Impact Factor
 Global reach, higher impact
Global reach, higher impact
Theranostics 2025; 15(17):9114-9130. doi:10.7150/thno.118958 This issue Cite
Research Paper
Macrophage-derived KIF13B interacts with USP9X to attenuate abdominal aortic aneurysm development by potentiating TFEB stability
Jingxuan Chen1†, Yitong Xu1†, Huahui Yu2, Yiran Liu3, Guolin Miao1,4, Yufei Han1, Liwen Zheng1, Zeyu Cai5, Zihao Zhou1, Jinxuan Chen1, Sijing Shi1, Pingping Lai1, Wenxi Zhang1, Lianxin Zhang1, Si Mei1, Yinqi Zhao1, Ling Zhang1, Wei Huang1, Yuhui Wang1, Dongyu Zhao3, Wei Kong5, Yanwen Qin2, Erdan Dong1,4,6, Xunde Xian1,7 ![]()
1. Institute of Cardiovascular Sciences, State Key Laboratory of Vascular Homeostasis and Remodeling, School of Basic Medical Sciences, Peking University, Beijing, China.
2. The Key Laboratory of Remodeling-Related Cardiovascular Diseases, Ministry of Education, Beijing Anzhen Hospital, Capital Medical University, Beijing Institute of Heart Lung and Blood Vessel Disease, Beijing, China.
3. Department of Biomedical Informatics, School of Basic Medical Sciences, State Key Laboratory of Vascular Homeostasis and Remodeling, Peking University, Beijing, China.
4. Department of Cardiology and Institute of Vascular Medicine, Peking University Third Hospital, Beijing, China.
5. Department of Physiology and Pathophysiology, School of Basic Medical Sciences, State Key Laboratory of Vascular Homeostasis and Remodeling, Peking University, Beijing, China.
6. Research Center for Cardiopulmonary Rehabilitation, University of Health and Rehabilitation Sciences Qingdao Hospital (Qingdao Municipal Hospital), School of Health and Life Sciences, University of Health and Rehabilitation Sciences, 369 Dengyun Road, Qingdao, China.
7. Beijing Key Laboratory of Cardiovascular Receptors Research, Peking University Third Hospital, Beijing, China.
†These authors contributed equally to this work.
Received 2025-6-5; Accepted 2025-8-7; Published 2025-8-16
Abstract

Rationale: Abdominal aortic aneurysm (AAA) is a highly lethal cardiovascular disorder for which there is no effective medication to date. Kinesin family member 13b (KIF13B), a vital motor protein, has been recently identified as a novel regulator of lipid metabolism. However, the role of KIF13B in AAA development has not been documented.
Methods: We determined the expression of KIF13B in aortic tissues from clinical patients and porcine pancreatic elastase (PPE) or angiotensin II (ANG II)-induced AAA mouse models. To investigate the influence of KIF13B on AAA expansion, we established global, myeloid cell-specific and vascular smooth muscle cell (VSMC)-specific conditional Kif13b-deficient mice in PPE and/or ANG II-induced AAA models.
Results: RNA-seq data from GEO database (GSE57691) revealed a significant decrease in KIF13B gene expression within the aortic tissues of patients with AAA. KIF13B protein levels were largely reduced in aortic tissue samples from patients and two mouse models with AAA. Complete inactivation of Kif13b or depleting Kif13b from myeloid cells but not smooth muscle cells (SMCs) exacerbated AAA development. Mechanistic studies identified transcription factor EB (TFEB) as a critical downstream target of KIF13B. KIF13B stabilized and upregulated TFEB by enhancing its deubiquitination through an interaction with deubiquitinase USP9X to maintain the proper function of lysosomes, thus inhibiting the senescence-associated secretory phenotype (SASP) and proinflammatory response of macrophages. Moreover, restoration of macrophage Kif13b or senolytic therapy dramatically mitigated AAA expansion in vivo.
Conclusions: In the present study, we provided a new insight into the pathogenesis of AAA and defined a KIF13B-USP9X-TFEB axis that is essential for the regulation of macrophage function, suggesting that macrophage-derived Kif13b is a beneficial regulator of vascular homeostasis and targeting KIF13B could be a potential therapeutic approach for the treatment of human AAA disease in future clinical trial.
Keywords: abdominal aortic aneurysm, KIF13B, macrophage, senescence, lysosome
Introduction
Abdominal aortic aneurysm (AAA) is a fatal vascular disease characterized by a localized, permanent dilatation of the abdominal aorta exceeding 1.5 times its normal diameter [1]. Major risk factors include hypertension, hypercholesterolemia, atherosclerosis, smoking, advanced age, and male sex [2]. Notably, AAA rupture carries a mortality rate exceeding 80% [1]; yet, no FDA-approved pharmacological therapies exist to slow AAA progression or reduce rupture risk. Current management relies on surgical repair or endovascular stenting, which is viable for fewer than 10% of patients [3], leaving the majority at significant risk. Thus, elucidating novel pathogenic mechanisms and identifying therapeutic targets for AAA remain urgent priorities.
The pathophysiology of AAA involves inflammatory cell infiltration into the vascular wall, vascular smooth muscle cell (VSMC) apoptosis, and degradation of extracellular matrix elastin [4]. Growing evidence implicates dysregulated lipid metabolism, particularly involving triglyceride-rich lipoproteins (TRLs), in AAA pathogenesis [4-8]. Genome-wide association studies have identified 121 AAA risk loci, including regulators of lipoprotein receptors such as PCSK9 [9]. Moreover, both clinical and preclinical studies further underscore the role of lipoprotein receptors (e.g., low-density lipoprotein receptor-associated protein 1, LRP1) in AAA development [10-14], highlighting the critical contribution of dysfunctional lipid metabolism to this devastating disease.
The kinesin-3 family, known for its role in intracellular transport of organelles and macromolecules [15], includes kinesin family member 13b (KIF13B), the largest family member. Emerging evidence demonstrates that KIF13B serves as a novel modulator of lipoprotein receptor function, specifically localized to lipid rafts in hepatocyte membranes where it colocalizes with LRP1 to regulate lipid metabolism [16]. While KIF13B with a high expression level in endothelial cells and metabolic organs has been reported to regulate vascular permeability and angiogenesis [17-20], its precise function in vascular homeostasis remains poorly understood. Moreover, our prior work has identified KIF13B as a novel modulator of lipid metabolism, essential for hepatic lipid homeostasis and protection against metabolic dysfunction-associated fatty liver disease (MAFLD) [21]. Given the established links between dyslipidemia, MAFLD, and cardiovascular disease (CVD) [22,23], we herein hypothesized that KIF13B might influence AAA pathogenesis. However, the fundamental role of KIF13B in AAA and its potential as a therapeutic target need to be further explored.
In this study, we investigate the involvement of KIF13B in AAA development and its underlying molecular mechanisms. Our key findings demonstrate that macrophage-specific KIF13B stabilizes transcription factor EB (TFEB), a master regulator of lysosomal biogenesis, via enhancing the deubiquitylation mediated by ubiquitin specific peptidase 9 x-linked (USP9X), to attenuate pro-inflammatory polarization and senescence of macrophages, ultimately suppressing AAA progression.
Methods
Human studies
All procedures involving human participants adhered to the principles of the Declaration of Helsinki and were approved by the Medical Ethics Committee of Beijing Anzhen Hospital (Beijing, China). Written informed consent was obtained from all patients prior to sample collection. Aortic tissue samples were collected from six AAA patients undergoing open surgical repair, with adjacent non-aneurysmal aortic tissue from the same patients serving as controls. Tissues were fixed in 10% neutral-buffered formalin, paraffin-embedded, and sectioned at 5 µm thickness [24].
Animal studies
All animal experiments complied with the National Institutes of Health Guide for the Care and Use of Laboratory Animals (8th edition, 2011) and were approved by the Animal Ethics Committee of Peking University (Protocol LA2023460). Global Kif13b knockout (Kif13b-/-) mice and floxed Kif13b (Kif13bf/f) mice were generated using CRISPR/Cas9 technology by the Institute of Laboratory Animal Science, Chinese Academy of Medical Sciences. Conditional myeloid cell-specific Kif13b knockout (Kif13bf/fLyz2-Cre, Lyz2Kif13bf/f) mice and VSMC-specific Kif13b knockout (Kif13bf/fSm22-Cre, Sm22Kif13bf/f) mice were generated by breeding Kif13bf/f mice with Lyz2-Cre and Sm22-Cre mice provided as a gift from Prof. Hongliang Li (Wuhan University), respectively. All mice were housed under specific pathogen-free conditions at the Peking University Health Science Center Animal Facility.
Statistical analysis
All statistical analysis was performed using GraphPad Prism version 9.0 (GraphPad Software). Continuous data are presented as mean ± SEM. Survival of AAA mice was examined using the Kaplan-Meier survival curve and differences were analyzed using the log-rank test (Mantel-Cox). Normality of data distribution was assessed by the Shapiro-Wilk test and equality of variance by the Brown-Forsythe test. For comparisons between two groups, differences were compared using unpaired Student's t-test (normal distribution and equal variance) or Mann-Whitney U-test (non-normal distribution) or Welch t-test (unequal variances). For multiple comparisons of equal variance (> 2 groups), one-way ANOVA analyses (single variable) or two-way ANOVA analyses (multiple variables) were used, followed by Tukey post hoc tests (equal variance). Kruskal-Wallis test and Bonferroni post hoc test were used for multiple tests of unequal variance. Pearson correlation analysis was used to assess the correlation between the expression levels of the 2 genes, values of P < 0.05 were considered statistically significant.
Detailed methodologies are provided in the Supplemental Material.
Results
KIF13B expression is decreased in aortic tissues of AAA patients and mouse models
To delineate the role of kinesin family member genes in AAA pathogenesis, we systematically performed differentially expressed genes (DEGs) analysis across multiple AAA models. Cross-model comparative analysis revealed that KIF13B gene was the sole kinesin family member exhibiting consistent and significant downregulation across all AAA variants (Figure 1A). To confirm the association between KIF13B and AAA, we analyzed bulk RNA-seq data from the GEO database (GSE57691), comprising aortic tissues from 44 AAA patients and 9 healthy controls. KIF13B mRNA levels were significantly downregulated in AAA tissues compared to controls (Figure 1B). Immunofluorescence staining and Western blot analysis corroborated these findings, showing reduced KIF13B protein expression in human AAA lesions, in which the positive staining content of α-SMA was markedly reduced in the AAA lesions, representing a loss of VSMC, whereas more CD68-positive macrophages were accumulated, indicating an enhanced inflammatory status, compared to adjacent normal aortas (ANAs) (Figure 1C-E). In addition, when compared to healthy control tissues, we also observed a significant decrease in KIF13B expression at both mRNA and protein levels in PPE-induced AAA mouse model as well as in the ANG II-induced AAA murine model in the context of hyperlipidemia by delivering adeno-associated virus 8 (AAV8) expressing PCSK9D337Y through tail vein, followed by a Western diet (WD) challenge (Figure 1F-I). Furthermore, to explore the functional relevance of KIF13B in AAA, we performed gene set enrichment analysis (GSEA) using the AAA-associated transcriptome. KIF13B expression strongly correlated with pathways linked to inflammation, lysosomal function, and cellular senescence, all established contributors to AAA pathogenesis [25] (Figure 1J). Taken together, these findings suggest a negative correlation between KIF13B expression levels and the incidence of AAA development.
KIF13B expression is decreased in aortic tissues of AAA patients and mouse models. (A) The Venn diagram illustrates the intersection of commonly downregulated genes and kinesin family member genes across different AAA samples and control tissues, including: the murine ANG II-AAA model (GSE265897), murine PPE-AAA model (GSE51228), and AAA patients (GSE57691). Genes with FDR < 0.05 and log2foldchange < 0 were included. (B) Normalized KIF13B signal in aortic samples comparing healthy controls (HC, n = 9) and abdominal aortic aneurysm (AAA, n = 44) patients within the GEO dataset (GSE57691). (C) Representative images of HE staining and co-localized immunofluorescence staining depict α-SMA (green), CD68 (grey), and KIF13B (red) protein levels in human AAA lesions versus adjacent normal aortas (ANAs) (n = 6/group). (D) Western blot analysis of KIF13B protein levels in human AAA lesions and ANAs (n = 6/group). (E) Quantitative analysis of KIF13B protein expression levels corresponding to C and D. (F) Representative images of HE staining, EVG staining, and immunofluorescence staining depict α-SMA (green), CD68 (red), and KIF13B (red) expression levels in WT mouse abdominal aortas treated with saline (n = 3) or porcine pancreatic elastase (PPE, n = 6). (G) qPCR analysis of Kif13b mRNA expression (n = 3/group) and western blot analysis of KIF13B protein expression in WT mouse abdominal aortas treated with saline or PPE (n = 6/group). (H) Representative images of HE staining, EVG staining, and immunofluorescence staining illustrate α-SMA (green), CD68 (red), and KIF13B (red) expression levels in WT mouse abdominal aortas perfused with saline (n = 3) or angiotensin II (ANG II, n = 6). (I) qPCR analysis of Kif13b mRNA expression (n = 3/group) and western blot analysis of KIF13B protein expression in WT mouse abdominal aortas perfused with saline or ANG II (n = 3/group). (J) Correlation analysis of TMM-normalized counts between KIF13B and other genes in KEGG pathways. Node sizes represent the absolute values of Pearson's correlation coefficients. Data are presented as mean ± SEM. B, E, G left, i were analyzed by unpaired Student's t-test; G right was analyzed by Welch's t-test.
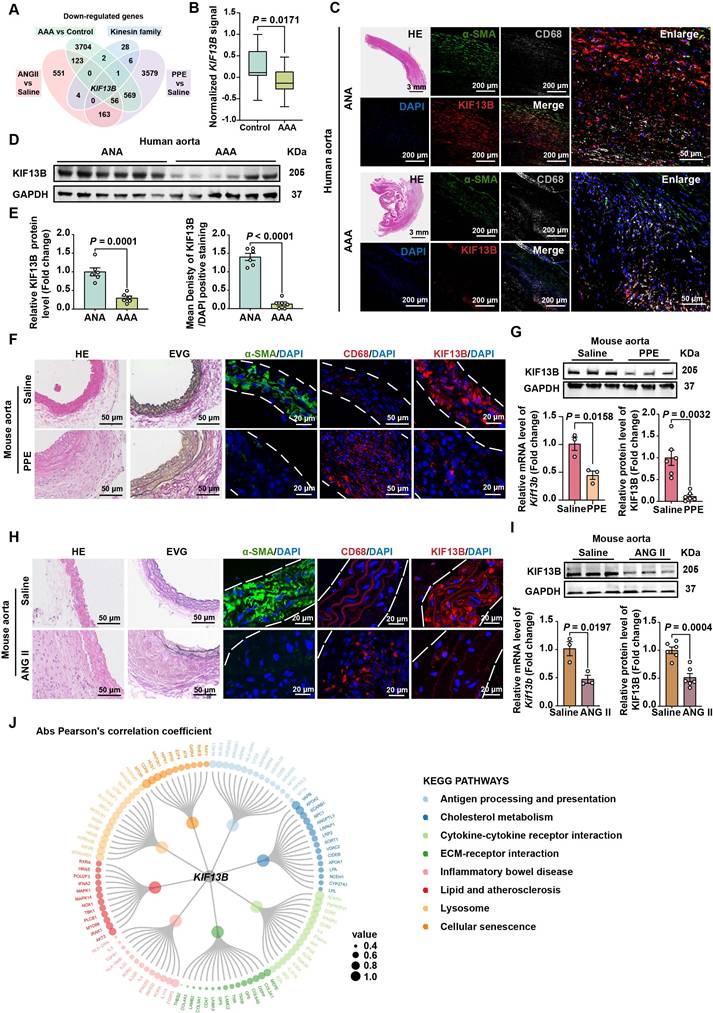
Global inactivation of Kif13b aggravates AAA development in vivo
To determine whether reduced KIF13B expression drives AAA pathogenesis or represents a compensatory response, we generated global Kif13b knockout (Kif13b-/-) mice. Following PPE induction, Kif13b-/- mice exhibited modestly elevated plasma cholesterol levels (Table S1) and severely aggravated AAA phenotypes showing the increased maximal diameter of the infrarenal abdominal aorta with mean value of 2.43 mm compared to wild-type (WT) mice with mean value of 1.85 mm (Figure 2A-B). Histological staining revealed an increase in vessel wall thickness and elastic fiber degradation in Kif13b-/- mice (Figure 2C-D), accompanied by reduced a-SMA-positive areas and more CD68 staining (Figure 2E). Since matrix metalloproteinase 2 (MMP2) and matrix metalloproteinase 9 (MMP9) are two key enzymes responsible for elastin destruction, we also examined the protein levels of MMP2 and MMP9 by immunofluorescence. Our result showed that loss of KIF13B led to the elevated levels of MMP2 and MMP9 (Figure 2E). Moreover, using terminal deoxynucleotidyl transferase dUTP nick-end labelling (TUNEL) staining, we found that depleting Kif13b promoted apoptosis in the aortic walls (Figure 2F).
Global inactivation of Kif13b aggravates AAA development in vivo. (A-F) PPE-Induced AAA Model: Eight-week-old male WT (n = 14) and Kif13b-/- (n = 15) mice were subjected to PPE-induced AAA. (A) Representative macroscopic images of AAA in each group. (B) Quantitative analysis of the maximal AAA diameter normalized to untreated aorta (Mann-Whitney U-test). (C) Residual elastic fiber area relative to vessel wall area (unpaired Student's t-test). (D) Representative HE and EVG staining of aortic sections. (E) Immunofluorescence staining of α-SMA (pink), CD68 (red), MMP2 (red), and MMP9 (red). (F) TUNEL staining (green) for apoptosis detection. (G-M) ANG II-Induced AAA Model with Hyperlipidemia: Eight-week-old male WT (n = 17) and Kif13b-/- (n = 14) mice underwent ANG II infusion combined with AAV8-PCSK9 injection and Western diet. (G-H) Plasma triglyceride (TG) and total cholesterol (TC) levels (n = 8/group; Mann-Whitney U-test for TG, unpaired t-test for TC). (I) Macroscopic AAA features. (J) Survival analysis (Kaplan-Meier method, log-rank test). (K) Maximal aortic diameter across segments (ascending [Asc], descending [Des], abdominal [Abd]; Welch's t-test for Asc, unpaired t-test for Des/Abd). (L) Elastin degradation severity (Mann-Whitney U-test). (M) Representative HE and EVG staining. Abbreviations: Asc, ascending aorta; Des, descending aorta; Abd, abdominal aorta. Data are mean ± SEM. Statistical methods are specified per panel.
To further confirm the deleterious effect of Kif13b deletion on AAA in a hyperlipidemic context, we also conducted a similar process to induce AAA mouse model using ANG II combined with AAV8-PCSK9D337Y injection and Western diet treatment in WT and Kif13b-/- mice. We found that Kif13b-/- mice exhibited markedly elevated plasma levels of triglyceride (TG) and total cholesterol (TC) (Figure 2G-H). ANG II infusion increased systolic and diastolic blood pressure to the same extent in WT and Kif13b-/- mice (Table S2). Survival curve analysis showed that the mortality rate of Kif13b-/- mice infused by ANG II was as high as 71.4%, which was three times higher than that of WT mice with mortality rate of 23.5 (Figure 2J). Next, we performed segmental analysis of aortic diameters in surviving mice and confirmed that Kif13b deletion resulted in significant enlargement of aortic root, ascending and abdominal aortic segments (Figure 2I-K) as well as increased vessel wall thickness and more disrupted elastic fibers (Figure 2L-M). These observations demonstrate that global Kif13b deletion promotes the development of AAA in vivo.
KIF13B deficiency exacerbates the enrichment of pro-inflammatory macrophages in AAA
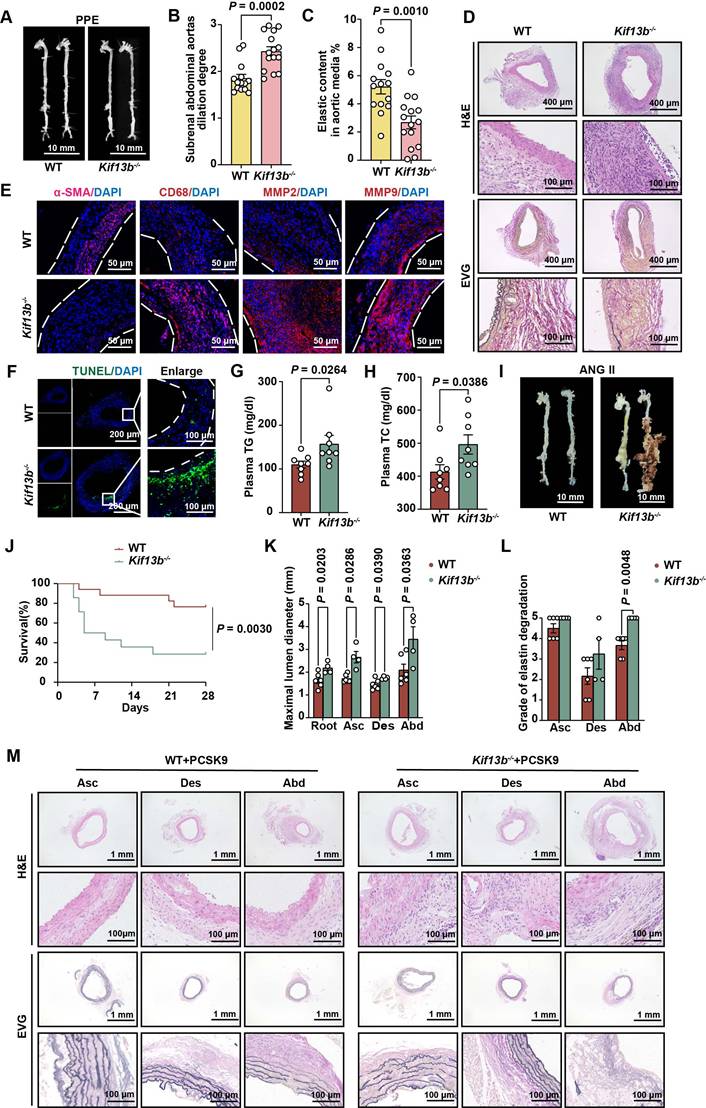
Of note that multiple cell types are involved in AAA formation. To delineate the cellular mechanisms underlying AAA exacerbation in Kif13b deficiency, we performed a single-cell sequencing (scRNA-seq) analysis on AAA tissues from 8-week-old male WT and Kif13b-/- mice subjected to PPE-induced AAA (Figure 3A). Unsupervised clustering of conserved gene expression profiles identified nine distinct cell populations, including macrophages, fibroblasts, smooth muscle cells (SMCs), and endothelial cells (Figure 3B), which were confirmed using canonical markers (Figure S1A). Strikingly, macrophages accounted for 50% of all cells in the AAA tissue of Kif13b-/- mice, a significant increase compared to WT controls (Figure 3C and Figure S1B), indicating that Kif13b deletion amplifies macrophage enrichment in AAA lesions.
KIF13B deficiency exacerbates the enrichment of pro-inflammatory macrophages in AAA. (A) Schematic of the single-cell sequencing experimental design. Aortic tissues from PPE-treated WT (n = 10) and Kif13b-/- (n = 9) mice were pooled into a single sample for sequencing. (B) Uniform Manifold Approximation and Projection (UMAP) plot depicting nine distinct cell clusters in AAA tissues, color-coded by cell type. (C) Proportional distribution of all cell types in each experimental group. (D) UMAP plot highlighting five macrophage subpopulations, color-labeled for differentiation. (E) Pseudotime analysis illustrating the differentiation trajectories of macrophage clusters shown in D. (F) Proportional distribution of macrophage subpopulations in each group. (G) Gene Set Variation Analysis (GSVA) of pro-inflammatory and anti-inflammatory gene signatures in each group. (H) Gene Set Enrichment Analysis (GSEA) of differentially expressed genes (DEGs) from D, using Hallmark and Reactome pathway databases. Pathways with a normalized enrichment score (NES) > 1.5 and P < 0.05 were considered significantly enriched.
scRNA-seq analysis further stratified macrophages into five distinct subpopulations, including three pro-inflammatory M1-like types marked by elevated CCR2, CD80, and CD86 and two anti-inflammatory M2-like types expressing CD206 and TREM2 (Figure 3D). Pseudotime trajectory analysis revealed a phenotypic shift from M2-like to M1-like macrophages during AAA progression (Figure 3E). Strikingly, Kif13b deletion skewed this balance, increasing the proportion of CD80-high pro-inflammatory macrophages while reducing CD206-high anti-inflammatory populations (Figure 3F, Figure S1C-E). Moreover, Gene Set Variation Analysis (GSVA) confirmed a pro-inflammatory state in Kif13b-deficient macrophages (Figure 3G). Consistently, complementary Gene Set Enrichment Analysis (GSEA) linked Kif13b deficiency to activation of oxidative phosphorylation, DNA repair, and cell cycle pathways (Figure 3H), suggesting KIF13B governs macrophage metabolic reprogramming and proliferative capacity to modulate polarization.
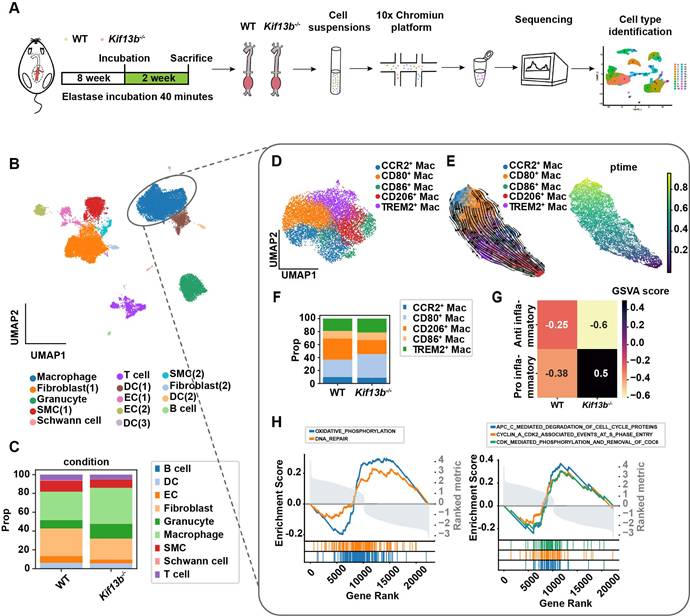
Loss of myeloid cell-specific KIF13B promotes AAA formation
To validate scRNA-seq findings, we constructed loxp flanked Kif13b (Kif13bf/f) mice, which were bred with the myeloid or smooth muscle cell-specific Cre transgenic mice to obtain macrophage (Lyz2Kif13bf/f) and smooth muscle cell (Sm22Kif13bf/f) specific-Kif13b conditional knockout mice, respectively. Both lines were subjected to PPE-induced AAA to evaluate the role of KIF13B in AAA development. Plasma lipid profiles remained unchanged across genotypes (Table S 1). As expected, mice lacking macrophage-derived Kif13b showed exacerbated AAA progression with increased aortic dilation, elastin degradation, and inflammatory infiltration; however, in contrast, depleting Kif13b from smooth muscle cells had no significant influence on AAA development (Figure 4A-G).
Loss of myeloid cell-specific KIF13B promotes AAA formation. (A-E) Eight-week-old male Kif13bf/f (n = 9), Lyz2CreKif13bf/f (n = 10), and Sm22αCreKif13bf/f (n = 10) mice were subjected to PPE-induced AAA. (A) Representative macroscopic images of AAA lesions. (B) Maximal AAA diameter normalized to untreated aorta. (C) Residual elastic fiber area relative to vessel wall area. (D-E) Representative HE and EVG staining of aortic sections. (F-G) Additional analyses in Kif13bf/f and Lyz2CreKif13bf/f mice. (F) Immunofluorescence staining of α-SMA (pink), CD68 (red), MMP2 (red), and MMP9 (red). (G) TUNEL staining (green) for apoptosis detection. Data are presented as mean ± SEM and analyzed by one-way ANOVA with Bonferroni post-hoc tests.
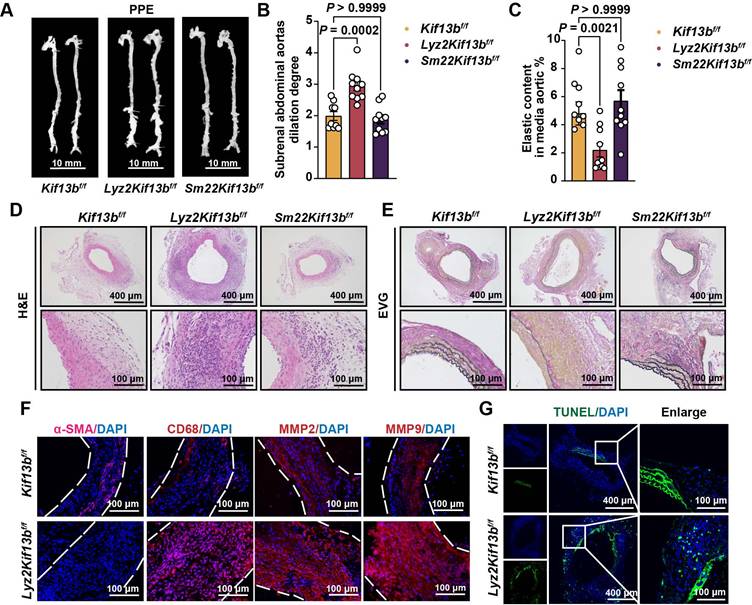
KIF13B maintains macrophage homeostasis by suppressing senescence
To investigate the relationship between KIF13B and macrophage inflammation, we analyzed LPS-induced pro-inflammatory polarization in vitro. LPS treatment significantly reduced KIF13B mRNA and protein levels in differentiated human acute monocytic leukemia cells (THP1 macrophages) (Figure S2A-C). Similarly, KIF13B expression was also significantly decreased in LPS-induced primary mouse bone marrow-derived macrophages (BMDMs) at both the mRNA and protein levels (Figure S2D-F).
We next defined the molecular mechanisms linking Kif13b deficiency to pro-inflammatory macrophage polarization by performing bulk RNA-seq on BMDMs from WT and Kif13b-/- mice (Figure 5A). KEGG and Gene Ontology (GO) analyses revealed pronounced activation of cellular senescence pathways in Kif13b-/- macrophages with an upregulation of pro-senescent genes and a downregulation of anti-aging genes (Figure 5B-C). Consistent with in vivo findings, Kif13b-/- BMDMs exhibited enhanced oxidative phosphorylation (OXPHOS), DNA repair, and cell cycle dysregulation (Figure 5D). Collectively, these data identify cellular senescence as a pivotal mechanism by which Kif13b deficiency promotes macrophage inflammation, then driving AAA progression.
KIF13B maintains macrophage homeostasis by suppressing senescence. (A) Volcano plot displaying differentially expressed genes (DEGs) in bone marrow-derived macrophages (BMDMs) from WT and Kif13b-/- mice (n = 3/group). Genes meeting significance thresholds (P < 0.05, |Fold change| > 1.2) are highlighted (1,925 upregulated in red; 1,816 downregulated in blue). (B) Bar plot showing significantly enriched KEGG and Gene Ontology (GO) pathways among DEGs. (C) Heatmap of senescence-related DEGs (pro- and anti-senescence genes) between genotypes. (D) Gene Set Enrichment Analysis (GSEA) of hallmark and Reactome pathways (significant enrichment: NES > 1.5, P < 0.05). (E-J) LPS-stimulated (10 ng/ml, 24h) BMDMs from 8-week-old male WT and Kif13b-/- mice. (E) SA-β-gal staining (representative images and quantification, n = 4). (F) ROS detection (fluorescence images and quantification, 3 independent experiments). (G) γ-H2A.X immunofluorescence (images and nuclear density quantification, n = 3). (H) qPCR analysis of SASP-related genes (n = 3). (I) Western blot of senescence markers (P21, P16, γ-H2A.X; n = 4). (J) qPCR analysis of cell cycle regulators (n = 3). (K-L) In vivo analysis of PPE-induced AAA. (K) SA-β-gal staining in aortic tissues (n = 6). (L) Immunofluorescence of CD68+ macrophages with γ-H2A.X (DNA damage marker). Data presented as mean ± SEM. E-G, H were analyzed by two-way ANOVA with Bonferroni post-hoc tests. I-J P19ARF and P21 were analyzed by unpaired Student's t-test. I-J P16INK4a was analyzed by Welch's t-test.
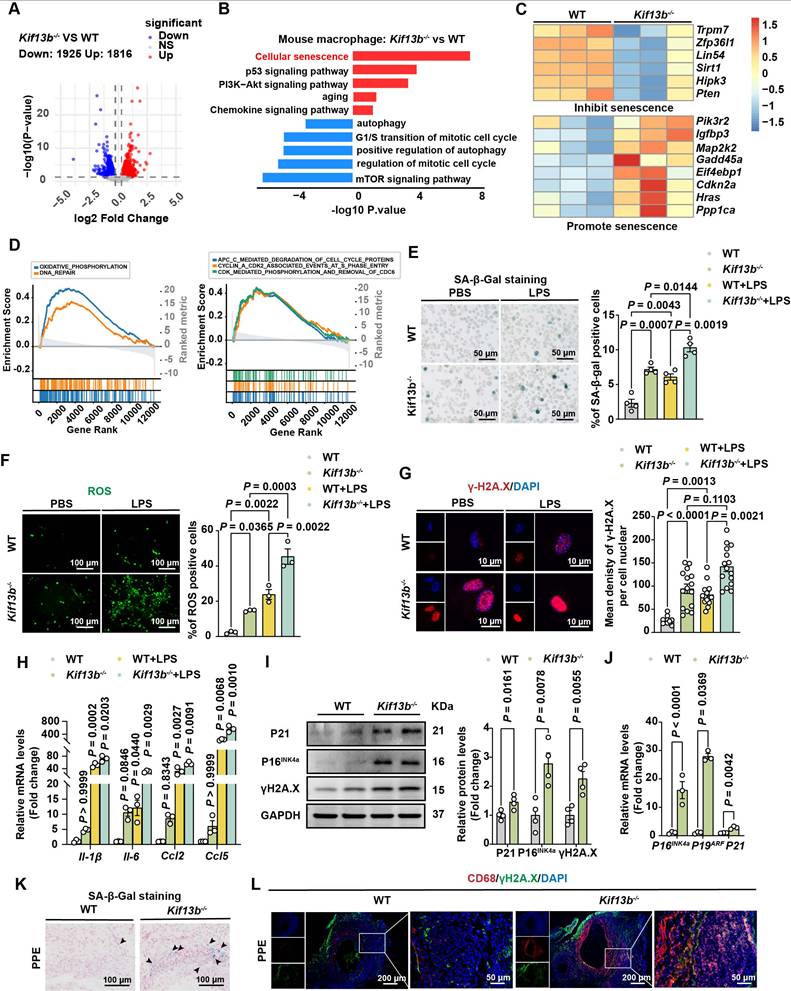
Given the established link between aging and AAA risk, we assessed whether Kif13b deletion promoted macrophage senescence using senescence-associated β-galactosidase (SA-β-gal) activity [26]. The result from SA-β-gal staining demonstrated that macrophage senescence was significantly driven by silencing Kif13b with or without LPS treatment (Figure 5E). In addition, ROS levels were elevated in Kif13b-/- macrophages under both basal and LPS-treated conditions (Figure 5F). Consistently, more γ-H2A. X, another hallmark of senescent cells, was prominently aggregated in the nucleus (Figure 5G). LPS-stimulated macrophages with Kif13b deficiency displayed worsened severity of senescence-associated secretory phenotype (SASP) with increased mRNA expression of Il-1β and Il-6, which can be used as a biomarker of senescence. The severity of SASP was worsened in Kif13b-deficient macrophages stimulated with LPS (Figure 5H), and upregulated expression of cell cycle regulators, including Cdkn1a (P21) and Cdkn2a (P16INK4a and P19ARF), at both mRNA and protein levels (Figure 5I-J). In accordance, SA-β-gal activity was elevated in aneurysmal tissue of Kif13b-/- mice deletion in vivo (Figure 5K). Immunofluorescence results further demonstrated that loss of Kif13b exacerbated macrophage senescence as evidenced by co-localization of CD68 and γ-H2A.X (Figure 5L).
Considering the therapeutic potential of senolytics in cardiovascular diseases, we tested quercetin, a well-established senolytic agent [27], in Kif13b-/- BMDMs. Our results revealed that quercetin effectively reduced the number of SA-β-gal positive cells in a dose-dependent manner and concurrently downregulated the expression of pro-inflammatory genes (Figure S3A-B). These findings confirm that Kif13b deficiency drives macrophage senescence and pro-inflammatory polarization and demonstrate that senolytic intervention mitigates these effects.
KIF13B maintains lysosomal function to inhibit macrophage senescence in vitro
Since KIF13B executed beneficial functions in cellular senescence and inflammation, to determine whether KIF13B overexpression could counteract senescence and inflammation, we constructed a lentiviral vector overexpressing Kif13b (LV- KIF13B-HA) to infect WT mouse BMDMs. Overexpression of KIF13B significantly reduced the level of senescence and decreased the mRNA expression of pro-inflammatory genes in macrophages, exclusively in LPS-induced pro-inflammatory macrophages (Figure 6A-B).
KIF13B maintains lysosomal function to inhibit macrophage senescence in vitro. (A-B) WT mouse BMDMs were infected with LV-GFP-HA (negative control, NC) or LV- KIF13B -HA overexpressing KIF13B (overexpression, OE), followed by treatment with PBS or 1 μg/ml LPS for 24 h ± chloroquine (CQ) pretreatment. (A) SA-β-gal staining (representative images, n = 6/group). (B) qPCR analysis of SASP-related genes (n= 3 /group). (C-E) Primary BMDMs from 8-week-old male WT and Kif13b-/- mice stimulated with PBS or 10 ng/ml LPS (n = 3 independent experiments/group). (C) Lysotracker (red) and DAPI (blue) staining (images and lysosomal density quantification). (D) Lysosensor staining (images and Yellow/Blue ratio quantification for pH measurement). (E) Bodipy-cholesterol uptake (images and Pearson's coefficient for lysosome-cholesterol colocalization). (F) qPCR and Western blot analysis of TFEB mRNA and protein expression levels in primary mouse BMDMs from WT and Kif13b-/- mice (n = 6/group), respectively. (G) Immunofluorescence of TFEB (red) with DAPI (images and quantification of cytoplasmic/nuclear density, n=6 experiments). (H-J) Primary mouse BMDMs were extracted from 8-week-old female WT and Kif13b-/- mice and then TFEB activator 1 (TA1) was used to treat Kif13b-/- macrophages. (H) SA-β-gal staining (n= 9 /group). (I) SASP gene expression (n= 3 /group). (J) γ-H2A.X staining (images and nuclear density quantification, n = 6 experiments). Data were presented as mean ± SEM. Data A-E and G were analyzed by two-way ANOVA. Data F were analyzed by unpaired Student's t-test. Data H-J were analyzed by one-way ANOVA with Bonferroni post-hoc tests.
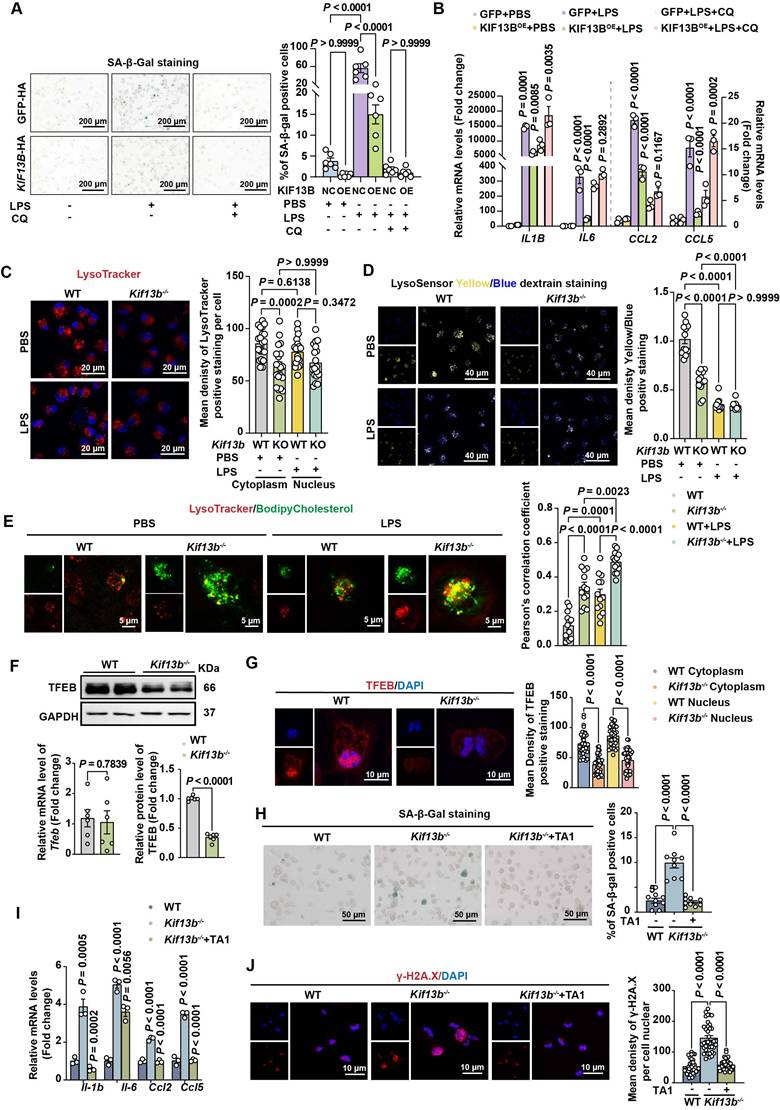
Emerging evidence implicates that lysosomal dysfunction in cellular senescence [28]. Our transcriptomic analysis revealed that genes associated with lysosomal pathway were highly enriched and their expression was upregulated upon Kif13b deletion (Figure S4A). To test whether KIF13B's beneficial effects depended on lysosomal activity, we employed the lysosomal inhibitor chloroquine. In WT mouse BMDMs pretreated with chloroquine, overexpression of KIF13B failed to suppress macrophage senescence and inflammation (Figure 6A-B). Moreover, Chloroquine-treated WT BMDMs showed no additional senescence exacerbation upon Kif13b deletion (Figure S4B), indicating that lysosomal function is a key mediator required for the regulatory role of KIF13B.
Next, to define the role of KIF13B in lysosomal regulation, we analyzed lysosomal integrity and activity in BMDMs. Lysotracker staining revealed a significant reduction in lysosome number in Kif13b-deficient cells compared to WT controls, whereas LPS stimulation failed to worsen this deficit in Kif13b-deficient cells (Figure 6C), suggesting that lysosomal dysfunction is a major consequence of Kif13b deficiency. In addition, we assessed lysosomal activity by monitoring lysosomal pH using Lysosensor and found that lysosomal activity was markedly decreased in Kif13b-deficient macrophages compared to controls treated with PBS (Figure 6D). However, LPS stimulation abolished this difference, likely due to lysosomal stress overriding KIF13B-dependent regulation (Figure 6D). Additionally, Kif13b deficiency diminished the mRNA abundance of vacuolar ATPases (V-ATPases), an essential regulator responsible for lysosomal proton transport (Figure S4C). Collectively, these findings demonstrate that knocking out Kif13b reduces the number of macrophage lysosomes and increases lysosomal pH, thus disrupting proper lysosomal functions.
Based on the evidence demonstrating a link between lysosomal cholesterol accumulation and SASP phenotype [29], we determined whether KIF13B regulated lysosomal cholesterol homeostasis. BODIPY-cholesterol labeling in BMDMs revealed that more lysosomal cholesterol content was detected in Kif13b-deficient cells under the PBS treatment condition, and Kif13b deletion further exacerbated lysosomal cholesterol accumulation in LPS-treated macrophages (Figure 6E).
TFEB is implicated in aging and age-related diseases [30]. We investigated whether TFEB mediated KIF13B's effects on lysosomal homeostasis. Interestingly, Tfeb mRNA expression remained unchanged in both WT and Kif13b-/- BMDMs (Figure 6F). However, using confocal technique and Western blot we found that TFEB protein levels, particularly in the nucleus, were dramatically reduced in Kif13b-deficient macrophages (Figure 6F-G), indicating impaired nuclear translocation. Moreover, we observed that TA1, a TFEB agonist, significantly reversed SASP phenotype in Kif13b-deficient macrophages, with fewer SA-β-gal-positive cells and less DNA damage, nearly close to WT control cells (Figure 6H-J). These data demonstrated TFEB as a critical downstream effector of KIF13B, mediating its anti-senescent and anti-inflammatory roles in macrophages.
KIF13B stabilizes TFEB via USP9X-mediated deubiquitination
As KIF13B localizes to the cytoplasm and does not directly regulate Tfeb transcription, we hypothesized it might modulate TFEB at the post-translational level. siRNA-mediated Kif13b knockdown in THP1-derived macrophages accelerated TFEB degradation under cycloheximide (CHX)-mediated translational blockade. By treating cells with the proteasome inhibitor MG132 or the lysosomal inhibitor chloroquine, respectively, we found that TFEB was mainly degraded through the proteasomal ubiquitination pathway (Figure 7A). Additionally, co-immunoprecipitation (co-IP) confirmed enhanced TFEB ubiquitination in Kif13b-/- macrophages (Figure 7B).
KIF13B stabilizes TFEB via USP9X-mediated deubiquitination. (A) TFEB protein stability was assessed by western blot in macrophages treated with PBS, 10 µM MG132 (proteasome inhibitor), or 10 µM chloroquine (lysosomal inhibitor) in the presence of 50 µg/ml cycloheximide (CHX) at indicated time points. (B) Ubiquitination status of TFEB was evaluated by co-immunoprecipitation in primary BMDMs (n = 3/group). (C) Integrated transcriptomic analysis identified six significantly downregulated deubiquitinating enzymes in both Kif13b-/- BMDMs and human AAA tissues (heatmap presentation). (D) The HDOCK database suggests that USP9X can bind to KIF13B. (E) Immunofluorescence imaging demonstrated co-localization of USP9X (green) and KIF13B (red) in human AAA lesions versus adjacent healthy tissue (n = 3/group). (F) USP9X expression was quantified by western blot in THP1-derived macrophages following KIF13B knockdown or overexpression, primary WT versus Kif13b-/- BMDMs (n = 4/group in each condition). (G-I) Protein interactions were confirmed through endogenous (G), semi-exogenous (H) co-IP assays in primary BMDMs and in vitro GST pull-down assays using recombinant proteins. (J) Ubiquitination status of USP9X was evaluated by co-immunoprecipitation in primary BMDMs (n = 3/group). (K) Schematic of human full-length KIF13B and its three functional domains (Motor, FHA, RLIP). (L) Protein interactions between USP9X and KIF13B or its different functional domains were confirmed through semi-exogenous co-IP assays in THP1-derived macrophages (M) TFEB ubiquitination was measured following treatment with 5 µM WP1130 (deubiquitinase inhibitor). (N-O) TFEB protein levels(N) and ubiquitination status (O) were analyzed in KIF13B -overexpressing BMDMs with DMSO or WP1130 treatment (n = 3/group). Data represent mean ± SEM. F was analyzed by unpaired Student's t-test. N was analyzed by one-way ANOVA with Bonferroni correction.
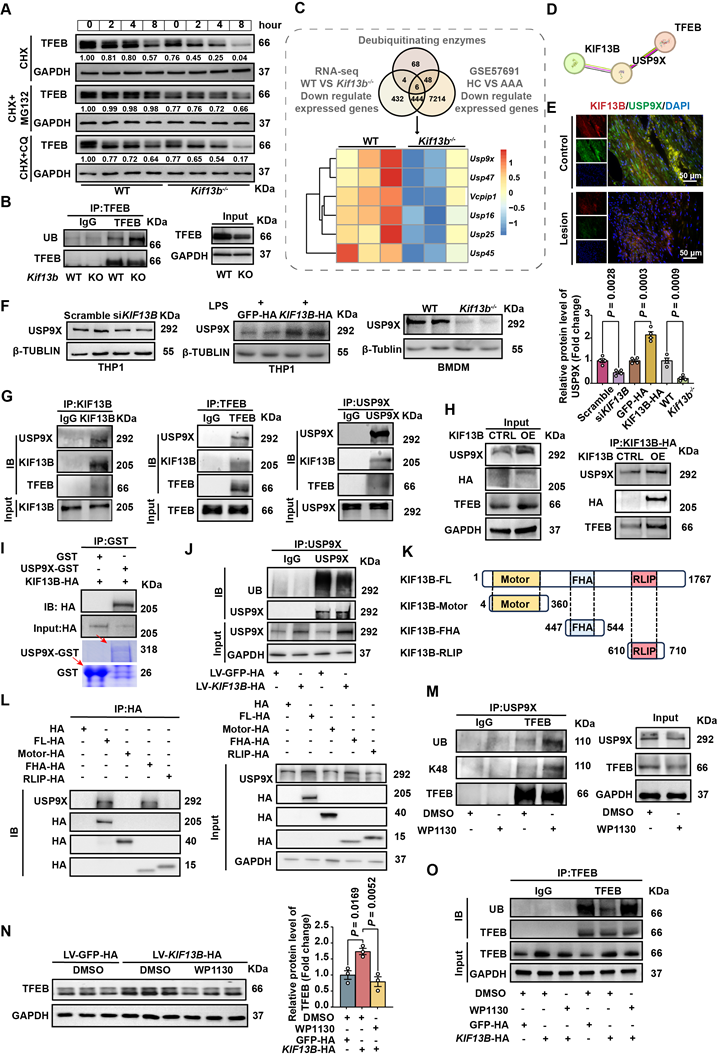
Next, to identify the potential ubiquitination enzymes responsible for TFEB degradation regulated by KIF13B, we integrated transcriptomic data primary mouse BMDMs and human AAA tissues, revealing six downregulated deubiquitinating enzymes (DUBs), including Usp9x, Usp45, Vcpip1, Usp47, Usp16 and Usp25. Among these, USP9X was the only candidate predicted by the HDOCK database to functionally interact with KIF13B with a high confidence score of 0.9186 (Figure 7C-D). Our immunofluorescence results showed a co-localization of KIF13B and USP9X in healthy aortic walls, which was abolished in AAA lesions (Figure 7E). Furthermore, Western blot data showed that siRNA-mediated Kif13b knockdown in THP1-derived macrophages reduced USP9X protein levels (Figure 7F). Conversely, LV-mediated overexpression of KIF13B in THP1-derived macrophages led to an increase in USP9X, which were consistently observed in BMDMs isolated from Kif13b-/- mice (Figure 7F). Importantly, co-IP revealed physical interaction between KIF13B, TFEB and USP9X (Figure 7G-H). Moreover, KIF13B was further validated to directly interact with USP9X through in vitro binding assays, demonstrating the effect on USP9X protein stability by suppressing ubiquitination-dependent degradation (Figure 7I-J).
To clarify the structure-function relationship of KIF13B and USP9X stabilization, a domain-specific transfection strategy was employed in THP1-derived macrophages by overexpressing full-length KIF13B and its discrete functional domains. It is noteworthy that USP9X only bound to fu full-length KIF13B or its FHA domain, concomitant with enhanced USP9X protein levels (Figures 7I-J). Consistently, the FHA domain alone possessed the anti-senescence capacity of full-length KIF13B, significantly reducing LPS-induced senescence markers in macrophages (Figure S5A). To confirm that USP9X may act as a deubiquitinating enzyme for TFEB, we treated mouse BMDM with the inhibitor of USP9X, WP1130 [31], which reduced USP9X and TFEB protein levels while increased K48-linked ubiquitination of TFEB (Figure 7M), consistent with proteasomal degradation. Additionally, the elevation of TFEB protein levels by KIF13B overexpression in BMDMs was abolished in the presence of WP1130 treatment, an effect that was mechanistically linked to altered ubiquitination dynamics (Figure 7N-O). Taken together, our data demonstrated that the FHA domain of KIF13B specifically mediates stabilization of USP9X protein, which subsequently deubiquitinates TFEB to suppress macrophage senescence and inflammatory responses.
Reconstitution of macrophage KIF13B or senolytic therapy effectively attenuates AAA progression in vivo
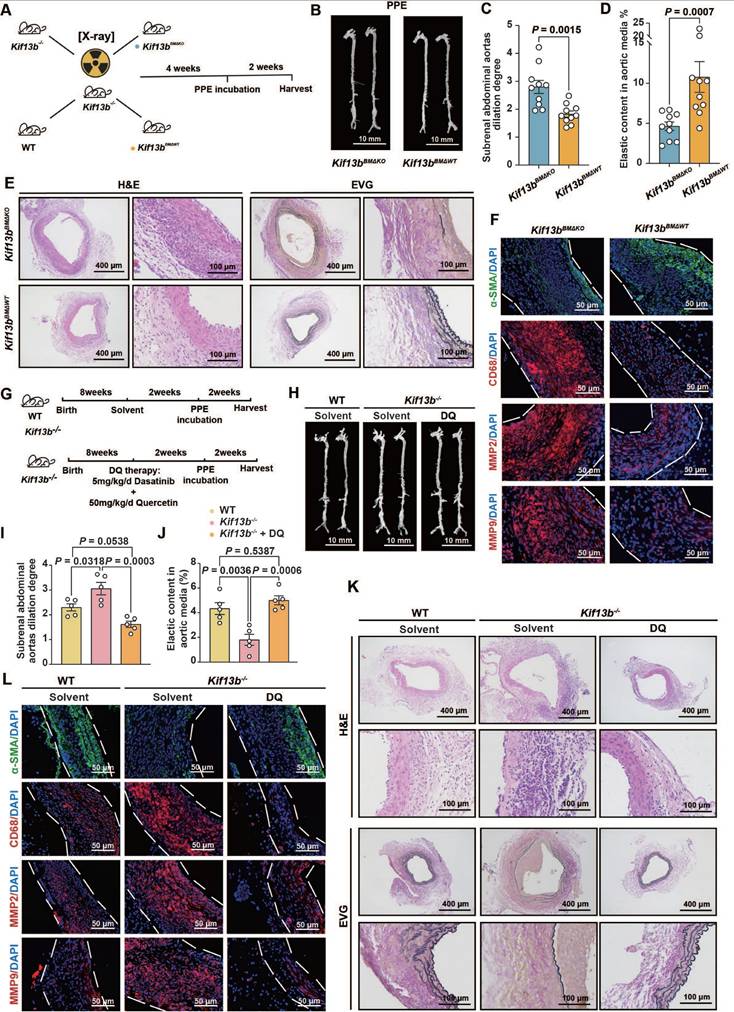
Based on the confirmed observations that Kif13b deficiency in macrophages triggered the exacerbation of AAA as described above, we sought to develop an intervention for mitigating the AAA process by reconstituting KIF13B in mouse macrophage. Kif13b-/- mice underwent bone marrow transplantation (BMT) and then received bone marrows isolated from either WT or Kif13b-deficient mice (Figure 8A). The results showed that transplantation of WT bone marrows significantly attenuated the severity degree of PPE-induced AAA expansion and elastic fiber degradation compared to Kif13b-/- mice transplanted with Kif13b-/- bone marrows (Figure 8B-D). Consistently, the phenotypes of AAA with accelerated extracellular matrix degradation, loss of contractile smooth muscle cells, macrophage infiltration and MMP secretion in PPE-infused Kif13b-/- mice were largely reversed by transplanting WT bone marrows (Figure 8E-F). Furthermore, to clarify the impact of senescence on AAA development in the context of Kif13b deficiency, we performed senolytic therapy on Kif13b-/- mice. In agreement with the senolytic results in vitro, oral administration of dasatinib and quercetin to Kif13b-/- mice significantly reversed AAA severity when compared to solvent-treated Kif13b-/- mice (Figure 8G-L), further suggesting that restoring myeloid KIF13B or eliminating senescent cells mitigates AAA progression caused by Kif13b deficiency, positioning KIF13B as a key protector against macrophage senescence-driven vascular pathology.
Reconstitution of macrophage KIF13B or senolytic therapy effectively attenuates AAA progression in vivo. (A-F) Eight-week-old male Kif13b-/- mice underwent bone marrow transplantation (BMT) from either Kif13b-/- or WT donors, followed by PPE-induced AAA modeling. (A) Study design (n = 10/group). (B) Representative images of the macroscopic features. (C) Quantitative analysis of the ratio of the maximal diameter of the AAA to the untreated normal aorta. (D) Residual elastic fiber area relative to vessel wall area. (E) Representative images of HE staining and EVG staining in each group. (F) Representative immunofluorescence staining images of α-SMA (green), CD68 (red), MMP2 (red) and MMP9 (red) expression levels in each group. (G-L) WT and Kif13b-/- mice received dasatinib (5mg/kg/d) + quercetin (50mg/kg/d) or vehicle for 2 weeks pre-PPE induction (n = 5/group). (G) Study design. (H) Representative images of the macroscopic features. (I) Quantitative analysis of the ratio of the maximal diameter of the AAA to the untreated normal aorta. (J) Residual elastic fiber area relative to vessel wall area. (K) Representative images of HE staining and EVG staining in each group. (L) Representative immunofluorescence staining images of α-SMA (green), CD68 (red), MMP2 (red) and MMP9 (red) expression levels in each group. Data were presented as mean ± SEM. Data C, D was analyzed by un-paired student t test. Data I, J was analyzed by one-way ANOVA with Bonferroni correction.
Discussion
AAA is a life-threatening disease with high morbidity and mortality worldwide; however, the therapeutic treatment is still lacking to date because the pathogenesis underlying this devastating disorder is complicated and remains elusive. In the present study, our results have elucidated pathogenic mechanisms linking the sustained senescence and proinflammation of macrophages to AAA formation. Meanwhile we also identified KIF13B as a key molecule responsible for maintaining proper lysosomal function to restrict senescence and inflammatory response in macrophages. First, our investigation revealed a significant reduction in KIF13B expression levels within aortic aneurysm lesions from both human patients and multiple mouse AAA models. Global inactivation of Kif13b or macrophage specific deletion of Kif13b exacerbated the development of aortic aneurysm in PPE or ANG II-infused mouse models. Furthermore, mechanistic studies revealed that depleting Kif13b elicited accelerated senescence of macrophages accompanied with a phenotypic switch of M2 to M1 due to abnormal cholesterol accumulation in lysosomes and impaired TFEB-dependent lysosomal biogenesis. KIF13B mediated the stability of TFEB at the protein level by interacting with USP9X, a critical deubiquitinating enzyme (DUB), thereby enhancing TFEB deubiquitination and elevating its cellular levels. Supplementation of TFEB agonist also reversed the deleterious effects caused by Kif13b deficiency in cultured macrophages in vitro. Finally, macrophage-specific restoration of KIF13B through BMT or senolytic therapy exhibited a pronounced protective effect on AAA progression in Kif13b-/- mice. These findings demonstrate that macrophage KIF13B acts as a protective factor of AAA pathogenesis.
KIF13B, the largest member of kinesin-3 family, possesses multiple functions, including cellular signaling transduction, receptor trafficking and cell division [15,32-34], and its deficits are associated with neurodegeneration, developmental defect and cancer [18,35,36]. Recently, our work discovered that hepatic KIF13B expression levels were negatively correlated with MAFLD progress and KIF13B orchestrated mitochondrial function and de novo lipogenesis (DNL) to maintain liver energy homeostasis by activating AMPK pathway, suggesting that KIF13B plays a critical role in hepatic lipid metabolism and the related metabolic disease [21]. To our knowledge, MAFLD has been reported to contribute to CVD through the mechanisms related to metabolic dysregulation, chronic inflammation, and oxidative stress [22]. Recent evidence from a UK-based prospective cohort study further supports a positive association between MAFLD and AAA [23]. Interestingly, while whole body Kif13b deficiency resulted in aggravated AAA expansion in both PPE and ANG II-infused mouse models, the latter caused more severity of AAA development and higher death rate than the former. This difference is likely due to the additional induction of hyperlipidemia in ANG II-infused mice through AAV8-PSCK9D337Y delivery and Western diet feeding. Consistently, two independent studies led by Roychowdhury and Lu have reported that PSCK9 and triglyceride rich lipoproteins could be potential therapeutic targets for the treatment of AAA [5,9], further supporting the concept that hyperlipidemia is a risk factor of AAA pathogenesis.
It is noteworthy that multiple cell types contribute to the pathogenesis of AAA, yet how Kif13b deficiency exacerbated AAA expansion remained unknown in the whole-body knockout mice. To address this, using single-cell RNA sequencing technology and bioinformatic analyses, we identified that macrophages were enriched and accumulated in the aortic lesions in Kif13b-/- mice, in which more M1-like cells marked with CCR2, CD80, and CD86 and less M2-like cells marked with CD206 and TREM2 were observed. These findings are consistent with the previous observations [37], suggesting that macrophages migrate into the injured sites and then contribute to the inflammatory response. Importantly, although VSMC phenotypic switching is a well-established characteristic of AAA, we unexpectedly found that VSMC-specific Kif13b depletion did not influence AAA formation. In contrast, macrophage-specific Kif13b knockout mice exhibited increased susceptibility to PPE-induced AAA. These results indicate that macrophages rather than VSMCs serve as the primary cellular drivers of AAA pathogenesis in the context of Kif13b deficiency. Therefore, loss of Kif13b in macrophages was sufficient to promote AAA development independently of plasma lipid level alterations.
Inflammation of the vascular wall has long been recognized as a key trigger for AAA [38]. Chronic inflammation, one of the twelve hallmarks of aging, is inextricably linked to the aging process [26,39]. Senescent cells that possess highly metabolic activities can secrete pro-inflammatory factors, termed SASP, to promote chronic inflammation, which in turn drives healthy cells into the senescence process [40]. Over the past decade, vascular cell senescence has been progressively linked to AAA pathogenesis, and significant progress has been made particularly in the field of the therapeutic targeting of smooth muscle cell senescence for the treatment of AAA disease [41-43]. However, the role of macrophage senescence in AAA pathogenesis remains poorly understood, despite linking immune dysfunction, which contributes to age-related and autoimmune diseases, to the aberrant accumulation of senescence-like macrophages [44]. Remarkably, our in vitro data demonstrated that Kif13b-deficient macrophages spontaneously developed senescent characteristics, with elevated SA-β-gal activity and activated SASP, even without proinflammatory intervention such as LPS stimulation. These cells also exhibited enhanced DNA damage responses and upregulated cyclin-dependent kinase inhibitors (CDKIs), resulting in G1 phase cell cycle arrest. Senescent macrophages lacking Kif13b expression migrated to and accumulated in the vascular wall, impairing vascular homeostasis through enhanced inflammatory responses, then leading to accelerated MMP-mediated extracellular matrix degradation and ultimately promoted AAA progression.
Importantly, accumulating evidence suggests that the integrity and function of lysosomes significantly decline in various diseases and during aging [45-47]. Ruckenstuhl et al. have positioned the lysosome as a central cellular hub for the control of aging [28]. A recent study has also confirmed that abnormal cholesterol accumulation in the lysosome is a hallmark of lysosomal dysfunction linked to aging-related inflammation [29]. Consistently, our results also showed that Kif13b-deficient BMDMs exhibited a reduced number of lysosomes, abnormally elevated lysosomal pH reflecting impaired enzymatic activity, and substantial cholesterol accumulation within lysosomes. Of note, it has been documented that when lysosomes are damaged, TFEB, a key transcription factor required for lysosomal biogenesis, undergoes nuclear translocation to activate the transcription of lysosome-associated genes [48]. Dysregulated TFEB has been detected in patients with aneurysms, which is highly associated with smooth muscle cell apoptosis and macrophage inflammation, but the underlying molecular mechanisms are still missing [49,50]. In the present study, TFEB was identified as a central downstream target of KIF13B in macrophages to mediate the regulation of macrophage senescence by KIF13B. Interestingly, rather than being able to promote nuclear translocation of TFEB, KIF13B also upregulated the protein level of TFEB by inhibiting its degradation via the proteasomal ubiquitination pathway. Through a comprehensive intersection analysis of transcriptomics data and GEO database combined with predicted functional interaction by STRING software, we identified USP9X, one of the deubiquitinating enzyme family members as a crucial modulator involved in KIF13B-mediated TFEB regulation. Interestingly, several studies have reported that USP9X mainly controls cell adhesion and polarity, which has been implicated in neurodegenerative disease and tumor [51,52]. However, recent investigation led by Wang and colleagues discovered that USP9X executed protective function in the development of atherosclerosis [53]. Similarly, our results also revealed a significant reduction in USP9X expression levels in the aortic lesions of AAA patients. We identified TFEB as a downstream target of USP9X in macrophages, wherein KIF13B suppresses TFEB ubiquitination and attenuates senescence through interacting with USP9X. Furthermore, BMT with WT bone marrows to Kif13b-/- mice or administration of senolytic therapy could largely reverse AAA progress in vivo, demonstrating that targeting macrophage KIF13B represents a novel therapeutic strategy for AAA. Given the intricate regulatory network governing the dynamic balance between ubiquitination and deubiquitination, our findings reported in the present study, while identifies that the FHA domain of KIF13B binds to USP9X and then maintains its protein stability, thereby enhancing USP9X protein expression, cannot fully elucidate the precise regulatory mechanism underlying this stabilization at the current stage and future investigations are warranted to solve this issue.
In summary, our current study provides the first evidence that KIF13B exerts a protective effect against AAA pathogenesis. We elucidated a novel molecular mechanism wherein KIF13B stabilizes TFEB through promoting USP9X-mediated deubiquitination to maintain lysosome homeostasis, thus mitigates macrophage senescence and inflammation. These findings not only identified an inhibitory role of macrophage-derived KIF13B in AAA progress, but also highlighted the potentially therapeutic strategies for the treatment of AAA disease.
Supplementary Material
Supplementary materials and methods, figures and tables.
Acknowledgements
The authors thank Drs. Hongliang Li and Zhigang She (Department of Cardiology, Renmin Hospital, School of Basic Medical Science, Institute of Model Animal of Wuhan University, Wuhan, China) for providing Lyz2-Cre and Sm22-Cre mice. We are indebted to Aiden Xian from Shepton High School for proofing the manuscript.
Funding
This work was supported by the National Natural Science Foundation of China (NSFC) 82270479, 82070460 and HY2021-1 to X.X.; the Beijing Natural Science Foundation 7242084 to X.X.; the Fundamental Research Funds for the Central Universities to X.X.; Peking University Medicine plus X Pilot Program-Platform Construction Project 2024YXXLHPT010 to X.X.; the National Key Research and Development Program of China from the Ministry of Science and Technology 2021YFF0702802 to Y.W.
Author contributions
X.X. provided funding, designed the study, directed data analysis, and reviewed and edited the article; J.C. played a central role in performing the experiments, analyzing the data, and writing the article; Y.X. constructed the viruses and was involved in completing the preparation of knockdown and overexpression of KIF13B in vitro; H.Y. and Y.Q. performed clinical sample collection; Y.L. and D.Z. were responsible for the bioinformatics analysis; G.M. was involved in subject direction; Y.H. bred and genotyped Kif13b smooth muscle-specific knockout mice; L.W. was involved in some of the pathological staining. Z.C. guided the modelling methodology of AAA.Y.X. and Z.Z. were involved in the design of the mechanistic mapping. J.C., S.S., P.L., W.Z., L.Z., S.M., Y.Z. participated in some of the molecular biology experiments. L.Z. was responsible for the procurement of laboratory consumables and reagents. W.H., Y.W., D.Z., W.K., Y.Q., E.D., and X.X. participated in the discussion of the experimental protocols and the interpretation of the data.
Resource availability
The data supporting the findings of this study are available from the corresponding author upon reasonable request.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Kent KC. Abdominal Aortic Aneurysms. Solomon CG, Ed. N Engl J Med. 2014;371:2101-8
2. Schanzer A, Oderich GS. Management of Abdominal Aortic Aneurysms. Solomon CG, Ed. N Engl J Med. 2021;385:1690-8
3. Chaikof EL, Dalman RL, Eskandari MK. et al. The Society for Vascular Surgery practice guidelines on the care of patients with an abdominal aortic aneurysm. J Vasc Surg. 2018;67:2-77.e2
4. Golledge J, Thanigaimani S, Powell JT, Tsao PS. Pathogenesis and management of abdominal aortic aneurysm. Eur Heart J. 2023;44:2682-97
5. Lu HS, Temel RE, Levin MG, Damrauer SM, Daugherty A. Research Advances in Abdominal Aortic Aneurysms: Triglyceride-Rich Lipoproteins as a Therapeutic Target. Arterioscler Thromb Vasc Biol. 2024;44:1171-4
6. Harrison SC, Holmes MV, Burgess S. et al. Genetic Association of Lipids and Lipid Drug Targets with Abdominal Aortic Aneurysm: A Meta-analysis. JAMA Cardiol. 2018;3:26
7. Nastasi DR, Norman R, Moxon JV. et al. The Potential Benefits and Costs of an Intensified Approach to Low Density Lipoprotein Cholesterol Lowering in People with Abdominal Aortic Aneurysm. Eur J Vasc Endovasc Surg. 2021;62:643-50
8. Thomas PE, Vedel-Krogh S, Nielsen SF, Nordestgaard BG, Kamstrup PR. Lipoprotein(a) and Risks of Peripheral Artery Disease, Abdominal Aortic Aneurysm, and Major Adverse Limb Events. J Am Coll Cardiol. 2023;82:2265-76
9. Roychowdhury T, Klarin D, Levin MG. et al. Genome-wide association meta-analysis identifies risk loci for abdominal aortic aneurysm and highlights PCSK9 as a therapeutic target. Nat Genet. 2023;55:1831-42
10. Chan CYT, Chan YC, Cheuk BL, Cheng SW. A Pilot Study on Low-density Lipoprotein Receptor-related Protein-1 in Chinese Patients with Abdominal Aortic Aneurysm. Eur J Vasc Endovasc Surg. 2013;46:549-56
11. Chan CYT, Chan YC, Cheuk BLY, Cheng SWK. Clearance of matrix metalloproteinase-9 is dependent on low-density lipoprotein receptor-related protein-1 expression downregulated by microRNA-205 in human abdominal aortic aneurysm. J Vasc Surg. 2017;65:509-20
12. Bown MJ, Jones GT, Harrison SC. et al. Abdominal Aortic Aneurysm Is Associated with a Variant in Low-Density Lipoprotein Receptor-Related Protein 1. Am J Hum Genet. 2011;89:619-27
13. Wild JB, Stather PW, Sylvius N, Choke E, Sayers RD, Bown MJ. Low Density Lipoprotein Receptor Related Protein 1 and Abdominal Aortic Aneurysms. Eur J Vasc Endovasc Surg. 2012;44:127-32
14. Munshaw S, Bruche S, Redpath AN. et al. Thymosin β4 protects against aortic aneurysm via endocytic regulation of growth factor signaling. J Clin Invest. 2021;131:e127884
15. Friel CT. The Kinesin Superfamily Handbook: Transporter, Creator, Destroyer [Internet]. 1st edn. Friel CT, Ed. First edition. | Boca Raton: CRC Press. 2020:CRC Press; 2020 [cited 10 June 2024]. Available at: https://www.taylorfrancis.com/books/9780429958250
16. Kanai Y, Wang D, Hirokawa N. KIF13B enhances the endocytosis of LRP1 by recruiting LRP1 to caveolae. J Cell Biol. 2014;204:395-408
17. Cho H-D, Nhàn NTT, Zhou C. et al. KIF13B mediates VEGFR2 recycling to modulate vascular permeability. Cell Mol Life Sci. 2023;80:91
18. Waters SB, Dominguez JR, Cho H-D, Sarich NA, Malik AB, Yamada KH. KIF13B-mediated VEGFR2 trafficking is essential for vascular leakage and metastasis in vivo. Life Sci Alliance. 2022;5:e202101170
19. Waters SB, Zhou C, Nguyen T. et al. VEGFR2 Trafficking by KIF13B Is a Novel Therapeutic Target for Wet Age-Related Macular Degeneration. Investig Opthalmology Vis Sci. 2021;62:5
20. Yamada KH, Nakajima Y, Geyer M. et al. KIF13B regulates angiogenesis through golgi-plasma membrane trafficking of VEGFR2. J Cell Sci. 2014 jcs.156109
21. Miao G-L, Zhang W-X, Xu Y-T. et al. Motor protein KIF13B orchestrates hepatic metabolism to prevent metabolic dysfunction-associated fatty liver disease. Mil Med Res. 2025;12:11
22. Badmus OO, Hinds TD, Stec DE. Mechanisms Linking Metabolic-Associated Fatty Liver Disease (MAFLD) to Cardiovascular Disease. Curr Hypertens Rep. 2023;25:151-62
23. Jia Y, Li Y, Yu J. et al. Association between metabolic dysfunction-associated fatty liver disease and abdominal aortic aneurysm. Nutr Metab Cardiovasc Dis. 2024;34:953-62
24. Yu H, Jiao X, Lv Q. et al. ATF4 Contributes to Abdominal Aortic Aneurysm Formation via Modulating M1 Macrophage Polarization and Inflammation. Aging Dis. 2025 16
25. Zheng S. Abdominal aortic aneurysm and cardiometabolic traits share strong genetic susceptibility to lipid metabolism and inflammation. Nat Commun. 2024;15:5652
26. Ogrodnik M, Carlos Acosta J, Adams PD. et al. Guidelines for minimal information on cellular senescence experimentation in vivo. Cell. 2024;187:4150-75
27. Di Micco R, Krizhanovsky V, Baker D, d'Adda Di Fagagna F. Cellular senescence in ageing: from mechanisms to therapeutic opportunities. Nat Rev Mol Cell Biol. 2021;22:75-95
28. Carmona-Gutierrez D, Hughes AL, Madeo F, Ruckenstuhl C. The crucial impact of lysosomes in aging and longevity. Ageing Res Rev. 2016;32:2-12
29. Roh K, Noh J, Kim Y. et al. Lysosomal control of senescence and inflammation through cholesterol partitioning. Nat Metab. 2023;5:398-413
30. Abokyi S, Ghartey-Kwansah G, Tse DY. TFEB is a central regulator of the aging process and age-related diseases. Ageing Res Rev. 2023;89:101985
31. Ma T, Chen W, Zhi X. et al. USP9X inhibition improves gemcitabine sensitivity in pancreatic cancer by inhibiting autophagy. Cancer Lett. 2018;436:129-38
32. Hirokawa N, Noda Y. Intracellular Transport and Kinesin Superfamily Proteins, KIFs: Structure, Function, and Dynamics. Physiol Rev. 2008;88:1089-118
33. Hirokawa N. Kinesin superfamily proteins (KIFs)_ Various functions and their relevance for important phenomena in life and diseases. Exp CELL Res. 2015;334:16-25
34. Kumari D, Ray K. Phosphoregulation of Kinesins Involved in Long-Range Intracellular Transport. Front Cell Dev Biol. 2022;10:873164
35. Noseda R, Guerrero-Valero M, Alberizzi V. et al. Kif13b Regulates PNS and CNS Myelination through the Dlg1 Scaffold. Barres BA, Ed. PLOS Biol. 2016;14:e1002440
36. Liao EH, Gray L, Tsurudome K. et al. Kinesin Khc-73/KIF13B modulates retrograde BMP signaling by influencing endosomal dynamics at the Drosophila neuromuscular junction. Rodal AA, Ed. PLOS Genet. 2018;14:e1007184
37. Zhao G, Lu H, Chang Z. et al. Single-cell RNA sequencing reveals the cellular heterogeneity of aneurysmal infrarenal abdominal aorta. Cardiovasc Res. 2021;117:1402-16
38. Yang G, Khan A, Liang W, Xiong Z, Stegbauer J. Aortic aneurysm: pathophysiology and therapeutic options. MedComm. 2024;5:e703
39. Kennedy BK, Berger SL, Brunet A. et al. Geroscience: Linking Aging to Chronic Disease. Cell. 2014;159:709-13
40. Li X, Li C, Zhang W, Wang Y, Qian P, Huang H. Inflammation and aging: signaling pathways and intervention therapies. Signal Transduct Target Ther. 2023;8:239
41. Chen H-Z, Wang F, Gao P. et al. Age-Associated Sirtuin 1 Reduction in Vascular Smooth Muscle Links Vascular Senescence and Inflammation to Abdominal Aortic Aneurysm. Circ Res. 2016;119:1076-88
42. Gao P, Gao P, Zhao J. et al. MKL1 cooperates with p38MAPK to promote vascular senescence, inflammation, and abdominal aortic aneurysm. Redox Biol. 2021;41:101903
43. Ouyang Y, Hong Y, Mai C. et al. Transcriptome analysis reveals therapeutic potential of NAMPT in protecting against abdominal aortic aneurysm in human and mouse. Bioact Mater. 2024;34:17-36
44. Wang L, Hong W, Zhu H. et al. Macrophage senescence in health and diseases. Acta Pharm Sin B. 2024;14:1508-24
45. Platt FM, d'Azzo A, Davidson BL, Neufeld EF, Tifft CJ. Lysosomal storage diseases. Nat Rev Dis Primer. 2018;4:27
46. Tan JX, Finkel T. Lysosomes in senescence and aging. EMBO Rep. 2023;24:e57265
47. Zhang Z, Yue P, Lu T, Wang Y, Wei Y, Wei X. Role of lysosomes in physiological activities, diseases, and therapy. J Hematol OncolJ Hematol Oncol. 2021;14:79
48. Tan A. Past, present, and future perspectives of transcription factor EB (TFEB): mechanisms of regulation and association with disease. Cell Death. 2022;29:1433-1449
49. Jia Y, Zhang L, Liu Z. et al. Targeting macrophage TFEB-14-3-3 epsilon Interface by naringenin inhibits abdominal aortic aneurysm. Cell Discov. 2022;8:21
50. Lu H, Sun J, Liang W. et al. Cyclodextrin Prevents Abdominal Aortic Aneurysm via Activation of Vascular Smooth Muscle Cell Transcription Factor EB. Circulation. 2020;142:483-98
51. Perurena N, Lock R, Davis RA. et al. USP9X mediates an acute adaptive response to MAPK suppression in pancreatic cancer but creates multiple actionable therapeutic vulnerabilities. Cell Rep Med. 2023;4:101007
52. Rott R, Szargel R, Haskin J. et al. α-Synuclein fate is determined by USP9X-regulated monoubiquitination. Proc Natl Acad Sci U S A. 2011;108:18666-71
53. Wang B, Tang X, Yao L. et al. Disruption of USP9X in macrophages promotes foam cell formation and atherosclerosis. J Clin Invest. 2022;132:e154217
Author contact
![]() Corresponding author: Xunde Xian (xianxundeedu.cn).
Corresponding author: Xunde Xian (xianxundeedu.cn).