Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2016; 6(11):1947-1962. doi:10.7150/thno.16139 This issue Cite
Research Paper
Depletion of γ-catenin by Histone Deacetylase Inhibition Confers Elimination of CML Stem Cells in Combination with Imatinib
Yanli Jin1, Yiwu Yao2, Li Chen3, Xiaohui Zhu3, Bei Jin1, Yingying Shen3, Juan Li4, Xin Du5, Yuhong Lu6, Sheng Jiang2 ![]() , Jingxuan Pan1, 7
, Jingxuan Pan1, 7 ![]()
1. Jinan University Institute of Tumor Pharmacology, Jinan University College of Pharmacy; State Key Laboratory of Ophthalmology, Zhongshan Ophthalmic Center, Sun Yat-sen University, Guangzhou, China;
2. Laboratory of Medicinal Chemistry, Guangzhou Institute of Biomedicine and Health, Chinese Academy of Sciences, Guangzhou, China;
3. Department of Pathophysiology, Zhongshan School of Medicine, Sun Yat-sen University, Guangzhou, China;
4. Department of Hematology, The First Affiliated Hospital, Sun Yat-sen University, Guangzhou, China;
5. Department of Hematology, Guangdong General Hospital/Guangdong Academy of Medical Sciences, Guangzhou, China;
6. Department of Hematology, The First Affiliated Hospital, Jinan University, Guangzhou, China;
7. Collaborative Innovation Center for Cancer Medicine, State Key Laboratory of Oncology in South China, Sun Yat-Sen University Cancer Center, Guangzhou, China.
Received 2016-5-12; Accepted 2016-7-12; Published 2016-8-12
Abstract

Quiescent leukemia stem cells (LSCs) that are insensitive to BCR-ABL tyrosine kinase inhibitors confer resistance to imatinib in chronic myelogenous leukemia (CML). Identifying proteins to regulate survival and stemness of LSCs is urgently needed. Although histone deacetylase inhibitors (HDACis) can eliminate quiescent LSCs in CML, little is known about the underlying mechanism that HDACis kill LSCs. By fishing with a biotin-labeled probe, we identified that HDACi JSL-1 bound to the protein γ-catenin. γ-Catenin expression was higher in LSCs from CML patients than normal hematopoietic stem cells. Silencing γ-catenin in human CML CD34+ bone-marrow (BM) cells sufficiently eliminated LSCs, which suggests that γ-catenin is required for survival of CML LSCs. Pharmacological inhibition of γ-catenin thwarted survival and self-renewal of human CML CD34+ cells in vitro, and of murine LSCs in BCR-ABL-driven CML mice. γ-Catenin inhibition reduced long-term engraftment of human CML CD34+ cells in NOD.Cg-Prkdcscid II2rgtm1Sug/JicCrl (NOG) mice. Silencing γ-catenin by shRNA in human primary CD34+ cells did not alter β-catenin, implying a β-catenin-independent role of γ-catenin in survival and self-renewal of CML LSCs. Taken together, our findings validate that γ-catenin may be a novel therapeutic target of LSCs, and suppression of γ-catenin by HDACi may explain elimination of CML LSCs.
Keywords: HDAC inhibitor, imatinib-resistance, leukemia stem cells, γ-catenin, BCR-ABL.
Introduction
Chronic myelogenous leukemia (CML) is derived from malignant transformation of hematopoietic stem cells (HSCs) caused by a fusion gene BCR-ABL because of a reciprocal chromosomal translocation t (9,22) (q34; q11) [1]. The encoding product BCR-ABL tyrosine kinase activates several signal pathways to drive the disease by phosphorylating wide substrates [2]. Imatinib (Gleevec, STI571), the first-line drug to inhibit BCR-ABL tyrosine kinase activity for CML, achieves complete cytogenetic response in most CML patients in chronic phase (CP) [3]. However, resistance to imatinib is an emerging problem, particularly in patients in advanced phases of accelerated phase (CP) and blast crisis (BC) [4]. Patients usually have poor prognosis once resistance appears.
Multiple mechanisms have been proposed to explain the acquired resistance. The most common mechanism accounting for ~50% clinical resistance is point mutation in BCR-ABL (e.g., T315I, G250E, Q252H, Y253H, and E255K/V) [2, 5]. However, the problem of mutant BCR-ABL resistance may hopefully be resolved the novel tyrosine kinase inhibitors (TKIs) such as ponatinib and DCC-2036 [6, 7]. The remaining half of clinical resistance to imatinib may be caused by other multiple factors, including leukemia stem cells (LSCs) or leukemia-initiating cells [6, 8], BCR-ABL-independent clones [4, 9, 10], and binding of imatinib to serum α-1 acid glycoprotein [11]. LSCs in CML possess common characteristics of cancer stem cells (CSCs) (e.g., self-renewal to maintain a pool of CSCs, proliferation to diverse-stage of leukemia cells, quiescence status) [12-14]. In addition, LSCs share several properties with normal HSCs, including the capacity for self-renewal and pluripotency and signaling pathways [15, 16]. Interestingly, although LSCs are believed to originate from malignant transformation of HSCs triggered by BCR-ABL, the continual maintenance of an LSC pool in CML may be BCR-ABL-independent probably because of gain of additional oncogenic hits [15, 17]. Compelling evidence has shown that LSCs cannot be eradicated by monotherapy of TKIs. Long-term follow-up observation revealed persistence of BCR-ABL-negative LSCs in even CML patients in remission with imatinib treatment [18, 19]. In this case, relapse frequently occurred once imatinib was discontinued. The STIM trial demonstrated relapse in most CML patients within 12 months of TKI discontinuation [20]. CML patients usually have to take TKI drugs for their rest of lives. Actually, CML primitive progenitor cells and bulk leukemia cells are sensitive to TKI-induced apoptosis, whereas CML LSCs are resistant to TKI-induced apoptosis [15]. Therefore, CML probably cannot be cured with TKIs alone, which just inactivate BCR-ABL enzyme activity, despite their effectiveness in controlling the progression of CP CML [8, 13, 17, 21].
CML LSCs are at least regulated at three layers [22]. First, like common types of cells, LSCs survival is regulated by apoptosis regulators (e.g., Bcl-2, survivin, Mcl-1) [23]. Second, the self-renewal of LSCs is regulated by HSC development-related pathways (e.g., Wnt/β-catenin, Hedgehog) [16], metabolism regulators (e.g., Alox5, Scd) [24], transcription factors (e.g., Foxo3a, HIF1α) and epigenetic regulators [8]. The third layer of regulations is from the microenvironment (also called niches). However, the precise regulation networks of CML LSCs are not completely understood.
Histone deacetylase inhibitors (HDACis) are a class of anti-tumor agents that loosen the transcription of repressed genes located on the surface of histone core by augmenting acetylation in lysine residues on histone proteins [25, 26]. HDACis can selectively kill malignant tumor cancer while sparing normal cells [12, 13]. Moreover, HDACis alone and combined with TKIs can eliminate quiescent LSCs in CML and acute myelogenous leukemia [12, 13]. However, little is known about the underlying mechanism that HDACis kill LSCs.
We recently synthesized a novel cyclic tetrapeptide class I-selective HDACi JSL-1 (Compound 12) [27]. We confirmed the effect of JSL-1 against the primitive LSCs from CML patients and CML mice transformed by retrovirus BCR-ABL transduction. The present study aimed at elucidating the underlying mechanism that HDACis kill LSCs. By using JSL-1 compound probe labeling with biotin, we identified its binding protein, γ-catenin. γ-Catenin level was higher in LSCs from CML patients than normal HSCs. Inhibition of γ-catenin by specific shRNA or by JSL-1 facilitated the elimination of CML LSCs. Taken together, our results revealed that γ-catenin regulates the stemness of CML LSCs, and that γ-catenin is targeted by HDACi. The findings improved the understanding of the molecular regulation of CML LSCs, and the mechanism by which HDACi selectively eliminates LSCs.
Materials and Methods
Primary cells
Peripheral blood or BM samples were obtained from patients with CML and from healthy adult donors in The First Affiliated Hospital of Sun Yat-sen University, Guangdong General Hospital/Guangdong Academy of Medical Sciences, and The First Affiliated Hospital of Jinan University. The characteristics of CML patients were described in Supplementary Table S1. The isolation and culture for primary CD34+ hematopoietic stem and progenitor cells were described as before [28]. Detailed information was in Supplementary materials and methods.
Western blot analysis
Whole cell lysates were prepared in radioimmunoprecipitation assay (RIPA) buffer [28, 29]. The detailed information for antibodies was summerized in Supplementary Table S2. The membranes were scanned by using the Odyssey infrared imaging system (LI-COR).
Lentivirus transduction of CML CD34+ cells
Lentivirus was produced by transient transfection in 293T cells with control shRNA (Scramble) or specific shRNA together with the pCMV-dR8.2 packing construct and the pCMV-VSVG envelope construct. Viral supernatants were collected 48 and 72 hr after transfection and purified with the 0.45-μm filter. CML CD34+ cells or healthy donor normal bone-marrow (NBM) CD34+ cells (1×106 cells/ml) were transduced by spinoculation (1,500 g, 90 min, 32oC) with virus-containing supernatants two rounds. Cells were harvested 48 hr later; the knockdown effect was examined by qRT-PCR analysis.
Analysis of cell viability, colony formation and apoptosis in CML cell lines
Cell viability was assessed by MTS assay (CellTiter 96 Aqueous One Solution reagent, Promega, Shanghai) [28, 30, 31].
For soft agar clonogenic assay, cells were treated with JSL-1 or diluent (DMSO, control) for 24 hr, then washed with phosphate buffered saline (PBS) and seeded in Iscove's medium containing 0.3% agar and 20% FBS in the absence of compound treatment [28, 30].
Apoptosis was evaluated by use of an Annexin V-fluorescein isothiocyanate (FITC)/PI apoptosis detection Kit (Sigma-Aldrich, Shanghai) and analyzed by use of a BD FACS Aria II flow cytometer [28, 30].
Analysis of cell apoptosis for primary CD34+ cells
CML or NBM CD34+ cells were labeled with Annexin V-FITC or CD38-PE antibody after treatment, then cell apoptosis (CD34+ CD38- Annexin V+) was dectected by use of the BD FACS Aria II flow cytometer [28].
For carboxyfluorescein diacetate succinimidyl ester (CFSE) assay, CFSE-labeled cells (CellTrace CFSE Proliferation Kit; Invitrogen) were cultured with JSL-1 for 96 hr, then harvested and stained with AnnexinV-PE; quiescent cell apoptosis (CD34+ CFSEmax Annexin V+) was determined by use of the BD FACS Aria II flow cytometer [12, 13].
Colony-forming cell (CFC)/replating and LTC-IC assay
CFC/replating and long-term culture-initiating cell (LTC-IC) assay were determined as decribed [12, 13].
For CFC/replating assay, CML or NBM CD34+ cells were treated with different doses of JSL-1 for 24 hr, washed with PBS and counted; 1,000 cells were plated in methylcellulose medium (H4434, StemCells Technologies, Vancouver, Canada). Colonies were counted on days 14. Cells were harvested and counted, and 1,000 cells were replated for the secondary and tertiary rounds, and colonies were counted on day 14 after each replating.
For LTC-IC assay, 2×106 CML or NBM cells were seeded in the LTC-IC medium (H5100, StemCells Technologies) on irradiated (80 Gy) M2-10B4 cells and treated with JSL-1 (1.0 μM) with or without imatinib (2.5 μM) for 1 week; compound-containing medium was removed, and cells were cultured for 5 weeks in drug-free LTC-IC medium by weekly half drug-free medium replacement. After 6 weeks, cells were harvested and plated into MethoCult medium (H4435, StemCells Technologies). LTC-IC-derived colonies were counted after 2 weeks.
CML mouse model
The retrovirus was produced by transient transfection with the MSCV-BCR-ABL-IRES-EGFP construct in Plat-E cells. Viral supernatants were collected 48 and 72 hr after transfection and purified with a 0.45-μm filter. BM cells from 6- to 8-week-old donor male C57BL/6J mice pretreated with 5-fluorouracil (5-FU) were stimulated with cytokines in vitro, then transplanted by tail-vein injection into irradiated (550 cGy) recipient female C57BL/6J mice after transduction two rounds with the MSCV-BCR-ABL-IRES-EGFP retrovirus [14, 32]. Two weeks after transplantation, 100% of the mice developed CML. The mice were then euthanized, and their splenic cells were injected into irradiated recipient C57BL/6J mice. Following transplantation, the mice were randomly divided into 4 groups and treated with imatinib (100 mg/kg/d by gavage), JSL-1 (25 mg/kg/d by intraperitoneal injection) or both imatinib and JSL-1 for 2 weeks.
For γ-catenin shRNA treatment in CML mice, splenic cells from CML mice were transduced with control shRNA or mouse γ-catenin shRNA lentivirus, then transplanted by tail-vein injection into sublethally irradiated recipient C57BL/6J mice 4 hr after the second round of transduction with mouse γ-catenin shRNA lentivirus. Following transplantation, the mice were randomly divided into 4 groups and treated with imatinib (100 mg/kg/d by gavage) for 2 weeks. Mice were euthanized and BM and splenic cells were collected, labeled with the indicated antibodies and analyzed by use of the BD FACS Aria II flow cytometer. The detailed information for the dectection of LSK, LT-HSC and ST-HSC was in Supplementary materials and methods.
Preparation of Cytoplasmic and Nuclear Fractions
Cytoplasmic and nuclear fractions were prepared as described previously [28].
Statistical analysis
All experiments were performed at least 3 times, and error bars are reported as mean ± SD, unless otherwise stated. GraphPad Prism 5.0 (GraphPad Software, San Diego, CA) was used for statistical analysis. Paired groups were analyzed by Student's t test. Multiple groups were analyzed by one-way ANOVA with post-hoc intergroup comparison with the Tukey's test. P < 0.05 was considered statistically significant. Kaplan-Meier survival curves were analyzed by log-rank test.
Results
JSL-1 inhibits growth of imatinib-sensitive and -resistant CML cells
We first confirmed the cellular inhibitory effect of JSL-1 (Fig. 1A) on HDAC in CML cells. Treatment of JSL-1 for 36 hr led to a dose-dependent increase in acetylated H3K9 and H4K16 in CML cells (Fig. 1B). We explored whether JSL-1 was active against CML cells harboring T315I BCR-ABL. Cell viability detected with MTS was decreased dose-dependently with JSL-1 regardless of the mutant or WT status of BCR-ABL (Fig. 1C). JSL-1 had striking inhibitory potency against the primary leukemia cells (Fig. 1D). Using soft agar or methylcellulose culture system, we discovered that JSL-1 inhibited the tumorigenicity of CML cells (Fig. 1E) and the clonogenicity derived from primary leukemia cells of CML patients (Fig. 1F).
JSL-1 potently inhibits the growth of imatinib-resistant chronic myelogenous leukemia (CML) cells expressing T315I BCR-ABL in mouse model. (A) Chemical structure of HDACi JSL-1. (B) Western blot analysis of protein levels of acetylated and total histone H3 and H4 in CML cells after treatment with JSL-1. (C-D) CML cells (C) or BM cells of CML patients (n=5) (D) were treated with various concentrations of JSL-1 for 72 hr; cell viability was measured by MTS assay. Dose-response curves were shown. (E-F) Clonogenicity of CML cells in soft agar (E) and BM cells from CML patients is methylcellulose (F) dose-dependently inhibited by JSL-1. (G-H) The growth curves of subcutaneous xenografts of CML cells. BALB/c nu/nu nude mice were subcutaneously inoculated with KBM5 (G) or KBM5-T315I (H) cells, then randomized into 2 or 3 groups. Mice were treated with JSL-1 alone, JSL-1 and imatinib, or vehicle during days 5-21 after inoculation of cells. The tumor growth curves were plotted. (I-J) Weights of tumors on days 21 or 14 after treatment. **, P<0.01 ***, P<0.0001. (K-L) Immnunohistochemical analysis with anti-Ki67 or c-ABL and H & E staining of xenograft tissues from mice.
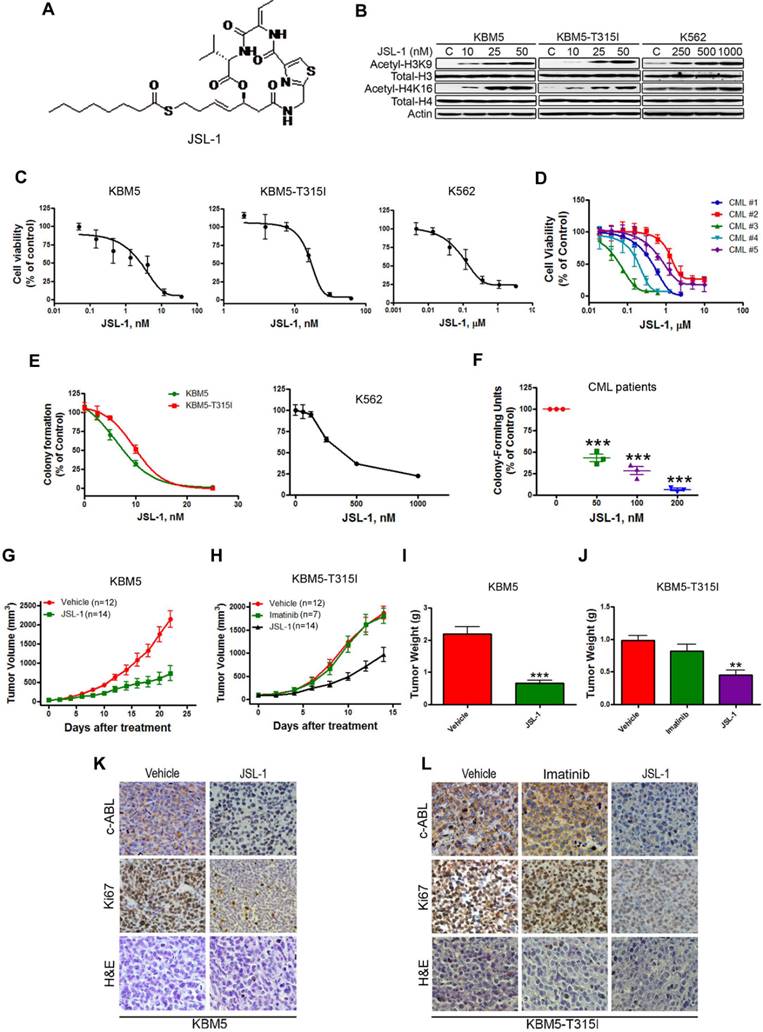
To assess the in vivo anti-tumor effect of JSL-1, four days after subcutaneous inoculation of KBM5 or KBM5-T315I cells in nude mice, when tumors were palpable, the mice were randomized to receive vehicle or JSL-1 for 14 days. Compared with vehicle treatment, JSL-1 treatment strikingly delayed the growth of tumors derived from KBM5 or KBM5-T315I cells (Fig. 1G-H). JSL-1 administration also elicited a tremendous decrease in tumor weights (Fig. 1I-J). Imatinib failed to inhibit the growth of KBM5-T315I xenografts in mice (Fig. 1H and 1J), suggesting their resistant to imatinib. Immnunohistochemical staining signals for c-ABL and Ki67 were less in tumors with JSL-1 than vehicle treatment (Fig. 1K-L).
γ-Catenin is important in JSL-1-mediated cell death of LSCs
The potent anti-leukemia activity of JSL-1 prompted us to further define potential targets other than HDAC. We first covalently labeled compound of JSL-1 with biotin (Fig. 2A) and confirmed the sustained biological activity similar to that of its corresponding parent compound JSL-1 (data not shown). We then screened potential target(s). Whole cell lysates from KBM5 cells were incubated with biotin-JSL-1, then precipitants with streptavidin agarose beads were separated on SDS-PAGE and viewed after silver staining (Fig. 2B). A consistently differential protein (Band 1, Fig. 2B) located at approximately 72 kDa underwent mass spectroscopy assay. Bioinformatics analysis suggested that this protein may be γ-catenin (plakoblobin). Western blot analysis of the immunoprecipitation pellets revealed γ-catenin rather than β-catenin in the biotin-JSL-1 lane (Fig. 2C), suggesting that γ-catenin may be a binding protein of JSL-1. We then examined γ-catenin in subsequent experiments.
JSL-1 inhibits γ-catenin in human leukemia stem cells (LSCs) in CML. (A) Chemical structure of Biotin-JSL-1. (B-C) KBM5 cell lysates were incubated with biotin-JSL-1 or biotin, then pulled down with streptavidin-agarose. The precipitates were resolved by SDS-PAGE, and the gel was stained with silver (B) or detected by Western blot analysis for γ-catenin and β-catenin (C). (D) The mRNA levels of γ-catenin in CP CML (n=6), AP CML (n=3) and BC CML (n=1) versus NBM (n=6) CD34+ cells were analyzed by qRT-PCR. (E) γ-Catenin protein levels detected by Western blot were overexpressed in human CML CD34+ cells relative to NBM CD34+ cells. (F) JSL-1 decreased γ-catenin and BCR-ABL protein levels but not β-catenin in human CML CD34+ cells (Western blot). (G) JSL-1 treatment time-dependently decreased levels of γ-catenin evaluated by Western blot in CML CD34+ cells after treated with JSL-1. (H) Treatment with JSL-1 or silencing γ-catenin did not alter the protein levels or cellular localization of β-catenin. K562 cells were treated with the indicated concentrtions of JSL-1 for 36 hr (left) or 72 hr after infected with lentiviral γ-catenin shRNA (right), the protein levels of γ-catenin and β-catenin in whole cell lysates (WCL), cytoplasma, and nuclear were detected by Western blot. Tubulin and PCNA were indicators of cytoplasma and nuclear extractions, respectively. (I-J) qRT-PCR analysis mRNA levels of c-Myc, CCND1 and LEF1 in human CML CD34+ cells after transfected with lentiviral shRNA against γ-catenin (I) or plasmid encoding γ-catenin (J). (K) TCF/LEF-dependent luciferase activity in human CML CD34+ cells after treated with JSL-1 abrogated. * P<0.05, *** P<0.0001.
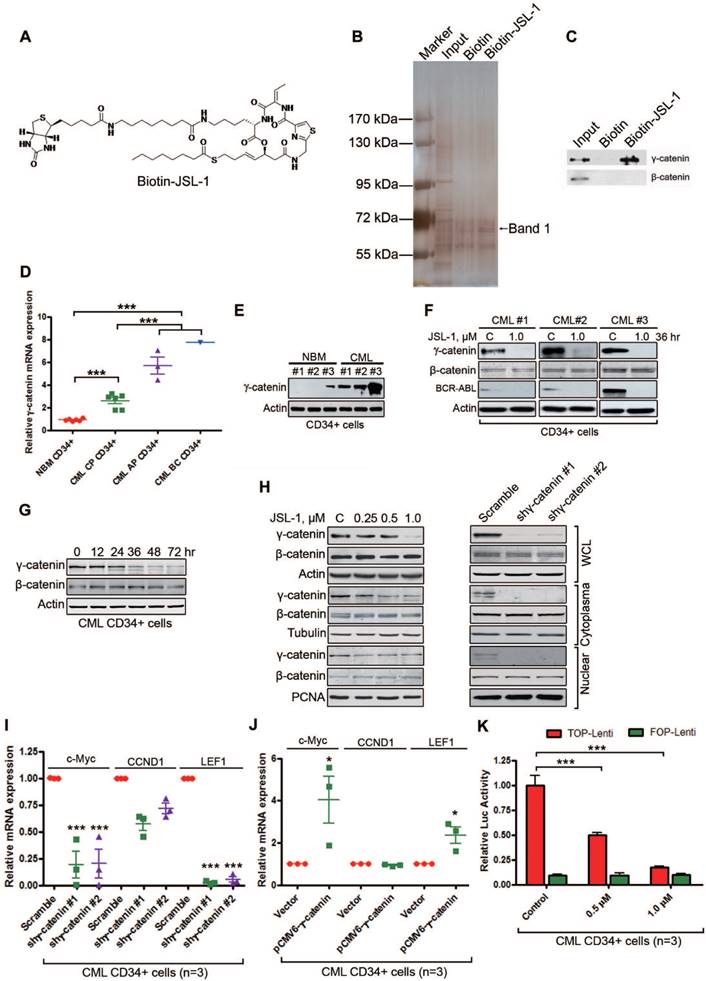
Because γ-catenin in the Wnt signaling pathway shares partially overlapping traits in regulating CSCs and is involved in myeloid leukemia with β-catenin [33, 34], we tested whether γ-catenin was overexpressed in CML LSCs. The mRNA levels of γ-catenin were higher in CML BM CD34+ cells than NBM CD34+ cells (Fig. 2D). Of note, the levels of γ-catenin in patients with AP-CML and BC-CML were much higher than those in patients with CP-CML (Fig. 2D). The protein levels of γ-catenin were higher in CML CD34+ cells than NBM CD34+ cells (Fig. 2E). To evaluate whether JSL-1 was a target of γ-catenin, primary CML CD34+ cells were treated with JSL-1. γ-Catenin and BCR-ABL protein levels were lower in JSL-1-treated CML CD34+ cells than control (Fig. 2F). Previous report demonstrated that overexpression of γ-catenin promotes the stabilization and nuclear localization of β-catenin in bulk leukemia cells [33]. To characterize whether the effect of γ-catenin inhibition be actually transmitted through changes in β-catenin levels or localization, we first conducted dynamic evaluation of β-catenin and γ-catenin in JSL-1 treated cells. The results showed that there was a remarkable reduction in γ-catenin, but not in β-catenin, as early as 36 hr after treatment with JSL-1. Levels of β-catenin were not markedly altered until 72 hr after treatment with JSL-1 (Fig. 2G). Further, inhibition of γ-catenin with JSL-1 treatment or silencing γ-catenin by shRNA did not affect the cellular localization of β-catenin (Fig. 2H). Similar results were obtained in CML CD34+ cells (Fig. S1A). The findings further supported that γ-catenin might act in a β-catenin-independent manner. Conversely, β-catenin knockdown by siRNA in K562 cells had no effect on the cellular localization of γ-catenin (Fig. S1B).
We next determined the underlying mechanism of reduced γ-catenin expression with JSL-1 treatment. Pre-incubation with ubiquitin-proteasome inhibitor MG132 did not reverse the JSL-1-decreased γ-catenin levels in K562 cells (Fig. S2A, left). Of note, MG132 alone treatment did not increase γ-catenin protein levels as compared with Dvl3 (Fig. S2A, right), which suggests the relative independence of the ubiquitin-proteasome system during decay of γ-catenin protein. However, JSL-1 treatment dose-dependently repressed γ-catenin mRNA levels (Fig. S2B). In addition, SAHA and trichostatin A (TSA) were capable of downregulating levels of γ-catenin protein (Fig. S2C) and mRNA (Fig. S2D), which suggests that γ-catenin downregulation may be an universal phenomenon on treatment with diverse types of HDACis.
To elucidate the underlying mechanism of γ-catenin expression affected by HDACis, we searched and discovered two putative binding sites for FoxM1 transcriptional factor in the promoter region of γ-catenin gene. FoxM1 protein levels were decreased in HDACi-treated K562 cells (Fig. S2C). Conversely, forced overexpression of FoxM1 in 293T cells increased the transcription of γ-catenin gene (Fig. S2E). We further examined whether FoxM1 directly bound to the promoter sequence of γ-catenin gene by chromatin immunprecipitation (ChIP) assay. The results suggested a binding capability of FoxM1 to γ-catenin gene promoter and tremendous inhibitory effect of JSL-1, SAHA and TSA on the binding (Fig. S2F).
Next, we ascertained whether γ-catenin affected its downstream target genes (e.g., c-Myc, CCND1, and LEF1) in LSCs. Silencing γ-catenin gene by transducing human CML CD34+ cells with γ-catenin shRNA reduced the mRNA levels of c-Myc and LEF1 but not CCND1 (Fig. 2I). Conversely, ectopic expression of γ-catenin in CML CD34+ cells significantly increased the mRNA levels of c-Myc and LEF1 but not CCND1 (Fig. 2J). The results agreed with the notion of preferential dependency of c-Myc and LEF1 rather than CCND1 on γ-catenin versus β-catenin [21]. γ-Catenin in complex with TCF4/LEF1 transcription factors can activate target genes whose promoter contains the regulatory elements [21]. Therefore, we examined whether JSL-1 reduced TCF4/LEF1-dependent transcription. Luciferase assay indicated that JSL-1 inhibited TCF/LEF-mediated transcription in CML CD34+ cells (Fig. 2K).
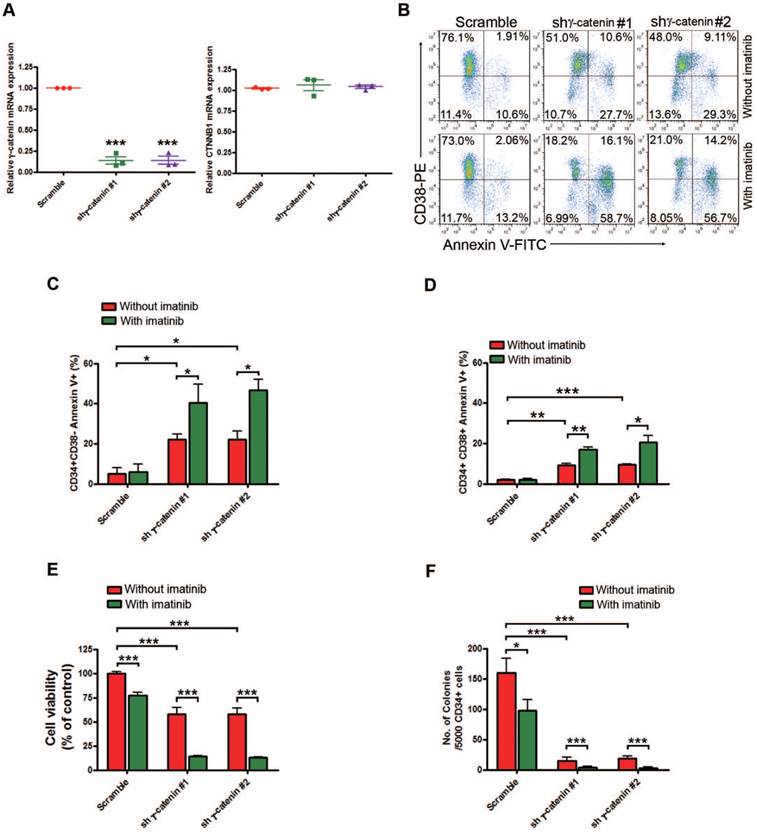
Given the potentially instrumental role of γ-catenin for self-renewal of cancer stem cells [21, 34, 35], we wondered whether JSL-1-decreased γ-catenin levels eliminated LSCs in CML. Silencing γ-catenin by lentiviral shRNA in primary CD34+ cells isolated from CML patients significantly increased the apoptotic cell percentage in the CD34+CD38- stem/primitive progenitor cells and CD34+CD38+ committed progenitor cells (Fig. 3A-D). Concomitantly, γ-catenin knockdown greatly counteracted the cell viability of CML CD34+ cells (Fig. 3E). The tumorigenicity of CD34+ CML cells, as reflected by the number of colony-forming cells (CFCs) in methylcellulose progenitor assays, was also significantly decreased by γ-catenin knockdown (Fig. 3F). Of importance, these effects of silencing γ-catenin were augmented in the presence of imatinib (Fig. 3B-F). Silencing γ-catenin did not markedly induce apoptosis in NBM CD34+ cells (Fig. S3A-D) and not significantly inhibit the cell viability of NBM CD34+ cells (Fig. S3E). However, the CFC growth in NBM CD34+ cells was decelerated with JSL-1, but the inhibition content of normal colony formation was much less as compared with CML CD34+ cells (Fig. S3F and Fig 3F). To identify the role of γ-catenin in JSL-1-induced cell growth inhibition, we first established KBM5 cells stably expressing either pCMV6-JUP (γ-catenin) or pCMV6 (empty vector) constructs by electrotransfection and examined their response to JSL-1. The results showed that forced overexpressing γ-catenin in CML cells led to a decreased sensitivity to JSL-1 treatment (Fig. S3G-H). Collectively, γ-catenin may be a binding protein of JSL-1 that is important in JSL-1-mediated cell survival or death in bulk CML cells and LSCs.
Silencing γ-catenin reduces proliferation, survival and colony formation in human CML LSCs. Human BM CML CD34+ cells (n=3) purified with immunomagnetic beads were transduced with lentiviral control shRNA (Scramble), shγ-catenin #1, or shγ-catenin #2 for 48 hr, then treated with imatinib (2.5 μM) for another 24 hr. (A) mRNA evaluation of γ-catenin (left) and CTNNB1 (β-catenin) (right) was performed by qRT-PCR. (B) Representative flow cytometry of apoptosis detected with Annexin V-FITC and CD38-PE was shown. (C-D) Apoptosis in CD34+CD38- stem/primitive progenitor cells (C) or CD34+CD38+ committed progenitor cells (D) was analyzed by flow cytometry after staining with Annexin V-FITC and CD38-PE labeling. (E) Cell viability of human CML CD34+ cells was determined by MTS assay. (F) Colony-formation assay of human CML CD34+ cells performed in drug free-methylcellulose medium (H4434) for 14 days. * P<0.05, ** P<0.01, *** P<0.0001.
γ-Catenin is required for maintaining self-renewal capacity of LSCs in CML mice
We further defined the in vivo role of γ-catenin in serial transplantational leukemogenicity of CML LSCs by using a BCR-ABL-driven mouse model mimicking human CML [14, 36]. C57BL/6 mice were irradiated and intravenously received 5-FU-enriched donor BM cells that were transduced with retroviral MSCV-IRES-EGFP carrying the p210 BCR-ABL cDNA. CML developed in recipient mice over 2 weeks. LSCs in BM and spleen in this model will recapitulate leukemia if they are intravenously transplanted into secondarily irradiated recipient mice (Fig. 4A).
Silencing γ-catenin reduces CML LSCs growth in CML mice. (A) Schematic strategy to murine CML experiment procedure. (B) Western blot analysis of the effect of γ-catenin shRNA in splenic mononuclear cells. (C) γ-Catenin knockdown alone or in combination with imatinib significantly prolonged survival of CML mice. Kaplan-Meier survival curves were shown, * P=0.0178, *** P<0.0001, log-rank test. (D-G) Analysis of LSCs in the BM cells of CML mice received treatment with shγ-catenin ± imatinib. Control (n=13), imatinib (n=10), γ-catenin shRNA (n=7), combination (n=10). (D) Proportion of GFP+ cells (leukemia cells) in BM. (E) Proportion of GFP+ myeloid cells (Gr-1+ Mac-1+) in BM. (F) Representative flow cytometry plots of GMPs and CMPs in BM. (G) GMP cell proportion in BM. (H) Representative flow cytometry plots of LSK cells, LT-HSCs, and ST-HSCs in BM. (I-K) Results for the GFP+ population in BM: LSK cells (I), LT-HSCs (J), and ST-HSCs (K). ** P<0.01, *** P<0.0001.
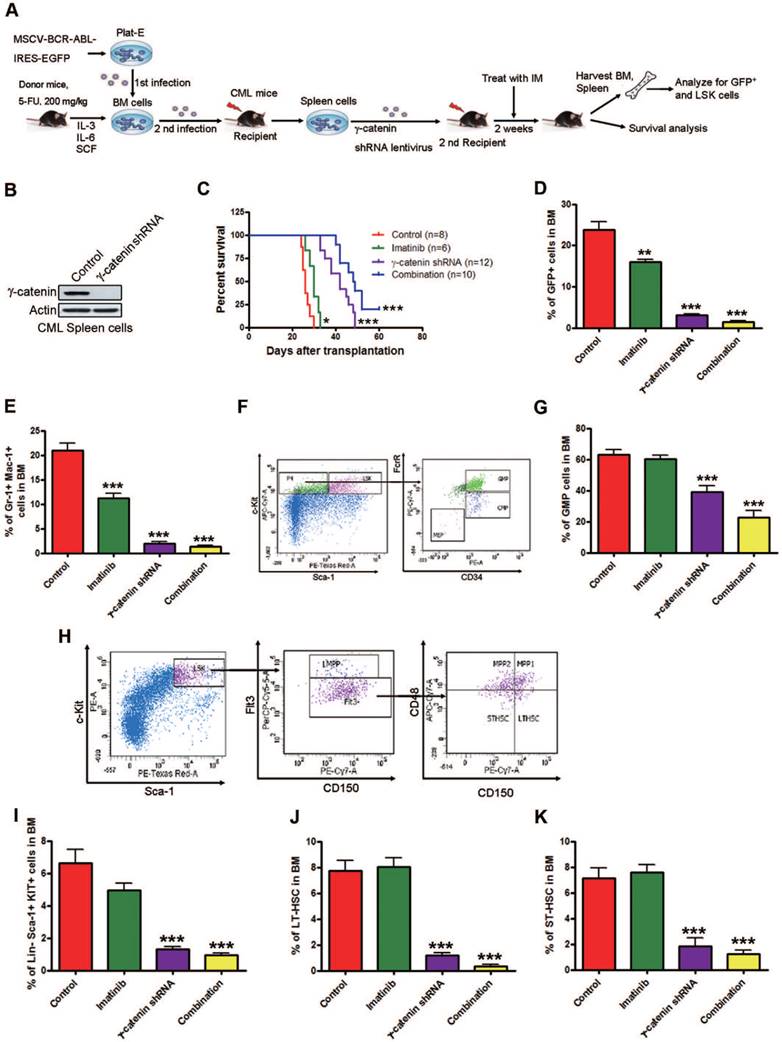
We knocked down γ-catenin by lentivirus shRNA transduction or scramble shRNA in splenic GFP+ cells collected from the first generation of CML mice as confirmed by Western blot (Fig. 4B). These splenic cells were then intravenously transplanted into sublethally irradiated (550 cGy) C57BL/6 mice. The recipient mice received ± imatinib. The mice were euthanized and BM and splenic mononuclear cells were harvested and analyzed by flow cytometer. γ-Catenin knockdown alone and with imatinib significantly prolonged the survival of CML mice (Fig. 4C). The populations of BCR-ABL-expressing leukemia cells (GFP+), myeloid cells (Gr-1+ Mac-1+) and granulocyte-macrophage progenitors (GMPs, Lin-Sca-1-c-Kit+CD34+FcγRII/IIIhig) in BM were significantly reduced in the γ-catenin-knockdown group and further reduced with imatinib treatment (Fig. 4D-G). Flow cytometry results revealed that γ-catenin knockdown alone or in combination with imatinib but not imatinib alone greatly decreased LSK cells (Lin-Sca-1+c-Kit+), LT-HSCs (LSK Flt3-CD150+ CD48-) and ST-HSCs (LSK Flt3-CD150-CD48-) in BM of CML mice (Fig. 4H-K). Knockdown of γ-catenin alone or in combination with imatinib significantly inhibited the splenomegaly of CML mice (Fig. S4A).
Splenic cells of γ-catenin-knockdown mice and in combination with imatinib but not imatinib alone showed markedly reduced proportion of BCR-ABL-expressing (GFP+) leukemia cells (Fig. S4B), myeloid cells (Gr-1+ Mac-1+) (Fig. S4C), GMPs (Fig. S4D-E), LSK cells (Fig. S4F-G), LT-HSCs (Fig. S4H) and ST-HSCs (Fig. S4I). These results suggest that silencing γ-catenin impairs the self-renewal capacity of CML LSCs and sensitizes them to imatinib in vivo; and that γ-catenin may be required for maintenance of self-renewal capacity in CML LSCs in vivo.
Pharmacological inhibition of γ-catenin by JSL-1 inhibits survival and self-renewal of human CML CD34+ cells
We next detected the effect of JSL-1 and imatinib alone or in combination on human CML stem cells. Given that JSL-1 and other HDACis (e.g. SAHA, TSA) potently inhibited γ-catenin in human CML K562 cells (Fig. S2). Then we determined whether JSL-1 affected survival of CML and normal stem/progenitor cells. CML or NBM CD34+ cells were incubated with a growth factor cocktail with JSL-1 ± imatinib for 24 hr, then labeled with Annexin V-FITC and CD38-PE. Apoptosis was analyzed by flow cytometer (Fig. 5A and Fig. S5A). JSL-1 treatment induced apoptosis in CML CD34+CD38- stem/primitive progenitor cells and CD34+CD38+ committed progenitor cells (Fig. 5B) but had limited effect on NBM CD34+CD38- cells (Fig. S5B). We also examined the effect of JSL-1 on apoptosis in CML and normal quiescent stem cells. CFSE-labeled CML or NBM CD34+ cells were incubated with JSL-1 for 96 hr. JSL-1 treatment increased the apoptosis of the CFSEmax undivided CML CD34+ cell fraction (Fig. 5C) but not the undivided counterparts in normal cells (Fig. S5C).
Pharmacological inhibition of γ-catenin induces enhanced sensitivity to apoptosis and reduced self-renewal capacity of CML CD34+ cells. JSL-1 treatment enhanced sensitivity of apoptosis to imatinib in human CML BM CD34+ cells. (A) Representative flow cytometry plots of apoptosis in CML CD34+ cells. (B) Quantitative analysis of apoptosis in CML CD34+ CD38- stem/primitive progenitor cells (left) and CD34+ CD38+ committed progenitor cells (right). (C) JSL-1 induced apoptosis of CML quiescent CD34+ cells. CML CD34+ cells were labeled with carboxyfluorescein diacetate succinimidyl ester (CFSE), then cultured with JSL-1 for 96 hr; cells were labeled with Annexin V-PE and analyzed by flow cytometry; results for CFSEmax and Annexin V+ cells were shown. (D) JSL-1 decreased long-term serial replating ability of human CML LSCs. (E-F) JSL-1 decreased long-term culture-initiating cell (LTC-IC) in human CML cells. The number (E) and frequency calculated by Poisson statistics (F) of LTC-IC in human CML cells treated with JSL-1 ± imatinib. * P<0.05, ** P<0.01, *** P<0.0001.
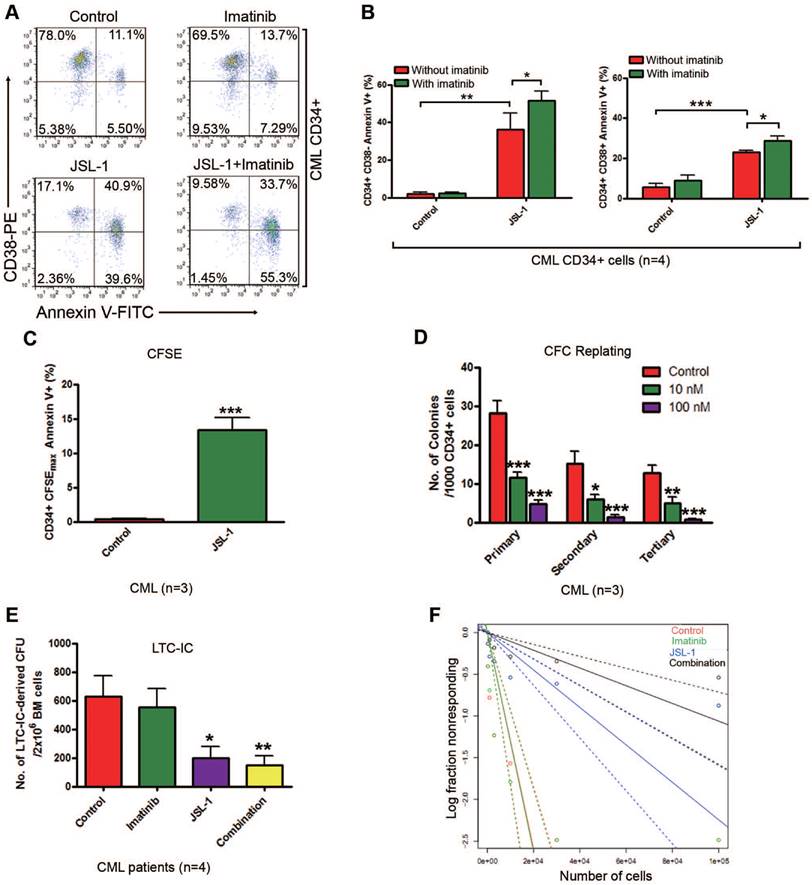
The capacity for self-renewal is the most important characteristic of LSCs. Therefore, we assessed whether JSL-1 treatment disabled the capacity for self-renewal in human CML LSCs by CFC/replating assay and LTC-IC assay. JSL-1 treatment at 10 and 100 nM significantly decreased the ability for CFC formation and serial plating capacity of CML CD34+ cells in methylcellulose (Fig. 5D) but did not affect the replating capacity of NBM CD34+ cells (Fig. S5D). Likewise, JSL-1 treatment greatly decreased the number (Fig. 5E) and frequency (Fig. 5F) of LTC-IC-derived colony-forming units (CFUs) in CML BM cells, with a limited effect on CFUs in NBM cells (Fig. S5E). The frequency of CML stem cells after treatment was shown in Supplementary Table S4. As anticipated, imatinib treatment had minimal effect on the number of LTC-IC-derived CFUs in CML and NBM cells (Fig. 5E and Fig. S5E). Thus, pharmacological inhibition of γ-catenin by JSL-1 may inhibit the survival and self-renewal capacity of quiescent human CML LSCs while sparing NBM HSCs.
γ-Catenin inhibition reduces long-term engraftment of human CML CD34+ cells in NOG mice
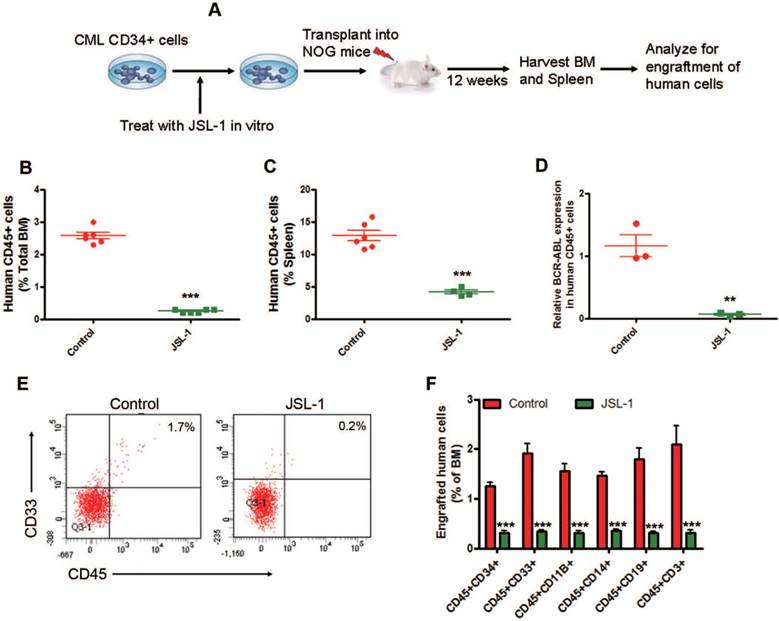
To investigate the long-term ex vivo effect of JSL-1 on engraftment of human CML CD34+ cells, MACS-purified human CML CD34+ cells were treated with JSL-1, then injected into NOD.Cg-Prkdcscid II2rgtm1Sug/JicCrl (NOG) mice by tail vein (Fig. 6A). JSL-1 treatment reduced the engraftment of human CML CD45+ (human leukocyte common antigen) cells in murine BM (Fig. 6B) and spleen (Fig. 6C) at 12 weeks after transplantation. JSL-1 treatment markedly decreased BCR-ABL mRNA levels in the engrafted human CML CD45+ cells sorted by flow cytometry from NOG murine BM cells (Fig. 6D). The proportion of engrafted human CML CD45+CD33+ (myeloid precursors), CD45+CD11B+ (myeloid cells or macrophages), CD45+CD14+ (monocytes), CD45+CD19+ (B lymphoid cells), and CD45+CD3+ (T lymphoid cells) in NOG murine BM cells was decreased with JSL-1 treatment (Fig. 6E-F). These results indicate that ex vivo JSL-1 treatment inhibites the long-term engraftment capacity of human CML CD34+ cells.
Pharmacological inhibition of γ-catenin inhibits long-term engraftment of human CML CD34+ cells in NOG mice. (A) A schematic strategy for long-term engraftment of human CML CD34+ cells in NOG mice. (B-C) JSL-1 treatment decreased the percentage of human CD45+ cells. Long-term engraftment in nucleated cells of BM (B) and spleen (C) in NOG mice were analyzed by flow cytometry. (D) Human BCR-ABL mRNA levels in CD45+ cells engrafted in BM at 12 weeks were analyzed by qRT-PCR. (E) Representative flow cytometry plots of human CD45 and CD33 expression in mice with cells from one of the two CML patients (n=4). (F) JSL-1 treatment decreased the proportion of engraftment of human CD34, CD33, CD11B, CD14, CD19 and CD3 cells in nucleated cells of BM. ** P<0.01, *** P<0.0001, Student's t test.
Pharmacological inhibition of γ-catenin reduces growth of LSCs in CML mice
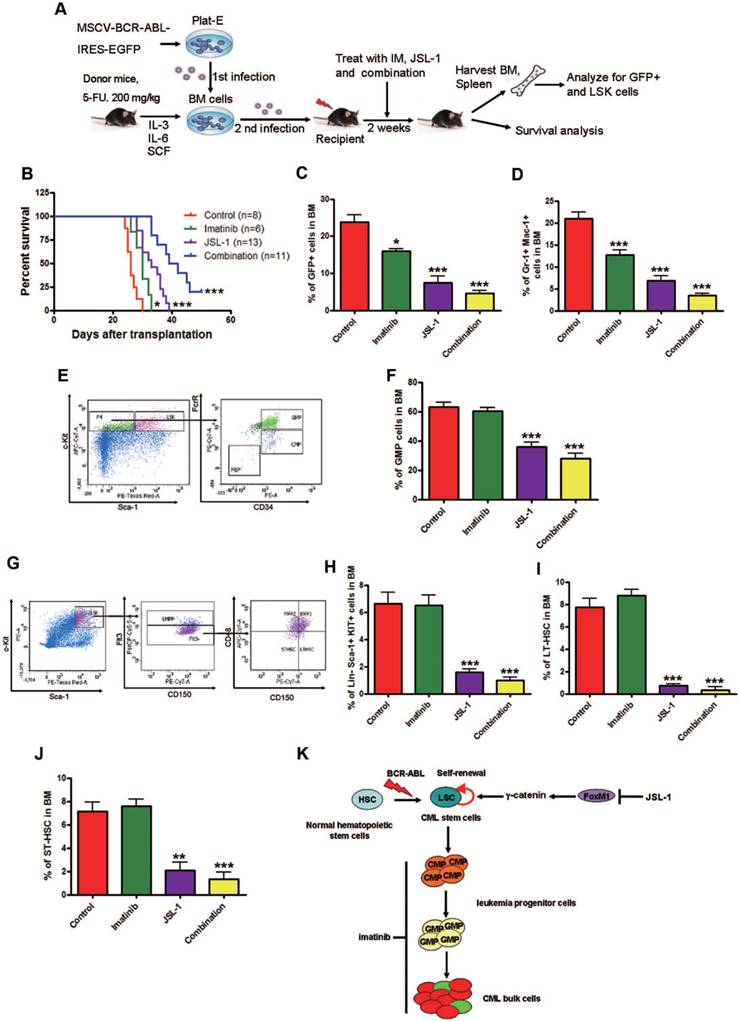
To evaluate the effect of JSL-1 on CML LSC growth in vivo, we used retroviral-BCR-ABL-driven CML mice model again. The CML mice were treated with JSL-1, imatinib, or both, for 2 weeks (Fig. 7A). JSL-1 treatment alone or in combination with imatinib significantly prolonged the survival of CML mice (Fig. 7B). The proportion of BCR-ABL-expressing leukemia cells, myeloid cells and GMPs in BM was reduced in mice with imatinib and JSL-1 treatment and further reduced with combined treatment (Fig. 7C-F). Flow cytometry results revealed that JSL-1 alone or in combination with imatinib but not imatinib alone greatly decreased the proportion of LSK cells, LT-HSCs and ST-HSCs in BM (Fig. 7G-J). JSL-1 treatment alone or in combination with imatinib greatly inhibited the splenomegaly of CML mice (Fig. S6A). Splenic mononuclear cells in CML mice treated with JSL-1 alone or in combination with imatinib but not imatinib alone showed markedly reduced the proportion of BCR-ABL-expressing leukemia cells (Fig. S6B), myeloid cells (Fig. S6C), GMPs (Fig. S6D-E), LSK cells (Fig. S6F-G), LT-HSCs (Fig. S6H) and ST-HSCs (Fig. S6I) in spleen. Thus, pharmacological inhibition of γ-catenin significantly reduced the fraction of LSCs and sensitized CML LSCs to imatinib in vivo.
Pharmacological inhibition of γ-catenin reduces growth of CML LSCs in mice. (A) A schematic strategy to generate the retroviral BCR-ABL-driven CML mouse model and drug treatment. (B) JSL-1 treatment alone or in combination with imatinib significantly prolonged survival of CML mice. Kaplan-Meier survival curves were shown, * P=0.0178, *** P<0.0001, log-rank test. (C) GFP+ cells in BM. (D) GFP+ myeloid cells (Gr-1+ Mac-1+) cells in BM. (E) Representative flow cytometry plots GMP in BM of mice treated with imatinib and JSL-1. (F) GMP cells in BM. (G-J) Analysis of LSCs in the BM cells of CML mice received treatment with imatinib ± JSL-1. Control (n=13), imatinib (n=10), JSL-1 (n=5), and combination (n=5). (G) Representative flow cytometry plots of LSK cells, LT-HSCs and ST-HSCs in BM of mice treated with imatinib and JSL-1. (H-J) Results for the GFP+ population in BM: LSK cells (H), LT-HSCs (I), and ST-HSCs (J). * P<0.05, ** P<0.01, *** P<0.0001. (K) A proposed model of HDACi JSL-1 action against self-renewal of CML LSCs. Quiescent CML LSCs are insensitive to imatinib. γ-Catenin is required for survival and self-renewal of LSCs. Loss of γ-catenin in combination with imatinib eliminates LSCs in CML.
Discussion
LSCs remain an obstacle to cure of CML. There is a desperate need to understand the self-renewal regulatory mechanism or develop small molecule agents capable of killing LSCs. In the present study, we discovered that γ-catenin is required for survival and self-renewal, which implies its important function as a novel therapeutic intervention target for LSCs. We validated a universal phenomenon of HDACis decreasing γ-catenin level and eliminating CML LSCs. Our results improve the current understanding of the molecular basis of HDACis eliminating LSCs.
Our in vivo and in vitro studies showed that JSL-1 potently eradicated LSCs. In addition, JSL-1 sensitized LSC cells to imatinib. Overexpressing γ-catenin in CML cells leads to a decreased sensitivity to JSL-1 treatment. These results agree with previous reports that inhibition of HDACs by Tenovin-6 and LAQ824 combined with imatinib enhanced the elimination of CML LSCs [12, 13]. So far, little is known about the mechanism of HDACis preferentially killing LSCs. Our results highlight that disturbing the FoxM1-γ-catenin-c-myc axis by HDAC inhibition in LSCs may shed light on the mechanism of HDACi selectively eliminating LSCs (Fig. 7K). However, JSL-1 may also exert its antitumor activity through γ-catenin-independent mechanisms (e.g., inactivating HDACs).
γ-Catenin is overexpressed in various types of cancer. However, its functions in tumor initiation and progression have been controversial. Earlier immunohistochemistry investigations with limited patient cases showed reduced γ-catenin expression in prostate, lung and renal carcinomas [37-39]. The authors postulated that γ-catenin might possess tumor suppressor activity because of the reduced expression in these cancer cells, and forced expression inhibited cell growth.
However, compelling recent evidence demonstrates that γ-catenin is oncogenic and distinct from β-catenin. First, γ-catenin shares overlapping regulatory elements with β-catenin. γ-Catenin is governed by APC and activates TCF/LEF transcription activity [40]. Transfection of wild-type γ-catenin in immortalized epithelial RK3E cells led to malignant transformation with stronger activation of oncogene c-myc expression [21]. Second, in colon cancer, γ-catenin is upregulated and its expression is associated with higher recurrence and impaired survival [41]. Third, γ-catenin was shown to increase the expression of Bcl-2, a pivotal anti-apoptosis regulatory protein positively associated with poor prognosis and chemotherapy resistance. Finally, γ-catenin promotes genomic instability as well as tumor mobility and migration [29].
For leukemia, evidence to date has clarified that γ-catenin acts as an oncogenic protein. Oncogenic fusion proteins PML-retinoic acid receptor (PML-RARα) and acute myeloid leukemia (AML1-ETO) directly activate the γ-catenin promoter to contribute to leukemogenesis in AML [35]. In CML cell lines, downregulation of γ-catenin sensitized CML cells to imatinib-induced apoptosis [42]. However, whether γ-catenin helps regulate the stemness of LSCs remains unknown. Our in vitro and in vivo evidence support that γ-catenin is required for LSCs to maintain their survival and self-renewal. To our knowledge, this is the first demonstration of γ-catenin engagement in CSCs. Consistently, Aceto et al. reported that knockdown of γ-catenin in breast cancer decreased the number of circulating tumor cell (CTC) clusters, which have traits similar to those of CSCs of highly metastatic potential [43]. Evidence from us and others highlights that γ-catenin may function as an oncogenic versus tumor suppressor gene in the context of LSCs.
Although our study initially started with JSL-1, subsequent data revealed that γ-catenin is a universal target of HDACis, including SAHA, which have been approved by the US Food and Drug Administration to treat skin lymphoma. Regarding the underlying mechanism that HDACi inhibits γ-catenin, our results showed that pre-incubation with ubiquitin-proteasome inhibitor MG132 did not reverse the JSL-1-decreased γ-catenin level in K562 cells. One therefore could postulate that JSL-1 mediated decrease in γ-catenin in an ubiquitin-proteasome-independent manner. We therefore turned our attention to transcription regulation. The results did confirm that JSL-1 affected the transcription of γ-catenin. γ-Catenin gene transcription was hindered in LSCs and bulk CML cells with HDACi treatment. Elevated acetylation in histone proteins may change transcription patterns: transcription of tumor suppressor genes such as p21 and p27 are usually released, whereas γ-catenin may be among those genes whose transcription is repressed. We further identified that FoxM1 bound to the promoter region of γ-catenin gene to activate transcription. HDACis globally suppress the recruitment of FoxM1 to the promoter of the γ-catenin gene. In addition, HDACi treatment led to a concomitant decrease in FoxM1 and γ-catenin levels. Therefore, reduced FoxM1 expression may repress γ-catenin gene transcription with HDACi treatment.
Because of the established role of β-catenin in controling self-renewal capacity of CML LSCs [16], we asked whether the effect of inhibiting γ-catenin was actually through changes in β-catenin levels. However, our data supported that γ-catenin regulated CML stem cells in a β-catenin-independent manner. The critical role of γ-catenin in the JSL-1-mediated growth suppression of CML cells was further indicated by the findings that JSL-1 bound γ-catenin rather than β-catenin, JSL-1 treatment preferentially led to a remarkable decrease and subcellular distribution in γ-catenin rather than β-catenin at least in the earlier phase. However, the underlying mechanism is not clear at the present time, and remains to be explored in future.
The oncogene c-myc plays an important role in initiating cancer and maintenance of stemness of CSCs of distinct tissue origins [44-46]. C-myc is also part of the cocktail of 4 transcriptional factors (Oct4, Sox2, Klf4 and c-myc) that triggers fibroblast cells to reprogram into induced pluripotent stem cells [47]. C-myc expression is more dependent on γ-catenin than β-catenin and is pivotal during γ-catenin-induced malignant transformation [21]. Our results revealed that c-myc expression was considerably lowered in the γ-catenin-silenced LSCs. JSL-1-suppressed c-myc expression may also facilitate elimination of LSCs. In conclusion, γ-catenin is required for survival and self-renewal of CML LSCs. γ-Catenin is targeted by HDACi. These findings improve the understanding of the molecular regulation network of CML LSCs, and the mechanism by which HDACi selectively eliminates LSCs.
Supplementary Material
Supplementary tables and figures.
Acknowledgements
This study was supported by grants from the National Basic Research Program of China (973 Program grant no. 2009CB825506 to J. Pan), National Natural Science Funds (no. 81025021, no. U1301226, no. 81373434, and no. 91213304 to J. Pan; 21172220 and 21472191 to S. Jiang), the Research Foundation of Education Bureau of Guangdong Province, China (Grant cxzd1103 to J. Pan), the Research Foundation of Guangzhou Bureau of Science and Technology, and,the Natural Science Foundation of Guangdong province (2015A030312014 to J. Pan). The authors thank Dr. Sai-Ching J. Yeung (The University of Texas M. D. Anderson Cancer Center, Houston, TX, USA) for a critical reading of the manuscript.
Author contributions
YLJ, YWY, LC, XHZ, BJ, YYS, SJ and JXP designed, performed the study and analysed the data; JL, XD, and YHL provided CML patients samples; YLJ and JXP wrote the manuscript.
Competing Interests
Authors have no conflicts of interest.
References
1. Druker BJ. Translation of the Philadelphia chromosome into therapy for CML. Blood. 2008;112:4808-17
2. Sawyers CL. Shifting paradigms: the seeds of oncogene addiction. Nat Med. 2009;15:1158-61
3. Hochhaus A, O'Brien SG, Guilhot F, Druker BJ, Branford S, Foroni L. et al. Six-year follow-up of patients receiving imatinib for the first-line treatment of chronic myeloid leukemia. Leukemia. 2009;23:1054-61
4. Ma L, Shan Y, Bai R, Xue L, Eide CA, Ou J. et al. A therapeutically targetable mechanism of BCR-ABL-independent imatinib resistance in chronic myeloid leukemia. Sci Transl Med. 2014;6:252ra121
5. Shah NP, Kasap C, Weier C, Balbas M, Nicoll JM, Bleickardt E. et al. Transient potent BCR-ABL inhibition is sufficient to commit chronic myeloid leukemia cells irreversibly to apoptosis. Cancer Cell. 2008;14:485-93
6. Zabriskie MS, Eide CA, Tantravahi SK, Vellore NA, Estrada J, Nicolini FE. et al. BCR-ABL1 compound mutations combining key kinase domain positions confer clinical resistance to ponatinib in Ph chromosome-positive leukemia. Cancer Cell. 2014;26:428-42
7. Chan WW, Wise SC, Kaufman MD, Ahn YM, Ensinger CL, Haack T. et al. Conformational control inhibition of the BCR-ABL1 tyrosine kinase, including the gatekeeper T315I mutant, by the switch-control inhibitor DCC-2036. Cancer Cell. 2011;19:556-68
8. Chen Y, Li S. Molecular signatures of chronic myeloid leukemia stem cells. Biomark Res. 2013;1:21
9. Donato NJ, Wu JY, Stapley J, Gallick G, Lin H, Arlinghaus R. et al. BCR-ABL independence and LYN kinase overexpression in chronic myelogenous leukemia cells selected for resistance to STI571. Blood. 2003;101:690-8
10. Neviani P, Santhanam R, Oaks JJ, Eiring AM, Notari M, Blaser BW. et al. FTY720, a new alternative for treating blast crisis chronic myelogenous leukemia and Philadelphia chromosome-positive acute lymphocytic leukemia. J Clin Invest. 2007;117:2408-21
11. Gambacorti-Passerini C, le Coutre P, Zucchetti M, D'Incalci M. Binding of imatinib by alpha(1)-acid glycoprotein. Blood. 2002;100:367-8 author reply 8-9
12. Li L, Wang L, Wang Z, Ho Y, McDonald T, Holyoake TL. et al. Activation of p53 by SIRT1 inhibition enhances elimination of CML leukemia stem cells in combination with imatinib. Cancer Cell. 2012;21:266-81
13. Zhang B, Strauss AC, Chu S, Li M, Ho Y, Shiang KD. et al. Effective targeting of quiescent chronic myelogenous leukemia stem cells by histone deacetylase inhibitors in combination with imatinib mesylate. Cancer Cell. 2010;17:427-42
14. Zhang H, Peng C, Hu Y, Li H, Sheng Z, Chen Y. et al. The Blk pathway functions as a tumor suppressor in chronic myeloid leukemia stem cells. Nat Genet. 2012;44:861-71
15. Corbin AS, Agarwal A, Loriaux M, Cortes J, Deininger MW, Druker BJ. Human chronic myeloid leukemia stem cells are insensitive to imatinib despite inhibition of BCR-ABL activity. J Clin Invest. 2011;121:396-409
16. Zhao C, Blum J, Chen A, Kwon HY, Jung SH, Cook JM. et al. Loss of beta-catenin impairs the renewal of normal and CML stem cells in vivo. Cancer Cell. 2007;12:528-41
17. Valent P. Targeting of leukemia-initiating cells to develop curative drug therapies: straightforward but nontrivial concept. Curr Cancer Drug Targets. 2011;11:56-71
18. Bhatia R, Holtz M, Niu N, Gray R, Snyder DS, Sawyers CL. et al. Persistence of malignant hematopoietic progenitors in chronic myelogenous leukemia patients in complete cytogenetic remission following imatinib mesylate treatment. Blood. 2003;101:4701-7
19. Chu S, McDonald T, Lin A, Chakraborty S, Huang Q, Snyder DS. et al. Persistence of leukemia stem cells in chronic myelogenous leukemia patients in prolonged remission with imatinib treatment. Blood. 2011;118:5565-72
20. Mahon FX, Rea D, Guilhot J, Guilhot F, Huguet F, Nicolini F. et al. Discontinuation of imatinib in patients with chronic myeloid leukaemia who have maintained complete molecular remission for at least 2 years: the prospective, multicentre Stop Imatinib (STIM) trial. Lancet Oncol. 2010;11:1029-35
21. Kolligs FT, Kolligs B, Hajra KM, Hu G, Tani M, Cho KR. et al. gamma-catenin is regulated by the APC tumor suppressor and its oncogenic activity is distinct from that of beta-catenin. Genes Dev. 2000;14:1319-31
22. Crews LA, Jamieson CH. Selective elimination of leukemia stem cells: hitting a moving target. Cancer Lett. 2013;338:15-22
23. Konopleva M, Milella M, Ruvolo P, Watts JC, Ricciardi MR, Korchin B. et al. MEK inhibition enhances ABT-737-induced leukemia cell apoptosis via prevention of ERK-activated MCL-1 induction and modulation of MCL-1/BIM complex. Leukemia. 2012;26:778-87
24. Zhang H, Li H, Ho N, Li D, Li S. Scd1 plays a tumor-suppressive role in survival of leukemia stem cells and the development of chronic myeloid leukemia. Mol Cell Biol. 2012;32:1776-87
25. West AC, Johnstone RW. New and emerging HDAC inhibitors for cancer treatment. J Clin Invest. 2014;124:30-9
26. Hojfeldt JW, Agger K, Helin K. Histone lysine demethylases as targets for anticancer therapy. Nat Rev Drug Discov. 2013;12:917-30
27. Yao Y, Tu Z, Liao C, Wang Z, Li S, Yao H. et al. Discovery of Novel Class I Histone Deacetylase Inhibitors with Promising in Vitro and in Vivo Antitumor Activities. J Med Chem. 2015;58:7672-80
28. Jin Y, Lu Z, Ding K, Li J, Du X, Chen C. et al. Antineoplastic mechanisms of niclosamide in acute myelogenous leukemia stem cells: inactivation of the NF-kappaB pathway and generation of reactive oxygen species. Cancer Res. 2010;70:2516-27
29. Pan H, Gao F, Papageorgis P, Abdolmaleky HM, Faller DV, Thiagalingam S. Aberrant activation of gamma-catenin promotes genomic instability and oncogenic effects during tumor progression. Cancer Biol Ther. 2007;6:1638-43
30. Shi X, Jin Y, Cheng C, Zhang H, Zou W, Zheng Q. et al. Triptolide inhibits Bcr-Abl transcription and induces apoptosis in STI571-resistant chronic myelogenous leukemia cells harboring T315I mutation. Clin Cancer Res. 2009;15:1686-97
31. Hong J, Hu K, Yuan Y, Sang Y, Bu Q, Chen G. et al. CHK1 targets spleen tyrosine kinase (L) for proteolysis in hepatocellular carcinoma. J Clin Invest. 2012;122:2165-75
32. Hao SX, Ren R. Expression of interferon consensus sequence binding protein (ICSBP) is downregulated in Bcr-Abl-induced murine chronic myelogenous leukemia-like disease, and forced coexpression of ICSBP inhibits Bcr-Abl-induced myeloproliferative disorder. Mol Cell Biol. 2000;20:1149-61
33. Morgan RG, Pearn L, Liddiard K, Pumford SL, Burnett AK, Tonks A. et al. gamma-Catenin is overexpressed in acute myeloid leukemia and promotes the stabilization and nuclear localization of beta-catenin. Leukemia. 2013;27:336-43
34. Muller-Tidow C, Steffen B, Cauvet T, Tickenbrock L, Ji P, Diederichs S. et al. Translocation products in acute myeloid leukemia activate the Wnt signaling pathway in hematopoietic cells. Mol Cell Biol. 2004;24:2890-904
35. Zheng X, Beissert T, Kukoc-Zivojnov N, Puccetti E, Altschmied J, Strolz C. et al. Gamma-catenin contributes to leukemogenesis induced by AML-associated translocation products by increasing the self-renewal of very primitive progenitor cells. Blood. 2004;103:3535-43
36. Peng C, Li S. CML mouse model in translational research. Methods Mol Biol. 2010;602:253-66
37. Breault JE, Shiina H, Igawa M, Ribeiro-Filho LA, Deguchi M, Enokida H. et al. Methylation of the gamma-catenin gene is associated with poor prognosis of renal cell carcinoma. Clin Cancer Res. 2005;11:557-64
38. Shiina H, Breault JE, Basset WW, Enokida H, Urakami S, Li LC. et al. Functional Loss of the gamma-catenin gene through epigenetic and genetic pathways in human prostate cancer. Cancer Res. 2005;65:2130-8
39. Winn RA, Bremnes RM, Bemis L, Franklin WA, Miller YE, Cool C. et al. gamma-Catenin expression is reduced or absent in a subset of human lung cancers and re-expression inhibits transformed cell growth. Oncogene. 2002;21:7497-506
40. Caca K, Kolligs FT, Ji X, Hayes M, Qian J, Yahanda A. et al. Beta- and gamma-catenin mutations, but not E-cadherin inactivation, underlie T-cell factor/lymphoid enhancer factor transcriptional deregulation in gastric and pancreatic cancer. Cell Growth Differ. 1999;10:369-76
41. Nagel JM, Kriegl L, Horst D, Engel J, Gautam S, Mantzoros CS. et al. gamma-Catenin is an independent prognostic marker in early stage colorectal cancer. Int J Colorectal Dis. 2010;25:1301-9
42. Niu CC, Zhao C, Yang ZD, Zhang XL, Wu WR, Pan J. et al. Downregulation of gamma-catenin inhibits CML cell growth and potentiates the response of CML cells to imatinib through beta-catenin inhibition. Int J Mol Med. 2013;31:453-8
43. Aceto N, Bardia A, Miyamoto DT, Donaldson MC, Wittner BS, Spencer JA. et al. Circulating tumor cell clusters are oligoclonal precursors of breast cancer metastasis. Cell. 2014;158:1110-22
44. Akita H, Marquardt JU, Durkin ME, Kitade M, Seo D, Conner EA. et al. MYC activates stem-like cell potential in hepatocarcinoma by a p53-dependent mechanism. Cancer Res. 2014;74:5903-13
45. Civenni G, Malek A, Albino D, Garcia-Escudero R, Napoli S, Di Marco S. et al. RNAi-mediated silencing of Myc transcription inhibits stem-like cell maintenance and tumorigenicity in prostate cancer. Cancer Res. 2013;73:6816-27
46. Pei Y, Moore CE, Wang J, Tewari AK, Eroshkin A, Cho YJ. et al. An animal model of MYC-driven medulloblastoma. Cancer Cell. 2012;21:155-67
47. Di Stefano B, Sardina JL, van Oevelen C, Collombet S, Kallin EM, Vicent GP. et al. C/EBPalpha poises B cells for rapid reprogramming into induced pluripotent stem cells. Nature. 2014;506:235-9
Author contact
![]() Corresponding authors: Jingxuan Pan, MD, Ph.D, Jinan University Institute of Tumor Pharmacology, Jinan University College of Pharmacy; State Key Laboratory of Ophthalmology, Zhongshan Ophthalmic Center, Sun Yat-sen University, 54 South Xianlie Road, Guangzhou 510060, People's Republic of China, Phone: +86-20-37628262, Email: panjx2sysu.edu.cn Or Sheng Jiang, Ph.D, Guangzhou Institute of Biomedicine and Health, Chinese Academy of Sciences, Guangzhou, China. Email: jiang_shengac.cn.
Corresponding authors: Jingxuan Pan, MD, Ph.D, Jinan University Institute of Tumor Pharmacology, Jinan University College of Pharmacy; State Key Laboratory of Ophthalmology, Zhongshan Ophthalmic Center, Sun Yat-sen University, 54 South Xianlie Road, Guangzhou 510060, People's Republic of China, Phone: +86-20-37628262, Email: panjx2sysu.edu.cn Or Sheng Jiang, Ph.D, Guangzhou Institute of Biomedicine and Health, Chinese Academy of Sciences, Guangzhou, China. Email: jiang_shengac.cn.