Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
1. Introduction
2. Mitochondria-Mediated...
3. Pharmacological Therapies for...
4. Nanotechnology-Based...
5. Discussion
6. Conclusion and Future...
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2026; 16(1):272-297. doi:10.7150/thno.121956 This issue Cite
Review
The application of nanotechnology in regulating mitochondrial function in tumor microenvironment for cancer therapy
Dazhong Wang2,#, Meng Yuan2,#, Ji Liu2,#, Ming Zhao1, ![]() , Ting Fang1,
, Ting Fang1, ![]()
1. Department of Pharmaceutics, School of Pharmacy, Shenyang Pharmaceutical University, No. 103, Wenhua Road, Shenyang, 110016, P. R. China.
2. Cancer Hospital of Dalian University of Technology, Liaoning Cancer Hospital & Institute No. 44 Xiaoheyan Road, Dadong District, Shenyang, 110042, Liaoning Province, P. R. China.
# These authors contributed to the work equally.
Received 2025-4-1; Accepted 2025-9-1; Published 2026-1-1
Abstract

Mitochondria are involved in energy production, signal conduction, and cellular differentiation in the human body, and they determine the direction of tumorigenesis and development. Mitochondria-targeted therapy in cancer cells has been reported since researchers discovered the relationship between mitochondria and cancer. However, the complexity of the tumor microenvironment (TME) can impair the therapeutic effect. Understanding the mechanisms of mitochondrial function in various cells of TME (e.g., tumor-associated macrophages (TAMs), cancer-associated fibroblasts (CAFs), cancer stem cells (CSCs), T cells, natural killer (NK) cells, tumor-associated neutrophils (TANs)), as well as mediated crosstalk with cancer cells, would be beneficial for accelerating these therapeutic strategies into clinical practice and leading to more effective disease treatment. Subsequently, we summarized representative small-molecule drugs targeting mitochondrial homeostasis, energy metabolism, and mitochondrial DNA (mtDNA) and evaluated their limitations. Building on this foundation, we reviewed the latest multifunctional nanomedicines. These agents leverage TME responsiveness, surface-targeting engineering, and multimodal synergy (combining chemotherapy, photodynamic therapy (PDT), sonodynamic therapy (SDT), radiodynamic therapy (RDT), and immunotherapy) to precisely deliver drugs, ions, genetic material, and even whole mitochondria to target organelles. This approach simultaneously remodels the immunosuppressive microenvironment and induces immunogenic cell death (ICD).
Keywords: tumor treatment, tumor microenvironment, mitochondrial function, nanodrugs
1. Introduction
There are several leading causes of cancer in all countries, as well as a number of barriers to increasing life expectancy because of cancer [1]. Current treatments for tumors include surgery, chemotherapeutic drugs, radiotherapy, and immunotherapy, administered alone or in combination. However, issues are emerging, such as multidrug resistance (MDR), stem-like cell growth, and an immunosuppressive tumor microenvironment (TME), due to the non-specific targeting of tumor cells and low cytotoxicity [2, 3]. Mitochondria have long been recognized as the metabolic engines of eukaryotic cells, and in the context of cancer, they operate as decision-making hubs that integrate nutrient availability, redox status, and death-survival signals [4-6]. Mitochondria are crucial for cancer cell survival through mechanisms like morphology, mitophagy, mitochondrial reactive oxygen species (mtROS) production, metabolism, mitochondrial DNA (mtDNA), and calcium signaling. They are essential for tumor development, invasion, metastasis, and treatment resistance, and also regulate cell growth, differentiation, and apoptosis [5].
Mitochondria orchestrate oncogenesis through multiple, interconnected mechanisms. Dynamic cycles of fusion and fission dictate metabolic flexibility, allowing tumors to switch between glycolysis and oxidative phosphorylation (OXPHOS) in response to fluctuating nutrient and oxygen availability [7-9]. Excessive mtROS generated by a hyperactive electron transport chain (ETC) promotes genomic instability, epithelial-mesenchymal transition (EMT), and therapy resistance [10]. Importantly, these adaptations are not restricted to malignant cells; tumor-associated macrophages (TAMs), cancer-associated fibroblasts (CAFs), cancer stem cells (CSCs), tumor-infiltrating T cells (TILs), natural killer (NK) cells, and neutrophils all exhibit mitochondria-driven metabolic identities that collectively shape an immunosuppressive and metabolically hostile niche [11]. Emerging research highlights intercellular mitochondrial transfer as a critical focus, demonstrating that: tumor cells deliver mitochondria harboring mutated mtDNA to TILs, impairing their antitumor functionality [12]; mitochondrial transfer from CAFs to tumor cells promotes metastatic progression [13]; while pharmacologically blocking mitochondrial trafficking from immune cells to tumor cells effectively suppresses tumor growth in experimental models [14].
Strategies targeting mitochondrial modulation offer a promising solution. The high negative membrane potential of the mitochondrial inner membrane necessitates unique physicochemical properties for drug delivery [15]. Lipophilic cations exploit electrostatic attraction and hydrophobic interactions to accumulate within the mitochondrial matrix, with triphenylphosphonium (TPP) and dequalinium (DQA) serving as widely exploited mitochondrial-targeting moieties for small molecule design [16]. Although exhibiting high mitochondrial specificity, these cationic carriers frequently induce multidrug resistance, compromising therapeutic efficacy [17]. Although compounds like the Bcl-2 antagonist ABT-737 promote tumor apoptosis by altering mitochondrial membrane permeability, their therapeutic potential is limited by challenges such as poor oral bioavailability [18]. Biguanide antidiabetic drugs (e.g., metformin, phenformin, buformin), established mitochondrial ETC inhibitors, have yielded disappointing clinical outcomes to date [19]. Notably, IACS-010759 effectively suppresses proliferation in glycolysis-deficient hypoxic tumor cells. Paradoxically, OXPHOS inhibition by this compound induces mitochondrial transfer from bone marrow mesenchymal stem cells (MSCs) to acute myeloid leukemia (AML) cells via tunneling nanotubes (TNTs), while concurrently triggering mitochondrial fission and mitophagy in AML cells—mechanisms that collectively enhance cancer cell migration [20].
The complexity of the TME, however, has hindered the clinical translation of conventional small-molecule mitochondrial modulators [21]. Systemic administration often leads to off-target toxicities in highly metabolic organs, while heterogeneous intra-tumoral oxygenation gradients and enzymatic heterogeneity establish pharmacological sanctuaries that facilitate resistance development [22]. Nanotechnology offers an unprecedented opportunity to overcome these barriers. By exploiting pH gradients, enzymatic signatures, or receptor overexpression within the TME, nanodrug delivery systems (NDDS) can deliver drugs, metal complexes, nucleic acids, or even isolated mitochondria to defined subcellular locations [16, 23]. Furthermore, precise modulation of mitochondrial dysfunction can be achieved through either monotherapy or combination regimens incorporating chemotherapy, photodynamic therapy (PDT), sonodynamic therapy (SDT), radiodynamic therapy (RDT), and/or immunotherapy [24, 25]; concurrently inducing immunogenic cell death (ICD) characterized by damage-associated molecular pattern release, thereby initiating adaptive antitumor immunity [26].
In this review, we synthesize the latest advances in understanding mitochondria-mediated crosstalk between tumor cells and their microenvironmental partners. First, we dissect the metabolic reprogramming of each major TME component, highlighting how mitochondrial dynamics, biogenesis, mitophagy, and metabolite exchange dictate pro- or anti-tumor phenotypes. Second, we catalog emerging pharmacological agents that target mitochondrial balance, energy metabolism, or mtDNA integrity, assessing their therapeutic potential and limitations. Finally, we critically evaluate how multifunctional nanodrugs can be engineered to translate these insights into precision cancer therapy, and we outline the key translational hurdles that must be addressed to bring mitochondria-targeted nanomedicine to the clinic.
2. Mitochondria-Mediated Cellular Crosstalk within TME
Mitochondria in cancer cells are different from those in normal cells in terms of energy metabolism, increased mitochondrial membrane potential (MMP), ROS production, low oxygen (O2) consumption, and high glutathione (GSH) levels [23]. Similarly, the mitochondria of tumor-associated stromal cells, CSCs, and tumor-associated immune cells are also different from those of normal cells, promoting or inhibiting tumor progression. This section aims to investigate the mechanism of mitochondria-mediated crosstalk between cancer cells and various cancer-associated cells, including TAMs, CAFs, CSCs, lymphoid lineage immune cells, and TANs within the TME (Figure 1).
The role of mitochondria in TME. Mitochondria-mediated crosstalk between cancer cells and cancer-associated cells within TME, inducing the mitochondrial metabolic reprogramming and anti-/pro-tumor effect. Created with BioRender.com. (http://biorender.com).
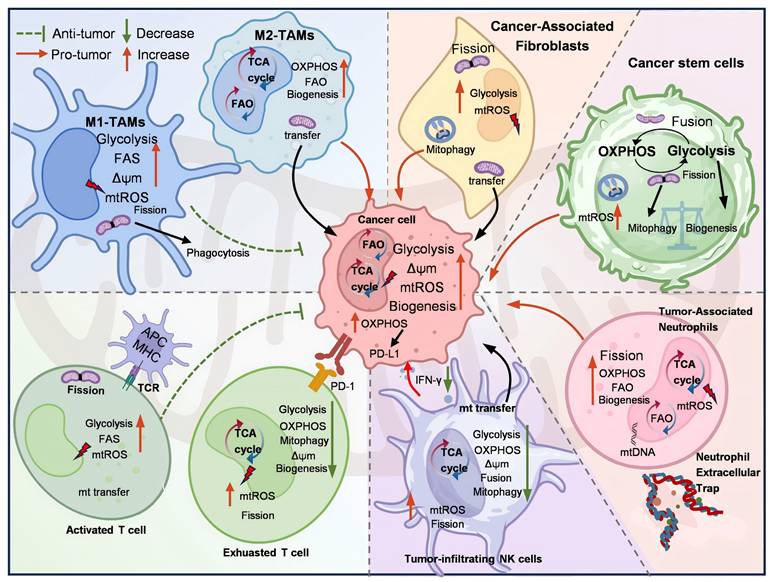
2.1 Mitochondria-mediated Interactions between TAMs and tumor cells
TAMs, defined as macrophages (MФ) that infiltrate and reside within TMEs, can be categorized into two distinct subsets. M1-TAMs are induced by T helper 1 (Th1) cytokines, including interferon-gamma (IFN-γ), granulocyte-macrophage colony-stimulating factor, and lipopolysaccharide. Conversely, M2-TAMs are activated by T helper 2 (Th2) cytokines, such as interleukin-4 (IL-4), interleukin-10 (IL-10), macrophage colony-stimulating factor (M-CSF), or tumor cell-surface molecules [27-29]. Notably, TAMs with an anti-inflammatory and pro-tumor M2 phenotype predominantly secrete immunosuppressive cytokines, such as IL-10 and transforming growth factor-beta (TGF-β), thereby promoting tumor proliferation and metastasis. In contrast, pro-inflammatory and anti-tumor M1-TAMs inhibit tumor progression and induce tissue damage by secreting IL-6, tumor necrosis factor-alpha (TNF-α), nitric oxide (NO), and ROS [30].
M1-TAMs predominantly use glycolysis for metabolism, increasing citrate and succinate levels. These metabolites fuel FAS and boost mitochondrial ROS and mtDNA production [31]. mtDNA activates the cGAS-STING pathway in TAMs, triggering IFN-I secretion (Figure 2). In contrast, IL-4 activates M2-TAMs, allowing FFA influx that stimulates STAT6. This promotes FAO, mitochondrial biogenesis, and TCA cycle activity to generate ATP, NADH, and FADH2 for OXPHOS [32]. M2-TAMs also upregulate ARG1, ARG2, and IDO1, diverting L-arginine to the urea cycle. These key metabolic changes in TME promote TGF-β secretion, thereby suppressing effector T cells. Additionally, TAM mitochondrial reprogramming releases cytokines (CCL2, IL-1α, IL-6, TNF-α) to effectively support colon cancer growth [33]. Activated STAT6 drives PPARδ and PPARγ expression, leading to the overexpression of anti-inflammatory genes.
Mitochondria metabolic reprogramming within cancer cells and TAMs and mitochondria-mediated crosstalk between them. Macrophage mitochondrial fission enhances phagocytosis, while fusion suppresses it. Beyond induction by Th1 cytokines, tumor-released damaged mtDNA activates cGAS-STING to drive IFN-I-mediated M1-TAM polarization and stimulates TLR9 to promote M2-TAM polarization. M1-TAMs shift from OXPHOS to glycolysis, increasing pyruvate flux toward acetyl-CoA; subsequent TCA cycle elevation boosts citrate (for FAS) and succinate (for ROS generation). Conversely, M2-TAMs exhibit increased free fatty acid influx fueling FAO, which enhances TCA cycling, OXPHOS, and mitochondrial biogenesis. Mitochondrial transfer from M2-TAMs to tumor cells induces ROS bursts that promote tumor proliferation. Created with BioRender.com. (http://biorender.com).
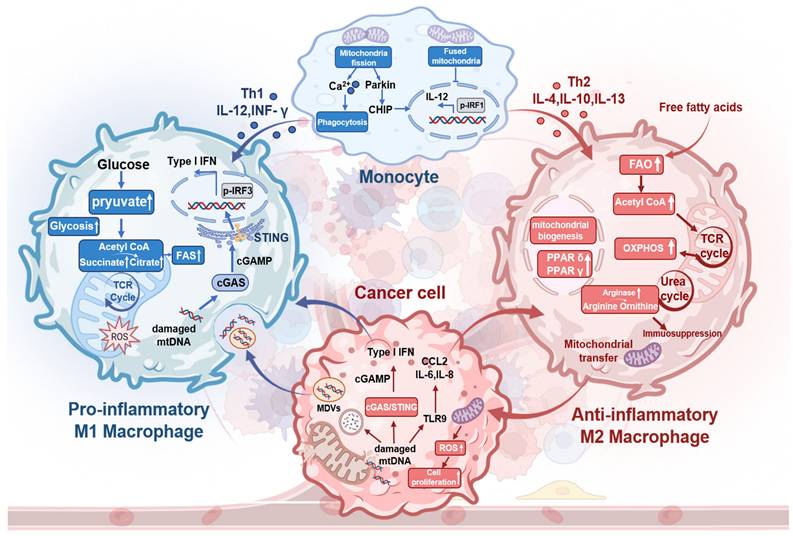
A continuous mitochondrial network maintained via fusion sustains high FAO and OXPHOS levels in M2-TAMs. Altering TAM mitochondrial dynamics impacts tumor biology: fusion suppresses M1-TAM polarization by inhibiting IRF1-mediated IL-12 production, while fragmentation activates downstream IRF1 through Parkin-dependent CHIP proteolysis [34]. Mitochondrial fission in MФ raises cytoplasmic calcium, enhancing phagocytosis via PKC-θ-mediated phosphorylation of WIP [35]. Fragmented MФ overexpresses ARG1 and shows increased phagocytic activity. M1-TAMs feature elongated mitochondria compared to M2 and unpolarized MФ, but tumors inhibit TAM fission to evade clearance. TAM-to-cancer cell mitochondrial transfer via tunneling nanotubes promotes cell motility, invasion, and EMT [36, 37]. This TAM-tumor mitochondrial crosstalk drives poor treatment outcomes.
2.2 Mitochondria-mediated Interactions between CAFs and tumor cells
CAFs are a heterogeneous population with different functions, mainly derived from resident fibroblasts that are induced by a variety of factors, such as TGF-β, IL-1β, and fibroblast growth factor (FGF). Mutual communication between CAFs and cancer cells involves the exchange of metabolites, the regulation of signaling pathways, and effects on tumor growth and metastasis. The activity of fibroblast mitochondria, which is involved in signaling pathways leading to metabolic switching and is often seen in CAFs, could either be a consequence of their activation by cancer cells or an active phenomenon favoring tumor development [38].
ROS and soluble factors (e.g., TGF-β) released by cancer cells mediate the transformation of normal fibroblasts into catabolic CAFs with reprogrammed metabolism from OXPHOS to aerobic glycolysis [39]. Overproduction of ROS not only damages the cells but also induces mitophagy in the mitochondria of CAFs [40]. Autophagy in CAFs inhibits tumor progression by promoting immunity and suppressing hypoxic conditions in the early stages. However, autophagy in CAFs promotes tumor development via providing nutrients (L-lactate, ketone bodies), cytokines (IL-1β, IL-6, IL-8), CXCL12, and α-SMA in advanced stages of tumorigenesis [41]. Mitochondrial transfer from normal oral fibroblasts (NOFs) to OSCC via TNTs is similar to what occurs between TAMs and cancer cells [40].
CAFs provide intermediate metabolites for mitochondrial activity in cancer cells. These metabolites reorganize cancer cells' metabolism to fuel the TCA cycle and/or to promote mitochondrial biogenesis, as well as supply three major nutrients, thereby providing resources for tumor growth and metastasis. In addition to substance transport, and accumulation of mitochondrial metabolites in tumors can induce the upregulation of mitochondrial activity in cancer cells [42]. Accumulation of succinate and fumarate induced HIF-1α stabilization and EMT, respectively [43]. When co-cultured with OSCC cells, NOFs and CAFs are the main sources of L-lactate, which is transported across membranes via monocarboxylate transporters [44]. The uptake of lactate by OSCC and prostate cancer cells shifts the NAD⁺/NADH ratio, leading to NADH accumulation. Subsequent oxidation of NADH by mitochondrial complex I drives ATP production, which fuels tumorigenic progression. Reciprocal interactions between CAFs and cancer cells have been shown to enhance the glycolytic capacity of CAFs, while cancer cells increase their mitochondrial function. Accumulation of mitochondrial metabolites induces EMT, disrupts redox balance, allowing for increased energy production by cancer cells, and stimulates tumor metastasis. The intricate dialogue between CAFs and cancer cells is a pivotal aspect of tumor progression and microenvironment modulation. In addition to the differences among these cancers, which depend on the tissues from which they originate, intra-tumor heterogeneity may be the source of interactions between different cancer cells and CAFs.
2.3 The mitochondria of CSCs
Treatment resistance in tumor cells and CSCs is a significant contributor to treatment failure and recurrence. CSCs, also termed as tumor-initiating cells, exhibit characteristics of tumor initiation, self-renewal, multidirectional differentiation, and plasticity within the TME [45, 46]. Maintenance of stemness and differentiation of CSCs depends on mitochondrial regulation, including mitochondrial dynamics, mitochondrial metabolic reprogramming, homeostatic imbalance, and mtDNA depletion [47, 48].
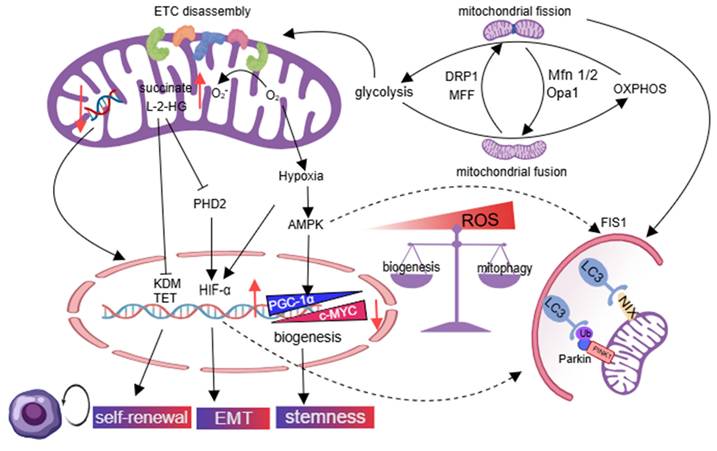
Glycolysis can serve as the main energy source for stem cells, which helps CSCs survive under hypoxic conditions [49]. The fragmented mitochondria are associated with overactivation of DRP1 and mitochondrial fission factor (MFF), which promote a metabolic transition from OXPHOS to glycolysis and attenuate ROS production in tamoxifen-resistant breast cancer, brain tumor-initiating cells, and liver CSCs, thereby preserving the self-renewal potential of CSCs [50]. The compromised aerobic oxidation function of fragmented mitochondria leads to the buildup of succinate and L-2-hydroxyglutarate (L-2-HG), which impedes the activity of ten-eleven translocation and histone lysine demethylase enzymes, ultimately promoting the self-renewal capacity of CSCs [51]. Succinate inhibits hydroxylation and subsequent degradation of HIF-1α by blocking α-ketoglutarate-dependent prolyl hydroxylases, and triggers the EMT, resulting in the emergence of CSCs. CSCs use mitophagy and mitochondrial biogenesis to maintain aggressive phenotypes and promote tumor relapse. AMPK/AP-1 pathway phosphorylates peroxisome proliferator-activated receptor γ co-activator 1α (PGC-1α), leading to improper mitochondria biogenesis, which supports the stemness of CSCs and their capacity for carcinogenesis [52, 53]. The MYC/PGC-1α ratio determines metabolic phenotype in pancreatic CSCs, with MYC promoting a glycolytic phenotype and inhibiting OXPHOS, while PGC-1α promotes mitochondrial biogenesis. PGC-1α, with its antioxidant properties, maintains CSC self-renewal by controlling low ROS levels, while increasing ROS levels can induce CSC death [54]. Fragmented mitochondria trigger mitophagy through BNIP3L in CSCs with glycolytic phenotypes, while adenosine monophosphate-activated protein kinase (AMPK) activation induces mitochondrial fission 1 (FIS1)-mediated mitophagy in leukemia stem cells (LSCs) to increase ROS levels and promote cell proliferation [55, 56]. The balance between mitophagy and mitochondrial biogenesis is controlled by CSCs, promoting aggressive CSC phenotypes and tumor relapse. CSCs in leukemia, colon cancer, and cholangiocarcinoma prefer OXPHOS [45, 57, 58]. Overexpression of mitochondrial fusion proteins (MFN1/2) and optic atrophy 1 (OPA1) upregulates mitochondrial mass, as well as enhances OXPHOS and NAD+ levels. Decreasing mtDNA copy number triggers retrograde signaling, leading to changes in nuclear gene expression that help cancer cells adapt to the tumor microenvironment and maintain their stemness [59] (Figure 3).
Mitochondrial metabolic reprogramming and homogeneity balance are disrupted in CSCs. Mitochondrial hyper-fission in cancer shifts metabolism from OXPHOS to glycolysis. Resultant mitochondrial dysfunction promotes CSC self-renewal, EMT, and stemness via altered nuclear proteins. Impaired mitochondrial biogenesis sustains CSCs through low ROS, while AMPK-activated mitophagy in LSCs elevates ROS to drive proliferation. Conversely, fusion-enriched tumors favor OXPHOS as a novel CSC trait. Created with Medpeer.com. (https://www.medpeer.cn).
2.4 The mitochondria of T and B lymphocytes
The T cell population includes naïve (TN), effector (TE), memory (TM), and regulatory (Treg) cells. Mitochondrial structure and function change during T cell development, influencing their function. TN cells have round mitochondria that depend on OXPHOS and FAO for metabolic quiescence, while TE cells have fragmented mitochondria with increased glycolysis, FAS, and OXPHOS linked to their proliferative state [60] (Figure 4). Asymmetric division leads to TM/Treg cells, with TM cells showing long, tubular mitochondria through Opa1-dependent fusion. Memory T cells revert to a quiescent metabolic state supported by FAO and OXPHOS [61]. CD8+ T cells with low mitochondrial membrane potential (ΔΨm) are more effective in vivo [62]. Tregs, which inhibit T cell activation and proliferation, prefer the FAO pathway for energy efficiency [63].
Mitochondria control the developmental and differentiation state of T cells. Naïve T cells primarily rely on OXPHOS and FAO for energy. Upon activation by APCs, T cells undergo Drp1-dependent mitochondrial fission and shift their metabolism toward aerobic glycolysis. However, within tumor microenvironments, exhausted T cells (TEX) exhibit high PD-1 expression. This suppresses OXPHOS, increases oxidative stress, and impairs mitochondrial biogenesis and function. Reduced mitophagy leads to accumulated depolarized mitochondria, while mitochondrial transfer further compromises T cell migration and activation - collectively accelerating TEX development. Created with Medpeer.com. (https://www.medpeer.cn).
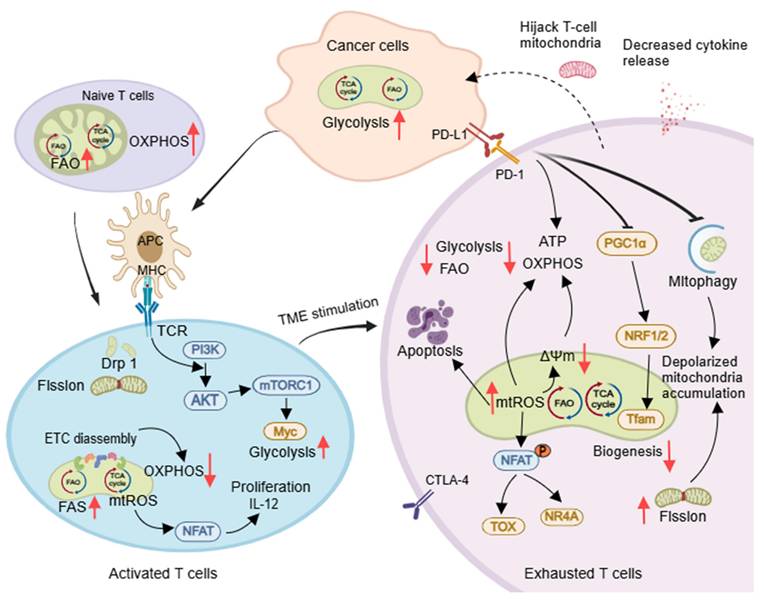
The activation of expansion of T cells is supported by DRP1-dependent mitochondrial fission, which promotes aerobic glycolysis and increases mitochondrial numbers and cristae loosening. This process occurs at immune synapses between the T-cell receptor (TCR) and antigen-presenting cells (APC) [64]. TCR also enhances glycolysis both in activated B lymphocytes [65]and T cells [66]via PI3K/AKT/mTOR pathway, and generates mtROS to activate the expression of transcription factor nuclear factor of activated T cells and IL-2, essential for maturation and proliferation of T cells [67]. Mitochondrial cristae loosening can reduce ETC supercomplex efficiency, promoting aerobic glycolysis [68]. Activated CD8+ T cells release perforin and granzyme and induce apoptosis mediated by death ligands/death receptors, leading to direct destruction of tumor cells (Figure 4). Defective mitochondrial fission in CD8+ T cells can reduce infiltration in solid cancer models [69].
T exhausted cells (TEX) within tumors are no longer able to respond to stimuli, and the onset of their exhaustion is driven by multiple tumor microenvironmental factors. These include persistent tumor antigenic stimulation, nutrient deprivation (such as glucose competition), metabolite accumulation (e.g., lactate), and hypoxia, as well as high expression of inhibitory receptors such as programmed cell death protein 1 (PD-1), TIM-3, LAG-3, and CTLA4 [70, 71]. These factors decrease mitochondrial quantity and alter function, restricting glycolysis due to glucose deficiency. Therapeutic strategies for TEX involve reducing ROS, lowering tumor hypoxia to prevent T cell exhaustion [71], inhibiting glycolysis to enhance CD8+ TM cell function [72], and promoting FAO to regulate mitochondrial CD8+ TILs (e.g., PGC-1α agonist bezafibrate) [73]. Further research is needed to understand how tumor-infiltrating B lymphocytes change their mitochondrial metabolism and how this impacts antitumor immune responses [74].
2.5 The mitochondria of NK cells
NK cells are innate cytotoxic lymphocytes that induce cell death through direct cytotoxicity and modulate a multicellular protective immune response by secreting cytokines and chemokines [75]. Additionally, NK cells can interact with dendritic cells, macrophages, and T cells to elicit an immune response [76]. An increase in mitochondrial mass and glycolysis occurs in NK cells upon activation, accompanied by higher basal OXPHOS rates. Furthermore, redistribution and accumulation of mitochondria at NK cell immune synapses formed during interactions with tumor cells have been observed [77].
Mitochondrial changes in tumor-infiltrating NK (ITNK) cells impair NK cellular functions in the TME with increased mitochondrial fragmentation, lower ΔΨm, and increased mtROS [78]. Metabolic deficits in peripheral blood NK cells from metastatic breast cancer patients can be rescued by blocking TGF-β [79]. Mitochondrial dynamics control the mature NK cell fitness. Circulating human CD56 dimCD16+NK (NKDim) cells have fused mitochondria and higher toxicity than CD56Br. Whereas mitochondrial fission and depolarization in NKDim impaired IFN-γ production after IL-2/12 stimulation, resulting in non-specific toxicity, this can be rescued by inhibiting mitochondrial division [80]. However, Terren et al. showed that cytokine stimulation does not significantly depolarize CD56bright and CD56dim NK cell subsets. IL-12/15/18 stimulation reduces mitochondrial cristae density, possibly through decreased OPA1 expression. Failure to upregulate mitophagy upon stimulation could lead to the accumulation of defective mitochondria, excessive ROS, and cell death. Altered mitochondrial morphology caused by hypoxia promotes tumor immune escape, proliferation, and metastasis [81]. NK cells can induce mitochondrial apoptosis in cancer cells, affecting their sensitivity to NK-mediated killing. Preactivated NK cells are resistant to BH3 mimetics, but combining these agents with NK cells can effectively kill cancer cells [82].
2.6 The mitochondria of TANs
Neutrophils, derived from bone marrow granulocyte-monocyte progenitor, exhibit anti-tumor activity by antibody-dependent cellular cytotoxicity, direct cytotoxic, and activation of adaptive immunity [83]. TANs display both anti-tumor (TAN1) and pro-tumor (TAN2) phenotypes within TME, similar to TAMs [84, 85].
Conventionally, mature neutrophils predominantly rely on glycolysis for energy, although mitochondria regulate apoptosis and neutrophil extracellular traps (NETs) formation. Neutrophils regulate mitochondrial function through glycolysis to produce more mtROS in hypoxia and support the production of pro-inflammatory NETs [86, 87]. Neutrophil elastase released from NETs activates TLR-4 on cancer cells, upregulates PGC-1α expression, increases mitochondrial biogenesis, mitochondrial density, ATP production, and oxygen consumption rate, but reduces ΔΨm, thereby promoting tumor growth through metabolic reprogramming of cancer cells [88]. This process also promotes tumor growth and metastasis by creating a chronic inflammatory environment. Resting neutrophils have elongated, tubular mitochondria that become fragmented and translocate upon stimulation [89]. TANs have more mitochondria and use them to support immune suppression and tumor growth by maintaining NADPH levels and producing ROS [90]. Both tumor cells and neutrophils increase glucose uptake, leading to higher lactate levels, which contribute to immunosuppression in the TME.
3. Pharmacological Therapies for Regulating Mitochondrial Function in the Treatment of Cancer
Mitochondria play a crucial role in regulating cell metabolism, redox balance, signaling, cell survival, and death of eukaryotic cells [89]. Numerous therapeutic approaches have been devised to regulate mitochondrial biological function for precise tumor treatment and recovery of compromised anti-tumor immune responses, ultimately benefiting the prognosis of cancer patients [60, 91]. We summarized the anti-tumor pharmacological drugs that regulate the biological function of mitochondria within TME from three aspects: mitochondrial balance (Table 1), energy metabolism, and mtDNA.
Summary of pharmacological agents targeting mitochondrial homeostasis regulation
| Regulating mitochondrial function | Tumor model/Cell model | Pharmacological agents | Mechanism of action | Ref. |
|---|---|---|---|---|
| Mitochondrial dynamics | Thyroid cancer cells, breast cancer stem cells (BCSCs), and pancreatic CSCs. | Mdivi-1 | Selective inhibitor of fission protein DRP1, inducing apoptosis in the tumor. | [94] [146] [147] |
| Lung carcinoma cells | Drpitor1 Drpitor1a | Inhibited the GTPase activity of DRP1, inducing apoptosis. | [148] | |
| Nasopharyngeal carcinoma (NPC) | Resveratrol | An inhibitor of Cyclooxygenase-2 (COX-2)/inhibition of DRP1 activation. | [149] | |
| Colon carcinoma | Celecoxib | Nonsteroidal anti-inflammatory drug, an inhibitor of COX-2/inhibition of DRP1 activation. | [150] | |
| liver cancer-initiating cells (LCICs) | Furamidine | Inhibited mitochondrial fission. | [50] | |
| BCSCs | Capivasertib (AZD5363) | The MFN1 inhibitor induced apoptosis in the tumor. | [151] | |
| Lung cancer cells | Baicalein | Induced mitochondrial fission, apoptosis, and autophagy. | [152] | |
| Mitophagy | Hepatocellular carcinoma (HCC) | Ketoconazole | COX2 inhibitor, pro-mitophagy via PINK1/Parkin pathway, induced apoptosis in HCC. | [150] |
| Lung cancer cells | Dihydroergotamide tartrate | Pro-mitophagy via the PINK1/Parkin pathway induced apoptosis and mitophagy. | [153] | |
| Triple negative breast cancer (TNBC) | 33c (ADTL-SA1215) | Sirtuin-3 inhibitor inhibits autophagy/mitophagy. | [99] | |
| TNBC | Flubendazole | DRP1-mediated mitophagy via targeting EVA1A. | [100] | |
| Cervical cancer cells | Sorafenib | Inhibited complexes II and III of the ETC to induce PINK1-Parkin-mediated mitophagy. | [154] | |
| Glioma cells | AT101 | BH3-mimetic, Pro-mitophagy | [155] | |
| Pancreas ductal adenocarcinoma | WJ460 | Increased mitophagy and ROS accumulation. | [156] | |
| Hepatic Cancer Stem Cells | Mdivi-1 | The DRP1 inhibitor inhibited mitophagy. | [157] | |
| Breast cancer cells | Liensinine | Blocked autophagosome-lysosome fusion at late stages of autophagy/mitophagy. | [158] | |
| Colorectal cancer cells | Analogues of Strigolactones | Autophagy/mitophagy inhibitor, blocking the fusion of autophagosomes and lysosomes. | [159] | |
| Cervical cancer cells | Melatonin | Inhibited mitophagy by downregulating Parkin, leading to apoptosis | [160] | |
| Leukemic stem cells | THZ-P1-2 | Inhibitor of phosphatidylinositol-5-phosphate 4-kinase type 2 protein, impaired autophagic flux. | [101] | |
| Bladder cancer cells | Nitazoxanide | Impaired autophagic flux, leading to apoptosis. | [161] | |
| Apoptosis | Small-cell lung cancer and acute lymphocytic leukemia, CAFs in Cholangiocarcinoma | Navitoclax (ABT-263) | BH-3 mimetics, an inhibitor of BCL-2, induce apoptosis. | [162] [163] |
| Chronic lymphocytic leukemia, Cholangiocarcinoma-associated fibroblasts, Colorectal cancer cells | Venetoclax (ABT-199) | Inhibited OXPHOS and TCA cycle independent of BCL-2 inhibition. | [164] [165] [166] | |
| Luminal breast cancers and breast cancer-associated fibroblasts | A-1210477 | BH-3 mimetics, an inhibitor of antiapoptotic MCL-1, induce apoptosis. | [104] | |
| TNBC | CPSI-1306 | Macrophage migration inhibitory factor (MIF) inhibitor, inducing apoptosis. | [167] | |
| gastric cancer | PIP | Increased ROS production and decreased ΔΨm, inducing intrinsic apoptosis. | [105] | |
| Gastric cancer (GC) Ovarian cancer | Cisplatin | Activated the mitochondrial apoptosis pathway, increasing the intracellular ROS, decreasing the MMP, and regulating the related proteins | [106, 107] | |
| gastric cancer (GC) | Ethyl acetate extract of COE | Inhibited Prohibitin (PHB) expression, | [108] | |
| glioma stem-like cells (GSCs) Pancreatic ductal adenocarcinoma cells | Trifluoperazine | Dopamine receptor antagonist, decreased OXPHOS, and increased ROS, inducing apoptosis. | [109] [168] | |
| glioma stem-like cells (GSCs) | mitoxantrone | Induced cleavage of Caspase9/3 and apoptosis. | [109] | |
| glioma stem-like cells (GSCs) | Pyrvinium pamoate | Inhibited mitochondrial complex I, inducing apoptotic death. | [109] | |
| Prostate-associated fibroblasts, colon adenocarcinoma cell line MDSCs. | Cinnamaldehyde | Enhanced proapoptotic activity through anti-topoisomerase I and II, or inhibiting NF-κB and activating protein 1. | [110] [169] | |
| BCSCs, hepatocellular carcinoma stem cells (HCSCs). | LND | LND, as a glycolysis inhibitor, induced apoptosis of BSCSs and HCSCs. | [111] | |
| Redox balance | Lymphoma, hepatocellular xenograft tumors. | SkQ1 | Mitochondria-targeted antioxidants reduce ROS, inhibit tumorigenesis, and reverse EMT. | [114] |
| Breast and ovarian cancers | Extracts from Olea europaea leaves | Increased mtROS and induction of apoptosis. | [115] | |
| Colorectal carcinoma | Sesamol | Induced the mitochondrial apoptosis pathway through pro-oxidant effect. | [170] | |
| Glioma cells | TPP-Demethoxycurcumin (DMC-TPP) | Induced mitochondria-dependent apoptosis by activation of caspase. | [117] | |
| Breast cancer cells | Baicalein | Production of ROS through the Fenton-like reaction leads to apoptosis. | [171] | |
| glioblastoma CSCs | N-heterocyclic carbene (NHC)-Ir(III) | ROS production and necroptosis-like cell death independent of caspase-dependent signaling. | [172] | |
| B16 melanoma-infiltrating T cells | N-acetylcysteine (NAC) | Reduced mtROS in CD8+ T cells and enhanced immune responses. | [173] | |
| Lymphoma and melanoma-infiltrating T cells | Nicotinamide ribose (NR) | Decreased mtROS in CD8+T cell, restoring mitophagy and reversing T cell exhaustion. | [174] | |
| Human blood monocytes | nanoshutter-1 | The NADPH oxidase-2 (NOX2) inhibitor attenuated macrophage differentiation via suppression of NOX2-mediated ROS production. | [175] | |
| Human blood monocytes Breast cancer cell | MnTE-2-PyP5+ (BMX-010) | The manganese porphyrin family of redox-active drugs inhibited M2 polarization by scavenging ROS. | [176] | |
| Breast cancer cell | Kaempferol | Inhibited neutrophil extracellular traps formation by ROS inhibition, and reduced tumor metastasis. | [177] | |
| Biogenesis | BCSCs Acute myeloid leukemia Pancreatic cancer (PC) | XCT790 | Inversed agonist of ERRα, reduces Oxygen consumption rate (OCR) and inhibits mitochondrial biogenesis. | [128] [129] [130] |
| Prostate Cancer Cells | SLU-PP-1072 | Decreased mitochondrial biogenesis. | [131] | |
| LLC infiltrating-CD8+ T cells Melanoma mice model-specific infiltrating-CD8+ T cells | bezafibrate | PGC-1α agonist, increased expression of ROS and PGC-1α in CD8+ T cells, and increased mitochondrial biogenesis. Increased FAO and mitochondrial respiratory capacity. | [178] [179] | |
| Melanoma mice model-specific CD8+ T cells | fenofibrate | Agonists of PPAR-α, increased CD8+ T cell function. | [132] | |
| B6 SJL mice lymphoma model | Idelalisib | PI3K inhibitor enhanced the anti-tumor activity of OT-T cells. | [180] | |
| Protein regulation | Acute myeloid leukemia (AML) chronic myeloid leukemia renal cell carcinoma ovarian cancer | Tigecycline | Inhibition of Mitochondrial protein translation. | [181] |
| Hepatocyte-derived carcinoma cells | Phenyl ester compounds | ClpP inhibitor induced apoptosis. | [182] | |
| Lung squamous cell carcinoma | ZK53 | ClpP activator inhibited cancer cells through degradation of ETC subunits and impaired OXPHOS. | [138] | |
| AML Lymphoma | ONC201, ONC212 | ClpP agonist, decreased activity of ETC, and increased ROS. | [183] | |
| TAMs in ovarian cancer cell | ONC201 | Dopamine receptor D2 antagonist; loss of mitochondrial integrity, switching to pro-inflammatory macrophages. | [140] | |
| LLC, colon carcinoma cell infiltrating-CD8+ T cells | SPD | Enhanced mitochondrial respiratory function in CD8+ T cells and anticancer immunotherapy. | [141] | |
| Mitochondrial Transfer | mesenchymal stem cells (MSCs) / T cell acute lymphoblastic leukemia (T-ALL) cells CAFs / Breast cancer cell | Cytochalasin D Anti-ICAM-1 | The combination of an actin inhibitor and an anti-adhesion molecule antibody inhibited mitochondrial transfer. | [184] [13] |
| MSCs / ALL | vincristine (VCR) | Microtubule inhibitors interfered with TNTs formation to reduce mitochondrial translocation. | [185] | |
| NK T cells or CD3+/CD8+ T | L-778123 +α-PD1 | Farnesyltransferase and geranylgeranyltransferase 1 inhibitors inhibited TNTs formation and mitochondrial transfer. | [14] |
3.1 Drugs regulating mitochondrial homeostasis
3.1.1 Mitochondrial Dynamics
Mitochondria exhibit morphological heterogeneity through a dynamic equilibrium established between their fusion and fission processes. Alterations in mitochondrial morphology serve as critical indicators of distinct functional states. Mitochondrial fusion is a highly regulated process, mediated by Mitofusin 1 and 2 (MFN1/2) located on the outer mitochondrial membrane, as well as OPA1 situated on the inner mitochondrial membrane [92]. Upon upregulation of OPA1, mitochondrial cristae undergo remodeling, becoming denser. This morphological change is accompanied by an increase in OXPHOS activity and a concomitant decrease in glycolysis levels. In contrast, mitochondrial fission, mediated by DRP1 and FIS1, induces an opposing metabolic shift [93].
As mentioned above, mitochondria in CAFs, CSCs, and/or other cancer-associated cells are more fragmented compared with their normal counterparts. Also, the activation process of M1-TAMs, T cells, and NK cells is associated with increased mitochondrial fission, such as T-cell activation mediated by DRP1 [60]. The selective inhibitor of DRP1, Mitochondrial division inhibitor-1 (Mdivi-1), inhibits proliferation and EMT in thyroid cancer cells by suppressing mitochondrial division [94]. Mitochondrial morphology changes to support tumor cell survival or death, highlighting the need for further research on mitochondrial dynamics.
3.1.2 Mitophagy
Mitophagy is a process that removes damaged mitochondria caused by ROS and hypoxia in cancers, promoting aerobic glycolysis and even enhancing drug resistance [95, 96]. Receptor-mediated mitophagy, involving proteins such as BCL2-interacting protein 3 (BNIP3), BCL2-interacting protein 3-like (BNIP3L), and FUN14 domain-containing 1, can promote ubiquitin-dependent mitophagy through the PINK1/Parkin pathway [97].
Ketoconazole promoted mitophagy and apoptosis through the accumulation of PINK1 in Hepatocellular carcinoma (HCC) [98]. Sirtuin-3, a protein primarily located in the mitochondria that relies on NAD+ and promotes mitophagy, was activated by compound 33c (ADTL-SA1215), thereby inhibiting the growth and migration of human breast cancer cells [99]. Flubendazole increased DRP1 expression, leading to PINK1/Parkin-mediated mitophagy [100]. On the contrary, inhibiting mitophagy may be effective in cancer treatment. THZ-P1-2 induced DNA damage and impaired autophagic flux in leukemic stem cells [101]. Treatment with Nitazoxanide induced mtROS production in bladder cancer cells, thereby triggering mitophagy initiation and concurrently inhibiting lysosomal degradation activity. This dual effect subsequently led to impaired mitophagic flux, resulting in the accumulation of damaged mitochondria. Consequently, mitochondrial ROS overload activated apoptosis signaling molecules [102]. Therefore, induction or inhibition of mitophagy, a mitochondrial quality control mechanism, is widely regarded as a promising anticancer therapeutic approach.
3.1.3 Apoptosis
Apoptosis, or programmed cell death (PCD), is an 'active death' behavior of cells mediated by gene regulation to maintain the steady state of the intracellular environment [103]. Induction of mitochondrial apoptosis has been exploited in clinical oncology to trigger cancer cell death. Myeloid cell leukemia-1 (MCL-1) specific inhibitor A-1210477 efficiently blocks the protective cross-talk between CAFs and cancer cells. Combination treatment of ABT-737 with A-1210477 significantly increases the cellular mortality of both cancer cells and CAFs [104]. Piperine (PIP) [105] and Cisplatin [106, 107], and Ethyl acetate extract from Celastrus orbiculatus (COE) [108] caused gastric cancer apoptotic cell death via the mtROS-associated signaling pathway. Trifluoperazine, Mitoxantrone, and Pyrvinium pamoate are mitochondrial inhibitors approved by the FDA and can serve as adjuvant therapies for Temozolomide-resistant Glioblastoma Multiforme (GBM) by inducing mitochondrial intrinsic apoptosis [109]. Cinnamaldehyde could not only induce apoptosis in MDSCs but also cause apoptosis in prostate-associated fibroblasts via the intrinsic pathway [110]. Wang et al. [111] designed and synthesized a novel mitochondrial-targeting compound, HYL001, consisting of Lonidamine (LND), lipophilic triphenylphosphonium (TPP+), and an aromatic ring, which serves as the hinge. HYL001 disrupts mitochondria morphology via decreasing the MMP to induce the apoptosis of BCSCs and hepatocellular carcinoma stem cells (HCSCs) by blocking Gln metabolism to amplify mitochondrial oxidative stress.
3.1.4 Redox balance
Maintaining mitochondrial redox balance between ROS and antioxidants is crucial for physiological and pathological processes, and the regulation of this balance can also be used to treat cancer [112]. Compared with normal cells, the mitochondria of tumor cells maintain a higher ROS level, causing genomic instability and metastasis [113].
The mitochondria-targeted antioxidant SkQ1 inhibited tumorigenesis in p53 knockout mice at an early stage. Additionally, SkQ1 reverses EMT by inhibiting the expression of mesenchymal N-cadherin [114]. Plant-derived polyphenolic compounds are known to act as antioxidants, including Extracts from Olea europaea leaves [115], Sesamol [116], TPP-modified Demethoxycurcumin [117]. Apoptosis, DNA fragmentation, and mitochondrial dysfunction are all effects of these compounds, which exhibit strong anticancer activity. The natural flavonoid Baicalein [118], which contains a phenolic hydroxyl group, produces hydroxyl radicals directly through a Fenton-like reaction with copper in cells, generating ROS and leading to genomic DNA damage [119]. Furthermore, the presence of excessive ROS in the TME, potentially generated by CAFs or stromal cells [120], can trigger the activation of immunosuppressive cells such as T cell exhaustion, macrophage M2 polarization, and neutrophil formation of NETs. Targeting these cells to diminish their ROS production may be a promising approach for therapeutic intervention. MtROS play significant roles in immune signaling pathways affecting T cell activation, proliferation, and apoptosis, as well as B cell activation [121]. Alleviating T cell exhaustion and retaining responsiveness to checkpoint blockade can be achieved by mitigating ROS or hypoxia [71]. Inducing ROS and blocking antioxidants may produce side effects in normal cells, so these agents must be more specific and efficient against tumor cells.
3.1.5 Biogenesis
Biogenesis of mitochondria is a dynamic process of continuous mitochondrial production that maintains the structure, number, and function of mitochondria in response to stress [52]. Nuclear transcriptional coactivator PGC-1α is essential for mitochondrial biogenesis and binds to a variety of nuclear receptors, including PPARs, the estrogen-related receptor (ERRα), and NRF1/NRF2 [122]. Increasing PGC-1α activates transcription factors, promotes mitochondrial gene expression, and enhances OXPHOS [123]. PGC1α has varying effects in different cancers, with lower levels associated with reduced survival rates for breast cancer [124], HCC [125], and osteosarcoma [126]. Boosting mitochondrial biogenesis through PGC1α may enhance cancer cell apoptosis. However, PGC-1α-mediated FAO and OXPHOS contribute to stem cell generation in hepatocellular carcinoma [127]. XCT790 [128-130] and SLU-PP-1072 [131], which are selective ERRα/γ inverse agonists, enhance chemosensitivity and suppress mitochondrial biogenesis in cancer cells. Fenofibrate [132] increases mitochondrial fatty acid catabolism, OXPHOS, and glycolysis to boost the proliferation of TN and CD8+ T cells. The mammalian target of rapamycin (mTOR) pathway, regulated by the PI3K-PKB (AKT)-mTOR, supports TN differentiation to TE. PI3K inhibitor Idelalisib [133] keeps CAR-T cells less differentiated, resulting in prolonged survival and improved antitumor abilities. Chemotherapy combined with mitophagy inhibitors (e.g., Mdivi-1 and 188Re-Liposome) sensitizes tumor cells to apoptosis and inhibits metastasis, resulting in tumor eradication [48].
3.1.6 Mitochondrial protein synthesis and degradation
Mitochondria contain over 1,000 proteins, with most being encoded by nuclear DNA. However, 13 key proteins are encoded by mitochondrial DNA and translated within the mitochondria. Several cancer cell lines have shown that Tigecycline reduces mitochondrial ATP production, resulting in slowed cell proliferation [134]. Mitochondria have a distinctive protein hydrolysis system involving more than 45 proteases, including respiratory complexes subunits and translocases, to maintain mitochondrial protein homeostasis by eliminating impaired and misfolded proteins [135].
Caseinolytic protease P (ClpP) is upregulated in human tumors, supporting proliferation and desensitizing cells to cisplatin [136]. Phenyl ester compounds (AV167, TG42, and TG43) inhibit ClpP activity and induce apoptosis in hepatocyte-derived carcinoma cells [137]. Conversely, ZK53, a selective activator of mitochondrial ClpP, inhibits lung squamous cell carcinoma by inhibiting OXPHOS and degrading ETC subunits [138]. ONC201 and its more potent analog ONC212, induce cancer cell death by suppressing dopamine receptor D2 and promoting activation of ClpP, leading to decreased ETC activity and increased ROS production [139]. ONC201 also switches macrophages to a pro-inflammatory state and inhibits OXPHOS in macrophages, enhancing the glycolytic ATP production and altering glutamate transport [140]. Spermidine (SPD) can enhance mitochondrial respiratory function in CD8+ T cells and potentially improve cancer immunotherapy [141]. Thus, mitochondrial proteins could be a potential target for treatment.
3.1.7 Mitochondrial transfer
Mitochondrial transfer can not only maintain metabolic homeostasis in the tissue microenvironment but also ensure intercellular communication under physiological conditions, whereas it can be impaired by cancer or other diseases. Researchers have described cancer cells absorbing mitochondria from non-malignant TME cells as a novel way to enhance tumor activity and immune escape [142]. Mitochondria transfer takes place between cells mediated by intercellular nanotubes, macrovesicles, cell junctions, and fusion [143]. Mitochondria transportation potentially contributes to cancer cell migration via reprogramming the metabolism of cancer cells.
Human breast cancer cells (MDA-MB-231) that received mitochondria from NK cells and CD3+/CD8+ T cells showed higher basal and spare respiratory capacities and lower metabolic mitogenic capacities. An aggressive immunocompetent breast cancer model treated with L-778123 and α-PD1 exhibited reduced nanotube formation and mitochondrial transfer, as well as improved antitumor activity [14]. Mitochondrial transfer from neural cells emerges as a key mechanism linking neural density to cancer progression. Astrocytes donate mitochondria to glioblastoma, fueling tumor growth via enhanced respiration and metabolic reprogramming [144]. Similarly, breast cancer cells (e.g., 4T1) acquire neuronal mitochondria via TNTs, augmenting OXPHOS, stemness, and stress resistance to drive metastasis [145]. These findings define the mechanistic basis of the association between neural density and poor prognosis, establishing a rationale for therapies targeting nerve-cancer metabolic crosstalk, such as local nerve blockade.
3.2 Drugs targeting mitochondrial energy metabolism
3.2.1 TCA cycle
The TCA cycle is a central hub that regulates cellular energy metabolism in the mitochondrial matrix. It converts metabolites of glucose, fatty acids, and amino acids into carbon dioxide, generating NADH and FADH2 as electron donors for ATP production through OXPHOS. Mutations in TCA enzymes (e.g., SDH, isocitrate dehydrogenase (IDH), and fumarate hydrogenase (FH)) have been reported in a variety of cancers. SDH and FH mutations lead to the accumulation of succinate and fumarate, respectively, and induce a glycolytic shift, causing a shift to glycolysis [186]. In contrast, IDH mutations convert α-ketoglutarate (α-KG) to 2-hydroxyglutarate (2-HG), a byproduct that inhibits α-KG-dependent enzymes, contributing to tumorigenesis. Devimistat (CPI-613), a lipoic acid analog, inhibits pyruvate dehydrogenase (PHD) and α-ketoglutarate dehydrogenase (α-KGDH) [187]. Lactate levels gradually rise over time and shift from promoting T cells' function to disrupting them in the tumor microenvironment. Dichloroacetate (DCA) specifically activates PDH, thereby promoting oxidative metabolism and glucose carbon flux through the TCA cycle. Tumors undergoing DCA treatment have their metabolism changed from glycolysis to OXPHOS, significantly increasing T cell function and cytokine production and rescuing them from apoptosis [188].
3.2.2 OXPHOS
The higher energy demand by the cancer cell is achieved by adjusting its metabolism, relying on aerobic glycolysis for energy instead of OXPHOS, as normal cells do. Biguanide antidiabetic drugs and IACS-010759, as mitochondrial complex I inhibitors, can be used for cancer treatment; however, their efficacy is limited due to the activation of metabolic compensation [19]. However, inhibition of OXPHOS induces mitochondria to transfer from bone marrow MSCs to AML cells via TNTs; the inhibitor also induces mitochondria division and mitophagy in AML cells, thereby promoting cancer cell migration [20]. The activity of mitochondrial complex I in the mitochondrial ETC decreases after Fenofibrate treatment, inhibiting gastric cancer cells' growth in vivo through reversing metabolic reprogramming and inducing apoptosis [189]. The Mito-FFa immunogenic death inducer, synthesized by TPP+ and Fenofibric Acid (FFa), targets mitochondria to increase the mtROS level of tumor cells, also inducing CD8+ T cell-mediated antitumor immunity [190].
New studies validate novel immunomodulatory effects of TPP+-dependent mitochondria-targeted drugs [191]. Mito-magnolol (Mito-MGN), a mitochondria-targeted derivative of magnolol, inhibits melanoma cell proliferation and OXPHOS more effectively than untargeted magnolol [192]. Mito-MGN, along with others like Mito-honokiol and Mito-metformin, inhibited pro-tumorigenic factors in the tumor immune microenvironment (TIME). Hydroxyurea (HU) modified with TPP+ inhibited more mitochondrial oxygen consumption than HU, potently inhibited tumor cell proliferation, M-MDSCs differentiation, and suppressor neutrophil activation, and stimulated TE response [193].
3.2.3 FAO
Cancers such as lymphomas and leukemias require elevated FAO for ATP production to survive [194]. Overexpression of Carnitine palmitoyltransferase-1 (CPT-1) is linked to cancer progression in breast, prostate, and lymphoma cancers [195]. The CPT-1 inhibitor Etomoxir has been shown to decrease proliferation in TNBC [196]. However, Etomoxir has been discontinued from clinical use due to toxicity at high doses. Melanomas drive FAO in DCs by upregulating CPT1A fatty acid transporter, inducing Treg generation. Etomoxir blocks this pathway, enhancing anti-PD-1 antibody immunotherapy activity [197]. A selective and reversible CPT-1A inhibitor, also known as Teglicar, has been demonstrated to repress FAO in leukemia cells, leading to mitochondrial damage as well as apoptosis [198]. An inhibitor of CPT-1 with a higher specificity, perhexiline, which inhibits both CPT1 and CPT2, has been investigated in mouse studies for numerous cancer types, such as colorectal cancer cells [199].
3.2.4 Glutamine metabolism
Reprogramming glutamine metabolism is crucial for tumor and immune cell survival in the TME. Tumor cells and immune cells compete for glutamine uptake, limiting its availability for T lymphocytes and impacting anti-tumor immune responses in TNBC. By converting glutamine into glutamate via glutaminase (GLS), glutamate is further converted by glutamate dehydrogenase (GDH) into α-KG, which can be used as an intermediate in the TCA cycle. Human malignancies suffer from high levels of GLS1, including breast, prostate, esophageal, and liver cancers [200]. Thus, inhibitors of GLS, such as BPTES and CB-839, demonstrate remarkable antiproliferative activity in tumor cells [201]. The anabolic pathway of glutamine is upregulated in CAFs compared to NOFs, providing nutrients for cancer cell growth. Glutamine metabolism induces the differentiation of M2-TAMs, and its inhibition may cause tumor shrinkage. On the contrary, inhibition of glutamine metabolism in NK cells is accompanied by suppression of their anti-tumor function [202].
3.3 mtDNA damage
The mitochondrial genome is capable of encoding 13 ETC subunits, mitochondrial rRNAs, and tRNAs, independently of nuclear genes. MtDNA has a high mutation rate and affects tumorigenesis [203]. Decreased levels of the mtDNA common deletion have been found in certain types of skin cancer and CAFs, which correlate with increased expression of mtDNA-encoded genes and improved mitochondrial function [204]. Mutations in ATP synthase gene reduce apoptosis and promote tumor growth. Mutations in mitochondrial Complex I promote ROS production, leading to metastasis of Lewis lung cancer mouse cells. Cancer cells may accumulate abnormal amounts of mtDNA in the cytoplasm as a result of oxidative stress and mitochondrial dysfunction [205]. MtDNA exhibits a dual function in modulating the TME; while mutations in mtDNA facilitate cancer progression and resistance to therapy [55], damaged mtDNA taken up by DCs stimulates IFN-I response and improves antigen presentation. Additionally, radiotherapy-induced damaged mtDNA enhances the anti-tumor function of CD8+ T cells [206]. Certain chemotherapeutic agents (cisplatin, paclitaxel), metal ions (Mn²⁺, Zn²⁺, Ca²⁺), and ROS act as pyroptosis inducers in tumor cells. This process concurrently releases damaged mtDNA, which activates the cGAS-STING pathway to enhance immunity [207]. Yu et al. employed cobalt fluoride (CoF₂) nanozymes to induce pyroptosis under ultrasound, generating ROS and damaged mtDNA that activate cGAS-STING [208]. Alternatively, gas therapy combined with Mn²⁺ induces significant mitochondrial dysfunction, thereby impeding cancer proliferation [209].
Shen et al. [210] found significantly reduced mtDNA content in most Pediatric high-grade gliomas (pHGGs) compared with normal brain, resulting in the metabolic transition toward aerobic glycolysis. They adopted a combination therapy in which dichloroacetate and metformin synergized with radiotherapy (RT), leading to a reversal of the metabolic phenotype, primarily by inducting oxidative stress. DCA is used to shift glucose metabolism toward mitochondrial oxidation. In contrast, metformin targets mitochondrial function while disrupting the tumor's energy balance, thereby increasing DNA damage and apoptosis. OXPHOS-dependent CSCs are targeted with first-in-class, specific mitochondrial transcription inhibitors (IMTs), targeting human mitochondrial RNA polymerase (POLRMT), as an enzyme that plays a crucial role in its biogenesis [211].
4. Nanotechnology-Based Therapeutics for Regulating the Mitochondria of Tumors or Tumor-Associated Cells
As previously discussed, despite substantial research on pharmacological agents to modulate mitochondrial function in tumor and tumor-associated cells, precise mitochondrial drug targeting remains challenging due to the dual barriers of the mitochondrial double-membrane structure and TME complexity [212]. The rapid advancement of nanotechnology continues to revolutionize conventional pharmacology. NDDS confer multiple advantages, including enhanced stability, superior targeting capability, and improved bioavailability for highly potent therapeutic agents [213, 214]. Recent years have witnessed remarkable progress in mitochondria-targeted nanotherapeutics designed for both malignant and stromal cells. These tailored nanoplatforms enable precise mitochondrial accumulation and potentiated regulation of mitochondrial functions. More significantly, synergistic integration of mitochondria-targeted nanodrugs with chemotherapy, PDT, SDT, RDT, magnetic hyperthermia therapy (MHT), photothermal therapy (PTT), and immunotherapy has demonstrated enhanced efficacy in combating tumor progression [24, 215] (Table 3).
Nanotechnological regulation of mitochondrial function in diverse cells
| Cell type | Nanotechnology | Drug | Regulating mitochondrial function | Therapeutics strategies | Ref. |
|---|---|---|---|---|---|
| Tumor cells | Polymer (SHC4H) | Hydroxychloroquine + Type I photosensitizer (SMNB) | Disrupted mitophagy to amplify oxidative stress. | Chemotherapy PDT | [237] |
| Micelles (A6-dMP-VP) | Oncolytic peptide + Type I photosensitizer (verteporfin) | Induced oxidative stress. | PDT Immunotherapy | [238] | |
| Micelles (Mito-CMPN) | Cisplatin + Mitoxantrone | Induced mitochondrial stress. | Chemotherapy PDT | [217] | |
| Micelles (CAT/CPT-TPP/PEG-Ce6) | Cinnamaldehyde + Catalase + Photosensitizer (Chlorin e6) | Induced oxidative stress and apoptosis. | PDT Immunotherapy | [239] | |
| Semiconduction polymer (TCa/SPN/a) | Ca2+ + cGAMP | Induced oxidative stress. | RDT Immunotherapy | [236] | |
| Metal NPs (Zn-LDH@Mg) | Ca2+ + Mg2+ | Induced oxidative stress. | MHT Immunotherapy | [219] | |
| metal oxide NPs (TiO2-xFx) | TiO2-xFx | Induced mitochondrial stress. | SDT Immunotherapy | [30] | |
| Metal oxide NPs (ZnCo-Fe3O4 CSNCs) | Zn2+ + Fe2+ | Disrupted morphology of mitochondria, depolarization of ΔΨm. | TAE MHT Immunotherapy | [220] | |
| Metal NPs (Sb2Se3@Pt) | Sb2Se3 + Pt | Induced oxidative stress. | SCT Immunotherapy | [221] | |
| Micelles (VB12-sericin-PBLG-IR780) | IR780 | Inducing pyroptosis via damaged mtDNA. | PTT PDT Immunotherapy | [222] | |
| TAMCs | Polymer (mPEI/M1mt) | Mitochondria | Transplanted mitochondria. | Immunotherapy | [240] |
| Dendrimer (D-DPA) | hydroxyl-terminated polyamidoamine (PAMAM) dendrimers | Induced immunosuppressive TAMs apoptosis. | Immunotherapy | [224] | |
| Micelles (Man-PEI-PCL/PEG-DMMA) | Mitofusin 1 shRNA (shMFN1) + Celastrol | Repolarized M2-TAMs to M1 phenotype. | Chemotherapy Gene therapy Immunotherapy | [225] | |
| CAFs | Mesoporous Silica (Se@A&F) | AMD3100 | Ruptured mitochondrial structure. | Chemotherapy Immunotherapy | [241] |
| Nanocages (HNav-FAP) | Navitoclax | Induced CAFs apoptosis. | Chemotherapy | [242] | |
| CSCs | Liposomes (RTB) | β-lapachone (LAP) + pDNAiNOS | Decreased mitochondrial OXPHOS. | Radio-immunotherapy Gene therapy | [227] |
| Polymer (CuET@PHF) | CuET nanocrystals | Induced CSCs apoptosis. | Chemotherapy PTP Immunotherapy | [243] | |
| Polymer (CuET/ICG) | CuET nanocrystals + ICG | Induced oxidative stress. | Chemotherapy PDT Immunotherapy | [232] | |
| T cell | Metal NPs (mitoNIDs) | Gold NPs | Induced mitochondria degradation via mitophagy. | Chemotherapy Immunotherapy | [233] |
| Biometic NPs (O-TMV@ABP) | Axitinib + PF-06446846 | Promoted T cell mitochondrial biogenesis. | Chemotherapy Immunotherapy | [234] | |
| Biometic NPs (PmMN@Om&As) | oxymatrine (Om) + Astragaloside IV (As) | Upregulated T cell biogenesis. | Chemotherapy Immunotherapy | [235] | |
| Liposome (L@Mn@SPD) | Mn + Spermidine | Increased mitochondrial respiration. | Chemotherapy Immunotherapy | [236] |
4.1 Nanodrugs regulating mitochondria of tumor cells and immunogenic cell death effect
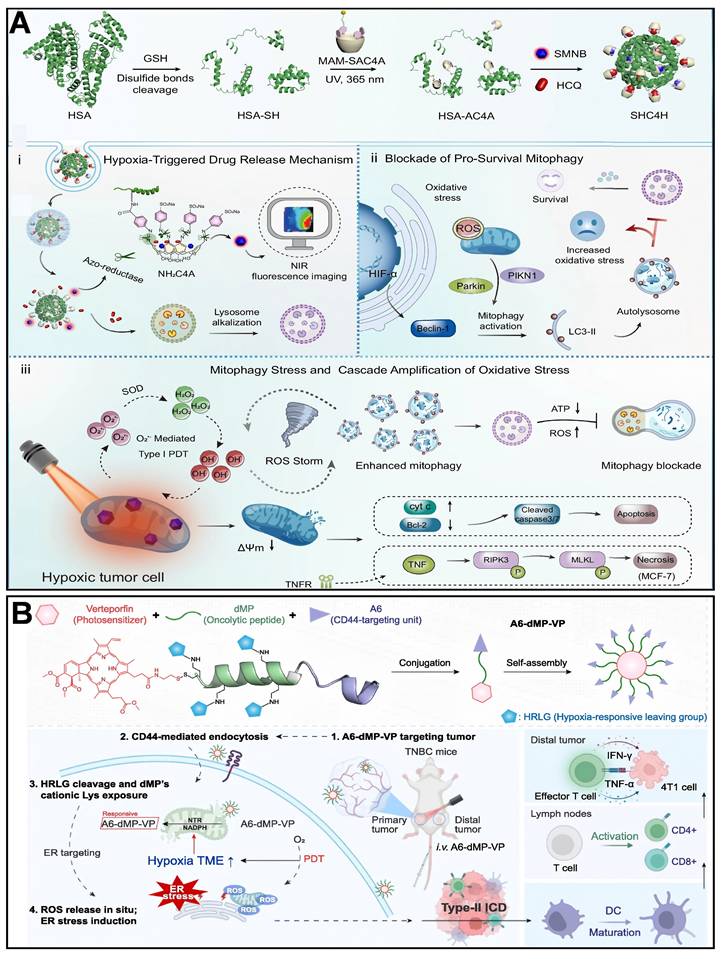
Wang et al. [216] developed a novel SHC4H nanoparticle, which combines chemotherapy and mitochondria-targeting photodynamic therapy (M-PDT). Azocalix [4] arene (AC4A) was modified onto human serum albumin (HSA) to form the HSA-AC4A carrier, which was co-loaded with hydroxychloroquine (HCQ) and a mitochondria-targeting Type I photosensitizer (SMNB). Utilizing the hypoxia-responsive property of AC4A, azobenzene bonds were reductively cleaved in the hypoxic tumor microenvironment, thereby releasing HCQ and SMNB. HCQ blocked mitophagic flux by alkalinizing lysosomes, while SMNB generated ROS via an electron transfer mechanism under 660 nm laser irradiation, damaging mitochondria and activating mitophagy. In hypoxic tumor cells, the dual attack of HCQ-induced mitophagic flux blockade and SMNB-triggered mitochondrial damage was achieved (Figure 5A). Growing evidence suggests that inducing mitochondrial stress is often more effective than triggering large-scale induction of ICD via endoplasmic reticulum (ER) stress. Ren et al. [27] designed and synthesized a tumor-targeting and hypoxia-responsive peptide-photosensitizer coupling, A6-dMP-VP, which consists of a CD44 targeting motif, A6, a tumor lysing peptide, dMP, a hypoxia-responsive motif, and a photosensitizer, vitexoporfin (VP), which self-assembles into nanoparticles and is preferentially enriched in 4T1 tumors. In the hypoxic tumor microenvironment, mitochondrial nitroreductase (NTR) cleaves the hypoxia-responsive motifs. It releases the cationic dMP, which binds to and destroys the mitochondrial membrane through electrostatic interaction. At the same time, VP generates ROS by light exposure, which exacerbates endoplasmic reticulum stress, and the two synergistically induce type II immunogenic cell death (Figure 5B). Zhang et al. [217] designed mitochondria-targeted multi-premedicine nanoparticles, Mito-CMPN, which target mitochondria via rhodamine B (RhB) and piggyback on cisplatin and mitoxantrone (MTO). Under laser irradiation, MTO generates ROS to trigger photodynamic therapy, while the nanoparticles release drugs in response to ROS and GSH in the tumor microenvironment, synergistically inducing mitochondrial stress. This process prompts tumor cells to release immunogenic molecules (e.g., CRT, HMGB1, ATP), activates dendritic cells, polarizes M2-type macrophages to M1-type, and enhances CD8+ T-cell infiltration, reversing the immunosuppressive microenvironment (Figure 6A). Hu et al. [218] developed ROS-responsive mitochondria-targeted micelles (CTC) co-delivering CPT, Ce6, and CAT to mitochondria via TPP. The triple synergy of CPT-induced ROS, Ce6-mediated photodynamic ROS, and CAT-catalyzed O₂ generation created a sustained ROS cascade (domino effect), leading to mitochondrial membrane collapse, ER stress activation, and immunogenic cell death (Figure 6B). Cheng et al. [30] developed a semiconductor polymer nanomessenger (TCa/SPN/a) co-loaded with Ca²⁺ and cGAMP. Under X-ray irradiation, it generated ROS via a radiodynamic effect to induce ICD, while simultaneously releasing mitochondria-targeted Ca²⁺ to trigger calcium overload and mitochondrial damage. This cascade activated the STING pathway, enhancing the immunotherapeutic effect and significantly suppressing tumor growth and metastasis in the breast cancer model.
(A) Design and hypoxia-activated antitumor action of SHC4H. (B) Schematic design and mechanism of action of the hypoxia-responsive lysosomal coupling A6-dMP-VP. (A) Reproduced with permission from [216], copyright 2025, Springer Nature Limited. (B) Reproduced with permission from [27], copyright 2025, Elsevier B.V.
(A) Mito-CMPNs-mediated mitochondria-targeted photodynamic therapy (PDT)/chemotherapy and its unique ability to improve cancer treatment in terms of immunostimulation. (B) Schematic illustration of ROS-responsive, mitochondria-targeted micelles for enhanced ICD via a domino effect of amplified oxidative stress. (C) Diagram showing the synthesis of Sb2Se3@Pt with a Schottky junction, and the mechanism of SCT therapy in modifying the tumor microenvironment to boost pyroptosis-mediated immune response against cancer. (A) Reproduced with permission from [217], copyright 2024, Elsevier B.V. (B) Reproduced with permission from [218], copyright 2024, Elsevier B.V. (C) Reproduced with permission from [221], copyright 2024, Wiley-VCH GmbH.
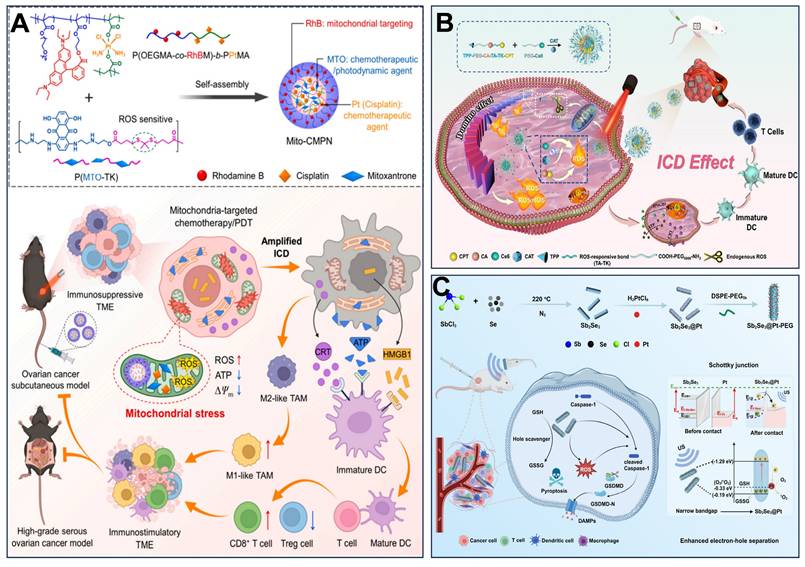
Furthermore, pyroptosis can also induce ICD. This process is characterized by transmembrane pore formation, cellular swelling, and lysis, culminating in the release of inflammatory factors and intracellular contents. Cheng's team investigated the link between mitochondrial damage and pyroptosis. They developed nanoparticle platforms combining metals or metal oxides with therapies such as MHT, SDT, immunotherapy, transarterial embolization (TAE), and sonocatalytic therapy (SCT) to induce tumor mitochondrial damage, pyroptosis, and antitumor immunity [30, 219-221]. Han et al. [219] synthesized Zn-LDH@Mg implants by a hydrothermal method, which had rough surfaces with Zn-layered double hydroxides (LDH) on the Mg implants. Zn-LDH@Mg induced Caspase/Gasdermin D (GSDMD)-dependent pyroptosis in hepatoma cells, enhanced by increased Zn²⁺ uptake amplifying mitochondrial oxidative stress. Resultant pyroptosis triggered ICD via “eat me” signals (e.g., calreticulin exposure). Sun et al. [30] constructed fluorinated titanium dioxide (TiO₂-ₓFₓ) sonosensitizers. Under ultrasound, these nanoparticles generated excessive ROS that induced mitochondrial damage and oxidative homeostasis disruption, ultimately triggering significant pyroptosis. Notably, SDT-facilitated pyroptosis prevented tumor recurrence by initiating a potent immune memory effect. Wang et al. [220] designed an innovative magnetic metallo-immunotherapeutic platform based on Zn-Fe₃O₄@Co-Fe₃O₄ core-shell nanocubes (ZnCo-Fe₃O₄ CSNCs). This platform integrates TAE, MHT, and immunotherapy. Under an alternating magnetic field (AMF), temperature-dependent release of Fe²⁺ and Zn²⁺ ions disrupted mitochondrial morphology, causing extensive vacuolation, membrane potential depolarization, and reduced ATP levels. Crucially, by suppressing ATP synthesis, this process also lowered heat shock protein (HSP) expression, thereby inducing GSDMD-dependent pyroptosis in hepatocellular carcinoma cells and subsequent immune activation. Nie et al. [221] developed a sonocatalytic platform, Sb2Se3@Pt core-shell heterostructures, that initiates pyroptosis to enhance SDT-immunotherapy. Under ultrasound (US) irradiation, Sb2Se3@Pt generates excessive ROS, inducing mitochondrial dysfunction and pyroptosis (Figure 6C). Guo et al. [222] prepared VB12-sericin-PBLG-IR780 nanomicelles to induce pyroptosis and stimulate antitumor immunity. Combined PTT and PDT, ATP synthase expression could be inhibited, and mitochondria were damaged by nanomicelles, leading to mtROS-induced mtDNA damage and pyroptosis through NLRP3/Caspase-1/gasdermin D pathway and DCs maturation. These studies demonstrate that mitochondrial dysfunction accompanies, and may even promote, the induction of tumor pyroptosis.
4.2 Nanodrugs regulating mitochondria of tumor-associated myeloid cells (TAMCs)
TAMCs are heterogeneous cells that infiltrate tumors and influence tumor growth profoundly, including TAMs, DCs, and neutrophils. Targeting mitochondria in TAMs reprogrammed TAMs from pro-tumor to anti-tumor, as a potential therapeutic strategy. Zhang et al. [223] designed a mannose-modified mitochondria-targeted delivery system (mPEI/M1mt), in which M1-type macrophage-derived mitochondria were electrostatically encapsulated in mannose-modified polyethyleneimine, and targeted uptake was achieved by utilizing the mannose receptor, which is highly expressed on the surface of M2-TAMs. mPEI/M1mt promoted M2-TAMs' metabolism through induction of a shift in the M2-TAMs' metabolic By inducing the metabolic pattern of M2-TAMs to shift from OXPHOS to glycolysis, mPEI/M1mt elevates the intracellular ROS level and activates the NF-κB p65, MAPK p38, and JNK signaling pathways, which in turn promote the polarization of M2-TAMs toward the pro-inflammatory and anti-tumor M1 phenotype (Figure 7A).
(A) Schematic representation of engineered mitochondria reprogramming M2 TAM to produce inflammatory TME and enhance cancer. (B) Illustration of RTB synthesis and its mechanism for eliminating CSCs by enhancing radio-immunotherapy. (A) Reproduced with permission from [223], copyright 2024, Wiley-VCH GmbH. (B) Reproduced with permission from [227], copyright 2025, Elsevier B.V.
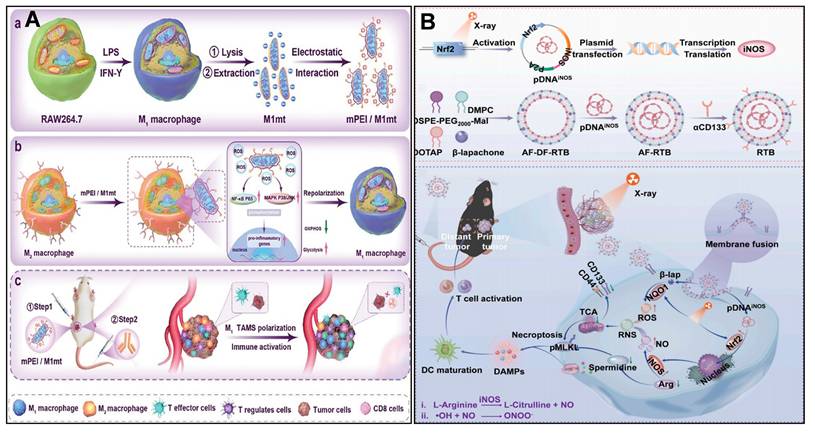
Sharma et al. [224] presented a dendrimer-mediated nanomedicine (D-DPA) that targets TAMs in glioblastoma. Generation four hydroxyl-terminated polyamidoamine (PAMAM) dendrimer was clicked by 5,7-dimethylpyrazolo[1,5-α] pyrimidin-3-ylacetamide (DPA), which can specifically target translocator protein (TPSO). D-DPA targeted specifically to immunosuppressed TAMs due to high TPSO expression. D-DPA showed stronger TPSO affinity than the free TPSO ligand PK11195, and was more co-located with mitochondria than the unmodified dendrimers. D-DPA upregulates the antitumor immune signaling of TNF-α and IL-1β, as well as inhibits the expression of protumor signal of Arg-1. Zhao et al. [225, 226] used P/shMFN1 NPs to repolarize pro-tumor TAMs to an anti-tumor phenotype by down-regulating the expression of the mitochondrial fusion gene. Mitofusin 1 shRNA (shMFN1) was loaded with Man-PEI-PCL, then coated with 2, 3-dimethylmaleic anhydride modified polyethylene glycol shell (PEG-DMMA) to form P/shMFN1. P/shMFN1 targeted M2-TAMs with mannose guidance to shMFN1, resulting in mitochondrial fragmentation and increased ROS generation. Consequently, M2-TAMs (F4/80+CD206+) shifted to M1-TAMs (F4/80+CD80+) through mitochondrial fission. They also designed another nanoparticle, P-aCD24/CEL, to target tumor cells, inducing ICD of tumor cells and reactivating the immune regulation of macrophages. Enhancing antitumor immune responses by targeting tumor cells and TAMs, respectively.
4.3 Nanodrugs regulating mitochondria of CAFs
Tumor stroma is dominated by CAFs, which secrete pro-tumor growth factors that enhance solid tumor pressure and interstitial fluid pressure (IFP). Targeting receptors overexpressed on CAFs can improve the effectiveness of nanotherapy, including fibroblast activation protein (FAP), sigma receptor, angiotensin II type I receptor, and tenascin C protein. Chemotherapy-induced ROS stimulates CAFs that confer chemoresistance via mitochondrial transfer through TNTs. Tumor-derived vesicles additionally activate CAF mitophagy, boosting mtDNA release, which drives metastasis in therapy-damaged lung cancer cells [228].
Li et al. [229] developed a diselenium-bonded organosilicon nanodelivery system (Se@A&F) targeting FAP, which specifically acts on CAFs in prostate cancer by carrying CXCR4 antagonist AMD3100. Se@A&F induced CAFs' inactivation and remodeled the tumor microenvironment by down-regulating CXCR4 expression on CAFs' surface and inhibiting the CXCL12/CXCR4 signaling axis. Se@A&F induces CAFs' inactivation and remodels the tumor microenvironment by down-regulating CXCR4 expression on CAFs' surface and inhibiting the CXCL12/CXCR4 signaling axis. Meanwhile, selenium released from the nanosystem induces mitochondrial dysfunction in tumor cells, triggering apoptosis of RM-1 tumor cells by generating ROS and disrupting MMP. Sitia et al. [242] developed a CAF-targeted pro-apoptotic Antibody-Drug Conjugate (ADC). Firstly, Navitoclax (Nav) was coupled with anhydrous CuSO4 (II), incubated with H-ferritin (HFn) to form HNav due to the affinity between metal ions and protein. Then, functionalized HNav-Fab consisted of HNav nanocages with the Fab fragment of an anti-FAP antibody (Fab@FAP) via a heterobifunctional linker NHS-PEG-Mal. Functionalizing HFn with FAP antibody fragments can significantly enhance the CAF-specific delivery of the drug and compete with the TfR1 receptor, thereby decreasing off-target distribution in tumor cells. The results showed that HNav-FAP targeted and induced apoptotic cell death by activation of the mitochondria pathway in FAP+ breast cancer cells. Therefore, HNav-FAP, when combined with chemotherapeutics, could be a promising therapeutic strategy to reduce the formation of metastases.
Mitochondrial double-stranded RNA (mt-dsRNA) from B-cell precursor acute lymphoblastic leukemia (B-ALL) cells induces CAF differentiation from MSCs through interferon activation. Resulting CAFs support chemoresistance by mitochondrial transfer via TNTs. Disrupting dsRNA prevents CAF generation, offering therapeutic potential [230].
4.4 Nanodrugs regulating mitochondria of CSCs
CSCs are implicated in tumorigenesis, recurrence, metastasis, and drug resistance. Of particular significance is the pivotal role of mitochondria in CSCs, prompting the exploration of anti-tumor stem cell therapeutic approaches that target mitochondria. Several aspects of mitochondrial morphology and structure, subcellular localization, mtDNA, metabolic status, and mitophagy activity determine the function and fate of CSCs.
CSCs depend on mitochondrial OXPHOS to maintain their stemness and therapy resistance, and arginine metabolism plays a key role in this process. Yao et al. [227] investigated a nano-integrated radiation-triggered bioreactor (RTB). This RTB system depletes arginine within CSC through radiation-induced iNOS expression, while generating NO to enhance the cellular toxicity and disrupt mitochondrial function in concert with β-lapachone (LAP)-generated ROS. This dual action drives the transition of CSC to a differentiated state and induces immunogenic cell death through the activation of RIPK1-dependent necrotic apoptosis, thereby stimulating a durable anti-tumor immune response (Figure 7B). Xiao et al. [231] investigated and developed the ROS-responsive targeting nanodrug CuET@PHF, which stabilizes the copper ion carrier CuET nanocrystals by dopamine and hydroxyethyl starch, and utilizes folic acid for active targeting. The drug induces copper death in cancer cells and CSCs by targeted delivery of copper ions to mitochondria, thereby disrupting tricarboxylic acid cycle proteins and triggering cell death. Zhang et al. [232] designed a bis(diethyldithiocarbamate)-copper/indocyanine green nanoparticles (CuET/ICG NPs) prepared by one-pot method, which could alleviate tumor hypoxia by inhibiting mitochondrial oxygen depletion and enhance PDT-induced immunogenic cell death, and at the same time activate the AMPK pathway by triggering energetic stress through mitochondrial dysfunction to down-regulate PD-L1 expression on the surface of CSCs and tumor activate AMPK pathway through mitochondrial dysfunction, down-regulate PD-L1 expression on the surface of CSCs and tumor cells, and relieve immunosuppression.
4.5 Nanodrugs regulating mitochondria of T cells
Mitochondrial dysfunction in CD8+ T cells leads to T cell exhaustion, which weakens their antitumor effects. Modulation of energy metabolism, redox balance, and dynamics of mitochondria in CD8+ T cells can remodel their antitumor functions. Tumor cell mitochondrial content is positively correlated with their resistance to CD8+ T cell killing, and tumor cells with high mitochondrial content are more likely to evade attack. mitoNIDs developed by Pan et al. were constructed by using gold nanoparticles (GNPs) as carriers, and modifying the mitochondrial membrane-bound peptide (KALKALKKKALKALKC) and LC3-targeting peptide (DDDWTHLSC) by gold-sulfur bonding to create a nano-agent with both mitochondrial targeting and autophagy activation functions. DDDWTHLSC). It not only enhances the recognition and activation of CD8+ T cells on tumor cells, but also improves the sensitivity of tumor cells to CD8+ T cell killing, which provides a new strategy to enhance the effect of tumor immunotherapy [233] (Figure 8A).
(A) Schematic overview of how mitoNID promotes mitochondrial degradation for optimized CD8+ T cell immunotherapy. (B) Schematic illustration of the preparation of O-TMV@ABP injectable hydrogel and its role in T cell recruitment, alleviation of T cell exhaustion, and overcoming MHC I-mediated immune evasion. (A) Reproduced with permission from [233], copyright 2025, Springer Nature Limited. (B) Reproduced with permission from [234], copyright 2022, Wiley-VCH GmbH.
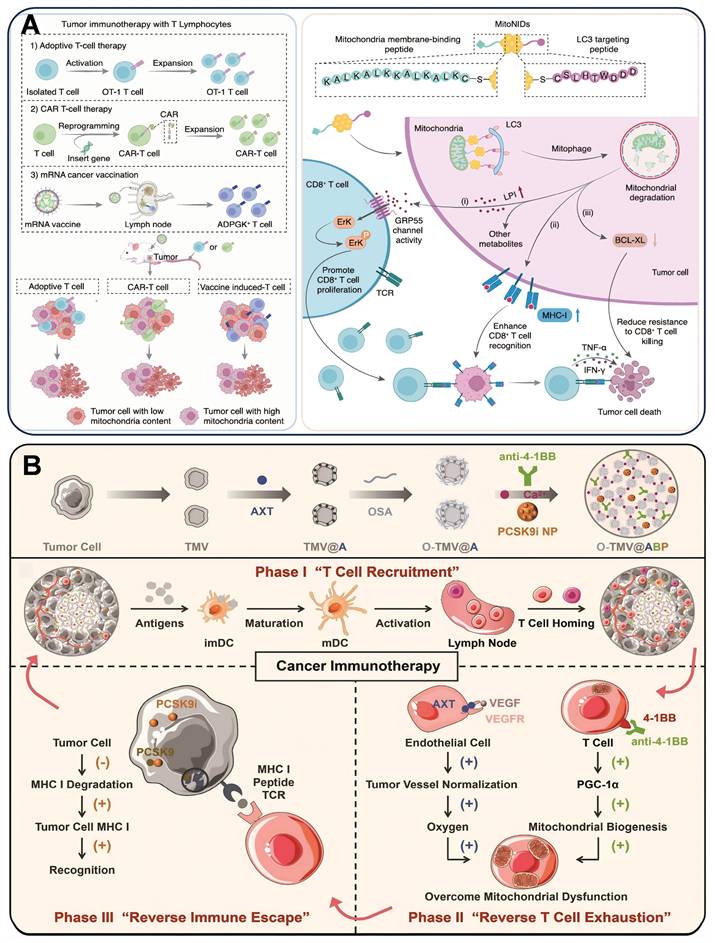
Insufficient major histocompatibility complex class I (MHC I) on tumor cells due to subtilisin/kexin type 9 (PCSK9) degradation hinders T cell recognition, reducing therapeutic effectiveness. Zhang et al. [234] designed an injectable hydrogel O-TMV@ABP that encapsulated axitinib (AXT), 4-1BB antibody, and PF-06446846 (PCSK9 inhibitor) in gelator oxidized sodium alginate-modified cancer cell membrane vesicles (O-TMV). The hydrogel worked in three steps. In the first stage, O-TMV triggered an immune response at the tumor site. In the second phase, the 4-1BB antibody and AXT reverse TEX. In the third phase, PF-06446846 enhanced MHC I expression on the tumor and promoted T cell recognition of tumor cells. This injectable hydrogel reprogrammed T cells to recognize tumor cells, thereby facilitating T cell-based cancer immunotherapy (Figure 8B). CAFs create a barrier to prevent TILs from entering the tumor by secreting collagen, FAP, and chemokine. Platelet membrane (Pm)-encapsulated magnetic metal-organic framework nanoplatforms composed of PmMN@Om&As. They simultaneously deliver oxymatrine (Om) and astragaloside IV (As) at the tumor site to inactivate CAFs and augment the level of TILs. The magnetic field and Pm coating help deliver Om and As to HCC cells. Inhibiting CAFs activation with Om can increase TIL levels and enhance mitochondrial function by upregulating PGC-1α. Combining PmMN@Om&As with α-PD-1 improves anti-HCC effects, prolonging survival. Enhancing TIL activity with As is an effective strategy [235]. Chen et al. [236] developed a liposomal nanoreactor (L@Mn@SPD) that forms Mn(OH)₂ precipitates within liposomes, co-delivering Mn²⁺, SPD, and O₂ to the TME. This nanoreactor alleviates TME hypoxia, simultaneously activating Mn-based STING immunostimulation and countering T cell exhaustion by enhancing mitochondrial respiration and ATP production. This approach overcomes the limitation of T cell mitochondrial dysfunction that compromises Mn-based cancer immunotherapy.
5. Discussion
Our analysis compares pharmacological agents and nanomedicines targeting mitochondria in tumor and tumor-associated cells, revealing fundamentally distinct design paradigms despite a shared therapeutic goal. Small molecules inherently rely on systemic exposure, prioritizing strategies like lipophilic cations (e.g., TPP⁺, DQA) for membrane potential-driven accumulation or substrate analogs (e.g., DCA, CPI-613) for enzymatic affinity to achieve sufficient mitochondrial residence [111, 117, 188]. Conversely, nanomedicines actively circumvent systemic exposure (a major source of side effects), leveraging the TME (pH, enzymes, hypoxia, or receptor overexpression) to achieve tumor-specific localization first [244]. This enables hierarchical targeting: to first confine the carrier within the tumor region. Subsequently, secondary release of drugs or ions enables hierarchical targeting: achieving "tumor-first" accumulation followed by “mitochondria-second” delivery.
Further distinctions lie in specificity, controllability, scope, and overcoming limitations. Small molecules typically pursue single-target interventions (e.g., DRP1, BCL-2, CPT-1) with fixed pharmacokinetic profiles, limiting spatiotemporal control. Nanomedicines embrace multimodal synergy (e.g., combining chemotherapy, PDT, SDT, PTT, immunotherapy, via mechanisms like ROS cascades, Ca²⁺ overload, ICD, STING pathway activation) and provide dynamic “on-demand” controllability using external/internal triggers (light, US, X-ray, H₂O₂, hypoxia/ROS/pH-sensitive linkers) [71, 215]. Critically, while small molecules primarily induce autonomous tumor cell mitochondrial death constrained by chemical draggability (solubility, stability, toxicity), nanomedicines exploit their engineerable nature to overcome these barriers. They deliver “undruggable” payloads (ions like Zn²⁺/Cu²⁺/Ca²⁺, proteins, shRNA, whole mitochondria) and actively reprogram intercellular mitochondrial transfer (e.g., between TAM/CAFs/ T cells and tumor cells), reshaping the immune-stromal network [23]. Through material selection (polymers, lipids, metal-organic frameworks, cell membrane coatings) and surface engineering (targeting peptides, antibodies, glycosylation), they transform “undruggable” payloads into “deliverable” therapeutics. In essence, small molecule strategies maximize mitochondrial affinity within the confines of limited chemical space, whereas nanomedicine leverages virtually infinite engineering space to achieve spatiotemporally precise, multi-modal mitochondrial intervention with diverse payloads.
6. Conclusion and Future Perspectives
This review summarizes the metabolic reprogramming of tumor-associated cellular mitochondria (TAMs, CAFs, CSCs, T cells, NK cells, TANs) in tumor tissues under the influence of tumor cells. Mitochondria, as the cellular powerhouses, play a key role in adapting to these metabolic changes. Modulation of mitochondrial function in tumor cells and associated cells can effectively impede tumor growth and metastasis, offering novel approaches to combat drug resistance and cancer recurrence. Additionally, targeting the mitochondria of tumors or CAFs, CSCs, TAMCs, and tumor-infiltrating T cells within the TME with nanomedicines can mitigate the immunosuppressive effects and enhance the anti-tumor response, thereby shifting the balance towards anti-tumor immunity.
Despite the significant progress made in current research, therapeutic strategies targeting mitochondria still face many challenges in clinical applications. Future research needs to delve deeper into the specific mechanisms of mitochondrial action in different types of tumors and how to precisely regulate mitochondrial function to optimize therapeutic outcomes. Additionally, the development of novel nanodrug delivery systems to achieve targeted therapy of mitochondria in tumor cells and tumor-associated cells will be an important direction for future research. Combining immunotherapy and metabolic therapy may lead to more significant clinical effects in cancer treatment. Lastly, the advancement of personalized medicine requires a better comprehension of the heterogeneity of the TME and how to tailor mitochondrial-targeted therapeutic strategies according to the specific circumstances of patients. With the continuous progress of science and technology, cancer therapies will increasingly target mitochondria in the future.
The following aspects should be addressed in future research to achieve these goals:
(1) Gain a comprehensive understanding of the mechanisms underlying mitochondrial function in various tumor-associated cells within the TME and their impact on tumor development and treatment responses. Investigate the interplay between mitochondria and other cell types in the TME, and assess how these interactions influence tumor progression and treatment outcomes.
(2) Develop and optimize nanotechnologies to improve the targeting and efficiency of drug delivery, reducing side effects.
(3) Explore the synergistic effects of mitochondria-targeted therapy in combination with other treatment modalities (including PTT, immunotherapy, chemotherapy, and radiotherapy) to optimize therapeutic efficacy.
(4) Validate the safety and effectiveness of mitochondria-targeted therapy through clinical trials and develop individual treatment plans according to the genetic background and tumor characteristics of patients.
Acknowledgements
This work was grateful to the financial support from the science and technology plan joint project of Liaoning Province (2024-MSLH-436).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Schapira L, Duffy CM. Cancer treatment and survivorship statistics, 2025: An urgent call to optimize health after cancer. CA Cancer J Clin. 2025;75:277-279
2. Xiong X, Zheng L-W, Ding Y, Chen Y-F, Cai Y-W, Wang L-P. et al. Breast cancer: pathogenesis and treatments. Signal Transduct Target Ther. 2025;10:49
3. Verde L, Galasso M, Savastano S, Colao A, Barrea L, Muscogiuri G. "Time" for obesity-related cancers: The role of chrononutrition in cancer prevention and treatment. Semin Cancer Biol. 2025;1:15-28
4. Teng Y. Remodeling of Mitochondria in Cancer and Other Diseases. Int J Mol Sci. 2023;24:7693
5. Genovese I, Carinci M, Modesti L, Aguiari G, Pinton P, Giorgi C. Mitochondria: Insights into Crucial Features to Overcome Cancer Chemoresistance. Int J Mol Sci. 2021;22:4770
6. Altieri DC. Mitochondria in cancer: clean windmills or stressed tinkerers? Trends Cell Biol. 2023;33:293-299
7. Yapa NMB, Lisnyak V, Reljic B, Ryan MT. Mitochondrial dynamics in health and disease. FEBS Lett. 2021;595:1184-1204
8. Ding H, Wei J, Fang L, Feng L, Gai S, He F. et al. A Multichannel Metabolic Pathway Interference Strategy for Complete Energy Depletion-Mediated Cancer Therapy. Adv Funct Mater. 2024;34:2312429
9. Jiang H, Ye J. The Warburg effect: The hacked mitochondrial-nuclear communication in cancer. Semin Cancer Biol. 2025;112:93-111
10. Kuo CL, Ponneri Babuharisankar A, Lin YC, Lien HW, Lo YK, Chou HY. et al. Mitochondrial oxidative stress in the tumor microenvironment and cancer immunoescape: foe or friend? J Biomed Sci. 2022;29:74
11. Allam J, Estrella J, Higbie V, Le P, Bent AH, Kotlov N. et al. Uncovering the tumor microenvironment (TME): Exploring survival and immunotherapy (IO) response in cancer of unknown primary (CUP). J Clin Oncol. 2025;43:4198-4198
12. Ikeda H, Kawase K, Nishi T, Watanabe T, Takenaga K, Inozume T. et al. Immune evasion through mitochondrial transfer in the tumour microenvironment. Nature. 2025;638:225-236
13. Goliwas KF, Libring S, Berestesky E, Gholizadeh S, Schwager SC, Frost AR. et al. Mitochondrial transfer from cancer-associated fibroblasts increases migration in aggressive breast cancer. J Cell Sci. 2023;136:1-16
14. Saha T, Dash C, Jayabalan R, Khiste S, Kulkarni A, Kurmi K. et al. Intercellular nanotubes mediate mitochondrial trafficking between cancer and immune cells. Nat Nanotechnol. 2022;17:98-106
15. Long X, Liu M, Nan Y, Chen Q, Xiao Z, Xiang Y. et al. Revitalizing Ancient Mitochondria with Nano-Strategies: Mitochondria-Remedying Nanodrugs Concentrate on Disease Control. Adv Mater. 2024;36:e2308239
16. Chen L, He Y, Lan J, Li Z, Gu D, Nie W. et al. Advancements in nano drug delivery system for liver cancer therapy based on mitochondria-targeting. Biomed Pharmacother. 2024;180:117520
17. Bailly C. Medicinal applications and molecular targets of dequalinium chloride. Biochem Pharmacol. 2021;186:114467
18. Beltzig L, Christmann M, Kaina B. Abrogation of Cellular Senescence Induced by Temozolomide in Glioblastoma Cells: Search for Senolytics. Cells. 2022;11:2588
19. Lord SR, Harris AL. Is it still worth pursuing the repurposing of metformin as a cancer therapeutic? Br J Cancer. 2023;128:958-966
20. Saito K, Zhang Q, Yang H, Yamatani K, Ai T, Ruvolo V. et al. Exogenous mitochondrial transfer and endogenous mitochondrial fission facilitate AML resistance to OxPhos inhibition. Blood Adv. 2021;5:4233-4255
21. El-Tanani M, Rabbani SA, Babiker R, Rangraze I, Kapre S, Palakurthi SS. et al. Unraveling the tumor microenvironment: Insights into cancer metastasis and therapeutic strategies. Cancer Lett. 2024;591:216894
22. Iaconisi GN, Ahmed A, Lauria G, Gallo N, Fiermonte G, Cowman MK. et al. Targeting mitochondria in Cancer therapy: Machine learning analysis of hyaluronic acid-based drug delivery systems. Int J Biol Macromol. 2024;283:137840
23. Buchke S, Sharma M, Bora A, Relekar M, Bhanu P, Kumar J. Mitochondria-Targeted, Nanoparticle-Based Drug-Delivery Systems: Therapeutics for Mitochondrial Disorders. Life (Basel). 2022;12:657
24. Ganji C, Muppala V, Khan M, Nagaraju GP, Farran B. Mitochondrial-targeted nanoparticles: Delivery and therapeutic agents in cancer. Drug Discov Today. 2023;28:103469
25. Wang C, Peng J, Xiao Y, Zhang Z, Yang X, Liang X. et al. Advances in nanotherapeutics for tumor treatment by targeting calcium overload. Colloids Surf B Biointerfaces. 2025;245:114190
26. Chu T, Huang Z, Ma W. Mitophagy: A double-edged sword in tumor cell death regulation and therapeutic response. Biochem Biophys Res Commun. 2025;777:152254
27. Kundu M, Butti R, Panda VK, Malhotra D, Das S, Mitra T. et al. Modulation of the tumor microenvironment and mechanism of immunotherapy-based drug resistance in breast cancer. Mol Cancer. 2024;23:92
28. Malikova I, Worth A, Aliyeva D, Khassenova M, Kriajevska MV, Tulchinsky E. Proteolysis of TAM receptors in autoimmune diseases and cancer: what does it say to us? Cell Death Dis. 2025;16:155
29. Masone MC. A distinct TAM subset mediates immunotherapy resistance in prostate cancer. Nat Rev Urol. 2025;22:70
30. Sun S, Huang X, Yang N, Lei H, Pei Z, Han Z. et al. Fluorinated Titanium Oxide (TiO(2-x)F(x)) Nanospindles as Ultrasound-Triggered Pyroptosis Inducers to Boost Sonodynamic Immunotherapy. ACS Nano. 2024;18:19756-19770
31. Hasan MN, Capuk O, Patel SM, Sun D. The Role of Metabolic Plasticity of Tumor-Associated Macrophages in Shaping the Tumor Microenvironment Immunity. Cancers (Basel). 2022;14:3331
32. Dubey S, Ghosh S, Goswami D, Ghatak D, De R. Immunometabolic attributes and mitochondria-associated signaling of Tumor-Associated Macrophages in tumor microenvironment modulate cancer progression. Biochem Pharmacol. 2023;208:115369
33. Wang X, Yang K, Yang B, Wang R, Zhu Y, Pan T. ANKRD22 participates in the proinflammatory activities of macrophages in the colon cancer tumor microenvironment. Cancer Immunol Immunother. 2025;74:86
34. Clark ML, Simeonov KP, Mowel WK, Michieletto MF, Joannas L, Wright JM. et al. Mitochondrial complex IV remodeling in tumor-associated macrophages amplifies interferon signaling and promotes anti-tumor immunity. Immunity. 2025;58:1670-1687
35. Li J, Ye Y, Liu Z, Zhang G, Dai H, Li J. et al. Macrophage mitochondrial fission improves cancer cell phagocytosis induced by therapeutic antibodies and is impaired by glutamine competition. Nat Cancer. 2022;3:453-470
36. Kidwell CU, Casalini JR, Pradeep S, Scherer SD, Greiner D, Bayik D. et al. Transferred mitochondria accumulate reactive oxygen species, promoting proliferation. Elife. 2023;12:e85494
37. Lee CW, Kuo CC, Liang CJ, Pan HJ, Shen CN, Lee CH. Effects of the media conditioned by various macrophage subtypes derived from THP-1 cells on tunneling nanotube formation in pancreatic cancer cells. BMC Mol Cell Biol. 2022;23:26
38. Zheng H, An M, Luo Y, Diao X, Zhong W, Pang M. et al. PDGFRα+ITGA11+ fibroblasts foster early-stage cancer lymphovascular invasion and lymphatic metastasis via ITGA11-SELE interplay. Cancer Cell. 2024;42:682-700
39. Magesh P, Thankachan S, Venkatesh T, Suresh PS. Breast cancer fibroblasts and cross-talk. Clin Chim Acta. 2021;521:158-169
40. Bordanaba-Florit G, Royo F, Albóniga OE, Clayton A, Falcón-Pérez JM, Webber J. Integration of proteomic and metabolomic analysis reveal distinct metabolic alterations of prostate cancer-associated fibroblasts compared to normal fibroblasts from patient's stroma samples. Biochim Biophys Acta Mol Basis Dis. 2024;1870:167229
41. Chen Y, Zhang X, Yang H, Liang T, Bai X. The "Self-eating" of cancer-associated fibroblast: A potential target for cancer. Biomed Pharmacother. 2023;163:114762
42. Sekine S, Sekine Y. Metabolite tunes MDV pathway to maintain mitochondrial fitness. Mol Cell. 2025;85:1253-1255
43. Gao A, Zhang M, Zhu SQ, Zou S, Chen H, Li X. et al. DNA polymerase iota promotes EMT and metastasis of esophageal squamous cell carcinoma by interacting with USP7 to stabilize HIF-1α. Cell Death Dis. 2024;15:171
44. Affinito A, Quintavalle C, Chianese RV, Roscigno G, Fiore D, D'Argenio V. et al. MCT4-driven CAF-mediated metabolic reprogramming in breast cancer microenvironment is a vulnerability targetable by miR-425-5p. Cell Death Discov. 2024;10:140
45. Zheng XX, Chen JJ, Sun YB, Chen TQ, Wang J, Yu SC. Mitochondria in cancer stem cells: Achilles heel or hard armor. Trends Cell Biol. 2023;33:708-727
46. Loh JJ, Ma S. Hallmarks of cancer stemness. Cell Stem Cell. 2024;31:617-639
47. Chakrabarty RP, Chandel NS. Mitochondria as Signaling Organelles Control Mammalian Stem Cell Fate. Cell Stem Cell. 2021;28:394-408
48. Fan M, Shi Y, Zhao J, Li L. Cancer stem cell fate determination: mito-nuclear communication. Cell Commun Signal. 2023;21:159
49. Shumin S, Nailin Y, Zifan P, Fei G, Liang C. Biomedical engineering targeting cancer stem cells to reinforce cancer therapy. Coord Chem Rev. 2025;530:216494
50. Tang M, Yang M, Wu G, Mo S, Wu X, Zhang S. et al. Epigenetic Induction of Mitochondrial Fission Is Required for Maintenance of Liver Cancer-Initiating Cells. Cancer Res. 2021;81:3835-3848
51. Xiao C, Li J, Hua A, Wang X, Li S, Li Z. et al. Hyperbaric Oxygen Boosts Antitumor Efficacy of Copper-Diethyldithiocarbamate Nanoparticles against Pancreatic Ductal Adenocarcinoma by Regulating Cancer Stem Cell Metabolism. Research. 2024:458-476
52. Popov LD. Mitochondrial biogenesis: An update. J Cell Mol Med. 2020;24:4892-4899
53. Wang H, Zhang W, Sun Y, Xu X, Chen X, Zhao K. et al. Nanotherapeutic strategies exploiting biological traits of cancer stem cells. Bioact Mater. 2025;50:61-94
54. Culp-Hill R, D'Alessandro A, Pietras EM. Extinguishing the Embers: Targeting AML Metabolism. Trends Mol Med. 2021;27:332-344
55. Mukherjee S, Bhatti GK, Chhabra R, Reddy PH, Bhatti JS. Targeting mitochondria as a potential therapeutic strategy against chemoresistance in cancer. Biomed Pharmacother. 2023;160:114398
56. Praharaj PP, Panigrahi DP, Bhol CS, Patra S, Mishra SR, Mahapatra KK. et al. Mitochondrial rewiring through mitophagy and mitochondrial biogenesis in cancer stem cells: A potential target for anti-CSC cancer therapy. Cancer Lett. 2021;498:217-228
57. de Beauchamp L, Himonas E, Helgason GV. Mitochondrial metabolism as a potential therapeutic target in myeloid leukaemia. Leukemia. 2022;36:1-12
58. Peiris-Pagès M, Martinez-Outschoorn UE, Pestell RG, Sotgia F, Lisanti MP. Cancer stem cell metabolism. Breast Cancer Res. 2016;18:55
59. Kim M, Gorelick AN, Vàzquez-García I, Williams MJ, Salehi S, Shi H. et al. Single-cell mtDNA dynamics in tumors is driven by coregulation of nuclear and mitochondrial genomes. Nat Genet. 2024;56:889-899
60. Bai R, Cui J. Mitochondrial immune regulation and anti-tumor immunotherapy strategies targeting mitochondria. Cancer Lett. 2023;564:216223
61. Corrado M, Pearce EL. Targeting memory T cell metabolism to improve immunity. J Clin Invest. 2022;132:1-11
62. Chen X, Lin P, Lu Y, Zheng J, Lin Y, Zhou Z. et al. Mitochondrial Regulation of CD8⁺ T Cells: Mechanisms and Therapeutic Modulation. Adv Sci. 2025: e03095.
63. Qin C, Gong S, Liang T, Zhang Z, Thomas J, Deng J. et al. HADHA Regulates Respiratory Complex Assembly and Couples FAO and OXPHOS. Adv Sci. 2024;11:e2405147
64. Yang W, Li Y, Feng R, Liang P, Tian K, Hu L. et al. PFOS causes lysosomes-regulated mitochondrial fission through TRPML1-VDAC1 and oligomerization of MCU/ATP5J2. J Hazard Mater. 2025;489:137685
65. Mukhopadhyay M. Learning the immunological repertoire. Nat Methods. 2025;22:651
66. Saillard M, Cenerenti M, Reichenbach P, Guillaume P, Su Z, Hafezi M. et al. Engineered CD4 TCR T cells with conserved high-affinity TCRs targeting NY-ESO-1 for advanced cellular therapies in cancer. Sci Adv. 2025;11:eadu5754
67. Lisci M, Griffiths GM. Arming a killer: mitochondrial regulation of CD8(+) T cell cytotoxicity. Trends Cell Biol. 2023;33:138-147
68. She H, Zheng J, Zhao G, Du Y, Tan L, Chen Z-S. et al. Arginase 1 drives mitochondrial cristae remodeling and PANoptosis in ischemia/hypoxia-induced vascular dysfunction. Signal Transduct Target Ther. 2025;10:167
69. Xie JH, Li YY, Jin J. The essential functions of mitochondrial dynamics in immune cells. Cell Mol Immunol. 2020;17:712-721
70. Liu X, Peng G. Mitochondria orchestrate T cell fate and function. Nat Immunol. 2021;22:276-278
71. Scharping NE, Rivadeneira DB, Menk AV, Vignali PDA, Ford BR, Rittenhouse NL. et al. Mitochondrial stress induced by continuous stimulation under hypoxia rapidly drives T cell exhaustion. Nat Immunol. 2021;22:205-215
72. Tang T, Wang W, Gan L, Bai J, Tan D, Jiang Y. et al. TIGIT expression in extrahepatic cholangiocarcinoma and its impact on CD8 + T cell exhaustion: implications for immunotherapy. Cell Death Dis. 2025;16:90
73. Hou Y, Shi P, Du H, Zhu C, Tang C, Que L. et al. HNF4α ubiquitination mediated by Peli1 impairs FAO and accelerates pressure overload-induced myocardial hypertrophy. Cell Death Dis. 2024;15:135
74. Samiea A, Celis G, Yadav R, Rodda LB, Moreau JM. B cells in non-lymphoid tissues. Nat Rev Immunol. 2025;25:483-496
75. Myers JA, Miller JS. Exploring the NK cell platform for cancer immunotherapy. Nat Rev Clin Oncol. 2021;18:85-100
76. Kim JE, Kim HS, Kim W, Lee EH, Kim S, Kim T. et al. Isoxazole-based molecules restore NK cell immune surveillance in hepatocarcinogenesis by targeting TM4SF5 and SLAMF7 linkage. Signal Transduct Target Ther. 2025;10:15
77. Xie J-H, Li Y-Y, Jin J. The essential functions of mitochondrial dynamics in immune cells. Cell Mol Immunol. 2020;17:712-721
78. CRISPR screens of tumor-infiltrating NK cells identify genetic checkpoints for CAR-NK therapy. Nat Biotechnol. 2024; 43: 696-697.
79. Slattery K, Woods E, Zaiatz-Bittencourt V, Marks S, Chew S, Conroy M. et al. TGFβ drives NK cell metabolic dysfunction in human metastatic breast cancer. J Immunother Cancer. 2021;9:e002044
80. Surace L, Doisne JM, Escoll P, Marie S, Dardalhon V, Croft C. et al. Polarized mitochondria as guardians of NK cell fitness. Blood Adv. 2021;5:26-38
81. Terrén I, Sandá V, Amarilla-Irusta A, Lopez-Pardo A, Sevilla A, Astarloa-Pando G. et al. IL-12/15/18-induced cell death and mitochondrial dynamics of human NK cells. Front Immunol. 2023;14:1211839
82. Pan R, Ryan J, Pan D, Wucherpfennig KW, Letai A. Augmenting NK cell-based immunotherapy by targeting mitochondrial apoptosis. Cell. 2022;185:1521-1538.e1518
83. Koenderman L, Tesselaar K, Vrisekoop N. On the origin of neutrophils. Cell Mol Immunol. 2025;22:459-460
84. Jaillon S, Ponzetta A, Di Mitri D, Santoni A, Bonecchi R, Mantovani A. Neutrophil diversity and plasticity in tumour progression and therapy. Nat Rev Cancer. 2020;20:485-503
85. Raftopoulou S, Valadez-Cosmes P, Mihalic ZN, Schicho R, Kargl J. Tumor-Mediated Neutrophil Polarization and Therapeutic Implications. Int J Mol Sci. 2022;23:3218
86. Willson JA, Arienti S, Sadiku P, Reyes L, Coelho P, Morrison T. et al. Neutrophil HIF-1α stabilization is augmented by mitochondrial ROS produced via the glycerol 3-phosphate shuttle. Blood. 2022;139:281-286
87. Zhao H, Wu L, Yan G, Chen Y, Zhou M, Wu Y. et al. Inflammation and tumor progression: signaling pathways and targeted intervention. Signal Transduct Target Ther. 2021;6:263
88. Zhang J, Miao C, Zhang H. Targeting neutrophil extracellular traps in cancer progression and metastasis. Theranostics. 2025;15:5846-5869
89. Peng S, Gao J, Stojkov D, Yousefi S, Simon HU. Established and emerging roles for mitochondria in neutrophils. Immunol Rev. 2023;314:413-426
90. Zhao R, Wang H, Li S, Wang L, Lei X, Peng P. et al. Tanshinone IIA@β-cyclodextrin encapsulated with Eucommia ulmoides rubber/acetylated starch film as novel oral delivery system for therapy of orthotopic colon cancer. Carbohydr Polym. 2025;353:123295
91. Ma Z, Han H, Zhao Y. Mitochondrial dysfunction-targeted nanosystems for precise tumor therapeutics. Biomaterials. 2023;293:121947
92. Quintana-Cabrera R, Scorrano L. Determinants and outcomes of mitochondrial dynamics. Mol Cell. 2023;83:857-876
93. Rochon K, Bauer BL, Roethler NA, Buckley Y, Su C-C, Huang W. et al. Structural basis for regulated assembly of the mitochondrial fission GTPase Drp1. Nat Commun. 2024;15:11328
94. Zhang L, Sun L, Wang L, Wang J, Wang D, Jiang J. et al. Mitochondrial division inhibitor (mdivi-1) inhibits proliferation and epithelial-mesenchymal transition via the NF-κB pathway in thyroid cancer cells. Toxicol In Vitro. 2023;88:105552
95. Panigrahi DP, Praharaj PP, Bhol CS, Mahapatra KK, Patra S, Behera BP. et al. The emerging, multifaceted role of mitophagy in cancer and cancer therapeutics. Semin Cancer Biol. 2020;66:45-58
96. Lu Y, Li Z, Zhang S, Zhang T, Liu Y, Zhang L. Cellular mitophagy: Mechanism, roles in diseases and small molecule pharmacological regulation. Theranostics. 2023;13:736-766
97. Denisenko TV, Gogvadze V, Zhivotovsky B. Mitophagy in carcinogenesis and cancer treatment. Discov Oncol. 2021;12:58
98. Liu S, Tian H, Ming H, Zhang T, Gao Y, Liu R. et al. Mitochondrial-Targeted CS@KET/P780 Nanoplatform for Site-Specific Delivery and High-Efficiency Cancer Immunotherapy in Hepatocellular Carcinoma. Adv Sci. 2024;11:e2308027
99. Zhang J, Zou L, Shi DF, Liu J, Zhang JF, Zhao RY. et al. Structure-Guided Design of a Small-Molecule Activator of Sirtuin-3 that Modulates Autophagy in Triple Negative Breast Cancer. J Med Chem. 2021;64:14192-14216
100. Zhen YQ, Yuan ZX, Zhang JH, Chen Y, Fu YN, Liu Y. et al. Flubendazole induces mitochondrial dysfunction and DRP1-mediated mitophagy by targeting EVA1A in breast cancer. Cell Death Dis. 2022;13:375
101. Lima K, Pereira-Martins DA, de Miranda LBL, Coelho-Silva JL, Leandro GDS, Weinhäuser I. et al. The PIP4K2 inhibitor THZ-P1-2 exhibits antileukemia activity by disruption of mitochondrial homeostasis and autophagy. Blood Cancer J. 2022;12:151
102. Chang M, Zhang L, Zhang T, Duan Y, Feng W, Yang S. et al. Ultrasound-augmented enzyodynamic-Ca2+ overload synergetic tumor nanotherapy. Biomaterials. 2024;307:122513
103. Zheng J, Zhang A, Du Q, Li C, Zhao Z, Li L. et al. Synergistic photoinduction of ferroptosis and apoptosis by a mitochondria-targeted iridium complex for bladder cancer therapy. J Colloid Interface Sci. 2024;683:420-431
104. Louault K, Bonneaud TL, Séveno C, Gomez-Bougie P, Nguyen F, Gautier F. et al. Interactions between cancer-associated fibroblasts and tumor cells promote MCL-1 dependency in estrogen receptor-positive breast cancers. Oncogene. 2019;38:3261-3273
105. Guo L, Yang Y, Sheng Y, Wang J, Ruan S, Han C. Mechanism of piperine in affecting apoptosis and proliferation of gastric cancer cells via ROS-mitochondria-associated signalling pathway. J Cell Mol Med. 2021;25:9513-9522
106. Li Y, He P, Liu Y, Qi M, Dong W. Combining Sodium Butyrate With Cisplatin Increases the Apoptosis of Gastric Cancer In Vivo and In Vitro via the Mitochondrial Apoptosis Pathway. Front Pharmacol. 2021;12:708093
107. Kleih M, Böpple K, Dong M, Gaißler A, Heine S, Olayioye MA. et al. Direct impact of cisplatin on mitochondria induces ROS production that dictates cell fate of ovarian cancer cells. Cell Death Dis. 2019;10:851
108. Tao L, Yin Z, Ni T, Chu Z, Hao S, Wang Z. et al. The Ethyl Acetate Extract From Celastrus orbiculatus Promotes Apoptosis of Gastric Cancer Cells Through Mitochondria Regulation by PHB. Front Pharmacol. 2021;12:635467
109. Datta S, Sears T, Cortopassi G, Woolard K, Angelastro JM. Repurposing FDA approved drugs inhibiting mitochondrial function for targeting glioma-stem like cells. Biomed Pharmacother. 2021;133:111058
110. Han L, Mei J, Ma J, Wang F, Gu Z, Li J. et al. Cinnamaldehyde induces endogenous apoptosis of the prostate cancer-associated fibroblasts via interfering the Glutathione-associated mitochondria function. Med Oncol. 2020;37:91
111. Wang Q, Li S, Xu C, Hua A, Wang C, Xiong Y. et al. A novel lonidamine derivative targeting mitochondria to eliminate cancer stem cells by blocking glutamine metabolism. Pharmacol Res. 2023;190:106740
112. Cheng X, Xu HD, Ran HH, Liang G, Wu FG. Glutathione-Depleting Nanomedicines for Synergistic Cancer Therapy. ACS Nano. 2021;15:8039-8068
113. Perillo B, Di Donato M, Pezone A, Di Zazzo E, Giovannelli P, Galasso G. et al. ROS in cancer therapy: the bright side of the moon. Exp Mol Med. 2020;52:192-203
114. Zinovkin RA, Lyamzaev KG, Chernyak BV. Current perspectives of mitochondria-targeted antioxidants in cancer prevention and treatment. Front Cell Dev Biol. 2023;11:1048177
115. Benot-Dominguez R, Tupone MG, Castelli V, d'Angelo M, Benedetti E, Quintiliani M. et al. Olive leaf extract impairs mitochondria by pro-oxidant activity in MDA-MB-231 and OVCAR-3 cancer cells. Biomed Pharmacother. 2021;134:111139
116. Han P, Ma F, Wang Z, Zhou X, Zheng K, Lu S. et al. Characteristics of ultrasound-induced sesamol-soybean peptide composite particles (SSPPs) and SSPP-stabilized Pickering emulsions. Int J Biol Macromol. 2025: 145367.
117. Shi L, Gao LL, Cai SZ, Xiong QW, Ma ZR. A novel selective mitochondrial-targeted curcumin analog with remarkable cytotoxicity in glioma cells. Eur J Med Chem. 2021;221:113528
118. Qin D, Hu W, Guo Y, Cheng R, Hao F, Zhao B. Baicalein based nano-delivery system restores mitochondrial homeostasis through PPAR signaling pathway to promote wound healing in diabetes. J Nanobiotechnology. 2025;23:360
119. Farhan M, Rizvi A. Understanding the Prooxidant Action of Plant Polyphenols in the Cellular Microenvironment of Malignant Cells: Role of Copper and Therapeutic Implications. Front Pharmacol. 2022;13:929853
120. Li Y, Zhang X, Wang Z, Li B, Zhu H. Modulation of redox homeostasis: A strategy to overcome cancer drug resistance. Front Pharmacol. 2023;14:1156538
121. Fan Y, Niu Z, Yin L, Yao L, Ding S, Tong Y. et al. Membrane biomimetic nanoenzyme-incorporated hybrid glycyrrhizic acid hydrogel for precise mitochondrial ROS scavenging for osteoarthritis treatment. Materials Today Bio. 2025;32:101778
122. Abu Shelbayeh O, Arroum T, Morris S, Busch KB. PGC-1α Is a Master Regulator of Mitochondrial Lifecycle and ROS Stress Response. Antioxidants (Basel). 2023;12:1075
123. Zeng Q-G, Li L, Chang T, Sun Y, Zheng B, Xue L-N. et al. Phosphorylation of POU3F3 Mediated Nuclear Translocation Promotes Proliferation in Non-Small Cell Lung Cancer through Accelerating ATP5PF Transcription and ATP Production. Adv Sci. 2025;12:e2411503
124. Zu Y, Chen XF, Li Q, Zhang ST, Si LN. PGC-1α activates SIRT3 to modulate cell proliferation and glycolytic metabolism in breast cancer. Neoplasma. 2021;68:352-361
125. Wang C, Dong L, Li X, Li Y, Zhang B, Wu H. et al. The PGC1α/NRF1-MPC1 axis suppresses tumor progression and enhances the sensitivity to sorafenib/doxorubicin treatment in hepatocellular carcinoma. Free Radic Biol Med. 2021;163:141-152
126. Liu W, Li L, Bai X, Zhang M, Lv W, Ma Y. et al. Osteosarcoma Cell-Derived Migrasomes Promote Macrophage M2 Polarization to Aggravate Osteosarcoma Proliferation and Metastasis. Adv Sci. 2025;12:e2409870
127. Yang T, Liang N, Zhang J, Bai Y, Li Y, Zhao Z. et al. OCTN2 enhances PGC-1α-mediated fatty acid oxidation and OXPHOS to support stemness in hepatocellular carcinoma. Metabolism. 2023;147:155628
128. De Luca A, Fiorillo M, Peiris-Pagès M, Ozsvari B, Smith DL, Sanchez-Alvarez R. et al. Mitochondrial biogenesis is required for the anchorage-independent survival and propagation of stem-like cancer cells. Oncotarget. 2015;6:14777-14795
129. Seo W, Yoo S, Zhong Y, Lee SH, Woo SY, Choi HS. et al. Targeting ERRα promotes cytotoxic effects against acute myeloid leukemia through suppressing mitochondrial oxidative phosphorylation. J Hematol Oncol. 2022;15:156
130. Liu SL, Liang HB, Yang ZY, Cai C, Wu ZY, Wu XS. et al. Gemcitabine and XCT790, an ERRα inverse agonist, display a synergistic anticancer effect in pancreatic cancer. Int J Med Sci. 2022;19:286-298
131. Schoepke E, Billon C, Haynes KM, Avdagic A, Sitaula S, Sanders R. et al. A Selective ERRα/γ Inverse Agonist, SLU-PP-1072, Inhibits the Warburg Effect and Induces Apoptosis in Prostate Cancer Cells. ACS Chem Biol. 2020;15:2338-2345
132. Chekaoui A, Ertl HCJ. PPARα Agonist Fenofibrate Enhances Cancer Vaccine Efficacy. Cancer Res. 2021;81:4431-4440
133. Zhang Y, Ma J, Li P, Lu K, Han Y, Hu X. et al. Fatty acid metabolism shapes immune responses in chronic lymphocytic leukemia. Biomark Res. 2025;13:42
134. Li X, Wang M, Denk T, Buschauer R, Li Y, Beckmann R. et al. Structural basis for differential inhibition of eukaryotic ribosomes by tigecycline. Nat Commun. 2024;15:5481
135. Zhang J, Qiao W, Luo Y. Mitochondrial quality control proteases and their modulation for cancer therapy. Med Res Rev. 2023;43:399-436
136. Cormio A, Sanguedolce F, Pesce V, Musicco C. Mitochondrial Caseinolytic Protease P: A Possible Novel Prognostic Marker and Therapeutic Target in Cancer. Int J Mol Sci. 2021;22:6228
137. Choi S-E, Hwang Y, Lee S-J, Jung H, Shin TH, Son Y. et al. Mitochondrial protease ClpP supplementation ameliorates diet-induced NASH in mice. J Hepatol. 2022;77:735-747
138. Zhou LL, Zhang T, Xue Y, Yue C, Pan Y, Wang P. et al. Selective activator of human ClpP triggers cell cycle arrest to inhibit lung squamous cell carcinoma. Nat Commun. 2023;14:7069
139. Odia Y, Hall MD, Cloughesy TF, Wen PY, Arrillaga-Romany I, Daghistani D. et al. Selective DRD2 antagonist and ClpP agonist ONC201 in a recurrent non-midline H3 K27M-mutant glioma cohort. Neuro Oncol. 2024;26:noae021
140. Geiß C, Witzler C, Poschet G, Ruf W, Régnier-Vigouroux A. Metabolic and inflammatory reprogramming of macrophages by ONC201 translates in a pro-inflammatory environment even in presence of glioblastoma cells. Eur J Immunol. 2021;51:1246-1261
141. Al-Habsi M, Chamoto K, Matsumoto K, Nomura N, Zhang B, Sugiura Y. et al. Spermidine activates mitochondrial trifunctional protein and improves antitumor immunity in mice. Science. 2022;378:eabj3510
142. Cai Q, Cai X, Shubhra QTH. Mitochondrial transfer drives immune evasion in tumor microenvironment. Trends Cancer. 2025;11:424-426
143. Wang ZH, Chen L, Li W, Chen L, Wang YP. Mitochondria transfer and transplantation in human health and diseases. Mitochondrion. 2022;65:80-87
144. Watson DC, Bayik D, Storevik S, Moreino SS, Sprowls SA, Han J. et al. GAP43-dependent mitochondria transfer from astrocytes enhances glioblastoma tumorigenicity. Nat Cancer. 2023;4:648-664
145. Hoover G, Gilbert S, Curley O, Obellianne C, Lin MT, Hixson W. et al. Nerve-to-cancer transfer of mitochondria during cancer metastasis. Nature. 2025;644:252-262
146. Ye Z, Deng M, Yang Y, Song Y, Weng L, Qi W. et al. Epithelial mitochondrial fission-mediated PANoptosis is crucial for ulcerative colitis and its inhibition by saquinavir through Drp1. Pharmacol Res. 2024;210:107538
147. Courtois S, de Luxán-Delgado B, Penin-Peyta L, Royo-García A, Parejo-Alonso B, Jagust P. et al. Inhibition of Mitochondrial Dynamics Preferentially Targets Pancreatic Cancer Cells with Enhanced Tumorigenic and Invasive Potential. Cancers (Basel). 2021;13:698
148. Wu D, Dasgupta A, Chen KH, Neuber-Hess M, Patel J, Hurst TE. et al. Identification of novel dynamin-related protein 1 (Drp1) GTPase inhibitors: Therapeutic potential of Drpitor1 and Drpitor1a in cancer and cardiac ischemia-reperfusion injury. FASEB J. 2020;34:1447-1464
149. Zhang P, Zhong D, Yu Y, Wang L, Li Y, Liang Y. et al. Integration of STING activation and COX-2 inhibition via steric-hindrance effect tuned nanoreactors for cancer chemoimmunotherapy. Biomaterials. 2024;311:122695
150. Sun B, Zhao Y, Yang S, Li X, Li N, Wang Y. et al. Celecoxib as a potential treatment for hepatocellular carcinoma in populations exposed to high PFAS levels. J Hazard Mater. 2025;489:137613
151. Fu Y, Dong W, Xu Y, Li L, Yu X, Pang Y. et al. Targeting mitochondrial dynamics by AZD5363 in triple-negative breast cancer MDA-MB-231 cell-derived spheres. Naunyn Schmiedebergs Arch Pharmacol. 2023;396:2545-2553
152. Deng X, Liu J, Liu L, Sun X, Huang J, Dong J. Drp1-mediated mitochondrial fission contributes to baicalein-induced apoptosis and autophagy in lung cancer via activation of AMPK signaling pathway. Int J Biol Sci. 2020;16:1403-1416
153. Tao H, Li L, Li H, Ma T, Zhu X, Cao J. et al. The anti-tumor effects of main component (benzethonium chloride) of butorphanol tartrate injection in non-small cell lung cancer. Sci Rep. 2024;14:30194
154. Zhang D, Zhao M, Jiang P, Zhou Y, Yan X, Zhou C. et al. RPL22L1 fosters malignant features of cervical cancer via the modulation of DUSP6-ERK axis. J Transl Med. 2025;23:244
155. Meyer N, Zielke S, Michaelis JB, Linder B, Warnsmann V, Rakel S. et al. AT 101 induces early mitochondrial dysfunction and HMOX1 (heme oxygenase 1) to trigger mitophagic cell death in glioma cells. Autophagy. 2018;14:1693-1709
156. Rademaker G, Boumahd Y, Peiffer R, Anania S, Wissocq T, Liégeois M. et al. Myoferlin targeting triggers mitophagy and primes ferroptosis in pancreatic cancer cells. Redox Biol. 2022;53:102324
157. Liu K, Lee J, Kim JY, Wang L, Tian Y, Chan ST. et al. Mitophagy Controls the Activities of Tumor Suppressor p53 to Regulate Hepatic Cancer Stem Cells. Mol Cell. 2017;68:281-292.e285
158. Zhou J, Li G, Zheng Y, Shen HM, Hu X, Ming QL. et al. A novel autophagy/mitophagy inhibitor liensinine sensitizes breast cancer cells to chemotherapy through DNM1L-mediated mitochondrial fission. Autophagy. 2015;11:1259-1279
159. Yang ST, Fan JB, Liu TT, Ning S, Xu JH, Zhou YJ. et al. Development of Strigolactones as Novel Autophagy/Mitophagy Inhibitors against Colorectal Cancer Cells by Blocking the Autophagosome-Lysosome Fusion. J Med Chem. 2022;65:9706-9717
160. Hosseinzadeh A, Alinaghian N, Sheibani M, Seirafianpour F, Naeini AJ, Mehrzadi S. Melatonin: Current evidence on protective and therapeutic roles in gynecological diseases. Life Sci. 2024;344:122557
161. Sun H, Ou T, Hu J, Yang Z, Lei Q, Li Y. et al. Nitazoxanide impairs mitophagy flux through ROS-mediated mitophagy initiation and lysosomal dysfunction in bladder cancer. Biochem Pharmacol. 2021;190:114588
162. Basu V, Shabnam n, Murghai Y, Ali M, Sahu S, Verma BK. et al. ONC212, alone or in synergistic conjunction with Navitoclax (ABT-263), promotes cancer cell apoptosis via unconventional mitochondrial-independent caspase-3 activation. Cell Commun Signal. 2024;22:441
163. Jia Y, Han L, Ramage CL, Wang Z, Weng CC, Yang L. et al. Co-targeting BCL-XL and BCL-2 by PROTAC 753B eliminates leukemia cells and enhances efficacy of chemotherapy by targeting senescent cells. Haematologica. 2023;108:2626-2638
164. Cui Y, Shao X, Yang H, Xin J, Liu Y, Zhang M. et al. MDM2 inhibitor APG-115 synergizes with ABT-199 to induce cell apoptosis in chronic lymphocytic leukemia. Front Pharmacol. 2024;15:1441383
165. Rizvi S, Mertens JC, Bronk SF, Hirsova P, Dai HM, Roberts LR. et al. Platelet-derived Growth Factor Primes Cancer-associated Fibroblasts for Apoptosis. J Biol Chem. 2014;289:22835-22849
166. Roca-Portoles A, Rodriguez-Blanco G, Sumpton D, Cloix C, Mullin M, Mackay GM. et al. Venetoclax causes metabolic reprogramming independent of BCL-2 inhibition. Cell Death Dis. 2020;11:616
167. Pracha SH, Shrestha S, Ryan N, Upadhaya P, Lamenza FF, Jagadeesha S. et al. Targeting macrophage migration inhibitory factor to inhibit T cell immunosuppression in the tumor microenvironment and improve cancer outcomes in head and neck squamous cell carcinoma. Oral Oncology. 2024;160:107126
168. Huang C, Lan W, Fraunhoffer N, Meilerman A, Iovanna J, Santofimia-Castaño P. Dissecting the Anticancer Mechanism of Trifluoperazine on Pancreatic Ductal Adenocarcinoma. Cancers (Basel). 2019;11:1869
169. He W, Zhang W, Zheng Q, Wei Z, Wang Y, Hu M. et al. Cinnamaldehyde causes apoptosis of myeloid-derived suppressor cells through the activation of TLR4. Oncol Lett. 2019;18:2420-2426
170. Khamphio M, Barusrux S, Weerapreeyakul N. Sesamol induces mitochondrial apoptosis pathway in HCT116 human colon cancer cells via pro-oxidant effect. Life Sci. 2016;158:46-56
171. Liu ZH, Yang CX, Zhang L, Yang CY, Xu XQ. Baicalein, as a Prooxidant, Triggers Mitochondrial Apoptosis in MCF-7 Human Breast Cancer Cells Through Mobilization of Intracellular Copper and Reactive Oxygen Species Generation. Onco Targets Ther. 2019;12:10749-10761
172. McCartin C, Mathieu E, Dontenwill M, Herold-Mende C, Idbaih A, Bonfiglio A. et al. An N-heterocyclic carbene iridium(III) complex as a potent anti-cancer stem cell therapeutic. Chem Biol Interact. 2022;367:110167
173. Vardhana SA, Hwee MA, Berisa M, Wells DK, Yost KE, King B. et al. Impaired mitochondrial oxidative phosphorylation limits the self-renewal of T cells exposed to persistent antigen. Nat Immunol. 2020;21:1022-1033
174. Yu YR, Imrichova H, Wang H, Chao T, Xiao Z, Gao M. et al. Disturbed mitochondrial dynamics in CD8(+) TILs reinforce T cell exhaustion. Nat Immunol. 2020;21:1540-1551
175. Dilly S, Romero M, Solier S, Feron O, Dessy C, Slama Schwok A. Targeting M2 Macrophages with a Novel NADPH Oxidase Inhibitor. Antioxidants (Basel). 2023;12:440
176. Griess B, Mir S, Datta K, Teoh-Fitzgerald M. Scavenging reactive oxygen species selectively inhibits M2 macrophage polarization and their pro-tumorigenic function in part, via Stat3 suppression. Free Radic Biol Med. 2020;147:48-60
177. Zeng J, Xu H, Fan PZ, Xie J, He J, Yu J. et al. Kaempferol blocks neutrophil extracellular traps formation and reduces tumour metastasis by inhibiting ROS-PAD4 pathway. J Cell Mol Med. 2020;24:7590-7599
178. Wan H, Xu B, Zhu N, Ren B. PGC-1α activator-induced fatty acid oxidation in tumor-infiltrating CTLs enhances effects of PD-1 blockade therapy in lung cancer. Tumori. 2020;106:55-63
179. Jin M, Liu S, Zhan M, Huang JD. Engineered Genetic Circuits Activated by Bezafibrate Improve ESC-Based TAA Cancer Vaccine Efficacy and PD-L1 Nanobody Therapy. Adv Sci. 2025;12:e2500272
180. Hu D, Cao J, Yu H, Ding N, Mi L, Ye Y. et al. PI3K inhibitor idelalisib enhances the anti-tumor effects of CDK4/6 inhibitor palbociclib via PLK1 in B-cell lymphoma. Cancer Lett. 2024;597:216996
181. Dong Z, Abbas MN, Kausar S, Yang J, Li L, Tan L. et al. Biological Functions and Molecular Mechanisms of Antibiotic Tigecycline in the Treatment of Cancers. Int J Mol Sci. 2019;20:3577
182. Gronauer TF, Mandl MM, Lakemeyer M, Hackl MW, Meßner M, Korotkov VS. et al. Design and synthesis of tailored human caseinolytic protease P inhibitors. Chem Commun (Camb). 2018;54:9833-9836
183. Ishizawa J, Zarabi SF, Davis RE, Halgas O, Nii T, Jitkova Y. et al. Mitochondrial ClpP-Mediated Proteolysis Induces Selective Cancer Cell Lethality. Cancer Cell. 2019;35:721-737.e729
184. Wang J, Liu X, Qiu Y, Shi Y, Cai J, Wang B. et al. Cell adhesion-mediated mitochondria transfer contributes to mesenchymal stem cell-induced chemoresistance on T cell acute lymphoblastic leukemia cells. J Hematol Oncol. 2018;11:11
185. Burt R, Dey A, Aref S, Aguiar M, Akarca A, Bailey K. et al. Activated stromal cells transfer mitochondria to rescue acute lymphoblastic leukemia cells from oxidative stress. Blood. 2019;134:1415-1429
186. Eniafe J, Jiang S. The functional roles of TCA cycle metabolites in cancer. Oncogene. 2021;40:3351-3363
187. Reddy VB, Boteju L, Boteju A, Shen L, Kassahun K, Reddy N. et al. In Vitro and In Vivo Metabolism of a Novel Antimitochondrial Cancer Metabolism Agent, CPI-613, in Rat and Human. Drug Metab Dispos. 2022;50:361-373
188. Rostamian H, Khakpoor-Koosheh M, Jafarzadeh L, Masoumi E, Fallah-Mehrjardi K, Tavassolifar MJ. et al. Restricting tumor lactic acid metabolism using dichloroacetate improves T cell functions. BMC Cancer. 2022;22:39
189. Chen L, Peng J, Wang Y, Jiang H, Wang W, Dai J. et al. Fenofibrate-induced mitochondrial dysfunction and metabolic reprogramming reversal: the anti-tumor effects in gastric carcinoma cells mediated by the PPAR pathway. Am J Transl Res. 2020;12:428-446
190. Wang Y, Wang W, Gu R, Chen J, Chen Q, Lin T. et al. In Situ Vaccination with Mitochondria-Targeting Immunogenic Death Inducer Elicits CD8(+) T Cell-Dependent Antitumor Immunity to Boost Tumor Immunotherapy. Adv Sci (Weinh). 2023;10:e2300286
191. Kalyanaraman B. Exploiting the tumor immune microenvironment and immunometabolism using mitochondria-targeted drugs: Challenges and opportunities in racial disparity and cancer outcome research. FASEB J. 2022;36:e22226
192. Cheng G, Hardy M, Zielonka J, Weh K, Zielonka M, Boyle KA. et al. Mitochondria-targeted magnolol inhibits OXPHOS, proliferation, and tumor growth via modulation of energetics and autophagy in melanoma cells. Cancer Treat Res Commun. 2020;25:100210
193. Cheng G, Hardy M, Topchyan P, Zander R, Volberding P, Cui W. et al. Mitochondria-targeted hydroxyurea inhibits OXPHOS and induces antiproliferative and immunomodulatory effects. iScience. 2021;24:102673
194. Gao Y, Song Z, Gan W, Zou X, Bai Y, Zhao X. et al. Selective and iron-independent ferroptosis in cancer cells induced by manipulation of mitochondrial fatty acid oxidation. Biomaterials. 2025;320:123259
195. Das M, Giannoudis A, Sharma V. The role of CPT1A as a biomarker of breast cancer progression: a bioinformatic approach. Sci Rep. 2022;12:16441
196. Wang S, Li X, Xu Z, Zhang K, Huang C, Luan Y. et al. Biomimetic nanoparticles targeting metabolic reprogramming of tumor and immune cells to enhance colon cancer therapy. Chem Eng J. 2025;519:165328
197. Tang L, Shi Y, Liao Q, Wang F, Wu H, Ren H. et al. Reversing metabolic reprogramming by CPT1 inhibition with etomoxir promotes cardiomyocyte proliferation and heart regeneration via DUSP1 ADP-ribosylation-mediated p38 MAPK phosphorylation. Acta Pharm Sin B. 2024;15:256-277
198. Al-Husein BA, Hashem J, Alawi SS, Alqudah MAY, Al-Azayzih A. Synergistic effect of venetoclax and teglicar in multiple myeloma cell lines. Pharmacia. 2024;7:1-9
199. Dhakal B, Li CMY, Li R, Yeo K, Wright JA, Gieniec KA. et al. The Antianginal Drug Perhexiline Displays Cytotoxicity against Colorectal Cancer Cells In Vitro: A Potential for Drug Repurposing. Cancers (Basel). 2022;14:1043
200. Liu HY, Zhang HS, Liu MY, Li HM, Wang XY, Wang M. GLS1 depletion inhibited colorectal cancer proliferation and migration via redox/Nrf2/autophagy-dependent pathway. Arch Biochem Biophys. 2021;708:108964
201. Usart M, Hansen N, Stetka J, Almeida Fonseca T, Guy A, Kimmerlin Q. et al. The glutaminase inhibitor CB-839 targets metabolic dependencies of JAK2-mutant hematopoiesis in MPN. Blood Adv. 2024;8:2312-2325
202. Ma G, Zhang Z, Li P, Zhang Z, Zeng M, Liang Z. et al. Reprogramming of glutamine metabolism and its impact on immune response in the tumor microenvironment. Cell Commun Signal. 2022;20:114
203. Huang G, Li H, Zhang H. Abnormal Expression of Mitochondrial Ribosomal Proteins and Their Encoding Genes with Cell Apoptosis and Diseases. Int J Mol Sci. 2020;21:8879
204. Fontana GA, MacArthur MR, Rotankova N, Di Filippo M, Beer HD, Gahlon HL. The mitochondrial DNA common deletion as a potential biomarker of cancer-associated fibroblasts from skin basal and squamous cell carcinomas. Sci Rep. 2024;14:553
205. Egan G, Khan DH, Lee JB, Mirali S, Zhang L, Schimmer AD. Mitochondrial and Metabolic Pathways Regulate Nuclear Gene Expression to Control Differentiation, Stem Cell Function, and Immune Response in Leukemia. Cancer Discov. 2021;11:1052-1066
206. Fang C, Mo F, Liu L, Du J, Luo M, Men K. et al. Oxidized mitochondrial DNA sensing by STING signaling promotes the antitumor effect of an irradiated immunogenic cancer cell vaccine. Cell Mol Immunol. 2021;18:2211-2223
207. Rao Z, Zhu Y, Yang P, Chen Z, Xia Y, Qiao C. et al. Pyroptosis in inflammatory diseases and cancer. Theranostics. 2022;12:4310-4329
208. Yu Q, Sun S, Yang N, Pei Z, Chen Y, Nie J. et al. Self-Cascaded Pyroptosis-STING Initiators for Catalytic Metalloimmunotherapy. J Am Chem Soc. 2025;147:3161-3173
209. Yang N, Sun S, Xu J, Gong F, Lei H, Hao Y. et al. Manganese Galvanic Cells Intervene in Tumor Metabolism to Reinforce cGAS-STING Activation for Bidirectional Synergistic Hydrogen-Immunotherapy. Adv Mater Processes. 2025;37:2414929
210. Shen H, Yu M, Tsoli M, Chang C, Joshi S, Liu J. et al. Targeting reduced mitochondrial DNA quantity as a therapeutic approach in pediatric high-grade gliomas. Neuro Oncol. 2020;22:139-151
211. Bonekamp NA, Peter B, Hillen HS, Felser A, Bergbrede T, Choidas A. et al. Small-molecule inhibitors of human mitochondrial DNA transcription. Nature. 2020;588:712-716
212. Antonicka H, Lin ZY, Janer A, Aaltonen MJ, Weraarpachai W, Gingras AC. et al. A High-Density Human Mitochondrial Proximity Interaction Network. Cell Metab. 2020;32:479-497.e479
213. Yang Y, Huang J, Liu M, Qiu Y, Chen Q, Zhao T. et al. Emerging Sonodynamic Therapy-Based Nanomedicines for Cancer Immunotherapy. Adv Sci (Weinh). 2023;10:e2204365
214. Kumar N, Mangla M. Nanotechnology and nanobots unleashed: pioneering a new era in gynecological cancer management - a comprehensive review. Cancer Chemother Pharmacol. 2025;95:18
215. Tabish TA, Hamblin MR. Mitochondria-targeted nanoparticles (mitoNANO): An emerging therapeutic shortcut for cancer. Biomater Biosyst. 2021;3:100023
216. Wang W, Yao S-Y, Luo J, Ding C, Huang Q, Yang Y. et al. Engineered hypoxia-responsive albumin nanoparticles mediating mitophagy regulation for cancer therapy. Nat Commun. 2025;16:596
217. Zhang W, Chen G, Chen Z, Yang X, Zhang B, Wang S. et al. Mitochondria-targeted polyprodrug nanoparticles induce mitochondrial stress for immunogenic chemo-photodynamic therapy of ovarian cancer. J Controlled Release. 2024;371:470-483
218. Hu X, Zhang M, Quan C, Ren S, Chen W, Wang J. ROS-responsive and triple-synergistic mitochondria-targeted polymer micelles for efficient induction of ICD in tumor therapeutics. Bioact Mater. 2024;36:490-507
219. Han X, Sun S, Yang N, Han Z, Pei Z, Yu Q. et al. Nano-Engineered Magnesium Implants for Magnetothermal Enhanced Pyroptosis to Boost Immunotherapy. Adv Funct Mater. 2024;34:2405836
220. Wang D, Zhang L, Nie J, Yang N, Sun S, Wang C. et al. Engineered Nano-Micro Pyroptosis Generators: A Magnetic-Metallo-Immunotherapeutic Strategy to Reinforce Transarterial Embolization. J Am Chem Soc. 2025;147:25536-25552
221. Nie J, Yang N, Sun S, Wang L, Pei Z, Wu J. et al. Antimony Component Schottky Nanoheterojunctions as Ultrasound-Heightened Pyroptosis Initiators for Sonocatalytic Immunotherapy. Angew Chem Int Ed Engl. 2025;64:e202416426
222. Guo W, Li Z, Huang H, Xu Z, Chen Z, Shen G. et al. VB12-Sericin-PBLG-IR780 Nanomicelles for Programming Cell Pyroptosis via Photothermal (PTT)/Photodynamic (PDT) Effect-Induced Mitochondrial DNA (mitoDNA) Oxidative Damage. ACS Appl Mater Interfaces. 2022;14:17008-17021
223. Zhang C-J, Li J-M, Xu D, Wang D-D, Qi M-H, Chen F. et al. Surface Molecularly Engineered Mitochondria Conduct Immunophenotype Repolarization of Tumor-Associated Macrophages to Potentiate Cancer Immunotherapy. Adv Sci. 2024;11:e2403044
224. Sharma A, Liaw K, Sharma R, Thomas AG, Slusher BS, Kannan S. et al. Targeting Mitochondria in Tumor-Associated Macrophages using a Dendrimer-Conjugated TSPO Ligand that Stimulates Antitumor Signaling in Glioblastoma. Biomacromolecules. 2020;21:3909-3922
225. Zhao M, Li J, Chen F, Han Y, Chen D, Hu H. Engineering nanoparticles boost TNBC therapy by CD24 blockade and mitochondrial dynamics regulation. J Control Release. 2023;355:211-227
226. Zhao M, Li J, Liu J, Xu M, Ji H, Wu S. et al. Charge-switchable nanoparticles enhance Cancer immunotherapy based on mitochondrial dynamic regulation and immunogenic cell death induction. J Control Release. 2021;335:320-332
227. Yao X, Yang H, Guo S, Liu Y, Zhang Q, Zhou Z. et al. Radiation-triggerable bioreactors enable bioenergetic reprograming of cancer stem cell plasticity via targeted arginine metabolism disruption for augmented radio-immunotherapy. Biomaterials. 2025;322:123391
228. Zhou Z, Qu C, Zhou P, Zhou Q, Li D, Wu X. et al. Extracellular vesicles activated cancer-associated fibroblasts promote lung cancer metastasis through mitophagy and mtDNA transfer. J Exp Clin Cancer Res. 2024;43:1-19
229. Li J, Lei T, Ouyang W, Ye Z, Li L, Li G. et al. Reshape tumor microenvironment by modulating CXCR4 with FAP-targeted diselenide-organosilica delivery system for prostate cancer immunotherapy. Chem Eng J. 2024;503:158308
230. Burt RJ, Dey A, Akarca A, Allen H, Amerikanou R, Atkinson S. et al. Mitochondrial dsRNA from B-ALL cells stimulates mesenchymal stromal cells to become cancer-associated fibroblasts. Blood Adv. 2024;8:5696-5709
231. Xiao C, Wang X, Li S, Zhang Z, Li J, Deng Q. et al. A cuproptosis-based nanomedicine suppresses triple negative breast cancers by regulating tumor microenvironment and eliminating cancer stem cells. Biomaterials. 2024;313:122763
232. Zhang Z, Zhao Q, Xu Q, Deng Q, Hua A, Wang X. et al. A mitochondria-interfering nanocomplex cooperates with photodynamic therapy to boost antitumor immunity. Biomaterials. 2025;317:123094
233. Pan X, Wang Z, Tan M, Fu Z, Nie G, Wang H. Nanoinducer-mediated mitochondria-selective degradation enhances T cell immunotherapy against multiple cancers. Nat Nanotechnol. 2025;20:947-958
234. Zhang D, Li Q, Chen X, Nie X, Xue F, Xu W. et al. An Injectable Hydrogel to Modulate T Cells for Cancer Immunotherapy. Small. 2022;18:e2202663
235. Guo H, Liu Y, Li X, Wang H, Mao D, Wei L. et al. Magnetic Metal-Organic Framework-Based Nanoplatform with Platelet Membrane Coating as a Synergistic Programmed Cell Death Protein 1 Inhibitor against Hepatocellular Carcinoma. ACS Nano. 2023;17:23829-23849
236. Cheng D, Luo L, Zhang Q, Song Z, Zhan Y, Tu W. et al. Ca2+- and cGAMP-Contained Semiconducting Polymer Nanomessengers for Radiodynamic-Activated Calcium Overload and Immunotherapy. Adv Sci. 2025;12:2411739
237. Wang W, Yao SY, Luo J, Ding C, Huang Q, Yang Y. et al. Engineered hypoxia-responsive albumin nanoparticles mediating mitophagy regulation for cancer therapy. Nat Commun. 2025;16:596
238. Ren M, Sun X, Lin J, Kan Y, Lu S, Zhang X. et al. Hypoxia-responsive oncolytic conjugate triggers type-II immunogenic cell death for enhanced photodynamic immunotherapy. J Controlled Release. 2025;382:113717
239. Hu X, Zhang M, Quan C, Ren S, Chen W, Wang J. ROS-responsive and triple-synergistic mitochondria-targeted polymer micelles for efficient induction of ICD in tumor therapeutics. Bioactive materials. 2024;36:490-507
240. Zhang CJ, Li JM, Xu D, Wang DD, Qi MH, Chen F. et al. Surface Molecularly Engineered Mitochondria Conduct Immunophenotype Repolarization of Tumor-Associated Macrophages to Potentiate Cancer Immunotherapy. Adv Sci (Weinh). 2024;11:e2403044
241. Li J, Lei T, Ouyang W, Ye Z, Li L, Li G. et al. Reshape tumor microenvironment by modulating CXCR4 with FAP-targeted diselenide-organosilica delivery system for prostate cancer immunotherapy. Chem Eng J. 2025;503:158308
242. Sitia L, Bonizzi A, Mazzucchelli S, Negri S, Sottani C, Grignani E. et al. Selective Targeting of Cancer-Associated Fibroblasts by Engineered H-Ferritin Nanocages Loaded with Navitoclax. Cells. 2021;10:328
243. Xiao C, Li J, Wang X, Li S, Xu C, Zhang Z. et al. Hydroxyethyl starch stabilized copper-diethyldithiocarbamate nanocrystals for cancer therapy. J Control Release. 2023;356:288-305
244. Wei D, Sun Y, Zhu H, Fu Q. Stimuli-Responsive Polymer-Based Nanosystems for Cancer Theranostics. ACS Nano. 2023;17:23223-23261
Author contact
![]() Corresponding authors: Ming Zhao (syphuzmcom), Ting Fang (fangtingtinacom).
Corresponding authors: Ming Zhao (syphuzmcom), Ting Fang (fangtingtinacom).