Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
1. Introduction
2. Aging, Immunosenescence, and...
3. Tumor Cell Senescence: Friend...
4. Innate Immunity Senescence:...
5. Adaptive Immunity Senescence:...
6. Stromal Senescence: The...
7. Role of SASP Within the TME:...
8. Novel Therapies Targeting...
9. Conclusion and Perspectives
Abbreviations
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2025; 15(16):8675-8703. doi:10.7150/thno.112633 This issue Cite
Review
The Role of Senescence, its Therapeutic Relevance and Clinical Implications in the Tumor Microenvironment
Hanzhe Shi1,2,3,4,5†, Mingming Xiao1,2,3,4,5†, Yangyi Li1,2,3,4,5†, Xiyu Liu6,7, Jintong Na6,7, Chen Liang1,2,3,4,5, Jie Hua1,2,3,4,5, Qingcai Meng1,2,3,4,5, Miaoyan Wei1,2,3,4,5, Wei Wang1,2,3,4,5, Jin Xu1,2,3,4,5, Xianjun Yu1,2,3,4,5 ![]() , Si Shi1,2,3,4,5
, Si Shi1,2,3,4,5 ![]()
1. Department of Pancreatic Surgery, Fudan University Shanghai Cancer Center, Shanghai, 200032, China.
2. Department of Oncology, Shanghai Medical College, Fudan University, Shanghai, 200032, China.
3. Shanghai Pancreatic Cancer Institute, Shanghai, 200032, China.
4. Shanghai Key Laboratory of Precision Medicine for Pancreatic Cancer, Shanghai, 200032, China.
5. Pancreatic Cancer Institute, Fudan University, Shanghai, 200032, China.
6. State Key Laboratory of Targeting Oncology, Guangxi Medical University, Nanning, Guangxi, 530021, China.
7. National Center for International Research of Bio-targeting Theranostics, Guangxi Medical University, Nanning, Guangxi, 530021, China.
†These authors contribute equally to this work.
Received 2025-2-21; Accepted 2025-7-15; Published 2025-7-28
Abstract

Cellular senescence is characterized by cell cycle arrest, resistance to apoptosis, the expression of senescence markers, and the acquisition of senescence-associated secretory phenotype (SASP). In this review, we discuss the role of cellular senescence within the tumor microenvironment. Some senescent innate immune cells fail to sustain their antitumor function and may even promote tumor progression. Senescent CD8+ and CD4+ T cells become dysfunctional and are implicated in immunosuppression, angiogenesis, and resistance to immunotherapy. Research on stromal senescence primarily focuses on the SASP. The SASP functions as a double-edged sword. It promotes immune surveillance in the early stages of a tumor while inhibiting tumor immunity in its advanced stages. Strategies to target senescence in cancer therapies include four main approaches: inducing senescence, inhibiting tumor-promoting SASP, clearing senescent cells, and reversing senescence. Although not yet in clinical practice, these approaches hold promise for future cancer treatments.
Keywords: cellular senescence, aging, tumor microenvironment, senescence-associated secretory phenotype, cancer treatment
1. Introduction
Aging is an inevitable biological process that all humans experience. Given the global impact of aging, advancing research into aging and age-related diseases is crucial. Numerous studies widely acknowledge that cancer is associated with age, demonstrating increased susceptibility among older individuals [1]. Indeed, the hallmarks of aging and cancer share remarkable similarities. Kroemer et al. have identified meta-hallmarks common to both aging and cancer, including genomic instability, epigenetic alterations, dysbiosis, and chronic inflammation [2]. Cellular senescence was first described in the 1960s when human fibroblasts exhibited a decline in proliferative capacity after numerous cell cycles in vitro [3]. Arne N Akbar and Sian M Henson have outlined the three phases of senescence induction: induction by stimuli, DNA damage response, and growth arrest [4]. In 2022, Douglas Hanahan introduced four new hallmarks to the previously established ten hallmarks of cancers [5], including the presence of senescent cells [6]. This underscores the critical importance of research on cellular senescence within the TME.
The tumor microenvironment (TME) is the habitat in which tumor cells live and proliferate. Beyond neoplastic cells, the TME encompasses a heterogeneous assemblage of innate and adaptive immune populations, cancer-associated fibroblasts, endothelial cells, mesenchymal stromal cells, and resident stem-like cells. Numerous reviews have established the link between the senescence of tumor cells and the onset and progression of cancers [41-45]. Therefore, our work will mainly focus on the senescence of non-cancerous components within the tumor microenvironment. In particular, accumulating evidence highlights the induction of senescence in both immune cells and stromal compartments [46-50]. Senescence exerts multifaceted effects on antitumor immunity. On the one hand, senescent cells secrete chemokines and surface ligands that recruit and activate immune surveillance [7-9]; on the other, senescent immune cells may become dysfunctional. Senescence may assist in evading immune clearance through transient cell-cycle re-entry or the release of immunosuppressive factors, thereby fostering resistance to therapy and adverse clinical outcomes. Deconvoluting this paradox is essential for a better understanding of senescence's dualistic roles within TME. Table 1 summarizes the common inductions of cellular senescence within the TME, their triggers, senescent cells involved, and their roles within TME (Table 1). Here, we review the changes in senescence within innate immunity, adaptive immunity, and stroma. We will elaborate on their contributions to tumor progression and cancer therapies, and the extent to which patients may benefit from targeting senescent cells within TME.
Types of senescence and their roles within the tumor microenvironment
| Type of senescence | Triggers | Role in TME | Mechanism | Senescent cell | Ref. |
|---|---|---|---|---|---|
| TIS | Chemotherapy Radiotherapy Targeted therapy | Anti-tumor | Immune surveillance by NK cells and macrophages | Tumor cell | [7-9] |
| Complement activation | Tumor cell | [10] | |||
| Recruitment of DCs and T cells | Tumor cell | [7, 9] | |||
| Sensitization of chemotherapy and ICB in PDAC | Tumor cell | [11] | |||
| Pro-tumor | Metastasis promotion | Tumor cell Fibroblast EC | [8, 12, 13] | ||
| Invasion promotion | EC | [12] | |||
| Stemness induction | Tumor cell | [8] | |||
| Immunosuppression | CD8+ T cell Fibroblast | [14-16] | |||
| Chemoresistance and EMT | Neutrophil Fibroblast | [8, 17] | |||
| ICB resistance | Macrophage CD8+ T cell | [18, 19] | |||
| OIS | Oncogene activation | Anti-tumor | Recruitment of CD4+ T cells | Tumor cell EC | [8, 20, 21] |
| Macrophage polarization towards M1 | Fibroblast | [22] | |||
| Pro-tumor | Tumorigenesis promotion | Macrophage Fibroblast | [8, 23] | ||
| Metastasis promotion | Tumor cell EC | [8, 13] | |||
| Invasion promotion | Tumor cell | [8] | |||
| Chemoresistance | Tumor cell | [8] | |||
| Immunosuppression | Fibroblast | [8] | |||
| SIPS | Stress signals | Pro-tumor | Tumorigenesis promotion(x2) | Fibroblast | [24, 25] |
| Immunosuppression | Tumor cell | [26] | |||
| RS | Shortened telomere length | Pro-tumor | Angiogenesis | EC | [27, 28] |
| Tumorigenesis promotion | Fibroblast | [29] | |||
| Impaired immune surveillance | CD8+ T cell | [30, 31] | |||
| Anti-tumor | Growth arrest | Tumor cell | [32] | ||
| Age-related immune dysfunction | Physiological aging | Pro-tumor | Macrophage polarization towards M2 | Macrophage | [33, 34] |
| Impaired immune surveillance | NK cell | [35] | |||
| Metastasis promotion | Neutrophil | [36] | |||
| Impaired antigen presentation | DC | [37-39] | |||
| ICB adverse events | CD4+ T cell | [40] |
TIS, therapy-induced senescence; OIS, oncogene-induced senescence; EC, endothelial cell; DC, dendritic cell; NK cell, natural killer cell; ICB, immune checkpoint blockade; PDAC, pancreatic ductal adenocarcinoma; EMT, epithelial-mesenchymal transition.
2. Aging, Immunosenescence, and Cellular Senescence
In 2013, Kroemer et al. proposed nine molecular hallmarks of aging, with cellular senescence as one of them [51]. This underscores the link between physiological aging and cellular senescence. Cellular senescence denotes a state of permanent proliferative arrest that cells enter following extended in vitro replication or upon exposure to sublethal stressors or oncogenic stimuli [52]. Senescent cells exhibit several characteristics: morphological abnormalities, irreversible cell cycle arrest, apoptosis resistance, expression of senescence markers, mitochondrial dysfunction, metabolic alterations, and the acquisition of senescence-associated secretory phenotype (SASP) [52, 53]. The onset of senescence is triggered by a variety of insults—irreparable DNA damage, telomere attrition, mitochondrial perturbations, metabolic derangements, and oncogene activation—all of which accrue with chronological aging [54]. Consequently, cells subjected either to a finite replicative lifespan or to diverse stressors during organismal aging undergo senescence, which in turn contributes to the pathogenesis of multiple age-related disorders. Specifically, metabolism, mitochondrial function, and senescence are interrelated in a bidirectional manner, each influencing and being influenced by the others. Senescent cells exhibit hallmark metabolic alterations, such as heightened aerobic glycolysis, sustained tricarboxylic acid (TCA) cycle activity, increased glutaminolysis, and lipid accumulation [55, 56]. For example, glycogen overload elevates reactive oxygen species (ROS), precipitating senescence [57], whereas methionine deprivation induces DNA damage-mediated senescence [58]. On the other hand, mitochondrial dysfunction—manifested as reduced respiratory capacity and membrane potential, aberrant organelle biogenesis, and mtDNA mutations—drives cells into senescence [59].
Senescence resulting from repeated cell divisions is termed replicative senescence (RS) [3], driven by telomere shortening. Stress-induced premature senescence (SIPS) encompasses senescence triggered by stress signals such as oncogene activation, hypoxia, and DNA damage [60-62]. Specifically, cellular senescence induced by treatments such as radiation, conventional chemotherapies, or targeted therapies is termed therapy-induced senescence (TIS) [16, 43, 63]. Senescence induced by the aberrant activation of oncogenic signaling is termed oncogene-induced senescence (OIS) [64]. TIS and OIS are both categorized into SIPS.
Differentiating cellular senescence from immunosenescence is critical, as these interrelated yet distinct phenomena both drive organismal aging and age-related pathology. Cellular senescence denotes a cell-intrinsic, irreversible proliferative arrest, whereas immunosenescence refers to the age-associated, systemic decline of immune competence across both innate and adaptive immunity. Immunosenescence can result from thymic involution, persistent antigen exposure, chronic inflammation, etc. [49, 65-68]. Importantly, cellular senescence of immune cells partly contributes to immunosenescence [48, 49]. Among these factors, thymic involution represents the most prominent and specific change associated with immunosenescence [69, 70]. As individuals age, thymic involution leads to thymic atrophy, reduction in thymocytes, and a decreased output of naïve T cells [69, 70]. Subsequently, older individuals may experience an altered phenotype of peripheral T cells, replicative senescence, and ultimately dysfunction in adaptive immunity [71], potentially leading to a higher mortality [72]. Concurrently, 'inflammaging'—a state of sterile, chronic, low-grade inflammation driven predominantly by innate immune cells—both contributes to and is exacerbated by immunosenescence [73]. Some researchers believe that inflammaging is a component of physiological aging. Once influenced by frail gene variants, it may lead to age-related diseases, termed as 'Second hit theory' [74]. Some may view inflammaging as the counterpart to immunosenescence [75], with each promoting the other. Although senescent cells potentiate inflammaging via pro-inflammatory SASP factors, they represent only one facet of this multifactorial process, which also encompasses accrual of cellular debris, accumulation of damage-associated molecular patterns (DAMPs), and a decline in proteasomal and autophagic clearance mechanisms [41, 76]. Moreover, immunosenescence will drive systemic aging [77]. Researchers have modeled physiological immunosenescence by knocking out Ercc1, a gene encoding a specific DNA repair protein, to reveal the senescence of non-lymphoid organs [77], highlighting the interaction between immunosenescence and aging.
3. Tumor Cell Senescence: Friend or Foe?
3.1. Senescence and Cancer Prior to Oncogenesis
Aging elevates oncogenic risk through chronic inflammation, genomic instability, dysbiosis, and epigenetic drift [78]. Under genotoxic stress—such as DNA damage or aberrant oncogene activation—normal cells either undergo apoptosis or enter a permanent growth arrest termed senescence, thereby acting as a potent tumor-suppressive barrier [79]. Mitochondrial pyruvate dehydrogenase (PDH) activation acts as a pivotal metabolic mechanism driving oncogene-induced senescence (OIS), linking enhanced mitochondrial respiration and redox stress to OIS-driven tumor suppression. This indicates the significant role of mitochondrial function in cellular senescence. Moreover, senescent cells propagate senescence to neighboring cells via paracrine SASP factors and juxtacrine signals [44], and are cleared by immune cells, a process called senescence surveillance [44, 45, 80]. For example, pre-malignant hepatocytes undergoing OIS are cleared by macrophages recruited through CD4⁺ T-cell-derived SASP chemokines, and both CD4⁺ and CD8⁺ T cells can mediate senescent-cell clearance [9, 20, 81]. Thus, senescence serves as a physiological barrier to oncogenesis.
With advancing age, two factors conspire to weaken this barrier. First, cells accrue senescence-inducing insults—telomere attrition, oxidative damage, and oncogenic mutations—at a higher frequency [53, 64, 82]. Second, immunosenescence compromises surveillance: macrophage phagocytic capacity wanes, antigen-presenting cell function declines, naïve T-cell output diminishes, and T-cell receptor diversity contracts [48, 75]. As a result, the presence of abundant senescent cells becomes a chronic feature of elderly people. Their chronic SASP secretion fosters a pro-tumor microenvironment by sustaining inflammation, promoting malignant transformation, suppressing immune clearance, and remodeling local stroma [44, 45].
3.2. Senescence and Cancer After Tumor Formation
Within established tumors, therapy-induced senescence (TIS) is prevalent [63]. DNA-damaging chemotherapies and radiotherapy induce senescence in malignant cells [83-85]. TIS was also observed in breast cancer, Ewing sarcoma, and neuroblastoma following treatment with CDK4/6 inhibitors [86], or in lung cancers and pancreatic cancers following treatment with MEK and CDK4/6 inhibitors [11, 87-90].
Senescent tumor cells do undergo growth arrest comparable to that of normal senescent cells. In various cancer models, senescent tumor cells uniformly exhibit cell-cycle arrest or markedly reduced proliferation [91]. Does this mean that senescence of tumor cells facilitates effective tumor suppression? Indeed, therapy-induced senescent tumor cells can attract NK cells and DCs into tumor sites via the upregulation of MHC-I and IL-15/IL-15RA complex [7, 9]. In lymphoma models with TIS, NK cells accumulate with enhanced response to tumor cells [92]. Similarly, in another metastatic melanoma model with OIS, the senescence-induced infiltration of myeloid cells inhibited tumor growth [93]. Preclinical evidence has also shown that therapy-induced senescent tumor cells induce complement activation and increase C3 expression [10]. It seems that senescence brings hope to tumor suppression.
However, senescent tumor cells may paradoxically fuel disease progression. First, the growth arrest of senescent tumor cells is not stable. The re-entry into the cell cycle of therapy-induced or oncogene-induced senescent tumor cells has been demonstrated in mice and patients with breast cancers, colorectal cancers, and acute myelogenous leukemia [63]. Mechanistically, increasing replication stress and DNA damage leads to genomic instability of oncogene-induced senescent tumor cells, enabling escape from growth arrest through various mutations [94]. Therapy-induced senescent tumor cells, on the other hand, escape from cell-cycle arrest through multiple mechanisms, including metabolic reprogramming, chromatin remodeling, and signaling pathway rewiring [94]. Second, preclinical and clinical observations have suggested that TIS may be detrimental. TIS has been associated with chemotherapy-induced cardiotoxicity, peripheral neuropathy, and ovarian damage in mice [16]. Using four immunohistochemical markers, including lipofuscin, p16INK4a, p21WAF1/Cip1, and Ki67, researchers have found that the tumoral senescence signature significantly affected overall survival (OS) in 155 NSCLC patients [95]. Single-cell analysis also revealed worse prognosis in patients with higher senescence signature [96]. Third, senescent tumor cells foster an immunosuppressive tumor microenvironment. A higher senescence signature correlates with increased crosstalk between tumor cells and immune cells [96]. This is not only attributed to SASP factors secretion, but also to the metabolic alterations of senescent tumor cells. While senescence-associated secretory phenotype (SASP) factors critically establish a protumoral TME, these will be addressed subsequently. Similar to non-malignant senescent cells, senescent tumor cells exhibit enhanced glycolysis [55]. Such a metabolic shift not only promotes tumor invasion but also exacerbates the Warburg effect, driving lactate accumulation that impairs T cell and macrophage function [55, 97]. Senescent tumor cells further display increased lipid uptake and diminished catabolism [55, 56], alterations that correlate with poorer clinical prognosis and immunotherapy resistance in cancer patients [98, 99]. Cellular senescence additionally heightens tumor cell dependence on glutamine metabolism, facilitating cell cycle re-entry [100, 101]. Notably, myeloid-derived suppressor cells (MDSCs) within the TME acquire mitochondrial DNA (mtDNA) released by senescent tumor cells, reinforcing their immunosuppressive activity [102]. Collectively, this evidence underscores the therapeutic potential of ablating senescent cells. Consequently, senescence-targeting strategies in oncology broadly fall into two categories: inducing senescence to potentiate immune-mediated clearance or eliminating senescent cells to mitigate their chronic protumoral effects on the TME, which will be discussed in the following section.
4. Innate Immunity Senescence: From Bad to Worse?
Innate immunity acts as the first line of defense against pathogens. Recently, interest has grown in the role of innate immune cells within the TME [103, 104]. Our focus will be on their induction, functional and phenotypic changes, and contributions to tumor progression.
4.1. Neutrophils
Neutrophils, pivotal components of the innate immune response, primarily originate from the bone marrow (BM) [105]. Although neutrophils are short-lived, they can undergo senescence with functional consequences. One reason for their prolonged survival is impaired GM-CSF-induced apoptosis [106]. In addition to physiological aging, it has been found that apolipoprotein E (APOE) secreted by tumor cells induces a subset of senescent neutrophils expressing the triggering receptor expressed on myeloid cells 2 (TREM2), which correlates with poor prognosis [107]. Patients with breast cancer receiving chemotherapy harbor highly senescent neutrophils [17]. This indicates that both tumors and cancer therapies can induce the senescence of neutrophils. Neutrophils become dysfunctional in killing microbes. Although neutrophil counts remain stable with age [108, 109], their defense against infection declines [110, 111]. However, this does not necessarily mean that senescent neutrophils are dysfunctional within the TME. We will separately discuss the effects of senescence on the anti-tumor and pro-tumor functions of neutrophils.
Senescent neutrophils are defined as CXCR4+CD62Llow neutrophils [36, 112, 113]. Neutrophils can exert antitumoral effects through various mechanisms [114], which can be potentially influenced by senescence. First, neutrophils can kill tumor cells opsonized with IgA or IgG via Fcγ- or Fcα-receptors [115], a process known as antibody-dependent cellular cytotoxicity (ADCC). Diminished Fcγ-mediated ADCC has been observed in senescent human neutrophils in both sexes, resulting from impaired free radical production [116]. However, FcαR1 (CD89) is the principal receptor mediating neutrophil cytotoxicity against cancer cells [117, 118]; therefore, it cannot yet be concluded that the overall ADCC capacity is reduced in senescent neutrophils. Neutrophils also exert antitumoral functions by secreting ROS and neutrophil extracellular traps (NETs) in certain scenarios. These functions will be focused on below. Moreover, neutrophils have been demonstrated to acquire antigen-presenting capabilities, bridging innate and adaptive immunity in lung cancer [119, 120]. Their potential as antigen-presenting cells is further supported by recent profiling [121]. However, further investigation is required to understand the influence of aging on this capacity. Overall, there is not yet sufficient evidence to definitively assess the impact of senescent neutrophils on tumor development. This may be due to the relatively recent association of neutrophils with tumors. Continued efforts are necessary to unravel the complexities surrounding neutrophil senescence.
Neutrophils exert a protumoral effect throughout the development of tumors (Figure 1A). ROS, though observed to kill tumor cells in some research [122], has been demonstrated to promote chronic inflammation and carcinogenesis via nitric oxide (NO) production [123] and cause severe T cell immunosuppression [124]. Given the elevated ROS levels in the elderly group [125], this increase may further promote tumorigenesis. NETs represent another age-associated mechanism that contributes to tumor promotion. NETs consist of DNA, histones, neutrophil elastase, matrix metalloproteinases, etc. [126]. The impact of NETs on the tumor is complex. Certain components of NETs, including myeloperoxidase and defensins, can directly kill tumor cells [114, 126]. The DNA structure of NETs is capable of capturing tumor cells, thereby preventing tumor metastasis [114]. However, NETs can facilitate tumor proliferation, invasion, angiogenesis, and the formation of immunosuppressive TME [126]. Therefore, it can be hypothesized that impaired NETs function in senescent neutrophils [127] may attenuate the aforementioned process, yet their overall impact on tumor development remains uncertain. Furthermore, neutrophils facilitate tumor metastasis (Table 1) [36, 113]. Adoptive transfer of a subset of CXCR4highCD62Llow senescent neutrophils promotes tumor metastasis of breast and melanoma cancer cells to the liver [113]. Accumulation of CXCR4+CD62Llow senescent neutrophils has also been found in the lung premetastatic niche at early stages of breast cancers, characterized by the expression of a specialized transcription factor SIRT1 [36, 128]. Finally, senescent neutrophils promote resistance to chemotherapy [17]. Senescent neutrophil-derived exosomal piRNA-17560 stimulates the expression of fat mass and obesity-associated protein (FTO) in breast cancer cells, leading to chemoresistance and epithelial-mesenchymal transition (EMT) [17]. Altogether, neutrophil senescence favors tumor progression, making senescent neutrophils a potential therapeutic target.
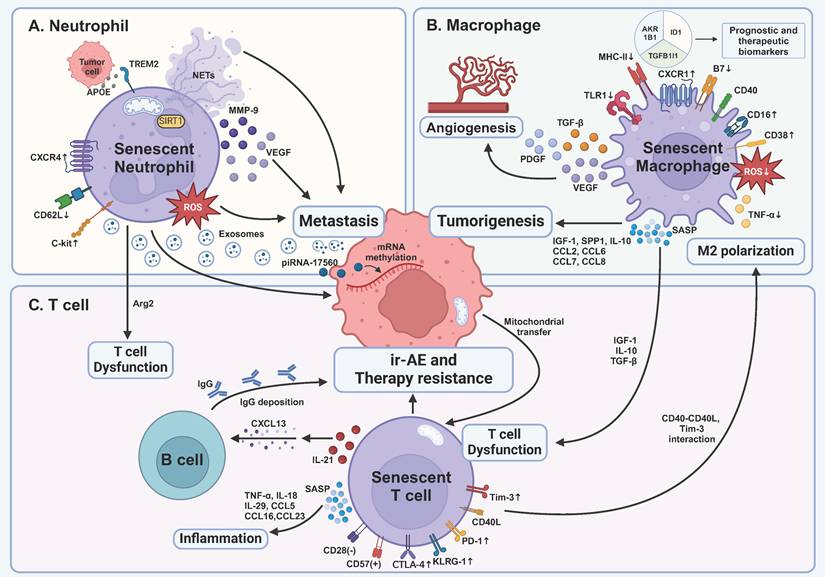
Impact of senescent immune cells on tumor development and treatment within the tumor microenvironment. A TREM2-expressing senescent neutrophils are induced by APOE secreted by prostate tumor cells, correlating with a poor prognosis. Senescent neutrophils promote cancer metastasis via distinct pathways, including ROS, mitochondria-dependent NETs, and cytokines. They also produce exosomes containing piRNA-17560, which causes chemotherapy resistance by RNA methylation of tumor cells. Senescent neutrophils lead to T cell dysfunction by Arg2 production. B Senescent macrophages' capability of killing tumor cells is inhibited, proven by decreased expression of MHC-II, B7, and impaired production of ROS and TNF-α. Senescent macrophages are another main force of SASP factors, leading to early tumorigenesis, angiogenesis, and immunosuppression. C Senescent T cells become dysfunctional as demonstrated by the expression of inhibitory receptors, including PD-1, Tim-3, and CTLA-4, and inhibitory SASP factors produced by senescent macrophages. In turn, senescent T cells enhance M2 polarization through CD40L and Tim-3 interaction. Moreover, senescent T cells secrete SASP factors to cause age-associated inflammation and deteriorate immune cell-related adverse events of ICB via IL-21-CXCL13-B cell-IgG axis. TNF-α, tumor necrosis factor-α; IL, interleukin; CTLA-4, cytotoxic T lymphocyte-associated antigen-4; Tim-3, T cell immunoglobulin and mucin domain-containing protein 3; PD-1, programmed death 1; KLRG-1, killer cell lectin-like receptor subfamily G 1; CXCL, C-X-C motif ligand; CCL, C-C chemokine motif ligand; SASP, senescence-associated secretory phenotype; ir-AE, immune cell-related adverse event; IGF-1, insulin-like growth factor 1; TGF-β, transforming growth factor-β; piRNA, PIWI-interacting RNA; NETs, neutrophil extracellular traps; TREM2, triggering receptor expressed on myeloid cells 2; APOE, apolipoprotein E; Arg2, arginase 2; SIRT1, silent mating type information regulation 2 homolog-1; MMP-9, matrix metallopeptidase 9; VEGF, vascular endothelial growth factor; ROS, reactive oxygen species; SPP1, secreted phosphoprotein 1; PDGF, platelet derived growth factor; TLR1, Toll-like receptor 1; MHC-II, major histocompatibility complex II. This figure was created with BioRender (https://biorender.com/).
Indeed, emerging efforts to target neutrophils in cancer therapy are showing promise [114], although the influence of senescent neutrophils on these therapies remains unclear. Interestingly, researchers have recently trained neutrophils to eliminate tumor cells in a ROS-dependent manner [129], highlighting the ROS's potential in defending against tumors. Though increased levels of ROS have been found in older populations [125], in patients with breast cancers, resistance to chemotherapy has been attributed to senescent neutrophils [17]. The efficacy of such approaches in elderly patients requires further exploration.
4.2. Macrophages
Macrophages are critical TME components, categorized as classically activated M1 macrophages and alternatively activated M2 macrophages based on their activation pathways [130]. M1 macrophages, predominantly antitumoral, are identified by CD14highCD16-MHC-IIhigh expression, while M2 macrophages, which are protumoral, exhibit CD14lowCD16high MHC-IIlow expression [130, 131]. Senescent macrophages comprise a heterogeneous subset characterized by elevated CD38 expression [132]. The senescence of macrophages can be induced by tumors. In a glioblastoma model, the 8B cells induced a senescence-like state of macrophages by the production of IL-6 [133], typical components of SASP [8]. This subset of macrophages, similar to M2 macrophages, produced high levels of Arginase-1 and inhibited T cell function within the TME [133, 134]. Radiotherapy has also been found to induce senescence of myeloid cells in MC38 colon cancer models [19].
M1 macrophages are activated by Th1 cells or IFN-γ and kill tumor cells with mechanisms similar to those employed during infections, including ROS, lysosomal enzymes, and NO. Recruited by CD4+ T cells, M1 macrophages acquire the ability to eliminate pre-malignant senescent hepatocytes [20], thereby preventing tumor initiation. They also serve as antigen-presenting cells (APCs) to activate adaptive immunity. However, the antitumoral capacity of senescent M1 macrophages is compromised in several ways (Figure 1B). Firstly, reductions in CD14+CD16- macrophages, representing M1 subsets, have been observed in both aged humans and mouse models in the peripheral blood [33, 34]. Using single-cell techniques, the M2 expansion in aged humans was also supported [135]. In liver models with chronic damage, however, p53-expressing senescent hepatic satellite cells have been proved to polarize M2 subsets into M1 subsets [22], indicating the number of M1 macrophages may be organ-dependent. Secondly, the production of cytokines such as TNF-α, as well as the expression of TLR1, was impaired [33]. The underlying mechanisms remain to be elucidated. Additionally, reduced ROS production in senescent macrophages was observed [8], weakening tumor immune surveillance. Furthermore, decreased expression of MHC-II and B7 costimulators in senescent macrophages indicates a diminished response to vaccination [136, 137]. These findings demonstrate significant impairments in the antitumoral functions of senescent M1 macrophages.
M2 macrophages, driven by Th2 cells or Tregs, promote tumor growth through various mechanisms (Figure 1B). As previously mentioned, increased M2 subsets are observed in elderly humans and mouse models [33, 34, 135]. The accumulation of senescent macrophages further promotes tumor progression [23, 138]. Prieto et al. have found that senescent alveolar macrophages expressing p16INK4a and Cxcr1 increased in the lungs with aging in human and Kras-driven mice models, and their removal attenuated the tumor development [138]. These studies underscore the significant role of senescent macrophages in early tumor initiation. M2 macrophages further promote tumor progression by inhibiting adaptive immunity and NK cells through the production of IL-10, TGF-β, and the expression of PD-L1 [130]. In aged mice, senescent macrophages produce elevated levels of IL-10 in the lungs, further suppressing the IL-12 axis, which is crucial for NK cell functionality [139]. In an MC38 colon cancer model, radiotherapy-induced senescent M2 macrophages were sufficient to inhibit T cell functionality. The clearance of senescent cells reversed the proliferation of T cells, suggesting that senescence may foster an immunosuppressive TME regulated by M2 subsets [19]. Finally, M2 macrophages promote angiogenesis through the production of VEGF, PDGF, and TGF-β [134], although the anti-angiogenic effect of senescent macrophages is compromised by the loss of Fas ligand (FasL) [140]. Overall, these findings suggest that senescence makes macrophages more likely to promote tumor progression.
4.3. MDSCs
Myeloid-derived suppressor cells (MDSCs) comprise a heterogeneous group of myeloid progenitor cells and immature myeloid cells (IMCs) [141]. They are categorized into monocytic (Mo-MDSCs) and polymorphonuclear MDSCs (PMN-MDSCs) [142], analogous to macrophages and neutrophils, respectively. MDSCs suppress tumor immunity through various mechanisms [141] while aging further enhances these immunosuppressive effects. First, aged mouse models exhibit expanded MDSC populations that produces IL-6 relevant to inflammaging [143-145]. In the bone marrow of aged mice, MDSCs make up the majority of the NF-κB-expressing cells, suggesting NF-κB's role in their increase [145]. Secondly, with aging, SASP factors can enhance the proliferation and functionality of MDSCs [146]. p16Ink4a and p21Cip1/Waf1 are highly expressed in Mo-MDSCs and stimulate CX3CR1 chemokine receptor expression, leading to the accumulation of Mo-MDSCs at tumor sites [147]. Third, single-cell analysis revealed that, in the high-senescence-signature group, malignant cells exhibited a greater degree of interaction with MDSCs across many human cancers, indicating an enhanced immunosuppressive capacity of MDSCs [96]. This evidence strongly suggests that aging reinforces the immunosuppressive role of MDSCs.
4.4. NK cells
NK cells are crucial for immune surveillance against cancers, consisting of immature CD3-CD56bright NK cells and mature CD3-CD56dim NK cells, with the latter accounting for the majority [148]. Mature NK cells directly eliminate tumor cells via the production of perforin and granzyme B, or expression of FasL and TNF-related apoptosis-inducing ligand (TRAIL) [148], while immature NK cells contribute to tumor cell elimination by producing cytokines such as IFN-γ, TNF-α, and GM-CSF [149]. Furthermore, NK cells play a critical role in neutralizing senescent cells, thereby preventing early tumorigenesis [8]. For example, uterine NK cells clear senescent decidual cells following the induction of IL-15 [150]. Senescence impacts both subsets of NK cells. In a study examining changes in aged NK cells, an increase in NK cell numbers with age was observed in 11 out of 13 studies [151]. The absolute number of immature CD3-CD56bright NK cells has been shown to decrease with aging [152-154]. Additionally, the response of NK cells to IL-2 was impaired [152], and their ability to produce IFN-γ and IL-8 was significantly inhibited [155, 156]. In mature CD3-CD56dim NK cells, age-related declines in perforin lead to reduced NK cell cytotoxicity (NKCC) [153]. Additionally, the expression of NK cell activating receptors like NKp30 and NKp46 was reduced in elderly groups [154]. Consistent with the reduction in NKCC, compromised tumor immunosurveillance of senescent NK cells against acute myeloid leukemia has been found [35]. Overall, this evidence suggests that the senescence of NK cells leads to a diminished antitumoral effect.
4.5. Dendritic cells
Dendritic cells (DCs), as the quintessential APCs, play a critical role in bridging innate and adaptive immunity. DCs are classified as conventional DCs (cDC1 and cDC2) or plasmacytoid DCs (pDCs). cDC1 and cDC2 are respectively tasked with antigen presentation to CD8+ T cells via MHC-I and CD4+ T cells via MHC-II, while pDCs are dedicated to antiviral and antitumor immunity through the production of type I interferons [157]. Aging impacts both cDCs and pDCs through several mechanisms. For cDCs, their absolute number remains unchanged with aging [37], and research has reported a diminished capacity for phagocytosis, migration, and T cell stimulation [37, 38]. In mouse models with B16-ovalbumin (OVA) melanomas, senescent DCs failed to effectively stimulate T cells due to defective CCR7 signaling, despite an unchanged capacity of antigen presentation [38], which led to tumor progression. In aged humans, a decreased expression of MHC peptide and CD40 in cDCs was observed, subsequently impairing CD4+ T cell expansion [39]. In pDCs, impaired production of type I and III interferon has been observed, resulting in reduced CD8+ T cell cytotoxicity [158]. Moreover, NK cells were unable to activate and eradicate lymphoma tumor cells due to a deficiency in IL-15, IL-18, and IFN-α production by pDCs [159]. Therefore, both DC subsets exhibit a functional decline in tumor immunity during aging.
5. Adaptive Immunity Senescence: The Main Force of Immunosenescence
Adaptive immunity plays a central role in tumor defense, with CD8+ cytotoxic T lymphocytes (CTLs) serving as the primary effector cells. Besides, CD4+ T cells, regulatory T cells (Tregs), and B cells all participate in the interaction of tumors and the TME. Thus, exploring the senescence of adaptive immunity is essential in discussions of immunosenescence and tumor progression. Our focus will primarily be on the role of senescent T cells within the TME, with a brief overview of senescent B cells, whose role in the TME remains less defined.
5.1. CD8+ T and CD4+ T Cells
Following cross-presentation and costimulation primarily by cDC1 in secondary lymphoid organs, naïve CD8+ T cells become activated and migrate to tumor sites, where they directly eliminate tumor cells through perforin/granzyme-mediated or FAS/FASL-mediated cytotoxicity. CD4+ T cells, particularly Th1 cells, enhance antitumor immune responses by augmenting CD8+ T cell activity and activating M1 macrophages by producing IFN-γ [160]. Like other innate immune cells, both CD8+ and CD4+ T cells can eliminate senescent cells [9, 20, 81]. Oncogene-induced pre-malignant senescent hepatocytes were cleared by macrophages, which were recruited by CD4+ T cells through the secretion of SASP [20]. Furthermore, senescent cancer cells are highly immunogenic, facilitating their recognition and elimination by DCs and CD8+ T cells [9]. T cells are crucial in the clearance of senescent cells.
T cells are the most extensively studied immune cells in the context of immunosenescence. Multiple pathways contribute to T cell senescence, with p38 and p53 being the most studied [4]. Senescent CD8+ T cells were induced in LCMV-infected mice and aged CMV-infected patients [161, 162], characterized by expression of killer cell lectin-like receptor subfamily G 1 (KLRG-1) and impaired proliferation [162]. TIS is also observed in T cells. In non-small cell lung cancer (NSCLC) patients, chemotherapy induces T cell senescence [163]. Lately, researchers have found that chemoradiotherapy induced senescence of CD8+ T cells in human cervical cancers [14]. Mechanistically, concurrent chemoradiotherapy triggers expression of atypical chemokine receptor 2 (ACKR2) on tumor cells, thus increasing the production of TGF-β and driving T cell senescence [14]. Peripheral phospholipids were also responsible for T cell senescence [164]. Furthermore, in various cancers including breast cancers, melanomas, colon cancers, prostate cancers, ovarian cancers and head and neck cancers [165-167], tumor-derived immunoglobulin-like transcript 4 (ILT4) and PD-L1 in EVs reprogrammed lipid metabolism and induced CD4+ T cell senescence via MAPK ERK1/2 signaling, leading to tumor progression and a poor prognosis [165, 168]. Tumor-T cell contact can activate cAMP pathways to trigger CD4+ T cell senescence, a process reversed by tumor cell TLR8 activation [166]. Recently, emerging evidence indicates that tumor cells further promote T cell senescence via mitochondrial transfer [169]. Mechanistically, T cells internalize tumor-derived mutated mtDNA, promoting cellular senescence and compromising effector functions and memory formation [169]. These findings underscore the previously underappreciated role of mitochondrial dysfunction in driving T cell senescence.
Senescence affects T cells in several ways (Figure 1C). First, regarding surface markers, senescent T cells are typically characterized as CD28-CD57+CD4+/CD8+ T cells [170-172], which is observed in many types of cancer, including lung cancer, ovarian cancer, head and neck cancer, and glioblastoma, as mentioned above [173-176]. Additionally, senescent T cells possess an increased expression of Tim-3, KLRG-1, and re-expression of the naïve T cell marker CD45RA [177, 178]. Expression of PD-1 and CTLA-4 was also observed in patients with acute myeloid leukemia (AML) and visceral adipose tissue of obese mice [179, 180], suggesting potential immunosuppression. Second, the cytotoxicity of senescent CD8+ T cells is reduced, as evidenced by lower levels of perforins and granzyme B [30, 31, 181], which leads to impaired antitumor immunity [181]. In contrast, senescent CD4+ T cells maintain their cytotoxic potential, with unchanged levels of perforins and granzyme B [182]. Third, senescent T cells acquire SASP, which is related to age-associated inflammation [183]. However, its role within the TME remains unclear. Fourth, senescent T cells modulate monocytes/macrophages through upregulated surface markers Tim-3 and CD40L [177]. This leads to the production of pro-inflammatory cytokines and angiogenic factors, including TNF, IL-1β, IL-6, MMP-9, VEGF-A, and IL-8 [184]. Interestingly, when co-cultured with senescent T cells, monocytes/macrophages exhibit increased CD16 expression, a characteristic of M2 macrophages [130, 131, 184]. It can be hypothesized that senescent T cells promote the polarization of macrophages from M1 subsets to M2 subsets. Fifth, senescent T cells undergo metabolic reprogramming akin to that of senescent somatic cells, characterized by enhanced glycolysis, mitochondrial biogenesis, and upregulated lipid metabolism [185, 186]. Accumulation of lipid droplets in these cells impairs effector functions and diminishes the efficacy of T-cell-based immunotherapies [187], while increased glycolytic flux further amplifies SASP secretion [185]. Overall, the evidence suggests that T cell senescence promotes a shift towards an immunosuppressive TME.
Accurate discrimination between senescent and exhausted T-cell phenotypes is essential, as both states are marked by functional impairment and co-express inhibitory receptors such as PD-1 and CTLA-4 [179, 180]. First, exhausted T cells are induced by constant stimulation of antigen, including chronic infection and cancer [188], wherein naïve T cells exhibit impaired differentiation into effector/memory subsets. Instead, they progress through precursor exhausted to terminally exhausted states [188]. Conversely, senescent T cells derive from effector or memory T cells [189]. Second, senescent T cells are typically regarded as CD28-CD57+CD4+/CD8+ T cells. Early T cell exhaustion is identified by expression of PD-1, TCF-1, and low expression of EOMES, while terminal T cell exhaustion is identified by high expression of PD-1, EOMES, and loss of TCF-1 [188]. On the contrary, senescent T cells exhibit far lower levels of PD-1 and CTLA-4 compared to exhausted T cells [161]. Third, while immune checkpoint blockade (ICB) can rejuvenate exhausted T cells, it has little effect on senescent T cells [177]. This phenomenon can be attributed to the differential expression of inhibitory receptors [161]. Currently, there are no viable approaches to reverse T cell senescence. Moreover, an optimal therapeutic effect from ICB requires the coreceptor CD28, which is absent in senescent T cells [190, 191]. Fourth, senescent T cells display a highly differentiated phenotype marked by the loss of CD27 and CD28 [4], whereas exhausted T cells can be categorized into several subsets based on their differentiation [192]. Thus, T cell senescence appears to be an irreversible endpoint, whereas T cell exhaustion may represent a reversible process.
Clinically, both senescent CD4+ T and CD8+ T cells were associated with poor survival rates and immunotherapy response in cancer patients [193-195], indicating that they may pose a barrier to effective cancer therapies. In metastatic breast cancer, patients undergoing chemotherapy exhibited a correlation between the increased number of senescent CD28-CD57+ T cells and shorter progression-free survival (PFS) [196]. This correlation may be due to the elevated levels of IL-6 and IL-10 [196], yet the mechanisms by which senescent T cells impact chemotherapy outcomes remain unclear. Regarding ICB, though it has minimal effects on senescent T cells, T cell senescence has been correlated with a lack of ICB benefit in elderly patients with distinct cancers [18, 194]. Furthermore, aged mice experienced more ICB-induced adverse events compared to young mice, mediated by the IL-21-CXCL13-auto-antibody axis in CD4+ T cells [40], highlighting senescence as a risk factor for ICB. Nonetheless, a multicenter study found that elderly patients with melanoma responded more efficiently to anti-PD-1 therapy [197]. This paradoxical finding warrants further investigation. It may be that senescent tumor cells become more susceptible to T cell immunity following PD-1-PD-L1 interaction blockade [198]. Together, senescent T cells become dysfunctional and contribute to an immunosuppressive TME, with their clinical implications necessitating further investigation.
5.2. Tregs
Regulatory T cells (Tregs), identified as CD4+FOXP3+CD25high T cells, play an important role in regulating tumor immunity. Tregs suppress tumor immunity through five primary mechanisms [199]. In addition, Tregs are capable of inducing immunosenescence [200, 201]. Firstly, Tregs induce DNA damage in T cells via glucose competition, subsequently leading to T cell senescence via p38, ERK1/2, and STAT pathways [200, 201]. Furthermore, a subset of Tregs, known as γδ regulatory T cells, can induce senescence of T cells and DCs in breast cancer models [202]. Interestingly, similar to tumor cells, activation of TLR8 with TLR8 ligands has been found to inhibit Treg-induced senescence by abrogation of Treg activity [201]. Tregs are also influenced by aging. Studies have demonstrated that aged mice exhibit increased numbers of Tregs and higher FOXP3 expression. This subset of Tregs produces elevated levels of IL-10 and suppresses T cells and DCs more effectively compared to their younger counterparts [203]. Single-cell analysis similarly revealed that, in cancers exhibiting a high senescence signature, there was increased infiltration of regulatory T cells (Tregs), which facilitated immune evasion and consequently promoted tumor progression [96]. Together, aged Tregs in the TME exhibit enhanced immunosuppressive capabilities.
5.3. B cells
Historically, B cells were considered minor contributors to tumor immunity, but recent studies have challenged this view [204]. It is now clear that B cells contribute to antitumor immunity through multiple mechanisms. First, activated B cells differentiate into plasma cells that secrete antibodies. IgGs have been found to coat tumor cells, facilitating their internalization by DCs and subsequent T cell activation [205]. Moreover, IgG-secreting B cells can inhibit cancer cell growth in early-stage NSCLC [206]. Different from IgGs, IgAs eliminate tumor cells in ovarian cancers through transcytosis [207]. Antibodies also indirectly enhance antitumor immunity through mechanisms including ADCC, antibody-dependent cellular phagocytosis (ADCP), and complement-dependent cytotoxicity (CDC) [204]. Second, B cells have been observed presenting antigens to CD4+ T cells by MHC-II or cross-presenting antigens to CD8+ T cells by MHC-I, thereby activating T cells [204]. Third, recent studies have demonstrated an association between B cells and tertiary lymphoid structures (TLSs) [208]. TLSs are ectopic lymphoid organs beyond classical lymphoid organs, which develop at sites with chronic inflammation [209]. The formation of TLSs relies on interactions between lymphoid tissue inducer cells (LTi cells) and stromal cells mediated by IL-7 and CXCL13 [209]. Subsequently, the production of VEGF, chemokines, and adhesion molecules facilitates the formation of high endothelial venules (HEVs) and the recruitment of lymphocytes [209]. In injured kidney models, TLS formation was observed in aged but not young mice [210]. Moreover, in aged tumor-bearing mice, IL-21 produced by CD4+ T cells induced CXCL13 secretion, thereby promoting TLS formation [40]. This suggests that aging may drive the formation of TLSs. Mature TLSs create an environment that allows B cells to exert antitumor immunity, while B cells act as 'administrators' of these structures [204, 208]. Interestingly, though TLSs are generally associated with a favorable prognosis in some cancers [204], in aged mice, TLSs promoted by CD4+ T cells led to ICB resistance [40]. Currently, there is insufficient evidence to fully understand the impact of senescent TLSs on tumor development, highlighting the need for further research.
Aging influences B cells in several ways. Specifically, gut microbiota has been shown to induce B cell senescence [211]. With aging, there is a significant decrease in B cells among peripheral blood mononuclear cells (PBMCs) due to reduced B lymphopoiesis in the bone marrow [212]. Despite the decreased number of B cells, an age-related increase in IgG and IgA levels was observed in elderly groups, along with a decrease in IgD and IgM levels [213]. Interestingly, these changes vary between genders [213]. Moreover, inflammaging in aged groups leads to a reduction in B cell progenitors and an accumulation of oncogenic mutations [214]. Although research focuses on the senescence of B cells, the link between senescent B cells and tumor immunity remains to be explored.
6. Stromal Senescence: The Supportive Structure of the TME
We have sequentially introduced the senescence of immune cells, but it is far from illuminating the complexities of the entire tumor microenvironment. The stroma is important in providing support and structure, promoting angiogenesis, regulating immunity, facilitating metastasis, and conferring chemoresistance during tumor progression, especially in cancers such as pancreatic cancer. It primarily comprises fibroblasts, endothelial cells, pericytes, and adipocytes, together with the extracellular matrix (ECM). Our discussion will focus primarily on the first two types of senescent stromal cells—fibroblasts and endothelial cells—and their roles within the TME.
6.1. Fibroblasts
Fibroblasts play a primary role in stromal formation, with cancer-associated fibroblasts (CAFs) receiving significant attention for their role in tumor progression. CAFs segregate into inflammatory CAFs (iCAFs) and myogenic CAFs (myCAFs), differentiated by their spatial localization [215]. iCAFs are located away from tumor cells, whereas myCAFs are adjuvant to tumor sites [215]. By secreting cytokines, chemokines, and other effector molecules, CAFs directly or indirectly remodel the TME, which involves crosstalk with immune cells, including polarization of immune cells, regulation of immunity, reduction of cytotoxic cytokines, upregulation of inhibitory receptors, and remodeling of the extracellular matrix (ECM) [216].
Various factors can induce fibroblast senescence. Radiotherapy causes DNA damage in fibroblasts, thereby triggering DDR and inducing senescence [217]. Novel therapies, such as CDK4/6 inhibitors, induced senescence of fibroblasts through the downregulation of Mdm2 in a melanoma model [15]. Histone deacetylase (HDAC) inhibitors, used to treat various tumors including T cell lymphoma and multiple myeloma, induce fibroblast senescence without DNA damage [218]. Interestingly, obesity increases the levels of deoxycholic acid in the enterohepatic circulation, which in turn drives the senescence of hepatic stellate cells through DDR [219], highlighting obesity as a significant contributor to stromal senescence.
The tumor-promoting nature of senescent fibroblasts was first suggested by A. Krtolica et al. in 2001, demonstrating their role in tumorigenesis in aged organisms [29]. Subsequent research has reinforced this finding. During tumor initiation, senescent fibroblasts promoted ovarian tumorigenesis, as evidenced by reduced tumor growth following the abrogation of the senescence program [220]. Further studies indicate that IL-4 or IL-10-mediated Th2 immunity, which is activated by NF-κB, predisposes aged H-Ras-activated mice to squamous cell carcinoma compared to younger counterparts [221]. MMP-3, secreted by senescent fibroblasts, leads to the dedifferentiation of premalignant epithelial cells, thereby increasing tumorigenesis risk [222]. Moreover, stroma-derived osteopontin (OPN), a component of the ECM, facilitated premalignant cell growth in elderly groups [223]. Interestingly, beyond endocrine effects, senescent fibroblast also stimulates neoplastic epithelial cell proliferation through the production of amphiregulin (AREG) in prostate models [224]. Together, stromal senescence robustly induces tumorigenesis through multiple mechanisms.
Although senescent fibroblasts are often tumor-promoting, some studies indicate that during early stages, stromal senescence aids in recruiting immune cells (Figure 2A), thereby facilitating the clearance of senescent cells and reducing cancer risk. In fibrotic murine livers, senescent HSCs exhibited increased ECM degradation, coupled with enhanced immune surveillance mediated by NK cells [225]. In another murine liver fibrosis model, p53-induced senescence of HSCs resulted in macrophage polarization towards M1 subsets, mediated by SASP, including IL6 and IFN-γ [22]. M1 macrophages, in turn, eliminate senescent HSCs, thereby limiting tumorigenesis [22]. Though evidence has shown that senescent tumor cells can induce immune surveillance in several models, in addition to livers such as multiple myeloma and lung cancers [8], data on similar properties in senescent fibroblasts outside the liver are limited and warrant further investigation.
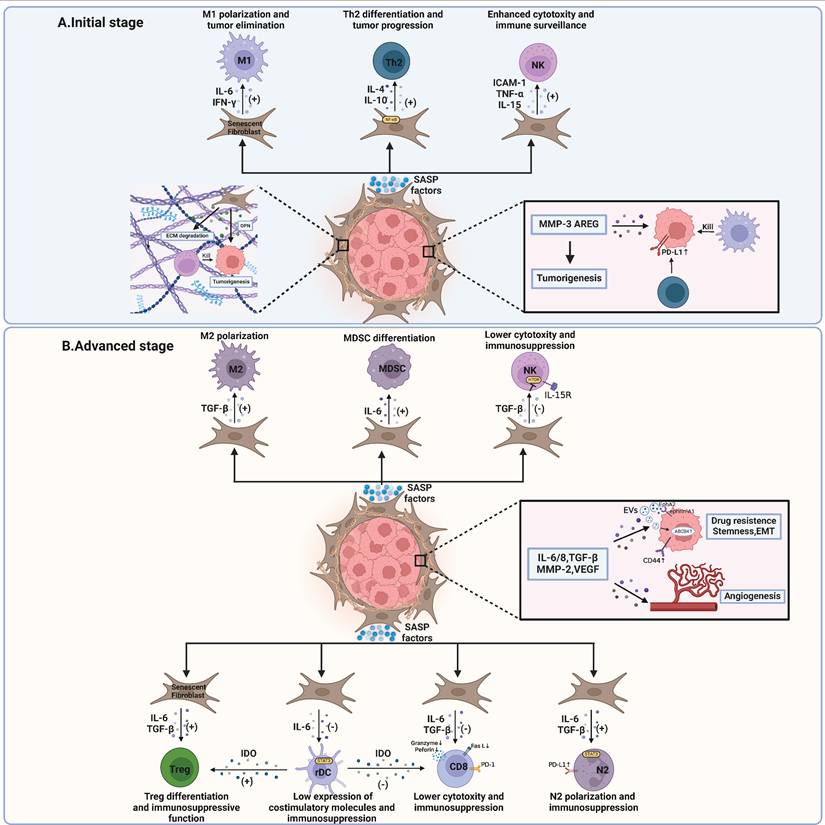
Impacts of SASP produced by senescent fibroblasts within the tumor microenvironment. A At the initial stage, senescent fibroblasts release SASP factors that encourage antitumoral immune responses. M1 macrophage polarization and NK cell-mediated cytotoxicity are bolstered by these factors. Additionally, ECM degradation by SASP factors facilitates enhanced immunosurveillance by NK cells. Conversely, other SASP factors may promote tumorigenesis through interactions with Th2 cells, which upregulate the expression of PD-L1 on tumor cells. ECM components like OPN can aid in tumor growth. B At advanced stages, SASP factors play a pivotal role in tumor progression. On one hand, they can induce cancer stemness, promote epithelial-mesenchymal transition (EMT), confer chemotherapy resistance, and stimulate angiogenesis. On the other hand, SASP factors from senescent fibroblasts contribute to an immunosuppressive microenvironment. This includes the recruitment of MDSCs, M2 and N2 polarization, inhibition of NK cell cytotoxicity, and Treg cell enhancement, which collectively inhibit effective anti-tumor immune responses. The interactions between PD-1 on T cells and PD-L1 on tumor cells further facilitate immune evasion by the tumor. MMP, matrix metalloproteinase; OPN, osteopontin; ECM, extracellular matrix; AREG, amphiregulin; ICAM-1, intercellular adhesion molecule-1; TNF-α, tumor necrosis factor-α; IL, interleukin; IFN-γ, interferon-γ; TGF-β, transforming growth factor-β; EVs, extracellular vesicles; VEGF, vascular endothelial growth factor; PD-1, programmed cell death protein 1; PD-L1, programmed cell death protein ligand 1; EMT, epithelial-mesenchymal transition; NK, natural killer; Th2, helper T cell 2; rDC, regulatory dendritic cell; IDO, indoleamine 2,3-dioxygenase; ABCB4, ATP-binding cassette subfamily B member 4; EphA2, erythropoietin-producing hepatocellular A2; ephrin-A1, recombinant human Ephrin A receptor 1. This figure was created with BioRender (https://biorender.com/).
During advanced stages of tumors, senescent fibroblasts are pivotal in tumor invasion, metastasis, angiogenesis, and a poor prognosis (Figure 2B). First, senescent fibroblast-derived MMP-2 and TGF-β induced keratinocyte invasion in squamous cell carcinoma models [226]. Second, excessive IL-8 secreted by senescent fibroblasts enhanced invasion and metastasis in pancreatic cancer [227]. The levels of IL-8 and stromal senescence, as represented by expression of p16INK4a, were associated with a poor prognosis of patients with pancreatic cancer [227]. In another research, IL-6 and IL-8 induced EMT and stemness of breast cancer cells, as demonstrated by fibroblastoid morphology, increased expression of CD44, and enhanced self-renewal capabilities in tumor cells, making them more aggressive [228]. Third, regarding angiogenesis, while early studies suggested reduced vascularization in aged tumor-bearing mice [229], subsequent research supports the idea that stromal senescence promotes vascularization via increased production of VEGF and TGF-β [27, 28]. Fourth, extracellular vesicles (EVs), as heterogeneous types of membrane vesicles important for intracellular communication, were secreted by senescent fibroblasts [230, 231]. Exosome, as a special category of EVs, was also found to be released in prostate cancers [232]. Not only did EVs promote tumor proliferation through EphA2-ephrin-A1 interaction [231], but they also resulted in drug resistance via inducing expression of ATP-binding cassette subfamily B member 4 (ABCB4) [230]. Interestingly, although traditional approaches emphasize inhibiting tumor angiogenesis [233], senescence-induced angiogenesis could be therapeutically employed [89, 90]. Induced by MEK and CDK4/6 inhibitors trametinib and palbociclib (T/P), senescence successfully triggers SASP, including a series of pro-angiogenesis factors, which surprisingly enhances the therapeutic effect of chemotherapy and ICB in KRAS mutant pancreatic ductal adenocarcinoma (PDAC) [89, 90]. This approach capitalizes on the desmoplastic nature of PDAC, which impedes drug delivery to tumor sites [234, 235]. However, the viability of promoting angiogenesis through senescence in other tumor types remains uncertain. Finally, senescent fibroblasts upregulated gene expression relating to immune regulation and SASP, resulting in impaired CD8+ T cell cytotoxicity and poor responsiveness to immunotherapy and chemotherapy [236-239]. The presence of senescent fibroblasts is correlated with a poor survival outcome using machine learning [238, 239].
6.2. Endothelial Cells
It is important to note that the stroma consists of more than just fibroblasts. Tumor-associated endothelial cells (ECs) also significantly impact the TME as a crucial stromal component. Analysis across various cancer types reveals that ECs exhibit the highest rate of cellular senescence among all cell types in the vascular compartment of cancers [240]. In liver sinusoids, the majority of p16INK4a-expressing senescent cells are ECs [241]. Indeed, ECs are particularly susceptible to senescence, being the first cell types affected by metabolites and senescence stimuli [46]. Due to their critical location, various factors contribute to the senescence of ECs. Metabolites and hormones, including insulin, glucose, triglycerides, cholesterol, amino acids, ROS, endothelin I, and angiotensin II, can induce EC senescence. Senescent ECs, in turn, produce higher levels of ROS, endothelin I, and angiotensin II, creating a vicious cycle [46]. Specifically, nitric oxide, crucial for vasodilation, is believed to attenuate EC senescence [47, 242]. Conversely, the endothelial nitric oxide synthase (eNOS) is impaired in senescent ECs [243], indicating the interplay between NO and senescence. Cytokines such as TNF-α and TGF-β can induce senescence of ECs [47, 244]. Moreover, like other cells, conventional cancer therapies [47, 245-247], targeted therapies including receptor tyrosine kinase inhibitors, VEGF inhibitors, and CDK4/6 inhibitors can all induce senescence of ECs [47, 248, 249]. Interestingly, kisspeptin-10 (KP-10), a member of multifunctional peptides inhibiting metastasis of cancers, can induce endothelial senescence [250]. In melanoma models, ECs exhibit upregulation of Krüppel-like factor 4 (KLF4), which induces senescence of ECs [13]. This suggests indirect tumor cell involvement in EC senescence.
Senescent ECs have a dual role in tumor development (Figure 3). On one hand, senescent ECs induce self-elimination by immune surveillance to evade tumorigenesis [21], with impaired angiogenesis capacity demonstrated by reduced proliferation and VEGF levels [247, 251]. The benefit of this for cancer patients remains to be determined. On the other hand, senescent ECs promote tumor metastasis and treatment resistance via secretion of SASP [12, 13, 246]. Moreover, the sustained activity of Notch1 receptors is observed in senescent ECs, which further promotes cancer metastasis through the production of VCAM-1 [249, 252]. Interestingly, though impaired angiogenesis was observed in senescent ECs, tumor-derived EVs can inhibit the senescence of ECs, thereby counteracting such effects [253]. Moreover, senescent EC-derived EVs can promote the proliferation and migration of tumor cells [253]. The pro-inflammatory profile of senescent ECs offers potential for survival prognostication and immunotherapy efficacy prediction using machine learning [240, 254], promising avenues for targeting or using senescent ECs as biomarkers.
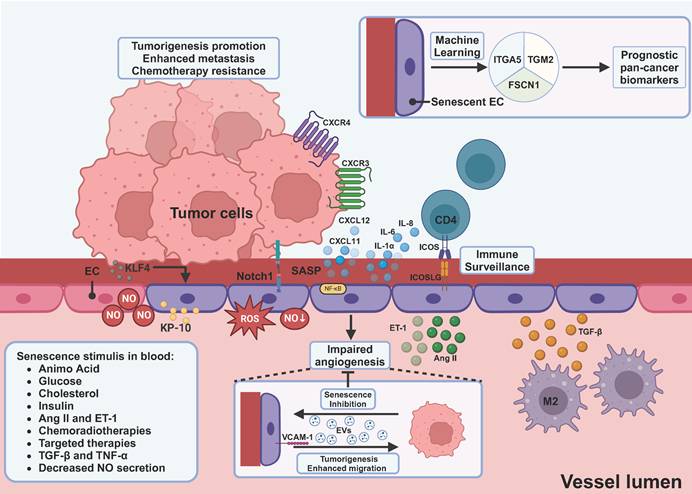
Induction and impact of endothelial cell senescence within the tumor microenvironment. Endothelial cells, as another important component of stroma, specifically become senescent when they encounter metabolites and hormones, including glucose, cholesterol, insulin, etc. M2-derived TGF-β can be another source of induction. Senescent endothelial cells produce increased levels of angiotensin II, endothelin 1, ROS, and decreased levels of NO, which in turn induce senescence of endothelial cells. Tumor-secreting KP-10 and upregulation of KLF4 on ECs can induce senescence of ECs. SASP factors produced by senescent endothelial cells have dual effects. On one hand, they lead to self-immunosurveillance mediated by CD4+ T cells. On the other hand, CXCL12 and CXCL11 can promote tumor cell metastasis and resistance to chemotherapy. Tumor-derived EVs are able to inhibit senescence of ECs, thereby counteracting the impaired angiogenesis of senescent ECs. Moreover, senescent EC-derived EVs and upregulation of VCAM-1 can promote proliferation, and migration of tumor cells. Using machine learning, ITGA5, TGM2, and FSCN1 were screened to be the potential prognostic pan-cancer biomarkers. EC, endothelial cell; NO, nitro oxide; Ang II, angiotensin II; ET-1, endothelin 1; KLF4, Kruppel-like factor 4; KP-10, kisspeptin-10; CXCL, C-X-C motif ligand; CXCR, C-X-C motif receptor; ICOS, inducible T cell co-stimulator; ICOSLG, inducible T cell co-stimulator ligand; VCAM-1, vascular cell adhesion molecule-1; ITGA5, integrin subunit alpha 5; TGM2, transglutaminase 2; FSCN1, fascin actin-bundling protein 1. This figure was created with BioRender (https://biorender.com/).
7. Role of SASP Within the TME: A Double-Edged Sword
In the previous section, we have detailed the senescent immune and stromal cells within the TME. Notably, SASP is increasingly recognized as a key mediator of cellular senescence. Earlier perspectives suggested that senescent cells acquire SASP only when cellular senescence is triggered by DNA damage or the DNA damage response (DDR) [54, 255]. However, current research suggests that SASP induction is a complex process mediated by multiple pathways [8, 41]. Four primary pathways are now identified as mediators of SASP induction: p53-p21/p16-Rb, DDR-NF-κB, p38 MAPK, as well as mTOR and cytoplasmic DNA-cGAS-STING pathways [8, 256]. Additionally, SASP is regulated by epigenetic mechanisms and oxylipins, such as dihomo-15d-PGJ2 [256]. The heterogeneity of SASP is influenced by the cell type and the causes of senescence, with IL-6 and IL-8 being commonly identified SASP factors [8]. In this section, we will concentrate on the dual role of SASP in tumor progression. Research indicates that SASP secretion is influenced by tissue type, cell type, and stage of progression. Specifically, SASP dynamics within the tumor microenvironment can be categorized into two distinct stages.
During tumor initiation, SASP factors help eliminate potential pre-malignant cells. Senescent hepatocytes contribute to tumor surveillance through SASP-mediated senescence surveillance [80, 257], which relies on the participation of immune cells. These two studies underscore the importance of timely senescence surveillance in the liver. This has also been demonstrated in other cancers, including lymphoma, melanoma, and osteosarcoma, where innate immunity-mediated clearance of senescent cells provides tumor-suppressive effects [8, 45]. Specifically, senescent pre-malignant cells may give rise to cancer if not cleared promptly. Senescent fibroblasts within the TME, as previously described, also exhibit anti-tumor activity during the early stages of tumor development. An exception arises in KRAS-driven lung cancer, where senescent macrophage SASP unexpectedly promotes early tumorigenesis [23], underscoring the need for deeper investigation into immune-derived SASP.
In established tumors, SASP fosters invasion, metastasis, and neovascularization, which we have elaborated on in the section on senescent fibroblasts. Senescent ECs produce SASP factors fostering metastasis in breast cancer [12] and melanoma models [13] and contributing to chemotherapy resistance [246]. Moreover, the immunosuppressive microenvironment created by SASP factors should be emphasized. IL-6 regulates both innate and adaptive immunity. In innate immunity, through IL-6-STAT3 signaling, HCC-derived CAFs activate and maintain PD-L1+ neutrophils, thus impairing T cell function via PD-1-PD-L1 interaction [258]. What's more, HCC-derived CAFs secreted IL-6 to generate regulatory DCs, which contribute to the dysfunction of T cells and the promotion of Treg activity via indoleamine 2,3-dioxygenase (IDO) upregulation [259]. CAF-derived IL-6 promotes the differentiation of monocytes into myeloid-derived suppressor cells (MDSCs), thereby mediating immune dysfunction, which has been observed in HCC [260], pancreatic cancer [261], and esophageal squamous cell carcinoma [262]. The extracellular matrix secreted by senescent fibroblasts was also able to limit NK cell cytotoxicity [263]. In adaptive immunity, CAFs can directly enhance Treg function while inhibiting T cell proliferation through IL-6 production [264]. Meanwhile, TGF-β, as another component of SASP [54], also acts as a regulator in tumor immunity. TGF-β not only promotes the transformation of monocytes into M2 macrophages [216] but also induces N2 neutrophil polarization in HCC [265]. Moreover, TGF-β blocks IL-15-induced activation of mTOR, which is essential for cytotoxicity and proliferation of NK cells [266]. Suppression of TGF-β successfully abrogated metastases in two mouse models [266]. TGF-β derived from CAFs also promotes both Th17 differentiation and the conversion of CD4+ naïve T cells into Tregs [267, 268] while inhibiting the production of perforin, granzyme B, FasL, and IFN-γ by CD8+ T cells [269]. Collectively, SASP factors produced by senescent cells are broadly immunosuppressive in advanced stages of tumors.
8. Novel Therapies Targeting Senescence: Next Hope for Cancer Treatment?
The advent of novel immunotherapies, including ICB, engineered chimeric antigen receptor (CAR) T cells, and cancer vaccines, has ushered in a new era in cancer treatment. Despite its success, ICB faces resistance driven by genetic and epigenetic aberrations in tumor cells, T cell exhaustion, cancer-associated fibroblasts (CAFs), and immunosuppressive mechanisms [270]. Consequently, there is an urgent need to overcome these obstacles. Emerging evidence highlights the promising potential of targeting senescence to enhance the efficacy of ICB. These approaches fall into four categories—induction of senescence, regulation of SASP, clearance of senescence, and senescence reprogramming (Figure 4).
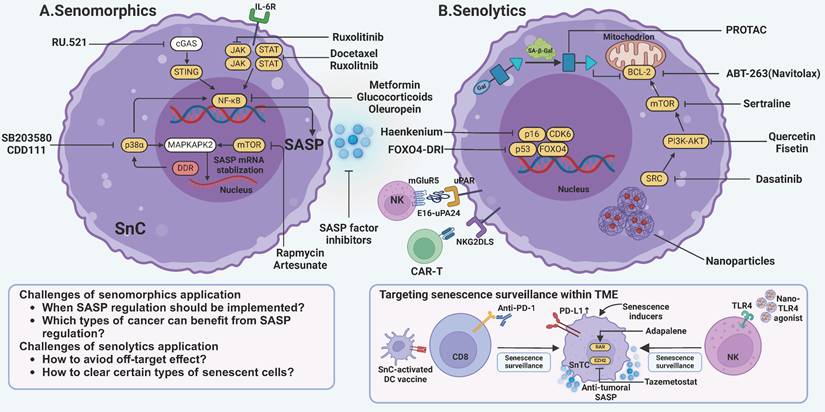
Targeting senescence by modulation of SASP or clearance of senescent cells. A One strategy to target senescence involves preventing senescent cells from producing tumor-promoting SASP factors. These drugs, termed senomorphics, primarily act on pathways such as cGAS-STING, JAK-STAT, and p38α-MAPKAPK2. Key mechanisms include NF-κB and mTOR to inhibit SASP production. Drugs such as metformin and rapamycin are among those used to modulate these pathways and mitigate the detrimental effects of SASP. B Another widely used strategy is to eliminate senescent cells with senolytics. The first-generation senolytics target anti-apoptotic pathways intrinsic to senescent cells, such as those involving BCL-2, PI3K-AKT, and mTOR. Second-generation senolytics find a new path by targeting specific surface markers on senescent cells. It utilizes innovative techniques like CAR-T cells, chimeric polypeptides, and vaccines. Notably, efforts are being made to enhance senescence surveillance mediated by T cells and NK cells. JAK, janus kinase; STAT, signal transducer and activator of transcription; NF-κB, nuclear factor-κB; cGAS, cyclic guanosine monophosphate-adenosine monophosphate synthase; STING, stimulator of interferon genes; mTOR, mammalian target of rapamycin; MAPKAPK2, mitogen-activated protein kinase-activated protein kinase 2; DDR, DNA damage response; BCL-2, B-cell lymphoma-2; PI3K, phosphatidylinositide 3-kinases; CAR-T, chimeric antigen receptor-T; GPNMB, glycoprotein nonmetastatic melanoma protein B; uPAR, urokinase-type plasminogen activator receptor; NKG2DLS, Natural killer group 2 member D ligands; SnC, senescent cell; SnTC, senescent tumor cell. This figure was created with BioRender (https://biorender.com/).
8.1. Induction of Senescence
In the early stages of tumors, senescence exerts antitumoral effects through several mechanisms, including clearance of senescent cells, activation of tumor immunity, and promotion of proper angiogenesis, as discussed above [89, 90, 225, 270]. Indeed, inducing senescence can improve the effect of cancer treatment (Figure 4). T/P-induced senescence fosters the accumulation of CD8+ T cells, leading to increased sensitivity to ICB and chemotherapy in human PDAC models [89, 90]. Interestingly, the desmoplastic nature of PDAC, which is typically resistant to drug treatment, was shown to benefit from T/P-induced SASP factor production, which promoted vascularization and improved drug delivery and chemotherapy response [89, 90]. Additionally, induction of senescence stimulated the production of antitumoral SASP factors, leading to NK cell-mediated tumor clearance [271]. EZH2 is a key gene regulating SASP secretion, whose blockade combined with ICB has successfully promoted the production of SASP chemokines, including CCL2 and CXCL9/10, leading to T cells and NK cells-mediated tumor immunity [11]. Nanoparticles co-delivering senescence inducers and TLR4 agonists extend survival in PDAC by activating T cells and NK cells [272]. Ali and JAK2 inhibitor ruxolitinib could also recruit T cells and NK cells within TME by inducing SASP secretion [273]. DC vaccines loaded with senescent tumor antigens or PD-1 blockade further potentiate T cell responses [198, 274]. Conclusively, T cells and NK cells are emerging as the primary force in eliminating senescent cells with the support of SASP. Beyond preclinical studies, clinical trials have explored inducing senescence with dexamethasone to re-sensitize response to ICB in patients with NSCLC (NCT04037462).
To harness the antitumor benefits of TIS while mitigating its deleterious immunosuppressive effects, two principles may guide clinical implementation. First, the temporal window for senolytic intervention is critical. Extended persistence of senescent cells within the TME fosters immunosuppression, angiogenesis, and metastatic niche formation as discussed above. Thus, senolytics should be deployed once TIS has maximally engaged immune-mediated tumor clearance but prior to the onset of a full-blown SASP or escape from growth arrest by senescent cells [275]. Second, current senescence-inducing modalities—chemotherapy, radiotherapy, and kinase inhibitors—lack specificity and can inadvertently drive senescence in immune effector populations, exacerbating immune dysfunctions [14, 19]. To obviate this, agents that selectively target tumor-cell senescence are required. For example, pharmacological inhibition of the replication origin kinase CDC7 induces senescence specifically in hepatocellular carcinoma cells without impairing normal immune cells [276]. Similarly, the natural alkaloid tryptanthrin (TRYP) rapidly triggers senescence in liver cancer cells, arresting proliferation while sparing systemic immunity [277].
8.2. Regulation of SASP
Conversely, inhibiting the tumor-promoting SASP factors also emerges as a plausible alternative. (Figure 4A). Drugs targeting SASP pathways are referred to as senomorphics. Key intervention points include transcriptional regulators, signal transduction cascades, metabolic nodes, and the SASP factors themselves [8, 41, 48]. For instance, inhibiting the JAK2/STAT3 pathway, which is involved in SASP-associated tumor growth and chemoresistance, induces robust immune surveillance in Ptennull tumors with docetaxel-induced senescence [278]. Targeting PTBP1 via RNA interference prevented the protumoral effects of SASP factors in tumor-bearing mice [279]. NF-κB and mTOR have emerged as prominent targets for mitigating senescence [8, 92, 93]. Metabolic reprogramming in senescent cells further dictates SASP output: in pancreatic cancer models, elevated NAD⁺ flux enhances NF-κB-dependent proinflammatory SASP [280], whereas inhibition of nicotinamide phosphoribosyltransferase (NAMPT)—the rate-limiting enzyme of the NAD⁺ salvage pathway—dampens SASP release and suppresses tumor growth [280]. In the hepatic niche, loss of the gluconeogenic enzyme fructose-1,6-bisphosphatase 1 (FBP1) in hepatocytes triggers secondary senescence of hepatic stellate cells via HMGB1 signaling; neutralization of extracellular HMGB1 attenuates HSC-derived SASP and impairs tumor progression [281]. Interestingly, metformin, a common medication for type II diabetes, can suppress NF-κB pathways [282]. It can also inhibit T cell senescence while maintaining its cytotoxicity [283]. Though metformin has shown potential in attenuating aging [284], NF-κB suppression unfortunately led to drug resistance and a poor prognosis in murine lymphoma and melanoma models [92, 93]. Epidemiological studies suggest a decreased incidence of cancer in individuals receiving metformin [285], suggesting its potential role in cancer prevention rather than treatment.
The mTOR-MK2 pathway also plays a crucial role in SASP production [286-289]. mTOR inhibitors like rapamycin reduce the secretion of tumor-promoting SASP factors [288, 289]. In a phase IIa randomized controlled trial, the use of rapamycin enhanced the response to influenza vaccination [290], demonstrating its potential to boost immunity. Moreover, unlike other mTOR inhibitors, brief administration of rapamycin can produce a sustained anti-SASP effect, thereby reducing the risk of adverse events associated with long-term treatment [291]. To date, clinical studies evaluating rapamycin's efficacy in targeting senescence remain in the early stages (Table 2) [292].
Current clinical trials targeting senescence against cancers
| Category | Drug | Mechanism | Combination therapy | Condition | Design | Reference | Status |
|---|---|---|---|---|---|---|---|
| Regulation of senescence | Dexamethasone | Induction of senescence | Anti-PD-1 therapy | NSCLC | Phase I/II | NCT04037462 | Terminated |
| Rapamycin | Inhibition of SASP | Alisertib | Advanced Solid Tumors | Phase I | [292] | With result | |
| Clearance of senescence | D plus Q | 1st-generation senolytics | None | Childhood Cancer | Phase II | NCT04733534 | Recruiting |
| Anti-PD-1 therapy | Head and Neck Cancer | Phase II | NCT05724329 | Active | |||
| None | Triple-negative Breast Cancer | Phase II | NCT06355037 | Recruiting | |||
| Fisetin | 1st-generation senolytics | None | Childhood Cancer | Phase II | NCT04733534 | Recruiting | |
| None | Breast Cancer | Phase II | NCT05595499 | Recruiting | |||
| None | Breast Cancer | Phase II | NCT06113016 | Recruiting | |||
| ABT-263 (Navitoclax) | 1st-generation senolytics | Gemcitabine | Advanced solid tumors | Phase I | [293] | With result | |
| Docetaxel | Advanced solid tumors | Phase I | [294] | With result | |||
| None | Lymphoid malignancies | Phase IIa | [295] | With result | |||
| Rituximab | Chronic lymphocytic leukemia | Phase II | [296] | With result | |||
| ABT-737 | 1st-generation senolytics | None | Ovarian Cancer | Observational study | NCT01440504 | Completed | |
| “One-two” punch therapy | Induction of senescence plus senolytics | Decitabine plus navitoclax | Acute myeloid leukemia | Phase Ib | NCT05222984 | Active | |
| Olaparib plus navitoclax | Triple-negative Breast Cancer | Phase I | NCT05358639 | Active |
NSCLC, non-small-cell lung cancer; D, dasatinib; Q, quercetin; SCLC, small-cell lung cancer.
These findings raise the question: Can the regulation of SASP factors signify the next breakthrough in cancer therapy? The dual role of SASP in tumor development complicates its clinical application. Thus, balancing the antitumoral and protumoral effects of SASP factors is crucial. Based on current research on SASP so far, several key characteristics of SASP can be identified. The secretion of SASP factors is stage-dependent and tissue-dependent, which presents two major challenges. First, determining when SASP should be induced or inhibited remains a critical question. At present, it remains challenging to determine whether SASP is beneficial or detrimental for a particular patient. Nor can the exact point at which the role of SASP is reversed be identified. However, since many cancers are diagnosed at advanced stages, it may be more beneficial to inhibit the production of SASP factors to achieve improved clinical outcomes. Second, for certain tumor types, it remains unclear which strategy is optimal. The answer may lie in identifying which component of the SASP factors is dominant in regulating the TME as a whole. For instance, in pancreatic ductal adenocarcinoma (PDAC) models, pro-angiogenic factors produced by senescent cells can promote the formation of a more 'open' microenvironment, thereby enhancing the response to chemotherapy and immunotherapy [89, 90]. While in lymphoma models. In lymphoma models, IL-6 produced by senescent endothelial cells (ECs) has been shown to protect tumor cells from chemotherapy [246]. Overall, the future of regulating SASP as an effective cancer therapy is likely to be personalized.
8.3. Clearance of Senescence
Senescent cell clearance via senolytics, drugs that selectively ablate senescent cells, is a second therapeutic strategy (Figure 4B). Unlike SASP inhibitors, senolytics remove the SASP source and can be dosed intermittently [297]. Currently, there are two generations of senolytic drugs. First-generation senolytics target multiple antiapoptotic pathways (SCAPs) in senescent cells, such as BCL-2, SRC kinases, PI3K-AKT, etc. In contrast, targets of second-generation senolytics are discovered via high-throughput library screens, and include lysosome‑targeted agents, vaccine‑based approaches, nanoparticle delivery, and CAR‑T cell strategies [297].
Classic first-generation senolytics strategies include dasatinib (D) plus quercetin (Q) and navitoclax (ABT-263). D plus Q induces senescent cell death by inhibiting tyrosine kinase and PI3K signaling, respectively [298]. The combination of D and Q has been observed to alleviate symptoms and increase survival rates in various age-related diseases, including postmenopausal osteoporosis [299], intervertebral disc degeneration [300], diabetic kidney disease [301], and SARS-CoV-2 [302]. D plus Q can indirectly suppress tumor development and metastasis by mitigating stromal senescence [303, 304]. This effect is attributed to the inhibition of protumoral SASP secreted by senescent fibroblasts and stem cells [303, 304]. Clinical trials (NCT04733534, NCT05724329, NCT06355037) are ongoing to determine whether D plus Q can be a viable approach to reverse chemoresistance or to improve survival as an effective and safe adjuvant therapy. ABT-263, one of the BCL-2 inhibitors, has demonstrated greater success in the context of cancer therapy, with the capacity to eliminate therapy-induced senescent cells in cancer models such as lung cancer, breast cancer, melanoma, ovarian cancer, and prostate cancer [45, 298, 305]. In preclinical studies, ABT-263 reversed side effects associated with TIS, including bone marrow suppression, cardiac dysfunction, and cancer recurrence [306]. Clinical studies combining ABT‑263 with chemotherapy are in progress (Table 2) [45, 293-296, 307]. Finally, fisetin is another promising senolytic targeting senescence in cancers. Fisetin is extracted from vegetables and fruits, with a mechanism of action similar to that of quercetin [298, 308]. In patients with small-cell lung cancers, fisetin successfully reversed the chemotherapy resistance induced by cellular senescence [309]. Several phase II clinical trials (NCT04733534, NCT05595499, NCT06113016) are underway to evaluate its efficacy and safety targeting cancers.
Despite the promise, senolytics have drawbacks. First, patients receiving ABT-263 are at risk of developing thrombocytopenia and neutropenia [45, 297], raising concerns about its safety. Second, resistance to BCL inhibition in senescent tumor cells has been reported [310, 311], though efforts are underway to target mitochondrial apoptotic pathways or employ sensitizer proteins to restore sensitivity to senolysis [310, 312]. Third, D plus Q failed to directly kill senescent cells and even exhibited tumor-promoting effects when used alone in animal HCC models due to the poor penetration in tumor sites [313, 314]. The potential of D plus Q in cancer treatment may be realized through novel delivery approaches, such as extracellular vesicles and nanoparticles [314, 315]. Fourth, the elimination of certain senescent cells may cause adverse consequences [241]. For example, acute clearance of senescent ECs in livers will compromise blood-tissue barriers, potentially accelerating liver fibrosis [241]. In light of these limitations, targeting specific markers to clear senescent cells, or eliminating certain types of senescent cells, has emerged as an alternative approach, namely the second-generation senolytics.
Second-generation senolytics exhibit enhanced target specificity, exploiting senescence-associated pathways through integrated immunotherapeutic strategies—including cancer vaccines, CAR-T cells, and antibody-drug conjugates (ADCs)—to achieve selective clearance (Figure 4B) [297]. SA-β-Gal is overexpressed in senescent cells, which is the most commonly used senescence marker [11, 47]. Not only can it be applied to specifically deliver ABT-263 [316], but it can also be recognized by engineered proteolysis-targeting chimeras (PROTACs), thereby selectively eliminating senescent cells [317-321]. Composed of a galactose (Gal) moiety, PROTACs like ARV-771 and MS999 can effectively clear senescent tumor cells without inducing significant adverse events [315, 318]. Another PROTAC drug 753b targeted BCL-xL and BCL-2 dually to inhibit tumor progression [321]. Moreover, new senescence markers are being found. For instance, urokinase-type plasminogen activator receptor (uPAR), a surface protein broadly expressed in T/P induced senescent cells, can be targeted by CAR-T cells, and its elimination improved the prognosis of mice with lung adenocarcinoma [87, 88, 322]. Moreover, using a chimeric polypeptide, uPAR-expressing senescent cells can be cleared by NK cells [323]. Natural killer group 2 member D ligands (NKG2DLs), another surface marker widely expressed in senescent cells, can be targeted by CAR-T cells safely [324]. The effectiveness of this approach in cancer treatment requires further investigation. Notably, CAR-T cells, like conventional T cells, may also undergo senescence [48]. First, children and young adults with B-ALL have benefited most from CAR-T therapy [325], and current clinical trials have yet to report great benefits in elderly patients (NCT05523661, NCT04537442, NCT05707273, NCT04300998). Second, research has observed increased expression of CD57 on CAR-T cells in the highly malignant glioblastoma multiforme models [326], and modulating p53 signaling pathways helps enhance CAR-T therapy in patients with chronic lymphocytic leukemia [327], suggesting the potential for CAR-T cell senescence. Finally, targeting metabolic dependencies of senescent tumor cells offers an additional avenue for senolytic intervention, given their heightened reliance on glycolysis and glutaminolysis for survival [55, 56]. Indeed, inhibiting glucose uptake or metabolism induces apoptosis selectively in senescent tumor cells [328], while blockade of glutamine utilization suppresses their escape from growth arrest [100]. A potential of mitochondrial-targeted therapy is also suggested. Mitochondrial physiology likewise serves both as a vulnerability and a biomarker for senolytic sensitivity [329-331]. Specifically, pharmacologic inhibition of the TBK1-ATAD3A-Pink1 axis attenuates Pink1-mediated mitophagy, mitigates cellular senescence, and enhances chemotherapeutic efficacy [329]. Moreover, mitochondrial dependence on BCL-XL and MCL-1 has emerged as a robust biomarker for forecasting senescent cell responsiveness to ABT-263 [330, 331]. Collectively, these findings position mitochondrial targeting at the forefront of next-generation anti-senescence therapies.
Based on the success of immunotherapy in cancer treatment, there is growing interest in targeting senescence to reverse the immunosuppressive microenvironment, thereby resensitizing immunotherapy. Senescent tumor cell clearance has been proven to reverse the immunotherapy resistance associated with the accumulation of senescent cells [19]. Senolytics disrupted SASP-mediated PD-L1/TGF-β signaling axis and replenished intratumoral CD8+ T cells with restored granzyme B expression by normalizing TME arginine metabolism via arginase-1 suppression [19]. Neutralization of senescent-cell-derived mtDNA reverses PMN-MDSC-mediated immunosuppression, enhances T-cell function, and potentiates chemotherapy [102, 169, 332]. Beyond malignant cells, senescent immune and stromal populations are also amenable to targeting (Table 3). Inhibition of cholesterol biosynthesis and lipid droplet formation prevents T-cell senescence and restores checkpoint-inhibitor efficacy [168, 187]. CD153, highly expressed in senescent T cells, can be recognized and eliminated by the CD153-CpG vaccine in mice with obesity-induced senescence [333]. One year after the first vaccine was invented, another seno-antigen glycoprotein nonmetastatic melanoma protein B (GPNMB) was screened via analysis of the transcriptome, and vaccination against GPNMB on senescent ECs was also effective in clearing senescent cells in mice with obesity-induced senescence [334]. Moreover, the elimination of senescent fibroblasts with senolytics awakened T cells and NK cells-mediated tumor immunity and resensitized response to chemotherapy in breast cancers and pancreatic cancers [236, 237, 263]. Senescent macrophages can also serve as a target, with their elimination through senolytics ameliorating early tumor growth and facilitating ICB-based immunotherapy [19, 23, 138]. Together, second-generation senolytics hold promise for attenuating senescence. Further clinical trials are needed to determine their safety and efficacy as cancer therapies.
Preclinical studies clearing senescent immune and stromal cells within the tumor microenvironment.
| Target | Treatment | Condition | Outcome | Reference |
|---|---|---|---|---|
| Neutrophil | 3MRp16 model with ganciclovir suicide gene strategy | Prostate cancer | Suppressed tumor growth | [107] |
| Procyanidin C1 | Melanoma | Reduced tumor metastasis and restored T cell responses | [128] | |
| Macrophage | Diphtheria toxin targeting tagged cells | Lung cancer | Diminished lung tumor burden and prolonged survival | [23] |
| ABT-737 | ||||
| ABT-263 | Lung cancer | Suppressed early tumorigenesis | [138] | |
| ABT-263 | Colon cancer | Restored CD8+ T cell proliferation and response to immunotherapy | [19] | |
| Nicotinamide mononucleotide | Glioblastoma | Inhibited T-cell dysfunction and delayed tumor initiation | [133] | |
| IL-4 | Aging | Improved the health span of aged mice | [335] | |
| T cell | Metformin | NA | Lowered IFN-γ and IL-6 and increased TNF-α production | [283] |
| CD153-CpG vaccine | Obesity | Improved obesity-induced metabolic disorders | [333] | |
| Endothelial cell | Anti-Notch1/ VCAM1 antibody | Ovarian cancer | Reduced tumor cell adhesion and lowered lung metastasis | [252] |
| GPNMB vaccine | Atherosclerosis | Improved metabolic disorders | [334] | |
| Fibroblast | ABT-199 | Pancreatic cancer | Restored CD8+ T cell function and response to immunotherapy | [236] |
| ABT-737 | Breast cancer | Enhanced NK cell function and infiltration | [263] | |
| Anti-TSPAN8 antibody | Breast cancer | Resensitize the response to chemotherapy | [237] | |
| Q | Osteosarcoma | Reduced tumor invasiveness | [303] | |
| D plus Q | Ovarian cancer | Reduced tumor metastasis | [304] |
D, dasatinib; Q, quercetin; GPNMB, glycoprotein nonmetastatic melanoma protein B.
Finally, a therapeutic paradigm known as the “one-two punch therapy” has emerged, whereby induction of tumor-cell senescence is immediately followed by selective senolysis to maximize antitumor efficacy while limiting chronic SASP-driven toxicity [41, 43, 45]. In TP53-mutated liver cancer, inhibition of the DNA-replication kinase CDC7 specifically induced senescence of liver cancer cells, while subsequent treatment with mTOR inhibitors sertraline markedly reduced tumor growth [276]. The combination of 'one-two punch' therapy and immunotherapy has demonstrated potent inhibition of tumor growth in colorectal cancer and lung cancer [336, 337]. Ongoing efforts are identifying inducers and senolytics with enhanced specificity for senescent tumor cells to improve the therapeutic index [277, 337, 338]. Notably, perturbation of methionine catabolism precipitates DNA damage-mediated senescence in liver cancer cells, and follow-on senolytic therapy effectively attenuates hepatocarcinogenesis [58], suggesting that metabolic targeting may unlock a new frontier for precise, safe senescence induction.
8.4. Senescence Reprogramming
Finally, since T cell exhaustion is reversible by ICB [177], researchers are exploring methods to revert cellular senescence. Opinions once held that no viable strategy had been identified to achieve this reversal, but recently, approaches have emerged to reverse senescence [339, 340]. Gu et al. showed that FBP1 suppression bypasses senescence in HCC progenitors, restoring their proliferative capacity [340].
Bi et al. demonstrated that exosomes derived from human embryonic stem cells and their miR-302b content can reverse cellular senescence by targeting key cell cycle inhibitors, leading to rejuvenation in aging mice without safety concerns [339]. These two studies have unveiled the potential for reversing aging, raising anticipation for further research.
Moreover, the aged immune system can be reprogrammed to generate rejuvenated immune cells. First, one of the characteristics of natural immunosenescence is the involution of the thymus [48, 49], leading to reduced output of naive T cells. Efforts to rejuvenate the thymus have shown promise. Aged mice receiving IL-7 have shown enhanced adaptive immunity, as evidenced by lower viral load [341], while the thymostimulatory property of IL-21 was further demonstrated in the humanized mice model [342, 343]. Gene modulation targeting Foxn1 can also partially rescue thymic involution and reduction of peripheral CD4+ T cells via exogenous FoxN1-cDNA [344], although recent research has indicated that Foxn1 overexpression does not prevent thymic involution [345]. The second approach is implementing hematopoietic transplantation. Through intrathymic injection of hematopoietic progenitor cells from healthy mice, thymic reconstitution could be achieved in mice with severe combined immunodeficiency [346]. Umbilical cord blood (UCB) can be an alternative source of HSCs [347]. Finally, CD8+ T cells isolated from HIV patients can be reprogrammed to pluripotent stem cells, which subsequently re-differentiate into CD8+ T cells with enhanced cytotoxicity and proliferation [348].
9. Conclusion and Perspectives
In conclusion, senescence is a ubiquitous process affecting all components of the TME. In this review, we highlighted that senescence extends beyond chronological aging, representing the sum of diverse senescence triggers. Aging can be understood as the cumulative effect of senescence inducers. In clinical settings, conventional and novel cancer therapies, oncogene-induced senescence, and interactions within the TME are significant contributors [54, 83-85]. This underscores the need for senescence studies to extend beyond just elderly populations.
Next, we have reviewed the properties of senescent immune cells in both innate and adaptive immunity, as well as the impact of SASP factors produced by senescent stromal cells. While adaptive and stromal senescence are well characterized, innate immune senescence in cancer remains understudied. This knowledge gap reflects the recent recognition of senescence in neutrophils and macrophages [50, 349]. As increasing studies have elucidated the roles of neutrophils and macrophages in tumor immunity [114, 121, 130, 134, 350-352], it becomes imperative to investigate the controversial role of innate immunosenescence in tumors.
Finally, we outline existing therapies targeting senescence. Though great progress has been made in targeting SASP and senescent cells, their clinical application remains distant, as discussed in the corresponding section above. Moreover, barriers to the clinical implementation of senescence-targeted therapy can be partly attributed to the classification of senescent cells in human samples, since it is essential for understanding how senescence influences responses to cancer therapy and clinical outcomes, and to what extent senescence-targeting therapy can benefit from cancer treatment [63]. Saleh et al. reviewed 21 studies that aimed to identify therapy-induced senescent cells in patient samples [16] and highlighted current limitations, including limited approaches for senescence detection, the challenge of obtaining cancer samples from patients who have not undergone chemotherapy, the requirement for freshly frozen tissue for SA-β-gal staining, and variability in baseline expression of senescence markers across different cancer samples [16].
To address these challenges, it is urgently necessary to find solutions for the following issues: (a)To identify reliable inducers of senescence. Numerous factors can induce senescence, but identifying the most suitable one for laboratory and clinical conditions is crucial. Chemotherapy or radiation-induced senescence is not suitable for all tumor types, especially for those treated with chemotherapy after surgery [16]. Finding an inducer that works universally across cell types or matching various tumors with their viable inducers is essential. Typically, Scott W Lowe et al. have utilized T/P to induce senescence of pancreatic cancers and lung cancers [11, 87, 89]. However, a stable inducer for research on immunosenescence is still lacking. (b)To standardize existing markers. So far, SenNet has recommended senescence markers across different tissues of humans and mice [353]. Standard protocols exist for senescence research in vivo. According to the minimum information for cellular senescence experimentation in vivo (MICSE) published in 2024 [354], markers used to detect senescent cells should include at least three markers of different properties of cellular senescence, at least one of which should be increased p16INK4a or p21Cip1/Waf1 expression. However, no standard has been established for clinical trials since MICSE is not intended for clinical practice. In breast cancers, progress is being made toward standardizing senescence detection, including the establishment of baseline Lamin B1 expression and a three-marker signature approach to detect TIS, which involves downregulation of Lamin B1 and Ki-67 and upregulation of p16INK4a [355, 356]. (c)To discover emerging markers. Discovering specific markers will aid in understanding functional changes and targeting specific senescent cells. For instance, senescent cells can be isolated using flow cytometry by the differential presence of dipeptidyl peptidase 4 (DPP4) [357]. Anti-DPP4 antibodies enable natural killer (NK) cell-mediated elimination of senescent cells, offering new perspectives on senescence-targeted therapy [357]. CD153, differentially expressed in senescent T cells, could be applied as a vaccine to selectively clear senescent T cells in mice [333]. Moreover, advances in technology, such as artificial intelligence, high-throughput sequencing, and single-cell sequencing, offer new opportunities for studying senescence. For instance, uPAR, as one of the targets of CAR-T, was discovered with RNA-sequencing [87, 88]. Discovery of novel senolytics can now be achieved using machine learning [358]. Whether these new markers can be the next-generation markers in clinical practices remains to be validated. (d) To create novel detection approaches. Machine learning and artificial intelligence are gaining popularity in detecting senescence [353, 359], although identifying specific types of senescent cells remains challenging. Recently, Zhou et al. have introduced a brand-new approach to specifically trace certain types of senescent cells [360]. In this study, they generated pulse-chase tracing (Sn-pTracer), Cre-based tracing and ablation (Sn-cTracer), and gene manipulation combined with tracing (Sn-gTracer) to track p16INK4a macrophages and ECs, thereby enabling the clearance of specific types of senescent cells [360]. It is believed that targeting senescent cells will become a reliable cancer therapy in the near future.
Abbreviations
SASP: senescence-associated secretory phenotype; DDR: DNA damages response; HSCs: hepatic stellate cells; PGE2: prostaglandin E2; HCC: hepatocellular carcinoma; OIS: oncogene-induced senescence; TME: tumor microenvironment; SA-βGal: senescence-associated ß-galactosidase; ECM: extracellular matrix; BM: bone marrow; TREM2: triggering receptor expressed on myeloid cells 2; APOE: apolipoprotein E; ADCC: antibody-dependent cellular cytotoxicity; NETs: neutrophil extracellular traps; NO: nitric oxide; FTO: fat mass and obesity-associated protein; EMT: epithelial-mesenchymal transition; APCs: antigen-presenting cells; FasL: Fas ligand; MDSCs: myeloid-derived suppressor cells; IMCs: immature myeloid cells; MO-MDSCs: monocytic myeloid-derived suppressor cells; PMN-MDSCs: polymorphonuclear myeloid-derived suppressor cells; TRAIL: TNF-related apoptosis-inducing ligand; NKCC: NK cell cytotoxicity; cDCs: classical dendritic cells; pDCs: plasmacytoid dendritic cells; CTLs: cytotoxic T lymphocytes; NSCLC: non-small cell lung cancer; ACKR2: atypical chemokine receptor 2; KLRG1: killer cell lectin-like receptor subfamily G 1; ICB: immune checkpoint blockade; PFS: progression-free survival; ADCP: antibody-dependent cellular phagocytosis; CDC: complement-dependent cytotoxicity; TLSs: tertiary lymphoid structures; LTi cells: lymphoid tissue inducer cells; HEVs: high endothelial venules; PMBCs: peripheral blood mononuclear cells; CAFs: cancer-associated fibroblasts; iCAFs: inflammatory CAFs; myCAFs: myogenic CAFs; HDAC: histone deacetylase; PDAC: pancreatic ductal adenocarcinoma; ECs: endothelial cells; eNOS: endothelial nitric oxide synthase; CAR: engineered chimeric antigen receptor; SCAPs: senescent cells antiapoptotic pathways; D: dasatinib; Q: quercetin; UCB: umbilical cord blood; cAMP: cyclic adenosine monophosphate; ILT4: immunoglobulin-like transcript 4; TGF-β: transforming growth factor-β; HLA-G: human leukocyte antigen-G; PKA: protein kinase A; CREB: cAMP-response element binding protein; ATM: ataxia telangiectasia-mutated; ERK1/2: extracellular regulated protein kinases 1/2; STAT1/3: signal transducer and activator of transcription 1/3; CDK: cyclin-dependent kinases; Rb: retinoblastoma protein; TLR8: Toll-like receptors 8; GLUT: glucose transporter; MMP: matrix metalloproteinase; OPN: osteopontin; ECM: extracellular matrix; AREG: amphiregulin; ICAM-1: intercellular adhesion molecule-1; TNF-α: tumor necrosis factor-α; IL: interleukin; IFN-γ: interferon-γ; TGF-β: transforming growth factor-β; EVs: extracellular vesicles; VEGF: vascular endothelial growth factor; PD-1: programmed cell death protein 1; PD-L1: programmed cell death protein ligand 1; EMT: epithelial-mesenchymal transition; NK: natural killer; Th2: helper T cell 2; rDC: regulatory dendritic cell; IDO: indoleamine 2,3-dioxygenase; ABCB4: ATP-binding cassette subfamily B member 4; EphA2: erythropoietin-producing hepatocellular A2; ephrin-A1: recombinant human Ephrin A receptor 1; JAK: janus kinase; STAT: signal transducer and activator of transcription; NF-κB: nuclear factor-κB; cGAS: cyclic guanosine monophosphate-adenosine monophosphate synthase; STING: stimulator of interferon genes; mTOR: mammalian target of rapamycin; MAPKAPK2: mitogen-activated protein kinase-activated protein kinase 2; DDR: DNA damage response; BCL-2: B-cell lymphoma-2; PI3K: phosphatidylinositide 3-kinases; CAR-T: chimeric antigen receptor-T; GPNMB: glycoprotein nonmetastatic melanoma protein B; uPAR: urokinase-type plasminogen activator receptor; NKG2DLS: Natural killer group 2 member D ligands; T/P: trametinib and palbociclib.
Acknowledgements
Funding
This study was jointly supported by the National Natural Science Foundation of China (U21A20374), Science and Technology Commission of Shanghai Municipality (NO. YDZX20243100002003), National Natural Science Foundation of China (92374102), Shanghai Municipal Science and Technology Major Project (21JC1401500), and Natural Science Foundation of Shanghai (23ZR1479300).
Author contributions
HZS, MMX and YYL collected the related studies and drafted the manuscript. XYL, JTN, CL, JH, QCM, WYW, WW and JX participated in the design of the review. XJY and SS initiated the study and revised the manuscript. All authors have read and agreed on the published version of the manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. DePinho RA. The age of cancer. Nature. 2000;408:248-54
2. López-Otín C, Pietrocola F, Roiz-Valle D, Galluzzi L, Kroemer G. Meta-hallmarks of aging and cancer. Cell Metab. 2023;35:12-35
3. Hayflick L. THE LIMITED IN VITRO LIFETIME OF HUMAN DIPLOID CELL STRAINS. Exp Cell Res. 1965;37:614-36
4. Akbar AN, Henson SM. Are senescence and exhaustion intertwined or unrelated processes that compromise immunity? Nat Rev Immunol. 2011;11:289-95
5. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646-74
6. Hanahan D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022;12:31-46
7. Borrelli C, Ricci B, Vulpis E, Fionda C, Ricciardi MR, Petrucci MT. et al. Drug-Induced Senescent Multiple Myeloma Cells Elicit NK Cell Proliferation by Direct or Exosome-Mediated IL15 Trans-Presentation. Cancer Immunol Res. 2018;6:860-9
8. Faget DV, Ren QH, Stewart SA. Unmasking senescence: context-dependent effects of SASP in cancer. Nat Rev Cancer. 2019;19:439-53
9. Marin I, Boix O, Garcia-Garijo A, Sirois I, Caballe A, Zarzuela E. et al. Cellular Senescence Is Immunogenic and Promotes Antitumor Immunity. Cancer Discov. 2023;13:410-31
10. Abu-Humaidan AH, Ismail MA, Ahmad FM, Al Shboul S, Barham R, Tadros JS. et al. Therapy-induced senescent cancer cells exhibit complement activation and increased complement regulatory protein expression. Immunol Cell Biol. 2024;102:240-55
11. Chibaya L, Murphy KC, DeMarco KD, Gopalan S, Liu H, Parikh CN. et al. EZH2 inhibition remodels the inflammatory senescence-associated secretory phenotype to potentiate pancreatic cancer immune surveillance. Nat Cancer. 2023;4:872-92
12. Hwang HJ, Lee YR, Kang D, Lee HC, Seo HR, Ryu JK. et al. Endothelial cells under therapy-induced senescence secrete CXCL11, which increases aggressiveness of breast cancer cells. Cancer Lett. 2020;490:100-10
13. Ma L, He X, Fu Y, Ge S, Yang Z. Senescent endothelial cells promote liver metastasis of uveal melanoma in single-cell resolution. J Transl Med. 2024;22:605
14. Dai D, Pei Y, Zhu B, Wang D, Pei S, Huang H. et al. Chemoradiotherapy-induced ACKR2(+) tumor cells drive CD8(+) T cell senescence and cervical cancer recurrence. Cell Rep Med. 2024: 101550.
15. Guan X, LaPak KM, Hennessey RC, Yu CY, Shakya R, Zhang J. et al. Stromal Senescence By Prolonged CDK4/6 Inhibition Potentiates Tumor Growth. Mol Cancer Res. 2017;15:237-49
16. Saleh T, Bloukh S, Hasan M, Al Shboul S. Therapy-induced senescence as a component of tumor biology: Evidence from clinical cancer. Biochim Biophys Acta Rev Cancer. 2023;1878:188994
17. Ou B, Liu Y, Gao Z, Xu J, Yan Y, Li Y. et al. Senescent neutrophils-derived exosomal piRNA-17560 promotes chemoresistance and EMT of breast cancer via FTO-mediated m6A demethylation. Cell Death Dis. 2022;13:905
18. Ferrara R, Naigeon M, Auclin E, Duchemann B, Cassard L, Jouniaux JM. et al. Circulating T-cell Immunosenescence in Patients with Advanced Non-small Cell Lung Cancer Treated with Single-agent PD-1/PD-L1 Inhibitors or Platinum-based Chemotherapy. Clin Cancer Res. 2021;27:492-503
19. Maggiorani D, Le O, Lisi V, Landais S, Moquin-Beaudry G, Lavallée VP. et al. Senescence drives immunotherapy resistance by inducing an immunosuppressive tumor microenvironment. Nat Commun. 2024;15:2435
20. Kang TW, Yevsa T, Woller N, Hoenicke L, Wuestefeld T, Dauch D. et al. Senescence surveillance of pre-malignant hepatocytes limits liver cancer development. Nature. 2011;479:547-51
21. Yin K, Patten D, Gough S, de Barros Gonçalves S, Chan A, Olan I. et al. Senescence-induced endothelial phenotypes underpin immune-mediated senescence surveillance. Genes Dev. 2022;36:533-49
22. Lujambio A, Akkari L, Simon J, Grace D, Tschaharganeh DF, Bolden JE. et al. Non-cell-autonomous tumor suppression by p53. Cell. 2013;153:449-60
23. Haston S, Gonzalez-Gualda E, Morsli S, Ge J, Reen V, Calderwood A. et al. Clearance of senescent macrophages ameliorates tumorigenesis in KRAS-driven lung cancer. Cancer Cell. 2023;41:1242-60.e6
24. Liu D, Hornsby PJ. Senescent human fibroblasts increase the early growth of xenograft tumors via matrix metalloproteinase secretion. Cancer Res. 2007;67:3117-26
25. Volonte D, Zou H, Bartholomew JN, Liu Z, Morel PA, Galbiati F. Oxidative stress-induced inhibition of Sirt1 by caveolin-1 promotes p53-dependent premature senescence and stimulates the secretion of interleukin 6 (IL-6). J Biol Chem. 2015;290:4202-14
26. Wang J, Tao Q, Pan Y, Wanyan Z, Zhu F, Xu X. et al. Stress-induced premature senescence activated by the SENEX gene mediates apoptosis resistance of diffuse large B-cell lymphoma via promoting immunosuppressive cells and cytokines. Immun Inflamm Dis. 2020;8:672-83
27. Coppé JP, Kauser K, Campisi J, Beauséjour CM. Secretion of vascular endothelial growth factor by primary human fibroblasts at senescence. J Biol Chem. 2006;281:29568-74
28. Yang F, Tuxhorn JA, Ressler SJ, McAlhany SJ, Dang TD, Rowley DR. Stromal expression of connective tissue growth factor promotes angiogenesis and prostate cancer tumorigenesis. Cancer Res. 2005;65:8887-95
29. Krtolica A, Parrinello S, Lockett S, Desprez PY, Campisi J. Senescent fibroblasts promote epithelial cell growth and tumorigenesis: a link between cancer and aging. Proc Natl Acad Sci U S A. 2001;98:12072-7
30. Appay V, Nixon DF, Donahoe SM, Gillespie GM, Dong T, King A. et al. HIV-specific CD8(+) T cells produce antiviral cytokines but are impaired in cytolytic function. J Exp Med. 2000;192:63-75
31. Yang OO, Lin H, Dagarag M, Ng HL, Effros RB, Uittenbogaart CH. Decreased perforin and granzyme B expression in senescent HIV-1-specific cytotoxic T lymphocytes. Virology. 2005;332:16-9
32. Rodriguez-Brenes IA, Wodarz D, Komarova NL. Quantifying replicative senescence as a tumor suppressor pathway and a target for cancer therapy. Sci Rep. 2015;5:17660
33. Nyugen J, Agrawal S, Gollapudi S, Gupta S. Impaired functions of peripheral blood monocyte subpopulations in aged humans. J Clin Immunol. 2010;30:806-13
34. Wang Y, Wehling-Henricks M, Samengo G, Tidball JG. Increases of M2a macrophages and fibrosis in aging muscle are influenced by bone marrow aging and negatively regulated by muscle-derived nitric oxide. Aging Cell. 2015;14:678-88
35. Sanchez-Correa B, Morgado S, Gayoso I, Bergua JM, Casado JG, Arcos MJ. et al. Human NK cells in acute myeloid leukaemia patients: analysis of NK cell-activating receptors and their ligands. Cancer Immunol Immunother. 2011;60:1195-205
36. Yang C, Wang Z, Li L, Zhang Z, Jin X, Wu P. et al. Aged neutrophils form mitochondria-dependent vital NETs to promote breast cancer lung metastasis. J Immunother Cancer. 2021 9
37. Agrawal A, Agrawal S, Cao JN, Su H, Osann K, Gupta S. Altered innate immune functioning of dendritic cells in elderly humans: a role of phosphoinositide 3-kinase-signaling pathway. J Immunol. 2007;178:6912-22
38. Grolleau-Julius A, Harning EK, Abernathy LM, Yung RL. Impaired dendritic cell function in aging leads to defective antitumor immunity. Cancer Res. 2008;68:6341-9
39. Pereira LF, de Souza AP, Borges TJ, Bonorino C. Impaired in vivo CD4+ T cell expansion and differentiation in aged mice is not solely due to T cell defects: decreased stimulation by aged dendritic cells. Mech Ageing Dev. 2011;132:187-94
40. Tsukamoto H, Komohara Y, Tomita Y, Miura Y, Motoshima T, Imamura K. et al. Aging-associated and CD4 T-cell-dependent ectopic CXCL13 activation predisposes to anti-PD-1 therapy-induced adverse events. Proc Natl Acad Sci U S A. 2022;119:e2205378119
41. Birch J, Gil J. Senescence and the SASP: many therapeutic avenues. Genes Dev. 2020;34:1565-76
42. Hanna A, Balko JM. No rest for the wicked: Tumor cell senescence reshapes the immune microenvironment. Cancer Cell. 2023;41:831-3
43. Prasanna PG, Citrin DE, Hildesheim J, Ahmed MM, Venkatachalam S, Riscuta G. et al. Therapy-Induced Senescence: Opportunities to Improve Anticancer Therapy. J Natl Cancer Inst. 2021;113:1285-98
44. Schmitt CA, Wang B, Demaria M. Senescence and cancer - role and therapeutic opportunities. Nat Rev Clin Oncol. 2022;19:619-36
45. Wang L, Lankhorst L, Bernards R. Exploiting senescence for the treatment of cancer. Nat Rev Cancer. 2022;22:340-55
46. Bloom SI, Islam MT, Lesniewski LA, Donato AJ. Mechanisms and consequences of endothelial cell senescence. Nat Rev Cardiol. 2023;20:38-51
47. Gabai Y, Assouline B, Ben-Porath I. Senescent stromal cells: roles in the tumor microenvironment. Trends Cancer. 2023;9:28-41
48. Lian JY, Yue Y, Yu WN, Zhang Y. Immunosenescence: a key player in cancer development. J Hematol Oncol. 2020 13
49. Liu Z, Liang Q, Ren Y, Guo C, Ge X, Wang L. et al. Immunosenescence: molecular mechanisms and diseases. Signal Transduct Target Ther. 2023;8:200
50. Solana R, Tarazona R, Gayoso I, Lesur O, Dupuis G, Fulop T. Innate immunosenescence: Effect of aging on cells and receptors of the innate immune system in humans. Seminars in Immunology. 2012;24:331-41
51. López-Otín C, Blasco MA, Partridge L, Serrano M, Kroemer G. Hallmarks of aging: An expanding universe. Cell. 2023;186:243-78
52. Gorgoulis V, Adams PD, Alimonti A, Bennett DC, Bischof O, Bishop C. et al. Cellular Senescence: Defining a Path Forward. Cell. 2019;179:813-27
53. Campisi J, di Fagagna FD. Cellular senescence: when bad things happen to good cells. Nat Rev Mol Cell Bio. 2007;8:729-40
54. Coppé JP, Desprez PY, Krtolica A, Campisi J. The Senescence-Associated Secretory Phenotype: The Dark Side of Tumor Suppression. Annu Rev Pathol-Mech. 2010;5:99-118
55. Kim Y, Jang Y, Kim MS, Kang C. Metabolic remodeling in cancer and senescence and its therapeutic implications. Trends Endocrinol Metab. 2024;35:732-44
56. Zhang Y, Tang J, Jiang C, Yi H, Guang S, Yin G. et al. Metabolic reprogramming in cancer and senescence. MedComm (2020). 2025;6:e70055
57. Favaro E, Bensaad K, Chong MG, Tennant DA, Ferguson DJ, Snell C. et al. Glucose utilization via glycogen phosphorylase sustains proliferation and prevents premature senescence in cancer cells. Cell Metab. 2012;16:751-64
58. Li F, Liu P, Mi W, Li L, Anderson NM, Lesner NP. et al. Blocking methionine catabolism induces senescence and confers vulnerability to GSK3 inhibition in liver cancer. Nat Cancer. 2024;5:131-46
59. Wang Z, Gao J, Xu C. Targeting metabolism to influence cellular senescence a promising anti-cancer therapeutic strategy. Biomed Pharmacother. 2024;177:116962
60. de Magalhães JP, Passos JF. Stress, cell senescence and organismal ageing. Mech Ageing Dev. 2018;170:2-9
61. Hornsby PJ. Senescence as an anticancer mechanism. J Clin Oncol. 2007;25:1852-7
62. Mikuła-Pietrasik J, Niklas A, Uruski P, Tykarski A, Książek K. Mechanisms and significance of therapy-induced and spontaneous senescence of cancer cells. Cell Mol Life Sci. 2020;77:213-29
63. Elshazly AM, Shahin U, Al Shboul S, Gewirtz DA, Saleh T. A Conversation with ChatGPT on Contentious Issues in Senescence and Cancer Research. Mol Pharmacol. 2024;105:313-27
64. Liu XL, Ding J, Meng LH. Oncogene-induced senescence: a double edged sword in cancer. Acta Pharmacol Sin. 2018;39:1553-8
65. Almanzar G, Schwaiger S, Jenewein B, Keller M, Herndler-Brandstetter D, Würzner R. et al. Long-term cytomegalovirus infection leads to significant changes in the composition of the CD8 T-cell repertoire, which may be the basis for an imbalance in the cytokine production profile in elderly persons. J Virol. 2005;79:3675-83
66. Kuilman T, Michaloglou C, Vredeveld LCW, Douma S, van Doom R, Desmet CJ. et al. Oncogene-induced senescence relayed by an interleukin-dependent inflammatory network. Cell. 2008;133:1019-31
67. Nikolich-Zugich J. Ageing and life-long maintenance of T-cell subsets in the face of latent persistent infections. Nat Rev Immunol. 2008;8:512-22
68. Suen H, Brown R, Yang S, Weatherburn C, Ho PJ, Woodland N. et al. Multiple myeloma causes clonal T-cell immunosenescence: identification of potential novel targets for promoting tumour immunity and implications for checkpoint blockade. Leukemia. 2016;30:1716-24
69. Lynch HE, Goldberg GL, Chidgey A, Van den Brink MR, Boyd R, Sempowski GD. Thymic involution and immune reconstitution. Trends Immunol. 2009;30:366-73
70. Rezzani R, Nardo L, Favero G, Peroni M, Rodella LF. Thymus and aging: morphological, radiological, and functional overview. Age (Dordr). 2014;36:313-51
71. Hohensinner PJ, Goronzy JJ, Weyand CM. Telomere dysfunction, autoimmunity and aging. Aging Dis. 2011;2:524-37
72. Ferrando-Martínez S, Romero-Sánchez MC, Solana R, Delgado J, de la Rosa R, Muñoz-Fernández MA. et al. Thymic function failure and C-reactive protein levels are independent predictors of all-cause mortality in healthy elderly humans. Age. 2013;35:251-9
73. Franceschi C, Garagnani P, Parini P, Giuliani C, Santoro A. Inflammaging: a new immune-metabolic viewpoint for age-related diseases. Nat Rev Endocrinol. 2018;14:576-90
74. Franceschi C, Bonafè M, Valensin S, Olivieri F, De Luca M, Ottaviani E. et al. Inflamm-aging. An evolutionary perspective on immunosenescence. Ann N Y Acad Sci. 2000;908:244-54
75. Fulop T, Larbi A, Dupuis G, Le Page A, Frost EH, Cohen AA. et al. Immunosenescence and Inflamm-Aging As Two Sides of the Same Coin: Friends or Foes? Front Immunol. 2017;8:1960
76. Santoro A, Bientinesi E, Monti D. Immunosenescence and inflammaging in the aging process: age-related diseases or longevity? Ageing Res Rev. 2021;71:101422
77. Yousefzadeh MJ, Flores RR, Zhu Y, Schmiechen ZC, Brooks RW, Trussoni CE. et al. An aged immune system drives senescence and ageing of solid organs. Nature. 2021;594:100-5
78. Montégut L, López-Otín C, Kroemer G. Aging and cancer. Mol Cancer. 2024;23:106
79. de Magalhães JP. Cellular senescence in normal physiology. Science. 2024;384:1300-1
80. Burgess DJ. Senescence: Tumorigenesis under surveillance. Nat Rev Cancer. 2011;12:6
81. Cai X, Yin G, Chen S, Tacke F, Guillot A, Liu H. CDK4/6 inhibition enhances T-cell immunotherapy on hepatocellular carcinoma cells by rejuvenating immunogenicity. Cancer Cell Int. 2024;24:215
82. Ohtani N, Takahashi A, Mann DJ, Hara E. Cellular senescence: a double-edged sword in the fight against cancer. Exp Dermatol. 2012;21(Suppl 1):1-4
83. Ewald JA, Desotelle JA, Wilding G, Jarrard DF. Therapy-Induced Senescence in Cancer. Jnci-J Natl Cancer I. 2010;102:1536-46
84. Onyema OO, Decoster L, Njemini R, Forti LN, Bautmans I, De Waele M. et al. Chemotherapy-induced Changes and Immunosenescence of CD8 T-Cells in Patients with Breast Cancer. Anticancer Res. 2015;35:1481-9
85. Tabasso AFS, Jones DJL, Jones GDD, Macip S. Radiotherapy-Induced Senescence and its Effects on Responses to Treatment. Clin Oncol-Uk. 2019;31:283-9
86. Takasugi M, Yoshida Y, Ohtani N. Cellular senescence and the tumour microenvironment. Mol Oncol. 2022;16:3333-51
87. Amor C, Feucht J, Leibold J, Ho YJ, Zhu C, Alonso-Curbelo D. et al. Senolytic CAR T cells reverse senescence-associated pathologies. Nature. 2020;583:127-32
88. Amor C, Feucht J, Leibold J, Ho YJ, Zhu C, Alonso-Curbelo D. et al. Author Correction: Senolytic CAR T cells reverse senescence-associated pathologies. Nature. 2024;627:E9
89. Ruscetti M, Morris JPt, Mezzadra R, Russell J, Leibold J, Romesser PB. et al. Senescence-Induced Vascular Remodeling Creates Therapeutic Vulnerabilities in Pancreas Cancer. Cell. 2020;181:424-41.e21
90. Ruscetti M, Morris JPt, Mezzadra R, Russell J, Leibold J, Romesser PB. et al. Senescence-Induced Vascular Remodeling Creates Therapeutic Vulnerabilities in Pancreas Cancer. Cell. 2021;184:4838-9
91. Wang B, Kohli J, Demaria M. Senescent Cells in Cancer Therapy: Friends or Foes? Trends Cancer. 2020;6:838-57
92. Chien Y, Scuoppo C, Wang X, Fang X, Balgley B, Bolden JE. et al. Control of the senescence-associated secretory phenotype by NF-κB promotes senescence and enhances chemosensitivity. Genes Dev. 2011;25:2125-36
93. Liu Y, Hawkins OE, Su Y, Vilgelm AE, Sobolik T, Thu YM. et al. Targeting aurora kinases limits tumour growth through DNA damage-mediated senescence and blockade of NF-κB impairs this drug-induced senescence. EMBO Mol Med. 2013;5:149-66
94. Evangelou K, Belogiannis K, Papaspyropoulos A, Petty R, Gorgoulis VG. Escape from senescence: molecular basis and therapeutic ramifications. J Pathol. 2023;260:649-65
95. Domen A, Deben C, De Pauw I, Hermans C, Lambrechts H, Verswyvel J. et al. Prognostic implications of cellular senescence in resected non-small cell lung cancer. Transl Lung Cancer Res. 2022;11:1526-39
96. Wei Q, Chen R, He X, Qu Y, Yan C, Liu X. et al. Multi-omics and single cell characterization of cancer immunosenescence landscape. Sci Data. 2024;11:739
97. Dou X, Fu Q, Long Q, Liu S, Zou Y, Fu D. et al. PDK4-dependent hypercatabolism and lactate production of senescent cells promotes cancer malignancy. Nat Metab. 2023;5:1887-910
98. Fu D, Zhang B, Fan W, Zeng F, Feng J, Wang X. Fatty acid metabolism prognostic signature predicts tumor immune microenvironment and immunotherapy, and identifies tumorigenic role of MOGAT2 in lung adenocarcinoma. Front Immunol. 2024;15:1456719
99. Li Q, Liu H. Identification of Prognostic Genes Related to Cell Senescence and Lipid Metabolism in Glioblastoma Based on Transcriptome and Single-Cell RNA-Seq Data. Int J Mol Sci. 2025 26
100. Pacifico F, Mellone S, D'Incalci M, Stornaiuolo M, Leonardi A, Crescenzi E. Trabectedin suppresses escape from therapy-induced senescence in tumor cells by interfering with glutamine metabolism. Biochem Pharmacol. 2022;202:115159
101. Chen L, Zhang W, Chen D, Yang Q, Sun S, Dai Z. et al. RBM4 dictates ESCC cell fate switch from cellular senescence to glutamine-addiction survival through inhibiting LKB1-AMPK-axis. Signal Transduct Target Ther. 2023;8:159
102. Lai P, Liu L, Bancaro N, Troiani M, Calì B, Li Y. et al. Mitochondrial DNA released by senescent tumor cells enhances PMN-MDSC-driven immunosuppression through the cGAS-STING pathway. Immunity. 2025;58:811-25.e7
103. Cao LL, Kagan JC. Targeting innate immune pathways for cancer immunotherapy. Immunity. 2023;56:2206-17
104. Galassi C, Chan TA, Vitale I, Galluzzi L. The hallmarks of cancer immune evasion. Cancer Cell. 2024;42:1825-63
105. Liew PX, Kubes P. The Neutrophil's Role During Health and Disease. Physiol Rev. 2019;99:1223-48
106. Tortorella C, Simone O, Piazzolla G, Stella I, Cappiello V, Antonaci S. Role of phosphoinositide 3-kinase and extracellular signal-regulated kinase pathways in granulocyte macrophage-colony-stimulating factor failure to delay fas-induced neutrophil apoptosis in elderly humans. J Gerontol A Biol Sci Med Sci. 2006;61:1111-8
107. Bancaro N, Calì B, Troiani M, Elia AR, Arzola RA, Attanasio G. et al. Apolipoprotein E induces pathogenic senescent-like myeloid cells in prostate cancer. Cancer Cell. 2023;41:602-19.e11
108. Chatta GS, Andrews RG, Rodger E, Schrag M, Hammond WP, Dale DC. Hematopoietic progenitors and aging: alterations in granulocytic precursors and responsiveness to recombinant human G-CSF, GM-CSF, and IL-3. J Gerontol. 1993;48:M207-12
109. Chatta GS, Price TH, Stratton JR, Dale DC. Aging and marrow neutrophil reserves. J Am Geriatr Soc. 1994;42:77-81
110. Simell B, Vuorela A, Ekström N, Palmu A, Reunanen A, Meri S. et al. Aging reduces the functionality of anti-pneumococcal antibodies and the killing of Streptococcus pneumoniae by neutrophil phagocytosis. Vaccine. 2011;29:1929-34
111. Simmons SR, Tchalla EYI, Bhalla M, Bou Ghanem EN. The Age-Driven Decline in Neutrophil Function Contributes to the Reduced Efficacy of the Pneumococcal Conjugate Vaccine in Old Hosts. Front Cell Infect Microbiol. 2022;12:849224
112. Liu Y, Cao X. Characteristics and Significance of the Pre-metastatic Niche. Cancer Cell. 2016;30:668-81
113. Peng Z, Liu C, Victor AR, Cao DY, Veiras LC, Bernstein EA. et al. Tumors exploit CXCR4(hi)CD62L(lo) aged neutrophils to facilitate metastatic spread. Oncoimmunology. 2021;10:1870811
114. Que H, Fu Q, Lan T, Tian X, Wei X. Tumor-associated neutrophils and neutrophil-targeted cancer therapies. Biochim Biophys Acta Rev Cancer. 2022;1877:188762
115. Behrens LM, van Egmond M, van den Berg TK. Neutrophils as immune effector cells in antibody therapy in cancer. Immunol Rev. 2023;314:280-301
116. Fülöp T Jr, Fóris G, Wórum I, Leövey A. Age-dependent alterations of Fc gamma receptor-mediated effector functions of human polymorphonuclear leucocytes. Clin Exp Immunol. 1985;61:425-32
117. Guettinger Y, Barbin K, Peipp M, Bruenke J, Dechant M, Horner H. et al. A recombinant bispecific single-chain fragment variable specific for HLA class II and Fc alpha RI (CD89) recruits polymorphonuclear neutrophils for efficient lysis of malignant B lymphoid cells. J Immunol. 2010;184:1210-7
118. Stockmeyer B, Dechant M, van Egmond M, Tutt AL, Sundarapandiyan K, Graziano RF. et al. Triggering Fc alpha-receptor I (CD89) recruits neutrophils as effector cells for CD20-directed antibody therapy. J Immunol. 2000;165:5954-61
119. Eruslanov EB, Bhojnagarwala PS, Quatromoni JG, Stephen TL, Ranganathan A, Deshpande C. et al. Tumor-associated neutrophils stimulate T cell responses in early-stage human lung cancer. J Clin Invest. 2014;124:5466-80
120. Singhal S, Bhojnagarwala PS, O'Brien S, Moon EK, Garfall AL, Rao AS. et al. Origin and Role of a Subset of Tumor-Associated Neutrophils with Antigen-Presenting Cell Features in Early-Stage Human Lung Cancer. Cancer Cell. 2016;30:120-35
121. Wu Y, Ma J, Yang X, Nan F, Zhang T, Ji S. et al. Neutrophil profiling illuminates anti-tumor antigen-presenting potency. Cell. 2024;187:1422-39.e24
122. Gershkovitz M, Caspi Y, Fainsod-Levi T, Katz B, Michaeli J, Khawaled S. et al. TRPM2 Mediates Neutrophil Killing of Disseminated Tumor Cells. Cancer Res. 2018;78:2680-90
123. Erdman SE, Rao VP, Poutahidis T, Rogers AB, Taylor CL, Jackson EA. et al. Nitric oxide and TNF-alpha trigger colonic inflammation and carcinogenesis in Helicobacter hepaticus-infected, Rag2-deficient mice. Proc Natl Acad Sci U S A. 2009;106:1027-32
124. Schmielau J, Finn OJ. Activated granulocytes and granulocyte-derived hydrogen peroxide are the underlying mechanism of suppression of t-cell function in advanced cancer patients. Cancer Res. 2001;61:4756-60
125. Ogawa K, Suzuki K, Okutsu M, Yamazaki K, Shinkai S. The association of elevated reactive oxygen species levels from neutrophils with low-grade inflammation in the elderly. Immun Ageing. 2008;5:13
126. Adrover JM, McDowell SAC, He XY, Quail DF, Egeblad M. NETworking with cancer: The bidirectional interplay between cancer and neutrophil extracellular traps. Cancer Cell. 2023;41:505-26
127. Hazeldine J, Harris P, Chapple IL, Grant M, Greenwood H, Livesey A. et al. Impaired neutrophil extracellular trap formation: a novel defect in the innate immune system of aged individuals. Aging Cell. 2014;13:690-8
128. Duan R, Jiang L, Wang T, Li Z, Yu X, Gao Y. et al. Aging-induced immune microenvironment remodeling fosters melanoma in male mice via γδ17-Neutrophil-CD8 axis. Nat Commun. 2024;15:10860
129. Kalafati L, Kourtzelis I, Schulte-Schrepping J, Li X, Hatzioannou A, Grinenko T. et al. Innate Immune Training of Granulopoiesis Promotes Anti-tumor Activity. Cell. 2020;183:771-85.e12
130. Xiang X, Wang J, Lu D, Xu X. Targeting tumor-associated macrophages to synergize tumor immunotherapy. Signal Transduct Target Ther. 2021;6:75
131. Narasimhan PB, Marcovecchio P, Hamers AAJ, Hedrick CC. Nonclassical Monocytes in Health and Disease. Annu Rev Immunol. 2019;37:439-56
132. Gu M, Liu Y, Zheng W, Jing Z, Li X, Guo W. et al. Combined targeting of senescent cells and senescent macrophages: A new idea for integrated treatment of lung cancer. Semin Cancer Biol. 2024;106-107:43-57
133. Wada H, Otsuka R, Germeraad WTV, Murata T, Kondo T, Seino KI. Tumor cell-induced macrophage senescence plays a pivotal role in tumor initiation followed by stable growth in immunocompetent condition. J Immunother Cancer. 2023 11
134. Cassetta L, Pollard JW. A timeline of tumour-associated macrophage biology. Nat Rev Cancer. 2023;23:238-57
135. Angarola BL, Sharma S, Katiyar N, Kang HG, Nehar-Belaid D, Park S. et al. Comprehensive single-cell aging atlas of healthy mammary tissues reveals shared epigenomic and transcriptomic signatures of aging and cancer. Nat Aging. 2025;5:122-43
136. van Duin D, Allore HG, Mohanty S, Ginter S, Newman FK, Belshe RB. et al. Prevaccine determination of the expression of costimulatory B7 molecules in activated monocytes predicts influenza vaccine responses in young and older adults. J Infect Dis. 2007;195:1590-7
137. Villanueva JL, Solana R, Alonso MC, Peña J. Changes in the expression of HLA-class II antigens on peripheral blood monocytes from aged humans. Dis Markers. 1990;8:85-91
138. Prieto LI, Sturmlechner I, Graves SI, Zhang C, Goplen NP, Yi ES. et al. Senescent alveolar macrophages promote early-stage lung tumorigenesis. Cancer Cell. 2023;41:1261-75.e6
139. Chiu BC, Stolberg VR, Chensue SW. Mononuclear phagocyte-derived IL-10 suppresses the innate IL-12/IFN-gamma axis in lung-challenged aged mice. J Immunol. 2008;181:3156-66
140. Kelly J, Ali Khan A, Yin J, Ferguson TA, Apte RS. Senescence regulates macrophage activation and angiogenic fate at sites of tissue injury in mice. J Clin Invest. 2007;117:3421-6
141. Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol. 2009;9:162-74
142. Jackaman C, Tomay F, Duong L, Abdol Razak NB, Pixley FJ, Metharom P. et al. Aging and cancer: The role of macrophages and neutrophils. Ageing Res Rev. 2017;36:105-16
143. Ruhland MK, Loza AJ, Capietto AH, Luo X, Knolhoff BL, Flanagan KC. et al. Stromal senescence establishes an immunosuppressive microenvironment that drives tumorigenesis. Nat Commun. 2016;7:11762
144. Enioutina EY, Bareyan D, Daynes RA. A role for immature myeloid cells in immune senescence. J Immunol. 2011;186:697-707
145. Flores RR, Clauson CL, Cho J, Lee BC, McGowan SJ, Baker DJ. et al. Expansion of myeloid-derived suppressor cells with aging in the bone marrow of mice through a NF-κB-dependent mechanism. Aging Cell. 2017;16:480-7
146. Chandra D, Jahangir A, Quispe-Tintaya W, Einstein MH, Gravekamp C. Myeloid-derived suppressor cells have a central role in attenuated Listeria monocytogenes-based immunotherapy against metastatic breast cancer in young and old mice. Br J Cancer. 2013;108:2281-90
147. Okuma A, Hanyu A, Watanabe S, Hara E. p16(Ink4a) and p21(Cip1/Waf1) promote tumour growth by enhancing myeloid-derived suppressor cells chemotaxis. Nat Commun. 2017;8:2050
148. Zhou Y, Cheng L, Liu L, Li X. NK cells are never alone: crosstalk and communication in tumour microenvironments. Mol Cancer. 2023;22:34
149. Bruno A, Ferlazzo G, Albini A, Noonan DM. A think tank of TINK/TANKs: tumor-infiltrating/tumor-associated natural killer cells in tumor progression and angiogenesis. J Natl Cancer Inst. 2014;106:dju200
150. Brighton PJ, Maruyama Y, Fishwick K, Vrljicak P, Tewary S, Fujihara R. et al. Clearance of senescent decidual cells by uterine natural killer cells in cycling human endometrium. Elife. 2017 6
151. Brauning A, Rae M, Zhu G, Fulton E, Admasu TD, Stolzing A. et al. Aging of the Immune System: Focus on Natural Killer Cells Phenotype and Functions. Cells. 2022 11
152. Borrego F, Alonso MC, Galiani MD, Carracedo J, Ramirez R, Ostos B. et al. NK phenotypic markers and IL2 response in NK cells from elderly people. Exp Gerontol. 1999;34:253-65
153. Hazeldine J, Hampson P, Lord JM. Reduced release and binding of perforin at the immunological synapse underlies the age-related decline in natural killer cell cytotoxicity. Aging Cell. 2012;11:751-9
154. Le Garff-Tavernier M, Béziat V, Decocq J, Siguret V, Gandjbakhch F, Pautas E. et al. Human NK cells display major phenotypic and functional changes over the life span. Aging Cell. 2010;9:527-35
155. Krishnaraj R, Bhooma T. Cytokine sensitivity of human NK cells during immunosenescence. 2. IL2-induced interferon gamma secretion. Immunol Lett. 1996;50:59-63
156. Mariani E, Pulsatelli L, Meneghetti A, Dolzani P, Mazzetti I, Neri S. et al. Different IL-8 production by T and NK lymphocytes in elderly subjects. Mech Ageing Dev. 2001;122:1383-95
157. Wculek SK, Cueto FJ, Mujal AM, Melero I, Krummel MF, Sancho D. Dendritic cells in cancer immunology and immunotherapy. Nat Rev Immunol. 2020;20:7-24
158. Sridharan A, Esposo M, Kaushal K, Tay J, Osann K, Agrawal S. et al. Age-associated impaired plasmacytoid dendritic cell functions lead to decreased CD4 and CD8 T cell immunity. Age (Dordr). 2011;33:363-76
159. Guo Z, Tilburgs T, Wong B, Strominger JL. Dysfunction of dendritic cells in aged C57BL/6 mice leads to failure of natural killer cell activation and of tumor eradication. Proc Natl Acad Sci U S A. 2014;111:14199-204
160. Speiser DE, Chijioke O, Schaeuble K, Münz C. CD4(+) T cells in cancer. Nat Cancer. 2023;4:317-29
161. Han S, Georgiev P, Ringel AE, Sharpe AH, Haigis MC. Age-associated remodeling of T cell immunity and metabolism. Cell Metab. 2023;35:36-55
162. Voehringer D, Blaser C, Brawand P, Raulet DH, Hanke T, Pircher H. Viral infections induce abundant numbers of senescent CD8 T cells. J Immunol. 2001;167:4838-43
163. Saavedra D, García B, Lorenzo-Luaces P, González A, Popa X, Fuentes KP. et al. Biomarkers related to immunosenescence: relationships with therapy and survival in lung cancer patients. Cancer Immunol Immunother. 2016;65:37-45
164. Ma M, Yang Y, Chen Z, Li X, Yang Z, Wang K. et al. T-cell senescence induced by peripheral phospholipids. Cell Biol Toxicol. 2023;39:2937-52
165. Gao A, Liu X, Lin W, Wang J, Wang S, Si F. et al. Tumor-derived ILT4 induces T cell senescence and suppresses tumor immunity. J Immunother Cancer. 2021 9
166. Ye J, Ma C, Hsueh EC, Dou J, Mo W, Liu S. et al. TLR8 signaling enhances tumor immunity by preventing tumor-induced T-cell senescence. EMBO Mol Med. 2014;6:1294-311
167. Ye J, Peng G. Controlling T cell senescence in the tumor microenvironment for tumor immunotherapy. Oncoimmunology. 2015;4:e994398
168. Ma F, Liu X, Zhang Y, Tao Y, Zhao L, Abusalamah H. et al. Tumor extracellular vesicle-derived PD-L1 promotes T cell senescence through lipid metabolism reprogramming. Sci Transl Med. 2025;17:eadm7269
169. Ikeda H, Kawase K, Nishi T, Watanabe T, Takenaga K, Inozume T. et al. Immune evasion through mitochondrial transfer in the tumour microenvironment. Nature. 2025;638:225-36
170. Bandrés E, Merino J, Vázquez B, Inogés S, Moreno C, Subirá ML. et al. The increase of IFN-gamma production through aging correlates with the expanded CD8(+high)CD28(-)CD57(+) subpopulation. Clin Immunol. 2000;96:230-5
171. Brenchley JM, Karandikar NJ, Betts MR, Ambrozak DR, Hill BJ, Crotty LE. et al. Expression of CD57 defines replicative senescence and antigen-induced apoptotic death of CD8 T cells. Blood. 2003;101:2711-20
172. Montes CL, Chapoval AI, Nelson J, Orhue V, Zhang X, Schulze DH. et al. Tumor-induced senescent T cells with suppressor function: A potential form of tumor immune evasion. Cancer Res. 2008;68:870-9
173. Huff WX, Bam M, Shireman JM, Kwon JH, Song L, Newman S. et al. Aging- and Tumor-Mediated Increase in CD8(+)CD28(-) T Cells Might Impose a Strong Barrier to Success of Immunotherapy in Glioblastoma. Immunohorizons. 2021;5:395-409
174. Onyema OO, Decoster L, Njemini R, Forti LN, Bautmans I, De Waele M. et al. Shifts in subsets of CD8+T-cells as evidence of immunosenescence in patients with cancers affecting the lungs: an observational case-control study. Bmc Cancer. 2015 15
175. Tsukishiro T, Donnenberg AD, Whiteside TL. Rapid turnover of the CD8CD28 T-cell subset of effector cells in the circulation of patients with head and neck cancer. Cancer Immunol Immun. 2003;52:599-607
176. Zhang SP, Ke X, Zeng SY, Wu M, Lou JF, Wu L. et al. Analysis of CD8 Treg cells in patients with ovarian cancer: a possible mechanism for immune impairment. Cell Mol Immunol. 2015;12:580-91
177. Crespo J, Sun H, Welling TH, Tian Z, Zou W. T cell anergy, exhaustion, senescence, and stemness in the tumor microenvironment. Curr Opin Immunol. 2013;25:214-21
178. Henson SM, Lanna A, Riddell NE, Franzese O, Macaulay R, Griffiths SJ. et al. p38 signaling inhibits mTORC1-independent autophagy in senescent human CD8⁺ T cells. J Clin Invest. 2014;124:4004-16
179. Knaus HA, Berglund S, Hackl H, Blackford AL, Zeidner JF, Montiel-Esparza R. et al. Signatures of CD8+ T cell dysfunction in AML patients and their reversibility with response to chemotherapy. JCI Insight. 2018 3
180. Shirakawa K, Yan X, Shinmura K, Endo J, Kataoka M, Katsumata Y. et al. Obesity accelerates T cell senescence in murine visceral adipose tissue. J Clin Invest. 2016;126:4626-39
181. Ikeda H, Kawase K, Nishi T, Watanabe T, Takenaga K, Inozume T. et al. Immune evasion through mitochondrial transfer in the tumour microenvironment. Nature. 2025
182. Libri V, Azevedo RI, Jackson SE, Di Mitri D, Lachmann R, Fuhrmann S. et al. Cytomegalovirus infection induces the accumulation of short-lived, multifunctional CD4+CD45RA+CD27+ T cells: the potential involvement of interleukin-7 in this process. Immunology. 2011;132:326-39
183. Callender LA, Carroll EC, Beal RWJ, Chambers ES, Nourshargh S, Akbar AN. et al. Human CD8(+) EMRA T cells display a senescence-associated secretory phenotype regulated by p38 MAPK. Aging Cell. 2018 17
184. Ramello MC, Tosello Boari J, Canale FP, Mena HA, Negrotto S, Gastman B. et al. Tumor-induced senescent T cells promote the secretion of pro-inflammatory cytokines and angiogenic factors by human monocytes/macrophages through a mechanism that involves Tim-3 and CD40L. Cell Death Dis. 2014;5:e1507
185. Liu L, Hao Z, Yang X, Li Y, Wang S, Li L. Metabolic reprogramming in T cell senescence: a novel strategy for cancer immunotherapy. Cell Death Discov. 2025;11:161
186. Møller SH, Hsueh PC, Yu YR, Zhang L, Ho PC. Metabolic programs tailor T cell immunity in viral infection, cancer, and aging. Cell Metab. 2022;34:378-95
187. Liu X, Hartman CL, Li L, Albert CJ, Si F, Gao A. et al. Reprogramming lipid metabolism prevents effector T cell senescence and enhances tumor immunotherapy. Sci Transl Med. 2021 13
188. McLane LM, Abdel-Hakeem MS, Wherry EJ. CD8 T Cell Exhaustion During Chronic Viral Infection and Cancer. Annu Rev Immunol. 2019;37:457-95
189. Tedeschi V, Paldino G, Kunkl M, Paroli M, Sorrentino R, Tuosto L. et al. CD8(+) T Cell Senescence: Lights and Shadows in Viral Infections, Autoimmune Disorders and Cancer. Int J Mol Sci. 2022 23
190. Hui E, Cheung J, Zhu J, Su X, Taylor MJ, Wallweber HA. et al. T cell costimulatory receptor CD28 is a primary target for PD-1-mediated inhibition. Science. 2017;355:1428-33
191. Kamphorst AO, Wieland A, Nasti T, Yang S, Zhang R, Barber DL. et al. Rescue of exhausted CD8 T cells by PD-1-targeted therapies is CD28-dependent. Science. 2017;355:1423-7
192. Hudson WH, Gensheimer J, Hashimoto M, Wieland A, Valanparambil RM, Li P. et al. Proliferating Transitory T Cells with an Effector-like Transcriptional Signature Emerge from PD-1(+) Stem-like CD8(+) T Cells during Chronic Infection. Immunity. 2019;51:1043-58.e4
193. Fornara O, Odeberg J, Wolmer Solberg N, Tammik C, Skarman P, Peredo I. et al. Poor survival in glioblastoma patients is associated with early signs of immunosenescence in the CD4 T-cell compartment after surgery. Oncoimmunology. 2015;4:e1036211
194. Gonnin C, Leemans M, Canoui-Poitrine F, Lebraud M, Corneau A, Roquebert L. et al. CD57(+) EMRA CD8(+) T cells in cancer patients over 70: associations with prior chemotherapy and response to anti-PD-1/PD-L1 therapy. Immun Ageing. 2024;21:89
195. Kim KH, Pyo H, Lee H, Oh D, Noh JM, Ahn YC. et al. Association of T Cell Senescence with Radiation Pneumonitis in Patients with Non-small Cell Lung Cancer. Int J Radiat Oncol Biol Phys. 2023;115:464-75
196. Song G, Wang X, Jia J, Yuan Y, Wan F, Zhou X. et al. Elevated level of peripheral CD8(+)CD28(-) T lymphocytes are an independent predictor of progression-free survival in patients with metastatic breast cancer during the course of chemotherapy. Cancer Immunol Immunother. 2013;62:1123-30
197. Kugel CH 3rd, Douglass SM, Webster MR, Kaur A, Liu Q, Yin X. et al. Age Correlates with Response to Anti-PD1, Reflecting Age-Related Differences in Intratumoral Effector and Regulatory T-Cell Populations. Clin Cancer Res. 2018;24:5347-56
198. Wang TW, Johmura Y, Suzuki N, Omori S, Migita T, Yamaguchi K. et al. Blocking PD-L1-PD-1 improves senescence surveillance and ageing phenotypes. Nature. 2022;611:358-64
199. Li C, Jiang P, Wei S, Xu X, Wang J. Regulatory T cells in tumor microenvironment: new mechanisms, potential therapeutic strategies and future prospects. Mol Cancer. 2020;19:116
200. Liu X, Mo W, Ye J, Li L, Zhang Y, Hsueh EC. et al. Regulatory T cells trigger effector T cell DNA damage and senescence caused by metabolic competition. Nat Commun. 2018;9:249
201. Ye J, Huang X, Hsueh EC, Zhang Q, Ma C, Zhang Y. et al. Human regulatory T cells induce T-lymphocyte senescence. Blood. 2012;120:2021-31
202. Ye J, Ma C, Hsueh EC, Eickhoff CS, Zhang Y, Varvares MA. et al. Tumor-derived γδ regulatory T cells suppress innate and adaptive immunity through the induction of immunosenescence. J Immunol. 2013;190:2403-14
203. Garg SK, Delaney C, Toubai T, Ghosh A, Reddy P, Banerjee R. et al. Aging is associated with increased regulatory T-cell function. Aging Cell. 2014;13:441-8
204. Laumont CM, Nelson BH. B cells in the tumor microenvironment: Multi-faceted organizers, regulators, and effectors of anti-tumor immunity. Cancer Cell. 2023;41:466-89
205. Carmi Y, Spitzer MH, Linde IL, Burt BM, Prestwood TR, Perlman N. et al. Allogeneic IgG combined with dendritic cell stimuli induce antitumour T-cell immunity. Nature. 2015;521:99-104
206. Chen J, Tan Y, Sun F, Hou L, Zhang C, Ge T. et al. Single-cell transcriptome and antigen-immunoglobin analysis reveals the diversity of B cells in non-small cell lung cancer. Genome Biol. 2020;21:152
207. Biswas S, Mandal G, Payne KK, Anadon CM, Gatenbee CD, Chaurio RA. et al. IgA transcytosis and antigen recognition govern ovarian cancer immunity. Nature. 2021;591:464-70
208. Fridman WH, Meylan M, Petitprez F, Sun CM, Italiano A, Sautès-Fridman C. B cells and tertiary lymphoid structures as determinants of tumour immune contexture and clinical outcome. Nat Rev Clin Oncol. 2022;19:441-57
209. Sautès-Fridman C, Petitprez F, Calderaro J, Fridman WH. Tertiary lymphoid structures in the era of cancer immunotherapy. Nat Rev Cancer. 2019;19:307-25
210. Souders CA, Bowers SL, Baudino TA. Cardiac fibroblast: the renaissance cell. Circ Res. 2009;105:1164-76
211. Kawamoto S, Uemura K, Hori N, Takayasu L, Konishi Y, Katoh K. et al. Bacterial induction of B cell senescence promotes age-related changes in the gut microbiota. Nat Cell Biol. 2023;25:865-76
212. Bulati M, Caruso C, Colonna-Romano G. From lymphopoiesis to plasma cells differentiation, the age-related modifications of B cell compartment are influenced by "inflamm-ageing". Ageing Res Rev. 2017;36:125-36
213. Listì F, Candore G, Modica MA, Russo M, Di Lorenzo G, Esposito-Pellitteri M. et al. A study of serum immunoglobulin levels in elderly persons that provides new insights into B cell immunosenescence. Ann N Y Acad Sci. 2006;1089:487-95
214. Henry CJ, Casás-Selves M, Kim J, Zaberezhnyy V, Aghili L, Daniel AE. et al. Aging-associated inflammation promotes selection for adaptive oncogenic events in B cell progenitors. J Clin Invest. 2015;125:4666-80
215. Chen Y, McAndrews KM, Kalluri R. Clinical and therapeutic relevance of cancer-associated fibroblasts. Nat Rev Clin Oncol. 2021;18:792-804
216. Mao X, Xu J, Wang W, Liang C, Hua J, Liu J. et al. Crosstalk between cancer-associated fibroblasts and immune cells in the tumor microenvironment: new findings and future perspectives. Mol Cancer. 2021;20:131
217. Ragunathan K, Upfold NLE, Oksenych V. Interaction between Fibroblasts and Immune Cells Following DNA Damage Induced by Ionizing Radiation. Int J Mol Sci. 2020 21
218. Pazolli E, Alspach E, Milczarek A, Prior J, Piwnica-Worms D, Stewart SA. Chromatin remodeling underlies the senescence-associated secretory phenotype of tumor stromal fibroblasts that supports cancer progression. Cancer Res. 2012;72:2251-61
219. Yoshimoto S, Loo TM, Atarashi K, Kanda H, Sato S, Oyadomari S. et al. Obesity-induced gut microbial metabolite promotes liver cancer through senescence secretome. Nature. 2013;499:97-101
220. Yang G, Rosen DG, Zhang Z, Bast RC Jr, Mills GB, Colacino JA. et al. The chemokine growth-regulated oncogene 1 (Gro-1) links RAS signaling to the senescence of stromal fibroblasts and ovarian tumorigenesis. Proc Natl Acad Sci U S A. 2006;103:16472-7
221. Golomb L, Sagiv A, Pateras IS, Maly A, Krizhanovsky V, Gorgoulis VG. et al. Age-associated inflammation connects RAS-induced senescence to stem cell dysfunction and epidermal malignancy. Cell Death Differ. 2015;22:1764-74
222. Parrinello S, Coppe JP, Krtolica A, Campisi J. Stromal-epithelial interactions in aging and cancer: senescent fibroblasts alter epithelial cell differentiation. J Cell Sci. 2005;118:485-96
223. Pazolli E, Luo X, Brehm S, Carbery K, Chung JJ, Prior JL. et al. Senescent stromal-derived osteopontin promotes preneoplastic cell growth. Cancer Res. 2009;69:1230-9
224. Bavik C, Coleman I, Dean JP, Knudsen B, Plymate S, Nelson PS. The gene expression program of prostate fibroblast senescence modulates neoplastic epithelial cell proliferation through paracrine mechanisms. Cancer Res. 2006;66:794-802
225. Krizhanovsky V, Yon M, Dickins RA, Hearn S, Simon J, Miething C. et al. Senescence of activated stellate cells limits liver fibrosis. Cell. 2008;134:657-67
226. Hassona Y, Cirillo N, Heesom K, Parkinson EK, Prime SS. Senescent cancer-associated fibroblasts secrete active MMP-2 that promotes keratinocyte dis-cohesion and invasion. Br J Cancer. 2014;111:1230-7
227. Wang T, Notta F, Navab R, Joseph J, Ibrahimov E, Xu J. et al. Senescent Carcinoma-Associated Fibroblasts Upregulate IL8 to Enhance Prometastatic Phenotypes. Mol Cancer Res. 2017;15:3-14
228. Ortiz-Montero P, Londoño-Vallejo A, Vernot JP. Senescence-associated IL-6 and IL-8 cytokines induce a self- and cross-reinforced senescence/inflammatory milieu strengthening tumorigenic capabilities in the MCF-7 breast cancer cell line. Cell Commun Signal. 2017;15:17
229. Pili R, Guo Y, Chang J, Nakanishi H, Martin GR, Passaniti A. Altered angiogenesis underlying age-dependent changes in tumor growth. J Natl Cancer Inst. 1994;86:1303-14
230. Han L, Long Q, Li S, Xu Q, Zhang B, Dou X. et al. Senescent Stromal Cells Promote Cancer Resistance through SIRT1 Loss-Potentiated Overproduction of Small Extracellular Vesicles. Cancer Res. 2020;80:3383-98
231. Takasugi M, Okada R, Takahashi A, Virya Chen D, Watanabe S, Hara E. Small extracellular vesicles secreted from senescent cells promote cancer cell proliferation through EphA2. Nat Commun. 2017;8:15729
232. Lehmann BD, Paine MS, Brooks AM, McCubrey JA, Renegar RH, Wang R. et al. Senescence-associated exosome release from human prostate cancer cells. Cancer Res. 2008;68:7864-71
233. Jiang X, Wang J, Deng X, Xiong F, Zhang S, Gong Z. et al. The role of microenvironment in tumor angiogenesis. J Exp Clin Cancer Res. 2020;39:204
234. Olive KP, Jacobetz MA, Davidson CJ, Gopinathan A, McIntyre D, Honess D. et al. Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science. 2009;324:1457-61
235. Provenzano PP, Cuevas C, Chang AE, Goel VK, Von Hoff DD, Hingorani SR. Enzymatic targeting of the stroma ablates physical barriers to treatment of pancreatic ductal adenocarcinoma. Cancer Cell. 2012;21:418-29
236. Assouline B, Kahn R, Hodali L, Condiotti R, Engel Y, Elyada E. et al. Senescent cancer-associated fibroblasts in pancreatic adenocarcinoma restrict CD8(+) T cell activation and limit responsiveness to immunotherapy in mice. Nat Commun. 2024;15:6162
237. Fan G, Yu B, Tang L, Zhu R, Chen J, Zhu Y. et al. TSPAN8(+) myofibroblastic cancer-associated fibroblasts promote chemoresistance in patients with breast cancer. Sci Transl Med. 2024;16:eadj5705
238. Chen D, Liu P, Lin J, Zang L, Liu Y, Zhai S. et al. A Distinguished Roadmap of Fibroblast Senescence in Predicting Immunotherapy Response and Prognosis Across Human Cancers. Adv Sci (Weinh). 2024: e2406624.
239. Liu L, Huang H, Cheng B, Xie H, Peng W, Cui H. et al. Revealing the role of cancer-associated fibroblast senescence in prognosis and immune landscape in pancreatic cancer. iScience. 2025;28:111612
240. Wu Z, Uhl B, Gires O, Reichel CA. A transcriptomic pan-cancer signature for survival prognostication and prediction of immunotherapy response based on endothelial senescence. J Biomed Sci. 2023;30:21
241. Grosse L, Wagner N, Emelyanov A, Molina C, Lacas-Gervais S, Wagner KD. et al. Defined p16(High) Senescent Cell Types Are Indispensable for Mouse Healthspan. Cell Metab. 2020;32:87-99.e6
242. Hayashi T, Matsui-Hirai H, Miyazaki-Akita A, Fukatsu A, Funami J, Ding QF. et al. Endothelial cellular senescence is inhibited by nitric oxide: implications in atherosclerosis associated with menopause and diabetes. Proc Natl Acad Sci U S A. 2006;103:17018-23
243. Matsushita H, Chang E, Glassford AJ, Cooke JP, Chiu CP, Tsao PS. eNOS activity is reduced in senescent human endothelial cells: Preservation by hTERT immortalization. Circ Res. 2001;89:793-8
244. Zeng X, Wang TW, Yamaguchi K, Hatakeyama S, Yamazaki S, Shimizu E. et al. M2 macrophage-derived TGF-β induces age-associated loss of adipogenesis through progenitor cell senescence. Mol Metab. 2024;84:101943
245. Benadjaoud MA, Soysouvanh F, Tarlet G, Paget V, Buard V, Santos de Andrade H. et al. Deciphering the Dynamic Molecular Program of Radiation-Induced Endothelial Senescence. Int J Radiat Oncol Biol Phys. 2022;112:975-85
246. Bent EH, Gilbert LA, Hemann MT. A senescence secretory switch mediated by PI3K/AKT/mTOR activation controls chemoprotective endothelial secretory responses. Genes Dev. 2016;30:1811-21
247. Ungvari Z, Podlutsky A, Sosnowska D, Tucsek Z, Toth P, Deak F. et al. Ionizing radiation promotes the acquisition of a senescence-associated secretory phenotype and impairs angiogenic capacity in cerebromicrovascular endothelial cells: role of increased DNA damage and decreased DNA repair capacity in microvascular radiosensitivity. J Gerontol A Biol Sci Med Sci. 2013;68:1443-57
248. Mongiardi MP, Radice G, Piras M, Stagni V, Pacioni S, Re A. et al. Axitinib exposure triggers endothelial cells senescence through ROS accumulation and ATM activation. Oncogene. 2019;38:5413-24
249. Wang D, Xiao F, Feng Z, Li M, Kong L, Huang L. et al. Sunitinib facilitates metastatic breast cancer spreading by inducing endothelial cell senescence. Breast Cancer Res. 2020;22:103
250. Usui S, Iso Y, Sasai M, Mizukami T, Mori H, Watanabe T. et al. Kisspeptin-10 induces endothelial cellular senescence and impaired endothelial cell growth. Clin Sci (Lond). 2014;127:47-55
251. Chang H, Rha SY, Jeung HC, Park KH, Kim TS, Kim YB. et al. Telomerase- and angiogenesis-related gene responses to irradiation in human umbilical vein endothelial cells. Int J Mol Med. 2013;31:1202-8
252. Wieland E, Rodriguez-Vita J, Liebler SS, Mogler C, Moll I, Herberich SE. et al. Endothelial Notch1 Activity Facilitates Metastasis. Cancer Cell. 2017;31:355-67
253. Castellani G, Buccarelli M, D'Alessandris QG, Ilari R, Cappannini A, Pedini F. et al. Extracellular vesicles produced by irradiated endothelial or Glioblastoma stem cells promote tumor growth and vascularization modulating tumor microenvironment. Cancer Cell Int. 2024;24:72
254. Zhu Z, Cao Q, Chen J, Sun Y, Liu F, Li J. et al. Expression pattern of cancer-associated cellular senescence genes in clear cell renal cell carcinoma distinguishes tumor subclasses with clinical implications. Sci Rep. 2025;15:442
255. Coppé JP, Rodier F, Patil CK, Freund A, Desprez PY, Campisi J. Tumor suppressor and aging biomarker p16(INK4a) induces cellular senescence without the associated inflammatory secretory phenotype. J Biol Chem. 2011;286:36396-403
256. Wang B, Han J, Elisseeff JH, Demaria M. The senescence-associated secretory phenotype and its physiological and pathological implications. Nat Rev Mol Cell Biol. 2024
257. Eggert T, Wolter K, Ji JL, Ma C, Yevsa T, Klotz S. et al. Distinct Functions of Senescence-Associated Immune Responses in Liver Tumor Surveillance and Tumor Progression. Cancer Cell. 2016;30:533-47
258. Cheng Y, Li H, Deng Y, Tai Y, Zeng K, Zhang Y. et al. Cancer-associated fibroblasts induce PDL1+ neutrophils through the IL6-STAT3 pathway that foster immune suppression in hepatocellular carcinoma. Cell Death Dis. 2018;9:422
259. Cheng JT, Deng YN, Yi HM, Wang GY, Fu BS, Chen WJ. et al. Hepatic carcinoma-associated fibroblasts induce IDO-producing regulatory dendritic cells through IL-6-mediated STAT3 activation. Oncogenesis. 2016;5:e198
260. Deng Y, Cheng J, Fu B, Liu W, Chen G, Zhang Q. et al. Hepatic carcinoma-associated fibroblasts enhance immune suppression by facilitating the generation of myeloid-derived suppressor cells. Oncogene. 2017;36:1090-101
261. Mace TA, Ameen Z, Collins A, Wojcik S, Mair M, Young GS. et al. Pancreatic cancer-associated stellate cells promote differentiation of myeloid-derived suppressor cells in a STAT3-dependent manner. Cancer Res. 2013;73:3007-18
262. Zhao Q, Huang L, Qin G, Qiao Y, Ren F, Shen C. et al. Cancer-associated fibroblasts induce monocytic myeloid-derived suppressor cell generation via IL-6/exosomal miR-21-activated STAT3 signaling to promote cisplatin resistance in esophageal squamous cell carcinoma. Cancer Lett. 2021;518:35-48
263. Ye J, Baer JM, Faget DV, Morikis VA, Ren Q, Melam A. et al. Senescent CAFs Mediate Immunosuppression and Drive Breast Cancer Progression. Cancer Discov. 2024;14:1302-23
264. Kato T, Noma K, Ohara T, Kashima H, Katsura Y, Sato H. et al. Cancer-Associated Fibroblasts Affect Intratumoral CD8(+) and FoxP3(+) T Cells Via IL6 in the Tumor Microenvironment. Clin Cancer Res. 2018;24:4820-33
265. Song M, He J, Pan QZ, Yang J, Zhao J, Zhang YJ. et al. Cancer-Associated Fibroblast-Mediated Cellular Crosstalk Supports Hepatocellular Carcinoma Progression. Hepatology. 2021;73:1717-35
266. Viel S, Marçais A, Guimaraes FS, Loftus R, Rabilloud J, Grau M. et al. TGF-β inhibits the activation and functions of NK cells by repressing the mTOR pathway. Sci Signal. 2016;9:ra19
267. Chen W, Jin W, Hardegen N, Lei KJ, Li L, Marinos N. et al. Conversion of peripheral CD4+CD25- naive T cells to CD4+CD25+ regulatory T cells by TGF-beta induction of transcription factor Foxp3. J Exp Med. 2003;198:1875-86
268. Gutcher I, Donkor MK, Ma Q, Rudensky AY, Flavell RA, Li MO. Autocrine transforming growth factor-β1 promotes in vivo Th17 cell differentiation. Immunity. 2011;34:396-408
269. Thomas DA, Massagué J. TGF-beta directly targets cytotoxic T cell functions during tumor evasion of immune surveillance. Cancer Cell. 2005;8:369-80
270. Morad G, Helmink BA, Sharma P, Wargo JA. Hallmarks of response, resistance, and toxicity to immune checkpoint blockade. Cell. 2021;184:5309-37
271. Colucci M, Zumerle S, Bressan S, Gianfanti F, Troiani M, Valdata A. et al. Retinoic acid receptor activation reprograms senescence response and enhances anti-tumor activity of natural killer cells. Cancer Cell. 2024;42:646-61.e9
272. Chibaya L, DeMarco KD, Lusi CF, Kane GI, Brassil ML, Parikh CN. et al. Nanoparticle delivery of innate immune agonists combined with senescence-inducing agents promotes T cell control of pancreatic cancer. Sci Transl Med. 2024;16:eadj9366
273. Wang Z, Chen Y, Fang H, Xiao K, Wu Z, Xie X. et al. Reprogramming cellular senescence in the tumor microenvironment augments cancer immunotherapy through multifunctional nanocrystals. Sci Adv. 2024;10:eadp7022
274. Liu Y, Pagacz J, Wolfgeher DJ, Bromerg KD, Gorman JV, Kron SJ. Senescent cancer cell vaccines induce cytotoxic T cell responses targeting primary tumors and disseminated tumor cells. J Immunother Cancer. 2023 11
275. Mongiardi MP, Pellegrini M, Pallini R, Levi A, Falchetti ML. Cancer Response to Therapy-Induced Senescence: A Matter of Dose and Timing. Cancers (Basel). 2021 13
276. Wang C, Vegna S, Jin H, Benedict B, Lieftink C, Ramirez C. et al. Inducing and exploiting vulnerabilities for the treatment of liver cancer. Nature. 2019;574:268-72
277. Zhang Y, Xiao B, Yuan S, Ding L, Pan Y, Jiang Y. et al. Tryptanthrin targets GSTP1 to induce senescence and increases the susceptibility to apoptosis by senolytics in liver cancer cells. Redox Biol. 2024;76:103323
278. Toso A, Revandkar A, Di Mitri D, Guccini I, Proietti M, Sarti M. et al. Enhancing chemotherapy efficacy in Pten-deficient prostate tumors by activating the senescence-associated antitumor immunity. Cell Rep. 2014;9:75-89
279. Georgilis A, Klotz S, Hanley CJ, Herranz N, Weirich B, Morancho B. et al. PTBP1-Mediated Alternative Splicing Regulates the Inflammatory Secretome and the Pro-tumorigenic Effects of Senescent Cells. Cancer Cell. 2018;34:85-102.e9
280. Nacarelli T, Lau L, Fukumoto T, Zundell J, Fatkhutdinov N, Wu S. et al. NAD(+) metabolism governs the proinflammatory senescence-associated secretome. Nat Cell Biol. 2019;21:397-407
281. Li F, Huangyang P, Burrows M, Guo K, Riscal R, Godfrey J. et al. FBP1 loss disrupts liver metabolism and promotes tumorigenesis through a hepatic stellate cell senescence secretome. Nat Cell Biol. 2020;22:728-39
282. Pernicova I, Korbonits M. Metformin-mode of action and clinical implications for diabetes and cancer. Nat Rev Endocrinol. 2014;10:143-56
283. Yang J, Liu HC, Zhang JQ, Zou JY, Zhang X, Chen WM. et al. The effect of metformin on senescence of T lymphocytes. Immun Ageing. 2023;20:73
284. Kulkarni AS, Gubbi S, Barzilai N. Benefits of Metformin in Attenuating the Hallmarks of Aging. Cell Metab. 2020;32:15-30
285. Barzilai N, Crandall JP, Kritchevsky SB, Espeland MA. Metformin as a Tool to Target Aging. Cell Metab. 2016;23:1060-5
286. Herranz N, Gallage S, Mellone M, Wuestefeld T, Klotz S, Hanley CJ. et al. mTOR regulates MAPKAPK2 translation to control the senescence-associated secretory phenotype. Nat Cell Biol. 2015;17:1205-17
287. Herranz N, Gallage S, Mellone M, Wuestefeld T, Klotz S, Hanley CJ. et al. Erratum: mTOR regulates MAPKAPK2 translation to control the senescence-associated secretory phenotype. Nat Cell Biol. 2015;17:1370
288. Laberge RM, Sun Y, Orjalo AV, Patil CK, Freund A, Zhou L. et al. MTOR regulates the pro-tumorigenic senescence-associated secretory phenotype by promoting IL1A translation. Nat Cell Biol. 2015;17:1049-61
289. Laberge RM, Sun Y, Orjalo AV, Patil CK, Freund A, Zhou L. et al. Author Correction: MTOR regulates the pro-tumorigenic senescence-associated secretory phenotype by promoting IL1A translation. Nat Cell Biol. 2021;23:564-5
290. Mannick JB, Morris M, Hockey HP, Roma G, Beibel M, Kulmatycki K. et al. TORC1 inhibition enhances immune function and reduces infections in the elderly. Sci Transl Med. 2018 10
291. Bitto A, Ito TK, Pineda VV, LeTexier NJ, Huang HZ, Sutlief E. et al. Transient rapamycin treatment can increase lifespan and healthspan in middle-aged mice. Elife. 2016 5
292. Davis SL, Ionkina AA, Bagby SM, Orth JD, Gittleman B, Marcus JM. et al. Preclinical and Dose-Finding Phase I Trial Results of Combined Treatment with a TORC1/2 Inhibitor (TAK-228) and Aurora A Kinase Inhibitor (Alisertib) in Solid Tumors. Clin Cancer Res. 2020;26:4633-42
293. Cleary JM, Lima CM, Hurwitz HI, Montero AJ, Franklin C, Yang J. et al. A phase I clinical trial of navitoclax, a targeted high-affinity Bcl-2 family inhibitor, in combination with gemcitabine in patients with solid tumors. Invest New Drugs. 2014;32:937-45
294. Puglisi M, Molife LR, de Jonge MJ, Khan KH, Doorn LV, Forster MD. et al. A Phase I study of the safety, pharmacokinetics and efficacy of navitoclax plus docetaxel in patients with advanced solid tumors. Future Oncol. 2021;17:2747-58
295. de Vos S, Leonard JP, Friedberg JW, Zain J, Dunleavy K, Humerickhouse R. et al. Safety and efficacy of navitoclax, a BCL-2 and BCL-X(L) inhibitor, in patients with relapsed or refractory lymphoid malignancies: results from a phase 2a study. Leuk Lymphoma. 2021;62:810-8
296. Kipps TJ, Eradat H, Grosicki S, Catalano J, Cosolo W, Dyagil IS. et al. A phase 2 study of the BH3 mimetic BCL2 inhibitor navitoclax (ABT-263) with or without rituximab, in previously untreated B-cell chronic lymphocytic leukemia. Leuk Lymphoma. 2015;56:2826-33
297. Chaib S, Tchkonia T, Kirkland JL. Cellular senescence and senolytics: the path to the clinic. Nat Med. 2022;28:1556-68
298. Carpenter VJ, Saleh T, Gewirtz DA. Senolytics for Cancer Therapy: Is All That Glitters Really Gold? Cancers (Basel). 2021 13
299. Wang Y, Che L, Chen X, He Z, Song D, Yuan Y. et al. Repurpose dasatinib and quercetin: Targeting senescent cells ameliorates postmenopausal osteoporosis and rejuvenates bone regeneration. Bioact Mater. 2023;25:13-28
300. Novais EJ, Tran VA, Johnston SN, Darris KR, Roupas AJ, Sessions GA. et al. Long-term treatment with senolytic drugs Dasatinib and Quercetin ameliorates age-dependent intervertebral disc degeneration in mice. Nat Commun. 2021;12:5213
301. Hickson LJ, Langhi Prata LGP, Bobart SA, Evans TK, Giorgadze N, Hashmi SK. et al. Senolytics decrease senescent cells in humans: Preliminary report from a clinical trial of Dasatinib plus Quercetin in individuals with diabetic kidney disease. EBioMedicine. 2019;47:446-56
302. Pastor-Fernández A, Bertos AR, Sierra-Ramírez A, Del Moral-Salmoral J, Merino J, de Ávila AI. et al. Treatment with the senolytics dasatinib/quercetin reduces SARS-CoV-2-related mortality in mice. Aging Cell. 2023;22:e13771
303. Bientinesi E, Ristori S, Lulli M, Monti D. Quercetin induces senolysis of doxorubicin-induced senescent fibroblasts by reducing autophagy, preventing their pro-tumour effect on osteosarcoma cells. Mech Ageing Dev. 2024;220:111957
304. Wang L, Xiong B, Lu W, Cheng Y, Zhu J, Ai G. et al. Senolytic drugs dasatinib and quercetin combined with Carboplatin or Olaparib reduced the peritoneal and adipose tissue metastasis of ovarian cancer. Biomed Pharmacother. 2024;174:116474
305. Khan S, Cao L, Wiegand J, Zhang P, Zajac-Kaye M, Kaye FJ. et al. PROTAC-Mediated Dual Degradation of BCL-xL and BCL-2 Is a Highly Effective Therapeutic Strategy in Small-Cell Lung Cancer. Cells. 2024 13
306. Demaria M, O'Leary MN, Chang J, Shao L, Liu S, Alimirah F. et al. Cellular Senescence Promotes Adverse Effects of Chemotherapy and Cancer Relapse. Cancer Discov. 2017;7:165-76
307. Vlahovic G, Karantza V, Wang D, Cosgrove D, Rudersdorf N, Yang J. et al. A phase I safety and pharmacokinetic study of ABT-263 in combination with carboplatin/paclitaxel in the treatment of patients with solid tumors. Invest New Drugs. 2014;32:976-84
308. Szymczak J, Cielecka-Piontek J. Fisetin-In Search of Better Bioavailability-From Macro to Nano Modifications: A Review. Int J Mol Sci. 2023 24
309. Chen YX, Wang CJ, Xiao DS, He BM, Li M, Yi XP. et al. eIF3a R803K mutation mediates chemotherapy resistance by inducing cellular senescence in small cell lung cancer. Pharmacol Res. 2021;174:105934
310. Jochems F, Baltira C, MacDonald JA, Daniels V, Mathur A, de Gooijer MC. et al. Senolysis by ABT-263 is associated with inherent apoptotic dependence of cancer cells derived from the non-senescent state. Cell Death Differ. 2024
311. Jochems F, Thijssen B, De Conti G, Jansen R, Pogacar Z, Groot K. et al. The Cancer SENESCopedia: A delineation of cancer cell senescence. Cell Rep. 2021;36:109441
312. Alcon C, Kovatcheva M, Morales-Sánchez P, López-Polo V, Torres T, Puig S. et al. HRK downregulation and augmented BCL-xL binding to BAK confer apoptotic protection to therapy-induced senescent melanoma cells. Cell Death Differ. 2024
313. Kovacovicova K, Skolnaja M, Heinmaa M, Mistrik M, Pata P, Pata I. et al. Senolytic Cocktail Dasatinib+Quercetin (D+Q) Does Not Enhance the Efficacy of Senescence-Inducing Chemotherapy in Liver Cancer. Front Oncol. 2018;8:459
314. Ji P, Wang C, Liu Y, Guo X, Liang Y, Wei J. et al. Targeted Clearance of Senescent Cells Via Engineered Extracellular Vesicles Reprograms Tumor Immunosuppressive Microenvironment. Adv Healthc Mater. 2024;13:e2400945
315. Zhang H, Xu X, Shou X, Liao W, Jin C, Chen C. et al. Senolytic Therapy Enabled by Senescent Cell-Sensitive Biomimetic Melanin Nano-Senolytics. Adv Healthc Mater. 2024;13:e2401085
316. Parshad B, Baker AG, Ahmed I, Estepa-Fernández A, Muñoz-Espín D, Fruk L. Improved Therapeutic Efficiency of Senescent Cell-specific, Galactose-Functionalized Micelle Nanocarriers. Small. 2024: e2405732.
317. Chang M, Dong Y, Xu H, Cruickshank-Taylor AB, Kozora JS, Behpour B. et al. Senolysis Enabled by Senescent Cell-Sensitive Bioorthogonal Tetrazine Ligation. Angew Chem Int Ed Engl. 2024;63:e202315425
318. Chang M, Gao F, Gnawali G, Xu H, Dong Y, Meng X. et al. Selective Elimination of Senescent Cancer Cells by Galacto-Modified PROTACs. J Med Chem. 2024;67:7301-11
319. Peng Y, Liu D, Huang D, Inuzuka H, Liu J. PROTAC as a novel anti-cancer strategy by targeting aging-related signaling. Semin Cancer Biol. 2024;106-107:143-55
320. McHugh D, Durán I, Gil J. Senescence as a therapeutic target in cancer and age-related diseases. Nat Rev Drug Discov. 2025;24:57-71
321. Yang Y, Jn-Simon N, He Y, Sun C, Zhang P, Hu W. et al. A BCL-xL/BCL-2 PROTAC effectively clears senescent cells in the liver and reduces MASH-driven hepatocellular carcinoma in mice. Nat Aging. 2025
322. Amor C, Fernández-Maestre I, Chowdhury S, Ho YJ, Nadella S, Graham C. et al. Prophylactic and long-lasting efficacy of senolytic CAR T cells against age-related metabolic dysfunction. Nat Aging. 2024;4:336-49
323. Ming X, Yang Z, Huang Y, Wang Z, Zhang Q, Lu C. et al. A chimeric peptide promotes immune surveillance of senescent cells in injury, fibrosis, tumorigenesis and aging. Nat Aging. 2024
324. Yang D, Sun B, Li S, Wei W, Liu X, Cui X. et al. NKG2D-CAR T cells eliminate senescent cells in aged mice and nonhuman primates. Sci Transl Med. 2023;15:eadd1951
325. Stirrups R. CAR T-cells for relapsed B-cell ALL in children and young adults. Lancet Oncol. 2018;19:e144
326. Zhu X, Prasad S, Gaedicke S, Hettich M, Firat E, Niedermann G. Patient-derived glioblastoma stem cells are killed by CD133-specific CAR T cells but induce the T cell aging marker CD57. Oncotarget. 2015;6:171-84
327. Roselle C, Horikawa I, Chen L, Kelly AR, Gonzales D, Da T. et al. Enhancing chimeric antigen receptor T cell therapy by modulating the p53 signaling network with Δ133p53α. Proc Natl Acad Sci U S A. 2024;121:e2317735121
328. Yao GD, Yang J, Li XX, Song XY, Hayashi T, Tashiro SI. et al. Blocking the utilization of glucose induces the switch from senescence to apoptosis in pseudolaric acid B-treated human lung cancer cells in vitro. Acta Pharmacol Sin. 2017;38:1401-11
329. He Y, Liu Y, Zheng M, Zou Y, Huang M, Wang L. et al. Targeting ATAD3A Phosphorylation Mediated by TBK1 Ameliorates Senescence-Associated Pathologies. Adv Sci (Weinh). 2025;12:e2404109
330. Jochems F, Baltira C, MacDonald JA, Daniels V, Mathur A, de Gooijer MC. et al. Senolysis by ABT-263 is associated with inherent apoptotic dependence of cancer cells derived from the non-senescent state. Cell Death Differ. 2025;32:855-65
331. MacDonald JA, Bradshaw GA, Jochems F, Bernards R, Letai A. Apoptotic priming in senescence predicts specific senolysis by quantitative analysis of mitochondrial dependencies. Cell Death Differ. 2025;32:802-17
332. Baldwin JG, Heuser-Loy C, Saha T, Schelker RC, Slavkovic-Lukic D, Strieder N. et al. Intercellular nanotube-mediated mitochondrial transfer enhances T cell metabolic fitness and antitumor efficacy. Cell. 2024;187:6614-30.e21
333. Yoshida S, Nakagami H, Hayashi H, Ikeda Y, Sun J, Tenma A. et al. The CD153 vaccine is a senotherapeutic option for preventing the accumulation of senescent T cells in mice. Nat Commun. 2020;11:2482
334. Suda M, Shimizu I, Katsuumi G, Yoshida Y, Hayashi Y, Ikegami R. et al. Senolytic vaccination improves normal and pathological age-related phenotypes and increases lifespan in progeroid mice. Nat Aging. 2021;1:1117-26
335. Zhou Z, Yao J, Wu D, Huang X, Wang Y, Li X. et al. Type 2 cytokine signaling in macrophages protects from cellular senescence and organismal aging. Immunity. 2024;57:513-27.e6
336. Wang T, Liu W, Shen Q, Tao R, Li C, Shen Q. et al. Combination of PARP inhibitor and CDK4/6 inhibitor modulates cGAS/STING-dependent therapy-induced senescence and provides "one-two punch" opportunity with anti-PD-L1 therapy in colorectal cancer. Cancer Sci. 2023;114:4184-201
337. Zhang J, Zhang S, Cheng C, Zhu C, Wang T, Tang L. et al. Targeting senescence with radioactive (223)Ra/Ba SAzymes enables senolytics-unlocked One-Two punch strategy to boost anti-tumor immunotherapy. Biomaterials. 2025;315:122915
338. Wang H, Yuan S, Zheng Q, Zhang S, Zhang Q, Ji S. et al. Dual Inhibition of CDK4/6 and XPO1 Induces Senescence With Acquired Vulnerability to CRBN-Based PROTAC Drugs. Gastroenterology. 2024;166:1130-44.e8
339. Bi Y, Qiao X, Cai Z, Zhao H, Ye R, Liu Q. et al. Exosomal miR-302b rejuvenates aging mice by reversing the proliferative arrest of senescent cells. Cell Metab. 2025;37:527-41.e6
340. Gu L, Zhu Y, Nandi SP, Lee M, Watari K, Bareng B. et al. FBP1 controls liver cancer evolution from senescent MASH hepatocytes. Nature. 2025;637:461-9
341. Henson SM, Snelgrove R, Hussell T, Wells DJ, Aspinall R. An IL-7 fusion protein that shows increased thymopoietic ability. J Immunol. 2005;175:4112-8
342. Al-Chami E, Tormo A, Pasquin S, Kanjarawi R, Ziouani S, Rafei M. Interleukin-21 administration to aged mice rejuvenates their peripheral T-cell pool by triggering de novo thymopoiesis. Aging Cell. 2016;15:349-60
343. Tormo A, Khodayarian F, Cui Y, Al-Chami E, Kanjarawi R, Noé B. et al. Interleukin-21 promotes thymopoiesis recovery following hematopoietic stem cell transplantation. J Hematol Oncol. 2017;10:120
344. Sun L, Guo J, Brown R, Amagai T, Zhao Y, Su DM. Declining expression of a single epithelial cell-autonomous gene accelerates age-related thymic involution. Aging Cell. 2010;9:347-57
345. Li J, Wachsmuth LP, Xiao S, Condie BG, Manley NR. Foxn1 overexpression promotes thymic epithelial progenitor cell proliferation and mTEC maintenance, but does not prevent thymic involution. Development. 2023 150
346. Tuckett AZ, Thornton RH, O'Reilly RJ, van den Brink MRM, Zakrzewski JL. Intrathymic injection of hematopoietic progenitor cells establishes functional T cell development in a mouse model of severe combined immunodeficiency. J Hematol Oncol. 2017;10:109
347. Politikos I, Boussiotis VA. The role of the thymus in T-cell immune reconstitution after umbilical cord blood transplantation. Blood. 2014;124:3201-11
348. Nishimura T, Kaneko S, Kawana-Tachikawa A, Tajima Y, Goto H, Zhu D. et al. Generation of rejuvenated antigen-specific T cells by reprogramming to pluripotency and redifferentiation. Cell Stem Cell. 2013;12:114-26
349. Licastro F, Candore G, Lio D, Porcellini E, Colonna-Romano G, Franceschi C. et al. Innate immunity and inflammation in ageing: a key for understanding age-related diseases. Immun Ageing. 2005;2:8
350. Jaillon S, Ponzetta A, Di Mitri D, Santoni A, Bonecchi R, Mantovani A. Neutrophil diversity and plasticity in tumour progression and therapy. Nat Rev Cancer. 2020;20:485-503
351. Sun R, Luo J, Li D, Shu Y, Luo C, Wang SS. et al. Neutrophils with protumor potential could efficiently suppress tumor growth after cytokine priming and in presence of normal NK cells. Oncotarget. 2014;5:12621-34
352. Wang TT, Zhao YL, Peng LS, Chen N, Chen W, Lv YP. et al. Tumour-activated neutrophils in gastric cancer foster immune suppression and disease progression through GM-CSF-PD-L1 pathway. Gut. 2017;66:1900-11
353. Suryadevara V, Hudgins AD, Rajesh A, Pappalardo A, Karpova A, Dey AK. et al. SenNet recommendations for detecting senescent cells in different tissues. Nat Rev Mol Cell Biol. 2024;25:1001-23
354. Ogrodnik M, Carlos Acosta J, Adams PD, d'Adda di Fagagna F, Baker DJ, Bishop CL. et al. Guidelines for minimal information on cellular senescence experimentation in vivo. Cell. 2024;187:4150-75
355. El-Sadoni M, Shboul SA, Alhesa A, Shahin NA, Alsharaiah E, Ismail MA. et al. A three-marker signature identifies senescence in human breast cancer exposed to neoadjuvant chemotherapy. Cancer Chemother Pharmacol. 2023;91:345-60
356. Saleh T, Alhesa A, El-Sadoni M, Abu Shahin N, Alsharaiah E, Al Shboul S. et al. The Expression of the Senescence-Associated Biomarker Lamin B1 in Human Breast Cancer. Diagnostics (Basel). 2022 12
357. Kim KM, Noh JH, Bodogai M, Martindale JL, Yang X, Indig FE. et al. Identification of senescent cell surface targetable protein DPP4. Genes Dev. 2017;31:1529-34
358. Smer-Barreto V, Quintanilla A, Elliott RJR, Dawson JC, Sun J, Campa VM. et al. Discovery of senolytics using machine learning. Nat Commun. 2023;14:3445
359. Duran I, Pombo J, Sun B, Gallage S, Kudo H, McHugh D. et al. Detection of senescence using machine learning algorithms based on nuclear features. Nat Commun. 2024;15:1041
360. Zhao H, Liu Z, Chen H, Han M, Zhang M, Liu K. et al. Identifying specific functional roles for senescence across cell types. Cell. 2024
Author contact
![]() Corresponding authors: Xianjun Yu, PHD, MD, yuxianjunorg. Si Shi, MD, shisiorg.
Corresponding authors: Xianjun Yu, PHD, MD, yuxianjunorg. Si Shi, MD, shisiorg.