Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
1. Introduction
2. Mechanisms of action and...
3. The use of OGT-based drugs is...
4. Factors contributing to low...
5. Theoretical vs real world...
6. Concluding remarks
Abbreviations
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2026; 16(2):599-616. doi:10.7150/thno.121775 This issue Cite
Review
The efficacy of oligonucleotide-based gene therapeutics in gene silencing
Christina Patra1 ![]() , Zain Hussein1, Veronika D. Ace1, Elena V. Misnik1,2, Daria S. Rybalko1,3, Adeliia A. Salimova1,4, Daria S. Ereshko5, Mikhail V. Dubovichenko1, Moustapha A.Y. Nour1, Valeriia S. Drozd1, Dmitry M. Kolpashchikov6,7
, Zain Hussein1, Veronika D. Ace1, Elena V. Misnik1,2, Daria S. Rybalko1,3, Adeliia A. Salimova1,4, Daria S. Ereshko5, Mikhail V. Dubovichenko1, Moustapha A.Y. Nour1, Valeriia S. Drozd1, Dmitry M. Kolpashchikov6,7
1. Laboratory of DNA-nanosensor diagnostics, ITMO University, Saint-Petersburg, 191002, Russian Federation.
2. Institute of Gene Biology, Russian Academy of Sciences, Moscow, Russian Federation.
3. Pediatric Research and Clinical Center for Infectious Diseases, Saint Petersburg, Russian Federation.
4. Department of Biomedical Sciences, University of Padova, 35131 Padova, Italy.
5. Infochemistry Scientific Center, ITMO University, Saint Petersburg, 191002, Russian Federation.
6. Chemistry Department, University of Central Florida Orlando, FL 32816-2366, USA.
7. Burnett School of Biomedical Sciences, University of Central Florida, Orlando, FL 32816, USA.
Received 2025-7-16; Accepted 2025-9-6; Published 2026-1-1
Abstract

Oligonucleotide-based gene therapeutics (OGTs) have emerged as a promising strategy for treating a variety of diseases, offering a tool for gene modulation at the mRNA level. Despite significant progress in OGTs development, their efficacy in both experimental and clinical settings has often fallen short of expectations. Current estimates suggest that less than 1% of transfected OGTs are released into the cytosol, significantly limiting the interaction with target RNA. Moreover, data suggests that only about 2% of the tested siRNAs achieve the expected 70% target gene knockdown in vitro. Clinically approved OGTs appear to be effective only against genetic disorders that lack effective alternative treatment, and even in these cases their therapeutic contribution remains marginal. Notably, the majority of approved OGTs, as well as those currently in clinical trials, are antisense oligonucleotides (ASOs) despite cell culture data showing that small interfering RNAs (siRNAs) exhibit greater potency. The delayed commercialization of siRNAs, despite high research interest, may be attributed to passenger stand-dependent off target effect and the immaturity of their design and modification strategies. This review critically evaluates the factors influencing therapeutic efficacy of OGTs and highlights the persistent gap between theoretical promise and clinical reality.
Keywords: gene silencing, therapeutic oligonucleotides, efficacy, clinical relevance, cancer, antisense oligonucleotides, siRNA, miRNA, ribozymes, deoxyribozymes, CRISPR/Cas
1. Introduction
Gene therapy has been regarded as a transformative approach for treating hereditary or acquired diseases, including cancer. Its core principle is the introduction of genetic material into a patient's cells to replace defective genes with healthy counterparts, aiming to achieve therapeutic benefits [1,2]. Among various strategies, gene silencing, a technique which targets and suppresses disease promoting genes at the mRNA level, has emerged as a more refined approach to gene therapy. Within this context, oligonucleotide-based gene therapeutics (OGTs) have received a significant attention as potential game-changers [3].
The principles underlying OGTs rely on complementary base pairing between the therapeutic oligonucleotide (ON) and the target mRNA, leading to either mRNA degradation or steric hindrance of the translation machinery. OGTs include RNAi inducing agents, (e.g. siRNA), antisense oligonucleotides (ASOs), DNAzymes (Dzs) and RNAzymes (Rzs) as well as Crispr/Cas13-based systems. The therapeutic potential of an OGT is characterized by its efficacy in silencing the targeted genes and its specificity, which is also reflected by the extent of off-target effects. While a recent comprehensive review has addressed the specificity of OGT [4], this work focuses on the analysis of OGT efficacy.
In this review, OGT efficacy is defined as the degree to which an OGT can reduce the expression of a targeted gene. Efficiency, on the other hand, refers to the practical performance of OGT under specific conditions, including the amount of OGT required to achieve a biological effect, the speed of its action, and the conditions under which it operates. Thus, while efficacy measures the outcome, efficiency is about optimizing the conditions and resources to achieve this outcome. Both terms are used according to these definitions in this review.
Current research endeavors are focusing on refining the OGT design and delivery systems [5-7]. Although productive internalization and release are critical for achieving high efficiency, these aspects are beyond the scope of this review, as the diverse array of delivery systems has been extensively reviewed [8,9]. Instead, this review focuses on evaluating current knowledge on OGTs through the lens of the efficacy demonstrated in experimental and clinical contexts. We evaluate the contribution of various factors into OGT efficacy and examine how closely experimental efficacy aligns with theoretical expectations.
2. Mechanisms of action and their contribution to OGT efficacy
2.1 Antisense Oligonucleotides
ASOs are short, single-stranded DNA (ssDNA) or their analogs designed to hybridize with a specific mRNA sequence (Figure 1A). Their mechanism of ASO action involves binding to the target mRNA, which can lead to two outcomes - (1) RNase H mediated mRNA degradation or (2) steric blockage. In the first mechanism, the mRNA component of the ASO/RNA heteroduplexes is degraded by RNase H1 in the cytoplasm or by RNase H2 in the nucleus [10,11]. In steric blockage either induce translational arrest by hindering interactions with ribosomal subunits or modulates splicing by influencing exon skipping or inclusion [10].
Oligonucleotide-mediated regulation of gene expression. A) ASOs bind to complementary mRNA sequences, leading to either RNase H-mediated degradation of the mRNA or steric inhibition for translation or splicing. B) the RISC complex associated with endogenous miRNA degrades imperfectly complementary mRNA targets, while exogenous antagomirs bind to miRNAs to inhibit their functions, thereby preventing suppression of their targets. C) the RISC complex guided by siRNAs strand degrades complementary mRNA target. Unlike ASOs, siRNAs require enzymes for their activity. D) Dzs and Rzs OGTs are ssDNA and ssRNA molecules with RNA cleaving activity. Unlike RNAi, CRISPR/Cas13, and ASOs, their activity is protein enzyme independent. E) The CRISPR/Cas13 system targets mRNA using crRNAs.
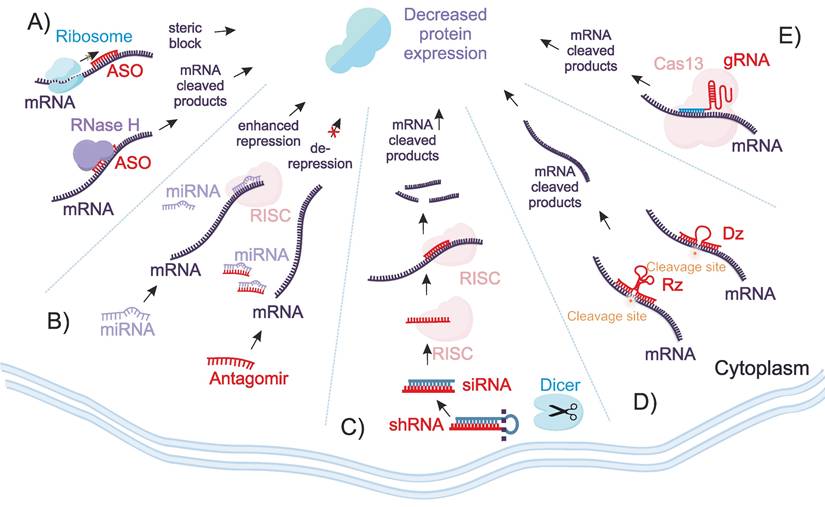
2.2 RNA Interference (RNAi) Agents
In 1998, Fire and Mello revealed the mechanism of RNA interference (RNAi) for the regulation of gene expression in the nematode Caenorhabditis elegans [12]. RNAi agents, including small interfering RNAs (siRNAs), short hairpin RNAs shRNAs and microRNAs (miRs), induce degradation of the specific mRNA target by harnessing the RNA-induced silencing complex (RISC) (Figure 1B-C). Guided by a short ssRNA template, RISC recognizes the complementary mRNA target via Watson-Crick base pairing, leading to mRNA degradation and inhibition of protein synthesis [13]. Unlike ASO, can induce silencing without involvement of endogenous enzymes, RNAi agents differ are double-stranded RNAi agents that require RISC for their activity [14].
2.3 DNAzymes (Dzs) and RNAzymes (RZs)
DZs and RZs are synthetic single stranded nucleic acids that possess catalytic activity [15-17]. Both RZs and DZs can be engineered to cleave particular mRNA sequences [18,19]. Both can be designed to recognize particular mRNA sequences with high specificity due to their short RNA binding [20,21]. However, their catalytic efficiency is often limited by the relatively low binding affinity of these short recognition arms [22]. Unlike RNAi, CRISPR/Cas agents and ASOs, DZs and RZs activity is completely protein enzyme-independent (Figure 1D).
2.4 CRISPR/Cas13
CRISPR/Cas13 is a member of the CRISPR/Cas family that has gained significant attention due to the unique ability to target RNA rather than DNA, thereby enabling gene manipulation at the transcriptional level [23]. Similar to other CRISPR/Cas systems, Cas13 proteins are guided by CRISPR RNAs (crRNAs) to complementary sequences of targeted RNAs [24,25] (Figure 1E). There are currently four main subtypes of Cas13 - Cas13a, Cas13b, Cas13c, and Cas13d [23]. Upon binding of the crRNA to the target RNA via Watson-Crick base pairing, Cas13 undergoes a conformational change that activates the catalytic site followed by cleavage of the target RNA [26]. Unlike other OGTs, CRISPR/Cas13 relies on the expression or co-delivery of the bacterial Cas 13 protein along with the crRNA, introducing an additional challenge in comparison with all other OGT.
Among all OGTs, ASOs and siRNAs remain at the forefront of successful clinical applications. In cell culture, siRNAs often demonstrate greater efficiency at lower concentrations compared to ASOs [27]. One major contributing factor is the RISC, which is more stable in cytoplasm, and processive [28], compared to the transient and less processive RNase H/ASO/mRNA complex [29]. In a direct comparison between siRNA and ASO efficiency against Influenza A viral RNA, Piasecka et al. showed that 8 nM siRNA achieved >84% decrease in viral RNA copies while ASO at the same concentration showed negligible activity [30]. Even at a much higher concentration of 750 nM ASO achieved only about 27% decrease in viral RNA level. Interestingly, despite the superior potency of siRNAs in vitro, ASOs have shown greater clinical success, with more approvals from Food and Drug Administration (FDA) and the European Medicines Agency (EMA) over the last two decades (Table 1). In contrast, siRNAs only entered the market in the last seven years [31].
FDA/EMA approved OGT formulations.
| Drug Name | Agent Type | Approval | Target Disease | Target Gene | Delivery Vehicle | Efficacy Data | Dosing Regimen |
|---|---|---|---|---|---|---|---|
| Fomivirsen (Vitravene) | ASO | FDA/EMA: 1998/1999 (Discontinued in 2004/2002) | Cytomegalovirus retinitis | CMV IE2 mRNA | N/A | Significant reduction in CMV replication in the eye | 330 µg once weekly for up to 3 weeks / IT injection |
| Mipomersen (Kynamro) | ASO | FDA/EMA: 2013 (Discontinued in 2019) | Homozygous familial hypercholesterolemia (HoFH) | ApoB | N/A | Reduced LDL-C levels by approximately 25% | 200 mg once weekly /SC injection |
| Nusinersen (Spinraza) | ASO | FDA/EMA: 2016 | Spinal muscular atrophy (SMA) | SMN2 | N/A | Improved motor function in SMA patients; achieved motor milestones in ~40% of treated patients | Initial: Loading dose of 12 mg on days 0, 14, and 28; Maintenance: every four months thereafter / IT injection |
| Milasen | ASO | compassionate use - 2018 | Batten disease | CLN7 | N/A | Improved motor function and cognitive abilities in case studies | 42 mg once every 3 months / ITH |
| Inotersen (Tegsedi) | ASO | FDA/EMA: 2018 | Hereditary transthyretin amyloidosis (hATTR) | TTR | N/A | Significant reduction in serum TTR levels (~80%) | 284 mg weekly for first three weeks / SC injection |
| Eteplirsen (Exondys 51) | ASO | FDA: 2016 | Duchenne muscular dystrophy (DMD) | DMD | N/A | Increased dystrophin levels by ~0.93% of normal after 180 weeks; improved motor function in some patients | 30 mg/kg once weekly / IV infusion |
| Golodirsen (Vyondys 53) | ASO | FDA: 2019 | Duchenne muscular dystrophy (DMD) | DMD | N/A | Increased dystrophin production in muscle tissue by ~60% | 30 mg/kg once weekly / IV infusion |
| Viltolarsen (Viltepso) | ASO | FDA: 2020 | Duchenne muscular dystrophy (DMD) | DMD | N/A | Increased dystrophin production in muscle tissue by ~60% | 80 mg/kg once weekly / IV infusion |
| Casimersen (Amondys 45) | ASO | FDA: 2021 | Duchenne muscular dystrophy (DMD) | DMD | N/A | Increased dystrophin production in muscle tissue by ~60% | 30 mg/kg once weekly / IV infusion |
| Tofersen (Qalsody) | ASO | FDA: 2023 | Amyotrophic lateral sclerosis (ALS) | SOD1 | N/A | Significant reduction in SOD1 levels; improved clinical outcomes in treated patients | Initial: Loading dose of 100 mg; Maintenance: every four weeks thereafter / IT injection |
| Volanesorsen (Waylivra) | ASO | EMA: 2019 | Familial chylomicronemia syndrome | ApoC-III | N/A | Reduces triglyceride levels significantly | Initial dose followed by maintenance dose every week |
| Patisiran (Onpattro) | siRNA | FDA/EMA: 2018 | Hereditary transthyretin amyloidosis (hATTR) polyneuropathy | TTR | Lipid nanoparticles (LNPs) | 80% reduction in serum TTR levels after 18 months | Initial: 0.3 mg/kg IV every 3 weeks (<100 kg); Maintenance: 30 mg IV every 3 weeks (≥100 kg) / IV infusion |
| Givosiran (Givlaari) | siRNA | FDA/EMA: 2019/2020 | Acute hepatic porphyria (AHP) | ALAS1 | GalNAc-conjugated siRNA | Significant reduction in ALA levels during acute attacks | 2.5 mg/kg once monthly / SC injection |
| Lumasiran (Oxlumo) | siRNA | FDA/EMA: 2020 | Primary hyperoxaluria type 1 (PH1) | HAO1 | GalNAc-conjugated siRNA | Significant reduction in urinary oxalate excretion by ~60% at 6 months | Initial: 3 mg/kg once monthly; Maintenance: 1 mg/kg SC once monthly / SC injection |
| Inclisiran (Leqvio) | siRNA | FDA/EMA: 2021/2020 | Heterozygous familial hypercholesterolemia and atherosclerotic cardiovascular disease | PCSK9 | GalNAc-conjugated siRNA | Significant reduction in LDL-C levels by ~50% at 6 months | Initial: 284 mg at day 1 and day 90; Maintenance: every 6 months / SC injection |
| Vutrisiran (AMVUTTRA) | siRNA | FDA/EMA: 2022 | Hereditary transthyretin amyloidosis (hATTR) cardiomyopathy | TTR | GalNAc-conjugated siRNA | Sustained reduction in serum TTR levels over dosing interval (up to ~80%) | Loading: 25 mg once monthly for three months; Maintenance: every three months / SC injection |
| Nedosiran (Rivfloza) | siRNA | FDA/EMA: 2023 | Primary hyperoxaluria type 1 (PH1) | GPD1L | GalXC™ RNAi platform | Significant reduction in urinary oxalate excretion by ~70% at month 6 | Initial: 3 mg/kg once every month; Maintenance: every three months / SC injection |
On the other hand, the therapeutic application of miRs is limited due to challenges with off-target effects and lack of specificity that stems directly from their natural function to regulate activity of multiple mRNA targets [32]. Similarly, CRISPR/Cas13 faces specificity challenges due to the collateral RNA cleavage, also known as collateral damage, which occurs due to the physical separation between the crRNA/RNA complex and the catalytic site of Cas13 [33-35]. Dzs and Rzs, while promising in vitro, have not yet achieved clinical translation mainly due to the stability issues, low affinity to the folded mRNA as discussed below and, possibly, due to the low intracellular concentration of Mg2+, a co-enzyme required for their catalytic activity. dependence.
3. The use of OGT-based drugs is limited by their low efficacy
3.1 The clinical efficacy of antiviral OGTs cannot yet compete with that of established treatments
While OGTs against infectious diseases have shown promising results in experimental research, their progress through clinical trials has been challenging [36]. Fomiversen, developed against Cytomegalovirus (CMV) retinitis, was the first antisense-RNA agent to gain FDA approval in 1998 and remains the only antiviral OGT to have passed clinical trials and reached the market. However, it was discontinued less than a decade later due to the low demand and market competition against the new and highly efficient antiretroviral treatments of the time [37].
Since then, several OGTs have entered clinical trials against viruses including Respiratory Syncytial Virus (RSV), Hepatitis B Virus (HBV), Ebola Virus and Human Immunodeficiency Virus (HIV). Among those, VIR-2218 and JNJ-3989 are currently in ongoing phase II trials for chronic HBV include. VIR-2218 is a N-Acetylgalactosamine (GalNAc) conjugated version of ALN-HBV - an earlier siRNA agent from the same company. VIR-2218 demonstrated improved safety: no elevation in liver inflammation markers was observed, compared to 28% elevation in those treated with ALN-HBV. The VIR-2218 efficiency was evident by dose-dependent reduction in Hepatitis B surface antigen (HbsAg) [38]. Treatment of HBV patients with JNJ-3989 provided sustained HBsAg reduction for up to 336 days after the last dose, although complete HBsAg clearance remained rare as demonstrated in the recent phase IIb study [39]. RG6346, a new agent against HBV in phase I trials, has also showed favorable safety and pharmacodynamic profile with reductions in HBV protein levels, although efficacy data are not yet available [40].
Other OGTs in phase II trials such as ALN-RSV01 against respiratory syncytial virus (RSV) and TKM-130803 against Ebola virus were discontinued due to the failure in meeting the target suppression efficacy and, in the case of Ebola, the lack of improvement in patient survival [36]. Interestingly, OGTs against HIV have been in phase I trials since their initiation in 2007, largely due to the complexity of HIV infection dynamic and lack of comprehensive pre-clinical studies [36].
Overall, the stagnation of antiviral OGT development can be attributed to persistent concerns regarding efficacy and safety. Moreover, the availability of established antiviral therapies presents a significant barrier to adopting OGTs. When existing treatments are effective, there may be less incentive for healthcare providers to switch to new therapies that have not yet demonstrated clear clinical superiority.
3.2 In mitigating genetic disorders, clinical efficacy is satisfactory due to the low competition from alternative treatments
For many genetic disorders, existing therapies are limited to symptom management and supportive care. The 16 OGTs approved for genetic disorders (Table 1) partially address the scarcity of disease-modifying treatment options. However, their perceived success is influenced by the lack of alternative effective treatments, making OGT the only treatment available. For example, Mipomersen, the first ASO approved for treating Homozygous familial hypercholesterolemia, was withdrawn due to market competition. Although Mipomersen could decrease low density lipoprotein cholesterol (LDL-C) by 28-36% and apolipoprotein B (ApoB) by 36-38%, it also carried a significant risk of hepatotoxicity [37,41]. Safer and more efficient pharmacological alternatives, such as monoclonal antibody-based PCSK9 inhibitors and statins that reduce LDL-C more than 50%, were preferred. Similarly, Volanesorsen for Familial Chylomicronemia Syndrome (FCS) faced FDA rejection due to safety concerns [42] and Tominersen for Huntington's Disease was discontinued due to lack of efficacy [43].
Furthermore, even the efficacy of FDA approved drugs, like Eteplirsen for some types of Duchenne muscular dystrophy, is under question. Patients receiving Eteplirsen weekly showed an increase in muscle dystrophin levels by only ~0.44% ± 0.43% of that of healthy individuals, up from a baseline of ~0.16% ± 0.12% with the median increase just 0.1% after 48 weeks. The EMA has not approved Eteplirsen, citing insufficient evidence of its efficacy [44]. This example highlights both differences in regulatory standards and the broader challenges of OGT in providing meaningful clinical outcomes.
A notable example of successful clinical OGT applications are the FDA and EMA approved gene therapies for hereditary transthyretin-mediated amyloidosis (hATTR). hATTR is caused by the deposit of both mutant and wild-type transthyretin (TTR) variants mainly in the nervous system, causing severe multisystem neurological manifestations. Traditional treatment strategies for hATTR include TTR stabilizers, liver transplantation, as well as neuropathy and cardiomyopathy management. All four approved OGTs, Patisiran [45], Vutrisiran [46], Inotersen [47], and Eplontersen (ASO currently under review) [48], target a specific genetically conserved region in the 3' untranslated region (3'-UTR) of all TTR isoforms. Each of these agents demonstrates the same significant efficacy of ~80-85% TTR knockdown. Notably, siRNA-based treatment requires lower and less frequent dosing compared to ASO (Table 1), supporting earlier observation that siRNAs are more potent than ASOs.
However, some siRNAs have encountered setbacks in clinical trials. One example is Revusiran targeting hATTR, which was withdrawn after the randomized, double-blind, placebo-controlled Phase III trial [49]. The trial was terminated in 2016 due to higher mortality observed in the treatment group compared with placebo [50]. Another failed RNAi agent is Fitusiran. It was developed to treat hemophilia A and B by targeting antithrombin to increase thrombin generation, thus promoting clot formation. However, during clinical trials, patients experienced serious thrombotic events, leading to a temporary suspension of the trials in 2017 and again in 2021. Although, trials have since resumed and completed, comprehensive safety and efficacy data have not been fully evaluated yet [51].
While not classified as OGT, Imetelstat is worth mentioning here due to its approval by the FDA in June 2024 and EMA in March 2025. Imetelstat is a 13-mer DNA oligonucleotide that acts as a first-in-class telomerase inhibitor, functioning not through gene silencing but via hybridization-dependent inhibition of enzyme active site. Clinical data suggest that Imetelstat restores normal haematopoiesis in patients with low-to-intermediate risk myelodysplastic syndromes and transfusion-dependent anaemia [52]. Other emerging modalities such as circular RNA and tRNA-derived fragments display significant regulatory properties that could be used for therapeutic purposes. However, their development is at an early stage with efficacy data being limited to date [53-55].
3.3 In cancer, clinical efficacy of OGTs is weak and relevant only as complementary treatment
The targets selected for cancer gene therapy encompass a wide range of molecular and genetic factors associated with cancer initiation, progression, and treatment response. These targets include oncomarkers such as dysregulated angiogenic factors, tumor suppressor genes, drug resistance genes like MDR1, and proteins such as survivin and VEGF, all of which underscore the potential of gene therapy to address diverse aspects of cancer biology. However, most currently utilized targets demonstrate insufficient efficacy in cancer cell elimination, poor treatment specificity, and are considered unsuitable as a monotherapy against cancer [56,57]. As of 2022, approximately 70 anticancer ASO and 20 of siRNA entered clinical trials. Of these, only two ASOs reached phase III [58], and none have received FDA approval to date.
Bcl-2 mRNA remains one of the most studied targets in anticancer OGT development. For example, BP1002 and PNT2258 entered phase I, while Oblimersen (G3139) passed phase III trials. Bcl-2 silencing by Oblimersen aimed to restore cancer cell sensitivity to chemotherapy. However, when combined with chemotherapeutic agents such as cisplatin and 5-fluorouracil, it achieved only slight improvement of 43% in overall survival compared to 40% in the control group [59]. Another study reported that AZD4785, which targeted mutated KRAS mRNA and successfully completed phase I clinical trials, but no updates have been published since 2017 [60]. Similarly, VEGF-targeting ASOs achieved therapeutic effect in combination with chemotherapeutics pemetrexed and cisplatin [61]. Phase I/II trials for VEGF-ASO were completed in 2011 without any further updates.
Except for approaches that seem to sensitize cancer cells to chemotherapeutics [62] other combination strategies under investigation include OGTs engineered to improve radiotherapy sensitivity [63], and dendritic cell immunotherapy response [64], although these approaches have not been tested in clinic yet. Collectively, these data support the view that monotherapy targeting traditional mRNA may not be sufficient to address the complexities of cancer thus highlighting the necessity of finding alternative approaches.
On the other hand, protein inhibitors for the same targets have been effective both as monotherapy and in combination with immunotherapy even against targets that were previously considered “undruggable”. The most popular Bcl-2 small molecule inhibitor, Venetoclax, was FDA approved in 2016 for the treatment of chronic and acute myeloid leukemia [65]. These results indicate that OGTs targeting Bcl-2 currently cannot compete with the small molecule inhibitors in terms of efficacy. Similarly, in 2022, the FDA approved Krazati, a KRAS inhibitor, for patients with non-small cell lung cancer [66].
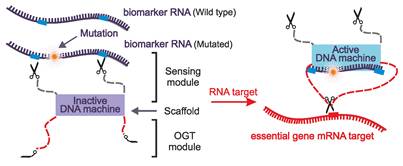
We believe that the idea of targeting cancer-related genes is fundamentally defective, as it merely suppresses malignant traits of the cancer cells rather than irradicates cancer cells, which is the ultimate therapeutic goal (Table 2) [67]. A more promising strategy could be the conditional activation of programmable agents triggered by cancer-specific genes, followed by downregulation of the vital genes that triggers cancer cell death. Several research groups have taken advantage of the programmability of nucleic acids to develop nucleic acid-based nanostructures that release the OGTs upon recognition of cancer markers. Examples include siRNA probes that are converted into Dicer substrate siRNA by an RNA trigger [68], reconfigurable nucleic acid nanoparticles that elicit a therapeutic siRNA response in the presence of mutated KRAS [69], RNA/DNA hybrids that re-assemble into active siRNA in the presence of their cognate hybrids [70], endogenous miRNA-triggered DNA nanostructures for the release of multiple, multifunctional siRNAs [71,72], and miRNA-triggered ASO release [73]. Our group has proposed targeting mRNA of vital genes exclusively in the presence of cancer-related mRNAs using marker-dependent Dz and ASO agents [67,74]. They include binary ASO that is active only in presence of KRAS RNA [75], binary Dz nanomachines [76], Dzs and ASOs that are activate at high but not low concentrations of miRs [77-79], and Dz-based logic gates that operate based on miRNA expression patterns [80]. The principle of DNA nanomachines for the conditional activation of OGTs is shown in Figure 3. The 'Cut' function of these nanomachines recognizes cancer-related mutations with high specificity and cleaves mRNA at two sites to release the cancer marker fragment. This fragment is retained by the DNA nanomachine and serves as an activator either the Dz or ASO function, which suppresses the targeted vital gene, ultimately triggering apoptosis in cancer cells.
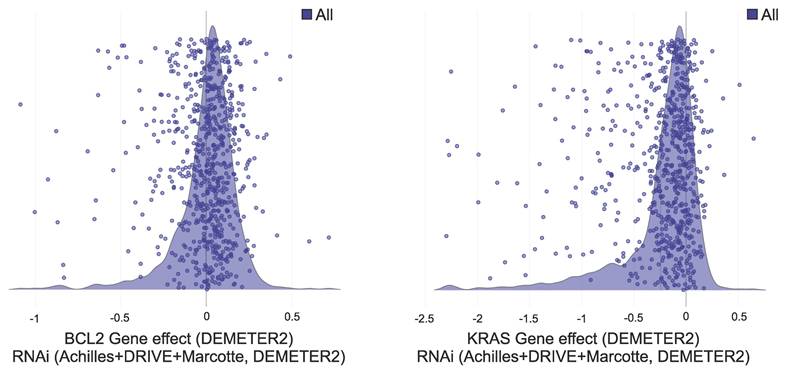
Essentiality of several cancer related genes that are commonly used as targets in cancer gene silencing research. The overall gene effect of each gene (DEMETER2 score) was calculated as the mean of its gene effect on each distinct cancel cell line included in the DepMap Portal database (as shown in Figure 2).
| Gene | Gene Effect RNAi (Achilles+DRIVE+Marcotte, DEMETER2) | Druggable Structure |
|---|---|---|
| KRAS | -0.06 | yes |
| BCL-2 | 0.04 | yes |
| MYC | -0.5 | yes |
| BMI1 | -0.03 | yes |
| EGFR | 0.05 | yes |
| BRCA1 | -0.1 | yes |
| CXCL14 | -0.1 | no |
| YAP | 0.03 | no |
| IDO1 | 0.1 | yes |
| TP53 | -0.1 | yes |
| mTOR | -0.5 | yes |
| STAT3 | -0.1 | yes |
| VEGFA | -0.1 | yes |
Graphs representing the gene effect (RNAi, Achilles+DRIVE+Marcotte, DEMETER2) of A) BCL2 and B) KRAS genes. Each purple dot on the graph represents a cell line on which the gene effect of the respective gene was tested by cell depletion assay. In the DEMETER2 scoring system, lower scores indicate higher gene essentiality; a score of 0 suggests the gene is non-essential in that cell line; a score of -1 approximates the median score for pan-essential genes, reflecting high essentiality. For both BCL2 and KRAS, most cell lines fall within the gene effect range of [-0.5, 0.5], suggesting low essentiality, while only a limited number of cell lines exhibit scores approaching or exceeding -1.
Conditionally activated OGT for the knockdown of vital genes [67,76]. The inactive DNA machine recognizes the mutated biomarker's RNA and cleaves it at predetermined sites. The cleavage product is then used to activate the DNA machine which acts as an OGT and silences the mRNA of the target essential gene, ultimately inducing apoptosis in cancer cells.
Considering the variability in vital gene expression across different tissues, target selection for such programmable systems should be tailored to specific cell type, taking into consideration environmental stressors including hormonal status [81,82]. Beyond selecting cancer-related activator genes chosen solely based on the individual cancer type and its dysregulations, this approach aligns with the principles of personalized medicine. The use of programmable OGTs capable of implementing the principles of Boolean logic could also offer a solution to cancer cell heterogeneity by enabling processing complex gene expression patterns and incorporating the synthetic lethality approaches.
4. Factors contributing to low OGTs' efficiency
4.1 Both the tissue and the cell specific delivery are not efficient
One of the major limitations of the clinical success of OGT is the efficient and tissue-specific delivery of these agents. Naked ONs face a short half-life when in the bloodstream owing to nuclease dependent rapid degradation [83]. Advances in chemical modifications and delivery platforms have partially improved this problem, optimizing the biodistribution of OGTs. Biodistribution, however, remains a critical factor influencing therapeutic efficacy. For example, after systemic administration, drugs often accumulate in the liver due to interactions with lipid transport proteins, reducing their chances to reach target tissues. Key physicochemical properties such as size, shape, and charge significantly influence biodistribution. For example, overly small or negatively charged nanoparticles have higher clearance rates [84,85]. Since systemic administration often results in poor extrahepatic targeting, alternative delivery routes can enhance tissue accumulation and therapeutic efficacy [86-88]. Local administration is generally preferred, as it requires lower doses and is less invasive [89].
Lipid-based delivery systems are currently among the most effective non-viral methods for in vivo nucleic acid delivery, with several products approved for clinical use (Table 1), while many candidates remain in trials. Although lipid-based systems exhibit passive targeting capabilities, their lipid composition influences the protein corona on their surface, affecting tissue preference [90]. Targeting efficiency can vary from approximately 5% to 90% depending on cell type [90], yet these systems still predominantly accumulate in the liver [91] due to association with ApoE and low density lipoprotein receptor (LDLR)-mediated endocytosis [92]. While this property favors hepatocyte targeting, it limits efficacy in other tissues and increases metabolic degradation in the liver [90]. Enhancing these systems with cell-specific penetrating proteins (CPPs) or antibodies, or direct bioconjugation with OGTs may help address these challenges [93].
Direct bioconjugation of OGT with lipids, cell penetrating peptides (CPPs), aptamers, antibodies, and sugars, enhances delivery by improving the specificity, cellular recognition, internalization and overall efficiency. CPPs have shown efficacy in targeting tissues such as skeletal muscle [94], heart [95], and the central nervous system [96], although transfection efficiencies varies depending on the cell type [97,98].
Bioconjugation with lipid moieties, such as cholesterol, enhances delivery by promoting endosomal escape than CPPs, primarily through facilitating membrane fusion and endosomal destabilization [93,99-102]. The endocytosis of cholesterol-OGTs is mediated by scavenger receptor type B1 (SCARB1, SR-B1) or LDLR, for HDL and LDL particles, respectively [103], leveraging the body's endogenous lipid transport and uptake system in vivo [104], though this approach favors hepatic accumulation over others tissue [105]. GalNAc conjugates, several of which are clinically approved (Table 1), are particularly effective for liver-targeted delivery. These conjugates bind to the asialoglycoprotein receptor (ASGR) for rapid internalization via clathrin-dependent endocytosis [106]. Experimentally, modifications such as 5′-(E)vinylphosphonate [107] and β-cyclodextrins [108] have been shown to enhance potency and stability of GalNAc-conjugated OGTs by 5-10 fold. Triantennary GalNAc, featuring three GalNAc sugars branching from a central core, significantly improves binding and efficacy [106,109].
Exosomes have also gained attention over the past decade due to their natural transport capabilities, long circulation time, and biocompatibility, making them promising vehicles for OGTs [110,111]. These lipid bilayer-encapsulated vesicles naturally facilitate intercellular communication and can traverse biological membranes [112], while evading phagocytosis and enhancing bioavailability [113,114]. Human exosomes are particularly promising due to their low immunogenicity and compatibility with RNA silencing applications [115]. They can be derived from patients to minimize immune responses [116] and exhibit natural tropism toward their parent cells, with potential for engineering enhanced targeting capabilities [117]. Exosomes release cargo through surface receptor interactions, fusion with the plasma membrane, or endocytosis via the endolysosomal pathway [118]. They effectively protect cargo from lysosomal degradation and facilitate release, demonstrating strong target gene silencing [119,120] and tumor size reduction [121,122]. Ongoing research focuses on optimizing exosome properties for therapeutic applications [123-125] with emphasis on improving cargo loading and interactions with sorting proteins [126].
Although non-viral methods generally exhibit lower transfection efficiencies compared to viral vectors, progress is being made in optimizing their formulations [127]. Specifically, transfection efficiencies up to ~10% have been reported for the most promising lipid-based systems [128], although this depends on various factors. The cell cycle phase plays a crucial role in intracellular delivery with mitotic with mitotic cells showing higher internalization rates [129,130]. The chemistry of both the delivery vehicle and the OGT itself also significantly impacts the productive uptake in different cell and tissue types [105]. These parameters complicate platform evaluation and raise concerns about reproducibility across different biological systems.
4.2 OGTs' concentrations affect the efficiency of cellular uptake
The concentration of OGTs is a critical determinant of their efficacy and safety in gene therapy applications. While higher concentrations may have higher efficacy, both in vitro [131] and in vivo [132], they can also induce stronger immune responses [132,133] and cellular toxicity [134], complicating data interpretation and reducing the clinical significance of such treatments. Conversely, lower concentrations may minimize toxicity and adverse effects but generally are expected to reduce gene silencing efficacy. Traditionally, in siRNA studies, concentrations as low as 1-10 nM are considered sufficient for effective target gene silencing in cell culture models [135]. Concentrations, such as 50 nM or 100 nM, are often considered high and may induce off-target effects and cellular toxicity without significantly improving silencing [136]. In ASO studies, concentrations between 10 - 100 nM are often found to be optimal, while concentrations 100 - 1 µM are typically categorized as high.
The concentration of OGTs directly affects the loading efficiency onto delivery vehicles and their ability to penetrate cell membranes and reach intracellular compartments. For example, Liu Yang et al. reported that the gene silencing efficiency improved from 10.9% to 79.5% as the nitrogen in lipids to phosphate in ASO ratio (N/P) increased from 2.5 to 15, highlighting that effective loading enhances cellular uptake and subsequent gene silencing. At the highest tested ASOs concentrations of 7.5, 15 and 30 nM, more than 50% target silencing was achieved, while cell viability remained above 75%. This data indicates that efficient liposome loading correlates with silencing efficacy, but also suggests a threshold beyond which further increases in concentration do not yield proportional improvement in gene silencing [137]. Similarly, Xie et al. reported that siRNA delivery via transferrin-polyethyleneimine (Tf-PEI) resulted in effective gene silencing in activated T cells. Tf-PEI polyplexes fully condensed 50 pmol of siRNA at an N/P of 7.5 but showed more productive internalization at higher N/P of 10, 15 and 20, with N/P of 15 reaching approximately 60% target gene downregulation [138]. These data underscore the importance of understanding the dose-response relationship and the biological mechanisms to advance the development of effective gene therapies.
4.3 The endosomal escape efficiency is low
In addition to tissue specificity, the efficient internalization and release of OGTs into cell cytoplasm is another critical factor of therapeutic success. Either naked or encapsulated in delivery vehicles, OGTs are typically internalized via endocytosis [139]. Endosomal escape is the process by which the endosomal cargo exits the endosome to reach its target site within intracellular compartments. However, it has been estimated that approximately 99% of OGTs remain entrapped within endosomes, hindering their cytosolic delivery and subsequently reducing treatment efficacy [140,141]. This entrapment may result in the two possible outcomes; i) natural maturation of early endosomes to late endosomes and lysosomes, where the cargo is eventually digested; or ii) the formation of a poorly defined so-called “depot endosome” in which the cargo remains intact for extended periods.
While the precise mechanistic process of endosomal escape still remains unclear, two main mechanisms have been suggested as potential escape routes from the depot endosomes; i) endosomal rupture, also known as the proton-sponge effect, where the endosome irrevocably ruptures releasing its whole cargo into the cytosol (Figure 4); and ii), or polyplex/polymer-mediated enhanced endosomal escape, in which small and reversible breaches in the endosomal lipid bilayer allow small amounts of cargo to escape into the cytosol (Figure 4) [140,142,143]. The latter mechanism may potentially explain the prolonged therapeutic effect observed in both experimental and clinical settings [144]. However, localized disruption of the endosomal lipid bilayer membrane is a rare and transient event that represents the rate-limiting step in the endosomal escape process [140]. In contrast, endosomal or lysosomal rupture can induce non-specific cytotoxicity as it is known to activate the inflammasome [145] that may lead to cell death [146]. The acidic environment within endosomes stimulates the endosomal escape of the entrapped cargo. The efficiency of the escape is also affected by several proteins of the endosomal and lysosomal systems [147-150]. A better understanding of these mechanisms could offer strategies for enhancing endosomal escape.
Possible fates of internalized OGTs. OGTs are not capable of diffusing through the cell membrane; thus they are taken up through endocytosis. Instead of undergoing maturation, “depot” endosomes can be formed, and release OGTs by the two mechanisms: (i) the proton sponge hypothesis suggests that the increased activity of membrane-bound ATPases causes increased influx of H+ and Cl- ions which is counteracted by water entry, resulting in swelling and rupture of the vesicles; (ii) the polyplex/polymer mediated escape theory suggests that the cationic polyplex/polymer is further protonated in the acidic environment (also impacted by ATPases activity), leading to enhanced direct interaction with the anionic lipids of the vesicle membrane, causing local destabilization of the membrane integrity and reversible pore formation through which the slow release of the cargo can occur [140,143].
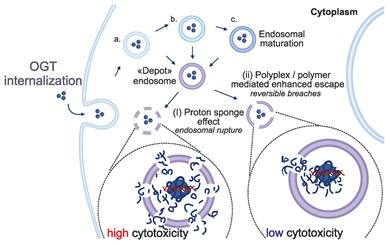
It is believed that cationic and hydrophobic delivery vesicles, nanoparticles and OGT conjugates can facilitate the rate-limiting step of endosomal escape. Cationic conjugates may electrostatically associate with the lumen anionic of endosomal bilayer, while hydrophobic conjugates may penetrate the lipid bilayer. Both processes can cause small transient breaches in the membrane that allow gradual release of OGTs [140]. Currently, several of these delivery systems are being actively investigated to increase internalization, enhance release and ultimately increased therapeutic efficacy [151,152].
4.4 The choice of the targeted sequence within mRNA affects efficacy
Efficient endosomal escape does not automatically guarantee effective target silencing. For therapeutic action to occur, the OGT must access and efficiently bind specific site within mRNA target. Proper target sequence selection requires consideration of several factors, including the identification of the disease-causing gene and its specific transcript, localization of the target mRNA, target sequence accessibility for OGT binding, conservation of the binding site across gene isoforms, possible nucleotide variations in the targeted sequence and unintended non-specific OGT interactions with the human transcriptome.
Conservation of the targeted site across multiple gene isoforms or even between healthy and mutated transcripts is of the utmost importance. The degree and nature of conservation should be evaluated in the context of the disease, the specific targeted gene, and the intended therapeutic strategy. For example, in the development of antiviral therapeutics, suppression of several essential and conserved genes could inhibit viral replication across multiple strains. In 2022, Yi-Chung Chang et al. demonstrated that siRNAs targeting conserved regions of vital Sars-Cov-2 genes such as RDRP, spike, and helicase, significantly inhibited multiple Sars-Cov-2 strains, including Delta. Viral replication decreased by 99% in vitro and in vivo when tested in infected mice receiving prophylactic treatment [153]. Similar findings have been reported for OGT-based antiviral against Sars-Cov-2 [154-157], influenza [30,155,156,158-160], HBV [161] and others [156,160,162]. Notably, antiviral activity can be elicited by targeting not only vRNA but also the host mRNA. For example, a study by Friefrich et al. in 2022 showed that an siRNA targeting exon1 of ACE2 mRNA, which serves as the entry receptor for SARS-CoV-2, reduced both ACE2 mRNA and protein levels by up to 90% for at least six days [163]. Perhaps the most successful example of proper target selection is the FDA approved RNAi agent Patisiran against hATTR amyloidosis, which targets so highly accessible and conserved region of the TTR gene that all the subsequently approved OGTs target the same exact site.
Target sequences must also be unique to the target gene to avoid unintended side effects. Computational tools [164] such as siDirect [165], DNAzyme builder [166] are available to create safe OGTs with minimal off-target effects. However, in practice, complete specificity is difficult to achieve. An OGT of approximately 20 nucleotides has a high potential of being partially complementary to multiple sites in the human transcriptome. Such off-target interactions can not only cause side effects but severely reduce on-target activity. Nevertheless, off-target effects are not always detrimental. Interestingly, in the same Yi-Chung Chang's study [153], one of their major off-target was CXCL5, a chemokine involved in humoral immunity. The authors hypothesized that this off-target interaction may have contributed to the antiviral effect of their OGT due to the CXCL5role in COVID-19-associated pathogenesis.
RNA secondary and tertiary structures influence hybridization thermodynamics and targeted site accessibility and thus determine OGT efficiency. It is well documented that ssRNA sites, such as loops, are more accessible and show better suppression efficiency when targeted than dsRNA regions such as stems [167,168]. For instance, Cas13 gRNAs targeting dsRNA regions of the long non-coding RNA XIST showed low gene silencing), which increased by almost five-fold when single-stranded regions of the same RNA were targeted instead [169]. At the same time, RNA binding domains (RBDs) that can be occupied by proteins should be considered during target site selection as RNA binding proteins (RBPs) can inhibit gene silencing via steric hindrance or enhance it via natural RBP interactions such as those observed for Ago/miRNA mediated gene regulation [170]. High ribosomal activity can also make the target site more accessible by unfolding its complex structure, facilitating RISC interactions and higher siRNA efficiency [171]. RNA secondary structure can be predicted by several tools, such as RNAstructure [172] or icSHAPE [173]. However, in vitro assays still offer a more accurate assessment by confirming the presence of stable, conserved structural elements, while also revealing many previously unknown structures [174]. For instance, Kierzek's group has extensively studied viral RNA structural motifs for OGT design. They identified regions in segment 5 of IAV mRNA that were accessible by ASO and RNAi agents [175,176], and achieved nearly 90% reduction of the viral titer [177] thus demonstrating how knowledge of secondary RNA structures can help in OGT design.
4.5 OGTs' design can increase efficiency by protecting from nuclease degradation, achieving affinity/specificity balance and enhancing OGT/protein interactions
To increase the probability of successful target binding, OGT design guidelines recommend avoiding sequences with strong internal secondary structures, maintaining a GC content of 30-50%, avoiding runs of four or more identical nucleotides, and excluding sequences that can target unintended genes due to partial sequence complementarity. Commonly, ~20 nt long OGT/target hybridization sites show the best knockdown efficiency, due to their sufficient affinity for the target under physiological conditions [178-181]. Lower affinity can reduce silencing efficacy due to unstable binding. However, the high affinity of the 20 nt OGT may cause non-specific binding to partially complementary fragments of unintended targets. The affinity/specificity dilemma suggests that the higher the affinity is, the lower the specificity of OGTs and vice versa [4,182]. The fragile OGT/mRNA complex stability should be fine-tuned by careful sequence design and/or by introducing chemical or structural alterations that can help attain an optimal affinity/specificity balance.
Advances in ON chemistry have improved ASO performance [183], with the phosphorothioate (PS) backbone remaining prevalent for its role in ON trafficking [184] and RNase H recruitment [185]. However, PS alone is insufficient to fully protect ASOs from nucleases and can increase cytotoxicity. This has led to the development of alternative backbones like mesylphosphoramidate (MsPA) [186,187]. For instance, Patutina et al. reported up to 90% target silencing in vitro after 72 h post-transfection with 100 nM MsPA ASOs, and up to 95% target silencing in vivo compared to approximately 50% silencing by PS-modified ASOs [188]. Sugar modifications such as 2'-O-methyl (2'OMe), 2'-methoxyethyl (2'MOe) [189,190] and locked nucleic acids (LNA) [191-193] have enhanced both stability and binding affinity but do not support RNase H activity. To address this problem, the 'gapmer' design was introduced. It combines central PS or native DNA to support RNase H activity with affinity-boosting modifications on the 5' and 3' ends [194]. LNA gapmers are among the most effective, allowing fine-tuning of affinity and demonstrating superior efficacy. Shin et al. have shown that an LNA gapmer achieved approximately 90% in vivo gene silencing in the lungs of mice two days after intratracheal administration, outperforming 2'MOe gapmers that achieved only 60% gene silencing [195]. Gapmers combining LNA, cEt, 2'-FANA, and 2'-5 linkages have been utilized for SNP-selective targeting [158,196,197]. Nonetheless, excessive ASO/target affinity can be counterproductive by limiting ASO recycling and depleting the ASO pool through off-target binding [198,199], a challenge shared by other nucleic acid systems guided by nucleases or ribozymes [200,201].
siRNA modifications should not affect Ago activity and not alter the agent's conformation, as an RNA-like conformation is essential for RISC assembly and activation. Extensive studies have shown that strategically placed modifications, like alternating 2'OMe and 2'-fluoro (2'F) or PS linkages at the edges of each strand, enhanced siRNA stability without compromising activity [142,202]. Hassler et al. reported that fully modified siRNA achieves efficient tissue accumulation, RISC loading, and gene silencing, with an IC50 of 0.9 nM versus 3.5 nM of native siRNA [203]. The 5'-phosphate of siRNA's guide strand is critical for RISC recognition. Chemical stabilization, such as introducing 5'-E-vinylphosphonate (5'-E-VP) and PS modifications, strengthens the 5'-end against nucleases without impeding RISC interaction, improving IC50 of 81 nM compared to 217 nM for native siRNA against PPIB mRNA, as shown by Haraszti et al. [204]. 5'-E-VP introduction was also shown to extend the siRNA-based silencing duration for over 30 days in rapidly dividing cells [205]. Special attention is given to the seed region of the siRNA guide strand. Fully or partially modified seed regions can alter both affinity and specificity to the intended target either positively, by reducing off-targets, or negatively, by disrupting structural integrity of the region thus inhibiting target binding [206-208]. It has been proposed, though, that the seed region should be considered a dual-functional domain and optimized to balance high target affinity and low off-target activity when modified with 2'OMe [209].
The role of the passenger strand must also be considered since it can be mistakenly loaded into the RISC, reducing efficacy and producing off-target effects. Some modifications such as LNA can significantly inhibit RISC activity if introduced in the guide strand. However, they may be useful for minimizing the unintentional passenger strand activity [208]. Likewise, bulky phosphoryl guanidine (PG) groups at the 5' end of the passenger strand can be used to reduce off-target effects, leading to approximately 0.5 fold change (FC) in on-target silencing compared to 0.9 FC of non-PG-modified siRNA [210]. Since 5' end modification can increase the RISC loading probability of a strand, modifying the 5' end of the guide strand can also help avoid unintended off-targets [211].
DZs and RZs, on the other hand, are better predisposed to address the affinity/specificity dilemma due to their use of two relatively short arms that cooperatively bind targeted RNA. The length of these arms can be adjusted to increase affinity to their target [212], increase specificity against SNVs [76], or be asymmetric to achieve efficiency/specificity balance [213,214]. DZs and RZs are governed by a well-characterized enzymatic cycle: 1) target binding, 2) catalytic reaction and 3) product release. Each step is crucial for achieving optimal reaction efficiency, and arm lengths can significantly influence both the initial binding and final release steps. Contrary to the earlier assumptions that high affinity Dz arms inhibit product release, recent findings suggest that longer high affinity arms increase Dz catalytic efficiency by facilitating RNA substrate binding, which is the limiting stage of the catalytic cycle. Very long arms with Tm > 65oC are not inhibited by the product release stage, as long as the cleavage products self-fold into stable secondary structures [22,182,215,216].
High affinity of Dz to RNA can be achieved by introducing LNA nucleotides in short flanking arms [217-219]. However, chemically modifying the catalytic core remains challenging, as even small chemical modifications often inhibit the catalytic activity [17,220,221]. This limitation impairs intracellular performance, as the unmodified core is vulnerable to nuclease degradation. To overcome these issues, researchers have explored strategic combinations of chemical modifications. These approaches have achieved up to a two-fold or greater reduction in target gene expression in cell-based studies [219]. In vitro selected chemically modified variants of Dzs and Rzs have shown promising results achieving more than fourfold increase in Dz activity and a twofold higher Rzs activity compared to unmodified equivalents [222-224].
The CRISPR/Cas13 system obeys several key rules for optimal activity: it targets an ssRNA region, requires perfect matching between gRNA and target sequences especially with the 'seed region' and avoids stable gRNA secondary structure. It has also been shown that fusion of gRNA with a nuclear localization signal (NLS) can increases efficacy of Cas13 systems [225]. Although the CRISPR/Cas13 system modifications are not yet thoroughly studied, some data suggests that 2'OMe and PS modifications placed at the 3' end of the gRNA can increase its transient target silencing in human T-cells from 40-45% to 60-65% [226].
Stereopure mixtures of OGTs have been proved to outperform the respective stereorandom counterparts [227,228]. Researchers use specific chemistries to tightly control the chiral configuration of the modified backbones. For example, chimeric PS/PN (phosphoryl guanidine) containing backbones developed by Kandasamy et al. [229], displayed enhanced activity both in vitro and in vivo with better pharmacological properties than stereorandom PS modified splice switching ONs. This improvement enabled a twofold reduction in dosage during in vivo testing [229]. Chiral chemical modifications have also been shown to affect siRNA agents [230,231], although other OGTs have yet to be studied in this context. However, this approach doesn't come without challenges-nuclease stability remains a significant concern and one of the major obstacles to its broader application [232,233].
Design can also affect the immunogenicity of OGTs. OGTs rich in guanosine (G) and uridine (U), and motifs such as CpG, GU and AU, are recognized as viral RNA mimics and can activate toll-like receptors 3, 7 and 8 (TLR3, TLR7 and TLR8 respectively) [234]. Pollak et al. have shown that native and modified OGTs, especially PS-modified, induce innate immune activation, by interacting with several extracellular proteins and TLR9 [235]. Such interactions ultimately lead to cytokine and interferon production which can interfere with the cellular uptake, trafficking, and processing of the oligonucleotides, thereby reducing their ability to engage target mRNA effectively. It can also lead to the activation of the complement system and opsonization of OGTs, which promotes their clearance by phagocytes, lowers their bioavailability and half-life in circulation and tissues, thus diminishing their effective concentration at the target site. To mitigate these effects design strategies such as reducing the PS content and strategic placement of 2'OMe have been proposed [235,236]. While well-documented adverse effects such as injection site reactions, fever, chills, and systemic inflammation can complicate clinical efficacy, many studies tend to underemphasize the immunogenic effect of OGTs.
5. Theoretical vs real world efficacy
The reported efficacy of OGTs in modern literature often falls short of theoretical expectations. The expected efficacy of OGTs is typically derived from computational and empirical models designed to predict how effectively OGTs will silence target genes. Such theoretical indexes include i) the Secondary Structure Score (Sscore), which estimates the strength of local mRNA secondary structures at the OGT target site; ii) the Duplex Score (Dscore), which estimates the stability of the OGT:mRNA duplex formation; and iii) the Competition Score (Cscore), which represents the difference between Dscore and Sscore [237]. These indices are incorporated into major designing tools such as SciTools suite by IDT [238] and Ufold [239]. Well-designed OGTs, with the help of these tools, are expected to achieve over 70% knockdown of the mRNA target under optimal conditions.
However, a 2016 study by Munkacsy et al., revealed a significant gap between theory and practice [240]. After evaluating 1643 samples across 429 experiments published in 207 siRNA studies, they found that 70% knockdown — was achieved in only 10 experiments (2.3%); 50% in 166 experiments (38.7%), and 30% in 79 experiments (18.5%) of the cases. Surprisingly, the unexpected upregulation was observed in 22 experiments (5.1%). The study concluded that the choice of the cell line and the validation method had the most impact on silencing efficiency. It also highlighted issues such as the improper use of controls and variability in experimental conditions, which contribute to inconsistencies and raise questions about reliability of the results. According to a recent study of Davis et al., target-specific features, such as exon presence, ribosome occupancy and so on, may likely be responsible for the low endogenous efficacy of siRNA [171]. This highlights the need for native expression assay inclusion in OGT development studies, that is often replaced by reporter-based expression assays.
Considering only ~1% of the internalized OGTs escape into the cytosol, the low efficiency of most OGTs might not look as disappointing as it initially seems. The inconsistency of theoretical prediction with experimental findings indicate that intrinsic aspects of OGT-assisted gene silencing are yet to be understood.
6. Concluding remarks
Although extensive research has led to considerable advancements, significant challenges still hinder the full potential of OGTs in medicine. The low efficiency of OGTs remains a primary barrier to their successful translation and widespread adoption in medical practice. Tissue specific delivery, efficient internalization and endosomal escape, are the major factors affecting OGT efficacy. Despite the growing use of cationic and lipid-based delivery systems, the percentage of OGTs that escape from endosomes is disappointingly low, estimated at less than ~1%. This result suggests that poor endosomal escape is a key factor limiting OGT efficiency and their overall therapeutic performance.
While experimental studies have characterized siRNA as the most potent among OGTs, evidence suggests that only 2.3% of siRNAs reach the theoretically predicted 70% target downregulation, while approximately 40% of the siRNAs fail to achieve 50% suppression level. These results highlight the inadequacy in siRNA design and the challenges associated with achieving consistent and effective gene silencing across different studies and experimental conditions. Inconsistent methodologies, and suboptimal controls further obscure accurate assessments of OGT performance, challenging their reliability as a therapeutic tool and halting the advancement of the field. Additionally, the lack of standardized reporting, such as varying concentration units (molarity vs. mass), different assay methodologies, and endpoint variability, including short-term vs. long-term effects, further impedes result validation and can confuse the reader ultimately eroding trust in the findings. Standardizing assays and reporting are ongoing and adopting existing (e.g. MIQE for PCR data), or developing new guidelines will help in comparative data analysis.
These broader challenges are also reflected by the performance of approved OGTs in clinic that often only marginally improve patient conditions. The limited clinical success of the approved OGTs is likely due to their suboptimal performance when compared with pre-existent therapeutics or a lack of alternative treatment options. Notably, ASOs, generally less efficient than the most potent siRNA, have higher success rates in FDA and EMA approval. Moreover, statistical data shows that more ASOs have entered clinical trials and moved to more advanced phases, than siRNA and miRs combined. Likely, challenges in siRNA design can be attributed to the greater complexity in designing and modifying them without reducing activity compared to ASO. On the other hand, miRs face challenges due to the inherently low selectivity making them less studied in both experimental and clinical settings. The Dz agents have not been successful in clinical trials due to the incorrect design of RNA binding arms, having too low affinity to targeted RNA, and the difficulty in chemical protection of their catalytic cores against nuclease degradation. CRISPR/Cas13 remains relatively new and not sufficiently studied tool that might be limited by greater challenges in delivery in comparison with ASO and siRNA, low specificity and collateral RNase activity. Notably the recent approval of Imetelstat, a first-in-class oligonucleotide agent that acts as a telomerase inhibitor, offers hope by restoring normal haematopoiesis for low to intermediate risk patients with anaemia.
Ultimately, while OGTs still face considerable hurdles, we are now at the stage when incremental improvements in delivery, design, or validation of OGT efficacy could unlock immense potential, revolutionizing gene therapy.
Abbreviations
ALS: amyotrophic lateral sclerosis; ALN-HBV: RNAi agent targeting hepatitis B virus; ApoB: apolipoprotein B; ApoC-III: apolipoprotein C-III; ASO: antisense oligonucleotides; ASGR: asialoglycoprotein receptor; AHP: acute hepatic porphyria; Bcl-2: B-cell lymphoma 2; BP1002: Bcl-2-targeting antisense oligonucleotide; cEt: constrained ethyl; CMV: cytomegalovirus; CPP: cell-penetrating peptide; CRISPR: clustered regularly interspaced short palindromic repeats; crRNA: CRISPR RNA; CXCL5: chemokine ligand 5; DMD: Duchenne muscular dystrophy; DNA: deoxyribonucleic acid; Dzs: DNAzymes; EMA: European Medicines Agency; FDA: United States Food and Drug Administration; FCS: familial chylomicronemia syndrome; GalNAc: N-Acetylgalactosamine; GPD1L: glycerol-3-phosphate dehydrogenase 1-like protein; gRNA: guide RNA; HBsAg: hepatitis B surface antigen; HBV: hepatitis B virus; HDL: high-density lipoprotein; HIV: human immunodeficiency virus; hATTR: hereditary transthyretin-mediated amyloidosis; IC50: half maximal inhibitory concentration; IE2: immediate-early protein 2; IT: intrathecal; IV: intravenous; KRAS: Kirsten rat sarcoma viral oncogene homolog; LDL: low-density lipoprotein; LDL-C: low-density lipoprotein cholesterol; LNA: locked nucleic acid; LNP: lipid nanoparticle; miR: microRNA; miRNA: microRNA; MOe: 2'-methoxyethyl; MsPA: mesylphosphoramidate; mRNA: messenger RNA; N/P: nitrogen to phosphate ratio; NLS: nuclear localization signal; OGT: oligonucleotide-based gene therapy/therapeutics; ON: oligonucleotide; ORF: open reading frame; PCSK9: proprotein convertase subtilisin/kexin type 9; PG: phosphoryl guanidine; PH1: primary hyperoxaluria type 1; PPIB: peptidylprolyl isomerase B; PS: phosphorothioate; qPCR: quantitative polymerase chain reaction; RISC: RNA-induced silencing complex; RNA: ribonucleic acid; RNAi: RNA interference; RNP: ribonucleoprotein; Rz: ribozyme; Rzs: RNAzymes; RSV: respiratory syncytial virus; SARS-CoV-2: severe acute respiratory syndrome coronavirus 2; SC: subcutaneous; SCORE: Secondary structure, duplex, and competition score index; siRNA: small interfering RNA; SMN2: survival motor neuron 2; SNV: single nucleotide variant; SOD1: superoxide dismutase 1; SR-B1: scavenger receptor class B member 1; ssDNA: single-stranded DNA; ssRNA: single-stranded RNA; TGF: transforming growth factor; Tf-PEI: transferrin-polyethyleneimine; Tm: melting temperature; TP53: tumor protein p53; TTR: transthyretin; UTR: untranslated region; VEGF: vascular endothelial growth factor; vRNA: viral RNA; ZFN: zinc finger nuclease.
Acknowledgements
All authors except DMK were supported by the FSER-2025-0019 grant of the Ministry of Education of Russian Federation.
Competing interests
The authors have declared that no competing interest exists.
References
1. Anderson WF. Prospects for human gene therapy. Science. 1984;226:401-9
2. Arjmand B, Larijani B, Sheikh Hosseini M, Payab M, Gilany K, Goodarzi P. et al. The horizon of gene therapy in modern medicine: advances and challenges. Adv Exp Med Biol. 2020;1247:33-64
3. Nóbrega C, Mendonça L, Matos CA. Gene therapy strategies: gene silencing. Handb Gene Cell Ther. 2020:127-46
4. Nedorezova DD, Dubovichenko MV, Belyaeva EP, Grigorieva ED, Peresadina AV, Kolpashchikov DM. Specificity of oligonucleotide gene therapy (ogt) agents. Theranostics. 2022;12:7132-57
5. Kulkarni JA, Witzigmann D, Thomson SB, Chen S, Leavitt BR, Cullis PR. et al. The current landscape of nucleic acid therapeutics. Nat Nanotechnol. 2021;16:630-43
6. Wu L, Zhou W, Lin L, Chen A, Feng J, Qu X. et al. Delivery of therapeutic oligonucleotides in nanoscale. Bioact Mater. 2022;7:292-323
7. Bost JP, Barriga H, Holme MN, Gallud A, Maugeri M, Gupta D. et al. Delivery of oligonucleotide therapeutics: chemical modifications, lipid nanoparticles, and extracellular vesicles. ACS Nano. 2021;15:13993-4021
8. Benizri S, Gissot A, Martin A, Vialet B, Grinstaff MW, Barthélémy P. Bioconjugated oligonucleotides: recent developments and therapeutic applications. Bioconjug Chem. 2019;30:366-83
9. Anwar S, Mir F, Yokota T. Enhancing the effectiveness of oligonucleotide therapeutics using cell-penetrating peptide conjugation, chemical modification, and carrier-based delivery strategies. Pharmaceutics. 2023;15:1130
10. Dhuri K, Bechtold C, Quijano E, Pham H, Gupta A, Vikram A. et al. Antisense oligonucleotides: an emerging area in drug discovery and development. J Clin Med. 2020;9:2004
11. Liang X-H, Sun H, Nichols JG, Crooke ST. RNase h1-dependent antisense oligonucleotides are robustly active in directing rna cleavage in both the cytoplasm and the nucleus. Mol Ther. 2017;25:2075-92
12. Fire A, Xu S, Montgomery MK, Kostas SA, Driver SE, Mello CC. Potent and specific genetic interference by double-stranded rna in caenorhabditis elegans. Nature. 1998;391:806-11
13. Dana H, Chalbatani GM, Mahmoodzadeh H, Karimloo R, Rezaiean O, Moradzadeh A. et al. Molecular mechanisms and biological functions of sirna. Int J Biomed Sci. 2017;13:48-57
14. Ohrt T, Merkle D, Birkenfeld K, Echeverri CJ, Schwille P. In situ fluorescence analysis demonstrates active sirna exclusion from the nucleus by exportin 5. Nucleic Acids Res. 2006;34:1369-80
15. Beger C, Krüger M, Wong-staal F. CHAPTER 5 - ribozymes in cancer gene therapy. Gene Ther Cancer Second Ed. 2002:95-108
16. Larcher LM, Pitout IL, Keegan NP, Veedu RN, Fletcher S. DNAzymes: expanding the potential of nucleic acid therapeutics. Nucleic Acid Ther. 2023;33:178-92
17. Micura R, Höbartner C. Fundamental studies of functional nucleic acids: aptamers, riboswitches, ribozymes and dnazymes. Chem Soc Rev. 2020;49:7331-53
18. Hudson AJ, Normand N, Ackroyd J, Akhtar S. Cellular delivery of hammerhead ribozymes conjugated to a transferrin receptor antibody. Int J Pharm. 1999;182:49-58
19. Aigner A, Fischer D, Merdan T, Brus C, Kissel T, Czubayko F. Delivery of unmodified bioactive ribozymes by an rna-stabilizing polyethylenimine (lmw-pei) efficiently down-regulates gene expression. Gene Ther. 2002;9:1700-7
20. Mahieu M, Deschuyteneer R, Forget D, Vandenbussche P, Content J. Construction of a ribozyme directed against human interleukin-6 mrna: evaluation of its catalytic activity in vitro and in vivo. Blood. 1994;84:3758-65
21. Cairns MJ, Sun L-Q. Target-site selection for the 10-23 dnazyme. Methods Mol Biol. 2004;252:267-77
22. Kolpashchikov DM, Gerasimova YV. Cleavage of structured rnas is accelerated by high affinity dnazyme agents. Chem Bio Chem. 2025;26:e202400950
23. Hu Y, Chen Y, Xu J, Wang X, Luo S, Mao B. et al. Metagenomic discovery of novel crispr-cas13 systems. Cell Discov. 2022;8:1-4
24. Shmakov S, Abudayyeh OO, Makarova KS, Wolf YI, Gootenberg JS, Semenova E. et al. Discovery and functional characterization of diverse class 2 crispr-cas systems. Mol Cell. 2015;60:385-97
25. Kordyś M, Sen R, Warkocki Z. Applications of the versatile crispr-cas13 rna targeting system. Wiley Interdiscip Rev RNA. 2022;13:e1694
26. Liu L, Li X, Ma J, Li Z, You L, Wang J. et al. The molecular architecture for rna-guided rna cleavage by cas13a. Cell. 2017;170:714-726.e10
27. Weiss A, Gilbert JW, Flores IVR, Belgrad J, Ferguson C, Dogan EO. et al. RNAi-mediated silencing of sod1 profoundly extends survival and functional outcomes in als mice. 2024. 2024 06.20.599943
28. Liu Y, Tan H, Tian H, Liang C, Chen S, Liu Q. Autoantigen la promotes efficient rnai, antiviral response, and transposon silencing by facilitating multiple-turnover risc catalysis. Mol Cell. 2011;44:502-8
29. Liang X-H, Nichols JG, Sun H, Crooke ST. Translation can affect the antisense activity of rnase h1-dependent oligonucleotides targeting mrnas. Nucleic Acids Res. 2018;46:293-313
30. Piasecka J, Lenartowicz E, Soszynska-Jozwiak M, Szutkowska B, Kierzek R, Kierzek E. RNA secondary structure motifs of the influenza a virus as targets for sirna-mediated rna interference. Mol Ther Nucleic Acids. 2020;19:627-42
31. Singh J, Saeedan AS, Kaithwas G, Ansari MN. Small interfering RNA: From designing to therapeutic in cancer. J Genet Eng Biotechnol. 2025;23:100484
32. Hanna J, Hossain GS, Kocerha J. The potential for microrna therapeutics and clinical research. Front Genet. 2019 10
33. Yan WX, Chong S, Zhang H, Makarova KS, Koonin EV, Cheng DR. et al. Cas13d is a compact rna-targeting type vi crispr effector positively modulated by a wyl domain-containing accessory protein. Mol Cell. 2018;70:327-339.e5
34. Smargon AA, Cox DBT, Pyzocha NK, Zheng K, Slaymaker IM, Gootenberg JS. et al. Cas13b is a type vi-b crispr-associated rna-guided rnase differentially regulated by accessory proteins csx27 and csx28. Mol Cell. 2017;65:618-630.e7
35. Abudayyeh OO, Gootenberg JS, Konermann S, Joung J, Slaymaker IM, Cox DBT. et al. C2c2 is a single-component programmable rna-guided rna-targeting crispr effector. Science. 2016;353:aaf5573
36. Kang H, Ga YJ, Kim SH, Cho YH, Kim JW, Kim C. et al. Small interfering rna (sirna)-based therapeutic applications against viruses: principles, potential, and challenges. J Biomed Sci. 2023;30:88
37. Alhamadani F, Zhang K, Parikh R, Wu H, Rasmussen TP, Bahal R. et al. Adverse drug reactions and toxicity of the food and drug administration-approved antisense oligonucleotide drugs. Drug Metab Dispos. 2022;50:879-87
38. Gane E, Lim Y-S, Kim JB, Jadhav V, Shen L, Bakardjiev AI. et al. Evaluation of rnai therapeutics vir-2218 and aln-hbv for chronic hepatitis b: results from randomized clinical trials. J Hepatol. 2023;79:924-32
39. Yuen M-F, Asselah T, Jacobson IM, Brunetto MR, Janssen HLA, Takehara T. et al. Efficacy and safety of the sirna jnj-73763989 and the capsid assembly modulator jnj-56136379 (bersacapavir) with nucleos(t)ide analogues for the treatment of chronic hepatitis b virus infection (reef-1): a multicentre, double-blind, active-controlled, randomised, phase 2b trial. Lancet Gastroenterol Hepatol. 2023;8:790-802
40. Gane EJ, Kim W, Lim TH, Tangkijvanich P, Yoon J-H, Sievert W. et al. First-in-human randomized study of rnai therapeutic rg6346 for chronic hepatitis b virus infection. J Hepatol. 2023;79:1139-49
41. Santos RD, Raal FJ, Catapano AL, Witztum JL, Steinhagen-Thiessen E, Tsimikas S. Mipomersen, an antisense oligonucleotide to apolipoprotein b-100, reduces lipoprotein(a) in various populations with hypercholesterolemia. Arterioscler Thromb Vasc Biol. 2015
42. Nature Biotechnology. Akcea's antisense drug rejection worries analysts. Nat Biotechnol. 2018;36:911-911
43. McColgan P, Thobhani A, Boak L, Schobel SA, Nicotra A, Palermo G. et al. Tominersen in adults with manifest huntington's disease. N Engl J Med. 2023;389:2203-5
44. Aartsma-Rus A, Goemans N. A sequel to the eteplirsen saga: eteplirsen is approved in the united states but was not approved in europe. Nucleic Acid Ther. 2019;29:13-5
45. Urits I, Swanson D, Swett MC, Patel A, Berardino K, Amgalan A. et al. A review of patisiran (onpattro®) for the treatment of polyneuropathy in people with hereditary transthyretin amyloidosis. Neurol Ther. 2020;9:301-15
46. Adams D, Tournev IL, Taylor MS, Coelho T, Planté-Bordeneuve V, Berk JL. et al. Efficacy and safety of vutrisiran for patients with hereditary transthyretin-mediated amyloidosis with polyneuropathy: a randomized clinical trial. Amyloid. 2023;30:18-26
47. Keam SJ. Inotersen: first global approval. Drugs. 2018;78:1371-6
48. Nie T. Eplontersen: first approval. Drugs. 2024;84:473-8
49. Butler JS, Chan A, Costelha S, Fishman S, Willoughby JLS, Borland TD. et al. Preclinical evaluation of rnai as a treatment for transthyretin-mediated amyloidosis. Amyloid. 2016;23:109-18
50. Judge DP, Kristen AV, Grogan M, Maurer MS, Falk RH, Hanna M. et al. Phase 3 multicenter study of revusiran in patients with hereditary transthyretin-mediated (hattr) amyloidosis with cardiomyopathy (endeavour). Cardiovasc Drugs Ther. 2020;34:357-70
51. Kenet G, Nolan B, Zulfikar B, Antmen B, Kampmann P, Matsushita T. et al. Fitusiran prophylaxis in people with hemophilia a or b who switched from prior bpa/cfc prophylaxis: the atlas-ppx trial. Blood. 2024;143:2256-69
52. Lennox AL, Huang F, Behrs MK, González-Sales M, Bhise N, Wan Y. et al. Imetelstat, a novel, first-in-class telomerase inhibitor: mechanism of action, clinical, and translational science. Clin Transl Sci. 2024;17:e70076
53. Jahns H, Degaonkar R, Podbevsek P, Gupta S, Bisbe A, Aluri K. et al. Small circular interfering rnas (scirnas) as a potent therapeutic platform for gene-silencing. Nucleic Acids Res. 2021;49:10250-64
54. He AT, Liu J, Li F, Yang BB. Targeting circular rnas as a therapeutic approach: current strategies and challenges. Signal Transduct Target Ther. 2021;6:185
55. Yu X, Xie Y, Zhang S, Song X, Xiao B, Yan Z. TRNA-derived fragments: mechanisms underlying their regulation of gene expression and potential applications as therapeutic targets in cancers and virus infections. Theranostics. 2021;11:461-9
56. Xie Z, Zeng X. DNA/rna-based formulations for treatment of breast cancer. Expert Opin Drug Deliv. 2017;14:1379-93
57. Wang Y, Minden A. Current molecular combination therapies used for the treatment of breast cancer. Int J Mol Sci. 2022;23:11046
58. Bartolucci D, Pession A, Hrelia P, Tonelli R. Precision anti-cancer medicines by oligonucleotide therapeutics in clinical research targeting undruggable proteins and non-coding rnas. Pharmaceutics. 2022;14:1453
59. Walker AR, Marcucci G, Yin J, Blum W, Stock W, Kohlschmidt J. et al. Phase 3 randomized trial of chemotherapy with or without oblimersen in older aml patients: calgb 10201 (alliance). Blood Adv. 2021;5:2775-87
60. AstraZeneca. A phase i, open-label, multicentre dose-escalation study to investigate the safety and pharmacokinetics of azd4785 in patients with advanced solid tumours where kras may be an important driver of tumour survival. 2019; NCT03101839.
61. Gitlitz DB. A phase i/ii study of vegf-antisense oligonucleotide (vegf-as, veglin) in combination with pemetrexed and cisplatin for the treatment of advanced malignant mesothelioma. 2014; NCT00668499.
62. Sousa C, Videira M. Dual Approaches in Oncology: The Promise of siRNA and Chemotherapy Combinations in Cancer Therapies. Onco. 2025;5:2
63. Yang Y, Yang C, Deng K, Xiao Y, Liu X, Du Z. Nucleic acid drugs in radiotherapy. ChemBioChem. 2025;26:e202400854
64. Wang J, Sun M, Zhu X, Zhao H, Mao D, Zhang Z. et al. Lentivirus-mediated rna interference targeting programmed death receptor ligand 1 increases the immunologic anti-tumor effect of dendritic cell vaccination against pancreatic cancer in scid-hu mice. Oncol Lett. 2019;18:1539-47
65. Vooght-Johnson RD. Venetoclax: evidence to date and clinical potential. Drugs Context. 2019
66. Dhillon S. Adagrasib: first approval. Drugs. 2023;83:275-85
67. Nedorezova DD, Fakhardo AF, Molden TA, Kolpashchikov DM. Deoxyribozyme-based dna machines for cancer therapy. ChemBioChem. 2020;21:607-11
68. Masu H, Narita A, Tokunaga T, Ohashi M, Aoyama Y, Sando S. An activatable sirna probe: trigger-rna-dependent activation of rnai function. Angew Chem Int Ed. 2009;48:9481-3
69. Avila YI, Ha A, Chandler MR. et al. Reconfigurable Nucleic Acid Nanoparticles with Therapeutic RNAi Responses to Intracellular Disease Markers. Adv Funct Mater. 2025 e08122
70. Afonin KA, Viard M, Martins AN, Lockett SJ, Maciag AE, Freed EO. et al. Activation of different split functionalities on re-association of rna-dna hybrids. Nat Nanotechnol. 2013;8:296-304
71. Jiang Q, Yue S, Yu K, Tian T, Zhang J, Chu H. et al. Endogenous microrna triggered enzyme-free dna logic self-assembly for amplified bioimaging and enhanced gene therapy via in situ generation of sirnas. J Nanobiotechnology. 2021;19:288
72. Zhong W, Huang L, Lin Y, Xing C, Lu C. Endogenous dual mirna-triggered dynamic assembly of dna nanostructures for in-situ dual sirna delivery. Sci China Mater. 2023;66:2938-46
73. Zhang J, Sharma R, Ryu K, Shen P, Salaita K, Jo H. Conditional antisense oligonucleotides triggered by mirna. ACS Chem Biol. 2021;16:2255-67
74. Nedorezova DD, Fakhardo AF, Nemirich DV, Bryushkova EA, Kolpashchikov DM. Towards dna nanomachines for cancer treatment: achieving selective and efficient cleavage of folded rna. Angew Chem Int Ed Engl. 2019;58:4654-8
75. Drozd VS, Eldeeb AA, Kolpashchikov DM, Nedorezova DD. Binary antisense oligonucleotide agent for cancer marker-dependent degradation of targeted rna. Nucleic Acid Ther. 2022;32:412-20
76. Molden TA, Niccum CT, Kolpashchikov DM. Cut and paste for cancer treatment: a dna nanodevice that cuts out an rna marker sequence to activate a therapeutic function. Angew Chem Int Ed. 2020;59:21190-4
77. Patra C, ElDeeb AA. Thresholding dna nanomachine as a highly cooperative and efficient enzyme-like system for controlled rna cleavage. ChemMedChem. 2023;18:e202300040
78. Nour MAY, Kulikova AV, Wanjohi J, El-Deeb AA. Distinguishing high from low concentrations of rna cancer marker: five-input dnazyme thresholding gate. ChemistrySelect. 2023;8:e202303011
79. Nour MAY, Drozd VS, Lemeshko EA, Tafran L, Salimova AA, Kulikova AV. et al. RNase h-dependent dna thresholder modulated by cancer marker concentration. Chem Commun. 2024;60:4427-30
80. Smirnov VV, Drozd VS, Patra CK, Hussein Z, Rybalko DS, Kozlova AV. et al. Towards the development of a dna automaton: modular rna-cleaving deoxyribozyme logic gates regulated by mirnas. Analyst. 2024
81. Nakao R, Yamamoto S, Yasumoto Y, Kadota K, Oishi K. Impact of denervation-induced muscle atrophy on housekeeping gene expression in mice. Muscle Nerve. 2015;51:276-81
82. Albuquerque APB, Balmaa M, Reis CA, Beltrão EIC. Identification of appropriate housekeeping genes for quantitative rt-pcr analysis in mda-mb-231 and nci-h460 human cancer cell lines under hypoxia and serum deprivation. J Mol Clin Med. 2018;1:127-34
83. Molitoris BA, Dagher PC, Sandoval RM, Campos SB, Ashush H, Fridman E. et al. SiRNA targeted to p53 attenuates ischemic and cisplatin-induced acute kidney injury. J Am Soc Nephrol. 2009;20:1754
84. Sadat SMA, Jahan ST, Haddadi A. Effects of size and surface charge of polymeric nanoparticles on in vitro and in vivo applications. J Biomater Nanobiotechnol. 2016;7:91-108
85. Xu M, Soliman MG, Sun X, Pelaz B, Feliu N, Parak WJ. et al. How entanglement of different physicochemical properties complicates the prediction of in vitro and in vivo interactions of gold nanoparticles. ACS Nano. 2018;12:10104-13
86. Fumoto S, Kawakami S, Hashida M, Nishida K, Fumoto S, Kawakami S. et al. Targeted gene delivery: importance of administration routes. Nov Gene Ther Approaches. 2013
87. Meryet-Figuière M, Lecerf C, Varin E, Coll J-L, Louis M-H, Dutoit S. et al. Atelocollagen-mediated in vivo sirna transfection in ovarian carcinoma is influenced by tumor site, sirna target and administration route. Oncol Rep. 2017;38:1949-58
88. Zhu C, Lee JY, Woo JZ, Xu L, Nguyenla X, Yamashiro LH. et al. An intranasal aso therapeutic targeting sars-cov-2. Nat Commun. 2022;13:4503
89. Antimisiaris SG, Marazioti A, Kannavou M, Natsaridis E, Gkartziou F, Kogkos G. et al. Overcoming barriers by local drug delivery with liposomes. Adv Drug Deliv Rev. 2021;174:53-86
90. Zhang T, Yin H, Li Y, Yang H, Ge K, Zhang J. et al. Optimized lipid nanoparticles (lnps) for organ-selective nucleic acids delivery in vivo. iScience. 2024 27
91. Samuelsson E, Shen H, Blanco E, Ferrari M, Wolfram J. Contribution of kupffer cells to liposome accumulation in the liver. Colloids Surf B Biointerfaces. 2017;158:356-62
92. Akinc A, Querbes W, De S, Qin J, Frank-Kamenetsky M, Jayaprakash KN. et al. Targeted delivery of rnai therapeutics with endogenous and exogenous ligand-based mechanisms. Mol Ther. 2010;18:1357-64
93. Qin B, Chen Z, Jin W, Cheng K. Development of cholesteryl peptide micelles for sirna delivery. J Control Release. 2013;172:159-68
94. Ezzat K, EL Andaloussi S, Zaghloul EM, Lehto T, Lindberg S, Moreno PMD. et al. PepFect 14, a novel cell-penetrating peptide for oligonucleotide delivery in solution and as solid formulation. Nucleic Acids Res. 2011;39:5284-98
95. Sahagun D, Zahid M. Cardiac-targeting peptide: from discovery to applications. Biomolecules. 2023;13:1690
96. Sela T, Mansø M, Siegel M, Marban-Doran C, Ducret A, Niewöhner J. et al. Diligent design enables antibody-aso conjugates with optimal pharmacokinetic properties. Bioconjug Chem. 2023;34:2096-111
97. Hao X, Li Q, Guo J, Ren X, Feng Y, Shi C. et al. Multifunctional gene carriers with enhanced specific penetration and nucleus accumulation to promote neovascularization of huvecs in vivo. ACS Appl Mater Interfaces. 2017;9:35613-27
98. Guo X, Chu X, Li W, Pan Q, You H. Chondrogenic effect of precartilaginous stem cells following nls-tat cell penetrating peptide-assisted transfection of eukaryotic htgfβ3. J Cell Biochem. 2013;114:2588-94
99. Yaghini E, Dondi R, Edler KJ, Loizidou M, MacRobert AJ, Eggleston IM. Codelivery of a cytotoxin and photosensitiser via a liposomal nanocarrier: a novel strategy for light-triggered cytosolic release. Nanoscale. 2018;10:20366-76
100. Østergaard ME, Jackson M, Low A, E Chappell A, G Lee R, Peralta RQ. et al. Conjugation of hydrophobic moieties enhances potency of antisense oligonucleotides in the muscle of rodents and non-human primates. Nucleic Acids Res. 2019;47:6045-58
101. Tran P, Weldemichael T, Liu Z, Li H. Delivery of oligonucleotides: efficiency with lipid conjugation and clinical outcome. Pharmaceutics. 2022;14:342
102. Wang S, Allen N, Liang X, Crooke ST. Membrane destabilization induced by lipid species increases activity of phosphorothioate-antisense oligonucleotides. Mol Ther Nucleic Acids. 2018;13:686-98
103. Ding Y, Wang W, Feng M, Wang Y, Zhou J, Ding X. et al. A biomimetic nanovector-mediated targeted cholesterol-conjugated sirna delivery for tumor gene therapy. Biomaterials. 2012;33:8893-905
104. Wolfrum C, Shi S, Jayaprakash KN, Jayaraman M, Wang G, Pandey RK. et al. Mechanisms and optimization of in vivo delivery of lipophilic sirnas. Nat Biotechnol. 2007;25:1149-57
105. Biscans A, Coles A, Haraszti R, Echeverria D, Hassler M, Osborn M. et al. Diverse lipid conjugates for functional extra-hepatic sirna delivery in vivo. Nucleic Acids Res. 2019;47:1082-96
106. Schmidt K, Prakash TP, Donner AJ, Kinberger GA, Gaus HJ, Low A. et al. Characterizing the effect of galnac and phosphorothioate backbone on binding of antisense oligonucleotides to the asialoglycoprotein receptor. Nucleic Acids Res. 2017;45:2294-306
107. Prakash TP, Kinberger GA, Murray HM, Chappell A, Riney S, Graham MJ. et al. Synergistic effect of phosphorothioate, 5′-vinylphosphonate and galnac modifications for enhancing activity of synthetic sirna. Bioorg Med Chem Lett. 2016;26:2817-20
108. Ohashi M, Tamura A, Yui N. Exploring receptor binding affinities and hepatic cell association of n-acetyl-d-galactosamine-modified β-cyclodextrin-based polyrotaxanes for liver-targeted therapies. Biomacromolecules. 2023;24:2327-41
109. Nair JK, Willoughby JLS, Chan A, Charisse K, Alam MdR, Wang Q. et al. Multivalent n-acetylgalactosamine-conjugated sirna localizes in hepatocytes and elicits robust rnai-mediated gene silencing. J Am Chem Soc. 2014;136:16958-61
110. Liu C, Su C. Design strategies and application progress of therapeutic exosomes. Theranostics. 2019;9:1015-28
111. Zhang Y, Liu Q, Zhang X, Huang H, Tang S, Chai Y. et al. Recent advances in exosome-mediated nucleic acid delivery for cancer therapy. J Nanobiotechnology. 2022;20:279
112. Abdelsalam M, Ahmed M, Osaid Z, Hamoudi R, Harati R. Insights into exosome transport through the blood-brain barrier and the potential therapeutical applications in brain diseases. Pharmaceuticals (Basel). 2023;16:571
113. Jung I, Shin S, Baek M-C, Yea K. Modification of immune cell-derived exosomes for enhanced cancer immunotherapy: current advances and therapeutic applications. Exp Mol Med. 2024;56:19-31
114. Samanta S, Rajasingh S, Drosos N, Zhou Z, Dawn B, Rajasingh J. Exosomes: new molecular targets of diseases. Acta Pharmacol Sin. 2018;39:501-13
115. Wahlgren J, Karlson TDL, Brisslert M, Vaziri Sani F, Telemo E, Sunnerhagen P. et al. Plasma exosomes can deliver exogenous short interfering rna to monocytes and lymphocytes. Nucleic Acids Res. 2012;40:e130
116. Barros FM, Carneiro F, Machado JC, Melo SA. Exosomes and immune response in cancer: friends or foes? Front Immunol. 2018 9
117. Limongi T, Susa F, Dumontel B, Racca L, Perrone Donnorso M, Debellis D. et al. Extracellular vesicles tropism: a comparative study between passive innate tropism and the active engineered targeting capability of lymphocyte-derived evs. Membranes (Basel). 2021;11:886
118. Krylova SV, Feng D. The machinery of exosomes: biogenesis, release, and uptake. Int J Mol Sci. 2023;24:1337
119. Pham TT, Chen H, Nguyen PHD, Jayasinghe MK, Le AH, Le MT. Endosomal escape of nucleic acids from extracellular vesicles mediates functional therapeutic delivery. Pharmacol Res. 2023;188:106665
120. Didiot M-C, Hall LM, Coles AH, Haraszti RA, Godinho BM, Chase K. et al. Exosome-mediated delivery of hydrophobically modified sirna for huntingtin mrna silencing. Mol Ther. 2016;24:1836-47
121. Aqil F, Munagala R, Jeyabalan J, Agrawal AK, Kyakulaga A-H, Wilcher SA. et al. Milk exosomes - natural nanoparticles for sirna delivery. Cancer Lett. 2019;449:186-95
122. Zhan Q, Yi K, Cui X, Li X, Yang S, Wang Q. et al. Blood exosomes-based targeted delivery of cpla2 sirna and metformin to modulate glioblastoma energy metabolism for tailoring personalized therapy. Neuro Oncol. 2022;24:1871-83
123. Zhang M, Hu S, Liu L, Dang P, Liu Y, Sun Z. et al. Engineered exosomes from different sources for cancer-targeted therapy. Signal Transduct Target Ther. 2023;8:1-20
124. Amiri A, Bagherifar R, Ansari Dezfouli E, Kiaie SH, Jafari R, Ramezani R. Exosomes as bio-inspired nanocarriers for rna delivery: preparation and applications. J Transl Med. 2022;20:125
125. Lönn P, Kacsinta AD, Cui X-S, Hamil AS, Kaulich M, Gogoi K. et al. Enhancing endosomal escape for intracellular delivery of macromolecular biologic therapeutics. Sci Rep. 2016;6:32301
126. Lee YJ, Shin KJ, Chae YC. Regulation of cargo selection in exosome biogenesis and its biomedical applications in cancer. Exp Mol Med. 2024;56:877-89
127. Cheng Y, Yumul RC, Pun SH. Virus-inspired polymer for efficient in vitro and in vivo gene delivery. Angew Chem Int Ed. 2016;55:12013-7
128. Alabdullah AA, Al-Abdulaziz B, Alsalem H, Magrashi A, Pulicat SM, Almzroua AA. et al. Estimating transfection efficiency in differentiated and undifferentiated neural cells. BMC Res Notes. 2019;12:225
129. Mortimer I, Tam P, MacLachlan I, Graham RW, Saravolac EG, Joshi PB. Cationic lipid-mediated transfection of cells in culture requires mitotic activity. Gene Ther. 1999;6:403-11
130. Hoffman EA, Poncius A, McKay BS, Stamer WD. Cell cycle synchronization and transfection efficiency of human trabecular meshwork cells. Invest Ophthalmol Vis Sci. 2004;45:4432
131. Zenke K, Okinaka Y. Establishing an effective gene knockdown system using cultured cells of the model fish medaka (oryzias latipes). Biol Methods Protoc. 2022;7:bpac011
132. Jiang Y, Hardie J, Liu Y, Ray M, Luo X, Das R. et al. Nanocapsule-mediated cytosolic sirna delivery for anti-inflammatory treatment. J Control Release. 2018;283:235-40
133. Feldmann DP, Xie Y, Jones SK, Yu D, Moszczynska A, Merkel OM. The impact of microfluidic mixing of triblock micelleplexes on in vitro/in vivo gene silencing and intracellular trafficking. Nanotechnology. 2017;28:224001
134. Yang D, Ruan Y, Chen Y. Safety and efficacy of gene therapy with onasemnogene abeparvovec in the treatment of spinal muscular atrophy: a systematic review and meta-analysis. J Paediatr Child Health. 2023;59:431-8
135. Hong SW, Jiang Y, Kim S, Li CJ, Lee D. Target gene abundance contributes to the efficiency of sirna-mediated gene silencing. Nucleic Acid Ther. 2014;24:192-8
136. Krueger U, Bergauer T, Kaufmann B, Wolter I, Pilk S, Heider-Fabian M. et al. Insights into effective rnai gained from large-scale sirna validation screening. Oligonucleotides. 2007;17:237-50
137. Yang L, Ma F, Liu F, Chen J, Zhao X, Xu Q. Efficient delivery of antisense oligonucleotides using bioreducible lipid nanoparticles in vitro and in vivo. Mol Ther Nucleic Acids. 2020;19:1357-67
138. Xie Y, Kim NH, Nadithe V, Schalk D, Thakur A, Kılıç A. et al. Targeted delivery of sirna to activated t cells via transferrin-polyethylenimine (tf-pei) as a potential therapy of asthma. J Control Release. 2016;229:120-9
139. Vocelle D, Chan C, Walton SP. Endocytosis controls sirna efficiency: implications for sirna delivery vehicle design and cell-specific targeting. Nucleic Acid Ther. 2020;30:22-32
140. Dowdy SF. Endosomal escape of rna therapeutics: how do we solve this rate-limiting problem? RNA. 2023;29:396-401
141. Gilleron J, Querbes W, Zeigerer A, Borodovsky A, Marsico G, Schubert U. et al. Image-based analysis of lipid nanoparticle-mediated sirna delivery, intracellular trafficking and endosomal escape. Nat Biotechnol. 2013;31:638-46
142. Dowdy SF. Overcoming cellular barriers for rna therapeutics. Nat Biotechnol. 2017;35:222-9
143. Bus T, Traeger A, Schubert US. The great escape: how cationic polyplexes overcome the endosomal barrier. J Mater Chem B. 2018;6:6904-18
144. Gene therapy needs a long-term approach. Nat Med. 2021;27:563-563
145. Dowling JK, O'Neill LAJ. Biochemical regulation of the inflammasome. Crit Rev Biochem Mol Biol. 2012;47:424-43
146. Zheng D, Liwinski T, Elinav E. Inflammasome activation and regulation: toward a better understanding of complex mechanisms. Cell Discov. 2020;6:1-22
147. Juliano RL. Intracellular trafficking and endosomal release of oligonucleotides: what we know and what we don't. Nucleic Acid Ther. 2018;28:166-77
148. Liang X, Sun H, Shen W, Crooke ST. Identification and characterization of intracellular proteins that bind oligonucleotides with phosphorothioate linkages. Nucleic Acids Res. 2015;43:2927-45
149. Liang X, Sun H, Hsu C-W, Nichols JG, Vickers TA, De Hoyos CL. et al. Golgi-endosome transport mediated by m6pr facilitates release of antisense oligonucleotides from endosomes. Nucleic Acids Res. 2020;48:1372-91
150. Crooke ST, Vickers TA, Liang X. Phosphorothioate modified oligonucleotide-protein interactions. Nucleic Acids Res. 2020;48:5235-53
151. Tenchov R, Bird R, Curtze AE, Zhou Q. Lipid nanoparticles─from liposomes to mrna vaccine delivery, a landscape of research diversity and advancement. ACS Nano. 2021
152. Tang H, Wang H, Gan Z, Ding Z, Yu Q. Engineering the hydrophilic-hydrophobic interface of polymeric micelles by cationic blocks for enhanced chemotherapy. ACS Appl Mater Interfaces. 2024
153. Chang Y, Yang C, Chen Y, Yang C, Chou Y, Chou H. et al. A sirna targets and inhibits a broad range of sars-cov-2 infections including delta variant. EMBO Mol Med. 2022;14:e15298
154. Lulla V, Wandel MP, Bandyra KJ, Ulferts R, Wu M, Dendooven T. et al. Targeting the conserved stem loop 2 motif in the sars-cov-2 genome. J Virol. 2021;95:10.1128 /jvi.00663-21
155. Xu C, Zhou Y, Xiao Q, He B, Geng G, Wang Z. et al. Programmable rna editing with compact crispr-cas13 systems from uncultivated microbes. Nat Methods. 2021;18:499-506
156. Abbott TR, Dhamdhere G, Liu Y, Lin X, Goudy L, Zeng L. et al. Development of crispr as an antiviral strategy to combat sars-cov-2 and influenza. Cell. 2020;181:865-876.e12
157. Dhorne-Pollet S, Fitzpatrick C, Da Costa B, Bourgon C, Eléouët J-F, Meunier N. et al. Antisense oligonucleotides targeting orf1b block replication of severe acute respiratory syndrome coronavirus 2 (sars-cov-2). Front Microbiol. 2022 13
158. Lenartowicz E, Nogales A, Kierzek E, Kierzek R, Martínez-Sobrido L, Turner DH. Antisense oligonucleotides targeting influenza a segment 8 genomic rna inhibit viral replication. Nucleic Acid Ther. 2016;26:277-85
159. Zhang Z, Zhang S, Wang S. DNAzymes dz13 target the c-jun possess antiviral activity against influenza a viruses. Microb Pathog. 2017;103:155-61
160. Freije CA, Myhrvold C, Boehm CK, Lin AE, Welch NL, Carter A. et al. Programmable inhibition and detection of rna viruses using cas13. Mol Cell. 2019;76:826-837.e11
161. Han K, Theodore D, McMullen G, Swayze E, McCaleb M, Billioud G. et al. Preclinical and phase 1 assessment of antisense oligonucleotide bepirovirsen in hepatitis b virus-transgenic mice and healthy human volunteers: support for clinical dose selection and evaluation of safety, tolerability, and pharmacokinetics of single and multiple doses. Clin Pharmacol Drug Dev. 2022;11:1191-202
162. Boyapalle S, Xu W, Raulji P, Mohapatra S, Mohapatra SS. A multiple sirna-based anti-hiv/shiv microbicide shows protection in both in vitro and in vivo models. PLoS One. 2015;10:e0135288
163. Friedrich M, Pfeifer G, Binder S, Aigner A, Vollmer Barbosa P, Makert GR. et al. Selection and validation of sirnas preventing uptake and replication of sars-cov-2. Front Bioeng Biotechnol. 2022 10
164. Niktab I, Haghparast M, Beigi M-H, Megraw TL, Kiani A, Ghaedi K. Design of advanced sirna therapeutics for the treatment of covid-19. Meta Gene. 2021;29:100910
165. Reynolds A, Leake D, Boese Q, Scaringe S, Marshall WS, Khvorova A. Rational sirna design for rna interference. Nat Biotechnol. 2004;22:326-30
166. Mohammadi-Arani R, Javadi-Zarnaghi F, Boccaletto P, Bujnicki JM, Ponce-Salvatierra A. DNAzymeBuilder, a web application for in situ generation of rna/dna-cleaving deoxyribozymes. Nucleic Acids Res. 2022;50:W261-5
167. Cairns MJ, Hopkins TM, Witherington C, Wang L, Sun L-Q. Target site selection for an rna-cleaving catalytic dna. Nat Biotechnol. 1999;17:480-6
168. Kurreck J, Bieber B, Jahnel R, Erdmann VA. Comparative study of dna enzymes and ribozymes against the same full-length messenger rna of the vanilloid receptor subtype i *. J Biol Chem. 2002;277:7099-107
169. Bandaru S, Tsuji MH, Shimizu Y, Usami K, Lee S, Takei NK. et al. Structure-based design of grna for cas13. Sci Rep. 2020;10:11610
170. Kakumani PK. AGO-rbp crosstalk on target mrnas: implications in mirna-guided gene silencing and cancer. Transl Oncol. 2022;21:101434
171. Davis SM, Hildebrand S, MacMillan HJ. et al. Systematic analysis of siRNA and mRNA features impacting fully chemically modified siRNA efficacy. Nucleic Acids Res. 2025;53:gkaf479
172. Reuter JS, Mathews DH. RNAstructure: software for rna secondary structure prediction and analysis. BMC Bioinformatics. 2010;11:129
173. Flynn RA, Zhang QC, Spitale RC, Lee B, Mumbach MR, Chang HY. Transcriptome-wide interrogation of rna secondary structure in living cells with icshape. Nat Protoc. 2016;11:273-90
174. Sun L, Li P, Ju X, Rao J, Huang W, Ren L. et al. In vivo structural characterization of the sars-cov-2 rna genome identifies host proteins vulnerable to repurposed drugs. Cell. 2021;184:1865-1883.e20
175. Soszynska-Jozwiak M, Michalak P, Moss WN, Kierzek R, Kesy J, Kierzek E. Influenza virus segment 5 (+)rna - secondary structure and new targets for antiviral strategies. Sci Rep. 2017;7:15041
176. Szabat M, Lorent D, Czapik T, Tomaszewska M, Kierzek E, Kierzek R. RNA secondary structure as a first step for rational design of the oligonucleotides towards inhibition of influenza a virus replication. Pathogens. 2020;9:925
177. Michalak P, Soszynska-Jozwiak M, Biala E, Moss WN, Kesy J, Szutkowska B. et al. Secondary structure of the segment 5 genomic rna of influenza a virus and its application for designing antisense oligonucleotides. Sci Rep. 2019;9:3801
178. Di Fusco D, Dinallo V, Marafini I, Figliuzzi MM, Romano B, Monteleone G. Antisense oligonucleotide: basic concepts and therapeutic application in inflammatory bowel disease. Front Pharmacol. 2019 10
179. Grimpe B. Deoxyribozymes and bioinformatics: complementary tools to investigate axon regeneration. Cell Tissue Res. 2012;349:181-200
180. Jarvis TC, Wincott FE, Alby LJ, McSwiggen JA, Beigelman L, Gustofson J. et al. Optimizing the cell efficacy of synthetic ribozymes. site selection and chemical modifications of ribozymes targeting the proto-oncogene c-myb. J Biol Chem. 1996;271:29107-12
181. Liu Y, Jing P, Zhou Y, Zhang J, Shi J, Zhang M. et al. The effects of length and sequence of grna on cas13b and cas13d activity in vitro and in vivo. Biotechnol J. 2023;18:2300002
182. Nedorezova DD, Dubovichenko MV, Kalnin AJ, Nour MAY, Eldeeb AA, Ashmarova AI. et al. Cleaving folded rna with dnazyme agents. ChemBioChem. 2024;25:e202300637
183. Wan WB, Seth PP. The medicinal chemistry of therapeutic oligonucleotides. J Med Chem. 2016;59:9645-67
184. Wang S, Sun H, Tanowitz M, Liang X, Crooke ST. Intra-endosomal trafficking mediated by lysobisphosphatidic acid contributes to intracellular release of phosphorothioate-modified antisense oligonucleotides. Nucleic Acids Res. 2017;45:5309-22
185. Beltinger C, Saragovi HU, Smith RM, LeSauteur L, Shah N, DeDionisio L. et al. Binding, uptake, and intracellular trafficking of phosphorothioate-modified oligodeoxynucleotides. J Clin Invest. 1995;95:1814-23
186. Miroshnichenko SK, Patutina OA, Burakova EA, Chelobanov BP, Fokina AA, Vlassov VV. et al. Mesyl phosphoramidate antisense oligonucleotides as an alternative to phosphorothioates with improved biochemical and biological properties. Proc Natl Acad Sci U S A. 2019;116:1229-34
187. Anderson BA, Freestone GC, Low A, De-Hoyos CL, III WJD, Østergaard ME. et al. Towards next generation antisense oligonucleotides: mesylphosphoramidate modification improves therapeutic index and duration of effect of gapmer antisense oligonucleotides. Nucleic Acids Res. 2021;49:9026-41
188. Patutina OA, Gaponova (Miroshnichenko) SK, Sen'kova AV, Savin IA, Gladkikh DV, Burakova EA. et al. Mesyl phosphoramidate backbone modified antisense oligonucleotides targeting mir-21 with enhanced in vivo therapeutic potency. Proc Natl Acad Sci U S A. 2020;117:32370-9
189. Goyal N, Narayanaswami P. Making sense of antisense oligonucleotides: a narrative review. Muscle Nerve. 2018;57:356-70
190. Aoki Y, Rocha CSJ, Lehto T, Miyatake S, Johansson H, Hashimoto Y. et al. Fine tuning of phosphorothioate inclusion in 2′-o-methyl oligonucleotides contributes to specific cell targeting for splice-switching modulation. Front Physiol. 2021 12
191. Kauppinen S, Vester B, Wengel J. Locked nucleic acid (lna): high affinity targeting of rna for diagnostics and therapeutics. Drug Discov Today Technol. 2005;2:287-90
192. Papargyri N, Pontoppidan M, Andersen MR, Koch T, Hagedorn PH. Chemical diversity of locked nucleic acid-modified antisense oligonucleotides allows optimization of pharmaceutical properties. Mol Ther Nucleic Acids. 2020;19:706-17
193. Linnane E, Davey P, Zhang P, Puri S, Edbrooke M, Chiarparin E. et al. Differential uptake, kinetics and mechanisms of intracellular trafficking of next-generation antisense oligonucleotides across human cancer cell lines. Nucleic Acids Res. 2019;47:4375-92
194. Yanagidaira M, Yoshioka K, Nagata T, Nakao S, Miyata K, Yokota T. Effects of combinations of gapmer antisense oligonucleotides on the target reduction. Mol Biol Rep. 2023;50:3539-46
195. Shin M, Chan IL, Cao Y, Gruntman AM, Lee J, Sousa J. et al. Intratracheally administered lna gapmer antisense oligonucleotides induce robust gene silencing in mouse lung fibroblasts. Nucleic Acids Res. 2022;50:8418-30
196. Danielsen MB, Lou C, Lisowiec-Wachnicka J, Pasternak A, Jørgensen PT, Wengel J. Gapmer antisense oligonucleotides containing 2′,3′-dideoxy-2′-fluoro-3′-c-hydroxymethyl-β-d-lyxofuranosyl nucleotides display site-specific rnase h cleavage and induce gene silencing. Chem Eur J. 2020;26:1368-79
197. Prakash TP, Yu J, Shen W, De Hoyos CL, Berdeja A, Gaus H. et al. Site-specific incorporation of 2′,5′-linked nucleic acids enhances therapeutic profile of antisense oligonucleotides. ACS Med Chem Lett. 2021;12:922-7
198. Pedersen L, Hagedorn PH, Lindholm MW, Lindow M. A kinetic model explains why shorter and less affine enzyme-recruiting oligonucleotides can be more potent. Mol Ther Nucleic Acids. 2014 3
199. Hagedorn PH, Hansen BR, Koch T, Lindow M. Managing the sequence-specificity of antisense oligonucleotides in drug discovery. Nucleic Acids Res. 2017;45:2262-82
200. Bisaria N, Jarmoskaite I, Herschlag D. Lessons from enzyme kinetics reveal specificity principles for rna-guided nucleases in rna interference and crispr-based genome editing. Cell Syst. 2017;4:21-9
201. Herschlag D, Piccirilli JA, Cech TR. Ribozyme-catalyzed and nonenzymic reactions of phosphate diesters: rate effects upon substitution of sulfur for a nonbridging phosphoryl oxygen atom. Biochemistry. 1991;30:4844-54
202. Czauderna F, Fechtner M, Dames S, Aygün H, Klippel A, Pronk GJ. et al. Structural variations and stabilising modifications of synthetic sirnas in mammalian cells. Nucleic Acids Res. 2003;31:2705-16
203. Hassler MR, Turanov AA, Alterman JF, Haraszti RA, Coles AH, Osborn MF. et al. Comparison of partially and fully chemically-modified sirna in conjugate-mediated delivery in vivo. Nucleic Acids Res. 2018;46:2185-96
204. Haraszti RA, Roux L, Coles AH, Turanov AA, Alterman JF, Echeverria D. et al. 5΄-vinylphosphonate improves tissue accumulation and efficacy of conjugated sirnas in vivo. Nucleic Acids Res. 2017;45:7581-92
205. Kremer A, Ryaykenen T, Segarra-Visent X, Sauer M, Breuer L, Wu Y. et al. Optimized chemical structure extends silencing duration of therapeutic siRNAs in dividing cancer and immune cells. Mol Ther. 2025
206. Dellafiore M, Aviñó A, Alagia A, Montserrat JM, Iribarren AM, Eritja R. SiRNA modified with 2′-deoxy-2′-c-methylpyrimidine nucleosides. ChemBioChem. 2018;19:1409-13
207. Schirle NT, Kinberger GA, Murray HF, Lima WF, Prakash TP, MacRae IJ. Structural analysis of human argonaute-2 bound to a modified sirna guide. J Am Chem Soc. 2016;138:8694-7
208. Iribe H, Miyamoto K, Takahashi T, Kobayashi Y, Leo J, Aida M. et al. Chemical modification of the sirna seed region suppresses off-target effects by steric hindrance to base-pairing with targets. ACS Omega. 2017;2:2055-64
209. Kobayashi Y, Fukuhara D, Akase D, Aida M, Ui-Tei K. SiRNA seed region is divided into two functionally different domains in rna interference in response to 2′-ome modifications. ACS Omega. 2022;7:2398-410
210. Pavlova AS, Yakovleva KI, Epanchitseva AV, Kupryushkin MS, Pyshnaya IA, Pyshnyi DV. et al. An influence of modification with phosphoryl guanidine combined with a 2′-o-methyl or 2′-fluoro group on the small-interfering-rna effect. Int J Mol Sci. 2021;22:9784
211. Shiohama Y, Fujita R, Sonokawa M, Hisano M, Kotake Y, Krstic-Demonacos M. et al. Elimination of off-target effect by chemical modification of 5′-end of sirna. Nucleic Acid Ther. 2022;32:438-47
212. Ackermann JM, Kanugula S, Pegg AE. DNAzyme-mediated silencing of ornithine decarboxylase. Biochemistry. 2005;44:2143-52
213. Abdelgany A, Uddin MK, Wood M, Taira K, Beeson D. Design of efficient dnazymes against muscle achr alpha-subunit crna in vitro and in hek 293 cells. J RNAi Gene Silencing. 2005;1:88-96
214. McCall MJ, Hendry P, Mir AA, Conaty J, Brown G, Lockett TJ. Small, efficient hammerhead ribozymes. Mol Biotechnol. 2000 14
215. Nedorezova DD, Dubovichenko MV, Eldeeb AA, Nur MAY, Bobkov GA, Ashmarova AI. et al. Cleaving folded rna by multifunctional dnazyme nanomachines. Chem Eur J. 2024;30:e202401580
216. Dubovichenko MV, Nedorezova DD, Patra C, Drozd VS, Andrianov VS, Ashmarova AI. et al. Marker-dependent cleavage of rna by binary (split) dnazyme (bidz) and binary dna machines (bidm). ChemBioChem. 2024;25:e202400665
217. Nguyen K, Malik TN, Chaput JC. Chemical evolution of an autonomous dnazyme with allele-specific gene silencing activity. Nat Commun. 2023;14:2413
218. Schubert S, Fürste JP, Werk D, Grunert H-P, Zeichhardt H, Erdmann VA. et al. Gaining target access for deoxyribozymes. J Mol Biol. 2004;339:355-63
219. Wang Y, Nguyen K, Spitale RC, Chaput JC. A biologically stable dnazyme that efficiently silences gene expression in cells. Nat Chem. 2021;13:319-26
220. Poudyal RR, Benslimane M, Lokugamage MP, Callaway MK, Staller S, Burke DH. Selective inactivation of functional rnas by ribozyme-catalyzed covalent modification. ACS Synth Biol. 2017;6:528-34
221. Liaqat A, Sednev MV, Stiller C, Höbartner C. RNA-cleaving deoxyribozymes differentiate methylated cytidine isomers in rna. Angew Chem Int Ed. 2021;60:19058-62
222. Wang Y, Liu E, Lam CH, Perrin DM. A densely modified m2+-independent dnazyme that cleaves rna efficiently with multiple catalytic turnover. Chem Sci. 2018;9:1813-21
223. Huang P-JJ, Liu J. In vitro selection of chemically modified dnazymes. ChemistryOpen. 2020;9:1046-59
224. Naghdi MR, Boutet E, Mucha C, Ouellet J, Perreault J. Single mutation in hammerhead ribozyme favors cleavage activity with manganese over magnesium. Noncoding RNA. 2020;6:14
225. Gruber C, Krautner L, Bergant V, Grass V, Ma Z, Rheinemann L. et al. Engineered, nucleocytoplasmic shuttling cas13d enables highly efficient cytosolic rna targeting. Cell Discov. 2024;10:1-4
226. Méndez-Mancilla A, Wessels H-H, Legut M, Kadina A, Mabuchi M, Walker J. et al. Chemically modified guide rnas enhance crispr-cas13 knockdown in human cells. Cell Chem Biol. 2022;29:321-327.e4
227. Byrne M, Vathipadiekal V, Apponi L, Iwamoto N, Kandasamy P, Longo K. et al. Stereochemistry enhances potency, efficacy, and durability of malat1 antisense oligonucleotides in vitro and in vivo in multiple species. Transl Vis Sci Technol. 2021;10:23
228. Liu Y, Dodart J-C, Tran H, Berkovitch S, Braun M, Byrne M. et al. Variant-selective stereopure oligonucleotides protect against pathologies associated with c9orf72-repeat expansion in preclinical models. Nat Commun. 2021;12:847
229. Kandasamy P, McClorey G, Shimizu M, Kothari N, Alam R, Iwamoto N. et al. Control of backbone chemistry and chirality boost oligonucleotide splice switching activity. Nucleic Acids Res. 2022;50:5443-66
230. Sakamuri S, Eltepu L, Liu D, Lam S, Meade BR, Liu B. et al. Impact of phosphorothioate chirality on double-stranded sirnas: a systematic evaluation of stereopure sirna designs. ChemBioChem. 2020;21:1304-8
231. Jahns H, Taneja N, Willoughby JLS, Akabane-Nakata M, Brown CR, Nguyen T. et al. Chirality matters: stereo-defined phosphorothioate linkages at the termini of small interfering rnas improve pharmacology in vivo. Nucleic Acids Res. 2022;50:1221-40
232. Yu D, Kandimalla ER, Roskey A, Zhao Q, Chen L, Chen J. et al. Stereo-enriched phosphorothioate oligodeoxynucleotides: synthesis, biophysical and biological properties. Bioorg Med Chem. 2000;8:275-84
233. Duschmalé J, Hansen HF, Duschmalé M, Koller E, Albaek N, Møller MR. et al. In vitro and in vivo properties of therapeutic oligonucleotides containing non-chiral 3′ and 5′ thiophosphate linkages. Nucleic Acids Res. 2020;48:63-74
234. Kaushal A. Innate immune regulations and various sirna modalities. Drug Deliv Transl Res. 2023:1-15
235. Pollak AJ, Zhao L, Vickers TA, Huggins IJ, Liang X-H, Crooke ST. Insights into innate immune activation via ps-aso-protein-tlr9 interactions. Nucleic Acids Res. 2022;50:8107-26
236. Shen W, De Hoyos CL, Migawa MT, Vickers TA, Sun H, Low A. et al. Chemical modification of ps-aso therapeutics reduces cellular protein-binding and improves the therapeutic index. Nat Biotechnol. 2019;37:640-50
237. Stull RA, Taylor LA, Szoka FC Jr. Predicting antisense oligonucleotide inhibitory efficacy: a computational approach using histograms and thermodynamic indices. Nucleic Acids Res. 1992;20:3501-8
238. SciTools web tools. Integr DNA Technol
239. Fu L, Cao Y, Wu J, Peng Q, Nie Q, Xie X. UFold: fast and accurate rna secondary structure prediction with deep learning. Nucleic Acids Res. 2022;50:e14
240. Munkácsy G, Sztupinszki Z, Herman P, Bán B, Pénzváltó Z, Szarvas N, et al. Validation of rnai silencing efficiency using gene array data shows 18.5% failure rate across 429 independent experiments. Mol Ther Nucleic Acids 2016; 5
Author contact
![]() Corresponding author: Mrs. Christina Patra, cpatraru.
Corresponding author: Mrs. Christina Patra, cpatraru.