Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and methods
Results
Discussion
Conclusions
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2019; 9(12):3425-3442. doi:10.7150/thno.33178 This issue Cite
Research Paper
Knockdown of NEAT1 induces tolerogenic phenotype in dendritic cells by inhibiting activation of NLRP3 inflammasome
Maomao Zhang1,2*, Yang Zheng1,2*, Yong Sun1,2, Shuang Li1,2, Liangqi Chen1,2, Xiangyuan Jin3, Xinyu Hou1,2, Xianglan Liu1,2, Qi Chen1,2, Jing Li1,2, Mingyang Liu1,2, Xianghui Zheng1,2, Yongxiang Zhang1,2, Jian Wu1,2 ![]() , Bo Yu1,2
, Bo Yu1,2
1. Department of Cardiology, The Second Affiliated Hospital of Harbin Medical University, Harbin, China
2. The Key Laboratory of Myocardial Ischemia, Harbin Medical University, Ministry of Education, Harbin, China
3. Department of Thoracic Surgery, Harbin Medical University Cancer Hospital, Harbin, China
*These authors contributed equally to this work.
Received 2019-1-5; Accepted 2019-4-29; Published 2019-5-18
Abstract

Rationale: Tolerogenic dendritic cells (tol-DCs) play essential roles in immune-related diseases and induce immune tolerance by shaping T-cell responses. Accumulating evidence suggests that long noncoding RNAs (lncRNAs) play important regulatory roles in the immune system. However, the potential roles and underlying mechanisms of lncRNAs in tol-DCs remain unclear.
Methods: RNA in-situ hybridization, histochemistry, and qRT-PCR were performed to determine the distribution and expression of NEAT1 in DCs. Flow cytometry was used to analyze the tolerogenic function of DCs. Small sequencing, followed by bioinformatic analysis, was performed to determine the target genes of NEAT1. The mechanism of NEAT1 was explored using a luciferase reporter, chromatin immunoprecipitation assays, and Immunofluorescence. In-vivo experiments were used to investigate the induction of immune tolerance via NEAT1-knockdown DCs.
Results: Our results show that lncRNA NEAT1 can induce tolerogenic phenotype in DCs. Mechanistically, small RNA-seq analysis revealed that NEAT1 knockdown preferentially affected the expression of miR-3076-3p. Furthermore, NEAT1 used the NLRP3 inflammasome as a molecular decoy for miR-3076-3p, thus facilitating the expression of tolerogenic phenotype in DCs. Moreover, the transcription factor E2F1 acted as a repressor of NEAT1 transcription via activity of H3K27ac. Our results also indicate that NEAT1 knockdown in DCs can induce immune tolerance in models of experimental autoimmune myocarditis and heart transplantation.
Conclusions: Taken together, our study shows the mechanism used by NEAT1 in inducing tol-DCs and highlights the therapeutic potential of targeting NEAT1 for the treatment of immune-related diseases.
Keywords: tolerogenic dendritic cells, immune tolerance, long non-coding RNA, NEAT1
Introduction
Diseases caused by dysfunction of immune regulation, such as autoimmune diseases, graft-versus-host disease (GVHD), and other chronic conditions, represent a growing medical challenge worldwide [1-3]. Growing evidence suggests that immune tolerance is an ideal state for patients with these diseases [4]. Immune tolerance is defined as unresponsiveness to a self or foreign antigen while maintaining reactivity to other antigens [5]. Dendritic cells (DCs), the most potent antigen-presenting cells (APCs), are central mediators in immune tolerance [6]. Tolerogenic DCs (Tol-DCs) are characterized by low expression of co-stimulatory molecules, production of immunomodulatory cytokines, and inhibition of T-cell proliferation [6]. Imbalance between Treg and Th17 cells is a new paradigm in our understanding of immune-related diseases [7,8]. Tol-DCs induce immune tolerance partially by regulating the differentiation of TH17 cells and regulatory T cells (Tregs) [9]. Tol-DCs have been used in cellular immune therapy for immune-related diseases such as type 1 diabetes and rheumatoid arthritis, and solid organ transplantation [4]. Improved understanding of molecular mechanisms involved in induction of tol-DCs will provide important targets for the treatment of immune-related diseases.
Recently discovered long noncoding RNAs (lncRNAs) play crucial roles in diverse processes and diseases [10,11]. Studying the functions of lncRNAs in immune cells has proven particularly advantageous. Numerous studies have shown that lncRNAs can regulate the development, differentiation, and activation of macrophages, T cells, and B cells [12-14]. However, our understanding of the functions of lncRNAs in DCs is still in its infancy. Wang et al. have shown that lnc-DC mediate DC differentiation and reduce the capacity of DCs to stimulate T-cell activation via initiation of the transcription factor STAT3 [15]. Our previous study has shown that lncRNA MALAT1 induces tol-DCs via the miRNA-155/DC-SIGH/IL-10 axis [16]. In the present study, we show that the lncRNA NEAT1 can induce tol-DCs. NEAT1 is localized to the nucleus and includes two transcripts: NEAT1 v1 (3.7 kb) and NEAT1 v2 (23 kb) [17]. NEAT1 v1 and NEAT1 v2 share the same promoter with different 3'-end—processing mechanisms [18]. NEAT1 v2 plays an important role in the formation of paraspeckles and function of NEAT1 [19]. Previous studies have shown that NEAT1 participates in immune regulation. Zhang et al. have shown that expression of NEAT1 is abnormally increased, and is positively correlated with clinical disease activity, in patients with systemic lupus erythematosus (SLE) [20]. Imamura et al. found that NEAT1 can activate the transcription of antiviral gene interleukin (IL)-8 by removing SFPQ (a NEAT1-binding paraspeckle protein) from the IL-8 promoter [21]. However, the exact molecular mechanism used by NEAT1 in induction of tol-DCs remains unclear.
The NACHT, LRR, and PYD domain-containing protein 3 (NLRP3) inflammasome is a key player in inflammation triggered by environmental or host-derived antigens [22]. Numerous studies have demonstrated that the NLRP3 inflammasome, which can be activated by endogenous or exogenous danger signals on Toll-like receptor 4 (TLR4), plays an important role in immune-related diseases [23-26]. NLRP3 is a multi-protein complex composed of NLRP3, the adaptor apoptosis-associated speck-like protein containing a caspase recruitment domain (ASC), and caspase-1 [27].The NLRP3 inflammasome triggers the transformation of procaspase-1 into caspase-1, and converts proIL-1β and proIL-18 into mature IL-1β and IL-18; these proteins are then secreted outside the cell and lead to the inflammatory immune cascade [28,29]. Several studies have shown that activation of the NLRP3 inflammasome can decrease the numbers of regulatory T cells (Treg) and boost the differentiation of Th17 cells [30,31]. The NLRP3 inflammasome is expressed by various immune cells including DCs, macrophages, T cells, and B cells [32-35]. Ghiringhelli et al. found that in DCs, the NLRP3 inflammasome induces IL-1β-dependent adaptive immunity against tumors [35]. Arnold et al. have shown that the NLRP3 inflammasome is required for the differentiation and/or recruitment of CD11b+ DCs during infection with H. pylori [36]. Currently, research on the NLRP3 inflammasome is focused on its role in activating DCs. However, the detailed DCs-suppressive function of the NLRP3 inflammasome has not yet been elucidated.
In this study, we found that NEAT1 can induce tol-DCs. NEAT1 was significantly upregulated in DCs treated with LPS. Knockdown of NEAT1 inhibited the expression of co-stimulatory molecules (CD80, CD86, MHCII), induced T cell hyporesponsiveness, expanded the populations of Tregs, and decreased the numbers of Th17 cells in vitro. In the cytoplasm, RNA-seq analysis revealed that NEAT1 could regulate the NLRP3 inflammasome by inhibiting the expression of miR-3076-3p. In the nucleus, miRNA let-7i regulated the expression of NEAT1 by binding to the transcription factor E2F1. E2F1 inhibited the acetylation of H3K27 by binding to the promoter of NEAT1. In vivo, transfusion with DCs, in which NEAT1 was knocked down, alleviated the inflammatory response in an experimental autoimmune model of myocarditis, and induced immune tolerance to a cardiac allograft in a heart transplantation model. Our results indicate that lncRNA NEAT1 contributed to the generation of tol-DCs.
Materials and methods
Animals
Balb/c or C57BL/6 mice (5 to 6-week-old males) were purchased from the Beijing Vital River Laboratory Animal Technology (Beijing, China). Mice were bred under specific pathogen-free (SPF) conditions at a 12 h light/dark cycle at the Second Affiliated Hospital of Harbin Medical University.
Ethics statement
All procedures involving animals were approved by the Institutional Animal Care and Use Committee of the Second Affiliated Hospital of Harbin Medical University. This study was conducted in accordance with the Guide for the Care and Use of Laboratory Animals (Institute of Laboratory Animal Resources/National Institutes of Health, Bethesda, MD, USA).
Culture and transfection of bone marrow-derived DCs
DCs were generated from the bone marrow cells of Balb/c mice as described previously [37]. Briefly, bone marrow was flushed from tibias and femurs. To remove fibrous tissue, the marrow was passed through a 200-pore sized mesh. After lysis of red blood cells, (Beyotime, China), the bone marrow cells were centrifuged, washed, and resuspended in Roswell Park Memorial Institute 1640 medium (Hyclone, South Logan, UT, USA) containing 10% fetal bovine serum (Sciencell, Carlsbad, CA, USA), 1% penicillin/streptomycin (Beyotime, Shanghai, China), 10 ng/ml IL-4, and 20 ng/ml granulocyte macrophage-colony stimulating factor (GM-CSF, PeproTech, Rocky Hill, NJ, USA). BM progenitors were then added to 6-well tissue culture plates (Corning, New York, USA) at 2 × 106 cells in 1 ml medium per well, and incubated at 37 °C and 5% CO2. Every other day, half of the medium was replaced with fresh medium.
lncRNA NEAT1 Smart Silencer (RiboBio, Guangzhou, China) was used to knock down the expression of NEAT1. NEAT1 Smart Silencer contains three siRNAs and three antisense oligonucleotides (Table S1). The siRNA for E2F1 was purchased from Sangon (Shanghai, China). The miRNA mimics, inhibitors, and plasmid DNA were purchased from GenePharma (Shanghai, China). All sequences are provided in Table S2. At day 5, cells were transfected with smart silencer, siRNA and miRNA mimics or inhibitors, or plasmid DNA using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) according to the manufacturer's protocol. At 6 h after transfection, the media were removed and replaced with fresh media; culturing was then continued for an additional 48 h. At day 7, the cells were treated for 12 h with the TLR ligand lipopolysaccharide (LPS; 200 ng/ml; Sigma-Aldrich, St. Louis, MO, USA), and then DCs were collected for further analysis.
RNA ISH (in situ hybridization histochemistry) for NEAT1
RNA ISH experiments were performed using a Fluorescent In situ Hybridization Kit (Ribobio, Guangzhou, China) according to the manufacturer's instructions. Briefly, DCs were fixed in 4% paraformaldehyde for 10 min and washed with phosphate buffered saline (PBS). The fixed cells were incubated with 20 μM FISH Probe in hybridization buffer at 37 °C overnight. The slides were washed and mounted using DAPI for detection. RNA ISH probes were designed and synthesized by Ribobio (Guangzhou, China). The images were captured using a confocal laser-scanning microscope (FluoView v5.0FV300; Olympus, Tokyo, Japan).
RNA extraction and qRT-PCR
Total RNA from each DCs subset was extracted using Trizol reagent (Invitrogen, Carlsbad, CA, USA). Nuclear and cytoplasmic RNA was isolated using a Cytoplasmic & Nuclear RNA Purification Kit (Norgen Biotek Corp., Thorold, ON, Canada). For analysis of lncRNA and mRNA, cDNA transcripts were amplified using a Transcriptor First Strand cDNA Synthesis Kit (Roche, Basel, Switzerland) according to the manufacturer's instructions. The incubation for reverse transcription was conducted for 60 min at 50 °C and for 5 min at 85 °C. PCR was conducted using Bestar Sybr Green qPCR Master Mix (DBI Bioscience, Germany) at the following settings: 40 cycles of 10 s at 95 °C, 30 s at 60 °C, and 30 s at 72 °C. For analysis of miRNAs, qRT-PCR was performed using Hairpin-it microRNA and U6 snRNA Normalization RT-PCR Quantitation Kit (GenePharma, Shanghai, China). Reverse transcription mixtures were incubated for 30 min at 25 °C, 30 min at 42 °C, and 5 min at 85 °C. PCR was performed at the following settings: 40 cycles of 12 s at 95 °C and 40 s at 62 °C. All reactions were performed in triplicate. All qRT-PCR data were analyzed using the 2-ΔΔCt method. The primers used are listed in Table S2.
Flow cytometry analysis
DC phenotypes were analyzed via surface expression of specific markers. DCs were immunolabeled with FITC-CD11C (clone N418), PE-CD80 (clone 16-10A1), PE-CD86 (clone GL1), or PE-MHC II (clone M5/114.15.2) (eBioscience, San Diego, CA, USA) at 4 °C for 30 min. The appropriate isotype-matched control was included for each immunolabeling procedure. The expression of CD4+, CD25+, and FoxP3+ was determined for Treg analysis. The following fluorochrome-conjugated antibodies were used: FITC-CD4 (clone RM4-5) (BD Biosciences, San Jose, CA, USA), APC-CD25 (clone PC61.5) (eBioscience, San Diego, CA, USA), and PE-FoxP3 (clone FJK-16s) (eBioscience, San Diego, CA, USA). To determine the percentage of Th17 cells, cells were stimulated with phorbol myristate acetate (PMA; 5 ng/ml) and ionomycin (50 ng/ml) for 4 h at 37 °C before incubation with FITC-CD4 (clone RM4-5) (BD Biosciences, San Jose, CA, USA) and PE-IL-17A (clone TC11-18H10.1) (BioLegend, San Diego, CA, USA). The cells were subjected to extracellular staining, then washed, fixed, and permeabilized using fixation/permeabilization agents (eBioscience, San Diego, CA, USA) for 30 min at 25 °C. The cells were then labeled using antibodies specific for IL-17A or FoxP3 for 30 min at 25 °C. Analysis was performed using FACSDiva Version 6.1.3 (BD Biosciences, San Jose, CA, USA) and FlowJo_V10 (TreeStar, Ashland, OR, USA).
Mixed lymphocyte reaction assay
DCs, plated at different densities (0-15 × 104), were used as stimulators for allogeneic T cells (2 × 105) harvested from the C57BL/6 mice. Isolated T cells were cultured for 5 d in 96-well round-bottom microplates in the presence of DCs pre-treated with mitomycin C (10 μg/ml, Sigma-Aldrich, St. Louis, MO, USA) at different DC/T cell ratios (1:10, 1:20, 1:50, or 1:100). In another mixed lymphocyte reaction (MLR) study, the responder cells were splenocytes isolated from transplantation mice or from the mice used to model experimental autoimmune myocarditis (EAM). Splenocytes from C57BL/6 mice, treated with 100 μg/ml mitomycin C, were used as stimulator cells. The responder cells (2.5 × 106/ml) and stimulator cells (5 × 106/ml) were cultured at 37 °C in plates (Corning, New York, USA) for 4 days. Then, these co-cultures were incubated with 10 μM BrdU for 24 h, and the proliferation of T cells was assessed using BrdU ELISA according to the manufacturer's instructions (Abcam, Cambridge, MA, USA). All proliferation assays were performed in triplicate.
ELISA
Culture supernatants were collected to measure the levels of cytokines secreted by DCs. The levels of IL-10 (Abcam, Cambridge, MA, USA), TGF-β (Abcam, Cambridge, MA, USA), IL-12 (Abcam, Cambridge, MA, USA), IL-17A (R&D Systems, Minneapolis, MN, USA), IL-1β (Abcam, Cambridge, MA, USA), and IL-6 (BOSTER, Wuhan, China) were determined by ELISA according to the manufacturer's instructions. The levels of IL-10, TGF-β, IL-12, and IL-17A were also determined in the plasma of mice that had undergone heart transplantation or induction of myocarditis. All the assays were performed in triplicate.
Small RNA sequencing and bioinformatics analysis
Approximately 1 μg of total RNA per sample was used to generate small RNA libraries using TruSeq Small RNA Sample Prep Kit protocol (Illumina, San Diego, CA, USA). Cluster generation and 50-bp single-end read sequencing were performed on an Illumina Hiseq 2500 platform at OE-Biotech (Shanghai, China). After quality assessment and post-processing of Fastq files, sequences were mapped to a reference genome. Non-coding RNAs were annotated as rRNAs, tRNAs, small nuclear RNAs (snRNAs), and small nucleolar RNAs (snoRNAs). These RNAs were aligned and then subjected to BLAST search against Rfam v.10.1 (http://www.sanger.ac.uk/software/Rfam) and GenBank databases (http://www.ncbi.nlm.nih.gov/genbank/). Known miRNAs were identified by aligning against the miRBase v.21 database (http://www.mirbase.org/). Unannotated small RNAs were analyzed by mirdeep2 to predict novel miRNAs. The differential levels of miRNA were determined using DESeq package in R statistical computing tool. The targets of differentially expressed miRNAs were predicted using MiRanda software with the parameters set as follows: S ≥ 150 ΔG ≤ -30 kcal/mol and demand strict 5' seed pairing. GO enrichment and KEGG pathway enrichment analyses of differently expressed miRNA target genes were performed using R based on hypergeometric distribution.
RNA-binding protein immunoprecipitation (RIP) assays
RIP experiments were performed using an EZ-Magna RIP RNA-Binding Protein kit (Merck Millipore, Billerica, MA, USA) according to the manufacturer's instructions. Briefly, DCs were cultured to 90% confluence and harvested using RIP lysis buffer containing a protease inhibitor and RNase inhibitor. Concurrently, an anti-Ago2 antibody (Abcam, Cambridge, MA, USA) containing magnetic beads was incubated in lysis buffer to conjugate the antibody to the magnetic beads. IgG (Merck Millipore, Billerica, MA, USA) was used as negative control (input group). The beads were attracted by a magnetic separator, and proteinase K was added to digest the protein. Then, the supernatants containing RNA were collected and extracted using Trizol reagent. Finally, reverse transcription and PCR analysis were performed. The primer sequences are provided in the Table S2.
Dual-luciferase assay
psiCHECK2[luc2P/TCF-LEF RE/Hygro] vector was purchased from Scbio (Guangzhou, China). The vector consisted of an NLRP3 or E2F1-3' untranslated region (UTR)-wild type (WT) and NLRP3 or E2F1-3' UTR-mutant. The cells were seeded into 24-well plates and co-transfected with 50 ng of the 3'-UTR reporter vector and 30 nM microRNA mimic, inhibitor, or negative control miRNA using Lipofectamine 2000 transfection reagent (Invitrogen, Carlsbad, CA, USA). After 48 h, the cells were lysed. Luciferase activity was measured via Dual-Luciferase Reporter Assay System (Promega, Madison, WI, USA) and normalized to the activity of Renilla luciferase.
Chromatin immunoprecipitation assays
Chromatin immunoprecipitation (CHIP) assays were performed using an EZ-Magna ChIP kit (Merck Millipore, Billerica, MA, USA) according to the manufacturer's instructions. Briefly, 2-3 × 105 cells were crosslinked using 1% formaldehyde for 10 min at 25 °C and then sonicated in lysis buffer. After centrifugation, 10 μl of the supernatant was used as input; the remaining lysate was subjected to a ChIP assay using antibodies specific for E2F1 (Abcam, San Diego, CA, USA) or H3K27ac (Cell Signaling Technology, Danvers, MA, USA). The primer sequences are provided in Table S2.
Western blot assay
Western blotting was performed according to standard protocols described previously [38,39]. Briefly, whole cell extracts were harvested using RIPA buffer (Beyotime, Shanghai, China) containing 1 mM PMSF and 1% protease inhibitor cocktail (Roche, Basel, Switzerland). The protein lysates were separated via 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to 0.22 μm Immobilon-NC (Merck Millipore, Billerica, MA, USA). Primary antibodies against E2F1 (clone EPR3818(3)) and anti-caspase-1 (clone EPR16883) were purchased from Abcam (Cambridge, MA, USA). anti-NLRP3 (clone D4D8T), anti-ASC (clone D2W8U), anti-GAPDH (clone D4C6R), and anti-β-actin (clone 13E5; used as loading control) were purchased from Cell Signaling Technology (Danvers, MA, USA).
Immunofluorescence
DCs, grown on glass coverslips, were fixed with 4% paraformaldehyde for 30 min at 25 °C and blocked with goat serum. To characterize the expression of NLRP3 in DCs, cells were incubated at 4 °C overnight with primary antibodies against NLRP3 (1:300, Abcam, Cambridge, MA, USA). DCs were then washed and incubated with Cy3-labeled goat anti-rabbit IgG(H+L) (1:200, Beyotime, Shanghai, China) for 1 h at 37 °C in the dark. Nuclei were counterstained with DAPI (Sigma-Aldrich, St. Louis, MO, USA). Fluorescent images were acquired with a confocal laser-scanning microscope (Fluo View v5.0 FV300; Olympus, Tokyo, Japan).
Analysis of methylation of CpG islands
Methylation status of the NEAT1 promoter was determined using bisulfite sequencing PCR (BSP). DNA was extracted from DCs using the Ezup Column Animal Genomic DNA Purification Kit (Sangon, Shanghai, China) according to standard procedure. Genomic DNA was modified using sodium bisulfite. The transformed DNA was then amplified via PCR. Primers are listed in Table S2. PCR-amplified products were ligated to pUC18-T vectors (Sangon, Shanghai, China) and transformed into E. coli DH5α cells. IPTG/X-gal plates were used to select qualifying colonies. Ten colonies for each sample were selected and cultured separately in LBamp+ medium overnight. The obtained plasmids were sequenced by Sangon, Shanghai, China.
Induction and treatment of experimental autoimmune myocarditis
Myosin was dissolved in 0.01 M PBS and emulsified with an equal volume of complete Freund's adjuvant (CFA, Sigma-Aldrich, St. Louis, MO, USA). Experimental autoimmune myocarditis was induced in 6-week-old Balb/c mice by subcutaneous injection with 150 μg myosin on days 0 and 7. EAM mice were injected with NEAT1-knockdown DCs (5 × 106 cells/mouse) twice a week for 2 weeks following the first immunization with myosin. PBS was used as treatment in the control group.
Echocardiography
In EAM mice, echocardiography was performed 21 days after injection with myosin using a GE/Vingmed ultrasound Vivid7 (GE Healthcare, Little Chalfont, UK). Left ventricular end-diastolic diameter (LVEDd), end-systolic diameter (LVEDs), diastolic interventricular septum thickness (IVSTd), and diastolic posterior wall septum thickness (PWTd) were determined in M-mode recordings.
Heart transplantation
After induction of general anesthesia, Balb/c mice were injected into the caudal vein with 0.3 ml PBS only (negative control), 1.0 × 106 LPS-mDCs, or NEAT1-knockdown DCs (derived from Balb/c murine bone marrow). At 24 h after transfusion, the donor heart grafts were harvested from C57BL/6 mice and transplanted into the abdominal cavity of Balb/c recipient mice using microsurgical techniques. Graft survival was monitored post-operatively by daily trans-abdominal palpation. Graft rejection was defined as complete cessation of palpable heartbeats, and further verified by direct visualization and histologic examination of the graft.
Histologic studies and immunohistochemical analysis of the hearts
The hearts from EAM mice were removed 21 days after the mice were injected with myosin. Heart weight was measured, and the ratio of heart weight/body weight was evaluated. The hearts were stained with hematoxylin and eosin (H&E), and assayed according to a semi-quantitative scale as described previously [40]: 0, no inflammatory infiltrates; 1, small foci of inflammatory cells between myocytes; 2, larger foci of > 100 inflammatory cells; 3, <10% of a cross-section involved; 4, >30% of a cross-section involved. Fourteen days after heart transplantation, cardiac allografts were removed from Balb/c recipients. H&E staining was scored according to the 2005 classification of International Society for Heart and Lung Transplantation for acute cellular rejection: Grade 0 R-no rejection; Grade 1 R-mild rejection: interstitial and/or perivascular infiltrate with up to 1 focus of myocyte damage; Grade 2 R-moderate rejection: 2 or more foci of infiltrate with associated myocyte damage; Grade 3 R-severe rejection: diffuse infiltrate with multifocal myocyte damage with or without edema, hemorrhage, and/or vasculitis.
For immunohistochemical analysis, the sections were dewaxed, subjected to antigen retrieval, and incubated with an anti-FoxP3, anti-NLRP3, anti-Caspase-1 or anti-ASC antibody (Wanleibio, Shenyang, China), followed by incubation with a secondary antibody. All histological evaluations were performed in a double-blind manner by two professional staff. Images were captured using an Olympus BX4I microscope.
Statistical analysis
Data analysis was performed with SPSS 23.0 (SPSS, lnc., USA) and GraphPad Prism 6 (GraphPad Software, lnc., CA, USA). Data are expressed as means ± standard deviation (SD). Survival of cardiac allografts was compared using the Mann-Whitney U test. Statistical significance was analyzed by analysis of variance (ANOVA) with Pyan method or Student's t-test. P value <0.05 was considered statistically significant.
Results
The expression of lncRNA NEAT1 is upregulated in LPS-induced DC maturation
Previous studies have shown that NEAT1 is localized in the nucleus [21,41]. In the first series of experiments, we tested the distribution of NEAT1 in DCs. RNA FISH confirmed that NEAT1 was distributed both in the nucleus and cytoplasm (Figure 1A). We then separated the nuclear and cytosolic RNA. Subcellular fraction location assays demonstrated that in both immature DCs (iDCs) and mature DCs (mDCs), approximately 90% of NEAT1 was distributed in the nucleus, and 10% was distributed in the cytoplasm (Figure 1B). These results show that NEAT1 may play an important role in the nucleus and cytoplasm. Next, we used quantitative RT-PCR to examine the expression of NEAT1 in DCs responding to different stimuli. The level of NEAT1 was increased 1.7-fold in DCs treated with LPS (200 ng/ml) for 24 h (Figure S1A). However, there was no change in the expression of NEAT1 after stimulation with lipoteichoic acid (TLR2), polyinosinic-polycytidylic acid (TLR3), and a TLR7/8 agonist (R848) for 24 h compared with that in iDCs (Figure S1A). These results suggest that NEAT1 may stimulate the maturation of DCs via the TLR4 ligand LPS. The expression of total NEAT1v1 and NEAT1v2 was upregulated in DCs treated with LPS during LPS-induced DC maturation (Figure 1C and Figure S1B). Our results showed that the expression levels of NEAT1v1 and NEAT1v2 were the highest in stimulating 200 ng/ml at different time points (2, 4, 8, 12, and 24 h). In 200 ng/ml, the expression of NEAT1 was up-regulated in 2, 4, 8, 12 and 24 h compared with iDC. However, the expression of NEAT1 displayed no statistical significance among 2, 4, 8, 12 and 24 h. Therefore, to investigate the function of NEAT1 in DCs we selected 200 ng/ml LPS stimulating DCs for 24 h in the next experiments.
Knockdown of NEAT1 induces tolerogenic phenotype in dendritic cells (DCs). (A) DCs were stimulated with LPS (200 ng/ml) and labeled using FISH; 18S served as cytoplasmic control transcript, and U6 served as nuclear control transcript. 18S (red), U6 (red), NEAT1 (red), and nuclei stained with DAPI (blue) are shown. Scale bars correspond to 50 µm. (B) After RNA was separated into the nuclear and cytosolic fractions, RNA expression levels were measured by qRT-PCR. GAPDH was used as cytosolic marker, and U6 was used as nuclear marker. (C) DCs were stimulated with lipopolysaccharide (LPS; 10 ng/ml, 200 ng/ml, or 1 µg/ml). NEAT1 v2 expression was determined by qRT-PCR at different time points (0, 4, 8, 12, and 24 h). (D,E) DCs were transfected with NEAT1 smart silencer (siNEAT1) or negative control (NC) for 12 h before treatment with LPS (200 ng/ml). The co-stimulators (CD80, CD86, and MHC II) were detected by flow cytometry (D). Cytokine levels (IL10, TGF-β, IL12, and IL-17A) were analyzed by ELISA in DC supernatants (E). Data are expressed as mean ± SD; n = 4 biological replicates; *p<0.05, **p<0.01, ***p<0.001, derived by Student's t-test. (F-J) DCs were transfected with NEAT1 smart silencer (siNEAT1) or NC for 12 h, then treated with 200 ng/ml LPS (12 h) and 10 μg/ml mitomycin C (2 h). These DCs were cocultured with allogeneic T cells for 48 h. T-cell proliferation was assessed by BrdU ELISA at the ratios of 1:10, 1:20, 1:50, and 1:100 (DC/T cells) (F). The numbers of CD4+CD25+FoxP3+Tregs were assessed by flow cytometry (G). The percentage of Tregs is shown in (H). The numbers of TH17 (CD4+ and IL-17A+) cells were assessed by flow cytometry (I). The percentage of TH17 is shown in (J). Data are expressed as mean ± SD; n = 3 biological replicates; **p<0.01, ***p<0.001, derived by Student's t-test.
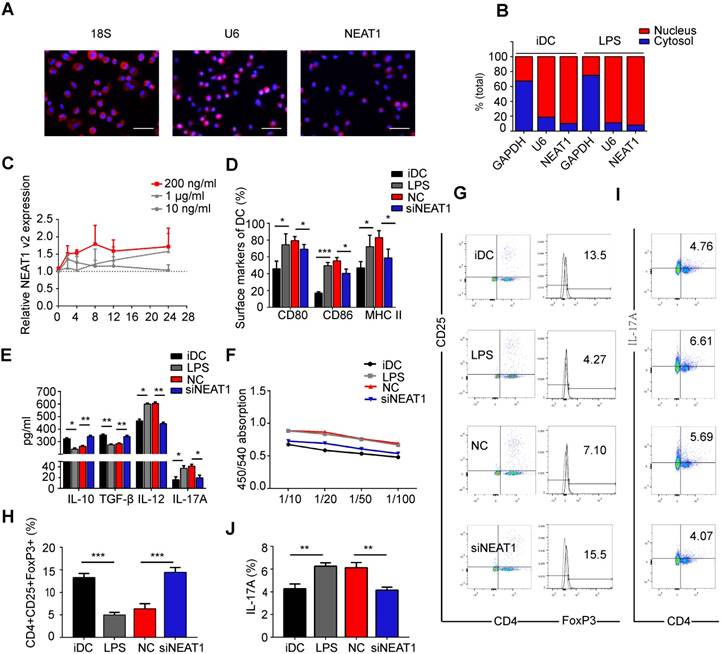
NEAT1 knockdown conditions DCs toward tolerogenic state
To investigate the influence of NEAT1 on the functional state of DCs, we used Lipofectamine 2000 to transfect DCs with a smart silencer of NEAT1 for 12 h before stimulation with LPS. We used flow cytometry to analyze the expression of co-stimulators CD80, CD86, and MHC II on the surface of DCs. Lipofectamine 2000, used for transfection of the smart silencer, did not affect activation of DCs or the expression of NEAT1 (Figure S1C-E). Expectedly, the knockdown of NEAT1 caused a decrease in the expression of NEAT1 (Figure S2A). After 24 h of stimulation with LPS, the expression of the co-stimulators was decreased in DCs with knockdown of NEAT1 compared with that in DCs transfected with a negative control (NC) (Figure 1D and Figure S2B). We then used an enzyme-linked immunosorbent assay (ELISA) to examine cytokine secretion in DCs. The levels of anti-inflammatory cytokines IL-10 and TGF-β were increased, whereas those of inflammatory cytokines IL-12, IL-17A, IL-1β, and IL-6 were decreased, in NEAT1-knockdown DCs compared with the levels of NC (Figures 1E and S2C-D).
We next examined how DCs affect the function of allogeneic T cells via mixed lymphocyte reaction (MLR). The proliferation of T cells was measured by BrdU ELISA. The ability of NEAT1-knockdown DCs to activate T-cell proliferation was decreased compared with that of NC (Figure 1F). To determine whether DCs affected the numbers of Treg and TH17 cells, we used flow cytometry to examine the expression of CD4, CD25, Foxp3, and IL-17A after 2 days of MLR. The numbers of Tregs co-cultured with NEAT1-knockdown DCs increased compared with those co-cultured with NC (Figure 1G and Figure 1H); the numbers of TH17 co-cultured with NEAT1-knockdown DCs decreased compared with those co-cultured with NC (Figure 1I and Figure 1J). NEAT1-knockdown DCs suppressed the expression of co-stimulators, inhibited the proliferation of T cells, induced the secretion of anti-inflammatory cytokines, and increased the numbers of Treg cells. These data suggest that knockdown of NEAT1 can induce tolerance in DCs.
NEAT1 regulates the expression of NLRP3 inflammasome by competing for miR-3076-3p
We used small RNA-sequencing to determine the NEAT1-mediated pathway in tol-DCs. For this, we used three samples of NEAT1-knockdown DCs and three samples of DCs treated with NC. Small RNA sequencing revealed that the expression of 31 miRNAs was increased, and that of 14 miRNAs was abolished, by NEAT1 knockdown (Figure 2A and Table S3). RT-qPCR confirmed that the expression of miR-3076-3p and miR-126b-5p was increased, while that of miR-130a-5p and miR-203-5p was decreased, in NEAT1-knockdown DCs (Figure S3A). In order to determine the biological function of differentially expressed miRNAs, we performed target prediction analysis using the software miRanda to analyze the function of possible target genes. Gene ontology (GO) analysis showed that biological processes were significantly enriched in regulation of transcription, DNA-templated, transcription, DNA-templated and positive regulation of transcription from RNA polymerase II promoter (Figure 2B). The most significantly pathways were enriched in glioma, proteoglycans in cancer, pathways in cancer, insulin secretion and Wnt signaling pathway (Figure 2C). We found that the inflammasome NLRP3 was the target gene of miR-3076-3p. miR-3076-3p was predicted to target both NEAT1 and NLRP3. The level of miR-3076-3p was decreased in mature DCs (Figure 2E); this was negatively correlated with the expression of NEAT1. We performed a RIP assay to assess whether NEAT1 can bind to miR-3076-3p. The results showed that NEAT1 and miR-3076-3p were more abundant in Ago2 than in IgG (Figure 2D). Knockdown of NEAT1 by a smart silencer increased the expression of miR-3076-3p, while the expression of miR-3076-3p was decreased in NEAT1-overexpressing DCs (Figure 2E). These results indicate that miR-3076-3p can serve as a target of NEAT1 in DCs. In addition, the expression of NEAT1 was inhibited by a miR-3076-3p mimic. However, miR-3076-3p inhibitor promoted the expression of NEAT1 (Figure S3B).
NEAT1 regulates the expression of NLRP3 by competing for miR-3076-3p. (A) Heat map image showing small RNA-seq analysis of microRNA (miRNA) expression in the NC group and siNEAT1 DCs group with three repeats. The red box was miR-3076-3p. (B) Gene ontology (GO) analysis conducted for all target genes of altered miRNAs. (C) Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis for all target genes of altered miRNAs. (D) Total cellular fractions were isolated from LPS-DCs, and immunoprecipitated using antibodies to Ago2 or IgG and RNA immunoprecipitation assays. Relative amounts of NEAT1 and miR-3076-3p bound to IgG or Ago2 were detected by qRT-PCR. Data are expressed as mean ± SD; n = 3 biological replicates; ***p<0.001, derived by Student's t-test. (E) The relative level of miR-3076-3p determined by qRT-PCR in iDCs, LPS-DCs, NC, siNEAT1 DCs, and DCs overexpressing NEAT1 (OE NEAT1). Data are expressed as mean ± SD; n = 3 biological replicates; **p<0.01, ***p<0.001, derived by Student's t-test. (F) A reporter construct with potential binding site for miR-3076-3p in the 3'-UTR region of NLRP3. 293T cells were transiently transfected with miR-3076-3p mimic or miR-3076-3p inhibitor for 24 h. Luciferase activities were measured and normalized to the level of Renilla luciferase used as control. Data are expressed as mean ± SD; n = 3 biological replicates; **p<0.01, ***p<0.001, ns, not significant, derived by Student's t-test. (G-I) DCs were transfected with miR-3076-3p mimic, miR-3076 inhibitor, or NC. Relative mRNA expression of NLRP3 was detected by qRT-PCR (G). Protein levels of NLRP3, caspase-1, and ASC were detected by western blotting (H) and immunofluorescence (I). NLRP3 (red) and nuclei stained with DAPI (blue) are shown. Scale bars correspond to 50 µm. The specific grouping shown in (H) is as follows: 1. iDCs; 2. LPS; 3. NC; 4. miR-3076-3p inhibitor; 5. miR-3076-3p mimic. Data are expressed as mean ± SD; n = 3 biological replicates; **p<0.01 derived by Student's t-test. (J-L) DCs transfected with siNEAT1, DCs overexpressing NEAT1 (OE NEAT1), DCs treated with siNEAT1+miR-3076-3p inhibitor or OE NEAT1+miR-3076-3p mimic, and negative control DCs. Relative mRNA expression of NLRP3 was detected by qRT-PCR (J). NLRP3. Protein levels of caspase-1 and ASC were detected by western blotting (K) and immunofluorescence (L). NLRP3 (red) and nuclei stained with DAPI (blue) are shown. Scale bars correspond to 50 µm. The specific grouping shown in (I) is as follows: 1. NC; 2. siNEAT1; 3. OE NEAT1; 4. siNEAT1+miR-3076-3p inhibitor; 5. OE NEAT1+miR-3076-3p mimic. Data are expressed as mean ± SD; n = 3 biological replicates; *p<0.05, ***p<0.001, ns, not significant, derived by Student's t-test.
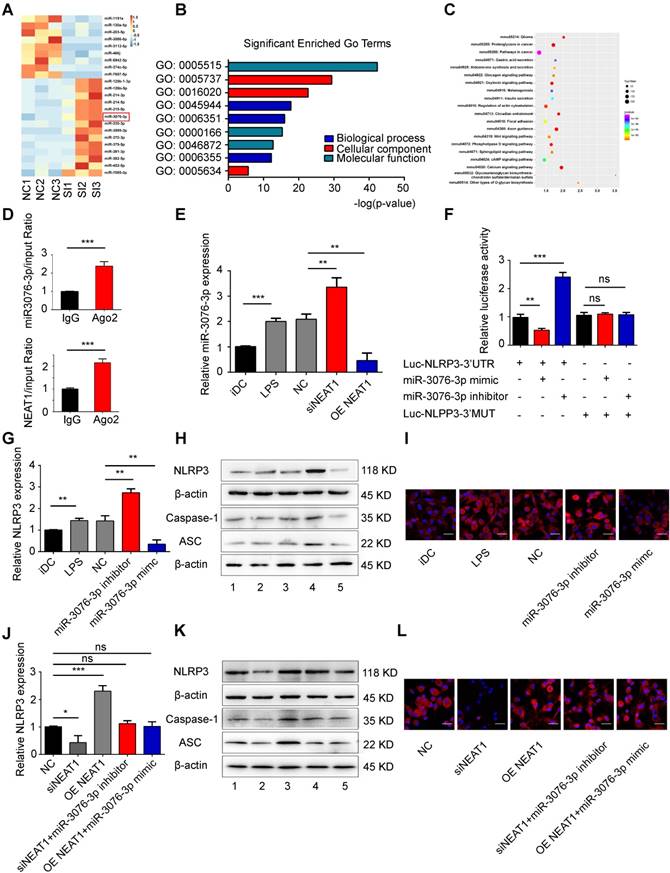
To confirm that direct binding occurred between miR-3076-3p and NLRP3, we cloned the 3'UTR of NLRP3 and mutant 3'UTR of NLRP3. These reporters were combined with miR-3076-3p mimics or with miR-3076-3p inhibitors and co-transfected into 293T cells. The miR-3076-3p mimic significantly reduced the luciferase activity of NLRP3-WT, but not that of NLRP3-Mut; the inhibitor of miR-3076-3p enhanced luciferase activity (Figure 2F). Treatment with a miR-3076-3p mimic significantly reduced the expression of NLRP3, caspase-1, and ASC mRNA and protein; the inhibitor of miR-3076-3p increased the levels of NLRP3, caspase-1, and ASC mRNA and protein (Figures 2G-I and S3C-G). These findings indicate that NLRP3 can serve as a direct target of miR-3076-3p. As expected, the increase in the expression of NLRP3, caspase-1, and ASC, promoted by NEAT1, was reversed by treatment with miR-3076-3p mimics. miR-3076-3p inhibitors reversed inhibition of NLRP3, caspase-1, and ASC induced by the knockdown of NEAT1 (Figures 2J-L and S3H-L). Taken together, these results indicate that NEAT1, acting as a ceRNA, regulated the expression of NLRP3 via the activity of miR-3076-3p in DCs.
Knockdown of NEAT1 induces DCs with tolerogenic phenotype via NLRP3 inflammasome
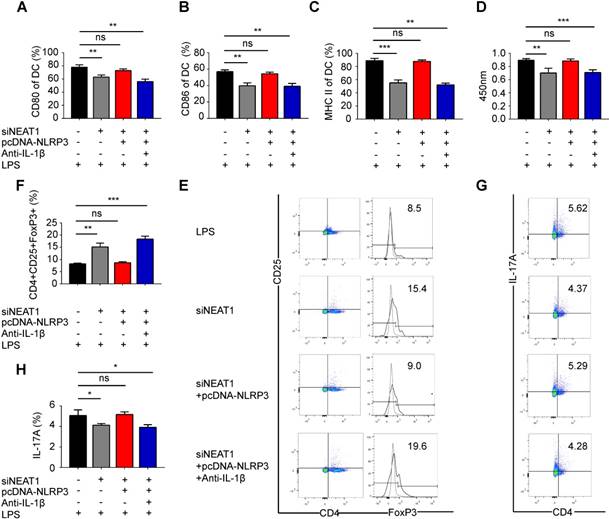
We have shown that the knockdown of NEAT1 can induce tol-DCs. We next examined whether the functions of NEAT1 are mediated by NLRP3 in tol-DCs. For this, we generated an NLRP3 expression vector, which was co-transfected with a NEAT1 smart silencer into DCs. As expected, the transfection of an NLRP3 expression vector restored the level of the NLRP3 protein in DCs (Figure S4A-B). We assessed the expression of the control protein GAPDH in DCs treated with NEAT1 smart silencer or NLRP3 expression vector. Our data indicate that overexpression of NLPR3 or siNEAT1 had no effect on the levels of GADPH in DCs (Figure S4C-D). Flow cytometry revealed that expression of exogenous NLRP3 reversed the siNEAT1-induced inhibition of co-stimulator molecules in DCs (Figures 3A-C and S4E). The results of the BrdU assay indicate that NLRP3 overexpression reversed inhibition of T-cell proliferation caused by the knockdown of NEAT1 in DCs (Figure 3D). Furthermore, the knockdown of NEAT1 induced increased numbers of Tregs; these numbers were decreased by overexpression of NLRP3 (Figure 3E and F). However, the numbers of TH17 cells were increased when co-cultured with NEAT1-knockdown DCs and the NLRP3 expression vector (Figure 3G and H). We found that the level of IL-1β was decreased in NEAT1-knockdown DCs (Figure S2C). We also co-transfected DCs with an NLRP3 overexpression vector and induced the knockdown of NEAT1 in the presence of antibodies that neutralize IL-1β. However, NLRP3 failed to reverse knockdown of NEAT1-induced tol-DCs by addition of IL-1β-specific antibodies (Figure 3A-H). These data indicate that NLPR3/IL1-1β axis is critically involved in the induction of tolerance in DCs via activity of NEAT1. Taken together, these data suggest that NEAT1 can induce tol-DCs via the NLRP3 axis.
Knockdown of NEAT1 induces tolerogenic DCs via regulation of the NLRP3 inflammasome. (A-C) DCs were transfected with siNEAT1, siNEAT1+pcDNA-NLRP3, or siNEAT1+pcDNA-NLRP3 in the presence of a neutralizing monoclonal antibody to IL-1β, then treated with 200 ng/ml LPS for 12 h. The co-stimulators (CD80, CD86, and MHC II) were detected by flow cytometry (A-C). Data are expressed as mean ± SD; n = 3 biological replicates; **p<0.01, ***p<0.001, ns, not significant, derived by Student's t-test. (D) DCs were transfected with siNEAT1, siNEAT1+pcDNA-NLRP3, or siNEAT1+pcDNA-NLRP3 in the presence of a neutralizing monoclonal antibody to IL-1β, then treated with 200 ng/ml LPS (for 12 h) and 10 μg/ml mitomycin C (for 2 h). Allogeneic T cells were cocultured with these DCs for 48 h. T-cell proliferation was assessed by BrdU ELISA. Data are expressed as mean ± SD; n = 4 biological replicates; **p<0.01, ***p<0.001, ns, not significant, derived by Student's t-test. (E-H) DCs were transfected with siNEAT1, siNEAT1+pcDNA-NLRP3, or siNEAT1+pcDNA-NLRP3 in the presence of a neutralizing monoclonal antibody to IL-1β, then treated with 200 ng/ml LPS (12 h) and 10 μg/ml mitomycin C (2 h). The numbers of CD4+CD25+FoxP3+Tregs were assessed by flow cytometry (E). The percentage of Tregs is shown in (F). The numbers of TH17 (CD4+ and IL-17A+) cells were assessed by flow cytometry (G). The percentage of TH17 is shown in (H). Data are expressed as mean ± SD; n = 4 biological replicates; *p<0.05, **p<0.01, ***p<0.001, ns, not significant, derived by Student's t-test
NEAT1 is regulated by transcription factor E2F1 via H3K27ac
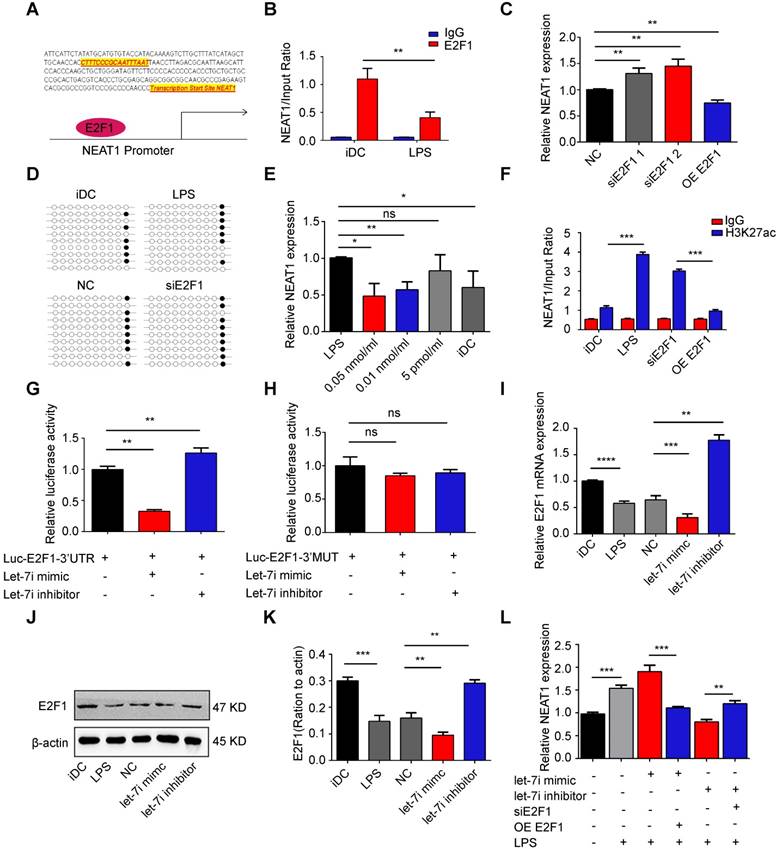
Several studies have shown that transcription factors can regulate the transcription of lncRNAs in the nucleus [42,43]. Therefore, we used CHIPBase (http://rna.sysu.edu.cn/chipbase/) to search for transcription factors that may regulate the transcription of NEAT1. We identified the transcription factor E2F1, which can bind to the promoter of NEAT1 (Figure 4A). To determine the effect of E2F1 on NEAT1, we monitored the E2F1 occupancy via chromatin immunoprecipitation (CHIP) assays. CHIP-PCR analysis revealed that E2F1 was enriched at the promoter of NEAT1 in immature DCs (iDCs; Figure 4B). Moreover, the binding of E2F1 was substantially reduced in mature DCs (Figure 4B). These results suggest that E2F1 can bind to the promoter of NEAT1. To further assess the importance of E2F1 in the regulation of NEAT1, we examined the effects of a knock in or knockdown of E2F1 in DCs. The efficiency of transfection is shown in Figure S5A-C. Deletion of E2F1, performed using two different excision strategies increased the levels of NEAT1. In contrast, overexpression of E2F1 (OE-E2F1) reduced the expression of NEAT1 (Figure 4C). These results strongly suggest that E2F1 negatively modulated the expression of NEAT1. To determine how E2F1 negatively controls the transcription of NEAT1, we used bisulfite sequencing (BSP) to assess the methylation status of a CpG island at the promoter of NEAT1 in DCs. We sequenced two CpG islands (Figure S5D) and showed that there were no differences in the total hypomethylation rate in imDCs, mature DCs, NC, and DCs with a knockdown of E2F1 (Figures 4D and S5E). These data indicate that the methylation status of CpG islands exerted no obvious effects on the expression of NEAT1. We then treated DCs with 5-aza-2'-deoxycytidine (a DNA demethylase) and Trichostatin A. The expression of NEAT1 was not significantly changed in demethylase-treated cells (Figure S5F). However, the expression of NEAT1 was downregulated in DCs treated with Trichostatin A (Figure 4E), suggesting that histone acetylation is closely associated with NEAT1 transcription. Subsequently, we used CHIP-PCR to examine whether E2F1 increases H3K27 acetylation (H3K27ac) at the promoter of NEAT1. We found that H3K27ac was enriched at the promoter of NEAT1 in DCs with a knockdown of E2F1 (Figure 4F). These results indicate that transcription factor E2F1 negatively controls the transcription of NEAT1 by regulating the binding of H3K27ac to the promoter of NEAT1.
NEAT1 was regulated by the let-7i/E2F1 axis. (A) The E2F1-binding elements were identified in the promoter region of NEAT1. (B) Chromatin immunoprecipitation assay was performed using E2F1 or IgG antibody in immature DCs (iDCs) and LPS-DCs. The relative expression of NEAT1 was detected by qRT-PCR. Data are expressed as mean ± SD; n = 3 biological replicates; **p<0.01, derived by Student's t-test. (C) DCs were transfected with E2F1 siRNA (siE2F1 1 or siE2F1 2), E2F1 pcDNA3.1 vector (OE E2F1), or negative control (NC) for 12 h before undergoing stimulation with LPS. The expression of NEAT1 was detected by qRT-PCR. Data are expressed as mean ± SD; n = 3 biological replicates; **p<0.01, derived by Student's t-test. (D) DCs were transfected with siE2F1 or NC for 12 h before undergoing stimulation with LPS. Methylation status of the CpG island was assessed by bisulfite sequencing in immature DCs (iDCs), LPS-DCs, NC, and siE2F1 DCs. Each row represents a single clone. (E) DCs were stimulated with Trichostatin A at different concentrations (0.05 nmol/ml, 0.01 nmol/ml, or 5 pmol/ml) for 12 h before undergoing stimulation with LPS. The expression of NEAT1 was detected by qRT-PCR. LPS-DCs were used as positive controls and iDCs were used as negative controls. Data are expressed as mean ± SD; n = 3 biological replicates; *p<0.05, **p<0.01, ns, not significant, derived using Student's t-test. (F) DCs were transfected with siE2F1 or OE E2F1 for 12 h before stimulation with LPS. Chromatin immunoprecipitation assay was performed using H3K27ac or IgG antibody in DCs. The relative expression of NEAT1 was detected by qRT-PCR. Data are expressed as mean ± SD; n = 3 biological replicates; ***p<0.001, derived using Student's t-test. (G-H) A reporter construct with potential binding site for let-7i in the 3'-UTR-wild type (WT) or 3'UTR-mutant type (MUT) region of E2F1 was generated. 293T cells were transiently transfected with miR-3076-3p mimic of miR-3076-3p inhibitor for 24 h. Luciferase activities were measured and normalized to the level of Renilla luciferase used as control (G-H). Data are expressed as mean ± SD; n = 3 biological replicates; **p<0.01 derived using Student's t-test. (I-K) DCs were transfected with a let-7i inhibitor or mimic for 12 h before stimulation with LPS. mRNA expression of E2F1 was detected by qRT-PCR (I). LRP3 protein level was detected by western blotting (J-K). Data are expressed as mean ± SD; n = 3 biological replicates; **p<0.01, ***p<0.001, derived using Student's t-test. (L) DCs were transfected with let-7i mimic, let-7i inhibitor, let-7i mimic+OE E2F1, or let-7i mimic+siE2F1 for 12 h before stimulation with LPS. The expression of NEAT1 was assessed by qRT-PCR. Data are expressed as mean ± SD; n = 3 biological replicates; **p<0.01, ***p<0.001, derived using Student's t-test.
The expression of NEAT1 is regulated by microRNA let-7i via binding to the 3'-UTR region of E2F1 mRNA
We have recently shown that microRNA let-7i suppresses the maturation of DCs and induces immune tolerance in a model of heart transplantation [38,39]. Interestingly, let-7i was predicted to target E2F1 using starBase v2.0 (http://starbase.sysu.edu.cn/). There were one-base target-microRNA matches for let-7i in the 3'-UTR of E2F1. Therefore, we performed a direct binding assay to assess the binding between let-7i and E2F1 in DCs. We generated a psiCHECK-2 luciferase construct containing 3'-UTR of E2F1 mRNA and the putative let-7i binding site. The dual-luciferase reporter assay revealed that co-transfection with the let-7i mimic significantly inhibited the translation of E2F1 luciferase reporter, which was markedly increased by the let-7i inhibitor (Figure 4G). However, luciferase activity was not significantly changed in cells transfected with the mutant reporter plasmid of E2F1 (Figure 4H). Consistently, the expression of E2F1 mRNA and protein was decreased in DCs transfected with the let-7i mimic, but increased in DCs transfected with the let-7i inhibitor (Figure 4I-K). Together, these results indicate that let-7i directly bound to E2F1 mRNA and regulated the expression of E2F1. We next used qRT-PCR to examine whether let-7i regulated NEAT1 via the transcription factor E2F1. Interestingly, the upregulation of NEAT1 in DCs transfected with the let-7i mimic was significantly reversed by overexpression E2F1. Meanwhile, the downregulation of NEAT1 in DCs transfected with the let-7i inhibitor was significantly reversed by E2F1 depletion (Figure 4L). These results show that let-7i can regulate the expression of NEAT1 via E2F1 activity in DCs.
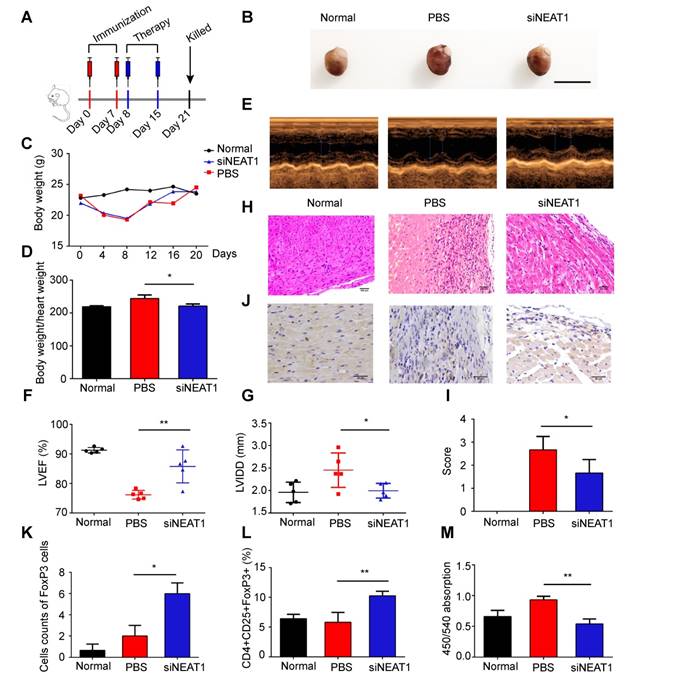
Knockdown of NEAT1 DCs prevents progression of experimental autoimmune myocarditis
Experimental autoimmune myocarditis (EAM) was established in Balb/c mice by subcutaneous injection with myosin on days 0 and 7 according to a previously published protocol [44]. To determine whether knockdown of NEAT1 in DCs can exert a therapeutic effect in EAM, NEAT1-knockdown DCs, PBS, or NC-transfected DCs were injected into mice via caudal vein on days 8 and 15 after the onset of EAM (Figure 5A). Mice were sacrificed on day 21, and the hearts were examined. Histological examination of the hearts showed no significant differences between the PBS-treated group and the NC-transfected group (Figure S6A-B). Therefore, we used PBS as the control treatment in our study. The hearts of EAM mice treated with NEAT1-knockdown DCs were smaller than those in the PBS-treated group (Figure 5B). The body weights of mice treated with NEAT1-knockdown DC and of those treated with PBS were decreased until day 8 (Figure 5C), indicating the development of myocarditis in these mice. The body weight/heart weight ratio in the group treated with siNETAT-transfected DCs was significantly lower than that in the PBS-treated group (Figure 5D). Echocardiographic analysis revealed that the function of the left ventricle was impaired in the PBS-treated group; however, treatment with NEAT1-knockdown DCs increased the left ventricular ejection fractions (LVEF) and decreased the left ventricular end diastolic dimension (LVDd) (Figure 5E-G). Histological examination of the hearts showed a slightly increased infiltration of inflammatory cells in the group treated with NEAT1-knockdown DCs (Figure 5H). The myocarditis score of the group treated with NEAT1-knockdown DCs was reduced compared with that of the PBS-treated group (Figure 5I). The expression of NLRP3, caspase-1, and ASC was decreased in the group treated with NEAT1-knockdown DCs (Figure S6C-E). Our immunohistochemical study showed that the hearts from the group treated with NEAT1-knockdown DCs contained more FoxP3+ cells than did those from the PBS-treated group (Figure 5J-K). The percentage of CD4+CD25+FoxP3+ cells was significantly increased in the spleens of mice treated with NEAT1-knockdown DCs (Figure 5L). To test the maximum proliferation of Balb/c (EAM) splenocytes (responder cells) against C57BL/6 splenocytes (stimulator cells), we performed the MLRs. On day 4 of MLRs, the proliferation of splenocytes, obtained from mice treated with NEAT1-knockdown DCs, was significantly suppressed compared with that of splenocytes obtained from the PBS-treated group (Figure 5M). Taken together, these data indicate that infusion of NEAT1-knockdown DCs protected mice from the development of EAM.
Knockdown of NEAT1 in DCs induces immune tolerance in experimental autoimmune myocarditis. (A) Injection schedule. BALB/C mice were injected with α-MyHC/CFA to induce experimental autoimmune myocarditis (EAM) on days 0 and 7. Then, siNEAT1 DCs or PBS were injected into mice on days 8 and 15. Mice were sacrificed on Day 21. (B) Panel shows images of hearts from three different mouse groups. Scale bars correspond to 1 cm. (C) Body weight (g) of siNEAT1 mice, EAM mice, and normal mice. (D) Body weight/heart weight ratio of siNEAT1 mice, EAM mice, and normal mice. Data are expressed as mean ± SD; n = 5 biological replicates; *p<0.05, derived using Student's t-test. (E-G) Representative images of left ventricular (LV) M-mode echocardiograms 21 days after injection (E). Left ventricular ejection fraction (LVEF) (F) and LV internal diastolic diameter (LVIDD) (G) are shown. Data are expressed as mean ± SD; n = 5 biological replicates; *p<0.05, **p<0.01, derived using Student's t-test. (H-I) Histologic studies of harvested hearts stained with hematoxylin and eosin (H). Assessment of hematoxylin and eosin staining by grading (I). Scale bars correspond to 100 µm.Data are expressed as mean ± SD; n = 5 biological replicates; *p<0.05, derived using Student's t-test. (J-K) Immunohistochemical labeling for FoxP3 (J) and counts of infiltrating FoxP3 cells (K) in hearts harvested from different groups. Scale bars correspond to 50 µm. Data are expressed as mean ± SD; n = 5 biological replicates; *p<0.05, derived using Student's t-test. (L) Expression of CD4+CD25+FoxP3+Treg in splenic T cells was assessed by flow cytometry in three different groups. Data are expressed as mean ± SD; n = 5 biological replicates; **p<0.01, derived using Student's t-test. (M) Results of T-cell proliferation assays of different groups assessed using a mixed lymphocyte reaction (MLR) by BrdU ELISA. The stimulator cells were C57BL/6 splenocytes treated with mitomycin C. Data are expressed as mean ± SD; n = 5 biological replicates; **p<0.01, derived using Student's t-test.
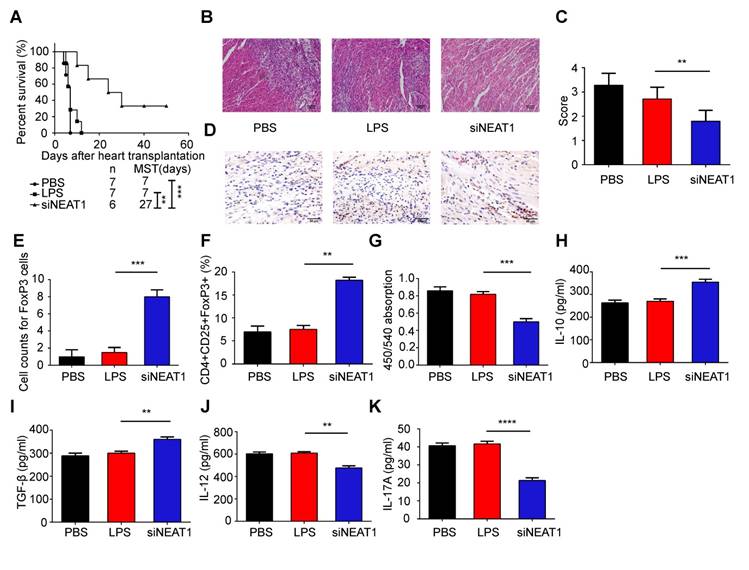
Transfusion with NEAT1-knockdown DCs induces immune tolerance in model of heart transplantation
We used the heart transplantation model to determine the role of NEAT1-knockdown DCs in transplant rejection. The heart of C57BL/6 mice was transplanted to Balb/C mice. The heart transplantation mice were injected with PBS or LPS-mDCs or knockdown of NEAT1 DCs. The median survival time (MST) of the PBS-treated and LPS-mDC-treated groups was 7 days. However, the mice transfused with NEAT1-knockdown DCs showed significantly prolonged allograft survival (MST of 27 days, Figure 6A). Hematoxylin and eosin (H&E) staining of the hearts, performed 1 week after transplantation, showed that infiltration of inflammatory cells was reduced in allografts obtained from recipients of NEAT1-knockdown DCs (Figure 6B). Treatment with NEAT1-knockdown DCs preserved allograft structure. Significant differences among the groups injected with PBS, LPS-mDCs, and NEAT1-knockdown DCs were determined by grading according to the 2005 classification of the International Society for Heart and Lung Transplantation (Figure 6C). T lymphocytes play a central role in the development of transplant rejection [45]. In this study, we used IHC to show that the numbers of FoxP3+ cells were increased in cardiac allografts from the recipients of NEAT1-knockdown DCs (Figure 6D-E). Results obtained using flow cytometry showed that the percentage of CD4+CD25+FoxP3+ cells was increased in the spleens of mice transfused with NEAT1-knockdown DCs (Figure 6F). We used MLRs to assay the proliferation of Balb/c splenocytes (responder cells) against that of C57BL/6 splenocytes (stimulator cells). The proliferation of splenocytes in the group treated with NEAT1-knockdown DCs was significantly suppressed compared with those of the PBS-treated group and LPS-mDC-treated group (Figure 6G). This suggests that transfusion with NEAT1-knockdown DCs induced suppressive cells in the recipients. The levels of anti-inflammatory cytokines IL-10 and TGF-β were increased in the plasma of mice transfused with NEAT1-knockdown DCs (Figure 6H-I), whereas the levels of inflammatory cytokines IL-12 and IL-17A were increased in mice transfused with PBS or LPS-mDCs (Figure 6J-K). Taken together, our results indicate that treatment with NEAT1-knockdown DCs significantly prolonged the survival of cardiac allografts, and induced the production of Tregs and anti-inflammatory cytokines. We conclude that knockdown of NEAT1 in DCs induced immune tolerance in mice that had undergone heart transplantation.
Knockdown of NEAT1 DCs induce immune tolerance in a model of heart transplantation. (A) Graft survival times of Balb/c mice transfused with PBS, LPS-DCs, or siNEAT1 DCs. MST, median survival time. Survival of cardiac allografts in groups of mice was compared using the Mann-Whitney U test. (B-C) Histologic studies of harvested cardiac allografts stained with hematoxylin and eosin 14 days after transplantation (B). Hematoxylin and eosin staining was graded according to the 2005 classification of International Society for Heart and Lung Transplantation for acute cellular rejection (C). Scale bars correspond to 100 µm.Data are expressed as mean ± SD; n = 6 biological replicates; **p<0.01, derived using Student's t-test. (D-E) Infiltration of FoxP3 cells into cardiac grafts of mice injected with PBS, LPS-DCs, or siNEAT1 DCs 14 days after grafting (D). Cell counts of infiltrating FoxP3 cells in cardiac allografts from each group (E). Scale bars correspond to 50 µm. Data are expressed as mean ± SD; n = 6 biological replicates; ***p<0.001, derived using Student's t-test. (F) Expression of CD4+CD25+FoxP3+Treg in splenocytes was assessed by flow cytometry in three different groups. Data are expressed as mean ± SD; n = 6 biological replicates; **p<0.01, derived using Student's t-test. (G) The proliferation of T cells in different groups was assessed using MLR by BrdU ELISA. The stimulator cells were C57BL/6 splenocytes treated with mitomycin C. Data are expressed as mean ± SD; n = 6 biological replicates; ***p<0.001, derived using Student's t-test. (H-K) The levels of cytokines (IL-10, TGF-β, IL-12, and IL-17A) were detected by ELISA in the plasma of recipient mice 14 days after heart transplantation. Data are expressed as mean ± SD; n = 6 biological replicates; **p<0.01, ***p<0.001, ****p<0.0001, derived using Student's t-test.
Discussion
Treatment with tol-DCs has been successfully implemented in various experimental animal models, but is considered a breakthrough in developing cell-based tolerance-inducing therapies (CTT) for patients with immune-related diseases [4,46]. Numerous clinical studies have shown that tol-DCs are safe and well-tolerated in the treatment of immune-related diseases [4]. The first phase I clinical trial using tol-DCs was performed in patients with type I diabetes (T1D). The results showed that tol-DCs are safely tolerated and increase the frequency of peripheral B220+CD11C-B cells [47]. Two trials have been published recently with respect to using tol-DCs in the treating rheumatoid arthritis (RA). Benham et al. found that patients with RA treated with tol-DCs show a reduction in effector T cells, increased ratio of regulatory to effector T cells, and reduced levels of serum IL-15 [48]. An AUTODECRA trial showed that tol-DCs are a safe and well-tolerated therapy with no target knee flares in patients with RA [49]. Patients with Crohn's disease, treated with tol-DCs, show low scores for Crohn's Disease Active Index and Crohn's Disease Endoscopic Index of Severity [50]. Dalia et al. found that tol-DCs induced stable antigen-specific hyporesponsiveness in myelin-reactive T cells collected from patients with multiple sclerosis [51]. These encouraging results of clinical trials show that tol-DCs can be a central factor in modulation of immune-related diseases.
Previous studies have shown that tol-DCs can be generated by various approaches such as those using pharmacological agents and genetic engineering [52]. Regardless of which methods are used for induction, the induced tol-DCs still show low expression of co-stimulatory molecules and reduced secretion of anti-inflammatory cytokines [53]. The expression of NEAT1 is upregulated in numerous human malignancies including prostate, lung, and breast cancers, and glioblastoma [42,54-56]. Previous studies have shown that NEAT1 can regulate the immune response in systemic lupus erythematosus (SLE), multiple sclerosis, and viral infections [20,21,57]. Imamura et al. have shown that NEAT1 can activate transcription of the antiviral gene IL-8 by relocating SFPQ from the IL-8 promoter after infection with a virus [21]. Although NEAT1 has been studied in immune response to various diseases, the function of NEAT1 in tol-DCs remains to be clarified. In this study, we first reported that knockdown of NEAT1 induced tol-DCs. Transfusion with NEAT1-knockdown DCs attenuated inflammation in mice with EAM and prolonged the survival of allografts in recipients of heart transplantation.
The mechanisms of action of lncRNAs are diverse and include RNA-RNA, RNA-DNA, and RNA-protein interactions [58]. Competing endogenous RNA (ceRNA) are used in the regulation of lncRNAs. ceRNA are used as decoys or sponges for miRNAs, hampering the targeting and repressive activity of miRNAs [59]. Acting as a ceRNA, NEAT1 regulates the target genes of miRNAs. NEAT1 induces the development of neuropathic pain in chronic constriction injury by targeting the miR-381/HMGB1 axis [60]. Jiang et al. demonstrated that NEAT1 serves as a ceRNA to modulate the activity of ZEB1 by sponging miR-448 in breast cancer [55]. In the present study, we performed small RNA sequencing in NEAT1-knockdown DCs. We found that NEAT1 regulated the function of DCs via miR-3076-3p/NLRP3 pathway. The NLRP3 inflammasome is a driving factor in many immune-related diseases such as type 2 diabetes, rheumatoid arthritis, atherosclerosis, and graft-versus-host disease [61-64]. NLRP3 plays a significant role in the development and activation of DCs. The development of CD11b+ DCs requires NLRP3 during steady-state and chronic infection [36]. Infection of DCs with the influenza A virus results in priming and activation of the NLRP3 inflammasome, and secretion of IL-1β and IL-18 [65]. IL-10-/- DCs inhibit the NLRP3 inflammasome via activity of P2X7R, thereby preventing DC apoptosis [66]. However, the role of the NLRP3 inflammasome in tol-DCs remains unclear. In this study, we observed that NEAT1 and NLRP3 were the target genes of miR-3076-3p. NEAT1 induced the expression of NLRP3 mRNA and protein, which was reversed by the administration of a miR-3076 mimic. Moreover, overexpression of NLRP3 reversed the knockdown of NEAT1 in NEAT1-knockdown DCs with a tolerogenic phenotype. Taken together, these results indicate that NEAT1 served as a ceRNA, modulating the expression of NLRP3 by sponging miR-3076-3p in tol-DCs.
Similar to coding RNA transcripts, lncRNAs can be regulated by transcription factors [67]. Li et al. demonstrated that transcription factor FOXN3 regulates the expression of NEAT1 in breast cancer [68]. Jen et al. revealed a novel mechanism by which the transcription factor Oct4 activates NEAT1 via the NEAT1 promoter in lung cancer [69]. In our study, we found that the transcription factor E2F1 could regulate the expression of NEAT1. E2F1 exerts a potent suppressive effect on the maturation of DCs. Fang et al. observed that E2F1 expression is transiently downregulated in DCs stimulated with LPS. DCs from E2F1-/- mice show a strongly enhanced maturation phenotype compared with that of DCs obtained from control mice [70]. We found that E2F1 could bind the promoter of NEAT1 and inhibit the expression of NEAT1. Previous studies have shown that transcription factors regulate the expression of target genes via epigenetic regulation [71]. However, the mechanism involved in epigenetic regulation of NEAT1 remains largely undetermined. DNA methylation is an essential epigenetic mechanism of regulating gene expression [72]. We examined the methylation status of a CpG island at the promoter of NEAT1 in DCs. Our results indicate that total hypomethylation levels did not change in iDCs, LPS-DCs, DCs with a knockdown of E2F1, or DCs with overexpression of E2F1. Similar to protein-coding RNAs, lncRNAs can be regulated by histone methylation or acetylation [43]. The expression of NEAT1 was not altered in DCs treated with a methyltransferase inhibitor. However, the expression of NEAT1 in DCs was downregulated by treatment with the histone deacetylase inhibitor Trichostatin A, suggesting that histone acetylation was involved in the regulation of NEAT1. Using CHIP-PCR, we found that the levels of H3K27ac decreased in LPS-DCs. However, E2F1 increased the binding of H3K27ac to the promoter of NEAT1. Our results show that NEAT1 can be regulated by the transcription factors E2F1 and H3K27ac. MicroRNA let-7i, a member of the let-7 family, is an inflammatory factor [73,74]. Our previous studies, as well as those of others, have shown that let-7i plays important roles in immune regulation, while the inhibitor of let-7i is a key suppressor of mature DCs [38,39,75]. Our previous studies have shown that downregulation of let-7i significantly impedes DC maturation, as evidenced by the reduced expression of CD80 and CD86 resulting from the suppressed expression of SOCS1 and IL-10 [38,39]. Taghikhani et al. have shown that overexpression of let-7i efficiently induces DC maturation [75].Transfusion with DCs treated with a let-7i inhibitor prolongs the survival of cardiac allografts and increases the numbers of Tregs in rats [39]. The results of our present study show that transcription factor E2F1 regulated the expression of NEAT1 via H3K27ac. Interestingly, we first predicted that E2F1 was the target gene of let-7i. Our results indicate that let-7i inhibited the expression of NEAT1 by targeting E2F1 in tol-DCs, and show the mechanism of upstream regulation of NEAT1 in tol-DCs.
Conclusions
We have shown that NEAT1 can induce immune tolerance in DCs. In the cytoplasm, NEAT1 served as a ceRNA, modulating NLRP3 by sponging miR-3076-3p. In the nucleus, let-7i regulated the expression of NEAT1 by binding to the transcription factor E2F1. E2F1 affected the distribution of H3K27ac in the promoter of NEAT1. Our results show that NEAT1 knockdown can induce immune tolerance in vivo. In the mouse models of EAM and heart transplantation, transfusion with NEAT1-knockdown DCs decreased inflammatory cell infiltration, inhibited the proliferation of T cells, and increased the numbers of Tregs. Treatment with NEAT1-knockdown DCs prolonged the survival of cardiac allografts in mice that had undergone heart transplantation. Our study shows that NEAT1 functioned via different regulatory mechanisms in tol-DCs. Our data indicate that the lncRNA NEAT1 may be a promising therapeutic target in the treatment of immune-related diseases.
Abbreviations
tol-DCs: tolerogenic dendritic cells; lncRNAs: long noncoding RNAs; GVHD: graft-versus-host disease; DCs: dendritic cells; APCs: antigen-presenting cells; SLE: systemic lupus erythematosus; NLRP3: NACHT, LRR, and PYD domains-containing protein 3; TLR4: toll-like receptor 4; SPF: specific pathogen-free; EAM: experimental autoimmune myocarditis; ISH: in situ hybridization histochemistry; MLR: mixed lymphocyte reaction; RIP: RNA-binding protein immunoprecipitation; UTR: untranslated region; WT: wild type; CHIP: chromatin immunoprecipitation; SDS-PAGE: sodium dodecyl sulfate-polyacrylamide gel electrophoresis; BSP: bisulfite sequencing PCR; LVEDs: left ventricular end-diastolic diameter; IVSTd: diastolic interventricular septum thickness; PWTd: posterior wall septum thickness; HE: hematoxylin and eosin; CTT: cell-based tolerance-inducing therapies; T1D: type I diabetes; RA: rheumatoid arthritis; ceRNA: competing endogenous RNA.
Supplementary Material
Table S1.
Supplementary figures and Table S2-S3.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant No. 81670373 to J.W., 81670459 to M.Z., and 81771846 to Y.S.), the National Key R&D Program of China (Grant No. 2016YFC1301100 to B.Y.), and the Key Laboratory of Myocardial Ischemia, Harbin Medical University, Ministry of Education (KF201806 to X.Z., KF201716 to Y.Z., KF201822 to L.C.).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Ramos-Casals M, Brito-Zeron P, Kostov B, Siso-Almirall A, Bosch X, Buss D. et al. Google-driven search for big data in autoimmune geoepidemiology: analysis of 394,827 patients with systemic autoimmune diseases. Autoimmunity reviews. 2015;14:670-9
2. Zeiser R, Blazar BR. Pathophysiology of Chronic Graft-versus-Host Disease and Therapeutic Targets. The New England journal of medicine. 2017;377:2565-79
3. Pothineni NVK, Subramany S, Kuriakose K, Shirazi LF, Romeo F, Shah PK. et al. Infections, atherosclerosis, and coronary heart disease. European heart journal. 2017;38:3195-201
4. Obregon C, Kumar R, Pascual MA, Vassalli G, Golshayan D. Update on Dendritic Cell-Induced Immunological and Clinical Tolerance. Frontiers in immunology. 2017;8:1514
5. Globinska A, Boonpiyathad T, Satitsuksanoa P, Kleuskens M, van de Veen W, Sokolowska M. et al. Mechanisms of allergen-specific immunotherapy: Diverse mechanisms of immune tolerance to allergens. Annals of allergy, asthma & immunology: official publication of the American College of Allergy, Asthma, & Immunology. 2018;121:306-12
6. Audiger C, Rahman MJ, Yun TJ, Tarbell KV, Lesage S. The Importance of Dendritic Cells in Maintaining Immune Tolerance. Journal of immunology. 2017;198:2223-31
7. Mo C, Zeng Z, Deng Q, Ding Y, Xiao R. Imbalance between T helper 17 and regulatory T cell subsets plays a significant role in the pathogenesis of systemic sclerosis. Biomedicine & pharmacotherapy = Biomedecine & pharmacotherapie. 2018;108:177-83
8. Han L, Jin H, Zhou L, Zhang X, Fan Z, Dai M. et al. Intestinal Microbiota at Engraftment Influence Acute Graft-Versus-Host Disease via the Treg/Th17 Balance in Allo-HSCT Recipients. Frontiers in immunology. 2018;9:669
9. Kleijwegt FS, Laban S, Duinkerken G, Joosten AM, Koeleman BP, Nikolic T. et al. Transfer of regulatory properties from tolerogenic to proinflammatory dendritic cells via induced autoreactive regulatory T cells. Journal of immunology. 2011;187:6357-64
10. Morlando M, Fatica A. Alteration of Epigenetic Regulation by Long Noncoding RNAs in Cancer. International journal of molecular sciences. 2018:19
11. Xu S, Kamato D, Little PJ, Nakagawa S, Pelisek J, Jin ZG. Targeting epigenetics and non-coding RNAs in atherosclerosis: From mechanisms to therapeutics. Pharmacology & therapeutics. 2018
12. Atianand MK, Caffrey DR, Fitzgerald KA. Immunobiology of Long Noncoding RNAs. Annual review of immunology. 2017;35:177-98
13. Sehgal L, Mathur R, Braun FK, Wise JF, Berkova Z, Neelapu S. et al. FAS-antisense 1 lncRNA and production of soluble versus membrane Fas in B-cell lymphoma. Leukemia. 2014;28:2376-87
14. Sallam T, Jones M, Thomas BJ, Wu X, Gilliland T, Qian K. et al. Transcriptional regulation of macrophage cholesterol efflux and atherogenesis by a long noncoding RNA. Nature medicine. 2018;24:304-12
15. Wang P, Xue Y, Han Y, Lin L, Wu C, Xu S. et al. The STAT3-binding long noncoding RNA lnc-DC controls human dendritic cell differentiation. Science. 2014;344:310-3
16. Wu J, Zhang H, Zheng Y, Jin X, Liu M, Li S. et al. The Long Noncoding RNA MALAT1 Induces Tolerogenic Dendritic Cells and Regulatory T Cells via miR155/Dendritic Cell-Specific Intercellular Adhesion Molecule-3 Grabbing Nonintegrin/IL10 Axis. Frontiers in immunology. 2018;9:1847
17. Hutchinson JN, Ensminger AW, Clemson CM, Lynch CR, Lawrence JB, Chess A. A screen for nuclear transcripts identifies two linked noncoding RNAs associated with SC35 splicing domains. BMC genomics. 2007;8:39
18. Zhang Q, Chen CY, Yedavalli VS, Jeang KT. NEAT1 long noncoding RNA and paraspeckle bodies modulate HIV-1 posttranscriptional expression. mBio. 2013;4:e00596-12
19. Clemson CM, Hutchinson JN, Sara SA, Ensminger AW, Fox AH, Chess A. et al. An architectural role for a nuclear noncoding RNA: NEAT1 RNA is essential for the structure of paraspeckles. Molecular cell. 2009;33:717-26
20. Zhang F, Wu L, Qian J, Qu B, Xia S, La T. et al. Identification of the long noncoding RNA NEAT1 as a novel inflammatory regulator acting through MAPK pathway in human lupus. Journal of autoimmunity. 2016;75:96-104
21. Imamura K, Imamachi N, Akizuki G, Kumakura M, Kawaguchi A, Nagata K. et al. Long noncoding RNA NEAT1-dependent SFPQ relocation from promoter region to paraspeckle mediates IL8 expression upon immune stimuli. Molecular cell. 2014;53:393-406
22. Shen HH, Yang YX, Meng X, Luo XY, Li XM, Shuai ZW. et al. NLRP3: A promising therapeutic target for autoimmune diseases. Autoimmunity reviews. 2018;17:694-702
23. Tate MD, Mansell A. An update on the NLRP3 inflammasome and influenza: the road to redemption or perdition? Current opinion in immunology. 2018;54:80-5
24. Jahan S, Kumar D, Chaturvedi S, Rashid M, Wahajuddin M, Khan YA. et al. Therapeutic Targeting of NLRP3 Inflammasomes by Natural Products and Pharmaceuticals: A Novel Mechanistic Approach for Inflammatory Diseases. Current medicinal chemistry. 2017;24:1645-70
25. Yazdi AS, Guarda G, Riteau N, Drexler SK, Tardivel A, Couillin I. et al. Nanoparticles activate the NLR pyrin domain containing 3 (Nlrp3) inflammasome and cause pulmonary inflammation through release of IL-1alpha and IL-1beta. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:19449-54
26. Dostert C, Petrilli V, Van Bruggen R, Steele C, Mossman BT, Tschopp J. Innate immune activation through Nalp3 inflammasome sensing of asbestos and silica. Science. 2008;320:674-7
27. Hughes MM, O'Neill LAJ. Metabolic regulation of NLRP3. Immunological reviews. 2018;281:88-98
28. Ebrahimi T, Rust M, Kaiser SN, Slowik A, Beyer C, Koczulla AR. et al. alpha1-antitrypsin mitigates NLRP3-inflammasome activation in amyloid beta1-42-stimulated murine astrocytes. Journal of neuroinflammation. 2018;15:282
29. Shao BZ, Cao Q, Liu C. Targeting NLRP3 Inflammasome in the Treatment of CNS Diseases. Frontiers in molecular neuroscience. 2018;11:320
30. Zhao C, Gu Y, Zeng X, Wang J. NLRP3 inflammasome regulates Th17 differentiation in rheumatoid arthritis. Clinical immunology. 2018;197:154-60
31. Patel D, Gaikwad S, Challagundla N, Nivsarkar M, Agrawal-Rajput R. Spleen tyrosine kinase inhibition ameliorates airway inflammation through modulation of NLRP3 inflammosome and Th17/Treg axis. International immunopharmacology. 2018;54:375-84
32. Okano T, Ashida H, Suzuki S, Shoji M, Nakayama K, Suzuki T. Porphyromonas gingivalis triggers NLRP3-mediated inflammasome activation in macrophages in a bacterial gingipains-independent manner. European journal of immunology. 2018
33. D'Addio F, Vergani A, Potena L, Maestroni A, Usuelli V, Ben Nasr M. et al. P2X7R mutation disrupts the NLRP3-mediated Th program and predicts poor cardiac allograft outcomes. The Journal of clinical investigation. 2018;128:3490-503
34. Ali MF, Dasari H, Van Keulen VP, Carmona EM. Canonical Stimulation of the NLRP3 Inflammasome by Fungal Antigens Links Innate and Adaptive B-Lymphocyte Responses by Modulating IL-1beta and IgM Production. Frontiers in immunology. 2017;8:1504
35. Ghiringhelli F, Apetoh L, Tesniere A, Aymeric L, Ma Y, Ortiz C. et al. Activation of the NLRP3 inflammasome in dendritic cells induces IL-1beta-dependent adaptive immunity against tumors. Nature medicine. 2009;15:1170-8
36. Arnold IC, Zhang X, Urban S, Artola-Boran M, Manz MG, Ottemann KM. et al. NLRP3 Controls the Development of Gastrointestinal CD11b(+) Dendritic Cells in the Steady State and during Chronic Bacterial Infection. Cell reports. 2017;21:3860-72
37. Son YI, Egawa S, Tatsumi T, Redlinger RE Jr, Kalinski P, Kanto T. A novel bulk-culture method for generating mature dendritic cells from mouse bone marrow cells. Journal of immunological methods. 2002;262:145-57
38. Zhang M, Liu F, Jia H, Zhang Q, Yin L, Liu W. et al. Inhibition of microRNA let-7i depresses maturation and functional state of dendritic cells in response to lipopolysaccharide stimulation via targeting suppressor of cytokine signaling 1. Journal of immunology. 2011;187:1674-83
39. Sun Y, Jin X, Liu X, Zhang M, Liu W, Li Z. et al. MicroRNA let-7i regulates dendritic cells maturation targeting interleukin-10 via the Janus kinase 1-signal transducer and activator of transcription 3 signal pathway subsequently induces prolonged cardiac allograft survival in rats. The Journal of heart and lung transplantation: the official publication of the International Society for Heart Transplantation. 2016;35:378-88
40. Eriksson U, Kurrer MO, Schmitz N, Marsch SC, Fontana A, Eugster HP. et al. Interleukin-6-deficient mice resist development of autoimmune myocarditis associated with impaired upregulation of complement C3. Circulation. 2003;107:320-5
41. Morchikh M, Cribier A, Raffel R, Amraoui S, Cau J, Severac D. et al. HEXIM1 and NEAT1 Long Non-coding RNA Form a Multi-subunit Complex that Regulates DNA-Mediated Innate Immune Response. Molecular cell. 2017;67:387-99 e5
42. Chen Q, Cai J, Wang Q, Wang Y, Liu M, Yang J. et al. Long Noncoding RNA NEAT1, Regulated by the EGFR Pathway, Contributes to Glioblastoma Progression Through the WNT/beta-Catenin Pathway by Scaffolding EZH2. Clinical cancer research: an official journal of the American Association for Cancer Research. 2018;24:684-95
43. Zhang E, Han L, Yin D, He X, Hong L, Si X. et al. H3K27 acetylation activated-long non-coding RNA CCAT1 affects cell proliferation and migration by regulating SPRY4 and HOXB13 expression in esophageal squamous cell carcinoma. Nucleic acids research. 2017;45:3086-101
44. Cai G, Wang H, Qin Q, Zhang J, Zhu Z, Liu M. et al. Amelioration of myocarditis by HVEM-overexpressing dendritic cells through induction of IL-10-producing cells. Cardiovascular research. 2009;84:425-33
45. DeWolf S, Sykes M. Alloimmune T cells in transplantation. The Journal of clinical investigation. 2017;127:2473-81
46. Suwandi JS, Toes RE, Nikolic T, Roep BO. Inducing tissue specific tolerance in autoimmune disease with tolerogenic dendritic cells. Clinical and experimental rheumatology. 2015;33:S97-103
47. Giannoukakis N, Phillips B, Finegold D, Harnaha J, Trucco M. Phase I (safety) study of autologous tolerogenic dendritic cells in type 1 diabetic patients. Diabetes care. 2011;34:2026-32
48. Benham H, Nel HJ, Law SC, Mehdi AM, Street S, Ramnoruth N. et al. Citrullinated peptide dendritic cell immunotherapy in HLA risk genotype-positive rheumatoid arthritis patients. Science translational medicine. 2015;7:290ra87
49. Bell GM, Anderson AE, Diboll J, Reece R, Eltherington O, Harry RA. et al. Autologous tolerogenic dendritic cells for rheumatoid and inflammatory arthritis. Annals of the rheumatic diseases. 2017;76:227-34
50. Jauregui-Amezaga A, Cabezon R, Ramirez-Morros A, Espana C, Rimola J, Bru C. et al. Intraperitoneal Administration of Autologous Tolerogenic Dendritic Cells for Refractory Crohn's Disease: A Phase I Study. Journal of Crohn's & colitis. 2015;9:1071-8
51. Raiotach-Regue D, Grau-Lopez L, Naranjo-Gomez M, Ramo-Tello C, Pujol-Borrell R, Martinez-Caceres E. et al. Stable antigen-specific T-cell hyporesponsiveness induced by tolerogenic dendritic cells from multiple sclerosis patients. European journal of immunology. 2012;42:771-82
52. Yoo S, Ha SJ. Generation of Tolerogenic Dendritic Cells and Their Therapeutic Applications. Immune network. 2016;16:52-60
53. Svajger U, Rozman P. Induction of Tolerogenic Dendritic Cells by Endogenous Biomolecules: An Update. Frontiers in immunology. 2018;9:2482
54. Wedge DC, Gundem G, Mitchell T, Woodcock DJ, Martincorena I, Ghori M. et al. Sequencing of prostate cancers identifies new cancer genes, routes of progression and drug targets. Nature genetics. 2018;50:682-92
55. Jiang X, Zhou Y, Sun AJ, Xue JL. NEAT1 contributes to breast cancer progression through modulating miR-448 and ZEB1. Journal of cellular physiology. 2018;233:8558-66
56. Wu F, Mo Q, Wan X, Dan J, Hu H. NEAT1/has-mir-98-5p/MAPK6 axis is involved in non-small-cell lung cancer (NSCLC) development. Journal of cellular biochemistry. 2017
57. Dastmalchi R, Ghafouri-Fard S, Omrani MD, Mazdeh M, Sayad A, Taheri M. Dysregulation of long non-coding RNA profile in peripheral blood of multiple sclerosis patients. Multiple sclerosis and related disorders. 2018;25:219-26
58. Zhang Y, Cao X. Long noncoding RNAs in innate immunity. Cellular & molecular immunology. 2016;13:138-47
59. Quinn JJ, Chang HY. Unique features of long non-coding RNA biogenesis and function. Nature reviews Genetics. 2016;17:47-62
60. Xia LX, Ke C, Lu JM. NEAT1 contributes to neuropathic pain development through targeting miR-381/HMGB1 axis in CCI rat models. Journal of cellular physiology. 2018;233:7103-11
61. Chimenti MS, Triggianese P, Conigliaro P, Candi E, Melino G, Perricone R. The interplay between inflammation and metabolism in rheumatoid arthritis. Cell death & disease. 2015;6:e1887
62. Chavez-Sanchez L, Espinosa-Luna JE, Chavez-Rueda K, Legorreta-Haquet MV, Montoya-Diaz E, Blanco-Favela F. Innate immune system cells in atherosclerosis. Archives of medical research. 2014;45:1-14
63. Jankovic D, Ganesan J, Bscheider M, Stickel N, Weber FC, Guarda G. et al. The Nlrp3 inflammasome regulates acute graft-versus-host disease. The Journal of experimental medicine. 2013;210:1899-910
64. Lee J. Adipose tissue macrophages in the development of obesity-induced inflammation, insulin resistance and type 2 diabetes. Archives of pharmacal research. 2013;36:208-22
65. Fernandez MV, Miller E, Krammer F, Gopal R, Greenbaum BD, Bhardwaj N. Ion efflux and influenza infection trigger NLRP3 inflammasome signaling in human dendritic cells. Journal of leukocyte biology. 2016;99:723-34
66. Omosun Y, McKeithen D, Ryans K, Kibakaya C, Blas-Machado U, Li D. et al. Interleukin-10 modulates antigen presentation by dendritic cells through regulation of NLRP3 inflammasome assembly during Chlamydia infection. Infection and immunity. 2015;83:4662-72
67. Jandura A, Krause HM. The New RNA World: Growing Evidence for Long Noncoding RNA Functionality. Trends in genetics: TIG. 2017;33:665-76
68. Li W, Zhang Z, Liu X, Cheng X, Zhang Y, Han X. et al. The FOXN3-NEAT1-SIN3A repressor complex promotes progression of hormonally responsive breast cancer. The Journal of clinical investigation. 2017;127:3421-40
69. Jen J, Tang YA, Lu YH, Lin CC, Lai WW, Wang YC. Oct4 transcriptionally regulates the expression of long non-coding RNAs NEAT1 and MALAT1 to promote lung cancer progression. Molecular cancer. 2017;16:104
70. Fang F, Wang Y, Li R, Zhao Y, Guo Y, Jiang M. et al. Transcription factor E2F1 suppresses dendritic cell maturation. Journal of immunology. 2010;184:6084-91
71. Firas J, Liu X, Lim SM, Polo JM. Transcription factor-mediated reprogramming: epigenetics and therapeutic potential. Immunology and cell biology. 2015;93:284-9
72. Schubeler D. Function and information content of DNA methylation. Nature. 2015;517:321-6
73. Taghikhani A, Hassan ZM, Ebrahimi M, Moazzeni SM. microRNA modified tumor-derived exosomes as novel tools for maturation of dendritic cells. Journal of cellular physiology. 2018
74. Satoh M, Tabuchi T, Minami Y, Takahashi Y, Itoh T, Nakamura M. Expression of let-7i is associated with Toll-like receptor 4 signal in coronary artery disease: effect of statins on let-7i and Toll-like receptor 4 signal. Immunobiology. 2012;217:533-9
75. Taghikhani A, Hassan ZM, Ebrahimi M, Moazzeni SM. microRNA modified tumor-derived exosomes as novel tools for maturation of dendritic cells. Journal of cellular physiology. 2019;234:9417-27
Author contact
![]() Corresponding author: Tel: +86-451-86605800; E-mail: wujian780805com
Corresponding author: Tel: +86-451-86605800; E-mail: wujian780805com