Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Conclusion
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2020; 10(22):10001-10015. doi:10.7150/thno.47491 This issue Cite
Research Paper
Dysregulated Kras/YY1/ZNF322A/Shh transcriptional axis enhances neo-angiogenesis to promote lung cancer progression
Che-Chung Lin1, I-Ying Kuo1, Li-Ting Wu1, Wen-Hui Kuan1, Sheng-You Liao2, Jayu Jen2, You-En Yang1, Cheng-Wei Tang1, Yi-Rong Chen3, Yi-Ching Wang1,2 ![]()
1. Department of Pharmacology, College of Medicine, National Cheng Kung University, Tainan, 70101, Taiwan.
2. Institute of Basic Medical Sciences, College of Medicine, National Cheng Kung University, Tainan, 70101, Taiwan.
3. Institute of Molecular and Genomic Medicine, National Health Research Institutes, Miaoli, 35053, Taiwan.
Received 2020-4-27; Accepted 2020-8-2; Published 2020-8-8
Abstract

Angiogenesis enhances cancer metastasis and progression, however, the roles of transcription regulation in angiogenesis are not fully defined. ZNF322A is an oncogenic zinc-finger transcription factor. Here, we demonstrate a new mechanism of Kras mutation-driven ZNF322A transcriptional activation and elucidate the interplay between ZNF322A and its upstream transcriptional regulators and downstream transcriptional targets in promoting neo-angiogenesis.
Methods: Luciferase activity, RT-qPCR and ChIP-qPCR assays were used to examine transcription regulation in cell models. In vitro and in vivo angiogenesis assays were conducted. Immunohistochemistry, Kaplan-Meier method and multivariate Cox regression assays were performed to examine the clinical correlation in tumor specimens from lung cancer patients.
Results: We validated that Yin Yang 1 (YY1) upregulated ZNF322A expression through targeting its promoter in the context of Kras mutation. Reconstitution experiments by knocking down YY1 under KrasG13V activation decreased KrasG13V-promoted cancer cell migration, proliferation and ZNF322A promoter activity. Knockdown of YY1 or ZNF322A attenuated angiogenesis in vitro and in vivo. Notably, we validated that ZNF322A upregulated the expression of sonic hedgehog (Shh) gene which encodes a secreted factor that activates pro-angiogenic responses in endothelial cells. Clinically, ZNF322A protein expression positively correlated with Shh and CD31, an endothelial cell marker, in 133 lung cancer patient samples determined using immunohistochemistry analysis. Notably, patients with concordantly high expression of ZNF322A, Shh and CD31 correlated with poor prognosis.
Conclusions: These findings highlight the mechanism by which dysregulation of Kras/YY1/ZNF322/Shh transcriptional axis enhances neo-angiogenesis and cancer progression in lung cancer. Therapeutic strategies that target Kras/YY1/ZNF322A/Shh signaling axis may provide new insight on targeted therapy for lung cancer patients.
Keywords: Lung cancer, Kras, Shh, transcription, angiogenesis
Introduction
Kras, encode by Kirsten rat sarcoma viral oncogene (chromosome 12p12.1), is one of the RAS small GTPase family proteins, which also include Hras and Nras [1,2]. Kras protein switches between GTP-bound (active) and GDP-bound (inactive) forms. Kras mutations, which predominately occur at codon 12, 13, or 61, can lead to Kras proteins with impaired GTPase activity, resulting in constitutive activation of downstream signaling pathways, and therefore contributes to tumor formation [3,4]. Importantly, integrative studies using clinical databases and genetically engineered mouse models showed that Kras mutation upregulated expression of FOSL1 to commit a transcriptional program including genes involved in mitosis progression to promote lung and pancreatic cancer progression [5,6]. In addition, Kras activation enhances NFκB (p65) expression and its transcription activity in endometrial cancer [7]. Importantly, Kras and NFκB concomitantly induce expression of Yin Yang 1 (YY1) transcription factor in pancreatic cancer [8]. Moreover, transcription of multiple effectors in the Kras pathway can be modulated by microRNAs [9,10]. It is important to unveil more transcription factors downstream of Kras pathway in lung cancer, a disease strongly associated with Kras dysfunctions. Of note, our transgenic mice model showed that mice harboring krasG12D/znf322a double transgenes possessed higher tumor initiating ability compared to those with krasG12D single transgene.
ZNF322A, also known as ZNF388 or ZNF489, is a zinc-finger transcription factor consisting of 11 Cys2His2 type krüppel-like zinc-finger motifs [11]. Our previous study showed that ZNF322A overexpression promotes lung tumor growth, metastasis and stemness properties partially through activating promoter activity of alpha-adducin and cyclin D1, while suppressing promoter activity of p53 and c-Myc [12,13]. In addition, we found that deregulation of CK1δ-GSK3β-FBXW7α protein degradation system or activation of EGFR-AKT signaling axis results in prolonged ZNF322A stability and transcription activity promoting lung cancer progression [14,15]. In our attempt to identify important transcriptional target genes of ZNF322A by integrating our chromatin-immunoprecipitation sequencing (ChIP-seq) and RNA sequencing (RNA-seq) datasets [13], we observed that ZNF322A downstream targets are significantly enriched in vasculature development and angiogenesis. However, the molecular basis for the interaction between ZNF322A and neo-angiogenesis in the context of Kras activation remain poorly defined.
Some well-known signaling axes and genes have been reported to participate in angiogenesis, such as interleukin-8 (IL-8)/CXC chemokine receptors1/2 pathway, NOTCH/delta-like-4 signaling axis and vascular endothelial growth factor (VEGF)/hypoxia induced factor 1 alpha (HIF1α) signaling axis [16-18]. Here we show that YY1 transcription factor is a crucial mediator between Kras and ZNF322A in enhancing lung cancer progression. Moreover, our data from lung cancer cell, animal and clinical models demonstrate that sonic hedgehog (Shh) is a downstream transcriptional target of ZNF322A for promoting angiogenesis, and imply Kras/YY1/ZNF322A/Shh transcriptional axis as a previously unknown mechanism contributing to neo-angiogenesis.
Materials and Methods
Generation of lung specific transgenic mice
All mouse studies were approved by the Institutional Animal Care and Use Committees of National Health Research Institutes (Permit Number: #101045A) and National Cheng Kung University (Permit Number: #106068) and were performed in accordance with relevant guidelines. To generate lung-specific znf322a transgenic mice, pcDNA4/TO/myc-His B expression vector (Invitrogen) was used as a backbone for the construction of the transgenic fragment. The order of the DNA fragments and corresponding franking enzyme sites on the transgenic construct are: Surfactant Protein A (SPA) promoter (Mlu I/Hind III), intron (Hind III/Kpn I), human ZNF322A (Kpn I/Xho I), and polyA sequence (Xba I/Sac II). The whole transgenic fragment was excised by Mlu I and Pme I digestion followed by purification and pronuclear injection of fertilized C57B/J6 mice oocytes. Znf322a-transgenic mice were identified by PCR analysis of genomic DNA isolated from tail biopsies. The presence of transgene was determined using the following primers: SPA-Forward primer (5'- TACAGCTCCTGGGCAACGTG -3') and SPA-Reverse (5'- TTGCTTGCATTCAAGGCACTG -3'), yielding a 292 bp PCR product.
Lung specific Tet-on KrasG12D C57B/J6 transgenic mice (Scgb1a1-rtTA/TetO-Kras4bG12D) was a gift from Dr. Ming-Derg Lai (Department of Biochemistry and Molecular Biology, National Cheng Kung University, Taiwan). Reverse tetracycline trans-activator (rtTA) protein was expressed under the control of Scgb1a1 (secretoglobin, family 1A, member 1) promoter. Doxycycline induces rtTA binding to tetracycline operator element, and subsequently promoted KrasG12D expression. KrasG12D mice were then crossed with znf322a lung-specific mice to create KrasG12D/znf322a double transgenic mice. All mice with a positive genotype and the control mice were maintained in the animal facility with continual observation till the appearance of disease phenotypes or at the indicated time points. Paraffin blocks of tumors were collected for hematoxylin and eosin (H&E) stain.
Cell lines and culture conditions
Human lung cancer cell lines H1299 and H460 cells were purchased from ATCC. Human umbilical vein endothelial cells (HUVECs) were kindly provided by Dr. Li-Wha Wu (Institute of Molecular Medicine, National Cheng Kung University, Taiwan). HUVECs seeded in dishes, which were coated with 0.1% gelatin for 1 h, were routinely maintained in endothelial cell growth medium-2 (EGM-2) with addition of growth factors (Lonza). All cell lines were authenticated by the Bioresource Collection and Research Center (Hsinchu, Taiwan) using short tandem repeat profiling (AmpFLSTR Identifiler Plus PCR Amplification Kit). Only mycoplasma negative cells were used.
Plasmids, RNAi and transfection
Plasmids used in this study are listed in Table S1. siGENOME SMARTpool siRNAs against ZNF322A were purchased from Dharmacon; siRNAs against Shh was purchased from Thermo Fisher; shRNA clones against YY1 (KH00440H, Qiagen) were obtained from Dr. Hsin-Ling Hsu (Institute of Molecular and Genomic Medicine, National Health Research Institutes, Taiwan). HA-tagged KrasG13V and KrasS17N plasmids were kindly provided by Dr. Hsiao-Sheng Liu (Department of Microbiology and Immunology, National Cheng Kung University, Tainan). Plasmid and siRNA transfections were carried out using TurboFect (Thermo Fisher) and Lipofectamine 2000 (Invitrogen) reagent according to manufacturer's protocol.
Promoter constructs and site-directed mutagenesis
ZNF322A promoter region (-529 to +223 of the transcriptional start site, TSS) was inserted into the KpnI and HindIII sites of pGL4.17 luciferase expression vector. Deletion of two YY1-binding sites within ZNF322A promoter (-129 to +223 of the TSS) and mutations of 3-mer of ZNFS22A-motif within Shh promoter regions (from TGAGGTCAGGAGTTCGAGACCAGCCTGCC to TGAGGTCAGGAACCCGAGACCAGCCTGCC; mutations are shown as underlined letters) were generated by site-directed mutagenesis using indicated wild-type promoter vectors and specific primers listed in Table S2.
Chromatin immunoprecipitation-quantitative polymerase chain reaction (ChIP-qPCR) and quantitative reverse transcriptase-polymerase chain reaction (RT-qPCR) assay
ChIP was performed in H1299, H1299 KrasG13V and H460 KrasQ61H lung cancer cells manipulated for YY1 or ZNF322A. Lung cancer cells (1.5 × 106 cells) seeded in a 10 cm dish were cross-linked followed by preparation of nuclear lysates using Magna ChIPTM protein G Kit (Millipore). Nuclear lysates were sonicated to shear DNA to around 200~300 bp followed by immunoprecipitation for 16 h at 4 °C using IgG, anti-YY1 or anti-HA-ZNF322A antibody listed in Table S3. Primers for PCR assay of ChIP samples and RT-qPCR reactions are listed in Table S2.
Transwell migration assay of lung cancer cells or HUVECs
For transwell migration assay of lung cancer cells, 5 × 105 cells were placed in the upper chamber of transwell (Falcon). DMEM medium containing 20% FBS was added to the lower chamber as chemoattractants and the cells were incubated at 37 °C for 12 h. For HUVECs migration, HUVECs (1 × 105) were placed in the upper chamber with serum-free EGM2 while the lower chamber was filled with conditioned medium derived from 1 × 105 lung cancer cells with EGM-2 medium at 1:1 ratio as chemoattractants and incubated at 37 °C for 24 h. The cells attached on the reverse side of the membrane were stained with crystal violet and counted under inverted microscope (Nikon E400, Tokyo, Japan) with randomly selected 10 fields.
Conditioned medium (CM) preparation, tube formation assay and in vivo Matrigel plug angiogenesis assay
Lung cancer cells expressing control, shYY1, siZNF322A, siShh, ZNF322A expression vector, or reconstitution of siShh in ZNF322A (ZNF322A/siShh) were used for CM preparation. Serum-free CM were prepared from culturing lung cancer cells (1 × 106 cells in each 10 cm dish) with 5 mL EBM-2 medium for 30 h. The cell viability was ascertained using the trypan blue dye exclusion assay and was > 98%. The media were collected and centrifuged using Amicon Ultra centrifugal filter units (Millipore) at 800 rpm for 5 min to remove cell debris and then at 3,000 rpm for 5 h at 4 °C to concentrate the CM.
HUVECs were seeded onto 48-well culture dishes coated with 100 μL of Matrigel (13.4 mg/mL; BD Biosciences) at a density of 1.2 × 104 per well. The seeded HUVECs were further treated with CM prepared from lung cancer cells at 37 °C for 6-8 h to allow tube formation. Six random views were photographed and quantified under an upright microscope (Nikon E400). The tube length was quantified using imaging software developed by Dr. Yung-Nien Sun (Department of Computer Science and Information Engineering, National Cheng Kung University, Taiwan).
Matrigel (9 mg/mL; 0.3 mL/mouse) alone or mixed with 50 μL CM derived from different lung cancer cells was injected subcutaneously into the flank of nude mice. On day 10, mice were sacrificed, plugs were removed and fixed in 3.7% formaldehyde/phosphate-buffered saline, paraffin embedded, and slides were immunohistochemically stained for CD31 (endothelial cells marker) and photographed. All mouse studies were approved by the National Cheng Kung University Institutional Animal Care and Use Committee (Permit Numbers: #106068).
Patient samples and clinical information
A total of 133 surgically resected lung cancer patients were recruited from National Cheng Kung University Hospital after obtaining appropriate institutional review board permission (#A-ER-104-075) and informed consent from the patients. These patients did not receive any anti-angiogenic therapy. The mean follow-up period for these patients was 74 months (range 9-169 months). The histological determinations, including tumor type and disease stage, were performed according to the World Health Organization classification and the TNM classification system, respectively. Information on the sex, age, and smoking history of the patients were obtained from hospital records. Paraffin blocks of tumors were collected for immunohistochemistry.
Immunohistochemistry assay
Immunohistochemistry was performed to detect protein expression of YY1, ZNF322A, Shh and CD31 in tumor sections from 133 lung cancer patients. Staining of YY1, ZNF322A and Shh was scored as 0 if no cells were stained positive; and scored as 1 if <10% tumor cells were immunostaining-positive; 2 for 10-25%; 3 f or 25-50%; and 4 for >50%. The staining was defined as “high expression” if the staining intensity score was ≥3. The surrounding normal tissue, which shows score 1 served as an internal positive control on each slide. CD31 staining was obtained and quantified using the TissueFax and HistoQuest software (TissueGnostics, Vienna, Austria). The mean staining positive area was calculated within the selected gates: 0.313 mm x 0.175 mm (100 X) for CD31. Six gates were selected in an individual tissue slide. The CD31 staining was graded as “high expression” if staining positive area is greater than 3%. Antibodies used and their experimental conditions are listed in Table S3.
Statistical analysis
Pearson's χ2 test was used to compare the correlation of YY1, ZNF322A, Shh and CD31 expression in lung cancer patients. Overall and progression-free survival curves were calculated according to the Kaplan-Meier method using the log-rank test. Cox regression comparison was performed to analyze the relative risk for the patient poor outcome. Quantification of the immunoblotting was analyzed using ImageJ software. Three independent experiments for cell studies and five mice per group for animal studies were analyzed unless indicated otherwise. The scripts used for the analysis are available upon request. Two-tailed Student's t-test was used in cell and animal studies. Data represent mean ± SEM. The levels of statistical significance were expressed as P-values, *P < 0.05; ** P < 0.01; *** P < 0.001.
Results
Kras mutation promotes ZNF322A expression at the transcription level
Our previous study identified ZNF322A as an oncogenic transcription factor, which promotes cancer progression by transcriptionally dysregulating downstream cancer-related genes. To further investigate the role of ZNF322A in lung tumorigenesis, we generated znf322a lung-specific transgenic mice using C57BL/6 mice. However, we did not observe obvious tumor initiation in the lung area of znf322a transgenic mice (Figure S1). Since Kras mutations predominantly occur at codon 12 and occasionally at codons 13 and 61 [19], we then crossed znf322a transgenic mice with KrasG12D lung-specific transgenic mice to generate KrasG12D/znf322a double transgenic mice. Notably, mice harboring KrasG12D/znf322a double transgenes possessed higher tumor initiating ability compared to those with KrasG12D single transgene after doxycycline-induced KrasG12D expression, i.e., precancerous adenomas at four months (Figure 1A) and advanced adenocarcinoma at six months (Figure 1B).
Oncogenic Kras upregulated ZNF322A mRNA expression and promoter activity. A and B, ZNF322A synergized Kras mutation-driven lung tumorigenesis in vivo in four months (A) or six months (B). The H&E stain results (Upper) and the scatter plot diagram (Lower) are shown. C and D, Constitutively-active KrasG13V promoted ZNF322A transcription in a dose-dependent manner. Immunoblots confirmed IPTG-induced ectopic overexpression of HA-KrasG13V in H1299 KrasWT cells (C). qRT-PCR revealed that ZNF322A mRNA expression was upregulated by KrasG13V overexpression (D). E and F, Stable KrasG13V expression promoted ZNF322A transcription in H1299 KrasG13V. Immunoblotting of KrasG13V (E) and ZNF322A mRNA expression (F) are shown. G, Promoter region (-529~+223) of ZNF322A. YY1 binding elements were identified by motif analysis using PWM software. The predicted sequences of the two YY1 binding sites in the ZNF322A promoter are as indicated below the map. H, Promoter activity assay was performed using ZNF322A-pGL4 promoter in H1299 KrasWT cells. I-K, Dominant-negative KrasS17N mutation attenuated ZNF322A transcription. Immunoblotting of KrasS17N (I), ZNF322A mRNA expression (J) and ZNF322A promoter activity (K) are shown. Data are presented as mean ± SEM and normalized to the control group (-). P-values determined using two-tailed Student's t-test. *P< 0.05; ** P< 0.01; *** P< 0.001.
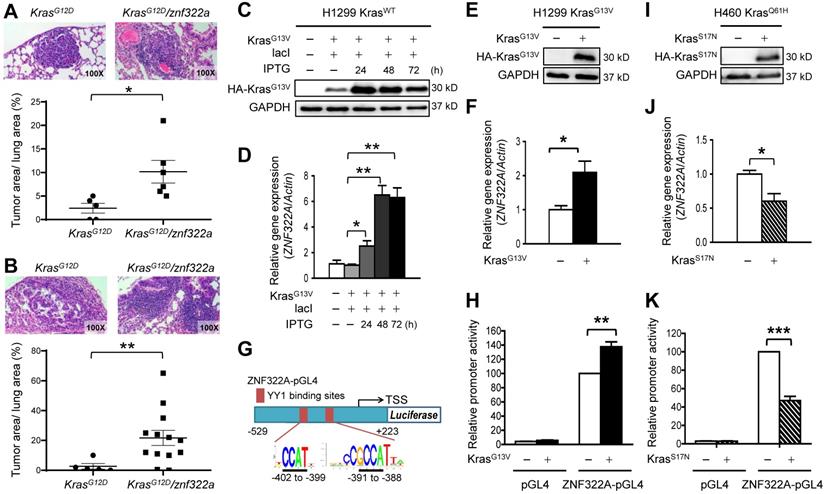
To confirm the positive correlation between Kras mutations and ZNF322A expression was not limited to KrasG12D mutation, we examined Kras-mediated ZNF322A expression in cell lines harboring Kras mutation at codon 13 or 61. To this end, we adapted cell culture systems with IPTG-induced constitutively active KrasG13V in H1299 cell line harboring wild-type Kras gene (H1299 KrasWT). Western blotting results confirmed that IPTG successfully induced ectopic overexpression of KrasG13V (Figure 1C). RT-qPCR revealed that ZNF322A mRNA expression was upregulated by KrasG13V activation (Figure 1D). We then established H1299 cell line stably expressing KrasG13V (H1299 KrasG13V) (Figure 1E) and found that ZNF322A mRNA expression was increased upon KrasG13V overexpression (Figure 1F). To further investigate whether Kras activation could drive ZNF322A transcription, we identified binding sites of YY1, which is the candidate mediator between Kras and ZNF322A (as described in next section), within the ZNF322A promoter. Two putative YY1 binding sites (5'-CCGCCATNTT-3') within the first 500 bp (-402 to -399; -391 to -388) of the ZNF322A promoter were identified using the PWM tool (available at https://ccg.epfl.ch/pwmtools/pwmscan.php). We thereby inserted ZNF322A promoter region [-529 to +223 of the TSS] into pGL4.17 vector to generate ZNF322A-pGL4 (Figure 1G). The luciferase reporter assay results confirmed that KrasG13V activation enhanced ZNF322A promoter activity (Figure 1H).
To further verify that Kras activated ZNF322A transcription, we overexpressed dominant negative KrasS17N in H1299 KrasWT and H460 KrasQ61H (endogenous KrasQ61H activated mutation) cell lines. Western blotting confirmed the overexpression of dominant negative KrasS17N (Figure 1I; Figure S2A). Notably, ZNF322A mRNA expression and promoter activity were reduced upon overexpression of dominant negative KrasS17N in H460 KrasQ61H and H1299 KrasWT cell lines (Figure 1J and 1K; Figure S2B-S2C). Collectively, these results of constitutively active and dominant negative Kras experiments suggested that active Kras mutation positively regulates ZNF322A transcription.
YY1 regulates ZNF322A transcription under Kras mutation
Next, we searched for the candidate mediators between Kras activation and ZNF322A transcription using the PROMO transcription factor (TF) prediction database (available at http://alggen.lsi.upc.es/cgi-bin/promo_v3/promo/promoinit.cgi?dirDB=TF_8.3), and 78 TFs that may bind to ZNF322A promoter were revealed. Those TFs were further mapped with Kras pathway (KEGG Mapper, available at http://www.genome.jp/kegg/). The 14 overlapping TFs were considered as the candidate transcription mediators downstream of Kras (Figure 2A). To investigate whether these candidate TFs could regulate ZNF322A transcription, we transfected H1299 or H460 cells with expression vectors of seven TFs available in our group. RT-qPCR analysis results revealed that overexpression of E2F1, ELK1, NFκB (p65), Oct4, Sp1 or STAT3 did not affect ZNF322A mRNA expression (Figure 2B; Figure S3A). Notably, RT-qPCR revealed that YY1 mRNA expression was dose-dependently upregulated by IPTG-induced constitutively active KrasG13V (Figure 2C; Figure S3B). Thus, we focused on YY1 transcription factor.
YY1 positively regulated ZNF322A transcription to promote cell migration and proliferation. A and B, Transcription factor (TF) prediction database PROMO revealed 78 TFs may bind to ZNF322A promoter. These TFs were further mapped with Kras pathway using KEGG Mapper (KEGG website) (A). The 14 overlapping TFs were considered as candidate transcriptional mediators between Kras and ZNF322A and seven of them were validated for ZNF322A transcription (colored, B). C, RT-qPCR revealed that YY1 mRNA expression was upregulated by KrasG13V in a dose-dependent manner. D-G, RT-qPCR analysis revealed that YY1 promoted ZNF322A mRNA expression in H1299 KrasG13V (D) and H460 KrasQ61H (E), while shYY1 reduced ZNF322A mRNA expression (F and G). H, ChIP-qPCR primers were designed in -462~-363 region of ZNF322A promoter as indicated below the map. Sequences of the wild-type and deletion (Del) promoters with deletion of two YY1 binding sites are shown. I and J, YY1 bound to ZNF322A promoter. ChIP assay was performed using anti-YY1 antibody in H1299 KrasG13V overexpressing YY1 (I) or knockdown of YY1 (J). IgG was used as negative control. K and L, YY1-mediated ZNF322A promoter activation was abolished using Del-ZNF322A-pGL4 promoter upon overexpression of YY1 (K) or knockdown of YY1 (L). M-N, K-ras/YY1 enhanced lung cancer cell proliferation and migration via promoting ZNF322A expression. KrasG13V promoted cancer cell migration (M), cell proliferation (N) and ZNF322A promoter activity (O) which were attenuated by shYY1 in H1299 cells. Data are presented as mean ± SEM and normalized to the control group (-). P-values determined using two-tailed Student's t-test. *P< 0.05; ** P< 0.01; *** P< 0.001.
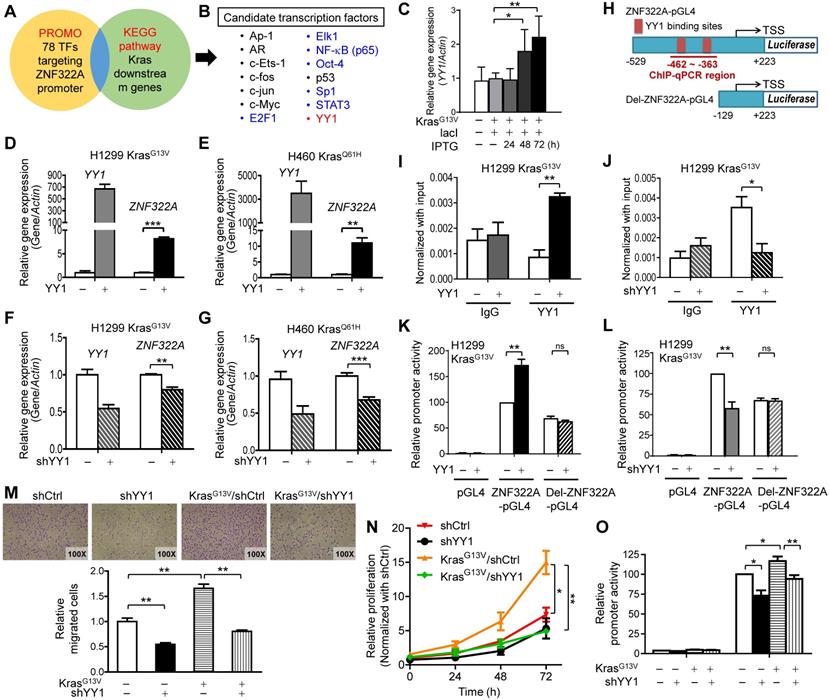
In order to determine whether YY1 regulated ZNF322A transcription, we ectopically overexpressed YY1 in H1299 KrasG13V and H460 KrasQ61H cell lines. Results of RT-qPCR analysis demonstrated that ZNF322A mRNA expression was significantly upregulated by YY1 (Figure 2D and 2E), while knockdown of YY1 (shYY1) reduced ZNF322A mRNA expression (Figure 2E and 2F) in H1299 KrasG13V and H460 KrasQ61H cells. The immunoblotting results confirmed the expression of YY1 protein upon overexpression or knockdown of YY1 in H1299 KrasWT, H1299 KrasG13V and H460 KrasQ61H cell lines (Figure S4A and S4B).
Next, we performed ChIP-qPCR at ZNF322A promoter region (-462~-363) which contained two YY1-binding sites (Figure 2H) to confirm that YY1 indeed binds to ZNF322A promoter region (Figure 2I). Knockdown of YY1 significantly attenuated its ability to bind the ZNF322A promoter, validating that the ChIP-qPCR results observed in Figure 2I was a true YY1 binding signal (Figure 2J). To further verify whether YY1 regulated the activity of ZNF322A promoter, luciferase promoter activity assay using ZNF322A-pGL4 (-529 to +223 of the TSS) and Del-ZNF322A-pGL4 (-129 ~ +223 with deletion of two YY1 binding sites at -402 ~ -388) (Figure 2H) were performed. As shown in Figure 2K and 2L, overexpression of YY1 increased promoter activity of the ZNF322A-pGL4 promoter, while YY1-mediated ZNF322A promoter activity was completely abolished when Del-ZNF322A-pGL4 promoter deleted for the two YY1 sites was used. In agreement, knockdown of YY1 reduced promoter activity of ZNF322A-pGL4, but not for Del-ZNF322A-pGL4 promoter. These results suggested that -462 ~ -363 regions in ZNF322A promoter contained the binding sites for YY1.
Kras/YY1 enhances lung cancer cell proliferation and migration via promoting ZNF322A transcription in vitro
Since we unveiled YY1 as a crucial mediator of Kras mutation-driven transcription of ZNF322A, we analyzed the role of YY1 in lung cancer cell proliferation and migration. Transwell migration assay showed that knockdown of YY1 significantly reduced cell migration promoted by KrasG13V (Figure 2M). Consistently, KrasG13V activation promoted cell proliferation, which was abolished by YY1 ablation (Figure 2N). Promoter activity assays validated that KrasG13V-activated ZNF322A promoter activity was attenuated by YY1 knockdown (Figure 2O). Collectively, these results supported that ZNF322A upregulation mediated by Kras/YY1 axis promotes proliferation and migration of lung cancer cells.
ZNF322A regulates mRNA expression of genes involved in angiogenesis
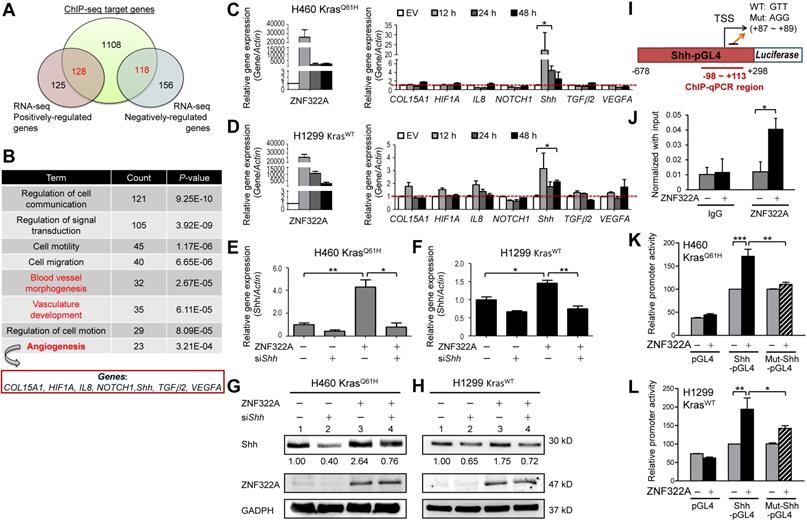
We further identified ZNF322A downstream genes by integrating our previous ChIP-seq and RNA-seq datasets (Figure 3A). Using DAVID Functional Annotation Clustering Tool (available at http://david.ncifcrf.gov/home.jsp), we found that many of the overlapped genes mapped to the angiogenesis-related pathways, including vasculature development, blood vessel morphogenesis and angiogenesis pathway (Figure 3B). Next, RT-qPCR was conducted to validate the mRNA expression level of seven angiogenic genes viz., COL15A1 (collagen, type XV, alpha 1), HIF1α, IL-8, NOTCH1, Shh, TGFβ2, and VEGFA in H460 KrasQ61H and H1299 KrasWT cells overexpressing ZNF322A. Among them, Shh mRNA level positively correlated with ZNF322A at all time points (12, 24, and 48 h) examined in both H460 KrasQ61H and H1299 KrasWT cell lines (Figure 3C and 3D).
ZNF322A transcriptionally activated Shh promoter. A and B, Integrated ChIP-seq and RNA-seq analysis revealed the novel role of ZNF322A in angiogenesis. ZNF322A-mediated transcriptome (A) and pathway analysis using DAVID software are shown. C and D, RT-qPCR validation of the seven angiogenic genes identified using DAVID. Only the Shh mRNA level was increased in ZNF322A-overexpressing H460 KrasQ61H (C) and H1299 KrasWT (D) lung cancer cells at 12, 24 and 48 h time points. E-H, Reconstitution experiments showed that Shh acted as a downstream factor of ZNF322A in lung cancer cells. RT-qPCR (E and F) and immunoblots (G and H) confirmed that Shh expression level was decreased in ZNF322A/siShh group compared with ZNF322A group (groups 4 vs. 3) in H460 KrasQ61H and H1299 KrasWT lung cancer cells. Normalized Shh protein fold changes are as indicated below the blots. I, Promoter region (-678~+298) of Shh. ChIP-qPCR primers designed in -98~+113 region of Shh promoter are labeled below the map. Sequences of the wild-type (WT) and mutated (Mut) promoters are shown (+87 ~ +89). J, ZNF322A bound to Shh promoter. ChIP assay was performed using anti-HA antibody in H460 KrasQ61H overexpressing ZNF322A. IgG was used as negative control. K and L, Overexpression of ZNF322A influenced promoter activity of Shh-pGL4 but not Mut-Shh-pGL4 in ZNF322A-overexpressing H460 KrasQ61H (K) and H1299 KrasWT (L) lung cancer cells. Data are presented as mean ± SEM. P-values determined using two-tailed Student's t-test. *P< 0.05; ** P< 0.01; *** P< 0.001.
ZNF322A transcriptionally activates the expression of Shh
In order to test whether Shh is a transcription target of ZNF322A, we determined Shh mRNA and protein expression levels in reconstitution experiments by knocking down Shh (siShh) in ZNF322A-overexpressed (ZNF322A) cancer cells. The siShh attenuated the ZNF322A-induced expression of Shh mRNA (bars 4 vs. 3, Figure 3E and 3F) and Shh protein (lanes 4 vs. 3, Figure 3G and 3H). These data suggested that Shh is a downstream effector of ZNF322A-mediated gene expression.
In our previous ChIP-seq study, we have revealed the ZNF322A binding DNA element using the MEME motif analysis [13]. As shown in Figure 3I, ZNF322A binding sequences were found at +87 ~ +89 on the Shh promoter. We then performed ChIP-qPCR to confirm ZNF322A binding at Shh promoter region (-98 ~ +113) in H460 KrasQ61H lung cancer cells (Figure 3J). Next, we examined whether ZNF322A binding enhanced Shh promoter activity by inserting Shh promoter region (-678 to +298 of the TSS) into pGL4.17 vector (Shh-pGL4) and then performed luciferase reporter assay combined with site-directed mutagenesis at +87 ~ +89 region by changing GTT to AGG sequences (Mut-Shh-pGL4, Figure 3I). Our results showed that ZNF322A overexpression activated Shh-pGL4 promoter activity but marginally changed Mut-Shh-pGL4 promoter activity in both H460 KrasQ61H and H1299 KrasWT lung cancer cells (Figure 3K and 3L). The results confirmed that ZNF322A enhances Shh promoter activity and +87 ~ +89 region in Shh promoter contains the binding site for ZNF322A.
Reconstitution experiments showed that ZNF322A/Shh axis increased endothelial cell migration and tube formation in vitro
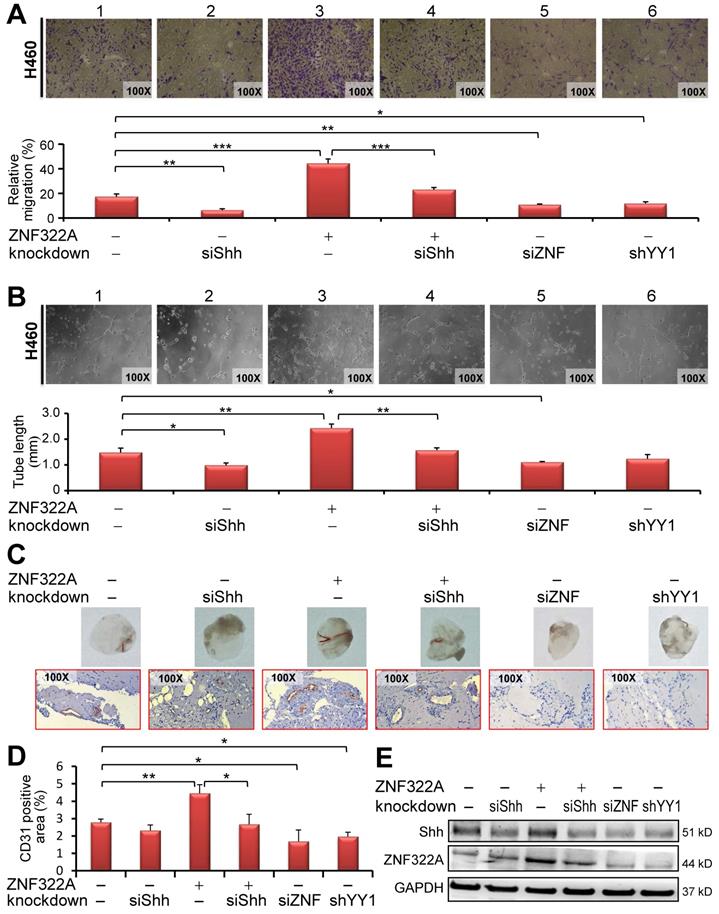
Our data so far suggest that ZNF322A transcriptionally activates the pro-angiogenesis gene Shh and that YY1 regulates ZNF322A gene expression. Therefore, we examined whether activation of YY1/ZNF322A/Shh exerted pro-angiogenesis effects. We performed transwell migration assay and tube formation of HUVECs cultured with the conditioned medium (CM) derived from cancer cells manipulated for YY1, ZNF322A and/or Shh expression level. In vitro HUVEC transwell migration (Figure 4A) and tube formation (Figure 4B) assays showed that si-Shh in H460 KrasQ61H lung cancer cells inhibited HUVECs migration (panel 2), whereas overexpression of ZNF322A promoted HUVECs migration (panel 3) compared with control group (panel 1, Figure 4A and 4B). Importantly, migration and tube formation abilities of HUVECs were indeed attenuated when treated with CM derived from reconstituted ZNF322A/siShh lung cancer cells (panel 4) compared with those from ZNF322A overexpressing lung cancer cells (panel 3, Figure 4A and 4B), suggesting that Shh is a downstream effector of ZNF322A-mediated pro-angiogenesis. In addition, knockdown of ZNF322A or YY1 attenuated HUVECs migration ability in CM from H460 KrasQ61H lung cancer cells (panels 5 and 6, Figure 4A and 4B). Similar results were observed in CM from H1299 KrasWT lung cancer cells (Figure S5). These results showed that ZNF322A/Shh axis promotes migration and tumor formation abilities of HUVECs.
YY1/ZNF322A/Shh axis regulated angiogenesis in vitro and in vivo. A and B, Transwell migration assay (A) and tumor formation assay (B) showed that CM derived from siShh in ZNF322A overexpressing (group 4) cells inhibited HUVECs migration ability or tumor formation ability compared with CM from ZNF322A-overexpressed (group 3) H460 KrasQ61H lung cancer cells. Knockdown of ZNF322A (group 5) or YY1 (group 6) in H460 KrasQ61H lung cancer cells also attenuated HUVECs migration and tumor formation abilities (panels 5 and 6). siShh was included for comparison (group 2). Migration ability was monitored at 24 h with each group quantified by comparison with initial seeding number of HUVECs. The tube formation was monitored at 6-8 h with each group quantified for the tube length. C-E, Knockdown of Shh inhibited angiogenesis mediated by ZNF322A overexpression in H460 KrasQ61H lung cancer cells using in vivo Matrigel plug angiogenesis assay. (C) Matrigel plug images (Upper) and IHC stains (Lower) showed that angiogenesis was decreased in Matrigel implants with CM from siShh, ZNF322A/siShh, siZNF322A or shYY1-H460 KrasQ61H cells but not with CM from ZNF322A-overexpressing H460 lung cancer cells compared with those from EV/siCtrl-H460 cells. (D) The angiogenesis of each group was measured by the area of CD31-positive stained cells. (E) Western blot confirmed that Shh and ZNF322A were successfully manipulated in H460 KrasQ61H lung cancer cells before CM collection. P values were calculated by two-tailed t-test. Data were mean ± SEM. *, P<0.05; **, P<0.01; ***, P<0.001.
ZNF322A/Shh axis enhanced in vivo angiogenesis
We further performed in vivo Matrigel plug angiogenesis assay. Mixtures of Matrigel with CM from H460 KrasQ61H lung cancer cells manipulated for expression of YY1, ZNF322A and/or Shh were injected subcutaneously into nude mice and then the Matrigel plugs were collected on day 10 for macroscopic analysis and IHC staining of CD31 to reveal blood vessel infiltration. As shown in Figure 4C (upper panel), Matrigel plugs from CM of siShh (panel 2), siZNF322A (panel 5) or shYY1 (panel 6) H460 cells showed a decrease of blood vessel-like structure while those from CM of ZNF322A (panel 3) showed an increase of infiltrated blood vessel-like structure. Notably, mice group injected with CM from ZNF322A/siShh-H460 cells (panel 4) showed less angiogenesis than those from ZNF322A-H460 cells (panel 3). In addition, we used IHC to determine the presence of CD31, an endothelial cells marker. CD31-positive infiltration signals were increased in ZNF322A group (panel 3) while less endothelial infiltration was observed for the ZNF322A/siShh, siShh, siZNF322A and shYY1 groups (lower panel, Figure 4C). Quantitative results are shown in Figure 4D. Western blot confirmed that ZNF322A and siShh were successfully manipulated in lung cancer cells before CM collection (Figure 4E). These in vivo results corroborated with the in vitro data, indicating that ZNF322A/Shh axis enhances angiogenesis.
Moreover, we examined whether CD31 endothelial cells marker was increased in tumor xenograft derived from H460 KrasQ61H lung cancer cells manipulated for ZNF322A expression level. IHC data revealed that CD31 signal was increased in ZNF322A overexpression group, and decreased in ZNF322A knockdown group compared to control group (Figure S6). Altogether, the results were consistent with the scenario that endothelial migration and angiogenesis abilities were promoted by ZNF322A, in part through promoting the expression of Shh, a pro-angiogenesis factor.
Positive correlations of ZNF322A, Shh and CD31 expression in lung cancer patients
Next, we confirmed our proposed YY1/ZNF322/Shh/CD31 axis in clinical samples. We performed IHC analysis to examine the expression of YY1, ZNF322A, Shh and CD31 in surgically resected tumor specimens from 133 lung cancer patients (Figure 5A; Table 1). The results demonstrated that 67.7% of patients showed high ZNF322A expression which correlated with advanced tumor stage (P=0.002; Table 1). High Shh expression was found in 72.2% of patients and was also associated with tumor stage (P=0.001; Table 1). Notably, high ZNF322A expression showed concordantly increased Shh expression and positive CD31 staining (P<0.001; Table 1). However, high YY1 expression only correlated with squamous cell carcinoma (SCC) patients, while high expression of ZNF322A, Shh, and CD31 (ZNF322Ahigh/Shhhigh/CD31high) tended to occur more frequently in adenocarcinoma (ADC) patients than in SCC patients (Table 1). In addition, we previously reported a positive correlation between high expression of ZNF322A and phosphorylated AKT in more than 80% of lung cancer patients [15]. Phosphorylated AKT has been postulated as a secondary event of oncogenic Kras in lung cancer [20,21], indicating Kras activation in this cohort. These clinical correlation data suggested that the ZNF322A/Shh/CD31 axis induced neo-angiogenesis in tumor and associated with advanced lung cancer.
Alteration of ZNF322A, YY1, Shh and CD31 expression in relation to clinicopathological parameters in 133 lung cancer patient patientsa
| Clinicopathological parameters | ZNF322A protein | YY1 protein | Shh protein | CD31 expression | |||||
|---|---|---|---|---|---|---|---|---|---|
| N=133 | N=43 (32.3%) | N=90 (67.7%) | N=45 (33.8%) | N=88 (66.2%) | N=37 (27.8%) | N=96 (72.2%) | N=60 (45.1%) | N=73 (54.9%) | |
| Total | Low (%) | High (%) | Low (%) | High (%) | Low (%) | High (%) | Low (%) | High (%) | |
| Age | |||||||||
| <65 | 71 | 20 (28.2) | 51 (71.8) | 26 (36.6) | 45 (63.4) | 15 (21.1) | 56 (78.9), P=0.05 | 31 (43.7) | 40 (56.3) |
| ≥65 | 62 | 23 (37.1) | 39 (62.9) | 19 (30.6) | 43 (69.4) | 22 (35.5) | 40 (64.5) | 29 (46.8) | 33 (53.2) |
| Sex | |||||||||
| Male | 76 | 23 (30.3) | 53 (69.7) | 22 (28.9) | 54 (71.1) | 25 (21.1) | 51 (78.9) | 33 (43.4) | 43 (56.6) |
| Female | 57 | 20 (35.1) | 37 (64.9) | 23 (40.4) | 34 (59.6) | 12 (32.9) | 45 (67.1) | 27 (47.4) | 30 (52.6) |
| Stage | |||||||||
| I-II | 81 | 34 (42.0) | 47 (58.0), P=0.002 | 25 (30.9) | 56 (69.1) | 31 (38.3) | 50 (61.7), P=0.001 | 42 (51.9) | 36 (48.1), P=0.038 |
| III-IV | 52 | 9 (17.3) | 43 (82.7) | 20 (38.5) | 32 (61.5) | 6 (11.5) | 46 (88.5) | 18 (34.6) | 34 (65.4) |
| Smoker | |||||||||
| No | 65 | 21 (32.3) | 44 (67.7) | 23 (35.4) | 42 (64.6) | 15 (18.5) | 29 (81.5) | 29 (44.6) | 36 (55.4) |
| Yes | 44 | 14 (31.8) | 30 (68.2) | 15 (34.1) | 29 (65.9) | 14 (34.1) | 21 (65.9) | 20 (45.5) | 24 (54.5) |
| Typeb | |||||||||
| SCC | 17 | 7 (41.2) | 10 (58.8) | 2 (11.8) | 15 (88.2), P=0.045 | 6 (35.3) | 11 (64.7) | 9 (52.9) | 8 (47.1) |
| ADC | 113 | 34 (30.1) | 79 (69.9) | 41 (36.3) | 72 (63.7) | 30 (26.5) | 83 (73.5) | 50 (44.2) | 63 (55.8) |
| Typec | |||||||||
| I-II | 84 | 28 (33.3) | 56 (66.7) | 28 (33.3) | 56 (66.7) | 24 (28.6) | 60 (71.4) | 43 (51.2) | 41 (48.8), P=0.048 |
| III-IV | 49 | 15 (30.6) | 34 (69.4) | 17 (34.7) | 32 (65.3) | 13 (26.5) | 36 (73.5) | 17 (34.7) | 36 (65.3) |
| N staged | |||||||||
| 0 | 66 | 29 (43.9) | 37 (56.1), P=0.004 | 19 (28.8) | 47 (71.2) | 26 (39.4) | 40 (60.6), P=0.003 | 34 (51.5) | 32 (48.5) |
| 1-2 | 67 | 14 (20.9) | 53 (79.1) | 26 (38.8) | 41 (61.2) | 11 (16.4) | 56 (83.6) | 26 (38.8) | 41 (61.2) |
| M stagee | |||||||||
| 0 | 121 | 43 (35.5) | 78 (64.5), P=0.011 | 40 (33.1) | 81 (66.9) | 36 (29.8) | 85 (70.2) | 56 (46.3) | 65 (53.7) |
| 1 | 11 | 0 (0.00) | 11 (100.0) | 4 (36.4) | 7 (63.6) | 1 (9.10) | 10 (90.9) | 4 (36.4) | 7 (63.6) |
| Shh | |||||||||
| Low | 37 | 21 (56.8) | 16 (43.2), P<0.001 | ||||||
| High | 96 | 22 (22.9) | 74 (77.1) | ||||||
| CD31 | |||||||||
| Low | 60 | 30 (50.0) | 30 (50.0), P<0.001 | 18 (30.0) | 42 (70.0) | 24 (40.0) | 36 (60.0), P=0.004 | ||
| High | 73 | 13 (17.8) | 60 (82.9) | 27 (36.5) | 47 (63.5) | 13 (17.8) | 60 (82.2) | ||
| YY1 | |||||||||
| Low | 45 | 16 (35.6) | 29 (64.4) | ||||||
| High | 88 | 31 (35.2) | 57 (64.8) | ||||||
aaThe protein expression pattern was defined as low cancer baseline expression (low) or high expression (high). The data were analyzed by Pearson χ2 test. P values with significance are shown as superscripts (P < 0.05). bADC, adenocarcinoma; SCC, squamous cell carcinoma; cT Stage: tumor size; d N Stage: lymph node metastasis; eM Stage: distant metastasis.
Lung cancer patients with concordant ZNF322Ahigh/Shhhigh/CD31high expression profile were associated with poor overall survival and progression-free survival. A, IHC images of tumor specimens from two representative lung cancer patients showed that YY1, ZNF322A, Shh and CD31 displayed a concordant expression pattern, -, low cancer baseline expression; +, high expression. B and C, Kaplan-Meier survival analysis showed that patients with ZNF322Ahigh and CD31high expression had poor overall survival (OS, B) and progression-free survival (PFS, C) in 133 lung cancer patients. D and E, Lung cancer patients with concordant ZNF322Ahigh/Shhhigh/CD31high expression profile were associated with worse OS (D) and PFS (E). P values were determined using log-rank test.
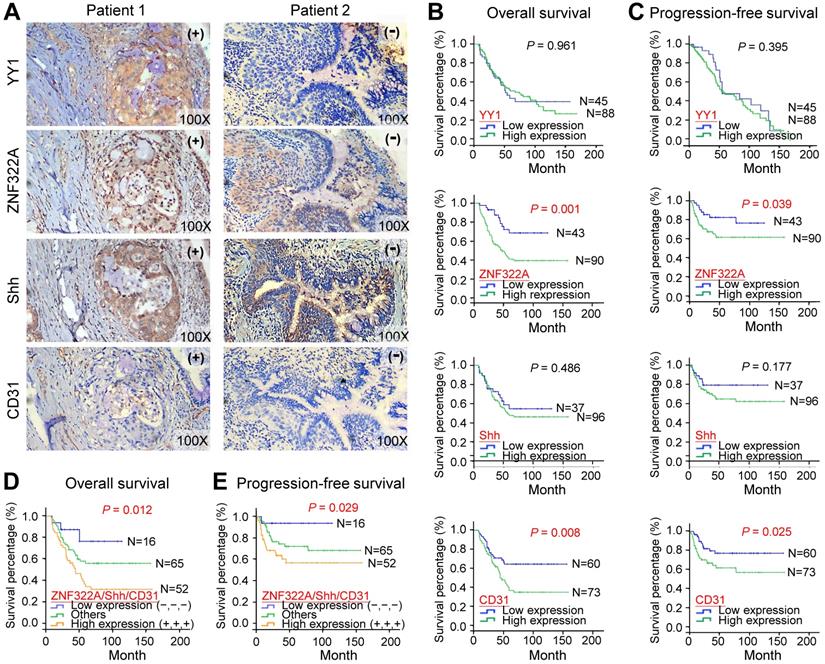
ZNF322Ahigh/Shhhigh/CD31high in lung cancer patients were associated with poor overall survival and progression-free survival
To determine whether the ZNF322A/Shh/CD31 axis was associated with prognosis in human lung cancer, we analyzed overall survival (OS) and progression-free survival (PFS) using the Kaplan-Meier method in 133 patients. Although YY1 and Shh did not show survival prediction potential, overexpression of ZNF322A correlated with poor OS (P=0.001; Figure 5B) and PFS (P=0.039; Figure 5C) in lung cancer patients. Moreover, lung cancer patients with concordantly high expression of ZNF322A, Shh, and CD31 (ZNF322Ahigh/Shhhigh/CD31high) showed the worse OS (P=0.012; Figure 5D) and PFS (P=0.029; Figure 5E).
Next, we performed univariate and multivariate Cox regression analyses in this cohort of 133 lung cancer patients. Univariate Cox regression analysis revealed that patients with ZNF322Ahigh, CD31high expression profile, late stage, or lymph node metastasis had poor survival outcome (Table 2). Importantly, multivariate Cox regression analysis indicated that patients with ZNF322Ahigh/CD31high expression profile showed significantly high risk of death (hazard ratio = 3.952, P = 0.012; Table 2) even after adjusting for the clinical parameters exhibiting potential risk in univariate analysis. These results indicated that the combination of high ZNF322A, high Shh and high CD31 expression could be used as an independent factor in predicting the clinical outcome in lung cancer patients.
Cox regression analysis of risk factors for cancer-related death in 133 lung cancer patients
| Characteristics | Univariate analysis | Multivariate analysis | ||
|---|---|---|---|---|
| HRa (95% CIb) | P-valuec | HRa (95% CIb) | P-valuec | |
| ZNF322A expression | ||||
| Low expression | 1.00 | - i | ||
| High expression | 2.781 (1.449-5.339) | 0.002 | - i | - i |
| YY1 expression | ||||
| Low expression | 1.00 | - i | ||
| High expression | 0.914 (0.536-1.559) | 0.741 | - i | - i |
| Shh expression | ||||
| Low expression | 1.00 | - i | ||
| High expression | 1.227 (0.686-2.195) | 0.490 | - i | - i |
| CD31 | ||||
| Low expression | 1.00 | - i | ||
| High expression | 2.021 (1.185-3.445) | 0.010 | - i | - i |
| ZNF322A/CD31d | ||||
| Low expression (-/-) | 1.00 | 1.00 | ||
| Others (-/+; +/-) | 4.915 (1.699-14.22) | 0.003 | 3.962 (1.352-11.60) | 0.012 |
| High expression (+/+) | 5.869 (2.084-16.52) | 0.001 | 3.952 (1.358-11.50) | 0.012 |
| Age expression | ||||
| <65 year-old | 1.00 | - i | ||
| >65 year-old | 0.797 (0.481-1.319) | 0.377 | - i | - i |
| Gender | ||||
| Female | 1.00 | - i | ||
| Male | 1.421 (0.852-2.370) | 0.178 | - i | - i |
| Smoking habit | ||||
| Non-smoker | 1.00 | - i | ||
| Smoker | 1.665 (0.948-2.922) | 0.076 | - i | - i |
| Typee | ||||
| SCC | 1.00 | - i | ||
| ADC | 0.636 (0.323-1.254) | 0.191 | - i | - i |
| Stage | ||||
| Stage I-II | 1.00 | 1.00 | ||
| Stage III-IV | 2.753 (1.656-4.577) | <0.001 | 1.364 (0.620-2.998) | 0.440 |
| T stagef | ||||
| Stage 1-2 | 1.00 | - i | ||
| Stage 3-4 | 1.345 (0.803-2.254) | 0.260 | - i | - i |
| N stageg | ||||
| N0 | 1.00 | 1.00 | ||
| ≥N1 | 2.681 (1.559-4.610) | <0.001 | 1.532 (0.688-3.410) | 0.296 |
| M stageh | ||||
| M0 | 1.00 | 1.00 | ||
| ≥M1 | 3.503 (1.752-7.003) | <0.001 | 1.969 (0.933-4.156) | 0.075 |
aHR, Hazard ratio. bCI, Confidence interval. cBold values indicate statistical significance (P < 0.05). dZNF322A expression is shown before the slash followed by CD31 expression. -, low expression; +, high expression. eADC, Adenocarcinoma; SCC, Squamous cell carcinoma. fT Stage: tumor size. gN Stage: lymph node metastasis. hM Stage: distant metastasis. iThe variables without significant HR in the univariate analysis were not included in the multivariate analysis.
Discussion
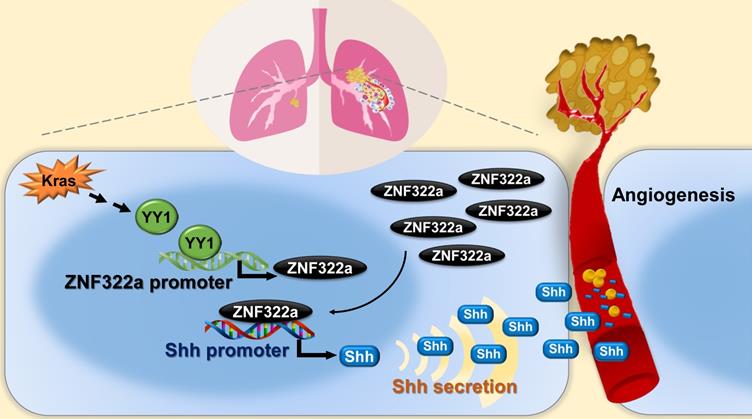
In this study, we identify Kras/YY1/ZNF322A/Shh transcriptional axis as part of an important mechanism underlining neo-angiogenesis and lung cancer metastasis. Mechanistically, oncogenic Kras signaling enhances expression of YY1, the transcription factor that directly activates ZNF322A transcription. Subsequently, overexpressed ZNF322A transcription factor binds to Shh promoter and enhances its expression. Furthermore, we demonstrate that Kras/YY1/ZNF322A-mediated Shh activation promotes angiogenesis abilities in vitro/vivo. Clinically, a positive correlation between ZNF322Ahigh/Shhhigh/CD31high is found in tumors derived from lung cancer patients with poor prognosis. Our findings not only present a previously undefined regulatory mechanism by which ZNF322A synergizes KrasG12D-induced lung tumorigenesis but also indicate that dysregulation of YY1/ZNF322A transcriptional axis promotes expression of angiogenic factor Shh and cancer progression in lung cancer (Figure 6).
Schematic diagram of Kras/YY1/ZNF322A/Shh transcription axis contributing to neo-angiogenesis in lung cancer model. Oncogenic Kras signaling enhances expression of YY1, the transcription factor that directly activates ZNF322A transcription. Subsequently, overexpressed ZNF322A transcription factor binds to Shh promoter and enhances its expression. Kras/YY1/ZNF322A-mediated Shh activation promotes angiogenesis abilities in vitro/vivo. Clinically, a positive correlation between ZNF322Ahigh/Shhhigh/CD31high is found in tumors derived from lung cancer patients with poor prognosis.
We discovered that YY1 positively regulated ZNF322A expression at the transcriptional level. Overexpression of YY1 enhanced ZNF322A mRNA expression and promoter activity of ZNF322A-pGL4, while knockdown of YY1 showed the reverse effects. Such YY1-mediated ZNF322A transcription regulation was abolished when the Del-ZNF322A-pGL4 promoter with deletion of two YY1 binding sites at -462 ~ -363 were used for promoter activity assay. YY1 can act either as an oncogene or a tumor suppressor depending on the cell context because of the multiple roles played by YY1 in regulation of transcription [22-24]. Previous studies have demonstrated that YY1 interacts with p300, AP-1 or TET-catalyzed chromatin complex to cooperatively regulate downstream gene transcription [25-27]. Our RT-qPCR results showed that overexpression of candidate transcription factors E2F1, ELK1, NFκB, Oct4, Sp1 or STAT3 did not influence ZNF322A mRNA expression. However, it is still possible that other TF candidates, for example c-jun or c-myc, which has been shown to cooperate with mutated Kras [28-30], may play a role in regulating ZNF322A transcription. Whether YY1 need additional TF or transcription co-regulators to drive ZNF322A transcription is worthy of further investigation.
The activity of the hedgehog (Hh) pathway is characterized by its dependence on Hh ligands which are produced in secretory cells such as cancer epithelial cell, mural cell, and stromal cell [31-33]. These ligands activate downstream signaling in receiving cells such as cancer epithelial cell, fibroblast, and endothelial cell [33, 34, 35]. Cancer cells have been shown to express Shh ligands and drive canonical signaling in tumor-associated fibroblasts to promote tumor angiogenesis through paracrine Shh signal to adjacent endothelial cells [36]. Although Shh is the most-studied hedgehog so far, only a few studies investigate transcription regulation of Shh gene. For example, NF-κB transcriptionally upregulates Shh expression in pancreatic carcinoma cells [37]. In addition, p63 directly targets and positively regulates the transcription of Shh signaling components such as Shh, Gli2 and Ptch1 to modulate the Shh signaling pathway [38]. Recent report shows that nuclear factor (erythroid-derived 2)-like 2 (NRF2) binds to the promoter of Shh to upregulate Shh mRNA and protein levels, which leads to activation of the Shh pathway and resistance to sorafenib in hepatocellular carcinoma [39]. Our study revealed a novel mechanism by which ZNF322A enhanced neo-angiogenesis in part by activating Shh expression at the transcription level.
Reconstitution experiments demonstrated that ZNF322A/Shh axis was important for angiogenic activity in vitro and in vivo in H460 lung cancer cells with endogenous Kras mutation. Interestingly, our clinical data indicated significant positive correlations between ZNF322A, Shh and CD31. In addition, ZNF322A, Shh and CD31 were all associated with T-N-M stage. These data suggested that ZNF322A, Shh and CD31 played important roles in lung tumor angiogenesis and metastasis. However, Shh or YY1 overexpression alone did not significantly correlate with poor OS and PFS rates. Since Shh expression pattern examined by IHC showed staining in both cytosolic and extracellular compartments in tumor specimens from lung cancer patients, it is possible that the cytosolic immature Shh proteins were also scored in our IHC result. Similarly, a ubiquitous immunoreactivity of YY1 in tumor samples may account for the absence of correlation with clinical parameters or ZNF322A expression pattern. Nevertheless, the expression profile of ZNF322Ahigh/Shhhigh/CD31high is a potential prognostic biomarker for lung cancer and may be for other cancers.
Conclusion
This study provides new mechanistic insights into how oncogenic Kras-induced YY1/ZNF322A transcriptional axis promotes lung cancer progression. We also demonstrate interplay between ZNF322A/Shh axis by lung cancer epithelial cells and endothelial cells in regulation of neo-angiogenesis. Mechanistically, YY1 transcription activation induced overexpression of oncoprotein ZNF322A, ZNF322A then bound to Shh promoter and enhanced its expression. Kras has been considered to be undruggable thus far. Therefore, therapeutic strategies have shifted toward Kras downstream signaling. We proposed that lung cancer patients with the expression profile of ZNF322Ahigh/Shhhigh/CD31high may be selected for further treatment with Shh neutralizing antibodies, although targeting Shh via antibodies has not reached human trials [40,41]. Alternatively, these patients may be treated with drugs targeting Shh signaling effectors such as the SMO antagonists or VEGFR2 inhibitor already approved by the US Food and Drug Administration [42-44]. Therapeutic strategies that target Kras/YY1/ZNF322A/Shh signaling axis may provide new insight on targeted therapy for lung cancer patients.
Abbreviations
ADC: adenocarcinoma; ChIP: chromatin-immunoprecipitation sequencing; CM: conditioned medium; HUVEC: human umbilical vein endothelial cell; OS: overall survival; PFS: progression-free survival; SCC: squamous cell carcinoma; Shh: sonic hedgehog; TF: transcription factor; YY1: Yin Yang 1.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
This work was supported by the Taiwan Ministry of Science and Technology grants (MOST109-2320-B-006-038-MY3). We thank National Health Research Institutes Transgenic Animal Core for assistance in the mouse oocyte pronuclear injection and are grateful for the support from the Human Biobank, Research Center of Clinical Medicine, National Cheng Kung University Hospital.
Contributions
YCW conceived the project. CCL, IYK, and LTW designed experiments. All authors contributed to performance of the experiments or data analysis. YCW wrote the manuscript. All authors read and approved the final manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Chang EH, Gonda MA, Ellis RW, Scolnick EM, Lowy DR. Human genome contains four genes homologous to transforming genes of Harvey and Kirsten murine sarcoma viruses. Proc Natl Acad Sci U S A. 1982;79:4848-52
2. Hall A, Marshall CJ, Spurr NK, Weiss RA. Identification of transforming gene in two human sarcoma cell lines as a new member of the ras gene family located on chromosome 1. Nature. 1983;303:396-400
3. Prior IA, Lewis PD, Mattos C. A comprehensive survey of Ras mutations in cancer. Cancer Res. 2012;72:2457-67
4. Westcott PM, Halliwill KD, To MD, Rashid M, Rust AG, Keane TM. et al. The mutational landscapes of genetic and chemical models of Kras-driven lung cancer. Nature. 2015;517:489-92
5. Román M, López I, Guruceaga E, Baraibar I, Ecay M, Collantes M. et al. Inhibitor of differentiation-1 sustains mutant KRAS-driven progression, maintenance, and metastasis of lung adenocarcinoma via regulation of a FOSL1 network. Cancer Res. 2019;79:625-38
6. Vallejo A, Perurena N, Guruceaga E, Mazur PK, Martinez-Canarias S, Zandueta C. et al. An integrative approach unveils FOSL1 as an oncogene vulnerability in KRAS-driven lung and pancreatic cancer. Nat Commun. 2017;8:14294
7. Mizumoto Y, Kyo S, Kiyono T, Takakura M, Nakamura M, Maida Y. et al. Activation of NF-kappaB is a novel target of KRAS-induced endometrial carcinogenesis. Clin Cancer Res. 2011;17:1341-50
8. Yuan P, He XH, Rong YF, Cao J, Li Y, Hu YP. et al. KRAS/NF-κB/YY1/miR-489 signaling axis controls pancreatic cancer metastasis. Cancer Res. 2017;77:100-11
9. Du F, Cao T, Xie H, Li T, Sun L, Liu H. et al. KRAS mutation-responsive miR-139-5p inhibits colorectal cancer progression and is repressed by Wnt signaling. Theranostics. 2020;10:7335-50
10. Chen K, Liu MX, Mak CS, Yung MM, Leung TH, Xu D. et al. Methylation-associated silencing of miR-193a-3p promotes ovarian cancer aggressiveness by targeting GRB7 and MAPK/ERK pathways. Theranostics. 2018;8:423-36
11. Li Y, Wang Y, Zhang C, Yuan W, Wang J, Zhu C. et al. ZNF322, a novel human C2H2 Kruppel-like zinc-finger protein, regulates transcriptional activation in MAPK signaling pathways. Biochem Biophys Res Commun. 2004;325:1383-92
12. Jen J, Lin LL, Lo FY, Chen HT, Liao SY, Tang YA. et al. Oncoprotein ZNF322A transcriptionally deregulates alpha-adducin, cyclin D1 and p53 to promote tumor growth and metastasis in lung cancer. Oncogene. 2016;35:2357-69
13. Jen J, Liu CY, Chen YT, Wu LT, Shieh YC, Lai WW. et al. Oncogenic zinc finger protein ZNF322A promotes stem cell-like properties in lung cancer through transcriptional suppression of c-Myc expression. Cell Death Differ. 2019;26:1283-98
14. Liao SY, Chiang CW, Hsu CH, Chen YT, Jen J, Juan HF. et al. CK1δ/GSK3β/FBXW7α axis promotes degradation of the ZNF322A oncoprotein to suppress lung cancer progression. Oncogene. 2017;36:5722-33
15. Liao SY, Kuo IY, Chen YT, Liao PC, Liu YF, Wu HY. et al. AKT-mediated phosphorylation enhances protein stability and transcription activity of ZNF322A to promote lung cancer progression. Oncogene. 2019;38:6723-36
16. Liu Q, Li A, Tian Y, Wu JD, Liu Y, Li T. et al. The CXCL8-CXCR1/2 pathways in cancer. Cytokine Growth Factor Rev. 2016;31:61-71
17. Spuul P, Daubon T, Pitter B, Alonso F, Fremaux I, Kramer I. et al. VEGF-A/Notch-induced podosomes proteolyse basement membrane Collagen-IV during retinal sprouting angiogenesis. Cell Rep. 2016;17:484-500
18. Arany Z, Foo SY, Ma Y, Ruas JL, Bommi-Reddy A, Girnun G. et al. HIF-independent regulation of VEGF and angiogenesis by the transcriptional coactivator PGC-1alpha. Nature. 2008;451:1008-12
19. Prior IA, Lewis PD, Mattos C. A Comprehensive survey of Ras mutations in cancer. Cancer Res. 2012;72:2457-67
20. Kim JY, Welsh EA, Fang B, Bai Y, Kinose F, Eschrich SA. et al. Phosphoproteomics reveals MAPK inhibitors enhance MET- and EGFR-driven AKT signaling in KRAS-mutant lung cancer. Mol Cancer Res. 2016;14:1019-29
21. Lai H, Wang Y, Duan F, Li Y, Jiang Z, Luo L. et al. Krukovine suppresses KRAS-mutated lung cancer cell growth and proliferation by inhibiting the RAF-ERK pathway and inactivating AKT pathway. Front Pharmacol. 2018;9:958
22. Khachigian LM. The Yin and Yang of YY1 in tumor growth and suppression. Int J Cancer. 2018;143:460-5
23. Tseng HY, Chen YA, Jen J, Shen PC, Chen LM, Lin TD. et al. Oncogenic MCT-1 activation promotes YY1-EGFR-MnSOD signaling and tumor progression. Oncogenesis. 2017;6:e313
24. Meliala ITS, Hosea R, Kasim V, Wu S. The biological implications of Yin Yang 1 in the hallmarks of cancer. Theranostics. 2020;10:4183-200
25. Fang S, Li J, Xiao Y, Lee M, Guo L, Han W. et al. Tet inactivation disrupts YY1 binding and long-range chromatin interactions during embryonic heart development. Nat Commun. 2019;10:4297
26. Lee JS, Galvin KM, See RH, Eckner R, Livingston D, Moran E. et al. Relief of YY1 transcriptional repression by adenovirus E1A is mediated by E1A-associated protein p300. Genes Dev. 1995;9:1188-98
27. Wang CC, Tsai MF, Dai TH, Hong TM, Chan WK, Chen JJ. et al. Synergistic activation of the tumor suppressor, HLJ1, by the transcription factors YY1 and activator protein 1. Cancer Res. 2007;67:4816-26
28. Bell CM, Raffeiner P, Hart JR, Vogt PK. PIK3CA cooperates with KRAS to promote MYC activity and tumorigenesis via the bromodomain protein BRD9. Cancers (Basel). 2019;11:1634
29. Corcoran RB, Contino G, Deshpande V, Tzatsos A, Conrad C, Benes CH. et al. STAT3 plays a critical role in KRAS-induced pancreatic tumorigenesis. Cancer Res. 2011;71:5020-9
30. Sen M, Wang X, Hamdan FH, Rapp J, Eggert J, Kosinsky RL. et al. ARID1A facilitates KRAS signaling-regulated enhancer activity in an AP1-dependent manner in colorectal cancer cells. Clin Epigenetics. 2019;11:92
31. Maity G, Mehta S, Haque I, Dhar K, Sarkar S, Banerjee SK. et al. Pancreatic tumor cell secreted CCN1/Cyr61 promotes endothelial cell migration and aberrant neovascularization. Sci Rep. 2014;4:4995
32. Ok CY, Singh RR, Vega F. Aberrant activation of the hedgehog signaling pathway in malignant hematological neoplasms. Am J Pathol. 2012;180:2-11
33. Yao Q, Renault MA, Chapouly C, Vandierdonck S, Belloc I, Jaspard-Vinassa B. et al. Sonic hedgehog mediates a novel pathway of PDGF-BB-dependent vessel maturation. Blood. 2014;123:2429-37
34. Srivastava RK, Kaylani SZ, Edrees N, Li C, Talwelkar SS1, Xu J. et al. GLI inhibitor GANT-61 diminishes embryonal and alveolar rhabdomyosarcoma growth by inhibiting Shh/AKT-mTOR axis. Oncotarget. 2014;5:12151-65
35. Valenti G, Quinn HM, Heynen GJJE, Lan L, Holland JD, Vogel R. et al. Cancer stem cells regulate cancer-associated fibroblasts via activation of hedgehog signaling in mammary gland tumors. Cancer Res. 2017;77:2134-47
36. Chen W, Tang T, Eastham-Anderson J, Dunlap D, Alicke B, Nannini M. et al. Canonical hedgehog signaling augments tumor angiogenesis by induction of VEGF-A in stromal perivascular cells. Proc Natl Acad Sci U S A. 2011;108:9589-94
37. Kasperczyk H, Baumann B, Debatin KM, Fulda S. Characterization of sonic hedgehog as a novel NF-kappaB target gene that promotes NF-kappaB-mediated apoptosis resistance and tumor growth in vivo. FASEB J. 2009;23:21-33
38. Memmi EM, Sanarico AG, Giacobbe A, Peschiaroli A, Frezza V, Cicalese A. et al. p63 sustains self-renewal of mammary cancer stem cells through regulation of Sonic Hedgehog signaling. Proc Natl Acad Sci U S A. 2015;112:3499-504
39. Wing Leung H, Ting Lau EY, Ning Leung CO, Leng Lei MM, Kit Mok EH, Ma VWS. et al. NRF2/SHH signaling cascade promotes tumor-initiating cell lineage and drug resistance in hepatocellular carcinoma. Cancer Lett. 2020;476:48-56
40. Michaud NR, Wang Y, McEachern KA, Jordan JJ, Mazzola AM, Hernandez A. et al. Novel neutralizing hedgehog antibody MEDI-5304 exhibits antitumor activity by inhibiting paracrine hedgehog signaling. Mol Cancer Ther. 2014;13:386-98
41. Carballo GB, Honorato JR, de Lopes GPF, Spohr TCLSE. A highlight on Sonic hedgehog pathway. Cell Commun Signal. 2018;16:11
42. Casey D, Demko S, Shord S, Zhao H, Chen H, He K. et al. FDA approval summary: Sonidegib for locally advanced basal cell carcinoma. Clin Cancer Res. 2017;23:2377-81
43. Rimkus TK, Carpenter RL, Qasem S, Chan M, Lo HW. Targeting the Sonic hedgehog signaling pathway: review of Smoothened and GLI Inhibitors. Cancers (Basel). 2016;8:22
44. Wu F, Zhang Y, Sun B, McMahon AP, Wang Y. Hedgehog signaling: from basic biology to cancer therapy. Cell Chem Biol. 2017;24:252-80
Author contact
![]() Corresponding author: Yi-Ching Wang, PhD. Department of Pharmacology and Institute of Basic Medical Sciences, National Cheng Kung University, No.1, University Road, Tainan 70101, Taiwan, R. O. C., Phone: +886-6-2353535 ext.5502; FAX: +886-6-2749296; E-mail: ycw5798ncku.edu.tw.
Corresponding author: Yi-Ching Wang, PhD. Department of Pharmacology and Institute of Basic Medical Sciences, National Cheng Kung University, No.1, University Road, Tainan 70101, Taiwan, R. O. C., Phone: +886-6-2353535 ext.5502; FAX: +886-6-2749296; E-mail: ycw5798ncku.edu.tw.