Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and methods
Results
Discussion
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2019; 9(25):7566-7582. doi:10.7150/thno.36942 This issue Cite
Research Paper
The hepatocyte-specifically expressed lnc-HSER alleviates hepatic fibrosis by inhibiting hepatocyte apoptosis and epithelial-mesenchymal transition
Kun Zhang1#, Mengxia Zhang1#, Qingbin Yao1#, Xiaohui Han1#, Yanmian Zhao1, Lina Zheng1, Guantong Li2, Qi Liu1, Yanan Chang3, Peijun Zhang4, Hongmei Cui1, Zhemin Shi1, Ting Chen1, Zhi Yao5, Tao Han2, Wei Hong1 ![]()
1. Department of Histology and Embryology, Key Laboratory of Immune Microenvironment and Disease of Ministry of Education, Tianjin Key Laboratory of Cellular and Molecular Immunology, School of Basic Medical Sciences, Tianjin Medical University, Tianjin, China
2. The Third Central Clinical College of Tianjin Medical University, Department of Hepatology and Gastroenterology, Tianjin Third Central Hospital, Tianjin Key Laboratory of Artificial Cells, Artificial Cell Engineering Technology Research Center of Public Health Ministry, Tianjin, China
3. Department of Pathology, Institute of Hematology and Blood Diseases Hospital, Chinese Academy of Medical Sciences and Peking Union Medical College, Tianjin, China
4. Tianjin Central Hospital of Gynecology Obstetrics, Tianjin, China
5. Department of Immunology, Key Laboratory of Immune Microenvironment and Disease of Ministry of Education, Tianjin Key Laboratory of Cellular and Molecular Immunology, School of Basic Medical Sciences, Tianjin Medical University, Tianjin, China
#These authors contributed equally to this work.
Received 2019-5-23; Accepted 2019-9-2; Published 2019-10-12
Abstract

Liver fibrosis leading to cirrhosis is one of the major health burdens worldwide with currently limited therapeutic options available. Long noncoding RNAs (lncRNAs) play important roles in various biological and pathological processes in a cell- or tissue-specific manner. However, there is still an important gap in the understanding of the role of hepatocyte-specific lncRNAs in liver fibrosis.
Methods: The expressions of lnc-Hser in human and mice fibrotic livers as well as primary hepatocytes (HCs) of mice developing liver fibrosis were determined by real-time RT-PCR. The roles and mechanisms of lnc-Hser in HCs and liver fibrosis were determined in vitro and in vivo.
Results: In this study, we have identified a hepatocyte-specifically expressed lnc-Hser, which was reduced in human and mice fibrotic livers as well as primary HCs of mice developing liver fibrosis. We have shown that silencing lnc-Hser aggravated liver fibrosis both in vitro and in vivo through inducing the epithelial-mesenchymal transition (EMT) and the apoptosis of HCs. In addition, knockdown of lnc-Hser promoted hepatic stellate cells (HSCs) activation through the signals derived from injured HCs. Mechanistically, we have revealed that lnc-Hser inhibited HCs apoptosis via the C5AR1-Hippo-YAP pathway and suppressed HCs EMT via the Notch signaling.
Conclusions: Our work has identified a hepatocyte-specific lnc-HSER that regulates liver fibrosis, providing a proof that this molecule is a novel biomarker for damaged HCs and a potential target for anti-fibrotic therapy.
Keywords: lncRNA, liver fibrosis, hepatocyte, C5AR1, Notch
Introduction
Liver fibrosis is a common pathological consequence of a sustained wound healing response to chronic liver injury, including viral hepatitis, alcohol abuse and autoimmune disease, characterized by abnormal deposition of extracellular matrix (ECM) and diminished hepatocytes (HCs) regeneration that is responsible for compromised liver function accounting for death [1, 2]. Progressive liver fibrosis leads to cirrhosis and its associated complications include portal hypertension, liver failure, and the high risk of incident hepatocellular carcinoma [3, 4]. Current models of chronic liver injury of various etiologies emphasize HCs apoptosis as a critical initiator of fibrosis by triggering HSCs to trans-differentiate into myofibroblasts with an up-regulation of pro-fibrogenic cytokines such as TGFβ, and an increased production of ECM compounds [5]. In addition, HCs EMT is also an important cellular event propagating liver fibrosis progression [6]. Therefore, developing a new therapeutic approach to protect HCs from apoptosis and EMT is needed.
Many molecules and signalings have been documented to initiate HCs apoptosis and EMT in liver fibrosis, including TGFβ1, Notch and Hippo pathways. TGFβ1, a known pro-apoptotic cytokine in mature HCs, is capable of mediating phenotypic changes and plasticity in the process of EMT, resulting in ECM deposition [6]. The Notch signaling network is an evolutionarily conserved intercellular signaling that regulates a range of cellular functions including proliferation, apoptosis, cell fate and differentiation [7]. Triggering Notch receptors (Notch1-4) activates proteolytic cleavages catalyzed by γ-secretase, generating the Notch intracellular domain (NICD), which enters nucleus to activate transcription of canonical Notch targets including the Hairy enhancer of split (Hes) and Hes-related (Hey) family genes through the transcription factor RBPJ. Of note, increasing evidences suggest that Notch signaling also contributes to the progression of liver fibrosis via inducing the EMT [8]. Moreover, it is well established that isolated HCs in culture rapidly lose their epithelial and functional properties and spontaneously undergo a de-differentiation process towards a mesenchymal cell type. This process, which exerts features of the EMT, is blocked in the presence of several inhibitors including Notch inhibitor [9]. The Hippo pathway is an important modulator involved in the regulatory process of cell proliferation, differentiation and death. Central to the Hippo pathway is a kinase cascade wherein MST (the mammalian orthologue of the Drosophila Hippo) phosphorylates and activates the adaptor protein Mob and the protein kinase LATS. Activated LATS phosphorylates the transcriptional co-activator YAP/TAZ, binding to 14.3.3 proteins in the cytoplasm, which accelerates the sequestration and degradation of YAP/TAZ. Unphosphorylated YAP translocates into the nucleus, interacts with a number of transcriptional factors to mediate the transcription of genes that control cell proliferation and apoptosis [10]. Additionally, G-protein-coupled receptors (GPCRs), the largest family of cell surface receptors, have been reported to function upstream of the Hippo pathway through Rho GTPase and cytoskeleton remodeling [11]. Interestingly, several lines of evidence indicate that the Hippo/YAP signaling plays a critical role in liver fibrosis. Some studies including our preliminary study found that YAP is increased and undergoes a nuclear localization at an early stage during the activation of HSCs, indicating that YAP may function at the earliest stage of HSCs activation [12, 13]. However, the role of YAP in HCs during liver fibrogenesis is still unclear. It has been reported that YAP promotes HCs death and subsequently leads to liver fibrosis [14], while others revealed that YAP protects HCs from death [15, 16]. Hence, further understanding of the cellular and molecular mechanism of liver fibrosis may lead to the development of more effective treatments.
Long noncoding RNAs (lncRNAs) represent a group of transcripts that are above 200 nucleotides in length but without protein coding potential [17, 18]. Emerging evidences have demonstrated that lncRNAs are expressed in a cell- or tissue-specific manner and involved in a variety of biological and pathological processes including organ fibrosis [19-25]. We and others have revealed that lncRNAs were implicated in HSCs activation and thereby regulate liver fibrosis. For example, H19 plays a critical role in the progression of cholestatic liver fibrosis either by promoting HSCs differentiation and activation or by preventing ZEB1-mediated inhibition of epithelial cell adhesion molecule [23, 26]. Our group have recently demonstrated that a liver-enriched lncRNA LFAR1 promoted HSCs activation and subsequently led to liver fibrosis [22], and a nuclear-retained lncRNA SCARNA10 functioned as a novel positive regulator of TGFβ signaling in liver fibrogenesis by inhibiting the binding of PRC2 to the promoters of the genes associated with ECM and TGFβ pathway [27]. However, there is still an important gap in understanding the role of cell-specific lncRNAs that mainly affect HCs during liver fibrogenesis.
In the present study, we have identified a HC-specifically expressed lnc-Hser, which was reduced in human and mice fibrotic livers as well as primary HCs of mice developing liver fibrosis. We have shown that silencing lnc-Hser aggravated liver fibrosis both in vitro and in vivo through inducing the EMT and the apoptosis of HCs. In addition, knockdown of lnc-Hser promoted HSCs activation through the signals derived from damaged HCs. we have also revealed that lnc-Hser inhibited HCs apoptosis via the C5AR1-Hippo-YAP pathway and suppressed HCs EMT through the Notch signaling. All these data suggest that lnc-HSER is a novel biomarker for damaged HCs and a potential target for anti-fibrotic therapy.
Materials and methods
Cell culture and antibodies
The non-tumorigenic mouse hepatocyte cell line AML12 was maintained in Dulbecco's modified Eagle's medium (DMEM, Invitrogen, Camarillo, CA) supplemented with 10% fetal bovine serum (FBS, Gibco, Gaithersburg, MD, USA), 1 × insulin-transferrin-sodium selenite media supplement (ITS; Sigma-Aldrich), dexamethasone (40 ng/ml), penicillin (100 U/ml) and streptomycin (100 μg/ml). The human hepatocyte cell line L02 and HEK293T were cultured in DMEM supplemented with 10% FBS, penicillin (100 U/ml) and streptomycin (100 μg/ml). All cells were cultured at 37°C in an atmosphere containing 5% CO2. For co-culture experiment, lnc-Hser-silenced and lnc-Hser-overexpressed AML12 cells and the controls were washed with PBS after 24 - 48h of lentivirus infection. The cells were subsequently incubated in DMEM supplemented with 10% FBS for 48h and the supernatants were centrifuged at 1100 rpm for 5 min and mixed with DMEM containing 10% FBS at 1:1 ratio for preparing conditioned medium (CM). Cells were treated with the C5AR1 inhibitor PMX 205 (Med Chem Express, USA) or γ-secretase inhibitor RO4929097 (Med Chem Express, USA) for 24 hours at concentrations of 5 μM. The antibodies were α-SMA (rabbit polyclonal, Abcam, ab5694), Collagen1 (rabbit polyclonal, Abcam, ab34710; Millipore, #234167), TGFβ (rabbit polyclonal, Abcam, ab66043), MMP2 (rabbit monoclonal, Abcam, ab92536), TIMP1 (mouse monoclonal, Santa Cruz,sc-21734), Notch2 (rabbit monoclonal, Cell Signaling Technology, #5732), Notch3 (rabbit polyclonal, Abcam, ab23426), Hes1 (rabbit polyclonal, Abcam, ab71559), phospho-YAP (Ser-127) (rabbit monoclonal, Cell Signaling Technology, #13619), total YAP/TAZ (rabbit monoclonal, Cell Signaling Technology, #8418), phospho-MST1/2 (rabbit monoclonal, Cell Signaling Technology, #49332), total MST1 (rabbit monoclonal, Cell Signaling Technology, #3682), phospho-LATS (Ser-909) (rabbit polyclonal, Cell Signaling Technology, #9157), total LATS1 (rabbit monoclonal, Cell Signaling Technology, #3477), C5AR1 (rabbit monoclonal, Proteintech, #21316-1-AP), Ki67 (rabbit monoclonal, Abcam, ab16667), Cleaved Caspase3 (rabbit polyclonal, Cell Signaling Technology, #9661), Caspase3 (rabbit monoclonal, Cell Signaling Technology, #9662), BAX (rabbit polyclonal, Abcam, ab32503), N-Cadherin (rabbit monoclonal, Cell Signaling Technology, #13116; Mouse monoclonal, Abcam, ab98952), E-cadherin (rabbit monoclonal, Cell Signaling Technology, #3195), β-Catenin (rabbit monoclonal, Cell Signaling Technology, #8480), Vimentin (rabbit monoclonal, Cell Signaling Technology, #5741), Snail (rabbit monoclonal, Cell Signaling Technology, #3879), rabbit IgG (Millipore, PP64B), goat anti rabbit IgG (Invitrogen, Alexa Fluor 488/594), goat anti mouse IgG (Invitrogen, Alexa Fluor 594).
Construction of plasmids
gRNA design was based on CRISPR design (http://crispr.mit.edu/) or CHOPCHOP (https://chopchop.rc.fas.harvard.edu/) and cloned into lenti-CRISPRv2. Oligos encoding shRNA specific for lnc-Hser and the negative control shRNA were ligated into pSUPER.retro.puro, and the fragment containing the H1 promoter and hairpin sequences was subcloned into the lentiviral shuttle pCCL.PPT.hPGK.GFP.Wpre (lnc-Hser-shRNA and Negative Control (NC)). The full-length lnc-Hser cDNA was sequentially amplified by PCR and ligated into the lentiviral shuttle pCCL.PPT.hPGK.IRES.eGFP/pre to generate the over-expression plasmid (LV-lnc-Hser and the empty plasmid as the LV-Control). These plasmids were used to produce lentivirus in HEK-293T cells with the packaging plasmids pMD2.BSBG, pMDLg/pRRE and pRSV-REV. Infectious lentiviruses were harvested at 36 h and 60 h after transfection and filtered through 0.45 μm PVDF filters. Recombinant lentiviruses used in vivo were concentrated 100-fold by ultracentrifugation (2 h at 120,000 g). The virus-containing pellet was dissolved in PBS and injected in mice within 48 h. The primer sets used are shown in Table S1.
Animals in vivo study
Animal protocols were approved by Tianjin Medical University Animal Care and Use Committee. The methods were carried out in accordance with the approved guidelines. All Balb/c mice aged at 8 weeks were obtained from Institute of Laboratory Animal Sciences, CAMS & PUMC (Beijing, China). For bile duct ligation (BDL) -induced mouse liver fibrosis model, mice were treated with sham operation or BDL operation for 3 to 21 days and all of mice were sacrificed under anesthesia with 3% sodium pentobarbital (45 mg/kg, ip). For carbon tetrachloride (CCl4)-induced mouse liver fibrosis model, forty Balb/c mice were randomly divided into four groups: Mice were treated with olive oil in combination with injection of lenti-NC (NC; n = 10), CCl4 in combination with injection of lenti-NC (NC + CCl4, n = 10), oil in combination with injection of lenti-lnc-Hser-shRNA (lnc-Hser-shRNA, n = 10) and CCl4 in combination with injection of lenti-lnc-Hser-shRNA (lnc-Hser-shRNA + CCl4, n = 10). The lentivirus was injected via the tail vein 2 weeks after the first injection of CCl4 (1 × 109 pfu/mouse). Mice in NC + CCl4 group and lnc-Hser-shRNA + CCl4 group were administered 5% CCl4 (v/v) dissolved in olive oil (0.2 ml/kg body weight) twice per week for additional 4 weeks via intraperitoneal (ip) injection after the lentivirus was injected. NC and lnc-Hser-shRNA group animals were injected with an equivalent volume of olive oil. After treatment with CCl4 for 6 weeks, all of mice were sacrificed under anesthesia with 3% sodium pentobarbital (45 mg/kg, ip). Liver specimens and serums were obtained for analyses.
Histology and immunohistochemistry
The specimens were sequentially fixed in 10% formalin for two days, transferred to ethanol of different concentration and embedded in paraffin in preparation for histopathological analysis. Thin sections (5 μm) were stained with H&E, Sirius red and Masson's trichrome staining for histopathological study. According to the above results, three sections were chosen from each group for immunohistochemical analysis. Briefly, sections prepared on slides were firstly submitted to antigen retrieval by incubation in citrate buffer (pH 6.0) for 5 min at 108 °C and pretreated with 3% H2O2 for 15 min at room temperature followed by washing with PBS. Slides were subsequently incubated in normal goat serum for 20 min to block the nonspecific immunoreactivity. Next, the slides were treated with primary antibody α-SMA (1:50), Col1α1 (1:1000) or TGFβ (1:50) overnight at 4°C. In addition, tissue sections were processed omitting the primary antibody as the negative control. The slides were incubated with secondary antibody (1:500) (HRP-conjugated anti-rabbit IgG) and the reaction products were visualized using diaminobenzidine (DAB) and monitored by microscopy.
ELISA and liver enzyme measurement
The TGFβ level in cell culture media and the alanine aminotransferase (ALT) and aspartate aminotransferase (AST) levels in the serum were assessed using commercial assay kits (Nanjing Jiancheng Corp., Nanjing, China) according to the manufacturer's protocols.
Hydroxyproline assay
Total collagen content was tested by measuring the amount of hydroxyproline in liver tissue using commercially available hydroxyproline detection kits (Nanjing Jiancheng Corp., Nanjing, China) according to the manufacturer's instructions.
Isolation and culture of primary HCs, HSCs, liver sinusoidal endothelial cell (LSECs) and Kupffer cell (KCs)
Primary mouse HCs, HSCs, LSECs and KCs were isolated by pronase/collagenase perfusion digestion followed by subsequent density gradient centrifugation, as previously described [22]. Magnetic cells sorting (MACS) and/or selective adhesion was further employed to isolate LSECs (CD146) and KCs (F4/80). In brief, primary HCs were isolated from the 6-week-old Balb/c mice by in situ perfusion with 30 ml SC1 solution and 30 ml 0.05% Collagenase IV solution sequentially. HCs were then pelleted by centrifugation 50 g for 4 minutes three times. Cell viability was determined by the trypan blue exclusion method.
TUNEL assay
For TUNEL staining, we used an in-situ cell detection kit (Roche) according to the manufacturer's protocol. After dewaxing and rehydration, we pretreated tissue sections with 3% H2O2 and subsequent proteinase K permeation. Pretreatment with DNase I served as a positive control and TUNEL reaction mixture lacking terminal transferase (TdT) as a negative control. Samples were analyzed by light microscopy (×400 magnification).
Nuclear-cytoplasmic fractionation
Cytoplasmic and nuclear RNA isolation were performed with PARIS™ Kit (Invitrogen, Grand Island, NY, USA) following the manufacturer's instruction, as previously described [22].
5' and 3' rapid amplification of cDNA ends (RACE)
We used the 5'-RACE and 3'-RACE analyses to determine the transcriptional initiation and termination sites of lnc-Hser using a SMARTer™ RACE cDNA Amplification Kit (Clontech, Palo Alto, CA) according to the manufacturer's instructions. In brief, RNA was isolated from primary HCs and 3'- and 5'-RACE-Ready cDNA were synthesized using SMART Scribe Reverse Transcriptase. The obtained band was gel purified and cloned with the lineareized pRACE vector. The obtained band was then sequenced. The gene-specific primers used for the PCR of the RACE analysis are provided in Table S1.
Apoptosis assay
The apoptosis of cells with different treatment was analyzed using a FITC Annexin V Apoptosis Detection Kit I (BD Biosciences) according to the manufacturer's instruction, as previously described [22].
Quantitative real-time polymerase chain reaction
Total RNA was extracted from livers or cells with Trizol reagent (Takara, Dalian, China) and nuclear and cytoplasmic RNA were prepared using PARIS™ Kit (Invitrogen, Grand Island, NY, USA). All RNAs were digested with DNase I (Takara, Dalian, China). Briefly, the first-strand cDNA was synthesized using AMV Reverse Transcriptase (Thermo Fisher Scientific, Basingstoke, UK). For real-time PCR, all reactions were performed in triplicate with SYBR Green master mix (Takara, Dalian, China) according to the manufacturer's instructions. The expression level of housekeeping gene GAPDH was used to normalize the expression level of the genes-of-interest. The sequences of primers for real-time PCR are listed in Table S1.
Western blot analysis
Immunoblotting analysis was performed as described previously [28]. The antibodies against α-SMA (1:1000), Collagen1 (1:1000), TGFβ (1:1000), MMP2 (1:1000), TIMP1(1:1000), Notch2 (1:1000), Notch3 (1:1000), Hes1 (1:1000), phospho-YAP (Ser-127) (1:1000), total YAP/TAZ (1:1000), phospho-MST1/2 (1:1000), total MST1 (1:1000), phospho-LATS (Ser-909) (1:1000), total LATS1 (1:1000), C5AR1 (1:1000), Caspase3 (1:1000), BAX (1:1000), N-Cadherin (1:1000), E-cadherin (1:1000), β-Catenin (1:1000), Vimentin (1:1000), Snail (1:1000) and GAPDH (1:8000) were diluted in TBS containing 5% milk and 0.1% Tween 20. Signal was detected using the chemiluminescence (ECL) system (Millipore).
Confocal microscopy
Primary HSCs, HCs and AML12 cells were re-plated on poly-lysine pre-coated glass coverslips, and were sequentially fixed with 4% paraformaldehyde in PBS overnight at 4 °C, permeabilized with 1% Triton X-100 in PBS for 0-60 min and blocked using 5% bovine serum albumin (BSA) in TBST with 0.1% Tween-20 for 30-60 min at room temperature. Next, the cells were incubated with primary antibodies against α-SMA (1:300), Col1α1 (1:500), Cleaved Caspase3 (1:400), N-Cadherin (Mouse monoclonal; 1:200) and Ki67 (1:250), overnight at 4 °C and an irrelevant isotype rabbit IgG was used as a negative control. Cells were then incubated with Alexa Fluor 488 (1:400 for Cleaved Caspase3) or Alexa Fluor 594 (1:400 for α-SMA, Col1α1, N-Cadherin and Ki67) in PBS away from light for 1 h at room temperature and the nuclei were stained with DAPI (5 μg/ml). Finally, the slides were washed with PBS and the coverslips were mounted with an anti-fade Mounting Medium (P0126, Beyotime, Shanghai, China). The immunofluorescence was then visualized by a confocal microscope (LSM 700) or a fluorescence microscope.
Study population
In total, 28 human fibrotic livers and 6 human healthy livers from patients with hepatic haemangioma were obtained from surgical resections without preoperative treatment at Tianjin Third Central Hospital (Tianjin, China). For histological scoring of liver fibrosis, we stained paraffin embedded 5 μm liver sections with Sirius red, H&E and Masson's trichrome staining. Hepatic fibrosis was scored (stages F0-F4) according to the METAVIR fibrosis staging system by three hepatopathologists blinded to the study protocol and stratified as normal liver (F0), mild fibrosis (F1-F2) or advanced fibrosis (F3-F4). All subjects were of the same ethnicity. Clinical and pathological characteristics including age, gender, ALT, AST and etiologies were recorded and summarized in Table S2. The study has been approved by the local Ethical Committee of Tianjin Third Central Hospital (Tianjin, China). Written informed consent was obtained from each patient according to the policies of the committee. The study methodologies were conformed to the standards set by the Declaration of Helsinki.
Statistical analysis
Data were expressed as mean ± SD. All the statistical analyses were performed with the SPSS 13.0 (IBM, Armonk, NY, USA). Statistical analyses were performed using either Student's t-test (two-group comparison) or one-way analysis of variance (more than two groups) followed by post hoc comparison, and differences with p<0.05 were considered significantly.
Results
lncRNA-Hser is specifically expressed in hepatocyte of normal liver
We have previously identified systemic variations in the expression of lncRNAs between mouse fibrotic and normal livers using a microarray analysis [22]. From that study, we noted that lncRNA ENSMUST00000154817 was significantly down-regulated in liver fibrosis according to the microarray data. (The microarray data discussed in that article have been deposited in NCBI Gene Expression Omnibus and are accessible through GEO Series accession number GSE80601). To explore the role of this molecule in liver fibrosis, we have firstly examined its expression patterns in various tissues of normal and fibrotic mice. As shown in Figure 1A, normal livers express highest level ENSMUST00000154817, which is down-regulated in fibrotic livers. In addition, we have isolated four types of primary cells including HCs, HSCs, LSECs and KCs from livers of healthy mice, and found ENSMUST00000154817 was mainly expressed in primary HCs rather than HSCs, LSECs and KCs (Figure 1B). These data demonstrated ENSMUST00000154817 was a HCs-specifically expressed lncRNA, and thus was named as lnc-Hser (hepatocyte specific expressed lncRNA). We have subsequently performed 5'- and 3'- RACE assay with primary HCs and found lnc-Hser was a 588-nucleotide transcript with poly (A) tail, inconsistent with the ensemble database (Figure S1A). The discrepancy may be due to the existence of different isoforms in various tissues. Cell fractionation followed by qRT-PCR showed that lnc-Hser was mainly located in the nuclei of primary HCs and AML12 cells (Figure S1B, C). As expected, the full-length transcript of lnc-Hser has no protein coding potential according to the coding potential calculator 2 (CPC2) and coding potential assessment tool (CPAT). The expression of lnc-Hser was then measured in total RNA extracts from fibrotic livers of mice treated with CCl4 or BDL for various time periods, and the results showed that the transcript of lnc-Hser was significantly down-regulated with persistent injury, correlating with gradual enhancement of α-SMA (Acta2) (Figure 1C, D). We have also isolated primary HCs from livers of mice treated with CCl4 for 6 weeks and found lnc-Hser exhibited a clear down-regulation compared with control, correlating with an up-regulation of Col1α1, Tnf-α and Mcp1 (Figure 1E). Notably, stimulation of primary HCs and AML12 cells with recombinant TGFβ1 resulted in a significantly reduced level of lnc-Hser (Figure 1F, G). Collectively, these data demonstrate that lnc-Hser is decreased in the livers and HCs of fibrotic mice, leading us to explore the possibility that lnc-Hser may be a regulator to the progression of liver fibrosis.
lnc-Hser is specifically expressed in HCs and down-regulated in fibrotic HCs and livers. (A) qRT-PCR analysis of lnc-Hser in various tissues from normal and fibrotic mice. (B) HCs, HSCs, KCs and LSECs were isolated from livers of Balb/c mice, and lnc-Hser expression was analyzed by qRT-PCR. (C, D) qRT-PCR analysis of lnc-Hser and Acta2 (α-SMA) in livers from mice treated with CCl4 (C) or undergone BDL (D) for indicated times. (E) Primary HCs were isolated from livers of mice treated with CCl4 or oil for 6 weeks, and the transcript of lnc-Hser, Col1α1, Tnf-α and Mcp1 was determined by qRT-PCR. (F, G) Primary HCs (F) and AML12 cells (G) were stimulated with TGFβ for 24 h and lnc-Hser, Col1α1, Tnf-α and Ctgf was determined by qRT-PCR. The data are expressed as the mean ± SD for at least triplicate experiments, *p< 0.05.
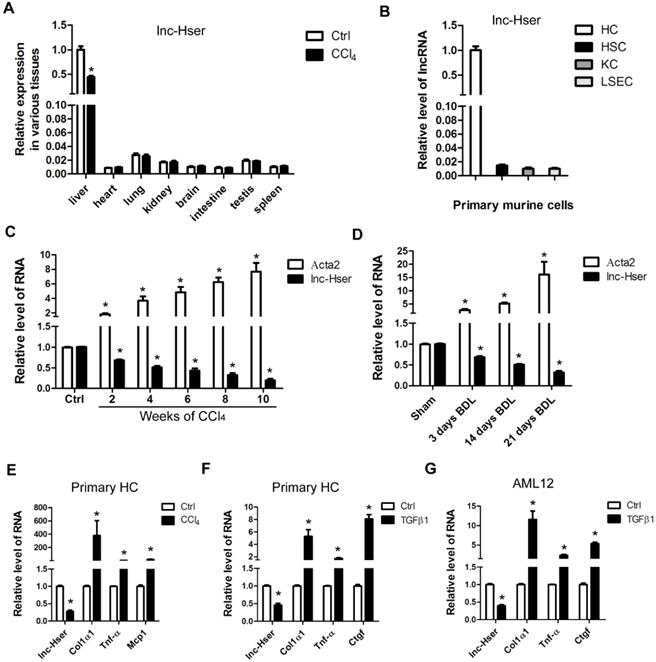
Knockdown of lnc-Hser facilitates the progression of liver fibrosis
To elucidate the in vivo function of lnc-Hser in hepatic fibrogenesis, we generated lnc-Hser down-expressed mouse models via tail vein injection of lenti-lnc-Hser-shRNA (lnc-Hser-shRNA) or lenti-negative control virus (NC) 2 weeks after the first injection of CCl4. After 6 weeks of CCl4 treatment, lnc-Hser silencing was confirmed by qRT-PCR in whole liver extracts (Figure S2A), and the CCl4 group mice infected with NC (NC-CCl4) developed liver fibrosis. However, administration of lnc-Hser-shRNA in CCl4 group (lnc-Hser-shRNA-CCl4) developed more severe phenotypes of fibrosis as demonstrated by both the staining of H&E, Sirius red, Masson's trichrome and the IHC for α-SMA, Collagen1 and TGFβ (Figure 2A and Figure S2B). The content of liver hydroxyproline in lnc-Hser-shRNA-CCl4 mice was also significantly higher in comparison with the NC-CCl4 mice (Figure 2B). Moreover, the expression of the predominant fibrotic genes including α-SMA, Col1α1, TGFβ, TIMP1, MMP2 and MMP9 was noticeably increased by CCl4 and was further enhanced in lnc-Hser-shRNA-CCl4 mice (Figure 2C, D). Notably, increased TUNEL positive HCs and serum level of ALT and AST were observed in lnc-Hser-shRNA-CCl4 versus NC-CCl4 mice, suggesting lnc-Hser silencing worsened CCl4-induced HCs injury (Figure S2B, C and Table S3). Consistent with these observations, knockdown of lnc-Hser exacerbated CCl4-induced HCs apoptosis assessed by IHC-Frozen, western blot and qRT-PCR (Figure S2D-F). In addition, the expression of the genes related to hepatic inflammation including Tnf-α, Il-1β, Mcp1 and Il-6, the mesenchymal markers N-Cadherin, β-Catenin, Vimentin and Snail1 was dramatically enhanced in lnc-Hser-shRNA-CCl4 versus NC-CCl4 mice (Figure S2D-F). Taken together, our results suggest that silencing lnc-Hser aggravates CCl4-induced liver fibrosis in vivo through inducing the EMT and the apoptosis of HCs.
Knockdown of lnc-Hser aggravates CCl4-induced liver fibrosis in vivo. Mice were treated with oil in combination with injection of lenti-NC (Negative Control, n = 10), or CCl4 in combination with injection of lenti-NC (NC + CCl4, n = 10), or oil in combination with injection of lenti-lnc-Hser-shRNA (lnc-Hser-shRNA, n = 10), or CCl4 in combination with injection of lenti-lnc-Hser-shRNA (lnc-Hser-shRNA + CCl4, n = 10). (A) Liver fibrosis was evaluated by stainings of H&E, Sirius red, Masson's trichrome, IHC for Collagen1 and α-SMA; scale bar = 400 μm for 10× and 100 μm for 40×. (B) Quantification of hepatic hydroxyproline content. The data are expressed as hydroxyproline (μg)/liver wet weight (g). (C) The protein level of α-SMA, Collagen1, TGFβ, MMP2 and TIMP1 was determined by western blot. GAPDH was used as an internal control. (D) The mRNA level of the predominant fibrotic genes, including α-SMA, Col1α1, Col1α2, Col3α1, Col4α5, Tgfβ, Timp1, Mmp2, Mmp9 and Ctgf, was determined by qRT-PCR. The data are expressed as the mean ± SD for at least triplicate experiments, *p<0.05 stands for NC + CCl4 or lnc-Hser-shRNA vs NC. #p<0.05 stands for lnc-Hser-shRNA + CCl4 vs NC + CCl4.
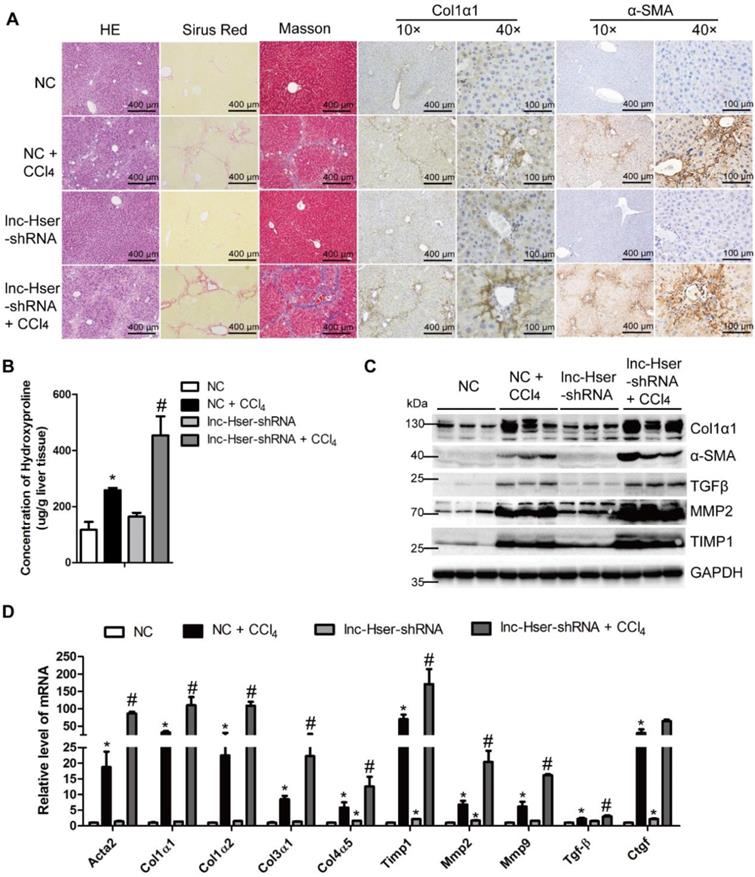
lnc-Hser ameliorates apoptosis and EMT of HCs in vitro
TGFβ1, a known pro-apoptotic cytokine in mature HCs, is also a major inducer of EMT [6]. To investigate whether lnc-Hser is required for the apoptosis and EMT of HCs in vitro, we firstly used lentiviral lnc-Hser (LV-lnc-Hser) for over-expression in primary HCs and AML12 cells. Recombinant TGFβ was then used to treat the cells after infection with LV-lnc-Hser or LV-Control, and total RNA was extracted for detecting the expression of apoptotic and EMT-related genes. The results showed that the expression of the pro-fibrotic genes α-SMA, Col1α1 and MMP2, the pro-apoptosis genes BAX and BAD, and the mesenchymal marker genes N-Cadherin, Vimentin, Fibronectin, Twist and Snail1 was markedly increased, whereas the expression of the anti-apoptosis gene Bcl2, the pro-proliferation genes Ki67 and Pcna, and the epithelial marker gene E-cadherin was significantly decreased upon TGFβ treatment. However, forced lnc-Hser expression abrogated TGFβ-induced dysregulation of these genes in primary HCs and AML12 cells assessed by qRT-PCR and western blot (Figure 3A-C and Figure S3A-D). Flow cytometry (FACS) analysis further confirmed that TGFβ stimulation dramatically induced AML12 apoptosis, however, forced lnc-Hser expression markedly suppressed TGFβ-induced apoptosis (Figure S3E). On the other hand, knockdown of lnc-Hser obviously up-regulated the expression of the pro-fibrotic genes, the pro-apoptosis genes, and the mesenchymal marker genes, while down-regulated the expression of the anti-apoptosis gene, pro-proliferation genes, and the epithelial marker gene in primary HCs and AML12 cells (Figure 3D-F and Figure S4A-E). Moreover, we generated stable knockdown of lnc-Hser using the CRISPR/Cas9 system with guide RNA pairs targeted to the promoter region and intron 1 of lnc-Hser in AML12 cells. As expected, the above results could be confirmed in lnc-Hser-silenced AML12 cells (Figure S5A-E). Additionally, the expression of the pro-apoptosis genes and the mesenchymal marker genes in the primary HCs isolated from lnc-Hser-shRNA-CCl4 mice exhibited a profound enhancement, but the expression of the anti-apoptosis gene and the pro-proliferation genes demonstrated a remarkable reduction, in comparison with the NC-CCl4 mice (Figure S6A). Taken together, these results suggest that silencing lnc-Hser induces the apoptosis and EMT of HCs.
lnc-Hser ameliorates apoptosis and EMT of HCs in vitro. (A-C) Primary HCs were infected with LV-lnc-Hser for 72 h and further treated with 10 ng/ml TGFβ for additional 24 h. The expression of lnc-Hser, pro-fibrogenic genes (Acta2, Col1α1 and Mmp2), apoptosis and proliferation -related genes (Bax, Bad, Bcl2, Ki67 and Pcna) and EMT-related genes (N-Cadherin, E-cadherin, Vimentin, Fibronectin, Twist and Snail1) was detected by qRT-PCR (A, B). The protein level of α-SMA, Col1α1, MMP2, total and cleaved Caspase3, BAX, N-Cadherin, E-cadherin, Vimentin, β-Catenin and SNAIL1 was detected by western blot. GAPDH was used as an internal control (C). (D, E) The expression of lnc-Hser, pro-fibrogenic genes, apoptosis-related genes and EMT-related genes was detected in primary HCs infected with lenti-lnc-Hser-shRNA or lenti-NC by qRT-PCR (D) and western blot. GAPDH was used as an internal control (E). (F) Primary HCs were transfected with siRNA-lnc-Hser or si-NC for 36 h, the expression and location of cleaved Caspase3 and N-Cadherin was determined by confocal microscopy. DAPI stained nuclei blue; scale bar = 50 μm. The data are expressed as the mean ± SD for at least triplicate experiments. *p<0.05 stands for vs LV-Control or NC. #p<0.05 stands for vs LV-Control + TGFβ or NC+ CCl4.
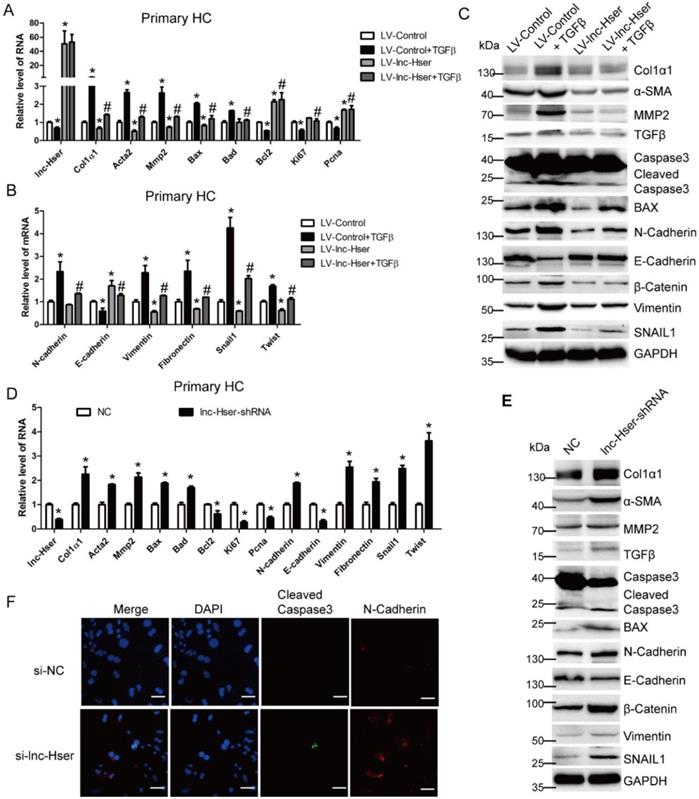
lnc-Hser is indirectly involved in the activation of HSCs
Activation of HSCs is the most principal cellular player promoting synthesis and deposition of ECM proteins in response to accumulated levels of inflammatory signals derived from damaged parenchymal cells [5]. The in vivo data showed that knockdown of lnc-Hser aggravated CCl4-induced liver fibrosis. Therefore, we investigated the effect of lnc-Hser on the activation of HSCs. Since the expression of lnc-Hser in primary HSCs is quite low, we over-expressed lnc-Hser using LV-lnc-Hser in the cells and subsequently treated them with TGFβ. The results showed that the expression of the predominant fibrotic genes including α-SMA, Col1α1, Col1α2, TIMP1 and MMP2 was up-regulated upon TGFβ treatment. However, over-expression of lnc-Hser neither directly suppressed nor abrogated TGFβ-induced up-regulation of these fibrotic genes in primary HSCs assessed by western blot and qRT-PCR (Figure 4A, B), suggesting that lnc-Hser is not directly involved in the regulation of the activation of HSCs. We therefore hypothesized that down-regulated lnc-Hser in HCs during liver fibrogenesis results in the apoptosis of HCs which subsequently acts on HSCs to promote the expression of pro-fibrotic genes.
lnc-Hser is indirectly involved in the activation of HSCs. (A, B) Primary HSCs were infected with LV-lnc-Hser for 72 h and further treated with 10 ng/ml TGFβ for additional 24 h. The protein level of α-SMA and Col1α1 was detected by western blot. GAPDH was used as an internal control (A). The RNA level of lnc-Hser, Acta2, Col1α1, Col1α2, Mmp2 and Timp1 was detected by qRT-PCR (B). (C-E) The conditioned medium from control, lnc-Hser -silenced or -over-expressed AML12 cells was used to treat primary HSC at day 2. After 48 hours incubation, the protein level of α-SMA and Col1α1 was detected by western blot. GAPDH was used as an internal control (C). The RNA level of lnc-Hser, Acta2, Col1α1, Col1α2, Timp1, Mmp2, Mmp9, Tgfβ, Pdgfβ and Ctgf was detected by qRT-PCR (D). The expression of α-SMA and Col1α1 was determined by confocal microscopy. DAPI stained nuclei blue; scale bar = 50 μm. The data are expressed as the mean ± SD for at least triplicate experiments, * p<0.05.
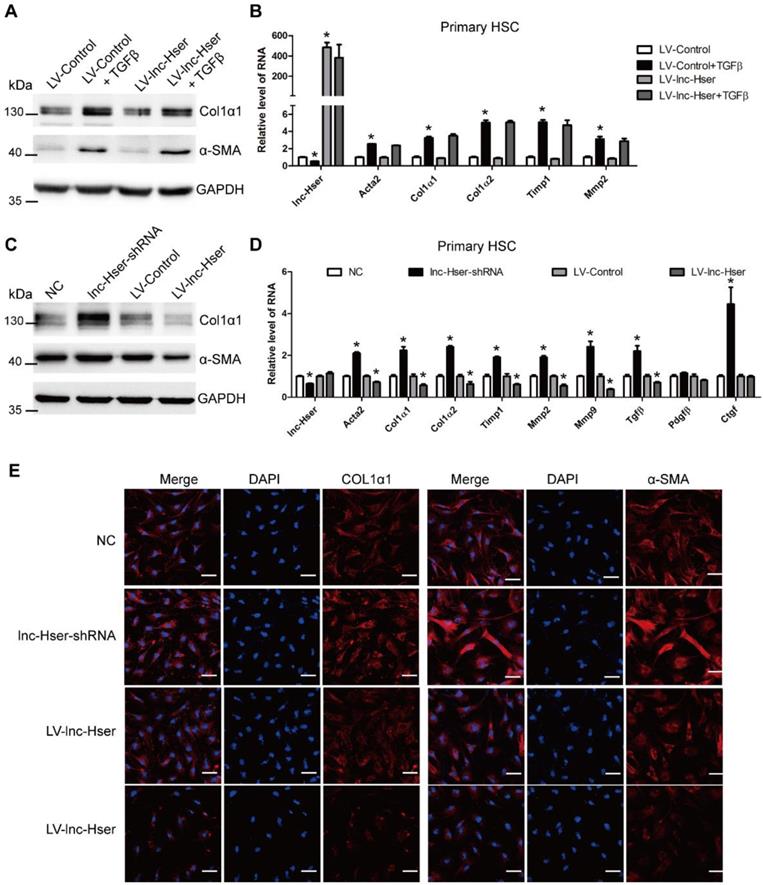
Hence, we used conditioned medium (CM) from control, lnc-Hser-silenced or lnc-Hser-over-expressed AML12 cells to treat primary HSCs. Compared with the control cells, treatment with the CM from lnc-Hser-silenced AML12 cells significantly promoted the expression of the fibrotic genes including α-SMA and Col1α1, but the administration with CM from lnc-Hser-over-expressed AML12 cells repressed the expression of these genes assessed by western blot, qRT-PCR and confocal microscopy (Figure 4C-E). Moreover, the level of TGFβ in supernatant from the cultured lnc-Hser-silenced AML12 cells and primary HCs was markedly increased (Figure S6B, C), suggesting that TGFβ is one of the factors derived from injured HCs that promotes HSCs activation. Taken together, these results clearly suggest that silencing lnc-Hser promotes HSCs activation through the signals derived from damaged HCs.
lnc-Hser inhibits HCs apoptosis via the C5AR1-Hippo-YAP pathway
It is well known that lncRNAs work in cis when their effects are restricted to the chromosome from which they are transcribed, and work in trans when they affect genes on other chromosomes [29]. To investigate the mechanism underlying lnc-Hser expression, we firstly examined whether lnc-Hser regulates its nearby genes in cis. The expression of Cntrl and C5, both of which are lnc-Hser neighboring genes, and C5AR1 which is a member of GPCRs family and plays an essential role in the early events leading to HCs proliferation and liver fibrosis [30, 31], was determined in either lnc-Hser over-expressed or silenced primary HCs and AML12 cells. We noticed that forced lnc-Hser expression down-regulated the expression of C5 and C5AR1 rather than Cntrl, whereas knockdown of lnc-Hser promoted the expression of both factors assessed by western blot and qRT-PCR (Figure 5A-D and Figure S7A-F). Similar results were observed in livers of the in vivo models (Figure 5E and Figure S7G). Since the GPCRs have been reported to function upstream of the Hippo pathway that modulates cell proliferation, differentiation and death, we then sought to explore whether lnc-Hser regulates the activation of the Hippo pathway. As shown in Figures S8 and S9, although over-expression of lnc-Hser slightly decreased the TGFβ-induced up-regulation of the compounds of the Hippo pathway including Lats1, Yap and Taz in primary HCs, Mst1 and Lats1 in AML12 cells, lnc-Hser had no effect on the mRNA level of these compounds. However, the phosphorylation of MST, LATS and YAP was significantly decreased in lnc-Hser-over-expressed cells, while markedly increased in lnc-Hser-silenced cells, suggesting that lnc-Hser inactivates the canonical Hippo pathway. Consistently, these results were also confirmed in the livers and primary HCs isolated from the in vivo model mice (Figure S10A-C). We then asked whether lnc-Hser ameliorates the apoptosis or EMT of HCs via C5AR1. PMX205, the specific inhibitor of C5AR1, was applied to treat the lnc-Hser-silenced AML12 cells and the expression of the pro-fibrotic, apoptosis and EMT associated genes was subsequently detected by western blot and qRT-PCR. Strikingly, PMX205 abrogated lnc-Hser silencing -induced up-regulation of both the pro-fibrotic and the pro-apoptotic genes but not the mesenchymal marker genes, suggesting that the inhibition of C5AR1 by lnc-Hser could suppress the apoptosis of HCs (Figure 5F-G). Moreover, FACS analysis revealed that the apoptosis induced by lnc-Hser silencing was abrogated by PMX205 (Figure S10D). Taken together, these results clearly suggest that lnc-Hser silencing promotes HCs apoptosis via the C5AR1-Hippo-YAP pathway.
lnc-Hser inhibits HCs apoptosis via C5AR1. (A, B) Primary HCs were infected with LV-lnc-Hser for 72 h and further treated with 10 ng/ml TGFβ for additional 24 h. The protein level of C5AR1 was detected by western blot. GAPDH was used as an internal control (A). The mRNA level of C5, C5ar1 and Cntrl was detected by qRT-PCR (B). (C) The protein level of C5AR1 in primary HCs infected with lenti-lnc-Hser-shRNA or lenti-NC was detected by western blot. GAPDH was used as an internal control. (D) The mRNA level of C5, C5ar1 and Cntrl was detected in primary HCs infected with lenti-lnc-Hser-shRNA or lenti-NC by qRT-PCR. (E) Mice were treated with oil in combination with injection of lenti-NC (Negative Control, n = 10), or CCl4 in combination with injection of lenti-NC (NC + CCl4, n = 10), or oil in combination with injection of lenti-lnc-Hser-shRNA (lnc-Hser-shRNA, n = 10), or CCl4 in combination with injection of lenti-lnc-Hser-shRNA (lnc-Hser-shRNA + CCl4, n = 10). qRT-PCR analysis of C5, C5ar1 and Cntrl in livers of mice in each group. (F-H) lnc-Hser was stably knocked down by the CRISPR/Cas9 system with guide RNA pairs in AML12 cells. PMX205, a specific inhibitor of C5AR1, was used to treat lnc-Hser-silenced AML12 cells for 24 h. The expression of lnc-Hser, pro-fibrogenic, apoptosis-related and EMT-related genes was detected by qRT-PCR (F, G) and western blot. GAPDH was used as an internal control (H). The data are expressed as the mean ± SD for at least triplicate experiments. *p<0.05 stands for vs LV-Control, NC or Control gRNA. #p<0.05 stands for vs LV-Control + TGFβ, NC+ CCl4 or lnc-Hser-gRNAs.
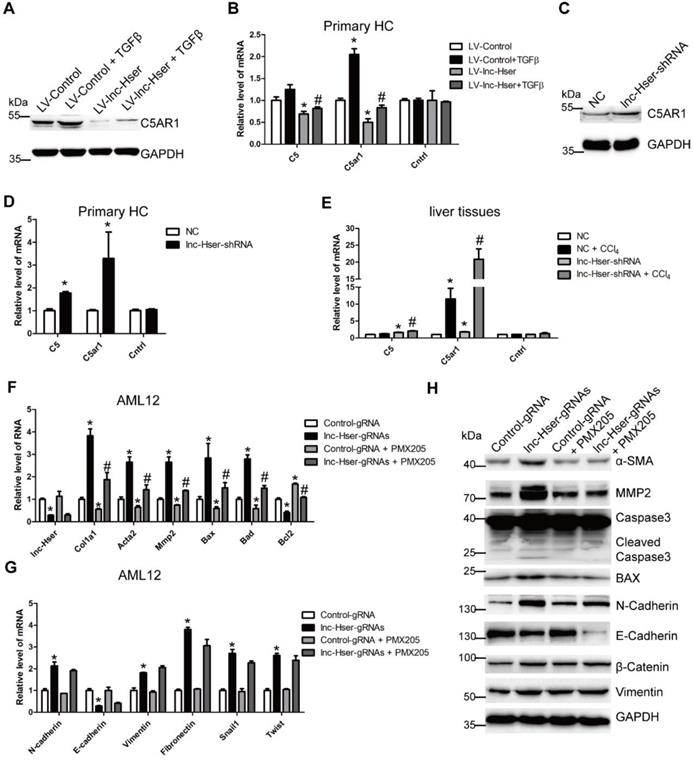
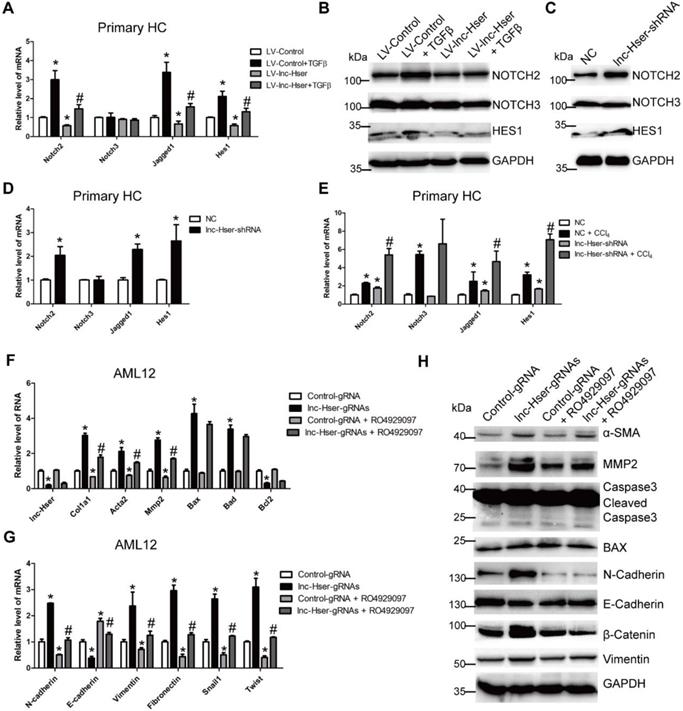
lnc-Hser inhibits HCs EMT via the Notch pathway
Increasing evidences suggest that Notch signaling contributes to the progression of liver fibrosis via inducing the EMT [7, 8]. We therefore considered the hypothesis that lnc-Hser may inhibit the EMT of HCs through the Notch pathway. The level of Notch2, Notch3, Jagged1 and Hes1 was detected in lnc-Hser-over-expressed and lnc-Hser-silenced primary HCs and AML12 cells. The results showed that forced lnc-Hser expression inhibited the expression of Notch2, Jagged1 and Hes1 rather than Notch3, whereas lnc-Hser silencing promoted the expression of these genes (Figure 6A-D and Figure S11A-F). These results were also confirmed in the livers and primary HCs isolated from the in vivo model mice (Figure 6E and Figure S12A, B). To further investigate whether lnc-Hser suppresses the EMT of HCs through the Notch pathway, the signaling was blocked by a γ-secretase inhibitor RO4929097 in lnc-Hser-silenced AML12 cells. Subsequently, the expression of the genes related to fibrosis, apoptosis and EMT was detected by qRT-PCR and western blot. Interestingly, blocking the Notch pathway abrogated lnc-Hser silencing -induced up-regulation of both the pro-fibrotic and the mesenchymal marker genes but not the pro-apoptosis genes (Figure 6F-G). Taken together, our data demonstrate that lnc-Hser inhibits HCs EMT via the Notch pathway.
lnc-Hser inhibits HCs EMT via the Notch pathway. (A, B) Primary HCs were infected with LV-lnc-Hser for 72 h and further treated with 10 ng/ml TGFβ for additional 24 h. The mRNA level of Notch2, Notch3, Jagged1 and Hes1 was detected by qRT-PCR (A). The protein level of NOTCH2, NOTCH3 and HES1 was detected by western blot. GAPDH was used as an internal control (B). (C) The protein level of NOTCH2, NOTCH3 and HES1 in primary HCs infected with lenti-lnc-Hser-shRNA or lenti-NC was detected by western blot. GAPDH was used as an internal control. (D) The mRNA level of Notch2, Notch3, Jagged1 and Hes1 in primary HCs infected with lenti-lnc-Hser-shRNA or lenti-NC was detected by qRT-PCR. (E) Mice were treated with oil in combination with injection of lenti-NC (Negative Control, n = 10), or CCl4 in combination with injection of lenti-NC (NC + CCl4, n = 10), or oil in combination with injection of lenti-lnc-Hser-shRNA (lnc-Hser-shRNA, n = 10), or CCl4 in combination with injection of lenti-lnc-Hser-shRNA (lnc-Hser-shRNA + CCl4, n = 10). qRT-PCR analysis of Notch2, Notch3, Jagged1 and Hes1 in the primary HCs isolated from mice in each group. (F-H) lnc-Hser was stably knocked down by the CRISPR/Cas9 system with guide RNA pairs in AML12 cells. The γ-secretase inhibitor RO4929097 was used to treat lnc-Hser-silenced AML12 cells for 24 h. The expression of lnc-Hser, pro-fibrogenic, apoptosis-related and EMT-related genes was detected by qRT-PCR (F, G) and western blot. GAPDH was used as an internal control (H). The data are expressed as the mean ± SD for at least triplicate experiments. *p<0.05 for stands vs LV-Control, NC or Control gRNA. #p<0.05 stands for vs LV-Control + TGFβ, NC + CCl4 or lnc-Hser-gRNAs.
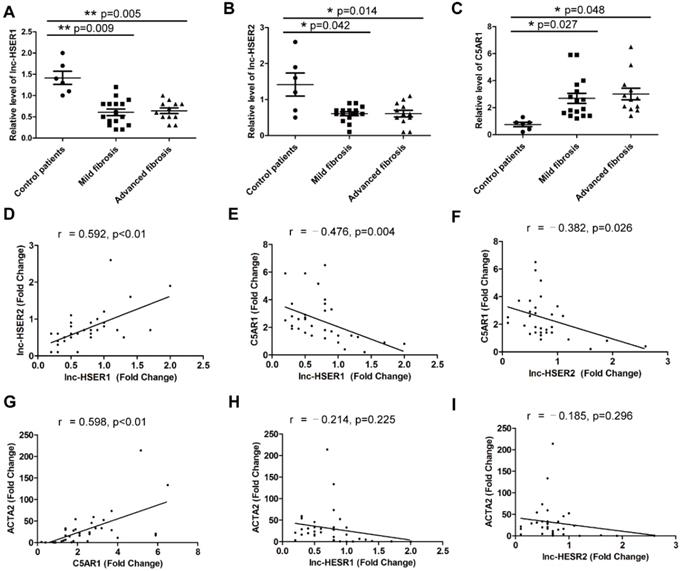
Potential role of lnc-HSER in human liver fibrosis
Although the function of lncRNA is important, the rapid sequence evolution of lncRNAs presents a challenge to identify functional counterparts between species. To address the question if our results with mice can be transferable to the human situation, the human ortholog lnc-HSER is needed to be identified. However, sequence analysis indicated that the full length of lnc-Hser was poorly conserved across species. Taking advantage of the presence of the conserved neighboring genes Cntrl and C5, we found that ENST00000466280, ENST00000460578, ENST00000489802, ENST00000480188 and NR_148450 were most likely to be the human homolog of lnc-Hser, all of which are located on human chromosome 9. Since the level of lnc-Hser was quite high in normal livers and was down-regulated in fibrotic livers, we next measured the expression of these five human lncRNAs in 6 healthy livers, 16 mild fibrotic livers (F1-F2) and 12 advanced fibrotic livers (F3-F4). The results showed that the transcript of ENST00000466280, ENST00000460578 and ENST00000480188 was enriched in healthy livers, but the level of ENST00000460578 and ENST00000480188 was significantly reduced in fibrotic livers (Figure 7A, B). Thus, we referred to ENST00000460578 as lnc-HSER1 and ENST00000480188 as lnc-HSER2. Moreover, the expression of C5AR1 was markedly increased in fibrotic livers, but not further increased with the progression of fibrosis (Figure 7C). In addition, lnc-HSER1 was found to be positively correlated with lnc-HSER2 (Figure 7D). A correlation of C5AR1 with lnc-HSER1, lnc-HSER2 and ACTA2 was also observed (Figure 7E-G). However, neither lnc-HSER1 nor lnc-HSER2 was correlated with ACTA2 (Figure 7H, I). To further investigate the role of lnc-HSER1 and lnc-HSER2, we over-expressed both lncRNAs in L02 cells and subsequently treated the cells with TNF-α. The results showed that the expression of the pro-inflammation genes IL-6, IL-1β and TNF-α, and the pro-apoptosis genes BAX and BAD was significantly up-regulated, whereas that of the mesenchymal marker genes N-Cadherin, Vimentin, Fibronectin, Twist and Snail1 was slightly up-regulated upon TNF-α treatment. Strikingly, forced lnc-HSER1 or lnc-HSER2 expression abrogated TNF-α-induced dysregulation of these genes (Figure S13A-D). Taken together, these data support the notion that lnc-HSER1/2 are significantly enriched in normal human livers but decreased in fibrotic livers and display a potential anti-fibrosis role.
Potential role of lnc-HSER in human liver fibrosis. (A-C) qRT-PCR analysis of lnc-HSER1, lnc-HSER2 and C5AR1 in liver samples from healthy (n = 6), mild fibrosis (n = 16) and advanced fibrosis (n = 12) patients. (D-I) The correlations of lnc-HSER1, lnc-HSER2, C5AR1 and ACTA2 were assessed using Pearson correlation analysis, n = 34. The data are expressed as the mean ± SD, *p<0.05.
Discussion
Liver fibrosis is a progressive pathologic process that involves deposition of ECM leading to distorted architecture and culminating in cirrhosis, which is one of the major health burdens worldwide [2]. However, the molecular basis of liver fibrosis is incompletely understood, which has limited the identification of therapeutic targets. In the current study, we identified a HCs-specifically expressed lnc-Hser that was decreased in human and mice fibrotic livers as well as primary HCs of mice developing liver fibrosis. We showed that knockdown of lnc-Hser induced the apoptosis and EMT of HCs in vitro, and aggravated CCl4-induced liver fibrosis in vivo. Although the in vivo data showed that simply knockdown of lnc-Hser did not induce liver fibrosis, the expression of genes related to liver fibrosis including Col4α5, Timp1 and Mmp2, the hepatic inflammation including Tnf-α, Mcp1 and Il-6, the mesenchymal markers N-Cadherin and β-Catenin was increased in livers of lnc-Hser-shRNA mice versus NC mice. The reason for lnc-Hser silencing did not induce significant liver fibrosis may be due to the fact that liver fibrogenesis is a complex process involved in many key factors, and the time period for hepatic fibrogenesis of the mice was only four weeks during which the liver had significant regeneration and self-renewal capacity to counteract the injury caused by silencing lnc-Hser. In addition, knockdown of lnc-Hser promoted HSCs activation through the signals derived from injured HCs. Mechanistically, we demonstrated that lnc-Hser inhibited HCs apoptosis via the C5AR1-Hippo-YAP pathway and suppressed HCs EMT via the Notch pathway. All these data support our conclusion that lnc-HSER is a novel biomarker for damaged HCs and a potential target for anti-fibrotic therapy.
It is well accepted that the apoptosis of HCs is a critical initiator of fibrosis by triggering HSCs activation either directly by the phagocytosis of the apoptotic bodies which promote the secretion of pro-inflammatory and pro-fibrogenic cytokines, or indirectly by the generation of damage-associated molecular patterns [2, 5]. However, the occurrence of HCs EMT in liver fibrosis remains controversial, for instance, Zeisberg et al. demonstrated that HCs undergo EMT in response to CCl4 in vivo [32], while Taura and colleagues employed a similar transgenic model and reported that HCs did not undergo EMT in CCl4-induced liver fibrosis [33]. Strikingly, several groups demonstrated that primary HCs and AML12 cells, when treated with the classical EMT inducer TGF-β, undergo EMT genotypic and phenotypic changes in vitro [8, 34, 35]. Consistently, our data showed that lnc-Hser silencing induced the apoptosis and EMT of HCs in vitro. On the other hand, the expression of the pro-apoptosis genes and the mesenchymal marker genes in the livers and primary HCs isolated from lnc-Hser-shRNA-CCl4 mice exhibited a profound increase, while the epithelial marker gene E-cadherin was not decreased. The reason for this could be when EMT initiates the epithelial cells to gain some phenotypes of the mesenchymal cells, for instance, up-regulated expression of N-cadherin and ECM, but still keeps some phenotypes of the epithelium. This is the so-called partial EMT. In this study, when EMT initiates during hepatic fibrogenesis, a reversible wound healing process, the HCs have to keep the cellular junctions to maintain the architecture of the hepatic lobule by normal expression of E-cadherin. However, the HCs have gained some mesenchymal phenotypes including over-expressed N-cadherin, β-catenin and Vimentin. Interestingly, our in vivo and in vitro results further revealed that the apoptosis and EMT of HCs were not co-localized, suggesting when lnc-Hser was silenced there are two populations of the cells among which one undergoes apoptosis and the other survived one undergoes EMT simultaneously. Hence, the inhibition of HCs apoptosis and EMT may be a therapeutic approach for the resolution of liver fibrosis.
Accumulating progresses have been made in better understanding the underlying mechanisms of liver fibrosis with recent major investigations focused on the pathways including TGFβ1, Notch and Hippo, involved in initiating HCs apoptosis and EMT in the pathogenesis of liver fibrosis. Notably, it has been reported that Notch signaling contributes to the progression of liver fibrosis via inducing the EMT [7, 8], and the EMT of in vitro cultured primary HCs could be blocked in the presence of several inhibitors including a Notch inhibitor [9]. Consistently, our results revealed that the up-regulated expression of the pro-fibrotic and the mesenchymal marker genes but not the pro-apoptosis genes induced by lnc-Hser silencing was abrogated by the γ-secretase inhibitor, suggesting that lnc-Hser inhibits HCs EMT via the Notch pathway. However, the role of YAP/TAZ in HCs apoptosis and proliferation remains controversial. We and others have demonstrated that the expression of YAP/TAZ was increased in fibrotic tissues and activated HSCs [12, 13], suggesting that YAP/TAZ activation is a critical driver of HSCs activation and subsequently induces liver fibrosis. Additionally, Lee et al. showed that YAP activation induced by inactivation of the Hippo pathway in HCs induced massive p53-dependent cell senescence/death [14]. Wang et al. demonstrated that silencing of HCs TAZ in murine models of nonalcoholic steatohepatitis (NASH) prevented or reversed hepatic inflammation, HCs death, and fibrosis [36]. However, it also has been reported that knockout of YAP in HCs resulted in increased HCs apoptosis and subsequently induced liver fibrosis in vivo and in vitro [15]. Similar results were also reported by Bai et al [16]. In addition, liver regeneration requires activated YAP and regenerative growth is blocked by preventing YAP activation, indicating that YAP promotes HCs proliferation [37]. In our study, we demonstrated that the canonical Hippo pathway was activated in damaged HCs and resulted in YAP inactivation.
LncRNAs represent a group of transcripts longer than 200 nucleotides that are 5' capped and polyadenylated like most mRNAs, yet this class of transcripts has limited coding potential. Emerging evidence has demonstrated that lncRNAs were expressed in a cell- or tissue-specific manner and involved in a variety of cellular processes including differentiation, proliferation, and apoptosis [22, 38-40]. In this study, we identified a novel lncRNA that was expressed with a highest level in livers and HCs but not in HSCs, LSECs and KCs, and was therefore named as lnc-Hser. However, the results of RACE showed that lnc-Hser is a 588-nucleotide transcript with three exons inconsistent with the sequence in the ensemble database. Both the transcripts share the same transcription start site and the first two exons, whereas the length of the third exon of lnc-Hser is longer than that of ENSMUST00000154817 displayed in database. The discrepancy may be due to the existence of different isoforms in various tissues. Although sequence analysis revealed no clear orthologous lnc-Hser in the human genome which is consistent with the idea that lncRNAs are less conserved than protein-coding genes, we found, by taking advantage of the presence of the conserved nearby genes Cntrl and C5, that five lncRNAs located on human chromosome 9 were most likely to be the human homologs of lnc-Hser. Additionally, we determined to regard the lncRNAs ENST00000460578 and ENST00000480188 as lnc-HSERs through detecting the transcripts of these lncRNAs in human livers and subsequently investigating the role of them in cells. Thus, it might be a way to discover homologous transcripts across species by the adjacent conserved genes, the expression profiles and the similar functions.
It is well known that lncRNAs work in cis when their effects are restricted to the chromosome from which they are transcribed, and work in trans when they affect genes on other chromosomes [29, 40]. In the current study, our data showed that lnc-Hser down-regulated the level of C5 rather than Cntrl in cis. Moreover, lnc-Hser could also inhibit the expression of Notch2, Jagged1, Hes1 and C5AR1 which is a GPCR family member and functions upstream of the Hippo pathway [11], but not Notch3, in trans. Since lnc-Hser was mainly located in the nucleus of HCs, we also performed RIP assay in AML12 cells and livers to pull down endogenous RNAs associated with SUZ12, an important subunit of PRC2, which physically associates with twenty percent of all human lncRNAs [37]. Our data also showed that a significant enrichment of lnc-Hser rather than β-actin with the SUZ12 (Figure S14A, B). Moreover, ChIP analysis was performed and the data demonstrated that over-expression of lnc-Hser increased the binding of SUZ12 across the promoters of C5ar1, Notch2 and Hes1 (Figure S14C), suggesting that lnc-Hser binds PRC2 and subsequently represses the transcription of the target genes. Additionally, ChIP results showed that TGFβ promoted the binding of SUZ12 across the lnc-Hser promoters (Figure S14D).
In conclusion, our data identified lnc-HSER as a novel regulator of C5AR1-Hippo-YAP and Notch pathways in liver fibrosis, suggesting that lnc-HSER might be a novel biomarker and a candidate of anti-fibrotic targets.
Abbreviations
lncRNAs: long non-coding RNAs; HC: hepatocyte; HSC: hepatic stellate cell; ECM: extracellular matrix; EMT: epithelial-mesenchymal transition; NICD: Notch intracellular domain; GPCRs: G-protein-coupled receptors; LSEC: liver sinusoidal endothelial cell; KC: Kupffer cell; CCl4: carbon tetrachloride; BDL: bile duct ligation; α-SMA: alpha-smooth muscle actin; FACS: flow cytometry; CM: conditioned medium; TGFβ: transforming growth factor-β.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (No.81670558; 81870429; 81800542; 81971331), the Natural Science Foundation of Tianjin (grant numbers 18JCQNJC81500; 18JCZDJC99000 and 19JCZDJC36700), the National 13th 5-year Plan for Hepatitis Research (No.2017ZX10203201-007), and the Science & Technology Development Fund of Tianjin Education Commission for Higher Education (No.2017KJ221).
Author contributions
K.Z., and W.H. conceived and designed the studies. K.Z., M.Z., Q.Y., and X.H., performed the majority of the experiments. Y.Z., L.Z., G.L., Q.L., Y.C., P.Z., H.C., Z.S., and T.C. helped to perform the experiments and collected the clinical samples. T.H., and Z.Y. helped to analyze the data. K.Z., and W.H. analyzed the data and wrote the manuscript. All authors critically reviewed and approved the final manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Lai M, Afdhal NH. Liver Fibrosis Determination. Gastroenterol Clin North Am. 2019;48:281-9
2. Parola M, Pinzani M. Liver fibrosis: Pathophysiology, pathogenetic targets and clinical issues. Mol Aspects Med. 2019;65:37-55
3. Zhang F, Lu S, He J, Jin H, Wang F, Wu L. et al. Ligand Activation of PPARgamma by Ligustrazine Suppresses Pericyte Functions of Hepatic Stellate Cells via SMRT-Mediated Transrepression of HIF-1alpha. Theranostics. 2018;8:610-26
4. Zhao Y, Wang Z, Feng D, Zhao H, Lin M, Hu Y. et al. p66Shc Contributes to Liver Fibrosis through the Regulation of Mitochondrial Reactive Oxygen Species. Theranostics. 2019;9:1510-22
5. Weiskirchen R, Weiskirchen S, Tacke F. Organ and tissue fibrosis: Molecular signals, cellular mechanisms and translational implications. Mol Aspects Med. 2019;65:2-15
6. Kaimori A, Potter J, Kaimori JY, Wang C, Mezey E, Koteish A. Transforming growth factor-beta1 induces an epithelial-to-mesenchymal transition state in mouse hepatocytes in vitro. J Biol Chem. 2007;282:22089-101
7. Hu B, Phan SH. Notch in fibrosis and as a target of anti-fibrotic therapy. Pharmacol Res. 2016;108:57-64
8. Chen Y, Zheng S, Qi D, Zheng S, Guo J, Zhang S. et al. Inhibition of Notch signaling by a gamma-secretase inhibitor attenuates hepatic fibrosis in rats. PLoS One. 2012;7:e46512
9. Xiang C, Du Y, Meng G, Soon Yi L, Sun S, Song N. et al. Long-term functional maintenance of primary human hepatocytes in vitro. Science. 2019;364:399-402
10. Zhang K, Qi HX, Hu ZM, Chang YN, Shi ZM, Han XH. et al. YAP and TAZ Take Center Stage in Cancer. Biochemistry. 2015;54:6555-66
11. Yu FX, Zhao B, Panupinthu N, Jewell JL, Lian I, Wang LH. et al. Regulation of the Hippo-YAP pathway by G-protein-coupled receptor signaling. Cell. 2012;150:780-91
12. Zhang K, Chang Y, Shi Z, Han X, Han Y, Yao Q. et al. omega-3 PUFAs ameliorate liver fibrosis and inhibit hepatic stellate cells proliferation and activation by promoting YAP/TAZ degradation. Sci Rep. 2016;6:30029
13. Mannaerts I, Leite SB, Verhulst S, Claerhout S, Eysackers N, Thoen LF. et al. The Hippo pathway effector YAP controls mouse hepatic stellate cell activation. J Hepatol. 2015;63:679-88
14. Lee DH, Park JO, Kim TS, Kim SK, Kim TH, Kim MC. et al. LATS-YAP/TAZ controls lineage specification by regulating TGFbeta signaling and Hnf4alpha expression during liver development. Nat Commun. 2016;7:11961
15. Zhang N, Bai H, David KK, Dong J, Zheng Y, Cai J. et al. The Merlin/NF2 tumor suppressor functions through the YAP oncoprotein to regulate tissue homeostasis in mammals. Dev Cell. 2010;19:27-38
16. Bai H, Zhang N, Xu Y, Chen Q, Khan M, Potter JJ. et al. Yes-associated protein regulates the hepatic response after bile duct ligation. Hepatology. 2012;56:1097-107
17. Li GY, Wang W, Sun JY, Xin B, Zhang X, Wang T. et al. Long non-coding RNAs AC026904.1 and UCA1: a "one-two punch" for TGF-beta-induced SNAI2 activation and epithelial-mesenchymal transition in breast cancer. Theranostics. 2018;8:2846-61
18. Wang H, Liang L, Dong Q, Huan L, He J, Li B. et al. Long noncoding RNA miR503HG, a prognostic indicator, inhibits tumor metastasis by regulating the HNRNPA2B1/NF-kappaB pathway in hepatocellular carcinoma. Theranostics. 2018;8:2814-29
19. Hitachi K, Nakatani M, Takasaki A, Ouchi Y, Uezumi A, Ageta H. et al. Myogenin promoter-associated lncRNA Myoparr is essential for myogenic differentiation. EMBO Rep. 2019:20
20. Wu XS, Wang F, Li HF, Hu YP, Jiang L, Zhang F. et al. LncRNA-PAGBC acts as a microRNA sponge and promotes gallbladder tumorigenesis. EMBO Rep. 2017;18:1837-53
21. Ziegler C, Graf J, Faderl S, Schedlbauer J, Strieder N, Forstl B. et al. The long non-coding RNA LINC00941 and SPRR5 are novel regulators of human epidermal homeostasis. EMBO Rep. 2019:20
22. Zhang K, Han X, Zhang Z, Zheng L, Hu Z, Yao Q. et al. The liver-enriched lnc-LFAR1 promotes liver fibrosis by activating TGFbeta and Notch pathways. Nat Commun. 2017;8:144
23. Song Y, Liu C, Liu X, Trottier J, Beaudoin M, Zhang L. et al. H19 promotes cholestatic liver fibrosis by preventing ZEB1-mediated inhibition of epithelial cell adhesion molecule. Hepatology. 2017;66:1183-96
24. Liang H, Pan Z, Zhao X, Liu L, Sun J, Su X. et al. LncRNA PFL contributes to cardiac fibrosis by acting as a competing endogenous RNA of let-7d. Theranostics. 2018;8:1180-94
25. Leisegang MS. LET's sponge: How the lncRNA PFL promotes cardiac fibrosis. Theranostics. 2018;8:874-7
26. Liu R, Li X, Zhu W, Wang Y, Zhao D, Wang X. et al. Cholangiocyte-derived exosomal LncRNA H19 promotes hepatic stellate cell activation and cholestatic liver fibrosis. Hepatology. 2019 [Epub ahead of print]
27. Zhang K, Han Y, Hu Z, Zhang Z, Shao S, Yao Q. et al. SCARNA10, a nuclear-retained long non-coding RNA, promotes liver fibrosis and serves as a potential biomarker. Theranostics. 2019;9:3622-38
28. Shi Z, Hong Y, Zhang K, Wang J, Zheng L, Zhang Z. et al. BAG-1M co-activates BACE1 transcription through NF-kappaB and accelerates Abeta production and memory deficit in Alzheimer's disease mouse model. Biochim Biophys Acta Mol Basis Dis. 2017;1863:2398-407
29. Kopp F, Mendell JT. Functional Classification and Experimental Dissection of Long Noncoding RNAs. Cell. 2018;172:393-407
30. Phieler J, Chung KJ, Chatzigeorgiou A, Klotzsche-von Ameln A, Garcia-Martin R, Sprott D. et al. The complement anaphylatoxin C5a receptor contributes to obese adipose tissue inflammation and insulin resistance. J Immunol. 2013;191:4367-74
31. Hillebrandt S, Wasmuth HE, Weiskirchen R, Hellerbrand C, Keppeler H, Werth A. et al. Complement factor 5 is a quantitative trait gene that modifies liver fibrogenesis in mice and humans. Nat Genet. 2005;37:835-43
32. Zeisberg M, Yang C, Martino M, Duncan MB, Rieder F, Tanjore H. et al. Fibroblasts derive from hepatocytes in liver fibrosis via epithelial to mesenchymal transition. J Biol Chem. 2007;282:23337-47
33. Taura K, Miura K, Iwaisako K, Osterreicher CH, Kodama Y, Penz-Osterreicher M. et al. Hepatocytes do not undergo epithelial-mesenchymal transition in liver fibrosis in mice. Hepatology. 2010;51:1027-36
34. Pinzani M. Epithelial-mesenchymal transition in chronic liver disease: fibrogenesis or escape from death? J Hepatol. 2011;55:459-65
35. Song J, Shi W. The concomitant apoptosis and EMT underlie the fundamental functions of TGF-beta. Acta Biochim Biophys Sin (Shanghai). 2018;50:91-7
36. Wang X, Zheng Z, Caviglia JM, Corey KE, Herfel TM, Cai B. et al. Hepatocyte TAZ/WWTR1 Promotes Inflammation and Fibrosis in Nonalcoholic Steatohepatitis. Cell Metab. 2016;24:848-62
37. Swiderska-Syn M, Xie G, Michelotti GA, Jewell ML, Premont RT, Syn WK. et al. Hedgehog regulates yes-associated protein 1 in regenerating mouse liver. Hepatology. 2016;64:232-44
38. Feng Y, Xu W, Zhang W, Wang W, Liu T, Zhou X. LncRNA DCRF regulates cardiomyocyte autophagy by targeting miR-551b-5p in diabetic cardiomyopathy. Theranostics. 2019;9:4558-66
39. Han X, Hong Y, Zhang K. TUG1 is involved in liver fibrosis and activation of HSCs by regulating miR-29b. Biochem Biophys Res Commun. 2018;503:1394-400
40. Zhang K, Shi ZM, Chang YN, Hu ZM, Qi HX, Hong W. The ways of action of long non-coding RNAs in cytoplasm and nucleus. Gene. 2014;547:1-9
Author contact
![]() Corresponding author: Wei Hong, Department of Histology and Embryology, School of Basic Medical Sciences, Tianjin Medical University, Tianjin, China. E-mail: hongweiedu.cn.
Corresponding author: Wei Hong, Department of Histology and Embryology, School of Basic Medical Sciences, Tianjin Medical University, Tianjin, China. E-mail: hongweiedu.cn.