Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and methods
Results and Discussion
Conclusion
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2017; 7(2):466-481. doi:10.7150/thno.17411 This issue Cite
Research Paper
Hyaluronic Acid Conjugated Magnetic Prussian Blue@Quantum Dot Nanoparticles for Cancer Theranostics
Yongbo Yang1, Lijia Jing1, Xiaoda Li1, Li Lin1, Xiuli Yue2 ![]() , Zhifei Dai3
, Zhifei Dai3 ![]()
1. School of Life Science and Technology, Harbin Institute of Technology, Harbin 150080, China;
2. School of Municipal and Environmental Engineering, Harbin Institute of Technology, Harbin 150080, China;
3. Department of Biomedical Engineering, College of Engineering, Peking University, Beijing 100871, China.
Received 2016-8-31; Accepted 2016-11-2; Published 2017-1-6
Abstract

A multifunctional nanotheranostic agent was developed by conjugating both hyaluronic acid and bovine serum albumin coated CuInS2-ZnS quantum dots onto the surface of magnetic Prussian blue nanoparticles. The obtained nanoagent could serve as an efficient contrast agent to simultaneously enhance near infrared (NIR) fluorescence and magnetic resonance (MR) imaging greatly. The coexistence of magnetic core and CD44 ligand hyaluronic acid was found to largely improve the specific uptake of the nanoagent by CD44 overexpressed HeLa cells upon applying an external magnetic field. Both NIR fluorescence and MR imaging in vivo proved high accumulation of the nanoagent at tumor site due to its excellent CD44 receptor/magnetic dual targeting capability. After intravenous injection of the nanoagent and treatment of external magnetic field, the tumor in nude mice was efficiently ablated upon NIR laser irradiation and the tumor growth inhibition was more than 89.95%. Such nanotheranostic agent is of crucial importance for accurately identifying the size and location of the tumor before therapy, monitoring the photothermal treatment procedure in real-time during therapy, assessing the effectiveness after therapy.
Keywords: Photothermal therapy, Bimodal Imaging, Prussian blue nanoparticles, Fe3O4 nanoparticles, Quantum Dots, Tumor targeting.
Introduction
Photothermal therapy (PTT) has received extensive interests as a promising non-invasive methodology to targetedly “burn” cancer cells with the aid of photoabsorbers in combination with near infrared (NIR) laser irradiation[1-4]. Until now, numerous of nanomaterials, such as Au-[5-9], copper sulfide-[10-15], and carbon-[16-18] nanostructures have been explored as potential photoabsorbers to ablate cancer cells due to their strong NIR absorption and excellent photothermal conversion property. In addition, a variety of theranostic agents have been developed by integrating the functions of medical imaging with PTT to identify the location and size of tumors before the therapy, and monitor the therapeutic procedure in real-time[19-24].
Prussian blue (PB) has been approved for treatment of radioactive exposure in clinic[25]. Owning to the excellent absorption in the NIR wavelength region, a number of PB based theranostic agents have been explored for imaging guided PTT of cancer[26-31]. Magnetic iron oxide nanoparticles (Fe3O4 NPs) have emerged as a versatile agent for magnetic resonance (MR) imaging and magnetic targeting of tumors due to their efficacy and safety[32-36]. In our previous report, magnetic Prussian blue (Fe3O4@PB) NPs were prepared by in situ growing PB shells on the surface of Fe3O4 NPs for MR imaging guided PTT of tumor[37]. Nevertheless, MR imaging has relatively long scanning time and low sensitivity in spite of high spatial resolution[38-40]. In recent years, NIR fluorescence cancer imaging is a growing field for both preclinical and clinical application to cancer patients because of high sensitivity and specificity, operational simplicity, real-time display, safety and cost effectiveness. As a result, the combination of NIR fluorescence and MR imaging shows great promise in enhancement of precisely diagnosis[41-44]. Therefore, we have pressing need for the development of nanoparticles for targeting cancer PTT combining fluorescence and magnetic resonance imaging.
Due to high NIR photoluminescence, deeper tissue penetration, and good biocompatibility, the cadmium-free CuInS2-ZnS (ZCIS) quantum dots (QDs) have been fabricated for in vivo imaging applications[45-47]. In this paper, hyaluronic acid (HA) conjugated magnetic Prussian blue@QD NPs were fabricated for targeted PTT under the guidance of NIR fluorescence/MR bimodal imaging. Fe3O4@PB (FP) NPs were prepared via in situ growth of PB shell on Fe3O4 core [37], followed by coating with polyethyleneimine (PEI). Then, both bovine serum albumin (BSA) coated ZCIS QDs (BQDs) and HA were conjugated to Fe3O4@PB@PEI (FPP) NPs via the coupling reaction between the amino of PEI and the carboxyl of both BQDs and HA to obtain Fe3O4@PB@PEI@BQDs-HA (FPPBH) NPs (Figure 1). In FPPBH NPs, each component has different function. Hyaluronic acid, a major constituent of extracellular matrix, has good biocompatibility[48-49], and capability to specifically bind CD44 receptor overexpressed in various cancer cells through receptor-ligand interaction[50-52], rendering the HA modified NPs attractable for cancer targeted imaging and therapy. The Fe3O4 core has the dual functions of magnetic targeting and MR imaging, while its external PB shell acts as an NIR photoabsorber for PTT. ZCIS QDs serves as an NIR fluorescent imaging agent. We characterized the physicochemical properties of the nanoagent including morphology, size distribution, photothermal capability. The in vitro experiments demonstrated the specificity of the obtained FPPBH NPs to tumor cells. In vivo tumor targeting of the FPPBH NPs was visualized in a mouse model by using fluorescence and MR imaging. Quantitative analysis was performed to evaluate the efficacy of MRI contrast enhancement. The photothermal cytotoxicity of the FPPBH NPs was evaluated using both cancer cells and tumor-bearing nude mice.
Schematic illustration of the FPPBH NPs: (A) Fabrication procedure; (B) NIR fluorescence/MR bimodal imaging guided cancer PTT through intravenous injection.
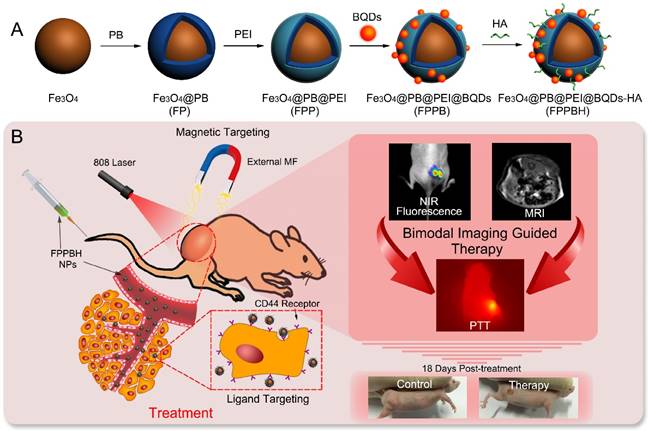
Materials and methods
Materials: Copper(I) iodide (99.995%) was purchased from J&K. Indium(III) acetate (99.99%) was purchased from Alfa Aesar. 1-dodecanethiol (98%), stearic acid (90%), zinc acetate (99%), octadecylamine (97%) and octadecene (90%) were purchased from Aladdin. FeSO4, FeCl3·6H2O and K4[Fe(CN)6] were purchased from Sinopharm Chemical Reagent Co., Ltd. HA of MW 5 805 Da (HA6k) and MW 31 200 Da (HA31k) were purchased from Zhenjiang Dong Yuan Biotech Company Limited. BSA was purchased from Roche. Polyethyleneimine, 1-Ethyl-3-[3-dimethylaminopropyl] carbodiimide hydrochloride (EDC), N-hydroxysuccinimide (NHS), 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) and calcein acetoxymethyl ester (Calcein AM) were purchased from Sigma. All other chemicals and reagents were of analytical grade.
Synthesis of ZCIS QDs: ZCIS QDs were prepared using the modified method according to our previous report[53]. Briefly, 8 mmol zinc acetate was completely dissolved in 6mL of decylamine and 14.0 mL of octadecene, followed by stirring at 160 °C under nitrogen flow for 10 min. The obtained Zn precursor solution was stored at 50 °C for use. Then, 0.1 mmol copper iodide, 0.1 mmol indium acetate, 1 mL of 1-dodecanethiol, 8 mL of octadecene and 0.3 mmol stearic acid were added into a 50 mL three-necked flask under vigorous magnetic stirring under the protection of nitrogen. When the solution was clear, the temperature was raised to 120 °C. After 30 min, the reaction mixture was heated to 230 °C and allowed for 20 min. Then, 1.25 mL Zn precursor was injected into the reaction mixture in 5 batches with a time interval of 15 min. Another 15 min later, the reaction mixture was naturally cooled to room temperature, and equal volume of chloroform was added thereafter. The obtained ZCIS QDs were precipitated by adding methanol and purified by repeated centrifugation and decantation. Zinc concentration was determined by Inductively Coupled Plasma Optical Emission Spectrometry (ICP-OES) and set as the concentration of ZCIS QDs.
Modification of ZCIS QDs with BSA: 1.2 μmol ZCIS QDs in chloroform were added into 50 mL of BSA aqueous solution (200 mg/mL), followed by probe sonication for 5 min. The mixture was stirred vigorously for 2 hours to remove remaining chloroform. Then, the obtained BQDs NPs were washed with deionized water using a Millipore ultrafiltration centrifuge tube (100 kDa cutoff size) for 30 min centrifugation at 5 000 rpm.
Preparation of Fe3O4 NPs and Fe3O4@PB NPs: Fe3O4 NPs and Fe3O4@PB NPs were prepared according to the reported method[37]. FeSO4 and FeCl3 (1:2 molar ratio) were dissolved in 20 mL of deionized water, followed by addition dropwise into 50 mL of NaOH aqueous solution (2.0 M) under vigorous stirring at 80 °C. 30 min later, the reaction mixture was cooled to room temperature. The obtained Fe3O4 NPs were washed and collected with an external magnetic field (MF).
50 mL of Fe3O4 NPs aqueous dispersion (1 mg/mL) were added dropwise into 100 mL of K4[Fe(CN)6] aqueous solution (2.0 mM, pH = 3.0) under mechanical stirring. After 30 min, 100 mL of FeCl3 aqueous solution (2.0 mM, pH = 3.0) was added dropwise into above solution, and stirred for 30 min. Then, the obtained Fe3O4@PB NPs were separated and washed 5 times by deionized water with an external magnetic field.
Preparation of Fe3O4@PB@PEI NPs: 100 mL of Fe3O4@PB NPs suspension was added dropwise into 50 mL of PEI aqueous solution (10 mg/mL, pH = 5.0), allowed for ultrasonication for 60 min. Fe3O4@PB@PEI NPs were washed 5 times and collected with an external magnetic field.
Preparation of Fe3O4@PB@PEI@BQDs NPs: 2 mL of deionized water containing 0.1 mmol EDC and 0.1 mmol NHS was added dropwise into 2 mL of BQDs aqueous solution (10 mg/mL) under mechanical stirring for 2 hours. Then, the carboxyl activated BQDs were added into 50 mL of FPP NPs aqueous solution at FPP/BQDs mass ratio of 20:1, 10:1, 5:1, 2:1 and 1:1. After stirring for 48h, FPPB NPs were washed 5 times to remove excess reactants with an external magnetic field.
Preparation of Fe3O4@PB@PEI@BQDs-HA (FPPBH) NPs: 0. 235 g HA6k (or 1.248 g HA31k) was dissolved in 3 mL of deionized water at 50 °C and cooled down to room temperature. Then, 2 mL of deionized water containing 0.1 mmol EDC and 0.1 mmol NHS were added dropwise into HA aqueous solution under stirring. After 2 h, the carboxyl activated HA was added into 50 mL of FPPB NPs (3 mg/mL) solution and kept stirring for 48 h. FPPBH NPs were washed 5 times to remove excess reactants with an external magnetic field.
Characterization techniques: UV-vis absorption and fluorescence spectra were collected by a UV-vis spectrophotometer (Varian Cary 4000, USA) and a fluorescence spectrophotometer (Varian Cary Eclipse, USA), respectively. The surface potentials of the NPs were determined with a PALS/90 Plus Particle Sizing and Potential Analyzer (Brookhaven, Holtsville, USA). The morphology of the NPs was characterized by a transmission electron microscope (TEM, Hitachi H-7650, Japan). Field-dependent magnetization measurement was used to study the room temperature magnetization curves of the NPs with a vibrating sample magnetometer (VSM, Lakeshore 7307, USA). The magnetic property of the NPs was characterized by placing the deionized water dispersed NPs nearby a magnet. The iron concentration of the NPs was measured with an inductively coupled plasma-optical emission spectroscope (ICP-OES, Perkin-Elmer model 3300 XL, USA). The elemental compositions of NPs were examined by an X-ray photoelectron spectroscopy (XPS, Thermo Fisher Scientific ESCALAB 250Xi, USA).
Cellular uptake efficiency study: The human cervical carcinoma cells (HeLa) and U87MG cells were seeded on cover slips at a density of 5×104 cells per well for 24 h at the environment of 37 °C and 5 vol.% CO2. Afterwards, the medium was discarded, and 1 mL fresh medium containing phosphate buffer solution (PBS) (control), FPPB NPs, FPPBH6k NPs and FPPBH31k NPs at the same iron concentration of 10 mg/L were added into each well and incubated at 37 °C and 5 vol.% CO2 for 4 h. To evaluate the role of HA6k or HA31k in cellular uptake of FPPBH6k NPs or FPPBH31k NPs, the HeLa cells and U87MG cells were pretreated with free HA6k or HA31k (0.1μM) for 2 h, followed by adding FPPBH6k NPs or FPPBH31k NPs and incubation for another 4 h. To further investigate the effect of external magnetic field on cellular uptake, a magnet was placed under the cell-growing plate bottom containing FPPBH6k NPs or FPPBH31k NPs for 15 min, followed by 4h incubation without external magnetic field. Then, the cells were rinsed with fresh PBS for 3 times, and the nuclei were strained with 4',6-diamidino-2-phenylindole (DAPI) for microscopic observation on a confocal laster scanning microscopy (CLSM, Carl Zeiss 510, Germany) at excition of 430 nm.
HeLa cells and U87MG cells were seeded in 24-well plates at a density of 1×105 cells per well. After 24 h incubation at 37 °C with 5 vol.% CO2, the cell medium was replaced by fresh medium contain FPPB NPs, FPPBH6k NPs or FPPBH31k NPs at the same iron concentration of 10 mg/L. After various treatments, the cells were rinsed with PBS for 3 times, centrifuged at 500 × g for 10 min after trypsinization, re-suspended in PBS, and measured by flow cytometer (FCM, BD FACSCalibur, USA).
Xenograft tumor models: All animal experiments were approved by institutional animal use committee and carried out ethically and humanely. BALB/c Nude mice were obtained from Beijing Vital River Laboratories. Tumors were induced by subcutaneous injection of 5×106 HeLa cells in 100µL PBS buffer. The tumor volume was monitored post-injection and calculated as the volume = (tumor length) × (tumor width)2/2. When the tumor volume reached to approximately 100 mm3, the tumor-bearing nude mice were used for NIR fluorescence imaging, MR imaging and PTT.
In vivo and ex vivo NIR fluorescence imaging: In vivo and ex vivo NIR fluorescence imaging of FPPB NPs and FPPBH31k NPs in tumor-bearing BALB/c nude mice was performed with NightOWL LB 983 in vivo Imaging System (Berthold Technologies GmbH & Co. KG, Germany) set at excitation 500 nm and emission 700 nm. Tumor-bearing nude mice were intravenously (i.v.) injected with 100 μL of FPPB NPs (1.6×103 mg/L) or FPPBH31k NPs (4.0×103 mg/L) at the same QDs concentration (n=6 for each group), respectively. For the magnetically targeted group, a permanent magnet (size = 20 mm × 20 mm × 5 mm, residual magnetism Br = 14.1-14.5 KGs, coercive force = 828-907 KOe, maximum magnetic energy product = 382-398 GOe) was pasted to tumor by a piece of scotch tape for 15 min before imaging. In vivo NIR fluorescence images were captured at various time points (0-48 h) post-injection. Then, nude mice were sacrificed, and major organs and tumor were taken out for determining the distribution of NPs ex vivo. All the NIR fluorescence intensity data were calculated by the region of interest (ROI) function of IndiGO Imaging Software (Berthold Technologies GmbH & Co. KG, Germany).
In vitro and in vivo MR imaging: In vitro and in vivo MR imaging were performed with a 3.0 T whole-body magnetic resonance imaging scanner (Philips Medical Systems). For in vitro MR imaging, the relaxivity value (r2, mM -1·s -1) was calculated from the slopes of relaxation time (T2) versus the concentration of iron, and T2-weighted images of FPPBH31k NPs at different iron concentration in PBS were obtained using the following parameters: repetition time (TR) = 3000 ms, echo time (TE) = 80 ms, slice thickness of 1.3 mm.
For in vitro MR imaging of cells, HeLa cells and U87MG cells were seeded in 24-well plates at a density of 1×105 cells per well. After 24 h incubation at 37 °C with 5 vol.% CO2, the cell medium was replaced by fresh medium containing FPPB NPs, FPPBH6k NPs or FPPBH31k NPs at the same iron concentration of 10 mg/L. After various treatments, the cells were rinsed with PBS for 3 times, centrifuged at 500 × g for 10 min after trypsinization, re-suspended in PBS, and measured.
For in vivo MR imaging, tumor-bearing BALB/c nude mice were intraveneously (i.v.) injected with 100 μL of FPPB NPs (1.6×103 mg/L) or FPPBH31k NPs (4.0×103 mg/L) at the iron concentration of 900 mg/L (n=6 for each group), respectively. For the magnetically targeted group, a permanent magnet was pasted to tumor by a piece of scotch tape for 15 min before imaging. T2-weighted images were obtained before and at the time points of 8 h and 24 h after injection.
Temperature elevation induced by NIR laser irradiation: FPPBH31k NPs with different iron concentration dispersed in RPMI-1640 medium were poured in quartz cuvettes (total volume of 3.0 mL), and irradiated by a continuous-wave diode NIR laser (T808D2W, Xi'an Minghui Optoelectronic Technology Co., China) with a center wavelength of 808 ± 10 nm at output power of 2 W for 10 min. The temperature of all samples was measured by a digital thermometer with a thermocouple probe every 10 s. RPMI-1640 medium was irradiated by NIR laser as control.
The photothermal stability of FPPBH31k NPs was also investigated. FPPBH31k NPs suspensions with iron concentration of 80 mg/L were irradiated with NIR laser for 10 min, followed by turn off the laser to cooling the sample to room temperature. This cycle repeated for 4 times, and the UV-vis absorption and fluorescence spectra of the irradiated FPPBH31k NPs suspensions were measured to evaluate the stability.
In vitro targeted photothermal ablation effect of FPPBH31k NPs: HeLa cells were seeded in 6-well plates at a density of 2×105 cells per well and incubated at 37 °C with 5 vol.% CO2 for 24h. Then, the cell medium was replaced by 1 mL FPPBH31k NPs or FPPB NPs suspension with the iron concentration of 40 mg/L. After 2h incubation, the culture medium with FPPBH31k NPs or FPPB NPs was removed and 1 mL fresh RPMI-1640 medium was added into each well. Afterwards, the cells were exposed to NIR laser (808 nm, 2 W/cm2, diameter of laser spot: 2mm) for 10 min. For the magnetically targeted photothermal group, a permanent magnet was placed under the cell-growing plate bottom for 15 min before the irradiation. After incubated for another 2 h, the cells were rinsed with PBS and stained with calcein acetoxymethyl ester (calcein AM) to verify the targeted photothermal effect of FPPBH31k NPs on cancer cells.
MTT assay was used to evaluate the targeted photothermal effect of FPPBH31k NPs in vitro. HeLa cells were seeded in 96-well plates at a density of 1×104 cells per well and incubated at 37 °C with 5 vol.% CO2 for 24h. The cell medium was replaced by different iron concentrations of FPPBH31k NPs or FPPB NPs suspension (200 μL per well) and incubated at 37 °C for 4h. Then, the cells were rinsed with PBS for 3 times, 200 μL of fresh RPMI-1640 medium was added into each well. Afterwards, the cells were exposed to NIR laser (808 nm, 2 W/cm2, diameter of laser spot: 2mm) for 10 min. For the magnetically targeted photothermal group, a magnet was placed under the plate bottom for 15 min before the irradiation. After the irradiation, the cells were incubated with fresh RPMI-1640 medium at 37 °C for 24 h. Finally, the cell viabilities were measured by standard MTT method, the optical density of untreated cells was set as 100%.
In vivo PTT efficacy evaluation: Tumor-bearing nude mice were randomly divided in to seven groups (n = 8). Three groups tumor-bearing nude mice were i.v. injected 100 μL of saline, FPPB NPs (1.6×103 mg/L) or FPPBH31k NPs (4.0×103 mg/L) at the same iron concentration, respectively. After 24h, the tumor of each mouse was irradiated by a continuous-wave diode NIR laser at 2 W/cm2 for 10 min. For the magnetically targeted group, 100 μL of FPPBH31k NPs (4 mg/mL) were i.v. injected into tumor-bearing nude mice, and a permanent magnet was pasted to tumor by a piece of scotch tape for 15 min before irradiation. The body surface temperature of mice were recorded by an infrared (IR) thermal camera during the irradiation every 2 min. The other three groups, tumor-bearing nude mice were i.v. injected saline, FPPB NPs or FPPBH31k NPs without NIR laser irradiation were used as the controls. The tumor volume of nude mice were measured every 3 days post irradiation to evaluate PTT efficacy.
Biocompatibility evaluation: Human umbilical vein endothelial (HUVEC) cells were seeded in 96-well plate at a density of 5×104 cells per well for 24 h. The cells were rinsed three times with PBS, followed by incubation with 200 μL different iron concentration of FPPBH31k NPs in RPMI-1640 medium at 37 °C and 5 vol.% CO2 for 28 h, 48h and 72h. Cell viabilities were measured by standard MTT method.
The in vivo biocompatibility of FPPBH31k NPs in mice was also investigated by histological analysis. Healthy BALB/c mice were i.v. injected double imaging/therapy dose of FPPBH31k NPs (100 μL, 8×103 mg/L) (n=6), major organs (heart, liver, spleen, lung and kidneys) were excised at day 1 and day 30 postinjection. Then, the organs were fixed with 10% neutral buffered formalin, made into paraffin embedded sections, stained with hematoxylin and eosin (H&E), and calculated under a digital microscope (Olympus IX, Japan). Healthy mice without FPPBH31k NPs injection were set as control. The body weight of mice were also monitored every 2 day. The tumor growth inhibition (TGI) was monitored post-injection and calculated as the TGI (%) = (1 - Te / Tc) × 100%, where Te = mean tumor volume of experimental groups at day 18, and Tc = mean tumor volume of untreat control groups at day 18.
Statistical analysis. Analysis of variance (ANOVA) and t-tests were performed to analyze the statistical significance of the experiment data. The level of significance in statistical analyses was defined as p< 0.05.
Results and Discussion
Preparation and characterization of FPPBH NPs
Highly photoluminescent ZCIS QDs were synthesized in 1-octadecene by using acetate salts of Cu, In, and Zn as cation precursor and dodecanethiol as the sulfur source according to the reported method[53]. Afterwards, BSA, which owns one free sulfhydryl and eight pairs of disulfide bounds inside the macromolecule, was used as surface capping agent to make hydrophobic QDs water soluble[54]. The potential toxicity of heavy metal elements has always limited the clinical application of QDs. It has been reported that BSA could be used to decrease the potential toxicity of QDs by alleviating the release of metal elements[55]. In this study, cadmium-free ZCIS QDs were modified with BSA to increase the biocompatibility of QDs. The obtained BQDs had a hydrodynamic diameter of 14.2 nm (particles distribution index, PDI = 0.149±0.09) (Figure S1A). BQDs displayed bright red fluorescence under a UV lamp (inset of Figure S1A). The TEM characterization clearly indicated that BQDs NPs were uniform and well dispersed (Figure S1B). As seen in Figure S1C and S1D, BQDs NPs had a strong adsorption in the ultraviolet region like ZCIS QDs, while the fluorescence intensity decreased slightly, accompanied by a visible red shift of the two fluorescence emission peak from 650 and 700 nm to 670 and 720 nm, respectively. It revealed that more defects on the ZCIS QDs surfaces were formed when native ligands were replaced with BSA, leading to a slight fluorescence intensity decrease[56].
The core-shell Fe3O4@PB NPs were prepared by in situ growing PB shells on superparamagnetic Fe3O4 NPs under acidic condition and purified by magnetic separation. The hydrodynamic diameter of Fe3O4 NPs and FP NPs was evaluated as 40.1±5.1 nm (PDI = 0.175±0.008) and 54.2±2.9 nm (PDI = 0.217±0.016), respectively (Figure S2). The zeta potential of FP NPs was -9.73±2.26 mV comparing to -7.95±3.11 mV of Fe3O4 NPs (Figure S3). Moreover, the UV-vis spectra of FP NPs exhibited a strong typical adsorption peak of PB NPs at NIR region (~700 nm) (Figure 2A), indicating that PB was successfully deposited on the Fe3O4 NPs surface. The thickness of PB shell on Fe3O4 NPs was evaluated to be 5 nm by transmission electronic microscopy (Figure 2D).
(A) UV-vis adsorption spectra of Fe3O4 NPs, FP NPs, FPP NPs, FPPBH6k NPs and FPPBH31k NPs; (B) Fluorescence spectra of BQDs NPs, FPPB NPs, FPPBH6k NPs and FPPBH31k NPs; TEM micrographs of (C) Fe3O4 NPs, (D) FP NPs, (E) FPP NPs, (F) FPPB NPs, (G) FPPBH6k NPs and (H) FPPBH31k NPs.
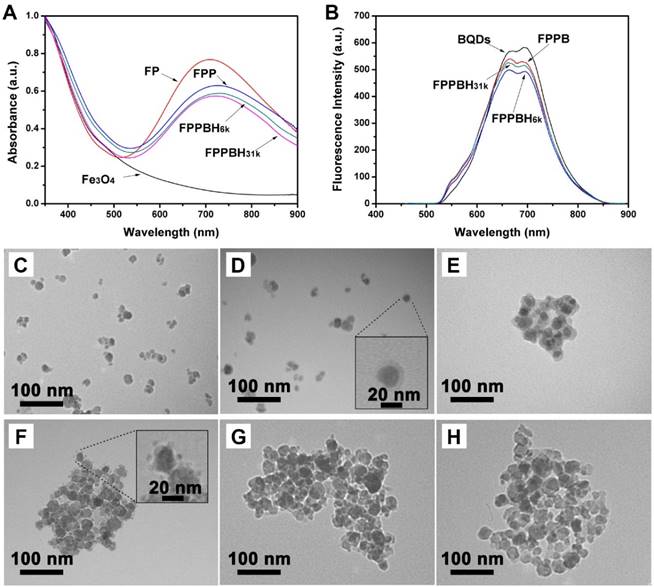
The Fe3O4@PB@PEI NPs were prepared via electrostatic self-assembly of PEI onto FP NPs. The potential of the NPs was altered into +24.75±3.53 mV after coating with PEI. The hydrodynamic diameter of FPP NPs was 102.9±16.1 nm (PDI = 0.282±0.025). The obviously increased hydrodynamic diameter could be due to aggregation of the NPs. Then, FPP NPs were modified with BQDs NPs through condensation reaction at different mass ratios of FPP to BQDs of 20:1, 10:1, 5:1, 2:1 and 1:1, respectively. The zeta potential of the obtained FPPB NPs at various mass ratios was monitored. As shown in Figure S4, the zeta potential of FPPB NPs decreased with reducing mass ratio of FPP/BQDs. The zeta potential was reduced to -6.07±4.58 mV at mass ratio of 2:1, indicating the free amino groups of FPP NPs were occupied by BQDs NPs. The FPPB NPs obtained at the mass ratio of 10:1 was selected to ensure sufficient amino groups for further conjugation of hyaluronic acid. At the FPP/BQDs mass ratio of 10:1, the hydrodynamic diameter of FPPB NPs was 121.0±7.4 nm (PDI = 0.296±0.014), and the zeta potential was +14.36±3.44 mV. Furthermore, as shown in Figure 2F, it could be observed that BQDs were conjugated onto the surface of FPP NPs.
The successful formation of FPPB NPs was also confirmed by observing typical fluorescence emission peaks of BQDs NPs in FPPB NPs aqueous solution (Figure 2B). The fluorescence of QDs can be quenched by fluorescence resonance energy transfer (FRET) when the emission spectrum of QDs overlaps with the absorption spectrum of another material at the distances less than 10 nm[57]. It has been reported to protect the fluorescence of QDs from being quenched by coating QDs with macromolecules or inorganic nonmetallic materials[58]. Recently, BSA was used for in situ ligand exchange of hydrophobic QDs under a ultrasound condition[54]. The surface modification of ZCIS QDs with BSA might reduce the fluorescence quenching through increasing the distance between ZCIS QDs and FPP NPs. Therefore, FPPB, FPPBH6k NPs and FPPBH31k NPs are highly fluorescent. Furthermore, BSA modification of ZCIS QDs could improve the biocompatability of ZCIS QDs.
The final FPPBH6k NPs and FPPBH31k NPs were prepared by modification of FPPB NPs with excess HA6k or HA31k. Compared with FPPB NPs, the zeta potential of FPPBH6k NPs and FPPBH31k NPs was changed to -4.35±2.35 mV and -6.17±2.71 mV, respectively (Figure S3). The hydrodynamic diameter of FPPBH6k NPs and FPPBH31k NPs was 139.9±12.3 nm (PDI = 0.371±0.019) and 148.3±13.4 nm (PDI = 0.356±0.009). These results indicated the successful preparation of FPPBH6k NPs and FPPBH31k NPs.
The magnetic property of FPPBH NPs was visually studied by placing FPPBH31k NPs aqueous dispersion near a magnet. As shown in Figure S5, FPPBH31k NPs could rapidly move to the magnet within 30 min. This result indicated that FPPBH31k NPs had a great potential for magnetic targeting. Moreover, the red fluorescent emitted from FPPBH31k NPs provided additional evidence that BQDs NPs can be firmly conjugated onto the surface of FPP NPs.
The XPS analysis has been completed. The results were shown in Figure S6 and Table S1. As expected for the in situ growing PB on Fe3O4 NPs, the N1s signal was detected at 398.88 eV. After PEI modification of FP NPs, the N1s signal was immensely strengthened. Due to the successful modification of BQDs on FPP NPs, new peaks at 1046.28 eV (Zn2p), 931.12 eV (Cu2p) and 443.50 eV (In3d) were detected. After modification with HA6k or HA31k, the XPS spectra showed an increase in the C1s signal and decrease in the N1s signal, which provide a further evidence of successful HA modification. Furthermore, the C1s signal of FPPBH31k NPs was stronger than that of FPPBH6k NPs due to the higher molecular weight of HA31k. The amount of BQDs on FPPB NPs has been quantified via XPS analysis. In Table S1, the atomic percentage of zinc and iron of FPPB were 0.58% and 2.73%, respectively. Hence, the molar ratio of zinc to iron in FPPB NPs was calculated to be 0.212. According to Table S1, the molar ratio of carbon to iron was calculated to be 26.63, 31.17 and 35.77 in FPPB NPs, FPPBH6k NPs and FPPBH31k NPs, respectively. The more amount of carbon on FPPBH31k NPs was found due to the higher molecular weight of HA31k than HA6k.
To evaluated the colloidal stability, FPPBH31k NPs were suspended in saline, plasma and RPMI-1640 medium. As shown in Figure S7, the FPPBH31k NPs could well dispersed in three kinds of physiologic medium and no aggregation or deposition was seen after stored at 4 °C for 7 days. The hydrodynamic diameter of the FPPBH31k NPs in saline, plasma and RPMI-1640 medium had no significant change after one week storage (p>0.05) (Table S2). The photo-stability study of FPPBH31k NPs has investigated. As shown in Figure S8, the fluorescence intensity of FPPBH31k NPs at 700 nm in saline, plasma and RPMI-1640 medium had no apparent change after one week storage (p>0.05). All these results indicated that FPPBH31k NPs possess good colloidal and photo stability.
Cellular uptake efficiency study
Cellular uptake of FPPB NPs, FPPBH6k NPs and FPPBH31k NPs was visualized by CLSM on HeLa cells (CD44+) and U87MG cells (CD44-)[59-60]. The iron concentration of these NPs was 10 mg/L. As shown in Figure 3, in the control groups, only blue fluorescence of the nuclei stained by DAPI could be observed when cells were treated with PBS. For the HeLa cells incubated with FPPBH6k NPs and FPPBH31k NPs, strong red fluorescence signals were observed, indicating the uptake of FPPBH6k NPs and FPPBH31k NPs by HeLa cells. In contrast, much weaker red fluorescence signals were observed in the FPPB NPs treated HeLa cells, and the U87MG cells treated with FPPB NPs, or FPPBH6k NPs, or FPPBH31k NPs. These results demonstrated that the HA surface modification could improve the internalization of the NPs by HeLa cells. To further evaluate the role of HA in the cellular uptake of FPPBH NPs, the CD44 receptor was blocked by pretreating the HeLa cells with free HA6k or HA31k for 2h, followed by incubation with FPPBH6k NPs or FPPBH31k NPs for 4 h. No prominent red fluorescence signals can be observed, which is similar to the control groups. Meanwhile, U87MG cells were pretreated with free HA6k or HA31k in the same method as HeLa cells. However, it is similar to the cells without the pretreatment of free HA that weak red fluorescence signals were observed. The above results showed that both FPPBH6k NPs and FPPBH31k NPs were able to be taken up by HeLa cells through the CD44 mediated receptor-ligand interaction. To evaluate the influence of external magnetic field on the cellular uptake, a magnet was placed under the cell-growing plate bottom during incubating cells with FPPBH6k NPs or FPPBH31k NPs for 15 min. The increase of fluorescence signals suggests that the external MF could enhance the cellular uptake.
CLSM micrographs of HeLa cells and U87MG cells treated with PBS for 4 h (A, I); FPPB NPs for 4 h (B, J); FPPBH6k NPs for 4 h (C, K); FPPBH31k NPs for 4 h (D, L); HA6k for 2 h followed by FPPBH6k NPs for 4 h (E, M); HA31k for 2 h followed by FPPBH31k NPs for 4 h (F, N), FPPBH6k NPs with external MF for 15 min followed by without external MF for 4 h (G, O); FPPBH31k NPs with external MF for 15 min followed by without external MF for 4 h (H, P). All these NPs have the iron concentration of 10 mg/L. Scale bar is 40 μm.
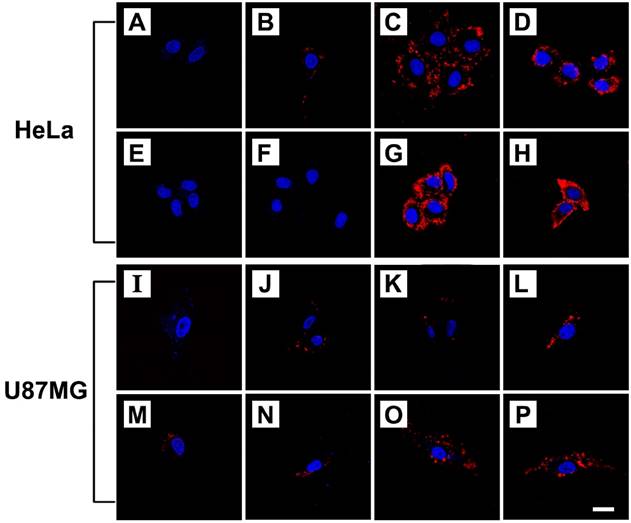
Flow cytometry analysis was used to quantitatively determine the cellular uptake of NPs. As shown in Figure S9, the fluorescence intensities of HeLa cells treated with either FPPBH6k NPs or FPPBH31k NPs were significantly higher than U87MG cells. At the two groups of HeLa cells pretreated with free HA, the fluorescence intensity was as low as the control group. In addition, no strong fluorescence intensities were detected in U87MG cells with various treatment. Moreover, the fluorescence intensity of HeLa cells treated with FPPBH31k NPs was 1.5 fold higher than that treated with FPPBH6k NPs, indicating that HA with higher MW had a better affinity to CD44 [52]. Therefore, FPPBH31k NPs was selected for further experiments.
On the other hand, the cellular uptakes of FPPBH6k NPs and FPPBH31k NPs in HeLa cells and U87MG cells were all significantly enhanced by applying an external MF. The flow cytometry analysis provided additional evidence that the enhancement of cellular uptake of FPPBH6k NPs and FPPBH31k NPs in HeLa cells was attributed to the conjugation of HA ligands which can specifically target to CD44 receptor overexpressed on HeLa cells, and the interaction between the cells surface and NPs was strengthened by the external MF.
CD44 is a principal cell surface receptor, which over expressed in most cancer cells and normal cells. However, the HA-binding domain of CD44 exists in low-affinity state on normal cells and high-affinity state on tumor cells[61-62]. In order to study tumor-specific targeting ability of FPPBH31k NPs, HUVEC cell line was selected as normal cell model. The cellular uptake of FPPBH31k NPs on HUVEC cells and HeLa cells was visualized by CLSM. The fluorescence intensities of HUVEC cells and HeLa cells were also measured by flow cytometer. As shown in Figure S10, HUVEC cells treated with FPPBH31k NPs, or FPPBH31k NPs with external MF display no red fluorescence signal, which is similar to the control group. The fluorescence intensities of HeLa cells treated with either FPPBH31k NPs or FPPBH31k NPs with external MF were significantly stronger than HUVEC cells (p<0.01). These results indicated that FPPBH31k NPs could specifically target to CD44 overexpressed tumor cells, instead of normal cells.
In vivo fluorescence imaging and biodistribution study
The tumor accumulation and biodistribution of FPPBH31k NPs were evaluated using NIR fluorescence imaging to investigating the in vivo dual-targeting efficacy. FPPB NPs and FPPBH31k NPs were intravenously (i.v.) injected into HeLa tumor-bearing BALB/c nude mice, respectively. A magnet was pasted on tumor site for 15 min before imaging. The variations of fluorescence intensity at tumor region were monitored within 48 h. As shown in Figure 4A and 4B, the fluorescence intensity gradually increased with increasing time, indicating the accumulation of NPs at tumor site. For the two groups without applying external MF, the fluorescence signal of the FPPBH31k NPs injected mice was stronger than that of FPPB NPs at 2h, 8h, 24h and 48h post-injection, respectively. The photon counts at region of interest (ROI) of FPPBH31k NPs injected group was about 1.3 times higher than that of FPPB NPs at 48 h post-injection. The enhanced accumulation of FPPBH31k NPs at tumor tissue could be attributed to the receptor-ligand interaction between HA and HeLa cells. By applying an external MF, the fluorescence intensity of FPPBH31k NPs injected group MF was 1.1 times higher than that of without MF. This result demonstrated that FPPBH31k NPs could actively accumulate at tumor site through ligand targeting, and the targeting effect was enhanced by the combination of an external MF.
In vivo Fluorescence images (A) and quantification analysis (B) of HeLa bearing nude mice at 0 h, 2h, 8h, 24h and 48h after tail vein injection of FPPB NPs, FPPBH31k NPs without and with MF; Ex vivo Fluorescence images (C) and quantification analysis (D) of main organs from HeLa bearing nude mice after 48h tail vein injection of FPPB NPs, FPPBH31k NPs without and with MF. Data shown as mean standard deviation (SD), n=6. (NS, non-significance, *p < 0.05, **p < 0.01)
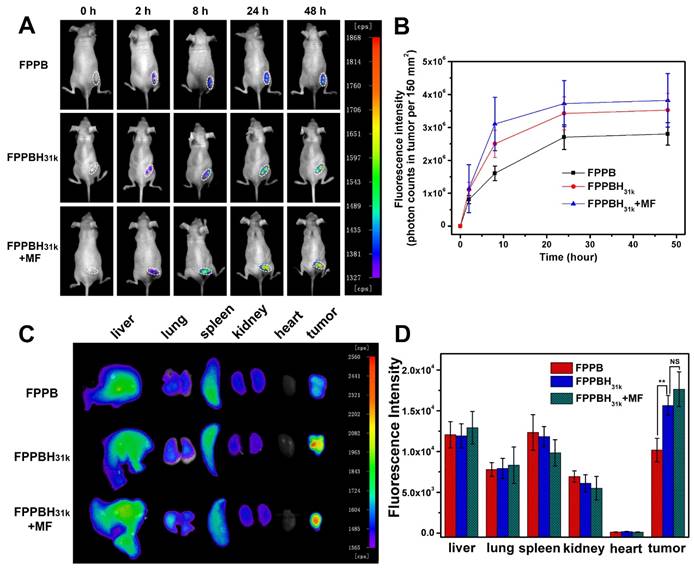
To further evaluate the biodistribution of the injected NPs, major organs (liver, lung, spleen, kidney, heart and tumor) were collected at 48 h post-injection. As shown in Figure 4C and 4D, the strong fluorescence signal in liver and spleen suggested that the foreign NPs were taken up by reticuloendothelial system (RES)[63]. The fluorescence intensity of excised tumor injected by FPPBH31k NPs was 1.5 times higher than that of FPPB NPs, and increased 10% upon applying an external MF treatment (Figure 4D).
The biodistribution of NPs was further confirmed via ICP-OES by analyzing the iron concentration in major organs and tumor after ex vivo fluorescence images study. As shown in Figure S11, after 48 h post-injection of NPs without and with MF, majority of iron was accumulated in liver, spleen and tumor by comparing with PBS injected HeLa tumor-bearing BALB/c nude mice. It was also found that the iron mass in each gram of tumor tissue was 20.8±2.2 μg (FPPB NPs), 27.2±1.9 μg (FPPBH31k NPs without MF) and 33.1±2.0 μg (FPPBH31k NPs with MF). Therefore, we were able to calculate the uptaken percentages of NPs by tumor tissue to be 23.3±2.5% (FPPB NPs), 30.5±2.1% (FPPBH31k NPs without MF) and 37.1±2.3% (FPPBH31k NPs with MF), respectively.
In vitro and in vivo MR imaging study
To evaluate the magnetic properties of the NPs, the room temperature magnetization curves of Fe3O4 NPs, FPPBH6k NPs and FPPBH31k NPs were obtained by a field-dependent magnetization measurement at 300 K (Figure 5A). No hysteresis was observed, and the saturation magnetization value (Ms) of Fe3O4 NPs, FPPBH6k NPs and FPPBH31k NPs was 61.3 emu/g, 53.4 emu/g and 50.1 emu/g, respectively, indicating that all these NPs possessed strong superparamagnetism.
(A) Magnetization curves of Fe3O4, FPPBH6k NPs and FPPBH31k NPs; (B) T2-weighted MR images of FPPBH31k NPs with various iron concentrations; (C) Linear relationship between T2 relaxation rate (1/T2) and iron concentrations for FPPBH31k NPs; in vivo T2-weighted MR images (D) and MR signal intensity (E) of HeLa bearing nude mice at 0 h, 8 h and 24 h after tail vein injection of FPPB NPs, FPPBH31k NPs without and with MF. Data shown as mean standard deviation (SD), n=6. (*p < 0.05, **p < 0.01)
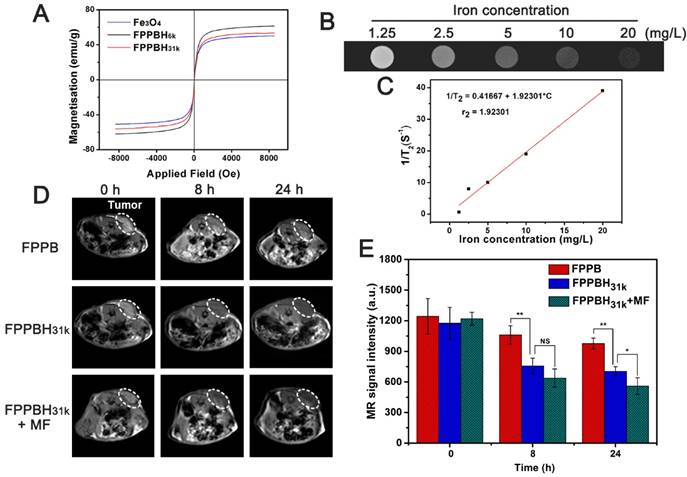
The in vitro MR imaging capability of FPPBH31k NPs was evaluated. The MR image became darker as the iron concentration of FPPBH31k NPs increased (Figure 5B). The T2 relaxation rate (1/T2) increased with the iron concentration linearly in the range of iron concentration from 1.25 to 20 mg/L (Figure 5C). These results indicated that FPPBH31k NPs exhibited the potential as a contrast agent for enhancing T2-weighted MR imaging.
The in vitro MR imaging of cells was evaluated too. As shown in Figure S12, the results were similar to that of cellular uptake efficiency study. In HeLa cells treated with FPPBH6k NPs and FPPBH31k NPs groups, the T2-weighted MR signal intensities were significantly lower than U87MG cells. Moreover, the MR signal intensity of HeLa cells treated with FPPBH31k NPs was lower than that treated with FPPBH6k NPs, and the signal intensity of these two groups has become much lower after treated with external MF. These results indicated that FPPBH31k NPs was better suited for targeted T2-weighted MR imaging of HeLa cells than FPPBH6k NPs.
The in vivo MR imaging of FPPB NPs and FPPBH31k NPs was then performed with the 3.0-T MR imaging system before and after intravenous injection at the time point of 8 h and 24 h (Figure 5D and 5E). The T2-weighted MR signals in the tumor region decreased gradually as the time increased. Moreover, the MR signal intensity of the FPPBH31k NPs injected mice decreased more obviously than that of FPPB NPs at 8 h and 24 h, and further reduced upon applying an external MF. Therefore, both the fluorescence and MR imaging results proved that HA modification and the use of an external MF could enhance significantly the accumulation of FPPBH31k NPs in tumor.
Photothermal effect of FPPBH31k NPs
The photothermal effect of FPPBH31k NPs was studied by monitoring the temperature of 3 mL RPMI-1640 medium containing FPPBH31k NPs of various iron concentrations (20, 40 and 80 mg/L) under NIR light irradiation (808 nm, 2W) (Figure 6A). After 10 min irradiation, the temperature of solution at the concentration of 20, 40 and 80 mg/L was elevated from 23.0 to 36.1, 40.0 and 45.9 °C, respectively. In contrast, the temperature of the RPMI-1640 medium without FPPBH31k NPs increased by only 3.1 °C. It suggested that FPPBH31k NPs could act as an efficient photothermal agent for tumor ablation.
(A) The temperature variations of RPMI-1640 medium suspensions of FPPBH31k NPs at different iron concentrations upon 808 nm laser irradiation for 10 min. (B) The temperature variations of FPPBH31k NPs suspensions (iron concentrations of 80 mg/L) during four irradiation cycles of LASER ON/OFF; UV-vis adsorption spectra (C) and Fluorescence spectra (D) of FPPBH31k NPs suspensions before and after four irradiation cycles of LASER ON/OFF.
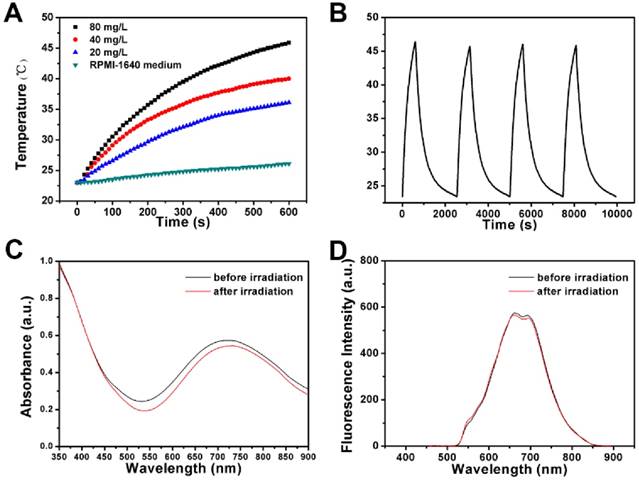
To investigate the NIR photostability, FPPBH31k NPs suspended in RPMI-1640 medium (iron concentration of 80 mg/L) was irradiated with 808 nm NIR laser for 10 min (LASER ON), followed by turning off the NIR laser and naturally cooling to the initial value at room temperature (LASER OFF). This cycle was repeated four times. As shown in Figure 6B, the temperature elevation of 22.9 °C was achieved after the first LASER ON, and no apparent decrease in the temperature elevation was observed at the second, the third and the fourth irradiation circle (p>0.05). Furthermore, only minor decrease of the absorbance at the NIR region was observed (Figure 6C), and no obvious change was observed in the fluorescence intensity (Figure 6D) after four irradiation cycles. It indicated the good photostability of FPPBH31k NPs after long period of NIR laser irradiation.
In vitro targeted photothermal ablation effect of FPPBH31k NPs
To study the targeted photothermal ablation effect in vitro, HeLa cells incubated with FPPBH31k NPs or FPPB NPs in 6-well plates were irradiated with an 808 nm NIR laser for 10 min with or without the use of the external MF at the bottom of the cell growing plate. Then, calcein AM was used to stain living HeLa cells to visualize the cell viability (Figure 7A-H). No apparent change in cell viability and density was observed when in presence of either the NPs or laser irradiation alone (Figure 7B-D). However, a small amount of HeLa cell death was observed in the co-presence of FPPBH31k NPs and laser irradiation without MF (Figure 7E). It might be because the concentration of the FPPBH31k was too low to achieve sufficient temperature elevation to induce cell death. Nevertheless, a significant cell death was observed in the laser irradiation region in the presence of FPPBH31k NPs combining an external MF (Figure 7F). Compared with FPPBH31k NPs combining NIR laser irradiation, FPPB NPs combining NIR laser irradiation induced less cell death no matter whether an external MF was used or not (Figure 7G and 7H).
Photothermal therapeutic efficacies of FPPBH31k NPs and FPPB NPs: (A) no agent, no MF, no laser; (B) FPPBH31k, no MF, no laser; (C) FPPBH31k, MF, no laser; (D) no agent, no MF, laser; (E) FPPBH31k, no MF, laser; (F) FPPBH31k, MF, laser; (G) FPPB, no MF, laser, and (H) FPPB, MF, laser; Scale bar is 40 μm. (I) Viabilities of HeLa cells incubated with different iron concentrations of FPPBH31k NPs and FPPB NPs under different treatments. Data shown as mean SD, n=3. (NS, non-significance, *p < 0.05, **p < 0.01)
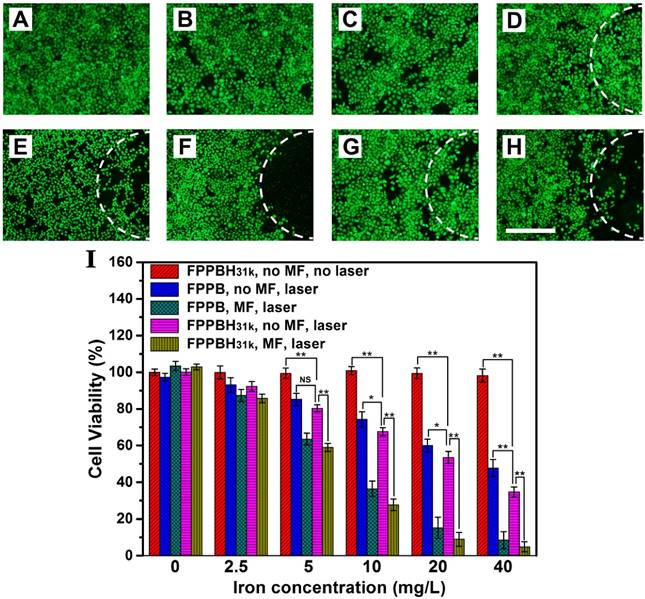
The photo-thermal ablation effect of the FPPBH31k NPs and FPPB NPs was further quantitatively evaluated by MTT assays. In Figure 7I, no obvious change in cell viability was seen at the iron concentration ranging from 0 to 40 mg/L without the use of NIR laser irradiation and external MF. However, FPPBH31k NPs combining external MF and laser showed significantly higher photothermal ablation effect than that no MF at various iron concentrations. Furthermore, the FPPB NPs treated HeLa cells showed slightly higher cell viability than FPPBH31k NPs upon NIR laser irradiation. All these results suggested that the the photothermal ablation effect of the NPs to HeLa cells could be greatly improved by the HA targeting and the use of an external MF, which can enhance cellular uptake of NPs.
In vivo PTT efficacy evaluation
The in vivo PTT was carried out on nude mice bearing HeLa cells with different treatments. When the volume of tumor reached approximately 100 mm3, the mice were randomly divided into seven groups: saline only, FPPB NPs only, FPPBH31kNPs only, saline + laser, FPPB NPs + laser, FPPBH31k NPs + laser and FPPBH31k NPs + laser + MF (n=6 for each group). According to the results of NIR fluorescence and MR imaging, NIR irradiation was carried out at 24 h after i.v. injection of saline and NPs duet to the high NPs accumulation. The body temperature of each mouse was recorded using an infrared thermal camera. As shown in Figure 8A, no obvious elevation of temperature on tumor region was observed in saline + laser group after 10 min irradiation. In contrast, the average temperature on tumor region of mice in FPPB NPs + laser, FPPBH31k NPs + laser and FPPBH31k NPs + laser + MF group was elevated to 40 °C, 46 °C and 49 °C, respectively. The higher temperature increment in FPPBH31k NPs + laser + MF group was ascribed to the HA targeting ability of FPPBH31k NPs to HeLa cells as well as magnetic targeting ability.
(A) Infrared thermal images of HeLa bearing nude mice under the 808 nm laser irradiation taken at different time intervals. (B) Therapeutic effectiveness expressed as tumor volume in each group after treatment in HeLa bearing nude mice. Data shown as mean SD, n=8. (C) Photographs of representative mice of the four different groups taken before and after treatment for 9 and 18 days.
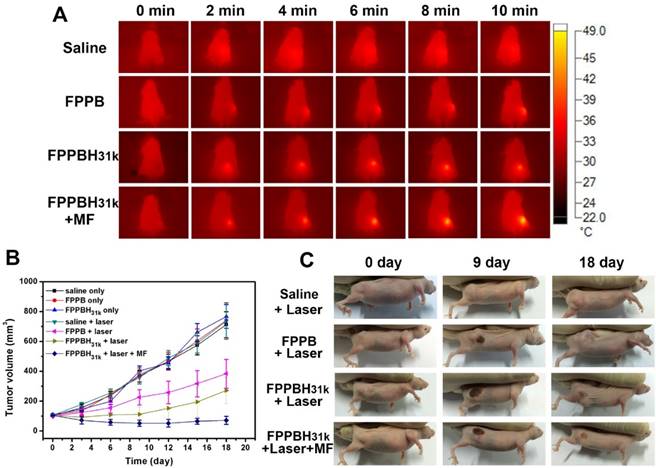
The in vivo PTT efficacy was evaluated by measuring the tumor volume each 3 days after treatment (Figure 8B and 8C). In mice treated with saline only, FPPB NPs only, FPPBH31k NPs only and saline + laser groups, the tumor volume increased from about 100 mm3 to approximately 710 mm3 over 18 days. For FPPB NPs + laser and FPPBH31k NPs + laser group, on the 18th day after treatment, the size of tumor was increased to 383.2 ± 96.5 mm3 and 271.6 ± 87.4 mm3, respectively. The tumor growth inhibition (TGI) was evaluated to be TGI: 46.18% and TGI: 61.85%. In marked contrast, the tumor volume was decreased to 71.6 ± 27.4 mm3 for FPPBH31k NPs + laser + MF group (TGI: 89.95%). Due to the CD44 receptor/magnetic dual targeting capability of FPPBH31k NPs, the tumor growth was effectively inhibited. Therefore, FPPBH31k NPs have a great potential to be used as a powerful agent for in vivo PTT of cancer.
Biocompatibility evaluation
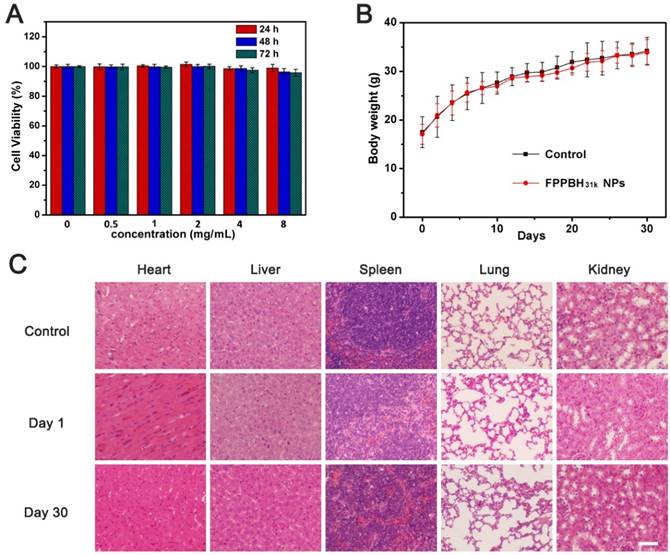
HUVEC cells were incubated with FPPBH31k NPs of different iron concentrations for 24, 48 and 72 h. Then, the viabilities of HUVEC cells were studied by MTT assays (Figure 9A). The cell viabilities of HUVEC cells were more than 95% at the iron concentration as high as 8×103 mg/mL after incubation for 72 h. It indicated that FPPBH31k NPs had good biocompatibility. We further evaluated the in vivo biocompatibility of FPPBH31k NPs by body weight change and histopathologic examination. No significant body weight loss was observed, and the body weight increase rate of FPPBH31k NPs injected group was similar to control (Figure 9B). Histological sections of vital organs excised at day 1 and day 30 demonstrated that neither apparent inflammation nor lesion in cellular structures was observed. These preliminary results indicated that FPPBH31k NPs could act as a secure theranostic agent for medical application.
(A) Viabilities of HUVEC cells incubated with different iron concentrations of FPPBH31k NPs for 24h, 48h and 72h. Data shown as mean SD, n=3. (B) Body weight change of mice after intravenous administration of FPPBH31k NPs; (C) Histological sections of heart, liver, spleen, lung and kidneys stained with hematoxylin and eosin (S&E) at day 1 and day 30 post-injection, untreated group was used as control. Scale bar is 40 μm. Data shown as mean SD, n=6.
Conclusion
A nanotheranostic agent was successfully developed by conjugation of HA and BSA modified ZCIS QDs onto the surface of PEI coated Fe3O4@PB NPs. The obtained FPPBH NPs showed good biocompatibility, great adsorption in the NIR region and strong NIR fluorescence. In vitro cellular uptake efficiency results indicated that the coexistence of magnetic core and HA could greatly improve the specific uptake of FPPBH NPs by CD44 overexpressed HeLa cells by applying an external magnetic field. Both NIR fluorescence and MR imaging in vivo proved high accumulation of FPPBH NPs at tumor site due to their excellent CD44 receptor/magnetic dual targeting capability. After intravenous injection of FPPBH NPs and treatment of external MF, the tumor in nude mice was efficiently ablated upon NIR light irradiation and the tumor growth inhibition was more than 89.95%. Thus, FPPBH NPs are a promising theranostic agent for targeted cancer photothermal therapy under the guidance of NIR fluorescence/MR bimodal imaging, providing an alternative methodology for noninvasive cancer diagnosis and therapy in the early future.
Supplementary Material
Figures S1-S12, Table S1-S2.
Acknowledgements
This work was financially supported by National Key Research and Development Program of China (No. 2016YFA0201400), State Key Program of National Natural Science of China (Grant No. 81230036), National Natural Science Foundation for Distinguished Young Scholars (No. 81225011), National Natural Science Foundation of China (No. 81371580) and the Foundation for Innovative Research Groups of the National Natural Science Foundation of China (No. 81421004).
Competing Interests
The authors have declared that no competing interest exists.
References
1. O'Neal DP, Hirsch LR, Halas NJ, Payne JD, West JL. Photo-thermal tumor ablation in mice using near infrared-absorbing nanoparticles. Cancer Lett. 2004;209:171-176
2. Chen JY, Wang DL, Xi JF, Au L, Siekkinen A, Warsen A. et al. Immuno gold nanocages with tailored optical properties for targeted photothermal destruction of cancer cells. Nano Lett. 2007;7:1318-1322
3. Wang CG, Chen JJ, Talavage T, Irudayaraj J. Gold nanorod/Fe3O4 nanoparticle "nano-pearl-necklaces" for simultaneous targeting, dual-mode imaging, and photothermal ablation of cancer cells. Angew Chem Int Ed Engl. 2009;48:2759-2763
4. Melancon MP, Lu W, Yang Z, Zhang R, Cheng Z, Elliot AM. et al. In vitro and in vivo targeting of hollow gold nanoshells directed at epidermal growth factor receptor for photothermal ablation therapy. Mol Cancer Ther. 2008;7:1730-1739
5. Nam J, Won N, Jin H, Chung H, Kim S. pH-Induced aggregation of gold nanoparticles for photothermal cancer therapy. J Am Chem Soc. 2009;131:13639-13645
6. Ke HT, Wang JR, Dai ZF, Jin YS, Qu EZ, Xing ZW. et al. Gold-nanoshelled microcapsules: a theranostic agent for ultrasound contrast imaging and photothermal therapy. Angew Chem Int Ed Engl. 2011;50:3017-3021
7. Ma Y, Liang XL, Tong S, Bao G, Ren QS, Dai ZF. Gold Nanoshell Nanomicelles for Potential Magnetic Resonance Imaging, Light-Triggered Drug Release, and Photothermal Therapy. Adv Funct Mater. 2013;23:815-822
8. Chen HL, Liu ZM, Li SY, Su CK, Qiu XJ, Zhong HQ. et al. Fabrication of Graphene and AuNP Core Polyaniline Shell Nanocomposites as Multifunctional Theranostic Platforms for SERS Real-time Monitoring and Chemo-photothermal Therapy. Theranostics. 2016;6:1096-1104
9. Zhao F, Hu B. Cancer therapy may get a boost from gold nanorods. Science Bulletin. 2015;60:279-280
10. Li YB, Lu W, Huang Q, Huang M, Li C, Chen W. Copper sulfide nanoparticles for photothermal ablation of tumor cells. Nanomedicine (Lond). 2010;5:1161-1171
11. Zhou M, Zhang R, Huang M, Lu W, Song SL, Melancon MP. et al. A chelator-free multifunctional [64Cu]CuS nanoparticle platform for simultaneous micro-PET/CT imaging and photothermal ablation therapy. J Am Chem Soc. 2010;132:15351-15358
12. Zha ZB, Wang JR, Zhang SH, Qu EZ, Ke HT, Dai ZF. Targeted delivery of CuS nanoparticles through ultrasound image-guided microbubble destruction for efficient photothermal therapy. Nanoscale. 2013;5:3216-3219
13. Zha ZB, Zhang SH, Deng ZJ, Li YY, Li CH, Dai ZF. Enzyme-responsive copper sulphide nanoparticles for combined photoacoustic imaging, tumor-selective chemotherapy and photothermal therapy. Chem Commun (Camb). 2013;49:3455-3457
14. Zhang SH, Zha ZB, Yue XL, Liang XL, Dai ZF. Gadolinium-chelate functionalized copper sulphide as a nanotheranostic agent for MR imaging and photothermal destruction of cancer cells. Chem Commun (Camb). 2013;49:6776-6778
15. Lin L, Li XD, Yang YB, Jing LJ, Yue XL, Chen XZ. et al. Chitosan Functionalized CuS Nanoparticles Boots Gene Transfection via Photothermal Effect. Curr Drug Deliv. 2016
16. Yang K, Zhang S, Zhang GX, Sun XM, Lee ST, Liu Z. Graphene in mice: ultrahigh in vivo tumor uptake and efficient photothermal therapy. Nano Lett. 2010;10:3318-3323
17. Thakare VS, Das M, Jain AK, Patil S, Jain S. Carbon nanotubes in cancer theragnosis. Nanomedicine (Lond). 2010;5:1277-1301
18. Jin YS, Wang JR, Ke HT, Wang SM, Dai ZF. Graphene oxide modified PLA microcapsules containing gold nanoparticles for ultrasonic/CT bimodal imaging guided photothermal tumor therapy. Biomaterials. 2013;34:4794-4802
19. Huang XH, El-Sayed IH, Qian W, El-Sayed MA. Cancer cell imaging and photothermal therapy in the near-infrared region by using gold nanorods. J Am Chem Soc. 2006;128:2115-2120
20. Seo WS, Lee JH, Sun XM, Suzuki Y, Mann D, Liu Z. et al. FeCo/graphitic-shell nanocrystals as advanced magnetic-resonance-imaging and near-infrared agents. Nat Mater. 2006;5:971-976
21. Yang K, Hu LL, Ma XX, Ye SQ, Cheng L, Shi XZ. et al. Multimodal imaging guided photothermal therapy using functionalized graphene nanosheets anchored with magnetic nanoparticles. Adv Mater. 2012;24:1868-1872
22. Chen M, Tang SH, Guo ZD, Wang XY, Mo SG, Huang XQ. et al. Core-shell Pd@Au nanoplates as theranostic agents for in-vivo photoacoustic imaging, CT imaging, and photothermal therapy. Adv Mater. 2014;26:8210-8216
23. Hu DH, Zhang JN, Gao GH, Sheng ZH, Cui HD, Cai LT. Indocyanine Green-Loaded Polydopamine-Reduced Graphene Oxide Nanocomposites with Amplifying Photoacoustic and Photothermal Effects for Cancer Theranostics. Theranostics. 2016;6:1043-1052
24. Meng H, Wei R. Use of smart designed nanoparticles to impact cancer surgery. Science Bulletin. 2015;60:142-143
25. Verzijl JM, Joore HC, van Dijk A, Wierckx FC, Savelkoul TJ, Glerum JH. In vitro cyanide release of four prussian blue salts used for the treatment of cesium contaminated persons. J Toxicol Clin Toxicol. 1993;31:553-562
26. Fu GL, Liu W, Feng SS, Yue XL. Prussian blue nanoparticles operate as a new generation of photothermal ablation agents for cancer therapy. Chem Commun (Camb). 2012;48:11567-11569
27. Jing LJ, Liang XL, Deng ZJ, Feng SS, Li XD, Huang MM. et al. Prussian blue coated gold nanoparticles for simultaneous photoacoustic/CT bimodal imaging and photothermal ablation of cancer. Biomaterials. 2014;35:5814-5821
28. Li XD, Liang XL, Ma F, Jing LJ, Lin L, Yang YB. et al. Chitosan stabilized Prussian blue nanoparticles for photothermally enhanced gene delivery. Colloids Surf B Biointerfaces. 2014;123:629-638
29. Cheng L, Gong H, Zhu WW, Liu JJ, Wang XY, Liu G. et al. PEGylated Prussian blue nanocubes as a theranostic agent for simultaneous cancer imaging and photothermal therapy. Biomaterials. 2014;35:9844-9852
30. Zhu WW, Liu K, Sun XQ, Wang X, Li YG, Cheng L. et al. Mn2+-Doped Prussian Blue Nanocubes for Bimodal Imaging and Photothermal Therapy with Enhanced Performance. ACS Applied Materials & Interfaces. 2015;7:11575-11582
31. Jing LJ, Shao SM, Wang Y, Yang YB, Yue XL, Dai ZF. Hyaluronic Acid Modified Hollow Prussian Blue Nanoparticles Loading 10-hydroxycamptothecin for Targeting Thermochemotherapy of Cancer. Theranostics. 2016;6:40-53
32. Petersein J, Saini S, Weissleder R. Liver. II: Iron oxide-based reticuloendothelial contrast agents for MR imaging. Clinical review. Magn Reson Imaging Clin N Am. 1996;4:53-60
33. Lee H, Lee E, Kim do K, Jang NK, Jeong YY, Jon S. Antibiofouling polymer-coated superparamagnetic iron oxide nanoparticles as potential magnetic resonance contrast agents for in vivo cancer imaging. J Am Chem Soc. 2006;128:7383-7389
34. Ke HT, Wang JR, Tong S, Jin YS, Wang SM, Qu EZ. et al. Gold nanoshelled liquid perfluorocarbon magnetic nanocapsules: a nanotheranostic platform for bimodal ultrasound/magnetic resonance imaging guided photothermal tumor ablation. Theranostics. 2013;4:12-23
35. Ma Y, Tong S, Bao G, Gao C, Dai ZF. Indocyanine green loaded SPIO nanoparticles with phospholipid-PEG coating for dual-modal imaging and photothermal therapy. Biomaterials. 2013;34:7706-7714
36. Cao Z, Yue XL, Li XD, Dai ZF. Stabilized magnetic cerasomes for drug delivery. Langmuir. 2013;29:14976-14983
37. Fu GL, Liu W, Li YY, Jin YS, Jiang LD, Liang XL. et al. Magnetic Prussian blue nanoparticles for targeted photothermal therapy under magnetic resonance imaging guidance. Bioconjug Chem. 2014;25:1655-1663
38. Cheng CY, Ou KL, Huang WT, Chen JK, Chang JY, Yang CH. Gadolinium-based CuInS2/ZnS nanoprobe for dual-modality magnetic resonance/optical imaging. ACS Appl Mater Interfaces. 2013;5:4389-4400
39. Sitbon G, Bouccara S, Tasso M, Francois A, Bezdetnaya L, Marchal F. et al. Multimodal Mn-doped I-III-VI quantum dots for near infrared fluorescence and magnetic resonance imaging: from synthesis to in vivo application. Nanoscale. 2014;6:9264-9272
40. Zheng XY, Sun LD, Zheng T, Dong H, Li Y, Wang YF. et al. PAA-capped GdF3 nanoplates as dual-mode MRI and CT contrast agents. Science Bulletin. 2015;60:1092-1100
41. Ding K, Jing LH, Liu CY, Hou Y, Gao MY. Magnetically engineered Cd-free quantum dots as dual-modality probes for fluorescence/magnetic resonance imaging of tumors. Biomaterials. 2014;35:1608-1617
42. Liu LW, Law WC, Yong KT, Roy I, Ding H, Erogbogbo F. et al. Multimodal imaging probes based on Gd-DOTA conjugated quantum dot nanomicelles. Analyst. 2011;136:1881-1886
43. Li JX. Nanotechnology-based platform for early diagnosis of cancer. Science Bulletin. 2015;60:488-490
44. Sreejith S, Huong TTM, Borah P, Zhao YL. Organic-inorganic nanohybrids for fluorescence, photoacoustic and Raman bioimaging. Science Bulletin. 2015;60:665-678
45. Yong K-T, Roy I, Hu R, Ding H, Cai HX, Zhu J. et al. Synthesis of ternary CuInS2/ZnS quantum dot bioconjugates and their applications for targeted cancer bioimaging. Integrative Biology. 2010;2:121-129
46. Deng DW, Chen YQ, Cao J, Tian JM, Qian ZY, Achilefu S. et al. High-Quality CuInS2/ZnS Quantum Dots for In vitro and In vivo Bioimaging. Chem Mater. 2012;24:3029-3037
47. Yu K, Ng P, Ouyang JY, Zaman MB, Abulrob A, Baral TN. et al. Low-temperature approach to highly emissive copper indium sulfide colloidal nanocrystals and their bioimaging applications. ACS Appl Mater Interfaces. 2013;5:2870-2880
48. Lapcik L Jr, Lapcik L, De Smedt S, Demeester J, Chabrecek P. Hyaluronan: Preparation, Structure, Properties, and Applications. Chem Rev. 1998;98:2663-2684
49. Kim H, Kim Y, Kim IH, Kim K, Choi Y. ROS-responsive activatable photosensitizing agent for imaging and photodynamic therapy of activated macrophages. Theranostics. 2013;4:1-11
50. Choi KY, Chung H, Min KH, Yoon HY, Kim K, Park JH. et al. Self-assembled hyaluronic acid nanoparticles for active tumor targeting. Biomaterials. 2010;31:106-114
51. Kamat M, El-Boubbou K, Zhu DC, Lansdell T, Lu XW, Li W. et al. Hyaluronic acid immobilized magnetic nanoparticles for active targeting and imaging of macrophages. Bioconjug Chem. 2010;21:2128-2135
52. Arpicco S, Lerda C, Dalla Pozza E, Costanzo C, Tsapis N, Stella B. et al. Hyaluronic acid-coated liposomes for active targeting of gemcitabine. Eur J Pharm Biopharm. 2013;85:373-380
53. Yang YB, Wang JR, Li XD, Lin L, Yue XL. A near infrared fluorescent/ultrasonic bimodal contrast agent for imaging guided pDNA delivery via ultrasound targeted microbubble destruction. RSC Advances. 2015;5:8404-8414
54. Zhang BB, Wang XH, Liu FJ, Cheng YS, Shi DL. Effective reduction of nonspecific binding by surface engineering of quantum dots with bovine serum albumin for cell-targeted imaging. Langmuir. 2012;28:16605-16613
55. Lai L, Jin JC, Xu ZQ, Ge YS, Jiang FL, Liu Y. Spectroscopic and Microscopic Studies on the Mechanism of Mitochondrial Toxicity Induced by CdTe QDs Modified with Different Ligands. J Membr Biol. 2015;248:727-740
56. Hartmann L, Kumar A, Welker M, Fiore A, Julien-Rabant C, Gromova M. et al. Quenching Dynamics in CdSe Nanoparticles: Surface-Induced Defects upon Dilution. ACS Nano. 2012;6:9033-9041
57. Guo JJ, Zhang Y, Luo YL, Shen F, Sun CY. Efficient fluorescence resonance energy transfer between oppositely charged CdTe quantum dots and gold nanoparticles for turn-on fluorescence detection of glyphosate. Talanta. 2014;125:385-392
58. Chen ML, He YJ, Chen XW, Wang JH. Quantum-dot-conjugated graphene as a probe for simultaneous cancer-targeted fluorescent imaging, tracking, and monitoring drug delivery. Bioconjug Chem. 2013;24:387-397
59. Alkhatib G, Broder CC, Berger EA. Cell type-specific fusion cofactors determine human immunodeficiency virus type 1 tropism for T-cell lines versus primary macrophages. J Virol. 1996;70:5487-5494
60. Upadhyay KK, Bhatt AN, Mishra AK, Dwarakanath BS, Jain S, Schatz C. et al. The intracellular drug delivery and anti tumor activity of doxorubicin loaded poly(gamma-benzyl L-glutamate)-b-hyaluronan polymersomes. Biomaterials. 2010;31:2882-2892
61. Ogino S, Nishida N, Umemoto R, Suzuki M, Takeda M, Terasawa H. et al. Two-state conformations in the hyaluronan-binding domain regulate CD44 adhesiveness under flow condition. Structure. 2010;18:649-656
62. Yang CX, He YQ, Zhang HZ, Liu YW, Wang WJ, Du Y. et al. Selective killing of breast cancer cells expressing activated CD44 using CD44 ligand-coated nanoparticles in vitro and in vivo. Oncotarget. 2015;6:15283-15296
63. Platt VM, Szoka FC Jr. Anticancer therapeutics: targeting macromolecules and nanocarriers to hyaluronan or CD44, a hyaluronan receptor. Mol Pharm. 2008;5:474-486
Author contact
![]() Corresponding authors: Xiuli Yue, Email: xiulidxcom, Zhifei Dai, Email: zhifei.daiedu.cn.
Corresponding authors: Xiuli Yue, Email: xiulidxcom, Zhifei Dai, Email: zhifei.daiedu.cn.