Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Results and Discussion
Conclusion
Materials and Methods
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2020; 10(24):11178-11196. doi:10.7150/thno.42587 This issue Cite
Research Paper
Synthesis and preliminary studies of 11C-labeled tetrahydro-1,7-naphthyridine-2-carboxamides for PET imaging of metabotropic glutamate receptor 2
Xiaofei Zhang1,2*, Yiding Zhang3*, Zhen Chen1, Tuo Shao1, Richard Van4, Katsushi Kumata3, Xiaoyun Deng1, Hualong Fu1, Tomoteru Yamasaki3, Jian Rong1, Kuan Hu3, Akiko Hatori3, Lin Xie3, Qingzhen Yu1, Weijian Ye5, Hao Xu5, Douglas J. Sheffler6, Nicholas D. P. Cosford6, Yihan Shao4, Pingping Tang2, Lu Wang1,5 ![]() , Ming-Rong Zhang3
, Ming-Rong Zhang3 ![]() , Steven H. Liang1
, Steven H. Liang1 ![]()
1. Division of Nuclear Medicine and Molecular Imaging, Massachusetts General Hospital & Department of Radiology, Harvard Medical School, Boston, MA, 02114, USA
2. State Key Laboratory and Institute of Elemento-Organic Chemistry, Collaborative Innovation Center of Chemical Science and Engineering, Nankai University, Tianjin 300071, China
3. Department of Radiopharmaceuticals Development, National Institute of Radiological Sciences, National Institutes for Quantum and Radiological Science and Technology, 4-9-1 Anagawa, Inage-ku, Chiba 263-8555, Japan
4. Department of Chemistry and Biochemistry, University of Oklahoma, Norman, Oklahoma 73019, United States
5. Department of Nuclear Medicine and PET/CT-MRI Center, The First Affiliated Hospital of Jinan University, 613 West Huangpu Road, Tianhe District, Guangzhou 510630, China
6. Cancer Metabolism and Signaling Networks Program and Conrad Prebys Center for Chemical Genomics, Sanford-Burnham Prebys Medical Discovery Institute, La Jolla, California 92037, United States.
*These two authors contributed equally to this work.
Received 2019-11-29; Accepted 2020-8-19; Published 2020-9-14
Abstract

Selective modulation of metabotropic glutamate receptor 2 (mGlu2) represents a novel therapeutic approach for treating brain disorders, including schizophrenia, depression, Parkinson's disease (PD), Alzheimer's disease (AD), drug abuse and addiction. Imaging mGlu2 using positron emission tomography (PET) would allow for in vivo quantification under physiological and pathological conditions and facilitate drug discovery by enabling target engagement studies. In this paper, we aimed to develop a novel specific radioligand derived from negative allosteric modulators (NAMs) for PET imaging of mGlu2.
Methods. A focused small molecule library of mGlu2 NAMs with tetrahydro naphthyridine scaffold was synthesized for pharmacology and physicochemical evaluation. GIRK dose-response assays and CNS panel binding selectivity assays were performed to study the affinity and selectivity of mGlu2 NAMs, among which compounds 14a and 14b were selected as PET ligand candidates. Autoradiography in SD rat brain sections was used to confirm the in vitro binding specificity and selectivity of [11C]14a and [11C]14b towards mGlu2. In vivo binding specificity was then studied by PET imaging. Whole body biodistribution study and radiometabolite analysis were conducted to demonstrate the pharmacokinetic properties of [11C]14b as most promising PET mGlu2 PET ligand.
Results. mGlu2 NAMs 14a-14g were synthesized in 14%-20% yields in five steps. NAMs 14a and 14b were selected to be the most promising ligands due to their high affinity in GIRK dose-response assays. [11C]14a and [11C]14b displayed similar heterogeneous distribution by autoradiography, consistent with mGlu2 expression in the brain. While PET imaging study showed good brain permeability for both tracers, compound [11C]14b demonstrated superior binding specificity compared to [11C]14a. Further radiometabolite analysis of [11C]14b showed excellent stability in the brain.
Conclusions. Compound 14b exhibited high affinity and excellent subtype selectivity, which was then evaluated by in vitro autoradiography and in vivo PET imaging study after labeling with carbon-11. Ligand [11C]14b, which we named [11C]MG2-1904, demonstrated high brain uptake and excellent in vitro/in vivo specific binding towards mGlu2 with high metabolic stability in the brain. As proof-of-concept, our preliminary work demonstrated a successful example of visualizing mGlu2 in vivo derived from NAMs, which represents a promising chemotype for further development and optimization aimed for clinical translation.
Keywords: positron emission tomography, metabotropic glutamate receptor 2, negative allosteric modulator, 11C, mGlu2
Introduction
In the central nervous system (CNS), L-glutamate is the major endogenous neurotransmitter that mediates a vast majority of synaptic excitations by interacting with two distinct types of receptors: the ionotropic glutamate receptors (iGluRs), which have voltage-gated cation channel activity, and the metabotropic glutamate receptors (mGlus), which are coupled to GTP-binding proteins to mediate intracellular second messenger systems such as phospholipase C (PLC) and adenylate cyclase (AC) [1-7]. Based on their pharmacology and signal transduction mechanism, mGlus consist of eight subtypes which are typically classified into three categories: Group I (mGlu1 and mGlu5) primarily activates PLC via Gαq coupling, leading to mobilization of intracellular Ca2+. Both group II (mGlu2 and mGlu3) and group III (mGlu4, mGlu6-8) preferentially couple to AC and Gαi, inhibiting the release of AC and suppressing cAMP production [5]. The mGlus modulate inhibitory activity within the CNS and the dysfunction of the glutamatergic system has been associated with the pathophysiology of numerous psychiatric and neurological disorders [8-11].
Group II mGlus negatively modulate the presynaptic release of glutamate and activation of potassium channels. As opposed to mGlu3 which is expressed throughout the CNS, mGlu2 has a more limited distribution and is localized extensively on presynaptic nerve terminals [8, 12]. In the brain, high expression of mGlu2 can be found in the striatum, cerebral cortex, hippocampus, amygdala and cerebellum [13-17]. It has been reported that mGlu2 dysfunction is related to several CNS disorders, thus attracting much attention as a promising therapeutic target [18, 19]. In particular, selective modulation of mGlu2 is a potential strategy for the treatment of schizophrenia [20-22], depression [23, 24], Parkinson's disease (PD) [25-28], Alzheimer's disease (AD) [29, 30], drug abuse and addiction [31-35].
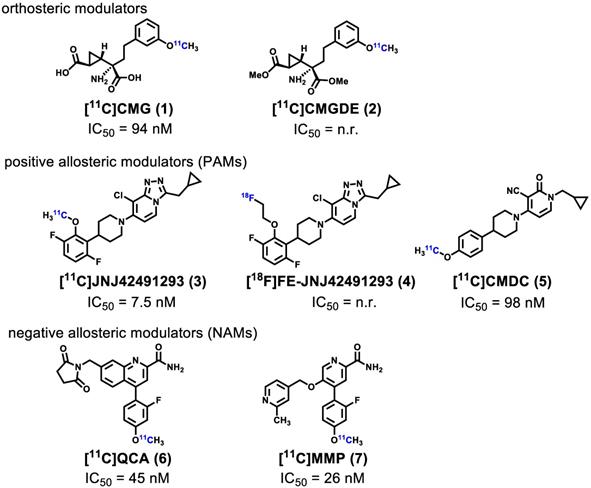
Noninvasive imaging of mGlu2 using positron emission tomography (PET) can visualize and provide quantitative measurement of the distribution and expression of this receptor under physiological and pathological conditions [36-40], further enabling a better understanding of the relationship between mGlu2 mediated glutamatergic signaling and CNS disorders. Furthermore, the development of high-affinity and selective mGlu2 PET tracers would enable clinical translation of mGlu2 modulators by providing a means to perform target engagement and dose occupancy studies. In the past several years, continuous research efforts have been invested in the development of PET ligands for imaging mGlu2, including [11C]CMG (1) [41], [11C]CMGDE (2) [41], [11C]JNJ42491293 (3) [42], [18F]FE-JNJ42491293 (4) [43], [11C]CMDC (5) [44], and our recently reported PET ligands namely [11C]QCA (6) [45] and [11C]MMP (7) [46] (Figure 1). As the first studied mGlu2 PET tracer, [11C]CMG (1) failed to cross blood-brain-barrier (BBB), which was then overcome by its ester derivative [11C]CMGDE (2) [41]. These two tracers showed low in vivo specificity and selectivity between mGlu2 and mGlu3 attributed to the conserved orthosteric binding sites. The first positive allosteric modulator (PAM)-based PET ligands 3 [42] and its fluoroethyl analog 4 [43], were subsequently reported in 2012 and 2013, and the radioligand 3 was advanced to human studies [47]. However, an unexpectedly high myocardial retention observed in humans along with off-target binding, confirmed by mGlu2 knockout rats, made these efforts of limited translational value. In addition, there are only limited preliminary mGlu2 imaging data in the human brain based on a mGlu2 ligand developed by Merck, Inc., the structure of which has not been disclosed [48]. During the preparation of this manuscript, a preliminary radiosynthesis of a potential mGlu2 radioligand [18F]JNJ-46356479 has been reported [49]. As a derivative of JNJ-40068782 [50], [11C]CMDC (5) was not further pursued due to limited BBB penetration [44]. In 2017, the first negative allosteric modulator (NAM)-based PET ligand, [11C]QCA (6) with good affinity (IC50 = 45 nM) and high selectivity for mGlu2 over mGlu3, was developed by our groups [45]. Although in vitro autoradiography (ARG) results showed excellent specific binding to mGlu2, low brain uptake (peak value ~ 0.3 SUV) caused by ATP binding cassette (ABC) efflux pump (PgP/Bcrp substrate) limited further investigation of [11C]QCA (6). Most recently, our research groups identified a new NAM PET ligand namely [11C]MMP (7) with an improved affinity (IC50 = 26 nM) with reasonable lipophilicity (LogD = 3.30) [46]. Unfortunately, animal PET imaging data demonstrated that [11C]MMP (7) exhibited moderate brain uptake (peak value ~ 0.6) and low levels of in vivo specific binding. To date, there is no NAM-based PET ligand that can visualize and quantify mGlu2 with sufficient brain penetration, high affinity and selectivity, and this represents an urgent and unmet need for drug discovery and clinical development.
Representative mGlu2 PET ligands that have been tested in animals and/or humans. n.r. = not reported.
In this study, we develop a focused array of NAMs with moderate to high affinity and selectivity for mGlu2 based on our continuous medicinal chemistry efforts. Herein, we describe our chemical syntheses, pharmacological screening and 11C-labeling of the most promising candidates. Comprehensive evaluations including brain permeability and specificity were conducted by in vitro autoradiography, ex vivo biodistribution and preliminary in vivo rodent PET imaging, all of which provide an excellent entry point for further mGlu2 PET ligand development aimed at clinical translation.
Results and Discussion
Medicinal Chemistry
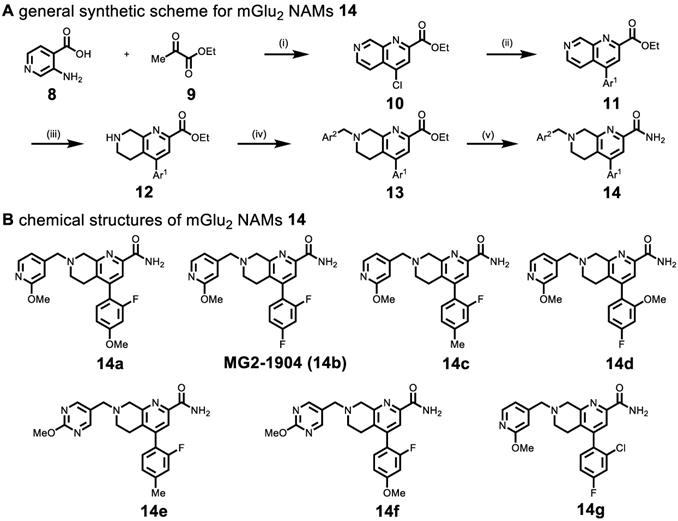
Based on recent advances in the development of mGlu2 NAMs (as therapeutic candidates) by scientists from Vanderbilt University[51] and Merck Research Laboratories (Patents WO/2018/063955, WO/2016/032921, WO/2013/066736), a reductionist approach was utilized to design the second-generation of potent, selective and brain penetrant compounds. For PET ligand development, we envisioned that a simple tetrahydro naphthyridine scaffold with suitably tethered Ar1 and Ar2 moieties could overcome the challenges associated with brain penetration and in vivo stability encountered in the previous series (Scheme 1). The optimization was carried out by truncation of the bicyclic core system in the first generation QCA 6. To obtain second-generation mGlu2 NAMs, we first employed acid-mediated cyclization between 3-aminoisonicotinic acid 8 and ethyl pyruvate 9 to synthesize naphthyridine ester 10 in 64% yield. Then aromatic motif Ar1 was installed via the Suzuki-Miyaura cross-coupling with different boronic acids in 72%-80% yields. Another aromatic moiety Ar2 was introduced by the reduction of naphthyridine moiety with NaBH3CN to provide tetrahydro naphthyridine 12, followed by SN2 displacement with a series of heteroarylmethyl chlorides in 42%-52% yields over two steps. The ensuing compounds 13a-13g were treated with ammonia in methanol to afford the desired mGlu2 NAMs 14a-14g in 70-80% yields. Together, we synthesized seven second-generation mGlu2 NAMs from starting material 8 in 14%-20% overall yields in five steps.
(A) Synthesis of mGlu2 NAMs 14a-14g. Reactions and conditions: (i) POCl3, 100 oC 1 h, 64% yield; (ii) Ar1-B(OH)2, Pd(PPh3)4, K2CO3, 1,4-Dioxane/H2O, 100 oC, overnight, 80%-72% yield; (iii) NaBH3CN, AcOH, 25oC, 5 min; (iv) Ar2-CH2Cl,K2CO3, MeCN, r.t., 4 h, 52%-42% yield; (v) ammonia 7 M in MeOH, 25 oC, 4 h, 80%-70% yield. (B) Chemical structures of mGlu2 NAMs 14a-14g.
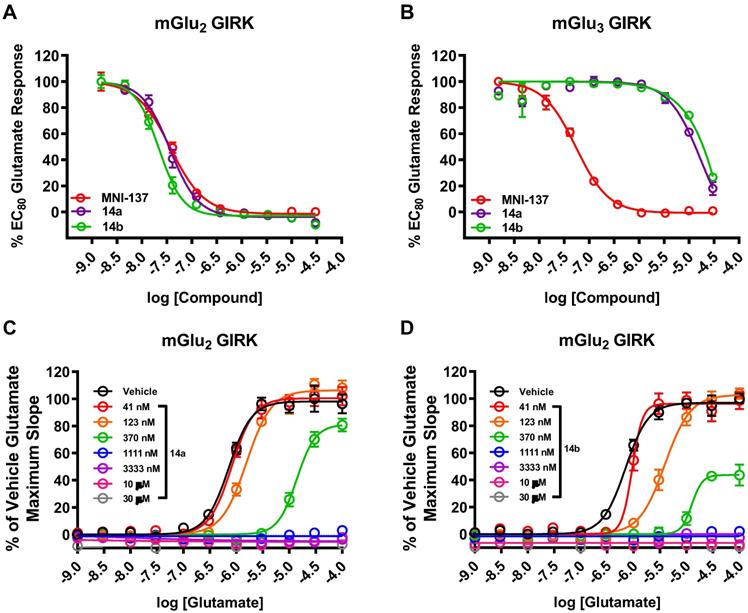
The pharmacological and physicochemical properties of these NAMs were investigated using our established procedures [45], and the results are depicted in Table 1. Representative concentration-response curves for determining the affinity and subtype selectivity (Figures 2A & 2B) as well as noncompetitive negative allosteric modulation (dose-dependently right shift and maximal response decrease towards increasing concentration of glutamate; Figures 2C & 2D) of compounds 14a and 14b are illustrated herein. For affinity and subtype selectivity evaluation of all NAMs 14, see details in Figure S1 and S10 in the supporting information. Various substituents at Ar1 and Ar2 groups were incorporated into the scaffold of tetrahydro naphthyridine carboxamide. Interestingly, the optimal Ar2 group, namely 2-fluoro-4-methylphenyl, in the first-generation mGlu2 NAMs failed to improve the ligand affinity, as reflected by unfavored results of IC50 129 nM for 14c, and 106 nM for 14e. Maintaining a 2-fluoro-4-mexothyphenyl group at the Ar2 position and changing the Ar2 group from pyridine to a pyrimidine moiety dramatically lowered affinity towards mGlu2 (IC50 39 nM for 14a vs. 318 nM for 14f). Varying the substituents on the Ar1 group also affected the affinity. The IC50 values decreased when the methoxy group was displaced by a fluorine at the para-position (14a vs. 14b), but increased when the fluoro substituent was displaced by methoxy or chloro substituents at the ortho-position (14b vs. 14d, 14g). All these NAMs showed excellent selectivity towards mGlu2 over mGlu3 (IC50 > 10-30 μM for mGlu3). Furthermore, we performed the binding selectivity assays of ligand 14b in a comprehensive CNS panel provided by the NIMH PDSP. The results indicated that 14b has no substantial interaction with related mGlu receptor family and no significant response with any other major brain targets. The compound also showed no activity in hERG safety assay (see details in Figure S10 and S11 in the supporting information). Compounds 14a-14g were slightly more lipophilic than the first-generation ligands, and tPSA values for this series were reduced, all of which may provide improved brain permeability compared with 6. As a result, NAMs 14a and 14b were selected to be the most promising ligands due to their high affinity (Figure 2A), excellent subtype selectivity (Figure 2B, inactive against mGlu3 up to 30 µM) and reasonable lipophilicity and tPSA in the second-generation. To our delight, methoxy substituents on these two compounds could be used as a labeling handle for carbon-11 [52], which could facilitate our preliminary in vivo evaluation on this scaffold with PET.
Pharmacology and physiochemical properties of mGlu2NAMs 14a-14g
| IC50 (nM) for mGlu2a | |||||
|---|---|---|---|---|---|
| compd. | mean | SEM | IC50 (μM) for mGlu3b | cLogDc | tPSAc |
| 14a | 39 | 10 | > 10 | 3.48 | 89.51 |
| 14b | 24 | 5 | > 10 | 3.54 | 80.28 |
| 14c | 129 | 24 | > 10 | 3.90 | 80.28 |
| 14d | 39 | 6 | > 10 | 2.92 | 89.51 |
| 14e | 106 | 14 | > 10 | 3.07 | 92.62 |
| 14f | 318 | 26 | > 10 | 2.66 | 101.87 |
| 14g | 87d | / | > 30e | 3.87 | 80.28 |
aValues of in vitro affinity were measured in triplicate assays in mGlu2 GIRK or bmGlu3 GIRK. cValues were calculated with ChemDraw 16.0 software. dValues were tested in duplicate assays in mGlu2 GIRK or emGlu3 GIRK.
Representative dose-response curves of 14a and 14b in GIRK assays using human HEK293 cells expressing mGlu2/3 receptors. In vitro affinity of 14a and 14b with (A) mGlu2 GIRK assay and (B) mGlu3 GIRK assay with the control mGlu2/3 NAM MNI-137. As a non-competitive mode of action, 14a (C) and 14b (D) showed right shift of mGlu2 dose-response curves and decreased maximal response with increasing glutamate concentration.
Radiochemistry
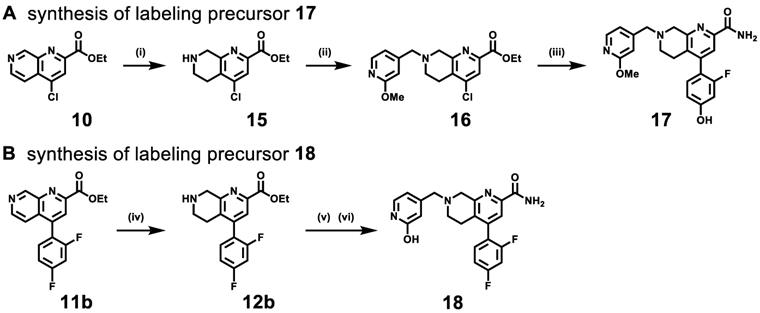
The hydroxyl group on (hetero)arenes is a feasible labeling site for 11C-labeled PET ligand development via methylation with [11C]CH3I under basic conditions [52]. As shown in Scheme 2, the syntheses of radiolabeling precursors 17 and 18 were conducted analogously to our synthetic route for second-generation mGlu2 NAMs. Compound 16 was obtained from intermediate 10 by NaBH3CN-mediated reduction and subsequent SN2 displacement with 2-methoxy-4-(chloromethyl)pyridine in 45% yield over two steps. The Suzuki cross-coupling with 2-fluoro-4-hydroxyphenylboronic acid and ammonolysis with 7N ammonia in methanol solution was employed to give radiolabeling precursor 17 in 44% yield over two steps. Amine alkylation between 2-hydroxy-4-(chloromethyl)pyridine and intermediate 12b (reduced from compound 11b) was conducted followed by ammonolysis fulfilled the synthesis of radiolabeling precursor 18 in 56% yield. In all, precursors 17 and 18 were synthesized from their corresponding 1,7-naphthyridine-2-carboxylates 10 and 11b in an overall yield of 20% and 22%, respectively, over three steps.
Syntheses of labeling precursors 17 (A) and 18 (B). Reactions and conditions: (i) NaBH3CN, AcOH, 25 ℃, 5 min; (ii) 2-methoxy-4-(chloromethyl)pyridine, K2CO3, MeCN, r.t., 4 h, 45% yield for two steps; (iii) (2-fluoro-4-hydroxyphenyl)boronic acid, Pd(dppf)Cl2, H2O/1,4-dixone, K2CO3, 100 ℃, 5 h then ammonia 7 M in MeOH, 25 ℃, 4 h, 42% yield for two steps; (iv) NaBH3CN, AcOH, 25 ℃, 5 min; (v) K2CO3, MeCN, r.t., 4 h; (vi) ammonia 7M in MeOH, 25 ℃, 4 h, 70% yield.
As shown in Scheme 3A, the radiosynthesis of [11C]14a was accomplished by passing gaseous [11C]CH3I through a solution of phenolic precursor 17 (1.0 mg) and NaOH (3-5 μL, 0.5 M) in DMF (300 μL). The reaction was conducted at 80 °C for 5 min, followed by purification using semi-preparative HPLC. The decay-corrected radiochemical yields of [11C]14a were 36.6 ± 7.3% (n = 7) based on the starting radioactivity of [11C]CO2. The radiosynthesis of [11C]14b was realized by 11C-methylation of hydroxypyridinyl precursor 18 (1.0 mg) in anhydrous DMF (300 μL; pre-saturated with 10-20 mg Cs2CO3) at 90 °C for 5 min (Scheme 3B), generating the desired product [11C]14b with decay-corrected radiochemical yields of 6.53 ± 1.5% (n = 10) after semipreparative HPLC purification. Both radiotracers were formulated in a saline solution containing 100 μL of 25% ascorbic acid in sterile water and 100 μL of 20% Tween 80 (see details in Methods). The radiochemical and chemical purities were greater than 99%, and molar activities were greater than 74 GBq/μmol (2 Ci/μmol). The overall synthesis time of both radiotracers was ca. 40 min, and no radiolysis was observed up to 90 min.
Radiosynthesis of [11C]14a (A) [11C]14b (B).
In Vitro Autoradiography
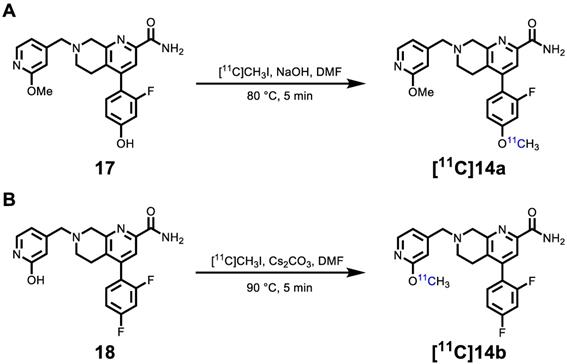
To confirm the in vitro binding specificity and selectivity of [11C]14a and [11C]14b towards mGlu2, in vitro autoradiography (ARG) studies were performed in the brain sections of Sprague-Dawley (SD) rats wild-type (Figures 3 & 4) and an image of the ROIs used for quantification is included in Figure S9 in the supporting information. Both ligands [11C]14a and [11C]14b (radioconcentration 1.25 μCi/mL each) displayed similar heterogeneous distribution with high radioactivity accumulated in the striatum and cerebral cortex, followed by hippocampus and cerebellum (Figure 3A & 3D; Figure 4A & 4D). Their heterogeneous patterns were in accordance with the biological expression of mGlu2 in rodents [13-17], as well as our previously published data for [11C]QCA [45]. Blocking studies were conducted by pretreatment with either the corresponding mGlu2 NAMs (10 μM of 14a for [11C]14a, Figure 3B; 10 μM of 14b for [11C]14b, Figure 4B) or QCA (10 μM of 6, Figure 3C and Figure 4C), which showed substantial radioactivity reduction in rat brain sections (vide infra). These results indicated that both ligands [11C]14a and [11C]14b exhibited high-level specific binding towards mGlu2 in vitro. These ARG results were further analyzed by the comparison with bound signals from brain regions of interest to that of the pons, in which the lowest uptake was observed. As shown in Figures 3E & 4E, higher contrast ratios were observed for [11C]14b, for example, 2.79 in the striatum, 2.61 in the cerebral cortex, 2.00 in the hippocampus and 1.78 in the cerebellum under baseline conditions. Under blocking conditions (preincubated with unlabeled compound QCA, 14b or 14a at 10 µM, respectively), these ratios were decreased by 45-69% for [11C]14b while only 17-54% for [11C]14a. These results demonstrated that [11C]14b had improved specific binding to mGlu2 compared with [11C]14a. Attributed to their excellent binding specificity, both radioligands were advanced to subsequent PET evaluation to study their potential for imaging of mGlu2 in vivo, although [11C]14b exhibited superior in vitro performance.
In vitro autoradiography of [11C]14a in rat brain sections. (A) Brain sections were treated with [11C]14a (1.25 μCi/mL); (B) Brain sections were pre-treated with 14a (10 μM), followed by [11C]14a (1.25 μCi/mL); (C) Brain sections were pre-treated with QCA (10 μM), followed by [11C]14a (1.25 μCi/mL); (D) Quantitative analysis of baseline and blocking experiments. The value is expressed as PSL per mm2; (E) Ratios of brain regions to the pons. Str = striatum; Ctx = cerebral cortex; HC = hippocampus; CB = cerebellum. Six serial brain sections were used for each conditions. Data are presented as mean ± SEM (n = 6) and analyzed by one-way ANOVA. Asterisks indicate statistical significance. *p < 0.05, ***p ≤ 0.001, and ****p ≤ 0.0001.
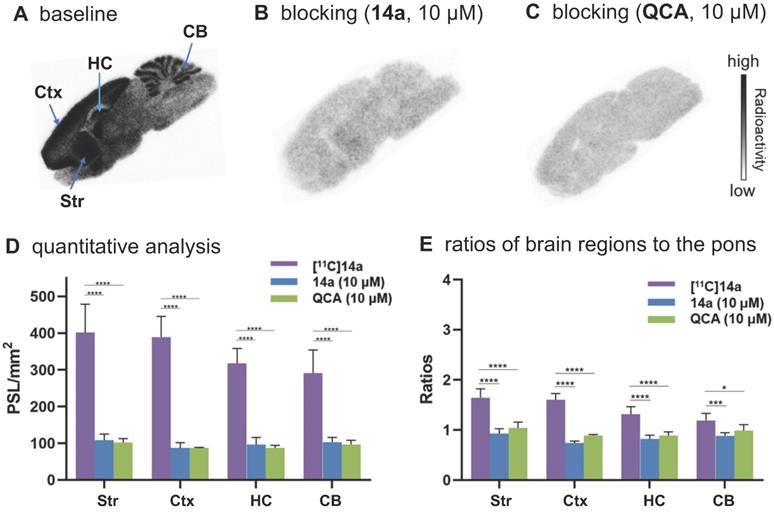
In vitro autoradiography of [11C]14b in rat brain sections. (A) Brain sections were treated with [11C]21b (1.25 μCi/mL); (B) Brain sections were pre-treated with 14b (10 μM), followed by [11C]21b (1.25 μCi/mL); (C) Brain sections were pre-treated with QCA (10 μM), followed by [11C]14b (1.25 μCi/mL); (D) Quantitative analysis of baseline and blocking experiments. The value is expressed as PSL per mm2; (E) Ratios of brain regions to the pons. Str = striatum; Ctx = cerebral cortex; HC = hippocampus; CB = cerebellum. Six serial brain sections were used for each conditions. Data are presented as mean ± SEM (n = 6) and analyzed by one-way ANOVA. Asterisks indicate statistical significance. *p < 0.05, ***p ≤ 0.001, and ****p ≤ 0.0001.
PET Imaging Studies in Rat Brain
Dynamic PET imaging studies were performed in the brain of Sprague-Dawley (SD) rats to assess the in vivo binding specificity and washout kinetics of our promising radioligands [11C]14a and [11C]14b. As shown in Figure S2A (supporting information), time-activity curves (TACs) of [11C]14a exhibited good brain permeability. Specifically, the radioactivity in all brain regions of interest increased rapidly, peaked at 2.5 min (for example, 1.7 SUV in the striatum), and then gradually washed out over 60 min. However, pretreatment with non-radioactive compound 14a (1 mg/kg) displayed marginal differences in major brain regions between baseline and blocking conditions (Figure S2B in supporting information), indicating low-to-modest in vivo specificity of [11C]14a.
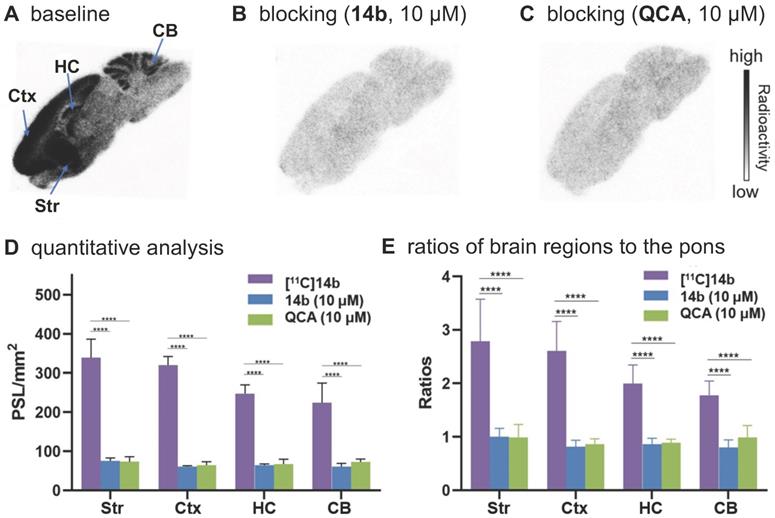
For PET imaging of [11C]14b in rat brains, representative PET images (summed coronal view at 0-10 min, 10-30 min and 30-60 min intervals; see Figure S12 for sagittal and horizontal views in the supporting information) in different brain regions, and the corresponding TACs are shown in Figure 5. The radioactivity in all brain regions reached a maximum level within 3 min, displayed heterogeneous distribution, which is consistent with mGlu2 distribution, and washed out gradually over 60 min (Figure 5A & 5D). Blocking studies with pretreatment of non-radioactive NAMs 14b and 14a successfully abolished heterogenous regional brain uptake, leading to a uniform distribution and reduced brain uptake (Figure 5B, 5C, 5E & 5F). Using the highest mGlu2 region, the striatum, as an example, we compared the regional TACs under baseline and blocking conditions in Figure 5G and found obvious reduction of the bound signals under blocking. These results indicated [11C]14b showed improved in vivo binding specificity to mGlu2 compared to [11C]14a.
Representative PET/MRI fused coronal images (summed at 0-10 min, 10-30 min and 30-60 min) and time-activity curves of [11C]14b under baseline and blocking conditions in SD rat brain. #Blocking conditions: 14b (1 mg/kg), 30 min i.v. before radioligand injection; †Blocking conditions: 14a (3 mg/kg), 30 min i.v. before radioligand injection. Data are presented as mean ± SEM (n = 3).
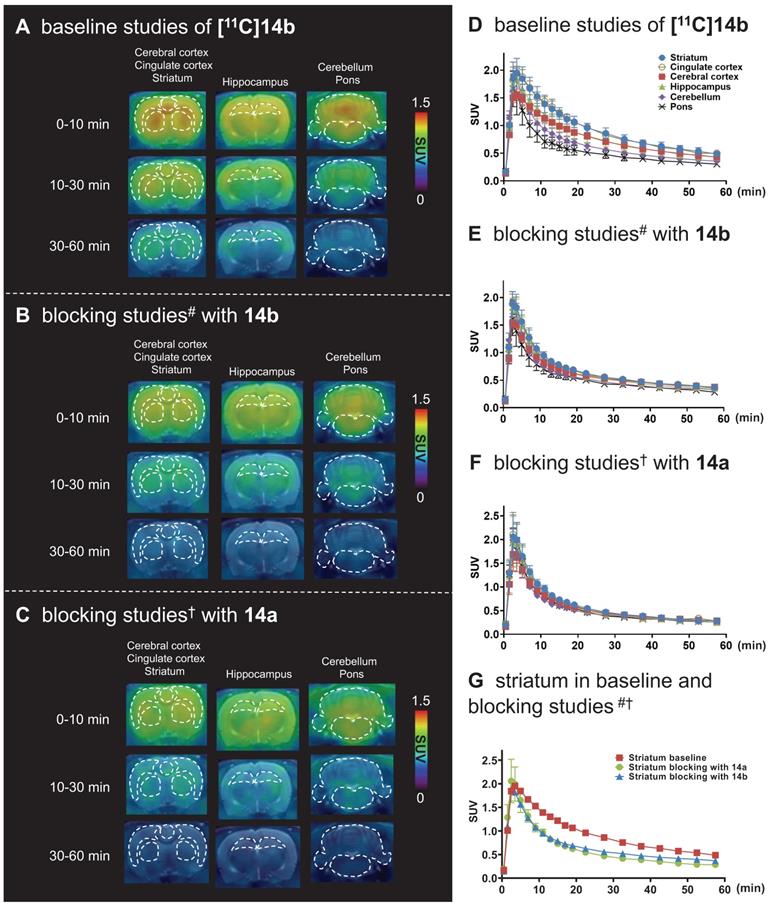
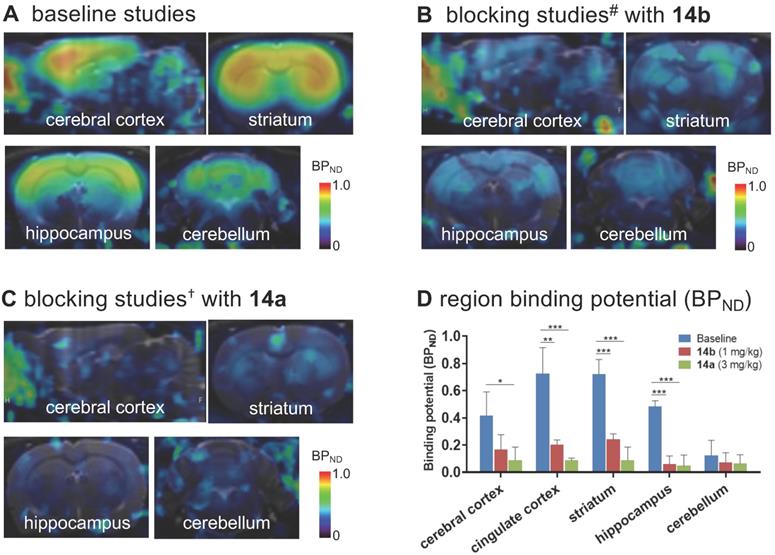
To further quantitatively estimate the specific binding of [11C]14b, the non-displaceable binding potential (BPND) values were analyzed by the simplified reference tissue model (SRTM) [53, 54], with the pons as the pseudo reference region (the lowest and consistent brain uptake between baseline and blocking conditions). As shown in Figure 6, the bound signal of [11C]14b in rat brain was found in a decreasing order of the striatum and cingulate cortex (0.72), followed by the cerebral cortex (0.42) and hippocampus (0.48), and the lowest BPND was identified in the cerebellum (0.12) under baseline conditions. The distribution pattern was consistent with the expression of mGlu2 in rat brain [13-17] as well as the in vitro ARG results in Figure 4. Under blocking conditions, the BPND values were decreased substantially by pretreatment with 14b (41-88% reduction, 1 mg/kg, Figure 6B and 6D) and 14a (48-90% reduction, 3 mg/kg, Figure 6C and 6D). The signal heterogeneity in the parametric brain mapping was also abolished under these conditions. In all, parametric PET images with the BPND scale could clearly visualize the uptake differences in detailed brain regions, and confirm the specific binding of [11C]14b in vivo between baseline and blocking conditions by PET.
Parametric mapping and binding potentials of [11C]14b in rat brains. #Blocking conditions: 14b (1 mg/kg), 30 min i.v. before radioligand injection; †blocking conditions: 14a (3 mg/kg), 30 min i.v. before radioligand injection. Data are presented as mean ± SEM (n = 3) and analyzed by one-way ANOVA. Asterisks indicate statistical significance. *p < 0.05, **p ≤ 0.01, and ***p ≤ 0.001.
Whole Body Biodistribution Studies and Radiometabolite Analysis of [11C]14b
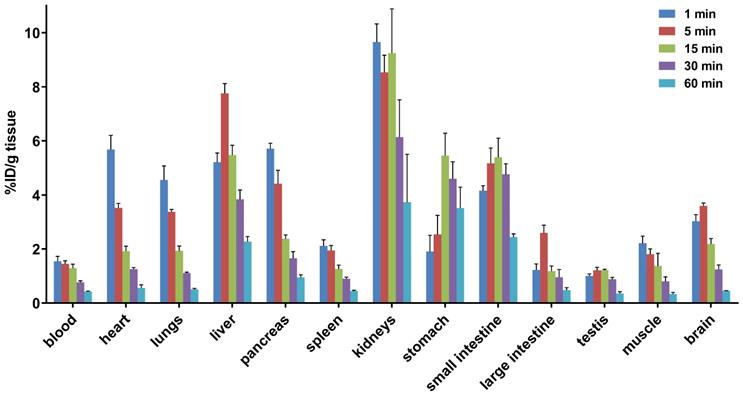
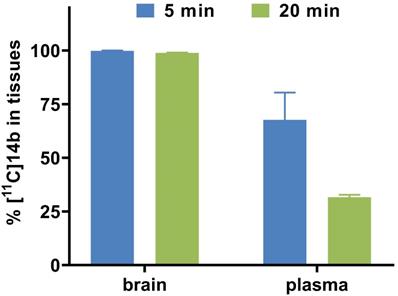
To study the pharmacokinetic properties of [11C]14b, the uptake, distribution, and clearance were studied in mice at five time points (1, 5, 15, 30, and 60 min) post tracer injection. The results were expressed as the percentage of the injected dose per gram of wet tissue (%ID/g) in Figure 7. The radioactivity of [11C]14b in blood was washed out rapidly, with the ratio of %ID/g(1min/60min) > 3.5. High radioactivity levels (> 4 %ID/g) were observed in several organs including the heart, lungs, liver, pancreas, kidneys and small intestine within the initial 1 min. After that, the signals in most organs decreased rapidly, while the radioactivity in the liver, large intestine and brain increased until 5 min and then washed out gradually. The signal in the stomach and small intestine reached a plateau after 15 min. The particular high uptake in the small intestine, kidney, and liver was probably due to the hepatobiliary and urinary excretion together with possible renal and intestinal reuptake pathways.[55] Notably, high brain uptake (ca. 3.6 %ID/g) was detected after 5 min post injection, which was consistent with in vivo imaging data obtained from the PET studies. Furthermore, to investigate the in vivo stability of [11C]14b, radiometabolites in the plasma and brain in SD rats were evaluated at two time points (5 and 20 min) post injection. The percentages of parent [11C]14b and corresponding radiometabolites which were determined by radio-HPLC are shown in Figure 8 (see details in Figure S6-S8 and Table S2 in the supporting information). The fraction corresponding to unchanged [11C]14b in plasma was decreased from 77% at 5 min to 21% at 20 min. On the other hand, [11C]14b showed excellent stability in the brain without any obvious 11C-labeled metabolite detected (unchanged fraction >99% at 20 min), which suggested the radiometabolites in the blood did not cross the BBB. It should be noted that, based on the radiometabolite analysis of [11C]14a, which has reasonable in vivo stability in the brain and plasma (see details in Figure S3-S5 and Table S1 in the supporting information), we have ruled out the possibility of high nonspecific binding of [11C]14a was attributed to in vivo metabolism. We postulated that the difference between IC50 values (39 nM of 14a versus 24 nM of 14b) and target selectivity may, in part, explain the increased nonspecific binding of [11C]14a in vitro (as seen in autoradiography) and in vivo (by PET study). Further in-depth pharmacological experiment is necessary to validate this hypothesis. As a result, the high in vivo stability of [11C]14b in the brain could facilitate further quantification of mGlu2 expression and target engagement studies by PET. Subsequent isotopologue-labeling of ligand 14b using 18F, saturation binding assay to determine Bmax and Kd, and further validation including PET imaging in mGlu2 knockout mice and non-human primate are underway to evaluate the suitability of this new chemotype for potential clinical translation.
Whole-body ex vivo biodistribution studies in mice at five different time points (1, 5, 15, 30 and 60 min) post injection of [11C]14b. Data are expressed as %ID/g (mean ± SD, n = 3). %ID/g = injected dose per gram of wet tissue.
Radiometabolite analysis of [11C]14b in rats (average two runs)
Conclusion
We have prepared a new array of tetrahydro-1,7-naphthyridine carboxamide-based mGlu2 NAMs with amenability for radiolabeling. The preliminary pharmacological and physicochemical evaluations were carried out to identify two most promising modulators 14a and 14b, the corresponding 11C-isotopologues of which were produced in good radiochemical yields and high radiochemical purities. The subsequent autoradiography, PET imaging, whole body distribution and radiometabolism studies demonstrated that [11C]14b (which we named [11C]MG2-1904) exhibited sufficient brain permeability, high specific binding, and suitable in vivo metabolic stability, which could be used for further quantitative measurement under different physiological and pathological conditions. Further validation including PET imaging in higher species are underway to evaluate the suitability of this new chemotype for potential clinical translation.
Materials and Methods
General Considerations. All the starting materials used in the syntheses were purchased from commercial vendors and used without further purification. Thin-layer chromatography (TLC) was conducted with 0.25 mm silica gel plates (60F254) and visualized by exposure to UV light (254 nm) or stained with potassium permanganate. Flash column chromatography was performed using silica gel (particle size 0.040-0.063 mm). 1H-Nuclear magnetic resonance (NMR) spectra were obtained on a 300 & 400 MHz on Bruker spectrometers. 13C NMR spectra were obtained at 75 & 100 MHz. Chemical shifts (δ) are reported in ppm and coupling constants are reported in Hertz. The multiplicities are abbreviated as follows: s = singlet, d = doublet, t = triplet, q = quartet, quint = quintet, sext = sextet, sept = setpet, m = multiplet, br = broad signal, dd = doublet of doublets. For all the HRMS measurements, the ionization method is ESI and the mass analyzer type is TOF on an AB SCIEX 500R Mass Spectrometer Systems. Lipophilicity (cLogD) and topological polar surface area (tPSA) were calculated by ChemDraw 16.0 software (PerkinElmer, USA). Carbon-11 (11C) was produced by 14N(p, α)11C nuclear reactions using a GE PETtrace cyclotron (16.5 MeV) or a Sumitomo CYPRIS HM-18 cyclotron. The animal experiments were approved by the Institutional Animal Care and Use Committee of Massachusetts General Hospital or the Animal Ethics Committee at the National Institute of Radiological Sciences. DdY mice (male; 7 weeks, 34-36 g) and SD rats (male; 7 weeks; 210-230 g) were kept on a 12 h light/12 h dark cycle and were allowed food and water ad libitum.
Medicinal Chemistry
Chemical syntheses of mGlu2 NAMs 14
Ethyl 4-chloro-1,7-naphthyridine-2-carboxylate (10). To 3-aminoisonicotinic acid (8) (5.24 g, 22.2 mmol) in a round-bottom flask was added ethyl pyruvate (9) (6 mL, 54.0 mmol, 2.4 equiv) and stirred for 10 mins before the addition of POCl3 (90 mL). The mixture was stirred at 100 oC for 1 h, then quenched by iced water (100 mL) and 1 N NaOH (300 mL) before extracted with dichloromethane (200 mL, three times). The combined organic layers were washed with saturated aqueous sodium chloride, dried over MgSO4 and concentrated in vacuo. The residue was purified by flash chromatography on silica gel (hexanes to ethyl acetate gradient column) to yield the compound (10) as brown solid (3.35 g, 64%). Rf = 0.3 (Hexanes/EtOAc = 10:1). 1H NMR (400 MHz, CDCl3) 9.75 (d, J = 0.9 Hz, 1H), 8.84 (d, J = 5.8 Hz, 1H), 8.47 (s, 1H), 8.08 (dd, J = 5.8, 1.0 Hz, 1H), 4.61 (q, J = 7.1 Hz, 2H), 1.53 (t, J = 7.1 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 163.8, 155.5, 149.7, 146.3, 143.3, 143.0, 130.8, 124.8, 116.1, 63.0, 14.3.
Ethyl 4-(2-fluoro-4-methoxyphenyl)-1,7-naphthyridine-2-carboxylate (11a). To a solution of ethyl 4-chloro-1,7-naphthyridine-2-carboxylate (10) (70.8 mg, 0.300 mmol), 2-fluoro-4-methoxyphenylboronic acid (51.0 mg, 0.300 mmol) and K2CO3 (82.8 mg, 0.600 mmol) in 1,4-dioxane/water (v/v, 10/1, 1.8 mL) was added Pd(PPh3)4 (34.6 mg, 0.03 mmol) under Ar. The mixture was stirred at 100 oC overnight, then quenched with water (3 mL) and extracted with ethyl acetate (5 mL, three times). The combined organic layers were washed with saturated aqueous sodium chloride, dried over MgSO4 and concentrated in vacuo. The residue was purified by flash chromatography on silica gel (hexanes to ethyl acetate gradient column) to yield the compound 11a as white solid (72 mg, 74%). Rf = 0.3 (Hexanes/EtOAc = 10:1). 1H NMR (300 MHz, CDCl3) δ 9.76 (s, 1H), 8.66 (d, J = 6.0 Hz, 1H), 8.35 (s, 1H), 7.67 (dd, J = 6.0, 2.7 Hz, 1H), 7.35 (t, J = 8.5 Hz, 1H), 7.00-6.77 (m, 2H), 4.59 (q, J = 7.1 Hz, 2H), 3.91 (s, 3H), 1.51 (t, J = 7.1 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 164.8 , 164.5 (d, J = 249.1 Hz), 157.9 (d, J = 10.0 Hz), 155.6 , 149.4 , 145.8 , 145.0 , 142.7 , 132.0 (d, J = 10.2 Hz), 128.5 (d, J = 12.1 Hz), 125.7 , 118.3 , 107.7 (d, J = 21.7 Hz), 99.7 (d, J = 26.0 Hz), 62.6 , 55.8 , 14.4.
Ethyl 4-(2,4-difluorophenyl)-1,7-naphthyridine-2-carboxylate (11b). Compound 11b was prepared in 80% yield as a white solid using a similar method that described for 11a. 1H NMR (300 MHz, CDCl3) δ 9.79 (d, J = 0.9 Hz, 1H), 8.69 (d, J = 5.9 Hz, 1H), 8.35 (s, 1H), 7.58 (d, J = 5.9 Hz, 1H), 7.44 (td, J = 8.3, 6.2 Hz, 1H), 7.17 - 7.03 (m, 2H), 4.60 (q, J = 7.1 Hz, 2H), 1.52 (t, J = 7.1 Hz, 3H). 13C NMR (75 MHz, CDCl3) δ 164.4, 163.7 (dd, J = 252.7, 11.7 Hz), 159.7 (dd, J = 252.2, 12.1 Hz), 155.7, 149.4, 145.6, 142.6, 142.4, 132.4 (dd, J = 9.8, 4.4 Hz), 131.6 (d, J = 10.8 Hz), 130.8, 128.5 (d, J = 12.7 Hz), 125.7, 119.5 (dd, J = 15.7, 4.0 Hz), 117.5 (d, J = 2.0 Hz), 112.3 (dd, J = 21.5, 3.8 Hz), 104.8 (t, J = 25.6 Hz), 62.6, 14.3.
Ethyl 4-(2-fluoro-4-methyphenyl)-1,7-naphthyridine-2-carboxylate (11c). Compound 11c was prepared in 72% yield as a white solid using a manner method that described for 11a. 1H NMR (300 MHz, CDCl3) δ 9.72 (s, 1H), 8.61 (d, J = 5.8 Hz, 1H), 8.29 (d, J = 0.6 Hz, 1H), 7.55 (ddd, J = 5.9, 2.6, 1.0 Hz, 1H), 7.27 (t, J = 7.7 Hz, 1H), 7.17 - 7.01 (m, 2H), 4.54 (q, J = 7.1 Hz, 2H), 2.43 (s, 3H), 1.45 (t, J = 7.1 Hz, 3H). 13C NMR (75 MHz, CDCl3) δ 164.6, 159.2 (d, J = 249.0 Hz), 155.6, 149.3, 145.3, 143.6, 142.7, 142.5 (d, J = 8.0 Hz), 134.8 (d, J = 10.4 Hz), 131.1, 131.0, 131.0, 125.6, 125.5, 125.5, 120.2 (d, J = 15.5 Hz), 117.9, 116.9, 116.6, 62.6, 21.3 (d, J = 1.7 Hz), 14.3.
Ethyl 4-(4-fluoro-2-methoxyphenyl)-1,7-naphthyridine-2-carboxylate (11d). Compound 11d was prepared in 80% yield as a white solid using a similar method that described for 11a. 1H NMR (300 MHz, CDCl3) δ 9.75 (s, 1H), 8.61 (d, J = 6.0 Hz, 1H), 8.30 (s, 1H), 7.54 (d, J = 5.9 Hz, 1H), 7.26 (s, 3H), 7.00 - 6.66 (m, 2H), 4.59 (q, J = 7.1 Hz, 2H), 3.72 (s, 3H), 1.50 (t, J = 7.1 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 164.8, 164.5 (d, J = 249.1 Hz), 157.9 (d, J = 10.1 Hz), 155.6, 149.4, 145.8, 145.0, 142.7, 138.4 (d, J = 10.2 Hz), 132.0 (d, J = 10.2 Hz), 131.6, 128.5 (d, J = 12.1 Hz), 125.7, 120.6, 118.3, 107.7 (d, J = 21.7 Hz), 99.7 (d, J = 26.0 Hz), 62.6, 55.8, 14.4.
Ethyl 4-(2-chloro-4-fluorophenyl)-1,7-naphthyridine-2-carboxylate (11g). Compound 11g was prepared in 75% yield as a white solid using a similar method that described for 11a. 1H NMR (400 MHz, CDCl3) δ 9.8 (s, 1H), 8.7 (d, J = 5.8 Hz, 1H), 8.3 (s, 1H), 7.4 - 7.3 (m, 3H), 7.2 (td, J = 8.2, 2.5 Hz, 1H), 4.6 (q, J = 7.5 Hz, 2H), 1.5 (t, J = 7.1 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 164.6, 163.0 (d, J = 253.1 Hz), 155.8, 149.4, 145.7, 145.6, 142.6, 134.2 (d, J = 10.5 Hz), 132.4 (d, J = 9.0 Hz), 131.0, 130.8 (d, J = 3.7 Hz), 125.6, 117.7 (d, J = 24.8 Hz), 117.6, 114.8 (d, J = 21.4 Hz), 62.8, 14.4.
Ethyl 4-(2-fluoro-4-methoxyphenyl)-4a,5,6,7,8,8a-hexahydro-1,7-naphthyridine-2-carboxylate (12a). To the solution of 11a (5.0 mmol) in AcOH (10 mL) was added NaBH3CN (0.94 g, 15.0 mmol, 3.0 equiv). The mixture was stirred for 5 mins at room temperature, then quenched by water (30 mL) and extracted with dichloromethane (200 mL, 3 times). The combined organic layers were washed with saturated aqueous sodium chloride, dried over MgSO4 and concentrated in vacuo. The residue 12a was used without further purification.
Compound 12b-g were prepared in a manner similar to that described for 12a and used without further purification.
Ethyl 4-(2-fluoro-4-methoxyphenyl)-7-((2-methoxypyridin-4-yl)methyl)-5,6,7,8-tetrahydro-1,7-naphthyridine-2-carboxylate (13a). To the residue 12a solution in MeCN (5 mL) was added K2CO3 (1.4 g, 10.0 mmol) before the addition of 4-(chloromethyl)-2-methoxypyridine (0.79 g, 5.0 mmol) in MeCN (5 mL). The mixture was stirred at room temperature for 4 h, then quenched with H2O and extracted with ethyl acetate (5 mL, three times). The combined organic layers were washed with saturated aqueous sodium chloride, dried over MgSO4 and concentrated in vacuo. The residue was purified by flash chromatography on silica gel (hexanes to ethyl acetate gradient column) to yield compound 13a as white solid (50% for two steps, 67.7 mg). Rf = 0.2 (Hexanes/EtOAc = 1:1). 1H NMR (300 MHz, CDCl3) δ 8.00 (d, J = 5.2 Hz, 1H), 7.76 (s, 1H), 7.06 (t, J = 8.5 Hz, 1H), 6.83 (d, J = 5.2 Hz, 1H), 6.76-6.58 (m, 3H), 4.36 (q, J = 7.1 Hz, 2H), 3.83 (s, 3H), 3.80 (s, 2H), 3.75 (s, 3H), 3.58 (s, 2H), 2.76-2.56 (m, 4H), 1.30 (t, J = 7.1 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 165.3, 164.6, 161.4 (d, J = 10.9 Hz), 159.8 (d, J = 247.5 Hz), 155.6, 149.9, 146.8, 145.1 (d, J = 53.5 Hz), 133.1, 132.1 (d, J = 9.9 Hz), 131.1 (d, J = 5.3 Hz), 128.5 (d, J = 12.1 Hz), 124.7, 117.3, 110.6, 110.4 (d, J = 2.9 Hz), 101.9 (d, J = 25.7 Hz), 61.8, 61.2, 58.9, 55.7, 53.4, 49.9, 27.2, 14.4.
Ethyl 4-(2,4-difluorophenyl)-7-((2-methoxypyridin-4-yl)methyl)-5,6,7,8-tetrahydro-1,7-naphthyridine-2-carboxylate (13b). Compound 13b was prepared in 45% yield as a white solid using a similar method that described for 13a. 1H NMR (300 MHz, CDCl3) δ 8.09 (dd, J = 5.3, 0.7 Hz, 1H), 7.82 (s, 1H), 7.21 (td, J = 8.3, 6.3 Hz, 1H), 7.08 - 6.87 (m, 3H), 6.76 (dt, J = 1.4, 0.7 Hz, 1H), 4.44 (qd, J = 7.1, 0.6 Hz, 2H), 3.91 (d, J = 0.7 Hz, 3H), 3.89 (s, 2H), 3.68 (s, 2H), 2.73 (s, 4H), 1.38 (td, J = 7.1, 0.6 Hz, 3H). 13C NMR (75 MHz, CDCl3) δ 165.1, 164.6, 155.7, 146.9, 145.5, 144.0, 132.8, 131.6, 131.5 (dd, J = 9.5, 5.0 Hz), 124.9, 124.5, 121.4, 117.3, 111.9 (d, J = 21.4 Hz), 110.7, 104.5 (t, J = 25.5 Hz), 61.9, 61.1, 58.7, 53.4, 49.8, 27.0, 14.3.
Ethyl 4-(2-fluoro-4-methyphenyl)-7-((2-methoxypyridin-4-yl)methyl)-5,6,7,8-tetrahydro-1,7-naphthyridine-2-carboxylate (13c). Compound 13c was prepared in 52% yield as a white solid using a similar method that described for 13a. 1H NMR (300 MHz, CDCl3) δ 8.09 (dd, J = 5.3, 0.7 Hz, 1H), 7.83 (s, 1H), 7.16-6.89 (m, 5H), 6.76 (s, 1H), 4.44 (q, J = 7.1 Hz, 2H), 3.92 (s, 3H), 3.89 (s, 2H), 3.68 (s, 2H), 2.74 (dd, J = 11.7, 4.9 Hz, 4H), 2.40 (s, 3H), 1.38 (t, J = 7.1 Hz, 3H). 13C NMR (75 MHz, CDCl3) δ 165.1, 164.6, 158.9 (d, J = 247.4 Hz), 147.6, 146.8, 145.3, 145.0, 141.4 (d, J = 7.9 Hz), 132.8, 130.2 (d, J = 3.8 Hz), 125.1 (d, J = 3.1 Hz), 124.5, 122.1, 117.2, 116.4 (d, J = 21.6 Hz), 110.6, 109.3, 61.8, 61.1, 58.6, 53.4, 49.8, 26.9, 21.1 (d, J = 1.6 Hz), 14.3.
4-((4-fluoro-2-methoxyphenyl))-7-((2-methoxypyridin-4-yl)methyl)-5,6,7,8-tetrahydro-1,7-naphthyridine-2-carboxylate (13d). Compound 13d was prepared in 48% yield as a white solid using a similar method that described for 13a. 1H NMR (400 MHz, CDCl3) δ 8.1 (d, J = 5.3 Hz, 1H), 7.8 (s, 1H), 7.1 (dd, J = 8.3, 6.6 Hz, 1H), 6.9 (dd, J = 5.2, 1.4 Hz, 1H), 6.8 - 6.7 (m, 3H), 4.4 (q, J = 7.1 Hz, 2H), 3.9 (s, 3H), 3.9 (s, 2H), 3.8 (s, 3H), 3.7 (s, 2H), 2.7 (s, 4H), 1.4 (t, J = 7.1 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 165.4, 164.6, 163.8 (d, J = 247.5 Hz), 157.4 (d, J = 9.9 Hz), 155.2, 149.9, 146.9, 146.8, 145.2, 133.2, 130.9 (d, J = 10.1 Hz), 124.8, 122.7 (d, J = 3.4 Hz), 117.3, 110.7, 107.2 (d, J = 21.4 Hz), 99.3 (d, J = 25.9 Hz), 61.8, 61.3, 58.8, 55.7, 53.4, 50.0, 26.9, 14.4.
Ethyl 4-(2-fluoro-4-methyphenyl)-7-((2-methoxypyrimidin-5-yl)methyl)-5,6,7,8-tetrahydro-1,7-naphthyridine-2-carboxylate (13e). Compound 13e was prepared in 45% yield as a white solid using a similar method that described for 13a. 1H NMR (300 MHz, CDCl3) δ 8.50 (s, 2H), 7.84 (s, 1H), 7.16 - 6.91 (m, 3H), 4.44 (q, J = 7.1 Hz, 2H), 4.01 (s, 3H), 3.88 (s, 2H), 3.66 (s, 2H), 2.40 (s, 3H), 1.39 (t, J = 7.1 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 164.8, 158.9 (d, J = 247.4 Hz), 152.6, 146.2, 145.7, 142.0, 141.9, 132.1, 130.2 (d, J = 3.6 Hz), 125.4 (d, J = 3.1 Hz), 125.0, 121.8, 121.6, 116.7, 116.5, 62.1, 50.1, 42.7, 36.1, 29.7, 26.8, 21.2, 14.3.
Ethyl 4-(2-fluoro-4-methoxyphenyl)-7-((2-methoxypyrimidin-5-yl)methyl)-5,6,7,8-tetrahydro-1,7-naphthyridine-2-carboxylate (13f). Compound 13f was prepared in 42% yield as a white solid using a similar method that described for 13a. 1H NMR (300 MHz, CDCl3) δ 8.49 (s, 2H), 7.76 (s, 1H), 7.04 (dd, J = 8.2, 6.6 Hz, 1H), 6.79 - 6.64 (m, 2H), 4.43 (q, J = 7.2 Hz, 2H), 3.99 (s, 3H), 3.85 (s, 2H), 3.74 (s, 3H), 3.64 (s, 2H), 1.37 (t, J = 7.1 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 165.3 (d, J = 1.9 Hz), 161.5 (d, J = 10.9 Hz), 159.9, 159.8 (d, J = 247.6 Hz), 155.3, 145.1 (d, J = 31.7 Hz), 133.1, 132.1 (d, J = 10.1 Hz), 131.0 (d, J = 5.3 Hz), 128.5 (d, J = 12.2 Hz), 124.8, 124.1, 117.3 (d, J = 16.8 Hz), 110.4 (d, J = 2.9 Hz), 101.9 (d, J = 25.7 Hz), 61.9, 58.6, 56.6, 55.7, 55.0, 49.6, 27.1, 14.3.
4-(2-chloro-4-fluoropheny)-7-((2-methoxypyrimidin-5-yl)methyl)-5,6,7,8-tetrahydro-1,7-naphthyridine-2-carboxylate (13g). Compound 13g was prepared in 42% yield as a white solid using a similar method that described for 13a. 1H NMR (400 MHz, CDCl3) δ 8.09 (d, J = 5.2 Hz, 1H), 7.77 (s, 1H), 7.30 - 7.20 (m, 1H), 7.20 - 7.12 (m, 1H), 7.09 (td, J = 8.2, 2.5 Hz, 1H), 6.90 (dd, J = 5.3, 1.4 Hz, 1H), 6.76 (s, 1H), 4.45 (qd, J = 7.1, 2.9 Hz, 2H), 3.92 (s, 3H), 3.91 - 3.86 (m, 2H), 3.67 (s, 2H), 2.80 - 2.54 (m, 4H), 1.40 (t, J = 7.1 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 165.09, 164.58, 162.42 (d, J = 251.6 Hz), 155.84, 149.75, 147.11, 146.83, 145.49, 133.33 (d, J = 10.3 Hz), 132.82 (d, J = 3.8 Hz), 132.65, 131.09 (d, J = 8.8 Hz), 124.04, 117.26 (d, J = 24.8 Hz), 117.23, 114.52 (d, J = 21.3 Hz), 110.63, 61.92, 61.14, 58.72, 53.38, 49.75, 26.98, 14.33.
4-(2-fluoro-4-methoxyphenyl)-7-((2-methoxypyridin-4-yl)methyl)-5,6,7,8-tetrahydro-1,7-naphthyridine-2-carboxamide (14a). To the solution of compound 13a (67.7 mg, 0.15 mmol) in MeOH (5 mL) was added ammonia in MeOH (7 N, 5 mL). The mixture was stirred for 4 h before the solvent was removed in vacuo. The residue was purified by flash chromatography on silica gel (hexanes to ethyl acetate gradient column) to yield compound 14a as white solid (72%, 45.6 mg). Rf = 0.2 (Hexanes/EtOAc = 1:2). Melting point 151-153 ºC. 1H NMR (300 MHz, CDCl3) δ 8.08 (d, J = 5.2 Hz, 1H), 7.89 (s, 1H), 7.78 (d, J = 4.5 Hz, 1H), 7.10 (t, J = 8.5 Hz, 1H), 6.90 (d, J = 5.2 Hz, 1H), 6.80 - 6.59 (m, 3H), 6.33 (s, 1H), 3.89 (s, 3H), 3.79 (s, 3H), 3.74 (s, 2H), 3.63 (s, 2H), 2.70 (dd, J = 14.4, 4.7 Hz, 4H). 13C NMR (75 MHz, CDCl3) δ 166.2 , 164.3 , 161.5 (d, J = 11.2 Hz), 159.6 (d, J = 244.8 Hz), 154.4, 131.9, 131.8 (d, J = 5.2 Hz), 121.6, 117.7, 117.3 (d, J = 16.4 Hz), 111.3 (d, J = 2.8 Hz), 110.3, 102.2 (d, J = 25.7 Hz), 60.4, 58.2, 56.2, 53.5, 50.1, 27.0. HRMS (ESI): calculated for C23H24FN4O3 [M + H], 423.1832; found, 423.1817.
4-(2,4-difluorophenyl)-7-((2-methoxypyridin-4-yl)methyl)-5,6,7,8-tetrahydro-1,7-naphthyridine-2- carboxamide (14b). Compound 14b was prepared in 80% yield as a white solid using a similar method that described for 14a. Melting point 128-130 ºC. 1H NMR (300 MHz, CDCl3) δ 8.11 (d, J = 5.2 Hz, 1H), 7.90 (s, 1H), 7.78 (s, 1H), 7.36 - 7.15 (m, 1H), 7.14 - 6.85 (m, 3H), 6.78 (s, 1H), 6.01 (d, J = 4.6 Hz, 1H), 3.92 (s, 3H), 3.79 (s, 2H), 3.68 (s, 2H), 2.72 (s, 4H). 13C NMR (75 MHz, CDCl3) δ 166.7, 164.6, 163.2 (dd, J = 249.9, 10.0 Hz), 159.3 (dd, J = 250.1, 11.9 Hz), 154.2, 145.0, 146.9, 146.6, 144.4, 132.4, 131.5 (dd, J = 9.5, 5.0 Hz), 121.8, 121.7 (dd, J = 16.8, 4.0 Hz), 117.2, 111.9 (dd, J = 21.2, 3.6 Hz), 110.5, 104.4 (t, J = 25.5 Hz), 61.1, 58.5, 53.4, 49.9, 26.9 (d, J = 3.0 Hz). HRMS (ESI): calculated for C22H21F2N4O2 [M + H], 411.1633; found, 411.1616
4-(2-fluoro-4-methyphenyl)-7-((2-methoxypyridin-4-yl)methyl)-5,6,7,8-tetrahydro-1,7-naphthyridine-2-carboxamide (14c). Compound 14c was prepared in 72% yield as a white solid using a similar method that described for 14a. 1H NMR (300 MHz, CDCl3) δ 8.12 (s, 1H), 7.93 (s, 1H), 7.78 (s, 1H), 7.20 - 6.86 (m, 4H), 6.79 (s, 1H), 5.89 (s, 1H), 3.93 (s, 3H), 3.79 (s, 2H), 3.69 (s, 2H), 2.74 (s, 4H), 2.39 (s, 3H). 13C NMR (75 MHz, CDCl3) δ 166.2 , 164.4 , 158.7 (d, J = 244.7 Hz), 154.4, 150.9, 147.9, 147.2, 144.8, 141.7 (d, J = 8.1 Hz), 131.7, 130.9 (d, J = 3.8 Hz), 126.0 (d, J = 2.9 Hz), 122.4 (d, J = 16.1 Hz), 121.4, 117.7, 116.6 (d, J = 21.7 Hz), 110.3, 60.4, 58.2, 53.5, 50.0, 26.9, 21.1 (d, J = 1.6 Hz). HRMS (ESI): calculated for C23H23FN4NaO3 [M + H], 407.1883; found, 407.1892.
4-(4-fluoro-2-methoxyphenyl)-7-((2-methoxypyridin-4-yl)methyl)-5,6,7,8-tetrahydro-1,7-naphthyridine-2- carboxamide (14d). Compound 14d was prepared in 75% yield as a white solid using a similar method that described for 14a. 1H NMR (300 MHz, CDCl3) δ 8.13 (d, J = 5.2 Hz, 1H), 7.87 (s, 1H), 7.77 (d, J = 4.1 Hz, 1H), 7.05 (dd, J = 8.2, 6.7 Hz, 1H), 6.97 (d, J = 5.0 Hz, 1H), 6.80 (s, 1H), 6.77 - 6.63 (m, 2H), 5.67 (d, J = 4.7 Hz, 1H), 3.94 (s, 3H), 3.81 (s, 2H), 3.75 (s, 3H), 3.72 (s, 2H), 2.74 (s, 4H). 13C NMR (75 MHz, CDCl3) δ 166.3, 164.3, 162.0, 157.7, 157.6, 152.4 (d, J = 223.4 Hz), 147.7, 147.2 (d, J = 7.0 Hz), 132.1, 131.4, 131.3, 123.0 (d, J = 2.9 Hz), 121.6, 117.8, 110.3, 107.4 (d, J = 21.2 Hz), 100.4 (d, J = 26.1 Hz), 60.5, 58.2, 56.4, 53.5, 50.1, 26.7. HRMS (ESI): calculated for C23H23FN4NaO3 [M + Na], 445.1652; found, 445.1641.
4-(2-fluoro-4-methyphenyl)-7-((2-methoxypyrimidin-5-yl)methyl)-5,6,7,8-tetrahydro-1,7-naphthyridine-2-carboxamide (14e). Compound 14e was prepared in 73% yield as a white solid using a similar method that described for 14a. 1H NMR (300 MHz, CDCl3) δ 8.53 (s, 2H), 7.93 (s, 1H), 7.77 (s, 1H), 7.15 - 6.93 (m, 3H), 5.56 (s, 1H), 4.02 (s, 3H), 3.78 (s, 2H), 3.67 (s, 2H), 2.73 (s, 4H), 2.40 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 166.4, 158.8 (d, J = 247.4 Hz), 151.0, 147.3, 146.2, 141.9 (d, J = 8.0 Hz), 131.5, 130.3 (d, J = 3.7 Hz), 125.4 (d, J = 3.0 Hz), 122.5, 121.8 (d, J = 16.3 Hz), 116.5 (d, J = 21.6 Hz), 49.7, 42.8, 36.2, 26.6, 26.6 , 21.2. HRMS (ESI): calculated for C22H23FN5O2 [M + H], 408.1836; found, 408.1826.
4-(2-fluoro-4-methoxyphenyl)-7-((2-methoxypyrimidin-5-yl)methyl)-5,6,7,8-tetrahydro-1,7-naphthyridine-2-carboxamide (14f). Compound 14f was prepared in 80% yield as a white solid using a similar method that described for 14a. 1H NMR (300 MHz, CDCl3) δ 8.54 (s, 2H), 7.93 (s, 1H), 7.78 (s, 1H), 7.13 (t, J = 8.6 Hz, 1H), 6.91 - 6.61 (m, 2H), 5.70 (s, 1H), 4.02 (s, 3H), 3.85 (s, 3H), 3.78 (s, 2H), 3.67 (s, 2H), 2.74 (s, 4H). 13C NMR (75 MHz, d6-DMSO) δ 166.2, 165.1, 161.5 (d, J = 11.2 Hz), 160.4, 159.6 (d, J = 244.8 Hz), 154.3, 147.9, 144.7, 131.9, 131.7 (d, J = 5.0 Hz), 124.9, 121.6, 117.3 (d, J = 16.4 Hz), 111.3, 102.2 (d, J = 25.7 Hz), 57.9, 56.2, 55.7, 55.0, 49.7, 26.9. HRMS (ESI): calculated for C22H23FN5O3 [M + H], 424.1785; found, 424.1785.
4-(2-chloro-4-fluorophenyl)-7-((2-methoxypyrimidin-5-yl)methyl)-5,6,7,8-tetrahydro-1,7-naphthyridine-2-carboxamide (14g). Compound 14g was prepared in 70% yield as a white solid using a similar method that described for 14a. 1H NMR (300 MHz, d6-DMSO) δ 8.12 (d, J = 5.2 Hz, 1H), 8.00 (s, 1H), 7.71 - 7.56 (m, 3H), 7.54 - 7.41 (m, 1H), 7.36 (td, J = 8.5, 2.5 Hz, 1H), 7.00 (d, J = 4.8 Hz, 1H), 6.80 (s, 1H), 3.84 (s, 3H), 3.71 (s, 4H), 2.78 - 2.63 (m, 2H), 2.65 - 2.50 (m, 2H). 13C NMR (75 MHz, d6-DMSO) δ 166.1, 164.3, 154.5, 150.9, 148.0, 147.2, 133.3 (d, J = 3.5 Hz), 132.8 (d, J = 11.6 Hz), 132.6, 132.3 (d, J = 9.1 Hz), 131.6, 121.0, 117.7, 117.4 (d, J = 24.7 Hz), 115.5, 115.2, 110.3, 60.4, 58.1, 53.5, 49.9, 26.7. HRMS (ESI): calculated for C22H21FN4O2 [M + H], 427.1337; found, 427.1325.
Chemical syntheses of radiolabeling precursors 17 and 18
Ethyl 4-chloro-7-((2-methoxypyridin-4-yl)methyl)-5,6,7,8-tetrahydro-1,7-naphthyridine-2-carboxylate (16). Compound 16 was prepared in 45% yield as a white solid using a similar method that described for 13a. 1H NMR (300 MHz, CDCl3) δ 8.10 (d, J = 5.2 Hz, 1H), 7.97 (s, 1H), 6.90 (d, J = 4.7 Hz, 1H), 6.84 - 6.62 (m, 1H), 4.44 (q, J = 7.1 Hz, 2H), 3.93 (s, 3H), 3.81 (s, 2H), 3.69 (s, 2H), 2.97 (t, J = 5.9 Hz, 2H), 2.82 (t, J = 5.9 Hz, 2H), 1.40 (t, J = 7.1 Hz, 3H). 13C NMR (75 MHz, CDCl3) δ 164.6, 164.2, 156.8, 149.4, 146.9, 146.2, 144.9, 132.4, 123.8, 117.1, 110.7, 62.1, 60.9, 58.2, 53.4, 49.6, 26.8, 14.2.
4-(2-fluoro-4-hydroxyphenyl)-7-((2-methoxypyridin-4-yl)methyl)-5,6,7,8-tetrahydro-1,7-naphthyridine-2-carboxamide (17). To a solution of ethyl 4-chloro-7-((2-methoxypyrimidin-5-yl)methyl)-5,6,7,8-tetrahydro-1,7-naphthyridine-2-carboxylate (16) (10.8 mg, 0.030 mmol), 2-fluoro-4-methoxyphenyl boronic acid (5.1 mg, 0.030 mmol) and K2CO3 (8.28 mg, 0.060 mmol) in 1,4-dioxane/water (v/v, 10/1, 1.8 mL) was added Pd(dppf)Cl2 (2.2 mg, 0.003 mmol) under Ar. The mixture was stirred at 100 oC for 4 h, then quenched with water (3 mL) and extracted with ethyl acetate (5 mL, three times). The combined organic layers were concentrated in vacuo. The residue was dissolved in 7 N ammonia methanol solution (2 mL) and stirred for 4h before quenched with ethyl acetate (5 mL) and concentrated in vacuo. The residue was purified by flash chromatography on silica gel (hexanes to ethyl acetate gradient column) to yield the compound 17 as white solid (5.3 mg, 44%). 1H NMR (300 MHz, CDCl3) δ 8.13 (d, J = 5.3 Hz, 1H), 7.87 (s, 2H), 7.08 - 6.93 (m, 2H), 6.84 - 6.65 (m, 3H), 5.68 (s, 1H), 3.95 (s, 3H), 3.81 (s, 2H), 3.71 (s, 2H), 2.77 (s, 4H).13C NMR (75 MHz, CDCl3) δ 165.2 , 164.6 , 159.7 (d, J = 247.2 Hz), 159.1 , 159.0 , 146.6 , 145.5 , 145.0 , 133.3 , 131.0 (d, J = 5.2 Hz), 124.9 , 117.4 , 116.3 (d, J = 16.4 Hz), 112.1 , 112.0 , 110.6 , 103.6 (d, J = 24.7 Hz), 61.0 , 58.4 , 53.6 , 49.7 , 27.0. HRMS (ESI): calculated for C22H22FN4O3 [M + H], 409.1676; found, 409.1691.
4-(2,4-difluorophenyl)-7-((2-hydroxypyridin-4-yl)methyl)-5,6,7,8-tetrahydro-1,7-naphthyridine-2-carboxamide (18) Precursor 18 was prepared in 56% yield as a white solid using a similar method that described for 14a. 1H NMR (400 MHz, CDCl3) δ 8.36 (d, J = 5.0 Hz, 1H), 7.94 (s, 1H), 7.80 (d, J = 4.6 Hz, 1H), 7.42 (d, J = 1.3 Hz, 1H), 7.32 - 7.28 (m, 1H), 7.24 (td, J = 8.4, 6.3 Hz, 1H), 7.04 - 6.89 (m, 2H), 6.13 (d, J = 4.6 Hz, 1H), 3.81 (s, 2H), 3.75 (s, 2H), 2.76 (s, 4H). 13C NMR (100 MHz, CDCl3) δ 166.7, 163.2 (dd, J = 250.8, 11.4 Hz), 159.2 (dd, J = 250.4, 11.8 Hz), 153.8, 151.9, 150.9, 149.8, 146.8, 144.4, 132.5, 131.5 (dd, J = 9.6, 4.9 Hz), 124.0, 122.4, 121.9, 121.6 (dd, J = 16.7, 3.8 Hz), 111.9 (dd, J = 21.1, 3.7 Hz), 104.4 (t, J = 25.5 Hz), 60.7, 58.5, 50.0, 26.9 (d, J = 3.5 Hz). HRMS (ESI): calculated for C22H19F2N4O2 [M + H], 397.1476; found, 397.1490.
Pharmacology
Cell Line Generation and Thallium Flux Assays. The general procedure for the preparation of human mGlu2 and mGlu3 was described previously [45, 56] with minor modifications in this work. The cloning sites were NheI/NotI for both receptors. HEK GIRK cells, generously provided by Lily Jan (University of California San Francisco, San Francisco, CA), were transfected with 24 μg of DNA using Fugene6 (Promega), stable transfectants were selected with 1 μg/mL puromycin dihydrochloride (Sigma-Aldrich, St. Louis, MO), and polyclonal human mGlu2 GIRK and mGlu3 GIRK cell lines were established. Cells were maintained following selection in 45% DMEM, 45% Ham's F12, 10% FBS, 100 units/mL penicillin/streptomycin, 20 mM HEPES, pH 7.3, 1 mM sodium pyruvate, 2 mM glutamine, 700 μg/mL G418 (Mediatech, Inc., Herndon, VA), and 600 μg/mL puromycin (growth media) at 37 °C in the presence of 5% CO2. All cell culture reagents were purchased from Invitrogen Corp. (Carlsbad, CA) unless otherwise noted.
Human mGlu2 and mGlu3 Thallium Flux in Vitro Assays. Potencies of these NAMs at both mGlu2 and mGlu3 were investigated by thallium flux through GIRK channels, which was disclosed in detail [57] as well as described [45] in our previous work. In particular, cells were plated into 384-well, black-walled, clear-bottomed poly(D-lysine_-coated plates at a density of 15 000 cells per well in 20 μL of DMEM containing 10% dialyzed FBS, 20 mM HEPES, and 100 units/mL penicillin/streptomycin (assay media). Plated cells were incubated overnight at 37 °C in the presence of 5% CO2. The next day, the medium was exchanged from the cells to assay buffer [Hanks' balanced salt solution (Invitrogen) containing 20 mM HEPES, pH 7.3] using an ELX405 microplate washer (BioTek), leaving 20 μL per well, followed by the addition of 20 μL per well of FluoZin2-AM (330 nM final concentration) indicator dye (Invitrogen; prepared as a stock in DMSO and mixed in a 1:1 ratio with Pluronic acid F-127) in assay buffer. Cells were incubated for 1 h at room temperature, and the dye was exchanged to assay buffer using an ELX405, leaving 20 μL per well. For concentration-response curve experiments, compounds were serially diluted 1:3 into 10 point concentration-response curves and were transferred to daughter plates using an Echo acoustic plate reformatter (Labcyte, Sunnyvale, CA). Test compounds were diluted to 2 times their final desired concentration in assay buffer (0.3% DMSO final concentration). Agonists were diluted in thallium buffer [125 mM sodium bicarbonate (added fresh the morning of the experiment), 1 mM magnesium sulfate, 1.8 mM calcium sulfate, 5 mM glucose, 12 mM thallium sulfate, and 10 mM HEPES, pH 7.3] at 5 times the final concentration to be assayed. Cell plates and compound plates were loaded onto a kinetic imaging plate reader (FDSS 6000 or 7000; Hamamatsu Corporation, Bridgewater, NJ). Appropriate baseline readings were taken (10 images at 1 Hz; excitation, 470 ± 20 nm; emission, 540 ± 30 nm), and test compounds were added in a 20 μL volume and incubated for approximately 1 h at room temperature before the addition of 10 μL of thallium buffer with or without an EC80 concentration of the agonist glutamate for affinity evaluation experiments or with a full concentration-response of glutamate for Schild analysis experiments. After the addition of agonist, data were collected for approximately an additional 2.5 min. Data were analyzed using Excel (Microsoft Corp, Redmond, WA). The slope of the fluorescence increase beginning 5 s after thallium/agonist addition and ending 15 s after thallium/agonist addition was calculated, corrected to vehicle and maximal agonist control slope values, and plotted using either XLfit (ID Business Solutions Ltd.) or Prism software (GraphPad Software, San Diego, CA) to generate concentration-response curves. Potencies were calculated from fits using a four-point parameter logistic equation.
Radiochemistry
Radiosynthesis of 4-(2-fluoro-4-(methoxy-11C)phenyl)-7-((2-methoxy-pyridin-4-yl)methyl)-5,6,7,8-tetrahydro-1,7-naphthyridine-2-carboxamide ([11C]14a). [11C]Methyl iodide ([11C]CH3I) was synthesized from cyclotron-produced [11C]CO2, which was produced by 14N(p, α)11C nuclear reaction. Briefly, [11C]CO2 was bubbled into a solution of LiAlH4 (0.4 M in THF, 300 μL). After evaporation, the remaining reaction mixture was treated with hydroiodic acid (57% aqueous solution, 300 μL). The resulting [11C]CH3I was transferred under helium gas with heating into a pre-cooled (-15 to -20 °C) reaction vessel containing precursor 17 (1.0 mg), NaOH (3-5 μL, 0.5 M) and anhydrous DMF (300 μL). After the radioactivity reached a plateau during transfer, the reaction vessel was warmed to 80 °C and maintained for 5 min. HPLC purification was completed on a Capcell Pak UG80 C18 column (10 mm ID × 250 mm) using a mobile phase of CH3CN / H2O (v/v, 55/45) at a flow rate of 5.0 mL/min. The retention time of [11C]14a was 9.0 min. The radioactive fraction corresponding to the desired product was collected in a sterile flask, evaporated to dryness in vacuo, and reformulated in a saline solution (3 mL) containing 100 µL of 25% ascorbic acid in sterile water and 100 µL of 20% Tween® 80 in ethanol. (Note: We added ascorbic acid to prevent potential radiolysis and Tween® 80 to improve aqueous solubility.) The synthesis time was ca. 40 min from end-of-bombardment. Radiochemical and chemical purity was measured by analytical HPLC (Capcell Pak UG80 C18, 4.6 mm ID × 250 mm). The identity of [11C]14a was confirmed by the co-injection with unlabeled 14a. Radiochemical yield was 36.6 ± 7.3% (n = 7, decay-corrected based on [11C]CO2) with >99% radiochemical purity and greater than 2 Ci/μmol molar activity.
Radiosynthesis of 4-(2,4-difluorophenyl)-7-((2-(methoxy-11C)pyridine-4-yl)methyl)-5,6,7,8-tetrahydro-1,7-naphthyridine-2-carboxamide ([11C]14b). Similar radiosynthesis of [11C]14a, [11C]CH3I was trapped in the reaction vessel containing precursor 18 (1.0 mg) and anhydrous DMF (300 μL, pre-saturated with 10-20 mg Cs2CO3). After the radioactivity reached a plateau during transfer, the reaction vessel was warmed to 90 °C and maintained for 5 min. HPLC purification was completed on a Capcell Pak UG80 C18 column (10 mm ID × 250 mm) using a mobile phase of CH3CN / H2O + 0.1% Et3N (v/v, 55/45) at a flow rate of 5.0 mL/min. The retention time of [11C]14b was 9.0 min. The radioactive fraction corresponding to the desired product was collected in a sterile flask, evaporated to dryness in vacuo, and reformulated in a saline solution (3 mL) containing 100 µL of 25% ascorbic acid in sterile water and 100 µL of 20% Tween® 80 in ethanol. The synthesis time was ca. 40 min from end-of-bombardment. Radiochemical and chemical purity was measured by analytical HPLC (Capcell Pak UG80 C18, 4.6 mm ID × 250 mm). The identity of [11C]14b was confirmed by the co-injection with unlabeled 14b. Radiochemical yield was 6.53 ± 1.5% (n = 10, decay-corrected based on [11C]CO2) with >99% radiochemical purity and greater than 2 Ci/μmol molar activity.
In Vitro Autoradiography
Sagittal rat brain slices were prepared into 20 μm sections with a cryostat (HM560; Carl Zeiss, Oberkochen, Germany), mounted on air plasma spray-coated glass slides, and stored at -80 °C before used for experiments. The sections were preincubated for 15 min in a glass tank containing 200 mL of Tris·HCl buffer (50 mM, pH 7.4) consisting of 2 mM of MgCl2 and 1.2 mM of CaCl2 at room temperature. After preincubation, the sections were incubated in a fresh buffer containing [11C]14a or [11C]14b (250 µCi in 200 mL buffer; molar activity 2.7 Ci/µmol of [11C]14a and 2.5 Ci/µmol of [11C]14b, respectively) in the glass tank for 30 min at room temperature. For blocking studies, non-radioactive 14a or 14b (10 μM) and QCA (10 μM) were chosen to determine the specificity of radiotracers for binding mGlu2. Six serial brain sections were used for each conditions. After incubation, brain sections were washed with cold buffer (3 × 5 min), immersed in cold distilled water, and then dried with cold air. The sections were placed in contact with imaging plates (BAS-MS2025, FUJIFILM, Tokyo, Japan). Autoradiograms were obtained and photostimulated luminescence (PSL) values in the ROIs were measured using a Bio-Imaging Analyzer System (BAS5000, FUJIFILM).
Small Animal PET Imaging Studies in Rat Brain
As we previously reported [45], PET scans were carried out by an Inveon PET scanner (Siemens Medical Solutions, Knoxville, TN, USA). During the scan, SD rats were anesthetized by oxygen mixed with 1-2% (v/v) isoflurane and kept body temperature at 40 °C by a commercially available circulation system (T/Pump TP401, Gaymar Industries, Orchard Park, NY). A 24-gauge catheter was inserted into the tail vein to facilitate a bolus injection. The radiotracer [11C]14a or [11C]14b (ca. 1 mCi / 150-200 μL) was injected into the rat via the preinstalled catheter, and the dynamic acquisition of PET signals in rat brain was started at the same time and lasted for 60 min in 3D list mode. For pretreatment studies, 14a (1 mg/kg for [11C]14a for self-blocking study in Figure S2; 3 mg/kg for [11C]14b for blocking study in Figure S12C) or 14b (1 mg/kg for [11C]14b for self-blocking study in Figure S12B), formulated in 300 μL of saline containing 10% ethanol and 5% Tween 80, was injected at 30 min via the tail vein catheter prior to the injection of [11C]14a or [11C]14b. The dynamic emission data were reconstructed by filtered back projection using Hanning's filter with a Nyquist cutoff of 0.5 cycle/pixel into 33 frames (10 s × twelve frames, 20 s × three frames, 30 s × three frames, 60 s × three frames, 150 s × three frames, and 300 s × nine frames). The TACs of [11C]14a or [11C]14b were analyzed from volumes of interest in the striatum, hippocampus, cortex, thalamus, pons and cerebellum normalized to a rat brain MRI template [58] using PMOD software (version 3.4; PMOD technology, Zurich, Switzerland). The radioactivity was decay-corrected to the injection time and expressed as the standardized uptake value (SUV) which equals to (radioactivity per milliliter of tissue per injected radioactivity) × (gram of body weight). To obtain non-displaceable binding potential (BPND), simplified reference tissue model (SRTM) [53, 54] was carried out on the basis of kinetic analysis using PMOD software. We acquired BP-parametric images using kinetic modeling without masking. Respective TACs in receptor-rich or -poor region were loaded for production of BP-map with default setting. Representative parametric images scaled with BPND were reconstructed by PMOD software using TACs in mGlu2-enriched and reference regions. Pons was selected as the reference region for mGlu2 (the lowest and consistent brain uptake and washout kinetics between baseline and blocking conditions). It should be noted that while there is a low-level expression of mGlu2 in the pons, it is not without existence. A reference region should be a region devoid of the target, but with similar tracer transport or diffusion to the other regions of interest with expression of the target. Therefore, there are some limitations when using pons as the reference region. For example, the partial volume effect in PET likely leads to spillover of activity from nearby higher uptake regions into the pons, artificially increasing the time-activity curve in the pons.
Whole Body ex vivo Biodistribution Studies in Mice
Each mouse was treated with a bolus injection of [11C]14b (50 μCi / 150 μL) via the tail vein. Three mice were sacrificed by cervical dislocation at each time point (1, 5, 15, 30 and 60 min) after injection. Major organs, including heart, lungs, liver, pancreas, spleen, kidneys, stomach (including contents), small intestine (including contents), large intestine (including contents), testes, muscle, whole brain and blood samples were quickly removed and weighed. The radioactivity remained in these organs was measured by a 2480 Wizard autogamma counter (PerkinElmer, USA). The results are expressed as the percentage of injected dose per gram of wet tissue (%ID/g) or standardized uptake value (SUV). All radioactivity measurements were decay-corrected to the time point of PET tracer injection based on half-life of 11C.
Radiometabolite Analysis
Sprague-Dawley rats were sacrificed by decapitation under anesthesia at 5 and 20 min (n = 2 each time point) after the intravenous injection of [11C]14b. Blood and whole brain samples were quickly harvested. Plasma was separated from the blood samples via 1) centrifuging at 15,000 g for 2 min at 4 °C, 2) mixing 0.5 mL of acetonitrile with 0.5 mL of supernatant, and then 3) vortexing for 15 s and centrifuging again at 15,000 g for 2 min for deproteinization. In terms of brain metabolite analysis, the removed rat brain was quickly placed on the ice, homogenized in an ice-cooled CH3CN/H2O (v/v, 1/1, 1 mL) solution, and then centrifuged at 15,000 g for 2 min at 4 °C. The supernatant was collected, whose radioactivity was >90% based on the total radioactivity in the brain homogenate. An aliquot of the supernatant (100 μL) obtained from the plasma or brain homogenate was injected into the radio-HPLC system and analyzed using a Capcell Pak UG80 C18 column (4.6 mm ID × 250 mm) in a mobile phase of CH3CN/H2O + 0.1% Et3N (v/v, 45/55) at a flow rate of 1.0 mL/min. The percentage of [11C]14b to total radioactivity (corrected for decay) on the HPLC charts was calculated as ([peak area for [11C]14b]/[total peak area]) × 100.
Statistical analysis
Statistical analysis is performed using the statistical computer package, GraphPad Prism (GraphPad Software Inc., San Diego, CA, USA). Results are expressed as means ± SEM. Statistical comparisons were made using one-way analysis of variance (ANOVA). Asterisks indicate statistical significance. *p < 0.05, ***p ≤ 0.001, and ****p ≤ 0.0001.
Abbreviations
mGlu2, metabotropic glutamate receptor 2; PD, Parkinson's disease; AD, Alzheimer's disease; PET, positron emission tomography; CNS, central nervous system; iGluRs, ionotropic glutamate receptors; mGlus, metabotropic glutamate receptors; PAMs, positive allosteric modulators; NAMs, negative allosteric modulators; BBB, blood-brain barrier; PgP, P-glycoprotein; Bcrp, breast cancer resistance protein; ARG, autoradiography; ABC, ATP binding cassette; NBS, N-bromosuccinimide; DMF, dimethylformamide; GTP, Guanosine triphosphate; cAMP, cyclic adenosine monophosphate; tPSA, topological polar surface area; GIRK, G protein-coupled inwardly rectifying potassium; BPND, non-displaceable binding potential; SUV, standardized uptake value; TAC, time-activity curve; %ID/g, the percentage of injected dose per gram of wet tissue.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
We thank Drs. Thomas J. Brady and Lei Zhang for their helpful discussion. Financial support from the NIH grants (MH106865 to N.D.P.C. and MH117125 to S.H.L.) and CSC scholarship to X.Z. (No. 201606200041) is gratefully acknowledged. We thank the National Institute of Mental Health's Psychoactive Drug Screening Program for in vitro CNS off-target binding screening and mGlu functional assays. The NIMH PDSP is Directed by Bryan L. Roth MD, PhD at the University of North Carolina at Chapel Hill and Project Officer Jamie Driscoll at NIMH, Bethesda MD, USA.
Author contributions
The manuscript was written through the contributions of all the authors. All authors have given approval to the final version of the manuscript. #X. Zhang and Y. Zhang contributed equally.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Nakanishi S. Molecular diversity of glutamate receptors and implications for brain function. Science. 1992;258:597-603
2. Niciu MJ, Kelmendi B, Sanacora G. Overview of glutamatergic neurotransmission in the nervous system. Pharmacol Biochem Behav. 2012;100:656-64
3. Lau A, Tymianski M. Glutamate receptors, neurotoxicity and neurodegeneration. Pflugers Arch. 2010;460:525-42
4. Traynelis SF, Wollmuth LP, McBain CJ, Menniti FS, Vance KM, Ogden KK. et al. Glutamate receptor ion channels: structure, regulation, and function. Pharmacol Rev. 2010;62:405-96
5. Kew JN, Kemp JA. Ionotropic and metabotropic glutamate receptor structure and pharmacology. Psychopharmacology (Berl). 2005;179:4-29
6. Niswender CM, Conn PJ. Metabotropic glutamate receptors: physiology, pharmacology, and disease. Annu Rev Pharmacool Toxicol. 2010;50:295-322
7. Reiner A, Levitz J. Glutamatergic Signaling in the Central Nervous System: Ionotropic and Metabotropic Receptors in Concert. Neuron. 2018;98:1080-98
8. Schoepp DD. Unveiling the functions of presynaptic metabotropic glutamate receptors in the central nervous system. J Pharmacol Exp Ther. 2001;299:12-20
9. Scheefhals N, MacGillavry HD. Functional organization of postsynaptic glutamate receptors. Mol Cell Neurosci. 2018;91:82-94
10. Gill SS, Pulido OM. Glutamate receptors in peripheral tissues: current knowledge, future research, and implications for toxicology. Toxicol Pathol. 2001;29:208-23
11. Mukherjee S, Manahan-Vaughan D. Role of metabotropic glutamate receptors in persistent forms of hippocampal plasticity and learning. Neuropharmacology. 2013;66:65-81
12. Testa CM, Friberg IK, Weiss SW, Standaert DG. Immunohistochemical localization of metabotropic glutamate receptors mGluR1a and mGluR2/3 in the rat basal ganglia. J Comp Neurol. 1998;390:5-19
13. Ohishi H, Neki A, Mizuno N. Distribution of a metabotropic glutamate receptor, mGluR2, in the central nervous system of the rat and mouse: an immunohistochemical study with a monoclonal antibody. Neurosci Res. 1998;30:65-82
14. Richards G, Messer J, Malherbe P, Pink R, Brockhaus M, Stadler H. et al. Distribution and abundance of metabotropic glutamate receptor subtype 2 in rat brain revealed by [3H]LY354740 binding in vitro and quantitative radioautography: Correlation with the sites of synthesis, expression, and agonist stimulation of [35S]GTPγs binding. J Comp Neurol. 2005;487:15-27
15. Wright RA, Johnson BG, Zhang C, Salhoff C, Kingston AE, Calligaro DO. et al. CNS distribution of metabotropic glutamate 2 and 3 receptors: transgenic mice and [3H]LY459477 autoradiography. Neuropharmacology. 2013;66:89-98
16. Ohishi H, Ogawa-Meguro R, Shigemoto R, Kaneko T, Nakanishi S, Mizuno N. Immunohistochemical localization of metabotropic glutamate receptors, mGluR2 and mGluR3, in rat cerebellar cortex. Neuron. 1994;13:55-66
17. Ohishi H, Shigemoto R, Nakanishi S, Mizuno N. Distribution of the messenger RNA for a metabotropic glutamate receptor, mGluR2, in the central nervous system of the rat. Neuroscience. 1993;53:1009-18
18. Vaidya A, Jain S, Jain AK, Agrawal A, Kashaw SK, Jain SK. et al. Metabotropic glutamate receptors: a review on prospectives and therapeutic aspects. Mini Rev Med Chem. 2013;13:1967-81
19. Golubeva AV, Moloney RD, O'Connor RM, Dinan TG, Cryan JF. Metabotropic Glutamate Receptors in Central Nervous System Diseases. Curr Drug Targets. 2016;17:538-616
20. Xing B, Han G, Wang MJ, Snyder MA, Gao WJ. Juvenile treatment with mGluR2/3 agonist prevents schizophrenia-like phenotypes in adult by acting through GSK3beta. Neuropharmacology. 2018;137:359-71
21. Maksymetz J, Moran SP, Conn PJ. Targeting metabotropic glutamate receptors for novel treatments of schizophrenia. Mol Brain. 2017;10:15
22. Conn PJ, Lindsley CW, Jones CK. Activation of metabotropic glutamate receptors as a novel approach for the treatment of schizophrenia. Trends Pharmacol Sci. 2009;30:25-31
23. Campo B, Kalinichev M, Lambeng N, El Yacoubi M, Royer-Urios I, Schneider M. et al. Characterization of an mGluR2/3 negative allosteric modulator in rodent models of depression. J Neurogenet. 2011;25:152-66
24. Dwyer JM, Lepack AE, Duman RS. mGluR2/3 blockade produces rapid and long-lasting reversal of anhedonia caused by chronic stress exposure. J Mol Psychiatry. 2013;1:15
25. Masilamoni GJ, Smith Y. Metabotropic glutamate receptors: targets for neuroprotective therapies in Parkinson disease. Curr Opin Pharmacol. 2018;38:72-80
26. Samadi P, Rajput A, Calon F, Grégoire L, Hornykiewicz O, Rajput AH. et al. Metabotropic Glutamate Receptor II in the Brains of Parkinsonian Patients. J Neuropathol Exp Neurol. 2009;68:374-82
27. Dickerson JW, Conn PJ. Therapeutic potential of targeting metabotropic glutamate receptors for Parkinson's disease. Neurodegener Dis Manag. 2012;2:221-32
28. Sebastianutto I, Cenci MA. mGlu receptors in the treatment of Parkinson's disease and L-DOPA-induced dyskinesia. Curr Opin Pharmacol. 2018;38:81-9
29. Lee HG, Zhu X, O'Neill MJ, Webber K, Casadesus G, Marlatt M. et al. The role of metabotropic glutamate receptors in Alzheimer's disease. Acta Neurobiol Exp (Wars). 2004;64:89-98
30. Caraci F, Nicoletti F, Copani A. Metabotropic glutamate receptors: the potential for therapeutic applications in Alzheimer's disease. Curr Opin Pharmacol. 2018;38:1-7
31. Caprioli D, Justinova Z, Venniro M, Shaham Y. Effect of Novel Allosteric Modulators of Metabotropic Glutamate Receptors on Drug Self-administration and Relapse: A Review of Preclinical Studies and Their Clinical Implications. Biol Psychiatry. 2018;84:180-92
32. Kalivas PW, Volkow ND. New medications for drug addiction hiding in glutamatergic neuroplasticity. Mol Psychiatry. 2011;16:974-86
33. Pomierny-Chamiolo L, Rup K, Pomierny B, Niedzielska E, Kalivas PW, Filip M. Metabotropic glutamatergic receptors and their ligands in drug addiction. Pharmacol Ther. 2014;142-305:281
34. Cleva RM, Olive MF. mGlu receptors and drug addiction. Wiley Interdiscip Rev Membr Transp Signal. 2012;1:281-95
35. Cross AJ, Anthenelli R, Li X. Metabotropic Glutamate Receptors 2 and 3 as Targets for Treating Nicotine Addiction. Biol Psychiatry. 2018;83:947-54
36. Phelps ME. Positron emission tomography provides molecular imaging of biological processes. Proc Natl Acad Sci U S A. 2000;97:9226-33
37. Kim K, Kim H, Bae S-H, Lee S-Y, Kim Y-H, Na J. et al. [18F]CB251 PET/MR imaging probe targeting translocator protein (TSPO) independent of its Polymorphism in a Neuroinflammation Model. Theranostics. 2020;10:9315-31
38. López-Picón FR, Kirjavainen AK, Forsback S, Takkinen JS, Peters D, Haaparanta-Solin M. et al. In vivo characterization of a novel norepinephrine transporter PET tracer [18F]NS12137 in adult and immature Sprague-Dawley rats. Theranostics. 2019;9:11-9
39. Tiwari AK, Ji B, Yui J, Fujinaga M, Yamasaki T, Xie L. et al. [18F]FEBMP: Positron Emission Tomography Imaging of TSPO in a Model of Neuroinflammation in Rats, and in vitro Autoradiograms of the Human Brain. Theranostics. 2015;5:961-9
40. Zinnhardt B, Belloy M, Fricke IB, Orije J, Guglielmetti C, Hermann S. et al. Molecular Imaging of Immune Cell Dynamics During De- and Remyelination in the Cuprizone Model of Multiple Sclerosis by [18F]DPA-714 PET and MRI. Theranostics. 2019;9:1523-37
41. Wang JQ, Zhang Z, Kuruppu D, Brownell AL. Radiosynthesis of PET radiotracer as a prodrug for imaging group II metabotropic glutamate receptors in vivo. Bioorg Med Chem Lett. 2012;22:1958-62
42. Andrés JI, Alcázar J, Cid JM, De Angelis M, Iturrino L, Langlois X. et al. Synthesis, Evaluation, and Radiolabeling of New Potent Positive Allosteric Modulators of the Metabotropic Glutamate Receptor 2 as Potential Tracers for Positron Emission Tomography Imaging. J Med Chem. 2012;55:8685-99
43. Majo V, Prabhakaran J, Simpson N, Arango V, Mann JJ, Kumar JD. Development of a [18F]-labeled positive allosteric modulator of the metabotropic glutamate receptor 2 (mGluR2) as a potential PET tracer. J Nucl Med. 2013;54:1072
44. Ma Y, Kumata K, Yui J, Zhang Y, Yamasaki T, Hatori A. et al. Synthesis and evaluation of 1-(cyclopropylmethyl)-4-(4-[11C]methoxyphenyl)-piperidin-1-yl-2-oxo-1,2-dihydropyridine-3-carbonitrile ([11C]CMDC) for PET imaging of metabotropic glutamate receptor 2 in the rat brain. Biorg Med Chem. 2017;25:1014-21
45. Zhang X, Kumata K, Yamasaki T, Cheng R, Hatori A, Ma L. et al. Synthesis and Preliminary Studies of a Novel Negative Allosteric Modulator, 7-((2,5-Dioxopyrrolidin-1-yl)methyl)-4-(2-fluoro-4-[11C]methoxyphenyl) quinoline-2-carboxamide, for Imaging of Metabotropic Glutamate Receptor 2. ACS Chem Neurosci. 2017;8:1937-48
46. Kumata K, Hatori A, Yamasaki T, Zhang Y, Mori W, Fujinaga M. et al. Synthesis and evaluation of 4-(2-fluoro-4-[11C]methoxyphenyl)-5-((2-methylpyridin-4-yl)methoxy)picolinamide for PET imaging of the metabotropic glutamate receptor 2 in the rat brain. Biorg Med Chem. 2019;27:483-91
47. Leurquin-Sterk G, Celen S, Van Laere K, Koole M, Bormans G, Langlois X. et al. What We Observe In Vivo Is Not Always What We See In Vitro: Development and Validation of 11C-JNJ-42491293, A Novel Radioligand for mGluR2. J Nucl Med. 2017;58:110-6
48. Lohith T, McQuade P, Salinas C, Anderson M, Reynders T, Bautmans A. et al. First-in-human PET imaging of mGluR2 receptors. J Nucl Med. 2016;57:213
49. Yuan G, Shoup TM, Moon S-H, Brownell A-L. A concise method for fully automated radiosyntheses of [18F]JNJ-46356479 and [18F]FITM via Cu-mediated 18F-fluorination of organoboranes. RSC Adv. 2020;10:25223-7
50. Lavreysen H, Langlois X, Ahnaou A, Drinkenburg W, te Riele P, Biesmans I. et al. Pharmacological characterization of JNJ-40068782, a new potent, selective, and systemically active positive allosteric modulator of the mGlu2 receptor and its radioligand [3H]JNJ-40068782. J Pharmacol Exp Ther. 2013;346:514-27
51. Bollinger KA, Felts AS, Brassard CJ, Engers JL, Rodriguez AL, Weiner RL. et al. Design and Synthesis of mGlu2 NAMs with Improved Potency and CNS Penetration Based on a Truncated Picolinamide Core. ACS Med Chem Lett. 2017;8:919-24
52. Deng X, Rong J, Wang L, Vasdev N, Zhang L, Josephson L. et al. Chemistry for Positron Emission Tomography: Recent Advances in 11C-, 18F-, 13N-, and 15O-Labeling Reactions. Angew Chem Int Ed. 2019;58:2580-605
53. Lammertsma AA, Hume SP. Simplified Reference Tissue Model for PET Receptor Studies. NeuroImage. 1996;4:153-8
54. Wang L, Cheng R, Fujinaga M, Yang J, Zhang Y, Hatori A. et al. A Facile Radiolabeling of [18F]FDPA via Spirocyclic Iodonium Ylides: Preliminary PET Imaging Studies in Preclinical Models of Neuroinflammation. J Med Chem. 2017;60:5222-7
55. Deutsch DG, Ueda N, Yamamoto S. The fatty acid amide hydrolase (FAAH). Prostaglandins Leukot Essent Fatty Acids. 2002;66:201-10
56. Dhanya RP, Sheffler DJ, Dahl R, Davis M, Lee PS, Yang L. et al. Design and synthesis of systemically active metabotropic glutamate subtype-2 and -3 (mGlu2/3) receptor positive allosteric modulators (PAMs): pharmacological characterization and assessment in a rat model of cocaine dependence. J Med Chem. 2014;57:4154-72
57. Niswender CM, Johnson KA, Luo Q, Ayala JE, Kim C, Conn PJ. et al. A novel assay of Gi/o-linked G protein-coupled receptor coupling to potassium channels provides new insights into the pharmacology of the group III metabotropic glutamate receptors. Mol Pharmacol. 2008;73:1213-24
58. Yui J, Hatori A, Kawamura K, Yanamoto K, Yamasaki T, Ogawa M. et al. Visualization of early infarction in rat brain after ischemia using a translocator protein (18kDa) PET ligand [11C]DAC with ultra-high specific activity. NeuroImage. 2011;54:123-30
Author contact
![]() Corresponding author: liang.stevenharvard.edu (S. H. Liang); zhang.ming-ronggo.jp (M.-R. Zhang); l_wang1009com (L. Wang).
Corresponding author: liang.stevenharvard.edu (S. H. Liang); zhang.ming-ronggo.jp (M.-R. Zhang); l_wang1009com (L. Wang).