Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
1. Copper
2. Iron
3. Zinc
4. Magnesium
5. Other essential metal elements
6. The overlooked orchestrators:...
7. Conclusion and perspectives
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2026; 16(3):1350-1373. doi:10.7150/thno.121988 This issue Cite
Review
Metal homeostasis as a therapeutic lever: advancing metalloimmunology to remodel the tumor microenvironment and enhance cancer immunotherapy
Xin Chen1, Qi Wang1, Hongxia Zhang1, Ye Wang1,2 ![]() , Haiying Zhu1
, Haiying Zhu1 ![]()
1. Department of Cell Biology, Naval Medical University (Second Military Medical University), Shanghai 200433, China.
2. Department of Urology, Chinese PLA General Hospital, Beijing 100853, China.
Received 2025-7-19; Accepted 2025-10-11; Published 2026-1-1
Abstract

Targeting the dysregulation of essential metal homeostasis represents a rapidly evolving frontier in cancer immunotherapy. The tumor microenvironment (TME) is a complex immunosuppressive ecosystem comprising tumor cells, immune cells, stromal components, extracellular matrix, and diverse cytokines/chemokines, characterized by hypoxia, acidosis, elevated redox stress, and metabolic dysregulation that drive tumor progression and immunotherapy resistance. Crucially, dysregulated homeostasis of essential metals (e.g., Cu, Fe, Zn, Mg, Mn, Ca, Cr, Na, K) pervades the TME, directly promoting tumorigenesis through oncogenic pathway activation and aberrant energy metabolism while facilitating immune evasion, amplifying immunosuppression, and undermining cancer immunotherapies. In response, recent strategies have focused on leveraging metalloimmunology to reprogram the TME via: (1) activation of innate/adaptive immunity, (2) disruption of tumor metabolism, (3) induction of programmed cell death, and (4) triggering of immunogenic cell death (ICD). These approaches synergize with existing immunotherapies to enhance efficacy, aided by nanotechnology-enabled precision delivery of metal-based agents. In conclusion, by mastering the intricate interplay between metal ions and the immunosuppressive TME, these strategies hold immense potential to remodel the TME, reinvigorate anti-tumor immunity, and ultimately enhance the efficacy of next-generation cancer immunotherapies. This review presents metalloimmunology as an integrative paradigm connecting metal biology, tumor immunology, and nanotechnology, providing a transformative outlook for immunotherapy.
Keywords: metal homeostasis, tumor microenvironment, nanomedicine, metalloimmunotherapy
Introduction
Cancer immunotherapy, as the fourth major cancer treatment modality following surgery, radiotherapy, and chemotherapy, achieves targeted tumor killing by activating or rebuilding the patient's own immune system, thus effectively overcoming the inherent limitations of traditional therapies such as non-selective killing and severe adverse reactions. Clinical evidence highlights the transformative impact of three principal immunotherapeutic approaches—immune checkpoint blockade (ICB), cellular therapies such as chimeric antigen receptor T-cell (CAR-T) therapy, and tumor vaccines—in managing aggressive malignancies. These immunotherapies have shown remarkable efficacy against diverse cancers, due to their unique strengths in precision targeting and broad clinical applicability [1, 2]. Nevertheless, current data reveals that over 70% of patients still do not benefit from ICB therapy [3], primarily due to immune tolerance, immunosuppressive TME, and off-target toxicities. Among these factors, the immunosuppressive TME stands as a critical barrier to immunotherapy efficacy. Consequently, strategies to remodel the TME and enhance the functionality of effector immune cells have emerged as pivotal research directions for improving the clinical outcomes of existing therapies.
The TME consists of tumor cells, surrounding immune cells, cancer-associated fibroblasts (CAFs), extracellular matrix, microvasculature, and diverse cytokines and chemokines. It constitutes a complex system characterized by hypoxia, low pH, elevated glutathione (GSH), reactive oxygen species (ROS) levels and adenosine triphosphate (ATP), and dysregulated enzyme expression. Recent studies have revealed that metal elements are also critical components of the TME, and that disruptions in metal homeostasis are one of its defining features.
The biological effects of metals have been recognized for millennia. Copper compounds were used for disinfection in ancient Egypt, and metal-based cancer treatments were recorded in the Compendium of Materia Medica. These empirical uses predate the discovery of underlying molecular mechanisms. In the 1960s, scientists discovered that cisplatin exerts antitumor effects by forming crosslinks with DNA guanine bases to block tumor cell replication, marking the advent of the "metal-based chemotherapy" era [4]. Since then, metals have not only been utilized for anti-inflammatory and antibacterial purposes but also as drug agents for targeted delivery or energy-converting components in nanoparticle systems for cancer treatment [5, 6]. Current research reveals that metal ions, as essential components of life, play critical roles in enzyme catalysis, electron transfer, signal transduction, and redox homeostasis. Consequently, dysregulation of essential metal can lead to functional abnormalities in tissues and organs [7]. The immune system is particularly sensitive to such dysregulation. The immunomodulatory functions of metal ions include but are not limited to the following: (1) Acting as second messengers or enzyme cofactors to initiate immune cell activation, (2) Influencing immune cell differentiation and polarization, and (3) Regulating immune cell metabolism and energy supply [8]. Thus, metal ions serve as pivotal "molecular switches" and "metabolic hubs" within the functional networks of immune cells.
Given these findings, the correlation between metal homeostasis in the TME and tumorigenesis, as well as its therapeutic applications, has attracted growing interest. Studies reveal that metals and their biomaterial derivatives in the TME can remodel the immunosuppressive TME through multiple mechanisms: inhibiting the activation of tumor-associated immunosuppressive cells, promoting the activation of tumor-suppressive immune cells [9], targeting tumor stroma and angiogenesis [10], and reversing tissue hypoxia and acidosis [11].
Recent advances in the integration of nanotechnology and metalloimmunology have rapidly expanded the development of metal-based nanomaterials. Metal-based nanomaterials are considered ideal therapeutic carriers due to their unique physical properties, enabling accurate tumor targeting and accumulation as well as stimuli-responsive drug release. Several current studies reveal that combining metal-based nanomaterials with immunotherapies can break through immunosuppressive barriers of the TME, establish long-lasting anti-tumor immune memory, and synergistically enhance therapeutic efficacy [12, 13].
In this review, we present a transformative perspective by establishing metalloimmunology as an integrative paradigm that bridges metal biology, tumor immunology, and therapeutic nanotechnology. Moving beyond a mere summary of individual metal effects, we construct a comprehensive framework illustrating how dysregulated homeostasis of essential metals such as copper, iron, zinc, and magnesium orchestrates immunosuppression within the TME. Importantly, we systematically explore nano-enabled therapeutic strategies—including engineered ion chelators, ionophores, and multimodal nanoplatforms—that enable precise spatiotemporal modulation of metal biology to remodel the TME and overcome resistance to immunotherapy. We further highlight cutting-edge applications where nanomaterials are designed not only for targeted metal delivery but also to synergistically induce ICD and amplify the efficacy of ICB, CAR-T cell therapy, and vaccines. By incorporating emerging insights into metalloproteins and metal crosstalk and extending the discussion to less-explored metals such as manganese and calcium, this review charts a forward-looking roadmap for precision metalloimmunotherapy, thereby defining a novel and rapidly evolving frontier in cancer treatment.
1. Copper
Clinical evidence strongly links dysregulated copper metabolism to malignant progression. Elevated copper and ceruloplasmin levels in serum and tumors, which are observed in hepatocellular carcinoma (HCC) [14], breast [15], lung [16], and hematological malignancies [17], are consistently associated with poor prognosis [18].
1.1 Mechanisms of copper homeostasis dysregulation driving immunosuppressive TME
Mechanistically, copper exerts paradoxical tumor-modulating effects. On the one hand, it promotes oncogenesis by enhancing mitochondrial metabolism, MAPK/ERK pathway activation, angiogenesis, immune evasion, and context-dependent cuproptosis induction. On the other hand, elevated copper suppresses tumors by triggering ICD and oxidative stress [19]. Notably, high cuproptosis signatures are closely linked immunosuppressive TME landscapes formation.
1.1.1 Copper homeostasis dysregulation drives tumor immune escape
Dysregulated copper homeostasis in the TME arises from concurrent dysregulation of metabolic fluxes, copper-binding protein dysfunction, and defective transporter machinery. Copper imbalance within the TME facilitates tumor immune escape primarily by modulating immune checkpoint networks, thereby weakening immunotherapeutic efficacy (Figure 1). Mechanistically, elevated TME copper levels upregulate programmed death-ligand 1 (PD-L1) expression on cancer cells via NF-κB, JAK/STAT, and PI3K/Akt/mTOR pathways [20]. PD-L1 binding to PD-1 on T cells directly suppresses their antitumor function. Furthermore, HCC cells utilize copper-dependent lysyl oxidase-like 4 (LOXL4) to polarize Kupffer cells toward an immunosuppressive phenotype and upregulate PD-L1, synergistically inhibiting CD8⁺ T cells [21]. Besides, the copper transporter SLC31A1 is overexpressed in multiple cancers and correlates with poor prognosis [22]. It shows strong positive correlation with PD-L1/CTLA-4 expression [23], demonstrating that copper accumulation actively shapes an immune-evasive microenvironment. Together, these findings indicate that copper homeostasis dysregulation critically imbalances immune checkpoint networks, making it a promising target for enhancing immunotherapy.
The representative mechanisms of four essential metal homeostasis imbalance within the TME driving the immunosuppressive microenvironment. Created in https://BioRender.com.
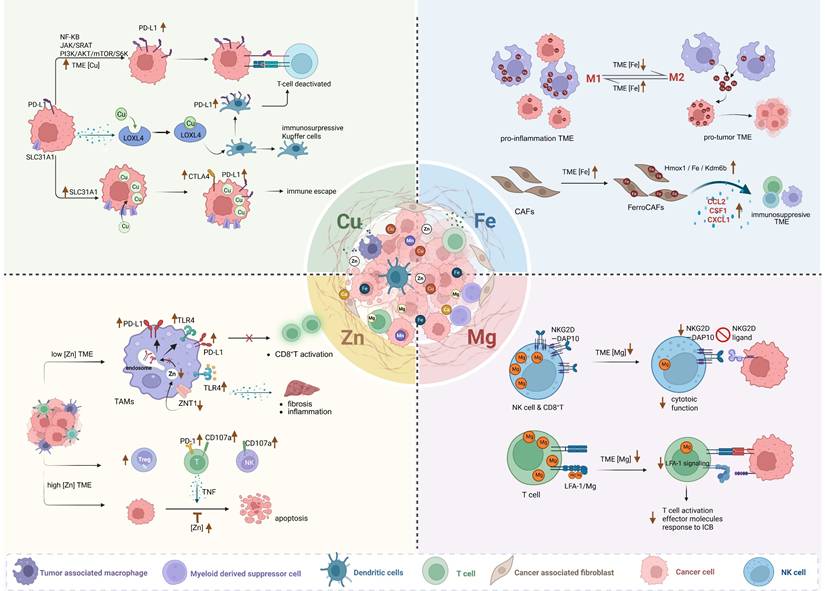
From the perspective of cellular interactions within the TME, the flow of copper between tumor cells and immune cells may alter the anti-tumor function of immune cells. For example, neuroblastoma cells drive tumor progression by excessively taking up copper. This sequestering behavior leads to depletion of copper in the TME, thereby suppressing the function of immune cells, particularly neutrophils [24].
Copper further drives vascular-immune crosstalk in the TME. Neovascular endothelial tip cells establish an immunosuppressive niche by inducing T cell exhaustion [25], while copper directly potentiates pro-angiogenic factors (e.g., vascular endothelial growth factor (VEGF), FGF) through enhanced ligand-receptor binding [26]. Mechanistically, the copper chaperone antioxidant 1 copper chaperone (ATOX1) activates PDGF signaling to drive smooth muscle migration and neointimal remodeling [27], and copper-mediated hypoxia-inducible factor-1α (HIF-1α) stabilization [28, 29] along with NF-κB activation upregulates VEGF expression [30]. This coordination of vascular induction and immune suppression reveals novel therapeutic vulnerabilities in the copper-modulated TME.
1.1.2 High cuproptosis signatures are closely associated with the formation of the immunosuppressive TME
Although copper accumulation triggers cuproptosis [31], tumor cells in hypoxic and copper-rich TME develop resistance to cuproptosis through copper binding to HIF-1α, which inhibits ubiquitin-mediated degradation of HIF-1α and amplifies hypoxia. This upregulating metallothionein 2A (MT2A) and pyruvate dehydrogenase kinase 1/3 (PDK1/3) while suppressing dihydrolipoamide S-acetyltransferase (DLAT) expression. Consequently, MT2A sequesters mitochondrial copper, conferring cuproptosis resistance and enabling tumor survival [32].
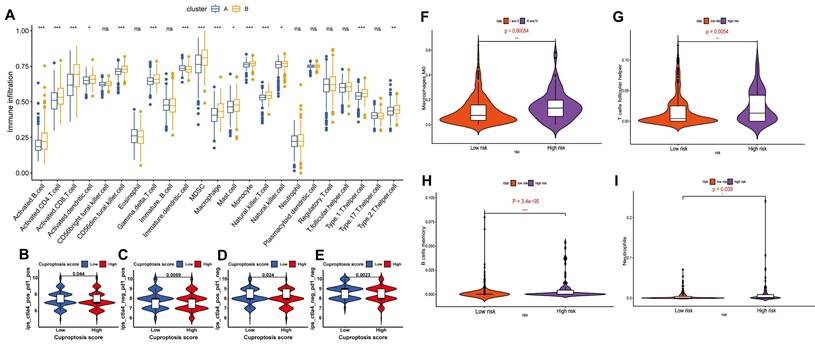
In addition to tumor cells evolving resistance to cuproptosis, high cuproptosis signatures in the TME are closely linked to the immunosuppressive TME as evidenced by three key observations: First, elevated cuproptosis-related genes (e.g., SLC31A1, FDX1) positively correlate with immunosuppressive cytokines, cells and immune checkpoint molecules while predicting poor prognosis. Second, in gliomas, elevated SLC31A1 expression is associated with M2 macrophage enrichment and poor prognosis but inversely links to anti-tumor immune cells (plasmacytoid dendritic cells (DCs), CD56ᵇʳiᵍʰᵗ NK cells, CD8⁺ T cells) [23, 33]. Third, multi-omics models confirm that high cuproptosis risk signatures strongly predict immunosuppressive TME profiles and ICB therapy failure [34-37] (Figure 2A-E). These findings establish cuproptosis-related genes as biomarkers for predicting ICB efficacy and guiding personalized immunotherapy. Given these understandings, patients with tumors characterized by high cuproptosis signatures and immunosuppressive TME—which typically respond poorly to immunotherapy—may benefit from therapeutic strategies that induce cuproptosis to inhibit tumor progression.
Cuproptosis and ferroptosis status affects the tumor immune microenvironment and the efficacy of immunotherapy. A) Tumor immune microenvironment analysis of two subtypes where cluster A has a better prognosis and shows higher expression of seven cuproptosis-promoting genes (FDX1, LIAS, LIPT1, DLD, DLAT, PDHA1 and PDHB) than cluster B (p <0.001). B-E) Analysis of the efficacy of 4 ICBs based on Cuproptosis score grouping predicted by IPS scores. ctla4_pos_pd1_pos (B), ctla4_neg_pd1_pos (C), ctla4_pos_pd1_neg (D) and ctla4_neg_pd1_neg (E); IPS, immune cell proportion score; pos, positive; neg, negative. Adapted from ref.[36]. Copyright 2022 Zhang, Chen, Fang, Tai, Chen and Cao. Licensed under CC BY 4.0. F-I) The landscape of immune infiltration in patients with HCC in the high-risk and low-risk ferroptosis groups. Adapted from ref.[70]. Licensed under CC BY 4.0. Copyright 2020 The Author(s).
1.2 Strategies targeting copper homeostasis dysregulation for cancer treatment
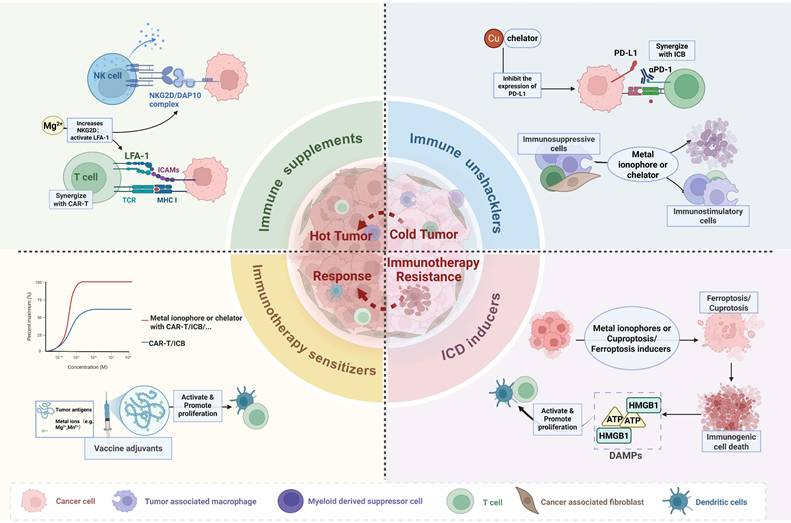
Current anti-tumor strategies targeting copper homeostasis primarily focus on two directions: (1) Copper chelators (e.g., D-penicillamine, ammonium tetrathiomolybdate (ATTM), Trientine) deplete copper in the TME, inhibiting copper-driven proliferation; (2) Copper ionophores (e.g., elesclomol, disulfiram (DSF)) elevate TME copper levels, inducing anti-tumor effects via ICD, cuproptosis and ROS burst (Table 1, Table S1). Both strategies remodel the TME and show potential for synergy with immunotherapy. Nanotechnology further enhances copper-based therapeutics by enabling targeted delivery, TME-responsive release, and photothermal capabilities, creating a multimodal synergistic effect (Figure 3).
Immunomodulatory effects of cancer treatments targeting copper dysregulation in the TME
| Practical application | Functions on immunity in TME | Correlation with immunotherapy |
|---|---|---|
| Copper chelators | 1. Inhibit PD-L1 expression in TME to suppress immune escape | Act like and synergize with ICB |
| 2. Modulate immune cell activity | Promote conversion to an immune-activated TME; enhance immunotherapy | |
| 3. Elevate pro-inflammatory cytokine levels in TME | TME-specific targeting (statistically significant) | |
| 4. Improve TME vascularization and extracellular matrix. | Facilitate immune cell infiltration and enhance immunotherapy response | |
| Copper ionophores | 1. Activate immune cells | Facilitate immune cell infiltration and enhance immunotherapy response |
| 2. Induce ICD via cuproptosis | Improve efficacy of TLR7 agonists and CD47 blockers | |
| 3. Enhance sensitivity and efficiency of immunotherapies | Enhance αPD-L1 blockers and CAR-T efficacy |
Strategies targeting metal homeostasis imbalance for cancer treatment. Therapeutic strategies targeting metal homeostasis imbalance in TME have significantly enhanced the efficacy and effectiveness of immunotherapy by converting immunologically "cold tumors" into "hot tumors", thereby transforming immunotherapy-resistant tumor microenvironments into immunotherapy-responsive one. Created in https://BioRender.com.
1.2.1 Copper chelators deplete TME copper to inhibit proliferation and remodel immunity
Copper chelators curb tumor progression by sustaining tumor dormancy, suppressing compensatory glycolysis in tumor cells, and synergizing with radio- and chemotherapy [38]. More importantly, they remodel the TME to enhance immunotherapeutic synergy. This involves downregulating immune checkpoints, activating antitumor immunity, and inhibiting pro-tumor immune cell infiltration and angiogenesis.
A key mechanism of copper chelators is the inhibition of PD-L1 expression and blockade of immune escape. Current research has revealed at least three mechanisms: (1) Downregulation of JAK/STAT signaling, preventing interferon-γ (IFN-γ)-induced PD-L1 upregulation [20]; (2) Suppression of EGFR signaling, promoting PD-L1 ubiquitination and degradation, thereby enhancing CD8+ T and natural killer (NK) cell infiltration and cytotoxicity [20]; (3) Targeting phosphatidylserine (PS) on cancer cells, suppressing PD-L1 and strengthening T cell-mediated immunity [39].
Secondly, copper chelators reshape the TME into an immunologically activated state through coordinated immunomodulatory effects. For example, copper chelators (e.g., TEPA) can redirect copper ions from "copper-rich" neuroblastoma cells to "copper-poor" neutrophils. Simultaneously, copper chelators reduce TGF-β levels, upregulate KC/CXCL1, increase infiltration of neutrophils and other immune cells, and polarize neutrophils towards the pro-inflammatory N1 phenotype. This enhances their migration and ADCC function, reshapes the immunosuppressive TME, and consequently improves the efficacy of anti-GD2 antibody therapy [24]. Moreover, copper chelators reduce infiltration of myeloid-derived suppressor cells (MDSCs) via Fas-FasL-mediated apoptosis [40, 41] while increasing infiltration of NK cells and CD4⁺ T cells [40, 42], redirecting low-reactive CD4⁺ T cells toward Th1-type differentiation [41] and amplifying T cell secretion of IFN-γ [40]. These observations provide a theoretical foundation for the clinical translation of copper chelators.
Collectively, these multidimensional changes convert immunosuppressive niches into immunologically active microenvironments. Notably, cytokine profiling reveals compartmentalized specificity: quantitative alterations of most cytokine induced by copper chelators (e.g., IFN-γ elevation, TGF-β reduction, N1-associated factors) reach statistical significance exclusively within the TME, indicating a precisely localized immunoregulatory mechanism rather than systemic effects [24].
Nanoparticle-based copper chelators, like LDNP@mSiO₂-DPA@FA-PEG, leverage nanoscale engineering for dual therapy. Their core-shell-shell lanthanide structure (NaYF4:Yb,Er@NaGdF₄@NaNdF₄) provides orthogonal functions: the NaNdF₄ outer shell for efficient 808 nm photothermal conversion and the NaYF4:Yb,Er core for NIR-IIb fluorescence under 980 nm excitation, enabling real-time tracking and optimal photothermal therapy (PTT) timing. The resulting photothermal ablation directly kills tumors and induces ICD. Crucially, the design synchronizes therapies: PTT-triggered ICD exposes antigens while locally released DPA depletes copper to downregulate PD-L1 and inhibit angiogenesis. This engineered synergy amplifies antitumor immunity and potently inhibits metastasis, as shown preclinically [43] (Table 5).
Synergistic anti-tumor effects of metal-based nanomaterials on immunotherapy
| Metal-based nanomaterials | Functions on immunity in TME | Correlation with immunotherapy | Extra functions of nanomaterials |
|---|---|---|---|
| Nano-copper chelators | |||
| LMDFP [43] | Inhibit immune escape by suppressing PD-L1 in TME; inhibit tumor angiogenesis | Enhance immunogenicity and immunotherapy response | PTT induces tumor ablation and ICD |
| RPTDH/R848 [133] | Inhibit tumor angiogenesis; activate/mature CAL-1 | / | pH-responsive; TLR7/8 agonist (R484) delivery |
| Nano-cuproptosis inducers | |||
| NP@ESCu [51] | Induce cuproptosis in cancer cells; reverse the immunosuppressive TME; upregulate PD-L1 expression in TME | Improve αPD-L1 treatment response rate | ROS-responsive release |
| CAT-ecSNA-Cu [134] | Reduce hypoxia in TME; enhance the sensitivity to cuproptosis; upregulate PD-L1; induce ICD via cuproptosis; activate immune response | Improve αPD-L1 treatment response rate | Loaded with catalase and immune activator |
| Nano-iron-based immunomodulators | |||
| Ferumoxytol [135] | Reverse the M2 polarization | / | / |
| Starch-coated iron oxide nanoparticles (IONPs) [136] ION [80] | Upregulate the TLR3-TRIF-IRF3 pathway and stimulate T cells | / | / |
| Promote macrophages to secrete ATP and HMGB1; enhance DCs and CTLs infiltration and activation | Enhance immunogenicity and immunotherapy response | / | |
| Nano-ferroptosis inducers | |||
| Fe3O4-SAS@PLT [78] | Reverse M2 polarization; induce ferroptosis and generate mild immunogenicity | Enhance immunotherapy response | Load SAS; PLT membrane enhances circulation time and tumor targeting; pH-responsive release Redox environment-responsive; SDT based on semiconductor polymers |
| SINM [137] | Induce ferroptosis, ICD, and polarization of M1 macrophages | Enhance immunogenicity and immunotherapy response | RSL3-loaded; PD-1 coating enhances targeting; pH-responsive release |
| PD-1@RSL3 NPs [138] | Promote ferroptosis in tumor cells; inhibit PD-1/PD-L1; enhance CD8+ T cells infiltration and DCs maturation | Improve αPD-L1 treatment response rate | |
| Zinc-based nanomaterials | |||
| DSF@Zn-DMSNs [96] | Enhance autophagy; trigger ICD; facilitate CTLs infiltration | Enhance immunogenicity and immunotherapy response | pH-responsive release |
| Magnesium-based nanomaterials | |||
| Mg-CaCO₃ [104] | Reduce ROS in CAFs reshapes immunosuppressive phenotypes to activated CD4⁺ T cells | / | pH-responsive release; hydrogen therapy directly kills tumor cells |
| R837-Pep@HM NPs [105] | Activate dendritic/T cells; enhance antigen presentation; trigger effector T proliferation | Enhance immunotherapy response | pH-responsive release; loaded with TLR7/8 agonist and melanoma neoantigen peptide |
| Manganese-based nanomaterials | |||
| MBMA-RGD in situ vaccine [113] | Induce ICD; activate STING; recruit and stimulate DCs; generate oxygen | Enhance immunogenicity and immunotherapy response | Actively targets tumor by binding integrin receptors; TME-responsive; trigger PDT and PTT |
| TMA NMs [114] | Activate STING pathway; induce ICD | Enhance immunogenicity and immunotherapy response | pH/GSH-responsive release; loaded with STING agonist |
| MnO₂ NPs [13] | Promote IFN-β secretion; repolarize neutrophils from N2 to N1; enhance CD8⁺ T cell infiltration and activation | Synergize with bacterial immunotherapies | / |
1.2.2 Copper ionophores elevate copper ion levels to remodel immunity, induce ICD and trigger cuproptosis
Copper ionophores combat tumors by elevating intracellular copper levels, employing multifaceted mechanisms including ROS generation, DNA damage, suppression of tumor-associated enzymes and proteasome activity, and targeting of cancer stem cells [38]. They concurrently enhance immunotherapy sensitivity through immune activation, ICD and cuproptosis triggering, and immune checkpoint molecules regulation.
In terms of mechanisms, Solier S. et al. revealed that chemically reactive copper(II) pools in mitochondria maintain NAD+ levels by promoting the NAD(H) redox cycle, which metabolically and epigenetically reprogramming macrophages toward the M1/pro-inflammatory state [44]. This suggests that copper ionophores targeting mitochondrial copper(II) pools may facilitate the conversion of M2 macrophages to the M1 phenotype, thereby reshaping the immunosuppressive TME. Besides, Zeng et al. demonstrated in colon cancer models that copper ionophores (elesclomol/CuCl₂) establish cuproptosis-promoting conditions that suppress WNT signaling and decrease PD-L1 expression. This dual action enhances CD8⁺ T cell cytotoxicity, increases TME immune infiltration, and ultimately impedes tumor progression [45].
Secondly, copper ionophore-induced cuproptosis, ICD, and pro-inflammatory TME remodeling demonstrate profound mechanistic interdependencies. In colorectal cancer (CRC) models, cuproptosis triggered by copper ionophores provokes endoplasmic reticulum (ER) stress, driving ICD through damage-associated molecular patterns (DAMPs) release—a process that orchestrates antigen-presenting cell maturation, macrophage M1 polarization, and cytotoxic T lymphocyte activation. Notably, this cuproptosis-ICD axis is synergistically amplified when copper ionophores combine with Toll-like receptor 7 (TLR7) agonists [46].
Parallel mechanisms operate in HCC, where the copper ionophore DSF combined with copper (DSF/Cu) promotes the intranuclear accumulation and aggregation of nuclear protein localization protein 4 (NPL4), inhibits the ubiquitin-proteasome system, and induces ER stress-mediated ICD. These events collectively stimulate DCs maturation and enhance CD8⁺ T cell cytotoxicity within the TME. Particularly significant is the observed synergy between DSF/Cu-induced ICD and CD47 blockade, which overcomes immune evasion mechanisms and potentiates ICB efficacy in HCC [47].
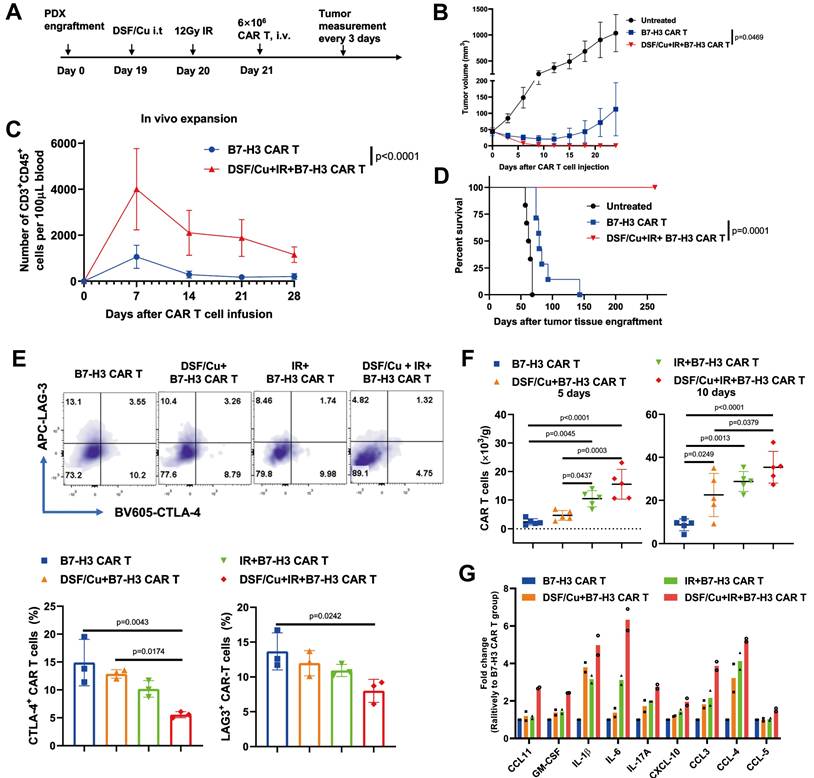
Furthermore, copper ionophores demonstrate significant potential for synergistic antitumor effects by sensitizing tumors to diverse immunotherapies. Mechanistically, elevated intracellular copper levels epigenetically upregulate PD-L1 membrane expression which is critical for overcoming resistance to anti-PD-L1 checkpoint blockade [20], as clinical evidence correlates low expression of PD-L1 with poor therapeutic response [48]. By pharmacologically increasing copper availability, ionophores establish a targetable PD-L1-positive state, enabling previously refractory tumors to respond to immunotherapy. Single-cell transcriptomics shows that intrahepatic cholangiocarcinoma (ICC) cells susceptible to cuproptosis are typically CD274 (PD-L1)-negative and reside in a TME with reduced monocytes and macrophages. These insights have led to the proposal of combining anti-PD-L1 therapy with cuproptosis-based treatment to achieve synergistic effects in ICB treatment [49]. In addition, combining disulfiram/copper (DSF/Cu) with ionizing radiation (IR) enables CAR-T cells with early memory-like properties, enhanced cytotoxicity, sustained anti-tumor responses, and improved in vivo expansion while reducing exhaustion. Mechanistically, it induces robust ICD in tumor cells, characterized by ER and oxidative stress, activation of the IRE1α-XBP1 axis, and release of DAMPs, which activates the JAK/STAT signaling pathway in CAR-T cells. Concurrently, the combination also converts the immunosuppressive TME into an immune-promoting state, significantly boosting CAR-T efficacy [50] (Table 5, Figure 4).
Tumors stressed by DSF/Cu and IR reverse immunosuppressive TME and activate robust and long-sustained therapeutic responses of CAR T against solid tumors. A-D) Therapeutic responses against solid tumors by RP B7-H3 CAR T (Reprogrammed B7-H3 Chimeric Antigen Receptor T cells) derived from PBMCs of patients with metastatic breast. A) Schema of TNBC-PDX (Triple-Negative Breast Cancer Patient-Derived Xenograft) mouse model. B) Tumor volumes (vehicle n = 6; the other two groups n = 7) of each group. C) Frequency of B7-H3 CAR T cells (CD3+ CD45+) in the blood of mice collected weekly (n = 7/group). D) Kaplan-Meier survival curve of mice (vehicle n = 6; the other two groups n = 7). E-G) Tumors stressed by DSF/Cu and IR reverse immunosuppressive TME in humanized mice. E) B7-H3 CAR T cells expressing markers associated with T-cell exhaustion (CD3+ CD45+ LAG-3+ or CD3+ CD45+ CTLA-4+) in tumor-infiltrating CAR T cells in tumor tissues 10 days after CAR T cell infusion (n = 5 mice/group). F) Number of B7-H3 CAR T cells in tumor tissues 5 days and 10 days after CAR T cell injection (n = 5 mice/group). G) Cytokine and chemokine levels in tumor tissues on day 5 after CAR T cell injection. Adapted from ref.[50]. Licensed under CC BY 4.0. Copyright 2023 The Author(s).
These findings above establish that copper-elevating agents functionally bridge cuproptosis, ICD induction, and immune activation, providing a compelling rationale for their synergy with existing immunotherapies. The nanoparticle NP@ESCu is designed for ROS-responsive release of elesclomol and copper ions within the TME. Its thioketal-based polymer backbone (PHPM) degrades under high ROS, triggering rapid cargo release and increasing intracellular copper by 4.2-fold. This copper overload downregulates Fe-S cluster proteins, inducing mitochondrial dysfunction and cuproptosis. RNA-seq analyses reveal activation of the cytokine-cytokine receptor interaction pathway, highlighting its immunomodulatory effects at the molecular level. Firstly, NP@ESCu induces cuproptosis in cancer cells while promoting DCs maturation and enhancing CD8⁺ T cell infiltration, thereby sensitizing them to immunotherapy. Secondly, NP@ESCu significantly upregulates PD-L1 expression on tumor cells which enhances tumor responsiveness to anti-PD-L1 checkpoint blockade, establishing potent synergy between cuproptosis induction and immunotherapy [51].
1.3 Current challenges and future directions
Targeting copper imbalance in the TME faces three key hurdles. First, copper biology is inherently double-edged: Copper chelators deplete TME copper to inhibit immunosuppression but may inadvertently impair cuproptosis-mediated antitumor effects. Conversely, copper ionophores induce cuproptosis yet risk exacerbating PD-L1 expression and immune evasion in resistant tumors. Second, systemic copper is tightly buffered, so prolonged manipulation may harm liver, kidney or brain. Third, tumors differ widely in cuproptosis sensitivity, making blind dosing unsafe.
Future work should therefore concentrate on precision delivery and real-time monitoring. First, utilizing nanoplatforms and subcellular mitochondrial targeting technology [52], specific copper chelators/ionophores need to be developed for precise tumor deliver, thus reducing systemic toxicity. Also, copper-64 PET [53] or activatable MRI agents can enable real-time imaging of copper flux to guide therapy and help stratify patients. Second, single-cell sequencing and high throughput screening of biopsies will identify tumors with high signatures that resist cuproptosis, allowing identifying responsive patients and personalized drug administration.
Besides, it should be mentioned that two redox-active states of copper, cuprous (Cu⁺) and cupric (Cu²⁺), within biological systems. The reversible cycling between these valence states underpins copper's role in redox reactions, contributing to both physiological enzymatic functions and pathological oxidative stress. Cu²⁺ can be reduced to Cu⁺ by cellular metalloreductases, facilitating its involvement in receptor activation and intracellular signaling. Conversely, Cu⁺ can participate in Fenton-like reactions, generating highly reactive hydroxyl radicals that promote oxidative damage to lipids, proteins, and DNA. This is a mechanism increasingly implicated in cancer progression and therapeutic resistance. In summary, the complex interconversion between Cu²⁺ and Cu⁺ and their influence on TME such as signaling pathways and redox reactions make it necessary for the design of nano-targeted drug delivery systems to take into account the issue of their valence state interconversion.
2. Iron
Similar to copper, iron plays a dual regulatory role in tumor biology, mediated largely by its redox activity and multiple valence states, ferrous (Fe²⁺) and ferric (Fe³⁺). On one hand, it promotes tumor proliferation and metastasis by participating in DNA replication, maintaining genomic integrity, and facilitating epigenetic regulation. More importantly, iron homeostasis dysregulation contributes to establishing an immunosuppressive TME. The redox cycling between Fe²⁺ and Fe³⁺ enables iron to participate in Fenton reactions, generating ROS that can induce ferroptosis, ICD, and oxidative stress.
2.1 Mechanisms of iron homeostasis dysregulation driving immunosuppressive TME
2.1.1 Iron homeostasis dysregulation drives immunosuppressive TME formation
Dysregulated iron metabolism is a hallmark of both cancer cells and the TME. To support high demand for rapid proliferation, tumor cells exhibit heightened iron demand, achieving a "high intracellular iron" phenotype through mechanisms like transferrin protein (TFR1) overexpression, enhanced iron import, and suppressed iron export [54]. Consequently, TFR1 serves as both a prognostic biomarker and therapeutic target [55].
Tumor cell iron sequestration within the TME creates iron-depleted conditions that alter immune cell phenotype and function. This environment fosters distinct metabolic phenotypes: cancer cells adopt an "iron-utilizing" state characterized by increased hepcidin and TFR1 expression alongside reduced ferritin levels, while lymphocytes and macrophages exhibit an "iron-donor" state marked by upregulated ferroportin 1 (FPN1) and ferritin expression, combined with elevated hepcidin and TFR1 levels [56]. This indicates that immune cells may serve as iron reservoirs to sustain tumor growth, potentially impairing their own function.
Critically, iron deprivation in the TME drives tumor-associated macrophages (TAMs) polarization from pro-inflammatory M1-like to pro-tumor M2-like phenotypes. Tumor cells exploit the transferrin-transferrin receptor (TFRC) axis to competitively limit macrophage iron uptake, lowering intracellular ferrous iron. This subsequently triggers HIF-1α-mediated upregulation of M2-associated genes, promoting M2 polarization [57]. M2 TAMs then adopt an "iron-releasing" phenotype by upregulating iron exporters, downregulating ferritin heavy chain 1, and enhancing heme oxygenase activity. They further supply iron to tumor cells via lipocalin 2-mediated efflux [58-60], thereby fueling tumor growth.
Conversely, increasing iron content in TAMs can reprogram them to pro-inflammatory M1-like macrophages. Research demonstrates that TAMs exposed to iron-enriched TME accumulate intracellular iron and shift toward the M1 phenotype, acquiring direct tumoricidal activity [61, 62]. Notably, M1 macrophages form an "iron-sequestering phenotype" by suppressing iron exporters and activating ferritin heavy chains, thereby retaining iron within the reticuloendothelial system to maintain high intracellular ferrous iron levels [58-60].
This dynamic equilibrium underscores a key immune evasion mechanism: tumor cells induce M2 polarization by competitively depriving macrophages of iron, while exploiting M2 macrophages' iron-release function to acquire growth-essential iron, which implies that elevating TME iron may reverse this process, promoting anti-tumor M1 polarization (Figure 1).
Beyond TAMs, iron also reshapes CAFs to foster immunosuppressive TME. In multiple cancers, activation of the Hmox1/iron/Kdm6b pathway transforms CAFs into "iron-loaded cancer-associated fibroblasts" (FerroCAFs). FerroCAFs recruit immunosuppressive myeloid cells via myeloid chemokines (e.g., CCL2, CSF1, CXCL1), contributing to immunosuppression and poor prognosis [63].
Taken together, these findings reveal the pivotal role of iron homeostasis dysregulation in tumor immune escape, creating a foundation for developing iron-targeted cancer treatments.
2.1.2 Ferroptosis in immune cells reshapes the TME to promote tumor progression
Ferroptosis, defined by Stockwell B. R. in 2012 as an iron-dependent, non-apoptotic cell death driven by lipid peroxidation [64], can be activated within the TME by dysregulated iron metabolism in both tumor and immune cells. Ferroptosis occurring in immune cells significantly reshapes the TME into an immunosuppressive state, thereby indirectly promoting tumor progression through two interconnected mechanisms. First, ferroptosis in immune cells directly depletes anti-tumor immune cell populations and impairs their function. For instance, CAFs promote ferroptosis in NK cells by upregulating iron-inducible regulatory genes Ferroportin1 and Hephaestin and enhancing Follistatin Like 1 (FSTL1) secretion, leading to elevated free iron, upregulated NCOA4, iron-dependent lipid peroxidation in NK cells, and ultimately suppressed their anti-tumor activity [65]. Similarly, in the iron-overloaded TME, ferroptosis of activated naive CD4⁺ T cells [66] and CD8⁺ T cells [67] leads to a decrease in their numbers and a decline in their anti-tumor functions. Second, the process of ferroptosis itself releases soluble mediators that actively suppress neighboring immune cells, particularly T cells (Figure 5). Ferroptotic neutrophils secrete prostaglandin E2 (PGE2), Indoleamine 2,3-dioxygenase (IDO), and oxidized lipids, which inhibit CD8⁺ T cell proliferation and cytotoxicity while enriching immunosuppressive ferroptosis-associated CD4⁺ T cells (Fer-CD4⁺ T). This creates a detrimental “neutrophil ferroptosis-Fer-CD4⁺ T cell activation” positive feedback loop that drives therapy resistance [68]. Analogously, pathologically activated polymorphonuclear myeloid-derived suppressor cells (PMN-MDSCs) undergo spontaneous ferroptosis, releasing oxidized lipids that potently inhibit T cell function [69]. Importantly, genetic or pharmacological inhibition of ferroptosis in PMN-MDSCs reverses this immunosuppression, delays tumor progression, and synergistically enhances immunotherapy efficacy in immunocompetent mouse models [69], underscoring the therapeutic relevance of this pathway.
Ferroptosis in immune cells drives the formation of TME and indirectly promotes tumor progression through the decrease in the quantity and function of immune cells and the various effective components released during ferroptosis which can affect the functions of other effector cells. Created in https://BioRender.com.
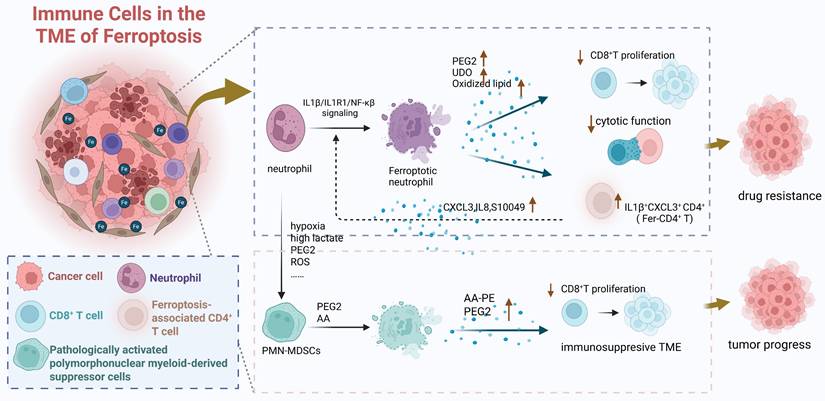
Multi-omics bioinformatics analyses provide strong validation for the close link between ferroptosis and an immunosuppressive TME. For example, diagnostic and prognostic models based on ferroptosis-related metabolism identify high-risk ferroptosis subgroups in HCC characterized by elevated tumor mutational burden and an immunosuppressive TME signature. This signature includes enrichment of immunosuppressive cell types (M0 macrophages, follicular helper T cells, memory B cells, neutrophils), increased expression of immune checkpoints (CD83, B7H3, OX40, CD134L), and strong associations with poor prognosis and reduced response to immunotherapy [70] (Figure 2F-I). Collectively, these findings integrate data on iron metabolism regulatory networks, immune cell dynamics, and key immune molecules, highlighting the critical role for immune cell ferroptosis in driving tumor immune escape. They provide a compelling rationale for developing personalized immunotherapy strategies targeting iron metabolism pathways.
2.2 Strategies targeting iron homeostasis dysregulation for cancer treatment
Antitumor strategies targeting iron imbalance primarily focus on two approaches: iron depletion or ferroptosis induction. Specifically, iron depletion utilizes chelators like deferoxamine (DFO), deferiprone (DFP), and deferasirox (DFX) to reduce iron levels within the TME and tumor cells, thereby inhibiting iron-dependent proliferation. Conversely, ferroptosis induction employs agents such as system Xc⁻ inhibitors and glutathione peroxidase 4 (GPX4) inhibitors to elevate TME iron levels and combat tumors through triggering ICD, inducing ferroptosis, and causing ROS bursts (Table 2, Figure 3, Table S1).
Immunomodulatory effects of cancer treatments targeting iron dysregulation in the TME
| Practical application | Functions on immunity in TME | Correlation with immunotherapy |
|---|---|---|
| Iron chelators | Activate immune effector cells | Promote conversion to an immune-activated TME |
| Ferroptosis inducers | 1. Enhance innate and adaptive immunity | Synergize with ICB |
| 2. Induce ICD via ferroptosis | Promote conversion to an immune-activated TME | |
| 3. Enhance sensitivity and efficiency of immunotherapies | Synergize with αPD-L1 blockers |
2.2.1 Iron chelators block tumor iron utilization and multidimensionally activate immunity
Iron chelators inhibit tumor progression by both suppressing cellular respiration to reduce energy production [71] and inhibiting proliferation/migration signaling pathways [72]. More importantly, their immunomodulatory effects significantly enhance immunotherapy synergy. Specifically, these agents remodel immune cell phenotypes, activate effector cells, and promote pro-inflammatory cytokine secretion. For instance, the intracellular chelator (TC3-S)₂ shifts macrophages from iron-releasing M2 to iron-sequestering M1 phenotypes, thereby reversing tumor growth and metastasis [73]. Similarly, in ovarian cancer models, DFP-mediated iron chelation activates the cGAS-STING-IRF3 axis to drive type I interferon production, concurrently upregulating NK cell-activating death receptor 5 (DR5) expression while promoting dendritic cell-derived IL-15 secretion to sustain NK responses—collectively impeding malignant progression through multidimensional immune activation [74].
2.2.2 Multiple mechanisms of ferroptosis inducers in exerting antitumor effects
Ferroptosis inducers not only directly trigger tumor cell death but also enhance immunotherapy sensitivity. Research indicates that ferroptosis is involved in T cell immunity and cancer immunotherapy. Immunotherapy-activated CD8+ T cells directly stimulate iron-dependent lipid peroxide accumulation in tumor cells, with amplified ferroptosis mechanism being functionally linked to improved tumor regression observed in immunotherapy protocols [75]. Also, ferroptosis induction reduces MDSCs and M2-like TAMs in the TME, while enhancing the activity and infiltration of tumor-infiltrating CD4⁺ and CD8⁺ T cells [76, 77]. Mechanistically, ferroptosis triggered by GPX4 inhibition leads to the exposure of calreticulin on the cell surface and release of high-mobility group box1 (HMGB1) which are key DAMPs that promote antigen presentation and myeloid cell maturation via TLR4 signaling. This shift in the immune landscape alleviates immunosuppression, thereby synergizing with ICB therapies and CXCR2 inhibition [77]. Thus, targeting ferroptosis metabolism directly enhances CD8⁺ T cell-mediated tumor killing and indirectly elevates overall cancer immunotherapy response rates.
Among cancer therapies targeting ferroptosis, iron-based nanomaterials have also been extensively studied. The biomimetic magnetic nanoparticle Fe3O4-SAS@PLT, constructed from mesoporous magnetic nanoparticles (Fe3O4) loaded with sulfasalazine (SAS) and coated with platelet (PLT) membranes. The mesoporous iron oxide core affords high drug-loading capacity and pH-responsive release, while the platelet membrane cloak provides immune evasion through surface-exposed CD47 and promotes tumor accumulation via P-selectin mediated binding to CD44 receptors on cancer cells. At the mechanistic level, the released Fe²⁺/Fe³⁺ ions catalyze hydroxyl radical generation through Fenton reactions in the TME, while SAS inhibits the system Xc‒ transporter, depleting glutathione and synergistically amplifying lipid peroxidation, thus culminating in potent ferroptotic cell death. Beyond direct cytotoxicity, this nanoparticle induces immunogenic remodeling by promoting DCs maturation and effector T-cell recruitment. It also drives macrophage polarization from M2 toward antitumoral M1 phenotypes, thereby alleviating microenvironmental suppression and significantly enhancing the efficacy of αPD-1 checkpoint blockade therapy against metastatic tumors [78] (Table 5).
Although traditional immunotherapy primarily induces regulated cell death (RCD), such death modalities are considered "immunologically silent" due to their lack of immunogenicity, making it difficult to effectively activate anti-tumor immune responses within the TME. In contrast, ferroptosis triggers ICD to amplify anti-tumor immunity. For instance, ferroptotic cancer cells promote bone marrow-derived dendritic cells (BMDCs) maturation and exert vaccine-like effects in mice, concurrently releasing ATP and HMGB1 as immune-related molecules to enhance antigen presentation [79]. Furthermore, iron oxide nanoparticles (IONs) increase ICD incidence by stimulating macrophage-derived ATP and HMGB1 secretion, thereby enhancing infiltration and activation of DCs and cytotoxic T cells (CTLs) [80] (Table 5). Collectively, these mechanisms provide a theoretical foundation for ferroptosis-immunotherapy synergy.
Besides, inducing ferroptosis can directly improve the efficiency and sensitivity of cancer immunotherapy. For example, the ferroptosis-inhibiting gene TYRO3 mediates resistance to anti-PD-1/PD-L1 therapy by suppressing innate immunity and ferroptosis. Conversely, inhibiting TYRO3 promotes tumor ferroptosis, sensitizing resistant tumors to αPD-1/PD-L1 therapy [81]. This mechanism offers a novel therapeutic target for overcoming immunotherapy resistance.
2.3 Current challenges and future directions
Despite significant progress in targeting iron dysregulation for cancer therapy, two interconnected challenges hinder clinical translation. First, iron's dual impact creates a therapeutic dilemma: Iron chelation inhibits tumors but risks impeding M1 polarization [61, 62], whereas ferroptosis inducers enhance immunogenicity yet damage immune effectors [66, 67]. Second, immunosuppressive cascades driven by ferroptosis (e.g., CAF-induced NK cell ferroptosis [65], neutrophil ferroptosis-Fer-CD4⁺ T cell axis [68]) remain inadequately targeted, with current approaches lacking selectivity in suppressing detrimental ferroptosis and preserving immunostimulatory cell death.
Future efforts should first prioritize cell-type-specific iron modulation. This requires designing nanocarriers—such as macrophage-targeted liposomes—to deliver iron chelators or loaders with spatial precision [82], while leveraging TME-responsive materials to differentially regulate iron pools in tumor versus immune cells. Second, optimizing ferroptosis-immunotherapy synergy requires developing combinatorial regimens that exploit ferroptosis-derived immunogenic signals while blocking immunosuppressive mediators like PGE₂ and oxidized lipids [68, 69]. Finally, advancing clinical biomarker stratification necessitates integrating multi-omics ferroptosis signatures with TME iron imaging to identify patients most likely to benefit from iron-centered therapies, especially those immunotherapy-resistant.
3. Zinc
Serum zinc levels typically decline in cancer patients, suggesting its potential as a diagnostic and prognostic biomarker [83]. Zinc homeostasis imbalance is common in TMEs, exhibiting tumor heterogeneity and local specificity [84]. Intratumoral zinc levels decrease in prostate, liver, pancreatic, and kidney cancers, but increase in breast, gastric, and lung cancers [85]. These alterations demonstrate a close link to tumorigenesis, highlighting the crucial roles of zinc homeostasis and its regulatory proteins.
3.1 Mechanisms of zinc homeostasis dysregulation driving immunosuppressive TME
Like iron, zinc promotes tumorigenesis by activating transcription factors and signaling pathways involved in cancer cell proliferation and migration [86]. Stromal and immune cells within the TME can act as "zinc reservoirs" for cancer cells. For instance, ZIP1-positive CAFs enhance gap junction formation in cancer cells via connexin-43 upregulation and function as Zn²⁺ reservoirs, absorbing and transferring Zn²⁺ to cancer cells, thereby promoting chemotherapy resistance in lung cancer [87].
Zinc homeostasis also directly modulates TAM phenotype and function (Figure 1). In zinc-deficient HCC TMEs, the zinc transporter ZNT1 is significantly downregulated, particularly in M2 TAMs. This reduces endosomal Zn²⁺ levels, impairing the endosomal clearance of Toll-like receptor 4 (TLR4) and PD-L1 from the cell membrane. Sustained TLR4 activation increases pro-inflammatory factor release, exacerbating chronic inflammation and liver fibrosis, while surface-retained PD-L1 suppresses CD8⁺ T cell activity, facilitating immune escape and tumorigenesis. Exogenous zinc supplementation restores ZNT1 function, reduces PD-L1 expression, enhances CD8⁺ T cell cytotoxicity, and synergizes with chemo- and immunotherapy to improve efficacy [88].
Conversely, zinc-elevated TMEs can also promote immune evasion and diminish immunotherapy response through: (1) Promoting immunosuppressive phenotypes: Zinc enhances the differentiation and function of Foxp3⁺ regulatory T cells (Tregs) via FOXO1-mediated transcriptional upregulation of Foxp3 [89], while simultaneously inhibiting Th1 cells, and altering CD4⁺ T cell subsets [90]. (2) Upregulating immunosuppressive checkpoints: Heightened PD-1 expression on CD4⁺ T, CD8⁺ T, and γδT cells [90]. (3) Reducing effector cell cytotoxicity: Decreased CD107a and lysosome-associated membrane protein 1 (LAMP1) expression on NK and CD8⁺ T cells [90]. (4) Enhancing tumor anti-apoptosis: Elevated zinc in cancer cells stabilizes inhibitor of apoptosis proteins (IAPs) and inhibits caspases activation, conferring resistance to T cell-derived TNF (tumor necrosis factor)-induced apoptosis [91] (Figure 1).
These findings highlight potential therapeutic targets: modulating zinc transporters; blocking STAT6/IRF4 signaling to reduce zinc flux in M2 macrophages; and antagonizing zinc-mediated anti-apoptotic pathways. Such strategies hold promises for improving the tumor immune microenvironment and enhancing therapeutic efficacy.
3.2 Strategies targeting zinc homeostasis dysregulation for cancer treatment
Currently, both zinc chelators and zinc ion supplements are applied to target zinc homeostasis imbalance, exerting anti-tumor effects through enhancing immunogenicity and alleviating inflammation (Table 3, Figure 3).
Immunomodulatory effects of cancer treatments targeting zinc dysregulation in the TME
| Practical application | Functions on immunity in TME | Correlation with immunotherapy |
|---|---|---|
| Zinc chelators | 1. Inhibit immunosuppressive cell phenotypes | Synergize with αPD-1 |
| 2. Increase tumor cells' sensitivity to immune killing | Enhance tumor cells' sensitivity to CAR-T cell-mediated killing | |
| Zinc supplements | 1. Reverse immunosuppressive TME | Synergize with αPD-L1 blockers |
| 2. Activate cGAS-STING immune pathway | Conversion to an immune-activated TME; enhance the efficacy of immunotherapy |
Mechanistically, Narayan S. et al. found that zinc chelators significantly reduce Foxp3+ Treg cell [89]. By depleting intracellular zinc in cancer cells, they rapidly degrade IAPs and restore the caspase pathway, sensitizing tumor cells to TNF-induced killing [91]. Besides, zinc chelators can synergize with αPD-1 [89] and CAR-T cells [91] to enhance anti-tumor effects in immunotherapy.
Conversely, zinc supplements enhance immunotherapy efficacy by reversing immunosuppressive TME. Studies have shown that intratumoral zinc injection directly induces tumor pyroptosis via both the caspase-1/GSDMD canonical pathway and the caspase-3/GSDME alternative pathway, triggered by zinc overload and ROS accumulation. This process promotes the release of pro-inflammatory cytokines (IFN-γ, IL-6, TNF-α) and increases TME infiltration of immune-promoting cells (mature DCs, CD4⁺, CD8⁺ T cells) [92] but reduces immunosuppressive cells (Tregs, MDSCs, M2 macrophages) [93]. Additionally, it facilitates the endocytosis and lysosomal degradation of surface PD-L1 and TLR4 by promoting endosomal Zn²⁺ influx via zinc transporter ZNT1 in macrophages, thus dampening TLR4/NF-κB-driven inflammation and reduces PD-L1-mediated T cell suppression, and accordingly decreasing IL-6 production and enhancing CD8⁺ T cell cytotoxicity [88].
Molecularly, zinc supplementation enhances cGAS-DNA binding affinity, thereby amplifying the stimulator of interferon genes (STING)-dependent interferon response [94]. Consequently, this promotes CD8⁺ T cell infiltration, activation, and effector function in the TME while suppressing tumor growth via interferon-α/β receptor (IFNAR) signaling—providing a novel strategy to boost immunotherapy response [95]. Simultaneously, STING activation upregulates tumor cell MHC-I expression and antigen-presentation genes, improving tumor antigen presentation. Besides, these zinc-induced immunomodulatory effects sensitize tumors to ICB therapy, as demonstrated by the optimal CD8⁺ T cell recruitment and maximal antitumor efficacy achieved through co-administration of zinc and αPD-1 [92].
In addition, leveraging zinc-based nano-drugs to chelate in situ within tumors and form toxic substances may artificially induce "zinc-induced death" in tumor cells. The TME-responsive DSF@Zn-DMSNs platform is constructed from zinc-doped dendritic mesoporous silica nanoparticles (Zn-DMSNs). This structure is further functionalized with DSF, which is loaded into the enlarged mesopores and stabilized via PEGylation. Upon exposure to the mildly acidic TME, the Zn-O bonds undergo hydrolysis, leading to biodegradation and concurrent release of Zn²⁺ and DSF, where they generate the highly toxic zinc-dithiocarbamate complex (ZnET) in situ. The highly toxic ZnET enhances autophagy while triggering ICD and facilitating CTLs infiltration. This reverses the immunosuppressive TME and achieves optimal therapeutic effects in colorectal cancer treatment [96] (Table 5).
3.3 Current challenges and future directions
Key challenges in targeting zinc dysregulation include significant intratumoral heterogeneity and zinc's context-dependent immunomodulation: deficiency drives PD-L1 retention and CD8⁺ T-cell suppression in TAMs [88], while excess promotes Treg expansion [89] and PD-1 upregulation [90]. Secondly, current systemic chelation/supplementation lacks spatial precision to resolve these dual roles.
To enable precision medicine, smart nano-delivery systems for localized zinc modulation. Further, transporters (ZIP1 in CAFs; ZNT1 in TAMs) in specific cells could be a promising target to disrupt pathological zinc flux. To overcome intratumoral heterogeneity of zinc level, integrating zinc-specific PET/MRI with zinc-relevant multi-omics signatures (e.g., transporter expression, immune cell profiling) will stratify patients for zinc-centered regimens, thereby bridging molecular imaging and targeted therapy.
4. Magnesium
Magnesium ions (Mg²⁺), the second most abundant cellular cation in cells, are critical cofactors and signaling molecules for normal physiology.
4.1 Mechanisms of magnesium homeostasis dysregulation driving immunosuppressive TME
Magnesium deficiency in the TME is associated with an increased incidence of cancers such as prostate [97] and colorectal cancer [98], as well as with poor prognosis. Magnesium deficiency promotes tumor progression by inducing genetic mutations, modulating inflammation, and activating epithelial-mesenchymal transition [98]. In response, higher magnesium intake correlates with improved prognosis and reduced cancer mortality [99]. Elevated serum Mg²⁺ levels were identified as an independent prognostic factor for better progression-free survival and overall survival in 1,441 ICB-treated patients within a multicenter study [100], suggesting magnesium supplementation could potentially enhance immunotherapy efficacy.
However, malignant colorectal tissues exhibit higher Mg²⁺ levels than adjacent non-cancerous tissues, and tumors with elevated magnesium content show increased mutation frequency in driver genes like TP53 and APC [98]. These seemingly conflicting findings highlight the complex and context-dependent relationship between TME magnesium imbalance and tumorigenesis.
Mechanistically, TME magnesium deficiency impairs anti-tumor immunity through multiple pathways (Figure 1). Firstly, low intracellular free Mg²⁺ destabilizes the critical activating receptor natural-killer group 2, member D (NKG2D) and its adaptor DAP10 in immune cells, impairing NK and CD8⁺ T cell recognition of tumor-expressed stress ligands and significantly reducing cytotoxicity. In vitro Mg²⁺ supplementation dose-dependently restores NKG2D expression and cytotoxic activity [101]. Secondly, low extracellular Mg²⁺ prevents lymphocyte function-associated antigen-1 (LFA-1) integrin on T cells from adopting its active conformation, weakening LFA-1-mediated co-stimulation. This impairs tumor-specific T cell activation, effector molecule secretion, and response to ICB. Local magnesium replenishment enhances LFA-1 signaling, increasing tumor-infiltrating CD8⁺ T cells and suppressing tumor growth in mice, highlighting the therapeutic potential of targeting the Mg²⁺-LFA-1 axis [102]. Thirdly, a low-Mg²⁺ TME exacerbates TAM polarization towards immunosuppression [103]. As a natural blocker of N-methyl-D-aspartate receptor (NMDAR) of the macrophages, Mg²⁺ inhibits its activation by tumor-secreted glutamate, thereby countering TAM transformation into an immunosuppressive phenotype [103]. Collectively, these findings underscore that restoring TME magnesium homeostasis represents a promising therapeutic strategy to enhance innate and adaptive anti-tumor immunity and ultimately improve the efficacy of cancer immunotherapies.
4.2 Strategies targeting magnesium homeostasis dysregulation for cancer treatment
Magnesium-targeted anti-tumor approaches focus on Mg2+ supplements and their derived nanomaterials. On one hand, these agents reshape the immunosuppressive microenvironment by activating immune cells. On the other hand, they directly enhance the efficacy and sensitivity of immunotherapies such as CAR-T cell function, ICB, and tumor vaccines (Table 4, Figure 3).
Immunomodulatory effects of cancer treatments targeting magnesium dysregulation on the TME
| Practical application | Functions on immunity in TME | Correlation with immunotherapy |
|---|---|---|
| Magnesium ion supplements | 1.Reverse the immunosuppressive TME | Synergize with αPD-L1 blockers |
| 2.Improve sensitivity and efficiency of immunotherapies | Enhance αPD-L1 blockers and CAR-T efficacy |
Mechanistically, Mg²⁺ supplementation inhibits TAM immunosuppressive polarization by downregulating Arg-1, PD-L1, and vascular endothelial growth factor A (VEGFa) while upregulating pro-inflammatory cytokines and CD80/CD86 co-stimulators. This in turn promotes CD8⁺ T-cell proliferation, IFN-γ secretion, and tumor-killing capacity [103].
As an activator of the LFA-1 integrin, Mg²⁺ binding to the metal ion-dependent adhesion site promotes conformational extension and headpiece opening of LFA-1. This leads to improved calcium flux, ERK phosphorylation, metabolic reprogramming, and cytokine production in CD8⁺ T cells. Consequently, Mg²⁺ supplementation potentiates the cytotoxic function, infiltration, and immune synapse maturation of pathogen- and tumor-specific T cells. It also significantly improves CAR-T cell function [102]. As noted in a previous multi-center retrospective study, serum Mg²⁺ levels are an independent prognostic factor for cancer patients undergoing ICB [100]. Other studies also indicate that combining magnesium ion supplements with αPD-1 antibodies significantly increases the complete remission rate in cancer models [103]. This may be related to the increased expression of PD-1 on CD8⁺ T cells following magnesium supplementation [102] (Table 5).
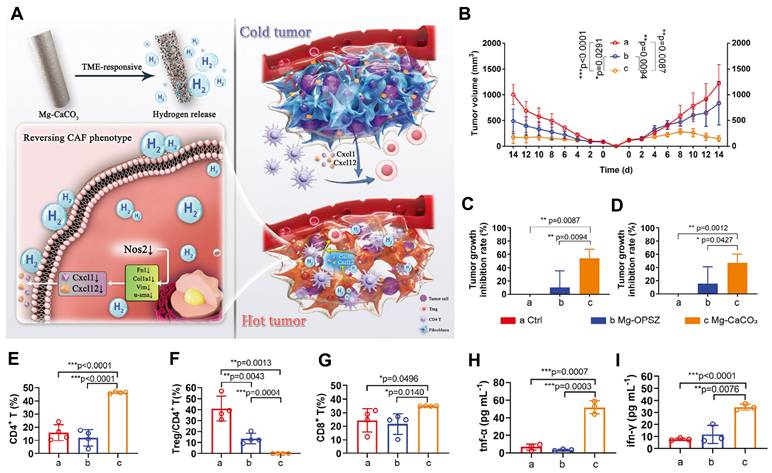
Magnesium-based nanomaterials exert anti-tumor effects by directly inducing cytotoxicity through hydrogen generation, while synergistically enhancing immunotherapy via immunosuppressive microenvironment remodeling, antigen presentation potentiation, and T-cell activation. For example, the TME-responsive Mg-CaCO₃ system, which consists of a magnesium core coated with CaCO₃ nanoparticles embedded in an organic polysilazane matrix. This structure enables controlled hydrogen release in response to the acidic TME, where CaCO₃ dissolution exposes the Mg surface, facilitating proton-driven hydrolysis and sustained H₂ production. Functionally, the released hydrogen directly induces redox imbalance in tumor cells, leading to apoptosis and reduces ROS in CAFs, activates CD4⁺ T cells—converting "cold" to "hot" tumors [104] (Figure 6). In tumor vaccine research, the R837-Pep@HM nanovaccine is composed of human serum albumin coordinated with magnesium ions and covalently conjugated with the TLR7/8 agonist imiquimod (R837) and melanoma neoantigen peptide. The human serum albumin scaffold serves as a biocompatible template that facilitates biomineralization with Mg²⁺, forming stable nanoparticles with enhanced lymphatic drainage and lymph node targeting due to optimal size and negative surface charge. The incorporation of Mg²⁺ augments DCs maturation via the Mg transporter MAGT1 and LFA-1-mediated T cell co-stimulation, while R837 activates TLR7/8 pathways, synergistically promoting neoantigen presentation and effector T-cell proliferation, thus prolonging survival in melanoma prevention models [105]. These findings confirm critical role of Mg²⁺ systemic immunotherapy synergy (Table 5).
Immunomodulatory effects of magnesium and hydrogen therapy reverse cancer-associated fibroblasts phenotypes and remodels TME to stimulate systematic anti-tumor immunity. A) Schematic illustration of TME remodeling and tumor inhibition mechanisms of Mg-CaCO3. B-I) Systematic immune activating effects of Mg-CaCO3. Tumor growth curves (B), tumor inhibition rate of primary tumors (C) and distant tumors (D) of Balb/c mice after different treatments (n = 5). E-G) Quantification of E) CD4+/CD45+ ratio, F) Treg/CD4+ ratio, and G) CD8+/CD45+ ratio (n = 4). H-I) Quantification of TNF-α(H) and IFN-γ(I) concentration in the serum of Balb/c mice after different treatments (n = 3). Adapted from ref.[104]. Licensed under CC BY 4.0. Copyright 2024 The Author(s).
4.3 Current challenges and future directions
Despite the established link between magnesium dysregulation and TME immunosuppression, significant challenges impede therapeutic translation. A central unresolved issue is the context-dependent duality of Mg²⁺'s role in cancer. While systemic deficiency correlates with increased cancer risk and poor prognosis, and TME deficiency demonstrably drives immunosuppression, elevated intratumoral Mg²⁺ levels have been observed in specific cancers and associated with increased oncogenic mutations. This paradox underscores the critical need to define the precise spatiotemporal dynamics and compartment-specific thresholds governing Mg²⁺'s pro- versus anti-tumor effects across different cancer types and stages. Resolving this duality is paramount to ensure the safety of Mg²⁺-modulating therapies. Secondly, achieving targeted delivery and sustained release of Mg²⁺ within the TME to maximize beneficial immune modulation while minimizing potential off-target effects remains a challenge. Furthermore, identifying robust predictive biomarkers beyond serum levels to select patients most likely to benefit from combining Mg²⁺ modulation with immunotherapies requires extensive clinical validation.
5. Other essential metal elements
Beyond the commonly discussed copper, iron, zinc, and magnesium, essential metals such as manganese, calcium, sodium, potassium, chromium, and molybdenum are vital for physiological functions, and their homeostasis imbalance is closely associated with tumorigenesis.
5.1 Mechanisms of metal homeostasis dysregulation driving immunosuppressive TME
Manganese, primarily present as Mn²⁺ in the TME, exhibits a dual role in tumor progression. First, Mn²⁺ enhances anti-tumor immunity. Manganese deficiency in melanoma, Lewis lung cancer, or T lymphoma mouse models significantly reduces TME infiltration of CD8⁺ T cells and accelerates tumor growth and metastasis [106]. Conversely, Mn²⁺ acts as an integrin activator in the TME: accumulation in primary Lewis lung cancer alters tumor cell surface molecules (e.g., syndecan-1, β1-integrin) to enhance migration and invasion, while tumor-secreted Mn-containing extracellular vesicles facilitate Mn accumulation in perivascular regions and metastatic sites [107]. This complex duality underscores the context-dependent impact of manganese on cancer biology.
Calcium ions (Ca²⁺), vital second messengers regulating cellular processes, significantly influence tumorigenesis, progression, and immunosuppressive TME formation. Calcium regulatory molecules mediate Ca²⁺ signaling and homeostasis, directly impacting immune cell function. For instance, deficiency of stromal interaction molecule 1 (STIM1) reduces Ca²⁺ concentration in BMDCs in mice, impairing antigen cross-presentation and migration [108]. In glioblastoma, a subset of cycling tumor cells drives TME remodeling through autonomous KCa3.1-mediated calcium oscillations. These signals propagate via the tumor microtubule network, activating MAPK/NF-κB to promote proliferation and survival. Targeted KCa3.1 inhibition disrupts this extracellular Ca²⁺ "signaling hub," reduces microglial activation, and suppresses tumor growth while extending survival [109].
However, current research predominantly focuses on cancer cells rather than the TME for modulating Ca²⁺ signaling. This gap underscores the critical need to systematically investigate calcium homeostasis and its associated pathways from an integrated TME perspective, which may reveal novel therapeutic opportunities.
Beyond these, sodium (Na⁺) accumulation, a hallmark of solid tumors, contributes to an immunosuppressive TME by modulating immune cell function. High extracellular Na⁺ can induce a pro-inflammatory, yet often pro-tumorigenic, polarization state in macrophages and T cells, exacerbating tumor progression and potentially impairing cytotoxic response [110]. Potassium (K⁺) shape T cell function and metabolism through membrane potential and signaling. Elevated extracellular K⁺, often resulting from tumor cell necrosis, impairs CD8⁺ T cell function by suppressing Akt-mTOR signaling and inhibiting effector responses [111].
5.2 Strategies targeting metal homeostasis dysregulation for cancer treatment
Mn²⁺ is a potent immune activator that enhances antitumor immunity mainly through cGAS-STING activation, improving antigen presentation and promoting CD8⁺ T cell differentiation, activation, and memory formation, along with NK cell activation and immune surveillance. The combination of Mn²⁺ with PD-1 antibodies, termed "manganese immunotherapy", significantly improves efficacy across tumor models while allowing reduced antibody doses [106]. Mn²⁺ also serves as a vaccine adjuvant by enhancing antigen uptake, presentation, and germinal center formation [112]. For instance, an MnO₂-based in situ vaccine (MBMA-RGD) induces ICD via photodynamic therapy (PDT) and PTT, activates STING, recruits and stimulates DCs, generates oxygen, and strongly suppresses metastasis and recurrence in mouse models without significant toxicity [113].
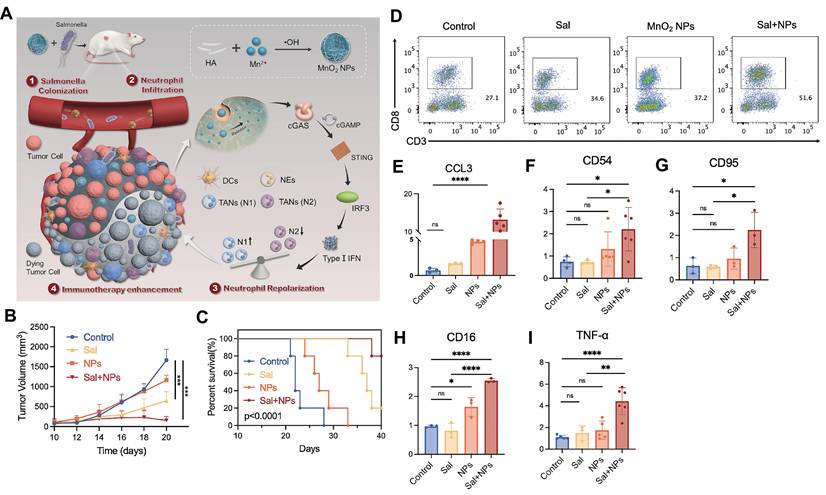
Recent advances in manganese-based nanoplatforms further demonstrate their immunomodulatory versatility. Mn-phenolic coordination networks (TMA NMs), synthesized using tannic acid and Mn²⁺ and stabilized with bovine serum albumin, disintegrate in response to pH/glutathione, releasing both Mn²⁺ and STING agonists to activate STING signaling, induce ICD, and remodel the immunosuppressive TME [114]. Similarly, MnO₂ nanoparticles synergize with bacterial immunotherapies like engineered Salmonella by activating STING within the TME. Released Mn²⁺ promotes IFN-β secretion, repolarizing tumor-associated neutrophils from N2 to N1 phenotype, which reshapes the immune landscape and enhances CD8⁺ T cell infiltration and activation [13] (Figure 7).
Repolarizing neutrophils via MnO2 nanoparticle-activated STING pathway enhances Salmonella-mediated tumor immunotherapy. A) Schematic illustration of the preparation of MnO2 NPs and the synergistic tumor therapy of salmonella and MnO2 NPs. B-I) The synergistic anti-tumor efficacy of salmonella and MnO2 NPs combinatorial therapy. (B) The tumor volume of mice and (C) the survival rate of mice post different treatments. (D) The number of CD8+T cells in tumor post different treatments detected by flow cytometry. (E-I) the level of CCL3, CD54, CD95, CD16, TNF-α in tumor tissues post different treatments detected by q-PCR. Adapted from ref. [13]. Licensed under CC BY 4.0. Copyright 2024 The Author(s).
Calcium-targeting strategies encompass both chelation and supplementation approaches to combat cancers. Specifically, EDTA-loaded layered double hydroxide (EDTA/LDH) nanomaterials enable acid-responsive EDTA release in tumors, depleting endoplasmic reticulum Ca²⁺ in innate immune cells. This subsequently drives anti-tumor phenotype polarization and immune cell infiltration into the TME [115]. On the other hand, calcium ions and their nano-derivatives amplify ICD [116], promote M1 macrophage polarization [117, 118], and DCs maturation [117] and activate immune cells [119]—thereby elevating tumor immunogenicity and potentiating anti-tumor immunity.
Furthermore, chromium ions (Cr³⁺) contribute to immune activation and synergistic immunotherapy for tumors. For example, Cr³⁺ can increase the expression of CXCL13 and CCL3 chemokines, thereby generating tertiary lymphoid structures in tumor, which promotes the infiltration, migration, and anti-tumor efficacy of CAR-T cells [120].
5.3 Current challenges and future directions
The therapeutic targeting of essential metals in the TME faces unresolved challenges. First, the context-dependent duality of metal actions complicates clinical translation. For instance, while Mn²⁺ bolsters cGAS-STING-mediated anti-tumor immunity [106], it concurrently enhances tumor migration and metastatic spread [107]. Second, another obstacle is the limited mechanistic findings on how other essential metal reprogram the TME and the near-absence of selective drug targets. A comprehensive TME-centric understanding of metal content dynamics and associated pathways is essential to uncover novel therapeutic nodes.
To address these, future studies should develop metal-specific delivery systems to spatially restrict actions and design imaging tags that report real-time metal ion flux signals. These systems can be combined with low-dose checkpoint inhibitors to define safe windows. Furthermore, integrating single-cell metallomics will map metal-responsive immune pathways and identify targets that favor anti-tumor activity. Finally, linking these imaging readouts to accessible biomarkers will allow clinicians to titrate metal-based immunotherapies on demand.
6. The overlooked orchestrators: metalloproteins in metal-immunity crosstalk
While our review has focused on the immunomodulatory roles of free metal ions, it is crucial to emphasize that their biological functions are predominantly executed through a vast array of metalloproteins. These proteins, which include metalloenzymes, metallotransporters, and metal-sensing transcription factors, are the fundamental effectors that translate metal availability into cellular signals, ultimately shaping the immune landscape of the TME.
A prime example is metallothionein (MT), a family of cysteine-rich, metal-binding proteins primarily known for regulating zinc and copper homeostasis and cellular redox balance [121]. MTs are not mere metal buffers; they are active players in anti-tumor immunity. In the immunosuppressive TME, MT overexpression is frequently induced by hypoxia [122] and metals themselves [123]. This overexpression sequesters zinc and copper, creating a dual effect: it confers resistance to metal-induced cell death (e.g., cuproptosis) in tumor cells, promoting their survival [124], and it enlarges the redox-mobilizable metal pool, enhancing p38 MAPK activation upon stimulation and promoting Tr1 cell differentiation, which may subsequently suppress anti-tumor immunity [125]. Furthermore, MTs can directly suppress the expression of pro-inflammatory cytokines and modulate the activation of NF-κB, a key transcription factor in immune responses, thereby dampening antitumor immunity [123, 126].
Beyond MTs, the zinc finger protein family, the largest class of transcription factors in humans, governs the expression of countless genes involved in immune cell differentiation and function [127]. Similarly, metalloenzymes like manganese-dependent superoxide dismutase (MnSOD) and copper-containing lysyl oxidases (LOXs) are indispensable for managing oxidative stress in the TME and remodeling the extracellular matrix to facilitate immune cell infiltration or exclusion [128, 129].
The immense diversity and specificity of metalloproteins make them a rich but underexplored source of therapeutic targets. Specific targeting of metalloproteins offers promising strategies for precision metalloimmunotherapy. For example, inhibiting MT can sensitize tumors to cuproptosis or release zinc to activate T-cells. Alternatively, designing activators for metal-dependent immunoenzymes represents another exciting direction. Future research focused on mapping the "metalloproteome" of the TME will be crucial to unravel the connections between metal homeostasis, protein function, and immune regulation. These insights will ultimately reveal new nodes for therapeutic intervention.
7. Conclusion and perspectives
Immunotherapy has revolutionized cancer treatment, yet its efficacy remains constrained by the immunosuppressive TME. In recent years, the critical role of dysregulated metal homeostasis in shaping the immunosuppressive TME has been gradually uncovered. By regulating immune cell functions, metabolic reprogramming, and immune signaling pathway networks, it profoundly influences tumor progression and immune escape.
Metal homeostasis imbalance fundamentally shapes the immunosuppressive TME through multidimensional mechanisms. Specifically, dysregulated copper metabolism not only drives immune escape via PD-L1 upregulation and angiogenesis but induces cuproptosis and ICD to release immune-activating signals and recruit effector cells as well. Iron homeostasis dysregulation is characterized by cancer cells "hijacking" iron resources, transforming immune cells into "iron reservoirs" that polarize macrophages toward immunosuppressive M2 phenotypes and suppress the activity of NK and T cells. Notably, ferroptosis induction simultaneously kills tumor cells and releases immunogenic signals to remodel the TME into an immune-activated state. For zinc, homeostasis disruption suppresses antitumor immunity, whereas supplementation restores immune cell function, activates signaling pathways (e.g., cGAS-STING), enhances antigen presentation, and reprograms tumor immunogenicity. Similarly, magnesium deficiency impairs immune cytotoxicity, while supplementation reverses TAM phenotype, potentiates CAR-T efficacy, and synergizes with ICB. Manganese, calcium, and chromium additionally contribute indispensable TME-regulatory roles. Although there are relatively few studies on the immune regulatory effects of these elements on the TME, the potential of them and their derived nanomaterials in combination with immunotherapy is considerable but remains to be developed. For instance, manganese overcomes ICB resistance by activating the STING pathway, serves as a vaccine adjuvant, and potentiates ICD when combined with radio- or chemotherapy. Manganese-based nanomaterials further facilitate multimodal therapy through integrated drug delivery and TME remodeling [13, 114]. Collectively, these findings underscore that metal homeostasis equilibrium is pivotal for sustaining antitumor immunity, thus targeting metal-immune networks offers novel therapeutic perspectives for overcoming immunotherapy resistance.
As summarized throughout this review, the intricate interplay between metal ions and immune regulation is mediated by their profound impact on key immune molecules and pathways. The regulatory relationships between the discussed metals and critical immune markers, such as PD-L1, CTLA-4, cGAS-STING, and various cytokines, are systematically cataloged in Table 6. This synthesis underscores that targeting metal homeostasis exerts its immunomodulatory effects not randomly, but through specific, molecularly definable circuits. Acknowledging these connections is therefore crucial for designing next-generation metalloimmunotherapies that precisely modulate the immune landscape of the TME.
Regulation of key immune markers by essential metals in the TME
| Immune marker | Category | Metal(s) | Effect | Mechanism | Ref.(s) |
|---|---|---|---|---|---|
| PD-L1 | Immune checkpoint | Copper | Upregulation | Activates NF-κB, JAK/STAT, PI3K pathways; promotes immune evasion. | [20], [21, 23] |
| Zinc | Downregulation in TAMs | ZNT1 mediates its endocytosis/lysosomal degradation in macrophages. | [88] | ||
| CTLA-4 | Immune checkpoint | Copper | Upregulation | Positively correlates with copper transporter SLC31A1 expression. | [23] |
| TLR4 | Pattern recognition receptor | Zinc | Downregulation | Zinc deficiency impairs endosomal clearance, sustaining TLR4 surface expression and chronic inflammation. | [88] |
| HIF-1α | Signaling molecule | Copper | Stabilization | Copper binding inhibits its ubiquitination, amplifying hypoxia and related immunosuppression. | [28, 29], [32] |
| cGAS-STING | Signaling pathway | Manganese, Zinc | Activation | Mn²⁺ and Zn²⁺ enhance cGAS-DNA binding, boosting IFN-I response and antitumor immunity. | [106] [94] |
| NKG2D | Activating receptor | Magnesium | Stabilization | Mg²⁺ is essential for NKG2D structure and function on NK/CD8⁺ T cells. | [101] |
| LFA-1 | Adhesion molecule | Magnesium | Activation | Mg²⁺ binds to LFA-1, promoting its active conformation for T cell co-stimulation. | [102] |
| Foxp3 | Transcription factor | Zinc | Upregulation | Zn²⁺ promotes Treg differentiation via FOXO1-mediated transcription, enhancing immunosuppression. | [89] |
| TGF-β | Immunosuppressive cytokine | Copper | Downregulation | Copper chelators reduce TGF-β levels, alleviating immunosuppression. | [24] |
| IFN-γ | Inflammatory cytokine | Copper, Iron, Zinc, Magnesium | Upregulation | A common endpoint of enhanced T/NK cell function via metal-specific mechanisms. | [40] [92] [103] |
| DAMPs (HMGB1, ATP) | Immunogenic signal | Iron, Copper | Release/Upregulation | Induced by ferroptosis/cuproptosis/ICD, activating dendritic cells and T cells. | [77] [79] |
Importantly, beyond their individual roles, a complex and synergistic crosstalk exists between different intracellular metals, forming an interconnected regulatory network that amplifies their impact on the TME. For instance, intracellular Zn²⁺ disorder can disrupt Ca²⁺ homeostasis, thereby inhibiting the electron transport chain and promoting the production of endogenous ROS, which assisted the killing of tumor cells [130]. The crosstalk between zinc and copper is multifaceted. The zinc transporter ZnT1, primarily known for zinc export, also facilitates Cu²⁺ entry into cells, directly linking the homeostasis of these two metals and potentially influencing copper-dependent cell death pathways [131]. Furthermore, this crosstalk can be harnessed therapeutically; as demonstrated by a recent study, a bimetallic peroxide nanoparticle co-delivering zinc and copper ions disrupts ion homeostasis to trigger PANoptosis. This process simultaneously activates multiple cell death pathways and robustly stimulates anti-tumor immune responses [132]. These examples underscore that the strategic co-modulation of essential metals, rather than targeting them in isolation, can yield superior immunotherapeutic outcomes by exploiting their inherent biological interconnectedness.
The strategic modulation of essential metal homeostasis has emerged as a transformative approach in cancer therapy. While compelling evidence links specific metal imbalances within the TME to profound immunosuppression and tumor progression, the path to clinical translation is paved with both promise and complexity. There are three critical general challenges in the application of these strategies to be addressed. First, most metals exhibit low selectivity and content-dependent duality. Consequently, overload or aggressive chelation risks off-target toxicity in healthy tissues or exerts pro-tumorigenic effects, demanding precise spatiotemporal and dosage control. Second, substantial heterogeneity exists in TME metal content and homeostasis regulation mechanisms across tumor types and stages, implying that metal-homeostasis-based therapies cannot adopt a "one-size-fits-all" approach. Third, existing strategies targeting TME metal homeostasis often suffer from a dearth of validated, tumor-specific biological targets. This lack of precision hinders therapeutic specificity and efficacy, necessitating more in-depth mechanistic studies to elucidate fundamental regulatory pathways and identify novel, exploitable targets. Moreover, the clinical translation of metal-based interventions faces additional challenges related to biocompatibility, long-term safety, and potential off-target effects, which must be thoroughly evaluated in preclinical and clinical settings.
Metal-based nanomaterials represent pivotal tools to overcome immunotherapy resistance due to their precise targeting capabilities, multimodal synergistic effects, and immunomodulatory properties. Nanoparticles can effectively utilize the enhanced permeability and retention (EPR) effect of tumor tissues, passively targeting tumor sites by penetrating the loose gaps in tumor vascular endothelium while avoiding renal clearance or hepatic/splenic capture. Through surface ligand modifications, nanoparticles are endowed with specific recognition ability. Moreover, TME-responsive release (pH/GSH/enzyme-triggered) ensures spatiotemporal control. Functionally, nanoparticles realize multimodal synergistic platforms by carrying multiple therapeutic agents, which can induce ICD through treatments such as chemotherapy, radiotherapy, PDT, PTT, and sonodynamic therapy (SDT), while intrinsic metal effects remodel the TME and amplify the metal-immune cycle. In short, the spatiotemporally ordered release of metal ions precisely targets immune network interactions, while multimodal synergistic platforms pioneer novel therapeutic dimensions in antitumor immunotherapy.
Current research on metal homeostasis in the TME remains relatively under-explored compared to that on metal-based therapeutic applications. Future progress demands integrating metallomics with genomics, proteomics, and metabolomics to systematically map metal-ion binding patterns and dynamic regulatory networks in the TME. A key focus should be on decoding the interactome of different metals, such as the Zn²⁺/Ca²⁺ and ZnT1-mediated Zn²⁺/Cu²⁺ axes, to build a holistic view of the metal regulatory landscape. Concurrently, developing highly sensitive detection technologies—including single-cell metallomics, in vivo metal tracking systems, and subcellular-targeted nanoprobes—will not only elucidate the dynamic roles of metals in TME evolution but also directly guide therapeutic interventions.
In summary, the interaction between metal homeostasis imbalance and the TME defines a novel therapeutic dimension for cancer treatment. Through advancements in nanotechnology, multi-omics integration, and interdisciplinary innovation, targeting the metal-immune network holds promise for overcoming current immunotherapy limitations and revolutionizing precision oncology.
Abbreviations
TME: tumor microenvironment;
ICD: immunogenic cell death;
ICB: immune checkpoint blockade;
CAR-T: chimeric antigen receptor T-cell;
CAFs: cancer-associated fibroblasts;
GSH: glutathione;
ROS: reactive oxygen species;
ATP: adenosine triphosphate;
HCC: hepatocellular carcinoma;
PD-L1: programmed death-ligand 1;
LOXL4: lysyl oxidase-like 4;
ADCC: antibody-dependent cellular cytotoxicity;
VEGF: vascular endothelial growth factor;
ATOX1: antioxidant 1 copper chaperone;
HIF-1α: hypoxia-inducible factor-1α;
MT2A: metallothionein 2A;
PDK1/3: pyruvate dehydrogenase kinase 1/3;
DLAT: dihydrolipoamide S-acetyltransferase;
DCs: dendritic cells;
ATTM: ammonium tetrathiomolybdate;
DSF: disulfiram;
IFN-γ: interferon-γ;
NK: natural killer;
PS: phosphatidylserine;
MDSCs: myeloid-derived suppressor cells;
PTT: photothermal therapy;
CRC: colorectal cancer;
ER: endoplasmic reticulum;
DAMPs: damage-associated molecular patterns;
TLR7: toll-like receptor 7;
NPL4: nuclear protein localization protein 4;
ICC: intrahepatic cholangiocarcinoma;
TFR1: transferrin protein;
FPN1: ferroportin 1;
TAMs: tumor-associated macrophages;
TFRC: transferrin-transferrin receptor;
FerroCAFs: iron-loaded cancer-associated fibroblasts;
FSTL1: follistatin Like 1;
PGE2: prostaglandin E2;
IDO: indoleamine 2,3-dioxygenase;
Fer-CD4+ T: ferroptosis-associated CD4+ T cell subsets;
PMN-MDSCs: polymorphonuclear myeloid-derived suppressor cells;
DFO: deferoxamine;
DFP: deferiprone;
DFX: deferasirox;
GPX4: glutathione peroxidase 4;
DR5: death receptor 5;
HMGB1: high-mobility group box 1;
SAS: sulfasalazine;
PLT: platelet;
RCD: regulated cell death;
BMDCs: bone marrow-derived dendritic cells;
IONs: iron oxide nanoparticles;
CTLs: cytotoxic T cells;
TLR4: toll-like receptor 4;
Tregs: regulatory T cells;
LAMP1: lysosome-associated membrane protein 1;
IAPs: inhibitor of apoptosis proteins;
TNF: tumor necrosis factor;
STING: stimulator of interferon genes;
IFNAR: interferon-α/β receptor;
NKG2D: natural-killer group 2, member D;
LFA-1: lymphocyte function-associated antigen 1;
NMDAR: N-methyl-D-aspartate receptor;
VEGFa: vascular endothelial growth factor A;
STIM1: stromal interaction molecule 1;
PDT: photodynamic therapy;
MT: metallothionein;
MnSOD: manganese-dependent superoxide dismutase;
LOXs: lysyl oxidases;
EPR: enhanced permeability and retention;
SDT: sonodynamic therapy
Supplementary Material
Supplementary table.
Acknowledgements
We would like to thank all members of our labs for fruitful scientific discussion and mutual supports. We also appreciate the supports from the National Natural Science Foundation of China (32171387), National Science and Technology Major Project (NO. 2024ZD0536000) and Beijing Natural Science Foundation (NO. 7254459).
Author contributions
H.Z. and Y.W. conceived and supervised the work. X.C. and Q.W. drafted the manuscript. X.C. designed figures and performed visualization. X.C., Q.W. and H.Z. edited the manuscript. H.Z. and Y.W. reviewed the manuscript and provided essential suggestions. Q.W. and X.C. participated in literature screening and analysis of relevant studies. H.Z. and Y.W. revised the manuscript. H.Z. made substantial contributions during the revision phase. All authors have read and approved the final manuscript.
Competing interests
The authors have declared that no competing interest exists.
References
1. Cha JH, Chan LC, Song MS, Hung MC. New approaches on cancer immunotherapy. Cold Spring Harb Perspect Med. 2020 10
2. Zhang Y, Zhang Z. The history and advances in cancer immunotherapy: understanding the characteristics of tumor-infiltrating immune cells and their therapeutic implications. Cell Mol Immunol. 2020;17:807-21
3. Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF. et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366:2443-54
4. Rosenberg B, VanCamp L, Trosko JE, Mansour VH. Platinum compounds: a new class of potent antitumour agents. Nature. 1969;222:385-6
5. Rogez B, Marmri Z, Thibaudau F, Baffou G. Thermoplasmonics of metal layers and nanoholes. APL Photonics. 2021 6
6. Kabir E, Noyon MROK, Hossain MA. Synthesis, biological and medicinal impacts of metallodrugs: a study. Results Chem. 2023;5:100935
7. Fouani L, Menezes SV, Paulson M, Richardson DR, Kovacevic Z. Metals and metastasis: exploiting the role of metals in cancer metastasis to develop novel anti-metastatic agents. Pharmacol Res. 2017;115:275-87
8. Wang C, Zhang R, Wei X, Lv M, Jiang Z. Metalloimmunology: The metal ion-controlled immunity. Adv Immunol. 2020;145:187-241
9. Guo X, Tu P, Wang X, Du C, Jiang W, Qiu X. et al. Decomposable nanoagonists enable NIR-elicited cGAS-STING activation for tandem-amplified photodynamic-metalloimmunotherapy. Adv Mater. 2024;36:e2313029
10. Zhan M, Yu X, Zhao W, Peng Y, Peng S, Li J. et al. Extracellular matrix-degrading STING nanoagonists for mild NIR-II photothermal-augmented chemodynamic-immunotherapy. J Nanobiotechnology. 2022;20:23
11. Dong Z, Liu Y, Wang C, Hao Y, Fan Q, Yang Z. et al. Tumor microenvironment modulating CaCO3-based colloidosomal microreactors can generally reinforce cancer immunotherapy. Adv Mater. 2024;36:e2308254
12. Zhao L, Gui Y, Cai J, Deng X. Biometallic ions and derivatives: a new direction for cancer immunotherapy. Mol Cancer. 2025;24:17
13. Lu S, Mi Z, Liu P, Ding J, Ma Y, Yang J. et al. Repolarizing neutrophils via MnO2 nanoparticle-activated STING pathway enhances Salmonella-mediated tumor immunotherapy. J Nanobiotechnology. 2024;22:443
14. Liu X, Zhang Y, Yishake D, Luo Y, Liu Z, Chen Y. et al. Dietary intake and serum levels of copper and zinc and risk of hepatocellular carcinoma: a matched case-control study. Chin Med J. 2024;137:596-603
15. Feng Y, Zeng JW, Ma Q, Zhang S, Tang J, Feng JF. Serum copper and zinc levels in breast cancer: a meta-analysis. J Trace Elem Med Biol. 2020;62:126629
16. Zhang X, Yang Q. Association between serum copper levels and lung cancer risk: a meta-analysis. J Int Med Res. 2018;46:4863-73
17. Li T, Shi L, Wei W, Xu J, Liu Q. The trace that is valuable: serum copper and copper to zinc ratio for survival prediction in younger patients with newly diagnosed acute myeloid leukaemia. BMC Cancer. 2023;23:14
18. Ge EJ, Bush AI, Casini A, Cobine PA, Cross JR, DeNicola GM. et al. Connecting copper and cancer: from transition metal signalling to metalloplasia. Nat Rev Cancer. 2022;22:102-13
19. Tang D, Kroemer G, Kang R. Targeting cuproplasia and cuproptosis in cancer. Nat Rev Clin Oncol. 2024;21:370-88
20. Voli F, Valli E, Lerra L, Kimpton K, Saletta F, Giorgi FM. et al. Intratumoral copper modulates PD-L1 expression and influences tumor immune evasion. Cancer Res. 2020;80:4129-44
21. Tan HY, Wang N, Zhang C, Chan YT, Yuen MF, Feng Y. Lysyl oxidase-like 4 fosters an immunosuppressive microenvironment during hepatocarcinogenesis. Hepatology. 2021;73:2326-41
22. Mi J, Luo J, Zeng H, Zhang H, Jamil M, Abdel-Maksoud MA. et al. Elucidating cuproptosis-related gene SLC31A1 diagnostic and prognostic values in cancer. Am J Transl Res. 2023;15:6026-41
23. Wu JH, Cheng TC, Zhu B, Gao HY, Zheng L, Chen WX. Identification of cuproptosis-related gene SLC31A1 and upstream LncRNA-miRNA regulatory axis in breast cancer. Sci Rep. 2023;13:18390
24. Rouaen JRC, Salerno A, Shai-Hee T, Murray JE, Castrogiovanni G, McHenry C. et al. Copper chelation redirects neutrophil function to enhance anti-GD2 antibody therapy in neuroblastoma. Nat Commun. 2024;15:10462
25. Pan X, Li X, Dong L, Liu T, Zhang M, Zhang L. et al. Tumour vasculature at single-cell resolution. Nature. 2024;632:429-36
26. Soncin F, Guitton JD, Cartwright T, Badet J. Interaction of human angiogenin with copper modulates angiogenin binding to endothelial cells. Biochem Biophys Res Commun. 1997;236:604-10
27. Kohno T, Urao N, Ashino T, Sudhahar V, McKinney RD, Hamakubo T. et al. Novel role of copper transport protein antioxidant-1 in neointimal formation after vascular injury. Arterioscler Thromb Vasc Biol. 2013;33:805-13
28. Rigiracciolo DC, Scarpelli A, Lappano R, Pisano A, Santolla MF, De Marco P. et al. Copper activates HIF-1α/GPER/VEGF signalling in cancer cells. Oncotarget. 2015;6:34158-77
29. Magar AG, Morya VK, Kwak MK, Oh JU, Noh KC. A molecular perspective on HIF-1α and angiogenic stimulator networks and their role in solid tumors: an update. Int J Mol Sci. 2024 25
30. Pan Q, Kleer CG, van Golen KL, Irani J, Bottema KM, Bias C. et al. Copper deficiency induced by tetrathiomolybdate suppresses tumor growth and angiogenesis. Cancer Res. 2002;62:4854-9
31. Tsvetkov P, Coy S, Petrova B, Dreishpoon M, Verma A, Abdusamad M. et al. Copper induces cell death by targeting lipoylated TCA cycle proteins. Science. 2022;375:1254-61
32. Yang Z, Su W, Wei X, Pan Y, Xing M, Niu L. et al. Hypoxia inducible factor-1α drives cancer resistance to cuproptosis. Cancer Cell. 2025
33. Wang J, Li S, Guo Y, Zhao C, Chen Y, Ning W. et al. Cuproptosis-related gene SLC31A1 expression correlates with the prognosis and tumor immune microenvironment in glioma. Funct Integr Genomics. 2023;23:279
34. Li H, Zu X, Hu J, Xiao Z, Cai Z, Gao N. et al. Cuproptosis depicts tumor microenvironment phenotypes and predicts precision immunotherapy and prognosis in bladder carcinoma. Front Immunol. 2022;13:964393
35. Zhu X, Zhang Z, Xiao Y, Wang H, Zhang J, Wang M. et al. A pan-cancer cuproptosis signature predicting immunotherapy response and prognosis. Heliyon. 2024;10:e35404
36. Zhang G, Chen X, Fang J, Tai P, Chen A, Cao K. Cuproptosis status affects treatment options about immunotherapy and targeted therapy for patients with kidney renal clear cell carcinoma. Front Immunol. 2022;13:954440
37. Feng S, Zhang Y, Zhu H, Jian Z, Zeng Z, Ye Y. et al. Cuproptosis facilitates immune activation but promotes immune escape, and a machine learning-based cuproptosis-related signature is identified for predicting prognosis and immunotherapy response of gliomas. CNS Neurosci Ther. 2024;30:e14380
38. Tang X, Yan Z, Miao Y, Ha W, Li Z, Yang L. et al. Copper in cancer: from limiting nutrient to therapeutic target. Front Oncol. 2023;13:1209156
39. Gao F, You W, Zhang L, Shen AZ, Chen G, Zhang Z. et al. Copper chelate targeting externalized phosphatidylserine inhibits PD-L1 expression and enhances cancer immunotherapy. J Am Chem Soc. 2025;147:5796-807
40. Liu YL, Bager CL, Willumsen N, Ramchandani D, Kornhauser N, Ling L. et al. Tetrathiomolybdate (TM)-associated copper depletion influences collagen remodeling and immune response in the pre-metastatic niche of breast cancer. NPJ Breast Cancer. 2021;7:108
41. Chakraborty P, Das S, Banerjee K, Sinha A, Roy S, Chatterjee M. et al. A copper chelate selectively triggers apoptosis in myeloid-derived suppressor cells in a drug-resistant tumor model and enhances antitumor immune response. Immunopharmacol Immunotoxicol. 2014;36:165-75
42. Crowe A, Jackaman C, Beddoes KM, Ricciardo B, Nelson DJ. Rapid copper acquisition by developing murine mesothelioma: decreasing bioavailable copper slows tumor growth, normalizes vessels and promotes T cell infiltration. PLoS One. 2013;8:e73684
43. Li W, Xin H, Gao W, Yuan P, Ni F, Ma J. et al. NIR-IIb fluorescence antiangiogenesis copper nano-reaper for enhanced synergistic cancer therapy. J Nanobiotechnology. 2024;22:73
44. Solier S, Müller S, Cañeque T, Versini A, Mansart A, Sindikubwabo F. et al. A druggable copper-signalling pathway that drives inflammation. Nature. 2023;617:386-94
45. Zeng J, Chen H, Liu X, Xia H, Chen L, Lin D. et al. Cuproptosis in microsatellite stable colon cancer cells affects the cytotoxicity of CD8(+)T through the WNT signaling pathway. Chem Biol Interact. 2024;403:111239
46. Li J, Zhang G, Sun Z, Jiang M, Jia G, Liu H. et al. Immunogenic cuproptosis in cancer immunotherapy via an in situ cuproptosis-inducing system. Biomaterials. 2025: 123201.
47. Gao X, Huang H, Pan C, Mei Z, Yin S, Zhou L. et al. Disulfiram/copper induces immunogenic cell death and enhances CD47 blockade in hepatocellular carcinoma. Cancers. 2022 14
48. Doroshow DB, Bhalla S, Beasley MB, Sholl LM, Kerr KM, Gnjatic S. et al. PD-L1 as a biomarker of response to immune-checkpoint inhibitors. Nat Rev Clin Oncol. 2021;18:345-62
49. Shen Z, Cai J, Tao L, Zheng J, Ye Z, Liu Y. et al. Exploration of a screening model for intrahepatic cholangiocarcinoma patients prone to cuproptosis and mechanisms of the susceptibility of CD274-knockdown intrahepatic cholangiocarcinoma cells to cuproptosis. Cancer Gene Ther. 2023;30:1663-78
50. Wang Y, Drum DL, Sun R, Zhang Y, Chen F, Sun F. et al. Stressed target cancer cells drive nongenetic reprogramming of CAR T cells and solid tumor microenvironment. Nat Commun. 2023;14:5727
51. Guo B, Yang F, Zhang L, Zhao Q, Wang W, Yin L. et al. Cuproptosis induced by ROS responsive nanoparticles with elesclomol and copper combined with αPD-L1 for enhanced cancer immunotherapy. Adv Mater. 2023;35:e2212267
52. Liu C, Guo L, Cheng Y, Gao J, Pan H, Zhu J. et al. A mitochondria-targeted nanozyme platform for multi-pathway tumor therapy via ferroptosis and cuproptosis regulation. Adv Sci. 2025: e2407425.
53. Yang N, Guo XY, Ding J, Wang F, Liu TL, Zhu H. et al. Copper-64 based PET-radiopharmaceuticals: ways to clinical translational. Semin Nucl Med. 2024;54:792-800
54. Guo Q, Li L, Hou S, Yuan Z, Li C, Zhang W. et al. The role of iron in cancer progression. Front Oncol. 2021;11:778492
55. Candelaria PV, Leoh LS, Penichet ML, Daniels-Wells TR. Antibodies targeting the transferrin receptor 1 (TfR1) as direct anti-cancer agents. Front Immunol. 2021;12:607692
56. Marques O, Porto G, Rêma A, Faria F, Cruz Paula A, Gomez-Lazaro M. et al. Local iron homeostasis in the breast ductal carcinoma microenvironment. BMC Cancer. 2016;16:187
57. Sun JL, Zhang NP, Xu RC, Zhang GC, Liu ZY, Abuduwaili W. et al. Tumor cell-imposed iron restriction drives immunosuppressive polarization of tumor-associated macrophages. J Transl Med. 2021;19:347
58. Recalcati S, Locati M, Marini A, Santambrogio P, Zaninotto F, De Pizzol M. et al. Differential regulation of iron homeostasis during human macrophage polarized activation. Eur J Immunol. 2010;40:824-35
59. Corna G, Campana L, Pignatti E, Castiglioni A, Tagliafico E, Bosurgi L. et al. Polarization dictates iron handling by inflammatory and alternatively activated macrophages. Haematologica. 2010;95:1814-22
60. Duan X, He K, Li J, Cheng M, Song H, Liu J. et al. Tumor associated macrophages deliver iron to tumor cells via Lcn2. Int J Physiol Pathophysiol Pharmacol. 2018;10:105-14
61. Costa da Silva M, Breckwoldt MO, Vinchi F, Correia MP, Stojanovic A, Thielmann CM. et al. Iron induces anti-tumor activity in tumor-associated macrophages. Front Immunol. 2017;8:1479
62. Thielmann CM, Costa da Silva M, Muley T, Meister M, Herpel E, Muckenthaler MU. Iron accumulation in tumor-associated macrophages marks an improved overall survival in patients with lung adenocarcinoma. Sci Rep. 2019;9:11326
63. Zhang K, Liu K, Hu B, Du G, Chen X, Xiao L. et al. Iron-loaded cancer-associated fibroblasts induce immunosuppression in prostate cancer. Nat Commun. 2024;15:9050
64. Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE. et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149:1060-72
65. Yao L, Hou J, Wu X, Lu Y, Jin Z, Yu Z. et al. Cancer-associated fibroblasts impair the cytotoxic function of NK cells in gastric cancer by inducing ferroptosis via iron regulation. Redox Biol. 2023;67:102923
66. Kumar A, Ye C, Nkansah A, Decoville T, Fogo GM, Sajjakulnukit P. et al. Iron regulates the quiescence of naive CD4 T cells by controlling mitochondria and cellular metabolism. Proc Natl Acad Sci U S A. 2024;121:e2318420121
67. Ma X, Xiao L, Liu L, Ye L, Su P, Bi E. et al. CD36-mediated ferroptosis dampens intratumoral CD8(+) T cell effector function and impairs their antitumor ability. Cell Metab. 2021;33:1001-12.e5
68. Zeng W, Zhang R, Huang P, Chen M, Chen H, Zeng X. et al. Ferroptotic neutrophils induce immunosuppression and chemoresistance in breast cancer. Cancer Res. 2025;85:477-96
69. Kim R, Hashimoto A, Markosyan N, Tyurin VA, Tyurina YY, Kar G. et al. Ferroptosis of tumour neutrophils causes immune suppression in cancer. Nature. 2022;612:338-46
70. Tang B, Zhu J, Li J, Fan K, Gao Y, Cheng S. et al. The ferroptosis and iron-metabolism signature robustly predicts clinical diagnosis, prognosis and immune microenvironment for hepatocellular carcinoma. Cell Commun Signal. 2020;18:174
71. Fryknäs M, Zhang X, Bremberg U, Senkowski W, Olofsson MH, Brandt P. et al. Iron chelators target both proliferating and quiescent cancer cells. Sci Rep. 2016;6:38343
72. Abdelaal G, Veuger S. Reversing oncogenic transformation with iron chelation. Oncotarget. 2021;12:106-24
73. Mertens C, Akam EA, Rehwald C, Brüne B, Tomat E, Jung M. Intracellular iron chelation modulates the macrophage iron phenotype with consequences on tumor progression. PLoS One. 2016;11:e0166164
74. Sandoval TA, Salvagno C, Chae CS, Awasthi D, Giovanelli P, Marin Falco M. et al. Iron chelation therapy elicits innate immune control of metastatic ovarian cancer. Cancer Discov. 2024;14:1901-21
75. Wang W, Green M, Choi JE, Gijón M, Kennedy PD, Johnson JK. et al. CD8(+) T cells regulate tumour ferroptosis during cancer immunotherapy. Nature. 2019;569:270-4
76. Zhao YY, Lian JX, Lan Z, Zou KL, Wang WM, Yu GT. Ferroptosis promotes anti-tumor immune response by inducing immunogenic exposure in HNSCC. Oral Dis. 2023;29:933-41
77. Conche C, Finkelmeier F, Pešić M, Nicolas AM, Böttger TW, Kennel KB. et al. Combining ferroptosis induction with MDSC blockade renders primary tumours and metastases in liver sensitive to immune checkpoint blockade. Gut. 2023;72:1774-82
78. Jiang Q, Wang K, Zhang X, Ouyang B, Liu H, Pang Z. et al. Platelet membrane-camouflaged magnetic nanoparticles for ferroptosis-enhanced cancer immunotherapy. Small. 2020;16:e2001704
79. Efimova I, Catanzaro E, Van der Meeren L, Turubanova VD, Hammad H, Mishchenko TA. et al. Vaccination with early ferroptotic cancer cells induces efficient antitumor immunity. J Immunother Cancer. 2020 8
80. Zhan S, Cao Z, Li J, Chen F, Lai X, Yang W. et al. Iron oxide nanoparticles induce macrophage secretion of ATP and HMGB1 to enhance irradiation-led immunogenic cell death. Bioconjug Chem. 2025;36:80-91
81. Jiang Z, Lim SO, Yan M, Hsu JL, Yao J, Wei Y. et al. TYRO3 induces anti-PD-1/PD-L1 therapy resistance by limiting innate immunity and tumoral ferroptosis. J Clin Invest. 2021 131
82. Galy B, Conrad M, Muckenthaler M. Mechanisms controlling cellular and systemic iron homeostasis. Nat Rev Mol Cell Biol. 2024;25:133-55
83. Chen R, Yang Y, Miao X, Qin Z, Ni C. Zinc dyshomeostasis: an emerging hallmark of cancer. Oncol Transl Med. 2025;11(3):p 101-111
84. Vieira D, Cruzado G, Harvey E, Merle G. Advanced sensing strategies for detecting zinc levels and zinc-related biomarkers in cancer pathogenesis. Sens Biosensing Res. 2025;47:100754
85. Wang J, Zhao H, Xu Z, Cheng X. Zinc dysregulation in cancers and its potential as a therapeutic target. Cancer Biol Med. 2020;17:612-25
86. Chen B, Yu P, Chan WN, Xie F, Zhang Y, Liang L. et al. Cellular zinc metabolism and zinc signaling: from biological functions to diseases and therapeutic targets. Signal Transduct Target Ther. 2024;9:6
87. Ni C, Lou X, Yao X, Wang L, Wan J, Duan X. et al. ZIP1(+) fibroblasts protect lung cancer against chemotherapy via connexin-43 mediated intercellular Zn(2+) transfer. Nat Commun. 2022;13:5919
88. Yang D, Tian T, Li X, Zhang B, Qi L, Zhang F. et al. ZNT1 and Zn 2+ control TLR4 and PD-L1 endocytosis in macrophages to improve chemotherapy efficacy against liver tumor. Hepatology. 2024;80:312-29
89. Narayan S, Dalal R, Rizvi ZA, Awasthi A. Zinc dampens antitumor immunity by promoting Foxp3(+) regulatory T cells. Front Immunol. 2024;15:1389387
90. Sugandha N, Rizvi ZA, Dalal R, Adhikari N, Awasthi A. Zinc mediates the interplay between Th1/Treg differentiation and regulates anti-tumor immunity. J Immunol. 2023;210:245.14-14
91. Lelliott EJ, Naddaf J, Ganio K, Michie J, Wang S, Liu L. et al. Intracellular zinc protects tumours from T cell-mediated cytotoxicity. Cell Death Differ. 2024;31:1707-16
92. Yang Y, Fan H, Xu X, Yao S, Yu W, Guo Z. Zinc ion-induced immune responses in antitumor immunotherapy. CCS Chem. 2024;6:2210-29
93. Tang W, Liu H, Li X, Ooi TC, Rajab NF, Cao H. et al. Efficacy of zinc carnosine in the treatment of colorectal cancer and its potential in combination with immunotherapy in vivo. Aging. 2022;14:8688-99
94. Du M, Chen ZJ. DNA-induced liquid phase condensation of cGAS activates innate immune signaling. Science. 2018;361:704-9
95. Zhang H, Wu J, Cui L, Wang T, Jin H, Guo H. et al. Pyrithione zinc alters mismatch repair to trigger tumor immunogenicity. Oncogene. 2025
96. Yang Y, Zhu Y, Wang K, Miao Y, Zhang Y, Gao J. et al. Activation of autophagy by in situ Zn(2+) chelation reaction for enhanced tumor chemoimmunotherapy. Bioact Mater. 2023;29:116-31
97. Gong H, Lin X, Huang S. Association between magnesium depletion score and prostate cancer. Sci Rep. 2025;15:4801
98. Zhang R, Hu M, Liu Y, Li W, Xu Z, He S. et al. Integrative omics uncovers low tumorous magnesium content as a driver factor of colorectal cancer. Genomics Proteomics Bioinformatics. 2024 22
99. Huang L, Lin R, Chen J, Qi Y, Lin L. Magnesium ion: a new switch in tumor treatment. Biomedicines. 2024 12
100. Feng Y, Gao M, Xu X, Liu H, Lu K, Song Z. et al. Elevated serum magnesium levels prompt favourable outcomes in cancer patients treated with immune checkpoint blockers. Eur J Cancer. 2024;213:115069
101. Chaigne-Delalande B, Li FY, O'Connor GM, Lukacs MJ, Jiang P, Zheng L. et al. Mg2+ regulates cytotoxic functions of NK and CD8 T cells in chronic EBV infection through NKG2D. Science. 2013;341:186-91
102. Lötscher J, Martí ILAA, Kirchhammer N, Cribioli E, Giordano Attianese GMP, Trefny MP. et al. Magnesium sensing via LFA-1 regulates CD8(+) T cell effector function. Cell. 2022;185:585-602.e29
103. Yuan D, Hu J, Ju X, Putz EM, Zheng S, Koda S. et al. NMDAR antagonists suppress tumor progression by regulating tumor-associated macrophages. Proc Natl Acad Sci U S A. 2023;120:e2302126120
104. Meng X, Liu Z, Deng L, Yang Y, Zhu Y, Sun X. et al. Hydrogen therapy reverses cancer-associated fibroblasts phenotypes and remodels stromal microenvironment to stimulate systematic anti-tumor immunity. Adv Sci. 2024;11:e2401269
105. Chen X, Cen L, Liu Q, Chu Y, Feng X, Ke Y. et al. A dual-adjuvant neoantigen nanovaccine loaded with imiquimod and magnesium enhances anti-tumor immune responses of melanoma. Biomater Sci. 2022;10:6740-8
106. Lv M, Chen M, Zhang R, Zhang W, Wang C, Zhang Y. et al. Manganese is critical for antitumor immune responses via cGAS-STING and improves the efficacy of clinical immunotherapy. Cell Res. 2020;30:966-79
107. Stelling MP, Soares MA, Cardoso SC, Motta JM, de Abreu JC, Antunes MJM. et al. Manganese systemic distribution is modulated in vivo during tumor progression and affects tumor cell migration and invasion in vitro. Sci Rep. 2021;11:15833
108. Nunes-Hasler P, Maschalidi S, Lippens C, Castelbou C, Bouvet S, Guido D. et al. STIM1 promotes migration, phagosomal maturation and antigen cross-presentation in dendritic cells. Nat Commun. 2017;8:1852
109. Hausmann D, Hoffmann DC, Venkataramani V, Jung E, Horschitz S, Tetzlaff SK. et al. Autonomous rhythmic activity in glioma networks drives brain tumour growth. Nature. 2023;613:179-86
110. Leslie TK, James AD, Zaccagna F, Grist JT, Deen S, Kennerley A. et al. Sodium homeostasis in the tumour microenvironment. Biochim Biophys Acta Rev Cancer. 2019;1872:188304
111. Gao Y, Liu S, Huang Y, Li F, Zhang Y. Regulation of anti-tumor immunity by metal ion in the tumor microenvironment. Front Immunol. 2024;15:1379365
112. Zhang R, Wang C, Guan Y, Wei X, Sha M, Yi M. et al. Manganese salts function as potent adjuvants. Cell Mol Immunol. 2021;18:1222-34
113. Peng P, Cao J, Cheng W, Ming H, He B, Duan X. et al. Manganese dioxide-based in situ vaccine boosts antitumor immunity via simultaneous activation of immunogenic cell death and the STING pathway. Acta Biomater. 2025;194:467-82
114. Meng Y, Huang J, Ding J, Zhou H, Li Y, Zhou W. Mn-phenolic networks as synergistic carrier for STING agonists in tumor immunotherapy. Mater Today Bio. 2024;26:101018
115. Bao Q, Fu H, Guo Y, Hu P, Shi J. Calcium-deprivation-activated immune responses for solid tumor regression. Chem. 2024;10:1175-95
116. Zheng P, Ding B, Jiang Z, Xu W, Li G, Ding J. et al. Ultrasound-augmented mitochondrial calcium ion overload by calcium nanomodulator to induce immunogenic cell death. Nano Lett. 2021;21:2088-93
117. Guo L, Ding J, Zhou W. Converting bacteria into autologous tumor vaccine via surface biomineralization of calcium carbonate for enhanced immunotherapy. Acta Pharm Sin B. 2023;13:5074-90
118. Kang H, Zhang K, Wong DSH, Han F, Li B, Bian L. Near-infrared light-controlled regulation of intracellular calcium to modulate macrophage polarization. Biomaterials. 2018;178:681-96
119. Yang W, Feng Z, Lai X, Li J, Cao Z, Jiang F. et al. Calcium nanoparticles target and activate T cells to enhance anti-tumor function. Nat Commun. 2024;15:10095
120. Zou Q, Liao K, Li G, Huang X, Zheng Y, Yang G. et al. Photo-metallo-immunotherapy: fabricating chromium-based nanocomposites to enhance CAR-T cell infiltration and cytotoxicity against solid tumors. Adv Mater. 2025;37:e2407425
121. Yang R, Roshani D, Gao B, Li P, Shang N. Metallothionein: a comprehensive review of its classification, structure, biological functions, and applications. Antioxidants. 2024 13
122. Kojima I, Tanaka T, Inagi R, Nishi H, Aburatani H, Kato H. et al. Metallothionein is upregulated by hypoxia and stabilizes hypoxia-inducible factor in the kidney. Kidney Int. 2009;75:268-77
123. Subramanian Vignesh K, Deepe GS Jr. Metallothioneins: emerging modulators in immunity and infection. Int J Mol Sci. 2017 18
124. Yang Z, Su W, Wei X, Pan Y, Xing M, Niu L. et al. Hypoxia inducible factor-1α drives cancer resistance to cuproptosis. Cancer Cell. 2025;43:937-54.e9
125. Rice JM, Zweifach A, Lynes MA. Metallothionein regulates intracellular zinc signaling during CD4(+) T cell activation. BMC Immunol. 2016;17:13
126. Sakurai A, Hara S, Okano N, Kondo Y, Inoue J-i, Imura N. Regulatory role of metallothionein in NF-κB activation. FEBS Lett. 1999;455:55-8
127. Scott CL, Omilusik KD. ZEBs: novel players in immune cell development and function. Trends Immunol. 2019;40:431-46
128. Grujicic J, Allen AR. Manganese superoxide dismutase: structure, function, and implications in human disease. 2025; 14: 848.
129. Tenti P, Vannucci L. Lysyl oxidases: linking structures and immunity in the tumor microenvironment. Cancer Immunol Immunother. 2020;69:223-35
130. Jiang Y, Shao K, Zhang F, Wang T, Han L, Kong X. et al. "Block and attack" strategy for tumor therapy through ZnO2/siRNA/NIR-mediating Zn2+-overload and amplified oxidative stress. 2023; 4: e321.
131. Li Y, Ma J, Wang R, Luo Y, Zheng S, Wang X. Zinc transporter 1 functions in copper uptake and cuproptosis. Cell Metab. 2024;36:2118-29.e6
132. Hou G, Chen Y, Lei H, Lu Y, Liu L, Han Z. et al. Bimetallic peroxide nanoparticles induce PANoptosis by disrupting ion homeostasis for enhanced immunotherapy. Sci Adv. 2024;10:eadp7160
133. Zhou P, Qin J, Zhou C, Wan G, Liu Y, Zhang M. et al. Multifunctional nanoparticles based on a polymeric copper chelator for combination treatment of metastatic breast cancer. Biomaterials. 2019;195:86-99
134. Huang Y, Liu X, Zhu J, Chen Z, Yu L, Huang X. et al. Enzyme core spherical nucleic acid that enables enhanced cuproptosis and antitumor immune response through alleviating tumor hypoxia. J Am Chem Soc. 2024;146:13805-16
135. Zanganeh S, Hutter G, Spitler R, Lenkov O, Mahmoudi M, Shaw A. et al. Iron oxide nanoparticles inhibit tumour growth by inducing pro-inflammatory macrophage polarization in tumour tissues. Nat Nanotechnol. 2016;11:986-94
136. Korangath P, Jin L, Yang CT, Healy S, Guo X, Ke S. et al. Iron oxide nanoparticles inhibit tumor progression and suppress lung metastases in mouse models of breast cancer. ACS Nano. 2024;18:10509-26
137. He S, Yu J, Xu M, Zhang C, Xu C, Cheng P. et al. A semiconducting iron-chelating nano-immunomodulator for specific and sensitized sono-metallo-immunotherapy of cancer. Angew Chem Int Ed Engl. 2023;62:e202310178
138. Mu Y, Fan Y, He L, Hu N, Xue H, Guan X. et al. Enhanced cancer immunotherapy through synergistic ferroptosis and immune checkpoint blockade using cell membrane-coated nanoparticles. Cancer Nanotechnol. 2023;14:83
Author contact
![]() Corresponding authors: Department of Urology, Chinese PLA General Hospital, Beijing 100853, China. Department of Cell Biology, Naval Medical University, Shanghai 200433, China. Email address: nature_wangyecom (Y. Wang); hyzhuedu.cn (H. Zhu).
Corresponding authors: Department of Urology, Chinese PLA General Hospital, Beijing 100853, China. Department of Cell Biology, Naval Medical University, Shanghai 200433, China. Email address: nature_wangyecom (Y. Wang); hyzhuedu.cn (H. Zhu).