Impact Factor
 Global reach, higher impact
Global reach, higher impact
Theranostics 2025; 15(18):9459-9485. doi:10.7150/thno.113940 This issue Cite
Review
Dissecting Tertiary Lymphoid Structures in Cancer: Maturation, Localization and Density
Guang-Liang Su1, Meng-Jie Zhang1 ![]() , Hao Li1,2
, Hao Li1,2 ![]() , Zhi-Jun Sun1,2
, Zhi-Jun Sun1,2 ![]()
1. State Key Laboratory of Oral & Maxillofacial Reconstruction and Regeneration, Key Laboratory of Oral Biomedicine Ministry of Education, Hubei Key Laboratory of Stomatology, School & Hospital of Stomatology, Frontier Science Center for Immunology and Metabolism, Taikang Center for Life and Medical Sciences, Wuhan University, Wuhan 430079, China.
2. Department of Oral Maxillofacial-Head Neck Oncology, School & Hospital of Stomatology, Wuhan University, Wuhan 430079, China.
Received 2025-3-18; Accepted 2025-7-20; Published 2025-8-30
Abstract

Tertiary lymphoid structures (TLSs) refer to ectopic lymphoid aggregates that form in non-lymphoid tissues at sites of chronic inflammation including cancers. TLSs have been recognized as significant predictors of the efficacy of immune checkpoint blockade (ICB) therapies and have the potential to elicit robust anti-tumor immune response. However, recent studies have revealed substantial heterogeneity in TLSs across different individuals and cancer types, which directly impacts the effectiveness of anti-tumor immunity. Concretely, the maturation status, localization, and density of TLSs profoundly influence the dynamic interactions among immune cells within these structures, potentially leading to adverse effects. This review provides an in-depth exploration of how the heterogeneity of TLSs influences cellular composition and immune dynamics, with the objective of influencing the efficacy of ICB therapies and modulating prognostic prediction accuracy. Additionally, the potential of combining TLSs with other biomarkers for predicting anti-tumor immunity outcomes is further investigated, alongside the introduction of advanced technologies for evaluating TLS heterogeneity. Collectively, these analyses aim to advance the understanding of TLS heterogeneity and facilitate its translation into clinical and translational medicine applications.
Keywords: Tertiary lymphoid structures, Heterogeneity, Maturation, Localization, Density
Introduction
Immune checkpoint blockade (ICB) therapy has unleashed anti-tumor immune response, leading to unprecedented durable response rates in various types of cancer [1]. However, due to primary and acquired resistance as well as toxicity associated with ICB, the number of patients benefiting from this treatment remains limited [2]. Consequently, there is an urgent need to develop diagnostic tools to identify patients who may benefit from ICB while seeking appropriate strategies to improve therapeutic outcomes. Programmed death ligand 1 (PD-L1) expression, microsatellite instability-high/defective mismatch repair (MSI-H/dMMR), and tumor mutation burden (TMB) are common predictive biomarkers in clinical practice [3-5]. Nevertheless, these biomarkers alone cannot fully predict immunotherapy responsiveness, underscoring the need to identify more precise biomarkers for therapeutic efficacy evaluation. Recent studies found that tertiary lymphoid structures (TLSs) demonstrate robust potential in improving prognosis and enhancing response to ICB therapy and may serve as a significant source of anti-tumor immunity within solid tumors [6-9].
TLSs are ectopic lymphoid aggregates that form within non-lymphoid tissues, capable of locally generating tumor-specific effector T cells, B cells, and antibodies, playing a crucial role in establishing an anti-tumor immune environment [10-12]. Current research indicates that the formation of TLSs is primarily driven by persistent antigenic stimulation within the chronic inflammatory microenvironment [13]. This encompasses a variety of conditions, including autoimmune disorders, persistent infections, and particularly cancer [14]. Under inflammation-induced conditions, lymphocytes accumulate and gradually organize into immune units similar to secondary lymphoid organs (SLOs), differentiating into B cell zones containing germinal centers (GCs) and surrounding T cell zones [15]. While the triggers and processes involved in the origin and development of TLSs have become increasingly clear [16, 17], several unresolved questions remain regarding their role in predicting and enhancing anti-tumor immune effects. Concretely, TLSs exhibit notable heterogeneity across patients and cancer types, involving differences in maturation status, localization, and density [10, 11, 18]. These variations significantly impact changes in the cellular composition and anti-tumor immunity of TLSs, which may lead to negative clinical prognosis [19, 20]. Thus, a deeper understanding of the mechanisms underlying the formation of TLS heterogeneity and its impact on anti-tumor immunity is essential. Combined with improvements in evaluation strategies and technologies, it will enhance the accuracy of ICB prognostic predictions and immunotherapy effectiveness.
This review summarizes the latest research on TLSs, focusing on immune dynamics in maturation, localization and density (Figure 1) [10, 11, 18-20]. First, by integrating existing evidence, this paper systematically explains the definition and influencing factors of TLS heterogeneity, as well as its significance for tumor immunity and clinical practice. Second, it explores the future development of combining TLS heterogeneity evaluation with other biomarkers to predict anti-tumor immune outcomes, while summarizing current advanced methods for assessing TLS heterogeneity. Finally, this paper outlines key directions for future research, including optimizing evaluation strategies, improving assessment technologies, and developing advanced intervention materials, aiming to further refine and enhance clinical transformation and application.
Definition of TLS heterogeneity. TLSs exhibit marked heterogeneity in maturation, localization and density across tumor types and individuals, profoundly impacting anti-tumor immune effects and clinical value. (A) Maturation status of TLSs. The maturation status of TLSs is classified into three categories: loosely aggregated lymphoid cells; primary follicles containing T cells, B cells, and FDCs; and mature polarized structures featuring GCs, HEVs, and a variety of immune cells, such as macrophages. (B) Localization of TLSs. TLSs are variably distributed within the body and can be categorized based on their localization as intra-tumoral, peri-tumoral, or within the tumor stroma. (C) Density of TLSs. The presence of TLSs indicates a robust anti-tumor immune response, capable of converting “cold” tumors into “hot” tumors. Moreover, higher density of TLSs are generally associated with better clinical outcomes. DC: dendritic cell; E-TLSs: early tertiary lymphoid structures; FDC: follicular dendritic cell; FRC: fibroblastic reticular cell; GC: germinal center; HEV: high endothelial venule; LV: lymphatic vessel; MDSC: myeloid-derived suppressor cell; NK: natural killer; PC: plasma cell; PFL-TLSs: primary follicle-like tertiary lymphoid structures; SFL-TLSs: secondary follicle-like tertiary lymphoid structures; Tfh: T follicular helper; Treg: regulatory T cell.
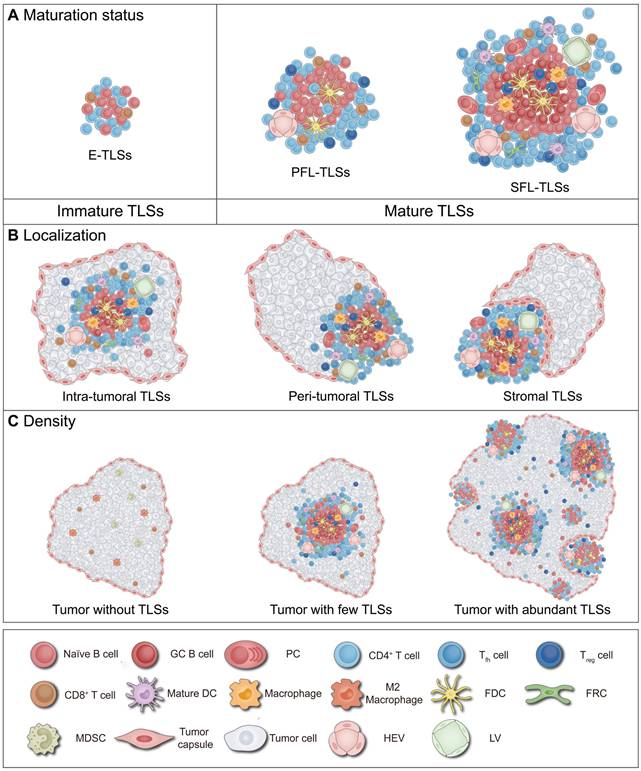
Structural characteristics and heterogeneity of TLSs
TLSs are temporary immune unit formed in chronic inflammation, featuring distinct T and B cell zones at their core [11]. The T cell region is dominated by CD4+ cells, which supports the activation of naïve B cells and the formation of GCs by facilitating antigen presentation [21]. Additionally, dendritic cells (DCs) expressing dendritic cell-lysosomal-associated membrane protein (DC-LAMP, also called LAMP3) located in this region can capture, process, and present antigens to activate initial CD8+ T cells [22]. In the TLSs, CD8+ T cells exert their anti-tumor immune effects by directly killing tumor cells and secreting effector molecules (e.g., granzyme B) [18]. CD20+ B cell follicles with GCs, supported by a network of follicular dendritic cells (FDCs), serve as vital sites for the proliferation of B cells and antibody class transformation [23]. Moreover, CD21+ FDCs located in this region play a critical role in the selection of memory B cells during GC reactions [24]. Besides serving as organized congregations of T and B cells, TLSs also encompass a diverse array of immune cells such as macrophages, natural killer (NK) cells, and neutrophils [23, 25, 26]. In addition, PNAd+ high endothelial venules (HEVs) constitute the vascular system of TLSs, and recruit circulating lymphocytes into TLSs by secreting chemokines such as CC motif chemokine ligand 19 (CCL19) and CCL21, along with adhesion molecules [10]. The dense stromal network constructed by fibroblastic reticular cells (FRCs) supports the entire TLS structure and anchors it at the inflamed tissue, ensuring effective local immune response [27]. In short, T/B cell compartments, other immune cells, HEVs, and the matrix network collectively constitute a dynamic and highly organized anti-tumor immune microenvironment (Figure 2).
Cellular composition of TLSs. The cellular composition of TLSs varies under different mature status, which affects anti-tumor immunity. Mature TLSs are composed of a diverse array of cell types, including T cells, B cells, DCs, FDCs, FRCs, macrophages, and HEVs et al. These cells perform distinct roles, collectively establishing and maintaining an immune niche within the tumor microenvironment that is either anti-tumoral or pro-tumoral. DC: dendritic cell; E-TLSs: early tertiary lymphoid structures; FDC: follicular dendritic cell; FRC: fibroblastic reticular cell; GC: germinal center; HEV: high endothelial venule; LV: Lymphatic vessel; MDSC: myeloid-derived suppressor cell; PC: plasma cell; PFL-TLSs: primary follicle-like tertiary lymphoid structures; SFL-TLSs: secondary follicle-like tertiary lymphoid structures; Tfh: T follicular helper; Treg: regulatory T cell.
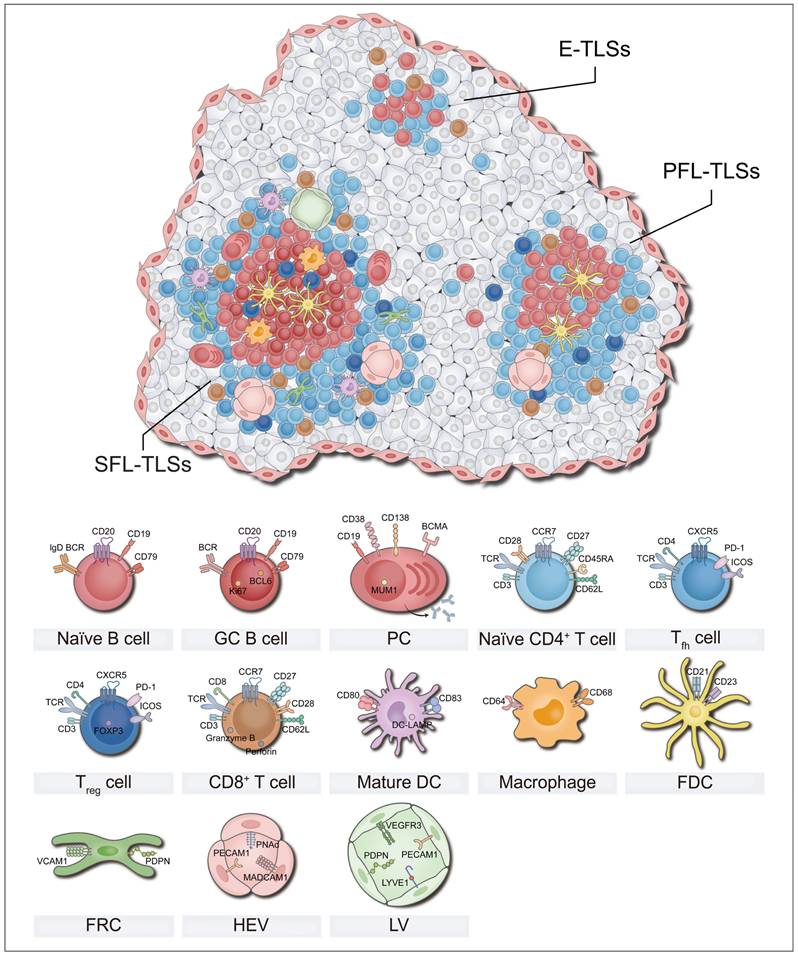
However, it is important to note that not all cancer patients possess TLSs, and not all TLSs exhibit complete structures [10, 11, 18]. Clinical and translational evidence has demonstrated substantial heterogeneity in TLS presence across cancer types, molecular subtypes, and disease stages [28-31]. Even when present, many TLSs display structural incompleteness—manifesting as lymphocyte aggregates lacking GCs or disorganized T/B cell compartmentalization [30, 32, 33]. These structurally impaired TLSs show functional limitations: the absence of GCs disrupts B cell affinity maturation, ultimately weakening responses to immunotherapy and correlating with reduced patient survival [6-9].
Formation and development of TLSs
Similar to SLOs, the formation of TLSs originates from the homing of lymphoid tissue inducer (LTi) cells or their substitute cells to inflammatory sites [10]. Extensive research has demonstrated that various persistent antigenic stimuli, including drugs, tobaccos, gut microbiota, and viruses, can induce the production of pro-inflammatory mediators, thereby promoting the recruitment of LTi cells and the development of lymphatic structures (Figure 3A) [34-37]. Under the influence of pro-inflammatory mediators, LTi cells interact with lymphoid tissue organizer (LTo) cells. This interaction activates signaling pathways such as lymphotoxin α1β2/lymphotoxin beta receptor (LTα1β2/LTβR), interleukin-7/ interleukin-7 receptor (IL-7/IL-7R), IL-17/IL-17R, and RANK/RANKL, which trigger downstream cytokine production (Figure 3B) [38-40]. Recently, one study found that in PDAC, IL-33 activates group 2 innate lymphoid cells (ILCs-2) expressing LT [41]. These ILCs-2 interact with LTβR+ myeloid organizer cells, contributing to the production of downstream factors and TLS formation [41]. Such cytokines intricately regulate cellular activity associated with TLS development at different status, involving stromal cell activation in conjunction with the LTi-LTo positive feedback loop, HEV expansion and compartmentalization of T/B cells, as well as GC formation with B cell differentiation [17]. This process is closely linked to the maturation of TLSs and the orchestration of immune responses (Figure 3C). However, it is important to note that in clinical observations, not all TLSs achieve full or optimal development, exhibiting variability in maturation status, localization, and density. Further studies have revealed that endogenous and exogenous factors influence signaling pathways and cellular activities during the induction, initiation, and maturation of TLSs. Current research has identified the STING pathway, LTα1β2/LIGHT pathway and other regulatory targets; however, the mechanisms underlying TLS heterogeneity remain to be fully elucidated.
Development of TLSs. The development of TLSs, from induction and initiation to maturation, is a highly orchestrated process. Under the influence of pro-inflammatory mediators, LTi cells interact with LTo cells. This interaction activates signaling pathways that trigger the production of downstream cytokines, which in turn regulate the maturation of TLSs. (A) Induction of TLSs. Various stimuli, including drugs, tobaccos, gut microbiota, and viruses, can induce the production of pro-inflammatory mediators, thereby promoting the recruitment of LTi cells and the development of TLSs. (B) Initiation of TLSs. LTi cells interact with LTo cells through various signaling axes, such as IL-7/IL-7R, IL-17/IL-17R, RANK/RANKL, and LTα1β2/LTβR, initiating cytokine expression and promoting the development of TLSs. These signaling pathways promote the secretion of VEGFA/C, adhesion molecules (e.g., VCAM1, ICAM1, MADCAM1 et al.) and chemokines (e.g., CXCL13, CXCL12, CCL21 et al.), aiding HEVs in the recruitment of lymphocytes into TLSs. (C) Maturation of TLSs. These cytokines intricately regulate cellular activities associated with TLS maturation at different status, including the recruitment of FDCs, compartmentalization of T/B cells, and the formation of GCs alongside B cell differentiation. This process is closely linked to the maturation of TLSs and the orchestration of immune responses. CAF: cancer-associated fibroblast; CCL21: CC motif chemokine ligand 21; CXCL13: CXC motif chemokine ligand 13; DC: dendritic cell; E-TLSs: early tertiary lymphoid structures; FDC: follicular dendritic cell; FRC: fibroblastic reticular cell; GC: germinal center; HEV: high endothelial venule; ICAM1: intercellular adhesion molecule 1; IL-17/IL-17R: interleukin 17/interleukin 17 receptor; IL-7/IL-7R: interleukin 7/interleukin 7 receptor; ILC: innate lymphoid cell; LTα1β2/LTβR: lymphotoxin α1β2/lymphotoxin beta receptor; LTi: lymphoid tissue-inducer; LTo: lymphoid tissue organizer; LV: Lymphatic vessel; MADCAM1: mucosal addressin cell adhesion molecule 1; PC: plasma cell; PFL-TLSs: primary follicle-like tertiary lymphoid structures; RANK/RANKL: receptor activator of nuclear factor κ B/receptor activator of nuclear factor κ B ligand; SFL-TLSs: secondary follicle-like tertiary lymphoid structures; Th 17: T helper 17; Tfh: T follicular helper; Treg: regulatory T cell; VCAM1: vascular cell adhesion molecule 1; VEGFA/C: vascular endothelial growth factors A/C; VSMC: vascular smooth muscle cell.
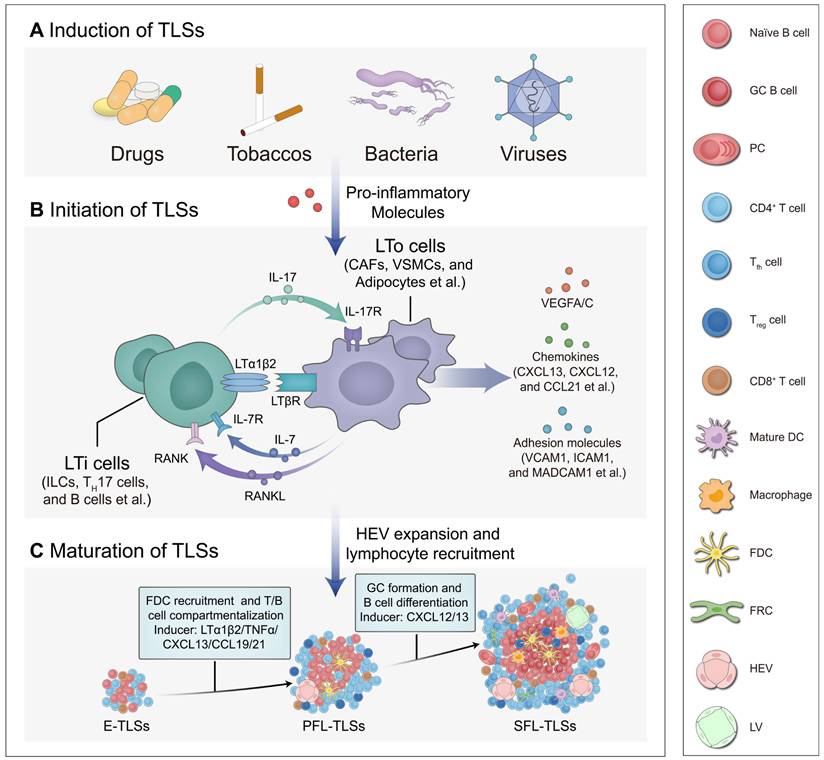
Maturation of TLSs
TLSs have been found to exhibit two distinct maturation status: mature and immature (Figure 1A) [10, 11, 42]. The critical distinction between these states mainly resides in the presence of organized GCs [10, 11, 42]. Previous clinical trials and animal studies have demonstrated that beyond the mere presence of TLSs, differences in their maturation status significantly influence prognoses for tumor patients and their responses to anticancer treatments [10, 11, 42]. Mature TLSs enhance immune responses through coordinated activation of T and B cells, correlating with better response to ICB (e.g., anti-PD-1/CTLA-4 therapies) and neoadjuvant chemotherapy (NAC) [9, 43, 44]. However, compared to mature TLS, immature TLS may inhibit effector cell function, potentially leading to poorer clinical outcomes in patients receiving the same treatments [9, 43, 44]. The following sections provide a detailed exploration of the definition of TLS maturation, factors influencing this process, and its implications for tumor immunity and clinical practice.
Definition of TLS maturation
The maturation of TLSs can be divided into several status characterized by the accumulation and development of FDCs and GCs [10, 11, 42]. In 1998, Wagner et al. first observed lymph node-like structures and GC presence in synovial tissues of rheumatoid arthritis patients [45]. Among the 9 studied patients, CD23⁺ FDCs participating in GCs reactions were identified in 4 cases, while absent in the remaining 5, indicating structural heterogeneity in TLS composition [45]. Subsequent studies revealed varying maturation status of TLSs in chronic inflammatory conditions and cancers, which may influence prognostic prediction in tumor therapy [6-11, 14, 18, 46]. In 2008, Dieu-Nosjean et al. first reported the explicit presence of TLSs in cancer contexts and noted that the infiltration of mature DCs within TLSs correlated with favorable patient outcomes [46]. Between 2020 and 2021, several groundbreaking studies further clarified that mature TLSs could predict the efficacy of ICB in solid tumors independently of PD-L1 expression [6-9]. Immature TLSs, also known as early tertiary lymphoid structures (E-TLSs), exhibit loosely organized aggregates of T cells and B cells with few DC infiltration [10, 11, 42, 47]. They are closely linked to T cell exhaustion, inflammatory activity, and immune suppression within the tumor microenvironment (TME) [47-49]. In contrast, mature TLSs represent highly organized lymphoid aggregates characterized by B cell follicles encircled by T cell zones [10, 11, 42, 47]. These structures can be classified as primary follicle-like (PFL) or secondary follicle-like (SFL) subtypes containing GCs [11, 49]. In mature TLS, PFL-TLS also has T follicular helper (Tfh) cells and FDCs networks that allow T cell immune activation and low-affinity antibody production [10, 11, 42, 49]. SFL-TLS are characterized by the presence of GCs with GC B cells and FDCs allowing the production of memory B cells and high-affinity antibody secreting plasma cells (PCs) [10, 11, 22, 42, 49]. Sometimes, TLS maturation represents not discrete status but a continuous evolution spanning immune activation to functional exhaustion. Recent studies employing spatial transcriptomics and pseudotime trajectory analysis have identified three distinct differentiation patterns of TLSs in hepatocellular carcinoma (HCC): mature, conforming, and deviating [50]. Mature TLSs possess fully developed GCs with high expression of key genes such as CXCL13, supporting the differentiation of B cells into antibody-secreting cells. Conforming TLSs, despite lacking classical GCs, demonstrate B cell differentiation trajectories highly aligned with those of mature TLSs. Mature and conforming TLSs display gradient expression continuity of genes like CXCL13 and AICDA. However, deviating TLSs exhibit interrupted B cell differentiation and defective HEV development, creating an immunosuppressive microenvironment.
The detection and classification of TLS maturation in pathology and clinical practice constitute a significant topic in immunology. In pathological examinations, hematoxylin and eosin/hematoxylin-eosin-saffron (H&E/HES) staining combined with CD3, CD20, and CD23 immunohistochemistry (IHC) are utilized to determine the TLS status [9, 30, 33, 42, 51-53]. Initial H&E/HES screening identifies visible lymphatic aggregates (≥50 cells) in viable tumor regions (excluding ulcerated/necrotic areas) [9, 53]. In most cases, further maturation classification of TLSs relied on IHC and multiplex immunohistochemistry/multiplex immunofluorescence (mIHC/mIF) techniques to characterize cellular components [9, 53, 54]. Concretely, for lymphoid aggregates without visible GCs, classification is determined by sequential CD20 and CD23 staining: CD20⁺ B cells with CD23⁺ FDCs indicate mature TLSs; CD20⁺ B cells without CD23⁺ FDCs suggest immature TLSs; CD20- B cells indicate non-TLSs [9, 53, 54]. Furthermore, lymphatic aggregates with visible GCs in initial H&E/HES staining can be directly classified as mature TLSs [53]. Notably, when dealing with dense TLSs, single CD20 and CD23 IHC staining exhibits higher sensitivity and accuracy compared to CD20/CD23 mIHC staining [53]. In clinical practice, the classic classification criteria for immature and mature TLSs summarized by Vanhersecke et al. represents a widely adopted strategy across multiple researches on TLSs [9, 31, 53, 54]. Concretely, if a sample contains only immature TLSs without mature TLSs, it is classified as immature TLSs. If a sample contains mature TLSs or both mature TLSs and immature TLSs (even when the mature TLSs proportion is lower), it is defined as mature TLSs [9, 31, 53, 54]. Additionally, mature TLSs undergo additional subclassification: cases containing PFL-TLSs with or without E-TLSs are classified as PFL-TLSs, while those demonstrating SFL-TLSs alone or coexisting PFL-TLSs and SFL-TLSs are uniformly categorized as SFL-TLSs [31, 33, 51, 55].
Statistical analyses reveal significant inter-individual and inter-tumoral heterogeneity in TLS maturation status [30, 32, 33]. Among hepatocellular carcinoma (HCC) cases, 47% of tumors exhibited TLSs, in which E-TLSs, PFL-TLSs and SFL-TLSs account for 26%, 16%, and 5% of patients, respectively [33]. Differently, in 90.8% esophageal squamous cell carcinoma (ESCC) patients, peri-tumor TLS was observed - including 74.7% E-TLSs, 54.1% PFL-TLSs, and 64.9% SFL-TLSs [30]. A multicancer studies (covering 20 cancer types with 477 samples) revealed two independent differentiation pathways for antibody-secreting cells (ASCs): classical GC-dependent and alternative extrafollicular (EF) pathways, showing cancer-type specificity [32]. In EF-dominant cancers (e.g., HCC, head and neck squamous cell carcinoma (HNSCC)), TLS lacks GC structure; whereas in GC-dominant cancers (e.g., colon adenocarcinoma (COAD), lung carcinoma (LC)), TLS typically presents mature structure [32].
Factors contributing to TLS maturation
The current approaches have facilitated the regulation of B cell migration and TLS development via the administration of cytokines such as CXC motif chemokine ligand 13 (CXCL13), LTα1β2/ tumor necrosis factor alpha (TNF-α), and CXCL12 [16, 17]. Nevertheless, although these interventions can induce the formation of TLSs, there is a notable paucity of GC B cell infiltration. The TME contains potential barriers to TLS maturation, such as tumor-draining lymph nodes (TDLNs) and epigenetics.
TDLNs are LNs that receive and process lymph fluid originating from the tumor area [56]. During the process of distant metastasis, tumors and their derivatives enter the TDLNs via afferent lymphatics, coinciding with vascular stroma remodeling and TME imbalance, which may influence the maturation of TLSs [56]. A study highlighted that Bregs preferentially accumulate in TDLNs and promote tumor growth [57]. However, unlike conventional Bregs, TDLN B cells exert immunosuppressive effects by inducing lymphangiogenesis in LNs rather than promoting IL-10 secretion or inducing regulatory T cell (Treg) differentiation [57]. In addition, a study involving 218 patients with radically resected lung adenocarcinoma indicated that patients with lymph node metastasis typically exhibited immature TLSs and insignificant lymphocyte infiltration, prompting further investigation into the influence of TDLNs on TLS maturation [58]. He et al. demonstrated that the immunosuppressive microenvironment of TDLNs blocked the maturation of TLSs and made TLSs lose its prognostic value [54]. The immunosuppressive factors enriched in TDLNs obstruct the formation of memory B cells and interfere with interferon gamma (IFN-γ) signal transduction in NK cells [54]. The interruption of IFN-γ signaling can synergize with immunosuppressive factors to affect the differentiation of memory B cells and the formation of GCs, thus limiting the development and maturation of TLSs [54].
Tumor cells adapt their metabolic pathways to meet the demands of rapid proliferation [59]. These metabolic changes can influence the activity of epigenetic modifiers, leading to changes in gene expression patterns and contributing to an immunosuppressive environment [59]. A pan-transcriptomic analysis involving 20 different cancer types indicated that increased glutamine metabolism in the TME promotes a bias in B cell differentiation towards an atypical memory (AtM) phenotype [32]. AtM B cells are localized centrally within immature TLSs, displaying exhausted and bystander phenotypes, and serve as key contributors to the immunosuppressive microenvironment within these structures [22, 32]. The study found that the glutamine-derived metabolite α-ketoglutarate facilitates the activation of AtM B cells by promoting the expression of transcription factors T-bet and BATF, as well as activating the mTORC1 signaling pathway [32]. Furthermore, Bessode et al. demonstrated that the accumulation of the tryptophan catabolizing enzyme indoleamine 2,3-dioxygenase 1 (IDO1) within non-small cell lung cancer (NSCLC) induces an immunosuppressive state by converting tryptophan into various immunosuppressive metabolites, such as L-kynurenine [60]. The study indicates that IDO1 is expressed in mature FDCs within TLSs and, through upregulating the transcription factors FOXP3 and Ki67, inhibits interactions between Tfh cells and B cells, thereby impairing plasmablast differentiation within mature TLSs [60]. Similarly, spatial transcriptomic analyses of HCC reveal that malignant cells impede the TLS maturation by modulating chromatin accessibility and transcriptional activity of tryptophan metabolism-related genes [50]. Malignant cells accumulate around immature TLSs, exhibiting increased promoter accessibility and upregulated expression of tryptophan metabolic enzymes, leading to the aberrant accumulation of tryptophan metabolites in the TME. These tryptophan metabolites inhibit the differentiation of B cells into GC B cells, thereby disrupting the normal maturation trajectory of TLSs.
Role of TLS maturation in tumor immunity
The effects of TLSs in tumor immunity varies significantly depending on its maturation. Mature TLSs are typically associated with robust anti-tumor immune responses in cancer. Research indicates that B cells within mature TLSs may be a critical determinant of the efficacy of anti-tumor immunity [61]. Within the GC of TLSs, naïve B cells differentiate into PCs that produce IgG and IgA, inducing macrophage/NK-cell-dependent tumor apoptosis [22, 62, 63]. Furthermore, there is potential cross-talk between B cells and T cells that can modulate the efficiency of anti-tumor immunity [64-66]. For example, CD86+ B cells clustered within TLS can present antigens to T cells, thereby inducing specific cellular immune responses [64]. In addition to antigen presentation, B cells also exhibit regulatory effects on T cell phenotypes [65, 67, 68]. A study on GC patients indicates that B cells within TLSs can promote the differentiation of naïve T cells into CD8+CD103+ resident memory T cells (Trms) rather than FOXP3+CD8+ Tregs [67, 68]. Importantly, the interaction between B cells and T cells may be bidirectional. Tfh cells secrete CXCL13, guiding B cell migration and promoting GC formation [66, 69]. A recent study has revealed that in HNSCC, progenitor exhausted CD4+ T cells, with features resembling Tfh cells, support these responses, by activating B cells to produce PCs in the GCs, and interacting with DC-LAMP+ DCs to support CD8+ T cell activation [12]. These findings indicate that mature TLS B cells and T cells work synergistically to enhance anti-tumor immune responses.
Immature TLS consist of loosely aggregated T cells, B cells, and stromal cells [10, 11, 42], potentially representing limited anti-tumor immune responses. In pancreatic ductal adenocarcinoma (PDAC), tumors with E-TLSs show significantly increased infiltration of CD3+ and CD8+ T cells compared to those without TLS [52]. Although both immature and mature TLSs share comparable T cell infiltration, mature TLSs exhibit enriched CD4+ memory T cells and naïve B cells, alongside increased TMB and major histocompatibility complex (MHC) I neoantigens [52]. Further research shows that E-TLSs tend to form an immunosuppressive microenvironment [48, 70]. For example, in breast cancer (BC), Bregs and Tregs accumulate in E-TLSs, thereby maintaining this immunosuppressive state [70]. In addition, pathological and gene expression profiles of 127 patients with early hepatopathy showed that the presence of E-TLSs was associated with increased expression of immunosuppression-related genes [48].
The prognostic and predictive value of TLS maturation
Currently, a substantial number of clinical studies have found that the maturation of TLSs is associated with prognosis and treatment response in cancer patients. Mature TLSs exhibits positive prognostic and predictive value in various solid tumors, such as ESCC, clear cell renal cell carcinoma (ccRCC), urothelial carcinoma (UCC), and renal cell carcinoma (RCC) [22, 30, 43, 51]. For example, in an analysis involving ESCC patients treated with the anti-PD-1 antibody nivolumab, mature TLSs was closely associated with better treatment responses and longer progression-free survival (PFS) [30]. Similarly, in ccRCC, the presence of mature TLSs and GCs is significantly associated with better overall survival (OS) and PFS [51]. Compared to E-TLSs, mature TLSs demonstrates higher infiltration of CD8+ T cells, CD20+ B cells, and DC-LAMP+ DCs [51]. In contrast, E-TLSs often indicate poorer prognostic outcomes [30, 42, 51, 52, 70]. In colorectal cancer (CRC), patients with a higher proportion of E-TLSs face increased risk of disease recurrence [42]. E-TLSs exhibit lower MSI and fail to effectively induce immune activation [42]. Similar findings have been reported in other solid tumors such as BC, ccRCC, ESCC, and PDAC [30, 51, 52, 70]. Notably, the immunological role of E-TLSs is not entirely negative in a pan-cancer context [30, 52]. For instance, one study on ESCC has indicated that the density of E-TLSs in the TME shows no clinical correlation with patient prognosis or responses to ICB therapy [30]. Additionally, in PDAC, the presence of E-TLSs is correlated with prolonged PFS and OS [52]. Tumors containing E-TLSs show higher levels of lymphocyte infiltration compared to those without any TLSs [52].
Furthermore, several key points about the clinical value of TLS maturation warrant emphasis. The maturation status of TLSs may be one of the most critical predictors of patient prognosis. TLS is commonly used as a parameter for predicting tumor patient survival [6, 71-73]. However, research shows that TLS maturation has a deeper association with tumor prognosis compared to TLS appearance alone [74]. When corticosteroids impair the formation of GCs, the predictive value of TLS presence is lost [74]. Additionally, further research is needed to explore the prognostic differences between PFL-TLSs and SFL-TLSs. A study involving 138 patients with lung squamous cell carcinoma (LSCC) found that only the number or proportion of SFL-TLSs was significantly associated with improved survival, while the prognostic value of E-TLSs and PFL-TLSs remains unconfirmed [74]. In intrahepatic cholangiocarcinoma (iCCA), mature TLSs shows a significant survival advantage over E-TLSs, but subdividing mature TLS into PFL-TLSs and SFL-TLSs revealed no additional prognostic differences [55].
Localization of TLSs
TLSs can be observed within the TME, which includes the tumor core and stroma [10, 75]. Based on their location within the TME, TLSs can be categorized into intra-tumoral, stromal, and peri-tumoral (i.e., junctional) regions, with the majority of TLSs being located at the peri-tumoral areas (Figure 1B) [11, 33, 51, 76-79]. Intra-tumoral TLSs are generally associated with enhanced responses to immunotherapies (e.g., ICB, cancer vaccines, and CAR-T therapies), characterized by intact vascular networks and robust immune cell infiltration [67, 77, 80, 81]. In contrast, peri-tumoral and stromal TLSs exhibit prognostic and predictive heterogeneity may due to vascular disruption and immunosuppressive microenvironments [33, 51, 76, 77, 82]. Herein, TLS localization including its definition, regulatory factors, and its impact on tumor immunity and patient prognosis will be studied.
Definition of TLS localization
TLSs localized within intra-tumoral, peri-tumoral, and stromal areas [11]. Nevertheless, there exists no universally accepted criterion for defining the localization of TLSs. Strictly speaking, TLSs at the invasive margin are termed peri-tumoral TLSs, while those situated within the tumor stroma and distinctly separated from the tumor parenchyma are defined as stromal TLSs [11]. Most studies do not distinguish between these two categories and commonly refer to both as peri-tumoral TLSs [30, 51, 76-79, 83]. Some studies have more specifically described the presence of stromal TLSs within TME [24, 33, 82, 84, 85]. Notably, the boundaries defining intra-tumoral versus peri-tumoral TLSs (including stromal TLSs) vary, with different studies setting distances from the invasive margin ranging from 0.5 millimeter to 10 millimeters [30, 51, 55, 78, 79, 83]. The precise boundary between peri-tumoral and stromal TLSs is rarely quantified in clinical practice. For this reason, if not otherwise indicated, stromal TLSs are covered by the peri-tumoral TLSs described below.
Quantification of TLS localization relies on the intra-tumoral (T-score) and peri-tumoral (P-score) grading systems [26, 55, 77]. The T-score employs a 0-3 grading scale based on the absolute count of TLSs within the tumor core [26, 55, 77]. While the P-score classifies grades (0-3) according to the proportional area coverage of TLS in the tumor peripheral zone [26, 55, 77]. However, no consensus exists for classifying cases simultaneously exhibiting intra-tumoral and peri-tumoral TLSs. Wu et al. proposed that if both intra-tumoral and peri-tumoral TLSs are observed, the patient should be considered intra-tumoral TLSs positive [76]. On the other hand, Xu et al. have chosen to discuss this situation separately [51]. Future studies need to standardize this debate. In clinical practice, combining T and P scores classifies patients into four immune subtypes (low-T/low-P, low-T/high-P, high-T/low-P, high-T/high-P), each featuring distinct TME and prognostic outcomes [26, 55, 79].
Existing evidence found that TLSs are more abundant in the peri-tumoral areas than in the core of tumors. Through pathological examination, in TLS-positive samples of various solid tumors (e.g. HCC, cSCC, ccRCC, iCCA, CRC), the proportion of intra-tumoral TLSs is approximately between 21% and 44%, while the proportion of peri-tumoral TLSs is roughly within the range of 56% to 79% [51, 76-79]. Interestingly, the localization of TLSs also influences their morphological characteristics. Research indicates that in the same tumor tissue, different localizations of TLSs can lead to heterogeneity in their morphology [20, 77, 78]. For example, Shang et al. showed that intra-tumoral TLSs in cholangiocarcinoma (CCA) were generally oval-shaped and well-developed, whereas peri-tumoral TLSs appeared squished, slender, or simply lymphatic aggregates [77]. Similar results have been reported in several studies concerning CRC [20, 78]. The specific mechanisms underlying these phenomena require further investigation.
Factors contributing to TLS localization
Existing evidence strongly indicates a close association between TLS localization and the state of tumor vasculature [55, 76, 86-88]. Under appropriate immune stimulation, TLSs tend to develop at the intersections of microvasculature within the tumor [87]. As the TME undergoes remodeling, functional vascular networks are disrupted, leading to the migration of TLSs along the invasive front to peri-tumoral or stromal regions [88]. A study involving 308 patients with pancreatic cancer (PC) has demonstrated that tumor tissues harboring intra-tumoral TLSs exhibit a higher number of CD31+ endothelial cells and exhibited elevated levels of vascular endothelial-cadherin expression [87]. Conversely, in tissues characterized by peri-tumoral TLSs or the absence of TLSs, vascular stability and maturation tend to be diminished [87]. Similarly, research across other malignancies, including iCCA, melanoma, and HNSCC, has confirmed that intra-tumoral TLSs possess a more intact vascular system compared to peri-tumoral TLSs [55, 76, 86]. Collectively, these data support the notion that vascular normalization facilitates the intra-tumoral localization of TLSs.
Given the pivotal role of vascular normalization, exploring potential factors that modulate this process such as STING and LIGHT signaling [89, 90], may reveal potential directions for regulating TLS localization. STING and LIGHT signaling pathways not only regulates the transcription of adhesion molecules like PNAd and MADCAM1 on endothelial cells but also involves the release of chemokines such as CXCL10, CXCL13 [91-94]. These adhesion molecules and chemokines work together to mediate the homing of lymphocytes and maintain the normal function of the vascular system [91-94]. For instance, a hydrogel platform for co-delivery of chitosan (a STING agonist) and CpG (a TLR9 agonist) to stimulate the development of vascular networks [89]. The study found that the synergistic activation of STING and TLR9 signaling significantly promotes the migration of immune cells to tumor sites and accelerates the formation of intra-tumoral TLSs [89]. In addition, the combination of anti-fibrotic drugs with LIGHT-coding plasmids represents a promising strategy for reshaping the vascular matrix and inducing intra-tumoral TLSs [90]. Concretely, antifibrotic drugs reversed the abnormal activation of fibroblasts and reduced collagen deposition in vessels; the LIGHT encoding plasmid upregulated adhesion molecules involved in endothelial-lymphocyte interactions, promoting the infiltration of cytotoxic T lymphocytes (CTLs) [90]. Notably, systemic STING and LIGHT application may pose risks of immune-related adverse events (irAEs) and immune cell off-target toxicity, highlighting the importance of precise control over signal expression [90, 95, 96].
Role of TLS localization in tumor immunity
The localization of TLSs directly correlates with their immunological efficacy in TME. When TLSs are located within the tumor, cancer patients generally exhibit enhanced immune response [26, 51, 76, 87], which might be associated with an intact vascular network. The vascular structures related to TLSs form physical barriers that effectively limit the invasion and metastasis of tumor cells [26, 55, 76, 77, 83]. For instance, in cSCC, intra-tumoral TLSs correlate with reduced subcutaneous fat penetration, decreased lymphatic vasculature, and reduced perineural invasion [76]. Furthermore, the vascular system associated with TLSs plays a significant role in promoting lymphocyte infiltration [26, 55, 78, 87]. Compared to peri-tumoral TLSs, the PDC tissues with intra-tumoral TLSs exhibit less vascular disruption, higher infiltration of T and B cells, as well as significantly higher expression of T helper 1 (Th 1)- and Th 17-related genes [87]. It should be noted that the vascular system associated with TLSs non-selectively recruits immune cells, including Tregs and M2 macrophages [26, 55, 78]. Additionally, intact vascular networks within intra-tumoral TLSs enhance immune cell recruitment, potentially contributing to TLS maturation processes [30, 33, 51, 55]. A retrospective analysis of 395 ccRCC patients indicated that proximal TLSs are mainly composed of SFL structures, while distal TLSs have a higher proportion of early TLSs [51]. Similar results have been observed in ESCC, iCCA, and HCC [30, 33, 55].
As the tumor invasion progresses, TLSs shift to the stromal regions farther from the tumor core and may be less affected by vascular immunity [33, 55, 83, 97]. In this case, the infiltrated immune cells in TME exhibit considerable heterogeneity. For example, in BC, the invasive margins accumulate higher density of CD163+ M2 macrophages compared to the tumor core [83]. These M2 macrophages contribute to abnormal angiogenesis and tumor metastasis by secreting factors such as IL-4, IL-10, and VEGF [98]. In addition, a study covering 170 HCC patients noted that the denser peri-tumoral TLSs were linked to an increased infiltration of neutrophils [97]. These neutrophils secrete mediators including α-defensins and transforming growth factor beta (TGF-β), which inhibit T cell activation and promote tumor cell proliferation [99, 100]. Notably, in patients with iCCA, increased peri-tumoral TLS density positively correlates with elevated Treg infiltration in intra-tumoral TLSs, implying immunological communication between distinct TLS niches [55]. However, some studies found that the presence of peri-tumoral TLSs can also favor anti-tumor immunity [30, 101]. In ESCC, mature peri-tumoral TLSs, particularly those characterized by GC B cells, are associated with a stronger anti-tumor immune response [30]. Moreover, CD83+DC-LAMP+ DC clusters show a peri-tumoral preference in positioning across various solid tumors, such as melanoma, BC [101, 102]. Mature DCs activate T cells through antigen presentation [101, 102]. T cells gather around mature DCs in the peri-tumor area, forming clusters of DC-T cells that resemble SLOs, which are characteristic of sustained immune response [101, 102]. Nevertheless, the exact mechanisms by which these critical immune cells are maintained during vascular invasion and TLS metastasis localization remain unclear.
The prognostic and predictive value of TLS localization
The therapeutic implications of TLSs in cancer are heavily influenced by their localization. The available evidence found that intra-tumoral TLSs may have better prognostic and predictive significance [26, 33, 76, 103]. In gastric carcinoma (GC), tumor resection specimens from responders exhibited a significantly higher number of intra-tumoral TLs compared to non-responders [67]. These intra-tumoral TLSs are characterized by enriched infiltration of CD8+ exhausted T cells, which exhibit responsiveness to anti-PD-1 therapy and can unleash their anti-tumor potential [67]. In NSCLC, the presence of intra-tumoral TLSs is closely associated with better DFS and OS [103]. The intra-tumoral TLSs showed a higher proportion of switched memory B cells and a lower proportion of naïve B cells, supporting specific humoral immunity [103]. Similarly, intra-tumoral TLS has also been shown to be a favorable prognostic and predictive predictor for other tumors, including CCA, CRC, HCC, and cSCC [33, 76, 77, 103]. In contrast, the presence of peri-tumoral TLS is generally associated with a higher risk of cancer recurrence and negative treatment response [51, 76, 77, 82]. For example, a study on CCA, a higher density of peri-tumoral TLSs was associated with a shorter 5-year OS in patients undergoing surgery or anti-PD-1 therapy [77]. Peri-tumoral TLSs in H&E stained sections usually appear as squished, slender, or simply lymphatic aggregates lacking mature structure [77]. Finkin et al. found that patients with a large number of hepatic stromal TLSs have a higher likelihood of late recurrence and mortality following HCC resection [82]. Study showed that stromal TLSs within HCC act as niches providing cytokines such as IL-6, LTα, and LTβ, supporting the survival and growth of tumor progenitor cells [82]. Notably, different prognostic and predictive value of peri-tumoral TLS have also been reported [30, 33]. In a study covering 34 ESCC cases, the density and maturation status of peri-tumoral TLSs emerged as valuable parameters for predicting long-term survival and anti-PD-1 therapy response [30]. However, another study involving 273 HCC patients reported no association between stromal TLSs and patient prognosis [33].
However, the clinical value of TLS localization may demonstrate heterogeneity across tumor types and disease progression stages. For instance, in HCC, the density of intra-tumoral TLSs is correlated with lower recurrence risk in early-stage patients, yet exhibits limited prognostic value in advanced stages [33]. Furthermore, the immunological classification based on T/P combined scores lacks a unified guideline in pan-cancer contexts [26, 55, 79, 87]. Several studies on PDAC and HCC have indicated that the combination of high T score and high P score is a key marker for optimal prognosis [79, 87]. This contrasts with the traditional view that a high T score coupled with a low P score is advantageous [26, 55]. Future studies should explore the practical significance of TLS localization across various cancers, to improve personalized prognosis in clinical practice. On the other hand, there was a significant association between the intra-tumoral localization of TLSs and its maturation status, both of which independently predicted positive clinical outcomes [30, 33, 51, 55]. Although there is currently a lack of systematic studies to verify their synergistic effects, integrative analysis may optimize the accuracy of prognostic models.
Density of TLSs
The presence and density of TLSs vary considerably across different cancer types and individual patients (Figure 1C) [10, 11, 18]. High TLS density is closely associated with the enrichment of mature DCs, effector T/B cells, and the development of HEVs [51, 104-106]. This correlation typically predicts better prognosis in cancer patients undergoing various treatments, including surgery, radiotherapy, chemotherapy, and immunotherapy [42, 73, 87]. But negative reports exist regarding the correlation between TLS density and patient prognosis [55, 69, 107], likely attributable to compositional heterogeneity in TLSs and the absence of standardized quantification criteria. Therefore, elucidating how TLS density is defined, what factors regulate it, and how it shapes both TME and clinical outcomes remains crucial.
Definition of TLS density
Currently, the definition of TLS density includes absolute TLS counting and the proportional area analysis of TLS. Absolute TLS counting involves characterizing the density of TLSs within a defined region by expressing the number of TLSs per square millimeter [30, 33, 78, 79]. This method is favored for its robustness and interpretability in the pan-cancer context. Furthermore, some studies opt for specific cell types, such as DC-LAMP+ DCs, as indicators of TLS presence, particularly in NSCLC research [46, 85]. Similarly, other studies have utilized B cell aggregates or HEVs as proxy markers for quantifying TLSs [108-110]. Despite this, these cell composition-based counting methods have not been sufficiently validated across a broader range of cancer types. The proportional area analysis involves normalizing the total area covered by TLSs relative to the entire tumor region to assess the distribution density of TLSs [24, 26, 87]. While this approach simplifies the evaluation of TLS density, it also compromises precision and reproducibility to a certain extent.
Multiple strategies provide feasible options for quantifying TLS density; however, determining the high and low density of TLSs remains a challenge. A widely applied strategy is the four-tier TLS scoring system [55, 77, 103]. This scoring system defines four distinct grades corresponding to the absence, minimal presence, moderate presence, and extensive presence of TLSs [55, 77, 103]. Nonetheless, this scoring system exhibits a degree of subjectivity, requiring the standardization of cutoff values for each grade and validation of their consistency across different tumor contexts. A more simplified alternative involves adopting a binary model to categorize TLS density as either high or low [30, 33, 72-74, 85]. In this approach, the threshold defining high and low TLS density varies across studies. Some studies opt to use the median total TLS density as the baseline for stratification [30, 33, 72, 73]. Others employ strategies such as the minimum p-value method or AUC-based ROC curve analysis to determine and validate the validity of the threshold [74, 85]. These approaches not only help mitigate biases arising from inter-individual variability but also enhance the consistency and comparability of results across different studies [74, 85].
Studies have shown that TLSs can be detected in most types of solid tumors [10, 11, 18], but their distribution characteristics vary significantly across cancer types and populations. For example, the TLS positivity rate in BC ranges from 37% to 39%, while CRC and ESCC exhibit much higher positivity rates of 80%-90% [28-31]. Distinct tumor types also demonstrate differences in TLS density. More aggressive cancers like UCC, ESCC show median TLS density of 0.16-0.36/mm², whereas low-infiltrative HCC maintains median density below 0.06/mm² [30, 33, 73]. Furthermore, the heterogeneity of TLS density was more prominent in metastatic lesions. TLS density in lung metastases vary widely, with CRC and prostate cancer (PCa) metastases exhibiting high levels and leiomyosarcom and osteosarcoma metastases showing minimal presence [10, 111]. Notably, TLSs remain undetectable in brain metastases of melanoma and BC [62, 112].
Factors contributing to TLS density
Several widely employed cancer therapies, including chemotherapy, radiotherapy, ICB, tumor vaccine and oncolytic virus (OV) have been shown to trigger TLS accumulation within TME [6, 37, 44, 113, 114]. Chemotherapy can promote the infiltration of immune cells into the tumor bed, induce immunogenic cell death, and exert beneficial effects on the accumulation and function of TLSs [115]. Zhang et al. found that bladder cancer (BCa) patient treated with chemotherapy exhibited a higher abundance of CD20+ B cells, Tfh cells, and TLSs compared to the treatment-naïve patient [44]. Similarly, Lu et al. reported that neoadjuvant chemotherapy in BC induces a subset of ICOS-L+ B cells expressing complement receptor CR2, which is associated with TLSs development and improved DFS and OS [116]. It merits emphasis that, while corticosteroids are frequently co-administered with chemotherapy to alleviate adverse reactions, their prescription demands caution. Research indicated that corticosteroids may induce the reduction of TLSs density in TME, potentially compromising the beneficial clinical outcomes associated with these structures [43, 74].
In contrast, the impact of radiotherapy on immune cells and TLSs is more complex. Local radiotherapy can stimulate the adaptive immune response crucial for TLS functionality by increasing the expression of MHC I and co-stimulatory molecules [117, 118]. However, some studies found that radiotherapy can transiently inhibit CD8+ T cells and enhance Treg infiltration, creating an immunosuppressive microenvironment [119]. Boivin et al. observed that hypofractionated radiotherapy initially led to a decrease in TLS density, which recovered within two weeks [113]. This finding further illustrates the dynamic influence of radiotherapy on TLS formation, with specific mechanisms warranting further investigation.
By targeting pathways such as PD-1/PD-L1 and CTLA-4, ICB therapy reinvigorates T cell-mediated anti-tumor responses and bolsters immune memory, providing a supportive TME for the formation and accumulation of TLSs [2]. Multiple studies confirmed that in a variety of solid tumors such as melanoma, UCC, and RCC, patients who responded to anti-PD-1 therapy showed denser tumor-infiltrating lymphocytes (TILs) and significant accumulation of TLSs [6, 43, 120]. Of note, Helmink et al. specifically focused on the impact of ICB on B cell populations in TLSs [6]. They revealed that TLSs-related B cells increased significantly in patients with high-risk melanoma and RCC who received ICB therapy. These B cells cooperate with other key immune components in TLSs to jointly optimize the immune efficacy of TLSs by altering T cell activation and function as well as through other mechanisms [6].
Tumor vaccines induce adaptive immune response through the use of tumor cells or their antigens, thereby inhibiting tumor growth, spread, and recurrence [121]. Recent studies have shown that therapeutic vaccination can promote the generation of TLSs in tumors with low immunogenicity [114, 122]. For instance, in patients with high-grade cervical intraepithelial neoplasia (CIN) treated with human papillomavirus oncoprotein vaccines, the regression of lesions correlates with the formation and clonal expansion of TLSs [114]. Similarly, Lutz et al. used a combination of irradiated allogeneic granulocyte-macrophage colony-stimulating factor-secreting PDAC vaccine with cyclophosphamide, successfully inducing T cell infiltration and the development of TLSs, transforming “cold” tumors into “hot” ones [122].
OV is a type of natural or genetically modified virus that can selectively infect and kill tumor cells, causing less damage to normal cells [123]. However, the role of OV in the TLS formation remains to be clarified. A recent study has pointed out that oncolytic herpes simplex virus-1 (oHSV) induces TLS formation in 4MOSC1 and MC38 subcutaneous tumor models, and increases B cell infiltration and TCF1+CD8+ T cell proliferation [37]. Mechanistically, oHSV increases the expression of TLS-related chemokines and simultaneously upregulates CXCL10/CXC motif chemokine receptor 3 (CXCR3) to promote TLS formation. Furthermore, oHSV-mediated TLS formation revealed superior response and survival rate when combined with aPD-1 treatment. Another study revealed that in ICB refractory HNSCC, oncolytic adenovirus induces TLS characteristics and enhances anti-tumor immunity [124]. The transcriptome analysis demonstrated that oncolytic adenovirus treatment induced TLS-associated gene signatures (e.g., CXCR5, LTA, LTB), increased B cell activation markers CD19 and immunoglobulin synthesis-related genes.
Role of TLS density in tumor immunity
In various cancers, such as HNSCC, GC, BC, BCa, an increase in TLS density correlates with enhanced TILs activity [44, 72, 86, 109, 110]. Compared with SLOs, TLSs, as non-encapsulated units, can more directly capture tumor antigens and pro-inflammatory mediators, thus accelerating local immune activation [11]. Notably, multiple studies on solid tumors such as LSCC, CRC, and ESCC have demonstrated that patients with different TLS density exhibit heterogeneity in TLS maturation [30, 42, 74]. Concretely, TLSslow tumor predominantly featuring E-TLS structures, whereas TLSshigh tissues show more mature TLSs [30, 42]. Mature TLSs may create a supportive immune microenvironment that promotes its accumulation, requiring experimental confirmation. While TLSs demonstrate potent anti-tumor immune potential, studies also report that TLSs can become immune-tolerant niches for malignant cells [70, 107, 125, 126]. For instance, in a CCL21-engineered melanoma model, FOXP3+ Tregs and myeloid-derived suppressor cells (MDSCs) were recruited to TLSs, promoting tumor growth [126]. Furthermore, other cell types such as M2 macrophages, Th 2 cells, and Bregs have been identified as contributors to the immunosuppressive microenvironment within TLSs [70, 107, 125]. Notably, several studies indicate that this immunosuppressive state within TLSs extends beyond the suppression of anti-tumor immunity and may directly support tumor growth and metastasis through the secretion of cytokines [28, 82].
Despite the presence of immunosuppressive cells, a higher density of TLSs is generally associated with enhanced anti-tumor immunity across most cancer types [51, 79, 85-87]. This may be attributed to the mature CD83+DC-LAMP+ DCs dominated TIL crosstalk. CD83+DC-LAMP+ DCs are ubiquitously present within TLSs, including those that are immature or located peripherally to the tumor core [30, 46, 48, 51, 74, 127]. These cells possess an immunomodulatory potential that does not vary with the heterogeneity of the TLSs. CD83+DC-LAMP+ DCs are localized within the T cell compartments of TLSs, where they activate the differentiation of naïve T cells via MHC-mediated antigen presentation [22]. In addition to T cell activation, several studies found a positive correlation between CD83+DC-LAMP+ DCs and GC B cells, NK cells, and HEVs [46, 68, 106, 111, 128, 129]. For instance, in murine models, CD11c+ DCs (specific mouse marker) can induce the development of the vascular system and promote TLS assembly by secreting LTβ or activating the STING signaling pathway [104, 105]. Furthermore, in BC, mature DCs and HEVs can co-develop despite the presence of Tregs [106], further substantiating the effectiveness of DC-mediated immune coordination in combating immune tolerance.
The prognostic and predictive value of TLS density
In various human cancers, higher TLS density is correlated with prolonged patient survival and improved response to ICB [6, 42, 74, 85, 87]. This finding has been extensively reviewed and summarized elsewhere [10, 11, 14, 18]. Despite this overall positive correlation, negative relationships between TLS density and patient outcomes have also been reported [55, 69, 107]. For instance, Ding et al. identified Treg -skewed TLSs in iCCA, where their density strongly correlated with poorer 5-year OS [55]. Tregs suppress anti-tumor immune responses by disrupting co-stimulatory signals between antigen-presenting cells (APCs) and effector T cells, and secreting immunosuppressive molecules [130]. One possible reason for the difference in the resulting data is a lack of consensus on what constitutes TLSs and how laboratory quantify them [20]. So far, the definition of TLSs has varied in each study [46, 76, 101, 102, 110]. Several studies have identified TLSs as DC-LAMP+ mature DC aggregates [46, 101, 102], while others define them as CD20+ B cell clusters or other immune aggregates [9, 76, 110]. TLSs are not functionally homogeneous immune aggregations, their cellular composition directly determines anti-tumor immune efficacy and clinical outcomes [6, 23, 55, 107, 125, 131]. For instance, in melanoma, elevated B cell infiltration and TLS density were observed in responsive tumors treated with anti-PD-1 alone or combined with anti-CTLA-4 [6]. Conversely, in a retrospective analysis of soft-tissue sarcomas (STS) samples from a phase II trial of the anti-PD-1 antibody pembrolizumab, high TLS-associated Treg infiltration was correlated with reduced objective response rates and poorer survival outcomes [131]. These findings demonstrate that the cellular composition of TLSs differentially modulates anti-tumor immune responses across cancer types (Table 1).
Immune cells in TLSs
| Cell types | Cell subsets | Markers | Functions | Cancer types | Cases | Prognostic value | Predictive value to ICB | Ref. |
|---|---|---|---|---|---|---|---|---|
| Lymphoid cells | Naïve T cells | CD3+CD27+CD28+CD45RA+CD45RO-CD62L+CCR7+ | Develop into functional T cells | Melanoma, LC | Mice | NA | NA | [160] |
| Th 1 cells | CD3+CD4+ Tbet+ | Promote CTL differentiation | MIBC | 153 patients | Association with longer OS | Positive | [44] | |
| GC | 82 patients | Association with longer RFS | NA | [161] | ||||
| PDC | 534 patients | Association with longer DFS, OS | NA | [87] | ||||
| Th 2 cells | CD3+CD4+BCL6+GATA3+ | Suppress Th 1 and Th 17 cell differentiation | CRC | 67 patients | Association with shorter RFS | NA | [107] | |
| Th 17 cells | CD3+CD4+RORγt+STAT3+ | Promote TLS formation | ESCC | 650 patients | Association with longer DFS, OS | NA | [162] | |
| PDC | 534 patients | Association with longer DFS, OS | NA | [87] | ||||
| Tfh cells | CD3+CD4+ICOS+CXCR5+PD-1++ | Activate GCs development | BC | 70 patients | Association with longer DFS | NA | [21] | |
| MIBC | 153 patients | Association with longer OS | Positive | [44] | ||||
| CRCLM | 603 patients | Association with longer RFS, OS | NA | [26] | ||||
| iCCA | 962 patients | Association with longer 5-year OS | NA | [55] | ||||
| CD3+CD4+TCF1+CXCL13+PD-1++ | HNSCC | 14 patients | NA | Positive | [12] | |||
| Tfr cells | CD3+CD4+ICOS+CXCR5+PD-1++FOXP3+ | Decrease GC B cells and CD8+ T cells infiltration | BC | 179 patients | Association with shorter DFS, RFS | NA | [69] | |
| Tregs | CD3+CD4+FOXP3++ CD127- | Block T cells reactivation; reduce the TLS abundance | iCCA | 962 patients | Association with shorter 5-year OS | NA | [55] | |
| GC | 82 patients | Association with shorter RFS | NA | [161] | ||||
| STS | 30 patients | Association with shorter RFS, OS | Negative | [131] | ||||
| T cells | CD3+CD8+CD28+CD39+CD45RA+CD45RO- | Secrete perforin and/or granzyme | ESCC | 31 patients | NA | Positive | [163] | |
| UCC | 45 patients | NA | Positive | [73] | ||||
| Memory T cells | CD3+CD45RA-CD45RO+CD62L+CCR7+ | Maintain immune memory | NSCLC | 458 patients | NA | NA | [85] | |
| Trms | CD3+CD69+CD103+CD62L-CCR7- | Maintain immune memory; secrete CXCL13 | GC | 53 patients | Association with longer DFS, OS | Positive | [67] | |
| LUAD | 49 patients | Association with longer DFS | NA | [164] | ||||
| Naïve B cells | CD20+IgM+IgD+CD27-CD38- | Develop into functional B cells | Melanoma | 46 patients | NA | Negative | [6] | |
| GC B cells | CD20+CD27-CD38+Ki67+AID+BCL6+ | Induce clonal expansion and somatic hypermutation; recruit T cells, DCs, macrophages, and NK cells | NSCLC | 196 patients | Association with longer OS | NA | [23] | |
| HGSOC | 570 patients | Association with longer OS | NA | [63] | ||||
| Melanoma | 46 patients | NA | Positive | [6] | ||||
| Memory B cells | CD20lowCD27+CD38+/- | Maintain immune memory | PDAC | 39 patients | Association with longer OS | NA | [52] | |
| Melanoma | 46 patients | NA | Positive | [6] | ||||
| Bregs | CD20+CD19+CD25+/IL-10+ | Induce Tregs and TAMs activation | BC | 489 patients | Association with shorter MFS | NA | [70] | |
| PCs | CD20+IgD-CD38++CD138+ | Secrete anti-tumor abs | HGSOC | 570 patients | Association with longer OS | NA | [63] | |
| RCC | 59 patients | Association with longer PFS, OS | Positive | [22] | ||||
| STS | 30 patients | Association with longer RFS, OS | Positive | [131] | ||||
| NK cells | CD56+NKp46+ | Execute ADCC | HGSOC | 167 patients | NA | NA | [129] | |
| ILCs-2 | CD45⁺CD127⁺CRTH2⁺ KLRG1⁺ | Induce TLS initiation | PDAC | 328 patients | Association with longer OS | NA | [41] | |
| ILCs-3 | NCR+ | Induce TLS initiation | NSCLC | 57 patients | NA | NA | [165] | |
| Myeloid cells | DCs | CD80+CD83+DC-LAMP+ | Promote Th 1 cells, CTLs, and NK cells infiltration | ccRCC | 186 patients | Association with longer DFS | NA | [127] |
| Melanoma | 82 patients | Association with longer OS | NA | [102] | ||||
| NSCLC | 74 patients | Association with longer DSS, DFS, and OS | NA | [46] | ||||
| CD11c+ | STS | 30 patients | Association with longer RFS, OS | NA | [131] | |||
| FDCs | CD21+ CD23+/- MHC II- | Support follicular B cells and GCs in TLSs | HGSOC | 570 patients | NA | NA | [63] | |
| NSCLC | 196 patients | NA | NA | [23] | ||||
| Neutrophils | MPO+COX2+ | Mediate inflammation; induce tumor angiogenesis | PC | 17 patients | NA | NA | [25] | |
| Macrophages | CD64+CD68+ CD163+CD169+ | Downregulate the GC reaction; recruit regulator cells | CRC | 67 patients | Association with shorter RFS | NA | [107] | |
| Other cells | FRCs | PDPN+VCAM1+ | Direct PCs dissemination; provide structural support | RCC | 59 patients | NA | NA | [22] |
| CAFs | PDPN+FAP- CCL19+ | Orchestrate TLS formation | Melanoma | Mice | NA | NA | [120] | |
| CRC | Mice | Association with longer OS | NA | [31] | ||||
| hMENAΔv6+ | Inhibit TLS formation | NSCLC | 2006 patients | Association with shorter OS | Negative | [103] | ||
| HEVs | PECAM1+PNAd+MADCAM1+ | Recruit lymphocytes | TNBC | 108 patients | Association with pCR | NA | [110] | |
| LECs | PECAM1+PDPN++LYVE1++VEGFR3+ | Recruit lymphocytes | Melanoma, LC | Mice | NA | NA | [160] |
ADCC: antibody-dependent cellular cytotoxicity; Bregs: regulatory B cells; BC: breast carcinoma; BCSS: breast cancer specific survival; CAFs: cancer-associated fibroblasts; ccRCC: clear cell renal cell carcinoma; CR: complete response; CRC: colorectal carcinoma; CRCLM: colorectal cancer liver metastases; CXCL13: CXC motif chemokine ligand 13; DCs: dendritic cells; DC-LAMP: dendritic cell-lysosomal-associated membrane protein; DFS: disease-free survival; DSS: disease-specific survival; ESCC: esophageal squamous cell carcinoma; FDCs: follicular dendritic cells; FRCs: fibroblastic reticular cells; GC: gastric carcinoma; HEVs: high endothelial venules; HGSOC: high-grade serous ovarian cancer; HNSCC: head and neck squamous cell carcinoma; ICB: immune checkpoint blockade; iCCA: intrahepatic cholangiocarcinoma; ILCs: innate lymphoid cells; LC: lung carcinoma; LECs: lymphatic endothelial cells; LUAD: lung adenocarcinoma; MFS: metastasis-free survival; MHC: major histocompatibility complex; MIBC: muscle invasive bladder carcinoma; NAC: neoadjuvant chemotherapy; NK cells: natural killer cells; NSCLC: non-small cell lung carcinoma; OS: overall survival; PC: pancreatic carcinoma; pCR: pathologic complete response; PCs: plasma cells; PDAC: pancreatic ductal adenocarcinoma; PDC: pancreatic ductal carcinoma; PR: partial response; RCC: renal cell carcinoma; RFI: recurrence-free interval; RFS: relapse-free survival; STS: soft-tissue sarcomas; TAMs: tumor-associated macrophages; Tfh cells: t follicular helper cells; Tfr cells: follicular regulatory t cells; TNBC: triple-negative breast carcinoma; Tregs: regulatory t cells; Trms: resident memory T cells.
Therefore, to establish the clinical value of TLS density in practice, it is essential to consider the heterogeneity and balance of cellular components within TLSs. Exploring the clinical significance of TLS classification based on their cellular components may represent a promising strategy [63, 107, 132]. A study on ovarian cancer (OC) categorized TLSs into four types based on size, cellular composition, and GC organization [63]. While investigators examined correlations between these TLS types and TIL (specifically PC) density, clinical outcome associations remained unexplored [63]. In BCa, TLSs are classified into C1-C5 subtypes based on the expression profiles of 39 TLS gene signatures [132]. These subtypes exhibit marked heterogeneity in TME and prognostic outcomes [132]. The C2 subtype, characterized by robust infiltration of B cells, CD8+ T cells, and Tfhs, is associated with optimal survival outcomes [132]. In contrast, the C4 subtype (with elevated CCL20 expression) and C3/C5 subtypes (dominated by Tregs or stromal cells) correlate with poorer prognosis, indicating immune escape or a cancer-promoting microenvironment [132]. Another research concerning CRC classified TLSs into five subtypes through quantitative analysis of six immune cell populations including Th cells, GC B cells and FDCs [107]. The GC-TLS, B cell-rich, and FDC-rich types have similar structural characteristics and favorable prognostic significance to mature TLS based on CD21/ CD23 classification [107]. Notably, Th 2-enriched TLSs exhibited skewed distribution in recurrent patients, indicating potential immunosuppressive imbalance in TME [107]. These advances indicate that establishing a unified standard across cancer types is important—requiring integration of TLS density and quantitative analysis of key functional cells, thereby achieving progress from precise prognostics to mechanistic intervention.
Evaluation of TLS heterogeneity
The efficacy heterogeneity of ICB has driven the exploration of more precise biomarkers. In recent years, TLSs, as critical hubs of anti-tumor immune responses within TME, have gradually garnered clinical attention [10, 11, 18]. However, the biological functions of TLSs and their synergistic or complementary relationships with other biomarkers have yet to be systematically elucidated. Moreover, the inherent heterogeneity of TLSs poses technical challenges to traditional assessment methods, necessitating the development of advanced analytical tools.
Joint evaluation of TLSs and other biomarkers
Evaluating the relationship between TLS metrics and other immunotherapy biomarkers (e.g., TMB, neutrophil-to-lymphocyte ratio (NLR), PD-L1 expression) can uncover whether TLSs possess synergistic or complementary value in predicting immunotherapy outcomes [71, 133, 134]. This approach may enhance response prediction compared to any single biomarker alone.
TMB alters protein structures on a genetic level, generating neoantigenic epitopes that may potentially promote immune activation and the development of TLSs [135]. TMB, defined as the total number of base substitution mutations and indels per megabase across the tumor genome, has shown broad correlations with the efficacy of various cancer immunotherapies [136]. Posch et al. revealed a positive association between BRAF mutations and the density and maturation of TLSs in 109 patients with stage II/III nmCRC [42]. This sparked deeper investigation into potential links between TMB and TLSs. Yet, several studies noted the absence of direct statistical correlation between TLS density and TMB in melanoma, BCa, and muscle-invasive bladder cancer (MIBC), both factors independently associated with patient survival [7, 132, 133]. Notably, Pagliarulo et al. confirmed the significant predictive value of combining TMB and TLSs assessment for patient prognosis [133]. The combination of high TLS density and elevated TMB was associated with the most favorable OS, indicating a potential synergistic biomarker effect [133]. Given the uncertainty of the association between TLSs and TMB in different cancer contexts, their relationship warrants further investigation.
Elevated levels of neutrophils can suppress lymphocyte and NK cell activation, potentially inhibiting the anti-tumor response mediated by TLSs [137]. NLR, established as a systemic inflammatory marker, has been confirmed as an independent prognostic factor for various malignant tumors [136]. Fukuhara et al. demonstrated a correlation between low blood NLR and high TLS expression in 147 NSCLC patients [71]. In addition, this potential relationship between NLR and TLS density has also been corroborated in GC patients [109]. However, some studies have indicated that there is no association between NLR and TLSs in uLMS and UCC [72, 138]. Despite ongoing debates regarding TLSs and NLR as independent prognostic factors, combined assessment has identified cancer patients with the most favorable prognosis. Multiple studies shown that Patients with high TLSs and low NLR have consistently shown a survival advantage over those assessed by single indicators [72, 109, 138].
Pre-existing expression of immune checkpoint molecules (e.g., PD-L1) within the TME establishes a biological foundation for ICB efficacy [3]. Extensive research indicates that TLSs independently predict ICB responsiveness, irrespective of checkpoint molecule expression levels [11]. This raises critical questions about potential synergistic interactions between TLSs and immune checkpoints. Deng et al. first proposed combining TLSs with PD-L1 status as a composite biomarker framework for immunotherapy in primary cardiac angiosarcoma (PCA) [134]. Their analysis demonstrated that even immature TLSs, when combined with PD-L1 positivity, guided anti-PD-1 therapy and correlated with transient metastatic LN regression [134]. However, research on TLS-immune checkpoint synergy remains preliminary, requiring further validation.
Cutting-edge technologies for TLS evaluation
Currently, multi-omics technologies including histopathology, genomics, and transcriptomics offer a range of options for the detection and quantification of TLSs (Table 2). Despite their practical value, these conventional methods still possess several limitations, such as insufficient depth of information provided, the necessity for destructive testing, and complex operational procedures, all of which highlight the pressing demand for innovative technologies to tackle TLS heterogeneity effectively. The development of advanced technologies, such as radiomics, deep learning models, and three-dimensional (3D) imaging, etc., has overcome the limitations of traditional methods and propelled the advancement of the TLS research field (Figure 4) [7, 139-141].
Current methods to detect and quantify TLSs
| Methods | Identified parameters | Advantages | Limitations | Ref | |
|---|---|---|---|---|---|
| Histopathology | H&E/HES staining | Cellular morphology, tissue structure | Low cost, simple operation, widely application | Cannot distinguish specific cells | [9, 28, 51, 53, 85, 87] |
| IHC staining | Specific protein localization/expression | High specificity, precise localization | Limited multiplexing capability, high antibody dependency | [44, 72, 76, 82] | |
| IF staining | Multiprotein colocalization (fluorescent labeling) | Multi-color labeling, high resolution (200 nm) | Autofluorescence interference | [7, 82, 120, 166] | |
| mIHC/mIF staining | Multiple protein markers | Multi-parametric (4-8 markers) | Antibody cross-reactivity | [26, 42, 44, 51, 54, 107] | |
| Traditional protein assay methods | Western blot | Specific protein expression | High specificity, semi-quantitative | Low throughput | [167, 168] |
| ELISA | Soluble protein concentration | High throughput, quantitative accuracy | Single-plex detection, antibody-dependent | [23, 167, 169] | |
| Flow cytometry | Cell surface/intracellular molecular markers | Multi-parametric (10+ markers), rapid | Limited to cell suspensions, high instrument cost | [6, 63, 120, 170] | |
| CyTOF | Metal-tagged proteins | Multi-parametric (40+ markers), no spectral overlap | High instrument cost, complex sample preparation | [6, 71] | |
| Genomics | MSI/MSS analysis | Microsatellite stability | Simple and cost-effective compared to whole-genome sequencing. | Low sensitivity | [24, 42, 78] |
| CGH analysis | Genome copy number variation | Genome-wide coverage, high resolution | Cannot detect balanced translocations or point mutations | [82] | |
| Transcriptomics | mRNA microarray analysis | Whole transcriptome expression profile | High throughput, moderate cost | Limited to known transcripts, narrow dynamic range | [6, 7, 21, 26] |
| RNA-seq | Whole transcriptome expression profile | Discovery of novel transcripts | High cost, complex data analysis | [6, 48, 52, 76] | |
| scRNA-seq | Single cell RNA expression profile | Single-cell resolution | Extremely high cost, technically demanding | [6, 7, 44, 47, 171] | |
| Spatial transcriptomics | GeoMx DSP | Spatial multi-omics (RNA/ protein) | Preserves spatial context, multi-target analysis | Limited resolution (10-100 μm) | [103, 142, 162, 172] |
| 10x Visium | Whole transcriptome spatial localization | Moderate resolution (55 μm, 2μm (HD version)), compatible with FFPE/fresh-frozen tissues | High sample preparation requirements | [172, 173] | |
| Stereo-seq | Subcellular spatial transcriptome | Ultra-high resolution (0.5 μm) | Massive data storage/computational demands | [173] | |
| Spatial proteomics | IMC | Metal-tagged proteins | Multi-parametric (40+ markers), no spectral overlap, compatible with FFPE sections | High instrument cost, complex metal-labeled antibody preparation | [47, 174-176] |
| GeoMx DSP | Spatial multi-omics (RNA/ protein) | Preserves spatial context, multi-target analysis | Limited resolution (10-100 μm) | [7, 142, 171, 177] | |
| CyCIF | Fluorescently labeled proteins | High throughput (4-5 cycles, 30+ markers), high sensitivity, compatible with standard microscopes | Photobleaching, time-consuming cycles | [155] | |
| CODEX | DNA-barcoded proteins | Ultra-high-parametric (50+ markers), no spectral unmixing | Complex decoding workflow, antibody validation challenges | [178, 179] | |
| Radiomics | CT | Anatomical structure, density difference | Rapid, widely application | Radiation exposure, low soft-tissue contrast | [139, 180, 181] |
| SPECT | Blood flow/metabolic distribution | Multi-target imaging | Low resolution, time-consuming | [144] | |
| MRI | Soft tissue contrast | No radiation, high resolution | Long scan times, high cost | [145] | |
| Deep learning model | Automatic image/data feature extraction | Handles complex data, high predictive power | Requires large annotated datasets | [140, 149-152] | |
| 3D imaging technique | 3D structure reconstruction | Enables 3D visualization | High equipment cost, complex data processing | [141, 154, 155] | |
3D: three-dimensional; CGH: comparative genomic hybridization; CODEX: co-detection by indexing; CT: computed tomography; CyCIF: cyclic immunofluorescence; CyTOF: cytometry by time-of-flight; ELISA: enzyme-linked immunosorbent assay; FFPE: formalin fixed paraffin embedded; GeoMx DSP: GeoMx digital spatial profiler; H&E: hematoxylin and eosin; HES: hematoxylin-eosin-saffron; IHC: immunohistochemistry; IF: immunofluorescence; IMC: imaging mass cytometry; mIHC: multiplex immunohistochemistry; mIF: multiplex immunofluorescence; MRI: magnetic resonance imaging; MSI/MSS: microsatellite instability or stability; PET: positron emission tomography; RNA-seq: RNA sequencing; scRNA-seq: single-cell RNA sequencing; SPECT: single-photon emission computed tomography.
Cutting-edge technologies for TLS evaluation. The development of advanced technologies, such as radiomics, deep learning models, and three-dimensional (3D) imaging, etc., has overcome the limitations of traditional methods and propelled the advancement of the TLS research field. Spatial omics: High-throughput sequencing, microscopic imaging localization, quantification of tissue molecular expression. Radiomics: Extract features from medical images, screen for disease-related indicators. Deep learning models: Preprocess datasets, design neural network architectures, and train models. 3D imaging technologies: Perform sample permeabilization, capture images, and reconstruct 3D models via software algorithms. 3D: three-dimensional.
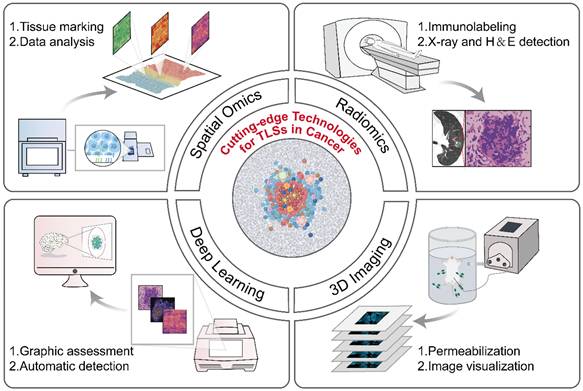
Spatial omics technologies, including spatial transcriptomics, proteomics, etc., have become key tools for elucidating the relationship between TLSs and tumor immunity [7, 142]. These technologies allow researchers to measure and map the expression of genes and proteins at specific localizations (i.e., “spatially”) within tissue sections, enhancing our understanding of cell interactions and their impact on treatment responses [143]. For instance, Cabrita et al. applied spatial proteomics to quantitatively analyze immune-related proteins in the TME [7]. Their analysis revealed that T cells in TLS-deficient regions exhibited dysfunctional phenotypes, whereas TLS-associated T cells showed increased CD4+ proportions and elevated BCL2 (an anti-apoptotic protein) levels, demonstrating the critical role of TLSs in maintaining functional T cell states [7]. In addition, a study using spatial multi-omics technologies unveiled the critical role of TLSs in the immune therapy response of HNSCC [142]. Spatial proteomic analysis revealed significant upregulation of T lymphocyte markers (CD3, CD8), as well as PD-L1, in patients who responded to ICB, indicating a close association between the “hot tumor” phenotype and immune response [142]. Further spatial transcriptomic analysis demonstrated that genes related to immune modulation, recruitment of various immune cells, and effective IFN response were markedly elevated in TLSs compared to normal GCs, indicating that TLSs harbor a distinct microenvironment for immune activation [142]. Despite representing a significant advance in characterizing TLSs, spatial omics still faces challenges such as high costs, low throughput, and limited spatial resolution, necessitating technological innovation and optimization [143].
Compared to invasive detection methods, radiomics techniques, including computed tomography (CT), positron emission tomography (PET), single-photon emission computed tomography (SPECT), and magnetic resonance imaging (MRI) emerge as a promising non-invasive approach for the detection and assessment of TLSs [16, 139, 144, 145]. This method utilizes computer technologies to extract and quantify high-level imaging features of tumors at high throughput and integrates this data with other clinical information to identify TLSs and their associations with tumor prognosis [146]. For example, in a recent study, CT scans revealed multiple small solid components within partial solid nodules in lung adenocarcinoma (LUAD), findings that correlated with the presence of TLSs in histopathological examination [139]. Using 99mTc-labeled albumin nanocolloid (99mTc-Nanocoll) as a tracer, Dorraji et al. successfully achieved non-invasive localization of TLSs in PC by SPECT [144]. Histopathological analysis further confirmed that these regions exhibited characteristic pathological features, including T/B cell compartmentalization and macrophage infiltration [144]. However, conventional imaging modalities (e.g., CT, SPECT) remain limited by radiation exposure risks and insufficient soft-tissue resolution, constraining their clinical applicability [145]. In contrast, MRI emerges as a superior alternative. For instance, a study on HCC developed an MRI-based predictive model by integrating intra- and peri-tumoral radiomic features [145]. This model not only non-invasively assesses peri-tumoral TLS density distribution but also stratifies patients' survival outcomes and immunotherapy response profiles, offering a reliable objective basis for clinical decision-making [145].
Although these technologies provide non-invasive and real-time alternatives for the detection of TLSs, they still face challenges in terms of accuracy and specificity. In recent years, materials for single-cell imaging have been developed to improve detection accuracy [147, 148]; however, whether imaging at the single-cell level can adequately characterize the presence of TLSs remains to be further investigated.
Deep learning models can automate the analysis and quantification of complex images and data related to TLSs, and demonstrate the potential to predict patient outcomes [140, 149-152]. Recently, several deep learning models have been developed towards automated segmentation and quantification of TLSs from H&E images in various tumor types [140, 149, 150]. Wang et al. devised an automated computational workflow to quantify the density of TLSs in routinely H&E-stained whole-slide images (WSIs) of LUAD tissue [140]. Additionally, a Cox proportional hazard regression model, incorporating clinicopathological variables and the TLS density, was established to assess its prognostic ability [140]. Similarly, deep learning models based on H&E images can be utilized to detect other parameters of TLSs, such as cellular composition and maturation, and these models have been extensively validated in different cancer settings [149, 150]. While deep learning models based on H&E images currently dominate, alternative data-driven approaches also exhibit distinct advantages [151, 152]. Unlike these studies that depended solely on pathologists' manual annotations of TLSs without mIHC guidance, Chen et al. leveraged mIHC markers—DAPI, CD3, and CD20—to identify TLSs, thus reducing the influence of subjective human judgment [151]. In addition, Li et al. developed a machine learning model that used spatial transcriptomic data to identify markers of TLSs and effectively predict TLS localization, holding the promising potential to impact cancer treatment strategies [152]. Particularly, the identified markers emphasize the significance of immunoglobulin genes in TLS detection, adding a novel perspective to existing knowledge [152]. Deep learning models offer powerful tools for the efficient and automated analysis of TLSs, yet they face challenges in accurately distinguishing between LNs and TLSs, highlighting the need for future research to further improve model accuracy and reliability [153].
3D imaging technology offers a novel perspective for analyzing the spatiotemporal heterogeneity of TLSs [141, 154, 155]. In CRC, whole-section 3D reconstruction has unveiled TLS networking features that conventional 2D pathology struggles to capture [155]. A single TLS network can span multiple tissue layers and extend across millimeters [155]. These 3D networks exhibit the gradient distribution of cellular structure (e.g. CD68⁺CD163⁺ macrophages, and B/T cells) and molecular markers (e.g. PD-L1, LAG-3, TIM-3), indicating that TLSs may play a dynamic coordinating role in anti-tumor immune response [155]. A study on Crohn disease utilizing 3D imaging revealed significant anatomical associations between TLSs and mesenteric lymphatics [154]. These B cell-enriched ectopic lymphoid structures are distributed along lymphatic pathways, especially in the fatty infiltrated mesenteric area, where TLSs embed directly into the lymphatic wall and trigger structural remodeling [154]. Notably, IL-33R⁺ ILCs detected within some TLSs imply their potential involvement in lymphatic wall remodeling through cellular interactions [154]. In addition, wildDISCO technology enables whole-body horizontal 3D imaging of mice through innovative whole-body transparency and multi-target immunolabeling strategies [141]. Using dual labeling of CD23 and CD3 antibodies, this technique reveals the fine distribution of TLSs in primary tumors and lung and intestinal metastases in BC metastasis models [141]. Subcellular-level imaging further demonstrates that even relatively large TLSs exhibit significantly smaller volumes compared to those in metastatic lesions [141].
Conclusions and prospects
TLSs exhibit marked heterogeneity in maturation, localization and density across tumor types and individuals, profoundly impacting anti-tumor immune effects and clinical value (Table 3) [10, 11]. Current evidence indicates that more mature, intra-tumoral, and higher-density TLSs serve as predictive biomarkers for enhanced response to ICB therapy and prolonged survival benefits [6, 18, 26, 30, 33, 132]. Some studies have indicated that mature TLSs can also be localized in the peri-tumoral area, predicting a stronger anti-tumor immune response [30, 78, 79]. Conversely, immature TLSs predominantly localized at the tumor periphery, low-density TLSs, or complete TLS absence are strongly associated with an immunosuppressive microenvironment, tumor progression, and unfavorable prognosis [6, 30, 33, 42, 55, 77]. Building on these clinical observations, current studies have identified TDLNs, the STING pathway, and tumor vaccines, along with other potential targets, as key regulators of TLS heterogeneity [54, 89, 114]. Significantly, the integrative analysis of TLS with other biomarkers such as TMB, NLR, and PD-L1 expression represents a novel prognostic prediction strategy [71, 133, 134]. Equally important, emerging computational-driven technologies like spatial omics and radiomics enable efficient and comprehensive dissection of TLS-immune microenvironment interactions [7, 139-141]. While the understanding of TLS heterogeneity has advanced considerably, translating these insights into clinical applications presents several challenges.
The prognostic and predictive value of different TLS characteristics in different cancer types
| TLS parameters | Cancer types | Cases | Received treatments | Prognostic value | Predictive value to ICB | Ref. | ||
|---|---|---|---|---|---|---|---|---|
| Maturation | Immature TLSs | E-TLSs | CRC | 109 patients | Surgical resection; chemotherapy | Association with higher risk of recurrence | NA | [42] |
| LSCC | 138 patients | Surgical resection; NAC | Association with shorter PFS, DFS, and OS | NA | [74] | |||
| PDAC | 63 patients | Surgical resection | Association with longer PFS, OS | NA | [52] | |||
| BC | 489 patients | Surgical resection | Association with shorter FS | NA | [70] | |||
| ESCC | 34 patients | ICB (αPD-1); surgical resection | No clinical association | No clinical association | [30] | |||
| MIBC | 153 patients | ICB (αPD-1); NAC | Association with shorter OS | Negative | [44] | |||
| ccRCC | 395 patients | Surgical resection | Association with shorter PFS, OS | NA | [51] | |||
| UCC | 24 patients | ICB (αPD-1 + αCTLA-4); surgical resection | NA | Negative | [43] | |||
| 6 tumor types and others | 328 patients | ICB (αPD-1); surgical resection | Association with shorter PFS, OS | Negative | [9] | |||
| Mature TLSs | PFL-TLSs | HCC | 273 patients | Surgical resection | Association with lower risk of recurrence | NA | [33] | |
| LSCC | 138 patients | Surgical resection; NAC | Association with shorter PFS, DFS, and OS | NA | [74] | |||
| CRC | 109 patients | Surgical resection; chemotherapy | Association with lower risk of recurrence | NA | [42] | |||
| ESCC | 34 patients | ICB (αPD-1); surgical resection | Association with longer PFS | Positive | [30] | |||
| MIBC | 153 patients | ICB (αPD-1); NAC | Association with shorter OS | Negative | [44] | |||
| UCC | 24 patients | ICB (αPD-1 + αCTLA-4); surgical resection | NA | Positive | [43] | |||
| ccRCC | 395 patients | Surgical resection | Association with shorter PFS, OS | NA | [51] | |||
| 6 tumor types and others | 328 patients | ICB (αPD-1); surgical resection | Association with shorter PFS, OS | Negative | [9] | |||
| SFL-TLSs | HCC | 273 patients | Surgical resection | Association with lower risk of recurrence | NA | [33] | ||
| LSCC | 138 patients | Surgical resection; NAC | Association with longer PFS, DFS, and OS | NA | [74] | |||
| CRC | 109 patients | Surgical resection; chemotherapy | Association with lower risk of recurrence | NA | [42] | |||
| PDAC | 63 patients | Surgical resection | Association with longer DFS | NA | [52] | |||
| RCC | 59 patients | ICB (αPD-1 + αCTLA-4); surgical resection | Association with longer PFS, OS | Positive | [22] | |||
| MIBC | 153 patients | ICB (αPD-1); NAC | Association with longer OS | Positive | [44] | |||
| ccRCC | 395 patients | Surgical resection | Association with longer PFS, OS | NA | [51] | |||
| BCa | 408 patients | ICB (αPD-1/αCTLA-4/αPD-1 + αCTLA-4); chemotherapy | Association with longer OS | Positive | [132] | |||
| ESCC | 34 patients | ICB (αPD-1); surgical resection | Association with longer PFS | Positive | [30] | |||
| UCC | 24 patients | ICB (αPD-1 + αCTLA-4); surgical resection | NA | Positive | [43] | |||
| 6 tumor types and others | 328 patients | ICB (αPD-1); surgical resection | Association with longer PFS, OS | Positive | [9] | |||
| Localization | Intra-tumoral TLSs | NSCLC | 74 patients | Surgical resection | Association with longer DSS, DFS, and OS | NA | [46] | |
| PDC | 534 patients | Surgical resection; chemotherapy; radiotherapy | Association with longer DFS, OS | NA | [87] | |||
| HCC | 273 patients | Surgical resection | Association with lower risk of recurrence | NA | [33] | |||
| HCC | 360 patients | Surgical resection | Association with lower risk of recurrence | NA | [79] | |||
| CCA | 471 patients | ICB (αPD-1); surgical resection; chemotherapy | Association with longer OS | Positive | [77] | |||
| CRCLM | 603 patients | Surgical resection; NAC | Association with longer RFS, OS | NA | [26] | |||
| GC | 53 patients | ICB (αPD-1); surgical resection | Association with longer OS | Positive | [67] | |||
| ccRCC | 395 patients | Surgical resection | Association with longer PFS, OS | NA | [51] | |||
| Peri-tumoral TLSs | HCC | 360 patients | Surgical resection | Association with longer RFS, OS | NA | [79] | ||
| iCCA | 962 patients | Surgical resection | Association with shorter 5-year OS | NA | [55] | |||
| CCA | 471 patients | ICB (αPD-1); surgical resection; chemotherapy | Association with shorter OS | Negative | [77] | |||
| CRC | 174 patients | Surgical resection | Association with longer RFS, OS | NA | [78] | |||
| CRCLM | 603 patients | Surgical resection; NAC | Association with shorter RFS, OS | NA | [26] | |||
| Cutaneous melanoma | 82 patients | Surgical resection; chemotherapy; radiotherapy | Association with longer OS | NA | [102] | |||
| ccRCC | 186 patients | Surgical resection | Association with longer DFS | NA | [127] | |||
| ccRCC | 395 patients | Surgical resection | Association with shorter PFS, OS | NA | [51] | |||
| GC | 53 patients | ICB (αPD-1); surgical resection | Association with longer OS | Positive | [67] | |||
| ESCC | 34 patients | ICB (αPD-1); surgical resection | Association with longer PFS | Positive | [30] | |||
| Stromal TLSs | HCC | 273 patients | Surgical resection | No clinical association | NA | [33] | ||
| HCC | 82 patients | Surgical resection | Association with higher risk of recurrence | NA | [82] | |||
| GC | 53 patients | ICB (αPD-1); surgical resection | Association with shorter OS | Negative | [67] | |||
| ccRCC | 395 patients | Surgical resection | Association with shorter PFS, OS | NA | [51] | |||
| Density | CRC | 109 patients | Surgical resection; chemotherapy | Association with lower risk of recurrence | NA | [42] | ||
| NSCLC | 458 patients | Surgical resection | Association with reduced risk of death | NA | [85] | |||
| NSCLC | 147 patients | Surgical resection | Association with longer DFS | NA | [71] | |||
| LSCC | 138 patients | Surgical resection; NAC | Association with longer PFS, DFS, and OS | NA | [74] | |||
| OSCC | 168 patients | NA | Association with longer RFS, 5-year OS | NA | [182] | |||
| Melanoma | 46 patients | ICB (αPD-1/αPD-1 + αCTLA-4); surgical resection | NA | Positive | [6] | |||
| Melanoma | Mice | ICB (αPD-1/αPD-1 + αCTLA-4) | Association with longer OS | Positive | [120] | |||
| MIBC | 153 patients | ICB (αPD-1); NAC | Association with longer OS | Positive | [44] | |||
| uLMS | 102 patients | Surgical resection | Association with longer OS | NA | [72] | |||
| ESCC | 34 patients | ICB (αPD-1); surgical resection | Association with longer PFS | Positive | [30] | |||
| PDC | 534 patients | Surgical resection; chemotherapy; radiotherapy | Association with longer DFS, OS | NA | [87] | |||
| UCC | 45 patients | ICB (αPD-1 + αCTLA-4); surgical resection | Association with longer RFS, OS | Positive | [73] | |||
| ccRCC | 395 patients | Surgical resection | No clinical association | NA | [51] | |||
BC: breast carcinoma; BCa: bladder cancer; CRC: colorectal carcinoma; CRCLM: colorectal cancer liver metastases; CTLA-4: cytotoxic T-lymphocyte-associated protein-4; ccRCC: clear cell renal cell carcinoma; DFS: disease-free survival; DSS: disease-specific survival; E-TLSs: early tertiary lymphoid structures; ESCC: esophageal squamous cell carcinoma; GC: gastric carcinoma; HCC: hepatocellular carcinoma; ICB: immune checkpoint blockade; iCCA: intrahepatic cholangiocarcinoma; LSCC: lung squamous cell carcinoma; MIBC: muscle invasive bladder carcinoma; NAC: neoadjuvant chemotherapy; NSCLC: non-small cell lung carcinoma; OS: overall survival; OSCC: oral squamous cell carcinoma; PD-1:programmed cell death protein 1; PDAC: pancreatic ductal adenocarcinoma; PDC: pancreatic ductal carcinoma; PFL-TLSs: primary follicle-like tertiary lymphoid structures; PFS: progression-free survival; RCC: renal cell carcinoma; RFS: relapse-free survival; SFL-TLSs: secondary follicle-like tertiary lymphoid structures; TNBC: triple-negative breast carcinoma; uLMS: uterine leiomyosarcoma; UCC: urothelial carcinoma.
Optimizing evaluation strategies for TLS heterogeneity
To effectively evaluate TLS heterogeneity, unified standards and comprehensive strategies must be established. Currently, there is a lack of standardized quantification criteria for TLSs across different patients and cancer types. Future studies should aim to define standardized criteria for spatial localization (e.g., distinguishing peritumoral from stromal regions) and establish uniform thresholds for classifying TLS density as high or low. For ambiguously defined spatial localization, an alternative strategy is to quantify TLS localization. This approach can be specifically manifested through calculating the precise distances from each target TLS margin to the tumor invasive margin or the interface between the tumor core and the tumor stroma. Through rigorous statistical analysis correlating continuous localization metrics with clinically meaningful endpoints (e.g., OS, PFS, RFS, and response rates to ICB), clinically relevant minimal thresholds or critical points can be identified. Furthermore, the distance-clinical association curves can serve as references to refine the boundary values of conventional localization dichotomization or trichotomization strategies. Moreover, the quantitative relationships between TLS maturation, density, characteristic molecular expression levels and their clinical significance also remain unclear. Therefore, it is necessary to standardize the quantification of TLSs and define statistically significant minimum thresholds. Furthermore, multi-parameter integrated evaluation may serve as an effective strategy to enhance prognostic prediction accuracy. For instance, in CRC, the proportion of SFL-TLSs demonstrates a positive correlation with overall TLS density [42]. A composite immune score incorporating both total TLS density and mature TLS density effectively stratifies patients with minimal recurrence risk [42]. Other favorable TLS features also tend to cluster, such as intra-tumoral TLSs with higher maturation status, warranting further exploration of their combined clinical utility [30, 33, 51, 55]. Moreover, integrating TLSs with other biomarkers such as TMB, NLR, and PD-L1 expression emerges as a promising prognostic prediction approach [42, 71, 134]. However, the statistical associations between TLSs and other biomarkers vary across cancer types, necessitating further research to define clinical applicability.
Advancing assessment technologies for TLS heterogeneity
Developing techniques suitable for assessing TLS heterogeneity represents a significant challenge. Recently, advanced technologies such as spatial omics, radiomics, deep learning models, and 3D imaging have emerged, overcoming the limitations of traditional methods and providing new options for the detection and evaluation of TLSs [6, 139-141]. However, these technologies still face issues such as high costs, low throughput, and insufficient precision, which limit their widespread application. In the future, developing new evaluation technologies or integrating existing ones could be key to addressing these challenges. For example, the integration of radiomics and deep learning models has been used to characterize the microenvironment of GC and predict treatment responses [156]. This approach maintains the advantages of non-invasiveness and high throughput, overcoming the limitations of single technologies.
Developing materials for precisely regulating TLS heterogeneity
Given the role of TLSs in anti-tumor immunity, developing biomaterials capable of modulating TLS heterogeneity to enhance anti-tumor immune response is crucial. Various materials, such as cytokine-loaded nanoparticles, oncolytic virus vaccines, and bioscaffolds, have been developed to induce TLS formation, demonstrating initial feasibility [157, 158]. However, current TLS induction methods are not yet mature, focusing primarily on the existence and density of TLSs rather than their precise maturation and localization. Moreover, some studies found that induced TLSs exhibit atypical states, requiring additional regulatory support [37, 158, 159]. Future research should focus on identifying and regulating key factors influencing TLS heterogeneity to achieve precise management of TLSs, thereby enhancing tumor immune response and improving patient outcomes.
Abbreviations
3D: three-dimensional; APCs: antigen-presenting cells; ASCs: antibody-secreting cells; AtM: atypical memory; BC: breast cancer; BCa: bladder cancer; Bregs: regulatory B cells; CCA: cholangiocarcinoma; CCL: CC motif chemokine ligand; CIN: cervical intraepithelial neoplasia; COAD: colon adenocarcinoma; CRC: colorectal cancer; cSCC: cutaneous squamous cell carcinoma; CT: computed tomography; CTLA-4: cytotoxic T-lymphocyte-associated protein-4; CTLs: cytotoxic T lymphocytes; CXCL: CXC motif chemokine ligand; CXCR: CXC motif chemokine receptor; ccRCC: clear cell renal cell carcinoma; DC-LAMP: dendritic cell-lysosomal-associated membrane protein; DCs: dendritic cells; DFS: disease-free survival; dMMR: defective mismatch repair; DSS: disease-specific survival; E-TLSs: early tertiary lymphoid structures; EF: extrafollicular; ESCC: esophageal squamous cell carcinoma; FDCs: follicular dendritic cells; FRCs: fibroblastic reticular cells; GC: gastric carcinoma; GCs: germinal centers; H&E: hematoxylin and eosin; HCC: hepatocellular carcinoma; HES: hematoxylin-eosin-saffron; HEVs: high endothelial venules; HNSCC: head and neck squamous cell carcinoma; ICI: immune checkpoint inhibitor; ICB: immune checkpoint blockade; IDO1: indoleamine 23-dioxygenase 1; IFN-γ: interferon gamma; IHC: immunohistochemistry; IL-7/IL-7R: interleukin-7/interleukin-7 receptor; ILCs: innate lymphoid cells; iCCA: intrahepatic cholangiocarcinoma; irAEs: immune-related adverse events; LC: lung carcinoma; LED: light-emitting diode; LNs: lymph nodes; LSCC: lung squamous cell carcinoma; LT α1β2/LTβR: lymphotoxin α1β2/lymphotoxin beta receptor; LTi: lymphoid tissue inducer; LTo: lymphoid tissue organizer; MDSCs: myeloid-derived suppressor cells; MHC: major histocompatibility complex; MIBC: muscle-invasive bladder cancer; MRI: magnetic resonance imaging; MSI-H: microsatellite instability-high; mIF: multiplex immunofluorescence; mIHC: multiplex immunohistochemistry; NAC: neoadjuvant chemotherapy; NK: natural killer; NLR: neutrophil-to-lymphocyte ratio; NSCLC: non-small cell lung cancer; OC: ovarian cancer; OS: overall survival; OV: oncolytic virus; oHSV: oncolytic herpes simplex virus-1; PCA: primary cardiac angiosarcoma; PC: pancreatic carcinoma; PCa: prostate cancer; PD-L1: programmed death-ligand 1; PDAC: pancreatic ductal adenocarcinoma; PCs: plasma cells; PET: positron emission tomography; PFL-TLSs: primary follicle-like tertiary lymphoid structures; PFS: progression-free survival; RCC: renal cell carcinoma; SFL-TLSs: secondary follicle-like tertiary lymphoid structures; SLOs: secondary lymphoid organs; SPECT: single-photon emission computed tomography; STS: soft-tissue sarcomas; TAMs: tumor-associated macrophages; TDLNs: tumor-draining lymph nodes; Tfh: T follicular helper; Tfrs: follicular regulatory T cells; TGF-β: transforming growth factor beta; Th: T follicular helper; Th 2: T helper 2; TILs: tumor-infiltrating lymphocytes; TLSs: tertiary lymphoid structures; TMB: tumor mutational burden; TME: tumor microenvironment; TNF-α: tumor necrosis factor alpha; Tregs: regulatory T cells; Trms: resident memory T cells; uLMS: uterine leiomyosarcoma; UCC: urothelial carcinoma; VSMCs: vascular smooth muscle cells.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (NFSC) 82303328 (H.L.), 82472818 (Z.J.S.), 82273202 (Z.J.S.), the Fundamental Research Funds for the Central Universities 2042023kf0141 (H.L.), and 2042022dx0003 (Z.J.S.), National Key Research and Development Program 2022YFC2504200 (Z.J.S.), the Natural Science Foundation of Wuhan 2023020201020516 (H.L.), and the Young Elite Scientist Support Program by CSA 2023PYRC001 (H.L.).
Author contributions
Zhi-Jun Sun, Hao Li, and Meng-Jie Zhang provided direction and guidance throughout the preparation of this manuscript. Guang-Liang Su drafted the manuscript and prepared the figures and tables. Zhi-Jun Sun, Hao Li, and Meng-Jie Zhang reviewed and revised the manuscript in great depth. All authors read and approved the final manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Ribas A, Wolchok JD. Cancer immunotherapy using checkpoint blockade. Science. 2018;359:1350-5
2. Morad G, Helmink BA, Sharma P, Wargo JA. Hallmarks of response, resistance, and toxicity to immune checkpoint blockade. Cell. 2021;184:5309-37
3. Taube JM, Klein A, Brahmer JR, Xu H, Pan X, Kim JH. et al. Association of PD-1, PD-1 ligands, and other features of the tumor immune microenvironment with response to anti-PD-1 therapy. Clin Cancer Res. 2014;20:5064-74
4. Bhamidipati D, Subbiah V. Tumor-agnostic drug development in dMMR/MSI-H solid tumors. Trends Cancer. 2023;9(10):828-39
5. Sha D, Jin Z, Budczies J, Kluck K, Stenzinger A, Sinicrope FA. Tumor mutational burden as a predictive biomarker in solid tumors. Cancer Discov. 2020;10:1808-25
6. Helmink BA, Reddy SM, Gao J, Zhang S, Basar R, Thakur R. et al. B cells and tertiary lymphoid structures promote immunotherapy response. Nature. 2020;577:549-55
7. Cabrita R, Lauss M, Sanna A, Donia M, Skaarup Larsen M, Mitra S. et al. Tertiary lymphoid structures improve immunotherapy and survival in melanoma. Nature. 2020;577:561-5
8. Petitprez F, de Reyniès A, Keung EZ, Chen TW, Sun CM, Calderaro J. et al. B cells are associated with survival and immunotherapy response in sarcoma. Nature. 2020;577:556-60
9. Vanhersecke L, Brunet M, Guégan JP, Rey C, Bougoüin A, Cousin S. et al. Mature tertiary lymphoid structures predict immune checkpoint inhibitor efficacy in solid tumors independently of PD-L1 expression. Nat Cancer. 2021;2(8):794-802
10. Sautès-Fridman C, Petitprez F, Calderaro J, Fridman WH. Tertiary lymphoid structures in the era of cancer immunotherapy. Nat Rev Cancer. 2019;19:307-25
11. Schumacher TN, Thommen DS. Tertiary lymphoid structures in cancer. Science. 2022;375:eabf9419
12. Li H, Zhang MJ, Zhang B, Lin WP, Li SJ, Xiong D. et al. Mature tertiary lymphoid structures evoke intra-tumoral T and B cell responses via progenitor exhausted CD4 + T cells in head and neck cancer. Nat Commun. 2025;16:4228
13. Pitzalis C, Jones GW, Bombardieri M, Jones SA. Ectopic lymphoid-like structures in infection, cancer and autoimmunity. Nat Rev Immunol. 2014;14:447-62
14. Zhao L, Jin S, Wang S, Zhang Z, Wang L, Chen Z. et al. Tertiary lymphoid structures in diseases: immune mechanisms and therapeutic advances. Signal Transduct Target Ther. 2024;9(1):225
15. Jones GW, Hill DG, Jones SA. Understanding immune cells in tertiary lymphoid organ development: it is all starting to come together. Front Immunol. 2016;7:401
16. Li H, Lin WP, Zhang ZN, Sun ZJ. Tailoring biomaterials for monitoring and evoking tertiary lymphoid structures. Acta Biomater. 2023;172:1-15
17. Li H, Ding JY, Zhang MJ, Yu HJ, Sun ZJ. Tertiary lymphoid structures and cytokines interconnections: The implication in cancer immunotherapy. Cancer Lett. 2023;568:216293
18. Teillaud JL, Houel A, Panouillot M, Riffard C, Dieu-Nosjean MC. Tertiary lymphoid structures in anticancer immunity. Nat Rev Cancer. 2024;24(9):629-46
19. Zhang Y, Xu M, Ren Y, Ba Y, Liu S, Zuo A. et al. Tertiary lymphoid structural heterogeneity determines tumour immunity and prospects for clinical application. Mol Cancer. 2024;23(1):75
20. Munoz-Erazo L, Rhodes JL, C Marion VC, Kemp RA. Tertiary lymphoid structures in cancer - considerations for patient prognosis. Cell Mol Immunol. 2020;17(6):570-5
21. Gu-Trantien C, Loi S, Garaud S, Equeter C, Libin M, de Wind A. et al. CD4⁺ follicular helper T cell infiltration predicts breast cancer survival. J Clin Invest. 2013;123:2873-92
22. Meylan M, Petitprez F, Becht É, Bougoüin A, Pupier G, Calvez A. et al. Tertiary lymphoid structures generate and propagate anti-tumor antibody-producing plasma cells in renal cell cancer. Immunity. 2022;55(3):527-41.e5
23. Germain C, Gnjatic S, Tamzalit F, Knockaert S, Remark R, Goc J. et al. Presence of B cells in tertiary lymphoid structures is associated with a protective immunity in patients with lung cancer. Am J Respir Crit Care Med. 2014;189(7):832-44
24. Di Caro G, Bergomas F, Grizzi F, Doni A, Bianchi P, Malesci A. et al. Occurrence of tertiary lymphoid tissue is associated with T-cell infiltration and predicts better prognosis in early-stage colorectal cancers. Clin Cancer Res. 2014;20:2147-58
25. García-Hernández ML, Uribe-Uribe NO, Espinosa-González R, Kast WM, Khader SA, Rangel-Moreno J. A unique cellular and molecular microenvironment is present in tertiary lymphoid organs of patients with spontaneous prostate cancer regression. Front Immunol. 2017;8:563
26. Zhang C, Wang XY, Zuo JL, Wang XF, Feng XW, Zhang B. et al. Localization and density of tertiary lymphoid structures associate with molecular subtype and clinical outcome in colorectal cancer liver metastases. J Immunother Cancer. 2023;11(2):e006425
27. Barone F, Gardner DH, Nayar S, Steinthal N, Buckley CD, Luther SA. Stromal fibroblasts in tertiary lymphoid structures: a novel target in chronic inflammation. Front Immunol. 2016;7:477
28. Liu X, Tsang JYS, Hlaing T, Hu J, Ni YB, Chan SK. et al. Distinct tertiary lymphoid structure associations and their prognostic relevance in HER2 positive and negative breast cancers. Oncologist. 2017;22:1316-24
29. Figenschau SL, Fismen S, Fenton KA, Fenton C, Mortensen ES. Tertiary lymphoid structures are associated with higher tumor grade in primary operable breast cancer patients. BMC Cancer. 2015;15:101
30. Hayashi Y, Makino T, Sato E, Ohshima K, Nogi Y, Kanemura T. et al. Density and maturity of peritumoral tertiary lymphoid structures in oesophageal squamous cell carcinoma predicts patient survival and response to immune checkpoint inhibitors. Br J Cancer. 2023;128:2175-85
31. Zhang Y, Liu G, Zeng Q, Wu W, Lei K, Zhang C. et al. CCL19-producing fibroblasts promote tertiary lymphoid structure formation enhancing anti-tumor IgG response in colorectal cancer liver metastasis. Cancer cell. 2024;42:1370-85.e9
32. Ma J, Wu Y, Ma L, Yang X, Zhang T, Song G. et al. A blueprint for tumor-infiltrating B cells across human cancers. Science. 2024;384:eadj4857
33. Calderaro J, Petitprez F, Becht E, Laurent A, Hirsch TZ, Rousseau B. et al. Intra-tumoral tertiary lymphoid structures are associated with a low risk of early recurrence of hepatocellular carcinoma. J Hepatol. 2019;70:58-65
34. van de Pavert SA, Olivier BJ, Goverse G, Vondenhoff MF, Greuter M, Beke P. et al. Chemokine CXCL13 is essential for lymph node initiation and is induced by retinoic acid and neuronal stimulation. Nat Immunol. 2009;10:1193-9
35. Morissette MC, Jobse BN, Thayaparan D, Nikota JK, Shen P, Labiris NR. et al. Persistence of pulmonary tertiary lymphoid tissues and anti-nuclear antibodies following cessation of cigarette smoke exposure. Respir Res. 2014;15(1):49
36. Genta RM, Hamner HW, Graham DY. Gastric lymphoid follicles in Helicobacter pylori infection: frequency, distribution, and response to triple therapy. Hum Pathol. 1993;24:577-83
37. Zhang MJ, Lin WP, Wang Q, Wang S, Song A, Wang YY. et al. Oncolytic herpes simplex virus propagates tertiary lymphoid structure formation via CXCL10/CXCR3 to boost antitumor immunity. Cell Prolif. 2024 null: e13740
38. A. Luther S, K. Mark A, G. Cyster J. Overlapping roles of CXCL13, interleukin 7 receptor α, and CCR7 ligands in lymph node development. J Exp Med. 2003;197(9):1191-8
39. Rangel-Moreno J, Carragher DM, de la Luz García-Hernández M, Hwang JY, Kusser K, Hartson L. et al. The development of inducible bronchus-associated lymphoid tissue depends on IL-17. Nat Immunol. 2011;12(7):639-46
40. Vondenhoff MF, Greuter M, Goverse G, Elewaut D, Dewint P, Ware CF. et al. LTβR signaling induces cytokine expression and up-regulates lymphangiogenic factors in lymph node anlagen. J Immunol. 2009;182(9):5439-45
41. Amisaki M, Zebboudj A, Yano H, Zhang SL, Payne G, Chandra AK. et al. IL-33-activated ILC2s induce tertiary lymphoid structures in pancreatic cancer. Nature. 2025;638:1076-84
42. Posch F, Silina K, Leibl S, Mündlein A, Moch H, Siebenhüner A. et al. Maturation of tertiary lymphoid structures and recurrence of stage II and III colorectal cancer. Oncoimmunology. 2018;7:e1378844
43. van Dijk N, Gil-Jimenez A, Siliņa K, Hendricksen K, Smit LA, de Feijter JM. et al. Preoperative ipilimumab plus nivolumab in locoregionally advanced urothelial cancer: the NABUCCO trial. Nat Med. 2020;26(12):1839-44
44. Zhang L, Zhang R, Jin D, Zhang T, Shahatiaili A, Zang J. et al. Synergistic induction of tertiary lymphoid structures by chemoimmunotherapy in bladder cancer. Br J Cancer. 2024;130:1221-31
45. Wagner UG, Kurtin PJ, Wahner A, Brackertz M, Berry DJ, Goronzy JJ. et al. The role of CD8+ CD40L+ T cells in the formation of germinal centers in rheumatoid synovitis. J Immunol. 1998;161:6390-7
46. Dieu-Nosjean MC, Antoine M, Danel C, Heudes D, Wislez M, Poulot V. et al. Long-term survival for patients with non-small-cell lung cancer with intratumoral lymphoid structures. J Clin Oncol. 2008;26(27):4410-7
47. Tietscher S, Wagner J, Anzeneder T, Langwieder C, Rees M, Sobottka B. et al. A comprehensive single-cell map of T cell exhaustion-associated immune environments in human breast cancer. Nat Commun. 2023;14:98
48. Meylan M, Petitprez F, Lacroix L, Di Tommaso L, Roncalli M, Bougoüin A. et al. Early hepatic lesions display immature tertiary lymphoid structures and show elevated expression of immune inhibitory and immunosuppressive molecules. Clin Cancer Res. 2020;26:4381-9
49. Fridman WH, Meylan M, Pupier G, Calvez A, Hernandez I, Sautès-Fridman C. Tertiary lymphoid structures and B cells: An intratumoral immunity cycle. Immunity. 2023;56:2254-69
50. Tang Z, Bai Y, Fang Q, Yuan Y, Zeng Q, Chen S. et al. Spatial transcriptomics reveals tryptophan metabolism restricting maturation of intratumoral tertiary lymphoid structures. Cancer Cell. 2025;43(6):1025-44.e14
51. Xu W, Lu J, Liu WR, Anwaier A, Wu Y, Tian X. et al. Heterogeneity in tertiary lymphoid structures predicts distinct prognosis and immune microenvironment characterizations of clear cell renal cell carcinoma. J Immunother Cancer. 2023;11(12):e006667
52. J. Gunderson A, Rajamanickam V, Bui C, Bernard B, Pucilowska J, Ballesteros-Merino C, et al. Germinal center reactions in tertiary lymphoid structures associate with neoantigen burden, humoral immunity and long-term survivorship in pancreatic cancer. Oncoimmunology. 2021;10(1):1900635
53. Vanhersecke L, Bougoüin A, Crombé A, Brunet M, Sofeu C, Parrens M. et al. Standardized pathology screening of mature tertiary lymphoid structures in cancers. Lab Invest. 2023;103(5):100063
54. He M, He Q, Cai X, Liu J, Deng H, Li F. et al. Intratumoral tertiary lymphoid structure (TLS) maturation is influenced by draining lymph nodes of lung cancer. J Immunother Cancer. 2023;11(4):e005539
55. Ding GY, Ma JQ, Yun JP, Chen X, Ling Y, Zhang S. et al. Distribution and density of tertiary lymphoid structures predict clinical outcome in intrahepatic cholangiocarcinoma. J Hepatol. 2022;76(3):608-618
56. du Bois H, Heim TA, Lund AW. Tumor-draining lymph nodes: At the crossroads of metastasis and immunity. Sci Immunol. 2021;6:eabg3551
57. Ganti SN, Albershardt TC, Iritani BM, Ruddell A. Regulatory B cells preferentially accumulate in tumor-draining lymph nodes and promote tumor growth. Sci Rep. 2015;5:12255
58. Wakasu S, Tagawa T, Haratake N, Kinoshita F, Oku Y, Ono Y. et al. Preventive effect of tertiary lymphoid structures on lymph node metastasis of lung adenocarcinoma. Cancer Immunol Immunother. 2023;72:1823-34
59. Sun L, Zhang H, Gao P. Metabolic reprogramming and epigenetic modifications on the path to cancer. Protein Cell. 2022;13(12):877-919
60. Bessede A, Peyraud F, Le Moulec S, Cousin S, Cabart M, Chomy F. et al. Upregulation of indoleamine 2,3-dioxygenase 1 in tumor cells and tertiary lymphoid structures is a hallmark of inflamed non-small cell lung cancer. Clin Cancer Res. 2023;29(23):4883-93
61. Fridman WH, Meylan M, Petitprez F, Sun CM, Italiano A, Sautès-Fridman C. B cells and tertiary lymphoid structures as determinants of tumour immune contexture and clinical outcome. Nat Rev Clin Oncol. 2022;19:441-57
62. Cipponi A, Mercier M, Seremet T, Baurain JF, Théate I, van den Oord J. et al. Neogenesis of lymphoid structures and antibody responses occur in human melanoma metastases. Cancer Res. 2012;72:3997-4007
63. Kroeger DR, Milne K, Nelson BH. Tumor-infiltrating plasma cells are associated with tertiary lymphoid structures, cytolytic t-cell responses, and superior prognosis in ovarian cancer. Clin Cancer Res. 2016;22:3005-15
64. Wennhold K, Thelen M, Lehmann J, Schran S, Preugszat E, García-Márquez M. et al. CD86+ antigen-presenting b cells are increased in cancer, localize in tertiary lymphoid structures, and induce specific T-cell responses. Cancer Immunol Res. 2021;9(9):1098-108
65. Kasikova L, Rakova J, Hensler M, Lanickova T, Tomankova J, Pasulka J. et al. Tertiary lymphoid structures and B cells determine clinically relevant T cell phenotypes in ovarian cancer. Nat Commun. 2024;15:2528
66. Gu-Trantien C, Migliori E, Buisseret L, de Wind A, Brohée S, Garaud S. et al. CXCL13-producing TFH cells link immune suppression and adaptive memory in human breast cancer. JCI insight. 2017;2(11):e91487
67. Hu C, Wang Y, Kong D, Huang Y, Lü J, Zhao M. et al. Tertiary lymphoid structure-associated B cells enhance CXCL13+CD103+CD8+Trm cell response to PD-1 blockade in gastric cancer. Gastroenterology. 2024;166(6):1069-84
68. Sun H, Liu Y, Cheng W, Xiong R, Gu W, Zhang X. et al. The distribution and maturation of tertiary lymphoid structures can predict clinical outcomes of patients with gastric adenocarcinoma. Front Immunol. 2024;15:1396808
69. Noël G, Fontsa ML, Garaud S, De Silva P, de Wind A, Van den Eynden GG. et al. Functional Th1-oriented T follicular helper cells that infiltrate human breast cancer promote effective adaptive immunity. J Clin Invest. 2021 131
70. Ishigami E, Sakakibara M, Sakakibara J, Masuda T, Fujimoto H, Hayama S. et al. Coexistence of regulatory B cells and regulatory T cells in tumor-infiltrating lymphocyte aggregates is a prognostic factor in patients with breast cancer. Breast cancer. 2019;26:180-9
71. Fukuhara M, Muto S, Inomata S, Yamaguchi H, Mine H, Takagi H. et al. The clinical significance of tertiary lymphoid structure and its relationship with peripheral blood characteristics in patients with surgically resected non-small cell lung cancer: a single-center, retrospective study. Cancer Immunol Immunother. 2022;71:1129-37
72. Matsuda N, Yamamoto H, Habu T, Iwata K, Matsubara K, Tanaka S. et al. Prognostic impact of tumor-infiltrating lymphocytes, tertiary lymphoid structures, and neutrophil-to-lymphocyte ratio in pulmonary metastases from uterine leiomyosarcoma. Ann Surg Oncol. 2023;30:8727-34
73. Gao J, Navai N, Alhalabi O, Siefker-Radtke A, Campbell MT, Tidwell RS. et al. Neoadjuvant PD-L1 plus CTLA-4 blockade in patients with cisplatin-ineligible operable high-risk urothelial carcinoma. Nat Med. 2020;26(12):1845-51
74. Siliņa K, Soltermann A, Attar FM, Casanova R, Uckeley ZM, Thut H. et al. Germinal centers determine the prognostic relevance of tertiary lymphoid structures and are impaired by corticosteroids in lung squamous cell carcinoma. Cancer Res. 2018;78:1308-20
75. de Visser KE, Joyce JA. The evolving tumor microenvironment: From cancer initiation to metastatic outgrowth. Cancer cell. 2023;41:374-403
76. Wu YH, Wu F, Yan GR, Zeng QY, Jia N, Zheng Z. et al. Features and clinical significance of tertiary lymphoid structure in cutaneous squamous cell carcinoma. J Eur Acad Dermatol Venereol. 2022;36:2043-50
77. Shang T, Jiang T, Lu T, Wang H, Cui X, Pan Y. et al. Tertiary lymphoid structures predict the prognosis and immunotherapy response of cholangiocarcinoma. Front Immunol. 2023;14:1166497
78. Wang Q, Shen X, An R, Bai J, Dong J, Cai H. et al. Peritumoral tertiary lymphoid structure and tumor stroma percentage predict the prognosis of patients with non-metastatic colorectal cancer. Front Immunol. 2022;13:962056
79. Li H, Liu H, Fu H, Li J, Xu L, Wang G. et al. Peritumoral tertiary lymphoid structures correlate with protective immunity and improved prognosis in patients with hepatocellular carcinoma. Front Immunol. 2021;12:648812
80. Zhang N, Liu X, Qin J, Sun Y, Xiong H, Lin B. et al. LIGHT/TNFSF14 promotes CAR-T cell trafficking and cytotoxicity through reversing immunosuppressive tumor microenvironment. Mol Ther. 2023;31(9):2575-90
81. Bai X, Zhou Y, Yokota Y, Matsumoto Y, Zhai B, Maarouf N. et al. Adaptive antitumor immune response stimulated by bio-nanoparticle based vaccine and checkpoint blockade. J Exp Clin Cancer Res. 2022;41(1):132
82. Finkin S, Yuan D, Stein I, Taniguchi K, Weber A, Unger K. et al. Ectopic lymphoid structures function as microniches for tumor progenitor cells in hepatocellular carcinoma. Nat Immunol. 2015;16:1235-44
83. Sofopoulos M, Fortis SP, Vaxevanis CK, Sotiriadou NN, Arnogiannaki N, Ardavanis A. et al. The prognostic significance of peritumoral tertiary lymphoid structures in breast cancer. Cancer Immunol Immunother. 2019;68:1733-45
84. Dieu-Nosjean MC, Giraldo NA, Kaplon H, Germain C, Fridman WH, Sautès-Fridman C. Tertiary lymphoid structures, drivers of the anti-tumor responses in human cancers. Immunol Rev. 2016;271:260-75
85. Goc J, Germain C, Vo-Bourgais TK, Lupo A, Klein C, Knockaert S. et al. Dendritic cells in tumor-associated tertiary lymphoid structures signal a Th1 cytotoxic immune contexture and license the positive prognostic value of infiltrating CD8+ T cells. Cancer Res. 2014;74:705-15
86. Liu Z, Meng X, Tang X, Zou W, He Y. Intratumoral tertiary lymphoid structures promote patient survival and immunotherapy response in head neck squamous cell carcinoma. Cancer Immunol Immunother. 2023;72(6):1505-21
87. Hiraoka N, Ino Y, Yamazaki-Itoh R, Kanai Y, Kosuge T, Shimada K. Intratumoral tertiary lymphoid organ is a favourable prognosticator in patients with pancreatic cancer. Br J Cancer. 2015;112(11):1782-90
88. Hiraoka N, Ino Y, Yamazaki-Itoh R. Tertiary lymphoid organs in cancer tissues. Front Immunol. 2016;7:244
89. Jin XK, Liang JL, Zhang SM, Ji P, Huang QX, Qin YT. et al. Engineering metal-based hydrogel-mediated tertiary lymphoid structure formation via activation of the STING pathway for enhanced immunotherapy. Mater Horiz. 2023;10:4365-79
90. Huang Y, Chen Y, Zhou S, Chen L, Wang J, Pei Y. et al. Dual-mechanism based CTLs infiltration enhancement initiated by Nano-sapper potentiates immunotherapy against immune-excluded tumors. Nat Commun. 2020;11:622
91. Remouchamps C, Boutaffala L, Ganeff C, Dejardin E. Biology and signal transduction pathways of the lymphotoxin-αβ/LTβR system. Cytokine Growth Factor Rev. 2011;22:301-10
92. Hua Y, Vella G, Rambow F, Allen E, Martinez AA, Duhamel M. et al. Cancer immunotherapies transition endothelial cells into HEVs that generate TCF1 + T lymphocyte niches through a feed-forward loop. Cancer Cell. 2022;40(12):1600-18.e10
93. Chen G, Zheng D, Zhou Y, Du S, Zeng Z. Olaparib enhances radiation-induced systemic anti-tumor effects via activating STING-chemokine signaling in hepatocellular carcinoma. Cancer Lett. 2024;582:216507
94. Li AQ, Fang JH. Anti-angiogenic therapy enhances cancer immunotherapy: Mechanism and clinical application. Interdiscip Med. 2024;2:e20230025
95. Amos SM, Duong CP, Westwood JA, Ritchie DS, Junghans RP, Darcy PK. et al. Autoimmunity associated with immunotherapy of cancer. Blood. 2011;118:499-509
96. Filderman JN, Appleman M, Chelvanambi M, Taylor JL, Storkus WJ. STINGing the tumor microenvironment to promote therapeutic tertiary lymphoid structure development. Front Immunol. 2021;12:690105
97. Zhang T, Lei X, Jia W, Li J, Nie Y, Mao Z. et al. Peritumor tertiary lymphoid structures are associated with infiltrating neutrophils and inferior prognosis in hepatocellular carcinoma. Cancer Med. 2023;12:3068-78
98. Cortés-Morales VA, Chávez-Sanchéz L, Rocha-Zavaleta L, Espíndola-Garibay S, Monroy-García A, Castro-Manrreza ME. et al. Mesenchymal stem/stromal cells derived from cervical cancer promote M2 macrophage polarization. Cells. 2023;12(7):1047
99. Tazzyman S, Lewis CE, Murdoch C. Neutrophils: key mediators of tumour angiogenesis. Int J Exp Pathol. 2009;90(3):222-31
100. Li Y, Qiu SJ, Fan JG, Zhou J, Gao Q, Ye X. et al. Intratumoral neutrophils: A poor prognostic factor for hepatocellular carcinoma following resection. J Hepatol. 2011;54(3):497-505
101. Bell D, Chomarat P, Broyles D, Netto G, Harb GM, Lebecque S. et al. In breast carcinoma tissue, immature dendritic cells reside within the tumor, whereas mature dendritic cells are located in peritumoral areas. J Exp Med. 1999;190:1417-26
102. Ladányi A, Kiss J, Somlai B, Gilde K, Fejos Z, Mohos A. et al. Density of DC-LAMP(+) mature dendritic cells in combination with activated T lymphocytes infiltrating primary cutaneous melanoma is a strong independent prognostic factor. Cancer Immunol Immunother. 2007;56:1459-69
103. Di Modugno F, Di Carlo A, Spada S, Palermo B, D'Ambrosio L, D'Andrea D. et al. Tumoral and stromal hMENA isoforms impact tertiary lymphoid structure localization in lung cancer and predict immune checkpoint blockade response in patients with cancer. EBioMedicine. 2024;101:105003
104. Moussion C, Girard JP. Dendritic cells control lymphocyte entry to lymph nodes through high endothelial venules. Nature. 2011;479:542-6
105. He T, Hao Z, Lin M, Xin Z, Chen Y, Ouyang W. et al. Oncolytic adenovirus promotes vascular normalization and nonclassical tertiary lymphoid structure formation through STING-mediated DC activation. OncoImmunology. 2022;11(1):2093054
106. Martinet L, Filleron T, Le Guellec S, Rochaix P, Garrido I, Girard JP. High endothelial venule blood vessels for tumor-infiltrating lymphocytes are associated with lymphotoxin β-producing dendritic cells in human breast cancer. J Immunol. 2013;191:2001-8
107. Yamaguchi K, Ito M, Ohmura H, Hanamura F, Nakano M, Tsuchihashi K. et al. Helper T cell-dominant tertiary lymphoid structures are associated with disease relapse of advanced colorectal cancer. Oncoimmunology. 2020;9:1724763
108. Sakimura C, Tanaka H, Okuno T, Hiramatsu S, Muguruma K, Hirakawa K. et al. B cells in tertiary lymphoid structures are associated with favorable prognosis in gastric cancer. J Surg Res. 2017;215:74-82
109. Yamakoshi Y, Tanaka H, Sakimura C, Mori T, Deguchi S, Yoshii M. et al. Association between the preoperative neutrophil-to-lymphocyte ratio and tertiary lymphoid structures surrounding tumor in gastric cancer. Mol Clin Oncol. 2021;14(4):76
110. Song IH, Heo SH, Bang WS, Park HS, Park IA, Kim YA. et al. Predictive value of tertiary lymphoid structures assessed by high endothelial venule counts in the neoadjuvant setting of triple-negative breast cancer. Cancer Res Treat. 2017;49:399-407
111. Remark R, Alifano M, Cremer I, Lupo A, Dieu-Nosjean MC, Riquet M. et al. Characteristics and clinical impacts of the immune environments in colorectal and renal cell carcinoma lung metastases: influence of tumor origin. Clin Cancer Res. 2013;19:4079-91
112. Lee M, Heo SH, Song IH, Rajayi H, Park HS, Park IA. et al. Presence of tertiary lymphoid structures determines the level of tumor-infiltrating lymphocytes in primary breast cancer and metastasis. Mod Pathol. 2019;32(1):70-80
113. Boivin G, Kalambaden P, Faget J, Rusakiewicz S, Montay-Gruel P, Meylan E. et al. Cellular composition and contribution of tertiary lymphoid structures to tumor immune infiltration and modulation by radiation therapy. Front Oncol. 2018;8:256
114. Maldonado L, Teague JE, Morrow MP, Jotova I, Wu TC, Wang C. et al. Intramuscular therapeutic vaccination targeting HPV16 induces T cell responses that localize in mucosal lesions. Sci Transl Med. 2014;6:221ra13
115. Zhu S, Zhang T, Zheng L, Liu H, Song W, Liu D. et al. Combination strategies to maximize the benefits of cancer immunotherapy. J Hematol Oncol. 2021;14:156
116. Lu Y, Zhao Q, Liao JY, Song E, Xia Q, Pan J. et al. Complement signals determine opposite effects of B cells in chemotherapy-induced immunity. Cell. 2020;180:1081-97.e24
117. Reits E, W Hodge J, Herberts C, A M Groothuis T, Chakraborty M, K Wansley E. et al. Radiation modulates the peptide repertoire, enhances MHC class I expression, and induces successful antitumor immunotherapy. J Exp Med. 2006;203(5):1259-71
118. Gameiro SR, Jammeh ML, Wattenberg MM, Tsang KY, Ferrone S, Hodge JW. Radiation-induced immunogenic modulation of tumor enhances antigen processing and calreticulin exposure, resulting in enhanced T-cell killing. Oncotarget. 2014;5(2):403-16
119. De Martino M, Daviaud C, Diamond JM, Kraynak J, Alard A, Formenti SC. et al. Activin A promotes regulatory T-cell-mediated immunosuppression in irradiated breast cancer. Cancer Immunol Res. 2021;9:89-102
120. Rodriguez AB, Peske JD, Woods AN, Leick KM, Mauldin IS, Meneveau MO. et al. Immune mechanisms orchestrate tertiary lymphoid structures in tumors via cancer-associated fibroblasts. Cell Rep. 2021;36:109422
121. Kaiser J. Personalized tumor vaccines keep cancer in check. Science. 2017;356:122
122. Lutz ER, Wu AA, Bigelow E, Sharma R, Mo G, Soares K. et al. Immunotherapy converts nonimmunogenic pancreatic tumors into immunogenic foci of immune regulation. Cancer Immunol Res. 2014;2:616-31
123. Shalhout SZ, Miller DM, Emerick KS, Kaufman HL. Therapy with oncolytic viruses: progress and challenges. Nat Rev Clin Oncol. 2023;20:160-77
124. Clubb JHA, Kudling TV, Heiniö C, Basnet S, Pakola S, Cervera Carrascón V. et al. Adenovirus encoding tumor necrosis factor alpha and interleukin 2 induces a tertiary lymphoid structure signature in immune checkpoint inhibitor refractory head and neck cancer. Front Immunol. 2022;13:794251
125. Gunnarsdóttir FB, Briem O, Lindgren AY, Källberg E, Andersen C, Grenthe R. et al. Breast cancer associated CD169+ macrophages possess broad immunosuppressive functions but enhance antibody secretion by activated B cells. Front Immunol. 2023;14:1180209
126. Shields JD, Kourtis IC, Tomei AA, Roberts JM, Swartz MA. Induction of lymphoidlike stroma and immune escape by tumors that express the chemokine CCL21. Science. 2010;328:749-52
127. Giraldo NA, Becht E, Pagès F, Skliris G, Verkarre V, Vano Y. et al. Orchestration and prognostic significance of immune checkpoints in the microenvironment of primary and metastatic renal cell cancer. Clin Cancer Res. 2015;21:3031-40
128. Suzuki A, Masuda A, Nagata H, Kameoka S, Kikawada Y, Yamakawa M. et al. Mature dendritic cells make clusters with T cells in the invasive margin of colorectal carcinoma. J Pathol. 2002;196(1):37-43
129. Truxova I, Kasikova L, Hensler M, Skapa P, Laco J, Pecen L. et al. Mature dendritic cells correlate with favorable immune infiltrate and improved prognosis in ovarian carcinoma patients. J Immunother Cancer. 2018;6:139
130. De Simone M, Arrigoni A, Rossetti G, Gruarin P, Ranzani V, Politano C. et al. Transcriptional landscape of human tissue lymphocytes unveils uniqueness of tumor-infiltrating T regulatory cells. Immunity. 2016;45:1135-47
131. Italiano A, Bessede A, Pulido M, Bompas E, Piperno-Neumann S, Chevreau C. et al. Pembrolizumab in soft-tissue sarcomas with tertiary lymphoid structures: a phase 2 PEMBROSARC trial cohort. Nat Med. 2022;28(6):1199-206
132. An Y, Sun JX, Xu MY, Xu JZ, Ma SY, Liu CQ. et al. Tertiary lymphoid structure patterns aid in identification of tumor microenvironment infiltration and selection of therapeutic agents in bladder cancer. Front Immunol. 2022;13:1049884
133. Pagliarulo F, Cheng PF, Brugger L, van Dijk N, van den Heijden M, Levesque MP. et al. Molecular, immunological, and clinical features associated with lymphoid neogenesis in muscle invasive bladder cancer. Front Immunol. 2021;12:793992
134. Deng S, Yang X, He L, Zhang Q, Zhao C, Meng H. Radiotherapy combined with anti-PD-1 and TKI for primary cardiac angiosarcoma considering the joint assessment of TLSs and PD-L1: a case report. J Cardiothorac Surg. 2024;19:194
135. Schumacher TN, Schreiber RD. Neoantigens in cancer immunotherapy. Science. 2015;348:69-74
136. Valero C, Lee M, Hoen D, Weiss K, Kelly DW, Adusumilli PS. et al. Pretreatment neutrophil-to-lymphocyte ratio and mutational burden as biomarkers of tumor response to immune checkpoint inhibitors. Nat Commun. 2021;12:729
137. Shaul ME, Fridlender ZG. Tumour-associated neutrophils in patients with cancer. Nat Rev Clin Oncol. 2019;16:601-20
138. Komura K, Tokushige S, Ishida M, Hirosuna K, Yamazaki S, Nishimura K. et al. Tertiary lymphoid structure and neutrophil-lymphocyte ratio coordinately predict outcome of pembrolizumab. Cancer Sci. 2023;114:4622-31
139. Xie M, Gao J, Ma X, Wu C, Zang X, Wang Y. et al. Consolidation radiographic morphology can be an indicator of the pathological basis and prognosis of partially solid nodules. BMC Pulm Med. 2022;22:369
140. Wang Y, Lin H, Yao N, Chen X, Qiu B, Cui Y. et al. Computerized tertiary lymphoid structures density on H&E-images is a prognostic biomarker in resectable lung adenocarcinoma. iScience. 2023;26:107635
141. Mai H, Luo J, Hoeher L, Al-Maskari R, Horvath I, Chen Y. et al. Whole-body cellular mapping in mouse using standard IgG antibodies. Nat Biotechnol. 2024;42(4):617-27
142. Sadeghirad H, Monkman J, Tan Chin W, Liu N, Yunis J, L Donovan M. et al. Spatial dynamics of tertiary lymphoid aggregates in head and neck cancer: insights into immunotherapy response. J Transl Med. 2024;22(1):677
143. Bressan D, Battistoni G, Hannon GJ. The dawn of spatial omics. Science. 2023;381:eabq4964
144. Dorraji ES, Oteiza A, Kuttner S, Martin-Armas M, Kanapathippillai P, Garbarino S. et al. Positron emission tomography and single photon emission computed tomography imaging of tertiary lymphoid structures during the development of lupus nephritis. Int J Immunopathol Pharmacol. 2021;35:20587384211033683
145. Long S, Li M, Chen J, Zhong L, Abudulimu A, Zhou L. et al. Spatial patterns and MRI-based radiomic prediction of high peritumoral tertiary lymphoid structure density in hepatocellular carcinoma: a multicenter study. J Immunother Cancer. 2024;12(12):e009879
146. Braman NM, Etesami M, Prasanna P, Dubchuk C, Gilmore H, Tiwari P. et al. Intratumoral and peritumoral radiomics for the pretreatment prediction of pathological complete response to neoadjuvant chemotherapy based on breast DCE-MRI. Breast Cancer Res. 2017;19(1):57
147. Beckford Vera DR, Smith CC, Bixby LM, Glatt DM, Dunn SS, Saito R. et al. Immuno-PET imaging of tumor-infiltrating lymphocytes using zirconium-89 radiolabeled anti-CD3 antibody in immune-competent mice bearing syngeneic tumors. PloS one. 2018;13:e0193832
148. Rashidian M, Ingram JR, Dougan M, Dongre A, Whang KA, LeGall C. et al. Predicting the response to CTLA-4 blockade by longitudinal noninvasive monitoring of CD8 T cells. J Exp Med. 2017;214:2243-55
149. van Rijthoven M, Balkenhol M, Siliņa K, van der Laak J, Ciompi F. HookNet: Multi-resolution convolutional neural networks for semantic segmentation in histopathology whole-slide images. Med Image Anal. 2021;68:101890
150. Li Z, Jiang Y, Li B, Han Z, Shen J, Xia Y. et al. Development and validation of a machine learning model for detection and classification of tertiary lymphoid structures in gastrointestinal cancers. JAMA network open. 2023;6:e2252553
151. Chen Z, Wang X, Jin Z, Li B, Jiang D, Wang Y. et al. Deep learning on tertiary lymphoid structures in hematoxylin-eosin predicts cancer prognosis and immunotherapy response. NPJ Precis Oncol. 2024;8(1):73
152. Li S, Wang Z, Huang HD, Lee TY. Machine learning-based characterization and identification of tertiary lymphoid structures using spatial transcriptomics data. Int J Mol Sci. 2024;25(7):3887
153. Wang B, Zou L, Chen J, Cao Y, Cai Z, Qiu Y. et al. A weakly supervised segmentation network embedding cross-scale attention guidance and noise-sensitive constraint for detecting tertiary lymphoid structures of pancreatic tumors. IEEE J Biomed Health Inform. 2024;28(2):988-99
154. Randolph GJ, Bala S, Rahier JF, Johnson MW, Wang PL, Nalbantoglu I. et al. Lymphoid aggregates remodel lymphatic collecting vessels that serve mesenteric lymph nodes in Crohn disease. Am J Pathol. 2016;186(12):3066-73
155. Lin JR, Wang S, Coy S, Yuan C, Yapp C, Tyler M. et al. Multiplexed 3D atlas of state transitions and immune interaction in colorectal cancer. Cell. 2023;186(2):363-81.e19
156. Jiang Y, Zhou K, Sun Z, Wang H, Xie J, Zhang T. et al. Non-invasive tumor microenvironment evaluation and treatment response prediction in gastric cancer using deep learning radiomics. Cell Rep Med. 2023;4(8):101146
157. Johansson-Percival A, He B, Li ZJ, Kjellén A, Russell K, Li J. et al. De novo induction of intratumoral lymphoid structures and vessel normalization enhances immunotherapy in resistant tumors. Nat Immunol. 2017;18:1207-17
158. Zhang Y, Xu J, Fei Z, Dai H, Fan Q, Yang Q. et al. 3D printing scaffold vaccine for antitumor immunity. Adv Mater. 2021;33:e2106768
159. Chelvanambi M, Fecek RJ, Taylor JL, Storkus WJ. STING agonist-based treatment promotes vascular normalization and tertiary lymphoid structure formation in the therapeutic melanoma microenvironment. J Immunother Cancer. 2021;9(2):e001906
160. Peske JD, Thompson ED, Gemta L, Baylis RA, Fu YX, Engelhard VH. Effector lymphocyte-induced lymph node-like vasculature enables naive T-cell entry into tumours and enhanced anti-tumour immunity. Nat Commun. 2015;6:7114
161. Hennequin A, Derangère V, Boidot R, Apetoh L, Vincent J, Orry D. et al. Tumor infiltration by Tbet+ effector T cells and CD20+ B cells is associated with survival in gastric cancer patients. Oncoimmunology. 2016;5:e1054598
162. Ling Y, Zhong J, Weng Z, Lin G, Liu C, Pan C. et al. The prognostic value and molecular properties of tertiary lymphoid structures in oesophageal squamous cell carcinoma. Clin Transl Med. 2022;12(10):e1074
163. Tanoue K, Ohmura H, Uehara K, Ito M, Yamaguchi K, Tsuchihashi K. et al. Spatial dynamics of CD39 + CD8 + exhausted T cell reveal tertiary lymphoid structures-mediated response to PD-1 blockade in esophageal cancer. Nat Commun. 2024;15:9033
164. Zhao H, Wang H, Zhao Y, Sun Q, Ren X. Tumor-resident T cells, associated with tertiary lymphoid structure maturity, improve survival in patients with stage III lung adenocarcinoma. Front Immunol. 2022;13:877689
165. Carrega P, Loiacono F, Di Carlo E, Scaramuccia A, Mora M, Conte R. et al. NCR(+)ILC3 concentrate in human lung cancer and associate with intratumoral lymphoid structures. Nat Commun. 2015;6:8280
166. Yuan H, Mao X, Yan Y, Huang R, Zhang Q, Zeng Y. et al. Single-cell sequencing reveals the heterogeneity of B cells and tertiary lymphoid structures in muscle-invasive bladder cancer. J Transl Med. 2024;22(1):48
167. Astorri E, Scrivo R, Bombardieri M, Picarelli G, Pecorella I, Porzia A. et al. CX3CL1 and CX3CR1 expression in tertiary lymphoid structures in salivary gland infiltrates: fractalkine contribution to lymphoid neogenesis in Sjogren's syndrome. Rheumatology. 2014;53:611-20
168. Wang J, Liang Y, Xue A, Xiao J, Zhao X, Cao S. et al. Intratumoral CXCL13 + CD160 + CD8 + T cells promote the formation of tertiary lymphoid structures to enhance the efficacy of immunotherapy in advanced gastric cancer. J Immunother Cancer. 2024;12(9):e009603
169. Koscsó B, Kurapati S, Rodrigues RR, Nedjic J, Gowda K, Shin C. et al. Gut-resident CX3CR1 hi macrophages induce tertiary lymphoid structures and IgA response in situ. Sci Immunol. 2020;5(46):eaax0062
170. Joshi NS, Akama-Garren EH, Lu Y, Lee DY, Chang GP, Li A. et al. Regulatory T cells in tumor-associated tertiary lymphoid structures suppress anti-tumor T cell responses. Immunity. 2015;43:579-90
171. Nayar S, Turner JD, Asam S, Fennell E, Pugh M, Colafrancesco S. et al. Molecular and spatial analysis of tertiary lymphoid structures in Sjogren's syndrome. Nat Commun. 2025;16:5
172. Yan Y, Sun D, Hu J, Chen Y, Sun L, Yu H. et al. Multi-omic profiling highlights factors associated with resistance to immuno-chemotherapy in non-small-cell lung cancer. Nat Genet. 2025;57(1):126-39
173. Liu Y, Ye S, He S, Chi D, Wang X, Wen YF. et al. Single-cell and spatial transcriptome analyses reveal tertiary lymphoid structures linked to tumour progression and immunotherapy response in nasopharyngeal carcinoma. Nat Commun. 2024;15(1):7713
174. Hoch T, Schulz D, Eling N, Gómez JM, Levesque MP, Bodenmiller B. Multiplexed imaging mass cytometry of the chemokine milieus in melanoma characterizes features of the response to immunotherapy. Sci Immunol. 2022;7:eabk1692
175. Gan X, Dong W, You W, Ding D, Yang Y, Sun D. et al. Spatial multimodal analysis revealed tertiary lymphoid structures as a risk stratification indicator in combined hepatocellular-cholangiocarcinoma. Cancer Lett. 2024;581:216513
176. Wu C, Zhang G, Wang L, Hu J, Ju Z, Tao H. et al. Spatial proteomic profiling elucidates immune determinants of neoadjuvant chemo-immunotherapy in esophageal squamous cell carcinoma. Oncogene. 2024;43:2751-67
177. Yolmo P, Rahimi S, Chenard S, Conseil G, Jenkins D, Sachdeva K. et al. Atypical B cells promote cancer progression and poor response to bacillus Calmette-Guérin in non-muscle invasive bladder cancer. Cancer Immunol Res. 2024;12:1320-39
178. L. Shiao Stephen, Gouin Kenneth, Ing Nathan, Y. Ho Alice, Basho Reva, Shah Aagam, et al. Single-cell and spatial profiling identify three response trajectories to pembrolizumab and radiation therapy in triple negative breast cancer. Cancer Cell. 2024;42(1):70-84.e8
179. Peng X, Smithy JW, Yosofvand M, Kostrzewa CE, Bleile M, Ehrich FD. et al. Decoding spatial tissue architecture: a scalable Bayesian topic model for multiplexed imaging analysis. bioRxiv: the preprint server for biology. 2024 null
180. Lu H, Lou H, Wengert G, Paudel R, Patel N, Desai S. et al. Tumor and local lymphoid tissue interaction determines prognosis in high-grade serous ovarian cancer. Cell Rep Med. 2023;4:101092
181. Li P, Liang Y, Zeng B, Yang G, Zhu C, Zhao K. et al. Preoperative prediction of intra-tumoral tertiary lymphoid structures based on CT in hepatocellular cancer. Eur J Radiol. 2022;151:110309
182. Li Q, Liu X, Wang D, Wang Y, Lu H, Wen S. et al. Prognostic value of tertiary lymphoid structure and tumour infiltrating lymphocytes in oral squamous cell carcinoma. Int J Oral Sci. 2020;12(1):24
Author contact
![]() Corresponding authors: Zhi-Jun Sun, School and Hospital of Stomatology, Wuhan University, 237 Luoyu Road, Wuhan 430079, Hubei Province, China, Email: sunzjedu.cn, Telephone: +86-27-8768-6215, ORCID: 0000-0003-0932-8013; Hao Li, School and Hospital of Stomatology, Wuhan University, 237 Luoyu Road, Wuhan 430079, Hubei Province, China, Email: lihaowhusosedu.cn, Telephone: +86-27-8768-6215, ORCID: 0000-0002-7890-6772; Meng-Jie Zhang, School and Hospital of Stomatology, Wuhan University, 237 Luoyu Road, Wuhan 430079, Hubei Province, China, Email: zhangmengjieedu.cn, Telephone: +86-27-8768-6215, ORCID: 0000-0002-6526-7086.
Corresponding authors: Zhi-Jun Sun, School and Hospital of Stomatology, Wuhan University, 237 Luoyu Road, Wuhan 430079, Hubei Province, China, Email: sunzjedu.cn, Telephone: +86-27-8768-6215, ORCID: 0000-0003-0932-8013; Hao Li, School and Hospital of Stomatology, Wuhan University, 237 Luoyu Road, Wuhan 430079, Hubei Province, China, Email: lihaowhusosedu.cn, Telephone: +86-27-8768-6215, ORCID: 0000-0002-7890-6772; Meng-Jie Zhang, School and Hospital of Stomatology, Wuhan University, 237 Luoyu Road, Wuhan 430079, Hubei Province, China, Email: zhangmengjieedu.cn, Telephone: +86-27-8768-6215, ORCID: 0000-0002-6526-7086.