Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Visualizing axonal myelin...
Informing drug development in...
Molecular imaging of...
Targeting neurodegeneration and...
Advanced magnetic resonance...
Concluding remarks
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2026; 16(4):1630-1657. doi:10.7150/thno.119559 This issue Cite
Review
Translational molecular imaging and drug development in multiple sclerosis
Daniel Tay1, Hazem Ahmed2, Alyaa Dawoud3, Mohamed Salam4, Luca Gobbi5, Uwe Grether5, Martin R. Edelmann5, Matthias B. Wittwer5, Ludovic Collin5, Kenneth Atz5, James Keaney5, Maude Giroud5, Alexia Rossi4, Antonio Giulio Gennari4, Gennaro Pagano5, Neil John Parrot5, Muhamed Barakovic5, Axel Rominger6, Catherine Gebhard7, Simon M. Ametamey2, Amit M. Saindane8, Steven H. Liang8 ![]() , Achi Haider5,9
, Achi Haider5,9 ![]()
1. Department of Biology and Applied Sciences, ETH Zurich, Otto-Stern-Weg 1, 8093 Zurich, Switzerland.
2. Center for Radiopharmaceutical Sciences ETH-PSI-USZ, Institute of Pharmaceutical Sciences ETH, Vladimir-Prelog-Weg 4, 8093 Zurich, Switzerland
3. Biochemistry Department, Faculty of Pharmacy and Biotechnology, German University in Cairo, 11835 Cairo, Egypt.
4. Department of Nuclear Medicine, University Hospital Zurich, University of Zurich, Zurich, Switzerland.
5. Pharma Research and Early Development, Roche Innovation Center Basel, F. Hoffmann-La Roche Ltd., Basel, Switzerland.
6. Department of Nuclear Medicine, Inselspital, Bern University Hospital, University of Bern, Freiburgstr. 18, 3010 Bern, Switzerland.
7. Department of Cardiology, University Hospital Bern, Bern, Switzerland.
8. Department of Radiology and Imaging Sciences, Emory University, 1364 Clifton Road, Atlanta, GA 30322, USA.
9. Department of Radiology, Division of Nuclear Medicine and Molecular Imaging Massachusetts General Hospital and Harvard Medical School, 55 Fruit Street, Boston, MA 02114, USA.
Received 2025-6-14; Accepted 2025-9-30; Published 2026-1-1
Abstract

Multiple sclerosis (MS) is a chronic inflammatory neurodegenerative disorder that typically affects young adults and is primarily characterized by demyelinating lesions in the central nervous system (CNS). According to the Revised McDonald Criteria, the clinical diagnosis of MS can be established based on a combination of clinical observations, the presence of focal lesions in at least two distinct CNS areas on magnetic resonance imaging (MRI) and the detection of specific oligoclonal bands in the cerebrospinal fluid. Conventional MRI remains a cornerstone of MS diagnosis and disease monitoring, providing high-resolution assessments of lesion burden and brain atrophy. In addition, advanced MRI methods are increasingly applied in research settings to probe myelin integrity, iron deposition, and biochemical changes, with the potential to complement established diagnostic workflows in the future. Despite remarkable advances in the management of MS over the past two decades, complex differential diagnoses and the lack of effective imaging tools for therapy monitoring remain major obstacles, thus channeling the development of innovative molecular imaging probes that can be harnessed in clinical practice. Indeed, positron emission tomography (PET) has a significant potential to advance the contemporary diagnosis and management of MS. Given the solid body of evidence implicating myelin dysfunction in the pathophysiology of MS, myelin-targeted imaging probes have been developed, and are currently under clinical evaluation for MS diagnosis and therapy monitoring. In parallel, ligands for the 18 kDa translocator protein (TSPO) and the cannabinoid receptor type 2 (CB2R) have been employed to capture neuroinflammatory processes by visualizing microglial activation, while other tracers allow the assessment of synaptic integrity across various disease stages of MS. Further, PET probes have been employed to delineate the role of activated microglia and facilitate the assessment of synaptic dysfunction across all disease stages of MS. This review discusses the challenges and opportunities of translational molecular imaging by highlighting key molecular concepts that are currently leveraged for diagnostic imaging, patient stratification, therapy monitoring and drug development in MS. Moreover, we shed light on potential future developments that hold promise to advance our understanding of MS pathophysiology, with the ultimate goal to provide the best possible patient care for every individual MS patient.
Keywords: translational molecular imaging, multiple sclerosis, demyelination, positron emission tomography (PET), neuroinflammation, tracer development, drug development
Introduction
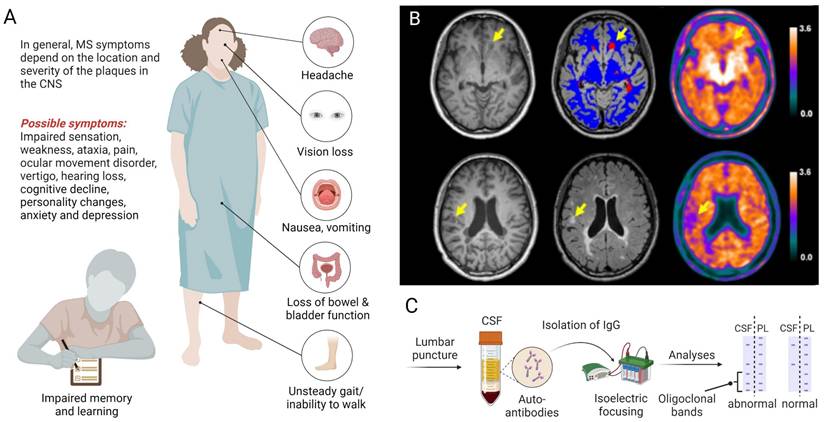
Multiple sclerosis (MS) is an inflammatory neurodegenerative disease that primarily affects the central nervous system (CNS) and is characterized by demyelinating lesions, gliosis and neuroaxonal degeneration, ultimately resulting in physical disability [1, 2]. Notably, MS affects young adults - with a disease onset between 20-40 years of age - initially manifesting as reversible episodes of neurological deficits (relapsing-remitting MS) in most patients [3]. Over the course of disease, however, patients are increasingly plagued by the development of persisting neurological deficits and permanent disability, a disease stage that is referred to as secondary progressive MS [4]. Notably, around 10% of patients present with a progressive disease course from the onset and fall into the category of primary progressive MS. While geographical differences in the relative prevalence of MS subtypes were not significant [5], several studies have corroborated a higher prevalence of MS in women [6-8]. Depending on the localization and severity of CNS lesions, clinical symptoms can include blurred vision with associated pain, impaired sensation in torso and extremities, weakness, ataxia, focal sensory disturbance, ocular movement dysfunction, vertigo, hearing loss and facial sensory disturbance (Figure 1A) [9]. Given the broad range of possible symptoms, the diagnosis of MS relies on a combination of clinical observations, spatiotemporal dissemination of focal lesions within the CNS on neuroimaging (Figure 1B) and laboratory findings, including the presence of specific oligoclonal bands in the cerebrospinal fluid (Figure 1C), as outlined in the Revised McDonald Criteria [10].
Clinical presentation and biomarker-guided diagnosis of multiple sclerosis (MS). A. The clinical manifestation of MS depends on the location and severity of lesions, involving a myriad of possible symptoms that affect several organs and ultimately lead to disability. B. Diagnostic imaging with T1-weighted (left panel), T2-weighted (middle panel) MRI and 18F-florbetapir PET (right panel) can be used to detect damaged white matter lesions (yellow arrows). Upper and lower rows represent examples from two distinct patients with relapsing-remitting MS according to the revised McDonalds criteria [10]. Figure 1B, Adapted with permission from Elsevier, Zhang et al., doi: 10.1016/j.eclinm.2021.100982, copyright 2021. C. The diagnosis of MS is supported by laboratory findings, including the presence of specific oligoclonal bands in the CSF following lumbar puncture. Abbreviations: CSF, cerebrospinal fluid; IgG, immunoglobulin G; MRI, magnetic resonance imaging; PET, positron emission tomography; PL, plasma.
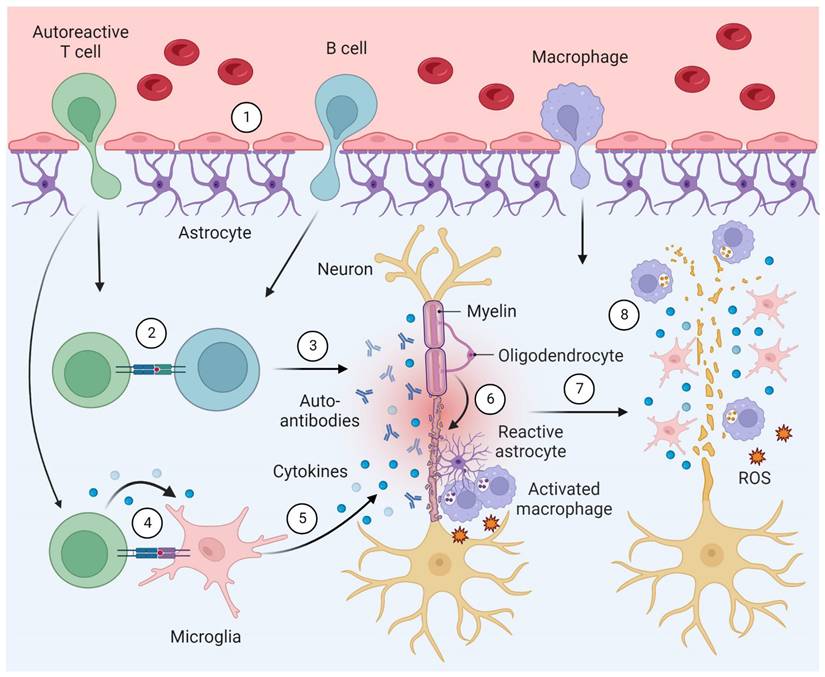
Neuroinflammation plays a fundamental role at all stages of MS, where the innate and adaptive immune system are both implicated in the pathophysiology [2]. Key hallmarks include CNS infiltration by macrophages and autoreactive T cells, facilitated by a leaky blood-brain barrier, as well as the production of antibodies against autoantigens. As the disease progresses, an excess release of pro-inflammatory mediators by activated microglia and astrocytes is observed, along with increasing numbers of autoantibody producing plasma cells (Figure 2) [3]. Notably, these processes involve pivotal bidirectional cell-to-cell interactions. Indeed, cell-to-cell interactions between T cells and microglia have been a topic of extensive research in recent years. The current body of evidence suggests that T cells play a crucial role in the activation of resident microglia, leading to the release of pro-inflammatory cytokines and the destruction of myelin in MS (Figure 2) [11-14]. Similarly, cell-to-cell interactions between T cells and B cells contribute to B cell differentiation, promoting the production of autoantibodies [11-13]. Collectively, these processes ultimately result in axonal demyelination and degeneration [2].
Simplified model of the pathophysiology in multiple sclerosis (MS). Pathophysiological hallmarks include (1) an impaired blood-brain barrier function, leading to infiltration of the central nervous system (CNS) by lymphocytes and macrophages. (2) Crosstalk between T and B lymphocytes prompts (3) the production of antibodies against oligodendrocyte antigens. (4) T cells interact with microglia, prompting (5) microglial activation and cytokine release. (6) Chronic inflammation ultimately leads to (6) axonal demyelination and the formation of glial scars by reactive astrocytes that are believed to exert protective functions by isolating damaged tissue. Macrophages are recruited to MS lesions, contributing to the phagocytosis of myelin debris - a process that triggers the release of pro-inflammatory cytokines and reactive oxygen species (ROS). (7) Axonal degeneration in MS lesions is driven by (8) activated microglial cells and macrophages.
To date, the treatment of MS can be divided into three main classes; 1) Short-term symptomatic therapies such as with nabiximols [15, 16] and fampridine [17, 18] to combat fatigue, pain and spasticity, 2) the management of acute MS relapses with high-dose corticosteroids, and 3) disease-modifying therapies (DMTs) that aim at attenuating the long-term clinical consequences of chronic inflammatory disease by attenuating peripheral immune cell activation and CNS infiltration [2, 3]. Strenuous efforts have been made to develop effective DMTs in the past two decades, which has led to the approval of various effective DMTs to date. These drugs provide a versatility of options to tailor MS therapy to individual patient's needs. For instance, traditional injectable DMTs (e.g. interferon β or glatiramer acetate) have traditionally been considered the first-line treatment option due to their favorable risk-benefit profiles. However, traditional injectable DMTs are linked to injection-related adverse effects - as opposed to oral DMTs such as fingolimod, teriflunomide and dimethyl fumarate - and elicit only limited efficacy in a significant subpopulation of MS patients, often triggering a switch to more potent DMTs [19, 20]. The concept of escalation therapy has proven particularly useful in clinical routine. The latter is based on an initial use of well-tolerated but moderately effective DMTs, followed by a switch to long-lasting immunosuppressive DMTs such as alemtuzumab or ocrelizumab along the disease course [21]. In patients with aggressive disease or indicators of poor prognosis, an alternative therapeutic strategy known as induction therapy has been suggested, in which potent DMTs are used at disease onset to preclude the accumulation of irreversible neuronal damage [3]. Regardless of the therapeutic strategy, there is a consensus that DMTs should be initiated as early as possible once the diagnosis of MS has been established. Despite substantial advances in the management of MS, differential diagnoses remain a challenge in contemporary clinical practice, at least in part, due to the broad range of diseases that mimic the clinical and radiological features of MS [22, 23]. Improved biomarkers that facilitate the early assessment of therapy response and progression of disease are urgently needed. In addition to routinely used magnetic resonance imaging (MRI), molecular imaging modalities such as positron emission tomography (PET) and optical coherence tomography (OCT) are emerging as valuable tools with potential to improve our understanding of MS pathophysiology and support patient management. Indeed, OCT of the retinal nerve fiber layer (RNFL) has advanced rapidly and currently allows the assessment of neurodegeneration in MS patients with no clinical history of optic neuritis [24]. While the annual thinning of RNFL is around 0.2 μm per year in healthy individuals, it can be up to ten times higher in MS patients [25, 26]. Nonetheless, a recent prospective multicenter study concluded that there was no correlation of OCT data with disability progression in patients with relapsing-remitting MS [27]. Additional studies are required to determine the diagnostic and prognostic value of OCT in relapsing-remitting MS.
While structural MRI is well established in the diagnosis and longitudinal monitoring of MS, its limited sensitivity for detecting early or diffuse neuroinflammatory changes, microglial activation, or evolving tissue damage in normal-appearing brain regions constrains its ability to fully capture disease activity and progression [28, 29]. Advanced techniques such as magnetization transfer imaging (MTI), diffusion tensor imaging (DTI), and magnetic resonance spectroscopy (MRS) provide additional insights into myelin integrity, axonal damage, and metabolic alterations, but they are limited by modest specificity, technical variability, and lack of standardization across centers. As a result, these approaches remain largely confined to research rather than clinical practice. These gaps highlight the need for complementary molecular imaging tools that can directly target disease-relevant biological processes and provide quantitative insights into MS pathophysiology. Applications of translational molecular imaging with PET have proven useful for the non-invasive quantification of biological processes in preclinical and clinical research, including the assessment of myelin homeostasis, neuroinflammation and neurodegeneration [30-36]. The principle of PET is based on the notion that PET radionuclides undergo radioactive decay by emitting a positron, which travels a certain distance (positron range) before it annihilates with an electron, generating a pair of 511 keV γ-rays that are concurrently released in opposite directions and can be captured by co-incidence detectors [37, 38]. Substantial efforts have been made to develop PET probes that would facilitate drug development by enabling the in vivo assessment of protein expression and drug occupancy for target validation, drug biodistribution and pharmacokinetics experiments, as well as target engagement and micro-dosing studies [30, 37-39]. These efforts have ultimately led to an arsenal of imaging tools to facilitate biomedical research from bench-to-bedside. PET harbors potential to monitor the in vivo efficacy of potential MS drug agents in the pipeline [34]. Indeed, by providing real-time information on biochemical processes in living patients, PET probes have been employed to study MS and were established as clinical tools that provide information on MS onset and progression [32]. Another critical advantage of PET imaging-guided drug development constitutes the capability to provide effective patient stratification during clinical MS trials. Despite the significant advances in MS-related imaging, the vast potential of PET is yet to be further elucidated and myelin-based imaging in MS patients has the potential to provide fundamental advances in the field. [40-42]. While MRI has traditionally been employed to support research into the pathophysiology of myelin, as well as to facilitate the diagnostic workup for MS patients, MRI is primarily suited to detect alterations in physiological and anatomical properties, which are typically preceded by molecular and biochemical changes. In contrast, molecular imaging with PET enables identification and quantification of biochemical processes that precede anatomical changes, thereby identifying key determinants that can be leveraged for diagnostic and therapeutic purposes. Despite its molecular precision, PET imaging remains constrained by radiation exposure, high costs, and the need for radiopharmaceutical infrastructure, which limit its applicability in routine clinical MS care. MRI, in contrast, is widely accessible, non-invasive, and forms the basis of diagnostic criteria and prognostic value in MS [43]. Nonetheless, PET imaging offers advantages in translational research by enabling high-sensitivity quantification of molecular targets and pharmacodynamic changes - parameters that are challenging to assess with conventional MRI techniques. As such, PET is best positioned as a complementary tool within the drug development continuum, particularly in early-phase clinical trials or mechanistic studies where target abundance and engagement, receptor occupancy, or in vivo biodistribution are critical endpoints. Moreover, post-mortem studies have significantly improved our understanding of molecular pathways that contribute to demyelination and axonal degeneration in MS. However, these studies are hampered by the lack of chronological information. Non-invasive in vivo imaging has the potential to overcome this limitation by providing a systematic reconstruction of the sequence of events in longitudinal studies, as well as by allowing for adequately powered clinical trials that encompass younger patients with prodromal and early stages of MS.
This work discusses the opportunities and limitations of available translational molecular imaging probes for MS, thereby focusing on molecular pathways that are harnessed to facilitate diagnostic imaging, patient stratification, therapy monitoring and drug development. Further, this review will provide a critical discussion of contemporary molecular concepts to visualize myelin dynamics, neuroinflammation and neurodegeneration, thereby exploring potential future trends in the field.
Visualizing axonal myelin dynamics
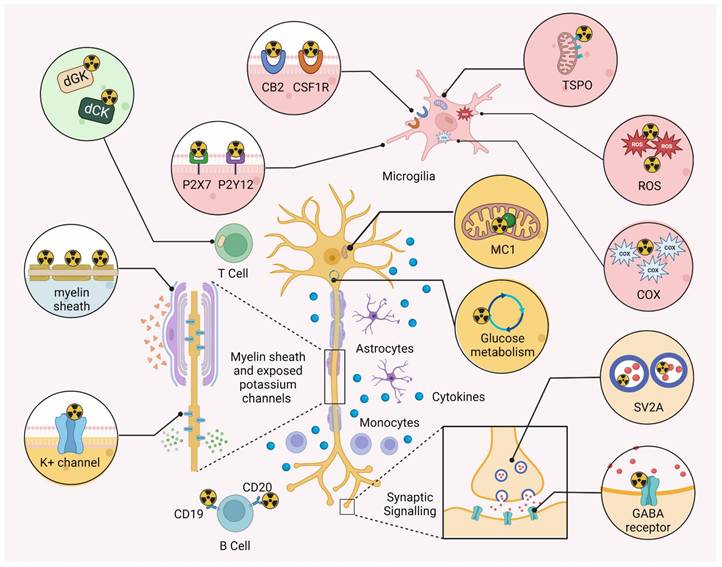
Myelin is characterized by a high lipid content, which changes the relaxation properties of adjacent water molecules, rendering MRI a suitable modality to detect advanced demyelinated lesions [44]. Hence, MRI is typically used to support the diagnosis of MS in contemporary clinical practice [10]. Despite the evident advantages, conventional MRI is limited due to its poor histopathological specificity. Indeed, while T1-weighted imaging shows some correlation with axonal density [45], it is difficult to discriminate between different processes such as demyelination, neuro-axonal damage, re-myelination and neuroinflammatory conditions [46]. Similarly, it is difficult to distinguish between distinct states of MS plaques (e.g. a completely demyelinated, a demyelinated “shadow plaque” and a remyelinated lesion) [31]. Along this line, the lack of histopathological specificity complicates the quantitative analysis of demyelination with MRI [31, 44]. Quantitative MRI (qMRI) techniques such as MTI [47, 48] and DTI [49-51] are able to improve histopathological specificity to a limited extent, especially with the use of novel hardware for ultra-high gradient strength MRI. However, these qMRI techhniques are not currently used for myelin imaging in clinical routine. Accordingly, the non-invasive assessment of myelin dynamics constitutes an unmet medical need in contemporary management of MS and the development of improved non-invasive imaging tools are critical to improve patient care [52]. There are three main classes of myelin-related radionuclide imaging; 1) Probes that accumulate in myelin-rich areas due to interactions with the β-sheet backbone in myelin basic protein (MBP), such as diamino stilbene (DAS) derivatives and probes that were initially developed for amyloid β (Aβ) imaging, 2) K+ channel-targeted tracers that specifically bind to K+ channels in exposed demyelinated axons and 3) coumarin-based probes that are believed to be capable of H-bonding with myelin. It should be noted, however, that the majority of reported myelin-targeted tracers primarily associate with myelin via hydrophobic interactions [53]. An overview of the different classes of myelin PET ligands is provided in Figure 3.
Translational molecular imaging concepts in multiple sclerosis (MS). Selected biological targets and processes that have been leveraged for positron emission tomography (PET) imaging in MS. PET can be used to improve diagnostic imaging and to facilitate drug discovery & development, and several probes have been successfully developed to non-invasively visualize myelination/demyelination by myelin-binding tracers and probes that image the potassium (K+) channel, which is exposed upon demyelination. Neuroinflammation imaging has traditionally relied on the visualization of biological targets that are overexpressed in activated microglia, including the 18-kDa translocator protein (TSPO), reactive oxygen species (ROS), cyclooxygenase (COX) enzymes, the cannabinoid type 2 receptor (CB2), colony stimulating factor 1 receptor (CSF1R) and purinergic receptors, P2X7 and P2Y12. Attempts to visualize the adaptive immune response have channeled the development of PET tracer that bind to CD19 and CD20 on B cells as well as probes that preferentially accumulate in T cells via a retention mechanism that involves binding to deoxyguanosine and deoxycytidine kinases (dGK and dCK). Neurodegeneration can be visualized by targeting glucose metabolism and mitochondrial complex I (MC-I), while synaptic integrity can be assessed with probes targeting the synaptic vesicle glycoprotein 2A (SV2A) or GABA receptors.
Targeting the β-sheet backbone in myelin
Despite the compelling rational of visualizing demyelinated lesions, a major challenge remains the presence of small areas of demyelination, which are typically masked due to the high abundance of surrounding intact myelin. In addition, since myelin is mostly composed of lipids, tracers for myelin are highly lipophilic, which typically results in high background radioactivity due to nonspecific tissue binding [44]. Notwithstanding these challenges, significant efforts have resulted in the development of promising probes to visualize MS lesions by interacting with the β-sheet backbone in MBP. Most stilbene-based tracers share a common DAS pharmacophore, which facilitates interactions with MBP. It has been hypothesized that the planar structure of DAS favors interactions with the unique β-sheet assembly present in MBP [54], although the exact binding mode remains to be fully elucidated [55]. The Congo red derivative, 11C-labeled 1, 4-bis (p-aminostyryl)-2-methoxybenzene (11C-BMB), was initially used for PET imaging of myelin [56], however, concerns have been raised about the histopathological specificity of 11C-BMB. The low histopathological specificity, along with a slow brain kinetics of the tracer and the short half-life of carbon-11 (20 min) have largely hampered the wider use of 11C-BMB. To overcome these limitations, a structurally optimized derivative of BMB, 11C-case imaging compound (11C-CIC) [57], was designed and synthesized based on an effective and more reliable radiolabeling procedure [57]. In a first-in-human study, 11C-CIC was deemed sensitive in detecting differences of myelin density within MS lesions [58]. Another lipophilic PET tracer, designated 11C-MeDAS (11C-N-methyl-4, 4′-diaminostilbene), was evaluated by small-animal PET in transgenic hyper-myelinated mice (Plp-AktDD) and respective control animals [59]. 11C-MeDAS exhibited favorable pharmacokinetics, target binding and blood-brain barrier penetration [59]. Demyelination and remyelination in the spinal cord of experimental autoimmune encephalitis (EAE) rat models of multiple sclerosis was successfully visualized by 11C-MeDAS PET [60], whereas the tracer demonstrated selectively for white matter regions in the brain and in spinal cord. Notably, 11C-MeDAS uptake was attenuated in the presence of inflammation-induced demyelination in EAE rats and progressively increased during the subsequent spontaneous remyelination, which was in concert with histopathological findings [54, 60]. In recent years, significant research efforts have been devoted to validating the quantitative accuracy of 11C-MeDAS and identifying suitable radiofluorinated analogs with a longer physical half-life to allow their distribution to nuclear medicine facilities without an on-site cyclotron. Quantitative PET refers to the use of kinetic tracer modeling methods such as compartment modelling, Logan analysis [61] and parametric quantification in order to break down the PET signal into perfusion- and myelin-related tracer uptake [58, 62].
A recently reported first-in-human study assessing quantitative outcome measures demonstrated that 11C-MeDAS PET can be used for accurate quantification of myelin density in MS patients [58]. The authors suggested that the reason for the improved efficacy of 11C-MeDAS over other lipophilic tracers is based upon its selectivity toward intact MBP. In particular, myelin damage was linked to a conformational change of MBP, resulting in the amelioration of the binding site for 11C-MeDAS [58].
Radiofluorinated derivatives of 11C-MeDAS, such as 18F-PENDAS [63], 18F-TAFDAS [64], or 18F-PEGMeDAS [65] were successfully synthesized and exhibited promising pharmacokinetic attributes by in vivo PET studies in rats. In conclusion, 11C-MeDAS and its fluorinated derivatives constitute promising stilbene-based tracers for the early delineation of MS lesions, with potential to facilitate diagnostic imaging in MS.
In a separate attempt to probe MBP, Aβ tracers - that emerged as invaluable tools to image amyloid deposition in neurodegenerative diseases such as Alzheimer's disease (AD) - have been repurposed for imaging white matter damage in MS [66, 67]. These Aβ tracers bind to the white matter, independently of the presence or absence of Aβ deposition in the brain [68]. Similar to the stilbene-based tracers, it is suggested that Aβ tracers bind to the white matter via hydrophobic interactions with MBP [69], although the detailed binding mechanism remains poorly understood. Notably, the β-sheet structure in both, amyloid peptide and MBP [70, 71], has been suggested as a common target for amyloid PET tracers [72]. Stankoff et al. [73] was the first to propose a potential utility of the Aβ PET ligand 11C-Pittsburgh compound (11C-PiB) for myelin imaging. 11C-PiB has been widely used and validated for Aβ plaques imaging in AD, However, given that the tracer also exhibited excess binding in white matter regions, independent of the presence of Aβ plaques [74], the binding observed in the grey matter was attributed to interactions with myelin [73]. A recent study demonstrated that 11C-PiB-based imaging was an effective tool for detecting demyelination in a non-human primate model of progressive MS [75]. This data provided initial evidence for the utility of 11C-PiB as a diagnostic marker in progressive MS. In recent years, much work has been done on advancing the quantitative use of 11C-PiB in MS [41, 42]. As such, quantitative studies revealed that 11C-PiB accumulation was up to 23% higher in myelin-rich white matter compared with grey matter regions [42]. While parametric maps were significantly correlated with the expression of genes encoding for abundant proteins in the myelin sheath, no correlation was found between quantitative 11C-PiB PET and non-myelin-associated genes [42]. Similarly, quantitative PET was employed to corroborate that 11C-PiB selectively binds to myelin in the white matter [41], including in the normal-appearing white matter [76]. In a recent study investigating the regional distribution of myelin repair in relapsing-remitting MS patients, a selective failure of remyelination in periventricular white matter lesions was detected, indicating that lesion proximity to ventricles is associated with a failure of myelin repair [77]. Notwithstanding the wide application and practical value, a key limitation of 11C-PiB PET is related to the sensitivity toward lysophosphatidylcholine (LPC) side effects - which may in some cases constitute a potential signal confounder [78]. Further, the lack of opportunities for satellite distribution due to the short half-life of carbon-11 confines the use of this probe to nuclear medicine facilities with an on-site cyclotron. These drawbacks have prompted calls for the use of suitable radiofluorinated amyloid tracer analogs.
Radiofluorinated amyloid PET ligands such as 18F-florbetapir and 18F-florbetaben, have recently been tested in patients with MS [79, 80]. In a study by Matías-Guiu et al. [79], twelve patients with established MS diagnosis and three healthy controls underwent MRI and 18F-florbetaben PET. 18F-Florbetaben uptake was measured in demyelinated plaques, normal-appearing white matter [80], and the grey matter [79]. Mean SUV relative to cerebellum was higher in normal-appearing white matter than in damaged WM and was significantly correlated with severity of patient disability [79]. Moreover, the latter study included a heterogeneous group of MS patients, including five individuals each with relapsing-remitting or secondary progressive MS, and two individuals with primary progressive MS. The authors found the most pronounced reduction of tracer uptake in the damaged white matter of individuals with relapsing-remitting MS [79]. Similarly, a study with 18F-florbetapir that encompassed twelve MS patients found a significantly reduced tracer uptake in damaged white matter lesions compared to intact white matter areas for all patients included [80]. The PET signal was significantly correlated with volumes of white matter regions on MRI, particularly in patients that were in the active phase of disease. These findings were further supported by the recent report of Zhang et al., in which the authors conducted a longitudinal study examining the utility of 18F-florbetapir for monitoring demyelination lesions [81]. In the latter study, it was observed that 18F-florbetapir uptake successfully differentiated between demyelination and inflammatory edema, which is typically indistinguishable on MRI. Moreover, results of the Expanded Disability Status Scale (EDSS) [82], a widely used clinical metric to assess MS-related disability, was correlated with the 18F-florbetapir PET signal, thus indicating that 18F-florbetapir may constitute a suitable tool for disease monitoring in MS [83]. In another 18F-florbetapir study encompassing 18 relapsing-remitting MS patients and 12 healthy controls, it was shown that demyelination in MS was not restricted to lesions detected through conventional MRI, but also involved the normal appearing white matter [40]. In addition to PET, SPECT probes have been suggested for myelin imaging in MS, with four diaryl oxadiazole derivatives that were evaluated as novel myelin-targeted probes [84]. Among these compounds, 123I-1,3,4-PODP-DM-based SPECT/CT was used to visualize LPC-induced demyelination in the mouse brain. While these results indicated a potential utility of 123I-1,3,4-PODP-DM as a SPECT probe for myelin imaging in MS, the available data stems from preclinical observations and clinical data are required to corroborate the utility of this probe to monitor disease progression as well as to predict response to therapy.
While the conventional wisdom implies that Aβ tracers predominantly target myelin in MS patients, this concept has been increasingly challenged by the notion that Aβ itself may constitute a biomarker of demyelination [80]. Indeed, various reports corroborated that the amyloid precursor protein (APP) accumulates in damaged axons [85-87], [88]. In particular, a high APP immunoreactivity was observed in actively demyelinating MS lesions but not in chronic lesions, suggesting a peculiar alteration of APP abundancy across disease stages [89]. Aβ may be involved in axonal remyelination, given that the β-site APP-cleaving enzyme 1 (BACE1) is involved in the cleavage of neuregulin 1, a protein that plays a critical role in oligodendrocyte differentiation and remyelination. Intriguingly, the genetic deletion of BACE1 during development leads to hypomyelination in mice [90]. While contemporary evidence points towards a potential protective role of Aβ in MS, CSF Aβ1-42 has been suggested as a putative biomarker of MS progression [91]. In conclusion, demyelinated lesions typically appear as areas of low tracer uptake on amyloid PET in MS patients. As such, contemporary evidence supports the concept that the attenuated 18F-florbetapir uptake in MS lesions is indeed driven by the reduced abundancy of myelin. Nonetheless, further studies are required to delineate the molecular mechanism by which amyloid PET ligands are capable of detecting demyelination. Despite the remaining knowledge gaps, commercial availability due to the widely established production sites for the use in AD, as well as encouraging preclinical and clinical MS studies to date render amyloid PET a valid option to be further elucidated for diagnostic applications in MS.
Probing demyelination via exposed K+ channels
An important consideration for probes that identify demyelinated lesions via a “negative” signal, which is characterized by attenuated tracer accumulation in the lesion compared to adjacent areas, is that spillover effects from adjacent areas hamper the quantitative assessment of myelin abundancy. Accordingly, a different approach has been proposed that leverages PET tracer selectively targeted toward proteins that become more accessible in demyelinated axons (Figure 3), ultimately resulting in an excess tracer uptake at sites of demyelination (“positive” signal) [44]. Upon demyelination, axonal K+ channels are upregulated and increasingly exposed to the surrounding microenvironment [92]. Notably, it was found that 4-aminopyridine (fampridine) and other aminopyridines act as K+ channel blockers by forming several hydrogen bonds with the K+ channel α-subunit [93]. Fampridine is the most extensively investigated drug in this class and is currently approved for symptomatic treatment for walking disability in patients with MS [94]. Radiolabeled derivatives of fampridine were suggested as potential PET tracers for imaging demyelinated lesions. Indeed, this concept has been leveraged in experimental models, and its successful application in humans could provide transformative insights; not only could it become a powerful tool for characterizing the extent of demyelination in the brains of MS patients, but also serve as a marker for therapy monitoring. Further, combined use of fampridine-based PET with MRI provides unique insights by enabling simultaneous quantification of surviving demyelinated axons and their subsequent myelin repair. Such a hybrid imaging approach may lay the foundation for a powerful outcome measure in clinical trials assessing the utility of novel remyelination therapies [31]. The development of fampridine analogues for PET imaging is presented as a case example in the drug development chapter to illustrate the role of molecular imaging in supporting lead identification and optimization during the drug development process for MS.
Myelin imaging with coumarin-based probes
Coumarins belong to a family of heterocyclic benzopyrones that can be naturally derived from plants and exhibit a wide array of biological activities, providing a versatile therapeutic profile [95]. Accordingly, coumarins have been harnessed as anticoagulant, antibacterial, anti-inflammatory, antioxidant, antitumor, antiviral and neuroprotective agents [96]. For instance, 3-(4-aminophenyl)-coumarin has been suggested as an exploratory therapy for AD [97-100]. Similarly, coumarin-based molecular imaging probes have been developed for fluorescence [101] and PET [102] imaging of myelin. Notably, coumarin-based probes are believed to exhibit superior binding properties toward myelin compared to stilbene-based tracers due to inherent structural characteristics, including the potential capability to undergo hydrogen binding [102]. It was hypothesized that interactions between coumarins and myelin were likely to take place at myelin sites that are enriched with nonpolar amino acids and fatty acids of myelin-associated lipids. Given that structure-activity relationship studies with coumarin derivatives revealed that the lack of a dimethylamino group substantially reduced the affinity for MBP, it is hypothesized that coumarins gain access to myelin binding sites by specific interactions through their dimethylamino groups, [102]. Further, studies unveiled that proteins in the myelin sheath have an overall positive charge at physiological pH due to an excess of lysine and arginine residues [103], potentially acting as hydrogen bond donors to the dimethylamino moiety in coumarins [102]. Given the promising pharmacological properties, Watanabe et al. recently reported on the development of several radioiodine labeled coumarin derivatives, of which radioiodine-labeled target compound 21 [104] has been deemed potentially useful for myelin-targeted SPECT imaging in MS. In addition to the suggested interactions with myelin, recent molecular modelling studies have raised the possibility that some coumarin derivatives may act as K+ channel blockers [105]. Such observations were made with several coumarin derivatives, including xanthotoxin [106], osthole [107], coumarsabin [108], and furochromones [109]. Nonetheless, it should be noted that the development of coumarin-based tracers for myelin imaging is still in its infancy and more work remains to be done to assess their potential for clinical applications in MS and beyond.
Comparison studies of myelin-targeted probes
To date, attempts to compare different classes of tracers for myelin imaging are limited. Nonetheless, a few comparison studies can be found in the literature. For instance, 11C-PiB uptake was compared with 11C-CIC and 11C‑MeDAS in rodents [110]. All tracers showed fast brain uptake and distribution volumes that were largely in accordance with myelin-rich regions, however, 11C-PiB revealed low uptake in some myelinated regions, including the cerebellum, midbrain and brain stem. Overall, 11C-MeDAS distribution seemed to correlate better with myelin density that the distributions of 11C-PiB and 11C-CIC, indicating that 11C-MeDAS was more suitable for quantitative myelin imaging [54, 110]. Another comparison study of 18F-florbetaben, 18F-florbetapir, 18F-flutemetamol, 11C-MeDAS, and 11C-PiB in baboons concluded that 18F-florbetapir and 18F-florbetaben exhibited superior tracer performance characteristics and indicated that 18F-florbetapir and 18F-florbetaben constitute the most suitable probes for myelin imaging [111]. Nonetheless, given the small number of comparison studies in this field, including the limited data in higher species, it seems premature to draw conclusions on the most suitable class of tracers for myelin imaging in patients. Hence, there is a need for more comparison studies where the efficiency and tracer performance characteristics are compared in non-human primates and humans.
Informing drug development in multiple sclerosis
Contemporary drug development constitutes an exceptionally costly and complex endeavor, with a strikingly low overall success rate. Between 2009 and 2018, the median research and development (R&D) cost per FDA-approved drug was estimated at $985 million [112]. Against this backdrop, translational molecular imaging has emerged as a valuable strategy to streamline drug development and enhance the probability of success. These efforts have led to the establishment of an extensive imaging toolkit that supports biomedical research from bench to bedside. Although the integration of PET imaging in drug development for MS is still in its early stages, a growing number of radioligands are currently being investigated in clinical trials (Tables 1 and 2). In this section, we delineate how molecular imaging can inform critical stages of drug development in MS, drawing on selected case studies to illustrate its potential [38].
Selected diagnostic clinical trials with radioligands used in multiple sclerosis. Molecular targets, radioligands used, primary and secondary endpoints - sorted by date in decreasing order. Abbreviations: TEAE: Treatment-emergent adverse event;. QSM: Quantitative susceptibility mapping; SUV: Standardized uptake values; EDSS: Expanded disability status scale; SPMS: Secondary progressive multiple sclerosis; rrMS: Relapsing and remitting MS; VT: Tissue volume of distribution; NfL: Neurofilament Light chain; TSPO: Translocator protein; NET: Norepinephrine transporter; GABAA: Gamma-aminobutyric acid receptor A; SV2A: Synaptic vesicle glycoprotein 2A.
| Clinical trial | Target | Radioligand | Primary end points | Secondary end points | Study dates |
|---|---|---|---|---|---|
| NCT01031199 (Phase I) | TSPO | 18F-FEDAA1106 (BAY85-8101) | Standard quantification variables from 3D PET and brain modeling. | Evaluated safety parameters, including TEAEs, electrocardiograms and vital signs | 2009 - 2009 |
| NCT01738347 (Phase I) | TSPO | 18F-GEH120714 | Evaluated safety of 18F-GEH120714 Injection in healthy subjects and subjects with rrMS | Determined biodistribution, internal radiation dosimetry and effective dose in healthy volunteers. | 2013 - 2016 |
| NCT01651520 (Early Phase) | GABAA | 11C-Flumazenil | Changes in neuronal atrophy and EDSS | n/a | 2013 - 2019 |
| NCT02207075 (Early Phase) | TSPO | 11C-PK-11195 | Baseline and changes of whole brain uptake of 11C-PK-11195 in SPMS subjects. | Relationship between T2-hyperintensities, conventional MRI measures and whole brain 11C-PK-11195 PET | 2014 - 2020 |
| NCT02649985 (Phase I & II) | TSPO | 18F-PBR06 and 11C-PBR28 | Measured VT levels | Measured SUVR values | 2016 - 2021 |
| NCT03207464 (Phase I & II) | NET | 11C-MRB | Measured VT levels | n/a | 2017 - 2021 |
| NCT04126772 (Early Phase) | TSPO and P2X7 | 11C-PK11195 11C-SMW139 | 11C-PK11195 binding, 11C-SMW139 binding and QSM signal in MS patient brains | Binding and QSM signal in brains of healthy controls, as well changes in clinical MS metrics | 2019 - 2025 |
| NCT04239820(Early Phase) | TSPO | 11C-PK11195 | Changes in microglia-activity in MS patients measured by 11C-PK11195 | MRI metrics, EDSS, MS composite score, changes in serum neurofilament light (NfL) and glial fibrillary acid protein (GFAP), QSM signal in MS patient brain. | 2020 - 2024 |
| NCT04634994 (Early Phase) | SV2A | 18F-SDM8 (18F-SynvesT-1) | Calculated VT over various brain regions | Calculated SUV over various brain regions | 2021 - 2022 |
| NCT04699747 (Phase I) | Potassium channels | 18F-3F4AP | Comparison of binding for 18F 3F4AP in the brain of healthy volunteers and MS subjects, as well as measure TEAEs. | Comparison of binding for 18F 3F4AP in brain lesions of multiple sclerosis subjects and determine variability in healthy controls and multiple sclerosis subjects | 2021 - 2024 |
| NCT04710550 (Phase I) | Potassium channels | 18F-3F4AP | Number of subjects with TEAEs | n/a | 2021 - 2025 |
| NCT03807973 (Phase I) | White blood cells | 89Zr-oxine | SUVs for the patients and healthy controls in various brain regions. | n/a | 2021 - 2025 |
| NCT05064436 (Phase I) | Bruton's tyrosine kinase | 11C-BMS-986196 | Incidence of TEAEs | Calculated SUV and VT in the brain | 2021- 2023 |
| NCT05147532 (Early Phase) | Myelin, TSPO | 18F-Florbetaben 18F-DPA-714 | Proportion of lesional demyelinated voxels at baseline that undergo remyelination during the relapsing and the progressive phases of MS | Percentage of voxels classified as significantly activated compared to control white matter, and the number/proportion of activated MS lesions measured via 18F-DPA-714 PET | 2022 - 2024 |
The past two decades have witnessed a remarkable expansion in the therapeutic strategies available for the treatment of MS, shifting the landscape from nonspecific immunomodulation toward more targeted strategies that suppress peripheral inflammation and increasingly targeting CNS pathology [113-115]. Disease-modifying therapies (DMTs) constitute the backbone of MS treatment and act at various stages of the immunopathological cascade, including immune cell differentiation, trafficking across the blood-brain barrier (BBB), microglial activation, and myelin repair (Figure 4A). First-generation DMTs such as interferon-β and glatiramer acetate remain widely used, particularly in patients with milder relapsing forms of MS, where they modulate T cell polarization and suppress antigen presentation by peripheral immune cells. These agents have been complemented by oral DMTs such as fumarates, which exert immunomodulatory and cytoprotective effects, at least in part, through activation of the nuclear factor erythroid 2-related factor 2 (Nrf2) pathway [116], and teriflunomide, which inhibits pyrimidine biosynthesis and reduces lymphocyte proliferation [117]. A significant advance in MS therapy has been the development of monoclonal antibodies targeting CD20 expressed on B cells [118]. Of note, ocrelizumab, ofatumumab, rituximab, and ublituximab mediate B cell depletion via antibody- or complement-dependent cytotoxicity, showing robust efficacy in reducing relapse rates, lesion activity, and disability progression in both relapsing-remitting and primary progressive MS [115]. Investigational anti-CD19 agents such as inebilizumab and CD19-targeted chimeric antigen receptor T (CAR-T) cells hold potential to extend the therapeutic reach by targeting earlier B cell lineages, as CD19 was shown to be abundantly expressed at early development stages of B cells [119]. Other established therapies include sphingosine-1-phosphate (S1P) receptor modulators, including fingolimod, siponimod, ozanimod, and ponesimod, prevent lymphocyte egress from lymphoid tissues, thereby limiting CNS infiltration [120, 121].
Interplay between imaging and disease modifying therapies (DMTs) in multiple sclerosis (MS). A. Mechanistic landscape of current and emerging (in blue colour) DMTs in MS. Primary sites of action of established and emerging DMTs (highlighted in blue) are shown across the MS disease continuum, spanning peripheral immune modulation, leukocyte trafficking and central nervous system (CNS) infiltration, microglial modulation, and remyelination therapy. B cell-depleting therapies include approved anti-CD20 monoclonal antibodies such as ocrelizumab, ofatumumab, rituximab, and ublituximab, as well as investigational anti-CD19 approaches (inebilizumab and CD19 chimeric antigen receptor T (CAR-T) cells). Bruton's tyrosine kinase (BTK) inhibitors such as tolebrutinib, evobrutinib, and fenebrutinib modulate signaling in B cells and myeloid lineage cells and are currently in late-stage development. T cell-modulating agents, including interferon-β (IFN-β), glatiramer acetate, fumarates (e.g. dimethyl fumarate), and teriflunomide, act by modulating the balance of pro- and anti-inflammatory T cell subsets, limiting T cell activation, and reducing the proliferation of lymphocytes. Sphingosine-1-phosphate (S1P) receptor modulators - including fingolimod, siponimod, ozanimod and ponesimod - block lymphocyte egress from secondary lymphoid tissues, thereby limiting CNS infiltration, while natalizumab prevents immune cell entry by inhibiting α4β1 integrin-mediated adhesion at the blood-brain barrier (BBB). DMTs such as cladribine, alemtuzumab, and mitoxantrone exert broader cytotoxic effects on proliferating immune cells. Within the CNS, masitinib and ibudilast inhibit the activation of microglia and mast cells, while temelimab, a monoclonal antibody targeting the envelope protein of the human endogenous retrovirus-W (HERV-W ENV), reduces innate immune activation and hold potential to preserve myelin integrity. Clemastine fumarate, a muscarinic receptor antagonist, enhances remyelination by promoting differentiation of oligodendrocyte precursor cells (OPCs), while metformin restores the regenerative capacity of aged OPCs. Elezanumab, an anti-repulsive guidance molecule-a (anti-RGMa) antibody, facilitates axonal regeneration and remyelination, and BTK inhibitors may additionally modulate CNS-resident microglia and macrophages. Pathways that can be probed with molecular imaging are denoted by a red star. Collectively, the therapeutic landscape of MS is rapidly evolving toward CNS-penetrant, pathway-specific agents that not only suppress peripheral inflammation but also directly modulate CNS disease processes and promote tissue repair. B. Receptor occupancy of PIPE-307. PIPE-307 displays dose-dependent M1 receptor occupancy in mouse brain following oral administration, with an ED₅₀ of 0.4 mg/kg. Total and nonspecific binding were determined using 3H-PIPE-307 in forebrain and cerebellum, respectively. Time course studies indicate that 30 mg/kg achieves rapid, full occupancy with ~50% occupancy persisting at 16 h, while 3 mg/kg maintains ~50% occupancy for approximately 10 h. This study exemplifies how tritiated radioligands can be used to quantify ex vivo target engagement and inform dose selection during early drug development. Figure. 4B was reprinted with permission from National Academy of Sciences, Poon et al. copyright (2024) doi: 10.1073/pnas.2407974121. C. Brain distribution of TSPO ligand 18F-DPA-714. Following intravenous administration of 304 MBq 18F-DPA-714, brain radioactivity was visualized at early (0-10 min) and late (30-90 min) post-injection time points. Parametric distribution volume ratio (DVR) maps were generated using the cerebellum as a reference region. This study illustrates the use of PET imaging to quantify TSPO expression as a surrogate for microglial activation in humans. Red star highlights imaging targets leveraged in MS drug development. Figure. 4B was reprinted with permission from Elsevier, Arlicot et al. copyright (2012) doi: 10.1016/j.nucmedbio.2011.10.012.
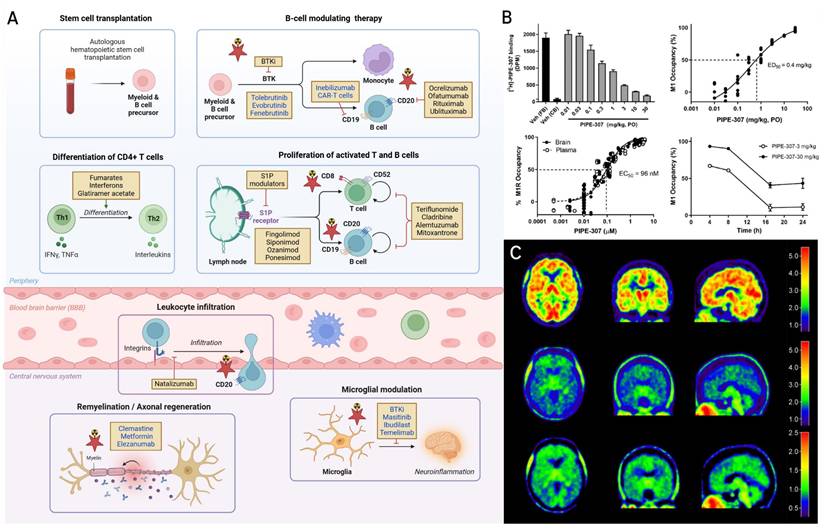
Natalizumab, a monoclonal antibody targeting the α4β1 integrin, blocks leukocyte adhesion and transmigration across the BBB and is associated with high efficacy in relapsing forms of MS, albeit with a suggested risk of progressive multifocal leukoencephalopathy in susceptible individuals [122, 123]. Beyond immune suppression, emerging therapies are being designed to address inflammation localized within the CNS and promote repair. Bruton's tyrosine kinase (BTK) inhibitors, including tolebrutinib, evobrutinib, and fenebrutinib, modulate B cell signaling and innate immune responses mediated by microglia [124, 125]. These agents are currently in late-phase development and offer the potential to cross the BBB and target CNS resident microglia [126-129]. In addition, strategies focused on neuroprotection and remyelination are gaining momentum. Masitinib, a tyrosine kinase inhibitor, and ibudilast, a phosphodiesterase inhibitor, have shown promise in reducing microglial activation and oxidative damage in progressive MS [130, 131]. Temelimab, a monoclonal antibody against the envelope protein of the human endogenous retrovirus-W (HERV-W ENV), may counteract microglia-mediated demyelination and support myelin preservation [132]. Several remyelination therapies are also under investigation. Clemastine fumarate, a muscarinic receptor antagonist, enhances oligodendrocyte precursor cell (OPC) differentiation and was among the first agents to demonstrate functional remyelination in a clinical trial [133]. Similarly, metformin has been shown to rejuvenate aged OPCs and restore their remyelination potential [134].
Finally, the anti-repulsive guidance molecule A (RGMa) antibody, elezanumab, facilitates axonal regeneration and remyelination and is currently being explored in relapsing forms of MS (NCT03737851) [135]. Collectively, the therapeutic pipeline is rapidly evolving beyond conventional immune modulation, toward mechanistically targeted, CNS-penetrant interventions that may ultimately transform treatment paradigms in MS. Several of the pathways modulated by DMTs can also be visualized with molecular imaging tools, creating opportunities to non-invasively monitor treatment effects and guide therapeutic decision-making. These intersections between imaging and pharmacotherapy are illustrated in Figure 4A, with pathways highlighted that are currently accessible by PET.
The integration of molecular imaging into drug development pipelines has opened new avenues to accelerate therapeutic innovation and refine clinical decision-making. While the diagnostic utility of PET lies in its capacity to detect and characterize disease-related processes, its value in drug development is centered around enabling in vivo assessments of drug biodistribution and pharmacokinetics, as well as in assessing pharmacodynamic response markers. Within the realm of MS, PET imaging serves as a transformative tool to non-invasively quantify target occupancy and monitor treatment-induced changes in neuroinflammation, demyelination, and neurodegeneration across different species [32]. Notably, such assessments provide key pharmacological insights, including central nervous system penetration, target specificity and selectivity, as well as the relationship between exposure in the target compartment and biological effect. A notable example is the use of 11C-PBR28 PET to quantify neuroinflammation by targeting the 18-kDa translocator protein (TSPO), which is upregulated on activated microglia. Indeed, studies have demonstrated significantly elevated TSPO levels in MS patients compared to healthy controls, underscoring the utility of TSPO PET as a biomarker of disease activity and a potential endpoint for therapeutic trials [136]. Collectively, PET imaging offers a powerful platform to de-risk early-stage drug development, enhance pharmacological precision, and support adaptive trial designs. As molecular imaging tools continue to evolve, their application in MS drug development is expected to expand - facilitating the emergence of tailored therapeutic strategies that address the complex pathophysiology of the disease while maximizing clinical benefit. In this chapter, we illustrate the interplay between molecular imaging and therapeutic development through selected case examples, spanning the continuum from target identification and validation to lead optimization, entry to human (EIH) studies and subsequent clinical development.
Target identification and validation
The target identification and validation stages focus on establishing the presence of a molecular target under pathophysiological conditions and verifying that its modulation may confer therapeutic benefit. Translational PET imaging enables non-invasive, quantitative validation of target expression in living subjects, providing direct evidence that a target is accessible and pharmacologically tractable in the human CNS. Of note, while traditional approaches to interrogating neurodegeneration or neuroimmune pathology in MS have largely relied on post-mortem analyses, these methods preclude longitudinal assessments in living patients and are limited by the instability of target proteins ex vivo - hence they are prone to bias that results from the post-mortem delay prior to tissue fixation. In contrast, non-invasive PET imaging provides an effective opportunity to capture dynamic biological processes in vivo. Two prominent strategies under evaluation are synaptic density imaging with synaptic vesicle glycoprotein 2A (SV2A)-targeted PET and immuno-PET approaches for visualizing immune cell populations.
The extent to which synaptic degeneration contributes to MS pathophysiology remains a subject of debate, in part due to conflicting post-mortem findings. Indeed, a systematic review by Mock et al. concluded that the link between synaptic loss and MS is still controversial, highlighting the need for in vivo techniques that circumvent the limitations of post-mortem tissue analyses [137]. Quantitative PET imaging offers a solution by allowing for the early live detection of synaptic dysfunction - before structural abnormalities become apparent on MRI [138]. SV2A is ubiquitously expressed in presynaptic terminals and serves as a robust biomarker of synaptic density. Several PET tracers have been developed for this purpose, including ligands from the UCB- and SDM-series. Notably, 11C-UCB-J [139] and 18F-labeled derivatives such as 18F-SDM-7 and 18F-SDM-8 [140, 141] have demonstrated high selectivity and brain penetration, with successful clinical application in neurodegenerative diseases such as PD, where significantly reduced SV2A binding was observed in the substantia nigra [142]. A clinical study assessing the utility of 18F-SDM-8 in a cohort of 30 MS patients (NCT04634994) has been initiated, and its results could shed light on the utility of SV2A PET as a biomarker for synaptic pathology in MS. Beyond neurodegeneration, molecular imaging can be leveraged for target validation by visualizing components of the adaptive immune response. Along this line, the capability to assess the involvement of B and T lymphocytes would be of crucial interest, given the body of evidence supporting the concept of autoimmune-like activity against CNS autoantigens in MS [1]. The breakdown of tolerance to autoantigens activates previously dormant autoreactive B and T cells in MS patients, although the underlying triggers are controversially discussed [143]. The availability of non-invasive imaging tools to visualize CNS infiltration by specific lymphocytes would substantially facilitate research into the origins of autoantigen generation, potentially opening up new avenues for therapeutic intervention. While activated B cells were visualized by targeting CD19 or CD20 with PET using 64Cu-CD19-mAb [144] or 64Cu-rituximab [145], respectively, activated T cells have been investigated in MS using a tracer known as 18F-F-AraG [146], which is a substrate of abundantly present kinases in T cells. Similarly, immuno-PET with 89Zr-labelled antibodies is increasingly popular due to the long half-life of 89Zr (t1/2 = 3.3 days) [147], as evidenced by its use in several existing clinical trials (Tables 1 and 2). For instance, 89Zr-Df-crefmirlimab, which shows strong affinity to CD8+ T-cells, has been used in clinical trials to identify the distribution of CD8+ T cells in the CNS of adults with MS and hence provide in-vivo imaging of the immune response, thereby enabling in vivo imaging of the adaptive immune response (NCT05849467). Notably, the ability to longitudinally track immune cell dynamics could be harnessed to identify and validate novel targets in MS. By capturing temporal shifts in B or T cell activity following intervention, immuno-PET offers a promising strategy to assess pharmacodynamic effects and establish the translational validity of a given therapeutic pathway.
Lead identification and optimization
Lead identification involves screening candidate molecules for their ability to modulate the intended biological target, followed by structure-activity relationship studies and chemical optimization. PET imaging plays a dual role: first, in the development of selective radiotracers as tool compounds to support lead selection and optimization via assessment of drug-target interactions, and second, by probing the pharmacodynamic effects of advanced molecules in preclinical animal models that include not only rodents but also non-human primates. When a lead structure is already available, the structural scaffold may serve as a basis to develop a suitable PET radioligand in parallel to the actual drug development efforts. As discussed before, PET tracers derived from fampridine for targeting demyelinated axons via exposed potassium channels constitutes a notable example in this regard. Potassium channel blockers represent a class of neuroprotective agents that mitigate the functional consequences of demyelination by inhibiting the pathological efflux of K⁺ ions through voltage-gated potassium (Kv) channels exposed on denuded axons (Figure 3) [148]. The molecular mechanism of fampridine action - through blockade of Kv channels via hydrogen bonding with the channel's α-subunit - was first elucidated through electrophysiological studies and later substantiated by molecular docking analyses [93, 149]. Despite its clinical success, fampridine exhibits dose-dependent off-target activity and lacks subtype-selectivity for Kv channels [150]. This underscores the need for more selective Kv channel modulators that preserve the therapeutic benefit while minimizing unwanted effects. Inspired by the story of fampridine, a series of structurally related analogues have been developed for PET imaging to visualize Kv channel exposure in demyelinated axons. These include 11C-3-methyl-4-aminopyridine (11C-3Me4AP) [151], 18F-3-fluoro-4-aminopyridine (18F-3F4AP) [152], and 11C-3-methoxy-4-aminopyridine (11C-3MeO4AP) [153]. Among these, 18F-3F4AP and 11C-3MeO4AP have shown favorable brain penetration, metabolic stability, and sensitivity to focal demyelination in non-human primates [152, 153]. Preliminary first-in-human images of 18F-3F4AP have recently been disclosed, and two clinical trials are underway to evaluate this tracer in patients with MS (NCT04699747) and other demyelinating disorders (NCT04710550) [154, 155]. If validated, 18F-3F4AP PET imaging could represent a paradigm shift in the non-invasive assessment of demyelination, providing spatially resolved, quantitative measures to inform both diagnostic and therapeutic strategies. Following clinical validation, these probes could serve as valuable tools to support the optimization of next-generation Kv channel blockers and accelerate their progression toward human translation.
Human translation and clinical development
Early clinical development focuses on demonstrating a suitable pharmacokinetic profile and pharmacological activity at safe doses in humans, thereby establishing a therapeutic window. A major advantage of molecular imaging lies in its versatility, allowing both purely diagnostic imaging for patient stratification as well as target occupancy and therapy monitoring to optimize the dosing regimen in patients. A prominent example of molecular imaging facilitating clinical development in MS is PIPE-307, a selective M1 receptor antagonist designed to promote remyelination, which was supported by PET-based receptor occupancy studies in the clinic. Emerging evidence suggests that the differentiation of oligodendrocyte precursor cells (OPCs) is negatively regulated by the M1 subtype of the muscarinic acetylcholine receptor - with studies demonstrating that pharmacological blockade or genetic deletion of M1 receptor promotes oligodendrocyte maturation and enhances remyelination in preclinical models [156-158]. Building on these mechanistic insights, a high-throughput screening campaign was conducted to identify small-molecule M1R antagonists using a novel combination of nanopillar array platforms and fluorescence-based readouts [157]. Among the resulting candidate compounds, PIPE-307 emerged as a highly potent and selective M1R antagonist, exhibiting nanomolar affinity in both mouse and human brain tissue homogenates, as determined using tritiated PIPE-307 (3H-PIPE-307, Figure 4B) [159]. Counter screening against other muscarinic receptor subtypes (M2-M5) revealed >10-fold functional selectivity, underscoring its utility for selective targeting of M1R. Of note, radiolabeling with tritium is a widely adopted strategy in the early stages of drug development, offering a minimally disruptive method to confirm target abundance and drug-target interactions in vitro or ex vivo [160]. Once target affinity is established, PET radiolabeling with carbon-11 or fluorine-18 is typically pursued to enable quantitative imaging in vivo. In the case of PIPE-307, its favorable binding profile justified the development of the PET-compatible analogue, 11C-PIPE-307, which was subsequently evaluated in a human receptor occupancy study (NCT04941781) - with results currently pending. In parallel, PIPE-307 has advanced into exploratory clinical development with promising outcomes. Proof of concept with PIPE-307 was obtained in the EAE mouse model, where treatment resulted in robust axonal remyelination, consistent with its proposed mechanism of action [159]. Notably, PIPE-307 has since successfully completed a Phase I clinical trial without evidence of cognitive adverse effects, and a Phase II trial (NCT06083753) is currently underway. The dual functionality of PIPE-307 - as both a therapeutic agent and a radiolabeled molecular imaging probe - exemplifies an effective paradigm shift in MS drug development that seamlessly integrates diagnostics and treatment efforts. Indeed, this approach could be extended to additional molecular targets. For instance, PIPE-791, a lysophosphatidic acid (LPA) receptor antagonist, has demonstrated the ability to modulate immunoinflammatory signaling and promote remyelination [161]. Receptor occupancy was confirmed using a tritiated analogue (3H-PIPE-791), supporting the potential for a theranostic application. However, translational studies employing a PET-compatible radioligand are warranted to evaluate the clinical feasibility and added value of this strategy. Beyond the established targets discussed above, additional clinical imaging trials are expanding the translational toolkit for MS. For example, a study investigating the combined use of the nasal anti-CD3 agent foralumab with 18F-PBR06 PET [162] to assess the capacity of this class of antibodies to attenuate microglial activation (NCT06292923). Another innovative study aims to assess remyelination of the optic nerve using 18F-florbetaben PET in conjunction with the N-methyl-D-aspartate (NMDA) receptor antagonist, ifenprodil, marking an effort to evaluate therapeutic efficacy of GluN2B-subtype selective NMDA inhibitors in MS (NCT06330077). These trials exemplify the increasing scope of PET imaging not only for mechanism-of-action studies, but also for guiding therapeutic interventions and individualized care.
Molecular imaging of neuroinflammation
For decades, TSPO-targeted PET has been the cornerstone of neuroinflammation imaging in the living human brain [32]. While TSPO is expressed in the outer mitochondrial membrane, a solid body of evidence suggests that it is highly abundant on activated microglia, rendering TSPO-targeted probes suitable tools to assess neuroinflammation in various neurological disorder, including MS (Figure 3) [163].
A series of TSPO PET ligands were successfully translated to humans, including the first-generation PET tracer, (R)-11C-PK11195, and subsequently developed analogs with improved signal-to-noise ratio. The latter include, but are not limited to, 11C-DPA-713, 18F-DPA-714 (Figure 4C), 11C-PBR28, 18F-PBR111 and 11C-ER176 [164]. Many of these probes were leveraged to study neuroinflammation in MS, providing crucial insights into the role of the immune system in the pathophysiology of MS. For instance, TSPO PET unveiled that activated microglia were involved across all stages of MS - with PET signal intensities that were closely linked to the severity of clinical symptoms [165-169]. Similarly, TSPO PET with 11C-PBR28 provided non-invasively quantification of immune cell infiltration in the cortico-meningeal compartment of MS patients, thus supporting the notion that localized inflammation within the meninges is implicated as a critical factor in the development of cortical demyelination in MS [170, 171]. Further, these studies suggested that neuroinflammation was present in brain areas that were classified as normal-appearing white matter by conventional MRI [48]. TSPO PET was capable of predicting later disease progression, pointing towards a potential prognostic role of tracers that allow early quantification of neuroinflammatory processes [172, 173]. In addition to the potential prognostic role, TSPO PET has been employed to assess the efficacy of immunotherapy in MS. Indeed, TSPO PET corroborated the reduction in microglial activity in response to natalizumab treatment in the white matter of MS patients after 1 year of treatment [174]. Similarly, the sphingosine l-phosphate receptor modulator, fingolimod, reduced microglial activation on TSPO PET in focal inflammatory MS lesions [175]. However, it should be noted that a limited number of ten MS patients was included in each of the two studies, and thus these findings are to be treated with caution. Additional clinical trials have evaluated alternative TSPO-targeted PET ligands, including 18F-FEDAA1106 (NCT01031199), 18F-GEH120714 (NCT01738347), and ¹¹C-PK11195 (NCT02207075, NCT04239820), with a focus on safety, biodistribution, and signal quantification in both relapsing-remitting and progressive MS populations (Table 1). In addition, recent interventional trials have investigated the modulation of TSPO PET signals in response to anti-CD20 therapy (Table 2). For example, two studies are evaluating the effect of ocrelizumab on microglial activation using 18F-DPA-714 [176] and 11C-PBR28 [177], providing a framework to assess treatment-related changes in TSPO signal intensity and link them to structural and clinical outcomes (NCT03691077 and NCT04230174). In a recent consensus paper by the North American Imaging in Multiple Sclerosis (NAIMS) Cooperative, it was concluded that while MRI and TSPO-positive PET are emerging as potential biomarkers of chronic active lesions in MS, these biomarkers do not have equivalent sensitivity and specificity to histopathological findings. The latter was attributed to the lack of standardization in the use of currently available imaging measures for identification, quantification, and monitoring of these lesions [178]. Nonetheless, preliminary studies have laid the foundation for standardized and adequately powered trials to come.
Selected target occupancy and therapy monitoring trials with probes used in multiple sclerosis. Molecular targets, radioligands used, primary and secondary endpoints - sorted by date in decreasing order. Abbreviations: TEAE: Treatment-emergent adverse events; MTR: Magnetization transfer ratio; QSM: Quantitative susceptibility mapping; SUV: Standardized uptake values; EDSS: Expanded disability status scale; VT: Tissue volume of distribution; WM: White matter; NfL: Neurofilament light chain; TSPO: Translocator protein; COX: Cyclooxygenase.
| Clinical trial | Target | Ligands | Primary end points | Secondary end points | Study dates |
|---|---|---|---|---|---|
| NCT03691077 (Phase III) | TSPO | Ocrelizumab, 18F-DPA714 and MRI | Determined if ocrelizumab treatment is associated with a decrease in the extent of microglial activation in WM | Decreased of microglial activation as measured by 18F-DPA-714 PET in various brain regions | 2018 - 2024 |
| NCT04230174 (Phase IV) | TSPO | 11C-PBR28, Ocrelizumab | Measured changes in 11C-PBR28 uptake in MS patient brains under Ocrelizumab therapy, incl. WM lesion load, cortical atrophy and demyelination in the cortex and in the NAWM as measured by MTR | Whether changes in 11C-PBR28 uptake or in structural imaging metrics correlate with measures of neurological disability and cognition | 2020 - 2022 |
| NCT04941781 (Phase I) | Muscarinic acetylcholine receptor M1 | 11C-PIPE-307 | M1AChR receptor occupancy determined from VT of 11C-PIPE-307 | n/a. | 2021 - 2021 |
| NCT05062083 (Phase II) | COX-1/ COX-2 | 11C-PS13 and 11C-MC1 | SUVR of Lesions with Injection of 11C-PS13 or 11C-MC1, before and after blockade with Ketoprofen or Celecoxib | SUVR of Chronic Active Lesions with Injection of 11C-PS13 and 11C-MC1 | 2022 - 2024 |
| NCT06292923 (Phase II) | CD3, TSPO | Foralumab and 18F-PBR06 | (i) Incidence of TEAEs in patients (ii) Changes in the Total Nasal Symptom Score (TNSS). Changes in microglial activation baseline measured via 18F-PBR06 PET. | (i) Changes in EDSS and other clinical scores. (ii) Changes in the mean number of gadolinium-enhancing lesions per T1-weighted MRI scan, and paramagnetic rim lesions via QSM. | 2023 - 2024 |
| NCT06083753 (Phase II) | Muscarinic acetylcholine receptor M1 | PIPE-307 MTR imaging | Number of patients with TEAE as well as Change in binocular 2.5% low contrast letter acuity (LCLA) | (i) Therapeutic efficiency via various tests of visual and physical impairment in MS patients. (ii) change in MRI measures of myelination and MS disease activity, NfL and blood concentration levels of PIPE-307 | 2023 - 2025 |
| NCT05849467 (Phase I) | CD8+ T cell | 89Zr-Df-crefmirlimab | Detected and localized infiltration of CD8+ T cells in the CNS of adults with MS and PML. | Assess safety of 89Zr-Dfcrefmirlimab in participants with CNS disease, and compare SUV before and after immune reconstitution, either spontaneous or facilitated | 2023 - 2025 |
| NCT05849467 (Phase I) | CD8+ T cell | 89Zr-Df-crefmirlimab | Detected and localized infiltration of CD8+ T cells in the CNS of adults with MS and PML via PET-CT scans | Assess safety of 89Zr-Dfcrefmirlimab, and compare SUV before and after immune reconstitution, either spontaneous or facilitated | 2023 - 2025 |
| NCT06330077 (Phase II) | myelin | Ifenprodil and 18F-florbetaben | Efficacy of ifenprodil on neural remyelination | Effect of ifenprodil treatment on remyelination levels and axonal damage in the visual pathway assessed by the amplitude of P100 and the blood concentration of NfL | 2024 - 2027 |
Despite the widespread use of TSPO PET, this class of probes is plagued by several shortcomings that are worthwhile mentioning. First, TSPO expression is not confined to microglia. Indeed, various studies have corroborated marked TSPO expression on perivascular, meningeal and choroid plexus macrophages and reactive astrocytes in the CNS [179-181]. In a few instances, TSPO expression was reported on epithelial and endothelial cells, however, more studies are needed to corroborate the latter observation [181, 182]. Another critical limitation of TSPO PET is that a number of clinically used probes are sensitive to a common polymorphism (rs6971) in the TSPO gene, imposing a need for genotyping and exclusion of the “low-affinity” variant prior to TSPO PET, which in itself may constitute a selection bias in clinical studies [163, 183, 184]. It is currently debated whether the TSPO PET ligand, 11C-ER176 [164], has the potential to allow for inclusion of all MS patients, independent of the TSPO gene variant. Another key limitation is the inability of TSPO-targeted probes to discriminate between pro- and anti-inflammatory microglia - information that can be crucial for instance to monitor response to anti-inflammatory therapy [185]. Collectively, the limitations of TSPO PET have spurred the identification of novel biomarkers for the assessment neuroinflammation in humans.
In the past two decades, a number of promising targets for neuroinflammation imaging with PET have been identified, some of which are in exploratory preclinical stages and others that have been validated in humans [185]. Among these targets, the cannabinoid receptor 2 (CB2), cyclooxygenase 1 and 2 (COX I and II), reactive oxygen species (ROS), colony stimulating factor 1 receptor (CSF1R) and purinergic receptors P2X7 and P2Y12, have been targeted with innovative PET probes that may be harnessed as MS-related biomarkers (Figure 3). Indeed, employing autoradiographic studies with brain sections of the EAE rat model and post-mortem human MS tissue, it was found that P2X7-targeted probes were associated with pro-inflammatory phenotype, while P2Y12 imaging was linked to an anti-inflammatory phenotype of innate immune cells in MS [186]. A dual-tracer clinical study combining 11C-PK11195 with the P2X7-targeted ligand 11C-SMW139 (NCT04126772) is currently ongoing, aiming to assess microglial phenotypes and complement PET readouts with quantitative susceptibility mapping. Another approach leverages the visualizing ROS as a strategy for neuroinflammation imaging due to the enhanced oxidative stress in MS [187]. ROS imaging with probes such as 11C-HM [188], 11C-DHQ1 [189], 18F-ROStrace [190] or 18F-4FN [191] constitutes an intriguing concept to further develop our understanding of the role of ROS in the pathophysiology of MS. In particular, the ability to identify inflammatory foci was demonstrated for the PET reporter, 18F-4FN, which was employed to assess innate immunity activation in murine models of neuroinflammation [191]. Along this line, CNS infiltration by myeloid cells was demonstrated on biopsy samples from patients with MS, which was corroborated by the ability of triggering receptor expressed on myeloid cells 1 (TREM1)-targeted PET with 64Cu-TREM1-mAb to monitor deleterious innate immune responses and disease progression in the EAE mouse model of MS [192]. Another important target for neuroinflammation imaging is CB2, which is only marginally expressed in healthy brain tissues and was shown to be upregulated upon microglial activation in MS [193]. Along this line, newly developed CB2 PET ligands including, but not limited to, thiazole-[194, 195], pyridine-[196-198], thiophen-[199] and oxoquinoline-[200-204]-based probes, could be of use in future studies assessing microglial activation in response to immunotherapy in MS. While the oxoquinoline derivative [11C]NE40 has been advanced to humans, clinical experience with [11C]NE40 confirmed brain penetration in humans, but did not detect the anticipated CB2R upregulation in Alzheimer's disease, highlighting challenges of CB2R-specificity and -selectivity in the CNS [205]. More recent generations of pyridine-based CB2 tracers, such as [18F]RoSMA-18-d6, have demonstrated markedly enhanced affinity and selectivity in preclinical models and postmortem human ALS tissue, with efforts underway to advance them toward clinical evaluation [185]. In parallel, the novel ligand [11C]MDTC has recently completed a first-in-human study, establishing safety, favorable dosimetry and test-retest reproducibility [206]. CB2 PET ligands remain at an experimental stage, but next-generation candidates may ultimately enable sensitive in vivo assessment of microglial activation in MS patients. CB2 PET ligands are at an experimental stage and it remains to be seen whether these tools are of added value for diagnostic applications in animal models of MS and MS patients. While the COX I and COX II-targeted probes, 11C-PS13 [207] and 11C-MC1 [208], respectively, are currently tested in a phase II trial involving 16 MS patients (NCT05062083), the CSF1R-targeted PET with 11C-CPPC proved to be highly selective for microglial cells in rodents and non-human primates and may constitute another promising candidate to further refine neuroinflammation imaging in MS [209]. Beyond the targets discussed above, additional clinical PET imaging studies are underway to explore alternative pathways in MS (Table 1). For instance, a clinical trial employing 11C-MRB (NCT03207464) is assessing norepinephrine transporter (NET) availability, which may reflect broader neuromodulatory disturbances [210]. Immune cell tracking using 89Zr-oxine to label white blood cells (NCT03807973) represents an innovative strategy to capture systemic immune cell trafficking into the CNS [211, 212]. Furthermore, a first-in-human study of 11C-BMS-986196 (NCT05064436) is exploring Bruton's tyrosine kinase (BTK) as a potential neuroinflammatory marker [213]. Lastly, a multimodal PET trial employing 18F-florbetaben and 18F-DPA-714 (NCT05147532) seeks to combine myelin and TSPO imaging to assess lesion repair and immune activation longitudinally. These studies exemplify the expanding frontiers of translational molecular imaging in MS, paving the way for new mechanistic insights and imaging-enabled trial designs.
“The sphingosine-1-phosphate (S1P) signaling axis plays a central role in regulating immune cell trafficking, vascular integrity, and CNS homeostasis, and has emerged as a clinically validated therapeutic pathway in MS [214]. Four oral disease-modifying therapies - fingolimod, siponimod, ozanimod, and ponesimod - are FDA-approved S1PR modulators that act by preventing lymphocyte egress from lymphoid tissues, thereby reducing CNS infiltration and neuroinflammation [120, 121]. Given this therapeutic advent, there has been increasing interest in developing molecular imaging tools that can non-invasively quantify S1P receptor expression and drug-target engagement in vivo, thereby advancing translational research and informing clinical decision-making. Of note, S1P receptor subtype 1 (S1PR1) is widely expressed in lymphocytes, endothelial cells, oligodendrocytes, and neurons, rendering it particularly interesting for MS pathophysiology [215]. Early PET tracers such as [11C]TZ3321 (also termed [11C]CS1P1) demonstrated selective binding to S1PR1 with high target-to-background contrast in preclinical models of vascular inflammation and EAE. Indeed, EAE rats showed increased uptake of [11C]TZ3321, which correlated with inflammatory lesions in the spinal cord, suggesting utility for monitoring neuroinflammation [216]. Notably, first-in-human studies with [11C]CS1P1 have confirmed its safety, favorable dosimetry, as well as reproducible brain uptake in healthy volunteers. Further, PET studies have indicated the ability of [11C]CS1P1 to visualize S1PR1 expression in MS lesions, aligning with MRI-detected abnormalities, thereby highlighting the potential of [11C]CS1P1 PET to serve as a biomarker for disease progression [217-220]. Building on these advances, a new generation of radiofluorinated S1PR1 tracers has been developed to overcome the logistical limitations of carbon-11. Radioligands such as [18F]TZ4877, [18F]FS1P1, and [18F]6h have demonstrated decent brain uptake and promising kinetics in vivo [221-224]. Among these, [18F]TZ4877 showed high uptake in rat spinal cord lesions, while [18F]6h exhibited robust brain penetration in primates, positioning them as potential candidates for further development. Collectively, these tracers represent an important step toward enabling quantitative, target-specific imaging of S1PR1 expression in the CNS. Although less advanced, imaging efforts targeting other S1PR subtypes are underway. [11C]5a, a S1PR2-targeted probe, has shown uptake in pancreatic and immune tissues but limited brain penetration, prompting the development of various analogs with the aim to enhance brain uptake [225, 226]. Novel radioligands for S1PR3 are at earlier stages of development, with preliminary fluorinated derivatives under evaluation. By contrast, S1PR5 is highly expressed on oligodendrocytes and natural killer cells, and has been implicated in promoting myelin preservation and neuroprotection [215]. Given the approval of siponimod, which exhibits functional selectivity for S1PR5, developing PET tracers for this receptor has become particularly interesting - to assess remyelination dynamics and immune cell trafficking in MS [227]. Taken together, S1PR-targeted molecular imaging represents a rapidly evolving field with high translational relevance for MS. PET radioligands for S1PR1 have progressed from preclinical validation to early human studies, while S1PR5-targeted imaging holds promise to monitor remyelination therapies. Looking ahead, integration of these approaches may not only refine patient stratification and therapeutic monitoring but also accelerate the development of next-generation S1P modulators with optimized CNS penetration and efficacy.”
Targeting neurodegeneration and reactive astrogliosis
Molecular imaging with PET has been suggested to probe neuroaxonal degeneration in MS. Of note, a link between low 18F-fluorodexyglucose (18F-FDG) uptake in the grey matter and cognitive dysfunction in MS patients was established around the onset of the 21 century [228]. The latter findings were later supported by a significant correlation of neuronal deterioration on magnetic resonance (MR) spectroscopy with attenuated 18F-FDG uptake in cortical and subcortical neural circuits that are relevant to cognitive function, including the hippocampus, cerebellum, caudate nucleus and prefrontal cortex [229]. However, it should be pointed out that 18F-FDG uptake is not specific to neurons and can be affected by immune cells, with significant ramifications on the reliability of 18F-FDG PET for neurodegeneration imaging. Indeed, 18F-FDG uptake has been associated with neuroinflammation and it is well established that activated microglia significantly contribute to 18F-FDG brain uptake due to their enhanced demand for glucose [230]. 18F-FDG PET in experimental autoimmune encephalomyelitis (EAE) - a common mouse model of MS - revealed that the 18F-FDG signal was associated with neuroinflammation. The authors observed that the 18F-FDG signal predicted the presence of neuroinflammatory lesions in the spinal cord and was sensitive to systemic immunosuppressive therapy [231]. Collectively, these results suggested that 18F-FDG PET images can be significantly affected by changes in leukocyte metabolism, prompting calls for more selective probes to image neurodegeneration.
Non-invasive assessment of neurodegeneration
As discussed above, attempts to visualize neurodegeneration involve probes that target SV2A. Indeed, given the ubiquitous expression of SV2A in presynaptic nerve terminals, these probes are capable of mapping synaptic density (Figure 3), providing valuable information on the progression of neurodegenerative disorders [139]. While SV2A-selective PET ligands have been successfully translated to human studies and explored in neurodegenerative disorders, their application in MS is plagued by potential limitations. In particular, post-mortem investigations have yielded inconsistent findings regarding the relationship between synaptic density and MS pathology, raising concerns about the interpretability of SV2A PET signals in this context [137]. Moreover, crude quantification of synaptic loss may lack specificity, as it does not discriminate between distinct synapse types that may be differentially affected in MS. To overcome these limitations, alternative molecular imaging strategies have been proposed to probe neurodegeneration with greater cellular or functional specificity. One such approach involves targeting gamma-aminobutyric acid (GABA) receptors [232]. GABA is a naturally occurring neurotransmitter that is widely found in the CNS and plays a critical role in the pathophysiology of various neuropathologies, including MS, schizophrenia, epilepsy, anxiety and sleep disorder [233, 234]. Notably, MR spectroscopy is not sensitive enough to distinguish between metabolic neurotransmitter concentrations and synaptic activity levels, whereas PET is more sensitive toward intra-synaptic changes, allowing for the quantification of GABA receptors across the entire brain [235]. In particular, benzodiazepines have attracted much interest among the PET community, whereas the GABAA receptor ligand flumazenil exhibited an outstanding target affinity and selectivity [236, 237]. The latter has spurred research into radiolabeled flumazenil, 11C-flumazenil (11C-FMZ) and derivatives thereof as potential PET ligands for MS [232]. Along this line, 11C-FMZ was evaluated in progressive and relapsing-remitting MS patients, displaying attenuated tracer uptake in both MS forms. Despite the small population size of 18 MS patients (9 progressive and 9 relapsing-remitting, these findings pointed toward a potential of 11C-FMZ to assess grey matter pathology in MS [238]. In concert with these observations, a recently reported multi-ligand study found that binding of 11C-FMZ correlated with that of the TSPO radioligand, 11C-PK11195, suggesting that immune-mediated GABAergic alterations may contribute to the pathogenesis of MS [239]. PET imaging was also used to show that neuronal damage is diffused throughout the cortical level in the grey matter of MS patients [238]. By mapping neuronal damage in the grey matter of patients at different MS stages, the authors observed that 11C-FMZ binding was significantly decreased in the cortical grey matter. While these findings are considered promising, the use of GABA receptor-targeted PET in MS is its infancy and further studies in larger study populations are warranted to establish tangible clinical benefits that may arise from this approach.
Evidence to date points toward inflammation-linked generation of nitric oxide (NO) and reactive oxygen species (ROS) in MS, which contributes to mitochondrial dysfunction and attenuates local ATP production in demyelinated axons [240]. Indeed, MS lesions are subject to a chronically reduced energy supply and enhanced energy demand by hyperexcitable demyelinated axons, prompting a state of energy deficiency and metabolic crisis [240-243]. Since cells generate energy primarily through the mitochondria, mitochondrial proteins have emerged as appealing target to assess the energy balance in affected neurons and hence reveal early insights about detrimental molecular variables that are capable of predicting neurodegeneration. Among the five mitochondrial complexes, mitochondrial complex I (MC-I) exhibits the lowest activity and thus constitutes the rate-limiting factor in the regulation of oxidative phosphorylation [244]. As such, MC-I targeted probes harbor potential to facilitate research into MS pathophysiology, potentially supporting the development of novel drug candidates that aim at restoring mitochondrial homeostasis in MS. To date, the most suitable MC-I PET ligand for brain imaging is 18F-BCPP-EF, which was first reported by Tsukada et al [244, 245]. While MC-I PET has been employed in a series of neurological diseases [244, 246-250], this concept has yet to be explored in MS. Nonetheless, given the rapid progress in the field, which has yielded novel PET probes with excellent performance characteristics for MC-I imaging, it is anticipated that MC-I PET ligands may find near-term applications in MS research [245].
Molecular imaging of reactive astrocytes
Astrocytes are involved at all stages of MS disease progression [251]. Astrogliosis is a process in which astrocytes are activated and undergo a spectrum of molecular, cellular, and functional changes that occur in response to all forms and severities of CNS injury and disease [252]. To date, astrogliosis constitutes an active ongoing field of research and molecular imaging of astrogliosis is at an exploratory stage. As such, applications of astrogliosis imaging in MS are limited. Of note, quantification of monoamine oxidase B (MAO-B), an enzyme located in the outer mitochondrial membrane, has been proposed as a potential target for visualizing astrogliosis [253]. In post mortem brain specimens of eleven AD patients and five control subjects, the spatial distribution of catalytic sites for MAO-B was investigated by quantitative autoradiography, suggesting preferential expression in mitochondria of reactive astrocytes that were involved in neurodegenerative remodeling [253]. Similarly, a recent first-in-human study with the highly selective MAO-B PET ligand [18F]SMBT-1 suggested that the tracer can potentially be used as a surrogate marker for astrogliosis [254]. Other MAO-B targeted PET ligands such as [18F]THK5351 and [11C]deuterium-L-deprenyl have been tested in MS patients and were found to add incremental value by identifying reactive astrocytes in a subset of inactive MS lesions [255, 256]. Beyond these ligands, early human studies with [11C]L-deprenyl and its structural analog [11C]DED confirmed retention in MAO-B-rich brain regions and increased uptake in early AD and MCI [257-259]. More recently, the reversible inhibitor [11C]SL25.1188 has been validated in first-in-human studies, offering improved quantification, favorable kinetics, and reduced susceptibility to confounding radiometabolites [260]. Collectively, these developments underscore the translational potential of MAO-B-targeted imaging biomarker of astrocyte reactivity in MS and related neurodegenerative conditions. Another strategy to image astrocytes involves imaging of metabolic or exocytotic mechanisms in reactive astrocytes. For instance, metabolic activity can be estimated with carbon-11 labeled acetate (11C-acetate). It is currently believed that 11C-acetate uptake in the brain can mainly be attributed to metabolism in astrocytes [261]. Along this line, a study found that 11C-acetate uptake was higher in the brains of MS patients, as compared to healthy controls, indicating an enhanced activation of astrocytes in the patient cohort [262, 263]. Another more exploratory concept toward imaging astrocytes includes targeting the adenosine A2A purinergic receptor (A2AR) with 11C-TMSX [264]. Indeed, 11C-TMSX uptake was increased in the normal-appearing white matter in secondary progressive MS patients and was correlated with neuroaxonal damage [265]. Further studies are warranted to assess the validity of A2AR as a target for astrogliosis imaging in MS. Overall, astrocyte PET is still in its infancy, however, the fundamental role of astrocytes in the pathophysiology of MS has been established from preclinical and post-mortem studies and the availability of suitable clinical PET ligands has potential to facilitate contemporary research and clinical care for MS patients. In particular, the concept of imaging astrogliosis via MAO-B seems promising based on the currently available body of literature.
Advanced magnetic resonance imaging techniques
Although PET imaging provides a decent combination of molecular specificity and quantitative accuracy, its broader clinical deployment remains limited by radiation burden to the patients and access to radiopharmaceutical skills and infrastructure. In contrast, advanced MRI techniques have emerged as non-radioactive alternatives in order to address the challenge of interrogating key molecular and microstructural changes in MS. One example is MTI [47, 48], which measures the exchange of magnetization between protons in free water and protons bound to macromolecules such as myelin. Another is DTI [49-51], which provides information on tissue microstructure by quantifying the local diffusion tensor of water. Diffusion MRI methods are increasingly being evaluated for clinical application in MS, particularly when combined with biophysical models such as neurite orientation dispersion and density imaging (NODDI), where the total diffusion signal is modeled as a sum of intra-neurite and free water compartments [266]. These approaches have not only focused on white matter but have also been extended to study cortical gray matter. Recently, a novel diffusion MRI model, soma and neurite density imaging (SANDI), has been applied to clinical data to investigate inflammation and degeneration in the cortical tissue [267]. Recent developments also include the introduction of novel hardware for ultra-high gradient strength diffusion MRI, such as the Connectome 2.0 scanner with gradients up to 500 mT/m [268], and the application of machine learning approaches to improve diffusion data fitting [269]. Quantitative T1 (qT1) mapping has also been explored as a sensitive measure of tissue microstructure, particularly in detecting cortical demyelination and remyelination. Recent postmortem studies demonstrated that qT1 correlates with histopathological markers of myelin and can differentiate between demyelinated, remyelinated, and normal-appearing cortical areas, underscoring its potential as a biomarker of tissue repair in MS [270]. Importantly, multimodal strategies that combine different qMRI techniques, for example, MTI with diffusion-based measures or MTI with quantitative susceptibility mapping (QSM), have shown promise in enhancing biological specificity, improving lesion characterization, and enabling a more comprehensive assessment of demyelination and repair dynamics [271]. It should be noted, however, that the reproducibility of some of these approaches remains to be validated, as studies are often conducted at different field strengths, with scanners from various vendors, as well as the use of non-standardized acquisition parameters. Another promising set of approaches are based on QSM, which measures local magnetic susceptibility to assess the spatial distribution of both paramagnetic sources (e.g., iron-containing compounds such as ferritin and hemoglobin) and diamagnetic sources (e.g., myelin and calcium) [272]. QSM has been particularly successful in identifying paramagnetic rim lesions (PRLs), a subset of chronic active lesions characterized by iron deposition in activated microglia/macrophages [273]. More recently, a technique known as χ-separation has been developed, enabling independent mapping of diamagnetic (myelin) and paramagnetic (iron) contributions to tissue susceptibility [274]. This method has been validated in relapsing-remitting MS patients, where concurrent monitoring of both demyelination and inflammation in the same voxel was demonstrated [275]. Building on this, it is now possible to acquire combined QSM and MTI data in a single MRI acquisition [276], further illustrating the versatility of these techniques. Nonetheless, it is important to note that most χ-separation studies to date have been performed on high-field research scanners, which are not widely available in routine clinical practice. In addition to these tools, sodium-23 MRI (23Na MRI) has also been explored in MS. The underlying principle is that sodium channels become abundantly available in demyelinated axons, leading to an abnormal accumulation of sodium within lesions. Indeed, increased total sodium concentration and intracellular sodium volume fraction have been shown to correlate with lesion size as well as with clinical measures of disability [277]. Moreover, 23Na MRI parameters have been reported to correlate with NODDI-derived diffusion metrics in the same patients [278]. Despite these advances, several challenges remain. The degree of histopathological specificity is still limited, complicating the quantitative analysis of demyelination with MRI [31, 44]. Reproducibility and widespread clinical availability also remain concerns, since many of these approaches rely on sophisticated biophysical modeling and state-of-the-art MRI equipment [51]. For 23Na MRI, additional hurdles include low signal-to-noise ratio due to the much lower gyromagnetic ratio of sodium compared to protons (3.8-fold lower), which leads to longer acquisition times and reduced spatial resolution. Another emerging concept relies of 19F-based MRI detection. Indeed, applications of 19F-MRI have demonstrated the ability to directly visualize and quantify fluorinated small molecules, including the FDA-approved S1PR modulator siponimod. In a proof-of-concept study, it was demonstrated that fluorine MRI is capable of mapping the biodistribution of siponimod in mice, revealing quantitative readout across the stomach, liver, kidney, thymus and the brain [279]. Using optimized ultrashort echo time sequences, siponimod was found to preferentially distribute to the cerebrum, with heterogeneous regional distribution patterns highlighting that 19F-MRI can capture not only CNS penetration but also spatial distribution patterns in drug accumulation. Importantly, sensitivity modeling indicated that acquisitions at standard 3.0 T clinical MRI systems could achieve ~2.75-fold higher relative sensitivity compared to preclinical 9.4 T setups. The study also emphasized that fluorine MRI offers the unique advantage of imaging unmodified drugs at clinically relevant doses without ionizing radiation, thereby distinguishing it from PET approaches that require radiolabeling. Collectively, these findings highlight a promising strategy for monitoring CNS biodistribution, pharmacokinetics, and open the avenues for theranostic applications of fluorinated MS therapies. While still at early stages, fluorine MRI represents an important addition to the molecular imaging arsenal, particularly for longitudinal safety studies and drug development pipelines where accessibility and non-radioactive biomarker options are critical. Taken together, these advanced MRI methods provide important insights into MS pathology. Similar to PET, MRI-based applications can interrogate physiological and pathophysiological processes, enabling quantitative assessment of molecular events. Given the diversity of applications, PET and MRI should be considered complementary modalities, with hybrid PET-MRI representing a particularly intriguing avenue for paired applications in clinical applications.
Concluding remarks
Notwithstanding the significant advances in the diagnosis and management of MS over the past two decades, limitations of contemporary clinical routine include a complex diagnostic workup that often requires sophisticated differential diagnoses, as well as the lack of effective tools to predict the course of disease and select the most effective treatment plan. Molecular imaging with PET is increasingly used in preclinical and clinical research - with the aim to overcome these limitations, however, these molecular imaging tools have not yet been implemented in clinical MS guidelines to date. While the potential of myelin-directed imaging probes for monitoring disease progression and response to therapy has been demonstrated, TSPO PET has shown initial encouraging results as a prognostic marker, which holds promise to improve clinical decision-making by allowing early clarification of the optimal therapeutic strategy, particularly for fast-progressing forms of MS. Other high-potential applications include the assessment of synaptic density and astrocyte reactivity in MS, however, more clinical data is required to corroborate the utility of these exploratory tools. Similarly, the non-invasive assessment of molecular alterations at the BBB holds potential to serve as an early marker in MS. These alterations may involve P-glycoprotein (P-gp) and multidrug resistance-associated proteins (MRPs), which are highly abundant at the BBB. Alterations in the composition of the BBB by such efflux proteins are typically reflected as shifts in the brain uptake kinetics of probes that are routinely used in the clinic [280]. The BBB integrity can further be assessed through advanced cerebral perfusion imaging techniques that utilize oxygen-15-labeled water, as well as a range of ionized PET and SPECT ligands. These radiolabeled ligands demonstrate a markedly increased uptake into brain tissues in response to compromised BBB permeability [281].
While structural and anatomical MRI remain firmly established as routine tools in clinical diagnosis and management of MS, thereby providing high-resolution images of CNS lesions, more advanced MRI techniques have been suggested to probe molecular and microstructural processes such as myelin integrity and iron deposition. These approaches are increasingly applied in research settings and may gradually transition into clinical use as protocols are standardized and validated. While inferior to MRI in terms of spatial resolution, PET offers high sensitivity and allows the use of tracer doses that are typically below a pharmacological threshold [30]. This makes PET particularly suitable for quantitative studies of drug biodistribution, receptor occupancy, and pharmacodynamic effects, which can directly inform dose selection in clinical development. Moreover, established modeling frameworks allow for absolute quantification when a metabolite-corrected input function or validated reference region is available. At the same time, it is important to note that PET comes with practical and regulatory challenges, including the need for on-site or nearby radiochemistry infrastructure, the limited half-lives of commonly used isotopes, moderate spatial resolution, and a radiation burden that may constrain repeated applications (Table 3). MRI does not share these limitations, which contributes to its wide availability and suitability for repeated longitudinal studies. Nonetheless, MRI often requires higher concentrations of contrast agents, and establishing modeling workflows for quantitative molecular imaging with MRI remains challenging. Thus, while MRI is expected to remain the clinical mainstay for structural and functional assessments, PET is likely to gain importance where highly sensitive and quantitative molecular readouts are warranted. Looking ahead, the convergence of PET with advanced MRI techniques, along with integrative analytic platforms, is poised to transform the diagnostic and therapeutic landscape of MS. The incorporation of multi-omics datasets (genomic, transcriptomic, and proteomic) with imaging-derived biomarkers, supported by artificial intelligence-driven analytical platforms, will enable more precise patient stratification, dynamic monitoring of disease progression, and prediction of therapeutic response. Such convergence is envisioned to support patient stratification, therapeutic monitoring, and biomarker-driven trial design, while also contributing to the future use of molecular imaging tools in the routine clinical management of MS.
Comparison of positron emission tomography (PET) and magnetic resonance imaging (MRI) with a specific focus on multiple sclerosis (MS). While each modality has distinct advantages and limitations. Hybrid PET/MRI applications hold potential to enhance trial design and therapy monitoring in MS.
| Positron Emission Tomography | Magnetic Resonance Imaging | |
|---|---|---|
| Sensitivity & resolution | Offers high sensitivity - typically enables tracer use at microdosing, which minimizes pharmacological interference, limited resolution compared to MRI. | Lower molecular sensitivity but widely used in MS for detecting lesions. Advanced protocols are being explored to probe inflammatory or myelin dynamics, high spatial resolution. |
| Quantification of molecular processes | Supported by established kinetic modeling frameworks that allow absolute quantification when appropriate input functions are available, directly applicable to MS drug development pipelines. | Quantitative MRI methods (e.g. myelin water imaging, quantitative susceptibility mapping) are increasingly applied in MS research to assess myelin integrity, iron deposition, and biochemical changes, though standardization remains a challenge. |
| Drug development utility | Particularly valuable in MS for visualizing myelin dynamics and neuroinflammatory targets (e.g. TSPO, P2X7, MAO-B), and for informing dose selection by quantifying CNS penetration and target occupancy of novel drug candidates. | Central to MS clinical trials as the established tool for lesion load, brain atrophy, and treatment monitoring. Advanced MRI applications (e.g. magnetization transfer, diffusion-based metrics) are being evaluated as novel biomarkers in MS. |
| Infrastructure & logistics | Requires radiochemistry facilities and radiotracer availability; the short half-life of commonly used isotopes limits broad accessibility. Radiation exposure may constrain repeated use, especially in younger MS populations. | No radiation burden and no need for tracer production, which has enabled MRI to become the routine imaging tool in MS. Longitudinal imaging of lesion evolution and cortical anatomy is readily feasible. |
| Clinical readiness | Molecular PET applications in MS remain mainly research-focused, though myelin-targeted tracers and receptor targeted ligands have entered clinical arena for MS-related applications. For instance, TSPO PET is widely used in MS research to study microglial activation. | Conventional MRI is standard of care for MS diagnosis and monitoring. Molecular MRI applications are largely investigational but may gradually transition into clinical use as validation increases. |
| Complementarity | Target-specific molecular readouts can complement MRI-derived lesion and atrophy measures. | Provides high-resolution anatomical, structural, and functional information that contextualizes PET signals and adds information on lesion burden and progression. |
| Integration | Hybrid PET/MRI allows simultaneous assessment of MS pathology at molecular, structural, and functional levels, exemplifying how multimodal imaging may support precision monitoring and biomarker-driven trial design. | |
Acknowledgements
Funding
CG was supported by grants from the Swiss National Science Foundation (SNSF), the Olga Mayenfisch Foundation, Switzerland, the OPO Foundation, Switzerland, the Novartis Foundation, Switzerland, the Swissheart Foundation, the Helmut Horten Foundation, Switzerland, the EMDO Foundation, the Iten-Kohaut Foundation, Switzerland, the University Hospital Zurich Foundation, the University of Zurich (UZH) Foundation, and the LOOP, Zurich.
Data availability
This is a literature review, data analyzed and summarized is included in the present manuscript.
Author contributions and consent
All authors contributed to the conception and design, material preparation, data collection and systematic analysis of the present review. All authors read and approved the final version of the manuscript.
Competing interests
CG has received research grants from the Novartis Foundation, Switzerland, Gerresheimer AG, Olten Switzerland, Switzerland, Bayer Pharmaceuticals, Switzerland, AMGEN, Switzerland. HA, SMA and AH are co-founders of Nemosia AG. Axel Rominger is an editor of the European Journal of Nuclear Medicine and Molecular Imaging.
References
1. Reich DS, Lucchinetti CF, Calabresi PA. Multiple sclerosis. N Engl J Med. 2018;378:169-80
2. Dendrou CA, Fugger L, Friese MA. Immunopathology of multiple sclerosis. Nat Rev Immunol. 2015;15:545-58
3. Filippi M, Bar-Or A, Piehl F, Preziosa P, Solari A, Vukusic S. et al. Multiple sclerosis. Nat Rev Dis Primers. 2018;4:43
4. Lublin FD, Reingold SC, Cohen JA, Cutter GR, Sørensen PS, Thompson AJ. et al. Defining the clinical course of multiple sclerosis: the 2013 revisions. Neurology. 2014;83:278-86
5. Compston A, Coles A. Multiple sclerosis. Lancet. 2008;372:1502-17
6. Walton C, King R, Rechtman L, Kaye W, Leray E, Marrie RA. et al. Rising prevalence of multiple sclerosis worldwide: Insights from the Atlas of MS, third edition. Mult Scler. 2020;26:1816-21
7. Alonso-Magdalena L, Zia E, Carmona ICO, Pessah-Rasmussen H, Sundström P. Incidence and Prevalence of Multiple Sclerosis in Malmö, Southern Sweden. Mult Scler Int. 2022;2022:5464370
8. Langer-Gould AM, Gonzales EG, Smith JB, Li BH, Nelson LM. Racial and Ethnic Disparities in Multiple Sclerosis Prevalence. Neurology. 2022;98:e1818-e27
9. McGinley MP, Goldschmidt CH, Rae-Grant AD. Diagnosis and Treatment of Multiple Sclerosis: A Review. Jama. 2021;325:765-79
10. Thompson AJ, Banwell BL, Barkhof F, Carroll WM, Coetzee T, Comi G. et al. Diagnosis of multiple sclerosis: 2017 revisions of the McDonald criteria. Lancet Neurol. 2018;17:162-73
11. Prinz M, Priller J. The role of peripheral immune cells in the CNS in steady state and disease. Nat Neurosc. 2017;20:136-44
12. Bielekova B, Sung MH, Kadom N, Simon R, McFarland H, Martin R. Expansion and functional relevance of high-avidity myelin-specific CD4+ T cells in multiple sclerosis. J Immunol. 2004;172:3893-904
13. van Zwam M, Huizinga R, Melief MJ, Wierenga-Wolf AF, van Meurs M, Voerman JS. et al. Brain antigens in functionally distinct antigen-presenting cell populations in cervical lymph nodes in MS and EAE. J Mol Med (Berl). 2009;87:273-86
14. Lassmann H. Multiple Sclerosis Pathology. Cold Spring Harb Perspect Med. 2018 8
15. Collin C, Ehler E, Waberzinek G, Alsindi Z, Davies P, Powell K. et al. A double-blind, randomized, placebo-controlled, parallel-group study of Sativex, in subjects with symptoms of spasticity due to multiple sclerosis. Neurol Res. 2010;32:451-9
16. Novotna A, Mares J, Ratcliffe S, Novakova I, Vachova M, Zapletalova O. et al. A randomized, double-blind, placebo-controlled, parallel-group, enriched-design study of nabiximols* (Sativex(®) ), as add-on therapy, in subjects with refractory spasticity caused by multiple sclerosis. Eur J Neurol. 2011;18:1122-31
17. Goodman AD, Brown TR, Edwards KR, Krupp LB, Schapiro RT, Cohen R. et al. A phase 3 trial of extended release oral dalfampridine in multiple sclerosis. Ann Neurol. 2010;68:494-502
18. Goodman AD, Brown TR, Krupp LB, Schapiro RT, Schwid SR, Cohen R. et al. Sustained-release oral fampridine in multiple sclerosis: a randomised, double-blind, controlled trial. Lancet. 2009;373:732-8
19. Granqvist M, Boremalm M, Poorghobad A, Svenningsson A, Salzer J, Frisell T. et al. Comparative Effectiveness of Rituximab and Other Initial Treatment Choices for Multiple Sclerosis. JAMA Neurol. 2018;75:320-7
20. Pardo G, Jones DE. The sequence of disease-modifying therapies in relapsing multiple sclerosis: safety and immunologic considerations. J Neurol. 2017;264:2351-74
21. Bowen J, Mehta R, Pelletier C, Tian M, Noxon V, Johnson BH. et al. Treatment Patterns Among Patients with Multiple Sclerosis Initiating Second-Line Disease-Modifying Therapy. Advances in Therapy. 2020;37:3163-77
22. Geraldes R, Ciccarelli O, Barkhof F, De Stefano N, Enzinger C, Filippi M. et al. The current role of MRI in differentiating multiple sclerosis from its imaging mimics. Nat Rev Neurol. 2018;14:199-213
23. Charil A, Yousry TA, Rovaris M, Barkhof F, De Stefano N, Fazekas F. et al. MRI and the diagnosis of multiple sclerosis: expanding the concept of "no better explanation". Lancet Neurol. 2006;5:841-52
24. Friese MA, Schattling B, Fugger L. Mechanisms of neurodegeneration and axonal dysfunction in multiple sclerosis. Nature Rev Neurol. 2014;10:225-38
25. Petzold A, de Boer JF, Schippling S, Vermersch P, Kardon R, Green A. et al. Optical coherence tomography in multiple sclerosis: a systematic review and meta-analysis. Lancet Neurol. 2010;9:921-32
26. Barkhof F, Calabresi PA, Miller DH, Reingold SC. Imaging outcomes for neuroprotection and repair in multiple sclerosis trials. Nat Rev Neurol. 2009;5:256-66
27. Paul F, Calabresi PA, Barkhof F, Green AJ, Kardon R, Sastre-Garriga J. et al. Optical coherence tomography in multiple sclerosis: A 3-year prospective multicenter study. Ann Clin Transl Neurol. 2021;8:2235-51
28. Filippi M, Rocca MA, De Stefano N, Enzinger C, Fisher E, Horsfield MA. et al. Magnetic resonance techniques in multiple sclerosis: the present and the future. Archives of neurol. 2011;68:1514-20
29. Kaunzner UW, Gauthier SA. MRI in the assessment and monitoring of multiple sclerosis: an update on best practice. Ther Adv Neurol Disord. 2017;10:247-61
30. Ametamey SM, Honer M, Schubiger PA. Molecular imaging with PET. Chem Rev. 2008;108:1501-16
31. Bauckneht M, Capitanio S, Raffa S, Roccatagliata L, Pardini M, Lapucci C. et al. Molecular imaging of multiple sclerosis: from the clinical demand to novel radiotracers. EJNMMI Radiopharm Chem. 2019;4:6
32. Bodini B, Tonietto M, Airas L, Stankoff B. Positron emission tomography in multiple sclerosis - straight to the target. Nat Rev Neurol. 2021;17:663-75
33. Ben-Shalom I, Karni A, Kolb H. The Role of Molecular Imaging as a Marker of Remyelination and Repair in Multiple Sclerosis. Int J Mol Sci. 2021 23
34. Thomas AM, Barkhof F, Bulte JWM. Opportunities for Molecular Imaging in Multiple Sclerosis Management: Linking Probe to Treatment. Radiology. 2022;303:486-97
35. Jain P, Chaney AM, Carlson ML, Jackson IM, Rao A, James ML. Neuroinflammation PET Imaging: Current Opinion and Future Directions. J Nucl Med. 2020;61:1107-12
36. Zhu L, Ploessl K, Kung HF. PET/SPECT imaging agents for neurodegenerative diseases. Chem Soc Rev. 2014;43:6683-91
37. Rudin M, Weissleder R. Molecular imaging in drug discovery and development. Nat Rev Drug Discov. 2003;2:123-31
38. Willmann JK, van Bruggen N, Dinkelborg LM, Gambhir SS. Molecular imaging in drug development. Nat Rev Drug Discov. 2008;7:591-607
39. Pimlott SL, Sutherland A. Molecular tracers for the PET and SPECT imaging of disease. Chem Soc Rev. 2011;40:149-62
40. Carotenuto A, Giordano B, Dervenoulas G, Wilson H, Veronese M, Chappell Z. et al. [(18)F]Florbetapir PET/MR imaging to assess demyelination in multiple sclerosis. Eur J Nucl Med Mol Imaging. 2020;47:366-78
41. Bodini B, Veronese M, García-Lorenzo D, Battaglini M, Poirion E, Chardain A. et al. Dynamic Imaging of Individual Remyelination Profiles in Multiple Sclerosis. Ann Neurol. 2016;79:726-38
42. Veronese M, Bodini B, García-Lorenzo D, Battaglini M, Bongarzone S, Comtat C. et al. Quantification of [(11)C]PIB PET for imaging myelin in the human brain: a test-retest reproducibility study in high-resolution research tomography. J Cereb Blood Flow Metab. 2015;35:1771-82
43. Rocca MA, Preziosa P, Barkhof F, Brownlee W, Calabrese M, De Stefano N. et al. Current and future role of MRI in the diagnosis and prognosis of multiple sclerosis. The Lancet regional health Europe. 2024;44:100978
44. Brugarolas P, Reich DS, Popko B. Detecting Demyelination by PET: The Lesion as Imaging Target. Mol Imaging. 2018;17:1536012118785471
45. van Waesberghe JH, Kamphorst W, De Groot CJ, van Walderveen MA, Castelijns JA, Ravid R. et al. Axonal loss in multiple sclerosis lesions: magnetic resonance imaging insights into substrates of disability. Ann Neurol. 1999;46:747-54
46. Filippi M, Rocca MA, Barkhof F, Brück W, Chen JT, Comi G. et al. Association between pathological and MRI findings in multiple sclerosis. Lancet Neurol. 2012;11:349-60
47. Chen JT, Collins DL, Atkins HL, Freedman MS, Arnold DL. Magnetization transfer ratio evolution with demyelination and remyelination in multiple sclerosis lesions. Ann Neurol. 2008;63:254-62
48. Datta G, Colasanti A, Rabiner EA, Gunn RN, Malik O, Ciccarelli O. et al. Neuroinflammation and its relationship to changes in brain volume and white matter lesions in multiple sclerosis. Brain. 2017;140:2927-38
49. Sbardella E, Tona F, Petsas N, Pantano P. DTI Measurements in Multiple Sclerosis: Evaluation of Brain Damage and Clinical Implications. Mult Scler Int. 2013;2013:671730
50. Krijnen EA, Lee H, Noteboom S, Chiang FL, Steenwijk MD, Schoonheim MM. et al. In vivo evidence for cell body loss in cortical lesions in people with multiple sclerosis. Ann Clin Transl Neurol. 2025;12:4-16
51. Granziera C, Wuerfel J, Barkhof F, Calabrese M, De Stefano N, Enzinger C. et al. Quantitative magnetic resonance imaging towards clinical application in multiple sclerosis. Brain. 2021;144:1296-311
52. Mallik S, Samson RS, Wheeler-Kingshott CA, Miller DH. Imaging outcomes for trials of remyelination in multiple sclerosis. J Neurol Neurosurg Psychiatry. 2014;85:1396-404
53. Faria Dde P, Copray S, Buchpiguel C, Dierckx R, de Vries E. PET imaging in multiple sclerosis. J Neuroimmune Pharmacol. 2014;9:468-82
54. de Paula Faria D, de Vries EF, Sijbesma JW, Dierckx RA, Buchpiguel CA, Copray S. PET imaging of demyelination and remyelination in the cuprizone mouse model for multiple sclerosis: a comparison between [11C]CIC and [11C]MeDAS. Neuroimage. 2014;87:395-402
55. Stankoff B, Poirion E, Tonietto M, Bodini B. Exploring the heterogeneity of MS lesions using positron emission tomography: a reappraisal of their contribution to disability. Brain Pathol. 2018;28:723-34
56. Stankoff B, Wang Y, Bottlaender M, Aigrot MS, Dolle F, Wu C. et al. Imaging of CNS myelin by positron-emission tomography. Proc Natl Acad Sci U S A. 2006;103:9304-9
57. Wang Y, Wu C, Caprariello AV, Somoza E, Zhu W, Wang C. et al. In vivo quantification of myelin changes in the vertebrate nervous system. J Neurosci. 2009;29:14663-9
58. van der Weijden CWJ, Meilof JF, van der Hoorn A, Zhu J, Wu C, Wang Y. et al. Quantitative assessment of myelin density using [(11)C]MeDAS PET in patients with multiple sclerosis: a first-in-human study. Eur J Nucl Med Mol Imaging. 2022;49:3492-507
59. Wu C, Wang C, Popescu DC, Zhu W, Somoza EA, Zhu J. et al. A novel PET marker for in vivo quantification of myelination. Bioorg Med Chem. 2010;18:8592-9
60. Wu C, Zhu J, Baeslack J, Zaremba A, Hecker J, Kraso J. et al. Longitudinal positron emission tomography imaging for monitoring myelin repair in the spinal cord. Ann Neurol. 2013;74:688-98
61. Logan J. Graphical analysis of PET data applied to reversible and irreversible tracers. Nucl Med Biol. 2000;27:661-70
62. Dimitrakopoulou-Strauss A, Pan L, Sachpekidis C. Kinetic modeling and parametric imaging with dynamic PET for oncological applications: general considerations, current clinical applications, and future perspectives. Eur J Nucl Med Mol Imaging. 2021;48:21-39
63. Tiwari AD, Zhu J, You J, Eck B, Zhu J, Wang X. et al. Novel (18)F-Labeled Radioligands for Positron Emission Tomography Imaging of Myelination in the Central Nervous System. J Med Chem. 2019;62:4902-14
64. Wu C, Eck B, Zhang S, Zhu J, Tiwari AD, Zhang Y. et al. Discovery of 1,2,3-Triazole Derivatives for Multimodality PET/CT/Cryoimaging of Myelination in the Central Nervous System. J Med Chem. 2017;60:987-99
65. Beuché S, Peyronneau MA, Jego B, Denis C, Bourbon P, Chauvière C. et al. Design, Radiosynthesis, and Evaluation of New Fluorinated Analogs of MeDAS for Myelin PET Imaging. J Med Chem. 2023;66:8030-42
66. Chandra A. Role of amyloid from a multiple sclerosis perspective: a literature review. Neuroimmunomodulation. 2015;22:343-6
67. Morbelli S, Bauckneht M, Capitanio S, Pardini M, Roccatagliata L, Nobili F. A new frontier for amyloid PET imaging: multiple sclerosis. Eur J Nucl Med Mol Imaging. 2019;46:276-9
68. Morbelli S, Bauckneht M. Amyloid PET Imaging: Standardization and Integration with Other Alzheimer's Disease Biomarkers. Methods Mol Biol. 2018;1750:203-12
69. Valentina G, Silvia M, Marco P. Dual-phase amyloid PET: hitting two birds with one stone. Eur J Nucl Med Mol Imaging. 2016;43:1300-3
70. Harauz G, Ishiyama N, Hill CM, Bates IR, Libich DS, Farès C. Myelin basic protein-diverse conformational states of an intrinsically unstructured protein and its roles in myelin assembly and multiple sclerosis. Micron. 2004;35:503-42
71. Stadelmann C, Timmler S, Barrantes-Freer A, Simons M. Myelin in the Central Nervous System: Structure, Function, and Pathology. Physiol Rev. 2019;99:1381-431
72. de Paula Faria D, Vlaming ML, Copray SC, Tielen F, Anthonijsz HJ, Sijbesma JW. et al. PET imaging of disease progression and treatment effects in the experimental autoimmune encephalomyelitis rat model. J Nucl Med. 2014;55:1330-5
73. Stankoff B, Freeman L, Aigrot MS, Chardain A, Dollé F, Williams A. et al. Imaging central nervous system myelin by positron emission tomography in multiple sclerosis using [methyl-¹¹C]-2-(4'-methylaminophenyl)- 6-hydroxybenzothiazole. Ann Neurol. 2011;69:673-80
74. Fodero-Tavoletti MT, Rowe CC, McLean CA, Leone L, Li QX, Masters CL. et al. Characterization of PiB binding to white matter in Alzheimer disease and other dementias. J Nucl Med. 2009;50:198-204
75. Carvalho RHF, Real CC, Cinini S, Garcez AT, Duran FLS, Marques FLN. et al. [(11)C]PIB PET imaging can detect white and grey matter demyelination in a non-human primate model of progressive multiple sclerosis. Mult Scler Relat Disord. 2019;35:108-15
76. Zeydan B, Schwarz CG, Lowe VJ, Reid RI, Przybelski SA, Lesnick TG. et al. Investigation of white matter PiB uptake as a marker of white matter integrity. Ann Clin Transl Neurol. 2019;6:678-88
77. Tonietto M, Poirion E, Lazzarotto A, Ricigliano V, Papeix C, Bottlaender M. et al. Periventricular remyelination failure in multiple sclerosis: a substrate for neurodegeneration. Brain. 2023;146:182-94
78. Zhang M, Hugon G, Bouillot C, Bolbos R, Langlois JB, Billard T. et al. Evaluation of Myelin Radiotracers in the Lysolecithin Rat Model of Focal Demyelination: Beware of Pitfalls!. Contrast Media Mol Imaging. 2019;2019:9294586
79. Matías-Guiu JA, Cabrera-Martín MN, Matías-Guiu J, Oreja-Guevara C, Riola-Parada C, Moreno-Ramos T. et al. Amyloid PET imaging in multiple sclerosis: an (18)F-florbetaben study. BMC Neurol. 2015;15:243
80. Pietroboni AM, Carandini T, Colombi A, Mercurio M, Ghezzi L, Giulietti G. et al. Amyloid PET as a marker of normal-appearing white matter early damage in multiple sclerosis: correlation with CSF β-amyloid levels and brain volumes. Eur J Nucl Med Mol Imaging. 2019;46:280-7
81. Zhang M, Ni Y, Zhou Q, He L, Meng H, Gao Y. et al. (18)F-florbetapir PET/MRI for quantitatively monitoring myelin loss and recovery in patients with multiple sclerosis: A longitudinal study. EClinicalMedicine. 2021;37:100982
82. Kurtzke JF. Rating neurologic impairment in multiple sclerosis: an expanded disability status scale (EDSS). Neurology. 1983;33:1444-52
83. Zhang M, Liu J, Li B, Chen S. (18)F-florbetapir PET/MRI for quantitatively monitoring demyelination and remyelination in acute disseminated encephalomyelitis. EJNMMI Res. 2019;9:96
84. Watanabe H, Maekawa R, Iikuni S, Kakae M, Matsuo N, Shirakawa H. et al. Characterization of Radioiodinated Diaryl Oxadiazole Derivatives as SPECT Probes for Detection of Myelin in Multiple Sclerosis. ACS Chem Neurosci. 2022;13:363-9
85. Mangiardi M, Crawford DK, Xia X, Du S, Simon-Freeman R, Voskuhl RR. et al. An animal model of cortical and callosal pathology in multiple sclerosis. Brain Pathol. 2011;21:263-78
86. Trapp BD, Peterson J, Ransohoff RM, Rudick R, Mörk S, Bö L. Axonal transection in the lesions of multiple sclerosis. N Engl J Med. 1998;338:278-85
87. Grant JL, Ghosn EE, Axtell RC, Herges K, Kuipers HF, Woodling NS. et al. Reversal of paralysis and reduced inflammation from peripheral administration of β-amyloid in TH1 and TH17 versions of experimental autoimmune encephalomyelitis. Sci Transl Med. 2012;4:145ra05
88. Jones R. Two sides to β-amyloid. Nat Rev Neurosc. 2012;13:666 -
89. Gehrmann J, Banati RB, Cuzner ML, Kreutzberg GW, Newcombe J. Amyloid precursor protein (APP) expression in multiple sclerosis lesions. Glia. 1995;15:141-51
90. Hu X, Hicks CW, He W, Wong P, Macklin WB, Trapp BD. et al. Bace1 modulates myelination in the central and peripheral nervous system. Nat Neurosci. 2006;9:1520-5
91. Pietroboni AM, Schiano di Cola F, Scarioni M, Fenoglio C, Spanò B, Arighi A. et al. CSF β-amyloid as a putative biomarker of disease progression in multiple sclerosis. Mult Scler. 2017;23:1085-91
92. Coman I, Aigrot MS, Seilhean D, Reynolds R, Girault JA, Zalc B. et al. Nodal, paranodal and juxtaparanodal axonal proteins during demyelination and remyelination in multiple sclerosis. Brain. 2006;129:3186-95
93. Caballero NA, Meléndez FJ, Niño A, Muñoz-Caro C. Molecular docking study of the binding of aminopyridines within the K+ channel. J Mol Model. 2007;13:579-86
94. Valet M, Quoilin M, Lejeune T, Stoquart G, Van Pesch V, El Sankari S. et al. Effects of Fampridine in People with Multiple Sclerosis: A Systematic Review and Meta-analysis. CNS Drugs. 2019;33:1087-99
95. Detsi A, Kontogiorgis C, Hadjipavlou-Litina D. Coumarin derivatives: an updated patent review (2015-2016). Expert Opin Ther Pat. 2017;27:1201-26
96. Garg SS, Gupta J, Sharma S, Sahu D. An insight into the therapeutic applications of coumarin compounds and their mechanisms of action. Eur J Pharm Sci. 2020;152:105424
97. Hu YH, Yang J, Zhang Y, Liu KC, Liu T, Sun J. et al. Synthesis and biological evaluation of 3-(4-aminophenyl)-coumarin derivatives as potential anti-Alzheimer's disease agents. J Enzyme Inhib Med Chem. 2019;34:1083-92
98. de Souza GA, da Silva SJ, Del Cistia CN, Pitasse-Santos P, Pires LO, Passos YM. et al. Discovery of novel dual-active 3-(4-(dimethylamino)phenyl)-7-aminoalcoxy-coumarin as potent and selective acetylcholinesterase inhibitor and antioxidant. J Enzyme Inhib Med Chem. 2019;34:631-7
99. Montanari S, Bartolini M, Neviani P, Belluti F, Gobbi S, Pruccoli L. et al. Multitarget Strategy to Address Alzheimer's Disease: Design, Synthesis, Biological Evaluation, and Computational Studies of Coumarin-Based Derivatives. ChemMedChem. 2016;11:1296-308
100. Abdshahzadeh H, Golshani M, Nadri H, Saberi Kia I, Abdolahi Z, Forootanfar H. et al. 3-Aryl Coumarin Derivatives Bearing Aminoalkoxy Moiety as Multi-Target-Directed Ligands against Alzheimer's Disease. Chem Biodivers. 2019;16:e1800436
101. Wang C, Popescu DC, Wu C, Zhu J, Macklin W, Wang Y. In situ fluorescence imaging of myelination. J Histochem Cytochem. 2010;58:611-21
102. Wang C, Wu C, Zhu J, Miller RH, Wang Y. Design, synthesis, and evaluation of coumarin-based molecular probes for imaging of myelination. J Med Chem. 2011;54:2331-40
103. Hu Y, Doudevski I, Wood D, Moscarello M, Husted C, Genain C. et al. Synergistic interactions of lipids and myelin basic protein. Proc Natl Acad Sci U S A. 2004;101:13466-71
104. Watanabe H, Sakai S, Iikuni S, Shimizu Y, Shirakawa H, Kaneko S. et al. Synthesis and biological evaluation of radioiodinated 3-phenylcoumarin derivatives targeting myelin in multiple sclerosis. Bioorg Med Chem Lett. 2020;30:127562
105. Zimin PI, Garic B, Bodendiek SB, Mahieux C, Wulff H, Zhorov BS. Potassium channel block by a tripartite complex of two cationophilic ligands and a potassium ion. Mol Pharmacol. 2010;78:588-99
106. Zagaja M, Pyrka D, Skalicka-Wozniak K, Glowniak K, Florek-Luszczki M, Glensk M. et al. Effect of xanthotoxin (8-methoxypsoralen) on the anticonvulsant activity of classical antiepileptic drugs against maximal electroshock-induced seizures in mice. Fitoterapia. 2015;105:1-6
107. Chen X, Pi R, Zou Y, Liu M, Ma X, Jiang Y. et al. Attenuation of experimental autoimmune encephalomyelitis in C57 BL/6 mice by osthole, a natural coumarin. Eur J Pharmacol. 2010;629:40-6
108. Chen YH, Wu KC, Yang CT, Tu YK, Gong CL, Chao CC. et al. Coumarsabin hastens C-type inactivation gating of voltage-gated K⁺ channels. Eur J Pharmacol. 2013;704:41-8
109. Bodendiek SB, Mahieux C, Hänsel W, Wulff H. 4-Phenoxybutoxy-substituted heterocycles-a structure-activity relationship study of blockers of the lymphocyte potassium channel Kv1.3. Eur J Med Chem. 2009;44:1838-52
110. Faria Dde P, Copray S, Sijbesma JW, Willemsen AT, Buchpiguel CA, Dierckx RA. et al. PET imaging of focal demyelination and remyelination in a rat model of multiple sclerosis: comparison of [11C]MeDAS, [11C]CIC and [11C]PIB. Eur J Nucl Med Mol Imaging. 2014;41:995-1003
111. Auvity S, Tonietto M, Caillé F, Bodini B, Bottlaender M, Tournier N. et al. Repurposing radiotracers for myelin imaging: a study comparing 18F-florbetaben, 18F-florbetapir, 18F-flutemetamol,11C-MeDAS, and 11C-PiB. Eur J Nucl Med Mol Imaging. 2020;47:490-501
112. Wouters OJ, McKee M, Luyten J. Estimated Research and Development Investment Needed to Bring a New Medicine to Market, 2009-2018. Jama. 2020 323
113. Oh J, Bar-Or A. Emerging therapies to target CNS pathophysiology in multiple sclerosis. Nat Rev Neurol. 2022;18:466-75
114. Olejnik P, Roszkowska Z, Adamus S, Kasarełło K. Multiple sclerosis: a narrative overview of current pharmacotherapies and emerging treatment prospects. Pharmacological reports: PR. 2024;76:926-43
115. Hauser SL, Cree BAC. Treatment of Multiple Sclerosis: A Review. Am J Med. 2020;133:1380-90.e2
116. Linker RA, Lee DH, Ryan S, van Dam AM, Conrad R, Bista P. et al. Fumaric acid esters exert neuroprotective effects in neuroinflammation via activation of the Nrf2 antioxidant pathway. Brain. 2011;134:678-92
117. O'Connor P, Wolinsky JS, Confavreux C, Comi G, Kappos L, Olsson TP. et al. Randomized trial of oral teriflunomide for relapsing multiple sclerosis. N Engl J Med. 2011;365:1293-303
118. Hauser SL, Bar-Or A, Comi G, Giovannoni G, Hartung HP, Hemmer B. et al. Ocrelizumab versus Interferon Beta-1a in Relapsing Multiple Sclerosis. N Engl J Med. 2017;376:221-34
119. LeBien TW, Tedder TF. B lymphocytes: how they develop and function. Blood. 2008;112:1570-80
120. McGinley MP, Cohen JA. Sphingosine 1-phosphate receptor modulators in multiple sclerosis and other conditions. Lancet. 2021;398:1184-94
121. Rolland WB, Lekic T, Krafft PR, Hasegawa Y, Altay O, Hartman R. et al. Fingolimod reduces cerebral lymphocyte infiltration in experimental models of rodent intracerebral hemorrhage. Exp Neurol. 2013;241:45-55
122. Maas RP, Muller-Hansma AH, Esselink RA, Murk JL, Warnke C, Killestein J. et al. Drug-associated progressive multifocal leukoencephalopathy: a clinical, radiological, and cerebrospinal fluid analysis of 326 cases. J Neurol. 2016;263:2004-21
123. Mahler C, Schumacher AM, Unterrainer M, Kaiser L, Höllbacher T, Lindner S. et al. TSPO PET imaging of natalizumab-associated progressive multifocal leukoencephalopathy. Brain. 2021;144:2683-95
124. Gruber RC, Wirak GS, Blazier AS, Lee L, Dufault MR, Hagan N. et al. BTK regulates microglial function and neuroinflammation in human stem cell models and mouse models of multiple sclerosis. Nat Commun. 2024;15:10116
125. Krämer J, Bar-Or A, Turner TJ, Wiendl H. Bruton tyrosine kinase inhibitors for multiple sclerosis. Nat Rev Neurol. 2023;19:289-304
126. Montalban X, Vermersch P, Arnold DL, Bar-Or A, Cree BAC, Cross AH. et al. Safety and efficacy of evobrutinib in relapsing multiple sclerosis (evolutionRMS1 and evolutionRMS2): two multicentre, randomised, double-blind, active-controlled, phase 3 trials. Lancet Neurol. 2024;23:1119-32
127. Reich DS, Arnold DL, Vermersch P, Bar-Or A, Fox RJ, Matta A. et al. Safety and efficacy of tolebrutinib, an oral brain-penetrant BTK inhibitor, in relapsing multiple sclerosis: a phase 2b, randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2021;20:729-38
128. Fox RJ, Bar-Or A, Traboulsee A, Oreja-Guevara C, Giovannoni G, Vermersch P. et al. Tolebrutinib in Nonrelapsing Secondary Progressive Multiple Sclerosis. N Engl J Med. 2025
129. Langlois J, Lange S, Ebeling M, Macnair W, Schmucki R, Li C. et al. Fenebrutinib, a Bruton's tyrosine kinase inhibitor, blocks distinct human microglial signaling pathways. J Neuroinflammation. 2024;21:276
130. Vermersch P, Brieva-Ruiz L, Fox RJ, Paul F, Ramio-Torrenta L, Schwab M. et al. Efficacy and Safety of Masitinib in Progressive Forms of Multiple Sclerosis: A Randomized, Phase 3, Clinical Trial. Neurol. neuroimmunol. neuroinflamm. 2022 9
131. Fox RJ, Coffey CS, Conwit R, Cudkowicz ME, Gleason T, Goodman A. et al. Phase 2 Trial of Ibudilast in Progressive Multiple Sclerosis. N Engl J Med. 2018;379:846-55
132. Hartung HP, Derfuss T, Cree BA, Sormani MP, Selmaj K, Stutters J. et al. Efficacy and safety of temelimab in multiple sclerosis: Results of a randomized phase 2b and extension study. Mult Scler. 2022;28:429-40
133. Green AJ, Gelfand JM, Cree BA, Bevan C, Boscardin WJ, Mei F. et al. Clemastine fumarate as a remyelinating therapy for multiple sclerosis (ReBUILD): a randomised, controlled, double-blind, crossover trial. Lancet. 2017;390:2481-9
134. Neumann B, Baror R, Zhao C, Segel M, Dietmann S, Rawji KS. et al. Metformin Restores CNS Remyelination Capacity by Rejuvenating Aged Stem Cells. Cell Stem Cell. 2019;25:473-85.e8
135. Huang L, Fung E, Bose S, Popp A, Böser P, Memmott J. et al. Elezanumab, a clinical stage human monoclonal antibody that selectively targets repulsive guidance molecule A to promote neuroregeneration and neuroprotection in neuronal injury and demyelination models. Neurobiol Dis. 2021;159:105492
136. Oh U, Fujita M, Ikonomidou VN, Evangelou IE, Matsuura E, Harberts E. et al. Translocator protein PET imaging for glial activation in multiple sclerosis. J Neuroimmune Pharmacol. 2011;6:354-61
137. Mock EEA, Honkonen E, Airas L. Synaptic Loss in Multiple Sclerosis: A Systematic Review of Human Post-mortem Studies. Front Neurol. 2021 12
138. Cai ZX, Li SY, Matuskey D, Nabulsi N, Huang YY. PET imaging of synaptic density: A new tool for investigation of neuropsychiatric diseases. Neurosci Lett. 2019;691:44-50
139. Finnema SJ, Nabulsi NB, Eid T, Detyniecki K, Lin SF, Chen MK. et al. Imaging synaptic density in the living human brain. Sci Transl Med. 2016;8:348ra96
140. Naganawa M, Li S, Nabulsi N, Henry S, Zheng MQ, Pracitto R. et al. First-in-Human Evaluation of (18)F-SynVesT-1, a Radioligand for PET Imaging of Synaptic Vesicle Glycoprotein 2A. J Nucl Med. 2021;62:561-7
141. Li S, Cai Z, Wu X, Holden D, Pracitto R, Kapinos M. et al. Synthesis and in vivo Evaluation of a Novel PET Radiotracer for Imaging of Synaptic Vesicle Glycoprotein 2A (SV2A) in Nonhuman Primates. ACS Chem Neurosci. 2019;10:1544-54
142. Matuskey D, Tinaz S, Wilcox KC, Naganawa M, Toyonaga T, Dias M. et al. Synaptic Changes in Parkinson Disease Assessed with in vivo Imaging. Ann Neurol. 2020;87:329-38
143. Bierhansl L, Hartung H-P, Aktas O, Ruck T, Roden M, Meuth SG. Thinking outside the box: non-canonical targets in multiple sclerosis. Nat Rev Drug Discov. 2022;21:578-600
144. Stevens MY, Cropper HC, Lucot KL, Chaney AM, Lechtenberg KJ, Jackson IM. et al. Development of a CD19 PET tracer for detecting B cells in a mouse model of multiple sclerosis. J Neuroinflammation. 2020;17:275
145. James ML, Hoehne A, Mayer AT, Lechtenberg K, Moreno M, Gowrishankar G. et al. Imaging B Cells in a Mouse Model of Multiple Sclerosis Using (64)Cu-Rituximab PET. J Nucl Med. 2017;58:1845-51
146. Guglielmetti C, Levi J, Huynh TL, Tiret B, Blecha J, Tang R. et al. Longitudinal Imaging of T Cells and Inflammatory Demyelination in a Preclinical Model of Multiple Sclerosis Using (18)F-FAraG PET and MRI. J Nucl Med. 2022;63:140-6
147. Yoon J-K, Park B-N, Ryu E-K, An Y-S, Lee S-J. Current Perspectives on 89Zr-PET Imaging. Int J Mol Sci. 2020 21
148. Bever CT Jr, Young D, Anderson PA, Krumholz A, Conway K, Leslie J. et al. The effects of 4-aminopyridine in multiple sclerosis patients: results of a randomized, placebo-controlled, double-blind, concentration-controlled, crossover trial. Neurology. 1994;44:1054-9
149. Bostock H, Sears TA, Sherratt RM. The effects of 4-aminopyridine and tetraethylammonium ions on normal and demyelinated mammalian nerve fibres. J Physiol. 1981;313:301-15
150. Fernandez O, Berger T, Hartung H-P, Putzki N. Historical overview of the rationale for the pharmacological use of prolonged-release fampridine in multiple sclerosis. Expert Rev Clin Pharmacol. 2014;5:649-65
151. Sun Y, Guehl NJ, Zhou YP, Takahashi K, Belov V, Dhaynaut M. et al. Radiochemical Synthesis and Evaluation of 3-[(11)C]Methyl-4-aminopyridine in Rodents and Nonhuman Primates for Imaging Potassium Channels in the CNS. ACS Chem Neurosci. 2022;13:3342-51
152. Guehl NJ, Ramos-Torres KM, Linnman C, Moon SH, Dhaynaut M, Wilks MQ. et al. Evaluation of the potassium channel tracer [(18)F]3F4AP in rhesus macaques. J Cereb Blood Flow Metab. 2021;41:1721-33
153. Guehl NJ, Neelamegam R, Zhou YP, Moon SH, Dhaynaut M, El Fakhri G. et al. Radiochemical Synthesis and Evaluation in Non-Human Primates of 3-[(11)C]methoxy-4-aminopyridine: A Novel PET Tracer for Imaging Potassium Channels in the CNS. ACS Chem Neurosci. 2021;12:756-65
154. Guehl N, Sun Y, Russo A, Ramos-Torres K, Dhaynaut M, Klawiter E. et al. <strong>First-in-human brain imaging with [18F]3F4AP, a PET tracer developed for imaging demyelination</strong>. J Nucl Med. 2022;63:2485 -
155. Brugarolas P, Wilks MQ, Noel J, Kaiser JA, Vesper DR, Ramos-Torres KM. et al. Human biodistribution and radiation dosimetry of the demyelination tracer [(18)F]3F4AP. Eur J Nucl Med Mol Imaging. 2023;50:344-51
156. Deshmukh VA, Tardif V, Lyssiotis CA, Green CC, Kerman B, Kim HJ. et al. A regenerative approach to the treatment of multiple sclerosis. Nature. 2013;502:327-32
157. Mei F, Fancy SPJ, Shen Y-AA, Niu J, Zhao C, Presley B. et al. Micropillar arrays as a high-throughput screening platform for therapeutics in multiple sclerosis. Nat Med. 2014;20:954-60
158. Mei F, Lehmann-Horn K, Shen Y-AA, Rankin KA, Stebbins KJ, Lorrain DS. et al. Accelerated remyelination during inflammatory demyelination prevents axonal loss and improves functional recovery. eLife. 2016 5
159. Poon MM, Lorrain KI, Stebbins KJ, Edu GC, Broadhead AR, Lorenzana AJ. et al. Targeting the muscarinic M1 receptor with a selective, brain-penetrant antagonist to promote remyelination in multiple sclerosis. Proc Natl Acad Sci U S A. 2024 121
160. Atzrodt J, Derdau V, Kerr WJ, Reid M. Deuterium- and Tritium-Labelled Compounds: Applications in the Life Sciences. Angew Chem Int Ed. 2018;57:1758-84
161. Poon MM, Lorrain KI, Stebbins KJ, Edu GC, Broadhead AR, Lorenzana AO. et al. Discovery of a brain penetrant small molecule antagonist targeting LPA1 receptors to reduce neuroinflammation and promote remyelination in multiple sclerosis. Sci Rep. 2024 14
162. Wang M, Gao M, Miller KD, Zheng QH. Synthesis of [¹¹C]PBR06 and [¹⁸F]PBR06 as agents for positron emission tomographic (PET) imaging of the translocator protein (TSPO). Steroids. 2011;76:1331-40
163. Kreisl WC, Kim MJ, Coughlin JM, Henter ID, Owen DR, Innis RB. PET imaging of neuroinflammation in neurological disorders. Lancet Neurol. 2020;19:940-50
164. Werry EL, Bright FM, Piguet O, Ittner LM, Halliday GM, Hodges JR. et al. Recent Developments in TSPO PET Imaging as A Biomarker of Neuroinflammation in Neurodegenerative Disorders. Int J Mol Sci. 2019 20
165. Rissanen E, Tuisku J, Rokka J, Paavilainen T, Parkkola R, Rinne JO. et al. In vivo Detection of Diffuse Inflammation in Secondary Progressive Multiple Sclerosis Using PET Imaging and the Radioligand ¹¹C-PK11195. J Nucl Med. 2014;55:939-44
166. Banati RB, Newcombe J, Gunn RN, Cagnin A, Turkheimer F, Heppner F. et al. The peripheral benzodiazepine binding site in the brain in multiple sclerosis: quantitative in vivo imaging of microglia as a measure of disease activity. Brain. 2000;123( Pt 11):2321-37
167. Giannetti P, Politis M, Su P, Turkheimer FE, Malik O, Keihaninejad S. et al. Increased PK11195-PET binding in normal-appearing white matter in clinically isolated syndrome. Brain. 2015;138:110-9
168. Politis M, Giannetti P, Su P, Turkheimer F, Keihaninejad S, Wu K. et al. Increased PK11195 PET binding in the cortex of patients with MS correlates with disability. Neurology. 2012;79:523-30
169. Airas L, Rissanen E, Rinne JO. Imaging neuroinflammation in multiple sclerosis using TSPO-PET. Clin Transl Imaging. 2015;3:461-73
170. Herranz E, Treaba CA, Barletta VT, Mehndiratta A, Ouellette R, Sloane JA. et al. Characterization of cortico-meningeal translocator protein expression in multiple sclerosis. Brain. 2024
171. Magliozzi R, Howell OW, Calabrese M, Reynolds R. Meningeal inflammation as a driver of cortical grey matter pathology and clinical progression in multiple sclerosis. Nat Rev Neurol. 2023;19:461-76
172. Sucksdorff M, Matilainen M, Tuisku J, Polvinen E, Vuorimaa A, Rokka J. et al. Brain TSPO-PET predicts later disease progression independent of relapses in multiple sclerosis. Brain. 2020;143:3318-30
173. Hamzaoui M, Garcia J, Boffa G, Lazzarotto A, Absinta M, Ricigliano VAG. et al. Positron Emission Tomography with [(18) F]-DPA-714 Unveils a Smoldering Component in Most Multiple Sclerosis Lesions which Drives Disease Progression. Ann Neurol. 2023;94:366-83
174. Sucksdorff M, Tuisku J, Matilainen M, Vuorimaa A, Smith S, Keitilä J. et al. Natalizumab treatment reduces microglial activation in the white matter of the MS brain. Neurol Neuroimmunol Neuroinflamm. 2019;6:e574
175. Sucksdorff M, Rissanen E, Tuisku J, Nuutinen S, Paavilainen T, Rokka J. et al. Evaluation of the Effect of Fingolimod Treatment on Microglial Activation Using Serial PET Imaging in Multiple Sclerosis. J Nucl Med. 2017;58:1646-51
176. Arlicot N, Vercouillie J, Ribeiro MJ, Tauber C, Venel Y, Baulieu JL. et al. Initial evaluation in healthy humans of [18F]DPA-714, a potential PET biomarker for neuroinflammation. Nucl Med Biol. 2012;39:570-8
177. Plavén-Sigray P, Schain M, Zanderigo F, Rabiner EA, Gunn RN, Ogden RT. et al. Accuracy and reliability of [(11)C]PBR28 specific binding estimated without the use of a reference region. Neuroimage. 2019;188:102-10
178. Bagnato F, Sati P, Hemond CC, Elliott C, Gauthier SA, Harrison DM. et al. Imaging chronic active lesions in multiple sclerosis: a consensus statement. Brain. 2024
179. Chechneva OV, Deng W. Mitochondrial translocator protein (TSPO), astrocytes and neuroinflammation. Neural Regen Res. 2016;11:1056-7
180. Lavisse S, Guillermier M, Hérard AS, Petit F, Delahaye M, Van Camp N. et al. Reactive astrocytes overexpress TSPO and are detected by TSPO positron emission tomography imaging. J Neurosci. 2012;32:10809-18
181. Guilarte TR, Rodichkin AN, McGlothan JL, Acanda De La Rocha AM, Azzam DJ. Imaging neuroinflammation with TSPO: A new perspective on the cellular sources and subcellular localization. Pharmacol Ther. 2022;234:108048
182. Bhoola NH, Mbita Z, Hull R, Dlamini Z. Translocator Protein (TSPO) as a Potential Biomarker in Human Cancers. Int J Mol Sci. 2018 19
183. Matthews PM. Chronic inflammation in multiple sclerosis - seeing what was always there. Nat Rev Neurol. 2019;15:582-93
184. Owen DR, Yeo AJ, Gunn RN, Song K, Wadsworth G, Lewis A. et al. An 18-kDa translocator protein (TSPO) polymorphism explains differences in binding affinity of the PET radioligand PBR28. J Cereb Blood Flow Metab. 2012;32:1-5
185. Chen Z, Haider A, Chen J, Xiao Z, Gobbi L, Honer M. et al. The Repertoire of Small-Molecule PET Probes for Neuroinflammation Imaging: Challenges and Opportunities beyond TSPO. J Med Chem. 2021;64:17656-89
186. Beaino W, Janssen B, Kooij G, van der Pol SMA, van Het Hof B, van Horssen J. et al. Purinergic receptors P2Y12R and P2X7R: potential targets for PET imaging of microglia phenotypes in multiple sclerosis. J Neuroinflammation. 2017;14:259
187. Ohl K, Tenbrock K, Kipp M. Oxidative stress in multiple sclerosis: Central and peripheral mode of action. Exp Neurol. 2016;277:58-67
188. Wilson AA, Sadovski O, Nobrega JN, Raymond RJ, Bambico FR, Nashed MG. et al. Evaluation of a novel radiotracer for positron emission tomography imaging of reactive oxygen species in the central nervous system. Nucl Med and Biol. 2017;53:14-20
189. Okamura T, Okada M, Kikuchi T, Wakizaka H, Zhang MR. A ¹¹C-labeled 1,4-dihydroquinoline derivative as a potential PET tracer for imaging of redox status in mouse brain. J Cereb Blood Flow Metab. 2015;35:1930-6
190. Hou C, Hsieh C-J, Li S, Lee H, Graham TJ, Xu K. et al. Development of a Positron Emission Tomography Radiotracer for Imaging Elevated Levels of Superoxide in Neuroinflammation. ACS Chem Neurosc. 2018;9:578-86
191. Pisaneschi F, Gammon ST, Paolillo V, Qureshy SA, Piwnica-Worms D. Imaging of innate immunity activation in vivo with a redox-tuned PET reporter. Nat Biotech. 2022;40:965-73
192. Chaney AM, Cropper HC, Jain P, Wilson E, Simonetta F, Johnson EM. et al. PET imaging of TREM1 identifies CNS-infiltrating myeloid cells in a mouse model of multiple sclerosis. Sci Transl Med. 2023;15:eabm6267
193. Docagne F, Mestre L, Loría F, Hernangómez M, Correa F, Guaza C. Therapeutic potential of CB2 targeting in multiple sclerosis. Expert Opin Ther Targets. 2008;12:185-95
194. Horti AG, Gao Y, Ravert HT, Finley P, Valentine H, Wong DF. et al. Synthesis and biodistribution of [11C]A-836339, a new potential radioligand for PET imaging of cannabinoid type 2 receptors (CB2). Bioorg Med Chem. 2010;18:5202-7
195. Moldovan R-P, Teodoro R, Gao Y, Deuther-Conrad W, Kranz M, Wang Y. et al. Development of a High-Affinity PET Radioligand for Imaging Cannabinoid Subtype 2 Receptor. J Med Chem. 2016;59:7840-55
196. Slavik R, Grether U, Müller Herde A, Gobbi L, Fingerle J, Ullmer C. et al. Discovery of a high affinity and selective pyridine analog as a potential positron emission tomography imaging agent for cannabinoid type 2 receptor. J Med Chem. 2015;58:4266-77
197. Haider A, Kretz J, Gobbi L, Ahmed H, Atz K, Bürkler M. et al. Structure-Activity Relationship Studies of Pyridine-Based Ligands and Identification of a Fluorinated Derivative for Positron Emission Tomography Imaging of Cannabinoid Type 2 Receptors. J Med Chem. 2019;62:11165-81
198. Haider A, Gobbi L, Kretz J, Ullmer C, Brink A, Honer M. et al. Identification and Preclinical Development of a 2,5,6-Trisubstituted Fluorinated Pyridine Derivative as a Radioligand for the Positron Emission Tomography Imaging of Cannabinoid Type 2 Receptors. J Med Chem. 2020;63:10287-306
199. Haider A, Müller Herde A, Slavik R, Weber M, Mugnaini C, Ligresti A. et al. Synthesis and Biological Evaluation of Thiophene-Based Cannabinoid Receptor Type 2 Radiotracers for PET Imaging. Front Neurosci. 2016;10:350
200. Slavik R, Müller Herde A, Haider A, Krämer SD, Weber M, Schibli R. et al. Discovery of a fluorinated 4-oxo-quinoline derivative as a potential positron emission tomography radiotracer for imaging cannabinoid receptor type 2. J Neurochem. 2016;138:874-86
201. Ahmad R, Koole M, Evens N, Serdons K, Verbruggen A, Bormans G. et al. Whole-Body Biodistribution and Radiation Dosimetry of the Cannabinoid Type 2 Receptor Ligand [11C]-NE40 in Healthy Subjects. Mol Imaging Biol. 2013;15:384-90
202. Raitio KH, Savinainen JR, Vepsäläinen J, Laitinen JT, Poso A, Järvinen T. et al. Synthesis and SAR Studies of 2-Oxoquinoline Derivatives as CB2 Receptor Inverse Agonists. J Med Chem. 2006;49:2022-7
203. Evens N, Muccioli GG, Houbrechts N, Lambert DM, Verbruggen AM, Van Laere K. et al. Synthesis and biological evaluation of carbon-11- and fluorine-18-labeled 2-oxoquinoline derivatives for type 2 cannabinoid receptor positron emission tomography imaging. Nucl Med and Biol. 2009;36:455-65
204. Haider A, Spinelli F, Herde AM, Mu B, Keller C, Margelisch M. et al. Evaluation of 4-oxo-quinoline-based CB2 PET radioligands in R6/2 chorea huntington mouse model and human ALS spinal cord tissue. Eur J Med Chem. 2018;145:746-59
205. Ahmad R, Postnov A, Bormans G, Versijpt J, Vandenbulcke M, Van Laere K. Decreased in vivo availability of the cannabinoid type 2 receptor in Alzheimer's disease. Eur J Nucl Med Mol Imaging. 2016;43:2219-27
206. Du Y, Coughlin JM, Brosnan MK, Chen A, Shinehouse LK, Abdallah R. et al. First-in-human imaging using [(11)C]MDTC: a radiotracer targeting the cannabinoid receptor type 2. Eur J Nucl Med Mol Imaging. 2023;50:2386-93
207. Kim MJ, Anaya FJ, Manly LS, Lee JH, Hong J, Shrestha S. et al. Whole-Body PET Imaging in Humans Shows That (11)C-PS13 Is Selective for Cyclooxygenase-1 and Can Measure the In vivo Potency of Nonsteroidal Antiinflammatory Drugs. J Nucl Med. 2023;64:159-64
208. Cortes M, Singh P, Morse C, Shrestha S, Jenko K, Kowalski A. et al. Synthesis of PET radioligands as potential probes for imaging COX-2 in neuroinflammation. J Nucl Med. 2015;56:1092 -
209. Horti AG, Naik R, Foss CA, Minn I, Misheneva V, Du Y. et al. PET imaging of microglia by targeting macrophage colony-stimulating factor 1 receptor (CSF1R). Proc Natl Acad Sci U S A. 2019;116:1686-91
210. Gallezot JD, Weinzimmer D, Nabulsi N, Lin SF, Fowles K, Sandiego C. et al. Evaluation of [(11)C]MRB for assessment of occupancy of norepinephrine transporters: Studies with atomoxetine in non-human primates. Neuroimage. 2011;56:268-79
211. Sato N, Wu H, Asiedu KO, Szajek LP, Griffiths GL, Choyke PL. (89)Zr-Oxine Complex PET Cell Imaging in Monitoring Cell-based Therapies. Radiology. 2015;275:490-500
212. Kahts M, Summers B, Ndlela AN, Gutta A, Nemutaduni P, More A. et al. First-in-human infection imaging with (89)Zr-labelled leukocytes and comparison of scan quality with [(99m)Tc]Tc-HMPAO-labelled leukocytes. Front Nucl Med. 2024;4:1426650
213. Watterson SH, Liu Q, Beaudoin Bertrand M, Batt DG, Li L, Pattoli MA. et al. Discovery of Branebrutinib (BMS-986195): A Strategy for Identifying a Highly Potent and Selective Covalent Inhibitor Providing Rapid in vivo Inactivation of Bruton's Tyrosine Kinase (BTK). J Med Chem. 2019;62:3228-50
214. Cartier A, Hla T. Sphingosine 1-phosphate: Lipid signaling in pathology and therapy. Science (New York, NY). 2019 366
215. Roggeri A, Schepers M, Tiane A, Rombaut B, van Veggel L, Hellings N. et al. Sphingosine-1-Phosphate Receptor Modulators and Oligodendroglial Cells: Beyond Immunomodulation. Int J Mol Sci. 2020 21
216. Jin H, Yang H, Liu H, Zhang Y, Zhang X, Rosenberg AJ. et al. A promising carbon-11-labeled sphingosine-1-phosphate receptor 1-specific PET tracer for imaging vascular injury. J Nucl Cardiol. 2017;24:558-70
217. Luo Z, Gu J, Dennett RC, Gaehle GG, Perlmutter JS, Chen DL. et al. Automated production of a sphingosine-1 phosphate receptor 1 (S1P1) PET radiopharmaceutical [(11)C]CS1P1 for human use. Appl Radiat Isot. 2019;152:30-6
218. Liu H, Laforest R, Gu J, Luo Z, Jones LA, Gropler RJ. et al. Acute Rodent Tolerability, Toxicity, and Radiation Dosimetry Estimates of the S1P1-Specific Radioligand [(11)C]CS1P1. Mol Imaging Biol. 2020;22:285-92
219. Qiu L, Jiang H, Cho K, Yu Y, Jones LA, Huang T. et al. Metabolite Study and Structural Authentication for the First-in-Human Use Sphingosine-1-phosphate Receptor 1 Radiotracer. ACS Chem Neurosci. 2024;15:1882-92
220. Jiang H, Joshi S, Liu H, Mansor S, Qiu L, Zhao H. et al. InVitro and In vivo Investigation of S1PR1 Expression in the Central Nervous System Using [(3)H]CS1P1 and [(11)C]CS1P1. ACS Chem Neurosci. 2021;12:3733-44
221. Gu J, Zheng MQ, Holden D, Fowles K, Qiu L, Felchner Z. et al. PET Imaging of Sphingosine-1-Phosphate Receptor 1 with [(18)F]TZ4877 in Nonhuman Primates. Mol Imaging Biol. 2025;27:54-63
222. Liu H, Luo Z, Gu J, Jiang H, Joshi S, Shoghi KI. et al. In vivo Characterization of Four (18)F-Labeled S1PR1 Tracers for Neuroinflammation. Mol Imaging Biol. 2020;22:1362-9
223. Qiu L, Jiang H, Yu Y, Gu J, Wang J, Zhao H. et al. Radiosynthesis and evaluation of a fluorine-18 radiotracer [(18)F]FS1P1 for imaging sphingosine-1-phosphate receptor 1. Org Biomol Chem. 2022;20:1041-52
224. Qiu L, Jiang H, Zhou C, Wang J, Yu Y, Zhao H. et al. Discovery of a Promising Fluorine-18 Positron Emission Tomography Radiotracer for Imaging Sphingosine-1-Phosphate Receptor 1 in the Brain. J Med Chem. 2023;66:4671-88
225. Yue X, Jin H, Liu H, Rosenberg AJ, Klein RS, Tu Z. A potent and selective C-11 labeled PET tracer for imaging sphingosine-1-phosphate receptor 2 in the CNS demonstrates sexually dimorphic expression. Org Biomol Chem. 2015;13:7928-39
226. Luo Z, Yue X, Yang H, Liu H, Klein RS, Tu Z. Design and synthesis of pyrazolopyridine derivatives as sphingosine 1-phosphate receptor 2 ligands. Bioorg Med Chem Lett. 2018;28:488-96
227. Ma B, Guckian KM, Liu XG, Yang C, Li B, Scannevin R. et al. Novel Potent Selective Orally Active S1P5 Receptor Antagonists. ACS Med Chem Lett. 2021;12:351-5
228. Blinkenberg M, Rune K, Jensen CV, Ravnborg M, Kyllingsbaek S, Holm S. et al. Cortical cerebral metabolism correlates with MRI lesion load and cognitive dysfunction in MS. Neurology. 2000;54:558-64
229. Blinkenberg M, Mathiesen HK, Tscherning T, Jønsson A, Svarer C, Holm S. et al. Cerebral metabolism, magnetic resonance spectroscopy and cognitive dysfunction in early multiple sclerosis: an exploratory study. Neurol Res. 2012;34:52-8
230. Xiang X, Wind K, Wiedemann T, Blume T, Shi Y, Briel N. et al. Microglial activation states drive glucose uptake and FDG-PET alterations in neurodegenerative diseases. Sci Transl Med. 2021;13:eabe5640
231. Radu CG, Shu CJ, Shelly SM, Phelps ME, Witte ON. Positron emission tomography with computed tomography imaging of neuroinflammation in experimental autoimmune encephalomyelitis. P Natl Acad Sci U S A. 2007;104:1937-42
232. Kumar P, Nagaraj C, Joshi R, Goud NS, Kumar D, Korann V. et al. Radiosynthesis of [F-18]flumazenil for imaging benzodiazepine receptors and its evaluation in human volunteers using simultaneous PET-MRI. J Radioanal Nucl Ch. 2021;329:581-9
233. Bhat R, Axtell R, Mitra A, Miranda M, Lock C, Tsien RW. et al. Inhibitory role for GABA in autoimmune inflammation. Proc Natl Acad Sci U S A. 2010;107:2580-5
234. Kim YK, Yang EJ, Cho K, Lim JY, Paik NJ. Functional Recovery After Ischemic Stroke Is Associated With Reduced GABAergic Inhibition in the Cerebral Cortex: A GABA PET Study. Neurorehabil Neural Repair. 2014;28:576-83
235. Andersson JD, Matuskey D, Finnema SJ. Positron emission tomography imaging of the gamma-aminobutyric acid system. Neurosci Lett. 2019;691:35-43
236. Sigel E, Ernst M. The Benzodiazepine Binding Sites of GABA(A) Receptors. Trends Pharmacol Sci. 2018;39:659-71
237. Hammers A. Flumazenil positron emission tomography and other ligands for functional imaging. Neuroimaging Clin N Am. 2004;14:537-51
238. Freeman L, Garcia-Lorenzo D, Bottin L, Leroy C, Louapre C, Bodini B. et al. The Neuronal Component of Gray Matter Damage in Multiple Sclerosis: A [C-11]Flumazenil Positron Emission Tomography Study. Ann Neurol. 2015;78:554-67
239. Kang Y, Rua SMH, Kaunzner UW, Perumal J, Nealon N, Qu WC. et al. A Multi-Ligand Imaging Study Exploring GABAergic Receptor Expression and Inflammation in Multiple Sclerosis. Mol Imaging Biol. 2020;22:1600-8
240. Halder SK, Milner R. Hypoxia in multiple sclerosis; is it the chicken or the egg? Brain. 2021;144:402-10
241. Trapp BD, Stys PK. Virtual hypoxia and chronic necrosis of demyelinated axons in multiple sclerosis. Lancet Neurol. 2009;8:280-91
242. Mahad DH, Trapp BD, Lassmann H. Pathological mechanisms in progressive multiple sclerosis. Lancet Neurol. 2015;14:183-93
243. Rosenberg G A. Molecular Physiology and Metabolism of the Nervous System: A Clinical Perspective. Oxford University Press; 2012. Chapter 12. Autoimmunity, Hypoxia, and Inflammation in Demyelinating Diseases. doi:10.1093/med/9780195394276.003.0012
244. Tsukada H. The Use of F-18-BCPP-EF as a PET Probe for Complex I Activity in the Brain. Method Enzymol. 2014;547:417-31
245. Tsukada H, Nishiyama S, Fukumoto D, Kanazawa M, Harada N. Novel PET probes 18F-BCPP-EF and 18F-BCPP-BF for mitochondrial complex I: a PET study in comparison with 18F-BMS-747158-02 in rat brain. J Nucl Med. 2014;55:473-80
246. Tsukada H, Kanazawa M, Ohba H, Nishiyama S, Harada N, Kakiuchi T. PET Imaging of Mitochondrial Complex I with 18F-BCPP-EF in the Brains of MPTP-Treated Monkeys. J Nucl Med. 2016;57:950-3
247. Terada T, Therriault J, Kang MS, Savard M, Pascoal TA, Lussier F. et al. Mitochondrial complex I abnormalities underlie neurodegeneration and cognitive decline in Alzheimer's disease. Eur J Neurol. 2022;29:1324-34
248. Terada T, Therriault J, Kang MSP, Savard M, Pascoal TA, Lussier F. et al. Mitochondrial complex I abnormalities is associated with tau and clinical symptoms in mild Alzheimer's disease. Mol Neurodegener. 2021;16:28
249. Venkataraman AV, Mansur A, Rizzo G, Bishop C, Lewis Y, Kocagoncu E. et al. Widespread cell stress and mitochondrial dysfunction occur in patients with early Alzheimer’s disease. Sci Transl Med. 2022;14:eabk1051
250. Terada T, Obi T, Bunai T, Matsudaira T, Yoshikawa E, Ando I. et al. In vivo mitochondrial and glycolytic impairments in patients with Alzheimer disease. Neurology. 2020;94:e1592-e604
251. Ponath G, Park C, Pitt D. The Role of Astrocytes in Multiple Sclerosis. Front Immunol. 2018;9:217
252. Sofroniew MV. Astrogliosis. Cold Spring Harb Perspect Biol. 2014;7:a020420
253. Saura J, Luque JM, Cesura AM, Da Prada M, Chan-Palay V, Huber G. et al. Increased monoamine oxidase B activity in plaque-associated astrocytes of Alzheimer brains revealed by quantitative enzyme radioautography. Neuroscience. 1994;62:15-30
254. Villemagne VL, Harada R, Doré V, Furumoto S, Mulligan R, Kudo Y. et al. First-in-Humans Evaluation of (18)F-SMBT-1, a Novel (18)F-Labeled Monoamine Oxidase-B PET Tracer for Imaging Reactive Astrogliosis. J Nucl Med. 2022;63:1551-9
255. Ishibashi K, Miura Y, Hirata K, Toyohara J, Ishii K. 18F-THK5351 PET Can Identify Astrogliosis in Multiple Sclerosis Plaques. Clin Nucl Med. 2020;45:e98-e100
256. Ng KP, Pascoal TA, Mathotaarachchi S, Therriault J, Kang MS, Shin M. et al. Monoamine oxidase B inhibitor, selegiline, reduces (18)F-THK5351 uptake in the human brain. Alzheimers Res Ther. 2017;9:25
257. Fowler JS, MacGregor RR, Wolf AP, Arnett CD, Dewey SL, Schlyer D. et al. Mapping human brain monoamine oxidase A and B with 11C-labeled suicide inactivators and PET. Science (New York, NY). 1987;235:481-5
258. Rodriguez-Vieitez E, Carter SF, Chiotis K, Saint-Aubert L, Leuzy A, Schöll M. et al. Comparison of Early-Phase 11C-Deuterium-l-Deprenyl and 11C-Pittsburgh Compound B PET for Assessing Brain Perfusion in Alzheimer Disease. J Nucl Med. 2016;57:1071-7
259. Meyer JH, Braga J. Development and Clinical Application of Positron Emission Tomography Imaging Agents for Monoamine Oxidase B. Front Neurosci. 2021;15:773404
260. Rusjan PM, Wilson AA, Miler L, Fan I, Mizrahi R, Houle S. et al. Kinetic modeling of the monoamine oxidase B radioligand [¹¹C]SL25.1188 in human brain with high-resolution positron emission tomography. J Cereb Blood Flow Metab. 2014;34:883-9
261. Waniewski RA, Martin DL. Preferential utilization of acetate by astrocytes is attributable to transport. J Neurosci. 1998;18:5225-33
262. Takata K, Kato H, Shimosegawa E, Okuno T, Koda T, Sugimoto T. et al. 11C-acetate PET imaging in patients with multiple sclerosis. PLoS One. 2014;9:e111598
263. Kato H, Okuno T, Isohashi K, Koda T, Shimizu M, Mochizuki H. et al. Astrocyte metabolism in multiple sclerosis investigated by 1-C-11 acetate PET. J Cereb Blood Flow Metab. 2021;41:369-79
264. Ishiwata K, Wang WF, Kimura Y, Kawamura K, Ishii K. Preclinical studies on [11C]TMSX for mapping adenosine A2A receptors by positron emission tomography. Ann Nucl Med. 2003;17:205-11
265. Rissanen E, Virta JR, Paavilainen T, Tuisku J, Helin S, Luoto P. et al. Adenosine A2A receptors in secondary progressive multiple sclerosis: a [(11)C]TMSX brain PET study. J Cereb Blood Flow Metab. 2013;33:1394-401
266. Kamiya K, Hori M, Aoki S. NODDI in clinical research. J Neurosc Methods. 2020;346:108908
267. Barakovic M, Weigel M, Cagol A, Schaedelin S, Galbusera R, Lu PJ. et al. A novel imaging marker of cortical "cellularity" in multiple sclerosis patients. Sci Rep. 2024;14:9848
268. Chan KS, Ma Y, Lee H, Marques JP, Olesen JL, Coelho S, et al. In vivo human neurite exchange time imaging at 500 mT/m diffusion gradients: Neurite exchange time imaging on Connectome 2.0. Imaging Neurosci (Camb). 2025; 3
269. Li Z, Li Z, Bilgic B, Lee HH, Ying K, Huang SY. et al. DIMOND: DIffusion Model OptimizatioN with Deep Learning. Adv Sci (Weinh). 2024;11:e2307965
270. Galbusera R, Weigel M, Bahn E, Schaedelin S, Cagol A, Lu PJ. et al. Quantitative T1 is sensitive to cortical remyelination in multiple sclerosis: A postmortem MRI study. Brain Pathol. 2025;35:e70010
271. Sanabria-Diaz G, Cagol A, Lu PJ, Barakovic M, Ocampo-Pineda M, Chen X. et al. Advanced MRI Measures of Myelin and Axon Volume Identify Repair in Multiple Sclerosis. Ann Neurol. 2024;97:134-48
272. Lee J, Ji S, Oh SH. So You Want to Image Myelin Using MRI: Magnetic Susceptibility Source Separation for Myelin Imaging. Magnetic resonance in medical sciences: MRMS. 2024;23:291-306
273. Gillen KM, Nguyen TD, Dimov A, Kovanlikaya I, Luu HM, Demmon E. et al. Quantitative susceptibility mapping is more sensitive and specific than phase imaging in detecting chronic active multiple sclerosis lesion rims: pathological validation. Brain Commun. 2025;7:fcaf011
274. Shin HG, Lee J, Yun YH, Yoo SH, Jang J, Oh SH. et al. χ-separation: Magnetic susceptibility source separation toward iron and myelin mapping in the brain. Neuroimage. 2021;240:118371
275. Zhu Z, Naji N, Esfahani JH, Snyder J, Seres P, Emery DJ. et al. MR Susceptibility Separation for Quantifying Lesion Paramagnetic and Diamagnetic Evolution in Relapsing-Remitting Multiple Sclerosis. Journal of magnetic resonance imaging: JMRI. 2024;60:1867-79
276. Jang A, Chan KS, Mareyam A, Stockmann J, Huang SY, Wang N. et al. Simultaneous 3D quantitative magnetization transfer imaging and susceptibility mapping. Magn Reson Med. 2025;94:735-44
277. Petracca M, Vancea RO, Fleysher L, Jonkman LE, Oesingmann N, Inglese M. Brain intra- and extracellular sodium concentration in multiple sclerosis: a 7 T MRI study. Brain. 2016;139:795-806
278. Cipriano E, Boffa G, Graziano N, Wigley C, Petracca M, Schiavi S. et al. Relationship between sodium and diffusion MRI metrics in multiple sclerosis. Brain Commun. 2025;7:fcae446
279. Starke L, Millward JM, Prinz C, Sherazi F, Waiczies H, Lippert C. et al. First in vivo fluorine-19 magnetic resonance imaging of the multiple sclerosis drug siponimod. Theranostics. 2023;13:1217-34
280. Pike VW. PET radiotracers: crossing the blood-brain barrier and surviving metabolism. Trends Pharmacol Sci. 2009;30:431-40
281. Moyaert P, Padrela BE, Morgan CA, Petr J, Versijpt J, Barkhof F. et al. Imaging blood-brain barrier dysfunction: A state-of-the-art review from a clinical perspective. Front Aging Neurosci. 2023;15:1132077
Author contact
![]() Corresponding authors: Achi Haider, Email: achi.haidercom; Steven H. Liang, Email: steven.liangedu.
Corresponding authors: Achi Haider, Email: achi.haidercom; Steven H. Liang, Email: steven.liangedu.