Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
1. Introduction
2. Acetate
3. Propionate
4. Butyrate
5. Other SCFAs
6. SCFAs and Cancer Treatment
7. Conclusions and Perspectives
Abbreviations
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2026; 16(3):1143-1163. doi:10.7150/thno.119304 This issue Cite
Review
Short-chain fatty acids in the tumor microenvironment: from molecular mechanisms to cancer therapy
Yan Xiang#, Ao Du#, Zhen Wang#, Hongyuan Pan, Kefei Yuan ![]()
Department of Liver Surgery, State Key Laboratory of Biotherapy and Cancer Center, West China Hospital, Sichuan University and Collaborative Innovation Center of Biotherapy, Chengdu, China.
# These authors contributed equally to this work.
Received 2025-6-10; Accepted 2025-10-1; Published 2026-1-1
Abstract

Short-chain fatty acids (SCFAs), including acetate, propionate, and butyrate, serve as pivotal metabolites within the tumor microenvironment (TME), playing essential roles in modulating tumor progression. Although the biological functions and mechanisms of SCFAs in the TME show some overlap, each SCFA also exerts some distinct regulatory effects on tumors and TME. Notably, even a single SCFA may exhibit pleiotropic effects across different cancer types or under varying conditions within the same malignancy. Consequently, according to the different metabolic microenvironment of patients, precise modulation of SCFA levels could effectively suppress tumor progression. Furthermore, SCFAs have been shown to potentiate the therapeutic efficacy of immunotherapy, radiotherapy, and chemotherapy. This review systematically outlines the sources, biological functions, and mechanisms of different SCFAs in the TME, while exploring potential therapeutic strategies based on SCFA modulation. These insights offer novel perspectives and directions for future research and clinical cancer therapy.
Keywords: short-chain fatty acids, tumor microenvironment, cancer therapy, molecular mechanisms, immune modulation
1. Introduction
Cancer is one of the leading causes of global mortality [1]. In recent years, with a deeper understanding of cancer pathogenesis, a growing number of therapeutic options have become available. However, most current cancer therapies primarily target tumor cells, often overlooking the role of the TME in cancer treatment. The TME is composed of various cellular and non-cellular components within the tumor niche [2]. In the past few decades, the importance of the TME in dynamically regulating cancer progression and influencing treatment outcomes has been widely recognized [3-5]. Paget compared the relationship between tumor cells and the TME to that of seeds and soil, emphasizing that tumor cell growth depends on the support of the TME [6,7]. As the “soil” for tumor growth, non-cellular components within the TME, such as chemokines cytokines, and growth factors, play crucial roles in regulating cancer progression [8]. Furthermore, nutrients, as essential non-cellular components for maintaining cellular functions within the TME, also perform highly complex roles in this microenvironment [9].
Glucose, amino acids, and lipids are currently regarded as the principal nutrients within the TME [10,11]. Unlike glucose and amino acids, lipid research has lagged due to their structural diversity and unique physicochemical properties [12,13]. However, advances in lipidomics have deepened our understanding of lipid functionality [14-16]. As the basic skeleton of lipids, fatty acids (FAs) exert diverse biological effects in the TME [17,18]. Compelling evidence indicates that FA synthesis, uptake, and metabolism profoundly influence cancer progression, and FAs also serve as signaling molecules to activate pathways governing cellular activities [13]. These findings highlight the necessity to explore the role of FA in tumor progression.
FAs are categorized by carbon chain length into short-chain fatty acids (SCFAs; <6 carbons), medium-chain fatty acids (MCFAs), and long-chain fatty acids (LCFAs) [19]. SCFAs, including acetate, propionate, and butyrate, are primarily generated through gut microbial fermentation of dietary fibers (e.g., inulin, pectin, resistant starch). Then they cross the intestinal epithelial cells, enter the bloodstream, and are transported to the TME via the circulatory system [20-22]. Their roles in the TME have attracted significant attention. Some studies have shown that SCFAs can directly either inhibit or promote tumor cell growth depending on the context [23,24]. Additionally, other research has indicated that SCFAs can modulate the function and fate of immune cells within the TME, thereby influencing tumor progression [25,26].
This review systematically delineates the origins, biological functions, and molecular mechanisms of SCFAs in the TME. We also explore SCFA-based therapeutic strategies, aiming to provide novel insights and translational directions for cancer treatment.
2. Acetate
2.1 Sources of Acetate
Acetate in the human body originates from exogenous uptake and endogenous synthesis (Figure 1). Exogenous acetate is primarily generated through the fermentation of dietary fibers by gut microbiota. Acetogenic bacteria are strict anaerobes. These bacteria generate acetate via two major pathways: (1) carbohydrate metabolism, where glucose is converted to pyruvate, which is then converted into acetyl-CoA and further transformed into acetate [27,28]; (2) the Wood-Ljungdahl pathway, where certain bacteria reduce CO₂ to generate acetate [29,30].
Schematic diagram of acetate production by gut microbiota and mammalian cells in the human body. (A) Exogenous acetate sources: Indigestible dietary fibers or polysaccharides are hydrolyzed into monosaccharides, which then enter the gut microbiota. These monosaccharides undergo glycolysis to produce pyruvate, which is subsequently converted to acetyl-CoA by pyruvate:ferredoxin oxidoreductase. Acetate is then synthesized via phosphotransacetylase and acetate kinase. Alternatively, the Wood-Ljungdahl pathway reduces CO₂ to acetate through a series of enzymatic reactions involving CODH/ACS, formate dehydrogenase, and 10-formyl-H4folate synthetase, ultimately leading to the formation of acetyl-CoA and acetate. (B) Endogenous acetate sources: Glucose-derived pyruvate undergoes decarboxylation, either through ROS-mediated or keto acid dehydrogenase-driven pathways, to produce acetate, which is subsequently converted to acetyl-CoA by ACSS for histone acetylation. Deacetylation of histones releases acetate, thereby completing the metabolic cycle. Abbreviations: ACSS, acetyl-CoA synthetase; CODH, carbon monoxide dehydrogenase; ROS, reactive oxygen species; THF, tetrahydrofolate.
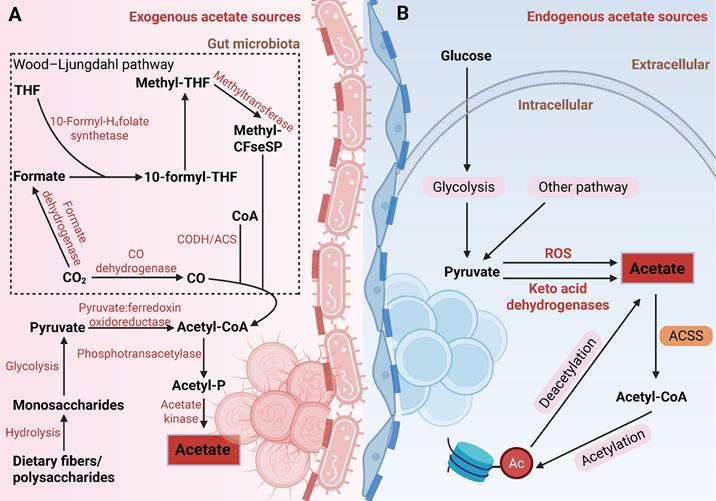
As research progresses, our understanding of the sources of acetate in both physiological and pathological states has become more comprehensive. Moving beyond exogenous uptake, the mechanisms of endogenous acetate production have also emerged as a growing research focus. In humans, endogenous acetate can be generated through pathways such as glucose metabolism and protein deacetylation. Liu et al. discovered that pyruvate, a product of glucose metabolism, can be decarboxylated to acetate through reactive oxygen species (ROS) or a novel enzymatic activity of alpha-keto acid dehydrogenase [31]. Additionally, chromatin serves as an acetate reservoir, releasing acetate through histone deacetylation [32,33]. These processes help alleviate metabolic stress in the TME, such as nutrient deprivation and acidosis, and are of significant pathological relevance. For instance, under nutrient-restricted conditions, glioma cells secrete acetate derived from glucose metabolism, promoting the proliferation of astrocytes [34]. In an acidic environment, cancer cells exhibit global deacetylation, and the released acetate anions are co-transported with protons via monocarboxylate transporters (MCTs), preventing further acidification of the intracellular pH [35]. Collectively, acetate in the TME originates from diverse metabolic pathways, highlighting its critical role as a key metabolite in the metabolic reprogramming of tumors.
2.2 Acetate and Cancer
Studies have shown that acetate can directly affect various tumors, including colorectal cancer (CRC), pancreatic ductal adenocarcinoma (PDAC), and hepatocellular carcinoma (HCC) [32,36,37]. The regulatory effects of acetate on tumors exhibit “double-edged sword” properties. For example, in non-alcoholic fatty liver disease-related hepatocellular carcinoma (NAFLD-HCC), acetate can inhibit tumor progression by activating the G protein-coupled receptor 43 (GPR43) [38]; whereas in glioblastoma, acetate supports tumor growth by sustaining the metabolic demands of cell proliferation [39]. Mechanistically, the antitumor effects of acetate are primarily mediated through two pathways: receptor-mediated signaling inhibition and metabolic interference-induced cell damage. Upon binding to GPR43, acetate inhibits the IL-6/JAK1/STAT3 signaling pathway, thereby blocking NAFLD-HCC initiation and growth [38]. Additionally, acetate can directly induce oxidative stress and mitochondrial dysfunction in CRC cells, activating caspase-dependent apoptosis pathways [40,41].
In comparison, the protumorigenic effects of acetate involve broader metabolic reprogramming and epigenetic regulation. Under metabolic stress, cancer cells efficiently take up acetate via monocarboxylate transporter 1 (MCT1) or sodium-coupled monocarboxylate transporter 1 (SMCT1) [28], which is then converted into acetyl-CoA by mitochondria-localized acetyl-CoA synthetase 1 (ACSS1) or nucleocytosol-localized acetyl-CoA synthetase 2 (ACSS2) [42,43]. This metabolic conversion serves multiple functions: energy supply (via the tricarboxylic acid cycle generating ATP), biosynthetic support (direct involvement in lipid synthesis), and histone acetylation promotion. Under hypoxic or glucose-limited conditions, acetate metabolism can account for up to 50% of the energy metabolism in glioblastoma (GBM), becoming a crucial alternative pathway for maintaining tumor energy homeostasis [39]. In the cytoplasm, acetate serves as a direct carbon source for fatty acid synthesis, thereby supporting tumor growth [44]. Furthermore, In low-oxygen or low-fat conditions, acetate uptake enhances the acetylation of lipid synthesis-related genes (ACACA and FASN), thus promoting lipid biosynthesis and aiding cancer cell survival and growth [45]. Under glucose limitation, acetate increases histone H3K27 acetylation, promoting the expression of SNAI1, facilitating renal cancer cell migration [46]. Recent studies have revealed that the metabolic reprogramming role of acetate in the TME is even more complex. In acidic TMEs, acetate does not regulate the expression of lipid synthesis genes (SREBF1, FASN, and ACACA) or total lipid content. Instead, it induces acetylation of the transcription factor SP1, alters polyamine metabolism, and enables pancreatic cancer cells to survive via the ACSS2-SP1-SAT1 axis [47]. Protein O-linked N-acetylglucosamine (O-GlcNAc) modification acts as an intracellular nutrient sensor, and high-glucose diets promote cancer progression by inducing O-GlcNAc modification [48-50]. As mentioned earlier, glucose is metabolized by the gut microbiome into acetate, and high-glucose diets increase acetate levels in the portal vein of HCC mouse models. After uptake by tumor cells, acetate upregulates glutamine and UDP-GlcNAc levels, enhancing the O-GlcNAc modification of eukaryotic elongation factor 1A1 (eEF1A1), thereby promoting tumor growth [23]. Furthermore, acetate produced by glucose metabolism in tumor cells is secreted into the TME, upregulating the expression of monoamine oxidase B (MAO-B) and MCT1, which stimulates reactive astrocyte proliferation. The proliferation of reactive astrocytes is closely associated with poor prognosis in glioblastoma patients [34,51] (Figure 2).
The biological functions and mechanisms of acetate in the TME. In the TME, acetate can regulate various molecular mechanisms by either binding to the GPR43 receptor or entering cells through monocarboxylate transporters MCT1 and SMCT1, or passive transport. Extracellular acetate binds to GPR43, activating the receptor and triggering a GPR43-mediated cellular signaling cascade. Acetate that enters the cell can replenish the intracellular acetyl-CoA pool, leading to increased histone acetylation and enhanced gene transcription. Additionally, acetate can serve as an alternative energy source for the tricarboxylic acid cycle or act as a key precursor for the biosynthesis of macromolecules such as lipids. Through these mechanisms, acetate regulates cancer cell proliferation, metastasis, and immunogenicity, while also affecting the proliferation, antitumor function, and tumor infiltration of immune cells in the TME. Abbreviations: ACSS1, acetyl-CoA synthetase 1; ACSS2, acetyl-CoA synthetase 2; DLAT, dihydrolipoamide S-acetyltransferase; GPR43, G protein-coupled receptor 43; ILC3, type 3 innate lymphoid cell; MCT1, monocarboxylate transporter 1; PD-1, programmed death-1; PD-L1, programmed death-ligand 1; SMCT1, sodium-coupled monocarboxylate transporter 1; TCA cycle, tricarboxylic acid cycle; TME, tumor microenvironment.
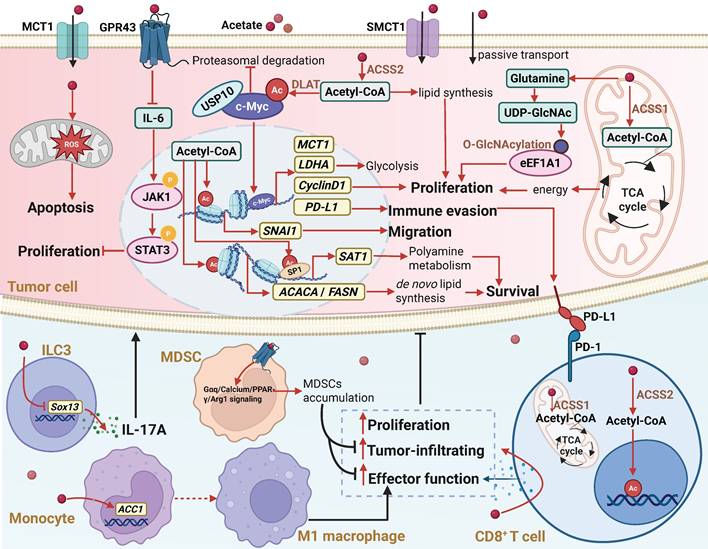
In the studies described above, acetate exhibits a dual role in tumors. It suppresses tumor progression in NAFLD-HCC and CRC [38,40,41], while promoting growth in other cancers, including GBM, renal cancer, and pancreatic cancer [39,46,47], suggesting that its dual effects may be attributed to tumor heterogeneity. Even within the same tumor type, acetate can exert opposite effects: it inhibits tumor growth in NAFLD-HCC but promotes progression in DEN+CCl4-induced HCC models [23,38]. The distinct effects of acetate may be associated with distinct metabolic patterns of the two aforementioned mouse models. Specifically, acetate may possess antitumor potential in lipid metabolism active NAFLD-HCC, while in glucose metabolism dominant HCC, it may function as a pro-tumor factor. Cellular stemness also influences the effects of acetate. Mashimo et al. reported that acetate, as an energy source, promotes the growth of GBM tumors [39]. In contrast, Long et al. found that acetate inhibits the proliferation of glioblastoma stem-like cells (GSCs) derived from GBM [52]. In animal models, GBM tumors generate energy by oxidizing acetate, supporting tumor growth. However, GSCs are more sensitive to acetate-induced epigenetic changes, which may interfere with their self-renewal capacity and suppress their proliferation. Furthermore, although acetate promotes tumor cell apoptosis in CRC [40,41], it reverses the anticancer effects of PINK1 overexpression in a mouse model, leading to tumor growth restoration [53]. This suggests that the effects of acetate are regulated by genetic background. PINK1 overexpression activates mitophagy and suppresses the production of acetyl-CoA in the glycolytic pathway. Acetate, on the other hand, supplements acetyl-CoA, partially counteracting the metabolic changes induced by PINK1 overexpression, thereby attenuating its tumor-suppressive effects. Collectively, these results suggest that the ultimate effect of acetate in tumors largely depends on the intrinsic characteristics of tumor cells.
2.3 Acetate and Tumor Immunity
Tumor immunity refers to the process by which the immune system recognizes and eliminates tumor cells. However, this process is frequently suppressed by the tumor and the TME [54]. Acetate exerts complex effects on tumor immunity. On the one hand, acetate is utilized by cancer cells to reduce their immunogenicity, thereby facilitating immune evasion. Studies have shown that acetate uptake via MCT1 promotes c-Myc acetylation, thereby enhancing Programmed death-ligand 1 (PD-L1) expression and subsequently suppressing the cytotoxic activity of immune cells against tumors [55]. On the other hand, acetate enhances the antitumor capacity of CD8+ T cells. Under glucose-limited conditions, acetate boosts metabolic activity in CD8+ T cells, promoting CD3/CD28 stimulation-induced degranulation and effector functions [56]. Furthermore, acetate increases histone acetylation and chromatin accessibility in an ACSS-dependent manner, enhancing transcription of the interferon gamma (IFN-γ) and cytokine production [57]. In co-culture systems with cancer cells, acetate restores acetyl-CoA pools in CD8+ T cells, further amplifying their activity and IFN-γ secretion [58]. However, within TME, cancer cells typically outcompete CD8+ T cells in acetate uptake and utilization due to their elevated expression of ACSS2 and MCT1 [59,55]. To overcome this metabolic competition and bolster CD8+ T cell immunity, several strategies have been explored: Depleting or inhibiting ACSS2 in cancer cells blocks acetate utilization, redirecting acetate to CD8+ T cells and enhancing their antitumor function [57]. In addition, upregulating ACSS1 expression in CD8+ T cells improves their acetate metabolic capacity, thereby boosting antitumor activity [60]. Alternatively, direct acetate supplementation elevates acetate levels in the TME, enhancing CD8+ T cell infiltration and IFN-γ production [58,61].
Furthermore, acetate also regulates other immune cells within the TME. In HCC, tumor-derived type 3 innate lymphoid cells (ILC3s) promote hepatic stellate cell (HSC) activation via an IL-17A-dependent pathway, driving liver fibrosis, which is associated with poor patient prognosis [62,63]. Hu et al. demonstrated that acetate inhibits histone deacetylase activity and increases acetylation of SRY-box transcription factor 13 (Sox13) at the Lys30, thereby reducing IL-17A production in tumor-derived ILC3s and suppressing HCC progression [62]. In the TME, M1 macrophages exert antitumor and immune-activating effects by secreting pro-inflammatory cytokines such as IL-6, IL-12, and TNF. In the meantime, M1 macrophages also play a crucial role in influencing the number and function of tumor-infiltrating CD8+ T cells [64,65]. Acetate enhances transcription of acetyl-CoA carboxylase 1 (ACC1) by increasing histone acetylation at the ACC1 promoter and induces M1 macrophage polarization, thereby enhancing CD8+ T cell antitumor activity in HCC patients [66]. Myeloid-derived suppressor cells (MDSCs), a heterogeneous myeloid cell population linked to chronic inflammation, exhibit potent immunosuppressive properties [67,68]. In lung adenocarcinoma (LUAD), acetate activates the Gαq/calcium/PPAR-γ/Arg1 signaling pathway via free fatty acid receptor 2 (FFAR2), enhancing MDSC-mediated immunosuppression and facilitating tumor immune evasion [69] (Figure 2).
Acetate also exhibits a dual role in antitumor immunity, which is regulated by multiple factors. First, the type of target cells determines the effect of acetate: when acting on CD8⁺ T cells, ILC3s, or macrophages, acetate can enhance immune responses and promote antitumor activity [58,61,66]; conversely, when acting on MDSCs, acetate can reinforce immunosuppressive signals, thereby facilitating tumor immune evasion [69]. In addition, the source of acetate also influences its immunomodulatory effects: acetate derived from probiotics tends to promote antitumor immunity, whereas acetate generated through host metabolism is more likely to suppress immune responses. This difference may reflect variations in the site of production and the local microenvironment. In HCC, acetate is largely enriched in the liver via the gut-liver axis, where it can modulate immune cells within a relatively immune-tolerant microenvironment to enhance antitumor effects [62]. In contrast, in tumors such as LUAD, characterized by hypoxia and lactate accumulation, cancer cell-derived acetate is more readily utilized as a metabolic substrate and can activate immunosuppressive pathways, thereby promoting tumor immune evasion [69]. Overall, the immunomodulatory effects of acetate result from the interplay among its source, the local microenvironment, and the target cell type.
3. Propionate
3.1 Sources of Propionate
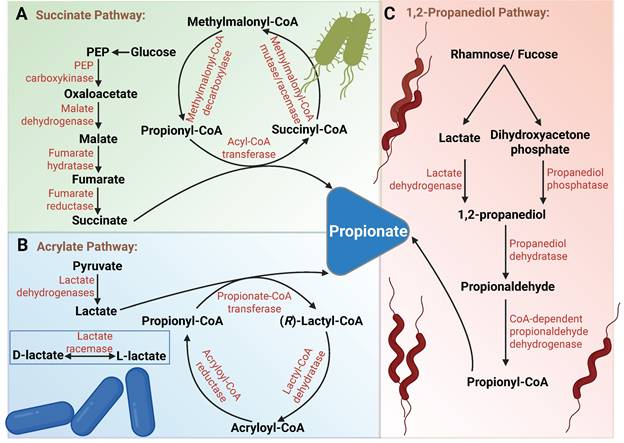
Propionate in humans primarily originates from fermentation by gut microbiota. Glucose and lactate serve as the main fermentative substrates, which are converted into propionate through three major pathways (Figure 3). (1) The genus Bacteroides can produce propionate via the succinate pathway using methylmalonyl-CoA [70]. (2) Soil bacterium Clostridium propionicum and Negativicutes, such as Megasphaera elsdenii in the rumen, can produce propionate via the acrylate pathway [71,72]. The succinate pathway and acrylate pathway can be distinguished by incubating with isotopically labeled substrates [73,74]. (3) Deoxysugars such as rhamnose can be converted into 1,2-propanediol through dihydroxyacetone phosphate or lactate, which is further metabolized to produce propionate. In the human gut, the anaerobic bacterium Roseburia inulinivorans has been found to generate propionate from fucose via the propanediol pathway [75,76]. The associations between the microbiome and cancer have been extensively studied. In this section, we will focus on the role and regulatory mechanisms of microbe-derived propionate within the TME.
Schematic illustration of propionate production by gut microbiota through fermentation of non-digestible carbohydrates. (A) Succinate Pathway: Glucose is metabolized through glycolysis to PEP, which is carboxylated to oxaloacetate by PEP carboxykinase. Oxaloacetate is then reduced to malate by malate dehydrogenase. Malate undergoes dehydration to form fumarate, which is subsequently reduced to succinate by membrane-bound fumarate reductase. Succinate is then further metabolized to propionate via the methylmalonyl-CoA pathway. (B) Acrylate Pathway: Pyruvate is reduced to lactate by lactate dehydrogenase (NAD-dependent). Notably, both L-lactate and D-lactate, as enantiomers, are present in microbial metabolism, and their interconversion is facilitated by lactate racemase. Lactate is then conjugated with coenzyme A by propionyl-CoA transferase to form lactyl-CoA. This intermediate undergoes reversible syn-dehydration catalyzed by an oxygen-sensitive dehydratase, resulting in the formation of acryloyl-CoA. Acryloyl-CoA is ultimately reduced to propionyl-CoA by acryloyl-CoA reductase, followed by decarboxylation via propionyl-CoA transferase to generate propionate. (C) 1,2-Propanediol Pathway: Rhamnose and fucose are catabolized by deoxy sugar lyases to produce lactate and DHAP. Lactate is reduced to 1,2-propanediol by lactate dehydrogenase, while DHAP is dephosphorylated to 1,2-propanediol by propanediol phosphatase. 1,2-propanediol is then dehydrated by propanediol dehydratase to form propionaldehyde, which is subsequently oxidized to propionyl-CoA by CoA-dependent propionaldehyde dehydrogenase. Finally, propionate is released from propionyl-CoA via catalysis by propionate kinase. Abbreviations: DHAP, dihydroxyacetone phosphate; PEP, phosphoenolpyruvate.
3.2 Propionate and Tumors
In comparison to acetate, propionate generally inhibits tumor progression (Figure 4). First, propionate regulates a series of molecular events to induce cancer cell death [77-80]. For example, propionate induces ROS generation and disrupts redox homeostasis, leading to mitochondrial fission, mitophagy, and subsequent ferroptosis and apoptosis [81,82]. Furthermore, propionate, as a histone deacetylase (HDAC) inhibitor, blocks histone deacetylation, leading to chromatin relaxation and the transcriptional activation of pro-apoptotic genes such as HECT domain E3 ubiquitin protein ligase 2 (HECTD2), ultimately inducing apoptosis in cancer cells. Specifically, propionate upregulates HECTD2, promotes the degradation of euchromatic histone-lysine N-methyltransferase 2 (EHMT2), and reduces H3K9me2 levels in the tumor necrosis factor alpha-induced protein 1 (TNFAIP1) promoter region, thereby increasing TNFAIP1 expression and inducing apoptosis in CRC cells [83]. Additionally, propionate suppresses cancer cell metastasis by activating the histone acetyltransferase p300, which mediates H3K27 acetylation and H3K4 methylation. This upregulates the transcription of epithelial genes, enhances intercellular contact and adhesion, and inhibits epithelial-to-mesenchymal transition (EMT), thereby impairing cancer cell migration and invasion [84]. Notably, activation of p300 by propionate represents a novel mechanism distinct from its classical HDAC inhibition, revealing an unrecognized pathway for histone acetyltransferase (HAT) activation by propionate [85]. Recent studies have found that propionate, by being converted to propionyl-CoA and modifying histones (such as H3K18pr and H4K12pr), significantly enhances chromatin accessibility, leading to the dysregulation of key cancer-associated genes, MYC, JUN, and AHNAK2, in CRC, thereby exhibiting antitumor effects [86]. However, in breast and lung cancers, ERK2 activation shifts the role of propionate from antitumor to pro-tumor via metabolic dysregulation. Specifically, ERK2 inhibits methylmalonyl-CoA epimerase (MCEE), reducing propionate-driven anaplerosis and increasing methylmalonic acid (MMA) production [87,88]. MMA accumulation in the TME then promotes tumor metastasis [89,90].
The biological functions and mechanisms of propionate in the TME. Similar to acetate, propionate plays a significant role in the TME through various mechanisms. Extracellular propionate can activate the GPR43 receptor, triggering a signaling cascade. Additionally, propionate enters cells, acting as an HDAC inhibitor or activating p300, which increases histone acetylation and gene transcription activity, accompanied by DNA damage. Propionate induces ROS generation or directly modulates multiple signaling pathways, leading to cell cycle arrest, inhibition of cancer cell proliferation and metastasis, and induction of apoptosis or ferroptosis. Moreover, propionate enhances the immunogenicity of cancer cells, thereby improving the recognition and killing ability of immune cells against cancer cells. Furthermore, propionate promotes pulmonary endothelial cells to secrete CCL20, recruiting Th17 cells to exert antitumor effects. However, there is currently no direct evidence that propionate regulates the function of immune cells in the TME. Notably, metabolic reprogramming in cancer cells inhibits MCEE activity, reducing propionate-driven anaerobic metabolism and increasing the production of MMA, which enhances the metastatic potential of tumors. Abbreviations: DC, dendritic cell; HDAC, histone deacetylase; MCEE, methylmalonyl-CoA epimerase; MMA, methylmalonic acid; ROS, reactive oxygen species; TME, tumor microenvironment.
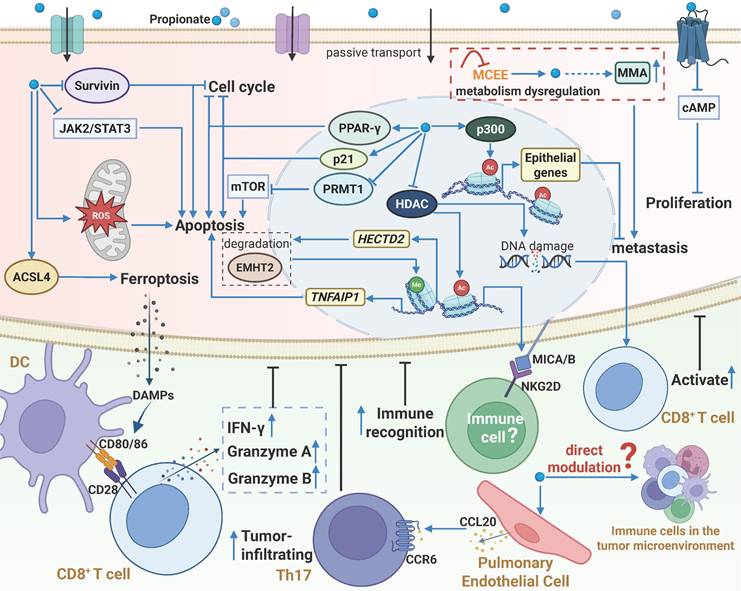
3.3 Propionate and Tumor Immunity
Beyond direct tumor suppression, propionate enhances cancer cell immunogenicity to activate antitumor immunity (Figure 4). Mowat et al. demonstrated that propionate directly stimulates CRC cells to enhance cytotoxic CD8+ T cell activation. Mechanistically, propionate inhibits histone deacetylation, causing DNA damage and upregulating chemokines, MHC-I, and antigen presentation genes in CRC cells. A feedback loop occurs: activated CD8+ T cells secrete IFN-γ, further stimulating cancer cells to amplify MHC-I expression and T cell activation. This loop is stronger in cancers with DNA mismatch repair defects and genomic instability [91]. NKG2D, an immune-activating receptor on NK and effector T cells, enhances cytotoxicity when its ligands are expressed [92]. In colon cancer, propionate induces metabolic reprogramming and histone acetylation/propionylation, leading to the upregulation of surface NKG2D ligands (MICA/B) on cancer cells, which activate immune responses [93]. Propionate also triggers immunogenic cell death (ICD) in acute myeloid leukemia (AML). Concretely, propionate enhances ACSL4-mediated mitophagy, thereby releasing DAMPs to promote dendritic cell (DC) maturation and antigen presentation[81]. In melanoma lung metastasis models, propionate upregulates CCL20 in pulmonary endothelial cells, recruiting Th17 cells via the CCL20/CCR6 axis to reduce metastasis [94]. However, the mechanism of CCL20 regulation remains unclear. Collectively, propionate shows broad potential in modulating cancer immunogenicity. In addition, studies have found that propionate can directly regulate γδ T cells [95], B cells [96], Tregs [97,98], and DCs [99] in other diseases [100]. However, how it regulates immune cell function in the TME requires further exploration.
4. Butyrate
4.1 Sources of Butyrate
Similar to propionate, butyrate in the human body is primarily synthesized through microbial metabolic activity. Microorganisms metabolize carbohydrates into pyruvate, which is converted to acetyl-CoA via the pyruvate dehydrogenase complex. Acetyl-CoA undergoes a series of enzymatic reactions to generate butyryl-CoA. Butyrate-producing bacteria then catalyze the conversion of butyryl-CoA to butyrate through two distinct pathways (Figure 5): (1) Phosphotransbutyrylase Pathway: Bacteria such as Coprococcus comes utilize butyryl-CoA:phosphate butyryltransferase and butyrate kinase to convert butyryl-CoA into butyrate [20,101]. (2) Butyryl-CoA:Acetyl-CoA Transferase Pathway: Species like Eubacterium rectale and Faecalibacterium prausnitzii employ butyryl-CoA:acetyl-CoA transferase to produce butyrate, a process requiring acetate consumption [102,103]. Notably, certain gut microbes (e.g., Eubacterium hallii, Anaerostipes spp.) can synthesize butyrate from both lactate and acetate, preventing lactate accumulation and maintaining intestinal homeostasis [20]. Beyond gut microbiota, Intratumor bacteria such as Roseburia have also been reported to produce butyrate [104]. As a key bacterial metabolite, butyrate not only plays a vital role in gut health but also exerts significant effects on tumorigenesis and progression [105]. This section will elaborate on the mechanisms and implications of butyrate in cancer.
Pathways of butyrate production by gut microbiota. Glucose and lactate are converted into pyruvate through glycolysis and oxidation, respectively. Pyruvate is converted into acetyl-CoA by the enzyme pyruvate: ferredoxin oxidoreductase. Acetyl-CoA is subsequently condensed into acetoacetyl-CoA via acetyl-CoA acetyltransferase. Acetoacetyl-CoA is reduced to β-hydroxybutyryl-CoA by β-hydroxybutyryl-CoA dehydrogenase. This intermediate undergoes dehydration by enoyl-CoA hydratase to form crotonyl-CoA, which is then reduced to butyryl-CoA by butyryl-CoA dehydrogenase. Two distinct pathways are involved in the conversion of butyryl-CoA to butyrate: (A) Butyryl-CoA:acetate CoA-transferase pathway: In this pathway, butyryl-CoA is converted into butyrate through the action of butyryl-CoA:acetate CoA-transferase, which transfers the CoA group to acetate, producing butyrate and releasing acetyl-CoA. (B) Butyrate kinase pathway: Butyryl-CoA is converted to butyryl-P by phosphotransbutyrylase. Subsequently, butyrate is generated by butyrate kinase. Unlike the butyryl-CoA:acetate CoA-transferase pathway, this pathway requires the conversion of butyryl-CoA to butyryl phosphate, but instead does not consume acetate to generate butyrate.
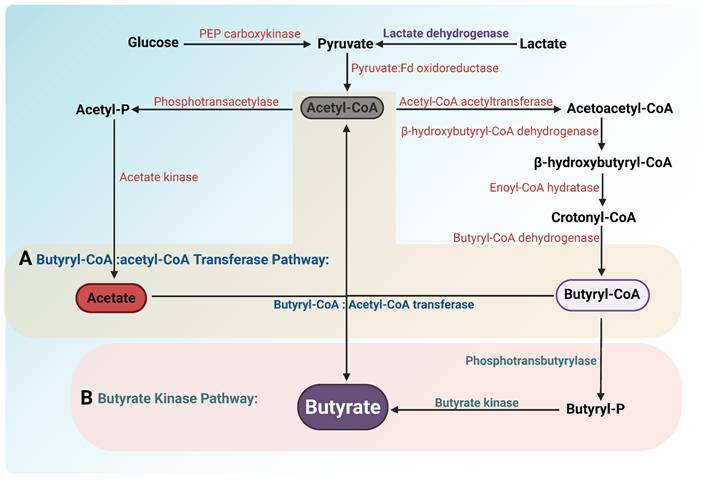
4.2 Butyrate and Cancer
Similar to acetate, butyrate exhibits a dual role in tumor initiation and progression (Figure 6), with its antitumor effects mediated through multiple mechanisms. As a signaling molecule, butyrate specifically activates G protein-coupled receptors (GPR41, GPR43, and the butyrate-specific receptor GPR109a) [106,107], thereby regulating cellular metabolism and key signaling pathways. In CRC, activation of the GPR109a-AKT axis by butyrate markedly reduces the membrane abundance of glucose transporter 1 (GLUT1) and glucose-6-phosphate dehydrogenase (G6PD), suppressing glucose uptake and glycolysis [108]. Activation of GPR43 and GPR109a further inhibits the Wnt/β-catenin pathway, blocking proliferative signaling [109], while GPR43 activation also downregulates SLC7A11 and GPX4 in a cAMP-PKA-dependent manner, resulting in lipid peroxidation and ferroptosis in CRC cells [110]. Within cells, butyrate functions as a HDAC inhibitor, increasing histone acetylation and thereby regulating gene expression and downstream signaling [111,112]. For example, it enhances the acetylation of genes associated with calcium signaling, disrupting intracellular calcium homeostasis and inducing ROS generation, which inhibits proliferation and metastasis in HCC [113]. HDAC inhibition by butyrate also modulates multiple pathways, including AKT/ERK and JAK2/STAT3, thereby suppressing invasion in CRC as well as proliferation and angiogenesis in myeloproliferative tumors [114,115]. Regarding cell death, butyrate promotes apoptosis by upregulating pro-apoptotic proteins (Bax, p53) and downregulating Bcl-2 [116,117], while also inducing ferroptosis in endometrial cancer, CRC, and PDAC cells through regulation of RBM3, CD44, and SLC7A11 expression balance as well as lipid metabolism [118-120]. Moreover, the antitumor effects of butyrate involve miRNA-mediated regulation [121,122]. In CRC, butyrate downregulates miR-106b, relieving repression of p21, leading to its overexpression and cell cycle arrest, thereby inhibiting cell proliferation [121]. It also suppresses c-Myc to inhibit miR-92a transcription, increasing p57 expression and further suppressing proliferation while promoting apoptosis [123]. In HCC, butyrate enhances miR-22 expression, inducing apoptosis and inhibiting cell proliferation [124].
The biological functions and mechanisms of butyrate in the TME. Extracellular butyrate can activate GPR43 and GPR109A receptors to regulate downstream signaling and glucose metabolism, thereby suppressing cancer cell proliferation. Once inside the cell, butyrate functions as an HDAC inhibitor to increase histone acetylation, activate gene transcription, and modulate associated signaling pathways, leading to inhibition of proliferation and metastasis, while also inducing DNA damage, apoptosis, or ferroptosis. In addition, butyrate can regulate miRNA expression and influence gene regulatory networks; however, it may promote proliferation through the IGF1 pathway. Moreover, butyrate enhances antitumor immunity by increasing tumor cell immunogenicity and activating immune effector cells. Abbreviations: CAF, cancer-associated fibroblast; GLUT, glucose transporter 1; GPR109A, G protein-coupled receptor 109 A; HAT, histone acetyltransferase; HDAC, histone deacetylase; PBMCs, peripheral blood mononuclear cells; PD-1, programmed death-1; PD-L1, programmed death-ligand 1; ROS, reactive oxygen species; TME, tumor microenvironment.
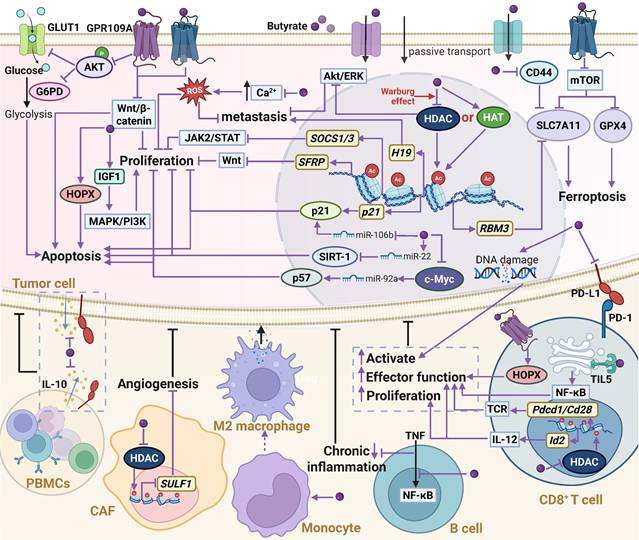
However, the pro-tumorigenic effects of butyrate have also been reported. For example, microbiota-derived butyrate can promote colorectal carcinogenesis by inducing senescence of colonic epithelial cells and driving abnormal proliferation and transformation in mouse colon epithelium [125,126]. This phenomenon, which contradicts the tumor-suppressive role of butyrate in CRC, is referred to as the “butyrate paradox”. Specifically, in normal colonic epithelial cells, butyrate is efficiently metabolized into acetyl-CoA, which enhances HAT activity and promotes cell proliferation. In contrast, in cancer cells, due to the Warburg effect, whereby cells preferentially undergo glycolysis rather than oxidative phosphorylation even in the presence of oxygen, butyrate metabolism is impaired, leading to its intracellular accumulation. Under these conditions, butyrate functions as a HDAC inhibitor to suppress tumor progression. Although both mechanisms ultimately increase histone acetylation, they target distinct sets of genes [127]. Therefore, the differential metabolic preferences between normal colonic epithelial cells and CRC cells are considered as a key determinant driving the dual roles of butyrate. Moreover, In prostate cancer, butyrate has also been shown to elevate circulating insulin-like growth factor-1 (IGF-1) levels, thereby activating the MAPK/PI3K pathway and promoting tumor progression [128]. The divergent outcomes observed in CRC, HCC, and prostate cancer are largely attributed to the dose-dependent effects of butyrate: at low concentrations, butyrate tends to promote tumor progression, whereas at high concentrations, it exerts inhibitory effects [129-131]. Because butyrate is extensively consumed in the intestine and liver, its systemic bioavailability is markedly reduced, resulting in relatively low concentrations within prostate tumors. Consequently, butyrate fails to accumulate intracellularly and instead acts as an extracellular signaling molecule that promotes tumor growth [128]. Nevertheless, this dose dependency is not absolute; under conditions of dysbiosis, cholestasis, or inflammation, even high concentrations of butyrate have been reported to facilitate HCC progression [132]. Therefore, the dual roles of butyrate cannot be explained by a single factor but are instead determined by the interplay among cellular metabolic states, local concentration gradients, and the host's physiological and pathological context.
4.3 Butyrate and Tumor Immunity
Butyrate is a key metabolite mediating crosstalk between the gut microbiome and the immune system. Many studies have reported that butyrate has the effect of promoting tumor immunity. In terms of immunogenic modulation, butyrate enhances CRC cells immunogenicity and potentiates their ability to activate CD8+ T cells [91,133]. Concurrently, it directly amplifies CD8+ T cell function. In gastric cancer(GC), butyrate acts as a GPR109A agonist and amplifies the cytotoxicity of CD8+ T cells and CAR-Claudin 18.2+ CD8+ T cells via the GPR109A/HOPX axis [134]. Interestingly, studies report that butyrate does not alter GPR41/43/109A expression [135], and GPR blockade fails to abrogate the effects of butyrate on CD8+ T cells [136], suggesting a GPR-independent mechanism. Kang et al. further identified TLR5 as a novel butyrate receptor, whose upregulation on CD8+ T cells activates the NF-κB pathway, enhancing cytotoxicity [135]. Additionally, butyrate modulates CD8+ T cells via HDAC inhibition. In B16-F0-bearing mice, it elevates H3K27ac at the Pdcd1 and Cd28 promoters in CD8+ T and Vδ2+ T cells, enhancing antitumor cytokine production through T-cell receptor (TCR) signaling pathway [137]. Butyrate also induces ID2 expression and enhances IL-12 signaling, thereby boosting cytotoxic CD8+ T cell responses [136].
Beyond T cells, butyrate augments NK cell activity and promotes liver-resident NK cell development [138]. Moreover, it activates macrophages to reinforce the intestinal mucus barrier [139] and suppresses DC antigen presentation, reducing pro-inflammatory cytokine release [140,141]. These findings suggest that butyrate has regulatory effects on innate immunity. For instance, in advanced GC patients, it was found that butyrate can reduce the expression of immunosuppressive factors PD-L1 and IL-10 in peripheral blood mononuclear cells (PBMCs), exerting antitumor effects [142]. Additionally, some butyrate-producing bacteria exhibit antitumor activity via butyrate secretion. Eubacterium rectale is a beneficial microbiota that helps prevent primary intestinal lymphoma. It produces butyrate to suppress the TNF/TLR4/MyD88 signaling pathway and inhibit the NF-κB pathway in B cells, thus reducing TNF-associated intestinal inflammation and the incidence of lymphoma in Eμ-Myc mice [143]. Despite the large body of research supporting the role of butyrate in promoting immunity in the TME, other studies reported its immunosuppressive effects. For example, in a lung cancer (LC) relapse model, butyrate derived from Roseburia inhibited HDAC2, increased H3K27 acetylation at the H19 promoter, and induced M2 macrophage polarization, thereby increasing the expression of H19 in tumor cells and promoting lung cancer metastasis [104]. In addition to regulating immune cells in TME, butyrate also modulates cancer-associated fibroblasts (CAFs), contributing to its antitumor effects. Specifically, Wang et al. identified a subset of CAFs expressing high levels of sulfatase 1 (SULF1), which is associated with poor prognosis in CRC patients. Butyrate suppresses SULF1 expression in CAFs by inhibiting HDAC activity, thereby attenuating SULF1-mediated angiogenesis [144] (Figure 6).
In contrast to its effects on tumor cells, butyrate enhances CD8⁺ T cell function across multiple tumor types. This effect is observed not only in the butyrate-rich intestinal environment but also in subcutaneous tumors with relatively low concentrations of butyrate, suggesting a broadly conserved regulatory role, albeit through distinct mechanisms [135,136]. Comparatively, its modulation of innate immune cells appears to be more tissue-specific: in the stomach and intestine, butyrate promotes antitumor immunity by acting on PBMCs and B cells [142,143], whereas in the lung it drives macrophage polarization toward an immunosuppressive phenotype, thereby attenuating antitumor responses. Such differential effects may be attributed to the lower butyrate concentrations in distal tissues [104]. Moreover, butyrate can inhibit angiogenesis by regulating CAF activity, further contributing to its antitumor properties [144]. Collectively, these findings highlight the cell-specific and tissue-dependent roles of butyrate in the regulation of tumor immunity.
5. Other SCFAs
5.1 Formate
Within the TME, formate originates not only from microbial secretion but also from host cell metabolism, primarily through the conversion of cellular serine mediated by serine hydroxymethyltransferase (SHMT) and methylenetetrahydrofolate dehydrogenase (MTHFD) [145]. As a key regulator of tumor metastasis, formate directly promotes migration and invasion in cancer cell lines [146-148]. For instance, formate derived from Fusobacterium nucleatum can activate aryl hydrocarbon receptor (AhR) signaling, promoting CRC invasion and enhancing cancer stemness [149]. Formate also reprograms lipid metabolism in glioblastoma cells, facilitating an invasive phenotype through MMP-mediated degradation of extracellular matrix (ECM) proteins [150]. Furthermore, as a nucleotide synthesis precursor, formate fuels purine and pyrimidine production to meet the high nucleotide demands of cancer cells [151]. In PDAC, cancer cells generate formate via an IDO1-dependent pathway. This formate enters the tetrahydrofolate (THF) cycle, where it is co-utilized by cancer cells and pancreatic stellate cells for purine nucleotide synthesis, driving proliferation [152]. Formate also modulates immune responses. Recent studies have shown that in melanoma, formate can enhance CD8⁺ T cell-mediated antitumor immunity via activation of the Nrf2 pathway [148]. In contrast, in CRC, formate promotes the expansion of Th17 cells in the mesenteric lymph nodes (MLN) and increases the release of pro-inflammatory cytokines, thereby accelerating tumor initiation and progression [149]. Overall, formate drives tumor progression by reprogramming cancer metabolism and perturbing immune regulation. Notably, with respect to CD8⁺ T cells, various SCFAs generally enhance their proliferation, activation, and cytotoxic function, thereby exerting broad antitumor immune effects.
5.2 Valerate
As a short-chain fatty acid with five carbon atoms, valerate predominantly exhibits antitumor properties within the TME. Lau et al. demonstrated that Lactobacillus acidophilus-derived valerate suppresses NAFLD-HCC development and enhances intestinal barrier integrity. Mechanistically, valerate binds to hepatocyte surface receptors GPR41/43, inhibiting the Rho-GTPase pathway and suppressing NAFLD-HCC initiation [153]. Valerate also inhibits proliferation of breast and liver cancer cells via epigenetic regulation [154,155], including HDAC inhibition and DNA methylation pattern alterations. Previous studies have confirmed its potent anti-inflammatory effects in inflammatory and autoimmune diseases [156], suggesting a potential role in modulating tumor immunity within the TME. In vitro, treatment of cytotoxic T lymphocytes (CTLs) with valerate enhances the expression of effector molecules (e.g., CD25, IFN-γ, TNF-α) through HDAC inhibition, thereby boosting their antitumor activity [25].
5.3 Isobutyrate and Isovalerate
Unlike straight-chain SCFAs, the formation of branched-chain SCFAs (such as isobutyrate and isovalerate) primarily depends on the metabolism of undigested proteins by gut microbiota [157]. These metabolites are present at relatively low concentrations in the body, and research on their regulatory effects on tumors has been limited. Notably, isobutyrate and isovalerate have been found to exhibit similar HDAC inhibitory effects on cancer cells [158]. Recent studies have demonstrated that isobutyrate exerts potent antitumor effects by modulating immune cell activity and tumor growth. Mechanistically, isobutyrate reduces the expression of programmed death-1 (PD-1) in T cells, increases the expression of MHC class II receptor HLA-DR, and activates T cells. Oral administration of isobutyrate enhances the antitumor effects of anti-PD-1 antibodies, reducing tumor volume and increasing the number of tumor-infiltrating T cells [159]. These findings highlight the potential of branched-chain short-chain fatty acids in tumor regulation, and further research into these metabolites will help us gain a more comprehensive understanding of the role of SCFAs in tumor progression.
6. SCFAs and Cancer Treatment
6.1 Dietary, Microbiota, and Metabolic Interventions Targeting SCFAs
Clinical studies demonstrate that cancer patients exhibit dysregulated levels of SCFA-producing microbiota and SCFAs compared to healthy individuals (Table 1). Microbes and SCFAs that are downregulated in cancer or upregulated after treatment are generally considered beneficial [160-162] (e.g., Bifidobacterium pseudolongum [38], Roseburia intestinalis [135], Lactobacillus acidophilus [153]). Therapeutic strategies to restore beneficial SCFAs include SCFA supplementation [163,164], fecal microbiota transplantation (FMT) [62,134,165], high-fiber diets [166], and probiotics [109,167]. Certain probiotics and bioactive metabolites described above have shown significant therapeutic effects in both clinical and preclinical studies. For instance, acetate produced by Bifidobacterium pseudolongum significantly downregulates progression in the NAFLD-HCC mouse model. Administration of Bifidobacterium pseudolongum or acetate can significantly inhibit NAFLD-HCC progression [38]. Likewise, FMT from high-SCFA donors significantly reduces tumor burden in HCC mice [62]. A clinical trial found that Alaska Native (AN) people have an increased risk of CRC due to a deficiency in colonic butyrate, caused by low dietary fiber intake. Supplementation with high-dose soluble fiber was shown to significantly reduce cancer risk in AN people (NCT03028831). Similarly, other clinical studies have reported comparable findings [168,169]. In CRC patients, oral administration of SCFA-producing probiotics or prebiotics-encapsulated probiotic spores can modulate the gut microbiota, increase the abundance of beneficial microorganisms, and significantly reduce the abundance of pathogenic microbes associated with CRC, highlighting the potential therapeutic benefits of probiotics in CRC treatment (NCT03072641) [170].
Changes in SCFAs levels and their properties in cancer patients.
| Cancer types | Compare groups | Levels of SCFAs and associated-bacteria | Properties | Ref. |
|---|---|---|---|---|
| CRC | CRC patients vs HCs (human fecal) | SCFAs↓ | Beneficial | [180] |
| CRC | CRC patients vs HCs (human fecal) | Butyrate/Roseburia intestinalis↓ | Beneficial | [135] |
| HCC | HCC patients vs HCs (human plasma) | Butyrate↓ | Beneficial | [113] |
| HCC | Recurrence vs non-recurrence patients (human plasma/ fecal) | Acetate/Bacteroides thetaiotaomicron↓ | Beneficial | [66] |
| GC | GC patients vs HCs (human fecal) | Butyrate↓ | Beneficial | [134] |
| GC | GC patients vs HCs (human fecal) | Butyrate/Faecalibacterium and Bifidobacterium↓ | Beneficial | [142] |
| BC | BC patients vs HCs (human fecal microbial compositions) | Propionate↓ | Beneficial | [202] |
| BC | BC patients with depression vs those without depression (human plasma) | Acetate↓ | Beneficial | [58] |
| AML | AML patients vs HCs (human fecal) | Propionate↓ | Beneficial | [81] |
| NSCLC | Human functional genomic | Propionate metabolism↓ | Beneficial | [84] |
| Multiple Myeloma | CRMM vs RRMM | Aropionate and butyrate/Agathobacter↓ | Beneficial | [203] |
| GC | GC vs IM vs CSG patients (human plasma) | Propionate and butyrate↓ | / | [204] |
| Pan cancer | Cachectic vs non-cachectic patients/ HCs (human fecal) | Acetate↓ | / | [205] |
| HCC | HCC mice vs WT mice (mice plasma) | Acetate↓(~50%) | Beneficial | [62] |
| NAFLD-HCC | NAFLD-HCC mice fed a HFHC diet vs control mice fed a HFLC diet (mice fecal) | Acetate/Bifidobacterium pseudolongum↓ | Beneficial | [38] |
| NAFLD-HCC | NAFLD-HCC mice fed a HFHC diet vs control mice fed a HFLC diet (mice fecal) | Valerate/ Lactobacillus acidophilus↓ | Beneficial | [153] |
| CRC | Responded vs nonresponded to ICB patients (human fecal) | Roseburia intestinalis↑ | Beneficial | [135] |
| CRC | Responded vs nonresponded to neoadjuvant radiochemotherapy patients (human fecal) | SCFAs↑ | Beneficial | [206] |
| NSCLC | Responded vs nonresponded to anti-PD-1 therapy patients (human plasma) | Butyrate↑ | Beneficial | [137] |
| Pan cancer | Responded vs nonresponded to oxaliplatin patients (human plasma) | Butyrate↑ | Beneficial | [136] |
| Pan cancer | Responded vs nonresponded to PD-1i therapy patients (human fecal) | SCFAs↑ | Beneficial | [162] |
| Pan cancer | CR-treated mice vs IF-treated mice /mice fed ad libitum(mice fecal) | Acetate↑ | Beneficial | [61] |
| CRC | CRC patients vs HCs (human fecal) | Formate/Fusobacterium nucleatum↑ | Harmful | [149] |
| CRC | Responded vs nonresponded to capecitabine patients (human fecal) | Isobutyrate↓ | Harmful | [207] |
| NSCLC | lung tumor tissues vs normal lung tissues | Acetate↑ | Harmful | [55] |
| LC | Recurrence vs non-recurrence patients (human tumor tissue) | Butyrate↑ | Harmful | [104] |
| LUAD | lung adenocarcinoma tissues vs normal lung tissues | Acetate↑ | Harmful | [69] |
| Metastatic Melanoma | MM patients resistance to anti-CTLA-4 blocking mAbs (Human plasma) | Propionate and butyrate↑ | Harmful | [189] |
↑, upregulate. ↓, downregulate. Abbreviations: CRC, colorectal cancer; HCC, hepatocellular carcinoma; GC, gastric cancer; BC, Breast cancer; AML, acute myeloid leukemia; NSCLC, non-small cell lung cancer; NAFLD-HCC, non-alcoholic fatty liver disease-related hepatocellular carcinoma; LC, lung cancer; LUAD, lung adenocarcinoma; HCs, healthy controls; CRMM, complete remission multiple myeloma; RRMM, refractory disease multiple myeloma; IM, intestinal metaplasia; CSG, chronic superficial gastritis; WT, wild type; HFHC, high-fat/high-cholesterol; HFLC, high-fat/low-cholesterol; ICB, immune checkpoint blockade; CR, calorie restriction; IF, intermittent fasting; MM, Metastatic Melanoma; SCFAs, short-chain fatty acids.
Conversely, microbes and SCFAs that are upregulated in cancer (e.g., Fusobacterium nucleatum [149], Rikenellaceae, Clostridiales [171]) are often pathogenic [69,104,172,173]. Therapeutic approaches targeting these harmful factors primarily rely on antibiotics and metabolic enzyme inhibition. In a high-fat diet-induced prostate cancer mouse model, treatment with a broad-spectrum antibiotic cocktail (Abx) significantly reduced pathogenic bacterial abundance and fecal SCFA levels, thereby suppressing tumor growth [171]. Blocking key metabolic pathways has also shown therapeutic promise. Inhibition of SHMT2/MTHFD1 to target formate metabolism induces a “folate trap” impeding nucleotide synthesis and promoting cancer cell death [174,175]. For instance, in Burkitt lymphoma (BL) cells, SHMT2 inhibitors deplete intracellular formate and trigger apoptosis [175]. Likewise, by inhibiting ACSS2 to block acetate utilization, the level of acetyl-CoA is reduced in cancer cells, thereby decreasing its ability for lipid synthesis and energy metabolism, effectively hindering tumor progression. In the preclinical breast cancer model, targeting ACSS2 using CRISPR-Cas9-guided editing or small molecule inhibitors (such as VY-3-135) to suppress acetate metabolism disrupts cancer cell adaptation to metabolic stress, leading to a significant reduction in tumor growth [56,176]. Additionally, the first human clinical trial of ACSS2 inhibitors for cancer therapy is currently underway (NCT04990739). Overall, whether to supplement SCFAs or inhibit their production to suppress tumor progression depends on the specific cancer context. Overall, whether supplementing or inhibiting the production or metabolism of SCFAs to suppress tumor progression depends on the specific cancer context and the composition of the microbiome. In some cases, SCFAs may exert beneficial effects by reshaping the microbiome or improving the TME, whereas in other cases, they may promote tumor growth by facilitating metabolic reprogramming. Inter-individual differences in the microbiome may be a key factor in these response variations [177]. Therefore, blindly supplementing SCFAs or inhibiting their production and metabolism may not consistently achieve the desired outcomes in clinical treatments.
6.2 SCFAs as Adjuvants in Radiation and Chemotherapy
Increasing evidence suggests that SCFAs can enhance the efficacy of cancer radiotherapy and chemotherapy while mitigating treatment-related toxicities (Figure 7). For instance, butyrate produced by three probiotic strains, Lactobacillus plantarum S2, L. pentosus S3, and L. rhamnosus 14E4, enhances the sensitivity of doxorubicin-resistant CRC cell lines (HT29-dx) to the drug [178]. Dong et al. demonstrated that FMT promotes the accumulation of Roseburia intestinalis in the gut, which secretes butyrate and activates radiation-induced autophagy via the OR51E1/RALB axis, thereby enhancing the radiotherapy sensitivity of CRC in mice [179]. Clinically, serum butyrate levels are significantly higher in oxaliplatin (OXA)-responsive CRC patients compared with non-responsive individuals [136]. Butyrate supplementation effectively overcomes resistance, resensitizing CRC cells to OXA [136,180,181]. Additionally, in lung cancer models, resistance to cisplatin, a first-line chemotherapeutic agent, is commonly mediated by EMT [182]. Treatment with propionate was found to upregulate epithelial transcriptional programs, strengthen cell-cell adhesion, and suppress chemoresistant EMT phenotypes, thereby sensitizing lung cancer cells to cisplatin [84]. Meanwhile, studies have shown that butyrate can also enhance the therapeutic efficacy of sorafenib in HCC mouse model [113].
Roles and clinical applications of SCFAs in various tumor types. The roles of acetate, propionate, butyrate, and other SCFAs in various tumor types and their potential clinical applications, including adjunctive roles in chemoradiotherapy, immunotherapy, and other therapeutic modalities. Abbreviations: ACT, adoptive T cell therapy; ICIs, immune checkpoint inhibitors; RA, retinoic acid.
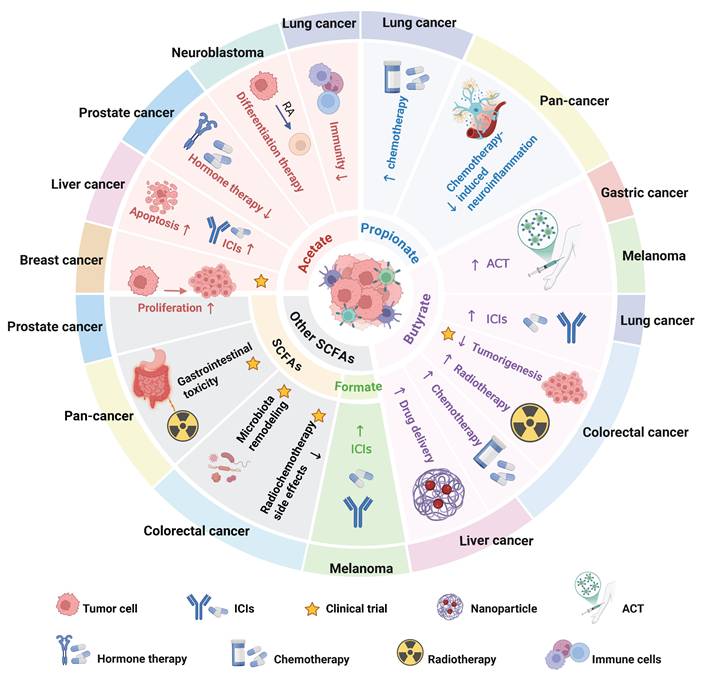
Moreover, SCFAs can alleviate the side effects caused by conventional therapies. In murine pancreatic cancer models, the combination of gemcitabine and butyrate not only significantly reduced cancer-associated stroma formation, but also preserved the integrity of the intestinal mucosal barrier, improved fecal microbiota composition, and alleviated chemotherapy-induced renal and hepatic injury [183]. Similarly, SCFAs have been shown to mitigate 5-fluorouracil (5-FU)-induced neuroinflammation [184]. Recent clinical studies revealed that lower plasma levels of SCFAs were significantly associated with more severe radiotherapy-related fatigue in patients with head and neck cancer [185]. Moreover, SCFAs were demonstrated to reduce gastrointestinal complications and microbiota dysbiosis in CRC patients undergoing chemotherapy (ChiCTR2000040916) [186]. Ongoing clinical studies are also investigating SCFA supplementation in patients receiving pelvic radiotherapy, with the aim of improving quality of life and reducing treatment-related toxicities (NCT04700527). Although SCFAs have shown potential in enhancing the efficacy of radiotherapy and chemotherapy while alleviating side effects, the current clinical studies have small sample sizes, and the assessment of dosage, safety, and individual variability remains insufficient. Excessive supplementation may lead to gut microbiota dysbiosis or other adverse effects. Therefore, large-scale clinical trials are needed in the future to systematically evaluate the safety and efficacy of SCFAs in chemotherapy and radiotherapy.
6.3 SCFAs as Adjuvants in Immunotherapy
As discussed in previous sections, SCFAs are recognized as effective modulators of immune responses, particularly in the context of cancer immunotherapy involving immune checkpoint inhibitors (ICIs) and adoptive T cell therapy (ACT) (Figure 7).
SCFAs have shown promising results in combination with ICIs. For example, in an HCC mouse model, treatment with PD-1 monoclonal antibody combined with acetate improved liver damage and inhibited tumor growth [62]. In a CRC mouse model, oral butyrate administration three days after tumor inoculation, followed by combination with anti-PD-1 therapy, increased the number of tumor-infiltrating CD8+ T cells and the production of their effector cytokines, significantly inhibiting tumor growth [135]. The enhancement of anti-PD-1 therapy by butyrate was also validated in an non-small cell lung cancer (NSCLC) mouse model [137]. Interestingly, formate, a short-chain fatty acid that has been reported to promote cancer, combined with anti-PD-1 therapy, also enhanced CD8+ T cell-mediated tumor control in a B16-OVA tumor model and improved mouse survival [187]. Additionally, Several clinical studies have shown that high concentrations of SCFAs in feces are significantly associated with longer progression-free survival and relatively higher response rates to PD-L1 inhibitors (ChiCTR2000032088) [162,188]. However, the role of SCFAs in immunotherapy is not without controversy. In mouse melanoma models and metastatic melanoma patients, elevated serum levels of butyrate and propionate were associated with resistance to CTLA-4 blockade and a higher proportion of Treg cells. Furthermore, in melanoma-bearing mice, supplementation with butyrate combined with α-CTLA-4 inhibited dendritic cell maturation and the accumulation of effector and memory tumor-specific CTLs [189]. Additionally, targeting the pro-cancer effects of acetate in lung adenocarcinoma and NSCLC by inhibiting acetate receptors (FFAR2) and downstream molecules regulated by acetate (USP10) slowed tumor growth and improved responses to ICI therapy [55,69]. While many studies have reported the beneficial role of SCFAs in enhancing cancer treatment, other studies have also highlighted their anti-inflammatory properties [190]. Therefore, the complex multifaceted effects of SCFAs are not surprising.
Given regulatory roles of SCFAs in T cell proliferation, cytotoxicity, and tumor infiltration, recent studies explore SCFAs as adjuvants for ACT. Prasad et al. found that SCFAs can enhance the metabolic adaptability of CAR-T cells, thereby improving their tumor-killing capacity [191]. In parallel, Luu et al. demonstrated that in vitro treatment of CTLs and chimeric antigen receptor (CAR) T cells with butyrate or valerate enhanced mTOR-mediated metabolic sensing, inhibited class I HDAC activity, and upregulated effector molecule expression, thereby improving antitumor efficacy in melanoma and pancreatic cancer models [25]. Similarly, Yu et al. reported that butyrate enhanced the cytotoxic activity of CD8+ T cells and CAR-Claudin 18.2+ CD8+ T cells against GC cells through GPR109A and homeobox protein HOPX signaling, as demonstrated by in vitro co-culture experiments and in vivo tumor-bearing mouse models [134]. Current findings suggest that SCFAs enhance T cell functionality and therapeutic efficacy in vivo, underscoring their clinical potential as ACT adjuvants. However, available data also suggest that the effects of SCFAs may be counterproductive due to the complexity of the TME [192]. Additionally, studies on SCFAs as adjuncts to ACT are still limited, underscoring the need for more extensive and in-depth research to fully explore their clinical potential.
6.4 Other Applications of SCFAs in Cancer Diagnosis and Therapy
Differentiation therapy is a treatment strategy that aims to alter the differentiation state of cancer cells using differentiation inducers, leading to the loss of malignant phenotypes. It has been successfully applied in the treatment of acute promyelocytic leukemia (APL) [193,194], although its efficacy in solid tumors has been less satisfactory [195]. Notably, Li et al. suggested that in a mouse neuroblastoma model, combining acetate with differentiation therapy (retinoic acid induction) not only restored histone acetylation under hypoxic conditions, but also reestablished the expression of neuronal differentiation markers and neuronal differentiation morphology, significantly improving the in vivo efficacy of retinoic acid [196]. Hormonal therapy regulates the body's endocrine balance to treat tumors, and it is considered a first-line treatment for prostate cancer. During prostate cancer treatment, acetate metabolism enhances c-MYC expression during neurodifferentiation (NED), thereby promoting resistance of primary adenocarcinoma prostate cancer (PCa) to hormonal therapy. Studies have shown that combining ACSS2 inhibitors with hormonal therapy can significantly enhance PCa sensitivity to the hormonal drug enzalutamide (ENZA), thereby effectively inhibiting tumor growth [197]. Furthermore, butyrate can be used to modify nanoparticles to enhance drug efficacy. Research indicates that oral butyrate-modified nanoparticles effectively and persistently promote trans-epithelial transport in the gut, drug accumulation in the liver, and drug uptake by HCC cells, thereby enhancing liver cancer treatment [198]. Additionally, acetate exhibits clinical potential in imaging technologies. 11C-acetate positron emission tomography (PET) has emerged as a promising imaging modality for prostate cancer diagnosis and therapeutic monitoring (NCT01144897) [34,199]. The combined application of SCFAs in other cancer therapies offers new hope for treatment and diagnostic strategies (Figure 7). However, several limitations currently exist, including insufficient translational research, significant variability in efficacy across different cancer types and patient populations, and constraints in clinical applications. Specifically, ensuring the precise delivery and targeted therapy of SCFAs remains a challenge, and there is a lack of unified mechanistic understanding. Furthermore, the choice of different dosages, combination regimens, and administration methods in clinical trials can all influence treatment outcomes. In the future, with advancements in precise delivery technologies, deeper mechanistic studies, and rigorous clinical validation, these strategies are expected to be optimized and more effectively integrated into individualized cancer treatment plans.
7. Conclusions and Perspectives
SCFAs, as key functional metabolites within the TME, profoundly influence tumor initiation, progression, and therapeutic response by orchestrating metabolic reprogramming, epigenetic modifications, and immune microenvironmental dynamics. SCFAs exhibit a 'double-edged sword' effect in tumor regulation—capable of both suppressing tumor growth and promoting malignant phenotypes. This duality highlights the biological complexity of SCFAs and underscores the necessity of precise modulation based on tumor heterogeneity, host metabolic status, and microbiota composition.
From a metabolic perspective, SCFAs can fuel tumor growth and survival by supplying acetyl-CoA or one-carbon units to regulate energy metabolism, lipid biosynthesis, and nucleotide synthesis. Conversely, under certain conditions, SCFAs may induce oxidative stress, mitochondrial dysfunction, and lipid peroxidation, thereby triggering tumor cell death. This 'metabolic switch' effect is largely dependent on pH, oxygen availability, and nutrient competition within the TME, suggesting that future interventions should incorporate metabolic imaging techniques (e.g., 11C-acetate PET) to dynamically monitor tumor metabolic profiles and optimize SCFA-based therapeutic strategies [200]. At the epigenetic level, SCFAs reshape chromatin accessibility and regulate gene expression by inhibiting HDACs, activating HATs, and providing acyl groups. These epigenetic modifications influence tumor cell proliferation, migration, and survival. Notably, the epigenetic effects of SCFAs are cell type-specific: their regulation of HDACs, HATs, and endogenous acyl donors differs between normal and cancer cells, leading to distinct target genes being modified and thereby producing divergent downstream phenotypes. This cell type-dependent regulation provides a theoretical rationale for developing precise combination therapies targeting epigenetic pathways. Immune regulation represents one of the key mechanisms by which SCFAs influence cancer therapy. On the one hand, SCFAs can enhance the metabolic adaptability and cytotoxicity of CD8⁺ T cells, promote M1 macrophage polarization, and modulate the function of ILCs to strengthen antitumor immunity. On the other hand, SCFAs may impair immune efficacy by activating regulatory Tregs or suppressing DCs antigen presentation. This bidirectional immunomodulation suggests that future studies should incorporate spatial profiling of immune cell subsets to delineate the spatiotemporal landscape of SCFA action within the TME, thereby enabling precision enhancement of immunotherapeutic outcomes [100,201].
As key metabolites of the gut microbiota, SCFAs exhibit substantial potential in tumor development and therapy, offering new avenues to improve cancer treatment outcomes. From a translational perspective, SCFAs have demonstrated value as adjuvants to chemotherapy, radiotherapy, and immunotherapy. Strategies such as increasing SCFA levels through dietary fiber, probiotics, or direct supplementation can enhance therapeutic sensitivity while alleviating toxic side effects. Moreover, targeting specific SCFA-metabolizing enzymes or receptors holds promise for reversing protumor metabolic phenotypes. However, the efficacy of SCFAs is constrained by concentration dependence, tissue specificity, and interindividual differences in gut microbiota, leading to considerable variability in therapeutic responses.
To overcome these limitations, future research should focus on three directions: (1) applying single-cell multi-omics to delineate the spatiotemporal networks and cell-type-specific effects of SCFAs; (2) developing precision delivery systems, such as engineered probiotics or nanoparticle-based carriers, to optimize tissue targeting and mitigate concentration dependence; and (3) tailoring SCFA-based interventions to individual metabolic and microbiota profiles, thereby advancing truly individualized cancer treatment.
Abbreviations
SCFAs: short-chain fatty acids; TME: tumor microenvironment; HDAC: histone deacetylase; HAT: histone acetyltransferase; GPR41/43/109 A: G protein-coupled receptor 41/43/109 A; FMT: fecal microbiota transplantation; Abx: antibiotic mixture; ICIs: immune checkpoint inhibitors; ACT: adoptive T cell therapy; FAs: lipids, fatty acids; MCFAs: medium-chain fatty acids; LCFAs: long-chain fatty acids; MCFAs: medium-chain fatty acids; ROS: reactive oxygen species; CAFs: cancer-associated fibroblasts; ACLY: ATP citrate lyase; PDAC: pancreatic ductal adenocarcinoma; CODH: carbon monoxide dehydrogenase; ACSS: acetyl-CoA synthetase; THF: tetrahydrofolate; CRC: colorectal cancer; HCC: hepatocellular carcinoma; NAFLD-HCC: non-alcoholic fatty liver disease-related hepatocellular carcinoma; MCT1: monocarboxylate transporter 1; SMCT1: sodium-coupled monocarboxylate transporter 1; eEF1A1: eukaryotic elongation factor 1A1; GBM: glioblastoma; MAO-B: monoamine oxidase B; GSC: glioma stem cell-like cell; DLAT: dihydrolipoamide S-acetyltransferase; USP10: ubiquitin-specific peptidase 10; PD-L1: programmed death-ligand 1; IFN-γ: interferon gamma; ILC3s: type 3 innate lymphoid cells; HSC: hepatic stellate cell; Sox13: SRY-box transcription factor 13; ACC1: acetyl-CoA carboxylase 1; MDSCs: myeloid-derived suppressor cells; LUAD: lung adenocarcinoma; FFAR2: free fatty acid receptor 2; TCA: tricarboxylic acid; PD-1: programmed death-1; PEP: phosphoenolpyruvate; DHAP: dihydroxyacetone phosphate; TNFAIP1: tumor necrosis factor alpha-induced protein 1; HECTD2: HECT domain E3 ubiquitin protein ligase 2; EHMT2: euchromatic histone-lysine N-methyltransferase 2; EMT: epithelial-to-mesenchymal transition; MCEE: methylmalonyl-CoA epimerase; MMA: methylmalonic acid; ICD: immunogenic cell death; AML: acute myeloid leukemia; DAMPs: damage associated molecular patterns; DC: dendritic cell; MPN: myeloproliferative neoplasms; GLUT1: glucose transporter 1; G6PD: glucose-6-phosphate dehydrogenase; IGF-1: insulin-like growth factor-1; TCR: T-cell receptor; PBMCs: peripheral blood mononuclear cells; LC: lung cancer; SULF1: sulfatase 1; AhR: aryl hydrocarbon receptor; ECM: extracellular matrix; THF: tetrahydrofolate; SHMT: serine hydroxymethyltransferase; MTHFD: methylenetetrahydrofolate dehydrogenase; MLNs: mesenteric lymph nodes; CTLs: cytotoxic T lymphocytes; AN: Alaska Native; GC: gastric cancer; BC: breast cancer; NSCLC: non-small cell lung cancer; HCs: healthy controls; CRMM: complete remission multiple myeloma; RRMM: refractory disease multiple myeloma; IM: intestinal metaplasia; CSG: chronic superficial gastritis; WT: wild type; HFHC: high-fat/high-cholesterol; HFLC: high-fat/low-cholesterol; ICB: immune checkpoint blockade; CR: calorie restriction; IF: intermittent fasting; MM: Metastatic Melanoma; DOXO: doxorubicin; OXA: oxaliplatin; 5-FU: 5-fluorouracil; CAR: chimeric antigen receptor; APL: acute promyelocytic leukemia; NED: neurodifferentiation; PCa: adenocarcinoma prostate cancer; ENZA: enzalutamide; PET: positron emission tomography; RA: retinoic acid.
Acknowledgements
The illustrations are created with BioRender.com. This work was supported by grants from the Natural Science Foundation of China (82472700, 82472745, 82472365, 82373400, 82372660, 82272685, 82202260 and 82173248), the Science and Technology Major Program of Sichuan Province (2022ZDZX0019), the Sichuan Science and Technology Program (2024NSFSC1894, 2023YFS0128, 2023NSFSC1874), the Project funded by China Postdoctoral Science Foundation (2022TQ0221), 1.3.5 project for disciplines of excellence, West China Hospital, Sichuan University (ZYGD22006), Scientific and Technological Innovation Ability Enhancement Project for Junior Faculties of Sichuan University (2024SCUQJTX043), the Sichuan University postdoctoral interdisciplinary Innovation Fund (10822041A2103).
Author contributions
Conceptualization: K.Y. Writing: Y.X., A.D., Z.W., H.P. Proofreading and revising: A.D. and Z.W. Supervision: K.Y. Figures: Y.X. All authors have read and approved the article.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A. et al. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J Clin. 2021;71:209-49
2. Toninelli M, Rossetti G, Pagani M. Charting the tumor microenvironment with spatial profiling technologies. Trends Cancer. 2023;9:1085-96
3. Wan M, Pan S, Shan B, Diao H, Jin H, Wang Z. et al. Lipid metabolic reprograming: the unsung hero in breast cancer progression and tumor microenvironment. Mol Cancer. 2025;24:61
4. Xiao Y, Yu D. Tumor microenvironment as a therapeutic target in cancer. Pharmacol Ther. 2021;221:107753
5. Jin M-Z, Jin W-L. The updated landscape of tumor microenvironment and drug repurposing. Signal Transduct Target Ther. 2020;5:166
6. Paget S. The distribution of secondary growths in cancer of the breast. 1889. Cancer Metastasis Rev. 1989;8:98-101
7. de Groot AE, Roy S, Brown JS, Pienta KJ, Amend SR. Revisiting Seed and Soil: Examining the Primary Tumor and Cancer Cell Foraging in Metastasis. Mol Cancer Res. 2017;15:361-70
8. Dong Z, Luo Y, Yuan Z, Tian Y, Jin T, Xu F. Cellular senescence and SASP in tumor progression and therapeutic opportunities. Mol Cancer. 2024;23:181
9. Kao K-C, Vilbois S, Tsai C-H, Ho P-C. Metabolic communication in the tumour-immune microenvironment. Nat Cell Biol. 2022;24:1574-83
10. Kanarek N, Petrova B, Sabatini DM. Dietary modifications for enhanced cancer therapy. Nature. 2020;579:507-17
11. Corn KC, Windham MA, Rafat M. Lipids in the tumor microenvironment: From cancer progression to treatment. Prog Lipid Res. 2020;80:101055
12. Lam SM, Wang Z, Li B, Shui G. High-coverage lipidomics for functional lipid and pathway analyses. Anal Chim Acta. 2021;1147:199-210
13. Du A, Wang Z, Huang T, Xue S, Jiang C, Qiu G. et al. Fatty acids in cancer: Metabolic functions and potential treatment. MedComm - Oncology. 2023;2:e25
14. Wu Z, Bagarolo GI, Thoröe-Boveleth S, Jankowski J. 'Lipidomics': Mass spectrometric and chemometric analyses of lipids. Adv Drug Deliv Rev. 2020;159:294-307
15. Jensen M, Liu S, Stepula E, Martella D, Birjandi AA, Farrell-Dillon K. et al. Opto-Lipidomics of Tissues. Adv Sci (Weinh). 2024;11:e2302962
16. Bogie JFJ, Haidar M, Kooij G, Hendriks JJA. Fatty acid metabolism in the progression and resolution of CNS disorders. Adv Drug Deliv Rev. 2020;159:198-213
17. Jeon YG, Kim YY, Lee G, Kim JB. Physiological and pathological roles of lipogenesis. Nat Metab. 2023;5:735-59
18. Lee H, Woo SM, Jang H, Kang M, Kim S-Y. Cancer depends on fatty acids for ATP production: A possible link between cancer and obesity. Semin Cancer Biol. 2022;86:347-57
19. Kimura I, Ichimura A, Ohue-Kitano R, Igarashi M. Free Fatty Acid Receptors in Health and Disease. Physiol Rev. 2020;100:171-210
20. Koh A, De Vadder F, Kovatcheva-Datchary P, Bäckhed F. From Dietary Fiber to Host Physiology: Short-Chain Fatty Acids as Key Bacterial Metabolites. Cell. 2016;165:1332-45
21. Elshahed MS, Miron A, Aprotosoaie AC, Farag MA. Pectin in diet: Interactions with the human microbiome, role in gut homeostasis, and nutrient-drug interactions. Carbohydr Polym. 2021;255:117388
22. Dalile B, Van Oudenhove L, Vervliet B, Verbeke K. The role of short-chain fatty acids in microbiota-gut-brain communication. Nat Rev Gastroenterol Hepatol. 2019;16:461-78
23. Zhou P, Chang W-Y, Gong D-A, Xia J, Chen W, Huang L-Y. et al. High dietary fructose promotes hepatocellular carcinoma progression by enhancing O-GlcNAcylation via microbiota-derived acetate. Cell Metab. 2023;35:1961-1975.e6
24. Ma H, Yu Y, Wang M, Li Z, Xu H, Tian C. et al. Correlation between microbes and colorectal cancer: tumor apoptosis is induced by sitosterols through promoting gut microbiota to produce short-chain fatty acids. Apoptosis. 2019;24:168-83
25. Luu M, Riester Z, Baldrich A, Reichardt N, Yuille S, Busetti A. et al. Microbial short-chain fatty acids modulate CD8+ T cell responses and improve adoptive immunotherapy for cancer. Nat Commun. 2021;12:4077
26. Yang W, Yu T, Huang X, Bilotta AJ, Xu L, Lu Y. et al. Intestinal microbiota-derived short-chain fatty acids regulation of immune cell IL-22 production and gut immunity. Nat Commun. 2020;11:4457
27. Schuchmann K, Müller V. Autotrophy at the thermodynamic limit of life: a model for energy conservation in acetogenic bacteria. Nat Rev Microbiol. 2014;12:809-21
28. Schug ZT, Vande Voorde J, Gottlieb E. The metabolic fate of acetate in cancer. Nat Rev Cancer. 2016;16:708-17
29. Ragsdale SW. Enzymology of the wood-Ljungdahl pathway of acetogenesis. Ann N Y Acad Sci. 2008;1125:129-36
30. Ragsdale SW, Pierce E. Acetogenesis and the Wood-Ljungdahl pathway of CO(2) fixation. Biochim Biophys Acta. 2008;1784:1873-98
31. Liu X, Cooper DE, Cluntun AA, Warmoes MO, Zhao S, Reid MA. et al. Acetate Production from Glucose and Coupling to Mitochondrial Metabolism in Mammals. Cell. 2018;175:502-513.e13
32. Comerford SA, Huang Z, Du X, Wang Y, Cai L, Witkiewicz AK. et al. Acetate dependence of tumors. Cell. 2014;159:1591-602
33. Kurdistani SK. Chromatin: a capacitor of acetate for integrated regulation of gene expression and cell physiology. Current Opinion in Genetics & Development. 2014;26:53-8
34. Kim D, Ko HY, Chung J-I, Park YM, Lee S, Kim SY. et al. Visualizing cancer-originating acetate uptake through monocarboxylate transporter 1 in reactive astrocytes in the glioblastoma tumor microenvironment. Neuro Oncol. 2024;26:843-57
35. McBrian MA, Behbahan IS, Ferrari R, Su T, Huang T-W, Li K. et al. Histone acetylation regulates intracellular pH. Mol Cell. 2013;49:310-21
36. Hosios AM, Vander Heiden MG. Acetate metabolism in cancer cells. Cancer Metab. 2014;2:27
37. Lyssiotis CA, Cantley LC. Acetate fuels the cancer engine. Cell. 2014;159:1492-4
38. Song Q, Zhang X, Liu W, Wei H, Liang W, Zhou Y. et al. Bifidobacterium pseudolongum-generated acetate suppresses non-alcoholic fatty liver disease-associated hepatocellular carcinoma. J Hepatol. 2023;79:1352-65
39. Mashimo T, Pichumani K, Vemireddy V, Hatanpaa KJ, Singh DK, Sirasanagandla S. et al. Acetate is a bioenergetic substrate for human glioblastoma and brain metastases. Cell. 2014;159:1603-14
40. Marques C, Oliveira CSF, Alves S, Chaves SR, Coutinho OP, Côrte-Real M. et al. Acetate-induced apoptosis in colorectal carcinoma cells involves lysosomal membrane permeabilization and cathepsin D release. Cell Death Dis. 2013;4:e507-e507
41. Oliveira CSF, Pereira H, Alves S, Castro L, Baltazar F, Chaves SR. et al. Cathepsin D protects colorectal cancer cells from acetate-induced apoptosis through autophagy-independent degradation of damaged mitochondria. Cell Death Dis. 2015;6:e1788
42. Hlavaty SI, Salcido KN, Pniewski KA, Mukha D, Ma W, Kannan T. et al. ACSS1-dependent acetate utilization rewires mitochondrial metabolism to support AML and melanoma tumor growth and metastasis. Cell Rep. 2024;43:114988
43. Schug ZT, Peck B, Jones DT, Zhang Q, Grosskurth S, Alam IS. et al. Acetyl-CoA synthetase 2 promotes acetate utilization and maintains cancer cell growth under metabolic stress. Cancer Cell. 2015;27:57-71
44. Li Z, Ji BW, Dixit PD, Tchourine K, Lien EC, Hosios AM. et al. Cancer cells depend on environmental lipids for proliferation when electron acceptors are limited. Nat Metab. 2022;4:711-23
45. Gao X, Lin S-H, Ren F, Li J-T, Chen J-J, Yao C-B. et al. Acetate functions as an epigenetic metabolite to promote lipid synthesis under hypoxia. Nat Commun. 2016;7:11960
46. Yao L, Jiang L, Zhang F, Li M, Yang B, Zhang F. et al. Acetate promotes SNAI1 expression by ACSS2-mediated histone acetylation under glucose limitation in renal cell carcinoma cell. Biosci Rep. 2020;40:BSR20200382
47. Murthy D, Attri KS, Shukla SK, Thakur R, Chaika NV, He C. et al. Cancer-associated fibroblast-derived acetate promotes pancreatic cancer development by altering polyamine metabolism via the ACSS2-SP1-SAT1 axis. Nat Cell Biol. 2024;26:613-27
48. Decourcelle A, Very N, Djouina M, Loison I, Thévenet J, Body-Malapel M. et al. O-GlcNAcylation Links Nutrition to the Epigenetic Downregulation of UNC5A during Colon Carcinogenesis. Cancers (Basel). 2020;12:3168
49. Jiang M, Wu N, Xu B, Chu Y, Li X, Su S. et al. Fatty acid-induced CD36 expression via O-GlcNAcylation drives gastric cancer metastasis. Theranostics. 2019;9:5359-73
50. Wong KKL, Liu T-W, Parker JM, Sinclair DAR, Chen Y-Y, Khoo K-H. et al. The nutrient sensor OGT regulates Hipk stability and tumorigenic-like activities in Drosophila. Proc Natl Acad Sci U S A. 2020;117:2004-13
51. Okolie O, Bago JR, Schmid RS, Irvin DM, Bash RE, Miller CR. et al. Reactive astrocytes potentiate tumor aggressiveness in a murine glioma resection and recurrence model. Neuro Oncol. 2016;18:1622-33
52. Long PM, Tighe SW, Driscoll HE, Fortner KA, Viapiano MS, Jaworski DM. Acetate supplementation as a means of inducing glioblastoma stem-like cell growth arrest. J Cell Physiol. 2015;230:1929-43
53. Yin K, Lee J, Liu Z, Kim H, Martin DR, Wu D. et al. Mitophagy protein PINK1 suppresses colon tumor growth by metabolic reprogramming via p53 activation and reducing acetyl-CoA production. Cell Death Differ. 2021;28:2421-35
54. Arner EN, Rathmell JC. Metabolic programming and immune suppression in the tumor microenvironment. Cancer Cell. 2023;41:421-33
55. Wang J, Yang Y, Shao F, Meng Y, Guo D, He J. et al. Acetate reprogrammes tumour metabolism and promotes PD-L1 expression and immune evasion by upregulating c-Myc. Nat Metab. 2024;6:914-32
56. Miller KD, O'Connor S, Pniewski KA, Kannan T, Acosta R, Mirji G. et al. Acetate acts as a metabolic immunomodulator by bolstering T-cell effector function and potentiating antitumor immunity in breast cancer. Nat Cancer. 2023;4:1491-507
57. Qiu J, Villa M, Sanin DE, Buck MD, O'Sullivan D, Ching R. et al. Acetate Promotes T Cell Effector Function during Glucose Restriction. Cell Rep. 2019;27:2063-2074.e5
58. Ye L, Hou Y, Hu W, Wang H, Yang R, Zhang Q. et al. Repressed Blautia-acetate immunological axis underlies breast cancer progression promoted by chronic stress. Nat Commun. 2023;14:6160
59. Li Z, Liu H, He J, Wang Z, Yin Z, You G. et al. Acetyl-CoA Synthetase 2: A Critical Linkage in Obesity-Induced Tumorigenesis in Myeloma. Cell Metab. 2021;33:78-93.e7
60. Vodnala SK, Eil R, Kishton RJ, Sukumar M, Yamamoto TN, Ha N-H. et al. T cell stemness and dysfunction in tumors are triggered by a common mechanism. Science. 2019;363:eaau0135
61. Mao Y-Q, Huang J-T, Zhang S-L, Kong C, Li Z-M, Jing H. et al. The antitumour effects of caloric restriction are mediated by the gut microbiome. Nat Metab. 2023;5:96-110
62. Hu C, Xu B, Wang X, Wan W-H, Lu J, Kong D. et al. Gut microbiota-derived short-chain fatty acids regulate group 3 innate lymphoid cells in HCC. Hepatology. 2023;77:48-64
63. Liu Y, Song Y, Lin D, Lei L, Mei Y, Jin Z. et al. NCR- group 3 innate lymphoid cells orchestrate IL-23/IL-17 axis to promote hepatocellular carcinoma development. EBioMedicine. 2019;41:333-44
64. Li M, Yang Y, Xiong L, Jiang P, Wang J, Li C. Metabolism, metabolites, and macrophages in cancer. J Hematol Oncol. 2023;16:80
65. Liu N, Wang X, Steer CJ, Song G. MicroRNA-206 promotes the recruitment of CD8+ T cells by driving M1 polarisation of Kupffer cells. Gut. 2022;71:1642-55
66. Ma H, Yang L, Liang Y, Liu F, Hu J, Zhang R. et al. B. thetaiotaomicron-derived acetic acid modulate immune microenvironment and tumor growth in hepatocellular carcinoma. Gut Microbes. 2024;16:2297846
67. Halaby MJ, Hezaveh K, Lamorte S, Ciudad MT, Kloetgen A, MacLeod BL. et al. GCN2 drives macrophage and MDSC function and immunosuppression in the tumor microenvironment. Sci Immunol. 2019;4:eaax8189
68. Skoulidis F, Araujo HA, Do MT, Qian Y, Sun X, Cobo AG. et al. CTLA4 blockade abrogates KEAP1/STK11-related resistance to PD-(L)1 inhibitors. Nature. 2024;635:462-71
69. Zhao Z, Qin J, Qian Y, Huang C, Liu X, Wang N. et al. FFAR2 expressing myeloid-derived suppressor cells drive cancer immunoevasion. J Hematol Oncol. 2024;17:9
70. Macy JM, Ljungdahl LG, Gottschalk G. Pathway of succinate and propionate formation in Bacteroides fragilis. J Bacteriol. 1978;134:84-91
71. Hetzel M, Brock M, Selmer T, Pierik AJ, Golding BT, Buckel W. Acryloyl-CoA reductase from Clostridium propionicum. An enzyme complex of propionyl-CoA dehydrogenase and electron-transferring flavoprotein. Eur J Biochem. 2003;270:902-10
72. Hino T, Kuroda S. Presence of lactate dehydrogenase and lactate racemase in Megasphaera elsdenii grown on glucose or lactate. Appl Environ Microbiol. 1993;59:255-9
73. Morrison DJ, Mackay WG, Edwards CA, Preston T, Dodson B, Weaver LT. Butyrate production from oligofructose fermentation by the human faecal flora: what is the contribution of extracellular acetate and lactate? Br J Nutr. 2006;96:570-7
74. Bourriaud C, Robins RJ, Martin L, Kozlowski F, Tenailleau E, Cherbut C. et al. Lactate is mainly fermented to butyrate by human intestinal microfloras but inter-individual variation is evident. J Appl Microbiol. 2005;99:201-12
75. Saxena RK, Anand P, Saran S, Isar J, Agarwal L. Microbial production and applications of 1,2-propanediol. Indian J Microbiol. 2010;50:2-11
76. Scott KP, Martin JC, Campbell G, Mayer C-D, Flint HJ. Whole-genome transcription profiling reveals genes up-regulated by growth on fucose in the human gut bacterium 'Roseburia inulinivorans'. J Bacteriol. 2006;188:4340-9
77. Ryu TY, Kim K, Son M-Y, Min J-K, Kim J, Han T-S. et al. Downregulation of PRMT1, a histone arginine methyltransferase, by sodium propionate induces cell apoptosis in colon cancer. Oncol Rep. 2019;41:1691-9
78. Kim K, Kwon O, Ryu TY, Jung C-R, Kim J, Min J-K. et al. Propionate of a microbiota metabolite induces cell apoptosis and cell cycle arrest in lung cancer. Mol Med Rep. 2019;20:1569-74
79. Semaan J, El-Hakim S, Ibrahim J-N, Safi R, Elnar AA, El Boustany C. Comparative effect of sodium butyrate and sodium propionate on proliferation, cell cycle and apoptosis in human breast cancer cells MCF-7. Breast Cancer. 2020;27:696-705
80. Filippone A, Casili G, Scuderi SA, Mannino D, Lanza M, Campolo M. et al. Sodium Propionate Contributes to Tumor Cell Growth Inhibition through PPAR-γ Signaling. Cancers (Basel). 2022;15:217
81. Wei Y, Liu W, Wang R, Chen Y, Liu J, Guo X. et al. Propionate promotes ferroptosis and apoptosis through mitophagy and ACSL4-mediated ferroptosis elicits anti-leukemia immunity. Free Radic Biol Med. 2024;213:36-51
82. Park H-S, Han J-H, Park JW, Lee D-H, Jang K-W, Lee M. et al. Sodium propionate exerts anticancer effect in mice bearing breast cancer cell xenograft by regulating JAK2/STAT3/ROS/p38 MAPK signaling. Acta Pharmacol Sin. 2021;42:1311-23
83. Ryu TY, Kim K, Han T-S, Lee M-O, Lee J, Choi J. et al. Human gut-microbiome-derived propionate coordinates proteasomal degradation via HECTD2 upregulation to target EHMT2 in colorectal cancer. ISME J. 2022;16:1205-21
84. Ramesh V, Gollavilli PN, Pinna L, Siddiqui MA, Turtos AM, Napoli F. et al. Propionate reinforces epithelial identity and reduces aggressiveness of lung carcinoma. EMBO Mol Med. 2023;15:e17836
85. Thomas SP, Denu JM. Short-chain fatty acids activate acetyltransferase p300. Elife. 2021;10:e72171
86. Nshanian M, Gruber JJ, Geller BS, Chleilat F, Lancaster SM, White SM. et al. Short-chain fatty acid metabolites propionate and butyrate are unique epigenetic regulatory elements linking diet, metabolism and gene expression. Nat Metab. 2025;7:196-211
87. Gomes AP, Ilter D, Low V, Drapela S, Schild T, Mullarky E. et al. Altered propionate metabolism contributes to tumour progression and aggressiveness. Nat Metab. 2022;4:435-43
88. Shafer M, Low V, Li Z, Blenis J. The emerging role of dysregulated propionate metabolism and methylmalonic acid in metabolic disease, aging, and cancer. Cell Metab. 2025;37:316-29
89. Gomes AP, Ilter D, Low V, Endress JE, Fernández-García J, Rosenzweig A. et al. Age-induced accumulation of methylmalonic acid promotes tumour progression. Nature. 2020;585:283-7
90. Li Z, Low V, Luga V, Sun J, Earlie E, Parang B. et al. Tumor-produced and aging-associated oncometabolite methylmalonic acid promotes cancer-associated fibroblast activation to drive metastatic progression. Nat Commun. 2022;13:6239
91. Mowat C, Dhatt J, Bhatti I, Hamie A, Baker K. Short chain fatty acids prime colorectal cancer cells to activate antitumor immunity. Front Immunol. 2023;14:1190810
92. Dhar P, Wu JD. NKG2D and its ligands in cancer. Curr Opin Immunol. 2018;51:55-61
93. Høgh RI, Møller SH, Jepsen SD, Mellergaard M, Lund A, Pejtersen M. et al. Metabolism of short-chain fatty acid propionate induces surface expression of NKG2D ligands on cancer cells. FASEB J. 2020;34:15531-46
94. Chen L, Zhou X, Wang Y, Wang D, Ke Y, Zeng X. Propionate and butyrate produced by gut microbiota after probiotic supplementation attenuate lung metastasis of melanoma cells in mice. Mol Nutr Food Res. 2021;65:e2100096
95. Dupraz L, Magniez A, Rolhion N, Richard ML, Da Costa G, Touch S. et al. Gut microbiota-derived short-chain fatty acids regulate IL-17 production by mouse and human intestinal γδ T cells. Cell Rep. 2021;36:109332
96. Sanchez HN, Moroney JB, Gan H, Shen T, Im JL, Li T. et al. B cell-intrinsic epigenetic modulation of antibody responses by dietary fiber-derived short-chain fatty acids. Nat Commun. 2020;11:60
97. Arpaia N, Campbell C, Fan X, Dikiy S, van der Veeken J, deRoos P. et al. Metabolites produced by commensal bacteria promote peripheral regulatory T-cell generation. Nature. 2013;504:451-5
98. Smith PM, Howitt MR, Panikov N, Michaud M, Gallini CA, Bohlooly-Y M. et al. The microbial metabolites, short-chain fatty acids, regulate colonic Treg cell homeostasis. Science. 2013;341:569-73
99. Nastasi C, Fredholm S, Willerslev-Olsen A, Hansen M, Bonefeld CM, Geisler C. et al. Butyrate and propionate inhibit antigen-specific CD8+ T cell activation by suppressing IL-12 production by antigen-presenting cells. Sci Rep. 2017;7:14516
100. Mann ER, Lam YK, Uhlig HH. Short-chain fatty acids: linking diet, the microbiome and immunity. Nat Rev Immunol. 2024;24:577-95
101. Louis P, Duncan SH, McCrae SI, Millar J, Jackson MS, Flint HJ. Restricted distribution of the butyrate kinase pathway among butyrate-producing bacteria from the human colon. J Bacteriol. 2004;186:2099-106
102. Duncan SH, Barcenilla A, Stewart CS, Pryde SE, Flint HJ. Acetate utilization and butyryl coenzyme A (CoA):acetate-CoA transferase in butyrate-producing bacteria from the human large intestine. Appl Environ Microbiol. 2002;68:5186-90
103. Louis P, Hold GL, Flint HJ. The gut microbiota, bacterial metabolites and colorectal cancer. Nat Rev Microbiol. 2014;12:661-72
104. Ma Y, Chen H, Li H, Zheng M, Zuo X, Wang W. et al. Intratumor microbiome-derived butyrate promotes lung cancer metastasis. Cell Rep Med. 2024;5:101488
105. Stoeva MK, Garcia-So J, Justice N, Myers J, Tyagi S, Nemchek M. et al. Butyrate-producing human gut symbiont, Clostridium butyricum, and its role in health and disease. Gut Microbes. 2021;13:1-28
106. Feitelson MA, Arzumanyan A, Medhat A, Spector I. Short-chain fatty acids in cancer pathogenesis. Cancer Metastasis Rev. 2023;42:677-98
107. Thangaraju M, Cresci GA, Liu K, Ananth S, Gnanaprakasam JP, Browning DD. et al. GPR109A is a G-protein-coupled receptor for the bacterial fermentation product butyrate and functions as a tumor suppressor in colon. Cancer Res. 2009;69:2826-32
108. Geng H-W, Yin F-Y, Zhang Z-F, Gong X, Yang Y. Butyrate Suppresses Glucose Metabolism of Colorectal Cancer Cells via GPR109a-AKT Signaling Pathway and Enhances Chemotherapy. Front Mol Biosci. 2021;8:634874
109. Chen D, Jin D, Huang S, Wu J, Xu M, Liu T. et al. Clostridium butyricum, a butyrate-producing probiotic, inhibits intestinal tumor development through modulating Wnt signaling and gut microbiota. Cancer Lett. 2020;469:456-67
110. Wang G, Qin S, Chen L, Geng H, Zheng Y, Xia C. et al. Butyrate dictates ferroptosis sensitivity through FFAR2-mTOR signaling. Cell Death Dis. 2023;14:292
111. Shin H, Kim J-H, Lee YS, Lee YC. Change in gene expression profiles of secreted frizzled-related proteins (SFRPs) by sodium butyrate in gastric cancers: induction of promoter demethylation and histone modification causing inhibition of Wnt signaling. Int J Oncol. 2012;40:1533-42
112. Yu DC, Waby JS, Chirakkal H, Staton CA, Corfe BM. Butyrate suppresses expression of neuropilin I in colorectal cell lines through inhibition of Sp1 transactivation. Mol Cancer. 2010;9:276
113. Che Y, Chen G, Guo Q, Duan Y, Feng H, Xia Q. Gut microbial metabolite butyrate improves anticancer therapy by regulating intracellular calcium homeostasis. Hepatology. 2023;78:88-102
114. Li Q, Ding C, Meng T, Lu W, Liu W, Hao H. et al. Butyrate suppresses motility of colorectal cancer cells via deactivating Akt/ERK signaling in histone deacetylase dependent manner. J Pharmacol Sci. 2017;135:148-55
115. Gao S, Chen C, Wang L, Hong L, Wu J, Dong P. et al. Histone deacetylases inhibitor sodium butyrate inhibits JAK2/STAT signaling through upregulation of SOCS1 and SOCS3 mediated by HDAC8 inhibition in myeloproliferative neoplasms. Exp Hematol. 2013;41:261-270.e4
116. Festa Ortega JF, Heidor R, Auriemo AP, Marques Affonso J, Pereira D' Amico T, Herz C. et al. Butyrate-containing structured lipids act on HDAC4, HDAC6, DNA damage and telomerase activity during promotion of experimental hepatocarcinogenesis. Carcinogenesis. 2021;42:1026-36
117. Zhang K, Ji X, Song Z, Song W, Huang Q, Yu T. et al. Butyrate inhibits the mitochondrial complex Ι to mediate mitochondria-dependent apoptosis of cervical cancer cells. BMC Complement Med Ther. 2023;23:212
118. Wang Z, Shu W, Zhao R, Liu Y, Wang H. Sodium butyrate induces ferroptosis in endometrial cancer cells via the RBM3/SLC7A11 axis. Apoptosis. 2023;28:1168-83
119. Bian Z, Sun X, Liu L, Qin Y, Zhang Q, Liu H. et al. Sodium Butyrate Induces CRC Cell Ferroptosis via the CD44/SLC7A11 Pathway and Exhibits a Synergistic Therapeutic Effect with Erastin. Cancers (Basel). 2023;15:423
120. Yang X, Zhang Z, Shen X, Xu J, Weng Y, Wang W. et al. Clostridium butyricum and its metabolite butyrate promote ferroptosis susceptibility in pancreatic ductal adenocarcinoma. Cell Oncol (Dordr). 2023;46:1645-58
121. Hu S, Dong TS, Dalal SR, Wu F, Bissonnette M, Kwon JH. et al. The microbe-derived short chain fatty acid butyrate targets miRNA-dependent p21 gene expression in human colon cancer. PLoS One. 2011;6:e16221
122. Xiao X, Cao Y, Chen H. Profiling and characterization of microRNAs responding to sodium butyrate treatment in A549 cells. J Cell Biochem. 2018;119:3563-73
123. Hu S, Liu L, Chang EB, Wang J-Y, Raufman J-P. Butyrate inhibits pro-proliferative miR-92a by diminishing c-Myc-induced miR-17-92a cluster transcription in human colon cancer cells. Mol Cancer. 2015;14:180
124. Pant K, Yadav AK, Gupta P, Islam R, Saraya A, Venugopal SK. Butyrate induces ROS-mediated apoptosis by modulating miR-22/SIRT-1 pathway in hepatic cancer cells. Redox Biol. 2017;12:340-9
125. Belcheva A, Irrazabal T, Robertson SJ, Streutker C, Maughan H, Rubino S. et al. Gut microbial metabolism drives transformation of MSH2-deficient colon epithelial cells. Cell. 2014;158:288-99
126. Okumura S, Konishi Y, Narukawa M, Sugiura Y, Yoshimoto S, Arai Y. et al. Gut bacteria identified in colorectal cancer patients promote tumourigenesis via butyrate secretion. Nat Commun. 2021;12:5674
127. Donohoe DR, Collins LB, Wali A, Bigler R, Sun W, Bultman SJ. The Warburg effect dictates the mechanism of butyrate-mediated histone acetylation and cell proliferation. Mol Cell. 2012;48:612-26
128. Matsushita M, Fujita K, Hayashi T, Kayama H, Motooka D, Hase H. et al. Gut Microbiota-Derived Short-Chain Fatty Acids Promote Prostate Cancer Growth via IGF1 Signaling. Cancer Res. 2021;81:4014-26
129. Donohoe DR, Garge N, Zhang X, Sun W, O'Connell TM, Bunger MK. et al. The microbiome and butyrate regulate energy metabolism and autophagy in the mammalian colon. Cell Metab. 2011;13:517-26
130. Irrazábal T, Belcheva A, Girardin SE, Martin A, Philpott DJ. The multifaceted role of the intestinal microbiota in colon cancer. Mol Cell. 2014;54:309-20
131. Burgess DJ. Metabolism: Warburg behind the butyrate paradox? Nat Rev Cancer. 2012;12:798
132. Singh V, Yeoh BS, Chassaing B, Xiao X, Saha P, Aguilera Olvera R. et al. Dysregulated Microbial Fermentation of Soluble Fiber Induces Cholestatic Liver Cancer. Cell. 2018;175:679-694.e22
133. Zhang Y, Tao Y, Gu Y, Ma Q. Butyrate facilitates immune clearance of colorectal cancer cells by suppressing STAT1-mediated PD-L1 expression. Clinics (Sao Paulo). 2023;78:100303
134. Yu X, Ou J, Wang L, Li Z, Ren Y, Xie L. et al. Gut microbiota modulate CD8+ T cell immunity in gastric cancer through Butyrate/GPR109A/HOPX. Gut Microbes. 2024;16:2307542
135. Kang X, Liu C, Ding Y, Ni Y, Ji F, Lau HCH. et al. Roseburia intestinalis generated butyrate boosts anti-PD-1 efficacy in colorectal cancer by activating cytotoxic CD8+ T cells. Gut. 2023;72:2112-22
136. He Y, Fu L, Li Y, Wang W, Gong M, Zhang J. et al. Gut microbial metabolites facilitate anticancer therapy efficacy by modulating cytotoxic CD8+ T cell immunity. Cell Metab. 2021;33:988-1000.e7
137. Zhu X, Li K, Liu G, Wu R, Zhang Y, Wang S. et al. Microbial metabolite butyrate promotes anti-PD-1 antitumor efficacy by modulating T cell receptor signaling of cytotoxic CD8 T cell. Gut Microbes. 2023;15:2249143
138. Tian P, Yang W, Guo X, Wang T, Tan S, Sun R. et al. Early life gut microbiota sustains liver-resident natural killer cells maturation via the butyrate-IL-18 axis. Nat Commun. 2023;14:1710
139. Liang L, Liu L, Zhou W, Yang C, Mai G, Li H. et al. Gut microbiota-derived butyrate regulates gut mucus barrier repair by activating the macrophage/WNT/ERK signaling pathway. Clin Sci (Lond). 2022;136:291-307
140. Nastasi C, Candela M, Bonefeld CM, Geisler C, Hansen M, Krejsgaard T. et al. The effect of short-chain fatty acids on human monocyte-derived dendritic cells. Sci Rep. 2015;5:16148
141. Uribe-Herranz M, Rafail S, Beghi S, Gil-de-Gómez L, Verginadis I, Bittinger K. et al. Gut microbiota modulate dendritic cell antigen presentation and radiotherapy-induced antitumor immune response. J Clin Invest. 2020;130:466-79
142. Lee SY, Jhun J, Woo JS, Lee KH, Hwang S-H, Moon J. et al. Gut microbiome-derived butyrate inhibits the immunosuppressive factors PD-L1 and IL-10 in tumor-associated macrophages in gastric cancer. Gut Microbes. 2024;16:2300846
143. Lu H, Xu X, Fu D, Gu Y, Fan R, Yi H. et al. Butyrate-producing Eubacterium rectale suppresses lymphomagenesis by alleviating the TNF-induced TLR4/MyD88/NF-κB axis. Cell Host Microbe. 2022;30:1139-1150.e7
144. Wang H, Chen J, Chen X, Liu Y, Wang J, Meng Q. et al. Cancer-Associated Fibroblasts Expressing Sulfatase 1 Facilitate VEGFA-Dependent Microenvironmental Remodeling to Support Colorectal Cancer. Cancer Res. 2024;84:3371-87
145. Tibbetts AS, Appling DR. Compartmentalization of Mammalian folate-mediated one-carbon metabolism. Annu Rev Nutr. 2010;30:57-81
146. Meiser J, Schuster A, Pietzke M, Vande Voorde J, Athineos D, Oizel K. et al. Increased formate overflow is a hallmark of oxidative cancer. Nat Commun. 2018;9:1368
147. Lee Y, Vousden KH, Hennequart M. Cycling back to folate metabolism in cancer. Nat Cancer. 2024;5:701-15
148. Hennequart M, Pilley SE, Labuschagne CF, Coomes J, Mervant L, Driscoll PC. et al. ALDH1L2 regulation of formate, formyl-methionine, and ROS controls cancer cell migration and metastasis. Cell Rep. 2023;42:112562
149. Ternes D, Tsenkova M, Pozdeev VI, Meyers M, Koncina E, Atatri S. et al. The gut microbial metabolite formate exacerbates colorectal cancer progression. Nat Metab. 2022;4:458-75
150. Delbrouck C, Kiweler N, Chen O, Pozdeev VI, Haase L, Neises L. et al. Formate promotes invasion and metastasis in reliance on lipid metabolism. Cell Rep. 2023;42:113034
151. Oizel K, Tait-Mulder J, Fernandez-de-Cossio-Diaz J, Pietzke M, Brunton H, Lilla S. et al. Formate induces a metabolic switch in nucleotide and energy metabolism. Cell Death Dis. 2020;11:310
152. Newman AC, Falcone M, Huerta Uribe A, Zhang T, Athineos D, Pietzke M. et al. Immune-regulated IDO1-dependent tryptophan metabolism is source of one-carbon units for pancreatic cancer and stellate cells. Mol Cell. 2021;81:2290-2302.e7
153. Lau HC-H, Zhang X, Ji F, Lin Y, Liang W, Li Q. et al. Lactobacillus acidophilus suppresses non-alcoholic fatty liver disease-associated hepatocellular carcinoma through producing valeric acid. EBioMedicine. 2024;100:104952
154. Han R, Nusbaum O, Chen X, Zhu Y. Valeric Acid Suppresses Liver Cancer Development by Acting as a Novel HDAC Inhibitor. Mol Ther Oncolytics. 2020;19:8-18
155. Shi F, Li Y, Han R, Fu A, Wang R, Nusbaum O. et al. Valerian and valeric acid inhibit growth of breast cancer cells possibly by mediating epigenetic modifications. Sci Rep. 2021;11:2519
156. Luu M, Pautz S, Kohl V, Singh R, Romero R, Lucas S. et al. The short-chain fatty acid pentanoate suppresses autoimmunity by modulating the metabolic-epigenetic crosstalk in lymphocytes. Nat Commun. 2019;10:760
157. Russell WR, Gratz SW, Duncan SH, Holtrop G, Ince J, Scobbie L. et al. High-protein, reduced-carbohydrate weight-loss diets promote metabolite profiles likely to be detrimental to colonic health. Am J Clin Nutr. 2011;93:1062-72
158. Waldecker M, Kautenburger T, Daumann H, Busch C, Schrenk D. Inhibition of histone-deacetylase activity by short-chain fatty acids and some polyphenol metabolites formed in the colon. J Nutr Biochem. 2008;19:587-93
159. Murayama M, Hosonuma M, Kuramasu A, Kobayashi S, Sasaki A, Baba Y. et al. Isobutyric acid enhances the anti-tumour effect of anti-PD-1 antibody. Sci Rep. 2024;14:11325
160. Hou H, Chen D, Zhang K, Zhang W, Liu T, Wang S. et al. Gut microbiota-derived short-chain fatty acids and colorectal cancer: Ready for clinical translation? Cancer Lett. 2022;526:225-35
161. Jia R, Shao S, Zhang P, Yuan Y, Rong W, An Z. et al. PRM1201 effectively inhibits colorectal cancer metastasis via shaping gut microbiota and short- chain fatty acids. Phytomedicine. 2024;132:155795
162. Nomura M, Nagatomo R, Doi K, Shimizu J, Baba K, Saito T. et al. Association of Short-Chain Fatty Acids in the Gut Microbiome With Clinical Response to Treatment With Nivolumab or Pembrolizumab in Patients With Solid Cancer Tumors. JAMA Netw Open. 2020;3:e202895
163. Shao X, Liu L, Zhou Y, Zhong K, Gu J, Hu T. et al. High-fat diet promotes colitis-associated tumorigenesis by altering gut microbial butyrate metabolism. Int J Biol Sci. 2023;19:5004-19
164. Shashni B, Nagasaki Y. Short-chain fatty acid-releasing nano-prodrugs for attenuating growth and metastasis of melanoma. Acta Biomater. 2023;159:226-36
165. Yan S, Chang J, Hao X, Liu J, Tan X, Geng Z. et al. Berberine regulates short-chain fatty acid metabolism and alleviates the colitis-associated colorectal tumorigenesis through remodeling intestinal flora. Phytomedicine. 2022;102:154217
166. Wu H, Van Der Pol WJ, Dubois LG, Morrow CD, Tollefsbol TO. Dietary Supplementation of Inulin Contributes to the Prevention of Estrogen Receptor-Negative Mammary Cancer by Alteration of Gut Microbial Communities and Epigenetic Regulations. Int J Mol Sci. 2023;24:9015
167. Li N, Niu L, Liu Y, Wang Y, Su X, Xu C. et al. Taking SCFAs produced by Lactobacillus reuteri orally reshapes gut microbiota and elicits antitumor responses. J Nanobiotechnology. 2024;22:241
168. Alvandi E, Wong WKM, Joglekar MV, Spring KJ, Hardikar AA. Short-chain fatty acid concentrations in the incidence and risk-stratification of colorectal cancer: a systematic review and meta-analysis. BMC Med. 2022;20:323
169. Watling CZ, Kelly RK, Murphy N, Gunter M, Piernas C, Bradbury KE. et al. Prospective Analysis Reveals Associations between Carbohydrate Intakes, Genetic Predictors of Short-Chain Fatty Acid Synthesis, and Colorectal Cancer Risk. Cancer Res. 2023;83:2066-76
170. Zheng D-W, Li R-Q, An J-X, Xie T-Q, Han Z-Y, Xu R. et al. Prebiotics-Encapsulated Probiotic Spores Regulate Gut Microbiota and Suppress Colon Cancer. Adv Mater. 2020;32:e2004529
171. Matsushita M, Fujita K, Hayashi T, Kayama H, Motooka D, Hase H. et al. Gut Microbiota-Derived Short-Chain Fatty Acids Promote Prostate Cancer Growth via IGF1 Signaling. Cancer Research. 2021;81:4014-26
172. Qu R, Zhang Y, Ma Y, Zhou X, Sun L, Jiang C. et al. Role of the Gut Microbiota and Its Metabolites in Tumorigenesis or Development of Colorectal Cancer. Adv Sci (Weinh). 2023;10:e2205563
173. Wang T, Cai G, Qiu Y, Fei N, Zhang M, Pang X. et al. Structural segregation of gut microbiota between colorectal cancer patients and healthy volunteers. ISME J. 2012;6:320-9
174. Green AC, Marttila P, Kiweler N, Chalkiadaki C, Wiita E, Cookson V. et al. Formate overflow drives toxic folate trapping in MTHFD1 inhibited cancer cells. Nat Metab. 2023;5:642-59
175. Wilke AC, Doebele C, Zindel A, Lee KS, Rieke SA, Ceribelli M. et al. SHMT2 inhibition disrupts the TCF3 transcriptional survival program in Burkitt lymphoma. Blood. 2022;139:538-53
176. Miller KD, Pniewski K, Perry CE, Papp SB, Shaffer JD, Velasco-Silva JN. et al. Targeting ACSS2 with a Transition-State Mimetic Inhibits Triple-Negative Breast Cancer Growth. Cancer Res. 2021;81:1252-64
177. Murga-Garrido SM, Hong Q, Cross T-WL, Hutchison ER, Han J, Thomas SP. et al. Gut microbiome variation modulates the effects of dietary fiber on host metabolism. Microbiome. 2021;9:117
178. Doublier S, Cirrincione S, Scardaci R, Botta C, Lamberti C, Giuseppe FD. et al. Putative probiotics decrease cell viability and enhance chemotherapy effectiveness in human cancer cells: role of butyrate and secreted proteins. Microbiol Res. 2022;260:127012
179. Dong J, Wang B, Xiao Y, Liu J, Wang Q, Xiao H. et al. Roseburia intestinalis sensitizes colorectal cancer to radiotherapy through the butyrate/OR51E1/RALB axis. Cell Rep. 2024;43:113846
180. He Y, Ling Y, Zhang Z, Mertens RT, Cao Q, Xu X. et al. Butyrate reverses ferroptosis resistance in colorectal cancer by inducing c-Fos-dependent xCT suppression. Redox Biol. 2023;65:102822
181. Chen L, Zhao R, Kang Z, Cao Z, Liu N, Shen J. et al. Delivery of short chain fatty acid butyrate to overcome Fusobacterium nucleatum-induced chemoresistance. J Control Release. 2023;363:43-56
182. De Las Rivas J, Brozovic A, Izraely S, Casas-Pais A, Witz IP, Figueroa A. Cancer drug resistance induced by EMT: novel therapeutic strategies. Arch Toxicol. 2021;95:2279-97
183. Panebianco C, Villani A, Pisati F, Orsenigo F, Ulaszewska M, Latiano TP. et al. Butyrate, a postbiotic of intestinal bacteria, affects pancreatic cancer and gemcitabine response in in vitro and in vivo models. Biomed Pharmacother. 2022;151:113163
184. Cross C, Davies M, Bateman E, Crame E, Joyce P, Wignall A. et al. Fibre-rich diet attenuates chemotherapy-related neuroinflammation in mice. Brain Behav Immun. 2024;115:13-25
185. Xiao C, Fedirko V, Claussen H, Richard Johnston H, Peng G, Paul S. et al. Circulating short chain fatty acids and fatigue in patients with head and neck cancer: A longitudinal prospective study. Brain Behav Immun. 2023;113:432-43
186. Huang F, Li S, Chen W, Han Y, Yao Y, Yang L. et al. Postoperative Probiotics Administration Attenuates Gastrointestinal Complications and Gut Microbiota Dysbiosis Caused by Chemotherapy in Colorectal Cancer Patients. Nutrients. 2023;15:356
187. Rowe JH, Elia I, Shahid O, Gaudiano EF, Sifnugel NE, Johnson S. et al. Formate Supplementation Enhances Antitumor CD8+ T-cell Fitness and Efficacy of PD-1 Blockade. Cancer Discov. 2023;13:2566-83
188. Liu X, Lu B, Tang H, Jia X, Zhou Q, Zeng Y. et al. Gut microbiome metabolites, molecular mimicry, and species-level variation drive long-term efficacy and adverse event outcomes in lung cancer survivors. EBioMedicine. 2024;109:105427
189. Coutzac C, Jouniaux J-M, Paci A, Schmidt J, Mallardo D, Seck A. et al. Systemic short chain fatty acids limit antitumor effect of CTLA-4 blockade in hosts with cancer. Nat Commun. 2020;11:2168
190. Rangan P, Mondino A. Microbial short-chain fatty acids: a strategy to tune adoptive T cell therapy. J Immunother Cancer. 2022;10:e004147
191. Prasad R, Rehman A, Rehman L, Darbaniyan F, Blumenberg V, Schubert M-L. et al. Antibiotic-induced loss of gut microbiome metabolic output correlates with clinical responses to CAR T-cell therapy. Blood. 2025;145:823-39
192. Wegner VD, Feile A, Alb M, Hudecek M, Hewitt P, Mosig AS. Short-Chain Fatty Acids Modulate Anti-ROR1 CAR T-Cell Function and Exhaustion in an Intestinal Adenocarcinoma-on-Chip Model. Adv Healthc Mater. 2025;14:e2405003
193. de Thé H. Differentiation therapy revisited. Nat Rev Cancer. 2018;18:117-27
194. Jin W, Dai Y, Chen L, Zhu H, Dong F, Zhu H. et al. Cellular hierarchy insights reveal leukemic stem-like cells and early death risk in acute promyelocytic leukemia. Nat Commun. 2024;15:1423
195. Leszczyniecka M, Roberts T, Dent P, Grant S, Fisher PB. Differentiation therapy of human cancer: basic science and clinical applications. Pharmacol Ther. 2001;90:105-56
196. Li Y, Gruber JJ, Litzenburger UM, Zhou Y, Miao YR, LaGory EL. et al. Acetate supplementation restores chromatin accessibility and promotes tumor cell differentiation under hypoxia. Cell Death Dis. 2020;11:102
197. Gao D, Shen Y, Xu L, Sun Y, Hu H, Xu B. et al. Acetate utilization promotes hormone therapy resistance in prostate cancer through neuroendocrine differentiation. Drug Resist Updat. 2024;77:101158
198. Yu Y, Shen X, Xiao X, Li L, Huang Y. Butyrate Modification Promotes Intestinal Absorption and Hepatic Cancer Cells Targeting of Ferroptosis Inducer Loaded Nanoparticle for Enhanced Hepatocellular Carcinoma Therapy. Small. 2023;19:e2301149
199. Fontana F, Ge X, Su X, Hathi D, Xiang J, Cenci S. et al. Evaluating Acetate Metabolism for Imaging and Targeting in Multiple Myeloma. Clin Cancer Res. 2017;23:416-29
200. Spick C, Herrmann K, Czernin J. Evaluation of Prostate Cancer with 11C-Acetate PET/CT. J Nucl Med. 2016;57:30S-37S
201. Dong Y, Zhang K, Wei J, Ding Y, Wang X, Hou H. et al. Gut microbiota-derived short-chain fatty acids regulate gastrointestinal tumor immunity: a novel therapeutic strategy? Front Immunol. 2023;14:1158200
202. Shrode RL, Knobbe JE, Cady N, Yadav M, Hoang J, Cherwin C. et al. Breast cancer patients from the Midwest region of the United States have reduced levels of short-chain fatty acid-producing gut bacteria. Sci Rep. 2023;13:526
203. Rodríguez-García A, Arroyo A, García-Vicente R, Morales ML, Gómez-Gordo R, Justo P. et al. Short-Chain Fatty Acid Production by Gut Microbiota Predicts Treatment Response in Multiple Myeloma. Clin Cancer Res. 2024;30:904-17
204. Kim YL, Lee W, Chung SH, Yu BM, Lee YC, Hong J. Metabolic alterations of short-chain fatty acids and TCA cycle intermediates in human plasma from patients with gastric cancer. Life Sci. 2022;309:121010
205. Ubachs J, Ziemons J, Soons Z, Aarnoutse R, van Dijk DPJ, Penders J. et al. Gut microbiota and short-chain fatty acid alterations in cachectic cancer patients. J Cachexia Sarcopenia Muscle. 2021;12:2007-21
206. Sánchez-Alcoholado L, Laborda-Illanes A, Otero A, Ordóñez R, González-González A, Plaza-Andrades I. et al. Relationships of Gut Microbiota Composition, Short-Chain Fatty Acids and Polyamines with the Pathological Response to Neoadjuvant Radiochemotherapy in Colorectal Cancer Patients. Int J Mol Sci. 2021;22:9549
207. Ziemons J, Aarnoutse R, Heuft A, Hillege L, Waelen J, de Vos-Geelen J. et al. Fecal levels of SCFA and BCFA during capecitabine in patients with metastatic or unresectable colorectal cancer. Clin Exp Med. 2023;23:3919-33
Author contact
![]() Corresponding author: Kefei Yuan, Department of Liver Surgery, State Key Laboratory of Biotherapy and Cancer Center, West China Hospital, Sichuan University and Collaborative Innovation Center of Biotherapy, Chengdu, 610041, China. E-mail address: ykf13com; Tel.: 17340135791.
Corresponding author: Kefei Yuan, Department of Liver Surgery, State Key Laboratory of Biotherapy and Cancer Center, West China Hospital, Sichuan University and Collaborative Innovation Center of Biotherapy, Chengdu, 610041, China. E-mail address: ykf13com; Tel.: 17340135791.