Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Results
Discussion
Materials and Methods
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2017; 7(19):4735-4752. doi:10.7150/thno.21662 This issue Cite
Research Paper
The cytotoxic effects of dimethyl sulfoxide in mouse preimplantation embryos: a mechanistic study
Min-Hee Kang1, Joydeep Das1, Sangiliyandi Gurunathan1, Hwan-Woo Park2, Hyuk Song1, Chankyu Park1, Jin-Hoi Kim1 ![]()
1. Department of Stem Cell and Regenerative Biotechnology, Humanized Pig Research Center (SRC), Konkuk University, Seoul, Korea.
2. Department of Cell Biology, College of Medicine, Konyang University, Daejeon, Korea
Received 2017-6-28; Accepted 2017-9-18; Published 2017-10-17
Abstract

Rationale: Dimethyl sulfoxide (DMSO) is commonly used as a solvent for water-insoluble substances, a vehicle for drug therapy, and a cryoprotectant for cultured cells. DMSO induced embryonic defects and its mechanism of action remains unclear. The rationale is based on the assumption that DMSO supplementation should induce long-term negative effects on both pre- and post-implantation embryo development.
Methods: DMSO induced oxidative stress, ER stress, autophagy, mitophagy, signaling responsible genes and proteins were determined by RT-qPCR, Western blotting, immunofluorescence, and confocal microscopy. DMSO induced mitochondrial dysfunction was measured by transmission electron microcopy and JC-1 assay. Apoptosis was estimated using TUNEL and comet assay. Post-implantation embryo developmental capability was estimated by implantation site and fetus numbers.
Results: Exposure to DMSO induced an early oxidative stress response within 0.5 to 2 h in 1-cell zygotes by disrupting the balance of pro- and anti-oxidants. Notably, DMSO-treated 2-cell embryos showed increased expression of unfolded protein response genes such as Hspa5, Hsp90b1, Ddit3, Atf4, and Xbp1. As a result, the development of many embryos is arrested at the 2-cell, 4-cell, or morula stages in a dose-dependent manner. Further, DMSO-induced endoplasmic reticulum stress increased mitochondrial Ca2+ levels, induced mitochondrial depolarization/dysfunction, and induced apoptotic cell death via the JNK/ATF2-dependent pathway. Consequently, treatment with DMSO increased the expression of autophagy initiation-, phagophore elongation-, and autophagosome formation-related genes, as well as localization of PINK1/Parkin, which are the main mediators of mitophagy, in mitochondria. Interestingly, DMSO causes cytotoxic effects in preimplantation embryos by inducing extensive mitophagy and autophagy. Especially, DMSO treatment decreased the inner cell mass and trophectoderm cell numbers as well as mRNA expression of B3gnt5 and Wnt3a in developed blastocysts, which decreased the implantation and developmental rates of full-term offspring after being transferred into pseudopregnant mice.
Conclusion: These results provide a significant contribution to finding effective protective agents to combat DMSO mediated reproductive toxicity for application in human embryos in the near future.
Keywords: Dimethyl Sulfoxide, Preimplantation embryos, Endoplasmic reticulum stress, Autophagy, Reactive oxygen species
Introduction
Dimethyl sulfoxide (DMSO) is a polar aprotic solvent that is easily miscible with both aqueous and organic media because of its amphipathic nature.[1] It is frequently used in a wide range of biological studies, and its function has been investigated in various contexts; however, its molecular mechanisms have not been clearly defined. DMSO is able to dissolve numerous weakly polar and nonpolar compounds, and is therefore considered as an efficient solvent of water-insoluble compounds in biological test media.[3] It is often employed as a vehicle for drug therapies.[4-6] In addition, it is frequently used as cryoprotectant for cultured cells owing to its membrane penetration and water displacement properties.[7] DMSO has been used in the medical field to treat various conditions, including rheumatologic diseases, pulmonary adenocarcinoma, chronic prostatitis, dermatologic diseases, musculoskeletal disorders, schizophrenia, traumatic brain edema, and gastrointestinal diseases.[8-16]
So far, there are limited studies demonstrating the toxicity of DMSO in embryonic development. Since DMSO is the most commonly used as a cryoprotectant for freezing of oocytes and embryos for storage purposes, we propose that it is imperative to understand its biological effects on the developmental competence of mammalian embryos both in vitro and in vivo; this remains a major concern in toxicological studies.[17-19] Recently, Zhou et al. demonstrated that DMSO treatment affected the asymmetric division of mouse oocytes.[20] Detrimental effects of DMSO have also been reported in the development of both rat and zebrafish embryos.[21-23] In another study, Pal et al. evaluated the effect of DMSO on the development and differentiation potential of human embryonic stem cells (hESCs).[24] They showed that DMSO induced a dose-dependent (0.01-1%) reduction in cell viability and down-regulation of key marker genes associated with stemness, ectoderm, mesoderm, and endoderm, indicating an aberrant and untimely differentiation trajectory.
Oxygen radicals play an important role in embryonic development, particularly low oxygen content and superoxide dismutase (SOD).[25] DMSO plays a dual role either as a pro-oxidant or anti-oxidant depending on the concentration and cell type.[26-28] For instance, DMSO reduced arsenite- or hydrogen peroxide (H2O2)-induced intracellular reactive oxygen species (ROS) production in human-hamster hybrid and mouse embryonic cells.[26, 27] On the other hand, Sadowska-Bartosz et al. reported that DMSO induced oxidative stress in yeast cells in a dose-dependent manner by enhancing the loss of succinate dehydrogenase and enhancing the formation of reactive oxygen species (ROS).[28]
Autophagy is a process by which cytoplasmic components, including macromolecules and organelles, are degraded by the lysosome; it is considered as a defense strategy against environmental stress.[29] The earliest autophagic event in mammalian development was observed in fertilized oocytes.[30] Autophagy occurs at low levels in unfertilized oocytes, but is massively induced within 4 h of fertilization; both of maternal mRNAs and proteins are rapidly degraded after the 2-cell stages.[30] For example, it is well documented that autophagy-related 5 (Atg5)-nulled embryos failed to develop beyond the 4- and 8-cell stages.[31] In addition, mitophagy is a critical process to maintain the quality of mitochondria and to regulate mitochondrial number; this is done by selective engulfment of damaged mitochondria by autophagosomes, which are subsequently catabolized by lysosomes in response to the loss of mitochondrial membrane potential.[32] Like autophagy, mitophagy serves a protective role in limiting cell death.
The present study aimed to investigate the impact of DMSO on mouse pre- and post-implantation embryos, as there are currently no reports investigating this. The first objective of our study was to investigate the effect of DMSO on oxidative stress responses and its mechanistic role in the embryo. The second objective was to evaluate the effect of DMSO on endoplasmic reticulum (ER) stress, mitochondrial calcium accumulation, and induction of autophagy and mitophagy. The final objective was to determine the adverse effects of DMSO on pre- and post-implantation embryos.
Results
Effects of DMSO on oxidative stress and calcium aggravation in mouse zygotes and embryos
First, we checked the effects of different concentrations of DMSO (0%, 0.5%, 1%, and 2% v/v) on the developmental competency of preimplantation embryos. Treatment with 0.5% DMSO did not significantly affect embryo development until the blastocyst stage. In 1% DMSO-treated groups, there was no significant change in embryo development until the 8-cell stages, but we observed ~31% inhibition at the morula stage and 59% inhibition at the early blastocyst stage compared to control groups. However, in 2% DMSO supplemented groups, significant developmental inhibition was observed from the 2-cell stages; no morulas or blastocysts were formed (Fig. 1A, Movie S1).
Effects of DMSO on oxidative stress and calcium accumulation during development from zygote to 2-cell stages. (A) Developmental rate of mice preimplantation embryos after incubation with 0%, 0.5%, 1%, and 2% DMSO. (B) Osmolarity of KSOM supplemented with DMSO at different concentrations. (C, D) ROS levels in 2-cell embryos treated with different concentrations of DMSO for 30 min and 2 h respectively, and analysis of the fluorescence pattern in the cytosol. Scale bar, 50 μm. (E) Gene expression analysis of pro- and anti-oxidants by RT-qPCR in 1-cell embryos treated with different concentrations of DMSO for 30 min. (F) ROS levels were determined in 2-cell embryos after 24 h exposure to DMSO at different concentrations. Fluorescence intensity in the nucleus and perinuclear region was compared between groups. Scale bar, 50 μm. (G, H) Calcium levels were determined by analyzing Rhod-2 AM fluorescence in 2-cell embryos after 24 h and 36 h exposure to DMSO at different concentrations. Scale bar, 50 μm. Fluorescence intensity in the nucleus, perinuclear region, and cortical region was compared between groups. *: p<0.05, **: p<0.01.
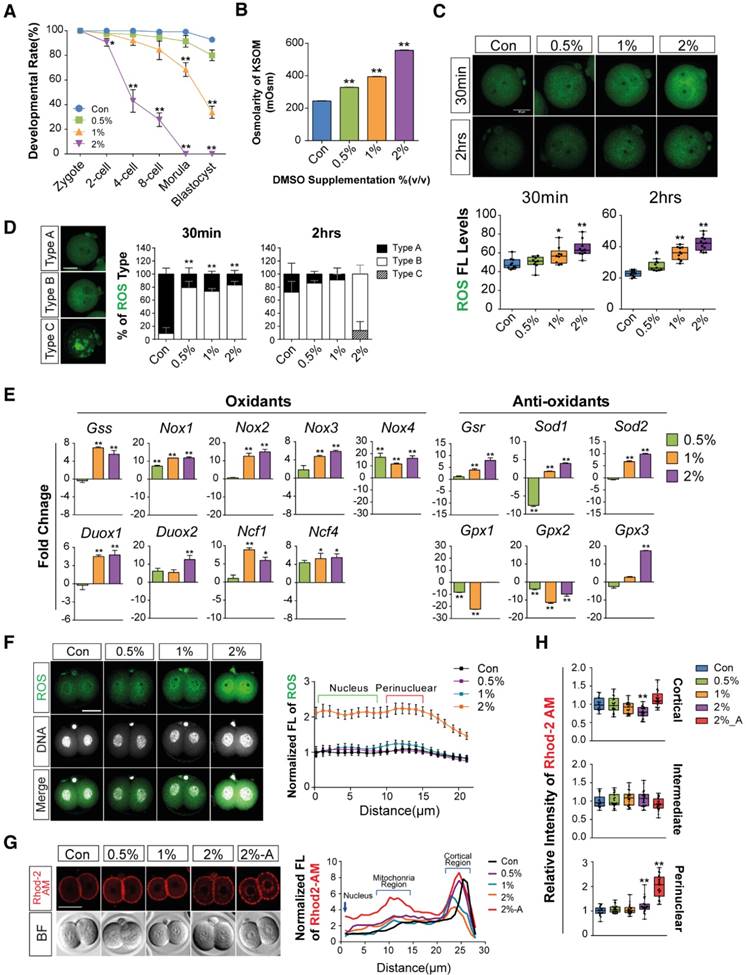
Mouse embryos are sensitive to the osmolarity of potassium simplex optimized medium (KSOM), which appears to be one of the major factors affecting developmental arrest at the 2-cells stage and survival of blastocysts.[33-35] We therefore investigated whether DMSO affects osmolarity of KSOM at each dosage of DMSO. As shown in Fig. 1B, the osmolarity of KSOM significantly increased from 224 to 328, 384, and 557 milliosmole (mOsm) when supplemented with 0%, 0.5%, 1%, and 2% DMSO, respectively. No morulas or blastocysts formed in the 2% DMSO-treated group, indicating that increased osmolarity of KSOM due to DMSO treatment might influence embryonic developmental capability.
Next, we checked the effects of DMSO on oxidative stress in zygotes. When zygotes were exposed to DMSO for 30 min, the production of intracellular ROS increased in a dose-dependent manner and was maintained for up to 2 h (Fig. 1C): ROS levels were significantly higher in DMSO-treated groups (Type B) than in control groups (Type A), especially in the cytosol and perinuclear region (Fig. 1D). Notably, ROS were found to accumulate in uncommonly high concentrations inside abnormally aggregated organelles of a few embryos from the 2% DMSO treated group (Type C) (Fig. 1D). At the same time, mRNA levels of pro-oxidant genes (Nox1, Nox2, Nox3, Nox4, Duox1, Duox2, Ncf1, Ncf4) were markedly up-regulated in DMSO-treated groups, while mRNA levels of some anti-oxidant genes (Gsr, Gpx3, Sod2) were also considerably up-regulated (Fig. 1E). On the other hand, the mRNA expressions of anti-oxidant genes such as Gpx1 and Gpx2 were down-regulated in DMSO-treated groups. In the case of Sod1, mRNA expression level decreased in the 0.5% DMSO-treated group, but increased when treated with higher doses of DMSO (Fig. 1E).
Most zygotes treated with DMSO for over 24 h developed to the 2-cell stages. ROS levels increased in the perinuclear region and nucleus in a dose-dependent manner (Fig. 1F). We also checked the mitochondrial calcium levels in the 2% DMSO-treated groups using Rhod-2 AM (a selective indicator for mitochondrial Ca2+). A substantial increase in fluorescence density was detected in the perinuclear region, where mitochondria are densely located. Interestingly, when the development of the 2-cell embryos was arrested after co-culture for 36 h with 2% DMSO, we observed a strong punctate accumulation of calcium in the perinuclear region (Fig. 1G & H). Taken together, these results suggest that 1- to 2-cell embryos exposed to DMSO show increased oxidative stress; this effect endured until development to 2-cell embryos. DMSO exposure especially disrupted the balance of pro- and anti-oxidant gene expression, leading to increased oxidative stress and Ca2+ levels in the mitochondria.
Effects of DMSO on mitochondria-mediated apoptosis, ER stress, and developmental arrest in embryos
Immunostaining analyses were performed to determine the effects of DMSO on mitochondria-mediated apoptosis in 2-cell embryos exposed to 2% DMSO for 24 h. DMSO exposure induced the translocation of phosphorylated p53 (a pro-apoptotic marker) from the cytosol to the nucleus (Fig. 2A), and decreased the expression of Bcl-2 (an anti-apoptotic protein) (Fig. 2B). On the other hand, the expression of BAX (a pro-apoptotic marker) was markedly increased in the mitochondrial location of the perinuclear area (Fig. 2C). As a result, DMSO exposure resulted in increased CYSC/cytochrome C expression (Fig. 2D). However, the mitochondrial membrane potential was decreased, as seen by the increased ratio of JC-1 aggregates to monomers in the 2-cell embryos and arrested embryos (Fig. 2E). The occurrence of cellular apoptosis was further supported by the comet assay, a technique frequently used to check DNA fragmentation in apoptotic cells by measuring the tail length. The tail length in 2% DMSO-treated embryos was significantly higher than in control groups, confirming apoptosis in 2-cell embryos (Fig. 2F). Taken together, the induction of apoptosis by DMSO is inextricably linked to mitochondrial disruption. Therefore, the increased CYSC/cytochrome C expression may induce apoptosis through a caspase protein cascade, a caspase-dependent pathway, indicating that DMSO induces translocation of phosphorylated p53 from the cytosol to the nucleus and increases expression of CYSC/cytochrome C in the perinuclear region and induces their translocation to the nucleus.
Effects of DMSO on mitochondria-mediated apoptosis, ER stress, and developmental arrest in 2-cell embryos. (A) Phosphorylated p53 immunostaining showing translocation of the signal from the cytosol to the nucleus in the 2% DMSO-treated group. (B, C) Bcl-2 and BAX protein expression was detected by immunocytochemistry; fluorescence signals around the nucleus were compared between the control and 2% DMSO-treated groups. (D) CYCS/Cytochrome C immunostaining in 2% DMSO-treated embryos. (E) Mitochondrial depolarized membrane status was analyzed using JC-1 dye. Right graph shows the JC-1 aggregate to monomer ratio, which indicates the membrane potential status of the treated groups. (F) Comet assay in 2-cell embryos treated with 2% DMSO. Tail lengths were analyzed (bottom-right panel). Enlarged nucleus-stained pictures are shown in the bottom-left panel. (G) The developmental arrest ratio from 2-cell to 8-cell stages after 48 h of exposure to DMSO at different concentrations. (H) The ER was immunostained using anti-PDI antibody and fluorescence intensity was determined using confocal microscopy at the 2-cell stages after 24 h exposure to DMSO at different concentrations. (I) ER stress-related gene expression was analyzed using RT-qPCR and normalized with respect to the control group. *: p<0.05, **: p<0.01. Scale bar, 50 μm.
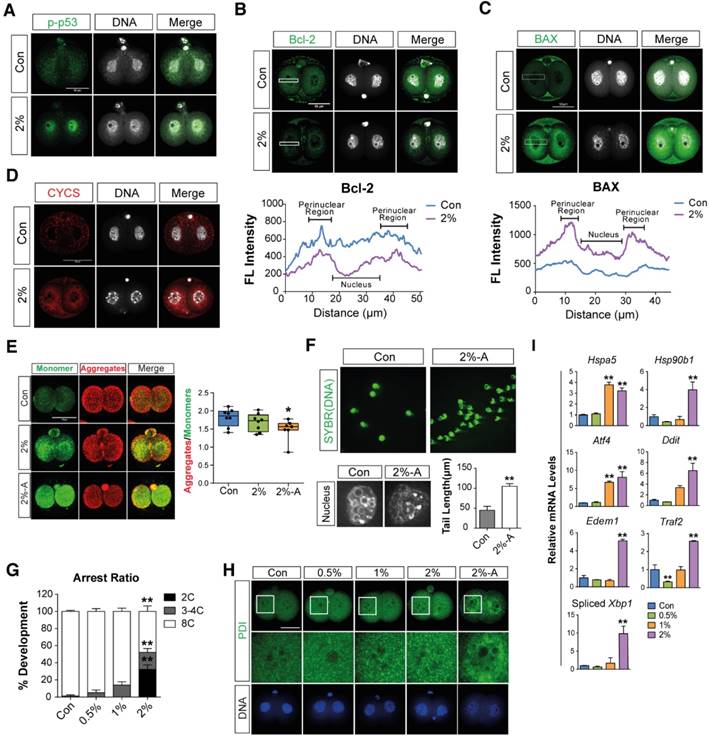
When we checked the developmental rate and cleavage of zygotes during in vitro culture after exposure to DMSO for 48 h, we found that embryo cleavage was significantly retarded in DMSO-treated groups in a dose-dependent manner, starting from 1% DMSO (Fig. 2G). Interestingly, approximately 32% and 20% of embryos in the 2% DMSO-treated groups are arrested at the 2- and 4-cell stages, respectively; however, in 0.5-1% DMSO-treated groups, significant developmental arrest was observed only at the 4-cell stages (Fig. 2G). Furthermore, immunostaining analysis with anti-PDI antibody showed that DMSO caused aggregation of the ER in the cytosol of 2-cell embryos that were arrested after co-culture with DMSO for 36 h (Fig. 2H). The effect of ER stress in embryos due to the exposure of DMSO was also analyzed by checking the expression levels of ER stress-related genes: all gene's expressions analyzed in this study were found to be significantly up-regulated in the 2% DMSO-treated group (Fig. 2I). Collectively, DMSO-triggered oxidative stress causes ER stress and induces apoptosis via mitochondrial membrane depolarization, and consequently inhibits development of the embryo from 2-cells to 8-cells.
Effects of DMSO on the induction of mitophagy and autophagy in 2-cell embryos
Mitophagy or autophagy can be formed as a defense strategy against environmental stress and serve a protective role in limiting cell death.[36, 37] We hypothesized that DMSO-induced cellular stress could activate mitophagy and autophagy in preimplantation embryos. In order to examine our hypothesis, we performed immunostaining of PINK1 and Parkin, which are the main mediators of mitophagy—i.e., autophagy of damaged mitochondria.[38-41] The embryos treated with 2% DMSO showed a considerable increase in mitophagy induction; this was evident from the increased localization of PINK1 and Parkin in the mitochondria, whereas untreated embryos exhibited stable expression of PINK1 and Parkin in the cytosol (Fig. 3A, B).
Effects of DMSO on the induction of mitophagy and autophagy in 2-cell embryos. (A, B) Immunostaining pattern of PINK1 and Parkin expression in 2-cell embryos exposed to 2% DMSO. The representative fluorescence intensities at the perinuclear region and nucleus are marked with a dotted-line box. (C) MAP1LC3B expression was detected by immunocytochemistry and co-localized with mitochondria using MitoTracker® Orange CMTMROS in DMSO-treated embryos. Bottom graph shows the comparison of punctate area per embryo among groups. (D) Lysosomes were immunostained using anti-LAMP1 antibody in 2% DMSO-treated groups. Fluorescence intensities due to LAMP1 in the cytoplasmic and perinuclear regions were analyzed in the right graph. (E) RT-qPCR analysis of autophagy-related genes in DMSO-treated embryos. **: p<0.01.
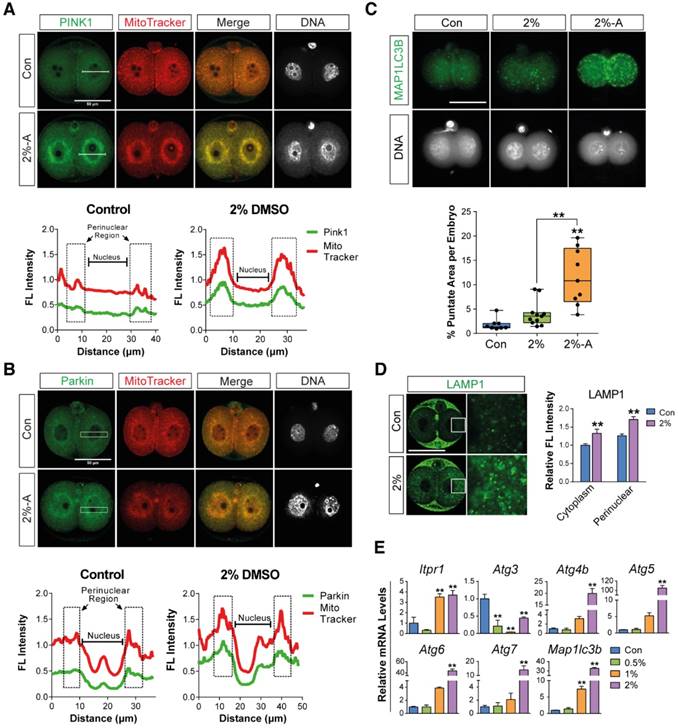
To further monitor autophagy and mitophagy induction, we followed the expression of MAP1LC3B, which is essential for mature autophagosome formation.[42] Embryos treated with 2% DMSO showed enhanced expression of MAP1LC3B in mitochondria in discrete, punctate structures, indicating increased autophagosome formation (Fig. 3C). Since autophagosomes containing abnormal intracellular proteins or damaged organelles are subject to fusion with the lysosome for hydrolysis, we measured the lysosomal content in the embryos by immunostaining with LAMP1, a type I transmembrane glycoprotein that is primarily localized in lysosomes and late endosomes. After treatment with 2% DMSO, LAMP1-positive signals were observed in high amounts, indicating the recruitment of large numbers of lysosomal vesicles (Fig. 3D). DMSO-triggered autophagy induction has also been confirmed by the dose-dependent up-regulation of various gene expressions involved in autophagy, such as Itpr1, Atg4, Atg5, Becn1, Atg7, and Map1lc3b (Fig. 3E). An increase in lysosomal vesicles and their contents might play a role in autolysis; our data demonstrate that the fusion of the autophagosome and lysosome in DMSO-treated embryos occurred frequently (Fig. S1), indicating that DMSO exposure causes mitophagy and extensive autophagy in 2-cell embryos.
Effects of DMSO-induced oxidative/ER stress and calcium accumulation on morula to blastocyst stage embryo development
To determine the further consequences of DMSO supplementation on the development of morulae to blastocyst stage embryos, zygotes were cultured in the presence or absence of 1% DMSO for 96 h. Representative photographs of blastocysts developed from morula stage embryos were taken by time-lapse photography under a microscope during in vitro culture (Fig. 4A). The addition of 1% DMSO delayed expanded blastocyst development and exhibited two distinct abnormal patterns: (i) cavities were formed, but not fully grown; and (ii) embryos were still arrested in the morula stage after 96 h. Therefore, we checked whether DMSO could induce oxidative/ER stress or calcium accumulation and mitochondrial dysfunction in morula stage embryos, as in 1- to 2-cell stage embryos. We observed that 1% DMSO supplementation caused a substantial increase in ROS levels in morula stage embryos (Fig. 4B).
Effects of DMSO-induced oxidative/ER stress and calcium accumulation on morula to blastocyst stage embryo development after zygotes were developed to morula stage embryos following 72 h DMSO exposure. (A) Time lapse images for morula to blastocyst transition with 4 h intervals. (B, C) In morulae, ROS levels and mitochondrial activities were measured and compared between DMSO-treated and control groups. Scale bar, 50 μm (D) TEM images of morulae in control and 1% DMSO-treated groups. Scale bar, 1 μm (E) Relative copy number of mitochondrial DNA in morulae treated with DMSO at different concentrations (F) RT-qPCR analysis of genes with a role in maintenance of mitochondrial morphology and distribution, as well as mitochondrial activity and function. (G) Calcium levels were checked after staining with Rhod-2 AM in morulae grown with different concentrations of DMSO. Scale bar, 100 μm (H) Immunoblot analysis of GRP78/BIP protein in morulae. *: p<0.05, **: p<0.01.
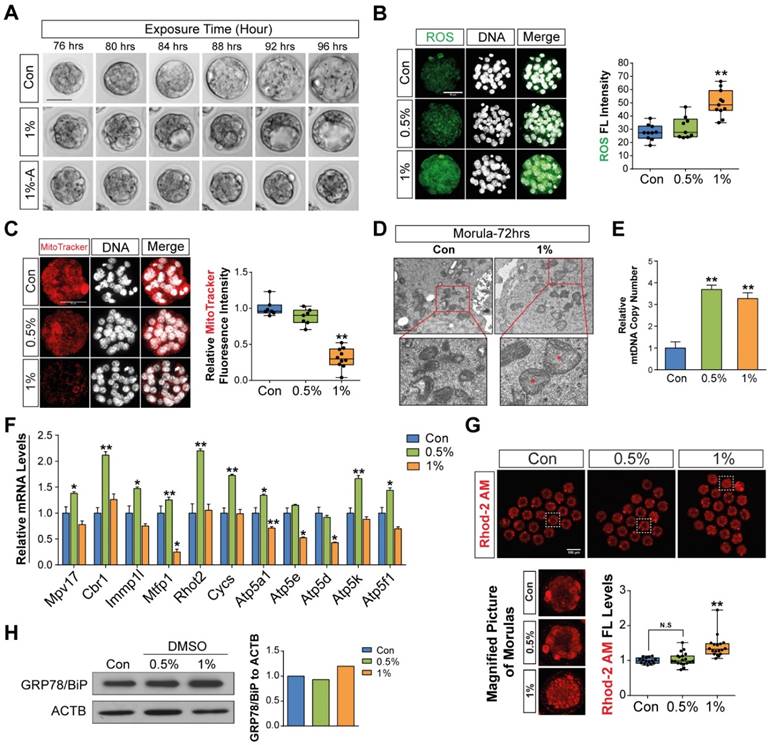
Next, we examined mitochondrial function using MitoTracker® Orange CMTMROS, which are effective tools to evaluate mitochondrial integrity. As shown in Fig. 4C, we observed a considerable decrease in fluorescence density in 1% DMSO-treated embryos; this was caused by decreased internalization of the MitoTracker® Orange CMTMROS into mitochondria, indicating mitochondrial dysfunction or loss of mitochondrial membrane potential. We also examined the ultra-structure of mitochondria in both control and 1% DMSO-treated morula stage embryos. DMSO-treated morulae showed an increased number of swollen mitochondria compared to control embryos (Fig. 4D). In addition, the mitochondrial DNA copy numbers in 0.5% and 1% DMSO-treated morulae were significantly increased (Fig. 4E). Notably, the expression of genes such as Mpv17, Cbr1, Immp1l, Mtfp1, Rhot2, Cycs, Atp5a1, Atp5e, Atp5d, Atp5k, and Atp5f1, which have roles in maintenance of mitochondrial morphology, distribution, activity, and function, were considerably dysregulated in 1% DMSO-treated embryos (Fig. 4F). However, the expression of these genes was substantially up-regulated in the 0.5% DMSO-treated group. This phenomenon may be transiently caused by a defense strategy to prevent embryo death due to environmental stress. Taken together, our results strongly indicated that exposure to 1% DMSO may result in severe mitochondrial damage and dysfunction, thus impeding or arresting further embryo development.
Mitochondrial Ca2+ levels in morula stage embryos were also significantly increased when exposed to 1% DMSO (Fig. 4G). We performed Western blot analysis to examine whether the onset of ER stress in morula stage embryos was caused by mitochondrial Ca2+ levels. As shown in Fig. 4H, Western blot analysis of ER stress marker proteins, including GRP78/BIP, showed a dose-dependent increase in protein expression after DMSO treatment. This clearly demonstrates that DMSO-induced early cellular stress responses, such as oxidative/ER stress and calcium accumulation, are still present in the morula stage embryos; as a result, these factors may result in developmental failure from morula stage embryos to blastocysts.
Effects of DMSO on mitophagic and autophagic responses in arrested morula stage embryos
To examine whether DMSO induced mitophagy and autophagy in arrested morula stage embryos, we analyzed the expression levels of known mitophagy marker proteins such as PINK1 and Parkin by immunostaining. Embryos treated with 1% DMSO showed an increase in mitophagy induction; this is evident from the increased localization of PINK1 and Parkin in mitochondria (Fig. 5A, B). We further investigated PINK1 and Parkin protein, and mRNA expression levels. As shown in Fig. 5C and D, the mRNA and protein expression levels of PINK1 and Parkin were significantly increased after exposure to 1% DMSO. Furthermore, TOM20 expression was increased in the 0.5% DMSO-treated group compared to the control group; however, the 1% treatment group showed a lesser increase in expression than the 0.5% treatment group. (Fig. 5D). Taking the data of Fig. 4F together, this phenomenon in the 0.5% treatment group may be transiently caused by regeneration of healthy mitochondria for survival against DMSO-induced acute cell stress. In addition, we examined p38 MAPK expression, which is an important regulator of mitophagy,[43] and found that p38 MAPK is increasingly phosphorylated in a dose-dependent manner after DMSO exposure (Fig. 5E). This indicates that embryos exposed to excessive doses of DMSO (1% or 2%) may be significantly damaged by severe mitochondrial dysfunction.
Effects of DMSO on mitophagic and autophagic responses in arrested morula stage embryos. (A, B) Immunostaining for PINK1 and Parkin in morulae exposed to 1% DMSO. Left and right panels show single and multiple projections, respectively. Scale bar, 20μm (C) RT-qPCR analysis of PINK1 and Parkin mRNA expression in arrested morulae treated with 1% DMSO. (D, E) Immunoblot analysis of PINK1, Parkin, TOM20, p38 and phospho-p38 proteins in DMSO-treated morulae. (F) Real Time RT-qPCR analysis on genes related to autophagic induction. (G) MAP1LC3B expression was detected by immunocytochemistry and co-localized with mitochondria in the DMSO-treated embryos. Left and right panels show single and multiple projections, respectively. Scale bar, 20μm (H) Lysosomes were immunostained using anti-LAMP1 antibody in 1% DMSO-treated groups and co-localized with mitochondria using MitoTracker® Orange CMTMROS in DMSO-treated embryos. Scale bar, 20 μm (I) Immunoblot analysis of ATG5, Beclin-1, and SQSTM1/p62 in morulae treated with DMSO at different concentrations. *: p<0.05, **: p<0.01. (J) MAP1LC3B-I and MAP1LC3B-II Western blotting analysis. Band intensities were quantified and then analyzed by Image J software. The experiments were independently repeated three times and β-Actin was used as a loading control. **: p<0.01.
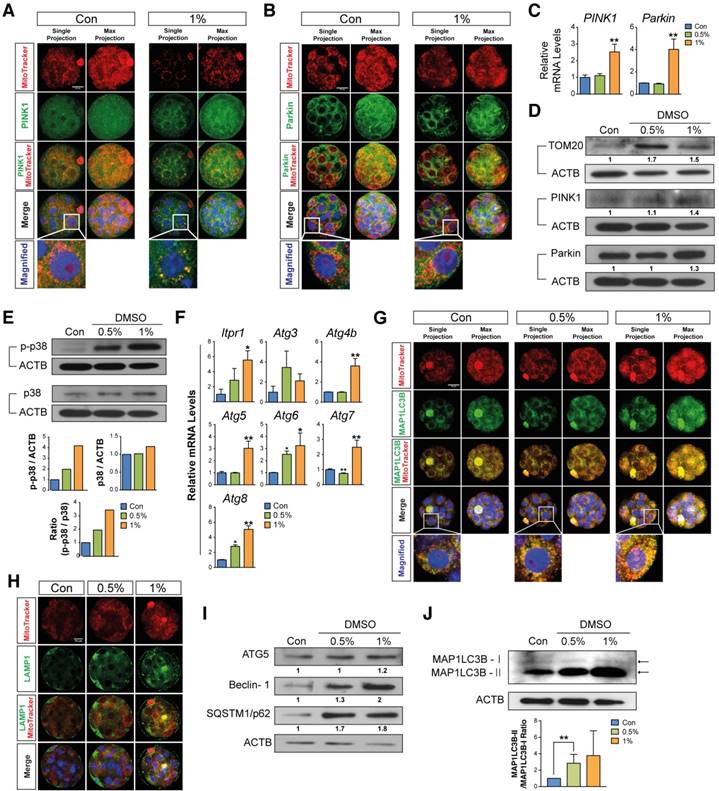
Next, we determined the expression levels of MAP1LC3B-I and MAP1LC3B-II by immunostaining and Western blot analysis. Both results demonstrated a dose-dependent increase in the expression of MAP1LC3B-II (Fig. 5G, J). Even though the expression levels of MAP1LC3B-I after long-term exposure could be detected, the expression levels of MAP1LC3B-I after short-term exposure (within 1 min) were not detected in both control and DMSO-treated groups. The lysosomal contents of the embryos were also measured by immunostaining with LAMP1. We found a dose-dependent increase in LAMP1-positive signals, indicating the recruitment of large numbers of lysosomal vesicles (Fig. 5H). Because autophagy can be induced by the up-regulation of IP3R1, ATG4, ATG5, Beclin-1, ATG7 and MAP1LC3B,[44] we performed Western blot and reverse transcription quantitative PCR (RT-qPCR) analysis of autophagy-regulated proteins including ATG5, Beclin-1, and SQSTM1/p62 in DMSO-treated morula stage embryos (Fig. 5F, I). The expression levels of these mRNAs and proteins were increased in a dose-dependent manner. These results indicate that developmental failure in morula stage embryos after exposure to DMSO may be caused by the induction of extensive autophagy and mitophagy.
Effects of DMSO on developmental competency of pre- and post-implantation embryos
When we checked the developmental rate of blastocysts after DMSO exposure (0%, 0.5%, and 1%) at 96 h, we found that some blastocysts developed to a fully expanded state (Fig. 6A). To investigate the quality of surviving blastocysts, we first checked the total number of cells—including the inner cell mass (ICM) and trophectoderm (TE) cells—in the blastocysts derived from zygotes, which were treated with 0.5% and 1% DMSO for 96 h. Immunoblotting with OCT3/4 and CDX2 showed that 1% DMSO treatment decreased the total number of cells—as well as ICM and TE cells—in blastocysts (Fig. 6B). Furthermore, the terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay showed a significant increase in the number of TUNEL-positive cells in blastocysts derived from 1% DMSO-treated zygotes (Fig. 6C).
Effects of DMSO on developmental competency of pre- and post-implantation embryos. (A) Images of blastocysts developed from zygotes supplemented with DMSO for 96 h. Scale bar, 100 μm (B) Inner cell mass (ICM)/trophectoderm (TE) specification in fully-grown blastocysts was analyzed using immunostaining with anti-OCT3/4 (green color) and anti-CDX2 (red color) antibodies. The number of cells in the ICM and TE are compared in the bottom graph. Scale bar, 50 μm (C) TUNEL assay of fully-grown blastocysts (96 h) after exposure to different concentrations of DMSO. The number of apoptosis-positive cells (red color) was compared between groups. Scale bar, 50 μm (D) RT-qPCR analysis of genes involved in the oxidative stress response and mitochondria-dependent apoptosis in fully grown blastocysts after exposure to different concentrations of DMSO for 96 h. (E) RT-qPCR analysis of genes that regulate implantation success in uterus. (F) Implantation rate in the uterus at 6.5 dpc after transferring blastocysts treated with DMSO at different concentrations. (G, H) Evaluation of the number of full-term pups at 18.5 dpc by cesarean section after transferring blastocysts to the uterus. Placenta weight, body weight, and size of the crown-rump of the pups. Scale bar, 1 cm (I) A representative hypothetical model for differential effects of DMSO on mouse preimplantation embryos. During in vitro growth of mouse zygotes to blastocyst stage embryos, exposure to different concentrations of DMSO greatly increased the osmolarity of KSOM, intracellular ROS levels, intracellular Ca2+ levels, ER stress, and autophagy/mitophagy numbers in developing preimplantation embryos; conversely, the mitochondrial function and developmental competency of preimplantation embryos were significantly decreased. After 1% or 2% DMSO supplementation, most embryos were arrested at the 2-cell, 4-cell, or morula stages. (J) Although some zygotes developed to the blastocyst stage after 1% DMSO supplementation, these blastocyst embryos showed decreased CDX2, OCT3/4, B3gnt5, and Wnt3a protein and gene expression. As a result, both implantation and full-term development rates were significantly reduced, compared to control group. *: p<0.05, **: p<0.01.
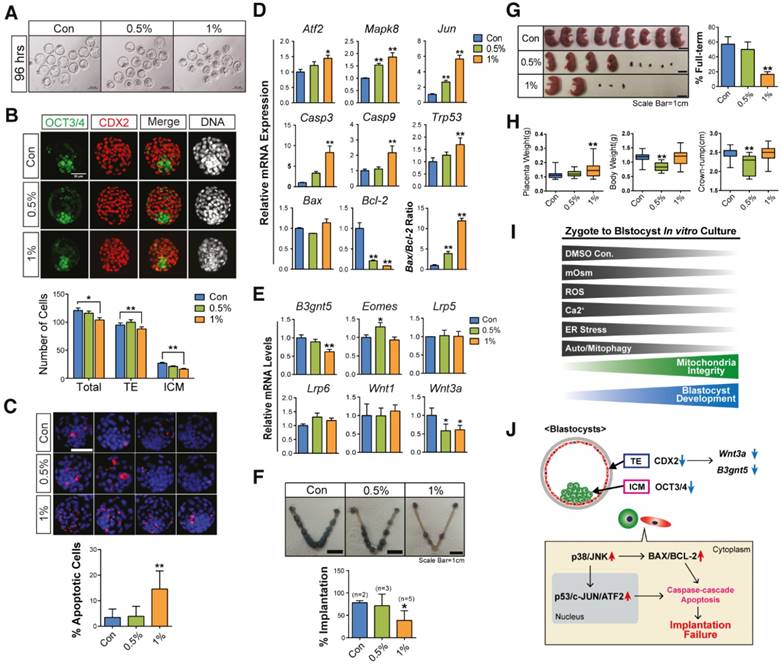
To determine the reasons behind developmental failure and subsequent poor quality blastocyst formation following DMSO exposure, we examined the expression levels of Jnk/Atf2 mRNAs, which are genes involved in oxidative stress-induced apoptosis, as well as other genes involved in mitochondria-dependent apoptosis.[45, 46] We observed that DMSO exposure increased the mRNA expression levels of Atf2, Mapk8, and Jun in a dose-dependent manner (Fig. 6D), indicating the activation of oxidative stress-responsive apoptosis signals. DMSO exposure also increased the mRNA expression levels of genes related to mitochondria-dependent apoptosis such as Casp9, Casp3, and Trp53, as well as the Bax/Bcl2 ratio, in a dose-dependent manner (Fig. 6D).
Next, we analyzed the expression levels of genes correlated with the implantation potential, such as B3gnt5, Eomes, and Wnt3a, in the developed blastocysts following DMSO exposure. Treatment with 1% DMSO significantly decreased the mRNA expression levels of B3gnt5, which is involved in cell differentiation and adhesion of blastocysts (Fig. 6E).[47] Treatment with 0.5% DMSO significantly increased the mRNA expression levels of Eomes, which is involved in TE differentiation;[48] however, treatment with 1% DMSO did not show any effect (Fig. 6E). It is well documented that the canonical WNT-CTNNB1 signaling pathway is critical for normal blastocyst function and implantation competency.[49, 50] Notably, DMSO exposure significantly decreased the mRNA expression levels of Wnt3a, but did not reduce the expression of Wnt1, Lrp5, or Lrp6 (Fig. 6E). The decrease in expression levels of both B3gnt5 and Wnt3a in DMSO-treated blastocysts might be at least partially associated with nonviable implantation or pregnancy loss following implantation.
The effects of DMSO treatment on post-implantation embryos were also investigated after transferring the DMSO-treated blastocysts into the uteri of pseudopregnant mice. We found that treatment with 1% DMSO significantly decreased the implantation rate, the number of full-term pups, and the placental weight compared with control blastocysts (Fig. 6F-H). The fetal length (crown-rump) and body weight were significantly decreased in 0.5% DMSO-treated groups (Fig. 6H), but no change was observed in 1% DMSO-treated groups. All these findings indicate that DMSO treatment has negative effects on the developmental capability of pre- and post-implantation embryos.
Discussion
The present study aimed to investigate the underlying mechanism of the cytotoxic effects of DMSO on mouse pre- and post-implantation embryos. For this purpose, mouse 1-cell zygotes were treated with DMSO at doses of 0%, 0.5%, 1%, or 2% (v/v) for 0.5-96 h to examine the effects of DMSO on (i) oxidative stress; (ii) ER stress, mitochondrial Ca2+ accumulation, mitochondrial dysfunction, autophagic and mitophagic responses; (iii) apoptosis-related signaling pathways; (iv) quality of blastocysts; (v) post-implantation development.
In this study, we determined that the osmolarity of KSOM supplemented with 0.5-2% DMSO increased to 328-557 mOsm, whereas the osmolarity of KSOM without DMSO was 224 mOsm (Fig. 1B). Osmolarity of the culture medium is an important parameter for regulating the fate of early embryo development: osmotic stress leads to an influx or efflux of water into the cell; hypoosmotic stress causes swelling, whereas hyperosmotic stress causes shrinkage.[51] Therefore, before beginning our detailed investigation into the cytotoxic mechanisms of DMSO, we checked the immediate effects of DMSO on KSOM. Preimplantation embryos cultured in medium with an osmolarity higher than 300 mOsm do not normally progress to the blastocyst stage in vitro;[52] therefore, DMSO supplementation of zygotes cultured in vitro may experience hyperosmotic stress. As a result, the development of many embryos may be arrested at the 2-cell, 4-cell, or morula stages.
Generally, mitochondria are the main endogenous ROS producer. Also, another important generator of ROS is NOX, a family of membrane-bound proteins that transfer electrons from NADPH to O2.[53] From the seven isoforms of the Nox genes family identified in mammalian cells, we examined Nox1, 2, 3, and 4 mRNA expression, and found their levels were significantly increased in DMSO-treated groups compared to control (Fig. 1E). In Nox4-overexpressing HEK293 cells and endothelial cells, NOX4 continuously produces H2O2.[54-56], Moreover, NOX 4 activity has been closely linked to elevated Nox 4 mRNA expression, indicating that NOX 4 directly targets Nox 4 gene itself to regulate NOX4 activity. Since the dual oxidases (DUOX) have specialized domains for the dismutation of O2-, and produce H2O2 as substrate for peroxidation and other oxidation reactions, [57] we also examined Duox1 and Duox2 mRNA expression as well as glutathione synthetase (Gss), p40, and p47 mRNA expression in DMSO-treated groups. Expression of these mRNAs, like the Nox mRNAs, was significantly increased in DMSO-treated groups. Even though Gpx1 and 2 mRNA expression in DMSO-treated groups was significantly decreased, expression of most antioxidant mRNAs, such as glutathione-disulfide reductase (Gsr), superoxide dismutase (Sod)1, Sod2, and glutathione peroxidase (Gpx)3, were significantly increased. Dichotomous effects of antioxidant mRNA expression in DMSO-treated groups may be transiently caused by excessive and uncontrolled production of oxygen radicals due to excessive DMSO exposure or by activities of all the enzymes increased in response to the oxidative challenges, because physiological cellular ROS production under various stress conditions is an essential component of normal antioxidant production. Taken together, we concluded that mRNA expression of these pro-oxidants and anti-oxidants are closely correlated with the level of DMSO-induced oxidative injury to embryos. As a result, increased ROS levels persisted in embryos until the morula stage.
Osmotic and oxidative stress responses generally differ, but they displayed overlapping symptoms in some cases.[51] Generally, oxidative stress is caused either by the disturbance of the cellular redox state, which is composed of both enzymatic and non-enzymatic defense mechanisms, or by the accumulation of ROS (such as superoxide, hydrogen peroxide, and hydroxyl radicals) within the cell, which may cause molecular damage to DNA, proteins, cell membranes, etc.[51] Therefore, the oxidative stress response in DMSO supplemented embryos may cause more direct harm to the cell than osmotic stress. In mammalian embryos, excess ROS concentration is often associated with developmental arrest or damage in vitro.[58, 59] Previous studies have reported that 0.1% or 0.5% DMSO exposure reduced arsenite- or H2O2-induced ROS production in human-hamster hybrid and mouse embryonic cells.[26, 27] On the other hand, Sadowska-Bartosz et al.[28] reported that 1% DMSO exposure induced oxidative stress in yeast cells. Therefore, we have concluded that DMSO supplementation for culturing mouse zygotes play dual roles: either as a pro- or anti-oxidant, depending on the concentration of DMSO.
There are numerous studies related to the detrimental role of ER stress in oocyte maturation and early mammalian embryonic development.[60-63] Because in vitro developmental competence of mammalian embryos is highly dependent on the coupled/cooperative response between oxidative and ER stress (i.e., stress plays a key role in protein structural modifications that are essential for embryos to function correctly),[59] we examined ER stress. In the present study, we observed abnormally aggregated ER-positive signals in the perinuclear region of 2-cell arrested embryos that were cultured with 2% DMSO for 36 h (Fig. 2H). Furthermore, the induction of ER stress in 2-cell or morula stage embryos following DMSO supplementation was confirmed by analyzing the expression of ER stress-activated genes and/or proteins. As shown in Fig. 3E, RT-qPCR analysis showed that Itpr1, Atg4b, Atg 5, Atg6, Atg7, and Atg 8 mRNA expression levels in 2 cell embryos treated with DMSO were significantly increased in a dose-dependent manner, compared with those of the control group, whereas Atg3 mRNA expression was significantly downregulated. MAP1LC3B-I can be converted to the MAP1LC3B-II form after being cleaved by a Cys-protease such as Atg-4, which is activated by ROS. This step is necessary for the action of Atg-7, which leads to Atg-5/Atg-12 conjugation and subsequent linkage to Atg16. These mRNA upregulations indicate that DMSO-induced autophagy required the classical autophagic machinery along with molecules involved in membrane extension of the autophagosomal membranes such as ATG7 and LC3. Even though it is unclear why Atg3 mRNA expression in DMSO-treated groups is downregulated, downregulation of Atg3 mRNA expression leads to accumulation of aberrant mitochondria, enhanced ROS generation, and defective ATP production (Fig. 1A, C, F, 4B-F). Therefore, we concluded that excessive accumulation of misfolded or unfolded proteins in the ER lumen generates ER stress, which ultimately results in substantial cellular damage, followed by apoptosis or degeneration.
The ER and mitochondria are physically and functionally closely linked. Therefore, Ca2+ release from the ER, which is the major intracellular Ca2+-storage organelle, has a direct effect on the physiological function of the mitochondria.[64] To assess the impact of DMSO-induced ER stress on mitochondria, we measured mitochondrial Ca2+ levels and the subsequent events leading to cell death in 2-cell or morula stage embryos. We observed that exposure to DMSO significantly increased the mitochondrial Ca2+ levels in early embryos, and also induced mitochondrial depolarization and dysfunction (Fig. 1G-H, 2E, 4C-G). Transmission electron microscopy (TEM) analysis demonstrated that most of 1% DMSO-treated morula stage embryos showed significantly swollen mitochondria when compared with control embryos (Fig. 4D), indicating severe mitochondrial damage. Furthermore, DMSO-treated 2-cell embryos showed a decrease in expression of the anti-apoptotic protein Bcl-2, translocation of phosphorylated p53 (a pro-apoptotic marker) from the cytosol to the nucleus, as well as a considerable increase in BAX expression, indicating that arrested 2-cell or morula stage embryos might experience mitochondria-dependent apoptosis.
Treatment with DMSO significantly reduced ICM and TE cell numbers in blastocysts, indicating a high likelihood of pregnancy loss following implantation. We also found that DMSO significantly decreased the expression of B3gnt5 and Wnt3a in the blastocysts (Fig. 6E). To determine the underlying mechanism, we counted fetus numbers after transferring the blastocysts into the uteri of pseudopregnant mice. DMSO treatment significantly decreased the implantation rate and the number of full-term pups compared to control blastocysts (Fig. 6F, G). Detrimental effects are linked to blastocysts with decreased Wnt3a and Eomes gene expression, which generally experienced spontaneous pregnancy loss (absorption), while blastocysts with decreased B3gnt5 and Eomes gene expression generally failed to implant.[65]
Another important adaptive response commonly induced to combat ER stress is autophagy.[66] Interestingly, disturbance of either ER function or ER calcium homeostasis triggers autophagy and apoptotic cell death.[67] However, the effect of autophagy on cell survival against ER stress is not always clear and depends on the tissue type. Ding et al.[68] demonstrated that ER-induced autophagy is important in mitigating ER stress-induced cell death in colon and prostate cancer cells; on the other hand, autophagy does not confer protection, but rather contributes to ER-induced apoptosis in non-transformed murine embryonic fibroblasts and in normal human colon cells. Our laboratory recently showed that treating mouse preimplantation embryos with the autophagy activator rapamycin significantly abrogated the chitosan-induced ER stress response and subsequent detrimental effects on development.[69] In our present study, we observed that DMSO-induced ER stress triggered mitophagy and autophagy in 2-cell or morula stage embryos (Fig. 2A-E, 5A-I). Mitophagy induction was confirmed by increased localization of PINK1 and Parkin, which are the main mediators of mitophagy, in mitochondria.[38-41]
Parkin promotes mitophagy in two different ways. In the first mechanism, Parkin causes the degradation of its substrates, Miro and Mitofusin, through ubiquitin-ligase activity.[70] Miro and Mitofusin are phosphorylated by PINK1 prior to Parkin-dependent degradation.[70] Proteasomal degradation of Miro and Mitofusin prevents mitochondrial movement, quarantines damaged mitochondria, and promotes their clearance by autophagosomal engulfment.[70] Alternatively, Parkin-mediated outer mitochondrial membrane protein ubiquitination is recognized by ubiquitin-binding adaptor proteins such as SQSTM1/p62, which accumulate on depolarized mitochondria and facilitate the recruitment of damaged mitochondria to autophagosomes through interactions with MAP1LC3B-II.[30, 69, 70] In this study, DMSO treatment caused a dose-dependent increase in the expression of several genes/proteins involved in autophagy initiation, phagophore elongation, and autophagosome formation, indicating an increase in autophagosome formation and accumulation (Fig. 3, 5). Furthermore, we investigated autolysosomal maturation and degradation by analyzing the expression levels of SQSTM1/p62, which is normally degraded by autolysosomes after incorporation into autophagosomes via direct binding to MAP1LC3B-II.[71, 72] Unsurprisingly, we observed that DMSO treatment resulted in a dose-dependent increase in the expression of SQSTM1/p62, indicating impaired autophagosomal maturation and degradation. The autophagosome accumulation observed in DMSO-treated embryos was mainly caused by blockage of autophagic flux, rather than by induction of autophagy. There have been several reports demonstrating that blockage of autophagic flux or impaired autophagic degradation is associated with cell death.[73, 74] Therefore, our results indicate that the cytotoxic effects of DMSO in preimplantation embryos are caused, at least in part, by the blockage of autophagic flux.
To assess whether withdrawing DMSO supplementation could rescue DMSO-mediated embryo toxicity, we transferred DMSO-exposed embryos into fresh KSOM medium. As shown in Fig. S2, we found that withdrawing 2% DMSO supplementation within 24 h significantly increased embryonic survival and hatching rates, decreased apoptosis, and ameliorated abnormal development of mouse embryos exposed to DMSO. However, embryos exposed to DMSO longer than 48 h showed markedly inhibited developmental competency at both 1% or 2% DMSO concentrations, indicating that both of exposure time and DMSO concentration are critical factors for embryonic developmental arrest.
In conclusion, DMSO treatment resulted in mitochondria-dependent apoptosis in mouse preimplantation embryos: this may be due to the induction of oxidative/ER stress, mitochondrial Ca2+ overload, or blockage of autophagic flux (Fig. 6I, J). DMSO also disrupted the expression of genes associated with blastocyst developmental competence and consequently decreased the implantation rate and the number of full-term pups. To the best of our knowledge, this is the first report to study the underlying mechanisms of DMSO-induced cytotoxicity in mouse preimplantation embryos. Therefore, our experimental findings will provide a significant contribution to finding effective protective agents to combat DMSO-mediated reproductive toxicity for application in human embryos in the near future.
Materials and Methods
Animals
Mice were housed in wire cages at 22±1 °C under a 12 h light-dark cycle with 50% humidity. All experiments were conducted in accordance with the Konkuk University Guide for the Care and Use of Laboratory Animals (IACUC approval number: KU15153).
Measurement of osmolarity in KSOM supplemented with DMSO
DMSO (Sigma-Aldrich, D2650) was added to KSOM at concentrations of 0%, 0.5%, 1%, and 2% (v/v). Osmolarity levels in each group were determined using the Advanced® Model 3320 Micro-Osmometer (Advanced Instrument, 3320) according to the manufacturer's instructions. Briefly, 20 µL of each KSOM-DMSO mixture was aspirated by the sampler tip, and placed on the instrument's cradle. Measurements were determined automatically using the freezing point of the mixtures; each measurement was independently repeated three times.
Zygote collection and in vitro culture with DMSO supplementation
BDF1 mice (8-12 weeks) were used for collection of zygotes. Female mice were intraperitoneally injected with pregnant mare's serum gonadotropin (PMSG; 5IU), followed by human chorionic gonadotropin (hCG; 5IU) after 48 h; they were then mated with stud BDF1 male mice.[75] At 18 h post hCG injection, the zygotes were isolated from oviducts and washed in HEPES-modified Chatot-Ziomek-Bavister (CZB) medium containing hyaluronidase. Ten zygotes per group were then cultured in 20 µL of KSOM supplemented with 0%, 0.5%, 1%, or 2% DMSO and subjected to further study.
RNA extraction and gene expression analysis
Total RNA was obtained from pools of ten embryos in each DMSO treatment group (0%, 0.5%, 1%, and 2%). RNA was added to 5 µL lysis buffer containing 0.3% IGEPAL® CA-630 (Sigma-Aldrich, I3021) and 0.1% BSA (Sigma-Aldrich, A3311), and then directly frozen-thawed in liquid nitrogen.[76] cDNA was synthesized from the RNA samples by using the QuantiTect® Reverse Transcription Kit (Qiagen, 205313) in a final volume of 20 µL, according to the manufacturer's instructions. RT-qPCR was run three times on 2 µL of cDNA from each reaction using the SensiFAST™ SYBR Hi-ROX Kit (Bioline, BIO-92005) in the ViiA™ 7 Real-Time PCR System (Thermo Fisher Scientific, ABI ViiA 7). Using the 2-ddCt method by normalizing with H2afz mRNA expression, relative quantification was analyzed. Detailed genes and primers information is listed in Table S1.
Mitochondrial activity measurement
Measurement of mitochondrial activity by fluorescence was performed as previously described.[77] Embryos were incubated in KSOM supplemented with 500 nM MitoTracker® Orange CMTMROS (Thermo Fisher Scientific, M7510) for 10 min, then washed three times in KSOM. Embryos were fixed in 4% paraformaldehyde for 30 min and were either mounted on glass slides for observation under a confocal microscope, or used for additional immunostaining. The intensity of fluorescence signals indicating mitochondrial activity was analyzed using Image J software (https://imagej.nih.gov/ij/); relative fluorescence was compared between DMSO-treated and control groups.
Relative quantification of mitochondria
To quantify the number of mitochondria, morula stage embryos, which were cultured with DMSO supplementation at concentrations of 0%, 0.5%, and 1% for 72 h from zygotes, were collected using 5 µL lysis buffer [125 μg/mL proteinase K, 100 mM Tris-HCl (pH 8.3), 100 mM KCl, 0.02% gelatin, 0.45% Tween 20] then lysed by snap-freezing.[78] The mtDNA content was determined by comparing the ratio of mtDNA to nuclear DNA, as measured by real time PCR.[79] Specific mitochondria primer sets are listed in Table S1.
Immunofluorescence
Immunostaining was performed as previously described.[80] Next, embryos were incubated with primary antibodies overnight at 4 °C; secondary antibodies labelled with Alexa488, 568, or 647 were then used to mark the primary antibodies. DNA was stained using TO-PRO®-3 Iodide (Thermo Fisher Scientific, T3605). The embryos were mounted on glass slides using Vectashield® mounting medium (Vector Laboratories, H-1000) and observed under laser confocal microscopes (Olympus, FV1000 and Zeiss, LSM510). All primary antibodies used are summarized in Table S2.
TUNEL assay
TUNEL assay was performed on the embryos using the In Situ Cell Death Detection Kit, TMR Red (Sigma-Aldrich, 12156792910) according to the manufacturer's instructions. After labeling, the embryos were washed three times with 1% BSA-PBS solution, and stained using 4 μM TO-PRO-3 (Thermo Fisher Scientific, T3605) marking DNA. Embryos mounted on glass slides were observed under a laser confocal microscope (Olympus, FV1000). The number of total cells and labeled dead cells were analyzed using Image J software.
Measurement of mitochondrial membrane potential
To measure the mitochondrial membrane potential, embryos were incubated in KSOM supplemented with 2 μg/mL JC-1 Dye (Thermo Fisher Scientific, T3168) for 30 min at 37 °C in a 5% CO2 atmosphere, according to the manufacturer's instructions. After washing in KSOM twice, embryos were placed in a drop of HEPES-CZB media on a glass-bottomed cell culture dish and observed under a laser confocal microscope (Zeiss, LSM 510). Detected fluorescence signals were analyzed using Image J software.
ROS measurement
To visualize ROS levels by fluorescence, embryos were incubated with KSOM supplemented with 5 μM CellRox® Green Reagent (Thermo Fisher Scientific, C10444) for 30 min at 37 °C in a 5% CO2 atmosphere, according to the manufacturer's instructions. Embryos were then washed in 0.1% PVA-PBS buffer three times and fixed in 4% paraformaldehyde. They were then permeabilized in a 1% BSA-PBS solution containing 0.5% Triton-X 100 for 30 min. DNA was stained using 4 μM TO-PRO®-3 Iodide (Thermo Fisher Scientific, T3605), and embryos were mounted on glass slides. All images were obtained using a laser confocal microscope (Zeiss, LSM 510) and analyzed using Image J software.
Calcium level measurement
Calcium levels were determined by loading 5 μM Rhod-2 AM (Thermo Fisher Scientific, R1244) onto embryos treated with 0%, 0.5%, 1%, and 2% DMSO for 30 min at 37 °C in a 5% CO2 atmosphere, according to the manufacturer's instructions. After washing in PBS three times, the embryos were placed in a drop of HEPES-CZB media covered with mineral oil on a glass-bottomed confocal dish and observed under a confocal microscope (Zeiss, LSM510).
Comet assay
Two zygotes were cultured with DMSO for 24 h and analyzed using the OxiSelect™ Comet Assay Kit (Cell Biolabs, STA-351) according to the manufacturer's instructions. Briefly, embryos were combined with OxiSelect™ Comet Agarose at 37 °C and placed onto OxiSelect™ Comet slides to harden. Comet slides with embryo-agarose material were transferred to a small container containing pre-chilled lysis buffer and immersed for 30 min at 4 °C in the dark. After aspirating the lysis buffer, electrophoresis was performed using pre-chilled TBE buffer for 10-15 min at 20 V. Finally, the slides were stained using Vista Green DNA dye, and fluorescence was observed under a microscope (Olympus, BX51). Tail length was analyzed by the Open Comet Plugin (http://www.cometbio.org/documentation.html) using Image J software.[81]
Immunoblotting
Morulas were developed from zygotes by incubating in KSOM supplemented with 0%, 0.5%, and 1% DMSO for 72 h. Thereafter, embryos were pooled in 10 µL of radioimmunoprecipitation assay buffer (RIPA) Cell Lysis Buffer (GenDEPOT, R4100). Lysed samples were denatured by adding 2X Laemmli sample buffer (Sigma-Aldrich, S3401), followed by boiling at 95 °C for 10 min. Immunoblotting was then performed as previously described.[82] HRP-conjugated secondary antibodies were applied to recognize primary antibodies; SuperSignal™ West Femto Maximum Sensitivity Substrate (Thermo Fisher Scientific, 34095) was used to detect HRP, followed by visualization of chemiluminescence using X-ray film. Densitometry analysis was conducted using Image J software. All blotting bands were normalized using ACTB. Detailed information on antibodies is listed in Table S2.
TEM analysis
Sample preparation of morulas for TEM was performed as previously described. [64] After embedding, the samples were sectioned using LKB-III ultratome (Leica, 8801A) with a serial ultrathin grade. Ultrathin sections were stained with uranyl acetate (Ted Pella, 19481) and lead citrate (Ted Pella, 19312) and examined using an electron microscope (Hitachi, H7600) at an accelerating voltage of 100 kV.
Embryo transfer and post-implantation analysis
Blastocysts were developed from zygote by incubating with 0%, 0.5%, and 1% DMSO for 96 h. Thereafter, embryos were transferred into the uteri of 2.5 days postcoitus (dpc) pseudopregnant ICR mice as previously described.[75] Two days after embryo transfer, the implantation rate was detected by sacrificing recipient ICR mice, followed by an intravenous injection with Chicago Sky Blue 6B (Sigma-Aldrich, C8679) as previously described.[75] At 18.5 dpc, the full-term development rate was checked after cesarean section of the recipient mice. Placenta weight, body weight, and crown-rump size were evaluated for individual pups.
Data analysis
All experimental data are presented as the mean ± SEM. Each experiment was performed at least three times and subjected to statistical analysis. Results from three representative experiments are presented in all figures. For statistical analysis, one-way ANOVA was first performed to determine any differences among groups (*: p<0.05; **: p<0.01). Fisher's post hoc test was then performed to determine significant differences between pairs. Values of p<0.05 and p<0.01 were considered significant. Statistical tests were performed using Graph Pad Prism Software (https://www.graphpad.com/scientific-software/prism/).
Abbreviations
ACTB: Actin Beta; Atf2: Activating Transcription Factor 2; Atf4: Activating Transcription Factor 4; Atg3: Autophagy Related 3; Atg4b: Autophagy Related 4B Cysteine Peptidase; Atg5: Autophagy Related 5; Atg6: Autophagy Related 6, Beclin-1; Atg7: Autophagy Related 7; Atp5a1: ATP Synthase, H+ Transporting, Mitochondrial F1 Complex, Alpha Subunit 1; Atp5d: ATP Synthase, H+ Transporting, Mitochondrial F1 Complex, Delta Subunit; Atp5e: ATP Synthase, H+ Transporting, Mitochondrial F1 Complex, Epsilon Subunit; Atp5f1: ATP Synthase, H+ Transporting, Mitochondrial Fo Complex Subunit B1; Atp5k: ATP synthase, H+ transporting, mitochondrial F1F0 complex, subunit E; B3gnt5: BetaGal Beta-1,3-N-Acetylglucosaminyltransferase 5; BAX: BCL2 Associated X, Apoptosis Regulator; BCL2: BCL2, Apoptosis Regulator; Casp3: Caspase 3; Casp9: Caspase 9; Cbr1: Carbonyl Reductase 1; CYCS: Cytochrome C, Somatic; DDIT3: DNA Damage Inducible Transcript 3; DMSO: Dimethyl Sulfoxide; Duox1: Dual Oxidase 1; Duox2: Dual Oxidase 2; Edem1: ER Degradation Enhancing Alpha-Mannosidase Like Protein 1; Eomes: Eomesodermin; ER: Endoplasmic-Reticulum; Gpx1: Glutathione Peroxidase 1; Gpx2: Glutathione Peroxidase 2; Gpx3: Glutathione Peroxidase 3; Gsr (or GR): Glutathione-Disulfide Reductase (or Glutathione reductase); Gss: Glutathione Synthetase; hESC: human embryonic stem cell; HSP90B1: Heat Shock Protein 90 Beta Family Member 1; HSPA5: Heat Shock Protein Family A (Hsp70) Member 5, GRP78, BIP; ICM: Inner cell mass; Immp1l: Inner Mitochondrial Membrane Peptidase Subunit 1; Itpr1: Inositol 1,4,5-Trisphosphate Receptor Type 1; JC-1: 5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazolylcarbocyanine iodide; JNK: JUN N-Terminal Kinase; Jun: Jun Proto-Oncogene, AP-1 Transcription Factor Subunit; LAMP1: Lysosomal Associated Membrane Protein 1; Lrp5: LDL Receptor Related Protein 5; Lrp6: LDL Receptor Related Protein 6; Mapk8: Mitogen-Activated Protein Kinase 8; Mpv17: MPV17, Mitochondrial Inner Membrane Protein; mtDNA: Mitochondrial DNA; Mtfp1: Mitochondrial Fission Process 1; Ncf1: Neutrophil Cytosolic Factor 1; Ncf4: Neutrophil Cytosolic Factor 4; Nox1: NADPH Oxidase 1; Nox2: NADPH Oxidase 2; Nox3: NADPH Oxidase 3; Nox4: NADPH Oxidase 2; p38: MAPK14, Mitogen-Activated Protein Kinase 14; p53: TP53, Tumore Protein 53; PARKIN: Parkin RBR E3 Ubiquitin Protein Ligase; Pink1: PTEN Induced Putative Kinase 1; Rhod2-AM: Dihydrorhod-2 Aetoxymethyl; Rhot2: Ras Homolog Family Member T2; ROS: Reactive Oxygen Species; Sod1: Superoxide Dismutase 1, Soluble; Sod2: Superoxide Dismutase 2, Mitochondrial; SQSTM/p62: Sequestosome 1, p62; SYBR: SYBR green; TE: Trophetoderm; TOM20: Translocase of Outer Mitochondrial Membrane 20; Traf2: Transferrin Receptor 2; Wnt1: Wnt Family Member 1; WNT3A: Wnt Family Member 3A; XBP1: X-Box Binding Protein 1.
Supplementary Material
Supplementary figures and tables.
Movie S1.
Acknowledgements
M.H. K. and J.H.K. designed the study; M.H.K. performed the research; J.H.K. supervised the study; J.D., G.S., H.W.P., H.S., and C.P. analyzed the data; K.M.H., S.G, J.D., and J.H.K wrote the manuscript. All authors reviewed the manuscript.
Grant support
This work was supported by the Science Research Center (2015R1A5A1009701) of the National Research Foundation of Korea, Republic of Korea.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Novak K. Drug facts and Comparisons. St. Louis, Missouri. Wolters Kluwer Health. 2002
2. Kelava T, Ćavar I, Čulo F. Biological actions of drug solvents. Periodicum biologorum. 2011;113:311-20
3. Santos NC, Figueira-Coelho J, Martins-Silva J, Saldanha C. Multidisciplinary utilization of dimethyl sulfoxide: pharmacological, cellular, and molecular aspects. Biochemical pharmacology. 2003;65:1035-41
4. Sankpal UT, Abdelrahim M, Connelly SF, Lee CM, Madero-Visbal R, Colon J. et al. Small molecule tolfenamic acid inhibits PC-3 cell proliferation and invasion in vitro, and tumor growth in orthotopic mouse model for prostate cancer. The Prostate. 2012;72:1648-58
5. Modesitt SC, Parsons SJ. In vitro and in vivo histone deacetylase inhibitor therapy with vorinostat and paclitaxel in ovarian cancer models: does timing matter? Gynecologic oncology. 2010;119:351-7
6. Li SY, Lo AC. Lutein protects RGC-5 cells against hypoxia and oxidative stress. International journal of molecular sciences. 2010;11:2109-17
7. Pegg DE. Principles of cryopreservation. Methods in molecular biology. 2007;368:39-57
8. Salim AS. Role of oxygen-derived free radical scavengers in the management of recurrent attacks of ulcerative colitis: a new approach. The Journal of laboratory and clinical medicine. 1992;119:710-7
9. Salim AS. Allopurinol and dimethyl sulfoxide improve treatment outcomes in smokers with peptic ulcer disease. The Journal of laboratory and clinical medicine. 1992;119:702-9
10. Rosenstein ED. Topical agents in the treatment of rheumatic disorders. Rheumatic diseases clinics of North America. 1999;25:899-918 viii
11. Goto I, Yamamoto-Yamaguchi Y, Honma Y. Enhancement of sensitivity of human lung adenocarcinoma cells to growth-inhibitory activity of interferon alpha by differentiation-inducing agents. British journal of cancer. 1996;74:546-54
12. Abdullaeva GK, Shakimova B. [An evaluation of the efficacy of treating rheumatoid arthritis with preparations for local use]. Revmatologiia. 1989:35-9
13. Shirley SW, Stewart BH, Mirelman S. Dimethyl sulfoxide in treatment of inflammatory genitourinary disorders. Urology. 1978;11:215-20
14. Swanson BN. Medical use of dimethyl sulfoxide (DMSO). Reviews in clinical & basic pharmacology. 1985;5:1-33
15. Smith RS. A comprehensive macrophage-T-lymphocyte theory of schizophrenia. Medical hypotheses. 1992;39:248-57
16. Ikeda Y, Long DM. Comparative effects of direct and indirect hydroxyl radical scavengers on traumatic brain oedema. Acta neurochirurgica Supplementum. 1990;51:74-6
17. Gupta MK, Uhm SJ, Lee HT. Cryopreservation of immature and in vitro matured porcine oocytes by solid surface vitrification. Theriogenology. 2007;67:238-48
18. Cuello C, Sanchez-Osorio J, Alminana C, Gil MA, Perals ML, Lucas X. et al. Effect of the cryoprotectant concentration on the in vitro embryo development and cell proliferation of OPS-vitrified porcine blastocysts. Cryobiology. 2008;56:189-94
19. Gook DA, Choo B, Bourne H, Lewis K, Edgar DH. Closed vitrification of human oocytes and blastocysts: outcomes from a series of clinical cases. Journal of assisted reproduction and genetics. 2016;33:1247-52
20. Zhou D, Shen X, Gu Y, Zhang N, Li T, Wu X. et al. Effects of dimethyl sulfoxide on asymmetric division and cytokinesis in mouse oocytes. BMC developmental biology. 2014;14:28
21. Kitchin KT, Ebron MT. Further development of rodent whole embryo culture: solvent toxicity and water insoluble compound delivery system. Toxicology. 1984;30:45-57
22. Maes J, Verlooy L, Buenafe OE, de Witte PA, Esguerra CV, Crawford AD. Evaluation of 14 organic solvents and carriers for screening applications in zebrafish embryos and larvae. PloS one. 2012;7:e43850
23. Hallare A, Nagel K, Kohler HR, Triebskorn R. Comparative embryotoxicity and proteotoxicity of three carrier solvents to zebrafish (Danio rerio) embryos. Ecotoxicology and environmental safety. 2006;63:378-88
24. Pal R, Mamidi MK, Das AK, Bhonde R. Diverse effects of dimethyl sulfoxide (DMSO) on the differentiation potential of human embryonic stem cells. Archives of toxicology. 2012;86:651-61
25. Goto Y, Noda Y, Mori T, Nakano M. Increased generation of reactive oxygen species in embryos cultured in vitro. Free radical biology & medicine. 1993;15:69-75
26. Liu SX, Athar M, Lippai I, Waldren C, Hei TK. Induction of oxyradicals by arsenic: implication for mechanism of genotoxicity. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:1643-8
27. Perez-Pasten R, Martinez-Galero E, Garduno-Siciliano L, Lara IC, Cevallos GC. Effects of dimethylsulphoxide on mice arsenite-induced dysmorphogenesis in embryo culture and cytotoxicity in embryo cells. Toxicology letters. 2006;161:167-73
28. Sadowska-Bartosz I, Paczka A, Molon M, Bartosz G. Dimethyl sulfoxide induces oxidative stress in the yeast Saccharomyces cerevisiae. FEMS yeast research. 2013;13:820-30
29. Mizushima N. Autophagy: process and function. Genes & development. 2007;21:2861-73
30. Tsukamoto S, Kuma A, Mizushima N. The role of autophagy during the oocyte-to-embryo transition. Autophagy. 2008;4:1076-8
31. Tsukamoto S, Kuma A, Murakami M, Kishi C, Yamamoto A, Mizushima N. Autophagy is essential for preimplantation development of mouse embryos. Science. 2008;321:117-20
32. Ashrafi G, Schwarz TL. The pathways of mitophagy for quality control and clearance of mitochondria. Cell death and differentiation. 2013;20:31-42
33. Dawson KM, Collins JL, Baltz JM. Osmolarity-dependent glycine accumulation indicates a role for glycine as an organic osmolyte in early preimplantation mouse embryos. Biology of reproduction. 1998;59:225-32
34. Hadi T, Hammer MA, Algire C, Richards T, Baltz JM. Similar effects of osmolarity, glucose, and phosphate on cleavage past the 2-cell stage in mouse embryos from outbred and F1 hybrid females. Biology of reproduction. 2005;72:179-87
35. Kamjoo M, Brison DR, Kimber SJ. Apoptosis in the preimplantation mouse embryo: effect of strain difference and in vitro culture. Molecular reproduction and development. 2002;61:67-77
36. Levine B. Eating oneself and uninvited guests: autophagy-related pathways in cellular defense. Cell. 2005;120:159-62
37. Lemasters JJ. Selective mitochondrial autophagy, or mitophagy, as a targeted defense against oxidative stress, mitochondrial dysfunction, and aging. Rejuvenation research. 2005;8:3-5
38. Narendra D, Tanaka A, Suen DF, Youle RJ. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. The Journal of cell biology. 2008;183:795-803
39. Geisler S, Holmstrom KM, Skujat D, Fiesel FC, Rothfuss OC, Kahle PJ. et al. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nature cell biology. 2010;12:119-31
40. Matsuda N, Sato S, Shiba K, Okatsu K, Saisho K, Gautier CA. et al. PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. The Journal of cell biology. 2010;189:211-21
41. Narendra D, Kane LA, Hauser DN, Fearnley IM, Youle RJ. p62/SQSTM1 is required for Parkin-induced mitochondrial clustering but not mitophagy; VDAC1 is dispensable for both. Autophagy. 2010;6:1090-106
42. Tanida I. Autophagy basics. Microbiology and immunology. 2011;55:1-11
43. Hirota Y, Yamashita S, Kurihara Y, Jin X, Aihara M, Saigusa T. et al. Mitophagy is primarily due to alternative autophagy and requires the MAPK1 and MAPK14 signaling pathways. Autophagy. 2015;11:332-43
44. He C, Klionsky DJ. Regulation mechanisms and signaling pathways of autophagy. Annual review of genetics. 2009;43:67-93
45. Zhang JY, Jiang H, Gao W, Wu J, Peng K, Shi YF. et al. The JNK/AP1/ATF2 pathway is involved in H2O2-induced acetylcholinesterase expression during apoptosis. Cellular and molecular life sciences: CMLS. 2008;65:1435-45
46. Lee HC, Sheu SH, Yen HW, Lai WT, Chang JG. JNK/ATF2 pathway is involved in iodinated contrast media-induced apoptosis. American journal of nephrology. 2010;31:125-33
47. Park MR, Gurunathan S, Choi YJ, Kwon DN, Han JW, Cho SG. et al. Chitosan nanoparticles cause pre- and postimplantation embryo complications in mice. Biology of reproduction. 2013;88:88
48. Strumpf D, Mao CA, Yamanaka Y, Ralston A, Chawengsaksophak K, Beck F. et al. Cdx2 is required for correct cell fate specification and differentiation of trophectoderm in the mouse blastocyst. Development. 2005;132:2093-102
49. Lade AG, Monga SP. Beta-catenin signaling in hepatic development and progenitors: which way does the WNT blow? Developmental dynamics: an official publication of the American Association of Anatomists. 2011;240:486-500
50. Tepekoy F, Akkoyunlu G, Demir R. The role of Wnt signaling members in the uterus and embryo during pre-implantation and implantation. Journal of assisted reproduction and genetics. 2015;32:337-46
51. Mager WH, de Boer AH, Siderius MH, Voss HP. Cellular responses to oxidative and osmotic stress. Cell stress & chaperones. 2000;5:73-5
52. Bell CE, Lariviere NM, Watson PH, Watson AJ. Mitogen-activated protein kinase (MAPK) pathways mediate embryonic responses to culture medium osmolarity by regulating Aquaporin 3 and 9 expression and localization, as well as embryonic apoptosis. Human reproduction. 2009;24:1373-86
53. Soberman RJ. Series Introduction: The expanding network of redox signaling: new observations, complexities, and perspectives. Journal of Clinical Investigation. 2003;111:571
54. Geiszt M, Kopp JB, Várnai P, Leto TL. Identification of renox, an NAD (P) H oxidase in kidney. Proceedings of the National Academy of Sciences. 2000;97:8010-4
55. Ago T, Kitazono T, Ooboshi H, Iyama T, Han YH, Takada J. et al. Nox4 as the major catalytic component of an endothelial NAD (P) H oxidase. Circulation. 2004;109:227-33
56. Shiose A, Kuroda J, Tsuruya K, Hirai M, Hirakata H, Naito S. et al. A novel superoxide-producing NAD (P) H oxidase in kidney. Journal of Biological Chemistry. 2001;276:1417-23
57. Dupuy C, Pomerance M, Ohayon R, Noël-Hudson M-S, Dème D, Chaaraoui M. et al. Thyroid oxidase (THOX2) gene expression in the rat thyroid cell line FRTL-5. Biochemical and biophysical research communications. 2000;277:287-92
58. Bain NT, Madan P, Betts DH. The early embryo response to intracellular reactive oxygen species is developmentally regulated. Reproduction, fertility, and development. 2011;23:561-75
59. Yoon SB, Choi SA, Sim BW, Kim JS, Mun SE, Jeong PS. et al. Developmental competence of bovine early embryos depends on the coupled response between oxidative and endoplasmic reticulum stress. Biology of reproduction. 2014;90:104
60. Wu LL, Russell DL, Norman RJ, Robker RL. Endoplasmic reticulum (ER) stress in cumulus-oocyte complexes impairs pentraxin-3 secretion, mitochondrial membrane potential (DeltaPsi m), and embryo development. Molecular endocrinology. 2012;26:562-73
61. Kim JS, Song BS, Lee KS, Kim DH, Kim SU, Choo YK. et al. Tauroursodeoxycholic acid enhances the pre-implantation embryo development by reducing apoptosis in pigs. Reproduction in domestic animals = Zuchthygiene. 2012;47:791-8
62. Zhang JY, Diao YF, Oqani RK, Han RX, Jin DI. Effect of endoplasmic reticulum stress on porcine oocyte maturation and parthenogenetic embryonic development in vitro. Biology of reproduction. 2012;86:128
63. Zhang JY, Diao YF, Kim HR, Jin DI. Inhibition of endoplasmic reticulum stress improves mouse embryo development. PloS one. 2012;7:e40433
64. La Rovere RM, Roest G, Bultynck G, Parys JB. Intracellular Ca(2+) signaling and Ca(2+) microdomains in the control of cell survival, apoptosis and autophagy. Cell calcium. 2016;60:74-87
65. Parks JC, McCallie BR, Janesch AM, Schoolcraft WB, Katz-Jaffe MG. Blastocyst gene expression correlates with implantation potential. Fertility and sterility. 2011;95:1367-72
66. Kroemer G, Mariño G, Levine B. Autophagy and the integrated stress response. Molecular cell. 2010;40:280-93
67. Nikoletopoulou V, Markaki M, Palikaras K, Tavernarakis N. Crosstalk between apoptosis, necrosis and autophagy. Biochimica et biophysica acta. 2013;1833:3448-59
68. Ding WX, Ni HM, Gao W, Hou YF, Melan MA, Chen X. et al. Differential effects of endoplasmic reticulum stress-induced autophagy on cell survival. The Journal of biological chemistry. 2007;282:4702-10
69. Choi YJ, Gurunathan S, Kim D, Seok JH, Park WJ, Cho SG. et al. Rapamycin ameliorates chitosan nanoparticle-induced developmental defects of preimplantation embryos in mice. Oncotarget. 2016
70. Wang X, Winter D, Ashrafi G, Schlehe J, Wong YL, Selkoe D. et al. PINK1 and Parkin target Miro for phosphorylation and degradation to arrest mitochondrial motility. Cell. 2011;147:893-906
71. Bjorkoy G, Lamark T, Brech A, Outzen H, Perander M, Overvatn A. et al. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. The Journal of cell biology. 2005;171:603-14
72. Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen H. et al. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. The Journal of biological chemistry. 2007;282:24131-45
73. Orecna M, De Paoli SH, Janouskova O, Tegegn TZ, Filipova M, Bonevich JE. et al. Toxicity of carboxylated carbon nanotubes in endothelial cells is attenuated by stimulation of the autophagic flux with the release of nanomaterial in autophagic vesicles. Nanomedicine: nanotechnology, biology, and medicine. 2014;10:939-48
74. Wan B, Wang ZX, Lv QY, Dong PX, Zhao LX, Yang Y. et al. Single-walled carbon nanotubes and graphene oxides induce autophagosome accumulation and lysosome impairment in primarily cultured murine peritoneal macrophages. Toxicology letters. 2013;221:118-27
75. Andras N, Marina G, Kristina V, Richard B. Manipulating the mouse embryo: a laboratory manual. Cold Spring Harbor Laboratory Press, Woodbury, NY, USA. 2003
76. Le AV-P, Huang D, Blick T, Thompson EW, Dobrovic A. An optimised direct lysis method for gene expression studies on low cell numbers. Scientific reports. 2015;5:12859
77. Manser RC, Houghton FD. Ca2+-linked upregulation and mitochondrial production of nitric oxide in the mouse preimplantation embryo. Journal of cell science. 2006;119:2048-55
78. Sakurai T, Watanabe S, Kamiyoshi A, Sato M, Shindo T. A single blastocyst assay optimized for detecting CRISPR/Cas9 system-induced indel mutations in mice. BMC biotechnology. 2014;14:69
79. Rooney JP, Ryde IT, Sanders LH, Howlett EH, Colton MD, Germ KE. et al. PCR based determination of mitochondrial DNA copy number in multiple species. Mitochondrial Regulation: Methods and Protocols. 2015:23-38
80. Bui H-T, Yamaoka E, Miyano T. Involvement of histone H3 (Ser10) phosphorylation in chromosome condensation without Cdc2 kinase and mitogen-activated protein kinase activation in pig oocytes. Biology of reproduction. 2004;70:1843-51
81. Gyori BM, Venkatachalam G, Thiagarajan P, Hsu D, Clement M-V. OpenComet: an automated tool for comet assay image analysis. Redox biology. 2014;2:457-65
82. Das J, Kang M-H, Kim E, Kwon D-N, Choi Y-J, Kim J-H. Hexavalent chromium induces apoptosis in male somatic and spermatogonial stem cells via redox imbalance. Scientific reports. 2015;5:13921
Author contact
![]() Corresponding author: Jin-Hoi Kim, PhD., Tel.: +82-2-450-3687, Fax: +82-2-458-5414, E-mail address: jhkim541ac.kr (J-H Kim)
Corresponding author: Jin-Hoi Kim, PhD., Tel.: +82-2-450-3687, Fax: +82-2-458-5414, E-mail address: jhkim541ac.kr (J-H Kim)