Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Conclusion
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2025; 15(18):9775-9792. doi:10.7150/thno.117743 This issue Cite
Research Paper
Enhancing glymphatic transport through angiotensin II type 2 receptor activation promotes neurological recovery after traumatic brain injury
Xiaoyu Zhang1, Bin Sun2, Wenzhong Li2, Tianyi Liu3, Wenchen Li1, Bo Chen1, Chuan He1, Qin Liu3, Shoujun Zhu2 ![]() , Haifeng Wang1
, Haifeng Wang1 ![]()
1. Department of Neurosurgery, The Second Hospital of Jilin University, Changchun, 130041, P. R. China.
2. Joint Laboratory of Opto-Functional Theranostics in Medicine and Chemistry, The First Hospital of Jilin University, Changchun, 130021, P. R. China.
3. Department of Neurosurgery, The First Hospital of Jilin University, Changchun, 130021, P. R. China.
Received 2025-5-17; Accepted 2025-8-19; Published 2025-9-3
Abstract

Background: Traumatic brain injury (TBI) may impair the function of the glymphatic system, leading to diminished metabolic waste clearance and aggravated neurological deficits. While angiotensin II type 2 receptor (AT2R) activation has demonstrated neuroprotective effects, its specific impact on the glymphatic system following TBI remains uncharacterized.
Methods: We utilized near-infrared II (NIR-II) probes with distinct protein-binding capacities to visualize glymphatic transport in TBI mice and investigate how compound 21 (C21)-mediated AT2R activation modulates post-traumatic glymphatic function. Perivascular aquaporin-4 (AQP4) polarization was analyzed by immunofluorescence. RNA sequencing was performed to explore the C21-induced dynamic immune modulation. β-amyloid clearance efficiency and phosphorylated tau accumulation were quantified in mouse brain tissue. Motor and cognitive functions were comprehensively evaluated through standardized behavioral tests.
Results: Our results demonstrate that C21-mediated AT2R activation enhanced glymphatic influx and promoted glymphatic clearance after TBI. Mechanistically, AT2R activation restored perivascular aquaporin-4 (AQP4) polarization and cerebral blood flow, suppressed astrogliosis and microglial activation, and attenuated neuroinflammatory responses. Furthermore, AT2R activation enhanced β-amyloid clearance efficiency and reduced phosphorylated tau accumulation, thereby promoting motor and cognitive functional recovery.
Conclusion: By employing non-invasive or minimally invasive NIR-II imaging, our study highlights the protective effects of AT2R activation on the glymphatic system following TBI, revealing its potential as a promising therapeutic strategy for mitigating TBI-induced damage and improving neurological outcomes.
Keywords: Traumatic brain injury, Glymphatic transport, AT2R, NIR-II, AQP4
Introduction
Traumatic brain injury (TBI) is a leading neurological disorder and the primary cause of disability and death in individuals under the age of 45 worldwide [1, 2], characterized by acute neurological dysfunction and long-term neurodegenerative pathologies, including cognitive decline, motor deficits, and heightened risks of neurodegenerative diseases [3-5]. A critical yet underexplored contributor to these outcomes is the dysregulation of the glymphatic system, a brain-wide perivascular network responsible for cerebrospinal fluid (CSF)-interstitial fluid (ISF) exchange and metabolic waste clearance [6-8]. Glymphatic dysfunction after TBI disrupts the removal of neurotoxic proteins such as β-amyloid and phosphorylated tau, exacerbating secondary injury cascades [9-11]. Despite its pathophysiological significance, therapeutic strategies targeting glymphatic restoration remain limited.
The glymphatic system, a brain-wide clearance network, operates through astrocyte-defined perivascular channels along penetrating arteries. Its function hinges on two phases: (1) the glymphatic influx phase, during which CSF moves from the subarachnoid space into the brain parenchyma via periarterial spaces penetrating the cortical surface, and (2) the glymphatic clearance phase, during which CSF-ISF mixtures carrying neurotoxic waste (e.g., β-amyloid, phosphorylated tau) exit via perivenous pathways [12-16]. Central to this process is the polarized expression of aquaporin-4 (AQP4) water channels at astrocytic end-feet surrounding perivascular spaces (PVS). This spatial polarization enables rapid CSF influx into the parenchyma while directing CSF-ISF efflux toward perivenous spaces, ensuring efficient waste clearance [17-19]. Following TBI, glial fibrillary acidic protein (GFAP)-positive reactive astrocytes exhibit aberrant proliferation and activation in the ipsilateral cerebral hemisphere, forming dense glial scars. These pathological alterations disrupt the polarized distribution of AQP4, leading to disorganized localization at perivascular end-feet [11, 20, 21]. Such pathological changes impair the glymphatic fluid transport, creating a vicious cycle: impaired waste clearance promotes neuroinflammation, which further damages glymphatic infrastructure. This dysfunction facilitates pathological accumulation of β-amyloid and phosphorylated tau, and ultimately results in chronic neurodegeneration and cognitive decline [10, 11, 21-23]. Consequently, enhancing glymphatic transport has emerged as a promising therapeutic target for mitigating TBI-induced neurological damage.
Given the pivotal role of the glymphatic system in TBI pathology, the exploration of potential therapeutic strategies is necessary. Emerging evidence suggests that the brain renin-angiotensin system (RAS), traditionally recognized for cardiovascular regulation, plays a pivotal role in neurological pathologies [24-26]. Among its components, the angiotensin II type 1 receptor (AT1R) and angiotensin II type 2 receptor (AT2R), both G protein-coupled receptors, are widely expressed in neurons, astrocytes, oligodendrocytes, and microglia within brain regions such as the cortex, hippocampus, and basal ganglia. During TBI, excessive AT1R activation induces detrimental effects such as reduced cerebral blood flow (CBF), elevated intracranial pressure, cerebral edema, exacerbated neuroinflammation, and long-term cognitive impairment [27-29]. Conversely, AT2R activation exerts neuroprotective effects in neurological diseases, promoting neurogenesis, attenuating pathological microglial/astrocytic activation, suppressing pro-inflammatory cytokine release (e.g., IL-1β and TNF-α), and reducing β-amyloid/phosphorylated tau accumulation [30-35]. Notably, compound 21 (C21), a highly selective nonpeptide AT2R agonist with 25,000-fold specificity for AT2R over AT1R [36], has demonstrated multifaceted neuroprotective effects in preclinical models of neurodegenerative, neuroinflammatory, and ischemic conditions. We therefore hypothesized that C21-mediated AT2R activation enhances glymphatic transport to alleviate TBI-induced neurological dysfunction.
Technological limitations have hindered real-time assessment of glymphatic system dynamics. Conventional imaging modalities lack the spatiotemporal resolution to capture rapid fluid transport or parenchymal clearance. Recent advances in near-infrared II (NIR-II) fluorescence imaging (1,000-1,700 nm) overcome these barriers, offering deep-tissue penetration, high resolution, and minimal autofluorescence[37-40]. This study developed NIR-II probes with adjustable albumin affinity to visualize distinct stages of glymphatic transport: BSA@IR-780 (albumin-bound cyanine) traces CSF influx into PVS and superficial cervical lymph nodes (CLNs); IR-808Ac (albumin-escaping) permits real-time visualization of parenchymal solute clearance; and IR-808 (albumin-seeking) quantifies clearance dynamics from the brain into the blood circulation. This advanced NIR-II imaging platform provided critical methodological support for investigating the effects of C21-mediated AT2R activation on post-TBI glymphatic transport. Our findings demonstrate that AT2R activation enhanced glymphatic transport in TBI mice, concurrent with reduced neuroinflammation, accelerated clearance of neurotoxic proteins, and improved neurological recovery.
Materials and Methods
Animals
All C57BL/6 male mice, aged 8-10 weeks, were purchased from Jilin Qianhe Model Biotechnology Co., Ltd. Their weights were maintained between 23 and 25 g. The mice were housed under a standard 12 h light/dark cycle in specific pathogen-free conditions, with controlled temperature (23 ± 1.5 °C) and humidity (50 ± 10%). They had unrestricted access to bedding, nesting materials, food, and water. All mouse experiments were conducted in accordance with the relevant guidelines and regulations of Jilin University and approved by the Animal Ethics Committee of the First Hospital of Jilin University (Scheme No.: 20240624-12). Mice were randomly allocated to different experimental groups, and individuals blinded to the group assignments performed experiments and data collection.
TBI
The closed-head TBI model was established using a modified weight drop device based on previously described methodologies [41, 42]. In contrast to other rodent models, this approach avoided the prolonged anesthesia, thereby more accurately replicating typical clinical conditions. Briefly, mice were anesthetized with 4% isoflurane (ISO) for 1 min, followed by maintenance anesthesia with 1% ISO. After exposing the skull and removing the periosteum, the impact point was marked at 2.0 mm posterior and 2.0 mm lateral to the bregma. The mice were then placed on a rubber surface (6 mm thick) with their heads positioned beneath a weight drop device. A 17.4-g striker (2 mm diameter) was placed perpendicular to the skull at the marked impact point. A 60-g weight was dropped from a height of 15 cm to induce TBI, after which the wound was sutured intermittently. In the TBI models used in this study, no macroscopic evidence of skull fractures or intracranial hemorrhage was observed upon gross examination. The sham group underwent anesthesia and surgical preparation but were not injured.
Drug administration and experimental design
C21 (HY-100113, MedChemExpress), a highly specific AT2R receptor agonist, was dissolved in phosphate buffered saline (PBS). Mice received intraperitoneal injection of C21 (0.03 mg/kg) or PBS 1 h after TBI, followed by two additional doses at 24 and 48 h post-injury. The selected dosage was determined according to previously established optimal efficacy parameters for stroke treatment [43]. The non-injury sham group underwent sham injury and received PBS injections (i.p.).
Behavioral tests
Behavioral assessments included the modified neurological severity score (mNSS), rotarod test, novel object recognition (NOR) test, open field test (OFT) and Morris water maze (MWM) test (methodological details in Supplementary Information).
Synthesis of probes
BSA@IR-780, IR-808, and IR-808Ac were synthesized following previously methods [40, 44].
Construction of the NIR-II fluorescence imaging system
For detailed NIR-II imaging setup procedures, refer to relevant classical literature [45, 46]. This study utilized a two-dimensional InGaAs array (Raptor Photonics) to collect NIR-II fluorescence signals. All imaging employed an 808 nm laser (power density ~65 mW/cm²) irradiated through an 850 nm short-pass filter. Emission from BSA@IR-780, IR-808, and IR-808Ac was isolated using 1000 nm + 1100 nm long-pass filters, ensuring signal capture within the NIR-II region. A short-wave infrared lens (35 mm focal length) mounted on the detector provided fields of view of ~2.8 × 2.3 cm². The imaged region remained consistently centered within the laser illumination area. Computer-controlled image exposure times are specified in the corresponding figure legends.
Intracisternal tracer infusions
Under ISO anesthesia, mice were fixed in a stereotactic frame. BSA@IR-780 was dissolved in artificial CSF at a concentration of 300 μM. After surgically exposing the cisterna magna (CM), a PE10 tube filled with BSA@IR-780 was inserted into the CM using a 30-gauge needle bridge. The inserted PE10 tube was first sealed with 3M vetbond tissue adhesive and reinforced with dental cement. Subsequently, dexmedetomidine (DEX) was dissolved in PBS and injected intraperitoneally (0.2 mg/kg). After 5 min, a syringe pump (Harvard Instruments) was used to deliver 7 μL of BSA@IR-780 at a rate of 1 μL/min. A heating pad was used to maintain the body temperature at 36.5-37 °C.
Visualization of BSA@IR-780 influx into brain
The influx of BSA@IR-780 into the mouse brain was monitored using an NIR-II fluorescence imaging system we had constructed. The brain was focused on the field view of ~2.8 × 2.3 cm². The distribution was monitored for 30 min with an interval of 5 min. After allowing the NIR-II tracer to circulate for 30 min, the brain was collected and the tracer distribution on the dorsal and ventral brain surfaces was imaged. The imaging conditions were 10 ms, 1200 nm long-pass filter, and 65 mW/cm2 power density.
The brain was then fixed with 4% paraformaldehyde (PFA) for 24 h. Coronal slices (100-μm thick) were prepared using a vibratome. A total of twelve slices were collected at 300 μm intervals, starting 1.6 mm anterior to bregma, to calculate the mean total tracer influx for the entire brain. The slices were imaged using a scanning imaging system (Azurespot).
Visualization of the BSA@IR-780 drainage into superficial CLNs
Mouse hair was shaved prior to imaging to expose the neck skin. Infusions were performed with mice in a prone position. After completing intracisternal infusion of BSA@IR-780, mice were placed supine for superficial CLNs imaging. The first image was acquired 10 min post-infusion start, with tracer movement monitored for 60 min at 5-min intervals. Imaging parameters: 10-ms exposure, 1200 nm long-pass filter, 65 mW/cm² power density.
Intra-parenchymal injections
To evaluate the clearance capacity of parenchymal metabolites, 2 μL of the NIR-II probe IR-808Ac (50 μM, dissolved in artificial CSF) or IR-808 (2.5 μM, dissolved in artificial CSF) was injected into the cerebral cortex of mice using stereotaxic technique. Mice were anesthetized with 4% ISO for 1 min, followed by maintenance with 1% ISO. The skull was exposed, and the periosteum was removed. A 33-gauge stainless-steel cannula (Hamilton) was inserted into the cerebral cortex at the following stereotactic coordinates: 2.00 mm posterior, 1.50 mm lateral to the bregma, and 2.00 mm below the brain surface. After 5 min of intubation, the injection was administered at 0.2 μL/min. After the injection, the needle was removed following a 5-min rest period, and NIR-II imaging of the mouse brain was conducted immediately.
FITC-labeled β-amyloid (1-42) (P2000022-FITC, PLLABS) was dissolved in artificial CSF at a concentration of 0.5 mg/mL. A 2-μL volume of the solution was injected into the brain parenchyma of mice using the same technique. The mice were then returned to their cages and allowed to move freely for 4 h. Subsequently, mice were anesthetized with chloral hydrate and perfused with cold PBS. The brains were harvested and fixed in 4% PFA at 4 °C for 24 h, sectioned into 100-μm thick slices, and the residual β-amyloid on the slices was observed under a confocal microscope (FV3000, Olympus).
Visualization of NIR-II probes clearance from brain
Using our custom NIR-II fluorescence imaging system, we monitored IR-808Ac clearance from the mouse brain within a field of view of ~2.8 × 2.3 cm². Immediately after injecting IR-808Ac into the parenchyma, we performed the initial NIR-II brain imaging. Mice were then returned to their cages without activity restrictions, with two additional imaging sessions conducted at 1-h intervals. All in vivo imaging used identical parameters: 15 ms exposure, 1200 nm long-pass filter, and 65 mW/cm² power density.
The percentage of IR-808Ac cleared from the mouse brain was calculated as clearance efficiency using the following formula:
Clearance efficiency (%) = [(initial fluorescence intensity - current fluorescence intensity) / initial fluorescence intensity] × 100
Then, the brain was collected. Both dorsal and ventral surfaces were imaged (dorsal: 10 ms, 1200 nm long-pass filter, 65 mW/cm²; ventral: 100 ms, 1200 nm long-pass filter, 65 mW/cm²) to assess the residual tracer. Brains were then fixed in 4% PFA for 24 h. Using a vibratome, we prepared 100-μm-thick coronal slices. Six slices, collected at 300-μm intervals starting 0.8 mm posterior to bregma, were imaged using an Azurespot scanning system.
After intraparenchymal injection of IR-808, the tracer was cleared into the peripheral circulation. IR-808 accumulation in the femoral vein was monitored using our NIR-II fluorescence imaging system within a field of view of ~2.8 × 2.3 cm². Prior to imaging, the mouse leg was shaved to expose the skin. Immediately following IR-808 injection, the mouse was positioned supine for imaging of the leg vasculature. Continuous monitoring was performed for 60 min at 5-min intervals under the following imaging parameters: 100 ms exposure, 1200 nm long-pass filter, and 65 mW/cm² power density.
Immunofluorescence
Mice were deeply anesthetized and perfused with cold PBS. Their brains were removed and fixed in 4% PFA at 4 °C overnight, then dehydrated in 15% and 30% sucrose solutions. After dehydration, the brains were immersed in optimal cutting temperature compound (Sakura Finetek USA, Torrance, California, USA) and sectioned into 30-μm slices using a cryostat microtome (CM1950, Leica Biosystems, Nurock, Germany). The tissue sections were washed with PBS and sealed with PBS containing 3% bovine serum albumin, 0.2% Triton X-100, and 0.05% Tween 20 at room temperature for 1.5 h. Sections were incubated overnight at 4 °C with primary antibodies, followed by a 2-h incubation at room temperature with secondary antibodies. The primary antibodies used were rabbit anti-AQP4 (1:500, CL488-16473, Proteintech), hamster (Armenian) anti-CD31 (1:400, MAB1398Z, Merck), chicken anti-GFAP (1:500, PA1-10004, Thermo Fisher Scientific), and rabbit anti-Iba1 (1:500, AB178846, Abcam). Stained sections were observed under a confocal microscope (FV3000, Olympus).
AQP4 polarization evaluation
To assess AQP4 polarization after TBI, we employed an established area-based method [23, 47]. Briefly, we measured the median AQP4 immunofluorescence intensity within the perivascular region. Using threshold analysis, we determined the percentage of area where AQP4 immunofluorescence intensity met or exceeded the levels observed around blood vessels (termed AQP4% area). Polarization was then calculated as: Polarization = 1 - AQP4% area. This value represents the percentage of area exhibiting AQP4 immunoreactivity lower than that in the perivascular astrocytic end-feet.
As an alternative approach, we quantified AQP4 polarization using immunofluorescence signals for AQP4 and the vascular marker CD31, following previous researches [48-50]. Briefly, lines perpendicular to blood vessels were randomly selected. AQP4 fluorescence intensity profiles were recorded along these lines, with signal within the vessel lumen representing the baseline intensity. Polarization was calculated as the ratio of peak AQP4 fluorescence intensity at perivascular end-feet to the average baseline intensity:
AQP4 polarization = perivascular peak intensity / average baseline intensity
Measurement of CBF
CBF was measured using a laser Doppler blood flow meter. Briefly, after anesthetizing the mice as described earlier, a midline scalp incision was made to expose the skull. The CBF of both hemispheres was then recorded using a laser speckle imager.
Measurement of mean arterial pressure (MAP)
MedLab non-invasive blood pressure analysis system (NJKEWBIO, Nanjing, China) was used to measure the MAP of mice. Place the mouse on a 37-39°C heating platform or in a thermostatic chamber for 5-10 min to promote tail vasodilation and enhance signal clarity. Gently restrain the mouse in a specialized holder, ensuring natural tail extension through the tail cuff sensor. Maintain unobstructed respiration by avoiding thoracic compression. Clean the tail with alcohol swabs to remove debris, then position the tail cuff approximately 1-2 cm from the tail base. Adjust cuff tension to achieve effective blood flow occlusion without tissue compression. Initialize the system and perform baseline calibration as per software instructions. The automated protocol initiates with cuff inflation to 150 mmHg (surpassing typical systolic pressure). Gradual deflation (2-3 mmHg/s) enables detection of hemodynamic parameters: systolic blood pressure (SBP) corresponds to the pulse wave reappearance point, while diastolic blood pressure (DBP) is identified at complete blood flow restoration. Conduct triplicate measurements, discarding outliers before calculating mean values. Derive MAP using the standard formula:
MAP = DBP + 1/3(SBP - DBP)
Heart rate and respiratory rate monitoring
The methods for measuring heart rate and respiratory rate in mice were consistent with those described in previous studies [40]. It is important to note that the mice used for physiological signal measurements and those used for imaging were separate cohorts within the same experimental group. For heart rate measurement, electrodes were attached to the limbs of the mice, and ECG signals were collected using a non-invasive small animal multifunctional physiological monitoring system (SftReplay). The average value over a 2-min recording period was used to calculate the heart rate of each mouse. For respiratory rate measurement, each mouse was placed in a prone position, and a respiratory sensor was positioned on the abdomen. Respiratory data were continuously collected for 2 min, and the average value was used to determine the respiratory rate of each mouse. To ensure accurate readings, the body temperature of the mice was maintained at 36.5-37.0°C using a thermostatically controlled heating pad, thereby minimizing potential confounding effects on physiological measurements.
RNA sequencing (RNA-seq)
Total RNA was extracted from the cortical tissue using TRIzol® Reagent according to the manufacturer's instructions (Magen). RNA samples were detected based on the A260/A280 absorbance ratio with a Nanodrop ND-2000 system (Thermo Scientific, USA), and the RIN of RNA was determined by an Agilent Bioanalyzer 4150 system (Agilent Technologies, CA, USA). Only qualified samples will be used for library construction. Paired-end libraries were prepared using ABclonal mRNA-seq Lib Prep Kit (ABclonal, China) following the manufacturer's instructions. The mRNA was purified from 1 μg total RNA using oligo (dT) magnetic beads followed by fragmentation carried out using divalent cations at elevated temperatures in ABclonal First Strand Synthesis Reaction Buffer. Subsequently, first-strand cDNAs were synthesized with random hexamer primers and Reverse Transcriptase (RNase H) using mRNA fragments as templates, followed by second-strand cDNA synthesis using DNA polymerase I, RNAseH, buffer, and dNTPs. The synthesized double stranded cDNA fragments were then adapter-ligated for preparation of the paired-end library. Adaptor-ligated cDNA was used for PCR amplification. PCR products were purified (AMPure XP system) and library quality was assessed on an Agilent Bioanalyzer 4150 system. Finally, sequencing was performed with an Illumina Novaseq 6000/MGISEQ-T7 instrument. The data generated from Illumina/BGI platform were used for bioinformatics analysis.
Western blot
Mice were perfused with cold PBS, and their brains were extracted. Tissue samples were collected from near the cerebral cortex around the injury site. Total proteins were extracted from each group using protease and phosphatase inhibitors (P1260, Beijing, China) and loaded onto 10% or 12% SDS-PAGE gels. Proteins were then transferred on polyvinylidene fluoride (PVDF) membranes using a wet electrotransfer device (Bio-Rad). The PVDF membranes were incubated overnight at 4 °C with specific primary antibodies, including: rabbit anti-pERK1/2 (Thr202/Tyr204) (1:3000, 28733-1-AP, Proteintech), rabbit anti-ERK1/2 (1:3000, A4782, ABclonal), mouse anti-Tau5 (1:2000, MA5-12808, Thermo Fisher Scientific), rabbit anti-pTau-Thr205 (1:2000, AP0168, ABclonal), rabbit anti-pTau-Ser396 (1:2000, 9632T, Cell Signaling Technology), and rabbit anti-pTau-Ser404 (1:2000, 44-758G, Thermo Fisher Scientific). After incubation, the membranes were washed thrice. After washing, membranes were incubated with horseradish peroxidase-conjugated secondary antibodies, and protein bands were detected using an enhanced chemiluminescence system (MilliporeSigma).
Statistical analysis
All image analyses were performed using ImageJ, and statistical analyses were conducted with GraphPad Prism 8. Data in all graphs are presented as mean ± standard deviation (SD), with individual data points and lines representing values from each mouse. Comparisons between two groups were performed using Student's t-test, whereas one-way ANOVA followed by Tukey's post-hoc test was applied for comparisons across three or more groups. Two-way ANOVA was employed to analyze interactions between continuous variables. For datasets violating assumptions of normality or homogeneity of variances, the nonparametric Kruskal-Wallis test was used to compare medians across three groups, followed by Dunn's test for multiple comparisons. Statistical significance was defined as *p* < 0.05.
Results
AT2R activation improved neurological recovery after TBI
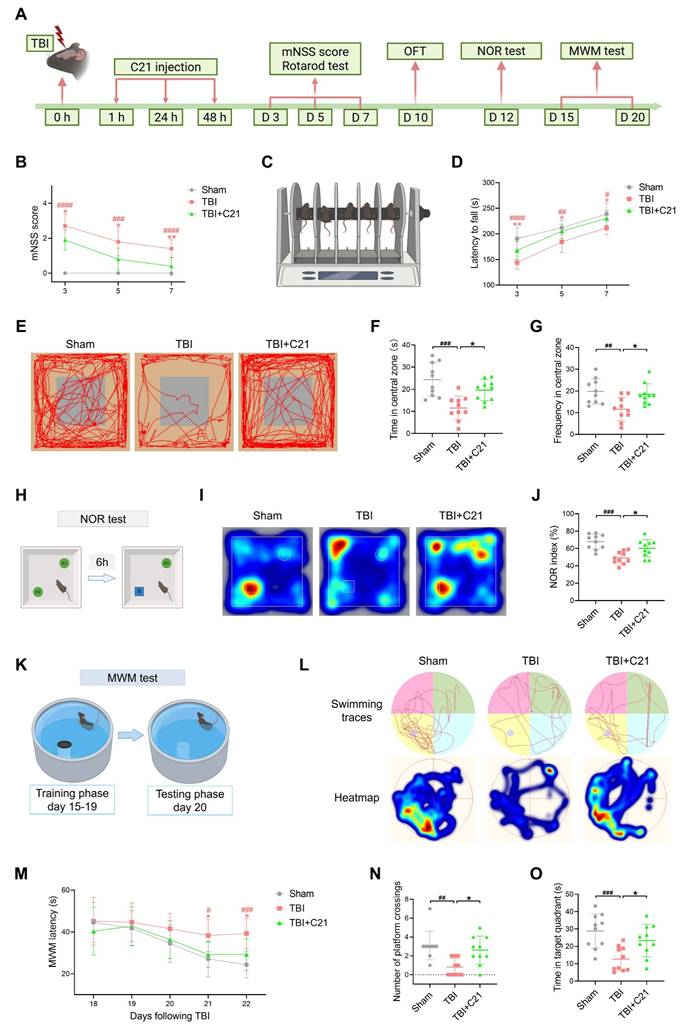
To investigate the impact of AT2R activation on neurological recovery after TBI, we performed behavioral tests assessing motor and cognitive functions (Figure 1A). Results from the mNSS and rotarod test revealed significant motor deficits post-TBI, while C21-treated mice exhibited progressive improvement in motor performance on days 3, 5, and 7 post-TBI (Figure 1B-D). For cognitive assessment, the OFT, NOR test, and MWM test were employed. The OFT was used to evaluate exploratory behavior in mice. Compared with the sham group, the TBI group spent less time in the central area and exhibited fewer crossings, indicating reduced exploratory activity. Treatment with C21 increased both the time spent in and crossings through the central area in TBI mice (Figure 1E-G). The NOR test was used to assess memory and object recognition. TBI mice spent less time near the new object compared with the sham group. Treatment with C21 increased the NOR index (Figure 1H-J). The MWM test assessed spatial learning and memory. During training sessions on days 18 and 19 post-injury, TBI+C21 mice showed shorter escape latencies than untreated TBI mice. (Figure 1K-M). On day 20, following platform removal, the TBI+C21 group crossed the former platform location more frequently and spent more time in the target quadrant (Figure 1N, O), suggesting enhanced spatial memory.
AT2R activation improved neurological recovery after TBI. (A) Workflow detailing the timeline for drug interventions and behavioral tests. (B) mNSS after TBI. (C, D) Latency to fall on the rotarod test after TBI. (E) Representative track sheets from the open-field test probe trial. (F, G) OFT results: Time spent in the central zone (F) and frequency of entries into the central zone (G). (H) Schematic diagram representation of the NOR test. The schematic diagram was created with BioRender.com. A1 and A2 represent old objects, while B represents the novel object. (I) Representative heatmaps from the NOR test probe trial. (J) NOR index for each group. (K) Schematic diagram of the MWM test. The schematic diagram was created with BioRender.com. (L) Representative swimming traces and heatmaps from the MWM test. (M) Learning curve during the training phase of the MWM test. (N, O) MWM probe trial results: number of platform crossings (N) and time spent in the target quadrant (O) for each group. All data are presented as mean ± SD (n = 10 per group). Statistical significance: #p < 0.05, ##p < 0.01, ###p < 0.001, ####p < 0.0001 versus sham group; *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 versus TBI+C21 group.
AT2R activation enhanced glymphatic influx in acute TBI mice
The restoration of glymphatic transport following TBI plays a critical role in neurological recovery [10]. To evaluate glymphatic influx in mice 3 days after TBI, 7 μL of BSA@IR-780 tracer (see Figure S1 for mass spectrometry) was injected into the CM (Figure 2A, B). Using NIR-II imaging, we dynamically tracked parenchymal penetration of BSA@IR-780 over 30 min. Consistent with TGN-020's known inhibition of AQP4-dependent glymphatic transport [19, 51], TGN-020 suppressed BSA@IR-780 influx (Figure S2). Compared to sham controls, TBI mice exhibited reduced BSA@IR-780 influx into PVS on the brain surface and diminished parenchymal diffusion, indicating TBI-induced glymphatic impairment. C21 treatment restored the influx of BSA@IR-780 into PVS on the brain surface and enhanced its diffusion into the brain parenchyma (Figure 2C-F).
AT2R activation enhanced glymphatic influx in acute TBI mice. (A, B) Experimental workflow (A) and schematic diagram (B) of transcranial NIR-II imaging to assess glymphatic influx. The schematic diagram was created with BioRender.com. (C, D) High-contrast transcranial imaging (C) and quantification of fluorescence intensity (D) for BSA@IR-780. Scale bar = 4 mm. (E, F) Representative images of BSA@IR-780 influx into the PVS (E) and corresponding fluorescence quantification (F). Scale bar = 2 mm. (G) Representative images of BSA@IR-780 distribution on dorsal and ventral brain surfaces. Scale bar = 4 mm. (H, I) Quantified fluorescence intensity on the dorsal (H) and ventral (I) surfaces. (J) Representative images of coronal slices at the indicated distances from bregma. Scale bar = 2 mm. (K, L) Quantified fluorescence intensity across all slices (K) and per slice (L). (M, N) Workflow (M) and schematic diagram (N) for non-invasive NIR-II imaging of superficial CLNs drainage in mice. The schematic diagram was created with BioRender.com. (O, P) Representative images (O) and fluorescence quantification (P) of superficial CLNs drainage. Scale bar = 4 mm. All data are presented as mean ± SD (n = 6 per group). Statistical significance: #p < 0.05, ##p < 0.01, ###p < 0.001, ####p < 0.0001 versus sham group; *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 versus TBI+C21 group.
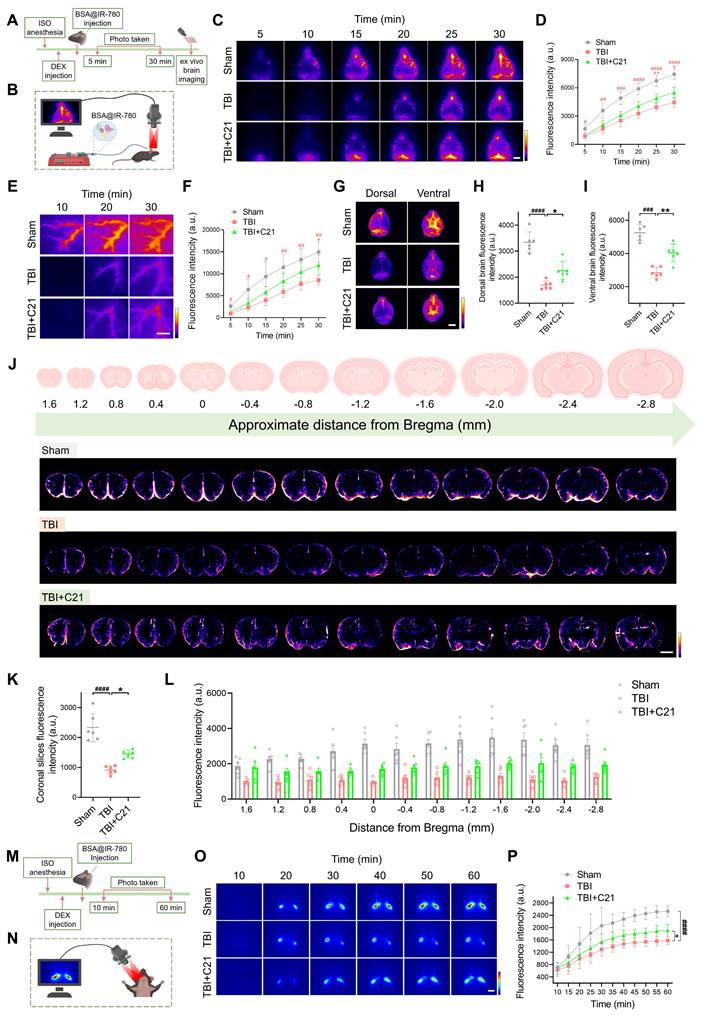
Following 30 minutes of in vivo NIR-II imaging, the brains were harvested and imaged ex vivo using the same modality. Imaging of the whole brains confirmed the in vivo findings. The fluorescence intensity of the BSA@IR-780 probe on both the dorsal and ventral sides of the brain of TBI mice was weaker than that of sham mice. However, treatment with C21 enhanced fluorescence intensity on both the dorsal and ventral sides of the brain, reflecting improved glymphatic influx (Figure 2G-I). To further verify these results, coronal slices were prepared from specific brain regions, and the distribution of BSA@IR-780 within the slices was analyzed (Figure 2J). The results revealed that C21 treatment increased the distribution of BSA@IR-780 in the brain parenchyma (Figure 2K, L).
After injection of BSA@IR-780, we used NIR-II imaging to quantitatively assess its drainage into the superficial CLNs (Figure 2M, N). The results showed that TBI mice exhibited reduced tracer drainage compared to sham-operated mice. However, C21 administration enhanced this drainage (Figure 2O, P).
AT2R activation promoted glymphatic clearance in acute TBI mice
To assess the effect of AT2R activation on glymphatic clearance in mice 3 days post-TBI, we first evaluated the parenchymal clearance of IR-808Ac (Figure 3A; ¹H NMR spectrum shown in Figure S3). This NIR-II probe enables transcranial imaging and is rapidly metabolized due to its albumin-escaping capacity (Figure 3B) [44]. After cortical injection of IR-808Ac, the parenchymal clearance efficiency was monitored by NIR-II imaging. Consistent with TGN-020's known disruption of glymphatic function via AQP4 inhibition [19, 51], TGN-020 suppressed parenchymal clearance of IR-808Ac (Figure S4). Compared with the sham group, TBI impaired IR-808Ac clearance, whereas C21 treatment improved parenchymal clearance efficiency (Figure 3C-E).
AT2R activation promoted glymphatic clearance in acute TBI mice. (A, B) Experimental workflow (A) and schematic diagram (B) for transcranial NIR-II imaging of parenchymal IR-808Ac clearance. The schematic diagram was created with BioRender.com. (C-E) High-contrast transcranial NIR-II imaging (C) and fluorescence intensity analysis of IR-808Ac (D, E). Scale bar = 4 mm. (F) Representative images of IR-808Ac distribution on dorsal and ventral brain surfaces. Scale bar = 4 mm. (G) Representative images of coronal slices at the indicated distances from bregma. Scale bar = 2 mm. (H, I) Quantified fluorescence intensity on the dorsal (H) and ventral (I) surfaces. (J, K) Quantified fluorescence intensity across all slices (J) and per slice (K). (L, M) Workflow (L) and schematic diagram (M) illustrating IR-808 delivery and detection in the femoral vein. The schematic diagram was created with BioRender.com. (N, O) Representative femoral vein images (N) and fluorescence quantification (O) of IR-808. Scale bar = 4 mm. All data are presented as mean ± SD (n = 6 per group). Statistical significance: #p < 0.05, ##p < 0.01, ###p < 0.001, ####p < 0.0001 versus sham group; *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 versus TBI+C21 group.
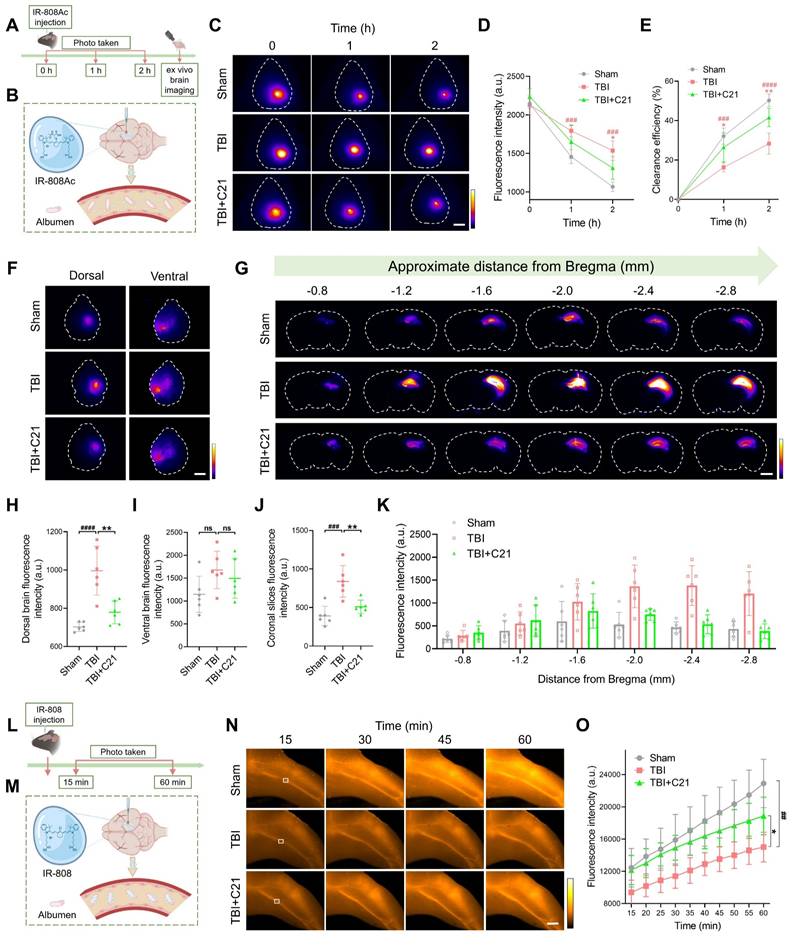
These findings were further validated using ex vivo brain imaging (Figure 3F). In TBI mice, the residual IR-808Ac in the dorsal side of the brain was higher than that in sham mice. However, treatment with C21 reduced the residual tracer (Figure 3H). Although C21 treatment also slightly reduced the residual tracer on the ventral side of the brain, the difference compared to TBI mice was not statistically significant (Figure 3I). Coronal slices imaging (Figure 3G) supported these observations, showing that C21 treatment effectively decreased residual IR-808Ac in the brain parenchyma compared to TBI mice (Figure 3J, K).
We further utilized IR-808 to assess glymphatic clearance (Figure 3L). Unlike IR-808Ac, IR-808 has strong albumin-seeking capacity, enabling it to bind to blood albumin and persist in systemic circulation for an extended period (Figure 3M) [44, 52]. This property makes IR-808 a suitable tracer for specifically monitoring its clearance from the brain parenchyma into the blood circulation. In this study, after IR-808 was injected into the cerebral cortex, NIR-II in vivo imaging was used to continuously monitor fluorescence intensity in the femoral vein. Images were captured from the same location in the femoral vein (Figure 3N), and fluorescence changes were analyzed. The results showed that the fluorescence intensity of IR-808 in the femoral vein of TBI mice was lower than that in sham-operated mice, suggesting impaired glymphatic clearance. However, treatment with C21 notably increased the fluorescence intensity of IR-808 in the femoral veins of TBI mice (Figure 3O), demonstrating improved glymphatic clearance.
AT2R activation restored AQP4 polarization and CBF in acute TBI mice
Decreased AQP4 polarization is a key mechanism underlying glymphatic system dysfunction after TBI [11]. To investigate the mechanism of AT2R activation on glymphatic transport in acute TBI mice, we analyzed AQP4 polarization in the perilesional cortex 3 days after TBI using double immunofluorescence staining for CD31 (a vascular endothelial marker) and AQP4. The results revealed that C21 treatment effectively reversed the TBI-induced decrease in AQP4 polarization (Figure 4A, B, E-G). Given that extensive reactive astrogliosis following TBI is closely associated with the loss of perivascular AQP4 polarization, we examined the expression of GFAP and AQP4 by immunofluorescence imaging. The results demonstrated increased GFAP expression in the TBI group compared to the sham-operated group. C21 treatment reduced GFAP expression, suggesting attenuation of reactive astrogliosis (Figure 4C, D).
AT2R activation restored AQP4 polarization and CBF in acute TBI mice. (A) Co-immunofluorescence staining of AQP4 (green) and CD31 (red) in the perilesional cortex. Scale bar = 100 μm. (B) Quantification of AQP4 polarization (AQP4 polarization = 1 - AQP4% area). (C) Co-immunofluorescence staining of AQP4 (green) and GFAP (red) in the perilesional cortex. Scale bar = 100 μm. (D) Quantification of the GFAP coverage area fraction. (E) Representative images of AQP4 (green) and CD31 (red) immunofluorescence staining. Scale bar = 50 μm. (F) Quantification of AQP4 fluorescence intensity profile along the white line in (E). (G) Quantification of AQP4 polarization (AQP4 polarization = vessel AQP4 peak intensity / average baseline intensity). (H) Representative Iba-1 immunofluorescence images and cytoskeletal morphology analysis in the perilesional cortex. Scale bar = 50 μm. (I-K) Quantitative analysis of microglial density (I), branch length (J), and endpoint voxels (K). (L, M) Representative images and quantification of CBF for all groups. (N, O) Measurements and quantification of MAP for all groups. (P) Schematic diagram of measuring heart rate and respiratory rate in mice. The schematic diagram was created with BioRender.com. (Q, R) Measurements of heart rate for all groups. (S, T) Measurements of respiratory rate for all groups. All data are presented as mean ± SD (n = 6-8 per group). Statistical significance: *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
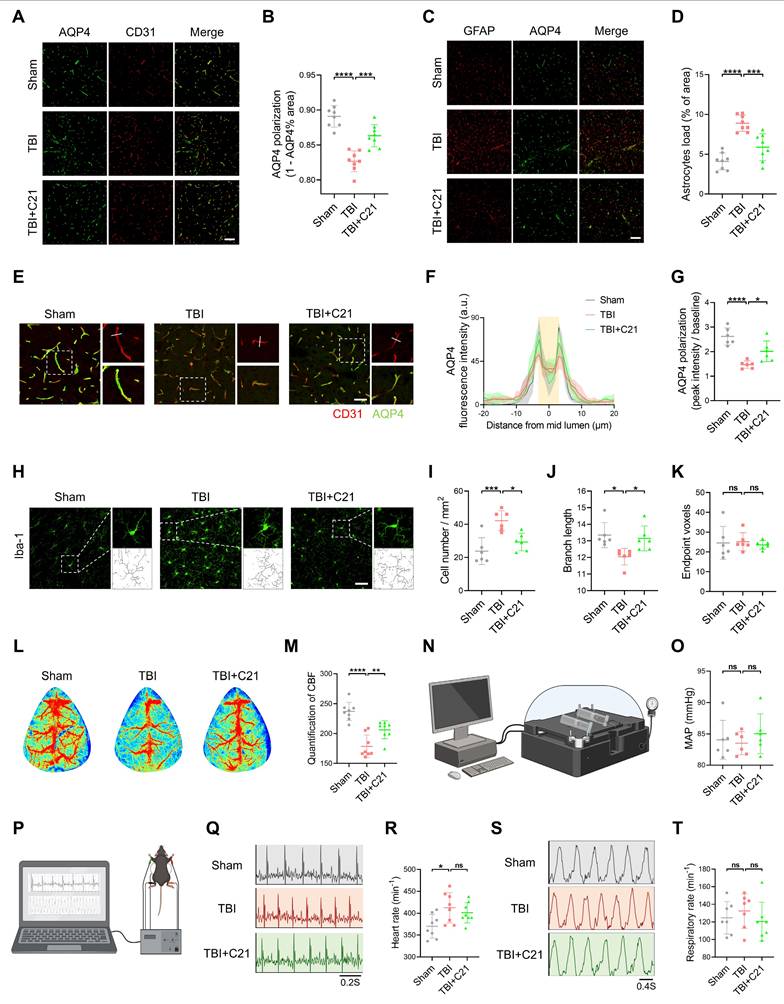
Under pathological conditions, the glymphatic system modulates microglial activation in the perilesional cortex [53]. Microglial activation was assessed by measuring the number of endpoint voxels and the average branch length of microglial cells, as activated microglia typically exhibit shortened branches and reduced endpoint voxels [54]. Using immunofluorescence staining for Iba1, we analyzed the number and morphology of microglia in brain tissue (Figure 4H; see supplementary methods for quantification of microglial activation). The results showed a significant increase in microglial density and a marked reduction in branch length in the TBI group compared with the sham group, reflecting heightened microglial activation. Treatment with C21 decreased microglial density and restored branch length, suggesting suppressed microglial activation (Figure 4I, J). However, there was no significant difference in endpoint voxel counts among these groups (Figure 4K).
The regulation of the glymphatic system involves dynamic interactions between blood pressure and CBF [55-58]. Additionally, emerging evidence suggests that cardiopulmonary rhythms (heart rate/respiratory rate) may modulate glymphatic drainage [59]. Therefore, we assessed cardiovascular parameter responses to TBI and C21 intervention. Our findings demonstrated that TBI significantly reduced CBF, whereas C21 treatment effectively restored post-TBI CBF levels (Figure 4L, M). Notably, C21 administration did not induce detectable alterations in MAP, heart rate, or respiratory rate in TBI mice (Figure 4N-T). Furthermore, C21 injection in healthy control mice elicited no significant changes in heart rate, respiratory rate, MAP, or CBF (Figure S5).
AT2R activation inhibited neuroinflammatory responses in acute TBI mice
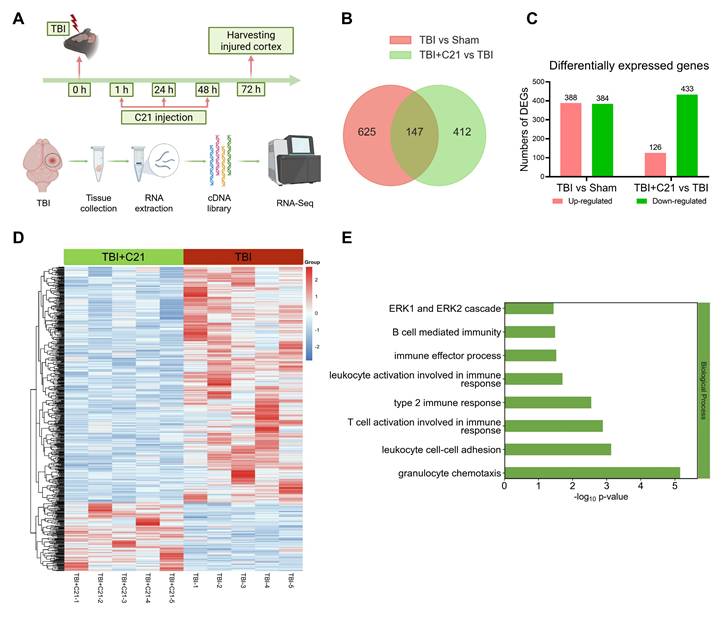
RNA-seq analysis of the perilesional cortex at 3 days post-TBI revealed a dynamic immune modulation induced by C21 treatment (Figure 5A). High inter-replicate correlation confirmed experimental reproducibility (Figure S6), while principal component analysis demonstrated clear disparities in gene expression profiles among Sham-operated, TBI, and TBI+C21 groups (Figure S7). Comparative analysis identified 772 differentially expressed genes (DEGs) between TBI and Sham-operated groups (388 upregulated, 384 downregulated; Figure 5B, C). The cluster map displayed the DEGs between the TBI and Sham-operated groups (Figure S8A). Gene ontology analysis of TBI-associated DEGs revealed marked enrichment in immune activation pathways, including B cell-mediated immunity, T cell activation involved in immune response, leukocyte activation involved in immune response, leukocyte cell-cell adhesion, granulocyte chemotaxis, and ERK1/2 cascade (Figure S8B). Notably, C21 administration substantially attenuated TBI-induced transcriptional alterations, with 559 DEGs distinguishing TBI+C21 from TBI groups (126 upregulated, 433 downregulated; Figure 5B, C). The cluster map displayed the DEGs between the TBI+C21 and TBI groups (Figure 5D). Crucially, C21 treatment reversed TBI-associated immune pathways, including B cell-mediated immunity, T cell activation involved in immune response, leukocyte activation involved in immune response, leukocyte cell-cell adhesion, granulocyte chemotaxis, and ERK1/2 cascade (Figure 5E). Together, these findings demonstrate that C21 treatment ameliorated neuroinflammatory responses initiated by TBI.
AT2R activation inhibited neuroinflammatory responses in acute TBI mice. (A) Flowchart of the RNA-seq analysis. (B) Venn diagram illustrating genetic variations. (C) Statistically up-regulated and down-regulated DEGs. (D) Cluster map of the DEGs among TBI+C21 group and TBI group. (E) Down-regulated biological process of gene ontology function enrichment of the DEGs in the TBI+C21 group compared with the TBI group. n = 5 per group.
In addition, RNA-seq analysis revealed that the AT2R signaling pathway involves the ERK1/2, a key regulator of brain injury and repair [60]. To investigate the potential signaling pathways mediating the protective effects of AT2R, we examined the effect of C21 on ERK1/2 activation in the injured cortex in mice 3 days after TBI using Western blot. Our results revealed that C21 led to a significant decrease in ERK1/2 (Thr202/Tyr204) phosphorylation (Figure S9), suggesting reduced activation of ERK1/2. Subsequently, we investigated whether inhibiting ERK activation would affect CBF and AQP4 polarization. The results revealed that administration of the ERK inhibitor U0126 promoted the restoration of both AQP4 polarization and CBF in mice 3 days post-TBI (Figure S10).
AT2R activation suppressed β-amyloid and phosphorylated tau accumulation in chronic TBI mice
TBI-induced impairment of the glymphatic system persists in the chronic phase [10]. 30 days after TBI, NIR-II imaging revealed persistent impairment of glymphatic influx in the TBI group. However, AT2R activation by C21 treatment restored glymphatic influx during the chronic TBI phase (Figure 6A, B). Further corroborating these findings, ex vivo imaging demonstrated a similar restoration of glymphatic influx (Figure 6C-F; see Figure S11 for detailed coronal slices analysis).
AT2R activation suppressed β-amyloid and phosphorylated tau accumulation in chronic TBI mice. (A, B) Representative transcranial NIR-II images (A) and quantified fluorescence intensity (B) of BSA@IR-780. Scale bar = 4 mm. (C) Representative images of BSA@IR-780 distribution on dorsal and ventral brain surfaces, as well as in coronal slices. Scale bars: 4 mm (surfaces), 2 mm (slices). (D-F) Quantified fluorescence intensity on dorsal (D) and ventral (E) surfaces, and across coronal slices (F). (G, H) Representative images (G) and quantification (H) of FITC-labeled β-amyloid (1-42) in brain parenchyma. Scale bar = 400 μm. (I-M) Western blots (I) and quantification of tau markers in perilesional cortex: pTauThr205 (J), pTauSer396 (K), pTauSer404 (L), and Tau5 (M). All data are presented as mean ± SD (n = 6 per group). Statistical significance: #p < 0.05, ##p < 0.01, ###p < 0.001, ####p < 0.0001 versus sham group; *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 versus TBI+C21 group.
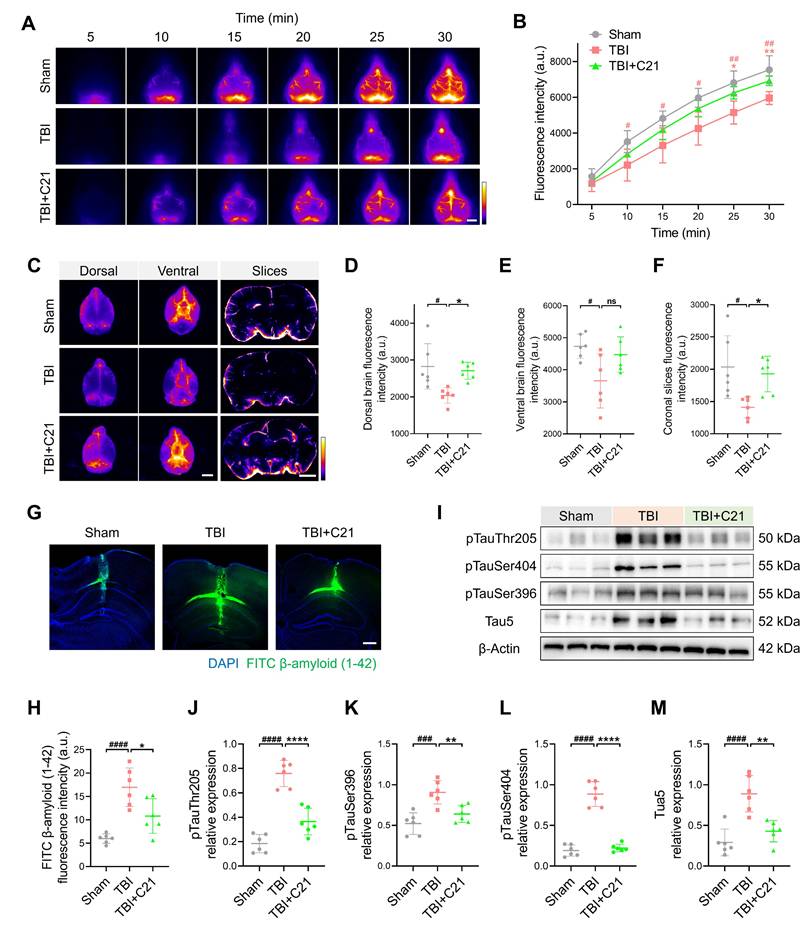
The accumulation of β-amyloid and phosphorylated tau following TBI has been identified as a direct contributor to the pathogenesis of neurodegenerative diseases [61, 62]. To assess β-amyloid clearance efficiency, FITC-labeled β-amyloid was injected into the cerebral cortex, and its residual levels were subsequently analyzed (Figure 6G). Quantitative analysis revealed increased β-amyloid retention in TBI mice compared to sham-operated controls. Notably, C21 treatment reduced this retention (Figure 6H). Additionally, Western blot was used to evaluate phosphorylated tau levels in the perilesional cortex (Figure 6I). C21 administration post-TBI effectively attenuated phosphorylated tau accumulation, particularly at the Thr205, Ser404, and Ser396 phosphorylation sites (Figure 6J-M).
Discussion
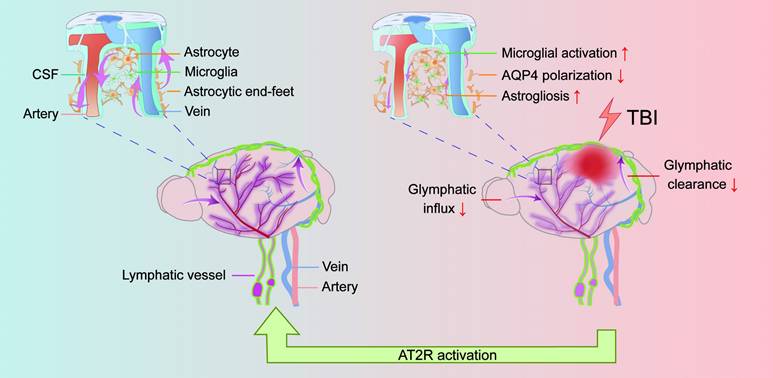
This study aimed to investigate the therapeutic potential of C21 in regulating glymphatic system function following TBI. Leveraging the unique physicochemical properties of NIR-II probes, we developed a multimodal NIR-II imaging platform to visualize both glymphatic influx and glymphatic clearance, enabling real-time dynamic monitoring of glymphatic transport in vivo. Our data demonstrated that C21-mediated AT2R activation not only effectively restored glymphatic transport but also significantly reduced neuroinflammation, β-amyloid and phosphorylated tau accumulation, and neurological functional impairments compared with untreated TBI mice (Figure 7).
TBI impairs glymphatic transport and is counteracted by AT2R activation. Under physiological conditions, CSF flows from the subarachnoid space into the brain parenchyma through periarterial spaces, exchanges solutes with ISF, and ultimately drains via perivenous pathways (left panel). However, TBI induces structural damage to the glymphatic system, impairing glymphatic influx and clearance. Mechanistically, post-TBI glymphatic dysfunction arises primarily from reactive astrogliosis-mediated perivascular structural remodeling and loss of AQP4 polarization at astrocytic end-feet (right panel). Critically, C21-mediated AT2R activation reverses these pathological changes, restoring glymphatic transport post-TBI. Furthermore, AT2R activation suppresses neuroinflammatory responses, reduces neurotoxic protein accumulation, and improves neurological recovery.
TBI impairs the function of glymphatic system through mechanical impact and neuroinflammatory pathways, manifesting as reduced AQP4 polarization and obstructed CSF-ISF circulation. These disruptions lead to diminished clearance of metabolic waste and neurotoxic proteins (e.g., β-amyloid, phosphorylated tau), ultimately contributing to neurological dysfunction [11, 21]. While the RAS primarily systemically regulates cardiovascular homeostasis, its dysregulation is implicated in hypertension and cardiovascular disorders. Notably, the localized brain RAS plays a pivotal role in neurological pathologies [63, 64]. In TBI, activation of the brain RAS via AT1R exacerbates injury and pathological progression. Conversely, AT2R, though minimally expressed in the adult brain under physiological conditions, demonstrates significant upregulation during ischemic, inflammatory, and neurodegenerative states, reflecting its adaptive role in injury repair. Studies have demonstrated that C21-mediated AT2R activation alleviates neuroinflammation, cerebral edema, and blood-brain barrier disruption in TBI mice, while concurrently improving myocardial fibrosis and increasing left ventricular ejection fraction [30, 31]. Furthermore, C21 treatment in stroke mice suppresses neuroinflammation, decreases phosphorylation of tau at Ser202/Thr205 and β-amyloid accumulation in ipsilateral hemispheres, and enhances cognitive recovery [33]. These neuroprotective mechanisms of AT2R activation provide a foundational rationale for its role in glymphatic restoration post-TBI. In this study, we employed NIR-II imaging technology to investigate AT2R-mediated regulation of glymphatic transport following TBI.
To investigate the AT2R modulation of post-TBI glymphatic influx, we first employed NIR-II imaging to track the CSF-mediated transport dynamics of BSA@IR-780 from the subarachnoid space to the brain parenchyma. The whole-brain macroscopic visualization capability of NIR-II imaging enabled precise mapping of tracer influx through the PVS across cortical surfaces. Our analyses revealed that TBI substantially compromises glymphatic influx, manifesting as a significantly delayed tracer influx. Crucially, C21-mediated AT2R activation effectively restored the influx of BSA@IR-780 and increased its flow speed in the PVS. NIR-II imaging was also employed to non-invasively monitor the drainage of BSA@IR-780 into the superficial CLNs. The results confirmed that AT2R activation enhances drainage into superficial CLNs. This dynamic macroscopic imaging approach provides clearer visualization of glymphatic transport impairment induced by TBI, while highlighting the protective effects of AT2R activation in restoring glymphatic drainage function post-TBI.
In addition, this study introduced an innovative approach using NIR-II probes to monitor the dynamic parenchymal clearance process in real-time, including albumin-escaping IR-808Ac capable of direct and real-time visualization of solute clearance in the brain parenchyma, and albumin-seeking IR-808 designed to track the dynamic of clearance solute from the brain into the blood circulation. This method provides a more precise and less invasive evaluation of glymphatic clearance in mice. Our results showed that the increased clearance efficiency of IR-808Ac in the brain parenchyma and the elevated fluorescence intensity of IR-808 in the femoral vein clearly indicated that AT2R activation enhanced glymphatic clearance following TBI. This innovative approach advances our understanding of the glymphatic clearance mechanism and highlights the therapeutic potential of AT2R activation in mitigating TBI-induced impairment of glymphatic clearance function.
Studies have established that TBI triggers diffuse reactive astrogliosis, culminating in hypertrophic glial scarring. This process disrupts perivascular AQP4 polarization and spatial distribution, representing a primary pathophysiological mechanism underlying post-TBI glymphatic system impairment [20]. Previous research indicated that AT2R activation suppresses post-stroke astrogliosis and ameliorates neuroinflammatory responses in 5XFAD murine models [33]. Furthermore, in angiotensin II-induced oxidative stress and inflammatory responses in astrocytes, AT2R activation attenuated reactive oxygen species overproduction, mitigated mitochondrial dysfunction, and inhibited NFκB-driven inflammatory cascades [65]. Our experimental findings demonstrated that AT2R activation restored AQP4 polarization and attenuated the reactive astrogliosis following TBI. These effects provide mechanistic insight into AT2R-mediated protection against TBI-induced glymphatic dysfunction.
Blood pressure regulation is crucial following TBI, as hemodynamic instability may disrupt cerebral arterial pulsations and subsequently impair the glymphatic system function [58]. Although preclinical studies indicate that C21 attenuates angiotensin II-induced hypertension [66], Timaru-Kast et al. reported that neither low nor high C21 doses affected blood pressure in TBI mice [67]. Our findings demonstrated that C21 administration did not markedly alter MAP, suggesting that its protective effects may not involve blood pressure modulation. In addition, studies have demonstrated an inverse correlation between glymphatic influx and heart rate, independent of respiratory rate [59]. Another study showed that CSF dynamics have been associated with respiratory rate [68]. Our study revealed that C21 administration did not significantly alter the heart rate or respiratory rate in TBI mice. These findings suggest that the restoration of the glymphatic system function by C21 in TBI occurs independently of alterations in heart rate or respiratory rate regulation.
The neuroinflammatory response following TBI involves complex interactions with glymphatic dysfunction. Post-TBI inflammatory factors impair AQP4 polarization, which exacerbating glymphatic transport impairment and amplifying neuroinflammation [11]. As key immune effectors in the central nervous system, microglia rapidly proliferate and display characteristic morphological activation in perilesional areas after TBI, characterized by shortened branches and reduced terminal endpoints [69]. Our cytoskeletal analysis demonstrated that C21 treatment significantly inhibited microglial activation in the perilesional cortex. Furthermore, RNA-seq analysis revealed that C21 administration effectively suppressed neuroinflammatory responses in the perilesional cortex, as evidenced by the downregulation of neuroinflammatory signaling pathways. In addition, RNA-seq analysis revealed that the protective mechanism of C21 in TBI involves the ERK pathway. Specifically, TBI induces sustained activation of the ERK pathway, leading to a dramatic increase in phosphorylated ERK levels that exacerbates secondary damage. Previous studies demonstrated that AT2R activation inhibits ERK activity via SHP1 tyrosine phosphatase activation [70]. Our results indicated that AT2R activation suppresses ERK1/2 phosphorylation in acute TBI mice. Crucially, ERK inhibition has been found protective in brain injury, suppressing neurotoxic astrocyte polarization, attenuating neuroinflammatory responses, reducing blood-brain barrier disruption, and promoting neurological function recovery [60, 71-73]. We found that ERK inhibition facilitated the restoration of AQP4 polarization and CBF in acute TBI mice. Collectively, these results suggested that the effects of AT2R activation on restoring AQP4 polarization and CBF might be mediated through ERK inhibition.
Evidence indicates that TBI induces persistent glymphatic system impairment in murine models, with functional deficits persisting up to 6 months post-injury [10]. Persistent dysfunction of glymphatic transport promotes β-amyloid and phosphorylated tau accumulation, thereby contributing to long-term neurological deficits [21]. Our experimental data corroborate these findings by revealing persistent impairment of glymphatic drainage efficiency at 30 days post-TBI. Crucially, C21-mediated AT2R activation maintained glymphatic transport function during the chronic phase. The mechanistic significance of these findings is further underscored by AT2R activation reducing β-amyloid and phosphorylated tau accumulation. Behavioral assessments substantiated the relevance of this intervention, demonstrating significant amelioration of TBI-induced motor coordination deficits and cognitive dysfunction. Thus, our findings demonstrate that AT2R-targeted intervention disrupts the pathological cascade of chronic glymphatic impairment following TBI, providing sustained protection against neuropathological progression and functional deterioration.
Conclusion
In this study, we employed NIR-II imaging to dynamically visualize glymphatic transport in mice, showing that C21-mediated AT2R activation enhanced glymphatic influx and promoted glymphatic clearance in TBI mice. Mechanistically, AT2R activation restored AQP4 polarization and simultaneously suppressed reactive astrogliosis, microglial activation, and neuroinflammatory responses. Furthermore, AT2R activation enhanced β-amyloid clearance efficiency and reduced phosphorylated tau accumulation, thereby contributing to improved post-traumatic neurological recovery. Collectively, these findings establish the critical role of AT2R in restoring post-TBI glymphatic function and identify it as a promising therapeutic target for TBI.
Abbreviations
TBI: traumatic brain injury; CSF: cerebrospinal fluid; ISF: interstitial fluid; AQP4: aquaporin-4; PVS: perivascular spaces; GFAP: glial fibrillary acidic protein; RAS: angiotensin system; AT1R: angiotensin II type 1 receptor; AT2R: angiotensin II type 2 receptor; CBF: cerebral blood flow; C21: compound 21; NIR-II: near-infrared II; CLNs: cervical lymph nodes; ISO: isoflurane; PBS: phosphate buffered saline; mNSS: modified neurological severity score; NOR: novel object recognition; OFT: open field test; MWM: Morris water maze; CM: cisterna magna; DEX: dexmedetomidine; PFA: paraformaldehyde; MAP: mean arterial pressure; SBP: systolic blood pressure; DBP: diastolic blood pressure; RNA-seq: RNA sequencing; SD: standard deviation; DEGs: differentially expressed genes.
Supplementary Material
Supplementary materials and methods, figures.
Acknowledgements
Western Blot was performed in the core facility of the First Hospital of Jilin University, the authors thank Nan Zhao for assistance. This work was supported by grants from the National Natural Science Foundation of China (Nos. 82372505), the Development and Reform Commission of Jilin Province (Nos. ZKJCFGW2023038), and Technology Development Project of Jilin Province (No. 20200603002SF and YDZJ202402072CXJD).
Author contributions
H.W., S.Z., and X.Z. conceived and designed the study. X.Z. performed all experiments, collected, and analyzed the data. B.S., W.L., and T.L. assisted with in vivo imaging in the NIR-II window. W.L., B.C., C.H., and Q.L. contributed to data collection and analysis. S.Z. and X.Z. drafted the manuscript, while W.H. reviewed and revised it. All authors reviewed and approved the final version of the paper.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Capizzi A, Woo J, Verduzco-Gutierrez M. Traumatic Brain Injury: An Overview of Epidemiology, Pathophysiology, and Medical Management. Med Clin North Am. 2020;104:213-38
2. Gao G, Wu X, Feng J, Hui J, Mao Q, Lecky F. et al. Clinical characteristics and outcomes in patients with traumatic brain injury in China: a prospective, multicentre, longitudinal, observational study. Lancet Neurol. 2020;19:670-7
3. Bieniek KF, Cairns NJ, Crary JF, Dickson DW, Folkerth RD, Keene CD. et al. The Second NINDS/NIBIB Consensus Meeting to Define Neuropathological Criteria for the Diagnosis of Chronic Traumatic Encephalopathy. J Neuropathol Exp Neurol. 2021;80:210-9
4. Graham NS, Sharp DJ. Understanding neurodegeneration after traumatic brain injury: from mechanisms to clinical trials in dementia. J Neurol Neurosurg Psychiatry. 2019;90:1221-33
5. LoBue C, Cullum CM, Didehbani N, Yeatman K, Jones B, Kraut MA. et al. Neurodegenerative Dementias After Traumatic Brain Injury. J Neuropsychiatry Clin Neurosci. 2018;30:7-13
6. Lohela TJ, Lilius TO, Nedergaard M. The glymphatic system: implications for drugs for central nervous system diseases. Nat Rev Drug Discov. 2022;21:763-79
7. Iliff JJ, Wang M, Liao Y, Plogg BA, Peng W, Gundersen GA. et al. A paravascular pathway facilitates CSF flow through the brain parenchyma and the clearance of interstitial solutes, including amyloid β. Sci Transl Med. 2012;4:147ra11
8. Tarasoff-Conway JM, Carare RO, Osorio RS, Glodzik L, Butler T, Fieremans E. et al. Clearance systems in the brain-implications for Alzheimer disease. Nat Rev Neurol. 2015;11:457-70
9. Peng S, Liu J, Liang C, Yang L, Wang G. Aquaporin-4 in glymphatic system, and its implication for central nervous system disorders. Neurobiol Dis. 2023;179:106035
10. Hussain R, Tithof J, Wang W, Cheetham-West A, Song W, Peng W. et al. Potentiating glymphatic drainage minimizes post-traumatic cerebral oedema. Nature. 2023;623:992-1000
11. Peters ME, Lyketsos CG. The glymphatic system's role in traumatic brain injury-related neurodegeneration. Mol Psychiatry. 2023;28:2707-15
12. Reeves BC, Karimy JK, Kundishora AJ, Mestre H, Cerci HM, Matouk C. et al. Glymphatic System Impairment in Alzheimer's Disease and Idiopathic Normal Pressure Hydrocephalus. Trends Mol Med. 2020;26:285-95
13. Beschorner N, Nedergaard M. Glymphatic system dysfunction in neurodegenerative diseases. Curr Opin Neurol. 2024;37:182-8
14. Plog BA, Nedergaard M. The Glymphatic System in Central Nervous System Health and Disease: Past, Present, and Future. Annu Rev Pathol. 2018;13:379-94
15. Mestre H, Mori Y, Nedergaard M. The Brain's Glymphatic System: Current Controversies. Trends Neurosci. 2020;43:458-66
16. Li W, Chen D, Liu N, Luan Y, Zhu S, Wang H. Modulation of lymphatic transport in the central nervous system. Theranostics. 2022;12:1117-31
17. Rasmussen MK, Mestre H, Nedergaard M. The glymphatic pathway in neurological disorders. Lancet Neurol. 2018;17:1016-24
18. Ishida K, Yamada K, Nishiyama R, Hashimoto T, Nishida I, Abe Y. et al. Glymphatic system clears extracellular tau and protects from tau aggregation and neurodegeneration. J Exp Med. 2022 219
19. Si X, Dai S, Fang Y, Tang J, Wang Z, Li Y. et al. Matrix metalloproteinase-9 inhibition prevents aquaporin-4 depolarization-mediated glymphatic dysfunction in Parkinson's disease. J Adv Res. 2024;56:125-36
20. Iliff JJ, Chen MJ, Plog BA, Zeppenfeld DM, Soltero M, Yang L. et al. Impairment of glymphatic pathway function promotes tau pathology after traumatic brain injury. J Neurosci. 2014;34:16180-93
21. Sullan MJ, Asken BM, Jaffee MS, DeKosky ST, Bauer RM. Glymphatic system disruption as a mediator of brain trauma and chronic traumatic encephalopathy. Neurosci Biobehav Rev. 2018;84:316-24
22. Lv C, Han S, Sha Z, Liu M, Dong S, Zhang C. et al. Cerebral glucagon-like peptide-1 receptor activation alleviates traumatic brain injury by glymphatic system regulation in mice. CNS Neurosci Ther. 2023;29:3876-88
23. Liu M, Huang J, Liu T, Yuan J, Lv C, Sha Z. et al. Exogenous interleukin 33 enhances the brain's lymphatic drainage and toxic protein clearance in acute traumatic brain injury mice. Acta Neuropathol Commun. 2023;11:61
24. Vadhan JD, Speth RC. The role of the brain renin-angiotensin system (RAS) in mild traumatic brain injury (TBI). Pharmacol Ther. 2021;218:107684
25. Villapol S, Janatpour ZC, Affram KO, Symes AJ. The Renin Angiotensin System as a Therapeutic Target in Traumatic Brain Injury. Neurotherapeutics. 2023;20:1565-91
26. Baron DH, Skrobot OA, Palmer JC, Sharma K, Kehoe PG. Renin Angiotensin System as a Potential Treatment Target for Traumatic Brain Injury: A Systematic Review and Meta-Analysis. J Neurotrauma. 2022;39:473-86
27. Higuchi S, Ohtsu H, Suzuki H, Shirai H, Frank GD, Eguchi S. Angiotensin II signal transduction through the AT1 receptor: novel insights into mechanisms and pathophysiology. Clin Sci (Lond). 2007;112:417-28
28. Chappell MC. Biochemical evaluation of the renin-angiotensin system: the good, bad, and absolute? Am J Physiol Heart Circ Physiol. 2016;310:H137-52
29. Forrester SJ, Booz GW, Sigmund CD, Coffman TM, Kawai T, Rizzo V. et al. Angiotensin II Signal Transduction: An Update on Mechanisms of Physiology and Pathophysiology. Physiol Rev. 2018;98:1627-738
30. Umschweif G, Liraz-Zaltsman S, Shabashov D, Alexandrovich A, Trembovler V, Horowitz M. et al. Angiotensin receptor type 2 activation induces neuroprotection and neurogenesis after traumatic brain injury. Neurotherapeutics. 2014;11:665-78
31. Qian Y, Dong S, Nie M, Tian Y, Liu M, Liu X. et al. Delayed Administration of an Angiotensin II Type 2 Receptor Agonist Promotes Functional Recovery of the Brain and Heart After Traumatic Brain Injury. J Neurotrauma. 2024;41:660-70
32. Jackson-Cowan L, Eldahshan W, Dumanli S, Dong G, Jamil S, Abdul Y. et al. Delayed Administration of Angiotensin Receptor (AT2R) Agonist C21 Improves Survival and Preserves Sensorimotor Outcomes in Female Diabetic Rats Post-Stroke through Modulation of Microglial Activation. Int J Mol Sci. 2021 22
33. Ahmed HA, Ismael S, Salman M, Devlin P, McDonald MP, Liao FF. et al. Direct AT2R Stimulation Slows Post-stroke Cognitive Decline in the 5XFAD Alzheimer's Disease Mice. Mol Neurobiol. 2022;59:4124-40
34. Jackson L, Dong G, Althomali W, Sayed MA, Eldahshan W, Baban B. et al. Delayed Administration of Angiotensin II Type 2 Receptor (AT2R) Agonist Compound 21 Prevents the Development of Post-stroke Cognitive Impairment in Diabetes Through the Modulation of Microglia Polarization. Transl Stroke Res. 2020;11:762-75
35. Alshammari A, Pillai B, Kamat P, Jones TW, Bosomtwi A, Khan MB. et al. Angiotensin II Type 2 Receptor Agonism Alleviates Progressive Post-stroke Cognitive Impairment in Aged Spontaneously Hypertensive Rats. Transl Stroke Res. 2025;16:584-99
36. Ranjit A, Khajehpour S, Aghazadeh-Habashi A. Update on Angiotensin II Subtype 2 Receptor: Focus on Peptide and Nonpeptide Agonists. Mol Pharmacol. 2021;99:469-87
37. Haque A, Alenezi KM, Alsukaibi AKD, Al-Otaibi AA, Wong WY. Water-Soluble Small Organic Fluorophores for Oncological Theragnostic Applications: Progress and Development. Top Curr Chem (Cham). 2024;382:14
38. Zhu N, Xu J, Su Q, Han T, Zhou D, Zhang Y. et al. Site-specific albumin tagging with NIR-II fluorogenic dye for high-performance and super-stable bioimaging. Theranostics. 2024;14:1860-72
39. Sun B, Fang D, Li W, Li M, Zhu S. NIR-II nanoprobes for investigating the glymphatic system function under anesthesia and stroke injury. J Nanobiotechnology. 2024;22:200
40. Li W, Sun B, Zhang X, Liu T, Zhu W, Liu X. et al. Near-Infrared-II Imaging Revealed Hypothermia Regulates Neuroinflammation Following Brain Injury by Increasing the Glymphatic Influx. ACS Nano. 2024;18:13836-48
41. Henninger N, Bouley J, Sikoglu EM, An J, Moore CM, King JA. et al. Attenuated traumatic axonal injury and improved functional outcome after traumatic brain injury in mice lacking Sarm1. Brain. 2016;139:1094-105
42. Bouley J, Chung DY, Ayata C, Brown RH Jr, Henninger N. Cortical Spreading Depression Denotes Concussion Injury. J Neurotrauma. 2019;36:1008-17
43. Ishrat T, Fouda AY, Pillai B, Eldahshan W, Ahmed H, Waller JL. et al. Dose-response, therapeutic time-window and tPA-combinatorial efficacy of compound 21: A randomized, blinded preclinical trial in a rat model of thromboembolic stroke. J Cereb Blood Flow Metab. 2019;39:1635-47
44. Du Y, Xu J, Zheng X, Dang Z, Zhu N, Jiang Z. et al. NIR-II Protein-Escaping Dyes Enable High-Contrast and Long-Term Prognosis Evaluation of Flap Transplantation. Adv Mater. 2024;36:e2311515
45. Zhu S, Herraiz S, Yue J, Zhang M, Wan H, Yang Q. et al. 3D NIR-II Molecular Imaging Distinguishes Targeted Organs with High-Performance NIR-II Bioconjugates. Adv Mater. 2018;30:e1705799
46. Tian R, Ma H, Zhu S, Lau J, Ma R, Liu Y. et al. Multiplexed NIR-II Probes for Lymph Node-Invaded Cancer Detection and Imaging-Guided Surgery. Adv Mater. 2020;32:e1907365
47. Kress BT, Iliff JJ, Xia M, Wang M, Wei HS, Zeppenfeld D. et al. Impairment of paravascular clearance pathways in the aging brain. Ann Neurol. 2014;76:845-61
48. Munk AS, Wang W, Bèchet NB, Eltanahy AM, Cheng AX, Sigurdsson B. et al. PDGF-B Is Required for Development of the Glymphatic System. Cell Rep. 2019;26:2955-69.e3
49. Yao D, Li R, Hao J, Huang H, Wang X, Ran L. et al. Melatonin alleviates depression-like behaviors and cognitive dysfunction in mice by regulating the circadian rhythm of AQP4 polarization. Transl Psychiatry. 2023;13:310
50. Sun Q, Peng S, Xu Q, Weikop P, Hussain R, Song W. et al. Enhancing glymphatic fluid transport by pan-adrenergic inhibition suppresses epileptogenesis in male mice. Nat Commun. 2024;15:9600
51. Harrison IF, Ismail O, Machhada A, Colgan N, Ohene Y, Nahavandi P. et al. Impaired glymphatic function and clearance of tau in an Alzheimer's disease model. Brain. 2020;143:2576-93
52. Zhu HJ, Sun YY, Du Y, Zhou SY, Qu Y, Pang SY. et al. Albumin-seeking near-infrared-II probe evaluating blood-brain barrier disruption in stroke. J Nanobiotechnology. 2024;22:742
53. Sugama S, Kakinuma Y. Noradrenaline as a key neurotransmitter in modulating microglial activation in stress response. Neurochem Int. 2021;143:104943
54. Hsu SJ, Zhang C, Jeong J, Lee SI, McConnell M, Utsumi T. et al. Enhanced Meningeal Lymphatic Drainage Ameliorates Neuroinflammation and Hepatic Encephalopathy in Cirrhotic Rats. Gastroenterology. 2021;160:1315-29.e13
55. Hauglund NL, Andersen M, Tokarska K, Radovanovic T, Kjaerby C, Sørensen FL. et al. Norepinephrine-mediated slow vasomotion drives glymphatic clearance during sleep. Cell. 2025;188:606-22.e17
56. Holstein-Rønsbo S, Gan Y, Giannetto MJ, Rasmussen MK, Sigurdsson B, Beinlich FRM. et al. Glymphatic influx and clearance are accelerated by neurovascular coupling. Nat Neurosci. 2023;26:1042-53
57. Andica C, Kamagata K, Takabayashi K, Kikuta J, Kaga H, Someya Y. et al. Neuroimaging findings related to glymphatic system alterations in older adults with metabolic syndrome. Neurobiol Dis. 2023;177:105990
58. Mestre H, Tithof J, Du T, Song W, Peng W, Sweeney AM. et al. Flow of cerebrospinal fluid is driven by arterial pulsations and is reduced in hypertension. Nat Commun. 2018;9:4878
59. Hablitz LM, Vinitsky HS, Sun Q, Stæger FF, Sigurdsson B, Mortensen KN. et al. Increased glymphatic influx is correlated with high EEG delta power and low heart rate in mice under anesthesia. Sci Adv. 2019;5:eaav5447
60. Wang X, Wang Q, Hou L, Wei G, He C, Li H. et al. Advances in ERK Signaling Pathway in Traumatic Brain Injury: Mechanisms and Therapeutic Potential. Neurochem Res. 2025;50:191
61. Lu KP, Kondo A, Albayram O, Herbert MK, Liu H, Zhou XZ. Potential of the Antibody Against cis-Phosphorylated Tau in the Early Diagnosis, Treatment, and Prevention of Alzheimer Disease and Brain Injury. JAMA Neurol. 2016;73:1356-62
62. Wu YC, Bogale TA, Koistinaho J, Pizzi M, Rolova T, Bellucci A. The contribution of β-amyloid, Tau and α-synuclein to blood-brain barrier damage in neurodegenerative disorders. Acta Neuropathol. 2024;147:39
63. Labandeira-Garcia JL, Labandeira CM, Guerra MJ, Rodriguez-Perez AI. The role of the brain renin-angiotensin system in Parkinson´s disease. Transl Neurodegener. 2024;13:22
64. Petek B, Villa-Lopez M, Loera-Valencia R, Gerenu G, Winblad B, Kramberger MG. et al. Connecting the brain cholesterol and renin-angiotensin systems: potential role of statins and RAS-modifying medications in dementia. J Intern Med. 2018;284:620-42
65. Bhat SA, Fatima Z, Sood A, Shukla R, Hanif K. The Protective Effects of AT2R Agonist, CGP42112A, Against Angiotensin II-Induced Oxidative Stress and Inflammatory Response in Astrocytes: Role of AT2R/PP2A/NFκB/ROS Signaling. Neurotox Res. 2021;39:1991-2006
66. Kemp BA, Howell NL, Keller SR, Gildea JJ, Padia SH, Carey RM. AT2 Receptor Activation Prevents Sodium Retention and Reduces Blood Pressure in Angiotensin II-Dependent Hypertension. Circ Res. 2016;119:532-43
67. Timaru-Kast R, Garcia Bardon A, Luh C, Coronel-Castello SP, Songarj P, Griemert EV. et al. AT2 activation does not influence brain damage in the early phase after experimental traumatic brain injury in male mice. Sci Rep. 2022;12:14280
68. Yamada S, Miyazaki M, Yamashita Y, Ouyang C, Yui M, Nakahashi M. et al. Influence of respiration on cerebrospinal fluid movement using magnetic resonance spin labeling. Fluids Barriers CNS. 2013;10:36
69. Borst K, Dumas AA, Prinz M. Microglia: Immune and non-immune functions. Immunity. 2021;54:2194-208
70. Bedecs K, Elbaz N, Sutren M, Masson M, Susini C, Strosberg AD. et al. Angiotensin II type 2 receptors mediate inhibition of mitogen-activated protein kinase cascade and functional activation of SHP-1 tyrosine phosphatase. Biochem J. 1997;325( Pt 2):449-54
71. Zhang ZH, Gu Y, Huang Z, Liu XY, Xu WT, Zhang XC. et al. Acupuncture regulates astrocyte neurotoxic polarization to protect blood-brain barrier integrity in delayed thrombolysis through mediating ERK1/2/Cx43 axis. IBRO Neurosci Rep. 2025;18:604-18
72. Pei X, Zhang L, Liu D, Wu Y, Li X, Cao Y. et al. Notoginsenoside R1 attenuates brain injury in rats with traumatic brain injury: Possible mediation of apoptosis via ERK1/2 signaling pathway. PLoS One. 2023;18:e0295903
73. Maddahi A, Edvinsson L. Cerebral ischemia induces microvascular pro-inflammatory cytokine expression via the MEK/ERK pathway. J Neuroinflammation. 2010;7:14
Author contact
![]() Corresponding authors: Shoujun Zhu, Email: sjzhuedu.cn, Haifeng Wang, Email: hfwangedu.cn.
Corresponding authors: Shoujun Zhu, Email: sjzhuedu.cn, Haifeng Wang, Email: hfwangedu.cn.