'Connor GM, Holmes A, Mulcahy F, Gardiner CM. Natural killer cells from long-term non-progressor HIV patients are characterized by altered phenotype and function. Clin Immunol. 2007;124:277-83
243. Alaoui L, Palomino G, Zurawski S, Zurawski G, Coindre S, Dereuddre-Bosquet N. et al. Early SIV and HIV infection promotes the LILRB2/MHC-I inhibitory axis in cDCs. Cell Mol Life Sci. 2017;75:1871-87
244. Huang J, Burke P, Yang Y, Seiss K, Beamon J, Cung T. et al. Soluble HLA-G inhibits myeloid dendritic cell function in HIV-1 infection by interacting with leukocyte immunoglobulin-like receptor B2. J Virol. 2010;84:10784-91
245. Blankson JN. Effector mechanisms in HIV-1 infected elite controllers: Highly active immune responses? Antiviral Res. 2010;85:295-302
246. Migueles SA, Osborne CM, Royce C, Compton AA, Joshi RP, Weeks KA. et al. Lytic granule loading of CD8+ T cells is required for HIV-infected cell elimination associated with immune control. Immunity. 2008;29:1009-21
247. Huang J, Burke PS, Cung TDH, Pereyra F, Toth I, Walker BD. et al. Leukocyte immunoglobulin-like receptors maintain unique antigen-presenting properties of circulating myeloid dendritic cells in HIV-1-infected elite controllers. J Virol. 2010;84:9463-71
248. Luque J, Lozano JM, García-Jurado G, Soriano-Sarabia N, González R, Vallejo A. et al. NK-associated regulatory receptors in a structured HAART interruption of HIV-1-positive individuals. AIDS Res Hum Retroviruses. 2008;24:1037-42
249. Halstead SB. Pathogenesis of dengue: Challenges to molecular biology. Science. 1988;239:476-81
250. Halstead SB, O'Rourke EJ. Antibody-enhanced dengue virus infection in primate leukocytes. Nature. 1977;265:739-41
251. Ong EZ, Zhang SL, Tan HC, Gan ES, Chan KR, Ooi EE. Dengue virus compartmentalization during antibody-enhanced infection. Sci Rep. 2017;7:40923
252. Upasani V, Vo HTM, Ung S, Heng S, Laurent D, Choeung R. et al. Impaired antibody-independent immune response of B cells in patients with acute dengue infection. Front Immunol. 2019;10:2500
253. Lu R, Zhao X, Li J, Niu P, Yang B, Wu H. et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: Implications for virus origins and receptor binding. Lancet. 2020;395:565-74
254. Patel H, Ashton NJ, Dobson RJB, Andersson L-M, Yilmaz A, Blennow K. et al. Proteomic blood profiling in mild, severe and critical covid-19 patients. Sci Rep. 2021;11:6357
255. Tomić S, Đokić J, Stevanović D, Ilić N, Gruden-Movsesijan A, Dinić M. et al. Reduced expression of autophagy markers and expansion of myeloid-derived suppressor cells correlate with poor T cell response in severe covid-19 patients. Front Immunol. 2021;12:614599
256. Manangeeswaran M, Ireland DDC, Thacker SG, Lee H-N, Kelley-Baker L, Lewkowicz AP. et al. BSL2-compliant lethal mouse model of SARS-CoV-2 and variants of concern to evaluate therapeutics targeting the spike protein. Front Immunol. 2022;13:919815
257. Anfossi N, Doisne JM, Peyrat MA, Ugolini S, Bonnaud O, Bossy D. et al. Coordinated expression of Ig-like inhibitory MHC class I receptors and acquisition of cytotoxic function in human CD8+ T cells. J Immunol. 2004;173:7223-9
258. Zhang Y, Tong S, Li S, Wang X, Ren H, Yin W. Increased ILT2 expression contributes to dysfunction of CD56dimCD16+NK cells in chronic hepatitis B virus infection. Antiviral Res. 2022;205:105385
259. Shang P, Simpson JD, Taylor GM, Sutherland DM, Welsh OL, Aravamudhan P. et al. Paired immunoglobulin-like receptor B is an entry receptor for mammalian orthoreovirus. Nat Commun. 2023;14:2615
260. Wang X, Meng X, Zheng Y, Jiang J, Yang B, Liu Y. et al. Increased frequency of ILT2-expressing CD56dimCD16+ NK cells correlates with disease severity of pulmonary tuberculosis. Tuberculosis. 2014;94:469-74
261. Singh VK, Khan A, Xu Y, Mai S, Zhang L, Mishra A. et al. Antibody-mediated LILRB2-receptor antagonism induces human myeloid-derived suppressor cells to kill mycobacterium tuberculosis. Front Immunol. 2022;13:865503
262. Petrilli JD, Araújo LE, da Silva LS, Laus AC, Müller I, Reis RM. et al. Whole blood mRNA expression-based targets to discriminate active tuberculosis from latent infection and other pulmonary diseases. Sci Rep. 2020;10:22072
263. Hogan LE, Jones DC, Allen RL. Expression of the innate immune receptor LILRB5 on monocytes is associated with mycobacteria exposure. Sci Rep. 2016;6:21780
264. Nakayama M, Underhill DM, Petersen TW, Li B, Kitamura T, Takai T. et al. Paired Ig-like receptors bind to bacteria and shape TLR-mediated cytokine production. J Immunol. 2007;178:4250-9
265. Munitz A, Cole ET, Beichler A, Groschwitz K, Ahrens R, Steinbrecher K. et al. Paired immunoglobulin-like receptor B (Pir-B) negatively regulates macrophage activation in experimental colitis. Gastroenterology. 2010;139:530-41
266. Bleharski JR, Li H, Meinken C, Graeber TG, Ochoa MT, Yamamura M. et al. Use of genetic profiling in leprosy to discriminate clinical forms of the disease. Science. 2003;301:1527-30
267. Baffari E, Fiume D, Caiazzo G, Sinistro A, Natoli S, Almerighi C. et al. Upregulation of the inhibitory receptor ILT4 in monocytes from septic patients. Hum Immunol. 2013;74:1244-50
268. Venet F, Schilling J, Cazalis MA, Demaret J, Poujol F, Girardot T. et al. Modulation of LILRB2 protein and mRNA expressions in septic shock patients and after ex vivo lipopolysaccharide stimulation. Hum Immunol. 2017;78:441-50
269. Ming S, Li M, Wu M, Zhang J, Zhong H, Chen J. et al. Immunoglobulin-like transcript 5 inhibits macrophage-mediated bacterial killing and antigen presentation during sepsis. J Infect Dis. 2019;220:1688-99
270. Smith CL, Dickinson P, Forster T, Craigon M, Ross A, Khondoker MR. et al. Identification of a human neonatal immune-metabolic network associated with bacterial infection. Nat Commun. 2014;5:4649
271. Langhorne J, Ndungu FM, Sponaas AM, Marsh K. Immunity to malaria: More questions than answers. Nat Immunol. 2008;9:725-32
272. Goel S, Palmkvist M, Moll K, Joannin N, Lara P, Akhouri RR. et al. RIFINs are adhesins implicated in severe plasmodium falciparum malaria. Nat Med. 2015;21:314-7
273. Ty M, Sun S, Callaway PC, Rek J, Press KD, van der Ploeg K. et al. Malaria-driven expansion of adaptive-like functional CD56-negative NK cells correlates with clinical immunity to malaria. Sci Transl Med. 2023;15:eadd9012
274. Kalmbach Y, Boldt AB, Fendel R, Mordmuller B, Kremsner PG, Kun JF. Increase in annexin v-positive B cells expressing LILRB1/ILT2/CD85j in malaria. Eur Cytokine Netw. 2006;17:175-80
275. Dechavanne C, Nouatin O, Adamou R, Edslev S, Hansen A, Meurisse F. et al. Placental malaria is associated with higher LILRB2 expression in monocyte subsets and lower anti-malarial IgG antibodies during infancy. Front Immunol. 2022;13:909831
276. Wang T, Liu M, Gao XJ, Zhao ZJ, Chen XG, Lun ZR. Toxoplasma gondii: The effects of infection at different stages of pregnancy on the offspring of mice. Exp Parasitol. 2011;127:107-12
277. Liu Y, Zhang L, Gao M, Zhang F, Xu X, Liu X. et al. Changes of inhibitory receptors on NK-92 cells and HLA-G on BeWo cells with toxoplasma gondii infection. Inflammation. 2013;36:1440-7
278. Li Y, Guo J, Zhang H, Li Z, Ren Y, Jiang Y. et al. LILRB4 regulates the function of decidual mdscs via the SHP-2/STAT6 pathway during toxoplasma gondii infection. Parasit Vectors. 2023;16:237
279. Basso AS, Argüello RJ, Albareda MC, Alvarez MG, Bertocchi G, Armenti AH. et al. Inhibitory receptors are expressed by trypanosoma cruzi-specific effector T cells and in hearts of subjects with chronic chagas disease. PLoS One. 2012;7:e35966
280. Song Y, Li J, Wu Y. Evolving understanding of autoimmune mechanisms and new therapeutic strategies of autoimmune disorders. Signal Transduct Target Ther. 2024;9:263
281. Doníz-Padilla L, Paniagua AE, Sandoval-Correa P, Monsiváis-Urenda A, Leskela S, Marazuela M. et al. Analysis of expression and function of the inhibitory receptor ILT2 in lymphocytes from patients with autoimmune thyroid disease. Eur J Endocrinol. 2011;165:129-36
282. Sinthuwiwat T, Buranapraditkun S, Kamolvisit W, Tongkobpetch S, Chetruengchai W, Srichomthong C. et al. A LILRB1 variant with a decreased ability to phosphorylate SHP-1 leads to autoimmune diseases. Sci Rep. 2022;12:15420
283. Rothe K, Quandt D, Schubert K, Rossol M, Klingner M, Jasinski-Bergner S. et al. Latent cytomegalovirus infection in rheumatoid arthritis and increased frequencies of cytolytic LIR-1+CD8+ T cells. Arthritis Rheumatol. 2016;68:337-46
284. Veit TD, Chies JA, Switala M, Wagner B, Horn PA, Busatto M. et al. The paradox of high availability and low recognition of soluble HLA-G by LILRB1 receptor in rheumatoid arthritis patients. PLoS One. 2015;10:e0123838
285. Monsiváis-Urenda A, Niño-Moreno P, Abud-Mendoza C, Baranda L, Layseca-Espinosa E, López-Botet M. et al. Analysis of expression and function of the inhibitory receptor ILT2 (CD85j/LILRB1/LIR-1) in peripheral blood mononuclear cells from patients with systemic lupus erythematosus (SLE). J Autoimmun. 2007;29:97-105
286. Inui M, Sugahara-Tobinai A, Fujii H, Itoh-Nakadai A, Fukuyama H, Kurosaki T. et al. Tolerogenic immunoreceptor ILT3/LILRB4 paradoxically marks pathogenic auto-antibody-producing plasmablasts and plasma cells in non-treated SLE. Int Immunol. 2016;28:597-604
287. Wiendl H, Feger U, Mittelbronn M, Jack C, Schreiner B, Stadelmann C. et al. Expression of the immune-tolerogenic major histocompatibility molecule HLA-G in multiple sclerosis: Implications for CNS immunity. Brain. 2005;128:2689-704
288. van der Touw W, Kang K, Luan Y, Ma G, Mai S, Qin L. et al. Glatiramer acetate enhances myeloid-derived suppressor cell function via recognition of paired Ig-like receptor B. J Immunol. 2018;201:1727-34
289. Xu Z, Lin CC, Ho S, Vlad G, Suciu-Foca N. Suppression of experimental autoimmune encephalomyelitis by ILT3.Fc. J Immunol. 2021;206:554-65
290. Scheltens P, De Strooper B, Kivipelto M, Holstege H, Chetelat G, Teunissen CE. et al. Alzheimer's disease. Lancet. 2021;397:1577-90
291. Kim T, Vidal GS, Djurisic M, William CM, Birnbaum ME, Garcia KC. et al. Human LilrB2 is a β-amyloid receptor and its murine homolog PirB regulates synaptic plasticity in an Alzheimer's model. Science. 2013;341:1399-404
292. Zhao P, Xu Y, Jiang LL, Fan X, Ku Z, Li L. et al. LILRB2-mediated TREM2 signaling inhibition suppresses microglia functions. Mol Neurodegener. 2022;17:44
293. Xu X, Zou P, Chen L, Jin G, Zhou H. IL-10 enhances promoter activity of ILT4 gene and up-regulates its expression in THP-1 cells. J Huazhong Univ Sci Technolog Med Sci. 2010;30:594-8
294. Dobrowolska H, Gill KZ, Serban G, Ivan E, Li Q, Qiao P. et al. Expression of immune inhibitory receptor ILT3 in acute myeloid leukemia with monocytic differentiation. Cytometry B Clin Cytom. 2012;84B:21-9
295. John S, Chen H, Deng M, Gui X, Wu G, Chen W. et al. A novel anti-LILRB4 CAR-T cell for the treatment of monocytic AML. Mol Ther. 2018;26:2487-95
296. Yang L, Feng Y, Wang S, Jiang S, Tao L, Li J. et al. Siglec-7 is an indicator of natural killer cell function in acute myeloid leukemia. Int Immunopharmacol. 2021;99:107965
297. Xian M, Wang Q, Xiao L, Zhong L, Xiong W, Ye L. et al. Leukocyte immunoglobulin-like receptor B1 (LILRB1) protects human multiple myeloma cells from ferroptosis by maintaining cholesterol homeostasis. Nat Commun. 2024;15:5767
298. Chen H, Chen Y, Deng M, John S, Gui X, Kansagra A. et al. Antagonistic anti-LILRB1 monoclonal antibody regulates antitumor functions of natural killer cells. J Immunother Cancer. 2020;8:e000515
299. Di Meo F, Iyer A, Akama K, Cheng R, Yu C, Cesarano A. et al. A target discovery pipeline identified ILT3 as a target for immunotherapy of multiple myeloma. Cell Rep Med. 2023;4:101110
300. Colovai AI, Tsao L, Wang S, Lin H, Wang C, Seki T. et al. Expression of inhibitory receptor ILT3 on neoplastic B cells is associated with lymphoid tissue involvement in chronic lymphocytic leukemia. Cytometry B Clin Cytom. 2007;72B:354-62
301. Zurli V, Wimmer G, Cattaneo F, Candi V, Cencini E, Gozzetti A. et al. Ectopic ILT3 controls BCR-dependent activation of AKT in B-cell chronic lymphocytic leukemia. Blood. 2017;130:2006-17
302. Castañeda-Partida L, Ocadiz-Delgado R, Sánchez-López JM, García-Villa E, Peñaloza-González JG, Velázquez-Aviña MM. et al. Global expression profiling of CD10+/CD19+ pre-B lymphoblasts from Hispanic B-ALL patients correlates with comparative target database analysis. Discov Oncol. 2022;13:28
303. Kamarashev J, Burg G, Mingari MC, Kempf W, Hofbauer G, Dummer R. Differential expression of cytotoxic molecules and killer cell inhibitory receptors in CD8+ and CD56+ cutaneous lymphomas. Am J Pathol. 2001;158:1593-8
304. Urosevic M, Kamarashev J, Burg Gn, Dummer R. Primary cutaneous CD8+ and CD56+ T-cell lymphomas express HLA-G and killer-cell inhibitory ligand, ILT2. Blood. 2004;103:1796-8
305. Nikolova M, Musette P, Bagot M, Boumsell L, Bensussan A. Engagement of ILT2/CD85j in Sezary syndrome cells inhibits their CD3/TCR signaling. Blood. 2002;100:1019-25
306. Harly C, Peyrat MA, Netzer S, Déchanet-Merville J, Bonneville M, Scotet E. Up-regulation of cytolytic functions of human vδ2-γδ T lymphocytes through engagement of ILT2 expressed by tumor target cells. Blood. 2011;117:2864-73
307. Naji A, Menier C, Maki G, Carosella ED, Rouas-Freiss N. Neoplastic B-cell growth is impaired by HLA-G/ILT2 interaction. Leukemia. 2012;26:1889-92
308. Pfistershammer K, Lawitschka A, Klauser C, Leitner J, Weigl R, Heemskerk MHM. et al. Allogeneic disparities in immunoglobulin-like transcript 5 induce potent antibody responses in hematopoietic stem cell transplant recipients. Blood. 2009;114:2323-32
309. Kuang Z, Tu J, Li X. Combined identification of novel markers for diagnosis and prognostic of classic hodgkin lymphoma. Int J Gen Med. 2021;14:9951-63
310. Carreras J, Ikoma H, Kikuti YY, Miyaoka M, Hiraiwa S, Tomita S. et al. Mutational, immune microenvironment, and clinicopathological profiles of diffuse large B-cell lymphoma and follicular lymphoma with BCL6 rearrangement. Virchows Arch. 2024;484:657-76
311. Cho J, Kim E, Yoon SE, Kim SJ, Kim WS. TET2 and LILRB1 mutations are frequent in Epstein-Barr virus-positive diffuse large B-cell lymphoma especially in elderly patients. Cancer. 2023;129:1502-12
312. Warnecke-Eberz U, Metzger R, Hölscher AH, Drebber U, Bollschweiler E. Diagnostic marker signature for esophageal cancer from transcriptome analysis. Tumour Biol. 2016;37:6349-58
313. Fan J, Wang L, Chen M, Zhang J, Li J, Song F. et al. Analysis of the expression and prognosis for leukocyte immunoglobulin-like receptor subfamily B in human liver cancer. World J Surg Oncol. 2022;20:92
314. Xu L, Li J, Li X, Wei X, Xu H, Sha Z. et al. Expression of leukocyte immunoglobulin-like receptor B2 in hepatocellular carcinoma and its clinical significance. J Cancer Res Ther. 2018;14:1655-9
315. Wang L, Fan J, Ye W, Han J, Zhang Y, Zhao L. et al. The expression of ILT4 in myeloid dendritic cells in patients with hepatocellular carcinoma. Immunol Invest. 2019;48:704-18
316. Fan J, Han J, Li J, Gu A, Yin D, Song F. et al. The expression and function of immunoglobulin-like transcript 4 in dendritic cells from patients with hepatocellular carcinoma. Hum Immunol. 2020;81:714-25
317. Zhao J, Li X, Li L, Chen B, Xu W, He Y. et al. Identification of neutrophil extracellular trap-driven gastric cancer heterogeneity and C5AR1 as a therapeutic target. Acta Biochim Biophys Sin (Shanghai). 2024;56:538-50
318. Zhang Y, Wang H, Xu X, Liu H, Hao T, Yin S. et al. Poor prognosis and therapeutic responses in LILRB1-expressing M2 macrophages-enriched gastric cancer patients. Front Oncol. 2021;11:668707
319. Carbone C, Piro G, Fassan M, Tamburrino A, Mina MM, Zanotto M. et al. An angiopoietin-like protein 2 autocrine signaling promotes EMT during pancreatic ductal carcinogenesis. Oncotarget. 2015;6:13822-34
320. Suciu-Foca N, Feirt N, Zhang QY, Vlad G, Liu Z, Lin H. et al. Soluble Ig-like transcript 3 inhibits tumor allograft rejection in humanized scid mice and T cell responses in cancer patients. J Immunol. 2007;178:7432-41
321. Gao Q, Mo S, Han C, Liao X, Yang C, Wang X. et al. Comprehensive analysis of LILR family genes expression and tumour-infiltrating immune cells in early-stage pancreatic ductal adenocarcinoma. IET Syst Biol. 2023;17:39-57
322. Cai Z, Wang L, Han Y, Gao W, Wei X, Gong R. et al. Immunoglobulin-like transcript 4 and human leukocyte antigen-G interaction promotes the progression of human colorectal cancer. Int J Oncol. 2019;54:1943-54
323. Chen QY, Chen YX, Han QY, Zhang JG, Zhou WJ, Zhang X. et al. Prognostic significance of immune checkpoints HLA-G/ILT-2/4 and PD-L1 in colorectal cancer. Front Immunol. 2021;12:679090
324. Shi W, Zhang F, Chen X, Wang S, Zhang H, Yang Z. et al. Tumor-derived immunoglobulin like transcript 5 induces suppressive immunocyte infiltration in colorectal cancer. Cancer Sci. 2022;113:1939-54
325. Liu J, Lu CX, Zhang F, Lv W, Liu C. Expression of ILT3 predicts poor prognosis and is inversely associated with infiltration of CD45RO+ T cells in patients with colorectal cancer. Pathol Res Pract. 2018;214:1621-5
326. Orsini G, Legitimo A, Failli A, Ferrari P, Nicolini A, Spisni R. et al. Quantification of blood dendritic cells in colorectal cancer patients during the course of disease. Pathol Oncol Res. 2014;20:267-76
327. Wang L, Geng T, Guo X, Liu J, Zhang P, Yang D. et al. Co-expression of immunoglobulin-like transcript 4 and angiopoietin-like proteins in human non-small cell lung cancer. Mol Med Rep. 2015;11:2789-96
328. Sun Y, Liu J, Gao P, Wang Y, Liu C. Expression of Ig-like transcript 4 inhibitory receptor in human non-small cell lung cancer. Chest. 2008;134:783-8
329. Li Q, Li J, Wang S, Wang J, Chen X, Zhou D. et al. Overexpressed immunoglobulin-like transcript (ILT) 4 in lung adenocarcinoma is correlated with immunosuppressive T cell subset infiltration and poor patient outcomes. Biomark Res. 2020;8:11
330. Kumata S, Notsuda H, Su MT, Saito-Koyama R, Tanaka R, Suzuki Y. et al. Prognostic impact of LILRB4 expression on tumor-infiltrating cells in resected non-small cell lung cancer. Thoracic Cancer. 2023;14:2057-68
331. de Goeje PL, Bezemer K, Heuvers ME, Dingemans A-MC, Groen HJ, Smit EF. et al. Immunoglobulin-like transcript 3 is expressed by myeloid-derived suppressor cells and correlates with survival in patients with non-small cell lung cancer. Oncoimmunology. 2015;4:e1014242
332. Su MT, Kumata S, Endo S, Okada Y, Takai T. LILRB4 promotes tumor metastasis by regulating MDSCs and inhibiting miR-1 family mirnas. Oncoimmunology. 2022;11:2060907
333. Lefebvre S, Antoine M, Uzan S, McMaster M, Dausset J, Carosella ED. et al. Specific activation of the non-classical class I histocompatibility HLA-G antigen and expression of the ILT2 inhibitory receptor in human breast cancer. J Pathol. 2001;196:266-74
334. Mamessier E, Sylvain A, Thibult ML, Houvenaeghel G, Jacquemier J, Castellano R. et al. Human breast cancer cells enhance self tolerance by promoting evasion from NK cell antitumor immunity. J Clin Invest. 2011;121:3609-22
335. Roberti MP, Juliá EP, Rocca YS, Amat M, Bravo AI, Loza J. et al. Overexpression of CD85j in TNBC patients inhibits cetuximab-mediated NK-cell adcc but can be restored with CD85j functional blockade. Eur J Immunol. 2015;45:1560-9
336. Liu J, Wang L, Gao W, Li L, Cui X, Yang H. et al. Inhibitory receptor immunoglobulin-like transcript 4 was highly expressed in primary ductal and lobular breast cancer and significantly correlated with IL-10. Diagn Pathol. 2014;9:85
337. Zhang H, Gao A, Liu Q, Zhang F, Wang S, Chen X. et al. ILT4 reprograms glucose metabolism to promote tumor progression in triple-negative breast cancer. J Cell Sci. 2023;136:jcs260964
338. Sharma N, Atolagbe OT, Ge Z, Allison JP. LILRB4 suppresses immunity in solid tumors and is a potential target for immunotherapy. J Exp Med. 2021;218:e20201811
339. Jacob JB, Wei KC, Bepler G, Reyes JD, Cani A, Polin L. et al. Identification of actionable targets for breast cancer intervention using a diversity outbred mouse model. iScience. 2023;26:106320
340. Oany AR, Mia M, Pervin T, Alyami SA, Moni MA. Integrative systems biology approaches to identify potential biomarkers and pathways of cervical cancer. J Pers Med. 2021;11:363
341. Dumont C, Jacquier A, Verine J, Noel F, Goujon A, Wu CL. et al. CD8+PD-1-ILT2+ T cells are an intratumoral cytotoxic population selectively inhibited by the immune-checkpoint HLA-G. Cancer Immunol Res. 2019;7:1619-32
342. Jacquier A, Lambert T, Delattre JF, Djouadou M, Vérine J, Dumont C. et al. Tumor infiltrating and peripheral CD4+ILT2+ T cells are a cytotoxic subset selectively inhibited by HLA-G in clear cell renal cell carcinoma patients. Cancer Lett. 2021;519:105-16
343. García M, Palma MB, Verine J, Miriuka S, Inda AM, Errecalde AL. et al. The immune-checkpoint HLA-G/ILT4 is involved in the regulation of VEGF expression in clear cell renal cell carcinoma. BMC Cancer. 2020;20:624
344. Cui K, Song H, Zhang H, Sun P. Bioinformatics screening of prognostic immune-related genes in renal clear cell carcinoma. J Appl Genet. 2024;66:311-22
345. Wu CL, Svendsen SG, Riviere A, Desgrandchamps F, Carosella ED, LeMaoult J. Multiplex bead-based immunoassay for the free soluble forms of the HLA-G receptors, ILT2 and ILT4. Hum Immunol. 2016;77:720-6
346. Pasero C, Gravis G, Guerin M, Granjeaud S, Thomassin-Piana J, Rocchi P. et al. Inherent and tumor-driven immune tolerance in the prostate microenvironment impairs natural killer cell antitumor activity. Cancer Res. 2016;76:2153-65
347. Vittrant B, Bergeron A, Molina OE, Leclercq M, Légaré X-P, Hovington H. et al. Immune-focused multi-omics analysis of prostate cancer: Leukocyte Ig-like receptors are associated with disease progression. Oncoimmunology. 2020;9:1851950
348. Young RM, Jamshidi A, Davis G, Sherman JH. Current trends in the surgical management and treatment of adult glioblastoma. Ann Transl Med. 2015;3:121
349. Lorenzo-Herrero S, Sordo-Bahamonde C, Martínez-Pérez A, Corte-Torres MD, Fernández-Vega I, Solís-Hernández MP. et al. Immunoglobulin-like transcript 2 blockade restores antitumor immune responses in glioblastoma. Cancer Sci. 2022;114:48-62
350. Wu P, Guo Y, Xiao L, Yuan J, Tang C, Dong J. et al. LILRB2-containing small extracellular vesicles from glioblastoma promote tumor progression by promoting the formation and expansion of myeloid-derived suppressor cells. Cancer Immunol Immunother. 2023;72:2179-93
351. Tabachnick-Cherny S, Pulliam T, Rodriguez HJ, Fan X, Hippe DS, Jones DC. et al. Characterization of immunosuppressive myeloid cells in merkel cell carcinoma: Correlation with resistance to PD-1 pathway blockade. Clin Cancer Res. 2024;30:1189-99
352. Czesnikiewicz-Guzik M, Lorkowska B, Zapala J, Czajka M, Szuta M, Loster B. et al. Nadph oxidase and uncoupled nitric oxide synthase are major sources of reactive oxygen species in oral squamous cell carcinoma. Potential implications for immune regulation in high oxidative stress conditions. J Physiol Pharmacol. 2008;59:139-52
353. Xiao M, Zhang X, Zhang D, Deng S, Zheng A, Du F. et al. Complex interaction and heterogeneity among cancer stem cells in head and neck squamous cell carcinoma revealed by single-cell sequencing. Front Immunol. 2022;13:1050951
354. Pan Z, Bao L, Lu X, Hu X, Li L, Chen J. et al. IL2RA+VSIG4+ tumor-associated macrophage is a key subpopulation of the immunosuppressive microenvironment in anaplastic thyroid cancer. Biochim Biophys Acta Mol Basis Dis. 2023;1869:166591
355. Sastow D, Syal A, Tremblay D. Contemporary CMML Risk Stratification and Management. Curr Hematol Malig Rep. 2025;20:8
356. Mannis GN, Aribi A, Dunavin N, Carraway HE, Saultz JN, Roboz GJ. et al. IO-202, a novel anti-LILRB4 antibody, with azacitidine for hypomethylating agent-naive chronic myelomonocytic leukemia: Phase 1b expansion cohort results. Blood. 2024;144:1008
357. Brudno JN, Maus MV, Hinrichs CS. CAR T cells and T-cell therapies for cancer: A translational science review. JAMA. 2024;332:1924-35
358. Gong LX, Sun H, Liu LT, Sun XY, Fang T, Yu Z. et al. LILRB4 represents a promising target for immunotherapy by dual targeting tumor cells and myeloid-derived suppressive cells in multiple myeloma. Haematologica. 2024;109:3650-69
359. Mandel I, Haves Ziv D, Goldshtein I, Peretz T, Alishekevitz D, Fridman Dror A. et al. BND-22, a first-in-class humanized ILT2-blocking antibody, promotes antitumor immunity and tumor regression. J Immunother Cancer. 2022;10:e004859
360. Boutros C, Tarhini A, Routier E, Lambotte O, Ladurie FL, Carbonnel F. et al. Safety profiles of anti-CTLA-4 and anti-PD-1 antibodies alone and in combination. Nat Rev Clin Oncol. 2016;13:473-86
361. Papadopoulos K, Li T, Lakhani N, Powderly J, George T, Teoh DGK. et al. 172P Phase I study of JTX-8064, a LILRB2 (ILT4) inhibitor, as monotherapy and combination with pimivalimab (pimi), a PD-1 inhibitor (PD-1i), in patients (pts) with advanced solid tumors. Immunooncol Technol. 2022 16
362. Umiker B, Hashambhoy-Ramsay Y, Smith J, Rahman T, Mueller A, Davidson R. et al. Inhibition of LILRB2 by a novel blocking antibody designed to reprogram immunosuppressive macrophages to drive T-cell activation in tumors. Mol Cancer Ther. 2023;22:471-84
363. Taylor MH, Naing A, Powderly J, Woodard P, Chung L, Lin WH. et al. Phase I dose escalation study of IO-108, an anti-LILRB2 antibody, in patients with advanced solid tumors. J Immunother Cancer. 2024;12:e010006
364. Siu LL, Wang D, Hilton J, Geva R, Rasco D, Perets R. et al. First-in-class anti-immunoglobulin-like transcript 4 myeloid-specific antibody MK-4830 abrogates a PD-1 resistance mechanism in patients with advanced solid tumors. Clin Cancer Res. 2022;28:57-70
365. Niu X, Wang C, Zhao J, Hu J, Hu Y, Sun J. et al. 1062 ES009, a LILRB2-specific blocking antibody, reprograms myeloid cells into pro-inflammation phenotype and potentiates T cell activation. J Immunother Cancer. 2022;10:A1104-A
366. Cheng LY, Chen LJ, Shi Y, Gu WY, Ding WD, Zheng X. et al. Efficacy and safety of bispecific antibodies vs. immune checkpoint blockade combination therapy in cancer: a real-world comparison. Mol Cancer. 2024;23:77
367. Murphy MB, Vitale L, Xia S, Peng Z, O'Neill T, Lillquist J. et al. CDX-585, a bispecific antibody with dual targeting of ILT4 and PD-1 checkpoint pathways. Immuno. 2023;3:273-88
368. Rafiei A, Gualandi M, Yang CL, Woods R, Kumar A, Brunner K. et al. IOS-1002, a stabilized HLA-B57 open format, exerts potent anti-tumor activity. Cancers (Basel). 2024;16:2902
369. Turner L, Lavstsen T, Berger SS, Wang CW, Petersen JE, Avril M. et al. Severe malaria is associated with parasite binding to endothelial protein C receptor. Nature. 2013;498:502-5
370. Frei AP, Jeon OY, Kilcher S, Moest H, Henning LM, Jost C. et al. Direct identification of ligand-receptor interactions on living cells and tissues. Nat Biotechnol. 2012;30:997-1001
371. Sun Z, Yi Z, Wei C, Wang W, Ren T, Cravedi P. et al. LILRB3 genetic variation is associated with kidney transplant failure in african american recipients. Nat Med. 2025;31:1677-87
372. Deng M, Chen H, Liu X, Huang R, He Y, Yoo B. et al. Leukocyte immunoglobulin-like receptor subfamily B: Therapeutic targets in cancer. Antib Ther. 2021;4:16-33
373. Marz W, Laufs U. Leucocyte immunoglobulin-like receptor subfamily-B5 (LILRB5) genetic variation and statin-associated muscle symptoms: Another piece in a puzzling puzzle. Eur Heart J. 2017;38:3576-8
374. Tedla N, Lee CW, Borges L, Geczy CL, Arm JP. Differential expression of leukocyte immunoglobulin-like receptors on cord-blood-derived human mast cell progenitors and mature mast cells. J Leukoc Biol. 2008;83:334-43
Author contact
![]() Corresponding authors: Anqi Li, E-mail: annlaqcom; laq12306com.cn; Telephone and fax numbers: 021-64370045. Yimin Li, E-mail: yimin_li_0107com; lym12999com.cn; Telephone and fax numbers: 021-64370045. Xiaodan Fu, E-mail: fuxiaodanedu.cn; jessicafu0225com; Telephone and fax numbers: 0731-89757291.
Corresponding authors: Anqi Li, E-mail: annlaqcom; laq12306com.cn; Telephone and fax numbers: 021-64370045. Yimin Li, E-mail: yimin_li_0107com; lym12999com.cn; Telephone and fax numbers: 021-64370045. Xiaodan Fu, E-mail: fuxiaodanedu.cn; jessicafu0225com; Telephone and fax numbers: 0731-89757291.