Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
1. Introduction
2. Molecular Characteristics and...
3. The Mechanistic Role of...
4. Translational Applications...
5. Take-Home Message and Future...
6. Concluding Remarks
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2025; 15(18):9944-9968. doi:10.7150/thno.121766 This issue Cite
Review
GM-CSF+ Th: a central player in autoimmunity
Sha-Sha Fan1,2,3,4,#, Xuan Xu5,#, Yu-Bin Luo1,3, Xiang-Yu Meng2,4 ![]() , Yi Liu1,3,6,‡,
, Yi Liu1,3,6,‡, ![]()
1. Department of Rheumatology and Immunology, Laboratory of Rheumatology and Immunology, West China Hospital, Sichuan University, Chengdu 610041, China.
2. Hubei Provincial Key Laboratory of Occurence and Intervention of Rheumatic Diseases, Hubei Minzu University, Enshi 445000, China.
3. Institute of Immunology and Inflammation, Frontiers Science Center for Disease-Related Molecular Network, West China Hospital, Chengdu 610041, China.
4. Health Science Center, Hubei Minzu University, Enshi 445000, China.
5. School of Life Sciences, Anhui Medical University, Hefei 230032, China.
6. West China Lecheng Hospital, Sichuan University, Boao 571435, Hainan, China.
‡Lead contact.
#Equal contribution.
Received 2025-7-16; Accepted 2025-8-10; Published 2025-9-12
Abstract

Autoimmune diseases are driven by a breach of self-tolerance, leading to chronic inflammation and organ damage. While the Th1 and Th17 paradigms have long dominated our understanding of T-cell-mediated pathology, a distinct subset of CD4+ T helper cells defined by the production of granulocyte-macrophage colony-stimulating factor (GM-CSF) has emerged as a central and non-redundant pathogenic driver. These GM-CSF-producing Th cells orchestrate autoimmune pathology across a spectrum of diseases, including multiple sclerosis, rheumatoid arthritis, and type 1 diabetes, among others. Mechanistically, they act as critical amplifiers of inflammation by activating and recruiting myeloid cells, which in turn mediate tissue destruction. Furthermore, they establish vicious feedback loops, enhancing their own differentiation and promoting the function of other effector T cells. Despite their established importance, significant heterogeneity and plasticity exist in their phenotype and regulatory networks, with context-dependent variations across different diseases and species. This review synthesizes our current understanding of these pivotal cells, critically evaluating their classification, molecular identity, and the signaling pathways governing their differentiation. We further dissect their specific roles in major autoimmune diseases and provide a forward-looking perspective on their immense translational potential, from novel therapeutic strategies targeting the GM-CSF axis to their use as biomarkers for patient stratification and monitoring.
Keywords: GM-CSF+ Th cells, autoimmune diseases, GM-CSF, differentiation regulation, pathogenic mechanisms, clinical applications
1. Introduction
Autoimmune diseases (AIDs) are chronic inflammatory conditions characterized by aberrant immune responses against self-tissues. They encompass a broad range of conditions, including multiple sclerosis (MS), rheumatoid arthritis (RA), type 1 diabetes (T1D), and inflammatory bowel disease (IBD). The pathogenesis of AIDs is driven by complex interactions between genetic susceptibility, environmental triggers, and immune dysregulation, although the precise molecular mechanisms remain incompletely understood [1]. Epidemiological evidence suggests that approximately 7.6%-10.2% of the global population suffers from AIDs, which significantly impairs quality of life and imposes a growing economic burden on healthcare systems [2]. The incidence of these diseases continues to rise, and the absence of effective early prevention strategies, combined with limited public awareness, further complicates disease management [3].
Within the immune regulatory network of AIDs, abnormal activation of CD4+ T helper (Th) cells represents a key driving factor. Naive CD4+ T cells differentiate into various subsets, including Th1, Th2, and Th17, as well as regulatory T cells (Tregs), under the influence of antigenic stimulation, changes in cytokine microenvironments, and transcription factor-driven regulation. These subsets mediate pro-inflammatory or anti-inflammatory responses by secreting specific cytokines (e.g., IFN-γ/TNF-α from Th1 cells, IL-17 from Th17 cells) [4, 5]. Disruption of this balance contributes to persistent inflammation and immune-mediated tissue damage, which are central features of autoimmune disease pathophysiology. In particular, hyperactivation of Th1 and/or Th17 cells has been implicated as a key mechanism of disease progression in several AIDs, including MS, RA, and T1D [6, 7].
However, recent research has revealed contradictory phenomena and challenges the long established Th1/Th17 dogma. In mouse models of AIDs such as MS and T1D, among others, knockout of key factors including IFN-γ, IL-17, T-bet, Rel, or RORγt does not consistently attenuate disease progression and may even exacerbate it. These findings suggest that disease pathogenesis is not solely dependent on canonical Th1 or Th17 pathways, but rather reflects a context-dependent interplay of effector T cell subsets with compensatory mechanisms and functional plasticity [8-18]. This finding suggests that the pathogenic role of the traditional Th1/Th17 axis may involve functional redundancy or microenvironment dependency, prompting researchers to reconsider the potential regulatory roles of other Th subsets in AIDs.
In recent years, the landscape of Th cell biology has been reshaped by the emergence of a distinct lineage defined by its production of GM-CSF. While GM-CSF can be secreted by various cell types, compelling evidence from multiple AIDs indicates that CD4+ T cells are the predominant source of this cytokine in both peripheral blood and inflamed tissues. This pathogenic Th subset was first formally identified in 2013 from in vitro differentiation experiments, which revealed a population characterized by abundant GM-CSF secretion but minimal expression of canonical lineage-defining cytokines (e.g., IFN-γ, IL-17) or transcription factors (e.g., T-bet, RORγt). This as a result has initially led to its designation as a potentially independent lineage, provisionally named “ThGM” [19].
However, the identity of these cells is complicated by their profound plasticity. In vivo, GM-CSF is often co-expressed with other pro-inflammatory molecules like TNF-α; and under the influence of cytokines such as IL-12, these cells can be induced to acquire a Th1-like, IFN-γ-producing phenotype. This functional flexibility, combined with a lack of unique surface markers or a stable transcriptional signature, has invoked a long-standing controversy regarding their precise definition: Are they a stable, independent lineage, or a transient, highly activated state that other Th subsets can adopt? This ambiguity has resulted in a plethora of overlapping terminologies in the literature (Table 1), necessitating a clear and unified nomenclature. For the purpose of this review, we will use the encompassing term “GM-CSF-producing Th cells” (i.e., GM-CSF+ Th cells) to refer to any CD4+ T cell for which GM-CSF production is a key functional feature.
Subtypes of GM-CSF+ Th cells, their cellular origin, molecular characteristics, driving and suppressing factors, down-stream recruited inflammatory cells, and associated autoimmune diseases.
| GM-CSF+ Th subtypes | Origin | Molecular characteristics | Drivers | Suppressors | Recruited infCells† | Associated Ads* | References | |||
|---|---|---|---|---|---|---|---|---|---|---|
| Human | Mouse | Human | Mouse | Human | Mouse | |||||
| GM-CSF+ only Th cells | naïve CD4+ T cells | Markers: GM-CSF+, IL-2+, TNF-α+, CCR4+, CCR10+, CXCR4+, CD154+; IL-4-, IL-17A-, IFN-γ-, CCR6-, CCR7‑, CXCR3-, CXCR5-, CD25‑, CD27-, CD137-; IL-3+/-, CCL20+/-; Transcriptional factors: ZNF35+, SP2+, TEF+, TWIST1+, MSC+, TRERF1+, PPARG+; GATA3-, RORγt-, T-bet- | Markers: GM-CSF+, IL-2+, IL-3+, IL-15Rβ, IL-23R+, TNF-α+, CCL5+, CXCL9+, CCR1+, CCR4+, CCR9+, CCR10+, CX3CR1+, CD11A+, CD11C+, CD49D+, CD54+, CD69+, CD95+, CD147+, α4β7+; IL-17A-, IL-4-, IFN-γ-, CCR7-, CCR8-, CD45-, PD-1-, LAG3-; Transcriptional factors: Bhlhe40+, TWIST1+, MSC+, TRERF1+, PPARG+, NFATC1+, AP-1+, RUNX1+, BATF+, ST2+; T-bet-, RORγt-, GATA3-; | IL-2, IL-7, IL-15, IL-6, IL-12, STAT5, ZNF35, SP2, TEF; | IL-2, IL-7, IL-23, CD5L, IL-1β, IL-1R1, GM-CSF, STAT4, STAT5, BATF, NLRP3, Rel; | TGF-β, IL-6, IL-12, IL-1β, IL-23, IFN-γ, IL-13, IL-21; 5A12, MTX, IFN-β, STAT3, RORγt, Treg, DMF; | IL-4, IL-12, IFN-γ, Treg | Eosinophils, ly6C+CCR2+MCs, Neutrophils, DCs | MS, EAE, EAU, RA, T1D, IBD, SpA, Psoriasis | [12], [14], [15], [19], [20], [21], [22], [23], [27], [84], [28], [30], [32], [35], [34], [38], [42], [50], [53], [86], [88], [109], [114], [112], [119], [120] |
| GM-CSF+ IFN-γ+ Th cells (GM-CSF+ Th1-like) | GM-CSF+ only Th cells | Markers: GM-CSF+, IFN-γ+, TNF-α+, IL-2+, IL-2RA+, CXCR4+, CD2+, CD154+, CD161+; IL-17A-, CCR7-, CD25-, CD27-, CD137-; IL-3+/-, CCL20+/-, Transcriptional factors: T-bet+, ZNF35+, SP2+, TEF+, RUNX3+, RORC2+, BHLHE40+, TNFRSF9+, RORγt-; | Markers: GM-CSF+, IFN-γ+, TNF-α+, CCR1+, CX3CR1+, CD11a+, CD49d+, CD54+; IL-17A-; Transcriptional factors: T-bet+, RUNX3+; RORγt-; | IL-12, T-bet; | IL-7, IL-12, RUNX3, T-bet, Bhlhe40; | IL-23, IL-1β, IL-6, TGF-β, RORγt; | IL-6, IL-12, IL-21, IL-23, IFN-γ, TGF-β; | ly6C+CCR2+MCs, lymphocytes, neutrophils, macrophages, DCs | MS, EAE, MS; RA, JIA, T1D, IBD, SpA, GN; | [11], [12], [13], [20], [21], [22], [23], [26], [27], [84], [31], [32], [33], [35], [37], [38], [42], [47], [50], [53], [71], [86], [88], [98], [109], [110], [113], [114], [118], [122] |
| Th1 | Markers: GM-CSF+, IFN-γ+, TNF-α+, CCR4+, CCR6+, CCR10+, CXCR3+, IL-17A-, Transcriptional factors: T-bet+, RORγt-, | Markers: GM-CSF+, IFN-γ+, TNF-α+, CXCR6+; IL-17A-; Transcriptional factors: T-bet+, Bhlhe40+; RORγt-; | IL-12, STAT4; | IL-7, IL-12, IFN-γ, T-bet, Bhelhe40; | IL-1β, IL-6, IL-23, IL-21, IL-27, TGF-β, RORγt, STAT3 | IL-10, IL-23; | ||||
| GM-CSF+ IL-17+ Th cells | Th17 | Markers: GM-CSF+, TNF-α+, IL-17A+, IFN-γ-; Transcriptional factors: RORγt+, T-bet-; | Markers: GM-CSF+, IL-17A+, IL-17F+, IL-21+, IL-22+, TNF-α+, CCL20+, CCR4+, GPR65+; IFN-γ-, Transcriptional factors: Bhlhe40+, RORγt+, BATF+, T-bet-; | IL-12, GRP65, IL-15, IL-23; | IL-12, IL-7, IL-15, IL-1β, IL-23, IL-17, RORγt, Bhelhe40; | IFN-γ, Ustekinumab | IL-10, IL-12, IL-27, IFN-γ, TGF-β; | neutrophils, ly6C+CCR2+MCs, DCs; | EAE, MS, MS Marburg, CIA, T1D, IBD, SpA; | [11], [12], [13], [14], [21], [22], [23], [26], [28], [30], [31], [32], [33], [35], [37], [38], [42], [71], [97], [98], [108], [109], [110], [113], [115], [119] |
| GM-CSF+ IL-17+IFN-γ+ Th cells | GM-CSF+ IL-17+ Th cells | Markers: GM-CSF+, IL-2+, IFN-γ+, TNF-α+, IL-17A+; Transcriptional factors, RORγt+, T-bet+, BHLHE40+ | Markers: GM-CSF+, IFN-γ+, IL-17A+, TNF-α+; Transcriptional factors: RORγt+, T-bet+, Bhlhe40+ | IL-12, BHELHE40 | IL-7, IFN-γ, IRF1, Bhlhe40 | Ustekinumab | neutrophils, ly6C+CCR2+MCs, DCs | EAE, MS, MS Marburg, CIA, T1D, IBD, SpA | [12], [21], [26], [33], [38], [42], [108], [110] | |
†, inflammatory cells recruited by GM-CSF+ Th cells by subtypes. DCs, dendritic cells; MCs, monocytes;
*, Autoimmune diseases associated with GM-CSF+ Th cells by subtypes. CIA, collagen-induced arthritis; EAE, experimental autoimmune encephalomyelitis; EAU, experimental autoimmune uveoretinitis; GN, glomerulonephritis; IBD, inflammatory bowel disease; JIA, juvenile idiopathic arthritis; RA, rheumatoid arthritis; SpA, spondyloarthritis; T1D, type 1 diabetes;
Regardless of their classification, their pathogenic importance is undisputed. Functional studies have revealed that GM-CSF+ Th cells act as powerful “amplifiers” of immune responses. They engage in autocrine feed-forward loops where GM-CSF promotes its own expression, and they participate in cross-regulatory networks that enhance the function of other effector T cell subsets [19, 20]. Their indispensable role was definitively demonstrated in preclinical models of autoimmunity, such as experimental autoimmune encephalomyelitis (EAE). Genetic deletion of Csf2 (the gene encoding GM-CSF) specifically in CD4+ T cells renders mice resistant to EAE, and this protection is reversed by the adoptive transfer of GM-CSF+ Th cells, confirming their non-redundant pathogenic function [21].
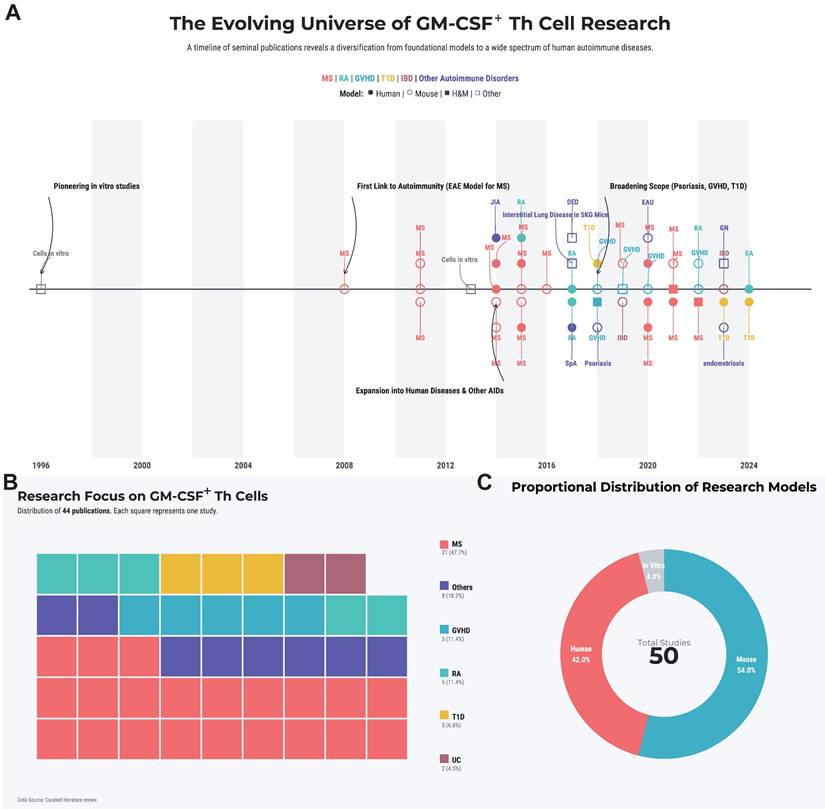
This review will systematically dissect the biology of these pivotal cells. We will synthesize the current understanding of their molecular identity and the complex regulatory networks governing their differentiation. Furthermore, we will analyze their specific pathogenic roles across the spectrum of major autoimmune diseases and, based on this evidence, critically evaluate emerging therapeutic strategies that target this axis. By integrating foundational research with translational prospects, we aim to provide a comprehensive theoretical framework for the future of precision intervention in autoimmunity. A landscape summary of the systematically retrieved literature is provided in Figure 1A-C and Table S1.
Literature landscape of GM-CSF+ Th cells in autoimmunity. (A) The historical trend of publications, with key landmark studies highlighted by black arrows. (B) A treemap illustrating the spectrum of autoimmune diseases studied in the literature. Each colored square denotes a single study, categorized by disease. (C) Distribution of study subject types. The area of each circle is proportional to the number of studies conducted on that subject type. Abbreviations: DED, dry eye disease; EAU, experimental autoimmune uveoretinitis; GN, glomerulonephritis; GVHD, graft-versus-host disease; ILD, interstitial lung disease; JIA, juvenile idiopathic arthritis; MS, mutiple sclerosis; RA, rheumatoid arthritis; SpA, spondyloarthritis; T1D, type 1 diabetes; UC, ulcerative colitis.
2. Molecular Characteristics and Differentiation Regulation of GM-CSF+ Th Cells
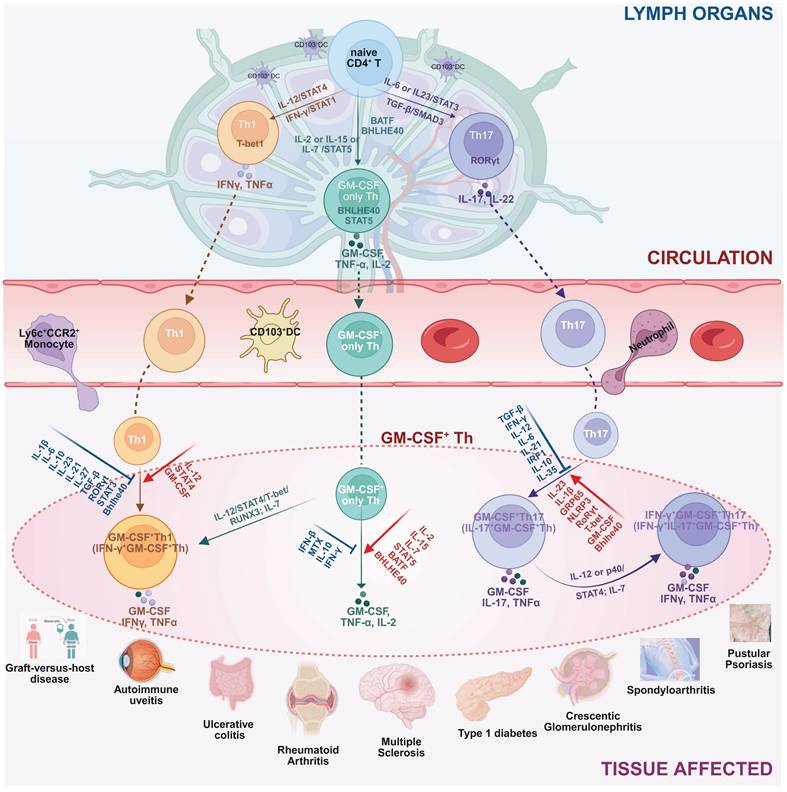
The complex molecular aspects of GM-CSF+ Th cells, including differentiation pathway, subtype heterogeneity, and plasticity of GM-CSF+ Th cells, plus a collection of related driving and suppressing factors, as well as associated diseases, are graphically summarized in Figure 2.
The differentiation pathway, subtype heterogeneity, and phenotype plasticity of GM-CSF+ Th cells, plus a collection of related driving and suppressing factors, as well as associated diseases. Illustrated in the diagram the generation and functional specialization of GM-CSF⁺ Th cells, beginning in the lymph organs where naive CD4⁺ T cells differentiate into Th1, Th17, or “GM-CSF only” Th lineages, driven by distinct cytokine signals (e.g., IL-12, IL-6, IL-2). After entering circulation, these cells migrate to peripheral tissues where they exhibit significant plasticity. In the inflammatory tissue microenvironment, Th1 and Th17 cells can be induced to produce GM-CSF, becoming pathogenic GM-CSF⁺ Th1 and GM-CSF⁺ Th17 cells. A further shift can occur where GM-CSF⁺ Th17 cells also gain the ability to produce IFN-γ, creating a highly inflammatory hybrid phenotype. Shown the specific cytokines and transcription factors that promote (red arrows) or suppress (blue T-bars) these states, and the resulting pathogenic cell subtypes were linked to a spectrum of human inflammatory and autoimmune diseases, including multiple sclerosis, rheumatoid arthritis, and psoriasis. Created in BioRender. Meng, X. (2025) https://BioRender.com/o6m32us.
2.1 Molecular diversity and disease specificity of GM-CSF+ Th cells
GM-CSF+ Th cells represent a highly heterogeneous subset of helper T cells whose molecular characteristics exhibit significant disease-specific characteristics despite sharing common features. All GM-CSF+ Th cell subsets can be defined by high expression of GM-CSF as their core characteristic, while frequently co-expressing TNF-α, IL-2, and IL-3. Some subsets may further co-express IFN-γ or IL-17A, indicating diverse pro-inflammatory effects when sub-grouped per common pathogenic factors [14, 22, 23]. Categorizations of GM-CSF+ Th cells vary across studies, but based on cellular characteristics, GM-CSF+ Th cells primarily fall within four subtypes, namely the GM-CSF+ only Th cells, GM-CSF+IFN-γ+ Th cells (i.e., the Th1-like), GM-CSF+IL-17+ Th cells (i.e., the Th17-like), and GM-CSF+IFN-γ+IL-17+ Th cells (i.e., the triple-positive or Th1/Th17-like).
In humans, Th1-like cells constitute the dominant GM-CSF⁺ Th subset. IL-12 promotes GM-CSF expression in these cells, while IL-1β, IL-6, and IL-23 may exert context-dependent suppressive effects on GM-CSF secretion, potentially as part of feedback or resolution mechanisms in inflammatory environments. Previous studies have revealed that the transcription factor regulatory network involved in autoimmune diseases exhibits several conserved features, prominently including members of the STAT family (e.g., STAT3, STAT4, and STAT5), the RUNX family (e.g., RUNX1 and RUNX3), and BHLHE40. Notably, BHLHE40 has been shown to directly bind to the CSF2 promoter, thereby promoting GM-CSF expression, while the RUNX family plays a key role in regulating cellular plasticity, enabling immune cells to transition between functional states relevant to autoimmune pathogenesis [24-26]. Notably, genome-wide chromatin accessibility analysis has revealed unique DNA accessibility regions and gene expression patterns in GM-CSF+ Th cells, suggesting epigenetic determinants in their differentiation [27]. Additionally, DNA methylation analysis shows that hypomethylation of the GM-CSF promoter region promotes GM-CSF secretion in GM-CSF+ Th cells, further suggesting the critical regulatory role of epigenetic modifications in determining the GM-CSF+ Th phenotype [27]. From a translational perspective, these findings provide a theoretical basis for developing targeted therapies through epigenetic regulation, suggesting possible effects of DNA methyltransferase inhibitors or histone deacetylase inhibitors.
Regarding disease specificity, GM-CSF+ Th cells may exhibit differential characteristics in various pathological backgrounds. For example, GM-CSF+ Th cells in MS and type 1 diabetes (T1D) often co-express both the Th1 (e.g., T-bet and CXCR3) and Th17 (e.g., RORγt and CCR6) markers, forming the so-called “Th1/Th17 hybrid subsets”, namely the triple-positive phenotype (IFN-γ+IL-17A+GM-CSF+) [13, 22, 28]. In contrast, GM-CSF+ Th cells in RA basically manifest the Th1-like characteristics, co-expressing IFN-γ+ and TNF-α+, while in certain cases become negative in Th1 and Th17 markers but exclusively secretes GM-CSF and IL-21 [29]. Particularly, GM-CSF+ Th cells in IBD predominantly exhibit a single GM-CSF+ phenotype without expressing any markers of the other Th subsets such as IL-17 or IFN-γ, but in certain cases may co-express TNF-α and IL-2, suggesting functional distinctiveness from traditional Th1/Th17 cells [12, 30]. Furthermore, evident cross-species differences are observed in MS-associated GM-CSF+ Th cells, with a Th1-biased phenotype in humans, while a Th17-like phenotype has been observed as predominant in mouse models [31, 32].
2.2 Conserved and species-specific regulation of GM-CSF+ Th differentiation
The differentiation of GM-CSF⁺ T helper cells follows a principle of “shared regulatory frameworks with disease- and species-specific modulation”. Core pro-differentiation pathways across contexts include the IL-12/STAT4, IL-23/RORγt, and IL-7/IL-2/STAT5 axes, which broadly support the generation and stabilization of GM-CSF-expressing subsets. Negative regulators such as TGF-β, IFN-γ, and IL-10 generally constrain GM-CSF production to prevent excessive inflammation [12, 13, 21]. For example, IL-23 and IL-1β can induce GM-CSF production in helper T cells in a dose-dependent manner, whereas IFN-γ, IL-12, and TGF-β exert inhibitory effects [13, 14, 19, 20]. The differentiation of GM-CSF⁺ T helper cells is governed by a conserved transcriptional network involving STAT3/4/5, BHLHE40, and RUNX1/3. STAT5 mediates IL-2/IL-7 signals to promote the GM-CSF-producing phenotype, while BHLHE40 directly binds the CSF2 promoter to enhance transcription. RUNX factors contribute to T cell plasticity and effector programming. In autoimmune diseases such as MS and T1D, GM-CSF⁺ Th cells frequently co-express T-bet and RORγt, forming hybrid Th1/Th17-like subsets with potent pro-inflammatory capacity [20, 21, 27, 31-33].
Nevertheless, disease-specific heterogeneity has been repeatedly reported. In MS, human cases rely more on the IL-12/STAT4 pathway driving the Th1-like phenotype (T-bet+), while mouse models frequently exhibit a Th17-like phenotype predominantly driven by IL-23/RORγt [11, 14, 28]. In contrast, GM-CSF+ Th cells in RA primarily differentiate into Th1-like phenotypes by IL-12/STAT4 or form a GM-CSF+IL-21+ subsets through CD147 activation [29]. The BHLHE40-driven triple-positive phenotype (IFN-γ+IL-17A+GM-CSF+) in T1D and the IL-1β-driven transdifferentiation from Th17 to GM-CSF+ Th in IBD further suggest the strong impact of disease microenvironments on differentiation pathways [30, 33].
Difference between humans and mice involves not only preferences for core pathways but also regulatory factor specificity. For example, IL-2RA polymorphisms directly affect GM-CSF secretion in human MS, while the synergistic effect of IL-7 and IL-2 plays a more critical role in mice [34, 35]. Additionally, the regulatory impact of Bhlhe40 on GM-CSF is significantly stronger in mouse models than in human subjects, suggesting possible species-specific compensatory mechanisms [26]. Notably, although the IL-7/IL-2/STAT5 axis is evolutionarily conserved, IL-2 alone can drive GM-CSF secretion in humans, whereas in mice it is prerequisite a synergistic action of both IL-7 and IL-2. This difference is potentially related to species difference in IL-2R expression levels and STAT5 phosphorylation efficiency [21, 36].
2.3 Interplay Between heterogeneity and plasticity in GM-CSF+ Th cells
The pathogenic identity of GM-CSF-producing Th cells is defined not by a static phenotype, but by a dynamic interplay between their inherent heterogeneity and profound plasticity. This heterogeneity is evident at multiple molecular levels. In terms of cytokine profiles, beyond their hallmark GM-CSF secretion, these cells can co-express a range of effector molecules including TNF-α, IL-2, and IL-3. Depending on the microenvironment, they polarize into distinct functional subsets, such as IFN-γ-producing (Th1-like), IL-17A-producing (Th17-like), or highly pro-inflammatory hybrid subsets expressing both [14, 32]. This functional diversity is underpinned by a flexible transcriptional network, where combinations of master regulators like T-bet and RORγt drive specific phenotypes in diseases like MS, while factors such as BHLHE40 can dictate unique triple-positive signatures in T1D [31, 33].
This spectrum of heterogeneity is not pre-determined but is actively and continuously shaped by the cells' remarkable plasticity. The core “rheostat” governing this plasticity is the dynamic balance of the IL-12 and IL-23 signaling axes. The IL-12/STAT4 pathway preferentially drives the Th1-like phenotype, prevalent in MS, while the IL-1β/IL-23/RORγt axis promotes the Th17-like phenotype often seen in other contexts [11, 37]. Furthermore, the IL-7/IL-2/STAT5 axis plays a crucial role in maintaining this plastic potential, enabling cells to adopt different fates, such as the GM-CSF single-positive subset in GVHD [20, 26, 38]. This foundational plasticity is then fine-tuned by disease-specific signals from the local microenvironment, for example, the IL-1β/NF-κB pathway in inflammatory bowel disease or CD147 signaling in rheumatoid arthritis, which ultimately sculpts the final pathogenic phenotype [29, 30].
While the foundational role of GM-CSF+ Th cells in autoimmunity is now established, several critical questions remain that will define the next phase of research.
First, moving beyond conserved pathways, how do disease-specific microenvironments, such as the gut microbiome in IBD, metabolic shifts in T1D, or the unique cytokine milieu of a tumor, precisely sculpt the phenotype and function of these cells? Understanding this context-dependent regulation is key to explaining clinical heterogeneity. Second, the significant discrepancies between human disease and murine models, particularly in the dominant Th1-like versus Th17-like polarization, represent a major translational hurdle. This necessitates not only large-scale clinical validation using patient samples but also the development of more sophisticated humanized models to bridge this gap. Finally, how do GM-CSF+ Th cells orchestrate the broader immune landscape? Elucidating their intricate crosstalk with other key players, such as regulatory T cells that restrain them and the diverse myeloid cell populations they activate, is essential to fully comprehend their net pathogenic impact.
In summary, GM-CSF+ Th cells function as highly adaptable “inflammatory amplifiers” within the autoimmune network. A deeper understanding of their heterogeneity and plasticity is the prerequisite for designing truly personalized therapies. The integration of multi-omics and single-cell analysis technologies will be instrumental in unraveling these complex dynamics, ultimately enabling us to target the right pathogenic subset, in the right patient, at the right time.
3. The Mechanistic Role of GM-CSF+ Th Cells in Pathogenesis of Various AIDs
3.1 Role of GM-CSF+ Th cells in multiple sclerosis
Multiple sclerosis (MS) is a chronic, immune-mediated demyelinating disease of the central nervous system (CNS), characterized by inflammation, myelin destruction, axonal loss, and neurodegeneration. Globally, MS affects an estimated 2.8 million individuals, with a prevalence of approximately 35.9 cases per 100,000 people, and shows striking geographical variation, being more common in high-latitude regions. Women are disproportionately affected, with a female-to-male ratio of approximately 3:1 [39]. The disease typically manifests in young adulthood (20-40 years of age), and its pathogenesis involves a complex interplay of genetic susceptibility, environmental triggers such as Epstein-Barr virus (EBV) infection and vitamin D deficiency, and aberrant immune regulation [40].
Historically, research into MS pathogenesis focused on the classic Th1 (IFN-γ+) and Th17 (IL-17+) lineages. However, their precise contributions were confounded by paradoxical observations. For instance, both genetic knockout of IFN-γ and antibody-mediated neutralization unexpectedly exacerbated experimental autoimmune encephalomyelitis (EAE), the primary mouse model for MS [8, 9]. Clinically, some MS patients experienced disease worsening following IFN-γ therapy [41]. Similarly, neither T-cell-specific IL-17A overexpression nor its complete deletion significantly altered susceptibility or the expansion of autoreactive T cells in murine EAE [42]. These findings strongly suggested that the traditional Th1/Th17 paradigm was incomplete and that other critical, non-redundant mediators were essential for driving CNS autoimmunity.
The pivotal role of GM-CSF emerged from foundational studies. As early as 2001, McQualter et al. discovered that mice genetically deficient in GM-CSF (i.e., by genetically Csf2-knockout) were completely resistant to myelin oligodendrocyte glycoprotein (MOG)-induced EAE. Furthermore, when anti-GM-CSF monoclonal antibody is administered at the time of antigen challenge (i.e., during immunization with MOG₃₅₋₅₅ peptide), it can prevent clinical disease onset during the treatment period and for 10 days after discontinuation. However, when intervention occurs after disease establishment (i.e., after clinical symptoms occur and the scoring reaches two), it only alleviates symptoms, allowing mice to fully recover within 20 days after treatment [43]. This foundational discovery implicated GM-CSF as a critical driver of disease initiation and an auxiliary factor in later stages. A subsequent landmark study in 2008 by Mark et al. directly linked this pathogenicity to T cells. They demonstrated that Th17-polarized cells required GM-CSF expression to become encephalitogenic, as adoptive transfer of GM-CSF+ Th17 cells was sufficient to induce EAE in naive recipients [44]. These studies collectively established GM-CSF as a key, non-redundant pathogenic factor in EAE. The cytokine GM-CSF thus emerged as a non-redundant driver of CNS autoimmunity. Supporting this, McQualter et al. had earlier shown that Csf2⁻/⁻ mice were entirely resistant to MOG-induced EAE, and anti-GM-CSF antibodies administered during immunization blocked disease initiation but were less effective after disease onset. The pathogenic role of GM-CSF appears to be dynamic across the course of MS. It is considered a critical driver of early disease initiation, while transitioning to an auxiliary yet persistent function in sustaining inflammation during later stages. This is strongly supported by clinical cohort studies, which consistently demonstrate a significant correlation between elevated GM-CSF levels and disease activity [14, 35, 45-50].
Subsequent research confirmed that CD4+ T cells are the predominant source of pathogenic GM-CSF in both MS and EAE. In MS patients, the frequency of GM-CSF+ Th cells is elevated in peripheral blood and the CNS during active disease and decreases during remission following IFN-β therapy, correlating strongly with disease activity [35, 47, 51]. This clinical observation was mechanistically validated in the FROG (fate-map and reporter of GM-CSF expression) mouse model, which enabled real-time tracking of GM-CSF-expressing cells. Komuczki et al. found that while various immune cells produced GM-CSF in peripheral lymph nodes post-immunization, CD4+ T cells were the dominant source within the CNS inflammatory milieu [20]. This finding, supported by numerous studies [20, 23, 43, 52]. solidifies that CNS pathology in EAE is primarily driven by GM-CSF produced by Th cells. Genetic evidence further reinforces this link, as an MS-associated polymorphism in the IL2RA gene (rs2104286) correlates with a higher proportion of GM-CSF+ Th cells [35], and IL-2 signaling is known to promote their differentiation.
Early studies described GM-CSF as a co-effector cytokine in Th1 and Th17 cells [31]. However, subsequent work demonstrated that GM-CSF expression, not IL-17 or IFN-γ, was essential for encephalitogenicity. El-Behi et al. showed that IL-1β and IL-23 induced GM-CSF, rather than IL-17, to confer pathogenicity to Th17 cells [11]. Similarly, Codarri et al. reported that RORγt promoted GM-CSF expression independently of IL-17 [14].
A paradigm shift occurred in 2014 when Sheng et al. identified GM-CSF+ Th cells as a distinct, independent pathogenic lineage regulated by an IL-7-STAT5 axis, which they termed “ThGM”. In their model, conditional deletion of STAT5 in CD4+ T cells abrogated EAE by specifically preventing the development of ThGM cells, without affecting Th1, Th17, or Treg populations [21]. Mice lacking STAT5 in CD4⁺ T cells failed to develop EAE despite intact Th1/Th17/Treg compartments, underscoring the indispensable role of GM-CSF⁺ Th cells.
Further studies identified GM-CSF⁺ Th subsets in MS patients that lacked IFN-γ and IL-17 but were dependent on IL-2/STAT5 signaling [13]. IL-12 could induce IFN-γ expression in these cells, producing a GM-CSF⁺IFN-γ⁺ Th1-like phenotype [53]. FROG mouse studies confirmed the presence of CNS-infiltrating ThGM cells in EAE, although many converted to IFN-γ⁺TNF-α⁺ “ex-GM-CSF⁺” cells over time [20, 23, 32]. Spath et al. validated the pathogenicity of GM-CSF⁺ Th cells directly, as mice receiving GM-CSF-overexpressing CD4⁺ T cells developed spontaneous CNS inflammation, unlike those receiving IL-17-overexpressing cells [49]. Research also found that the relatively EAE-resistant Albino Oxford (AO) rats showed restricted production, differentiation, and proliferative capacity of neural antigen-specific GM-CSF+ Th cells in their draining lymph nodes (dLNs), while EAE-susceptible Dark Agouti (DA) rats presented more pronounced GM-CSF+ Th cell-associated inflammatory responses in spinal cord (SC) tissues [54].
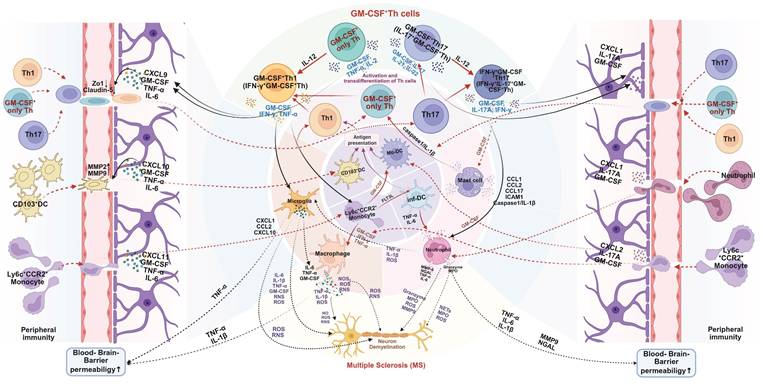
These studies offer significant insights into the immunopathogenesis of MS and its murine model-EAE. Importantly, it is not merely that Th1 or Th17 cells act as passive carriers of GM-CSF-mediated pathogenicity. Rather, the acquisition of GM-CSF-producing capacity is a prerequisite for these T cell subsets to exert their full pathogenic potential in MS or EAE [55-57]. In addition to Th1 and Th17 cells, independent CD4⁺ T cell subsets that produce high levels of GM-CSF, without expressing classic Th1 or Th17 markers, can also induce EAE, highlighting a broader spectrum of GM-CSF+ effector T cells. However, such findings from animal models should be interpreted with caution. Human MS pathology is multifaceted, and both Th1 and Th17 cells are widely distributed in MS lesions [58-60]. Furthermore, the plasticity of CD4⁺ T cells allow for dynamic phenotype switching. For instance, IL-12 can induce IFN-γ expression in previously GM-CSF-only T cells, while Th1 and Th17 cells can acquire GM-CSF-producing ability under specific inflammatory conditions. This suggests that the actual pathogenic network in vivo involves a flexible and interconvertible pool of effector T cells, rather than fixed Th lineages [61]. Despite the heterogeneity of contributing cell types, likely differing between individuals, GM-CSF secretion consistently emerges as a critical mediator of neuroinflammation. Thus, future research should prioritize elucidating the regulatory networks controlling GM-CSF production in CD4⁺ T cells. Special attention should be given to the distinctions between GM-CSF⁺ Th1, Th17, and non-classical effector subsets, and Treg, to identify novel therapeutic targets for preventing their differentiation or blocking GM-CSF secretion [62-64]. Mechanistically, GM-CSF+ Th cells orchestrate CNS destruction through a multi-pronged assault (Figure 3). After differentiating in peripheral lymphoid organs, they cross the blood-brain barrier (BBB). Their secreted GM-CSF compromises BBB integrity by downregulating tight junction proteins, facilitating the infiltration of pro-inflammatory myeloid cells (e.g., CCR2+Ly6Chi monocytes) and other lymphocytes [64-66]. Within the CNS, GM-CSF acts as a master regulator, promoting the differentiation of monocytes into inflammatory macrophages and activating microglia. These myeloid cells, in turn, release a barrage of destructive mediators, including pro-inflammatory cytokines (e.g., IL-1β, IL-6, TNF-α), reactive oxygen species (ROS), and reactive nitrogen species (RNS), which directly damage myelin and neurons [14, 43, 60, 67-72]. GM-CSF also supports DC activation, autoantigen presentation, and Th cell proliferation in the CNS [73]. This process creates a self-amplifying inflammatory loop, as GM-CSF also supports the survival and proliferation of the pathogenic Th cells themselves in an autocrine/paracrine manner, sustaining chronic inflammation [43, 67].
Mechanistic role of GM-CSF+ Th cells in pathogenesis of multiple sclerosis (MS). GM-CSF⁺ Th cells are pivotal in driving MS pathology. They initiate the disease process by increasing blood-brain barrier (BBB) permeability, allowing for the infiltration of peripheral immune cells into the central nervous system (CNS). Within the CNS, these Th cells orchestrate a self-amplifying neuroinflammatory cascade. They exhibit significant plasticity, such as IL-12-driven transdifferentiation into more pathogenic subtypes, and potently activate resident microglia and recruited myeloid cells (e.g., macrophages, neutrophils). This activated myeloid network releases a barrage of neurotoxic mediators, including cytokines, reactive oxygen species (ROS), and proteases, that directly cause the demyelination and neuronal damage characteristic of MS. Created in BioRender. Meng, X. (2025) https://BioRender.com/o6m32us.
Clinically, MS pathology predominantly features macrophages with accompanying neutrophils [74]. While pathophysiological heterogeneity is well-recognized across MS subtypes, such as Relapsing-Remitting (RRMS) and Progressive MS (PMS), a starkly different profile is observed in certain fulminant demyelinating disorders. For instance, Marburg's variant, historically misclassified as a rare MS subtype, is now predominantly considered a severe phenotype of MOG antibody-associated disease (MOGAD). It is pathologically characterized by extensive neutrophilic infiltration into lesions, leading to the release of matrix metalloproteinases (e.g., MMP-9) and ROS. This cascade results in widespread BBB disruption, fulminant demyelination, and an exceptionally rapid clinical progression driven by a hyperacute immune response [75, 76]. EAE induced by GM-CSF⁺IFN-γ⁺ Th1-like cells is macrophage-predominant, resembling classical MS, while GM-CSF⁺IL-17⁺ Th17-like cells induce neutrophil-rich inflammation similar to Marburg MS [44, 77]. This suggests that the specific sub-lineage of GM-CSF+ Th cells (Th1-like vs. Th17-like) can dictate the character of the resulting neuroinflammation. While GM-CSF+ Th1-like cells appear predominant in standard MOG-induced EAE models the balance between these subsets likely varies among human MS patients, contributing to clinical heterogeneity [20, 37].
A recent study reported that the frequency of GM-CSF⁺ Th17-like cells in C57BL/6 mice immunized with MOG positively correlates with EAE severity, suggesting that this specific Th subset may possess heightened encephalitogenic potential. These findings imply that the abundance of GM-CSF⁺ Th17-like cells could serve as a key determinant of disease intensity [11].
In clinical studies involving MS patients, the relative distribution of IFN-γ⁺GM-CSF⁺ versus IL-17⁺GM-CSF⁺ Th cells varies significantly across cohorts, indicating heterogeneity in the GM-CSF⁺ Th cell compartment among individuals. This observed diversity underscores the importance of conducting future clinical investigations using stratified analyses with adequately powered sample sizes to derive more definitive conclusions. Moreover, notable discrepancies exist between findings from EAE models and clinical observations in MS. For instance, while anti-IFN-γ monoclonal antibody treatment exacerbates disease in EAE models, administration of IFN-γ in some MS patients has been associated with clinical deterioration [11, 44]. These contrasting outcomes suggest that IFN-γ may exert divergent effects in murine versus human systems. One potential explanation lies in the differential composition of Th cell subsets and the context-dependent influence of cytokine milieu, particularly IL-12 and IL-23, on Th cell plasticity and GM-CSF expression. In EAE, where Th17 cells may be more prominent in the CNS microenvironment, IFN-γ may act as a suppressor of GM-CSF secretion in GM-CSF⁺ Th17-like cells. Therefore, neutralizing IFN-γ in this context may relieve this suppression, enhance GM-CSF production, and exacerbate disease severity. Conversely, in MS patients whose CNS pathology is dominated by GM-CSF⁺ Th1-like cells, exogenous IFN-γ may amplify inflammatory cascades. These mechanistic nuances highlight the necessity for personalized treatment strategies that account for patient-specific Th subset profiles and cytokine dynamics, rather than applying uniform therapeutic approaches [78-80].
As illustrated in Figure 3, GM-CSF+ Th act as central orchestrators of MS and EAE pathogenesis through a combination of BBB disruption, activation of myeloid cells, induction of oxidative stress, and self-sustaining autocrine loops. Unlike the conventional Th1/Th17 paradigm, GM-CSF⁺ Th cells constitute a non-redundant and independently regulated effector subset. This distinct immunological identity opens new avenues for therapeutic intervention, including the use of anti-GM-CSF monoclonal antibodies and STAT5 inhibitors. Additionally, these findings reinforce the critical importance of timing in clinical intervention, particularly the stage of disease progression at which GM-CSF signaling is targeted. Nevertheless, the inconsistencies between EAE models and human MS, such as the opposing roles of IFN-γ, underscore the need for caution when extrapolating preclinical data to clinical practice. Tailored therapeutic strategies, guided by in-depth immunophenotyping and molecular profiling, may ultimately yield more effective and individualized treatment paradigms for MS.
Future research could prioritize several key areas. First, applying single-cell multi-omics technologies to dissect the dynamic heterogeneity and transcriptional or epigenetic regulatory networks of GM-CSF⁺ CD4⁺ T cells at high resolution. Second, refining therapeutic strategies, such as developing dual-targeted approaches that simultaneously inhibit GM-CSF and other pro-inflammatory cytokines (e.g., IL-17 or IFN-γ) implicated in MS pathogenesis. Third, improving animal models by constructing humanized mouse models that more faithfully replicate the phenotypic and functional characteristics of Th cell subsets observed in MS patients.
3.2 Role of GM-CSF+ Th cells in rheumatoid arthritis
Rheumatoid arthritis (RA) is a chronic autoimmune disease characterized by persistent synovitis, joint destruction, and systemic immune dysregulation, with a global prevalence of approximately 0.5-1%. Women are affected two to three times more often than men. The pathogenesis of RA involves a multifaceted interplay among genetic predisposition, environmental triggers, and aberrant immune responses [81]. Although biologics and small-molecule inhibitors have substantially improved therapeutic outcomes, complete remission remains elusive, and long-term complications such as cardiovascular disease continue to threaten patient prognosis [82]. Anti-citrullinated protein antibodies (ACPAs) can be detected 5-10 years before RA onset, but specific preventive targets and effective preventive measures are still lacking. Therefore, identifying key pathogenic factors is crucial for RA prevention. Recently, increasing attention has been directed toward GM-CSF⁺ Th cells and their secreted cytokine GM-CSF.
In a pioneering study using the collagen-induced arthritis (CIA) model, Ian K et al. demonstrated that GM-CSF-deficient (GM-CSF⁻/⁻) mice were resistant to arthritis, where heterozygous mice exhibited mild disease and wild-type mice developed severe arthritis, suggesting a dose-dependent contribution of GM-CSF to disease severity. Moreover, exogenous GM-CSF injection exacerbated joint damage, confirming GM-CSF's role as an endogenous pathogenic factor [83]. In RA patients, elevated levels of GM-CSF are detected both in peripheral blood and synovial fluid, predominantly secreted by CD4⁺ Th cells. Peripheral GM-CSF⁺ Th cells exhibit minimal co-expression of other cytokines, whereas intra-articular GM-CSF⁺ Th cells often co-produce TNF-α and, in ~80% of cases, also express IFN-γ. IL-12 enhances the co-secretion of GM-CSF and IFN-γ, implicating the Th1 differentiation pathway [84]. However, GM-CSF⁺IFN-γ⁺ Th cells in joints may not represent bona fide Th1 cells, but could instead derive from GM-CSF⁺ single-positive precursors in peripheral blood that acquire IFN-γ expression locally under IL-12 influence. Despite similar cytokine profiles, these cells may differ in origin and function, warranting further lineage-tracing investigation. In vitro studies corroborate that IL-12 can promote GM-CSF and IFN-γ production in human CD4+ T cells, while Th17-polarizing factors inhibit GM-CSF+ Th cell differentiation and GM-CSF secretion [13]. Clinically, methotrexate (MTX) treatment leads to a significant reduction of GM-CSF⁺ Th cells in peripheral blood, correlating with disease activity and indicating their potential as dynamic biomarkers [85]. Additionally, higher baseline proportions of GM-CSF⁺ Th cells are associated with superior therapeutic responses to TNF-α inhibitors, suggesting a predictive value for treatment stratification [86].
In the synovial fluid of RA patients, CD4⁺ T cells are the predominant source of GM-CSF, with significantly higher secretion than in healthy controls. GM-CSF+ Th cells promote monocyte differentiation into CD1c+ inflammatory dendritic cells (infDCs), which secrete pro-inflammatory cytokines such as IL-6 and TNF-α, thereby perpetuating inflammation through a feed-forward loop [87, 88]. These infDCs contribute to T and B cell activation, maintain chronic inflammation, and promote joint damage by enhancing neutrophil gelatinase-associated lipocalin (NGAL), impairing chondrocyte function, and activating synoviocytes. Furthermore, monocyte-derived dendritic cells (MoDCs) differentiated by GM-CSF can process RA-associated autoantigens, the type II collagen (CII) and cartilage glycoprotein 39 (HCgp39), and present their immunodominant epitopes via MHC class II molecules, initiating autoreactive T cell responses [89-92]. Taken together, GM-CSF⁺ Th cells drive multiple RA pathomechanisms, including inflammation, immune activation, and tissue damage, and are central players in sustained synovial pathology [43, 88]. Their accumulation in inflamed joints offers mechanistic insight into the chronicity of RA and identifies them, along with GM-CSF itself, as potential therapeutic targets [93].
Despite encouraging preclinical results, clinical trials targeting GM-CSF have yielded mixed outcomes. While early-phase studies of GM-CSF-neutralizing antibodies (e.g., mavrilimumab) demonstrated reductions in inflammatory biomarkers, many patients failed to achieve primary or key secondary endpoints in phase III trials [94]. This limited efficacy may reflect disease chronicity and refractoriness, as participants often had poor responses to prior synthetic and biologic disease-modifying antirheumatic drugs (DMARDs), including JAK inhibitors. Notably, mavrilimumab demonstrated greater efficacy in early RA, consistent with CIA model findings where early anti-GM-CSF intervention prevented disease onset, whereas late intervention only mitigated disease severity [95].
It is also essential to recognize fundamental interspecies differences in the biology of GM-CSF⁺ Th cells. In the SKG mouse model, GM-CSF augments macrophage production of IL-1β and IL-6 and promotes the expansion of both Th17 and GM-CSF⁺ Th cell populations [88]. Notably, anti-GM-CSF therapy in this model yields greater suppression of arthritis severity compared to IL-17 blockade, underscoring a more central role for GM-CSF in disease progression [95]. Mechanistically, murine Th17 cells are capable of producing GM-CSF upon IL-23 stimulation, and the GM-CSF⁺ Th17 phenotype is well-documented in experimental models of inflammatory arthritis. However, this immunological landscape diverges in human RA. Synovial tissues from RA patients typically exhibit low frequencies of IL-17-producing Th cells, and GM-CSF is predominantly produced by Th1-like CD4⁺ T cells co-expressing IFN-γ [96, 97]. This Th1-skewed GM-CSF⁺ subset appears to be the dominant effector population in human RA, as supported by recent single-cell transcriptomic and mass cytometry studies [84, 88]. Interestingly, recent investigations in SKG mice have identified IL-17A⁺GM-CSF⁺ Th cells enriched within inflamed joints and lymphoid tissues. Treatment with CCR4-targeted antagonists effectively suppressed their function, resulting in reduced synovitis and joint damage [97, 98]. However, the actual abundance of these double-positive cells remains relatively low, and their IFN-γ expression status has not been systematically assessed, necessitating further investigation into their heterogeneity and relevance. The limited efficacy of anti-IL-17A monoclonal antibodies in RA clinical trials (e.g., secukinumab [99], brodalumab [100], CNTO6785 [101]) further underscores human-mouse differences, and suggests that IL-17-associated pathways, while prominent in murine arthritis, may play a subordinate role in human RA. These findings collectively highlight the need for cautious interpretation of Th cell-associated mechanisms across species and support a more targeted focus on Th1-like GM-CSF⁺ cells in human RA pathogenesis and therapy.
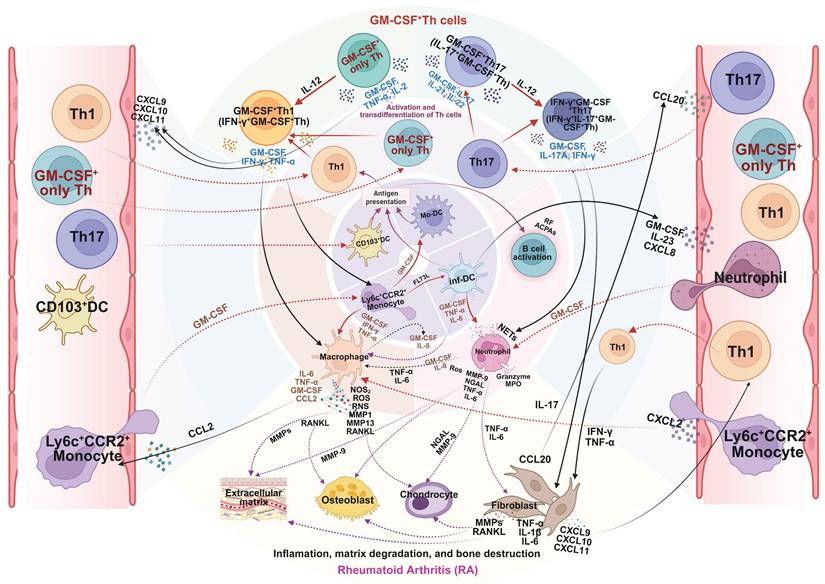
As summarized in Figure 4, GM-CSF+ Th cells function as a pivotal hub within the RA pathological network, amplifying inflammation, facilitating autoimmunity, and driving tissue destruction. Although current clinical strategies targeting GM-CSF are constrained by disease stage and patient heterogeneity, early intervention and combinatorial approaches merit further exploration. Future studies incorporating single-cell multi-omics and longitudinal tracking may refine patient stratification and optimize GM-CSF⁺ Th cell-centered therapeutic strategies.
Mechanistic role of GM-CSF+ Th cells in pathogenesis of rheumatoid arthritis (RA). GM-CSF⁺ Th cells are key drivers of RA pathology. After infiltrating the synovial joint, they undergo local reactivation and differentiation, orchestrating a destructive inflammatory network. By secreting GM-CSF and other cytokines, they activate macrophages, neutrophils, and synovial fibroblasts, while also promoting autoantibody production by B cells. This cellular crosstalk unleashes a cascade of pathogenic mediators such as NOS, ROS, MMPs, and RANKL, leading directly to the cartilage degradation and bone erosion damage of RA. Created in BioRender. Meng, X. (2025) https://BioRender.com/o6m32us.
3.3 Role of GM-CSF+ Th cells in miscellaneous autoimmune disorders
The critical role of GM-CSF+ Th cells in the pathogenesis of a wide variety of other autoimmune diseases, such as type 1 diabetes, ulcerative colitis, and spondyloarthritis, are graphically summarized in Figure 5.
The versatile mechanistic role of GM-CSF⁺ Th cells in the pathogenesis of miscellaneous autoimmune diseases. GM-CSF⁺ Th cells orchestrate a central inflammatory hub that drives a wide range of autoimmune diseases. By activating a common set of myeloid effector cells, including macrophages, neutrophils, and eosinophils, this pathway can induce shared while tissue-specific pathologies. The specific clinical manifestations, be it the β-cell destruction in type 1 diabetes, joint inflammation in spondylitis, epithelial damage in pustular psoriasis and ulcerative colitis, or renal failure in crescentic glomerulonephritis, is determined by the target organ and the precise downstream inflammatory mediators that are engaged. Created in BioRender. Meng, X. (2025) https://BioRender.com/o6m32us.
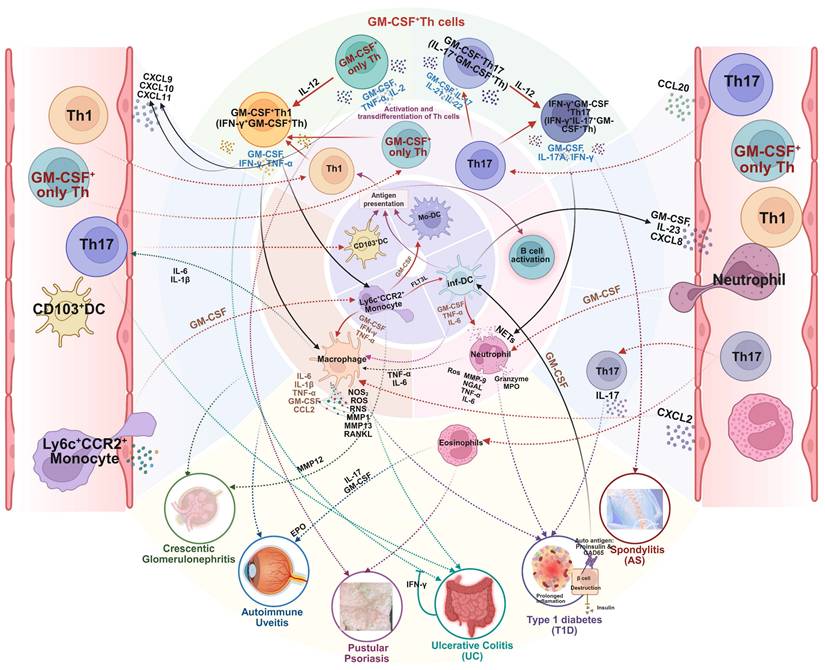
3.3.1 Role of GM-CSF+ Th cells in type 1 diabetes
Type 1 diabetes (T1D) is an autoimmune disorder characterized by progressive and irreversible destruction of pancreatic β cells, ultimately resulting in insufficient insulin production [102, 103]. CD4⁺ T cells are central to T1D pathogenesis, with early research identifying Th1 and Th17 subsets as key pathogenic drivers [104-107]. However, these studies were largely constrained by a limited understanding of CD4⁺ T cell heterogeneity, possibly overlooking functionally important subsets. Recent advances have highlighted GM-CSF and GM-CSF⁺ Th cells as critical contributors to autoimmune pathology, including T1D [33, 106-109].
Emerging evidence suggests that GM-CSF⁺ Th cells play a central role in mediating β cell destruction in T1D. In patients with T1D, autoreactive CD4⁺ T cells targeting self-antigens such as proinsulin and glutamic acid decarboxylase 65 (GAD65) are elevated, with a significant increase in GM-CSF⁺ Th cells among them. These cells contribute to disease progression by exerting multiple pro-inflammatory effects via GM-CSF secretion. On one hand, GM-CSF promotes recruitment of immune cells—including macrophages and neutrophils—into pancreatic islets, enhancing local immune-mediated injury. On the other hand, GM-CSF facilitates Th17 polarization and IL-17 production, amplifying chronic inflammation and accelerating β cell destruction [110].
Moreover, GM-CSF⁺ Th cells often co-express IL-17A and IFN-γ, forming a pathogenic feed-forward loop wherein these pro-inflammatory cytokines mutually reinforce their expression. This loop intensifies local inflammation and directly contributes to β cell dysfunction and insulin insufficiency. Clinical studies show that T1D patients exhibit significantly higher peripheral levels of GM-CSF⁺ Th cells compared to healthy controls, correlating positively with plasma IL-17A, IFN-γ, and IL-2 levels [33, 110].
Findings from animal models and clinical studies further support the pathogenic role of GM-CSF⁺ Th cells in T1D. In clinical trials, T1D patients responding to alefacept therapy showed a marked reduction in circulating GM-CSF⁺ Th cells. Notably, baseline levels of these cells inversely correlated with post-treatment C-peptide retention, suggesting their potential as predictive biomarkers for therapeutic response [33, 108]. Mechanistically, GM-CSF⁺ Th cells activate inflammatory dendritic cells (infDCs), enhancing their antigen-presenting capacity and breaking immune tolerance, ultimately leading to irreversible β cell destruction [110]. In addition to producing GM-CSF, these Th cells release high levels of IL-17A, IFN-γ, and IL-2, establishing a pro-inflammatory islet microenvironment that perpetuates autoimmunity.
Although the regulatory mechanisms underlying GM-CSF⁺ Th cell differentiation remain incompletely understood, IL-23 has been implicated in promoting their expansion through specific signaling pathways. Ustekinumab, a monoclonal antibody targeting the p40 subunit shared by IL-12 and IL-23, has demonstrated efficacy in reducing GM-CSF⁺ Th cell numbers and alleviating β cell loss in T1D models [108]. These findings indicate that ustekinumab may offer therapeutic benefit by modulating GM-CSF⁺ Th cell-mediated inflammation.
Further exploration of the transcriptional regulation and microenvironmental interactions of GM-CSF⁺ Th cells within pancreatic islets may uncover novel therapeutic targets. In particular, dual-targeted strategies that simultaneously address inflammatory signaling and immune cell migration hold promise for improving clinical outcomes in T1D. Despite ongoing challenges, GM-CSF⁺ Th cells are increasingly recognized as central amplifiers of inflammation and autoimmunity in T1D, offering a new immunological axis for therapeutic innovation.
3.3.2 Role of GM-CSF+ Th cells in ulcerative colitis
Ulcerative colitis (UC) is a chronic inflammatory bowel disease (IBD) affecting millions worldwide, significantly reducing quality of life and increasing the risk of colorectal cancer. While its immune network is complex, recent research has identified GM-CSF-producing Th cells as key pathogenic drivers that amplify intestinal inflammation. This view is supported by foundational evidence from dextran sodium sulfate (DSS)-induced colitis models, where mice genetically deficient in GM-CSF (Csf2-/-) exhibit markedly reduced intestinal inflammation and less severe disease, establishing the necessity of GM-CSF in this pathogenic process [12].
Mechanistically, GM-CSF+ Th cells orchestrate intestinal damage through at least two interconnected pathways. First, they directly activate myeloid cells, such as macrophages, through the secretion of GM-CSF. These activated macrophages then release a storm of pro-inflammatory cytokines, including IL-1β and IL-6, which in turn drive the expansion of Th17 cells. This creates a potent, self-amplifying inflammatory loop: GM-CSF+ Th cells → GM-CSF → Macrophages → IL-1β → Th17 expansion. Second, adoptive transfer experiments have provided direct proof of their pathogenicity, confirming that GM-CSF+ Th cells are sufficient on their own to induce severe colitis, while their depletion or the neutralization of GM-CSF significantly alleviates disease [30].
Despite this strong evidence, the narrative is not without complexity. An early study from 2014 reported that conditional knockout of GM-CSF in CD4+ T cells did not prevent colitis induction, suggesting a less critical role [21]. This discrepancy may be explained by the profound plasticity of T cells and the specific inflammatory context of different experimental models. More recent and detailed mechanistic work has clarified that in the gut, a unique IL-1β-driven pathway, rather than the canonical IL-23/RORγt axis, is responsible for inducing this pathogenic GM-CSF+ phenotype. This IL-1β signaling, acting via IRAK1/NF-κB, can also convert pre-existing Th17 cells into GM-CSF producers, explaining the frequent co-expression of IL-17 and GM-CSF observed in UC patients [30].
Perhaps the most unique aspect of UC immunopathology is the powerful regulatory role exerted by the intestinal epithelium itself. IFN-γ signaling in intestinal epithelial cells (IECs) has been found to be profoundly protective. In mice with an IEC-specific deletion of the IFN-γ receptor (IfngrΔIEC), GM-CSF+ Th cells accumulate uncontrollably in the gut [12]. Mechanistically, IFN-γ signaling in IECs enhances their ability to present antigen and promotes the expression of CD39 on adjacent T cells. CD39 is an ecto-enzyme that degrades extracellular ATP, a potent danger signal. By clearing ATP, this pathway prevents the activation of the NLRP3 inflammasome in myeloid cells, thereby shutting down the production of IL-1β, the very cytokine required to drive the pathogenic GM-CSF+ Th cell program. When this IFN-γ-mediated epithelial “brake” is lost, the NLRP3/IL-1β pathway becomes hyperactive, fueling a vicious cycle of GM-CSF+ Th cell expansion and neutrophil infiltration [12, 30].
This intricate regulatory network highlights the therapeutic potential of targeting this axis. While anti-GM-CSF antibodies can alleviate inflammation, their efficacy may be limited unless combined with other immunomodulators. The unique dependency on the IL-1β/NLRP3 pathway suggests that inhibitors of this pathway could be particularly effective in UC. Furthermore, strategies that enhance the protective IFN-γ signaling within the intestinal epithelium could offer a novel approach to restoring immune homeostasis in the gut. Unraveling the distinct functional characteristics of tissue-resident versus circulating GM-CSF+ Th cells remains a key area for future investigation.
3.3.3 Role of GM-CSF+ Th cells in less frequent autoimmune disorders
Beyond the major autoimmune diseases, the pathogenic footprint of GM-CSF-producing Th cells extends to a wide array of less common but severe disorders. While the specific clinical manifestations differ, a synthesis of these findings reveals a set of conserved, fundamental pathogenic principles that these cells employ to drive tissue damage.
A primary and universal mechanism is the recruitment and activation of myeloid cells. This principle is vividly illustrated across multiple organ systems. In the skin of patients with pustular psoriasis, a unique IL-23R+ subset of GM-CSF+ Th cells orchestrates the massive infiltration of neutrophils and monocytes, driving the formation of sterile pustules. Neutralizing GM-CSF in a preclinical model of this disease completely abrogates this myeloid influx and restores skin homeostasis [111]. Similarly, in experimental autoimmune uveitis (EAU), GM-CSF+ Th cells specifically recruit eosinophils to the retina. These activated eosinophils then release toxic enzymes like eosinophil peroxidase (EPO), causing severe retinal damage and vision loss, an effect that is blocked by anti-GM-CSF antibodies [112].
A second core principle is the induction of destructive enzymatic pathways in target tissues. This is exemplified in crescentic glomerulonephritis (cGN), where GM-CSF+ Th1-like cells accumulate in the kidneys. The GM-CSF they produce activates infiltrating monocyte-derived cells, inducing them to secrete matrix metalloproteinase 12 (MMP12). This enzyme directly degrades the glomerular basement membrane, leading to irreversible kidney damage and functional decline. This discovery positions the GM-CSF/MMP12 axis as a key therapeutic target for preventing renal destruction in T-cell-driven nephritis [113].
Third, GM-CSF+ Th cells often act as initiators of a broader, self-sustaining inflammatory network. In spondyloarthritis (SpA), GM-CSF+ Th cells in the synovial fluid interact with dendritic cells, enhancing their antigen-presenting capacity and creating a persistent local immune response that drives chronic joint inflammation [114]. A similar mechanism is observed in dry eye disease (DED), where GM-CSF+ Th17 cells in draining lymph nodes recruit and activate CD11b+ myeloid cells at the ocular surface, initiating a cycle of inflammation that can be disrupted by local administration of anti-GM-CSF antibodies [115].
Finally, the central role of these cells is underscored in the context of alloimmunity, such as in graft-versus-host disease (GVHD). Here, GM-CSF produced by donor-derived T cells is a critical factor that promotes myeloid cell production of IL-1β and ROS, driving multi-organ damage. The pathogenic importance of this axis is conserved across numerous other inflammatory conditions, including juvenile idiopathic arthritis, Behçet's disease, and endometriosis [34, 36, 116-122].
Moreover, as uncovered recently in an elegant piece of work by Gan and colleagues [123], GM-CSF-producing CD4+ T cells are integral to pro-inflammatory circuit that can sustain and amplify autoimmune responses through the signaling pathway involving GM-CSF, glucocorticoid-induced tumor necrosis factor receptor-related ligand (GITRL), and mTORC1, with the Sjögren's syndrome as a key example. In this autoimmune disease, the GM-CSF-GITRL-mTORC1 pathogenic loop has been shown to drive the differentiation and expansion of pathogenic Th17 cells secreting GM-CSF. Specifically, in this mechanism, GM-CSF stimulates monocytes to express GITRL, which then acts on CD4+ T cells through GITR to promote Th17 differentiation via mTORC1 pathway activation. The resulting pathogenic Th17 cells produce additional GM-CSF, creating a self-amplifying circuit that perpetuates inflammation. Flow cytometry analysis revealed that blocking GITRL significantly inhibited the expansion of Th17, Th17.1, and GM-CSF+ CD4+ T cells, while GM-CSF treatment dose-dependently enhanced GITRL expression on monocytes. Importantly, mechanistic studies confirmed that mTORC1 activation is essential for this process, as rapamycin treatment effectively inhibited GITRL-induced phosphorylation of S6 and STAT3 and suppressed Th17 expansion. The significance of this pathway was further validated in vivo, where administration of rAAV6-shRNA targeting GITRL alleviated disease progression in a NOD mouse model of Sjögren's Syndrome [123]. This GITRL-mTORC1-GM-CSF feedback loop represents a key mechanism by which GM-CSF+ T cells not only function as downstream effectors but also actively shape pathogenic T cell responses through intercellular communication with monocytes. These findings highlight the complex regulatory networks involving GM-CSF+ T cells in autoimmune conditions and suggest potential therapeutic approaches targeting this pathway.
In conclusion, regardless of the specific disease context, be it the joint, skin, eye, or kidney, GM-CSF+ Th cells consistently function as central pathogenic nodes. They do so by executing a core playbook of disease-driving mechanisms: recruiting myeloid cells, inducing tissue-degrading enzymes, enhancing pathogenic Th cell response, and orchestrating self-sustaining inflammatory networks. A deeper understanding of these shared principles, combined with the study of disease-specific nuances, will be essential for developing effective, targeted therapies for this broad spectrum of devastating disorders.
4. Translational Applications Targeting GM-CSF+ Th Cells
GM-CSF⁺ Th cells represent a highly heterogeneous and plastic subset of helper T cells. Their indispensable role in the pathogenesis of multiple autoimmune diseases has been confirmed in both clinical and experimental settings. Defined by their dependence on STAT5/BHLHE40 signaling, co-expression of pro-inflammatory cytokines (e.g., IL-17A, IFN-γ), and close interaction with myeloid cells, GM-CSF⁺ Th cells offer promising targets for next-generation immunotherapies.
4.1 Cytokine- and receptor-based therapeutic strategies
Several cytokine pathways crucial for GM-CSF⁺ Th cell differentiation have been exploited therapeutically. For instance, IL-2/IL-2R signaling is known to promote GM-CSF⁺ Th differentiation through STAT5 phosphorylation. Clinical use of daclizumab, an anti-IL-2Rα monoclonal antibody, showed efficacy in reducing inflammatory activity in relapsing multiple sclerosis (MS) by limiting GM-CSF secretion [124, 125]. However, its eventual market withdrawal due to severe immune-related adverse effects underscores the dual role of IL-2 signaling: promoting effector T cell activation while supporting Treg homeostasis [126]. This dichotomy highlights the need for more selective downstream targets in the GM-CSF axis to avoid collateral immunosuppression.
Similarly, IL-7 and IL-15 have also been implicated in the differentiation of GM-CSF⁺ Th cells via STAT5 activation. In vitro studies demonstrate that both cytokines drive naïve CD4⁺ T cells toward the GM-CSF⁺ phenotype through STAT5 phosphorylation. Lusvertikimab (anti-IL-7R) has shown promising results in phase II trials for ulcerative colitis, while HuMax-IL15 (anti-IL-15) has demonstrated tolerability and efficacy in early-phase RA trials [127]. These findings support the potential of IL-7 and IL-15 blockade to indirectly suppress GM-CSF⁺ Th cell expansion in autoimmune contexts. Ustekinumab, a monoclonal antibody targeting the p40 subunit shared by IL-12 and IL-23, has shown clinical efficacy in multiple autoimmune conditions—such as Crohn's disease [128], ulcerative colitis [129], plaque psoriasis (psoriasis vulgaris) [130], and type 1 diabetes [108]. Although not directly targeting GM-CSF, blockade of IL-23 may reduce GM-CSF⁺ Th differentiation by disrupting upstream cytokine signals. However, its mechanism involves broader immunosuppressive effects, and its relevance to GM-CSF⁺ Th biology remains partially indirect.
4.2 Direct GM-CSF pathway inhibition
Anti-GM-CSF monoclonal antibodies represent a more direct strategy. MOR103 showed favorable safety and tolerability in phase 1b trials for MS [131]. In RA, mavrilimumab (anti-GM-CSF receptor antibody) significantly reduced disease activity, particularly in early disease stages [132]. Agents antagonizing GM-CSF or its receptor demonstrated certain promising efficacy performance and basically favorable safety profiles [94, 133-148], as summarized in Table 2. However, therapeutic outcomes may vary across patients, likely due to T cell heterogeneity and irreversible tissue damage in late-stage disease.
Tabular summary of clinical trials of anti-GM-CSF agents in autoimmune disorders
| Target | Molecule | Drug Type | Indication | Phase | Status | ClinicalTrials.gov identifier | Efficacy outcomes | Safety profiles | Reference |
|---|---|---|---|---|---|---|---|---|---|
| GM-CSFR | Mavrilimumab KPL-301 (aka, CAM-3001) | Monoclonal antibody | RA | I | Completed | NCT00771420 | A significant reduction in disease activity was observed in patients with high baseline disease activity | Safety was favorable with most adverse events being mild to moderate, with no SAEs or DLTs. | [133] |
| RA | II | Completed | NCT01050998 | Mavrilimumab significantly increased the proportion of patients achieving a reduction in DAS28-CRP score at Week 12 (55.7% vs. 34.7%, P = 0.003). The 100 mg dose group demonstrated significant effects across multiple efficacy endpoints. | Mild to moderate adverse events such as infections and hypersensitivity reactions were observed. | [134] | |||
| RA | IIa | Completed | NCT01050998 | Mavrilimumab demonstrated significant efficacy across multiple dose groups, especially in the 30 mg and 100 mg dose groups. Improvements in DAS28-CRP and HAQ-DI scores were notable. | Most adverse events were mild to moderate. Overall tolerability was favorable. | [135] | |||
| RA | IIb | Completed | NCT01706926 | Mavrilimumab significantly reduced disease activity in RA patients with clinically meaningful responses observed as early as in Week 1. ACR20 response rates were significantly higher in all dose groups compared to placebo. | No treatment-related safety signals were identified, demonstrating good tolerability. | [136] | |||
| RA | IIb | Completed | NCT01706926 | Mavrilimumab significantly reduced inflammatory markers and improved RA symptoms. The efficacy was associated with the inhibition of myeloid and T-cell signaling pathways. | Overall tolerability was favorable. No serious adverse events were reported, demonstrating a favorable safety profile. | [137] | |||
| RA | IIb | Completed | NCT01715896 | Mavrilimumab demonstrated clinical efficacy in patients with refractory RA. | 51.4% of patients in the mavrilimumab treatment group reported treatment-related adverse events. No deaths or specific safety signals were observed. | [138] | |||
| RA | IIb | Completed | NCT01712399 | Mavrilimumab showed sustained efficacy in long-term treatment, significantly improving disease status. Biomarker analyses indicated effective control of inflammatory response. | Long-term treatment was well-tolerated, with no increase in treatment-related adverse events. Common adverse events were mild to moderate and transient in nature. | [139] | |||
| GM-CSF | GSK 3196165 Otilimab (aka, MOR103) | Monoclonal antibody | RA | Ib/IIa | Completed | NCT01023256 | Clinical efficacy included reductions in disease activity scores and joint counts with higher EULAR response rates. | MOR103 was well-tolerated in patients with moderate RA. No serious adverse events were reported. | [140] |
| RA | IIa | Completed | NCT02799472 | Some positive results were observed in target engagement and biomarkers, but significant improvements in synovitis were not achieved. | Otilimab was well-tolerated. No serious adverse events or deaths were reported. | [141] | |||
| RA | IIb | Completed | NCT02504671 | Although otilimab combined with methotrexate did not meet the primary endpoint of DAS28-CRP remission, improvements in disease activity scores were observed. Pain and physical function reported by patients improved significantly. | Otilimab was well-tolerated. No deaths or clinically significant pulmonary events were reported. | [94] | |||
| RA | III | Completed | NCT04134728 | Otilimab showed no superiority in efficacy compared to placebo. | Safety was acceptable. | [142] | |||
| RA | III | Completed | NCT03980483, NCT03970837 | In two trials, otilimab increased the proportion of patients achieving low disease activity per CDAI and reduced HAQ-DI scores compared to placebo. | The safety profile of otilimab was comparable to placebo and tofacitinib. No new safety concerns were identified. | [143] | |||
| RA | III | Completed | NCT04333147 | Otilimab demonstrated some efficacy, with stable response rates for patients achieving low disease activity per CDAI. | In long-term studies (<2.5 years) of otilimab for RA, safety was favorable. The incidence of adverse events was low. | [144] | |||
| Namilumab (previously MT203) | Monoclonal antibody | RA | Ib | Completed | NCT01317797 | A reduction in disease activity scores and improvement in joint symptoms were observed. | Namilumab was well-tolerated in patients with mild to moderate RA. Safety was comparable to placebo. | [145] | |
| PP | II | Completed | NCT02129777 | Namilumab showed no significant efficacy in reducing PASI scores compared to placebo. | Safety was favorable, with no serious adverse events or significant drug-related safety issues identified. | [146] | |||
| RA | II | Completed | NCT02379091 | The primary endpoint was achieved at Week 12, showing a clear dose-response effect. | Tolerability was favorable. No serious infections were reported. | [147] | |||
| axSpA | II | Completed | NCT03622658 | Namilumab showed no efficacy in patients with active axSpA compared to placebo. | Tolerability was favorable. | [148] |
RA, rheumatoid arthritis; PP, plaque psoriasis; axSpA, axial spondyloarthritis; DAS28-CRP, disease activity Score in 28 joints using C-reactive protein; ACR, American college of rheumatology; HAQ-DI, health assessment questionnaire disability Index; EULAR, European alliance of associations for rheumatology; CADI, clinical disease activity index; PASI, psoriasis area and severity index.
Notably, the novel anti-GM-CSF agent otilimab failed to meet primary and secondary endpoints in RA patients refractory to DMARDs or JAK inhibitors. Despite encouraging preclinical data, it showed no superiority over sarilumab, likely due to suboptimal patient selection and late-stage intervention [94]. These findings support the importance of biomarker-based stratification and early therapeutic timing. Nevertheless, those mixed clinical outcomes observed with GM-CSF-targeting therapies, particularly the failure of otilimab in rheumatoid arthritis trials, need cautious interpretation and warrant deeper critical analysis. Several factors likely contribute to these inconsistent results. Patient heterogeneity represents a significant challenge, as inflammatory diseases encompass diverse endotypes with varying pathophysiological mechanisms and GM-CSF dependency. The timing of therapeutic intervention could be crucial, because many trials enrolled patients with established disease where irreversible tissue damage had already occurred, potentially diminishing efficacy. Additionally, single-target approaches may be insufficient for conditions with redundant inflammatory pathways. Biomarker-based stratification was basically absent in most trials, preventing identification of GM-CSF-dependent patient subgroups who might benefit most from the targeted therapy. Furthermore, dosing regimens may have been suboptimal, with insufficient target engagement at affected tissue sites. These considerations highlight the need for refined patient selection strategies using predictive biomarkers, earlier therapeutic intervention before tissue damage becomes irreversible, and potentially combination approaches targeting complementary pathways. Future trials should address these factors to optimize the therapeutic potential of GM-CSF pathway modulation in inflammatory conditions.
4.3 Inflammasome and IL-1 axis modulation
The NLRP3 inflammasome, which can promote GM-CSF production via IL-1β amplification, is another relevant target. MCC950, a selective NLRP3 inhibitor, has shown anti-inflammatory efficacy in multiple autoimmune disease models—including MS [149, 150], RA [151], and IBD [152, 153]. However, its clinical application is hampered by pharmacokinetic and safety limitations. Moreover, disease-specific paradoxical effects, such as symptom exacerbation in Sjögren's syndrome [154], emphasize the complexity of inflammasome targeting.
Canakinumab, a monoclonal antibody against IL-1β, has demonstrated favorable safety and efficacy in autoimmune settings [155]. Nevertheless, heterogeneity in trial populations limits its interpretability in disease-specific contexts, emphasizing the need for more refined cohort definitions in future RA studies.
4.4 Precision biomarkers and stratified therapies
Advances in mass cytometry and ELISA-based serum profiling enable dynamic tracking of GM-CSF⁺ Th cells or circulating GM-CSF levels. For example, a ≥50% reduction in GM-CSF⁺ Th cells correlates with improved joint swelling index in RA [29]. Furthermore, IL2RA genetic polymorphisms (e.g., rs2104286) are associated with elevated GM-CSF⁺ Th frequencies [35], suggesting their utility in genotype-based treatment selection [27, 48, 156]. These patients may particularly benefit from STAT5-targeted therapies or CRISPR-mediated interventions.
4.5 Targeting STAT5 and transcriptional networks
STAT5 is a critical regulator of GM-CSF⁺ Th cell fate. IL-2, IL-7, and IL-15 converge on STAT5 phosphorylation to drive GM-CSF⁺ Th differentiation. Although several STAT5 inhibitors (e.g., pimozide) have demonstrated efficacy in oncology [157, 158], off-target effects and structural homology with other STAT family members limit their specificity in autoimmunity. Furthermore, STAT5 plays essential roles in hematopoiesis and Treg maintenance, raising concerns about long-term safety.
To improve specificity, cell surface markers such as CD69, CXCR4, and CX3CR1 may be used to guide GM-CSF⁺ Th cell-selective targeting. In addition, transcription factors including BHLHE40, Rel, RUNX1, NLRP3, and T-bet have also been implicated in GM-CSF⁺ Th cell regulation, offering new molecular targets for therapy.
4.6 CRISPR-Cas9 gene editing
Recent advances in CRISPR-Cas9 gene editing provide precise tools for disrupting key transcriptional regulators. Lin et al. developed mouse models harboring Y694F and Y699F mutations in STAT5A/B, which abrogate phosphorylation-dependent GM-CSF⁺ Th cell differentiation [159]. This method demonstrates greater specificity and durability than small-molecule inhibitors and allows for cell-type-restricted interventions. Similar approaches could target BHLHE40, NLRP3, and other GM-CSF-regulatory transcription factors. Nonetheless, CRISPR technology still faces hurdles, including off-target editing and delivery efficiency, which must be addressed for clinical translation.
Despite the immense promise of these strategies, significant translational hurdles remain before they can be widely implemented. The most formidable of these is the issue of cross-species differences. The markers used to identify GM-CSF+ Th cells, the specific cytokine cues that drive their differentiation, and even their dominant pathogenic phenotype can differ substantially between standard mouse models and human patients. For instance, while mouse EAE models often feature a prominent GM-CSF+ Th17-like response, the pathology in human MS is frequently dominated by a Th1-like counterpart. This chasm between preclinical models and human disease underscores the critical necessity of developing and validating therapeutic targets in more sophisticated systems, such as humanized mice or patient-derived organoid models.
Ultimately, effectively targeting these “inflammatory amplifiers” requires more than just better models; it demands a paradigm shift in our therapeutic philosophy. The future of treating autoimmunity lies in moving beyond broad, indiscriminate immunosuppression toward the goal of precision immune remodeling. This future will be built on a foundation of multi-dimensional strategies that leverage cutting-edge technologies like epigenetic editing to correct faulty gene programs, synthetic biology to create “smart” therapies (e.g., logic-gated CAR-T cells), and artificial intelligence to design patient-specific interventions based on complex multi-omics data. The ultimate goal is no longer simply to dampen the immune response, but to selectively erase pathogenic immune memory, restore self-tolerance, and rebuild a balanced and functional immune homeostasis, offering the potential for truly curative solutions for autoimmune diseases.
A summary of these current and future therapeutic strategies is provided in Table 3 and Figure 6.
Potential strategies for autoimmune disease management through targeting GM-CSF+ Th cells.
| Potential Targets | Drug types | Agents | Disease* & development stage | Adverse effects & limitations | Possible strategies for improvement |
|---|---|---|---|---|---|
| GM-CSF | anti-GM-CSF mAb | Otilimab | RA, phase III[94] | drug resistance, immunosuppression, lung injury (proteinosis) | precise patient stratification and biomarker - guided treatment; optimization of dosing regimens; enhancement of safety monitoring and management; combination therapy strategies synergizing with other immunosuppressive drugs; improvement of patient compliance; conducting additional real - world studies. |
| GM-CSF receptors | anti-GM-CSF receptor mAb | Mavrilimumab | RA, phase IIb[132] | limited response rate, immunosuppression | |
| IL-2 receptors | anti-IL-2Rα mAb | Daclizumab | MS[124], [125], approved (2016), withdrawn (2018) [126] | severe inflammatory brain disease, hepatic injury, drug-induced rash, allergic reactions | |
| IL-7 receptors | anti-IL-7Rα mAb | Lusvertikimab | UC, phase II (CoTikiS) | immunosuppression; impaired immune tolerance, formation of anti-drug antibodies | |
| IL-15 | anti-IL-15 mAb | HuMax-IL15 | RA, phase I/II[127] | mild to moderate infections, NK cell function impairment, possible tumorigenicity,inadequate efficacy | precision patient selection, e.g. overexpression of GM-CSF+ Th1/Th17 cells and IL-15 both; integrating disease staging criteria into treatment decisions, e.g focusing on early-stage disease; |
| STAT5 | small-molecule targeted inhibitors | Pimozide (repositioning) | inhibiting STAT5 phosphorylation in cells[157], [158] | mechanistic complexity, lack of human study, off-target effect; | targeted delivery of STAT5 inhibitor to GM-CSF-expressing Th cells with STAT5 overexpression; |
| IL-12 & IL-23 (P40) | anti-(IL-12 and IL-23) P40 mAb | Ustekinumab | clinical trial: Crohn's Disease[128], UC[128], Psoriasis[130], T1D[108] | limited clinical data, immunosuppression, individual variability, potential impacts on adolescent development | evaluating IL-12/IL-23 p40 as a biomarker for stratifying patients |
| NLRP3 | NLRP3 inflammasome inhibitor | MCC950 | preclinical studies in animal models: MS[149], [150], RA[151], UC[152], [153] | limited clinical data, hepatotoxicity, immunosuppression, off-target effects, tissue-specific variability | developing oral prodrugs and nanoparticle-delivered long-acting sustained-release formulations to improve bioavailability; stratifying patients using NLRP3 genetic markers; designing bispecific antibodies targeting NLRP3 and IL-1β to mitigate immunosuppression-related risks; engineering reversible inhibitors to preserve host defense during infections |
| IL-1β | anti-IL-1β mAb | Canakinumab | RA, phaseI/II[155] | immunosuppression, individual variability in efficacy, high cost | screening patient populations responsive to IL-1β inhibition; developing long-acting sustained-release and oral formulations. |
| STAT5/T-bet/BHLHE40/Rel | Conditional gene editing or Small-molecule inhibitor | CRISPR-mediated knockout of STAT5 phosphorylation sites | mouse model with tyrosine-to-phenylalanine mutations (Y694F and Y699F) in STAT5A and STAT5B[159] | o-target effects, Editing efficiency, cell or tissue specificity, potential immune response | optimizing sgRNA design targeting GM-CSF+ Th cells, using Cas9 variants, combining with other editing techniques, improving delivery systems, Inducible gene knockout, multiplex gene editing, functional validation and compensation mechanism studies |
* MS, multiple sclerosis; RA, rheumatoid arthritis; T1D, type 1 diabetes; UC, ulcerative colitis.
Possible approaches for targeting GM-CSF+ Th cells. This figure outlines a three-stage framework for therapeutically targeting the GM-CSF⁺ Th cell pathway, from initial development to final effector function. Stage 1 (Induction & Differentiation): This stage can be targeted by blocking the initial cytokine signals (e.g., IL-2, IL-7, IL-15) that drive the differentiation of naive T cells into the GM-CSF⁺ lineage. Stage 2 (Amplification & Stabilization): Intervention at this stage focuses on disrupting the transcriptional machinery and autocrine feedback loops (e.g., NLRP3/IL-1β) that reinforce and stabilize the pathogenic cell state. Stage 3 (Effector Function & Tissue Damage): The final stage offers therapeutic opportunities to neutralize the key effector molecule, GM-CSF (e.g., with Otilimab), or its receptor (e.g., with Mavrilimumab), thereby preventing the activation of myeloid cells and subsequent inflammatory tissue damage. Created in BioRender. Meng, X. (2025) https://BioRender.com/o6m32us.
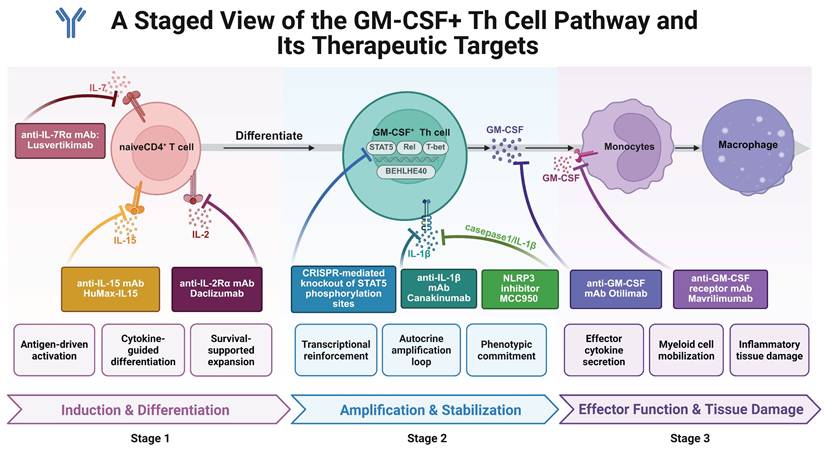
5. Take-Home Message and Future Perspectives
GM-CSF⁺ Th cells constitute a highly heterogeneous and plastic subset of CD4⁺ helper T cells that act as “inflammatory amplifiers” in autoimmune diseases. Through the secretion of GM-CSF, these cells activate myeloid populations such as macrophages and dendritic cells, disrupt tissue barriers (e.g., the blood-brain barrier, pancreatic islets, or synovial membranes), induce oxidative stress, and engage in feed-forward loops alongside pro-inflammatory cytokines like IL-17 and IFN-γ, thereby driving disease progression. Across different disease contexts, GM-CSF⁺ Th cells exhibit disease-specific phenotypes and regulatory pathways. For instance, a Th1-like profile predominates in RA, whereas in T1D, a notable enrichment of IFN-γ⁺IL-17⁺GM-CSF⁺ triple-positive cells is observed. Their differentiation is governed by a network of signaling pathways, including IL-23/STAT3/RORγt, IL-7/STAT5, as well as epigenetic mechanisms. Despite the promising outcomes observed in preclinical models using GM-CSF-targeted therapies, including monoclonal antibodies, STAT5 inhibitors, and epigenetic modifiers, clinical translation remains hampered by several challenges. These include inter-patient heterogeneity in therapeutic efficacy, unclear windows for optimal intervention, and biological discrepancies between human disease and animal models.
Future research should aim to achieve mechanistic and translational breakthroughs in multiple dimensions. First, the integration of single-cell RNA sequencing (scRNA-seq), spatial transcriptomics, and epigenomic profiling will be instrumental in mapping the spatiotemporal evolution and plasticity of GM-CSF⁺ Th subsets across tissues and disease stages. Elucidating regulatory circuits, such as BHLHE40-RUNX1 interactions, may uncover lineage-defining checkpoints. Second, dissecting the metabolic programs of GM-CSF⁺ Th cells, including glycolysis and glutaminolysis, could enable the design of metabolic reprogramming strategies as adjunct therapies. Third, combining genetic risk variants (e.g., IL2RA, ZNF35), epigenetic biomarkers (e.g., GM-CSF promoter methylation), and immunophenotypic data (e.g., from multi-omic profiling) will facilitate the development of predictive biomarker systems for therapeutic stratification.
Next-generation therapeutic designs, such as logic-gated CAR-Tregs or switchable STAT5 inhibitors, may achieve spatiotemporal control of GM-CSF⁺ Th cell activity while minimizing systemic immunosuppression. Combinatorial approaches targeting GM-CSF alongside key inflammatory pathways (e.g., IL-1β, IL-23, or CXCL10) may overcome the challenge of cytokine redundancy; for instance, combining anti-GM-CSF monoclonal antibodies with JAK inhibitors could yield synergistic effects. Multifunctional nanomedicines that simultaneously suppress inflammation and promote tissue regeneration (e.g., β-cell recovery or remyelination) may offer a comprehensive therapeutic paradigm.
Advances in translational modeling, including organoid-immune cell co-culture systems and humanized mouse models, can more accurately replicate human disease microenvironments and bridge cross-species gaps in drug efficacy. Finally, longitudinal cohort studies in high-risk populations (e.g., preclinical T1D or RA) may help define the critical threshold of GM-CSF⁺ Th cell expansion, guiding early and precisely timed interventions.
6. Concluding Remarks
Research on GM-CSF+ Th cells is catalyzing a paradigm shift of autoimmune disease management from broad-spectrum immunosuppression to precision-guided immune remodeling. In the coming decades, the integration of multi-omics technologies, gene editing, and artificial intelligence is expected to drive the development of targeted therapies capable of halting, or potentially reversing, autoimmune pathology by modulating this pathogenic subset. However, several key challenges must be addressed to facilitate successful clinical translation. These include elucidating the mechanistic basis of phenotypic heterogeneity and functional plasticity among GM-CSF⁺ Th subpopulations, establishing real-time monitoring systems to track their dynamics in vivo, and ensuring therapeutic safety across diverse patient populations. Ultimately, it is only through sustained interdisciplinary collaboration, bridging medical immunology, bioinformatics, bioengineering, and clinical rheumatology, that curative strategies can be realized to effectively confront the complexity of autoimmune diseases.
Supplementary Material
Supplementary table.
Acknowledgements
This work was supported by the Clinical Research Innovation Project, West China Hospital, Sichuan University (2019HXCX01). XYM is supported by National Natural Science Foundation of China (82303057), Natural Science Foundation of Hubei Province of China (2023AFB521), and “Chutian Scholars Program” of Hubei Province of China. SSF was supported by the Natural Science Fund and Innovation Horizontal Project of Hubei Minzu University (4178037 and H23123), the guiding Project of Hubei Provincial Department of Education (B2019090), the Youth Project of the Clinical Research Center of Minzu University Hospital (OIR202402Q). XX is supported by the Research Fund for the Doctoral Program of Anhui Medical University (0801059201). YBL is supported by the National Natural Science Foundation of China (No. 82271828) and scientific application and foundation project of Science and Technology Department of Guangxi Province (No. AB25069068).
Author contributions
SSF, XX, YBL, and XYM: writing - original draft; YL, XX, YXM, and YBL: writing - review and editing; YL: conceptualization, project administration, supervision, funding acquisition. All authors have read and approved the final manuscript.
Generative AI statement
Generative AI GPT-4o, Claude-Sonnet-3.7, Gemini-2.5-Pro, and Grok 3.0 were used for language assistance during writing but not for any content generation.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Conrad N, Misra S, Verbakel JY, Verbeke G, Molenberghs G, Taylor PN. et al. Incidence, prevalence, and co-occurrence of autoimmune disorders over time and by age, sex, and socioeconomic status: a population-based cohort study of 22 million individuals in the UK. Lancet. 2023;401:1878-90
2. Winthrop KL, Mease P, Kerschbaumer A, Voll RE, Breedveld FC, Smolen JS. et al. Unmet need in rheumatology: reports from the Advances in Targeted Therapies meeting, 2023. Ann Rheum Dis. 2024;83:409-16
3. Schett G, Mackensen A, Mougiakakos D. CAR T-cell therapy in autoimmune diseases. Lancet. 2023;402:2034-44
4. Zhu J, Yamane H, Paul WE. Differentiation of effector CD4 T cell populations (*). Annu Rev Immunol. 2010;28:445-89
5. Sun L, Su Y, Jiao A, Wang X, Zhang B. T cells in health and disease. Signal Transduct Target Ther. 2023;8:235
6. Jain A, Irizarry-Caro RA, McDaniel MM, Chawla AS, Carroll KR, Overcast GR. et al. T cells instruct myeloid cells to produce inflammasome-independent IL-1β and cause autoimmunity. Nat Immunol. 2020;21:65-74
7. Dardalhon V, Korn T, Kuchroo VK, Anderson AC. Role of Th1 and Th17 cells in organ-specific autoimmunity. J Autoimmun. 2008;31:252-6
8. Ferber IA, Brocke S, Taylor-Edwards C, Ridgway W, Dinisco C, Steinman L. et al. Mice with a disrupted IFN-gamma gene are susceptible to the induction of experimental autoimmune encephalomyelitis (EAE). J Immunol. 1996;156:5-7
9. Lublin FD, Knobler RL, Kalman B, Goldhaber M, Marini J, Perrault M. et al. Monoclonal anti-gamma interferon antibodies enhance experimental allergic encephalomyelitis. Autoimmunity. 1993;16:267-74
10. Mills KHG. IL-17 and IL-17-producing cells in protection versus pathology. Nat Rev Immunol. 2023;23:38-54
11. El-Behi M, Ciric B, Dai H, Yan Y, Cullimore M, Safavi F. et al. The encephalitogenicity of T(H)17 cells is dependent on IL-1- and IL-23-induced production of the cytokine GM-CSF. Nat Immunol. 2011;12:568-75
12. Malik A, Sharma D, Aguirre-Gamboa R, McGrath S, Zabala S, Weber C. et al. Epithelial IFNγ signalling and compartmentalized antigen presentation orchestrate gut immunity. Nature. 2023;623:1044-52
13. Noster R, Riedel R, Mashreghi MF, Radbruch H, Harms L, Haftmann C. et al. IL-17 and GM-CSF expression are antagonistically regulated by human T helper cells. Sci Transl Med. 2014;6:241ra80
14. Codarri L, Gyülvészi G, Tosevski V, Hesske L, Fontana A, Magnenat L. et al. RORγt drives production of the cytokine GM-CSF in helper T cells, which is essential for the effector phase of autoimmune neuroinflammation. Nat Immunol. 2011;12:560-7
15. Gerondakis S, Strasser A, Metcalf D, Grigoriadis G, Scheerlinck JY, Grumont RJ. Rel-deficient T cells exhibit defects in production of interleukin 3 and granulocyte-macrophage colony-stimulating factor. Proc Natl Acad Sci U S A. 1996;93:3405-9
16. Damsker JM, Hansen AM, Caspi RR. Th1 and Th17 cells: adversaries and collaborators. Ann N Y Acad Sci. 2010;1183:211-21
17. Kamali AN, Noorbakhsh SM, Hamedifar H, Jadidi-Niaragh F, Yazdani R, Bautista JM. et al. A role for Th1-like Th17 cells in the pathogenesis of inflammatory and autoimmune disorders. Mol Immunol. 2019;105:107-15
18. Chen Y, Dana R. Autoimmunity in dry eye disease - An updated review of evidence on effector and memory Th17 cells in disease pathogenicity. Autoimmun Rev. 2021;20:102933
19. Zhang J, Roberts AI, Liu C, Ren G, Xu G, Zhang L. et al. A novel subset of helper T cells promotes immune responses by secreting GM-CSF. Cell Death Differ. 2013;20:1731-41
20. Komuczki J, Tuzlak S, Friebel E, Hartwig T, Spath S, Rosenstiel P. et al. Fate-Mapping of GM-CSF Expression Identifies a Discrete Subset of Inflammation-Driving T Helper Cells Regulated by Cytokines IL-23 and IL-1β. Immunity. 2019;50:1289-304.e6
21. Sheng W, Yang F, Zhou Y, Yang H, Low PY, Kemeny DM. et al. STAT5 programs a distinct subset of GM-CSF-producing T helper cells that is essential for autoimmune neuroinflammation. Cell Res. 2014;24:1387-402
22. Laurent C, Deblois G, Clénet ML, Carmena Moratalla A, Farzam-Kia N, Girard M. et al. Interleukin-15 enhances proinflammatory T-cell responses in patients with MS and EAE. Neurol Neuroimmunol Neuroinflamm. 2021 8
23. Galli E, Hartmann FJ, Schreiner B, Ingelfinger F, Arvaniti E, Diebold M. et al. GM-CSF and CXCR4 define a T helper cell signature in multiple sclerosis. Nat Med. 2019;25:1290-300
24. Deng YN, Bellanti JA, Zheng SG. Essential Kinases and Transcriptional Regulators and Their Roles in Autoimmunity. Biomolecules. 2019 9
25. Yosef N, Shalek AK, Gaublomme JT, Jin H, Lee Y, Awasthi A. et al. Dynamic regulatory network controlling TH17 cell differentiation. Nature. 2013;496:461-8
26. Lin CC, Bradstreet TR, Schwarzkopf EA, Sim J, Carrero JA, Chou C. et al. Bhlhe40 controls cytokine production by T cells and is essential for pathogenicity in autoimmune neuroinflammation. Nat Commun. 2014;5:3551
27. Éliás S, Schmidt A, Gomez-Cabrero D, Tegnér J. Gene Regulatory Network of Human GM-CSF-Secreting T Helper Cells. J Immunol Res. 2021;2021:8880585
28. Hasegawa H, Mizoguchi I, Orii N, Inoue S, Katahira Y, Yoneto T. et al. IL-23p19 and CD5 antigen-like form a possible novel heterodimeric cytokine and contribute to experimental autoimmune encephalomyelitis development. Sci Rep. 2021;11:5266
29. Guo N, Ye S, Zhang K, Yu X, Cui H, Yang X. et al. A critical epitope in CD147 facilitates memory CD4(+) T-cell hyper-activation in rheumatoid arthritis. Cell Mol Immunol. 2019;16:568-79
30. Hu Y, Xu F, Zhang R, Legarda D, Dai J, Wang D. et al. Interleukin-1β-induced IRAK1 ubiquitination is required for T(H)-GM-CSF cell differentiation in T cell-mediated inflammation. J Autoimmun. 2019;102:50-64
31. Cravens PD, Hussain RZ, Zacharias TE, Ben LH, Herndon E, Vinnakota R. et al. Lymph node-derived donor encephalitogenic CD4+ T cells in C57BL/6 mice adoptive transfer experimental autoimmune encephalomyelitis highly express GM-CSF and T-bet. J Neuroinflammation. 2011;8:73
32. Rasouli J, Casella G, Yoshimura S, Zhang W, Xiao D, Garifallou J. et al. A distinct GM-CSF(+) T helper cell subset requires T-bet to adopt a T(H)1 phenotype and promote neuroinflammation. Sci Immunol. 2020 5
33. Balmas E, Chen J, Hu AK, DeBerg HA, Rosasco MG, Gersuk VH. et al. Islet-autoreactive CD4+ T cells are linked with response to alefacept in type 1 diabetes. JCI Insight. 2023 8
34. Piper C, Hainstock E, Yin-Yuan C, Chen Y, Khatun A, Kasmani MY. et al. Single-cell immune profiling reveals a developmentally distinct CD4+ GM-CSF+ T-cell lineage that induces GI tract GVHD. Blood Adv. 2022;6:2791-804
35. Hartmann FJ, Khademi M, Aram J, Ammann S, Kockum I, Constantinescu C. et al. Multiple sclerosis-associated IL2RA polymorphism controls GM-CSF production in human TH cells. Nat Commun. 2014;5:5056
36. Tugues S, Amorim A, Spath S, Martin-Blondel G, Schreiner B, De Feo D. et al. Graft-versus-host disease, but not graft-versus-leukemia immunity, is mediated by GM-CSF-licensed myeloid cells. Sci Transl Med. 2018 10
37. Grifka-Walk HM, Giles DA, Segal BM. IL-12-polarized Th1 cells produce GM-CSF and induce EAE independent of IL-23. Eur J Immunol. 2015;45:2780-6
38. Arbelaez CA, Glatigny S, Duhen R, Eberl G, Oukka M, Bettelli E. IL-7/IL-7 Receptor Signaling Differentially Affects Effector CD4+ T Cell Subsets Involved in Experimental Autoimmune Encephalomyelitis. J Immunol. 2015;195:1974-83
39. Koch-Henriksen N, Magyari M. Apparent changes in the epidemiology and severity of multiple sclerosis. Nat Rev Neurol. 2021;17:676-88
40. Lassmann H, van Horssen J, Mahad D. Progressive multiple sclerosis: pathology and pathogenesis. Nat Rev Neurol. 2012;8:647-56
41. Panitch HS, Hirsch RL, Haley AS, Johnson KP. Exacerbations of multiple sclerosis in patients treated with gamma interferon. Lancet. 1987;1:893-5
42. Haak S, Croxford AL, Kreymborg K, Heppner FL, Pouly S, Becher B. et al. IL-17A and IL-17F do not contribute vitally to autoimmune neuro-inflammation in mice. J Clin Invest. 2009;119:61-9
43. McQualter JL, Darwiche R, Ewing C, Onuki M, Kay TW, Hamilton JA. et al. Granulocyte macrophage colony-stimulating factor: a new putative therapeutic target in multiple sclerosis. J Exp Med. 2001;194:873-82
44. Kroenke MA, Carlson TJ, Andjelkovic AV, Segal BM. IL-12- and IL-23-modulated T cells induce distinct types of EAE based on histology, CNS chemokine profile, and response to cytokine inhibition. J Exp Med. 2008;205:1535-41
45. Carrieri PB, Provitera V, De Rosa T, Tartaglia G, Gorga F, Perrella O. Profile of cerebrospinal fluid and serum cytokines in patients with relapsing-remitting multiple sclerosis: a correlation with clinical activity. Immunopharmacol Immunotoxicol. 1998;20:373-82
46. Ghezzi L, Cantoni C, Cignarella F, Bollman B, Cross AH, Salter A. et al. T cells producing GM-CSF and IL-13 are enriched in the cerebrospinal fluid of relapsing MS patients. Mult Scler. 2020;26:1172-86
47. Rasouli J, Ciric B, Imitola J, Gonnella P, Hwang D, Mahajan K. et al. Expression of GM-CSF in T Cells Is Increased in Multiple Sclerosis and Suppressed by IFN-β Therapy. J Immunol. 2015;194:5085-93
48. Håkansson I, Tisell A, Cassel P, Blennow K, Zetterberg H, Lundberg P. et al. Neurofilament levels, disease activity and brain volume during follow-up in multiple sclerosis. J Neuroinflammation. 2018;15:209
49. Spath S, Komuczki J, Hermann M, Pelczar P, Mair F, Schreiner B. et al. Dysregulation of the Cytokine GM-CSF Induces Spontaneous Phagocyte Invasion and Immunopathology in the Central Nervous System. Immunity. 2017;46:245-60
50. Broux B, Mizee MR, Vanheusden M, van der Pol S, van Horssen J, Van Wijmeersch B. et al. IL-15 amplifies the pathogenic properties of CD4+CD28- T cells in multiple sclerosis. J Immunol. 2015;194:2099-109
51. Kaskow BJ, Baecher-Allan C. Effector T Cells in Multiple Sclerosis. Cold Spring Harb Perspect Med. 2018 8
52. Monaghan KL, Wan ECK. The Role of Granulocyte-Macrophage Colony-Stimulating Factor in Murine Models of Multiple Sclerosis. Cells. 2020 9
53. Rasouli J, Casella G, Zhang W, Xiao D, Kumar G, Fortina P. et al. Transcription Factor RUNX3 Mediates Plasticity of ThGM Cells Toward Th1 Phenotype. Front Immunol. 2022;13:912583
54. Stojić-Vukanić Z, Pilipović I, Vujnović I, Nacka-Aleksić M, Petrović R, Arsenović-Ranin N. et al. GM-CSF-Producing Th Cells in Rats Sensitive and Resistant to Experimental Autoimmune Encephalomyelitis. PLoS One. 2016;11:e0166498
55. Fletcher JM, Lalor SJ, Sweeney CM, Tubridy N, Mills KH. T cells in multiple sclerosis and experimental autoimmune encephalomyelitis. Clin Exp Immunol. 2010;162:1-11
56. Carbajal KS, Mironova Y, Ulrich-Lewis JT, Kulkarni D, Grifka-Walk HM, Huber AK. et al. Th Cell Diversity in Experimental Autoimmune Encephalomyelitis and Multiple Sclerosis. J Immunol. 2015;195:2552-9
57. Saligrama N, Zhao F, Sikora MJ, Serratelli WS, Fernandes RA, Louis DM. et al. Opposing T cell responses in experimental autoimmune encephalomyelitis. Nature. 2019;572:481-7
58. Restorick SM, Durant L, Kalra S, Hassan-Smith G, Rathbone E, Douglas MR. et al. CCR6(+) Th cells in the cerebrospinal fluid of persons with multiple sclerosis are dominated by pathogenic non-classic Th1 cells and GM-CSF-only-secreting Th cells. Brain Behav Immun. 2017;64:71-9
59. Ifergan I, Davidson TS, Kebir H, Xu D, Palacios-Macapagal D, Cann J. et al. Targeting the GM-CSF receptor for the treatment of CNS autoimmunity. J Autoimmun. 2017;84:1-11
60. Croxford AL, Spath S, Becher B. GM-CSF in Neuroinflammation: Licensing Myeloid Cells for Tissue Damage. Trends Immunol. 2015;36:651-62
61. Segal BM. The Diversity of Encephalitogenic CD4+ T Cells in Multiple Sclerosis and Its Animal Models. J Clin Med. 2019 8
62. Costa-Pereira S, Lanzinger M, Núñez N, Villar-Vesga J, Andreadou M, Prisco F. et al. Regulatory T cells suppress GM-CSF-producing T helper cells via IL-2 modulation to restrain immunopathology. Cell Rep. 2025;44:115642
63. Van Dis E, Fox DM, Morrison HM, Fines DM, Babirye JP, McCann LH. et al. IFN-γ-independent control of M. tuberculosis requires CD4 T cell-derived GM-CSF and activation of HIF-1α. PLoS Pathog. 2022;18:e1010721
64. McWilliams IL, Rajbhandari R, Nozell S, Benveniste E, Harrington LE. STAT4 controls GM-CSF production by both Th1 and Th17 cells during EAE. J Neuroinflammation. 2015;12:128
65. Zhang H, Zhang S, Zhang J, Liu D, Wei J, Fang W. et al. ZO-1 expression is suppressed by GM-CSF via miR-96/ERG in brain microvascular endothelial cells. J Cereb Blood Flow Metab. 2018;38:809-22
66. Croxford AL, Lanzinger M, Hartmann FJ, Schreiner B, Mair F, Pelczar P. et al. The Cytokine GM-CSF Drives the Inflammatory Signature of CCR2+ Monocytes and Licenses Autoimmunity. Immunity. 2015;43:502-14
67. Ponomarev ED, Shriver LP, Maresz K, Pedras-Vasconcelos J, Verthelyi D, Dittel BN. GM-CSF production by autoreactive T cells is required for the activation of microglial cells and the onset of experimental autoimmune encephalomyelitis. J Immunol. 2007;178:39-48
68. Feng R, Muraleedharan Saraswathy V, Mokalled MH, Cavalli V. Self-renewing macrophages in dorsal root ganglia contribute to promote nerve regeneration. Proc Natl Acad Sci U S A. 2023;120:e2215906120
69. Kobayashi SD, Voyich JM, Whitney AR, DeLeo FR. Spontaneous neutrophil apoptosis and regulation of cell survival by granulocyte macrophage-colony stimulating factor. J Leukoc Biol. 2005;78:1408-18
70. Martin SJ, Brand-Arzamendi K, Saab G, Muccilli A, Oh J, Schneider R. GM-CSF is a marker of compartmentalised intrathecal inflammation in multiple sclerosis. Mult Scler. 2023;29:1373-82
71. Russi AE, Walker-Caulfield ME, Guo Y, Lucchinetti CF, Brown MA. Meningeal mast cell-T cell crosstalk regulates T cell encephalitogenicity. J Autoimmun. 2016;73:100-10
72. Gilgun-Sherki Y, Melamed E, Offen D. The role of oxidative stress in the pathogenesis of multiple sclerosis: the need for effective antioxidant therapy. J Neurol. 2004;251:261-8
73. King IL, Kroenke MA, Segal BM. GM-CSF-dependent, CD103+ dermal dendritic cells play a critical role in Th effector cell differentiation after subcutaneous immunization. J Exp Med. 2010;207:953-61
74. Brück W, Sommermeier N, Bergmann M, Zettl U, Goebel HH, Kretzschmar HA. et al. Macrophages in multiple sclerosis. Immunobiology. 1996;195:588-600
75. Marignier R, Hacohen Y, Cobo-Calvo A, Pröbstel AK, Aktas O, Alexopoulos H. et al. Myelin-oligodendrocyte glycoprotein antibody-associated disease. Lancet Neurol. 2021;20:762-72
76. Nunes JC, Radbruch H, Walz R, Lin K, Stenzel W, Prokop S. et al. The most fulminant course of the Marburg variant of multiple sclerosis-autopsy findings. Mult Scler. 2015;21:485-7
77. Elenein RG, Sharer LR, Cook SD, Pachner AR, Michaels J, Hillen ME. A second case of Marburg's variant of multiple sclerosis with vasculitis and extensive demyelination. Mult Scler. 2011;17:1531-8
78. Pierson ER, Goverman JM. GM-CSF is not essential for experimental autoimmune encephalomyelitis but promotes brain-targeted disease. JCI Insight. 2017;2:e92362
79. Arellano G, Ottum PA, Reyes LI, Burgos PI, Naves R. Stage-Specific Role of Interferon-Gamma in Experimental Autoimmune Encephalomyelitis and Multiple Sclerosis. Front Immunol. 2015;6:492
80. Safavi F, Thome R, Li Z, Zhang GX, Rostami A. Dimethyl fumarate suppresses granulocyte macrophage colony-stimulating factor-producing Th1 cells in CNS neuroinflammation. Neurol Neuroimmunol Neuroinflamm. 2020 7
81. Di Matteo A, Bathon JM, Emery P. Rheumatoid arthritis. Lancet. 2023;402:2019-33
82. Takanashi S, Kaneko Y, Takeuchi T. Elderly patients with comorbidities in the definition of difficult-to-treat rheumatoid arthritis. Ann Rheum Dis. 2021;80:1491-3
83. Campbell IK, Rich MJ, Bischof RJ, Dunn AR, Grail D, Hamilton JA. Protection from collagen-induced arthritis in granulocyte-macrophage colony-stimulating factor-deficient mice. J Immunol. 1998;161:3639-44
84. Yamada H, Haraguchi A, Sakuraba K, Okazaki K, Fukushi JI, Mizu-Uchi H. et al. Th1 is the predominant helper T cell subset that produces GM-CSF in the joint of rheumatoid arthritis. RMD Open. 2017;3:e000487
85. Khullar A, Dhir V, Saikia B, Yadav AK, Leishangthem B, Prasad CB. et al. Rheumatoid arthritis synovial fluid shows enrichment of T-cells producing GMCSF which are polyfunctional for TNFα and IFNγ. Clin Exp Rheumatol. 2024;42:1435-41
86. Bystrom J, Clanchy FI, Taher TE, Al-Bogami MM, Muhammad HA, Alzabin S. et al. Response to Treatment with TNFα Inhibitors in Rheumatoid Arthritis Is Associated with High Levels of GM-CSF and GM-CSF(+) T Lymphocytes. Clin Rev Allergy Immunol. 2017;53:265-76
87. Cook AD, Braine EL, Campbell IK, Rich MJ, Hamilton JA. Blockade of collagen-induced arthritis post-onset by antibody to granulocyte-macrophage colony-stimulating factor (GM-CSF): requirement for GM-CSF in the effector phase of disease. Arthritis Res. 2001;3:293-8
88. Reynolds G, Gibbon JR, Pratt AG, Wood MJ, Coady D, Raftery G. et al. Synovial CD4+ T-cell-derived GM-CSF supports the differentiation of an inflammatory dendritic cell population in rheumatoid arthritis. Ann Rheum Dis. 2016;75:899-907
89. Tsark EC, Wang W, Teng YC, Arkfeld D, Dodge GR, Kovats S. Differential MHC class II-mediated presentation of rheumatoid arthritis autoantigens by human dendritic cells and macrophages. J Immunol. 2002;169:6625-33
90. Nakano H, Lin KL, Yanagita M, Charbonneau C, Cook DN, Kakiuchi T. et al. Blood-derived inflammatory dendritic cells in lymph nodes stimulate acute T helper type 1 immune responses. Nat Immunol. 2009;10:394-402
91. Serbina NV, Salazar-Mather TP, Biron CA, Kuziel WA, Pamer EG. TNF/iNOS-producing dendritic cells mediate innate immune defense against bacterial infection. Immunity. 2003;19:59-70
92. Rivollier A, He J, Kole A, Valatas V, Kelsall BL. Inflammation switches the differentiation program of Ly6Chi monocytes from antiinflammatory macrophages to inflammatory dendritic cells in the colon. J Exp Med. 2012;209:139-55
93. Gravallese EM, Firestein GS. Rheumatoid Arthritis - Common Origins, Divergent Mechanisms. N Engl J Med. 2023;388:529-42
94. Taylor PC, Weinblatt ME, McInnes IB, Atsumi T, Strand V, Takeuchi T. et al. Anti-GM-CSF otilimab versus sarilumab or placebo in patients with rheumatoid arthritis and inadequate response to targeted therapies: a phase III randomised trial (contRAst 3). Ann Rheum Dis. 2023;82:1527-37
95. Shiomi A, Usui T, Ishikawa Y, Shimizu M, Murakami K, Mimori T. GM-CSF but not IL-17 is critical for the development of severe interstitial lung disease in SKG mice. J Immunol. 2014;193:849-59
96. Becher B, Segal BM. T(H)17 cytokines in autoimmune neuro-inflammation. Curr Opin Immunol. 2011;23:707-12
97. Honzawa T, Matsuo K, Hosokawa S, Kamimura M, Kaibori Y, Hara Y. et al. CCR4 plays a pivotal role in Th17 cell recruitment and expansion in a mouse model of rheumatoid arthritis. Int Immunol. 2022;34:635-42
98. Sendo S, Saegusa J, Okano T, Takahashi S, Akashi K, Morinobu A. CD11b+Gr-1(dim) Tolerogenic Dendritic Cell-Like Cells Are Expanded in Interstitial Lung Disease in SKG Mice. Arthritis Rheumatol. 2017;69:2314-27
99. Blanco FJ, Möricke R, Dokoupilova E, Codding C, Neal J, Andersson M. et al. Secukinumab in Active Rheumatoid Arthritis: A Phase III Randomized, Double-Blind, Active Comparator- and Placebo-Controlled Study. Arthritis Rheumatol. 2017;69:1144-53
100. Pavelka K, Chon Y, Newmark R, Lin SL, Baumgartner S, Erondu N. A study to evaluate the safety, tolerability, and efficacy of brodalumab in subjects with rheumatoid arthritis and an inadequate response to methotrexate. J Rheumatol. 2015;42:912-9
101. Mease PJ, Jeka S, Jaller JJ, Kitumnuaypong T, Louthrenoo W, Mann H. et al. CNTO6785, a Fully Human Antiinterleukin 17 Monoclonal Antibody, in Patients with Rheumatoid Arthritis with Inadequate Response to Methotrexate: A Randomized, Placebo-controlled, Phase II, Dose-ranging Study. J Rheumatol. 2018;45:22-31
102. DiMeglio LA, Evans-Molina C, Oram RA. Type 1 diabetes. Lancet. 2018;391:2449-62
103. Wen X, Yang J, James E, Chow IT, Reijonen H, Kwok WW. Increased islet antigen-specific regulatory and effector CD4(+) T cells in healthy individuals with the type 1 diabetes-protective haplotype. Sci Immunol. 2020 5
104. Arif S, Tree TI, Astill TP, Tremble JM, Bishop AJ, Dayan CM. et al. Autoreactive T cell responses show proinflammatory polarization in diabetes but a regulatory phenotype in health. J Clin Invest. 2004;113:451-63
105. Honkanen J, Nieminen JK, Gao R, Luopajarvi K, Salo HM, Ilonen J. et al. IL-17 immunity in human type 1 diabetes. J Immunol. 2010;185:1959-67
106. Kenefeck R, Wang CJ, Kapadi T, Wardzinski L, Attridge K, Clough LE. et al. Follicular helper T cell signature in type 1 diabetes. J Clin Invest. 2015;125:292-303
107. Walker LS, von Herrath M. CD4 T cell differentiation in type 1 diabetes. Clin Exp Immunol. 2016;183:16-29
108. Tatovic D, Marwaha A, Taylor P, Hanna SJ, Carter K, Cheung WY. et al. Ustekinumab for type 1 diabetes in adolescents: a multicenter, double-blind, randomized phase 2 trial. Nat Med. 2024;30:2657-66
109. Mishra A, Jajodia A, Weston E, Jayavelu ND, Garcia M, Hossack D. et al. Identification of functional enhancer variants associated with type I diabetes in CD4+ T cells. Front Immunol. 2024;15:1387253
110. Knoop J, Gavrisan A, Kuehn D, Reinhardt J, Heinrich M, Hippich M. et al. GM-CSF producing autoreactive CD4(+) T cells in type 1 diabetes. Clin Immunol. 2018;188:23-30
111. Hartwig T, Zwicky P, Schreiner B, Yawalkar N, Cheng P, Navarini A. et al. Regulatory T Cells Restrain Pathogenic T Helper Cells during Skin Inflammation. Cell Rep. 2018;25:3564-72.e4
112. Bing SJ, Silver PB, Jittayasothorn Y, Mattapallil MJ, Chan CC, Horai R. et al. Autoimmunity to neuroretina in the concurrent absence of IFN-γ and IL-17A is mediated by a GM-CSF-driven eosinophilic inflammation. J Autoimmun. 2020;114:102507
113. Paust HJ, Song N, De Feo D, Asada N, Tuzlak S, Zhao Y. et al. CD4(+) T cells produce GM-CSF and drive immune-mediated glomerular disease by licensing monocyte-derived cells to produce MMP12. Sci Transl Med. 2023;15:eadd6137
114. Al-Mossawi MH, Chen L, Fang H, Ridley A, de Wit J, Yager N. et al. Unique transcriptome signatures and GM-CSF expression in lymphocytes from patients with spondyloarthritis. Nat Commun. 2017;8:1510
115. Dohlman TH, Ding J, Dana R, Chauhan SK. T Cell-Derived Granulocyte-Macrophage Colony-Stimulating Factor Contributes to Dry Eye Disease Pathogenesis by Promoting CD11b+ Myeloid Cell Maturation and Migration. Invest Ophthalmol Vis Sci. 2017;58:1330-6
116. Piper C, Pesenacker AM, Bending D, Thirugnanabalan B, Varsani H, Wedderburn LR. et al. T cell expression of granulocyte-macrophage colony-stimulating factor in juvenile arthritis is contingent upon Th17 plasticity. Arthritis Rheumatol. 2014;66:1955-60
117. Keller M, Spanou Z, Schaerli P, Britschgi M, Yawalkar N, Seitz M. et al. T cell-regulated neutrophilic inflammation in autoinflammatory diseases. J Immunol. 2005;175:7678-86
118. Lin F, Yu H, Zhang L, Zhou J, Cao Y, Wu S. et al. Differential expression of interleukin-35 receptor distinguishes different subsets of granulocyte-macrophage-colony-stimulating factor-producing T helper cells in a mouse endometriosis model. Mol Immunol. 2023;164:28-38
119. Gartlan KH, Koyama M, Lineburg KE, Chang K, Ensbey KS, Kuns RD. et al. Donor T-cell-derived GM-CSF drives alloantigen presentation by dendritic cells in the gastrointestinal tract. Blood Adv. 2019;3:2859-65
120. Ullrich E, Abendroth B, Rothamer J, Huber C, Büttner-Herold M, Buchele V. et al. BATF-dependent IL-7RhiGM-CSF+ T cells control intestinal graft-versus-host disease. J Clin Invest. 2018;128:916-30
121. Waller EK. A new role for an old cytokine: GM-CSF amplifies GVHD. Blood. 2020;135:520-1
122. Piper C, Zhou V, Komorowski R, Szabo A, Vincent B, Serody J. et al. Pathogenic Bhlhe40+ GM-CSF+ CD4+ T cells promote indirect alloantigen presentation in the GI tract during GVHD. Blood. 2020;135:568-81
123. Gan Y, Zhou H, Guo Y, Huang B, Liu H, Wang Z. et al. A GITRL-mTORC1-GM-CSF Positive Loop Promotes Pathogenic Th17 Response in Primary Sjögren Syndrome. Arthritis Rheumatol. 2024;76:1419-30
124. Baldassari LE, Rose JW. Daclizumab: Development, Clinical Trials, and Practical Aspects of Use in Multiple Sclerosis. Neurotherapeutics. 2017;14:842-58
125. Gold R, Giovannoni G, Selmaj K, Havrdova E, Montalban X, Radue EW. et al. Daclizumab high-yield process in relapsing-remitting multiple sclerosis (SELECT): a randomised, double-blind, placebo-controlled trial. Lancet. 2013;381:2167-75
126. ▼ Daclizumab withdrawn from the market worldwide. Drug Ther Bull. 2018; 56: 38.
127. Baslund B, Tvede N, Danneskiold-Samsoe B, Larsson P, Panayi G, Petersen J. et al. Targeting interleukin-15 in patients with rheumatoid arthritis: a proof-of-concept study. Arthritis Rheum. 2005;52:2686-92
128. Sandborn WJ, Rebuck R, Wang Y, Zou B, Adedokun OJ, Gasink C. et al. Five-Year Efficacy and Safety of Ustekinumab Treatment in Crohn's Disease: The IM-UNITI Trial. Clin Gastroenterol Hepatol. 2022;20:578-90.e4
129. Panaccione R, Danese S, Sandborn WJ, O'Brien CD, Zhou Y, Zhang H. et al. Ustekinumab is effective and safe for ulcerative colitis through 2 years of maintenance therapy. Aliment Pharmacol Ther. 2020;52:1658-75
130. Gordon KB, Strober B, Lebwohl M, Augustin M, Blauvelt A, Poulin Y. et al. Efficacy and safety of risankizumab in moderate-to-severe plaque psoriasis (UltIMMa-1 and UltIMMa-2): results from two double-blind, randomised, placebo-controlled and ustekinumab-controlled phase 3 trials. Lancet. 2018;392:650-61
131. Constantinescu CS, Asher A, Fryze W, Kozubski W, Wagner F, Aram J. et al. Randomized phase 1b trial of MOR103, a human antibody to GM-CSF, in multiple sclerosis. Neurol Neuroimmunol Neuroinflamm. 2015;2:e117
132. Crotti C, Biggioggero M, Becciolini A, Agape E, Favalli EG. Mavrilimumab: a unique insight and update on the current status in the treatment of rheumatoid arthritis. Expert Opin Investig Drugs. 2019;28:573-81
133. Burmester GR, Feist E, Sleeman MA, Wang B, White B, Magrini F. Mavrilimumab, a human monoclonal antibody targeting GM-CSF receptor-α, in subjects with rheumatoid arthritis: a randomised, double-blind, placebo-controlled, phase I, first-in-human study. Ann Rheum Dis. 2011;70:1542-9
134. Burmester GR, Weinblatt ME, McInnes IB, Porter D, Barbarash O, Vatutin M. et al. Efficacy and safety of mavrilimumab in subjects with rheumatoid arthritis. Ann Rheum Dis. 2013;72:1445-52
135. Takeuchi T, Tanaka Y, Close D, Godwood A, Wu CY, Saurigny D. Efficacy and safety of mavrilimumab in Japanese subjects with rheumatoid arthritis: findings from a Phase IIa study. Mod Rheumatol. 2015;25:21-30
136. Burmester GR, McInnes IB, Kremer J, Miranda P, Korkosz M, Vencovsky J. et al. A randomised phase IIb study of mavrilimumab, a novel GM-CSF receptor alpha monoclonal antibody, in the treatment of rheumatoid arthritis. Ann Rheum Dis. 2017;76:1020-30
137. Guo X, Higgs BW, Bay-Jensen AC, Wu Y, Karsdal MA, Kuziora M. et al. Blockade of GM-CSF pathway induced sustained suppression of myeloid and T cell activities in rheumatoid arthritis. Rheumatology (Oxford). 2018;57:175-84
138. Weinblatt ME, McInnes IB, Kremer JM, Miranda P, Vencovsky J, Guo X. et al. A Randomized Phase IIb Study of Mavrilimumab and Golimumab in Rheumatoid Arthritis. Arthritis Rheumatol. 2018;70:49-59
139. Burmester GR, McInnes IB, Kremer JM, Miranda P, Vencovský J, Godwood A. et al. Mavrilimumab, a Fully Human Granulocyte-Macrophage Colony-Stimulating Factor Receptor α Monoclonal Antibody: Long-Term Safety and Efficacy in Patients With Rheumatoid Arthritis. Arthritis Rheumatol. 2018;70:679-89
140. Behrens F, Tak PP, Østergaard M, Stoilov R, Wiland P, Huizinga TW. et al. MOR103, a human monoclonal antibody to granulocyte-macrophage colony-stimulating factor, in the treatment of patients with moderate rheumatoid arthritis: results of a phase Ib/IIa randomised, double-blind, placebo-controlled, dose-escalation trial. Ann Rheum Dis. 2015;74:1058-64
141. Genovese MC, Berkowitz M, Conaghan PG, Peterfy C, Davy K, Fisheleva E. et al. MRI of the joint and evaluation of the granulocyte-macrophage colony-stimulating factor-CCL17 axis in patients with rheumatoid arthritis receiving otilimab: a phase 2a randomised mechanistic study. Lancet Rheumatol. 2020;2:e666-e76
142. Buckley CD, Simón-Campos JA, Zhdan V, Becker B, Davy K, Fisheleva E. et al. Efficacy, patient-reported outcomes, and safety of the anti-granulocyte macrophage colony-stimulating factor antibody otilimab (GSK3196165) in patients with rheumatoid arthritis: a randomised, phase 2b, dose-ranging study. Lancet Rheumatol. 2020;2:e677-e88
143. Fleischmann RM, van der Heijde D, Strand V, Atsumi T, McInnes IB, Takeuchi T. et al. Anti-GM-CSF otilimab versus tofacitinib or placebo in patients with active rheumatoid arthritis and an inadequate response to conventional or biologic DMARDs: two phase 3 randomised trials (contRAst 1 and contRAst 2). Ann Rheum Dis. 2023;82:1516-26
144. Weinblatt ME, Taylor PC, McInnes IB, Atsumi T, Strand V, Takeuchi T. et al. Long-term safety and efficacy of anti-GM-CSF otilimab in patients with rheumatoid arthritis: long-term extension of three phase 3 randomised trials (contRAst X). BMJ Open. 2025;15:e088869
145. Huizinga TW, Batalov A, Stoilov R, Lloyd E, Wagner T, Saurigny D. et al. Phase 1b randomized, double-blind study of namilumab, an anti-granulocyte macrophage colony-stimulating factor monoclonal antibody, in mild-to-moderate rheumatoid arthritis. Arthritis Res Ther. 2017;19:53
146. Papp KA, Gooderham M, Jenkins R, Vender R, Szepietowski JC, Wagner T. et al. Granulocyte-macrophage colony-stimulating factor (GM-CSF) as a therapeutic target in psoriasis: randomized, controlled investigation using namilumab, a specific human anti-GM-CSF monoclonal antibody. Br J Dermatol. 2019;180:1352-60
147. Taylor PC, Saurigny D, Vencovsky J, Takeuchi T, Nakamura T, Matsievskaia G. et al. Efficacy and safety of namilumab, a human monoclonal antibody against granulocyte-macrophage colony-stimulating factor (GM-CSF) ligand in patients with rheumatoid arthritis (RA) with either an inadequate response to background methotrexate therapy or an inadequate response or intolerance to an anti-TNF (tumour necrosis factor) biologic therapy: a randomized, controlled trial. Arthritis Res Ther. 2019;21:101
148. Worth C, Al-Mossawi MH, Macdonald J, Fisher BA, Chan A, Sengupta R. et al. Granulocyte-macrophage colony-stimulating factor neutralisation in patients with axial spondyloarthritis in the UK (NAMASTE): a randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Rheumatol. 2024;6:e537-e45
149. Bakhshi S, Shamsi S. MCC950 in the treatment of NLRP3-mediated inflammatory diseases: Latest evidence and therapeutic outcomes. Int Immunopharmacol. 2022;106:108595
150. Coll RC, Hill JR, Day CJ, Zamoshnikova A, Boucher D, Massey NL. et al. MCC950 directly targets the NLRP3 ATP-hydrolysis motif for inflammasome inhibition. Nat Chem Biol. 2019;15:556-9
151. Guo C, Fu R, Wang S, Huang Y, Li X, Zhou M. et al. NLRP3 inflammasome activation contributes to the pathogenesis of rheumatoid arthritis. Clin Exp Immunol. 2018;194:231-43
152. Saber S, El-Kader EMA. Novel complementary coloprotective effects of metformin and MCC950 by modulating HSP90/NLRP3 interaction and inducing autophagy in rats. Inflammopharmacology. 2021;29:237-51
153. Perera AP, Fernando R, Shinde T, Gundamaraju R, Southam B, Sohal SS. et al. MCC950, a specific small molecule inhibitor of NLRP3 inflammasome attenuates colonic inflammation in spontaneous colitis mice. Sci Rep. 2018;8:8618
154. Zhou J, Onodera S, Yu Q. Inhibition of NLRP3 inflammasome activity by MCC950 leads to exacerbation of Sjӧgren's syndrome pathologies in non-obese diabetic mice. Immunology. 2023;168:697-708
155. Arnold DD, Yalamanoglu A, Boyman O. Systematic Review of Safety and Efficacy of IL-1-Targeted Biologics in Treating Immune-Mediated Disorders. Front Immunol. 2022;13:888392
156. Gregory SG, Schmidt S, Seth P, Oksenberg JR, Hart J, Prokop A. et al. Interleukin 7 receptor alpha chain (IL7R) shows allelic and functional association with multiple sclerosis. Nat Genet. 2007;39:1083-91
157. Nelson EA, Walker SR, Xiang M, Weisberg E, Bar-Natan M, Barrett R. et al. The STAT5 Inhibitor Pimozide Displays Efficacy in Models of Acute Myelogenous Leukemia Driven by FLT3 Mutations. Genes Cancer. 2012;3:503-11
158. Xiao Z, Liang J, Deng Q, Song C, Yang X, Liu Z. et al. Pimozide augments bromocriptine lethality in prolactinoma cells and in a xenograft model via the STAT5/cyclin D1 and STAT5/Bcl-xL signaling pathways. Int J Mol Med. 2021;47:113-24
159. Lin JX, Ge M, Liu CY, Holewinski R, Andresson T, Yu ZX. et al. Tyrosine phosphorylation of both STAT5A and STAT5B is necessary for maximal IL-2 signaling and T cell proliferation. Nat Commun. 2024;15:7372
Author contact
![]() Corresponding authors: Xiang-Yu Meng, Health Science Center, Hubei Minzu University, 39 Xueyuan Road, Enshi 445000, China. E-mail: mengxy_whucom. Yi Liu, Department of Rheumatology and Immunology, West China Hospital, Sichuan University, 37 Guoxue Lane, Chengdu 610041, Sichuan, China. E-mail: yiliu8999cn.
Corresponding authors: Xiang-Yu Meng, Health Science Center, Hubei Minzu University, 39 Xueyuan Road, Enshi 445000, China. E-mail: mengxy_whucom. Yi Liu, Department of Rheumatology and Immunology, West China Hospital, Sichuan University, 37 Guoxue Lane, Chengdu 610041, Sichuan, China. E-mail: yiliu8999cn.