Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Methods
Results
Discussion
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2025; 15(16):8360-8376. doi:10.7150/thno.117523 This issue Cite
Research Paper
PTEN-mediated senescence of lung epithelial cells drives ventilator-induced pulmonary fibrosis
Mengyu Li1,2,3,4,#, Yu Wang1,2,3,4,#, Zhiqiang Hu1,2,3,4, Shiqian Huang1,2,3,4, Pu Chen1,2,3,4, Lin Chen1,2,3,4, Jing Wu1,2,3,4, Zhouyang Wu1,2,3,4, Shanglong Yao1,2,3,4, ![]() , Yiyi Yang1,2,3,4,
, Yiyi Yang1,2,3,4, ![]()
1. Department of Anesthesiology, Union Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, 430022, China.
2. Institute of Anesthesia and Critical Care Medicine, Union Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, 430022, China.
3. Key Laboratory of Anesthesiology and Resuscitation (Huazhong University of Science and Technology), Ministry of Education, China.
4. Hubei Provincial Clinical Research Center for Anesthesiology, Wuhan 430022, China.
# Mengyu Li and Yu Wang contributed equally to this work as co-first authors.
Received 2025-5-14; Accepted 2025-7-14; Published 2025-7-25
Abstract

Rationale: Mechanical ventilation (MV), a life-saving intervention for acute respiratory distress syndrome (ARDS), may exacerbate pulmonary fibrosis (PF) through unclear mechanisms. Although Phosphatase and Tensin homolog (PTEN) suppresses chronic PF, its role in MV-induced PF remains unknown. This study will determine whether PTEN mediates MV-PF via lung epithelial cell senescence.
Methods: Human lung epithelial cells exposed to hydrochloric acid (HCl) and mechanical stretch (48 hours) and a murine "two-hit" (HCl+MV) model (14-day observation) were used. PTEN's role was assessed via siRNA (in vitro) and knockout (in vivo). Single-cell transcriptomics analyzed senescence-associated secretory phenotype (SASP) and pathway enrichment. RG7388 (MDM2-P53 inhibitor) was administered to PTEN knockout mice to evaluate P53-mediated senescence.
Results: HCl+MV induced epithelial-mesenchymal transition (EMT) and fibrosis in vitro and in vivo. PTEN knockout or knockdown attenuated these effects. Single-cell profiling indicated PTEN's role in EMT and fibrosis via cell senescence pathways, particularly in epithelial cells exhibiting imbalances in the SASP scores. Furthermore, our experiments confirmed that senescence activation during fibrosis was reversed by PTEN inhibition. RG7388 treatment in PTEN knockout mice implicated P53-mediated senescence in PTEN's regulatory role.
Conclusions: Our study demonstrates that PTEN plays a pivotal role in MV-PF, by mediating pulmonary epithelial cell senescence. Future studies may focus on developing strategies to modulate PTEN activity and cell senescence to prevent or treat this devastating disease.
Keywords: mechanical ventilation-pulmonary fibrosis, epithelial senescence, phosphatase and tensin homolog, P53 signaling
Introduction
Acute lung injury/acute respiratory distress syndrome (ALI/ARDS) represents a life-threatening progressive disorder characterized by a triphasic disease progression consisting of exudative, proliferative, and fibrotic phases [1,2]. The initial exudative phase (≤7 days post-injury) stems from endothelial dysfunction, while later stages feature collagen deposition and epithelial cells (ECs) damage leading to pulmonary fibrosis (PF), a form of interstitial lung disease (ILD) [3-5]. This fibrotic transformation, marked by irreversible extracellular matrix (ECM) accumulation, impairs gas exchange and drives poor outcomes [3,6], underscoring the critical need for effective interventions. Mechanical ventilation (MV) remains a critical intervention for managing ARDS, yet it paradoxically contributes to ventilator-induced lung injury and accelerates PF development [7,8]. Clinically, PF was detected in open-lung biopsies from 53% of patients ventilated ≥5 days [9]. Prolonged ventilation (>12 days) was associated with a 64% incidence of PF and 57% mortality [10]. Unlike chronic ILD (e.g., idiopathic pulmonary fibrosis [IPF]), which develops over years, MV-induced PF represents a rapidly progressive form of acute ILD driven by mechanical stress and resulting in accelerated ECM deposition.
Emerging evidence suggests that repetitive mechanical stress from cyclic alveolar inflation/deflation during MV initiates pathological cascades involving inflammatory infiltration and oxidative damage [11]. Of particular interest is epithelial-mesenchymal transition (EMT), a reparative mechanism whereby ECs acquire mesenchymal phenotypes [12]. While essential for wound healing, dysregulated EMT has been increasingly recognized as a driver of fibrotic remodeling in ventilator-associated lung injury [13,14]. The molecular pathways linking MV to EMT activation remain poorly understood. Our previous work identified Piezo1 mechanoreceptors [15] and lipid mediators like Resolvin D1 [16] as potential regulators of MV-induced EMT activation and fibrosis. Further investigation into these pathways may reveal therapeutic targets to mitigate ventilation-associated fibrotic complications and improve patient outcomes.
Phosphatase and tensin homolog deleted on chromosome 10 (PTEN), a recently characterized dual-specificity lipid/protein phosphatase, serves as a critical regulator of cellular growth, survival, and migration. Emerging evidence demonstrates its anti-fibrotic potential through negative regulation of the integrin-PI3K/Akt proliferative pathway, with documented therapeutic effects across multiple organ systems including hepatic [17] and renal fibrosis [18,19]. PTEN functions as an antifibrotic mediator in chronic IPF by stabilizing the alveolar basement membrane and suppressing fibroblast proliferation, differentiation, and collagen secretion [20]. Notably, the pathomechanisms underlying MV-associated pulmonary fibrosis (MV-PF) differ fundamentally from chronic IPF [20]. Despite PTEN's established protective roles in chronic fibrosis models, its functional dynamics in this distinct mechano-inflammatory fibrotic paradigm remain unexplored.
Our investigation into MV-PF revealed an unexpected protective effect of PTEN depletion, which markedly attenuated cellular senescence in pulmonary EC and subsequently ameliorated EMT progression and fibrotic remodeling. Intriguingly, emerging experimental evidence also demonstrated that PTEN exhibits pro-fibrotic effects in cardiac fibrosis by driving M2 macrophage polarization [21]. These paradoxical findings challenge the conventional understanding of PTEN as a known fibrosis suppressor. We therefore propose a novel regulatory paradigm: PTEN may exacerbate acute MV-PF through senescence potentiation in mechano-stressed epithelium. PTEN may serve as a potential "phase-specific regulator" of fibrogenesis, suppressing fibrosis in chronic microenvironments (e.g., IPF) while paradoxically accelerating acute fibrogenesis through senescence activation (e.g., MV-PF).
Recent preclinical studies have established a pathophysiologically-relevant "dual-hit" murine model (HCl aspiration-induced ARDS + MV) to investigate mechanotransduction-mediated EMT in MV-PF [22,23]. This experimental paradigm uniquely recapitulates the clinical progression from aspiration-induced alveolar injury to ventilation-aggravated fibrogenesis. Through integrated single-cell transcriptomics and conditional PTEN knockout models, we systematically characterized the phase-specific mechanoregulatory function of PTEN, employing in vivo and ex vivo analyses to dissect its therapeutic targetability in acute mechanical injury contexts.
Methods
Cell culture and Si-PTEN transfection
Human lung bronchial EC (BEAS-2B) were purchased from the American Type Culture Collection (ATCC) and cultured according to the manufacturer's instructions. The cells were passaged at a 1:3 ratio every other day, with the culture medium changed every two days. All cell lines used in this study were authenticated by Short Tandem Repeat (STR) analysis and tested for mycoplasma contamination.
BEAS-2B cells were seeded in a six-well plate and cultured until the cell density reached approximately 40% for transfection. The siRNA lyophilized powder was formulated into a 20 μM solution, followed by the preparation of an siRNA-Lipo2000 complex (5 μl of Lipo2000, 150 μl of Opti-Minimal Essential Medium (MEM), and 5 μl of siRNA). The control wells received 2 ml of Opti-MEM, while the transfection wells initially received 1.69 ml of Opti-MEM, followed by the addition of 310 μl of the siRNA-Lipo2000 complex. The six-well plate was gently agitated to mix the medium thoroughly. The plate was then incubated in a 37°C cell culture incubator with CO2 for 72 hours. After incubation, total RNA was extracted from the cells, and the knockdown efficiency of PTEN was verified using real-time quantitative PCR.
Mechanical stretch system and in vitro cell studies
HCl stimulation was used to mimic lung injury caused by aspiration of gastric contents in clinical patients, and the mechanical stretch system was used to simulate the cyclic stretching and contraction of cells, mimicking the use of ventilators in clinical treatment. Cells were seeded into both standard 6-well plates at densities of 4.0×10^5 cells/well. On the following day, once the cells reached a confluence of 40%, 2 ml of Dulbecco's Modified Eagle Medium (DMEM) with a pH of 4.0 was added. After a 30-minute treatment, the supernatant was immediately discarded, and the wells were gently washed twice with 2 ml of sterile PBS. Then, 2 ml of complete DMEM containing 10% serum was added, and the plates were placed in a 37°C CO2 incubator for subsequent stretching treatment.
The cell model was applied using the Flexcell FX500T cell strain loading system. The strain system, controlled by a vacuum pump, creates negative pressure on a collagen-coated elastic culture membrane, causing the attached cells to experience mechanical strain through expansion and contraction. The magnitude of the strain experienced by the cells is related to the degree of stretch applied to the culture membrane, known as the strain rate (%). In this experiment, a 20% strain rate was used, with a loading program controlled by FX-5000 computer software.
Ethics statement for animal experiments
All animal experiments were conducted with strict adherence to the revised Institute of Laboratory Animal Resources, Commission on Life Sciences, National Research Council "Guide for the Care and Use of Laboratory Animals" National Academy Press, Washington, D.C. 1996, and protocols endorsed by the Ethics Committee of Tongji Medical College, Huazhong University of Science and Technology (IACUC Number: 3733), ensuring the highest standards of animal welfare and research integrity.
Generation of conditional PTEN knockout mice
To generate male mice with tamoxifen-inducible PTEN deletion specifically in the lung EC (PTENCKO), PTENF/F mice (PTENtm2.1Apat/J; Jackson Laboratory) were crossed with Sftpc-CreERT mice (Jackson Laboratory). Genotyping was undertaken by PCR using primers specific for Cre and PTEN. To knock out PTEN, 3-week-old PTENF/F; Sftpc-CreERT mice were given a tamoxifen suspension (0.1 ml of 10 mg/ml) (HY-Y1888, MCE, USA) by intraperitoneal injection for 7 d. Control groups consisted of age- and weight-matched PTENF/F mice. The efficacy of PTEN knockout in lung EC was validated through genotyping and confirmed by immunofluorescent double labeling, ensuring the precision of our conditional knockout model.
Two-hit model of MV after acid aspiration-induced lung injury in mice
Mice were anesthetized by intraperitoneal injection of 2% pentobarbital. After induction of anesthesia, a 24G arterial catheter was used for tracheal intubation, with the needle tip cut to prevent damage to the oral cavity and trachea. Following successful intubation, 2 ml/kg of HCl with a pH of 1.2 was instilled into the trachea, and the chest was quickly compressed to induce coughing, ensuring the HCl diffused throughout the lungs to avoid localized inflammation. After extubation, mice were placed on an electric heating pad to recover and returned to their cages for continued housing. Twenty-four hours later, mice were re-anesthetized, intubated, and mechanically ventilated for 2 hours using a small animal ventilator with a respiratory rate of 120 breaths per minute, an inspiratory/expiratory ratio of 1:1, and a tidal volume of 3 ml/kg. After ventilation, mice were allowed to recover on a heating pad and then returned to their cages for a further 14 days of housing. Samples were collected on day 14 for analysis.
Single cell transcriptome analysis
The 10x Genomics platform was utilized to perform single-cell transcriptome analysis. This technology encapsulates cells with beads carrying unique Cell Barcodes in droplets, allowing for the collection of cell-containing droplets and subsequent lysis within the droplets. This process links mRNA within the cells to the Cell Barcodes on the beads, forming Single Cell GEMs. Reverse transcription reactions are carried out within the droplets to construct cDNA libraries, which are then sequenced to determine the absolute quantity of each transcript molecule within individual cells, utilizing UMI and Cell Barcode information.
Quality control of the samples was performed using the 10x genomics official software Cell Ranger, which integrates the STAR software for aligning reads to the reference genome. This process provides quality control metrics such as the number of high-quality cells, genes, and genome alignment rates, allowing for the assessment of sample quality.
Histology and scoring of lung injury
Lung tissue was stained with Hematoxylin and Eosin (HE) for histological examination of lung injury. The process involved fixing lung tissue in 4% paraformaldehyde for 24 hours, paraffin embedding, and sectioning at 5μm. Sections were deparaffinized and rehydrated through a series of xylene and ethanol washes, followed by staining with hematoxylin for nuclei and eosin for cytoplasm. After staining, sections were dehydrated, cleared, and mounted for microscopic examination and photography.
For Masson's trichrome staining to assess lung fibrosis, a similar deparaffinization and rehydration process was followed, with specific staining steps including hematoxylin, Masson's trichrome, phosphomolybdic acid, and aniline blue. After staining, sections were dehydrated, cleared, and mounted. Lung fibrosis was scored using the Ashcroft scale.
Western blot (WB) assay
Use SDS-PAGE gels to separate equal amounts of protein lysate. Transfer the proteins to a nitrocellulose filter membrane (Millipore, USA). Incubate with primary antibody and secondary antibody sequentially. Then, the membranes were treated using Western Quick Block Kit (PS108, Epizyme Biotech, China) in dark. Eventually, the western blot bands were captured using BioRID ChemiDoc XRS+ (Bio-Rad, Hercules, CA, USA). Antibodies are listed in Supplementary Table S1.
Imunofluorescence assay
Freshly prepared lung OCT sections or cell climbing films were incubated with 0.2% Triton X-100 in PBS for 15 min followed by 5% bovine serum albumin for 30 min at room temperature. The slides were then incubated with primary antibody respectively at 4°C overnight. After washing, the slides were incubated with FITC-labeled secondary antibody respectively for 60 min at room temperature. Slides were counterstained with DAPI, cover-slipped, and fluorescent images captured using a Nikon A1R Confocal Microscope (Tokyo, Japan). Antibodies are listed in Supplementary Table S1.
Quantitative real-time PCR
Novozymes RNA Extraction Kit (Invitrogen, Carlsbad, CA, USA) was used to extract total RNA. Novozymes Reverse Transcription Kit with gDNA Eraser (RR047A, TAKARA, Shiga, Japan) was used to reverse transcribe total RNA to cDNA. The quantitative reverse transcription PCR analysis was performed using FastStart Universal SYBR Green Master (4913914001, Roche, Basel, Switzerland) and the StepOne Real-time PCR System (Applied Biosystems, Foster City, CA, USA). Using the 2-ΔΔCt method to calculate relative folding changes. The primer sequences are detailed in Table S2.
Enzyme-linked-immunosorbent serologic assay (ELISA)
On day 14 post-modeling, bronchoalveolar lavage fluid (BALF) was collected for ELISA detection of TGF-β1, IL-1β, and IL-6. Lung tissue was also used for ELISA detection of Collagen-I and hydroxyproline. The assay methods were performed according to the manufacturer's instructions.
Statistical analysis
The data are presented as the mean ± SED. Comparisons between two groups were performed using independent sample t tests. One-way ANOVA and the Bonferroni post hoc test were used to compare more than two groups. All statistical analyses were performed using Prism 9 (GraphPad Software, USA), and a P value<0.05 indicated statistical significance.
Results
MV exacerbated PF and EMT, downregulated PTEN expression
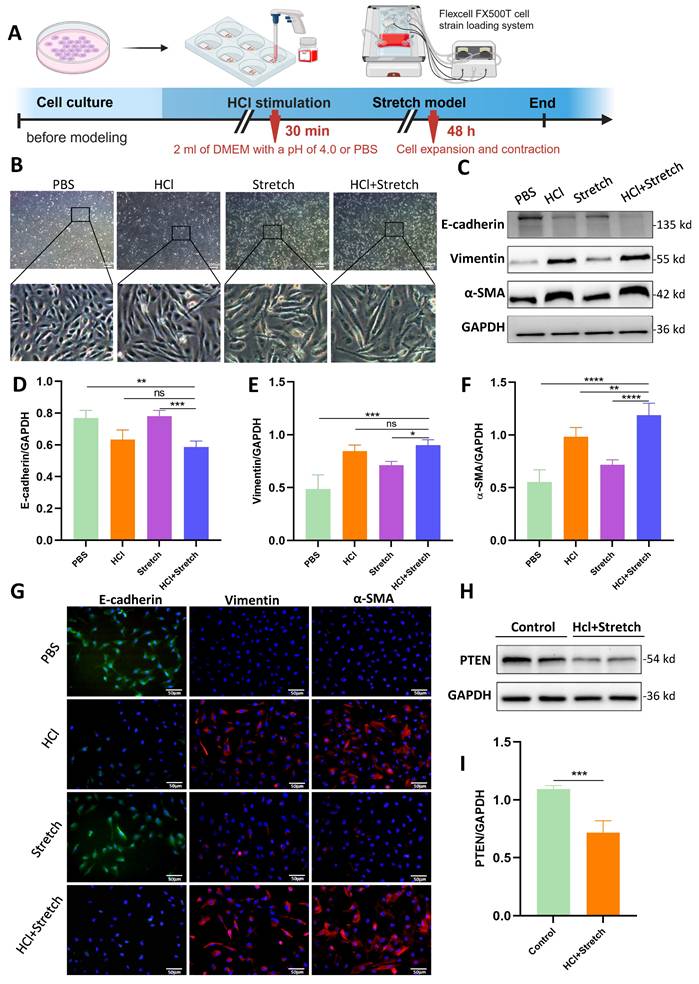
Acidic Stimulation and mechanical stretch induced spindle-like morphological changes in BEAS-2B cells, accompanied by increased intercellular spacing (Figure 1A, B). WB revealed significantly decreased E-cadherin (epithelial marker) and increased vimentin/α-SMA (mesenchymal markers) in HCl + stretch vs. control (PBS) cells (Figure 1C-F), consistent with immunofluorescence-confirmed EMT (Figure 1G). PTEN expression was notably reduced in HCl + stretch cells (Figure 1H, I).
In vitro effects of HCl and mechanical stretch on cellular morphology and epithelial mesenchymal transition (EMT). (A) Schematic of mechanical stretch and HCl injury model in BEAS-2B cells. Cells were cultured and subjected to HCl stimulation followed by mechanical stretch using the Flexcell FX500T cell strain loading system. (B) Representative phase-contrast images showing morphological changes (spindle shape, intercellular spacing) under different conditions: control (PBS), HCl stimulation, stretch, and HCl + stretch. (C) Western blot analysis of E-cadherin, Vimentin, and α-SMA protein expression in control (PBS), stretch, and HCl-treated cells with or without stretch. GAPDH served as a loading control. (D-F) Quantification of E-cadherin (epithelial marker), Vimentin, and α-SMA (mesenchymal markers) expression levels normalized to GAPDH. Data are presented as mean ± SEM. n = 4 /group. (G) Immunofluorescence staining for E-cadherin (green), Vimentin (red), and α-SMA (red) in control (PBS), stretch, and HCl-treated cells with or without stretch. Nuclei were counterstained with DAPI (blue). (H) Western blot analysis of PTEN protein expression in control (PBS) and HCl + stretched cells. GAPDH served as a loading control. (I) Quantification of PTEN expression levels normalized to GAPDH. Data are presented as mean ± SEM. n = 4 /group. Comparisons between two groups were performed using independent sample t tests. One-way ANOVA and the Bonferroni post hoc test were used to compare more than two groups. Not significant (ns): P > 0.05, *: P < 0.05, **: P < 0.01, ***: P < 0.001, ****: P < 0.0001.
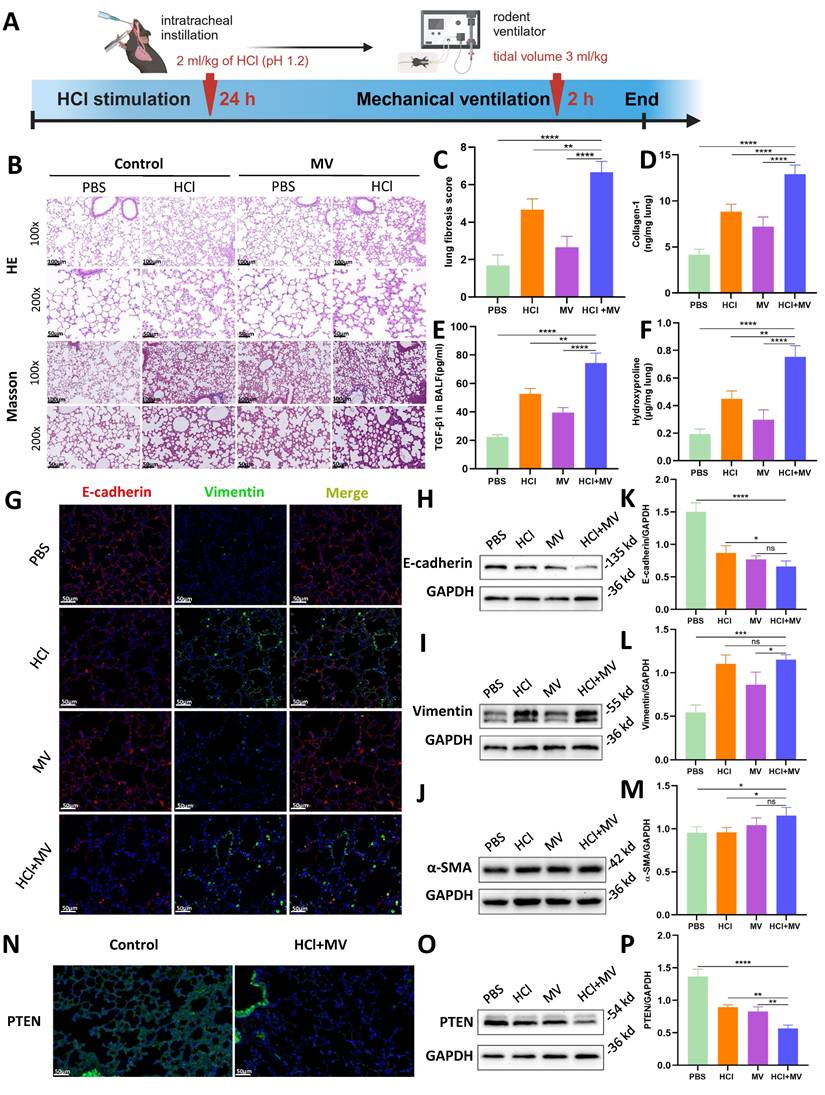
In vivo animal experiments found that HCl+MV treatment (Figure 2A) induced severe lung injury, characterized by inflammatory cell infiltration, alveolar wall thickening, and fibrotic cord formation (HE staining; Fig. 2B). Masson's trichrome staining revealed extensive collagen deposition (Fig. 2B,C), supported by elevated collagen I and hydroxyproline levels in lung tissue and increased TGF-β1 in BALF (Fig. 2D-F). EMT was confirmed by decreased E-cadherin and increased vimentin/α-SMA expression (Immunofluorescence/WB; Fig. 2G-M). Notably, PTEN expression was significantly reduced in HCl+MV-treated lungs (Fig. 2N-P).
In vivo effects of HCl and mechanical ventilation on lung fibrosis and epithelial mesenchymal transition (EMT). (A) Experimental timeline of "two-hit" (HCl + MV) murine model. Mice were subjected to HCl stimulation followed by mechanical ventilation (MV) for 24 hours and 2 hours, respectively. (B) Hematoxylin and Eosin (HE) and Masson's trichrome staining of lung tissue (14d post-injury) showing inflammatory cell infiltration, alveolar wall thickening, and fibrotic cord formation. Blue: collagen. (C) Ashcroft fibrosis score in control (PBS), MV, and HCl-treated mice with or without MV. Data are presented as mean ± SEM. n = 4 mice /group. (D-F) Quantification of Collagen-I, hydroxyproline in lung tissue, and TGF-β1 levels in bronchoalveolar lavage fluid (BALF). Data are presented as mean ± SEM. n = 4 mice /group. (G) Immunofluorescence staining for E-cadherin (red) and Vimentin (green) in lung sections under different conditions. Nuclei were counterstained with DAPI (blue). (H-J) Western blot analysis of E-cadherin, Vimentin, and α-SMA protein expression in lung tissue under different conditions. (K-M) Quantification of E-cadherin (epithelial marker), Vimentin, and α-SMA (mesenchymal markers) expression levels normalized to GAPDH. Data are presented as mean ± SEM. n = 4 mice /group. (N) Immunofluorescence staining for PTEN (green) in lung sections under different conditions. Nuclei were counterstained with DAPI (blue). (O) Western blot analysis of PTEN protein expression in lung tissue under different conditions. (P) Quantification of PTEN expression levels normalized to GAPDH. Data are presented as mean ± SEM. n = 4 mice /group. One-way ANOVA and the Bonferroni post hoc test were used to compare more than two groups. Not significant (ns): P > 0.05, *: P < 0.05, **: P < 0.01, ***: P < 0.001, ****: P < 0.0001.
Genetic deletion of PTEN protected against MV-PF
Although the role of PTEN in chronic IPF is increasingly recognized , its function in acute MV-PF remains unknown. In vitro, PTEN small interfering RNA (siRNA) was constructed and transfected into BEAS-2B cells using a transfection reagent (Figure 3A). WB analysis revealed a significant downregulation of PTEN protein expression levels (Figure 3B, C).
PTEN knockdown attenuates epithelial mesenchymal transition (EMT) and morphological changes in vitro. (A) Schematic representation of the experimental procedure. BEAS-2B cells were transfected with PTEN siRNA (Si-PTEN), followed by HCl stimulation and mechanical stretch using the Flexcell FX500T cell strain loading system. (B) Western blot analysis of PTEN protein expression in BEAS-2B cells transfected with Si-PTEN. GAPDH served as a loading control. (C) Quantification of PTEN expression levels normalized to GAPDH. Data are presented as mean ± SEM. n = 4 /group. (D) Relative mRNA expression of PTEN in BEAS-2B cells transfected with Si-PTEN. Data are presented as mean ± SEM. n = 4 /group. (E) Representative phase-contrast images showing cell morphology in wild-type (WT) and Si-PTEN transfected cells under HCl + Stretch condition. (F) Western blot analysis of E-cadherin, Vimentin, and α-SMA protein expression in WT and Si-PTEN transfected cells under HCl + Stretch condition. (G-I) Quantification of E-cadherin, Vimentin, α-SMA expression levels normalized to GAPDH. Data are presented as mean ± SEM. n = 4 /group. (J) Immunofluorescence staining for E-cadherin (green), Vimentin (red), and α-SMA (red) in WT and Si-PTEN transfected cells under HCl + Stretch condition. Nuclei were counterstained with DAPI (blue). (K) Sequence alignment of human and mouse PTEN showing 99.75% sequence homology (NCBI alignment score: 854; E-value ≈ 0). The table below the alignment score indicates the percentage identity, and the distribution graph shows the alignment scores of the top 1 Blast Hits on 1 subject sequence. Comparisons between two groups were performed using independent sample t tests. **: P < 0.01, ***: P < 0.001, ****: P < 0.0001.
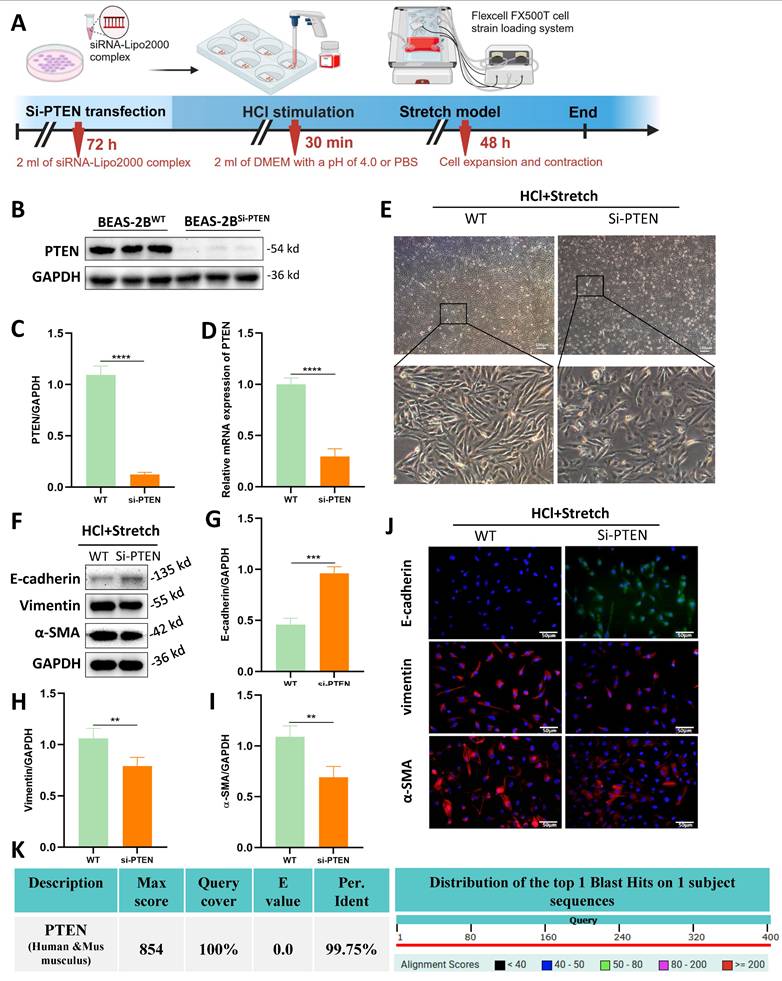
RT-qPCR was used to detect PTEN mRNA expression levels in transfected BEAS-2B cells (Figure 3D), showing a significant decrease in PTEN mRNA expression, indicating successful siRNA transfection. Transfection of PTEN siRNA reversed the morphological changes induced by mechanical stretching in BEAS-2B cells (Figure 3E). WB (Figure 3F-I) and immunofluorescence (Figure 3J) revealed that PTEN knockdown improved the EMT phenotype caused by HCl and mechanical stretching in BEAS-2B cells, characterized by increased expression of E-cadherin and decreased expression of Vimentin and α-SMA. Notably, human and mouse PTEN share 99.8% sequence homology (NCBI alignment score: 854; E-value≈0) (Fig. 3K), enabling translational studies in mouse lung tissue to investigate PTEN's role in MV-PF pathogenesis.
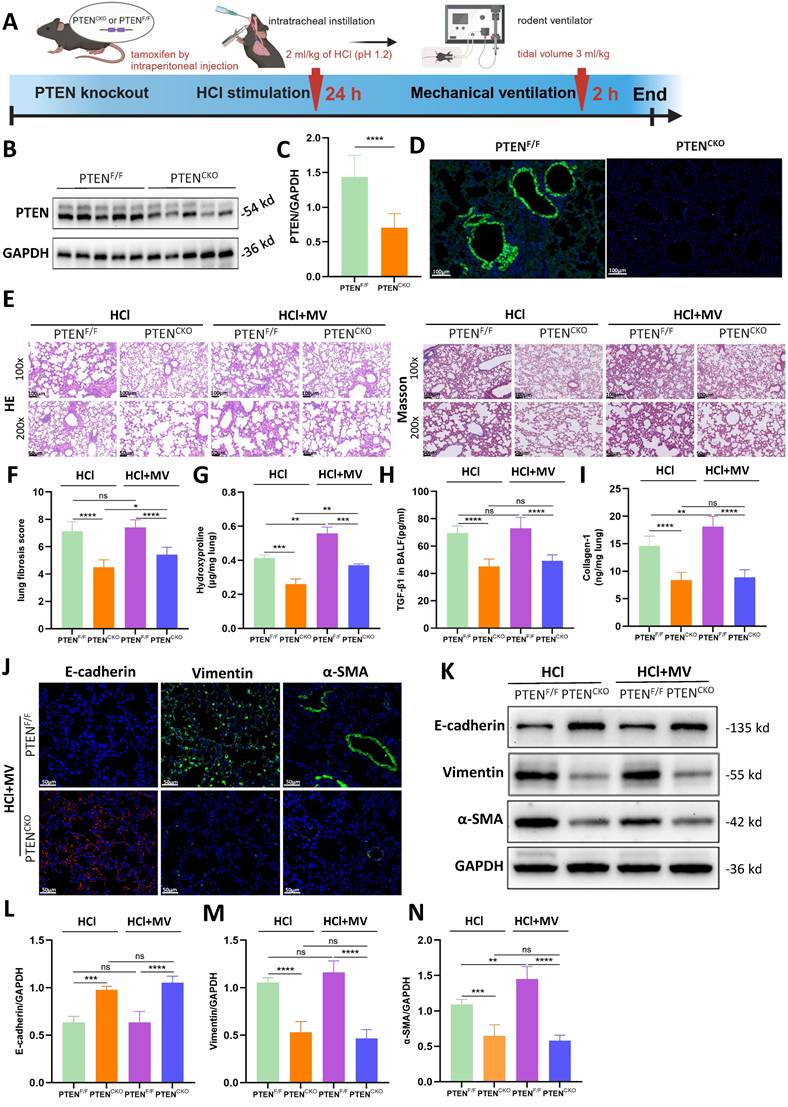
A lung EC-specific PTEN knockout mouse model using the Cre/Loxp system was established to generate PTEN-flox/Sftpc-cre mice. A comprehensive diagram outlines the experimental workflow starting from the induction of PF in PTEN knockout mice by HCl and MV (Figure 4A). Compared to PTENF/F mice, PTEN protein expression in the lung tissue of PTENCKO mice was significantly downregulated as detected by WB (Figure 4B, C). Immunofluorescence results were consistent with WB findings, showing that PTEN was extensively downregulated in the lung tissue of knockout mice, with almost no fluorescence intensity observed in the bronchial ECs (Figure 4D). As shown in Figure 4E, compared to positive control mice, the lung tissue damage in PTEN knockout mice after HCl and HCl+MV was less severe, with only a few fibrous cords and masses, and significantly reduced blue collagen deposition. Quantitative assessment showed PTEN-deficient mice had substantially lower fibrosis scores after combined HCl+MV challenge relative to controls (Figure 4F). Biochemical analyses confirmed these findings, with PTEN knockout animals exhibiting significantly reduced levels of Collagen-I, hydroxyproline, and TGF-β1 in BALF following either HCl alone or HCl+MV exposure (Figure 4G-I). Immunohistochemical and WB analyses further revealed that PTEN deletion reversed the characteristic EMT signature observed in control mice, evidenced by elevated E-cadherin expression and suppressed vimentin and α-SMA levels across HCl and HCl+MV injury conditions (Figure 4J-N).
PTEN knockout alleviates lung injury and epithelial mesenchymal transition (EMT) in vivo. (A) Schematic representation of the experimental procedure. PTENCKO mice were induced with pulmonary fibrosis using HCl and mechanical ventilation (MV). (B) Western blot analysis of PTEN protein expression in lung tissue from PTENF/F and PTENCKO mice. GAPDH served as a loading control. (C) Quantification of PTEN expression levels normalized to GAPDH. Data are presented as mean ± SEM. n = 4 mice/group. (D) Immunofluorescence staining for PTEN (green) in lung sections from PTENF/F and PTENCKO mice. Nuclei were counterstained with DAPI (blue). (E) Hematoxylin and Eosin (HE) and Masson's trichrome staining of lung sections showing reduced tissue damage and collagen deposition in PTENCKO mice compared to PTENF/F mice after HCl treatment with or without MV. (F) Quantification of Ashcroft fibrosis score in PTENCKO mice compared to PTENF/F mice after HCl treatment with or without MV. Data are presented as mean ± SEM. n = 4 mice/group. (G-I) Quantification of Collagen-I, hydroxyproline in lung tissue, and TGF-β1 levels in bronchoalveolar lavage fluid (BALF). Data are presented as mean ± SEM. n = 4 mice/group. (J) Immunofluorescence staining for E-cadherin (red), Vimentin (green), and α-SMA (green) in lung sections from PTENCKO and PTENF/F mice under HCl + MV condition. Nuclei were counterstained with DAPI (blue). (K) Western blot analysis of E-cadherin, Vimentin, α-SMA protein expression in lung tissue from PTENCKO and PTENF/F mice under different treatment conditions. (L-N) Quantification of E-cadherin, Vimentin, α-SMA expression levels normalized to GAPDH in lung tissue from PTENCKO and PTENF/F mice under different treatment conditions. Data are presented as mean ± SEM. n = 4 mice/group. Comparisons between two groups were performed using independent sample t tests. One-way ANOVA and the Bonferroni post hoc test were used to compare more than two groups. Not significant (ns): P > 0.05, *: P < 0.05, **: P < 0.01, ***: P < 0.001, ****: P < 0.0001.
scRNA-seq disclosed senescence pathway activation in lung EC following MV-PF
These findings establish PTEN knockout as pivotal for reversing MV-PF. To elucidate how PTEN promotes MV-PF, we utilized scRNA-seq to reveal changes in different lung cells in MV-PF model following PTEN knockout (Figure 5A). The measurement process includes lung tissue extraction, single-cell isolation, next-generation sequencing analysis using the 10x Genomics platform, detailed bioinformatics analysis, and subsequent validation of results. Using Uniform Manifold Approximation and Projection (UMAP), we identified cell populations based on the expression of known cell type-specific marker genes. Through these approaches, we delineated 11 distinct lung cell populations - including B cells, T cells, phagocytes, monocytes, endothelial cells, fibroblasts, neutrophils, Natural Killer (NK) cells, Dendritic cells (DCs), ECs, and mast cells (Figure 5B, C). Comparative analysis revealed PTEN knockout significantly altered cellular distribution patterns in the lung microenvironment (Figure 5D), demonstrating its broad impact on pulmonary cell ecology during fibrotic progression.
Single-cell transcriptomics implicates senescence pathways in PTEN-regulated mechanical ventilation-associated pulmonary fibrosis. (A) Schematic representation of the single-cell RNA sequencing (scRNA-seq) workflow. Following HCl and mechanical ventilation (MV), lung tissues were extracted, single cells were isolated, and next-generation sequencing was performed using the 10x Genomics platform. (B) Proportion of different cell types in lung tissue from PTENF/F and PTENCKO (knockout) mice under HCl+MV condition. (C) Uniform Manifold Approximation and Projection (UMAP) plot showing distinct cell populations in lung tissue from mice under HCl+MV condition. (D) UMAP plot comparing PTENF/F and PTENCKO mice under HCl+MV condition, highlighting changes in cellular distribution patterns. (E) Heatmap of the top 15 differentially expressed genes (DEGs) in lung epithelial cells (ECs) following PTEN knockout. (F) Pathway analysis in lung ECs showing significant enrichment in pathways related to cellular senescence. (G) Heatmap of the top 15 DEGs in immune cells (macrophages, monocytes, dendritic cells, mast cells, and neutrophils) following PTEN knockout. (H) Pathway analysis in immune cells showing significant enrichment in pathways related to interleukin-1 and interleukin-1 β signaling (cellular senescence-related pathways). (I) Senescence-associated secretory phenotype (SASP) scores in 11 major lung cell types (AddModuleScore), with the highest scores observed in mast cells and fibroblasts. Differences in SASP scores were observed only in lung ECs, indicating a protective effect of PTEN knockout on MV-PF through downregulation of cellular senescence in lung ECs. Comparisons between two groups were performed using independent sample t tests.
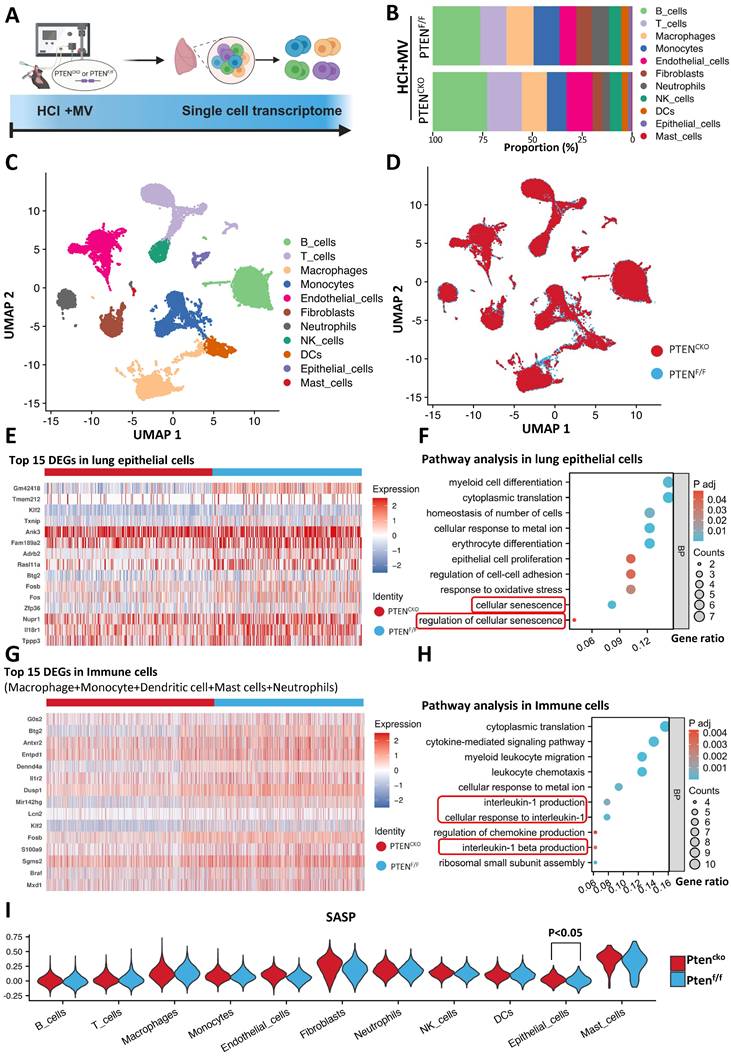
Heatmap analysis further confirmed the presence of numerous differentially expressed genes (DEGs) in lung tissues during MV-PF injury after PTEN knockout, particularly within lung ECs and the pulmonary immune cell microenvironment. Figure 5E shows the top 15 DEGs in MV-PF lung ECs following PTEN knockout. Additionally, GO pathways related to cellular oxidative damage and response to mechanical stimulation were significantly enriched. Notably, the cellular senescence signaling pathway was positively regulated in PTENF/F mice (Figure 5F). DEGs analysis and pathway enrichment analysis of pulmonary immune cell clusters revealed significant upregulation of senescence-associated secretory phenotype (SASP) pathways, such as IL-1 and IL-1β (Figures 5G, H), suggesting that PTEN expression activation can promote cellular senescence in the pulmonary microenvironment.
To further analyze the changes in SASP during MV-PF after PTEN knockout, we used the AddModuleScore function to score and visualize the senescence gene set in single cells. To systematically identify SASP-related genes, we conducted a PubMed search using the keywords 'Senescence-Associated Secretory Phenotype' and 'Review,' limiting results to studies published between 2017 and 2024 for relevance. This strategy focused on review articles that extensively covered SASP-related genes in both normal and disease states. From three selected reviews [24-26], we compiled a list of SASP genes, yielding 81 unique entries after deduplication (see Supplementary Table S3 for the complete list). Figures 5I shows the scores of the SASP gene set in the 11 major cell types of lung tissue, with the highest scores in mast cells and fibroblasts. Furthermore, differences in SASP scores were observed only in lung EC, indicating that PTEN knockout exerts a protective effect on MV-PF through the downregulating of the cellular senescence in lung EC.
PTEN knockdown ameliorated fibrosis-associated senescence
In vitro cellular experiments (Figure 6A), compared to the PBS control group, the HCl + stretch groups exhibited a marked increase in P53 and P21 fluorescence intensity (Figure 6B). WB analysis further confirmed that P53 and P21 protein expression was significantly elevated in the HCl + stretch groups compared to normal cells in the control group (Figure 6C-E). Additionally, both immunofluorescence and WB analyses revealed a significant decrease in P53 and P21 expression in BEAS-2B cells following PTEN knockdown (Figure 6F-I).
PTEN knockdown suppresses cell senescence in vitro and in vivo. (A) Schematic representation of the in vitro experimental procedure. BEAS-2B cells were transfected with or without PTEN siRNA and subjected to HCl + stretch. (B) Immunofluorescence staining for P53 (red) and P21 (red) in BEAS-2B cells under different conditions. Nuclei were counterstained with DAPI (blue). (C) Western blot analysis of P53 and P21 protein expression in BEAS-2B cells. (D-E) Quantification of P53 and P21 expression levels normalized to GAPDH. Data are presented as mean ± SEM. n = 4 /group. (F) Immunofluorescence staining for P53 (red) and P21 (red) in BEAS-2B cells following PTEN knockdown under HCl + Stretch condition. Nuclei were counterstained with DAPI (blue). (G) Western blot analysis of P53 and P21 protein expression in BEAS-2B cells following PTEN knockdown under HCl + Stretch condition. (H-I) Quantification of P53 and P21 expression levels normalized to GAPDH. Data are presented as mean ± SEM. n = 4 /group. (J) Schematic representation of the in vivo experimental procedure. Mice were subjected to HCl stimulation and mechanical ventilation (MV). (K) Immunofluorescence staining for P53 (red) and P21 (red) in lung tissue from mice under different conditions. Nuclei were counterstained with DAPI (blue). (L) Western blot analysis of P53 and P21 protein expression in lung tissue from mice under different conditions. (M-N) Quantification of P53 and P21 expression levels normalized to GAPDH. Data are presented as mean ± SEM. n = 4 mice/group. (O) Western blot analysis of P53 and P21 protein expression in lung tissue from PTENF/F and PTENCKO mice under HCl with or without MV conditions. (P-Q) Quantification of P53 and P21 expression levels normalized to GAPDH. Data are presented as mean ± SEM. n = 4 mice/group. (R) Quantification of IL-1β levels in bronchoalveolar lavage fluid (BALF) from mice under different conditions. Data are presented as mean ± SEM. n = 4 mice/group. (S) Quantification of IL-6 levels in BALF from mice under different conditions. Data are presented as mean ± SEM. n = 4 mice/group. Comparisons between two groups were performed using independent sample t tests. One-way ANOVA and the Bonferroni post hoc test were used to compare more than two groups. Not significant (ns): P > 0.05, *: P < 0.05, **: P < 0.01, ***: P < 0.001, ****: P < 0.0001.
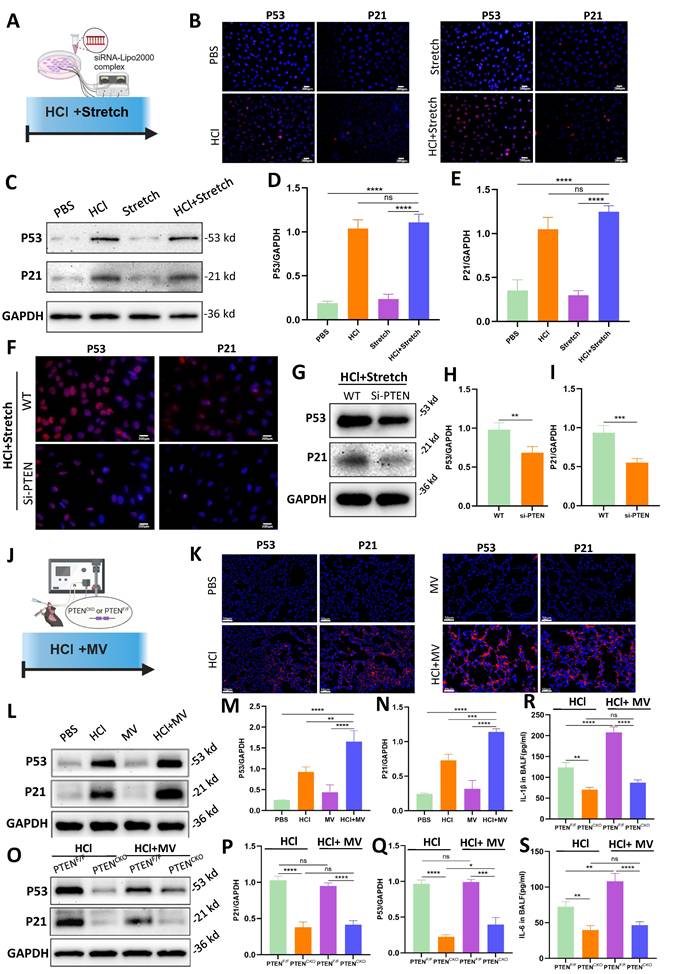
In vivo experiments (Figure 6J), compared to the PBS control group, mice treated with HCl + MV exhibited a significant increase in P53 and P21 fluorescence intensity in lung tissue (Figure 6K). WB analysis revealed that P53 and P21 expression increased in mice from the HCl + MV group compared to the control group (Figure 6L-N). In vivo PTEN knockout experiments showed that compared to the positive control group, the expression of P53 and P21 in the lung tissue of the PTEN-knockout mice group was significantly reduced (Figure 6O-Q). Furthermore, the expression of IL-1β and IL-6 in the BALF of mice was also significantly reduced in the PTEN-knockout group (Figure 6R, S).
PTEN regulated P53-mediated cellular senescence in MV-PF
RG7388 is an inhibitor of the MDM2-P53 complex, which reduces the degradation of P53 in the nucleus and increases its levels (Figure 7A). As shown in Figure 7B, histological staining with HE and Masson's trichrome revealed that in PTENCKO mice treated with HCl and MV, those administered RG7388 exhibited significantly more severe lung damage compared to the untreated PTENCKO group, with damage levels similar to those in PTENF/F mice. This included marked thickening of the alveolar walls, structural disruption, and extensive collagen deposition. Compared to PTENCKO mice, the fibrosis score in PTENCKO mice treated with RG7388 was significantly higher (Figure 7C). Compared to PTENCKO mice not treated with the inhibitor, those treated with RG7388 showed significant increases in Collagen-I, hydroxyproline, and TGF-β1 (Figure 7D-F). These results indicate that upregulation of P53 can exacerbate the fibrotic damage caused by HCl + MV in PTENCKO mice. Collectively, these findings suggest that PTEN knockout can mitigate the fibrotic damage of lung tissue in mice, but this protective effect is reversed upon P53 upregulation with RG7388, leading to increased lung damage.
PTEN regulates epithelial mesenchymal transition (EMT) via P53-senescence axis. (A) Schematic representation of the experimental design involving P53 activation using the MDM2-P53 complex inhibitor RG7388 in PTENCKO or PTENF/F mice subjected to HCl and mechanical ventilation (MV). (B) Hematoxylin and Eosin (HE) and Masson's trichrome staining of lung sections from PTENF/F, PTENCKO, and PTENCKO + RG7388 mice under HCl+MV condition. (C) Quantification of Ashcroft fibrosis score in PTENF/F, PTENCKO, and PTENCKO + RG7388 mice under HCl+MV condition. Data are presented as mean ± SEM. n = 4 mice/group. (D-F) Quantification of Collagen-I, hydroxyproline in lung tissue, and TGF-β1 levels in bronchoalveolar lavage fluid (BALF) under HCl+MV condition. Data are presented as mean ± SEM. n = 4 mice/group. (G) Western blot analysis of P53 and P21 protein expression in lung tissue from PTENF/F, PTENCKO, and PTENCKO + RG7388 mice under HCl+MV condition. (H-I) Quantification of P21 and P53 expression levels normalized to GAPDH. Data are presented as mean ± SEM. n = 4 mice/group. (J) Immunofluorescence staining for P53 (red) and P21 (red) in lung sections from PTENF/F, PTENCKO, and PTENCKO + RG7388 mice under HCl+MV condition. Nuclei were counterstained with DAPI (blue). (K) Western blot analysis of E-cadherin, Vimentin and α-SMA protein expression in lung tissue from PTENF/F, PTENCKO, and PTENCKO + RG7388 mice under HCl+MV condition. (L-N) Quantification of E-cadherin, Vimentin and α-SMA expression levels normalized to GAPDH. Data are presented as mean ± SEM. n = 4 mice/group. (O) Immunofluorescence staining for E-cadherin (red), Vimentin (green), and α-SMA (green) in lung sections from PTENF/F, PTENCKO, and PTENCKO + RG7388 mice under HCl+MV condition. Nuclei were counterstained with DAPI (blue). One-way ANOVA and the Bonferroni post hoc test were used to compare more than two groups. **: P < 0.01, ***: P < 0.001, ****: P < 0.0001.
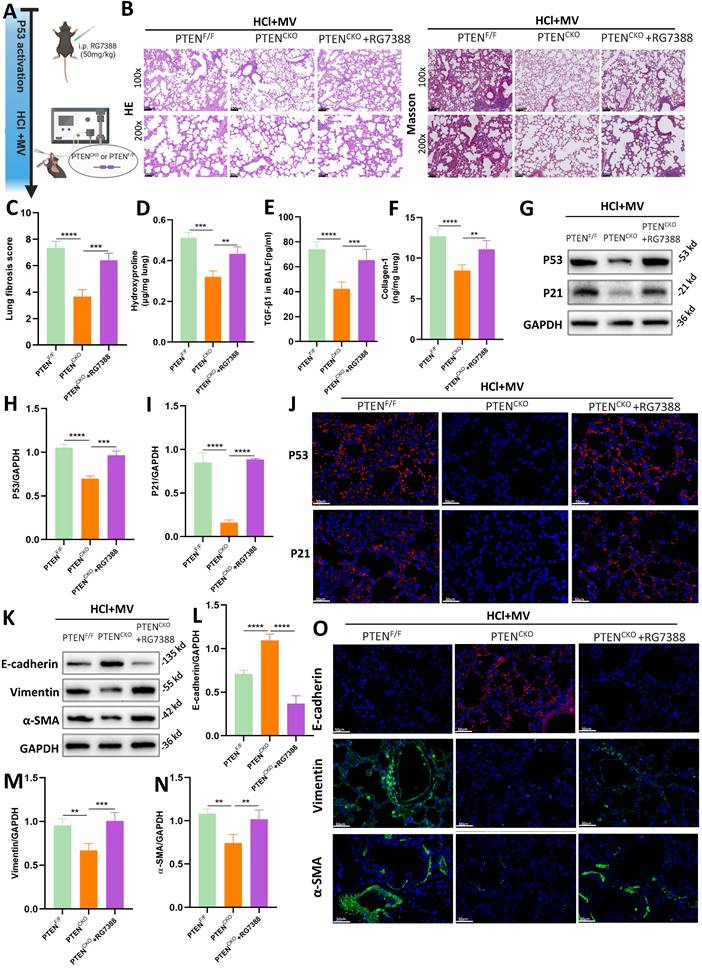
As illustrated in Figures 7G-I and J, WB and immunofluorescence analyses revealed that in PTENCKO mice treated with intraperitoneal injections of RG7388, there was a significant increase in the expression of P53 and P21 in lung tissue. This indicates that RG7388 effectively exerts its action, promoting the expression of cellular senescence markers. Furthermore, as shown in Figures 7K-O, the upregulation of P53 using the inhibitor RG7388 exacerbated EMT in PTENCKO mice.
Discussion
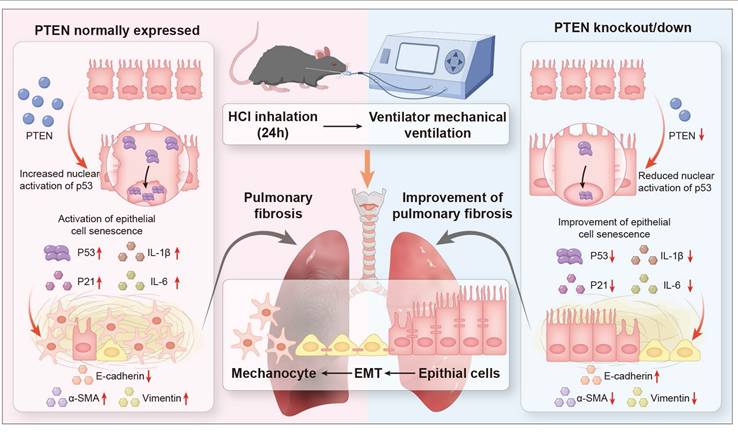
The progression of PF in the context of MV and acid aspiration, underscores the need for a deeper understanding of the underlying mechanisms. Current research predominantly focused on the roles of EMT and cellular senescence, yet the interplay between these processes and their upstream regulators remains underexplored, particularly in MV-PF. The findings reveal that PTEN, previously recognized as an anti-fibrotic molecule, may instead mediate EMT through the P53 cell senescence pathway in MV-PF. By integrating in vitro and in vivo approaches, using PTEN gene editing and RNA interference techniques, single-cell omics detection, etc., this research contributes valuable insights into the molecular dynamics of MV-PF and the pathological progression towards fibrosis, emphasizing the clinical relevance of PTEN in therapeutic strategies. In summary, our findings indicate that downregulating PTEN expression can inhibit P53-mediated senescence of lung ECs, thereby ameliorating EMT and MV-PF (Figure 8).
Schematic representation of the role of PTEN in mediating EMT and cellular senescence in MV-PF. Proposed model: PTEN upregulation in mechanical ventilation-associated pulmonary fibrosis promotes P53-mediated epithial cell senescence, driving epithelial mesenchymal transition (EMT) and fibrosis. PTEN knockout or inhibition ameliorates this cascade.
IPF carries a poor prognosis due to irreversible ECM deposition and lung tissue destruction [27]. Even for acute exacerbations of IPF, current anti-inflammatory agents and immunosuppressants fail to improve survival rates [28]. For MV-associated fibrosis, prolonged MV exposure (e.g., in COVID-19 patients) may induce accelerated EMT, contributing to treatment-resistant fibrosis [29]. However, experimental evidence suggests that acute-onset MV-PF may be partially reversible through early intervention [16]. Protective ventilation strategies reducing lung stretch have improved outcomes by minimizing ventilator-induced injury [30], highlighting the need for targeted therapies during this potentially reversible phase.
PTEN serves as an antifibrotic mediator in IPF by stabilizing the alveolar basement membrane and inhibiting fibroblast proliferation, differentiation, and collagen secretion [20]. Interestingly, our preliminary data suggest differential PTEN signaling patterns in MV-PF compared to traditional chronic PF, highlighting the necessity for mechanistic investigations into PTEN-regulated pathways. Specifically, PTEN downregulation was observed in both in vitro and in vivo MV-PF models. However, this downregulation may not exacerbate fibrotic progression; instead, it could reflect a self-protective compensatory mechanism in which reduced PTEN expression paradoxically attenuates fibrosis. Corroborating this hypothesis, prior studies report that PTEN inhibition ameliorates cardiac fibrosis through M2 macrophage polarization [21]. To clarify PTEN's role in MV-PF, we employed PTEN knockdown or knockout strategies across in vivo and vitro experimentals. Remarkably, these interventions reduced collagen deposition and significantly alleviated EMT, indicating that targeted PTEN might hold therapeutic potential for MV-driven fibrotic injury.
To explore the molecular mechanisms by which PTEN knockout alleviates EMT and MV-PF, we employed single-cell sequencing to generate a cellular atlas of MV-PF and to examine changes in the cellular microenvironment post-PTEN knockout. Our results indicated that while PTEN knockout had minimal impact on cell clustering and quantity, it led to a series of DEGs within lung ECs and immune cell clusters, which were enriched in pathways related to cellular senescence. SASP scoring of the major cell clusters showed differences primarily in lung ECs. Senescent cells, through their secretome known as the SASP, are a major driver of fibrotic diseases [31,32].
Senescence of alveolar type 2 ECs is sufficient to drive progressive fibrotic injury [33]. Early targeting senescence-related pathways in lung ECs, particularly type II ECs, represents a promising therapeutic approach for preventing IPF. For instance, researchers have found that YAP1, by targeting Prdx3, can improve mitochondrial function, inhibit the senescence of alveolar type II ECs, and thereby alleviate IPF [34]. Under physiological conditions, alveolar type II ECs in the injured lung activate self-repair programs, promoting their own proliferation and differentiation into alveolar type I ECs to maintain alveolar epithelial homeostasis [35]. Persistent lung injury can lead to the senescence of alveolar type II ECs and impaired lung repair, ultimately resulting in cronic PF [33]. However, the role and initiation mechanisms of lung ECs senescence in MV-PF have not yet been fully elucidated. Sftpc is a marker for alveolar type II ECs. In this study, PTENFL/FL mice were crossed with Sftpc-CreERT mice to establish a conditional PTEN knockout mouse model, primarily targeting the deletion of PTEN gene expression in alveolar type II ECs. The results showed an increased expression of senescence markers in response to HCl-MV, which diminishes upon PTEN knockout, suggests that PTEN may regulate EMT through the SASP that contributes to the fibrotic process.
Moreover, the interaction between the PTEN and P53 signaling pathways is particularly noteworthy. The downregulation of P53, a well-known regulator of cellular senescence [36,37], in the absence of PTEN suggests a mechanistic link between PTEN deficiency and the alleviation of senescence-related pathways. The observation that P53 activation can reverse the protective effects of PTEN knockout further supports this relationship, indicating that PTEN may influence fibrosis and EMT by modulating P53-mediated cellular responses. In cancers, concurrent loss of PTEN and P53 paradoxically induces tumor development and exacerbates poor prognosis, as seen in gliomas [38] and triple-negative breast cancer [39]. However, recent studies have unveiled a synergistic role of PTEN and P53 in DNA damage and cellular senescence. For instance, recent research reports that both P53 and PTEN are key cell fate regulators and that they have a synergistic role in the DNA damage response. Controlling the dynamics of PTEN and P53 may be a feasible strategy [40]. Studies have also revealed the significant roles of the PTEN and P53 signaling pathways in the oxidative stress and replicative senescence of mesenchymal stem cells [41].
Our findings position PTEN as a potential therapeutic target for acute MV-PF patients. The temporal specificity of PTEN's pro-fibrotic action suggests that inhibition strategies may require precise timing—early in MV-PF. In summary, the interplay between PTEN knockout and the resultant senescence phenotypic changes in lung EC provides a compelling framework for understanding the molecular underpinnings of PF. Targeting these pathways may offer novel therapeutic avenues to address the challenges posed by MV-related fibrotic injuries.
Abbreviations
ALI/ARDS: Acute lung injury/acute respiratory distress syndrome; ECs: epithelial cells; PF: pulmonary fibrosis; ILD: interstitial lung disease; ECM: extracellular matrix; MV: Mechanical ventilation; IPF: idiopathic pulmonary fibrosis; EMT: epithelial-mesenchymal transition; PTEN: Phosphatase and tensin homolog deleted on chromosome 10; MV-PF: MV-associated pulmonary fibrosis; ATCC: American Type Culture Collection; MEM: Minimal Essential Medium; DMEM: Dulbecco's Modified Eagle Medium; HE: Hematoxylin and Eosin; WB: Western blot; IF: Imunofluorescence; ELISA: Enzyme-linked-immunosorbent serologic assay; BALF: bronchoalveolar lavage fluid; STR: Short Tandem Repeat; UMAP: Using Uniform Manifold Approximation and Projection; NK: Natural Killer; DCs: Dendritic cells; DEGs: differentially expressed genes; SASP: senescence-associated secretory phenotype.
Supplementary Material
Supplementary methods and tables.
Acknowledgements
Funding
This research was supported by Hubei Technological Innovation Special Fund (2019ACA167), the National Natural Science Foundation of China (82002100, 81772123), the Bethune Medical Foundation (grant number: bnmr-2021-002) and the China International Medical Foundation (grant number: Z-2017-24-2421).
Availability of data and materials
The data used to support the results of this study are available from the corresponding author upon reasonable request.
Author contributions
Mengyu Li and Yu Wang contributed equally to this work. Conceptualization: Mengyu Li, Yu Wang, Yiyi Yang, Shanglong Yao; Methodology: Mengyu Li, Yu Wang, Zhiqiang Hu, Shiqian Huang; Investigation: Pu Chen, Lin Chen, Jing Wu, Zhouyang Wu; Formal Analysis: Pu Chen, Zhiqiang Hu, Shiqian Huang; Writing - Original Draft: Mengyu Li, Yu Wang; Writing - Review & Editing: All authors; Supervision: Yiyi Yang, Shanglong Yao; Funding Acquisition: Yu Wang, Jing Wu, Yiyi Yang, Shanglong Yao. All authors approved the final manuscript and agreed to be accountable for all aspects of the work.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Matthay MA, Arabi Y, Arroliga AC, Bernard G, Bersten AD, Brochard LJ. et al. A New Global Definition of Acute Respiratory Distress Syndrome. Am J Respir Crit Care Med. 2024;209:37-47
2. Song Y, Xu F, Seeley EJ, Ou J, Zhu X, Sun R. et al. Acute respiratory distress syndrome: emerging research in China. Am J Respir Crit Care Med. 2014;190:1090-3
3. Fan Y, Moser J, Jongman RM, Borghuis T, Vonk JM, Timens W. et al. Compositional changes of the lung extracellular matrix in acute respiratory distress syndrome. Am J Physiol Cell Physiol. 2025;328:C1279-C1292
4. Pezet MG, Torres JA, Thimraj TA, Matkovic I, Schrode N, Murray JW. et al. Human respiratory airway progenitors derived from pluripotent cells generate alveolar epithelial cells and model pulmonary fibrosis. Nat Biotechnol. 2025 doi: 10.1038/s41587-025-02569-0
5. Feng J, Huang X, Peng Y, Yang W, Yang X, Tang R. et al. Pyruvate kinase M2 modulates mitochondrial dynamics and EMT in alveolar epithelial cells during sepsis-associated pulmonary fibrosis. J Transl Med. 2025;23:205
6. Ma W, Tang S, Yao P, Zhou T, Niu Q, Liu P. et al. Advances in acute respiratory distress syndrome: focusing on heterogeneity, pathophysiology, and therapeutic strategies. Signal Transduct Target Ther. 2025;10:75
7. Mooney JJ, Raimundo K, Chang E, Broder MS. Mechanical ventilation in idiopathic pulmonary fibrosis: a nationwide analysis of ventilator use, outcomes, and resource burden. Bmc Pulm Med. 2017;17:84
8. Slutsky AS, Ranieri VM. Ventilator-induced lung injury. N Engl J Med. 2013;369:2126-36
9. Papazian L, Doddoli C, Chetaille B, Gernez Y, Thirion X, Roch A. et al. A contributive result of open-lung biopsy improves survival in acute respiratory distress syndrome patients. Crit Care Med. 2007;35:755-62
10. Martin C, Papazian L, Payan MJ, Saux P, Gouin F. Pulmonary fibrosis correlates with outcome in adult respiratory distress syndrome. A study in mechanically ventilated patients. Chest. 1995;107:196-200
11. Cabrera-Benitez NE, Laffey JG, Parotto M, Spieth PM, Villar J, Zhang H. et al. Mechanical ventilation-associated lung fibrosis in acute respiratory distress syndrome: a significant contributor to poor outcome. Anesthesiology. 2014;121:189-98
12. Heise RL, Stober V, Cheluvaraju C, Hollingsworth JW, Garantziotis S. Mechanical stretch induces epithelial-mesenchymal transition in alveolar epithelia via hyaluronan activation of innate immunity. J Biol Chem. 2011;286:17435-44
13. Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell. 2009;139:871-90
14. Cabrera-Benitez NE, Parotto M, Post M, Han B, Spieth PM, Cheng WE. et al. Mechanical stress induces lung fibrosis by epithelial-mesenchymal transition. Crit Care Med. 2012;40:510-7
15. Fang XZ, Li M, Wang YX, Zhang P, Sun MM, Xu JX. et al. Mechanosensitive ion channel Piezo1 mediates mechanical ventilation-exacerbated ARDS-associated pulmonary fibrosis. J Adv Res. 2023;53:175-86
16. Yang Y, Hu L, Xia H, Chen L, Cui S, Wang Y. et al. Resolvin D1 attenuates mechanical stretch-induced pulmonary fibrosis via epithelial-mesenchymal transition. Am J Physiol Lung Cell Mol Physiol. 2019;316:L1013-24
17. Ma L, Wei J, Zeng Y, Liu J, Xiao E, Kang Y. et al. Mesenchymal stem cell-originated exosomal circDIDO1 suppresses hepatic stellate cell activation by miR-141-3p/PTEN/AKT pathway in human liver fibrosis. Drug Deliv. 2022;29:440-53
18. An C, Jiao B, Du H, Tran M, Zhou D, Wang Y. Myeloid PTEN deficiency aggravates renal inflammation and fibrosis in angiotensin II-induced hypertension. J Cell Physiol. 2022;237:983-91
19. Zhao S, Li W, Yu W, Rao T, Li H, Ruan Y. et al. Exosomal miR-21 from tubular cells contributes to renal fibrosis by activating fibroblasts via targeting PTEN in obstructed kidneys. Theranostics. 2021;11:8660-73
20. Rackow AR, Judge JL, Woeller CF, Sime PJ, Kottmann RM. miR-338-3p blocks TGFbeta-induced myofibroblast differentiation through the induction of PTEN. Am J Physiol Lung Cell Mol Physiol. 2022;322:L385-400
21. Zhuang C, Guo Z, Zhu J, Wang W, Sun R, Qi M. et al. PTEN inhibitor attenuates cardiac fibrosis by regulating the M2 macrophage phenotype via the PI3K/AKT/TGF-beta/Smad 2/3 signaling pathway. Int J Cardiol. 2022;356:88-96
22. Zhang R, Pan Y, Fanelli V, Wu S, Luo AA, Islam D. et al. Mechanical Stress and the Induction of Lung Fibrosis via the Midkine Signaling Pathway. Am J Respir Crit Care Med. 2015;192:315-23
23. Mao P, Li J, Huang Y, Wu S, Pang X, He W. et al. MicroRNA-19b Mediates Lung Epithelial-Mesenchymal Transition via Phosphatidylinositol-3,4,5-Trisphosphate 3-Phosphatase in Response to Mechanical Stretch. Am J Respir Cell Mol Biol. 2017;56:11-9
24. Wang L, Lankhorst L, Bernards R. Exploiting senescence for the treatment of cancer. Nat Rev Cancer. 2022;22:340-55
25. He S, Sharpless NE. Senescence in Health and Disease. Cell. 2017;169:1000-11
26. Faget DV, Ren Q, Stewart SA. Unmasking senescence: context-dependent effects of SASP in cancer. Nat Rev Cancer. 2019;19:439-53
27. Moss BJ, Ryter SW, Rosas IO. Pathogenic Mechanisms Underlying Idiopathic Pulmonary Fibrosis. Annu Rev Pathol. 2022;17:515-46
28. Spagnolo P, Kropski JA, Jones MG, Lee JS, Rossi G, Karampitsakos T. et al. Idiopathic pulmonary fibrosis: Disease mechanisms and drug development. Pharmacol Ther. 2021;222:107798
29. Virseda-Berdices A, Behar-Lagares R, Martinez-Gonzalez O, Blancas R, Bueno-Bustos S, Brochado-Kith O. et al. Longer ICU stay and invasive mechanical ventilation accelerate telomere shortening in COVID-19 patients 1 year after recovery. Crit Care. 2024;28:267
30. Brower RG, Matthay MA, Morris A, Schoenfeld D, Thompson BT, Wheeler A. Ventilation with lower tidal volumes as compared with traditional tidal volumes for acute lung injury and the acute respiratory distress syndrome. N Engl J Med. 2000;342:1301-8
31. O'Reilly S, Tsou PS, Varga J. Senescence and tissue fibrosis: opportunities for therapeutic targeting. Trends Mol Med. 2024;30:1113-25
32. Maus M, Lopez-Polo V, Mateo L, Lafarga M, Aguilera M, De Lama E. et al. Iron accumulation drives fibrosis, senescence and the senescence-associated secretory phenotype. Nat Metab. 2023;5:2111-30
33. Yao C, Guan X, Carraro G, Parimon T, Liu X, Huang G. et al. Senescence of Alveolar Type 2 Cells Drives Progressive Pulmonary Fibrosis. Am J Respir Crit Care Med. 2021;203:707-17
34. Su W, Guo Y, Wang Q, Ma L, Zhang Q, Zhang Y. et al. YAP1 inhibits the senescence of alveolar epithelial cells by targeting Prdx3 to alleviate pulmonary fibrosis. Exp Mol Med. 2024;56:1643-54
35. Alysandratos KD, Herriges MJ, Kotton DN. Epithelial Stem and Progenitor Cells in Lung Repair and Regeneration. Annu Rev Physiol. 2021;83:529-50
36. Peng Y, Du J, Gunther S, Guo X, Wang S, Schneider A. et al. Mechano-signaling via Piezo1 prevents activation and P53-mediated senescence of muscle stem cells. Redox Biol. 2022;52:102309
37. Kang C, Xu Q, Martin TD, Li MZ, Demaria M, Aron L. et al. The DNA damage response induces inflammation and senescence by inhibiting autophagy of GATA4. Science. 2015;349:a5612
38. Verma R, Chen X, Xin D, Luo Z, Ogurek S, Xin M. et al. Olig1/2-Expressing Intermediate Lineage Progenitors Are Predisposed to PTEN/P53-Loss-Induced Gliomagenesis and Harbor Specific Therapeutic Vulnerabilities. Cancer Res. 2023;83:890-905
39. Gasparyan M, Lo MC, Jiang H, Lin CC, Sun D. Combined P53- and PTEN-deficiency activates expression of mesenchyme homeobox 1 (MEOX1) required for growth of triple-negative breast cancer. J Biol Chem. 2020;295:12188-202
40. Gupta S, Panda PK, Silveira DA, Ahuja R, Hashimoto RF. Quadra-Stable Dynamics of P53 and PTEN in the DNA Damage Response. Cells-Basel. 2023;12:1085
41. Matsuda S, Nakagawa Y, Kitagishi Y, Nakanishi A, Murai T. Reactive Oxygen Species, Superoxide Dimutases, and PTEN-P53-AKT-MDM2 Signaling Loop Network in Mesenchymal Stem/Stromal Cells Regulation. Cells-Basel. 2018;7:36
Author contact
![]() Corresponding authors: Dr. Yiyi Yang (Email: 2008yangyiyicom) and Shanglong Yao (yaoshanglongedu.cn); Department of Anesthesiology, Union Hospital, Tongji Medical College, Huazhong University of Science and Technology, No. 1277, Jiefang Avenue, Wuhan 430022, China.
Corresponding authors: Dr. Yiyi Yang (Email: 2008yangyiyicom) and Shanglong Yao (yaoshanglongedu.cn); Department of Anesthesiology, Union Hospital, Tongji Medical College, Huazhong University of Science and Technology, No. 1277, Jiefang Avenue, Wuhan 430022, China.