Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2018; 8(1):109-123. doi:10.7150/thno.20356 This issue Cite
Research Paper
PrLZ increases prostate cancer docetaxel resistance by inhibiting LKB1/AMPK-mediated autophagy
Jin Zeng*, Wei Liu*, Yi-Zeng Fan, Da-Lin He, Lei Li ![]()
Department of Urology, the First Affiliated Hospital of Xi'an Jiaotong University, Xi'an 710061, China
* These two authors contributed equally to this work.
Received 2017-3-31; Accepted 2017-9-25; Published 2018-1-1
Abstract

Rationale: Docetaxel-mediated chemotherapy is the first-line standard approach and has been determined to show a survival advantage for metastatic castration-resistant prostate cancer (mCRPC) patients. However, a substantial proportion of patients eventually becomes refractory due to drug resistance. The detailed mechanisms remain unclear. We have previously reported that Prostate Leucine Zipper (PrLZ), a specific oncogene of prostate cancer (PCa), promotes PCa cell growth at the castration-resistant stage, thus suggesting a vital role of PrLZ in the progression of CRPC. In this study, we aimed to investigate the role of PrLZ in docetaxel resistance in PCa, focusing on PrLZ-regulating autophagy pathway.
Methods: Human PCa PC3, LNCaP and C4-2 cell lines were used as the model system in vitro and PCa xenografts and PrLZ-knockout mice were used as the model system in vivo. Docetaxel-induced cell death and apoptosis in PCa were determined by MTT and flow cytometry assay. The role of PrLZ on the regulation of autophagy and liver kinase B1/AMP-activated protein kinase (LKB1/AMPK) signaling pathway was analyzed using immunoblotting, immunoprecipitation, siRNA silencing and plasmid overexpression.
Results: PrLZ increased docetaxel-mediated drug resistance both in vitro and in vivo. Mechanistic dissection revealed that PrLZ interacted with LKB1 and further inhibited the activation of LKB1/AMPK signals, which negatively contributed to the induction of autophagy. Moreover, PrLZ/LKB1-mediated autophagy conferred resistance to docetaxel-induced cell death and apoptosis both in vitro and in vivo.
Conclusion: These findings identify a novel role of PrLZ in autophagy manipulation and provide new insight into docetaxel chemoresistance in PCa, suggesting a new strategy for treating mCRPC by targeting this newly identified signaling pathway.
Keywords: PrLZ, Prostate cancer, Docetaxel, Autophagy, LKB1.
Introduction
Prostate cancer (PCa) is one of the most lethal malignancies, accounting for an estimated 26,120 deaths in the United States [1]. Chemotherapy with docetaxel, a member of the taxane family, is used as the first-line standard chemotherapy treatment and has been determined to show a survival advantage in metastatic castration-resistant prostate cancer (mCRPC) patients [2]. However, a substantial proportion of patients with mCRPC treated with docetaxel eventually become refractory and progress due to the development of drug resistance. Numerous factors, including the tumor microenvironment, have been recognized [3]. Nevertheless, it is controversial rather than conclusive. Hence, there is an urgent need to seek out the potential reason for docetaxel chemoresistance.
The factors contributing to chemoresistance are various, but one crucial factor is the capability of cancer cells to manage stress. Macroautophagy (hereafter referred to as autophagy), an evolutionarily conserved process for the degradation of aging organelles and damaged proteins in response to diverse stimuli, is considered to be a favored survival strategy employed by cancer cells to adapt to different types of stress [4]. Induction of autophagy has been identified to facilitate the resistance of cancer cells to chemotherapeutic drugs, and inhibition of autophagy could be therapeutically beneficial in some cancer patients [5]. However, it has also been proposed that autophagy functions as a cell death mechanism and may even have differential effects, depending on the cell type or the genetic background [6, 7]. Thus, defining the roles of autophagy in chemoresistance and the mechanisms involved is critical for enhancing the efficiency of chemotherapy and developing novel anticancer strategies.
Prostate Leucine Zipper (PrLZ), also known as PC-1, is a member of the tumor protein D52 family and is localized at chromosome 8q21.1, one of the most amplified regions in PCa. PrLZ is specifically expressed in prostate tissues, and it is frequently overexpressed in advanced PCa tissues [8]. Recent studies have demonstrated that PrLZ exhibits oncogenic properties, which might be correlated with PCa progression [9, 10]. Our previous study revealed that PrLZ expression was associated with the proliferation and invasion of PCa cells and could protect PCa cells from androgen deprivation-induced apoptosis [11]. Additionally, we found that increased PrLZ-mediated androgen receptor transactivation promoted PCa cell growth at the castration-resistant stage, suggesting a vital role of PrLZ in the progression of CRPC [12]. Recently, another group reported that suppression of PrLZ sensitized PCa cells to ionizing radiation by attenuating DNA damage repair and inducing autophagic cell death [13]. However, the regulatory molecular mechanism of PrLZ in autophagy, especially the role in PCa chemoresistance, has not been fully elucidated.
In the present study, we found that PrLZ suppressed autophagy by regulating liver kinase B1/AMP-activated protein kinase (LKB1/AMPK) signaling in PCa. Mechanistically, PrLZ-LKB1 interaction negatively contributed to the induction of autophagy. More importantly, PrLZ/LKB1-mediated inhibition of autophagy conferred resistance to docetaxel-induced apoptosis in PCa cells. A possible PrLZ-LKB1 interaction-mediated inhibition of the autophagy pathway is proposed, which may provide new insight into docetaxel chemoresistance in PCa.
Materials and Methods
Reagents, Antibodies, and Plasmids
Rabbit monoclonal antibodies against microtubule-associated protein 1 light chain 3 Ⅱ/Ⅰ (LC3-Ⅱ/Ⅰ) (4445), autophagy-associated gene 5 (ATG5) (4445), p62 (5114), phosphorylated AMPK (9957), total AMPK (9957), phosphorylated mammalian target of rapamycin (mTOR) (9862), total mTOR (9862), phosphorylated LKB1 (Ser428, 3482), phosphorylated transforming growth factor beta-activated kinase 1 (TAK1) (Thr 184/187, 4508), androgen receptor (AR) (5153) and green fluorescent protein (GFP) (2956) were purchased from Cell Signaling Technology. Mouse monoclonal Flag antibody (F1804), the Nuclei EZ Prep Nuclei Isolation Kit (NUC101), chloroquine diphosphate salt (CQ, C6628) and bafilomycin A1 (Baf A1, B1793) were purchased from Sigma. Mouse antibody against total-LKB1 (sc-32245) and rabbit antibody against Ca2+/calmodulin-dependent protein kinase kinase-β (CaMKKβ) (sc-9629) were purchased from Santa Cruz Biotechnology. Mouse monoclonal β-actin antibody (cw0096A) was purchased from CWBIO. Mouse monoclonal GFP antibody (12A6) was obtained from the Developmental Studies Hybridoma Bank. Rabbit polyclonal histone H1 antibody (17510-1-AP) was purchased from Proteintech. Dynabeads Protein G (10004D) and TRIzol reagent were purchased from Invitrogen. Docetaxel (ab141248) and antibodies against Ste20-related adaptor protein (STRAD, ab192879) and mouse protein 25 (MO25, ab51132) were purchased from Abcam. Rabbit polyclonal PrLZ antibody was kindly provided by Professor Ruoxiang Wang (Department of Medicine, Cedars-Sinai Medical Center, Los Angeles, CA, USA). The GFP-LKB1 plasmid was a kind gift from Junying Yuan (Addgene plasmid # 21147). GFP-red fluorescent protein (RFP)-LC3 adenovirus was purchased from HANBIO. pcDNA3-Flag-PrLZ plasmid was constructed according to standard protocols. The PrimerScript RT reagent kit and SYBR Green Master Mix were purchased from Takara.
Cell culture, siRNA transfection and establishment of stable clone cells
Human PCa C4-2, LNCaP and PC3 cell lines were maintained in RPMI-1640 medium (Gibco) supplemented with 10% fetal bovine serum (Gibco) at 37°C in a humidified atmosphere with 5% CO2. The 293T cell line was culture in Dulbecco's modified Eagle's medium (Gibco) supplemented with 10% fetal bovine serum (Gibco). PC3 stably transfected with PrLZ (PC3-PrLZ), C4-2 stably transfected with sh-PrLZ (C4-2-sh-PrLZ), and their respective corresponding empty vector control sublines (PC3-Vec, C4-2-Sc) were established as previously described [12]. C4-2 docetaxel-resistant (DTX-R) cells were generated by culturing C4-2 cells under increasing DTX concentrations from 10 μM to 30 μM (every 30 d) for 3 months. After generation, the cells were maintained in medium with 10 μM DTX. All cell lines were used for fewer than 6 months and were routinely screened for mycoplasma every 4 weeks. No genotypic authentication was conducted. Each cell line was used in early passages. ATG5 siRNA (si-ATG5) sequence: GAAGTTTGTCCTTCTGCTA, LKB1 siRNA (si-LKB1) sequence: GAAGTTTGTCCTTCTGCTA and the corresponding negative control (si-NC) were designed and synthesized by Ribobio. Cell transfection using Lipofectamine 2000 transfection reagent (Life Technologies, 11668-027) was performed according to the manufacturer's protocol.
Construction of PrLZ knockout mice and tissue extraction
All the mouse experiments were performed under protocols approved by the Institutional Animal Care and Use Committee of Xi'an Jiaotong University. All the animal experiments were performed in adherence with the NIH Guidelines on the Use of Laboratory Animals. We generated PrLZ knockout (KO) mice that lacked PrLZ in the prostate (PrLZ-CKO; C57/B6) by mating loxP site- PrLZ female transgene (PrLZ flox/flox; C57/B6) mice with PB-Cre+/+ male (C57BL/6). Wild-type and mutant KO male mice were treated under normal or fasting conditions for 24 h before being euthanized, and the prostate tissues of each group were isolated. Total protein of the prostate tissue was extracted and used for subsequent western blotting.
Western blot analysis
The cells were washed with phosphate-buffered saline (PBS) and lysed with Radioimmunoprecipitation assay (RIPA) buffer containing proteinase inhibitors and phosphatase inhibitors on ice. After centrifugation, 30 μg of clarified cell lysate was electrophoresed in sodium dodecyl sulfate (SDS)-polyacrylamide (10% or 15%) and transferred to polyvinylidene fluoride membranes. The membranes were blocked with 5% nonfat milk for 1 h at room temperature and incubated with primary antibody at 4°C overnight. After being washed with TBST (Tris-buffered saline with Tween) buffer, the membranes were incubated with horseradish peroxidase-conjugated secondary antibody for 1 h. The protein immunoreactive signals were detected using an ECL detection system, followed by exposure to X-ray film.
Transmission electron microscopy
Treated cells were washed with PBS buffer and fixed with glutaraldehyde (pH 7.4). The cells were then treated with 1% osmium tetroxide and dehydrated in a graded series of ethanol. Subsequently, the cells were embedded in Ultracut (LEICA ULTRACUT R, Bensheim, Germany) and cut into 60 nm sections, followed by uranyl acetate and lead citrate staining. Finally, the ultrathin sections were observed under a HITACHI H07650 transmission electron microscope (HITACHI, Ltd, Japan).
Confocal fluorescence microscopy and Immunofluorescence staining
Cells were cultured on slides, transiently transfected with GFP-RFP-LC3 adenovirus and treated as indicated. After being washed with PBS buffer, the cells were fixed with 4% paraformaldehyde. The slides were then blocked with glycerol, and the localization of LC3 puncta were visualized with a confocal fluorescence microscope.
Immunofluorescence staining of PrLZ and LKB1 was performed as previously described [11]. Briefly, after permeabilization with 0.1% Triton X-100, the cells were incubated with primary antibodies (diluted at 1:100) at 4°C overnight and subsequently with fluorescein isothiocyanate (FITC)/Cy3 secondary antibodies for 1 h at room temperature. Then, the cells were stained with DAPI dye for 5 min and blocked with glycerol. Finally, the cells were analyzed using a fluorescence microscope.
Quantitative real-time PCR
Total RNA of PCa cells was extracted using TRIzol reagent following the manufacturer's protocol. Subsequently, the complementary DNA (cDNA) was synthesized using a PrimerScript RT reagent kit. In addition, the relative levels of the target gene mRNA transcript were measured using qRT-PCR. The sequences of the primers for PrLZ and AR PCR amplification were 5'-GAGATGGACTTATATGAGGACTAC-3' (forward) and 5'-TTGCTGCTAACACTTGAGAC-3' (reverse) (PrLZ, 286 bp), and 5'-CCAGGGACCATGTTTTGCC-3' (forward) and 5'-CGAAGACGACAAGATGGACAA-3' (reverse) (AR, 226 bp), respectively. All the experiments were performed in triplicate. Human β-actin cDNA was amplified as an internal control.
Co-immunoprecipitation
The treated cells were harvested and disrupted in IP buffer (50 mM Tris HCl, 150 mM NaCl, 1 mM ethylenediaminetetraacetic acid (EDTA), 1% Triton X-100) with protease inhibitors and phosphatase inhibitors. The cell lysates (1 mg/mL) were then incubated with Flag or GFP antibody with gentle rocking overnight at 4°C, followed by incubation with Protein G Dynabeads for 3 h at 4°C. After washing twice with IP buffer, the proteins on the Dynabeads were eluted by boiling at 95°C for 5 min in SDS sample loading buffer and separated by SDS-PAGE for western blotting.
Nuclear protein extraction
After trypsinization, the cells were washed with PBS and collected using Nuclei EZ Prep Nuclei Isolation Kit according to the manufacturer's instructions. The protein concentration was normalized using the BCA assay. The remaining cytoplasmic and nuclear extracts were subjected to western blotting as previously described [12].
MTT assay
Briefly, 5×103 cells were seeded onto 96-well plates and treated with docetaxel for 24 h. After treatment, 20 μL of MTT (3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide) dye solution was added to each well with 180 μL of medium and incubated at 37°C for 4 h. Subsequently, the cells were lysed with dimethyl sulfoxide to dissolve the formazan crystals. Then, the optical density of each well was measured at a wavelength of 570 nm using a 96-well microplate reader (Bio-Rad, Hercules, CA, USA).
Flow cytometry analysis
The PCa cells were exposed to docetaxel for 24 h. After washing with PBS buffer, the cells were collected and stained with FITC-conjugated Annexin V and propidium iodide (PI) according to the manufacturer's protocol. The apoptotic cells were then analyzed using flow cytometry (BD FACScan flow cytometer, BD Biosciences). The experiments were performed in triplicate.
PCa xenograft animal model
Four-week-old male nude mice were purchased from the Laboratory Animal Center of Xi'an Jiaotong University. The administration of the animals was approved by the Institutional Animal Care and Use Committee of Xi'an Jiaotong University. Briefly, PC3-Vec or PC3-PrLZ cells (5×106) were subcutaneously injected into two flanks of nude mice. When the volume of the xenografts reached 150 mm3, the mice were randomly divided into four groups: PC3-Vec and PC3-PrLZ groups with solvent or docetaxel treatment (15 mg/kg/week via intraperitoneal injection, 5 mice per group) [14]. The body weights of the nude mice and tumor size were measured every other day, and the tumor volume was calculated using the following formula: tumor volume (mm3) = π/6 × (length) × (width)2. After one month, the mice were euthanized, and the tumors were weighed and prepared for subsequent experiments.
Immunohistochemistry
Briefly, fresh tissues were fixed with 4% paraformaldehyde, embedded with paraffin and cut into 4 μm sections. The sections were then dewaxed and rehydrated, followed by antigen retrieval for 5 min. The sections were blocked with endogenous peroxidase for 30 min and then incubated with primary antibody against PrLZ or LKB1 at 4°C overnight. Subsequently, the slides were washed with PBS and incubated with HRP-conjugated secondary antibody for 1 h. The slides were then colored with diaminobenzidine (DAB), followed by hematoxylin counterstaining. Finally, each slide was observed under a microscope (Olympus Optical Co, Tokyo, Japan).
Statistical analysis
The results are presented as the mean±SD. All statistical analyses were performed using GraphPad Prism 5.2 software. Differences in each group were analyzed by one-way ANOVA, followed by Dunnett's t-test for separate comparisons. When the comparison involved only two groups, Student's t-test was used. P < 0.05 was determined to represent a significant difference.
Data availability
All data are available within the articles as figure source data or Supplementary Information Files, or from the authors upon reasonable request.
Results
PrLZ inhibits autophagosome formation in PCa cells
To investigate the potential link between PrLZ and autophagy in PCa, the increasing ratio of cellular LC3-II to LC3-I was used as an indicator of autophagosome formation [15]. The basal level of PrLZ and autophagic-related proteins in PCa cells were first assayed (Supplementary Fig. S1A). The results revealed that LC3-Ⅱ levels in the prostate were significantly higher in PrLZ knockout mice than in wild-type mice following starvation (Fig. 1A). Exogenous overexpression of PrLZ in PCa PC3 cells decreased LC3-Ⅱ levels (Fig. 1B), while stable knockdown of PrLZ increased LC3-Ⅱ levels and the LC3-Ⅱ/ LC3-Ⅰ ratio when compared with control C4-2 cells (Fig. 1C), suggesting the possible regulation of autophagosome formation by PrLZ. To further confirm the regulation of autophagic flux by PrLZ, bafilomycin A1 (Baf A1), an inhibitor of lysosomal acidification, was used. As expected, knocking down PrLZ in the presence of Baf A1 resulted in higher levels of LC3-Ⅱ and a higher LC3-Ⅱ/ LC3-Ⅰ ratio compared with knocking down PrLZ alone or Baf A1 treatment alone (Fig. 1B and C). Similar results were obtained after pretreatment with chloroquine or NH4Cl, two other well-known lysosome inhibitors (Fig. 1D and E, Supplementary Fig. S1B and C), which indicated that the increase in LC3-II levels by knocking down PrLZ was due to an increase in production rather than decreased recycling of LC3-II. To examine autophagic clearance, the levels of p62, a classical macroautophagy substrate, were then assayed. Interestingly, neither PrLZ overexpression nor knockdown affected p62 protein levels compared with the respective negative controls (Supplementary Fig. S1B-G).
PrLZ inhibits autophagosome formation in PCa. (A) Wild-type (WT) or knockout (KO) PrLZ mice underwent food starvation (ST) for 24 h. Following euthanasia, the prostate tissues were analyzed by western blotting for levels of LC3-Ⅰ, LC3-Ⅱ, PrLZ and the LC3-Ⅱ/ LC3-Ⅰ ratio. The expressions of LC3-Ⅱ, LC3-Ⅰ, PrLZ and the LC3-Ⅱ/ LC3-Ⅰ ratio in PC3 cells stably transfected with PrLZ or vector (Vec) and C4-2 cells stably transfected with PrLZ shRNA (sh-PrLZ) or scramble (Sc) in the presence or absence of 10 nM bafilomycin A1 (Baf A1) (B, C) or 50 μM chloroquine (CQ) (D, E) were analyzed by western blotting. (F) Transmission electron microscopy images show the distinctive double membrane structure of autophagic vesicles in PC3 cells stably transfected with PrLZ or vector. The arrows indicate the autophagic vesicles. The scale bars represent 1 μm (low magnification) and 400 nm (high magnification). Representative phase contrast, fluorescence photomicrographs (G) and the quantification (H) of GFP-RFP-LC3 puncta in PC3 cells stably transfected with PrLZ are shown. Yellow puncta denote autophagic vesicle structures. The scale bars represent 10 μm. The results represent the mean ± S.D. of 3 independent experiments. *, P<0.05.
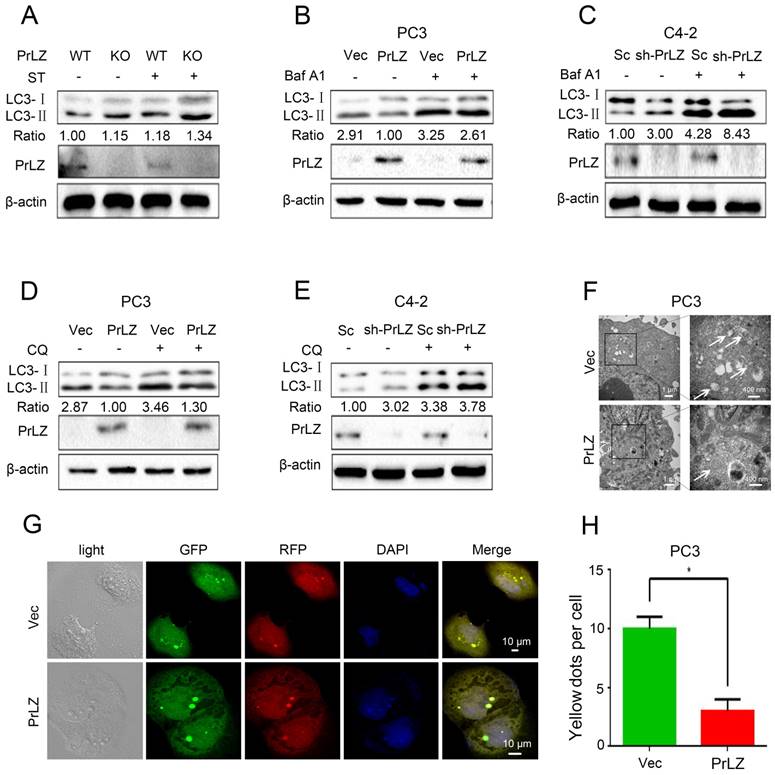
Transmission electron microscopy images consistently revealed a significant decrease in the autophagic double-membrane compartments containing lamellar structures in PrLZ-overexpressing PC3 cells compared with vector control cells (Fig. 1F). More importantly, a pronounced decrease in fluorescent GFP-RFP yellow LC-3 puncta was observed in PrLZ-overexpressing PC3 cells compared with vector control cells (Fig. 1G and H), while knocking down PrLZ significantly increased the number of GFP-RFP yellow LC-3 puncta (Supplementary Fig. S1H and I). Taken together, the results shown in Fig. 1A-H and Supplementary Fig. S1A-I indicated that PrLZ inhibited the autophagic flux.
PrLZ is involved in glucose starvation-mediated autophagy in PCa cells
Because PrLZ was potentially involved in the formation of autophagosomes, we next aimed to explore its role in autophagy. Glucose starvation, a classic trigger of autophagy, was thus used as the model system. As shown in Fig. 2A, glucose starvation resulted in the elevation of LC3-II levels in a time-dependent manner. Interestingly, significant downregulations of PrLZ and AR were observed after glucose starvation (Fig. 2A, Supplementary Fig. S2A). Furthermore, the mRNA level of PrLZ was also decreased after glucose starvation as determined by quantitative real-time polymerase chain reaction (qRT-PCR) (Fig. 2B). Autophagy is known to deliver cytoplasmic proteins and organelles to lysosomes for degradation. We next examined whether activation of lysosomal degradation contributed to the decrease in PrLZ protein. As shown in Fig. 2C, there was no significant difference in the expression of PrLZ in the presence or absence of chloroquine after glucose starvation. Additionally, cycloheximide, a well-known protein synthesis inhibitor, had no remarkable effect on the LC3-Ⅱ/ LC3-Ⅰ ratio and failed to restore the suppressive effect of PrLZ induced by glucose starvation (Fig. 2D). These observations demonstrated that PrLZ might be transcriptionally regulated by glucose starvation.
PrLZ is involved in glucose starvation-induced autophagy in PCa cells. (A) PCa C4-2 cells underwent glucose starvation (GS) treatment for the indicated time period (0, 6, 12, 24 h). The expression levels of LC3-Ⅰ, LC3-Ⅱ, PrLZ and the LC3-Ⅱ/ LC3-Ⅰ ratio were assayed by western blotting. (B) Under similar conditions, quantification of PrLZ mRNA levels were detected by real-time quantitative PCR. The results represent the mean ± S.D. of 3 independent experiments. *, P < 0.05 and **, P < 0.01. PCa C4-2 cells were treated with glucose starvation (GS) for 24 h in the presence or absence of 50 μM chloroquine (CQ) (C) or 100 nM cycloheximide (CHX) (D). PC3 cells with PrLZ or vector (E, F), and C4-2 cells with PrLZ shRNA (sh-PrLZ) or scramble (Sc) (G) underwent glucose starvation (GS) treatment for 24 h. The expression levels of LC3-Ⅰ, LC3-Ⅱ and PrLZ, and the LC3-Ⅱ/ LC3-Ⅰ ratio were detected by western blotting, and the quantification of PrLZ mRNA levels were detected by real-time quantitative PCR. The results represent the mean ± S.D. of 3 independent experiments. ****, P < 0.0001. (H) Quantification of GFP-RFP-LC3 puncta in PrLZ knockdown (sh-PrLZ) C4-2 cells that underwent glucose starvation (GS) for 24 h. The results represent the mean ± S.D. of 3 independent experiments. *, P < 0.05 and **, P < 0.01.
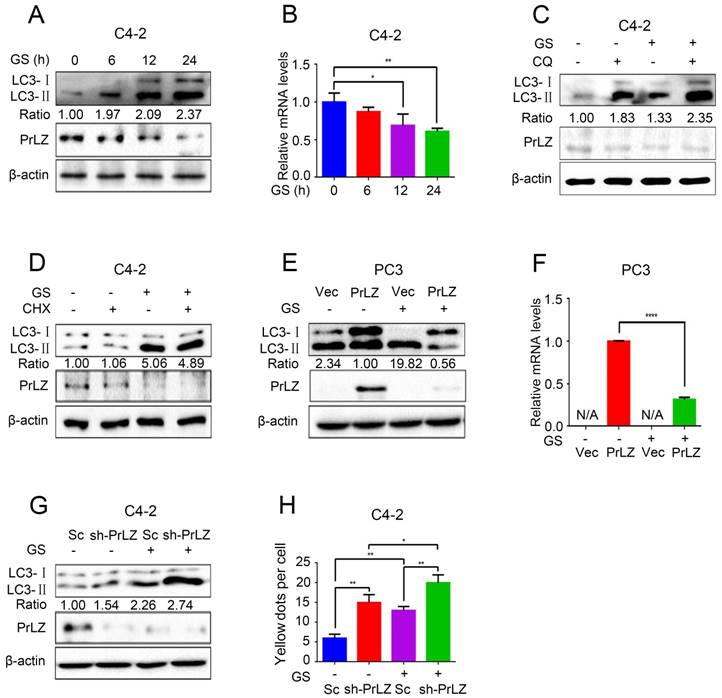
Furthermore, overexpression of exogenous PrLZ partially reversed the glucose starvation-induced increase in the LC3-Ⅱ level and the LC3-Ⅱ/ LC3-Ⅰ ratio (Fig. 2E), and the level of exogenous PrLZ mRNA significantly decreased after glucose starvation treatment (Fig. 2F). By contrast, PrLZ deficiency significantly potentiated the autophagic effect induced by glucose starvation (Fig. 2G and H). Collectively, these findings supported the view that glucose starvation-induced autophagy was partially mediated by the downregulation of PrLZ.
LKB1/AMPK signaling is required for PrLZ-mediated autophagy during glucose deprivation in PCa cells
Next, we aimed to explore the regulatory mechanisms of PrLZ in the autophagic process, focusing on several key autophagy upstream kinases. The mTOR signaling is crucial for autophagosome initiation, with inactivated mTOR inducing autophagy [16]. However, no significant changes in mTOR Ser2448 phosphorylation were observed after overexpressing or knocking down PrLZ (Fig. 3A). It has been reported that autophagy can also be induced by the activation of AMPK, which is a key energy sensor [17]. Our results indicated that overexpression of PrLZ significantly decreased the protein level of phosphorylated-AMPK (Thr172), while knocking down PrLZ activated AMPK (Fig. 3A). We then assayed three AMPK upstream kinases, LKB1, TAK1 and CaMKKβ, for activation upon manipulation of PrLZ. The results showed that phosphorylated-LKB1, instead of TAK1 or CaMKKβ, was inhibited in PrLZ-overexpressing PC-3 cells. Consistently, the phosphorylated-LKB1 was increased in PrLZ knockdown C4-2 cells (Fig. 3A, Supplementary Fig. S3A). The results indicated that PrLZ might negatively regulate LKB1/AMPK signaling. Consistently, a marked increase in the phosphorylation of LKB1 and AMPK and no difference in phosphorylated- and total-mTOR were observed after glucose deprivation in C4-2 cells (Fig. 3B).
LKB1/AMPK activation is required for PrLZ-mediated autophagy in PCa cells. (A) The expressions of LKB1, mTOR and AMPK signaling-related molecules were detected by western blotting in PC3 cells with PrLZ or vector (Vec) and C4-2 cells with PrLZ shRNA (sh-PrLZ) or scramble (Sc). (B) C4-2 cells underwent glucose starvation (GS) at the indicated time points (0, 6, 12, 24 h). The expression levels of LKB1, mTOR and AMPK signaling-related molecules were then detected by western blotting. PC3 cells with PrLZ or vector (C) and C4-2 cells with PrLZ shRNA (sh-PrLZ) or scramble (Sc) (D) underwent glucose starvation (GS) treatment for 24 h. The expressions of LKB1, mTOR and AMPK signaling-related molecules were then detected by western blotting. Small interference RNA against LKB1 (si-LKB1) was transfected into C4-2 cells for 72 h. The expression levels of p-LKB1, p-AMPK, LC3-Ⅰ, LC3-Ⅱ and PrLZ, and the LC3-Ⅱ/ LC3-Ⅰ ratio were assayed in PrLZ knockdown C4-2 cells (E) or C4-2 cells that underwent glucose starvation (GS) for 24 h (F).
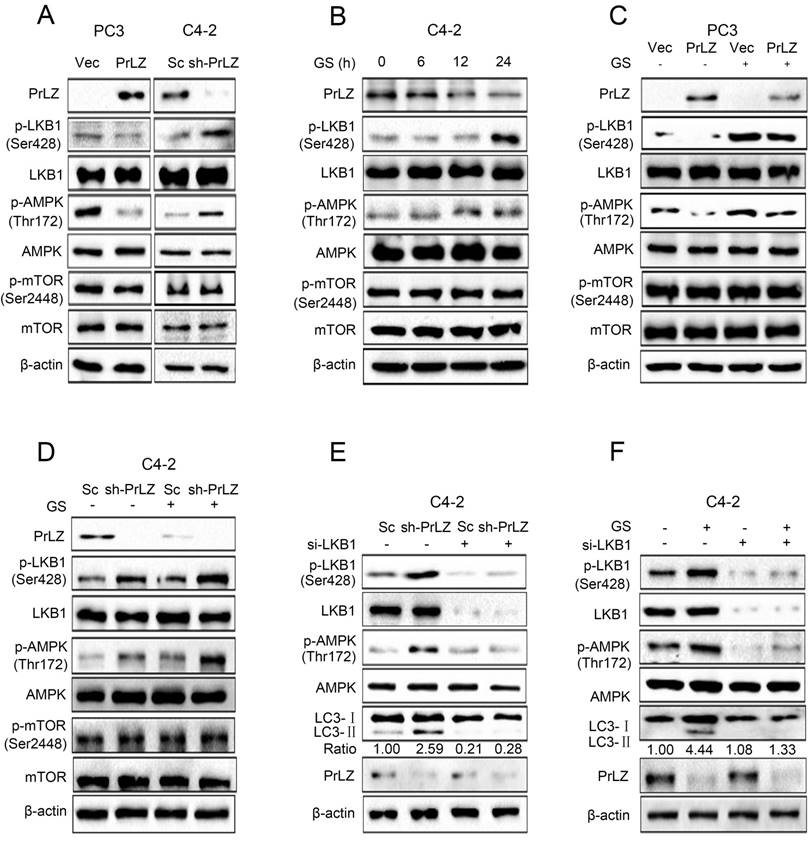
Furthermore, PrLZ overexpression was found to remarkably attenuate the activation of phosphorylated-LKB1 and phosphorylated-AMPK triggered by glucose starvation (Fig. 3C), while knocking down PrLZ activated LKB1/AMPK signaling upon glucose starvation stress (Fig. 3D). To confirm the role of LKB1 in PrLZ-mediated autophagy, we silenced LKB1 expression using small interfering RNA (siRNA) to explore the connections between LKB1 and the autophagic process. The results showed that activation of LKB1 and AMPK by either knocking down PrLZ (Fig. 3E) or glucose starvation (Fig. 3F) were completely inhibited by knocking down LKB1, thus suggesting a key role of the LKB1/AMPK axis in PrLZ-mediated autophagy under glucose starvation conditions.
PrLZ interacts with LKB1
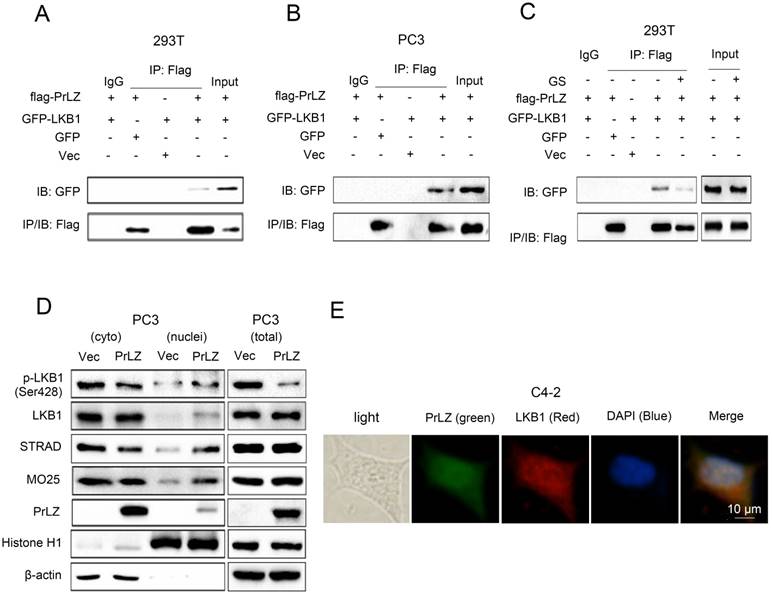
To gain further insight into the molecular mechanism between PrLZ and LKB1, we assayed the potential interaction between PrLZ and LKB1. The data presented in Fig. 4A and 4B show that immunoprecipitation of PrLZ resulted in co-precipitation of LKB1, suggesting a physical interaction between PrLZ and LKB1. More importantly, autophagy induction by glucose starvation significantly reduced the interaction of PrLZ and LKB1 (Fig. 4C), indicating that the physical interaction between PrLZ and LKB1 might exert negative regulatory effects on the autophagic process. In addition, we determined that overexpression of PrLZ resulted in the nuclear translocation of phosphorylated LKB1, total LKB1, STRAD and MO25, which was confirmed by subcellular fractionation analysis (Fig. 4D). In addition, immunofluorescence staining indicated that PrLZ and LKB1 were co-localized in both the cytoplasm and nucleus of C4-2 cells (Fig. 4E).
PrLZ interacts with LKB1. Co-immunoprecipitation of PrLZ and LKB1 was assayed in 293T and PC3 cells co-transfected with GFP, pcDNA3.0 empty vector (Vec), GFP-LKB1 or flag-tagged PrLZ plasmid under (A, B) normal or (C) glucose-starvation conditions (GS, 24 h). 10% of input was used. (D) Expression of p-LKB1 (Ser428), LKB1, STRAD, MO25 and PrLZ in both cytosolic (cyto) and nuclear (nuclei) fractions were detected by western blotting in PC3 cells stably transfected with PrLZ or vector. Histone H1 and β-actin were used as internal control markers for the nuclear and cytoplasmic fractions, respectively. (E) Representative phase contrast and fluorescence photomicrographs of the co-localization of PrLZ and LKB1 via confocal microscopy in C4-2 cells. Scale bars represent 10 μm.
PrLZ confers resistance to docetaxel-induced apoptosis via inhibition of autophagy in PCa cells
Docetaxel is the first-line chemotherapeutic drug for the treatment of metastatic PCa [18]. To explore whether PrLZ is involved in the inhibition of growth induced by docetaxel, the IC50 of docetaxel in PCa C4-2 and PC-3 cells was first determined using an MTT assay. Based on the IC50 of docetaxel in C4-2 cells (15 μM) and PC3 cells (18 μM) (Supplementary Fig. S3B), we chose 20 μM docetaxel as the representative dose in most of the in vitro experiments. Our findings showed that overexpression of PrLZ partially reversed the cytotoxic effect of docetaxel and impaired the apoptotic induction of docetaxel; however, loss of PrLZ sensitized the cytotoxic effect of docetaxel (Fig. 5A and B, Supplementary Fig. S3C and D). Furthermore, increasing expression of exogenous PrLZ partially restored the growth inhibition of docetaxel in C4-2 cells with low PrLZ expression (Supplementary Fig. S3E). These results strongly suggested a resistant effect of PrLZ on docetaxel-induced growth inhibition and apoptosis.
PrLZ confers resistance to docetaxel-induced apoptosis and autophagy in PCa cells. (A) The viability of PC3-PrLZ and PC3-Vec cells treated with 20 μM docetaxel (DTX) for 24 h was determined using a MTT assay. The results represent the mean ± S.D. of 3 independent experiments. **, P<0.01. (B) Under similar treatment conditions, apoptosis was determined using flow cytometry analysis. (C) Autophagy- and apoptosis-related molecules were detected by western blotting in C4-2 cells following treatment with different doses of DTX (0, 5, 10, 20 μM) for 24 h. PC3 cells with PrLZ (D) and C4-2 cells with sh-PrLZ (E) were treated with 20 μM DTX for 24 h. Autophagy- and apoptosis-related molecules were detected by western blotting. (F) The quantification of GFP-RFP-LC3 puncta in C4-2 cells with sh-PrLZ treated with docetaxel (20 μM, 24 h). The results represent the mean ± S.D. of 3 independent experiments. **, P<0.01. (G) Co-immunoprecipitation of PrLZ and LKB1 was assayed in 293T cells co-transfected with GFP, pcDNA3.0 empty vector, GFP-LKB1 or flag-tagged PrLZ plasmids under 20 μM docetaxel treatment for 24 h.
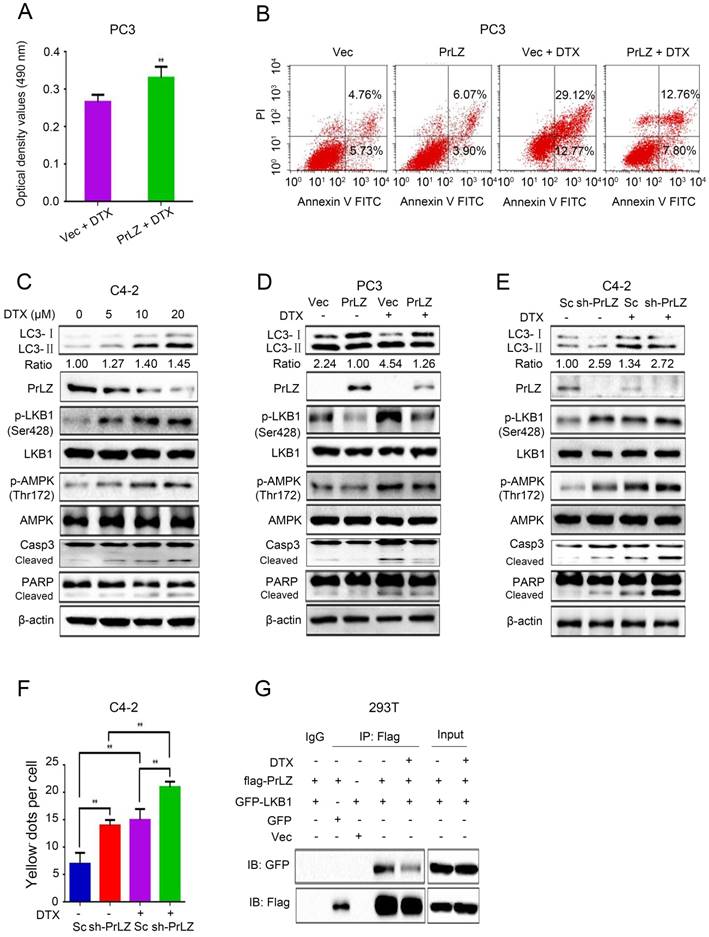
Additionally, docetaxel treatment resulted in a significant upregulation of LC3-Ⅱ, phosphorylated-LKB1, phosphorylated-AMPK, cleaved-caspase-3, cleaved-PARP and the LC3-Ⅱ/ LC3-Ⅰ ratio, and downregulation of the PrLZ and AR protein levels in C4-2 cell lines in a concentration-dependent manner (Fig. 5C, Supplementary Fig. S2B). Similar results were obtained in LNCaP and PC-3 cells (Supplementary Fig. S3F and G). PrLZ overexpression significantly reversed docetaxel-induced autophagy and apoptosis, while knocking down PrLZ reinforced the autophagic and apoptotic effects induced by docetaxel (Fig. 5D-F). Furthermore, docetaxel treatment also resulted in decreased binding between PrLZ and LKB1 (Fig. 5G). Additionally, our results revealed a significant downregulation of LC3-II, phosphorylated-LKB1, phosphorylated-AMPK and the LC3-II/LC3-I ratio, and upregulation of PrLZ and AR in the docetaxel-resistant C4-2 cells compared with the corresponding C4-2 parental cells (Supplementary Fig. S3H and I). Together, our results confirmed that PrLZ confers resistance to docetaxel-induced apoptosis and autophagy in PCa cells.
PrLZ/LKB1-mediated autophagy is involved in docetaxel-induced apoptosis in PCa cells
To further identify the role of LKB1 in docetaxel-induced cell apoptosis and autophagy, we knocked down LKB1 with siRNA. Docetaxel treatment resulted in significant activation of caspase-3 and PARP in PCa C4-2, LNCaP and PC3 cells (Fig. 6A; Supplementary Fig. S4A and B), suggesting that activation of the caspase cascade was involved in PCa docetaxel treatment. More importantly, the docetaxel-induced activation of the caspase cascade, increase in LC3-Ⅱ level and LC3 puncta (Fig. 6A and B, Supplementary Fig. S5A), inhibition of cell growth (Fig. 6C) and induction of apoptosis in C4-2 or PC3-PrLZ cells (Fig. 6D, Supplementary Fig. S4C) could be partially reversed after LKB1 siRNA treatment, suggesting a vital role of LKB1 in regulating docetaxel-induced PCa cell apoptosis and autophagy. Similarly, the knockdown of LKB1 significantly reversed PrLZ silencing-induced autophagy and apoptosis in C4-2 cells and reinforced PrLZ overexpressing-induced inhibition of autophagy in PC-3 cells (Fig. 6E, Supplementary Fig. S4D).
PrLZ/LKB1-mediated autophagy is involved in docetaxel-induced apoptosis in PCa cells. (A) Small interference RNA against LKB1 (si-LKB1) was transfected into C4-2 cells for 48 h. The expressions of autophagy- and apoptosis-related molecules were assayed by western blotting in C4-2 cells treated with docetaxel (DTX) (20 μM, 24 h). Under similar conditions, the GFP-RFP-LC3 puncta (B), cell viability (C) and percentage of apoptotic cells (D) were quantified in DTX-treated LKB1 knockdown (si-LKB1) and control C4-2 cells. The results represent the mean ± S.D. of 3 independent experiments. *, P<0.05, **, P<0.01, ***, P<0.001 and ****, P<0.0001. (E) The expression levels of autophagy- and apoptosis-related molecules were determined by western blotting in PrLZ knockdown (sh-PrLZ) cells transfected with LKB1 siRNA (si-LKB1). (F) Small interference RNA against ATG5 (si-ATG5) was transfected into C4-2 cells for 48 h. The expression levels of apoptosis markers and the LC3-Ⅱ/ LC3-Ⅰ ratio were determined by western blotting in C4-2 cells treated with docetaxel (DTX) (20 μM, 24 h). Under similar treatment conditions, the GFP-RFP-LC3 puncta (G), cell viability (H) and percentage of apoptotic cells (I) were quantified in docetaxel-treated C4-2 cells. The results represent the mean ± S.D. of 3 independent experiments. **, P<0.01, ***, P<0.001 and ****, P<0.0001.
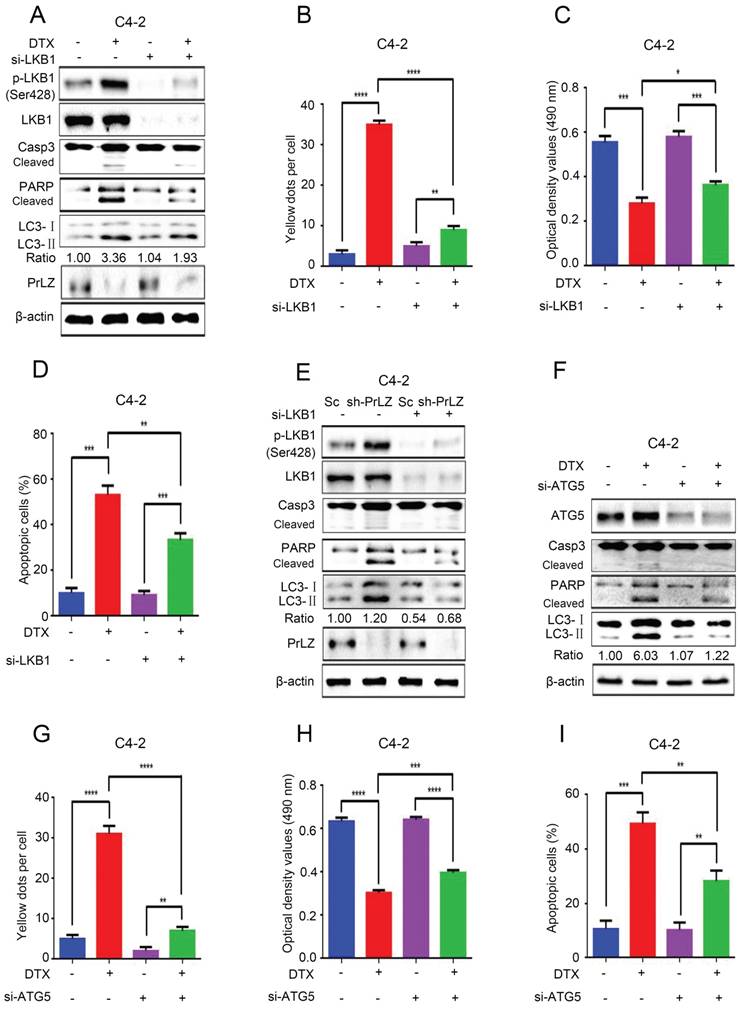
Both autophagy and apoptosis play multiple, essential roles in cellular homeostasis, and an extensive crosstalk between them has been widely studied. The autophagic process can be protective or lethal. Protective autophagy directs the degradation of cytoplasmic components and recycling of amino acids to overcome nutrient deprivation, whereas lethal autophagy leads to autophagic cell death [19]. To further clarify the relationship between docetaxel-induced cell apoptosis and autophagy, small interference RNA was applied to knockdown ATG5, which is a key ATG protein for autophagosome formation. Not surprisingly, knocking down ATG 5 by siRNA significantly inhibited docetaxel-induced autophagy in C4-2 cells (Fig. 6F and G, Supplementary Fig. S5B). More importantly, docetaxel-induced activation of the caspase cascade (Fig. 6F), inhibition of cell growth (Fig. 6H) and induction of apoptosis (Fig. 6I) could be abolished after ATG5 siRNA treatment, suggesting a lethal role of autophagy induced by docetaxel.
PrLZ protects PCa cells from docetaxel-mediated apoptosis via inhibition of autophagy in vivo
To verify our in vitro results and to evaluate the function of PrLZ in the anti-tumor activity of docetaxel in vivo, PCa xenografts in athymic nude mice were used as the in vivo model system. PrLZ played an oncogenic role in tumors to promote the growth of PCa xenografts and impeded docetaxel-mediated growth inhibition in vivo without decreasing the body weight of the mice (Fig. 7A, Supplementary Fig. S4E). PrLZ overexpression increased the tumor weight from 0.14 g per mouse in the control group to 0.4 g per mouse in the PrLZ group, which caused a 2-fold increase in tumor weight (P<0.05). A significant 5-fold increase in tumor weight was still observed in the PrLZ group under docetaxel treatment. Moreover, PC3 tumors displayed growth retardation after docetaxel administration (tumor volumes from 320 mm3 to 138 mm3 after 18 days of the treatment), whereas PC3/PrLZ tumors maintained a rapid growth rate in the xenograft mouse model (tumor volumes from 138 mm3 to 363 mm3 after 18 days of treatment), which indicated that PrLZ might contribute to the chemoresistance of PCa to docetaxel treatment (Fig. 7B and C).
PrLZ protects docetaxel-induced PCa cells from apoptosis via inhibition of autophagy in vivo. PC3-PrLZ or PC3-vector cells were subcutaneously injected into male nude mice, and mice were treated with docetaxel (DTX) (15 mg/kg/week) intraperitoneally. The mice were euthanized after 4 weeks. Tumor weights (A) and illustrative diagrams of isolated tumor xenografts (B) are shown. (C) Tumor volumes in the 4 groups (PC3-Vec + Vehicle, PC3-PrLZ + Vehicle, PC3-Vec + DTX, PC3-PrLZ + DTX, 5 mice per group). The results represent the mean ± S.D. of 3 independent experiments. *, P<0.05 and **, P<0.01. (D). Representative immunohistochemical photomicrographs and (E) quantitative results of the cytoplasmic and nuclear distribution of p-LKB1 in the different groups (PC3-Vec + Vehicle, PC3-PrLZ + Vehicle, PC3-Vec + DTX, PC3-PrLZ + DTX) are shown. The quantitative results represent the mean ± S.D. of 3 independent experiments. *, P<0.05, **, P<0.01 and ***, P<0.001. Scale bars represent 10 μm. (F) The expression levels of LC3 I/II, PrLZ, p-LKB1 (Ser428), p-AMPK (Thr172) and cleaved PARP were determined by western blotting in 3 representative mice from different groups (PC3-Vec + Vehicle, PC3-PrLZ + Vehicle, PC3-Vec + DTX, PC3-PrLZ + DTX). (G) Wild-type (WT) or knockout (KO) PrLZ mice underwent food starvation for 24 h. Following euthanasia, the prostate tissues were analyzed by western blotting for levels of p-LKB1 (Ser428) and p-AMPK (Thr172).
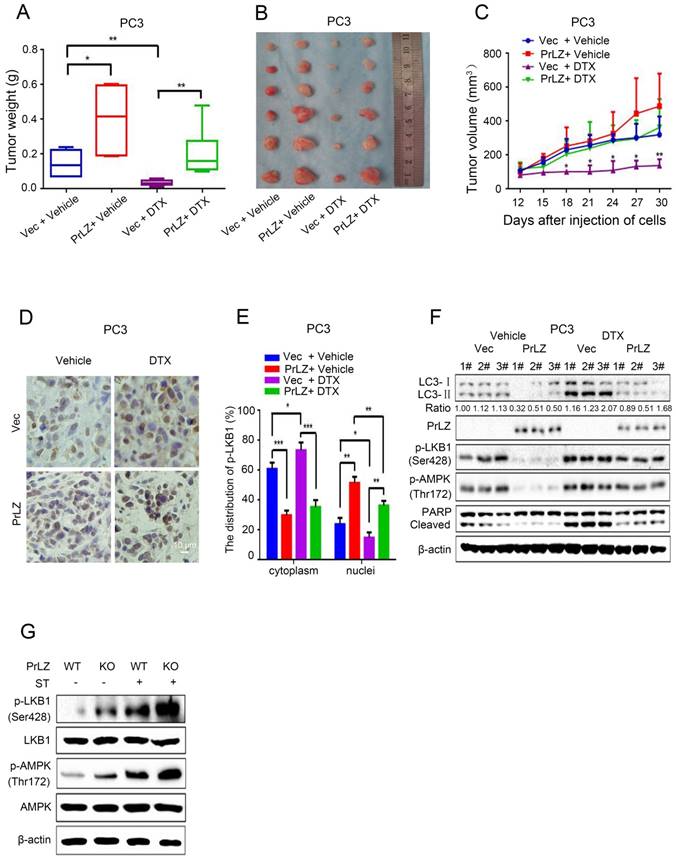
Immunohistochemical staining of tumor tissues demonstrated that overexpression of PrLZ could alter the distribution of LKB1, with increased accumulation in the nucleus and decreased expression in the cytoplasm in the presence or absence of docetaxel treatment (Fig. 7D and E). In addition, PrLZ overexpression significantly attenuated the docetaxel-induced increase in LC-3 Ⅱ levels, increase in cleaved subunits of PARP and activation of LKB1 and AMPK, which were consistent with our in vitro results (Fig. 7F). More importantly, PrLZ knockout further facilitated food starvation-induced activation of LKB1 and AMPK (Fig. 7G). Overall, the data from Fig. 7A-G show that docetaxel inhibited proliferation and induced autophagy in PCa by downregulating PrLZ in vivo.
Discussion
We and others have previously identified a vital role of PrLZ in the progression of CRPC, which was associated with an increased PrLZ/AR interaction [8, 11, 12]. Docetaxel is widely used as the standard first-line therapeutic strategy for CRPC patients [2, 20], but resistance eventually develops. In the present study, we demonstrate that overexpression of PrLZ confers resistance to docetaxel-induced apoptosis and autophagy in PCa cells. Mechanistically, the interaction between PrLZ and LKB1-mediated inhibition of autophagy is involved in resistance to docetaxel-induced apoptosis in PCa cells.
Accumulating evidence suggests that autophagy acts in both cancer progression and suppression [21]. Recent studies have shown that the autophagy and apoptosis pathways are regulated by common factors, share common components and exert overlapping functions [22]. There have been limited reports on the relationship between PrLZ and autophagy. It has been reported that PrLZ overexpression confers PCa cell resistance to rapamycin treatment by antagonizing rapamycin-induced cytostasis and autophagy [23]. Another report revealed that suppression of PrLZ sensitized PCa cells to ionizing radiation by attenuating DNA damage repair and inducing autophagic cell death [13]. However, there is almost a complete lack of evidence for a direct relationship between PrLZ and autophagy. More importantly, the upstream molecular regulatory mechanisms of PrLZ in autophagy have still not been fully elucidated. In the present study, we found that inhibition of PrLZ directly induced basal level autophagy, while overexpression of PrLZ impaired the autophagic flux, as determined by LC3 conversion, LC3 turnover and LC3 fluorescent puncta assays. However, p62 expression, a well-known autophagic substrates, remained unchanged upon overexpression or knockdown of PrLZ. One reasonable explanation is that p62 could be transcriptionally and post-translationally regulated by numerous factors [24], and thus, it is not a good autophagy marker candidate in our PCa cell model. In addition, starvation is a classic approach used to induce autophagy [25]. Here, we found that glucose starvation-induced autophagy was directly mediated by PrLZ in PCa cells, confirming the vital role of PrLZ in autophagy. Importantly, we also revealed that both PrLZ mRNA and AR protein were downregulated in PCa cells after autophagy induction. In accordance with our previous study results that AR could directly bind to the PrLZ promoter and lead to the increased expression of PrLZ in LNCaP and C4-2 cells at the CRPC stage, our data suggests that AR might manipulate PrLZ transcription upon autophagic induction. Further studies are needed to identify the related molecular mechanisms [12].
Several critical molecules and pathways have been shown to initiate and maintain autophagy; of these, the mTOR and AMPK pathways have been well characterized [26, 27]. AMPK, a key molecule involved in the maintenance of cellular energy homeostasis, plays an important role in the initiation of autophagy [27]. Our mechanistic studies revealed that PrLZ inhibited autophagy via regulation of AMPK signaling instead of mTOR signaling. Knockdown of PrLZ strongly increased Thr172 phosphorylation in AMPK, which is crucial for the initiation of autophagy. However, we failed to observe inactivation of mTOR. Phosphorylation of the Thr172 site in the AMPK α subunit can be mediated by LKB1 as well as CaMKKβ [28]. Moreover, studies have shown that TAK1 can also activate AMPK [29], but the detailed mechanism has remained elusive. Intriguingly, our results demonstrated that manipulation of PrLZ strongly affected the phosphorylation level of LKB1 instead of TAK1 and CaMKKβ under both basal and glucose starvation conditions.
Because we identified the regulatory role of PrLZ on LKB1, we further explored the mechanisms leading to the inhibition of LKB1 activation by PrLZ. A physical interaction between PrLZ and LKB1 was observed using an immunoprecipitation assay. More importantly, the binding between PrLZ and LKB1 was impaired by glucose deprivation, indicating that the physical interaction between PrLZ and LKB1 might exert negative regulatory effects on the autophagic process. Further dissection of the mechanisms showed that overexpression of PrLZ resulted in increased translocation of phosphorylated-LKB1, STRAD and MO25 to the nucleus, and the possible binding between LKB1 and STRAD/MO25 in the nucleus might limit the distribution of LKB1 in the cytoplasm, which may then inhibit its kinase activity. As initiation of autophagy occurs when a double membrane forms within the cytoplasm that engulfs proteins and organelles, most autophagy related-kinases are considered to exert their physiological functions in the cytoplasm [30]. Disruption of the cellular distribution of LKB1 by PrLZ may partially explain the inhibitory effects of PrLZ on LKB1 activity. However, further in-depth studies are needed to elucidate other molecular mechanisms that contribute to the inhibition of LKB1 activation by PrLZ.
Docetaxel resistance is a major obstacle in the context of PCa and, especially, CRPC. A variety of mechanisms underlying docetaxel resistance have been well established, such as DNA repair mechanisms [18], drug export transporters and resistance to apoptosis [31]. Because autophagy plays a dual role in cancer progression, the relationship between autophagy and docetaxel resistance has received great attention by cancer researchers. It was reported that inhibition of autophagy with 3-methyladenine partially reversed docetaxel-induced cytotoxic and apoptotic effects in PCa PC3 cells [32]. Autophagy may also be a potential mechanism of chemoresistance to docetaxel in PCa following pretreatment with pantoprazole [6]. Additionally, autophagy regulated by high-mobility group box 1, a highly conserved non-histone nuclear protein that binds DNA and promotes the assembly of proteins on specific DNA targets, could confer resistance to docetaxel in human lung adenocarcinoma [33]. However, whether autophagy plays a cytoprotective or a pro-apoptotic role in docetaxel-induced apoptosis in PCa is still unknown. In our studies, overexpression of PrLZ markedly impaired the efficacy of apoptosis induction and the cytostatic effects induced by docetaxel in vitro and in vivo, suggesting that PrLZ appears to be resistant to the therapeutic characteristics of docetaxel in PCa cells. Furthermore, the PrLZ/LKB1 interaction-mediated inhibition of autophagy promoted resistance to docetaxel in PCa cells. Taken together, these data provide supporting evidence that inhibition of autophagy by PrLZ may play a survival role in PCa cells undergoing apoptosis after docetaxel treatment (Fig. 8).
Schematic model for PrLZ-mediated LKB1-dependent autophagy on docetaxel resistance in prostate cancer. PrLZ could directly interact with LKB1. The physical interaction between PrLZ and LKB1 might exert negative regulatory effects on the phosphorylated AMPK and subsequent autophagic process, which confers resistance to docetaxel in prostate cancer.
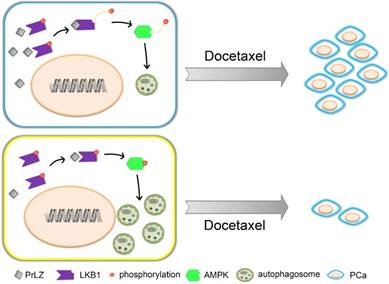
In summary, our data showed that overexpression of PrLZ protected PCa cells from apoptosis induced by docetaxel, which was mediated by the inhibition of autophagy. Mechanistically, negative regulation of autophagy by PrLZ was mediated by LKB1/AMPK signaling. Considering its prostate specificity and vital role in the progression of CRPC, PrLZ and the autophagy pathway may become attractive therapeutic targets for CRPC and, especially, docetaxel-resistant CRPC therapy.
Abbreviations
AMPK: AMP-activated protein kinase; ATG: autophagy-associated gene; AR: androgen receptor; CaMKKβ: Ca2+/calmodulin-dependent protein kinase kinase-beta; CHX: cycloheximide; CQ: chloroquine; DTX: docetaxel; GFP: green fluorescent protein; LKB1: liver kinase B1; mCRPC: metastatic castration-resistant prostate cancer; MO25: mouse protein 25; mTOR: mammalian target of rapamycin; PCa: prostate cancer; PrLZ: prostate leucine zipper; REP: red fluorescent protein (RFP); STRAD: Ste20-related adaptor protein; TAK1: transforming growth factor beta-activated kinase 1.
Supplementary Material
Supplementary figures.
Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (NO. 81072107, 81130041, 81472679, 81672538 and 81773206) and the National Key Research and Development Program (NO. 2016YFC0902603).
Author contributions
J.Z. and W.L. have contributed equally to this work. L.L. designed and supervised all experiments and contributed to the manuscript preparation. J.Z. and W.L. performed the experiments, analyzed the data and contributed to the manuscript preparation. Y.-Z.F. collected animal samples and participated in the animal experiments. D.-L.H. contributed to the data analysis and manuscript preparation.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA: a cancer journal for clinicians. 2016;66:7-30
2. Tsao CK, Galsky MD, Oh WK. Docetaxel for Metastatic Hormone-sensitive Prostate Cancer: Urgent Need to Minimize the Risk of Neutropenic Fever. European urology. 2016;70:707-8
3. Sun Y, Campisi J, Higano C, Beer TM, Porter P, Coleman I. et al. Treatment-induced damage to the tumor microenvironment promotes prostate cancer therapy resistance through WNT16B. Nature medicine. 2012;18:1359-68
4. Liu X, Klionsky DJ. The Atg17-Atg31-Atg29 complex and Atg11 regulate autophagosome-vacuole fusion. Autophagy. 2016;12:894-5
5. Galluzzi L, Pietrocola F, Levine B, Kroemer G. Metabolic control of autophagy. Cell. 2014;159:1263-76
6. Tan Q, Joshua AM, Saggar JK, Yu M, Wang M, Kanga N. et al. Effect of pantoprazole to enhance activity of docetaxel against human tumour xenografts by inhibiting autophagy. British journal of cancer. 2015;112:832-40
7. Kondo Y, Kanzawa T, Sawaya R, Kondo S. The role of autophagy in cancer development and response to therapy. Nature reviews Cancer. 2005;5:726-34
8. Wang R, Xu J, Saramaki O, Visakorpi T, Sutherland WM, Zhou J. et al. PrLZ, a novel prostate-specific and androgen-responsive gene of the TPD52 family, amplified in chromosome 8q21.1 and overexpressed in human prostate cancer. Cancer research. 2004;64:1589-94
9. Wu R, Wang H, Wang J, Wang P, Huang F, Xie B. et al. EphA3, induced by PC-1/PrLZ, contributes to the malignant progression of prostate cancer. Oncology reports. 2014;32:2657-65
10. Li L, Zhang D, Zhang L, Zhu G, Sun Y, Wu K. et al. PrLZ expression is associated with the progression of prostate cancer LNCaP cells. Mol Carcinog. 2009;48:432-40
11. Zhang D, He D, Xue Y, Wang R, Wu K, Xie H. et al. PrLZ protects prostate cancer cells from apoptosis induced by androgen deprivation via the activation of Stat3/Bcl-2 pathway. Cancer research. 2011;71:2193-202
12. Li L, Xie H, Liang L, Gao Y, Zhang D, Fang L. et al. Increased PrLZ-mediated androgen receptor transactivation promotes prostate cancer growth at castration-resistant stage. Carcinogenesis. 2013;34:257-67
13. Shang ZF, Wei Q, Yu L, Huang F, Xiao BB, Wang H. et al. Suppression of PC-1/PrLZ sensitizes prostate cancer cells to ionizing radiation by attenuating DNA damage repair and inducing autophagic cell death. Oncotarget. 2016;7:62340-51
14. Domingo-Domenech J, Vidal SJ, Rodriguez-Bravo V, Castillo-Martin M, Quinn SA, Rodriguez-Barrueco R. et al. Suppression of acquired docetaxel resistance in prostate cancer through depletion of notch- and hedgehog-dependent tumor-initiating cells. Cancer cell. 2012;22:373-88
15. Chang C, Su H, Zhang D, Wang Y, Shen Q, Liu B. et al. AMPK-Dependent Phosphorylation of GAPDH Triggers Sirt1 Activation and Is Necessary for Autophagy upon Glucose Starvation. Mol Cell. 2015;60:930-40
16. Lamb CA, Yoshimori T, Tooze SA. The autophagosome: origins unknown, biogenesis complex. Nature reviews Molecular cell biology. 2013;14:759-74
17. Russell RC, Yuan HX, Guan KL. Autophagy regulation by nutrient signaling. Cell Res. 2014;24:42-57
18. Tannock IF, de Wit R, Berry WR, Horti J, Pluzanska A, Chi KN. et al. Docetaxel plus prednisone or mitoxantrone plus prednisone for advanced prostate cancer. The New England journal of medicine. 2004;351:1502-12
19. Baehrecke EH. Autophagy: dual roles in life and death? Nature reviews Molecular cell biology. 2005;6:505-10
20. Hwang C. Overcoming docetaxel resistance in prostate cancer: a perspective review. Therapeutic advances in medical oncology. 2012;4:329-40
21. Pei J, Deng J, Ye Z, Wang J, Gou H, Liu W. et al. Absence of autophagy promotes apoptosis by modulating the ROS-dependent RLR signaling pathway in classical swine fever virus-infected cells. Autophagy. 2016;12:1738-58
22. Song S, Tan J, Miao Y, Li M, Zhang Q. Crosstalk of autophagy and apoptosis: Involvement of the dual role of autophagy under ER stress. Journal of cellular physiology. 2017;232:2977-84
23. Yu L, Shang ZF, Wang J, Wang H, Huang F, Zhang Z. et al. PC-1/PrLZ confers resistance to rapamycin in prostate cancer cells through increased 4E-BP1 stability. Oncotarget. 2015;6:20356-69
24. Cohen-Kaplan V, Livneh I, Avni N, Fabre B, Ziv T, Kwon YT. et al. p62- and ubiquitin-dependent stress-induced autophagy of the mammalian 26S proteasome. Proc Natl Acad Sci U S A. 2016;113:E7490-E9
25. Kuma A, Hatano M, Matsui M, Yamamoto A, Nakaya H, Yoshimori T. et al. The role of autophagy during the early neonatal starvation period. Nature. 2004;432:1032-6
26. Rodon J, Dienstmann R, Serra V, Tabernero J. Development of PI3K inhibitors: lessons learned from early clinical trials. Nature reviews Clinical oncology. 2013;10:143-53
27. Sui X, Chen R, Wang Z, Huang Z, Kong N, Zhang M. et al. Autophagy and chemotherapy resistance: a promising therapeutic target for cancer treatment. Cell death & disease. 2013;4:e838
28. Carling D, Sanders MJ, Woods A. The regulation of AMP-activated protein kinase by upstream kinases. International journal of obesity. 2008;32(Suppl 4):S55-9
29. Russo GL, Russo M, Ungaro P. AMP-activated protein kinase: a target for old drugs against diabetes and cancer. Biochemical pharmacology. 2013;86:339-50
30. He C, Klionsky DJ. Regulation mechanisms and signaling pathways of autophagy. Annual review of genetics. 2009;43:67-93
31. Kroon J, Kooijman S, Cho NJ, Storm G, van der Pluijm G. Improving Taxane-Based Chemotherapy in Castration-Resistant Prostate Cancer. Trends in pharmacological sciences. 2016;37:451-62
32. Pickard RD, Spencer BH, McFarland AJ, Bernaitis N, Davey AK, Perkins AV. et al. Paradoxical effects of the autophagy inhibitor 3-methyladenine on docetaxel-induced toxicity in PC-3 and LNCaP prostate cancer cells. Naunyn-Schmiedeberg's archives of pharmacology. 2015;388:793-9
33. Pan B, Chen D, Huang J, Wang R, Feng B, Song H. et al. HMGB1-mediated autophagy promotes docetaxel resistance in human lung adenocarcinoma. Molecular cancer. 2014;13:165-83
Author contact
![]() Corresponding author: L.L. (E-mail: lilydrcom)
Corresponding author: L.L. (E-mail: lilydrcom)