Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Supplementary Material
Abbreviations
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2018; 8(4):906-920. doi:10.7150/thno.20746 This issue Cite
Research Paper
Mesenchymal Stromal/stem Cell-derived Extracellular Vesicles Promote Human Cartilage Regeneration In Vitro
Lucienne A. Vonk3, Sanne F. J. van Dooremalen1,2, Nalan Liv1, Judith Klumperman1, Paul J. Coffer1,2, Daniël B.F. Saris3,4,5, Magdalena J. Lorenowicz1,2 ![]()
1. Center for Molecular Medicine, University Medical Center Utrecht, Utrecht University, Universiteitsweg 100, 3584 CG, Utrecht, The Netherlands;
2. Regenerative Medicine Center, Uppsalalaan 8, 3584 CT, Utrecht, The Netherlands;
3. Department of Orthopedics, University Medical Center Utrecht, Utrecht University, PO Box 85500, 3508 GA, Utrecht, The Netherlands;
4. MIRA institute, University of Twente, ME125, PO Box 217, 7500 AE, Enschede, The Netherlands;
5. Department of Orthopedics, Mayo Clinic College of Medicine, Rochester, MN 55905, USA.
Received 2017-4-26; Accepted 2017-10-8; Published 2018-1-1
Abstract

Osteoarthritis (OA) is a rheumatic disease leading to chronic pain and disability with no effective treatment available. Recently, allogeneic human mesenchymal stromal/stem cells (MSC) entered clinical trials as a novel therapy for OA. Increasing evidence suggests that therapeutic efficacy of MSC depends on paracrine signalling. Here we investigated the role of extracellular vesicles (EVs) secreted by human bone marrow derived MSC (BMMSC) in human OA cartilage repair.
Methods: To test the effect of BMMSC-EVs on OA cartilage inflammation, TNF-alpha-stimulated OA chondrocyte monolayer cultures were treated with BMMSC-EVs and pro-inflammatory gene expression was measured by qRT-PCR after 48 h. To assess the impact of BMMSC-EVs on cartilage regeneration, BMMSC-EVs were added to the regeneration cultures of human OA chondrocytes, which were analyzed after 4 weeks for glycosaminoglycan content by 1,9-dimethylmethylene blue (DMMB) assay. Furthermore, paraffin sections of the regenerated tissue were stained for proteoglycans (safranin-O) and type II collagen (immunostaining).
Results: We show that BMMSC-EVs inhibit the adverse effects of inflammatory mediators on cartilage homeostasis. When co-cultured with OA chondrocytes, BMMSC-EVs abrogated the TNF-alpha-mediated upregulation of COX2 and pro-inflammatory interleukins and inhibited TNF-alpha-induced collagenase activity. BMMSC-EVs also promoted cartilage regeneration in vitro. Addition of BMMSC-EVs to cultures of chondrocytes isolated from OA patients stimulated production of proteoglycans and type II collagen by these cells.
Conclusion: Our data demonstrate that BMMSC-EVs can be important mediators of cartilage repair and hold great promise as a novel therapeutic for cartilage regeneration and osteoarthritis.
Keywords: Mesenchymal stem/stromal cells, extracellular vesicles, cartilage regeneration, inflammation, osteoarthritis.
Introduction
Osteoarthritis (OA) is the most common form of joint disease, leading to chronic pain, stiffness, and disability (1). Several factors, such as mechanical stress and pro-inflammatory cytokines, are considered to contribute to disruption of cartilage homeostasis and initiation of cartilage damage in OA (2,3). Cartilage has a very limited ability for self-repair and although current cell therapies, such as autologous chondrocyte implantation, give satisfactory results for the treatment of focal cartilage defects, cell-based treatments for OA are more challenging due to disturbed joint homeostasis and ongoing cartilage degradation (4, 5). Therefore, at the moment we lack effective disease-modifying medical therapy for OA.
Recently, human multipotent mesenchymal stromal (stem) cells (MSC) have entered clinical trials as a novel, less invasive therapy for cartilage defects and OA (6, 7). MSC are a promising alternative for current therapies as they are more likely to control the imbalance between anabolic and catabolic processes in an OA joint, thanks to their immunomodulatory and regenerative capacities (8-10). Although the results of this novel treatment are promising, the fate of MSC in vivo and the molecular mechanism underlying their beneficial effects in cartilage repair remain unclear. Increasing evidence suggests that the therapeutic efficacy of MSC depends on paracrine signaling (11, 12) and more recently their therapeutic potential has been attributed to the secretion of extracellular vesicles (EVs) (13-15). EVs are secreted by all cell types and range in size from 40-100 nm (exosomes) and from 100-1000 nm (microvesicles). Exosomes are formed by the invagination of the limiting membrane of multivesicular bodies (MVBs), which are part of the cellular endo-lysosomal system. Upon fusion of MVBs with the plasma membrane, exosomes are released into the extracellular environment. Microvesicles bud directly off the plasma membrane. EVs exert many of their functions as an intercellular shuttle, carrying cargo such as protein and RNA to be transferred from one cell to another (16). Although long anticipated, it has only recently been reported that EVs can target specific cell types e.g. tumor-derived exosomes interacting with immune cells (17-19).
EVs released by MSC increasingly appear to play an important role in intercellular communication and tissue repair. MSC-EVs have been shown to exert immunomodulatory properties in vitro, and to some extent also possess regenerative properties in a mouse model of myocardial ischemia/reperfusion injury, a rat skin wound model and a rat osteochondral defect model (14, 15, 20).
In this study we investigated the regenerative and immunomodulatory potential of EVs secreted by human bone marrow derived MSC (BMMSC) in chondrocytes from OA patients using an in vitro human cartilage repair model (21). Our data show that BMMSC-EVs downregulate tumor necrosis factor alpha (TNF-alpha) induced expression of pro-inflammatory cyclooxygenase-2 (COX2), pro-inflammatory interleukins and collagenase activity in OA chondrocytes. The anti-inflammatory effect of BMMSC-EVs involves the inhibition of NFκB signaling, activation of which is an important component of OA pathology. We also demonstrate that treatment of OA chondrocytes with BMMSC-EVs from independent allogeneic BMMSC donors induces production of proteoglycans and type II collagen and promotes proliferation of these cells. Thus, our findings indicate that BMMSC-EVs have ability to promote human OA cartilage repair by reducing the inflammatory response and stimulation of OA chondrocytes to produce extracellular matrix, the essential processes for restoring and maintaining cartilage homeostasis.
Materials and Methods
Donors
Cartilage was obtained from redundant material from five female and three male patients (age 57 - 80, average 71 years) who had undergone total knee arthroplasty. The anonymous collection of this material was performed according to the Medical Ethics regulations of the University Medical Center Utrecht and the guideline ''good use of redundant tissue for research'' of the Dutch Federation of Medical Research Societies (22, 23).
Cell isolation and expansion
Cartilage samples were rinsed in phosphate-buffered saline (PBS), cut into small pieces and subjected to sequential treatments of Dulbecco's modified Eagle's medium (DMEM, Gibco, Paisley, UK) supplemented with 1% fetal bovine serum (FBS, HyClone, Logan, UT), 100 U/mL penicillin, 100 mg/mL streptomycin (all from Gibco) and 2.5% (w/v) Pronase E (Sigma, St. Louis, MO) for 1 h, then with DMEM supplemented with 25% FBS, 100 U/mL penicillin, 100 mg/mL streptomycin and 0.125% (w/v) collagenase (CLS-2, Worthington, Lakewood, NJ) for 16 h at 37°C. Chondrocytes were expanded in DMEM supplemented with 10% FBS (HyClone, Logan, UT), 100 U/mL penicillin, and 100 mg/mL streptomycin and used at passage two. The MSCs used are classified as ATMPs and manufactured in the GMP-licensed Cell Therapy Facility, Department of Clinical Pharmacy of the University Medical Center Utrecht. Briefly, bone marrow aspirates were obtained from third-party non-HLA-matched healthy donors as approved by the Dutch Central Committee on Research Involving Human Subjects (CCMO, Biobanking bone marrow for MSC expansion, NL41015.041.12). The bone marrow donor or the parent or legal guardian of the donor signed the informed consent approved by the CCMO. Bone marrow was separated using a density gradient centrifugation (Lymphoprep, Axis Shield). MSCs were isolated by plastic adherence and expanded using the MC3 systems and α-minimal essential medium (α-MEM) with l-glutamine from Macopharma supplemented with 5% platelet lysate and 3.3 IU/mL heparin up to passage 3 (7). Characterization of MSCs fits the internationally convened minimal criteria for these cells (41). The ATMP MSCs used for extracellular vesicles production in this study were obtained from surplus cells of one male and one female donor used for the IMPACT trial (NCT02037204) (7) and cultured for additional passages (passage 4-7) in α-MEM (Gibco Invitrogen, Carlsbad, CA, USA) supplemented with 5% human platelet lysate (PL), 100 U/mL penicillin and 100 μg/mL streptomycin (Gibco Invitrogen) and 10 U/mL heparin and maintained at 37 ºC and 5% CO2. The PL was depleted from PL-derived extracellular vesicles by overnight centrifugation at 100,000 × g.
In vitro cartilage regeneration assay
Cells were cultured in fibrin glue constructs. Per donor the following was performed: For the fibrin glue constructs, passage two chondrocytes were resuspended in a 1:15 diluted fibrinogen component of Beriplast (Baxter) using PBS and 50 µL was injected in a 96-well plate and combined with a 1:50 diluted component of thrombin, resulting in 100 µL constructs containing 0.25 x 106 cells per construct. Chondrocytes were subsequently cultured in 24-well plate in DMEM supplemented with 2% insulin-transferrin-selenium (ITS)-X (Invitrogen), 2% ASAP, 2% human serum albumin (Sanquin Blood Supply Foundation), and 1% penicillin/streptomycin (100 U/mL, 100 mg/mL).
In vitro inflammation assay
Chondrocytes were cultured in monolayer at 37°C and 5% CO2 at a seeding density of 25,000 cells/cm2 in medium consisting of DMEM supplemented with 10% FBS, 100 U/mL penicillin, and 100 mg/mL streptomycin. After pre-incubation for 24 h, cells were treated with 10 ng/mL TNF-alpha (Immunotools).
Glycosaminoglycan analysis
After 4 weeks of culture, samples were digested overnight in a papain digestion buffer (250 mg/mL papain (Sigma-Aldrich), 0.2 M NaH2PO4, 0.1 M EDTA, 0.01 M cysteine) at 60°C before quantification of the sulphated glycosaminoglycans (GAG) content with a 1,9-dimethylmethylene blue (DMMB) assay. The absorption ratio was set at 540-595 nm using chondroitin-6-sulfate (Sigma-Aldrich) as a standard. DNA content in the papain digests was determined using a Picogreen DNA assay (Invitrogen) according to the manufacturer's instructions.
Quantitative real-time PCR
Total RNA was isolated from the cells using TRIzol (Invitrogen) according to the manufacturer's instructions. Total RNA (200-500 ng) was reverse transcribed using the High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems). Real-time polymerase chain reactions (PCRs) were performed using FastStart Universal SYBR Green Master mix (Roche Diagnostics) in a LightCycler 96 (Roche Diagnostics) according to the manufacturer's instructions. Quantification was performed relative to the levels of the housekeeping gene 18S and normalized to control conditions. The data analysis was performed using the 2-ΔΔCT method (25). The primer sequences are listed in the table in Supplemental Material and Methods. The amplified PCR fragment extended over at least one exon border (except for 18S).
Proliferation
To test for proliferation in monolayer cultures, 10 μM 5-ethylnyl-2'-deoxyuridine (EdU, Invitrogen) was supplemented to the culture medium. After 5 days, medium was aspirated, cells were washed with PBS, formalin fixed for 15 min and permeabilized. EdU was visualized using Click-iT EdU Alexa Fluor 488 Imaging kit (Invitrogen) according to the manufacturer's instructions. Cells were photographed using an EVOS FLoid Cell Imaging microscope and analyzed in ImageJ.
For fibrin glue constructs in regeneration cultures, an AlamarBlue assay (10% (w/v) resazurin (Alfa Aesar, Thermo Scientific) was performed to measure metabolic activity according to manufacturer's protocol.
Collagenase activity assay
To analyze collagenase activity, the Enzchek Gelatinase/Collagenase Assay Kit (Invitrogen) was used according to the manufacturer's instructions. DQ Collagen Fluorescein conjugate was added to undiluted conditioned medium. This was incubated for 4 h at ambient temperature, protected from light. The fluorescence was measured at 480 nm.
Histological analysis
Samples were dehydrated using graded alcohol steps, immersed in xylene, embedded in paraffin, and cut into 5 mm sections. To evaluate the proteoglycan content, 0.125% safranin-O (Merck counterstained with Weigert's hematoxylin [Klinipath], 0.4% fast green [Merck]) staining was used. Type II collagen deposition was determined by immunohistochemistry. Antigen retrieval was performed by subjecting the sections to 1 mg/mL pronase (Sigma-Aldrich) for 30 min at 37°C, followed by 10 mg/mL hyaluronidase (Sigma-Aldrich) incubation for 30 min at 37°C. Subsequently, the sections were blocked using a 5% bovine serum albumin (BSA) in PBS solution for 1 h, followed by an overnight incubation at 4°C with a primary antibody against human type II collagen (mouse anti-human type II collagen, II-II6B3, 1:100 dilution in PBS-BSA-5%; Developmental Studies). After washing, the slides were incubated with a horseradish peroxidase-conjugated anti-mouse secondary antibody (1:100 dilution in PBS-BSA-5%) for 60 min at ambient temperature. Immunoreactivity was visualized using 3,3'-diaminobenzidine (DAB, Sigma-Aldrich). The sections were counterstained with Mayer's hematoxylin.
Extracellular vesicles isolation
To isolate extracellular vesicles, the conditioned medium from cultured human BMMSCs was subjected to differential centrifugation as described before (26). In short, cells were removed by two sequential centrifugations at 200 × g for 10 min. Collected supernatant was subsequently centrifuged two times at 500 × g for 10 min, followed by 10,000 × g for 45 min. Vesicles were finally pelleted by ultracentrifugation at 100,000 × g for 16 h in SW28 or SW32Ti rotor (Beckman) followed by washing in PBS containing 0.5% BSA and pelleting in SW40 or SW60 rotor (Beckman).
Sucrose density gradient and Western blotting
Extracellular vesicles isolated by differential centrifugation were suspended in 250 μL PBS-2.5M sucrose, loaded in a SW60 tube and overlaid with 15 successive 250 mL layers of 20 mM Tris pH 7.4 containing decreasing concentrations of sucrose (from 2 to 0.4 M). Tubes were centrifuged for 16 h at 200,000 × g at 4°C. Fractions of 250 μL were collected and sucrose density was measured using a refractometer. Fractions were mixed 1:1 with Laemmli sample buffer and incubated for 5 min at 95°C, followed by SDS-page and Western blotting analysis using standard procedures. In brief, proteins were transferred to polyvinylidene difluoride (PVDF) membrane (Millipore) and incubated with the following antibodies: mouse anti-CD9 (1:1000; Biolegend), mouse anti-CD63 (1:1000; Abcam) and rabbit anti-calnexin (1:1000; Abcam). Membranes were washed, incubated with appropriate peroxidase-conjugated secondary antibodies and developed by ELC (Amersham Pharmacia).
For assessment of IκBα phosphorylation, chondrocytes were cultured in 6-well plates in monolayer at a seeding density of 25,000 cells/cm2. After 24 h pre-incubation with BMMSC-EVs from BMMSC donor 3, cells were treated with 10 ng/mL TNF-alpha (Immunotools). Directly after treatment, cells were lysed with Laemmli sample buffer containing HaltTM Protease Inhibitor Cocktail and a HaltTM phosphatase inhibitor cocktail (Thermo Scientific) and incubated for 5 min at 95°C, followed by SDS-page and Western blotting analysis using standard procedures. For detection of phosphorylated IκBα, mouse anti- IκBα (ser32/36) (1:1000; Cell Signaling) was used, and for detection of tubulin, mouse anti-tubulin (Sigma-Aldrich (T9026)) was used, followed by incubation with appropriate peroxidase-conjugated secondary antibodies and developed by SuperSignal West Dura Extended Duration Substrate (ThermoFisher).
Nanoparticle Tracking Analysis
Size distribution of isolated EVs and EV quantification was determined by NTA using NanoSight NS500 instrument (Malvern Instruments Ltd, Malvern, UK), equipped with sCMOS camera. Data was analyzed with the NTA software version 3.1. (build 3.1.54), with detection threshold set to 5, and blur and Max Jump Distance set to auto. Samples were diluted 200- or 400-fold with PBS to reach optimal concentration for instrument linearity. Readings were taken at a camera level set to 13 and with manual monitoring of temperature.
Immuno-Electron microscopy
Immuno-EM analysis of whole-mount BMMSC-EVs was performed as previously described (26). BMMSC-EVs were isolated by differential centrifugation followed by sucrose density gradient purification. Fractions corresponding to the densities 1.072-1.0899 g/mL, 1.1082-1.972 g/mL and 1.2025-1.2575 g/mL were pooled and pelleted by centrifugation at 100,000 × g for 16 h. The pellets of purified EVs were fixed in 2% paraformaldehyde and mounted on formvar/carbon-coated TEM grids. EVs were labelled with mouse anti-CD9 (1:100; Biolegend) or mouse anti-human CD63 (1:300; DSHB Hybridoma Product H5C6, deposited by August, J.T. / Hildreth, J.E.K.) followed by rabbit anti-mouse (1:200, Rockland, 610-4120) and Protein A coupled to 10 nm colloidal gold (1:50, CMC, UMC Utrecht), contrasted with uranyl oxalate (pH 7), and then contrasted-embedded in a mixture of 2% methyl cellulose/4% uranyl acetate (pH 4). Grids were imaged at 80 kV with a Tecnai T-12 Transmission Electron Microscope (FEI). Size of individual vesicles was measured in at least 5 different frames using FIJI (27).
Uptake of BMMSC-EVs by OA chondrocytes
BMMSC-EVs were isolated from TERT-BMMSC previously generated from primary BMMSC in our laboratory (28), and were labeled with carboxyfluorescein diacetate succinimidyl-ester (CFSE) (Invitrogen) as previously described (29). Briefly, EVs collected after ultracentrifugation at 100,000 × g were incubated with 20 μM CFSE for 60 min at 37°C in a final volume of 30 μL PBS containing 0.5% BSA. Labeled EVs were further diluted with 95 μL PBS containing 0.5% BSA and subjected to sucrose density gradient purification. Subsequently, fractions corresponding to the densities 1.072-1.0899 g/ml, 1.1082-1.972 g/ml and 1.2025-1.2575 g/ml were pooled and pelleted by centrifugation at 100,000 × g for 16 h. Chondrocytes from OA patients were plated on glass coverslips and incubated with CFSE-labelled BMMSC-EVs in humidified chamber for 30 min at 37°C followed by fixation in 0.1 M phosphate buffer containing 4% paraformaldehyde for 15 min at room temperature and permeabilization with 0.1% Triton X-100 for 5 min. Thereafter, chondrocytes were analyzed by immunofluorescence and confocal microscopy.
Confocal microscopy
For BMMSC-EV uptake analysis, fixed OA chondrocytes were incubated with 2% BSA for 30 min followed by 1 h incubation with a rabbit anti-EEA1 antibody (1:200; Cell Signaling) and mouse anti-LAMP-1 (1:400, BD Pharmigen) followed by subsequent incubation with a donkey-anti-rabbit antibody labelled with DyLight 549 (Jackson) and a goat-anti-mouse antibody labelled with DyLight 649 (Jackson), respectively. For nuclear staining, DAPI was used. Images were recorded on a Zeiss LSM 700 confocal microscope (Germany). The collected z-stacks were 0.36 μm.
For NFκB p65 nuclear translocation analysis OA chondrocytes were cultured in 6-well plates and plated on glass coverslips, fixed directly after treatment in 0.1 M phosphate buffer containing 3.7% formaldehyde for 15 min at room temperature and permeabilized with 0.1% Triton X-100 for 5 min. Thereafter, cells were incubated with 2% BSA for 30 min followed by 1 h incubation with a rabbit anti-NFκB p65 antibody (1:100; Cell Signaling) and subsequent incubation with a goat-anti-rabbit-Ig antibody labelled with Alexa 681 (Molecular Probes). For nuclear staining, DAPI was used. Images were recorded on a Zeiss LSM 700 confocal microscope (Germany). The quantification of NFκB p65 positive nuclei was done using the ImageJ Plugin “Cell counter”. The NFκB p65 positive nuclei were calculated as percentage of total nuclei labelled with DAPI.
Statistical analysis
Unpaired or paired two-sided student's test was used to calculate statistical differences. A P-value of <0.05 was considered statistically significant.
Results
Bone marrow derived MSC from two independent healthy donors secrete extracellular vesicles positive for exosomal markers
Bone marrow derived MSC, similar to other cell types in the body, secrete different subsets of extracellular vesicles. So far the immunomodulatory and/or tissue repair properties of BMMSC-EVs have been attributed to the EV subset referred to as exosomes (13-15,20,30). We undertook a detailed characterization of BMMSC-EVs from two independent healthy BMMSC donors. The BMMSC-EVs were isolated by a well-established differential centrifugation method and our analyses were focused on the EV population pelleted by ultracentrifugation at 100,000 × g. Western blot analysis of BMMSC-EVs subjected to sucrose density gradient revealed that they are positive for exosomal markers such as tetraspanins CD9 and CD63 [Figure 1A]. BMMSC-EVs from both donors floated at the densities ranging from 1.14 g/mL to 1.22 g/mL, previously reported to contain exosomes. CD63 signal was more abundant in higher density fractions (1.18 g/mL to 1.22 g/mL), while CD9 signal was also detected with high intensity in fractions corresponding to densities of 1.15 g/mL and 1.16 g/mL. All fractions were negative for calnexin, an integral protein of endoplasmic reticulum, confirming the purity of the isolated EVs.
Characterization of extracellular vesicles derived from two bone marrow MSC donors. BMMSC-EVs are positive for exosomal markers CD63 and CD9. (A) EVs isolated from conditioned medium derived from primary bone marrow MSCs were subjected to sucrose density gradient followed by Western blot analysis for presence of CD63, CD9 or calnexin. Representative western-blots of 3 independent experiments are shown. (B) EVs isolated from conditioned medium derived from primary bone marrow MSCs and subjected to sucrose density gradient. The EVs residing in the fractions corresponding to densities 1.1082 g/mL to 1.972 g/mL were pooled and analyzed by immuno-electron microscopy. Representative micrographs of 3 independent experiments are shown. Scale bar is 50 nm. (See also Figure S1.) CL- cell lysate.
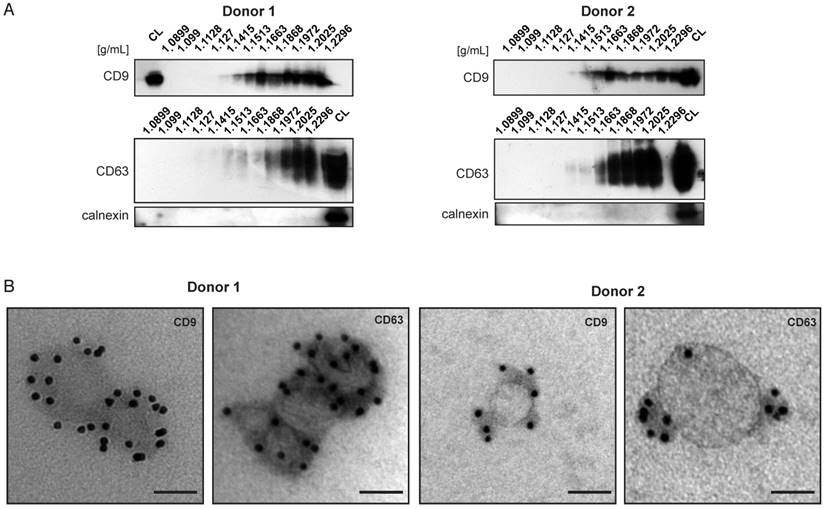
BMMSC-EVs were heterogeneous in size, as determined by electron microscopy and nanoparticle tracking analysis [Figure 1B and Figure S1A-C]. The sizes of EVs positive for CD9 and CD63 ranged between 40 nm and 150 nm, corresponding to sizes previously reported for exosomes. We also detected BMMSC-EVs, which were negative for exosomal markers [Figure S1C]. Some, but not all, of these EVs were larger than 150 nm.
Taken together, these data show that EVs isolated from BMMSC donors are heterogeneous in size and are positive for exosomal markers.
Osteoarthritic chondrocytes internalize bone marrow MSC-derived EVs
We hypothesized that BMMSC-EVs interact with chondrocytes from osteoarthritic patients to modulate cartilage regeneration. To test this, BMMSC-derived EVs were labeled with carboxyfluorescein diacetate succinimidyl-ester (CFSE), a membrane permeable nonfluorescent compound that becomes fluorescent after cleavage of its acetate groups by intracellular esterases (29), and incubated with OA chondrocytes. BMMSC-EVs not only interacted, but were taken up by OA chondrocytes after as short as 30 min of incubation, as shown by Z projections of the imaged cells [Figure 2]. CFSE-labeled BMMSC-EVs co-localized with late endosomal marker LAMP-1, but showed little or no co-localization with early endosomal marker EEA1, indicating that BMMSC-EVs were rapidly internalized by OA chondrocytes and already after 30 min of incubation were present in the late endosomal compartment of these cells.
BMMSC-EVs are taken up by chondrocytes derived from OA patients. CFSE labelled EVs derived from TERT-bone marrow MSC were incubated for 30 min with OA chondrocytes and the uptake of EVs was analyzed by confocal microscopy. Left panel: Representative images of co-localization analyses of CFSE labelled (green) BMMSC-EVs with late endosomal marker LAMP1 (red) in OA chondrocytes are shown. Arrow heads indicate the co-localization (yellow) of BMMSC-EVs with LAMP1. Right panel: Representative images of co-localization analyses of CFSE labelled (green) BMMSC-EVs with early endosomal marker EEA1 (red) in OA chondrocytes is shown. Images are representative of 3 independent experiments. Scale bar is 10 µm.
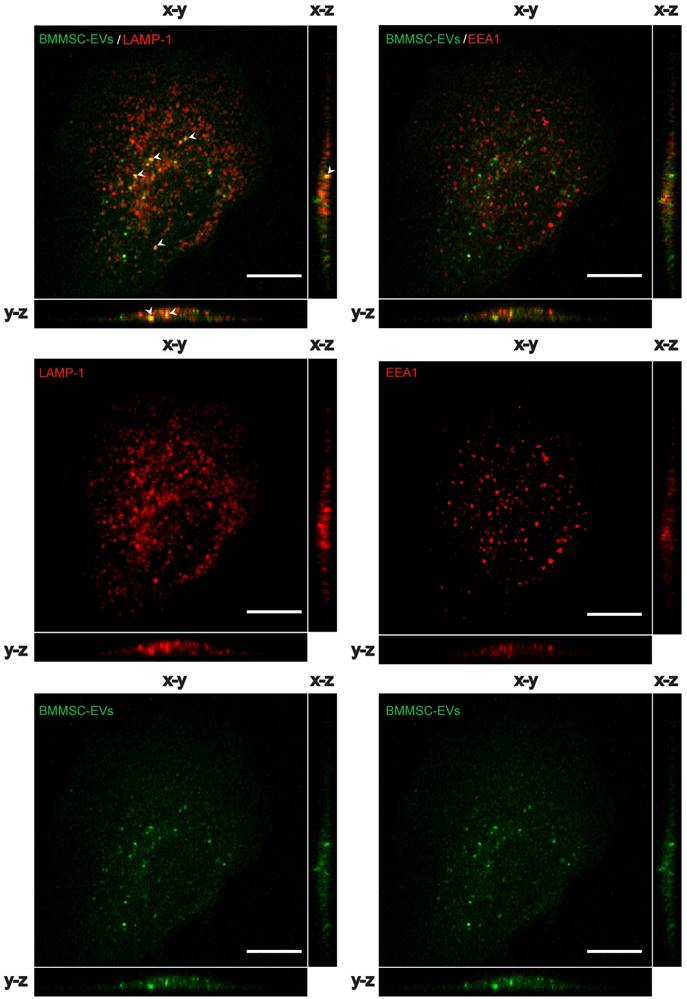
These data indicate that bone marrow MSC secrete EVs that interact and are rapidly taken up by chondrocytes that come from an osteoarthritic joint.
BMMSC-EVs inhibit TNF-alpha-induced inflammatory effects in chondrocytes derived from osteoarthritic patients
One of typical OA symptoms is synovial inflammation (31). Elevated levels of synovial pro-inflammatory cytokines, such as TNF-alpha, activate chondrocytes to produce pro-inflammatory mediators and stimulate continues cartilage degradation. An important pro-inflammatory factor present in the inflamed OA joint is prostaglandin E2, which is produced by cytokine-stimulated expression of cyclooxygenase 2 (COX2) (32). Thus, increased COX2 gene expression is a hallmark of OA chondrocytes.
To test whether BMMSC-EVs could inhibit progressing of OA cartilage inflammation, we mimicked the inflammatory conditions by stimulating OA chondrocytes with TNF-alpha. TNF-alpha treated cells were incubated for 48 h with BMMSC-EVs or with conditioned medium from BMMSC (BMMSC-CM), which was used for EV isolation, or with BMMSC conditioned medium depleted of EVs (BMMSC-EDCM) as a control, and COX2 gene expression was analyzed. Treatment with BMMSC-EVs from two independent donors significantly downregulated TNF-alpha-induced COX2 expression [Figure 3A]. The effect of MSC-EVs was less prominent then the downregulation of COX2 expression by BMMSC-CM, suggesting that, secondary to BMMSC-EVs, there are additional factors present in BMMSC-CM that contribute to its anti-inflammatory potential. Importantly, the depletion of EVs from BMMSC-CM resulted in significantly lower inhibition of COX2 expression, indicating that EVs are an important component of BMMSC anti-inflammatory paracrine signaling [Figure 3A]. Next, we tested whether BMMSC-EVs could also inhibit expression of other pro-inflammatory mediators previously demonstrated to be important for progressing cartilage degradation (33, 34). Indeed, BMMSC-EVs downregulated expression of pro-inflammatory interleukins, namely: IL-1 alpha, IL-1 beta, IL-6, IL-8 and IL-17, further corroborating the important anti-inflammatory role of BMMSC-EVs in OA chondrocytes [Figure 3B].
BMMSC-EVs inhibit TNF-alpha-induced inflammatory effects in chondrocytes derived from osteoarthritic patients. (A,B) Chondrocytes from six OA patients were treated with BMMSC-EVs, BMMSC conditioned medium (BMMSC-CM) or BMMSC conditioned medium depleted of EVs (BMMSC-EDCM) - all equivalent of 500x103 cells - from two healthy allogeneic BMMSC donors for 48 h and with 10 ng/mL TNF-alpha for 24 h. For BMMSC-EVs, equivalent of 500x103 cells equals: ~1.7x108 particles for BMMSC donor 1 and ~1.8x109 particles for BMMSC donor 2 as determined by NTA. Gene expression was analyzed in OA chondrocytes by qRT-PCR. Quantification of data from 3 independent experiments performed in duplicate is shown as mean ± SEM normalized for 18S. ****p<0.0001; *** p<0.001; ** p<0.01; * p<0.05. The data are presented as fold changes relative to TNF-alpha treated control. (C) Chondrocytes from four OA patients were treated with BMMSC-EVs from one allogeneic BMMSC donor for 48 h and with 10 ng/mL TNF-alpha for 24 h. BMMSC-EVs equivalent of 2.5 x106 cells was added every 24 h hours, which equals ~1.2x109 particles as determined by NTA for this donor. Collagenase activity in chondrocyte conditioned medium is shown. Data from 3 independent experiments are shown as mean ± SEM. * p<0.05. (D) BMMSC-EVs rescue proliferation of OA chondrocytes abrogated by TNF-alpha. OA chondrocytes were treated with BMMSC-EVs from one allogeneic BMMSC donor for 48 h and with 10 ng/mL TNF-alpha for 24 h. BMMSC-EVs equivalent of 500 x103 cells was added, which equals ~2.4x108 particles as determined by NTA for this donor. Cell proliferation was assessed by EdU assay. Data from 2 independent experiments performed in triplicates are shown as mean ± SEM. *** p<0.001; ** p<0.002
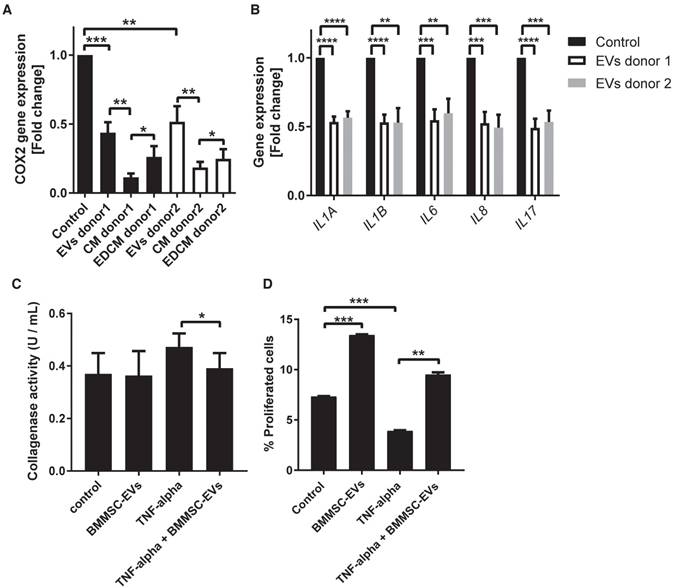
TNF-alpha upregulates collagenases, which play a major role in cartilage degradation in OA (35, 36). To test whether BMMSC-EVs also have an inhibitory effect on cartilage degradation, collagenase activity was measured in the conditioned medium of OA chondrocytes treated with TNF-alpha and incubated with BMMSC-EVs. TNF-alpha increased collagenase activity, which could be inhibited by the addition of BMMSC-EVs [Figure 3C].
An important consequence of progressing cartilage inflammation is reduced chondrocyte proliferation and increased apoptosis. To investigate whether BMMSC-EVs have an effect on OA chondrocyte proliferation, the TNF-alpha treated cells were incubated with BMMSC-EVs and their proliferation was assessed by EdU analysis. Treatment with BMMSC-EVs induced proliferation of OA chondrocytes and abrogated the inhibitory effect of TNF-alpha on this process [Figure 3D]. We have not observed a significant effect of BMMSC-EVs on TNF-alpha induced chondrocyte apoptosis, as determined by cleaved caspase-3 levels (data not shown).
Together, these data show that BMMSC-EVs have anti-inflammatory effect in TNF-alpha stimulated chondrocytes from OA patients.
BMMSC-EVs inhibit TNF-alpha-induced pro-inflammatory NFκB signaling in OA chondrocytes
NFκB signaling is the major signaling pathway activated by TNF-alpha in OA chondrocytes (37, 38). To understand whether the anti-inflammatory effect of BMMSC-EVs, could be mediated through the regulation of NFκB activity, we treated TNF-alpha-stimulated OA chondrocytes with BMMSC-EVs and analyzed the subcellular localization of the p65 subunit of NFκB. As expected, treatment of OA chondrocytes with TNF-alpha induced translocation of p65 from cytoplasm to nucleus suggesting NF κB activation, which could be abrogated by BMMSC-EVs [Figure 4A and B]. To further substantiate these data, we tested whether phosphorylation of IκBα, an inhibitory subunit of NFκB, that promotes the activation of NFκB transcriptional activity, could be blocked by BMMSC-EVs. Indeed, TNF-alpha-induced phosphorylation of IκBα in OA chondrocytes was inhibited by treatment with BMMSC-EVs [Figure 4C].
BMMSC-EVs inhibit TNF-alpha-induced pro-inflammatory NFκB signaling in OA chondrocytes. Chondrocytes from two OA patients were treated with BMMSC-EVs from one allogeneic BMMSC donor for 24 h and with 10 ng/mL TNF-alpha for 30 min. BMMSC-EVs equivalent of 2.5 x106 cells was added, which equals ~1.2x109 particles as determined by NTA for this donor. (A) BMMSC-EVs inhibit TNF-alpha-induced nuclear translocation of p65 subunit of NFκB. Nuclear translocation of p65 subunit of NFκB was assessed by immunofluorescence analysis and confocal microscopy. Representative images of two independent experiments are shown. (B) Quantification of data presented in A. Data from 2 independent experiments are shown as mean ± SEM. (C) BMMSC-EVs abrogate TNF-alpha-induced phosphorylation of IκBα. The OA chondrocytes were treated as in (A) and lysed in sample buffer directly after treatment. Cell lysates were analyzed by Western-blot for presence of pIκBα. Representative results of two independent experiments performed with chondrocytes of four OA donors are shown.
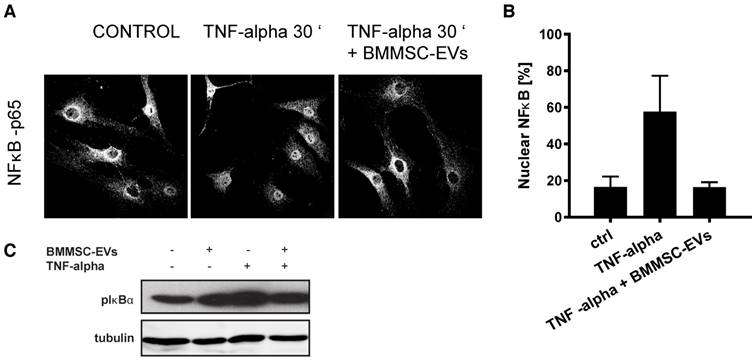
These data demonstrate that the anti-inflammatory effect of BMMSC-EVs involves inhibition of NFκB signaling pathway.
BMMSC-EVs promote proteoglycan and type II collagen production by osteoarthritic chondrocytes
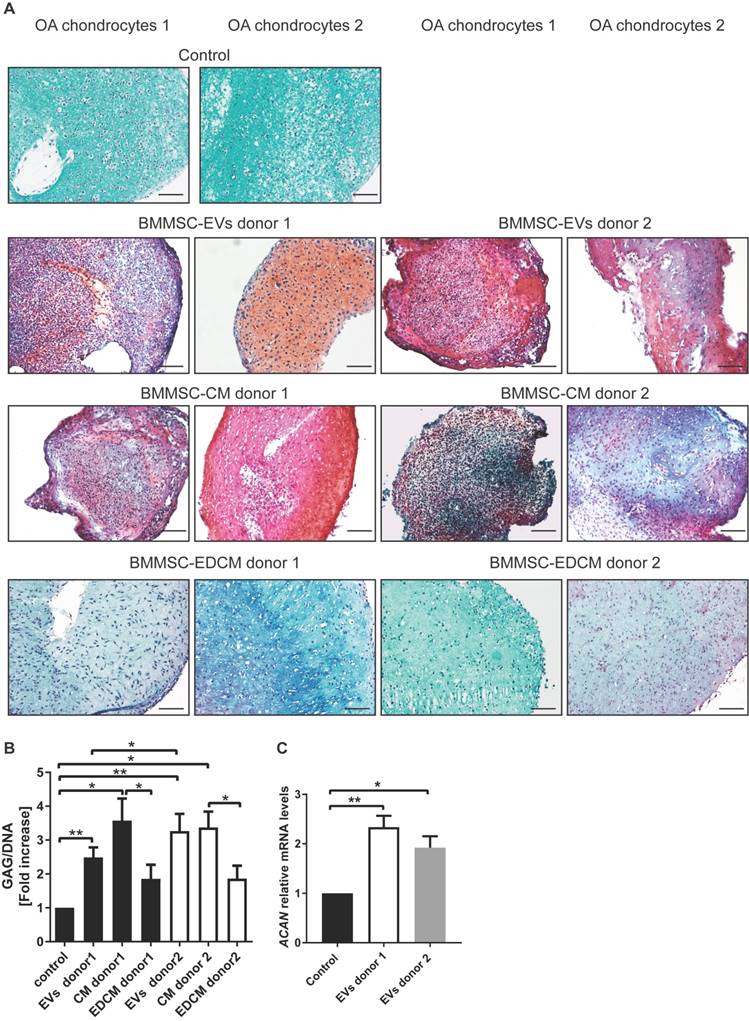
To investigate the effects of BMMSC-EVs on cartilage regeneration by chondrocytes from an osteoarthritic joint, the cells were cultured in fibrin constructs and treated with BMMSC-EVs for 28 days. Treatment with MSC-EVs was repeated every 5 days to ensure the presence of active extracellular vesicles in the cultures, as EVs stability at 37°C is limited (39). BMMSC-EVs significantly improved the content of proteoglycans in the newly formed tissue after 28 days, as shown by safranin-O stainings [Figure 5A], and as demonstrated by quantitative biochemical measurement of GAG amount per cell [Figure 5B and Figure S2]. Importantly, treatment of OA chondrocytes with BMMSC-EVs induced gene expression of aggrecan, the major proteoglycan in articular cartilage, further corroborating the beneficial effect of BMMSC-EVs on proteoglycan production in OA patient cells [Figure 5C]. The production of type II collagen, another essential component of articular cartilage, was also upregulated by treatment with BMMSC-EVs, as verified by immunohistochemistry analysis [Figure 6A] and COL2A1 gene expression in 28 day OA chondrocyte cultures [Figure 6B].
BMMSC-EVs promote proteoglycan production by chondrocytes derived from osteoarthritic patients. (A) BMMSC-EVs upregulate proteoglycan expression at the protein level in OA chondrocytes. Chondrocytes from OA patients were cultured for 28 days in fibrin glue. The BMMSC-EVs, BMMSC conditioned medium (BMMSC-CM), BMMSC conditioned medium depleted from EVs (BMMSC-EDCM) - all equivalent of 500x103 cells from two healthy allogeneic BMMSC donors - were added every 5 days. For BMMSC-EVs, equivalent of 500x103 cells equals ~1.7x108 particles for BMMSC donor 1 and ~1.8x109 particles for BMMSC donor 2 as determined by NTA. Images of Safranin-O proteoglycan staining of 28 day cultures of chondrocytes from two OA patients are shown. The images are representative of at least 3 independent experiments. Scale bar is 200 µm. (B) 28 day chondrocytes cultures from seven OA patients treated as in (A) were digested and analyzed for proteoglycan content (determined as glycosaminoglycans, normalized for DNA). Data of 3 independent experiments are presented as mean ± SEM. **p< 0.05, *p< 0.03. The data are presented as fold increases relative to untreated control. (C) The expression of ACAN is upregulated by BMMSC-EVs in OA chondrocytes. Gene expression was analyzed in 28 day culture of OA chondrocytes from 6 OA patients by qRT-PCR. Quantification of data from 3 independent experiments performed at least in duplicates are shown as mean ± SEM normalized for 18S. * p< 0.02, **p< 0.005. The data are presented as fold increases relative to untreated control.
BMMSC-EVs promote collagen II production by chondrocytes derived from osteoarthritic patients. (A) BMMSC-EVs upregulate collagen II protein expression in OA chondrocytes. Chondrocytes from OA patients were cultured for 28 days in fibrin glue. The BMMSC-EVs, BMMSC conditioned medium (BMMSC-CM), BMMSC conditioned medium depleted from EVs (BMMSC-EDCM) - all equivalent of 500x103 cells - from healthy allogeneic BMMSC donor were added every 5 days. For BMMSC-EVs, equivalent of 500x103 cells equals: ~1.7x108 particles for BMMSC donor 1 and ~1.8x109 particles for BMMSC donor 2. Images of collagen II staining of 28 day cultures of chondrocytes from two OA patients are shown. The images are representative of at least 3 independent experiments. Scale bar is 100 µm. (B) The expression of COLII is upregulated by BMMSC-EVs in OA chondrocytes. Gene expression was analyzed in 28 day culture of OA chondrocytes from 6 OA patients by qRT-PCR. Quantification of data from 3 independent experiments performed at least in duplicates are shown as mean ± SEM normalized for 18S. ** p< 0.01. The data are presented as fold increases relative to untreated control. (C) The expression of genes controlling chondrocyte regeneration is upregulated by BMMSC-EVs. Gene expression was analyzed in 28 day culture of OA chondrocytes from 2 OA patients. Quantification of data from 2 independent experiments performed at least in duplicates are shown as mean ± SEM normalized for 18S. ** p< 0.02; *p< 0.05. The data are presented as fold increases relative to untreated control. (D) BMMSC-EVs downregulate expression of genes involved in hypertrophic chondrocyte differentiation. Gene expression was analyzed in 28 day culture of OA chondrocytes from 2 OA patients. Quantification of data from 2 independent experiments performed at least in duplicates are shown as mean ± SEM normalized for 18S. ** p< 0.01; * p< 0.02. The data are presented as fold increases relative to untreated control. (E) BMMSC-EVs promote metabolic activity of OA chondrocytes. Chondrocytes from 2 OA patients were cultured for 5 days in fibrin glue. Where indicated, BMMSC-EVs from one allogeneic BMMSC donor were added to the cultures as equivalent of 500x103 cells (~2.4x108 particles). At day 5 AlamarBlue assay was performed to determine metabolic activity of the cells. Data of 2 independent experiments performed in triplicates are shown as mean ± SEM. ****p< 0.0001 (F) BMMSC-EVs promote OA chondrocyte proliferation. DNA content of 28 day chondrocytes cultures from 7 OA patients treated as in (A) is presented. Data of 3 independent experiments performed at least in duplicates are shown as mean ± SEM. ***p< 0.001, **p< 0.01, *p< 0.05. The data are presented as fold increases relative to untreated control.
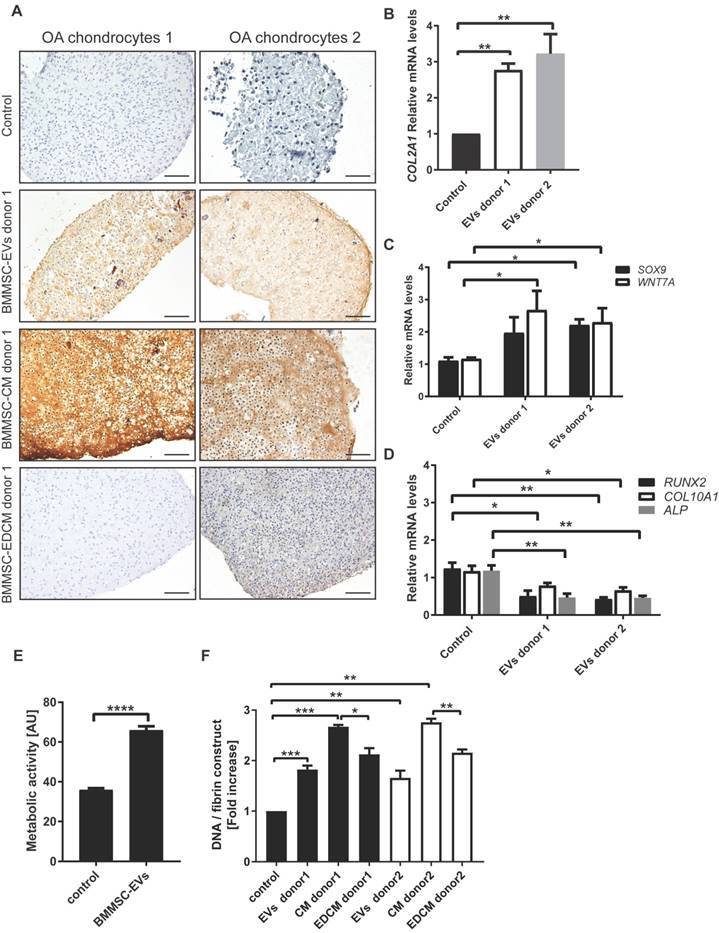
To address the specificity of BMMSC-EV effect, OA chondrocytes were treated with BMMSC-CM and BMMSC-EDCM. BMMSC-CM induced production of proteoglycans and type II collagen to a similar extent as BMMSC-EVs [Figure 5A, B; Figure S2A and Figure 6A], and this effect was significantly inhibited when EVs were removed from BMMSC-CM. These data indicate that BMMSC-EVs are an essential component of BMMSC paracrine signaling promoting proteoglycans and type II collagen production by OA chondrocytes.
To gain more insight into the mechanism by which BMMSC-EVs promote production of cartilage components, the expression levels of genes previously shown to play important role in chondrogenesis were measured (40). BMMSC-EVs from 2 different donors increased gene expression levels of both SRY-box 9 (SOX9) and Wnt family member 7A (WNT7A) [Figure 6C]. SOX9 is a chondrogenic transcription factor, whereas WNT7A is upregulated in transforming growth factor-beta (TGF-beta)-induced chondrogenesis (41). Importantly, treatment of OA chondrocyte with BMMSC-EVs resulted in downregulated expression of genes involved in hypertrophy, namely runt related transcription factor 2 (RUNX2), type X collagen (COL10A1) and alkaline phosphatase (ALP) [Figure 6D].
Next we asked whether treatment with BMMSC-EVs had an impact on proliferation of OA chondrocytes during regeneration. Indeed, BMMSC-EVs promoted proliferation of OA chondrocytes in monolayer culture [Figure 6E]. In regeneration cultures, this was indirectly supported by an increase in DNA content [Figure 6F]. BMMSC-EVs induced an increase in DNA content, albeit not as strong as they stimulated the production of extracellular matrix components of OA cartilage. The increase in proliferation of OA chondrocytes was also more dependent on other secreted factors present in BMMSC-CM, as depletion of BMMSC-EVs from BMMSC-CM led to a significant but small reduction of BMMSC-CM effect [Figure 6F]. Importantly, the overall metabolic activity was also increased in regeneration cultures treated with BMMSC-EVs [Figure 6E].
Taken together, these data demonstrate that treatment with BMMSC-EVs promotes OA cartilage regeneration.
Discussion
Osteoarthritis is one of the most prevalent joint diseases and a major public health problem. It is characterized by progressive articular cartilage destruction and synovitis (42,43). Current therapies attempt to relieve the symptoms, but they cannot stop or reverse the ongoing cartilage degeneration (4,44). The ideal treatment aiming for an optimal OA joint repair should promote regenerative properties of chondrocytes and fight destructive effects of inflammation. In this study we show for the first time that extracellular vesicles derived from BMMSC may fulfil these requirements. Our data demonstrate that BMMSC-EVs have an anti-inflammatory effect on TNF-alpha-stimulated OA chondrocytes. We also provide evidence that BMMSC-EVs induce production of crucial extracellular matrix components of OA chondrocytes, proteoglycans and type II collagen, a process that is essential for proper cartilage regeneration.
The BMMSC-EVs from two independent bone marrow MSC donors tested in our study showed similar characteristics regarding the presence of surface markers (CD63 and CD9), size and flotation density in sucrose gradient. All reported features of BMMSC-EVs from both donors corresponded to those previously shown for exosomes (45-47). In line with the absence of detected differences in the BMMSC-EVs derived from two BMMSC donors, we found no significant differences in both anti-inflammatory and pro-regenerative effects of BMMSC-EVs from these two donors on OA chondrocytes. The only exception was slightly stronger induction of proteoglycan production by BMMSC-EVs from BMMSC donor two in OA chondrocytes [Figure 5B]. Screening of more BMMSC-EVs donors should facilitate defining whether this minor difference in the BMMSC-EVs effect on proteoglycan production is a more general phenomenon that results in real functional differences between EVs from different BMMSC donors and may be important for future clinical application of BMMSC-EVs.
This study is the first to show the interaction between human BMMSC-EVs and chondrocytes from OA patients. Our data demonstrate that OA chondrocytes internalize BMMSC-EVs by endocytosis, as CFSE labeled vesicles co-localized with the late endocytic marker LAMP-1 after 30 min of co-incubation with the cells. This suggests that by BMMSC-EV uptake, chondrocytes might utilize protein, RNA or other types of cargo transferred by BMMSC-EVs and initiate specialized signaling to facilitate OA cartilage repair. Future experiments are necessary to identify the precise molecular mechanism governing BMMSC-EV-mediated effects on OA chondrocytes.
MSC-EVs have been shown to possess immunomodulatory properties in vitro and also in the first-in-man study, where these vesicles were used to treat severe therapy-refractory GvHD patient (13,48). Indeed, our data also demonstrate that BMMSC-EVs from two independent BMMSC donors inhibited TNF-alpha-induced expression of COX2 and expression of pro-inflammatory interleukins in OA chondrocytes, indicating their significant anti-inflammatory potential in cartilage cells. Our study also shows that treatment with BMMSC-EVs inhibits TNF-alpha-induced collagenase activity and promotes OA chondrocytes proliferation, the crucial processes for retaining cartilage homeostasis. Although, BMMSC-EVs stimulated an almost 2-fold reduction in COX2 expression, their relative contribution to the effect of the total MSC secretome was smaller compare to their strong regenerative properties. The other, as yet unidentified, secretory factors present in BMMSC-CM seem to play, next to EVs, an important role in BMMSC immunomodulatory effects on OA chondrocytes. However, analysis of BMMSC-EVs' impact on immune cells present in synovial fluid and immune cells penetrating joint synovial tissue of OA patients, such as macrophages, should give a more complete picture of BMMSC-EVs anti-inflammatory properties.
Our study is the first to shed light on the molecular mechanism behind the anti-inflammatory effects of BMMSC-EVs on OA chondrocytes. We show that BMMSC-EVs suppress the phosphorylation of IκBα, an inhibitory subunit of NFκB, and prevent its translocation to the nucleus, thus abrogating its transcriptional activity. This corresponds with BMMSC-EVs-mediated inhibition of collagenase activity and downregulation of expression of interleukins and COX2, which are established targets of NFκB signaling in OA chondrocytes (49).
Production of essential extracellular matrix components, such as proteoglycans and type II collagen, is crucial for proper cartilage regeneration (50-53). Treatment of OA chondrocytes with BMMSC-EVs induced the synthesis of these important cartilage constituents in our in vitro cartilage repair model (21), indicating that BMMSC-EVs can indeed promote cartilage regeneration. Importantly, this effect was specific to BMMSC-EVs as depletion of BMMSC-EVs from BMMSC conditioned medium strongly inhibited the BMMSC-CM-mediated proteoglycans and type II collagen production by OA chondrocytes. Our report is the first to show the specificity of BMMSC-EVs regenerative effects and our data are the first to assess the real contribution of BMMSC-EVs in the effect mediated by the total MSC secretome. Previous studies have solely focused on showing the regenerative potential of isolated MSC-derived vesicles alone in vivo (14,15,20,54,55). However, these reports have not investigated what is the impact of EVs with regard to other factors secreted by MSC.
Our data demonstrate that treatment with BMMSC-EVs upregulates SOX9 and WNT7A expression in OA chondrocytes, suggesting that these factors are involved in the effect of BMMSC-EVs on cartilage regeneration. In line with the induced SOX9 expression, BMMSC-EVs have downregulated genes involved in hypertrophic differentiation, namely RUNX2, COL10A1 and ALP (56, 57). This suggests that BMMSC-EVs not only promote chondrogenesis but also inhibit hyperthophic differentiation of chondrocytes.
Besides stimulation of extracellular matrix production, treatment of OA chondrocytes with BMMSC-EVs induced proliferation of these cells, an effect that was only partially inhibited by the depletion of EVs from BMMSC conditioned medium. This indicates that other factors in BMMSC secretome also contribute to the control of OA chondrocytes proliferation. Likewise, a recent study demonstrated that fibroblast growth factor 1 secreted by BMMSC can promote proliferation of chondrocytes (58).
Taken together, the data presented here demonstrate for the first time that BMMSC-EVs have both regenerative and immunoregulatory properties in human OA cartilage. The dual potential of BMMSC-EVs makes them a promising candidate for an optimal therapy for OA, which should promote cartilage repair and inhibit ongoing cartilage degradation. Additionally, BMMSC-EVs-based therapy seems to carry less safety risk, since EVs, in contrast to cells, cannot replicate or become transformed. Therefore, BMMSC-EVs could be administered at earlier stages of OA to improve joint homeostasis and prevent OA from further development. This would potentially lower or delay the necessity of surgery for these patients. In cases of advanced OA, BMMSC-EVs could be used to improve the joint condition and might need to be supplemented with other treatment approaches such as cell-transplantations or joint distraction, as there is hardly any cartilage left in late-stage OA joints. However, BMMSC-EV therapy has little precedence, thus its ethical status needs to be clearly defined before it can be introduced to clinics. This may require more in-depth characterization of BMMSC-EVs regarding their RNA and protein content.
Supplementary Material
Supplementary figures and table.
Abbreviations
α-MEM: α-minimal essential medium; BMMSC: bone marrow derived MSC; BMMSC-CM: conditioned medium from BMMSC; BMMSC-EDCM: BMMSC conditioned medium depleted from EVs; CFSE: carboxyfluorescein diacetate succinimidyl-ester; COX2: cyclooxygenase-2; DMMB: 1,9-dimethylmethylene blue; EVs: extracellular vesicles; GAG: glycosaminoglycan; MSC: mesenchymal stromal/stem cells; MVBs: multivesicular bodies; OA: osteoarthritis; PCR: polymerase chain reaction; TNF-alpha: tumor necrosis factor alpha.
Acknowledgements
L.A. Vonk and D.B.F. Saris were supported by ZonMw TAS grants, M.J. Lorenowicz was supported by a grant (13-3-306) of Dutch Arthritis Foundation, S.F.J. van Dooremalen was supported by grant of WKZ Foundation.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Cross M, Smith E, Hoy D, Nolte S, Ackerman I, Fransen M. et al. The global burden of hip and knee osteoarthritis: estimates from the Global Burden of Disease 2010 study. Ann Rheum Dis. 2014;73:1323-1330
2. Liu-Bryan R, Terkeltaub R. Emerging regulators of the inflammatory process in osteoarthritis. Nat Rev Rheumatol. 2015;11:35-44
3. Robinson WH, Lepus CM, Wang Q, Raghu H, Mao R, Lindstrom TM. et al. Low-grade inflammation as a key mediator of the pathogenesis of osteoarthritis. Nat Rev Rheumatol. 2016;12:580-592
4. Diekman BO, Guilak F. Stem cell-based therapies for osteoarthritis: challenges and opportunities. Curr Opin Rheumatol. 2013;25:119-26
5. Saris DBF, Dhert WJA, Verbout AJ. Joint homeostasis. The discrepancy between old and fresh defects in cartilage repair. J Bone Joint Surg Br. 2003;85:1067-76
6. Kristjánsson B, Honsawek S. Current Perspectives in Mesenchymal Stem Cell Therapies for Osteoarthritis. Stem Cells Int. 2014;2014:1-13
7. Windt TS de, Vonk LA, Slaper-Cortenbach ICM, Broek MPH van den, Nizak R, Rijen MHP van. et al. Allogeneic Mesenchymal Stem Cells Stimulate Cartilage Regeneration and Are Safe for Single-Stage Cartilage Repair in Humans upon Mixture with Recycled Autologous Chondrons. Stem Cells. 2017;35:256-264
8. Uccelli A, Rosbo NK de. The immunomodulatory function of mesenchymal stem cells: mode of action and pathways. Ann N Y Acad Sci. 2015;1351:114-126
9. Roelofs AJ, Rocke JPJ, Bari C De. Cell-based approaches to joint surface repair: a research perspective. Osteoarthr Cartil. 2013;21:892-900
10. Maumus M, Guérit D, Toupet K, Jorgensen C, Noël D. Mesenchymal stem cell-based therapies in regenerative medicine: applications in rheumatology. Stem Cell Res Ther. 2011;2:14
11. Bahr L von, Batsis I, Moll G, Hägg M, Szakos A, Sundberg B. et al. Analysis of tissues following mesenchymal stromal cell therapy in humans indicates limited long-term engraftment and no ectopic tissue formation. Stem Cells. 2012;30:1575-8
12. Gnecchi M, Danieli P, Malpasso G, Ciuffreda MC. Paracrine Mechanisms of Mesenchymal Stem Cells in Tissue Repair. Methods in molecular biology (Clifton, NJ). 2016;1416:123-146
13. Kordelas L, Rebmann V, Ludwig A-K, Radtke S, Ruesing J, Doeppner TR. et al. MSC-derived exosomes: a novel tool to treat therapy-refractory graft-versus-host disease. Leukemia. 2014;28:970-973
14. Lai RC, Arslan F, Lee MM, Sze NS, Choo A, Chen TS. et al. Exosome secreted by MSC reduces myocardial ischemia/reperfusion injury. Stem Cell Res. 2010;4:214-222
15. Zhang B, Wang M, Gong A, Zhang X, Wu X, Zhu Y. et al. HucMSC-exosome mediated -Wnt4 signaling is required for cutaneous wound healing. Stem Cells. 2014
16. Valadi H, Ekström K, Bossios A, Sjöstrand M, Lee JJ, Lötvall JO. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat Cell Biol. 2007;9:654-9
17. Hood JL, San RS, Wickline SA. Exosomes released by melanoma cells prepare sentinel lymph nodes for tumor metastasis. Cancer Res. 2011;71:3792-3801
18. Altevogt P, Bretz NP, Ridinger J, Utikal J, Umansky V. Novel insights into exosome-induced, tumor-associated inflammation and immunomodulation. Semin Cancer Biol. 2014;28C:51-57
19. Robbins PD, Morelli AE. Regulation of immune responses by extracellular vesicles. Nat Rev Immunol. 2014;14:195-208
20. Zhang S, Chu WC, Lai RC, Lim SK, Hui JHP, Toh WS. Exosomes derived from human embryonic mesenchymal stem cells promote osteochondral regeneration. Osteoarthr Cartil. 2016;24:2135-2140
21. Windt TS de, Saris DBF, Slaper-Cortenbach ICM, Rijen MHP van, Gawlitta D, Creemers LB. et al. Direct Cell-Cell Contact with Chondrocytes Is a Key Mechanism in Multipotent Mesenchymal Stromal Cell-Mediated Chondrogenesis. Tissue Eng Part A. 2015;21:2536-47
22. Diest PJ van. No consent should be needed for using leftover body material for scientific purposes. For. BMJ. 2002;325:648-51
23. Federa. Code Goed Gebruik van lichaamsmateriaal 2011No Title.
24. Dominici M, Blanc K Le, Mueller I, Slaper-Cortenbach I, Marini F, Krause DS. et al. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy. 2006;8:315-317
25. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402-8
26. Thery C, Amigorena S, Raposo G, Clayton A. Isolation and characterization of exosomes from cell culture supernatants and biological fluids. Curr Protoc Cell Biol. 2006 Chapter 3:Unit 3 22
27. Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T. et al. Fiji: an open-source platform for biological-image analysis. Nat Methods. 2012;9:676-82
28. Gómez-Puerto MC, Verhagen LP, Braat AK, Lam EW-F, Coffer PJ, Lorenowicz MJ. Activation of autophagy by FOXO3 regulates redox homeostasis during osteogenic differentiation. Autophagy. 2016:1-13
29. Keller S, König A-K, Marmé F, Runz S, Wolterink S, Koensgen D. et al. Systemic presence and tumor-growth promoting effect of ovarian carcinoma released exosomes. Cancer Lett. 2009;278:73-81
30. Zhang B, Yin Y, Lai RC, Tan SS, Choo ABH, Lim SK. Mesenchymal stem cells secrete immunologically active exosomes. Stem Cells Dev. 2014;23:1233-44
31. Liu-Bryan R, Terkeltaub R. Emerging regulators of the inflammatory process in osteoarthritis. Nat Rev Rheumatol. 2014;11:35-44
32. Fan HW, Liu GY, Zhao CF, Li XF, Yang XY. Differential expression of COX-2 in osteoarthritis and rheumatoid arthritis. Genet Mol Res. 2015;14:12872-12879
33. Rogers EL, Reynard LN, Loughlin J. The role of inflammation-related genes in osteoarthritis. Osteoarthr Cartil. 2015;23:1933-8
34. Honorati MC, Bovara M, Cattini L, Piacentini A, Facchini A. Contribution of interleukin 17 to human cartilage degradation and synovial inflammation in osteoarthritis. Osteoarthr Cartil. 2002;10:799-807
35. Salminen HJ, Säämänen A-MK, Vankemmelbeke MN, Auho PK, Perälä MP, Vuorio EI. Differential expression patterns of matrix metalloproteinases and their inhibitors during development of osteoarthritis in a transgenic mouse model. Ann Rheum Dis. 2002;61:591-7
36. Mitchell PG, Magna HA, Reeves LM, Lopresti-Morrow LL, Yocum SA, Rosner PJ. et al. Cloning, expression, and type II collagenolytic activity of matrix metalloproteinase-13 from human osteoarthritic cartilage. J Clin Invest. 1996;97:761-8
37. Li D, Wu Z, Duan Y, Hao D, Zhang X, Luo H. et al. TNFα-mediated apoptosis in human osteoarthritic chondrocytes sensitized by PI3K-NF-κB inhibitor, not mTOR inhibitor. Rheumatol Int. 2012;32:2017-22
38. Roman-Blas JA, Jimenez SA. NF-kappaB as a potential therapeutic target in osteoarthritis and rheumatoid arthritis. Osteoarthr Cartil. 2006;14:839-48
39. Sokolova V, Ludwig AK, Hornung S, Rotan O, Horn PA, Epple M. et al. Characterisation of exosomes derived from human cells by nanoparticle tracking analysis and scanning electron microscopy. Colloids Surf B Biointerfaces. 2011;87:146-150
40. Crombrugghe B de, Lefebvre V, Behringer RR, Bi W, Murakami S, Huang W. Transcriptional mechanisms of chondrocyte differentiation. Matrix Biol. 2000;19:389-94
41. Tuli R, Tuli S, Nandi S, Huang X, Manner PA, Hozack WJ. et al. Transforming growth factor-beta-mediated chondrogenesis of human mesenchymal progenitor cells involves N-cadherin and mitogen-activated protein kinase and Wnt signaling cross-talk. J Biol Chem. 2003;278:41227-36
42. Chen D, Shen J, Zhao W, Wang T, Han L, Hamilton JL. et al. Osteoarthritis: toward a comprehensive understanding of pathological mechanism. Bone Res. 2017;5:16044
43. Mathiessen A, Conaghan PG. Synovitis in osteoarthritis: current understanding with therapeutic implications. Arthritis Res Ther. 2017;19:18
44. Yu SP, Hunter DJ. Intra-articular therapies for osteoarthritis. Expert Opin Pharmacother. 2016;17:2057-2071
45. Raposo G, Stoorvogel W. Extracellular vesicles: Exosomes, microvesicles, and friends. J Cell Biol. 2013;200:373-383
46. Théry C, Boussac M, Véron P, Ricciardi-Castagnoli P, Raposo G, Garin J. et al. Proteomic analysis of dendritic cell-derived exosomes: a secreted subcellular compartment distinct from apoptotic vesicles. J Immunol. 2001;166:7309-18
47. Tauro BJ, Greening DW, Mathias RA, Ji H, Mathivanan S, Scott AM. et al. Comparison of ultracentrifugation, density gradient separation, and immunoaffinity capture methods for isolating human colon cancer cell line LIM1863-derived exosomes. Methods. 2012;56:293-304
48. Zhang B, Yin Y, Lai RC, Tan SS, Choo ABH, Lim SK. Mesenchymal stem cells secrete immunologically active exosomes. Stem Cells Dev. 2014;23:1233-44
49. Attur M, Dave M, Abramson SB, Amin A. Activation of diverse eicosanoid pathways in osteoarthritic cartilage: a lipidomic and genomic analysis. Bull NYU Hosp Jt Dis. 2012;70:99-108
50. Gao Y, Liu S, Huang J, Guo W, Chen J, Zhang L. et al. The ECM-Cell Interaction of Cartilage Extracellular Matrix on Chondrocytes. Biomed Res Int. 2014;2014:1-8
51. Demoor M, Ollitrault D, Gomez-Leduc T, Bouyoucef M, Hervieu M, Fabre H. et al. Cartilage tissue engineering: molecular control of chondrocyte differentiation for proper cartilage matrix reconstruction. Biochim Biophys Acta. 2014;1840:2414-40
52. Erggelet C, Mandelbaum BR. Eike H. Mrosek MD JMSM. Principles of cartilage repair. Springer Science & Business Media. 2008
53. Sophia Fox AJ, Bedi A, Rodeo SA. The Basic Science of Articular Cartilage: Structure, Composition, and Function. Sports Health. 2009;1:461-468
54. Tao S-C, Yuan T, Zhang Y-L, Yin W-J, Guo S-C, Zhang C-Q. Exosomes derived from miR-140-5p-overexpressing human synovial mesenchymal stem cells enhance cartilage tissue regeneration and prevent osteoarthritis of the knee in a rat model. Theranostics. 2017;7:180-195
55. Zhu Y, Wang Y, Zhao B, Niu X, Hu B, Li Q. et al. Comparison of exosomes secreted by induced pluripotent stem cell-derived mesenchymal stem cells and synovial membrane-derived mesenchymal stem cells for the treatment of osteoarthritis. Stem Cell Res Ther. 2017;8:64
56. Kronenberg HM. Developmental regulation of the growth plate. Nature. 2003;423:332-6
57. Mackie EJ, Ahmed YA, Tatarczuch L, Chen K-S, Mirams M. Endochondral ossification: How cartilage is converted into bone in the developing skeleton. Int J Biochem Cell Biol. 2008;40:46-62
58. Wu L, Leijten J, Blitterswijk CA van, Karperien M. Fibroblast Growth Factor-1 Is a Mesenchymal Stromal Cell-Secreted Factor Stimulating Proliferation of Osteoarthritic Chondrocytes in Co-Culture. Stem Cells Dev. 2013;22:2356-2367
Author contact
![]() Corresponding author: Magdalena Lorenowicz, PhD, Address: Regenerative Medicine Center, UMC Utrecht, Uppsalalaan 8, 3584CT Utrecht, The Netherlands Tel: +31- 88-755-67941 Email: m.j.lorenowicznl
Corresponding author: Magdalena Lorenowicz, PhD, Address: Regenerative Medicine Center, UMC Utrecht, Uppsalalaan 8, 3584CT Utrecht, The Netherlands Tel: +31- 88-755-67941 Email: m.j.lorenowicznl