Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Methods
Results
Discussion
Conclusion
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2026; 16(4):1959-1974. doi:10.7150/thno.122130 This issue Cite
Research Paper
MAP3K3 Contributes to Myocardial Ischemia/Reperfusion Injury by Promoting Myeloid Cell Diapedesis through TAL1/JAM-A Pathway
Shiyu Hu1,3,4,5,6,*, Jian Zhang1,3,4,5,6,*, Jingpu Wang1,3,4,5,6, Chenguang Li1,3,4,5,6, Yiwen Wang1,3,4,5,6, Jiayu Liang1,3,4,5,6, Rong Huang1,3,4,5,6, Ji'e Yang7, Yang Gao1,3,4,5,6, Yanan Qu1,3,4,5,6, Hongbo Yang1,3,4,5,6, Juying Qian1,3,4,5,6, Wenwen Tang2, ![]() , Jiatian Cao1,3,4,5,6,
, Jiatian Cao1,3,4,5,6, ![]() , Feng Zhang1,3,4,5,6,
, Feng Zhang1,3,4,5,6, ![]() , Junbo Ge1,3,4,5,6
, Junbo Ge1,3,4,5,6
1. Department of Cardiology, Shanghai Institute of Cardiovascular Diseases, Zhongshan Hospital, Fudan University, 200032 Shanghai, China.
2. Vascular Biology and Therapeutics Program, Department of Pharmacology, Yale University School of Medicine, New Haven, CT 06520, USA.
3. National Clinical Research Center for Interventional Medicine, 200032 Shanghai, China.
4. State Key Laboratory of Cardiovascular Diseases, Zhongshan Hospital, Fudan University, 200032 Shanghai, China.
5. NHC Key Laboratory of Ischemic Heart Diseases, 200032 Shanghai, China.
6. Key Laboratory of Viral Heart Diseases, Chinese Academy of Medical Sciences, 200032 Shanghai, China.
7. Department of Cardiology, The first Affiliated Hospital of University of Science and Technology of China (Anhui Provincial Hospital), 230000 Anhui, China.
*These authors contributed equally.
Received 2025-7-21; Accepted 2025-11-9; Published 2026-1-1
Abstract

Rationale: Extensive leukocyte diapedesis is a defining step in inflammation and contributes critically to myocardial ischemia/reperfusion injury (MI/RI). Infiltrating leukocytes amplify local inflammation and exacerbate myocardial damage. However, the upstream control of the trans-endothelial migration step remains incompletely understood.
Methods: Peripheral blood myeloid cells were isolated from MI/RI patients and healthy donors to examine MAP3K3 expression and its correlation with cardiac markers. Mouse MI/RI models were established to investigate MAP3K3 expression of myeloid cells in the heart. Myeloid-specific Map3k3 deficiency mice were used to evaluate the impact of MAP3K3 depletion on MI/RI severity and on myeloid cell diapedesis from the bone marrow. RNA sequencing and various manipulations of the MAP3K3/TAL1/JAM-A axis were used to elucidate its role in diapedesis. Finally, the therapeutic potential of pazopanib, a MAP3K3 inhibitor, was evaluated in the mouse MI/RI model.
Results: MAP3K3 expression was upregulated in both monocytes and neutrophils from MI/RI patients and was positively correlated with the severity of MI/RI. In mice, MAP3K3 in cardiac myeloid cells peaked at day 3 post-MI/RI. Myeloid cell-specific depletion of MAP3K3 alleviated MI/RI by reducing the infiltration of myeloid cells into cardiac tissue. Functionally, MAP3K3 facilitated myeloid cell de-adhesion and transmigration across endothelial barriers. Further mechanistic studies identified the MAP3K3/TAL1/JAM-A signaling pathway as a key regulator of myeloid cell diapedesis. MAP3K3 phosphorylates TAL1 at Ser-122, leading to its ubiquitination and attenuating its transcriptional repression of F11r (encoding JAM-A). Through JAM-A, MAP3K3 promotes integrin internalization, thereby enhancing de-adhesion and myeloid cell transmigration. Treatment with pazopanib, a MAP3K3 inhibitor, ameliorated MI/RI injury and reduced myeloid cell diapedesis into the heart by blocking MAP3K3 phosphorylation activity.
Conclusions: MAP3K3 orchestrates myeloid cell diapedesis via a TAL1/JAM-A dependent program during MI/RI. Targeting MAP3K3, exemplified by pazopanib, may offer a therapeutic strategy for MI/RI and related inflammatory conditions.
Keywords: myocardial ischemia/reperfusion injury, myeloid cells, diapedesis, MAP3K3, TAL1, JAM-A
Introduction
Although early restoration of blood supply reduces cardiomyocyte loss after myocardial infarction (MI) [1], secondary myocardial ischemia/reperfusion injury (MI/RI) still threatens the heart function [2]. Extensive leukocyte division and subsequent leukocyte diapedesis plays a central role in different inflammatory processes, including MI/RI. While leukocyte division (e.g., hematopoietic stem cell differentiation) sustains immune cell populations, leukocyte diapedesis is a dynamic process enabling monocytes, macrophages, and neutrophils to exit vasculature toward inflammatory sites [3]. Recruited leukocytes in the heart produce a large amount of pro-inflammatory factors, leading to an inflammatory cascade and myocardial injury [4].
The complete process of leukocyte diapedesis includes chemotaxis of leukocytes, leukocyte adhesion to vessels, and transmigration through vessels [5]. During reperfusion, dead or injured cardiomyocytes released damage-associated molecular patterns and produced chemokines such as monocyte chemoattractant protein 1 (MCP-1) and interleukin 8, which recruited leukocytes from bones to blood and tissues together with the activation of the complement system [4]. The elevation of several endothelial and cardiomyocyte-mediated adhesion factors such as P-selectin, E-selectin, intercellular adhesion molecule 1 (ICAM-1), and platelet endothelial cell adhesion molecule 1 (PECAM-1) promote the adhesion and rolling of peripheral leukocytes [6], which then infiltrate the ischemic myocardium through the vascular wall. Unveiling the key signaling pathway involved in leukocyte diapedesis offers valuable insights into the mechanisms underlying severe inflammation during MI/RI and presents potential therapeutic targets for its treatment.
Mitogen-activated protein kinase kinase kinase 3 (MAP3K3) is a highly conserved member of the MAP3K superfamily with Ser/Thr protein kinase activity [7]. MAP3K3 is involved in multiple important biological pathways: activation of ERK5 and p38 in the MAPK pathway [7-9], regulation of toll-like receptor pathway and NF-κB pathway [10, 11], lymphocyte differentiation [12], and development of the cardiovascular system and cerebrovascular disease [13-15]. MAP3K3 is reported as a key kinase in different cell types and inflammatory processes, such as neutrophils in acute lung injury [16], and platelets in MI [17, 18]. MAP3K3 in neutrophil mainly influences reactive oxygen species (ROS) formation [16], while MAP3K3 in platelets participates in thrombosis [17]. However, the role of MAP3K3 in myeloid cells, especially the biological process of diapedesis, is still unknown.
T cell acute lymphocytic leukemia (TAL1) is reported to play a vital role in the generation of erythroid and myeloid lineages [19], and its overexpression in lymphocytes can cause acute lymphoblastic leukemia [20]. Its function in the diapedesis of myeloid cells remains unclear.
Junctional adhesion molecule-A (JAM-A, encoded by F11r gene) is primally located in the tight junction between endothelial cells [21]. JAM-A on platelets has also been found to be involved in platelet-leukocyte aggregation [22, 23]. A few studies have found its role in leukocytes as a regulator of cell polarization [24] and transmigration through blood vessels [25]. JAM-A controls integrin internalization, promotes the de-adhesion of leukocytes from endothelial cells, and leads to the transmigration of leukocytes through blood vessels [25]. Deficiency of JAM-A can reduce leukocyte diapedesis during hepatic ischemia/reperfusion injury [26], MI/RI [27], acute lung injury [28], and atherosclerosis [29].
In this study, we disclosed the function of MAP3K3 on leukocyte diapedesis and unveiled the regulation function of MAP3K3 on JAM-A through the phosphorylation and ubiquitination of TAL1 on serine 122. Targeting MAP3K3 or JAM-A can both influence leukocyte diapedesis. Deficiency of MAP3K3 in myeloid cells or administration of pazopanib (an inhibitor of MAP3K3 [16]) attenuated MI/RI by decreasing leukocyte diapedesis. We provided a new insight into the MAP3K3/TAL1/JAM-A pathway in leukocyte diapedesis, highlighting its potential as a therapeutic target not only for MI/RI but also for other inflammatory diseases.
Methods
All data, study methods and materials that support the findings of this study are available from the corresponding authors on reasonable request. Detailed methods are provided in the Supplementary Material.
Patients
Peripheral blood of the MI/RI group was obtained 12 hours after PCI in 66 patients diagnosed with acute coronary syndrome (ACS) (clear diagnosis of myocardial infarction, STEMI and non-STEMI, and the presence of culprit vessels, stenosis greater than 90%, on coronary angiography). Peripheral blood of the control group was obtained from 6 donors with negative coronary angiography results (have coronary artery stenosis less than 50% and no other diseases that clearly affected coronary artery function) [30]. The study was approved by the Ethics Committee of the Fudan University Zhongshan Hospital, China (approval number: B2021-754). Informed consent was received from all patients as per the Declaration of Helsinki.
Animal study design and establishment of MI/RI models
Mouse MI/RI model was constructed with a 45-minute ligation time at the left anterior descending coronary artery [31].
The Map3k3fl/fl and Map3k3CKO mice were constructed by Cyagen Biosciences in C57BL/6 mice (Lyz2-Cre mice were from Jackson Laboratory Bar, Harbor, ME). Littermate mice with average age of 6-8 weeks were used for all experiments.
Sex-matched wild-type (WT) mice (Shanghai Jiesijie) aged 6-8 weeks were randomly divided into the saline group (200 μL saline) and pazopanib-treated group. Pazopanib-treated mice were injected intraperitoneally with 1.5 mg/kg pazopanib per mouse as previously described [16, 32]. The mice were injected immediately after surgery (after ligation, before reperfusion), 24 hours after surgery, and 48 hours after surgery.
All animal experiments were conducted following the ARRIVE guidelines and approved by the Animal Care and Use Committee (approval number: 2023-196, Zhongshan Hospital, Fudan University).
Statistical analysis
Statistical analyses were performed using GraphPad Prism software (version 8.4.2), following previously described methods [33]. For datasets involving two or more independent factors, comparisons were made using two-way ANOVA, while categorical variables were analyzed by chi-square test. A P value of less than 0.05 was considered statistically significant.
Results
MAP3K3 expression in myeloid cells increased after MI/RI
Through a bioinformatic transcriptome analysis on GSE123342 [34], we found MAP3K3 expression levels increased significantly in peripheral blood mononuclear cells (PBMCs) from MI patients compared to stable coronary artery disease (CAD) patients (Figure 1A). In human left ventricle tissue ischemic cardiomyopathy (ICM, GSE57338 [35]), MAP3K3 was also upregulated (Figure 1B). We validated this upregulation of MAP3K3 in neutrophils and monocytes from MI/RI patients respectively (Figure 1C-E). The age, sex, and morbidity of hypertension, diabetes, hyperlipidemia, and stroke between MI/RI patients and the Control group differed insignificantly, suggesting that the elevated expression level of MAP3K3 was mainly caused by MI/RI (detailed in Table 1).
MAP3K3 expression in myeloid cells increased and correlated with cardiac markers during MI/RI. (A) Heatmap of the differential expression genes (DEGs) in peripheral blood mononuclear cells (PBMCs) from myocardial infarction (MI) patients (n=45) compared to stable coronary artery disease (CAD) patients (n=14) (GSE123342). (B) MAP3K3 gene expression levels in human left ventricle tissue from ischemic cardiomyopathy (n=13) compared to non-failing donors (n=14) (GSE116250, unpaired t test). (C-D) Relative protein levels of MAP3K3 of monocytes (unpaired t test with Welch's correction) and neutrophils (unpaired t test) from myocardial ischemia/reperfusion injury (MI/RI) patients and healthy donors, respectively (n=6 each). (E) Relative mRNA levels of MAP3K3 of neutrophils and monocytes from MI/RI patients (n=66 each) and healthy donors (n=6 each), respectively (Mann-Whitney test for both). (F) Spearman correlation analysis of MAP3K3 mRNA levels in neutrophils with creatine kinase MB isoenzyme (CK-MB, n=66) and lactate dehydrogenase (LDH, n=52) levels in plasma of patients with MI/RI. (G) Pearson correlation analysis of MAP3K3 mRNA levels in monocytes with CK-MB (n=66), cardiac troponin T (cTnT, n=66), and LDH (n=52) levels in plasma of patients with MI/RI. (H) Flowcytometric dot plot analysis and staining strategy for neutrophils (CD45+Ly6G+) and monocytes (CD45+Ly6G-CD11b+Ly6C+) in MI/RI tissues and MAP3K3 expression levels in either myeloid cell. (I) MAP3K3 expression levels of neutrophils or monocytes after MI/RI in different time (n=4, one-way ANOVA test with Tukey's multiple comparisons test, each). All data were displayed as mean ± SEM. * P < 0.05; ** P < 0.01; *** P < 0.001; **** P < 0.0001.
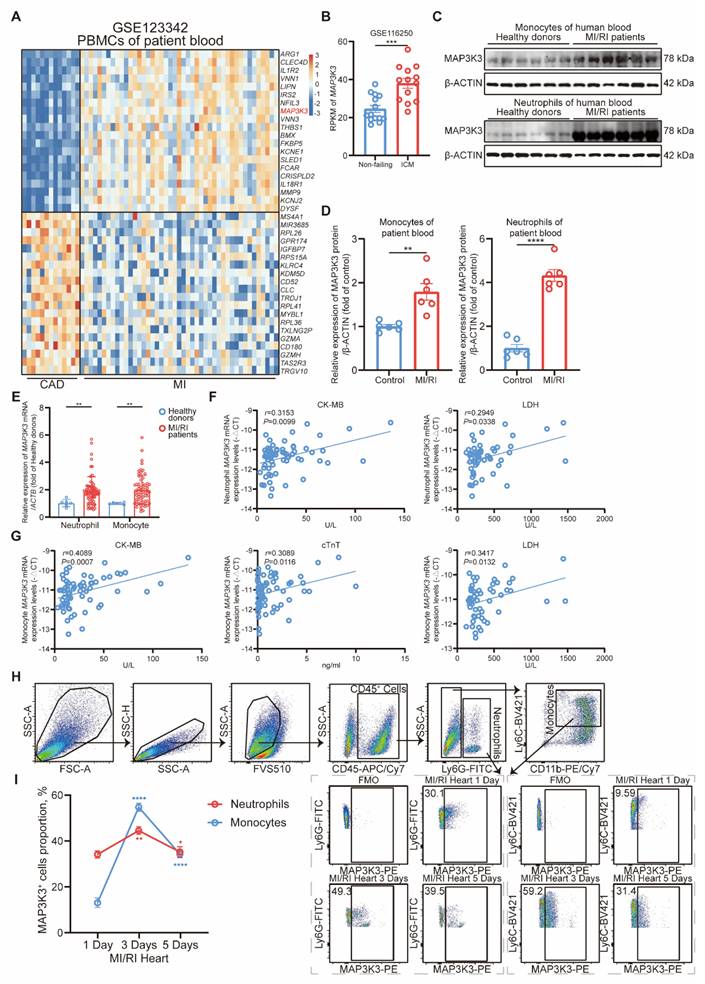
Clinical characteristics of 66 patients with MI/RI and 6 Control group donors.
| Characteristics | MI/RI (n=66) | Control (n=6) | P value |
|---|---|---|---|
| Age | 67.23±1.35 | 58.00±4.06 | 0.051 |
| Sex, male (%) | 42 (63.6) | 5 (83.3) | 0.332 |
| Hypertension (%) | 42 (63.6) | 5 (84.4) | 0.332 |
| Diabetes (%) | 32 (48.5) | 3 (50.0) | 0.943 |
| Hyperlipidemia (%) | 15 (33.7) | 2 (66.7) | 0.558 |
| Stroke (%) | 7 (10.6) | 0 (0.0) | 0.401 |
| STEMI (%) | 31 (47.0) | ||
| Degree of stenosis in the culprit vessel (%) | 93.56±1.05 | 25.00±8.06 | <0.0001 |
| Ejection fraction (%) | 53.66±1.26 | 59.67±3.08 | 0.145 |
| cTnT (ng/mL) | 1.41±0.25 | 0.01±0.00 | <0.0001 |
| CK-MB (U/L) | 27.83±3.11 | 15.33±1.26 | 0.0004 |
| Pro-BNP (pg/mL) | 2775.31±695.63 | 101.07±30.17 | <0.0001 |
| LDH (U/L) | 392.79±41.24 | 169.33±9.07 | 0.0017 |
| WBC count (109/L) | 8.48±0.34 | 6.48±1.14 | 0.1262 |
| Neutrophil percentage (%) | 68.82±1.01 | 57.55±3.31 | 0.002 |
| Lymphocyte percentage (%) | 20.53±0.93 | 30.20±3.03 | 0.004 |
| NLR% | 4.10±0.31 | 2.07±0.34 | 0.043 |
| Monocyte percentage (%) | 8.56±0.27 | 7.68±0.44 | 0.331 |
| Neutrophil count (109/L) | 5.91±0.31 | 3.82±0.74 | 0.049 |
| Lymphocyte count (109/L) | 1.63±0.83 | 2.66±0.70 | 0.135 |
| NLR | 4.11±0.32 | 1.85±0.33 | 0.001 |
| Monocyte count (109/L) | 0.77±0.60 | 0.49±0.80 | 0.069 |
| Glycosylated hemoglobin (%) | 6.84±0.25 | 5.82±0.25 | 0.108 |
| CRP (mg/L) | 21.12±3.40 | 0.97±0.33 | <0.0001 |
| Previous MI episodes (%) | 11 (16.7) | 0 (0.0) | |
| Dual-antiplatelet therapy, aspirin + clopidogrel or ticagrelor (%) | 23 (34.8) | ||
| Statins (%) | 66 (100) | ||
| β-blocker (%) | 40 (60.6) | 2 (66.7) | 0.193 |
| Anti-hypertension therapy (%) | 42 (63.6) | 5 (84.4) | 0.332 |
| Anti-diabetes therapy (%) | 32 (48.5) | 3 (50.0) | 0.943 |
Data presented as mean ± standard error of the mean (SEM) or number (percentage). Statistical analysis was described in methods. STEMI, ST-elevated myocardial infarction; cTnT, cardiac troponin T; CK-MB, creatine kinase MB isoenzyme; pro-BNP, pro B-type natriuretic peptide; LDH, lactate dehydrogenase; WBC, white blood cell; NLR, neutrophil-to-lymphocyte ratio; CRP, C-reactive protein.
To further explore the relationship between MAP3K3 and the severity of MI/RI, we assessed the relationship between MAP3K3 expression level in neutrophils and monocytes of MI/RI patients and serum cardiac markers [36, 37], including CK-MB (creatine kinase M-type), cTnT (cardiac troponin T), LDH (lactate dehydrogenase), pro-BNP (pro B-type natriuretic peptide), CRP (C-reactive protein), IL-6 (interleukin-6), and complete blood count. We found that mRNA expression levels of MAP3K3 in neutrophils and monocytes had a positive correlation with CK-MB, and LDH, while MAP3K3 in monocytes also had a positive correlation with cTnT (Figure 1F-G). The mRNA expression levels of MAP3K3 in neutrophils and monocytes also had a positive correlation with mRNA expression levels of IL-6, indicating that MAP3K3 might aggravate inflammation in MI/RI patients (Figure S1A-B).
To explore the expression level of MAP3K3 in infiltrating myeloid cells in cardiac tissue after MI/RI, we conducted flowcytometric dot plot analysis on cardiac tissue after MI/RI at different time points (Figure 1H and Figure S1C). We found that MAP3K3 expression levels peaked at 3 days after MI/RI in myeloid cells (Figure 1I). Immunofluorescence staining of MI/RI tissue sections also showed large amounts of infiltrating myeloid cells in heart tissue with high expression levels of MAP3K3 (Figure S1D).
Myeloid-specific Map3k3 deficiency alleviates MI/RI through dysfunction of diapedesis from bone marrow
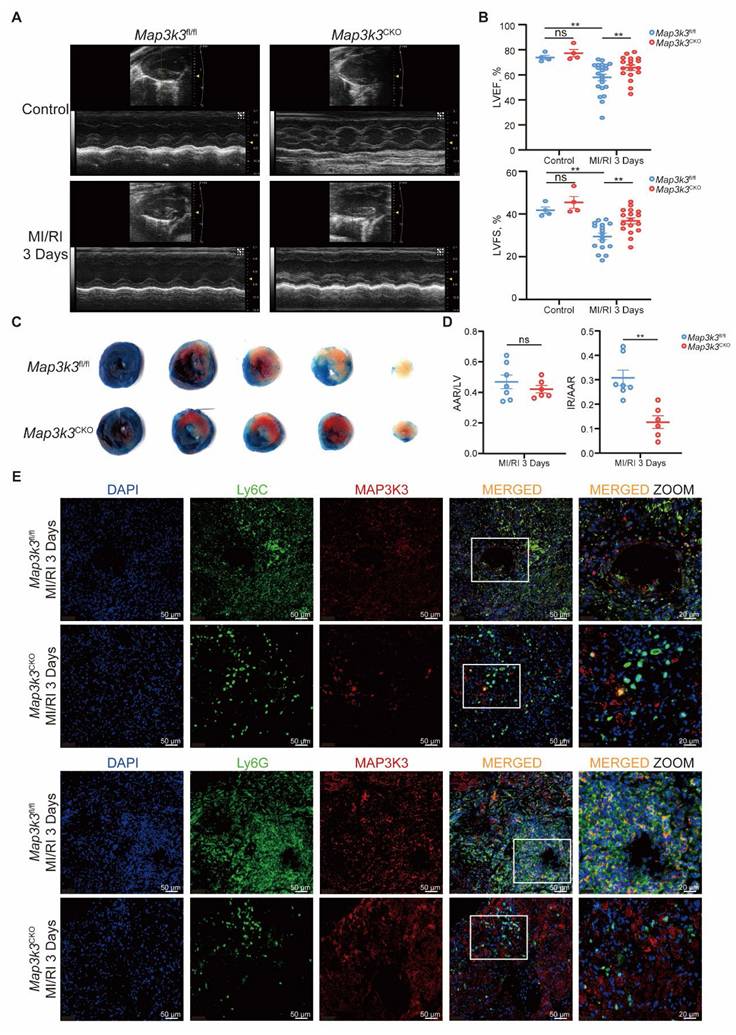
We next generated myeloid-specific Map3k3 deficiency (Map3k3CKO) mice and conducted MI/RI models in Map3k3CKO mice compared to Map3k3fl/fl mice. Deficiency of Map3k3 in myeloid cells preserved heart function reflected by elevated left ventricular ejection fraction (LVEF) and left ventricular fraction shortening (LVFS) (Figure 2A-B) and reduced infarcted size (Figure 2C-D). Expression levels of apoptotic protein BAX and cleaved CASPASE-3 also decreased in Map3k3CKO mice (Figure S2A-B). Deficiency of Map3k3 in myeloid cells also reduced apoptosis of cardiomyocytes stained by TdT-mediated dUTP nick end labeling (TUNEL, Figure S2C).
Myeloid-specific Map3k3 deficiency alleviated MI/RI and decreased myeloid cell infiltration to heart. (A) Representative images of echocardiography of Map3k3CKO mice and Map3k3fl/fl mice undergoing MI/RI. (B) Left ventricular ejection fraction (LVEF, top) and left ventricular fractional shortening (LVFS, bottom, one-way ANOVA test with Tukey's multiple comparisons test for both) of control + Map3k3fl/fl mice (n=4), control + Map3k3CKO mice (n=4), MI/RI + Map3k3fl/fl mice (n=17), and MI/RI + Map3k3CKO mice (n=17). (C) Representative image of left ventricular tissue sections stained with Evans blue and 2,3,5-triphenyl tetrazolium chloride at 3 days after MI/RI to delineate the area at risk (AAR, red) and the infarcted area (IR, white). (D) The ratios of AAR/LV (left) and IR/AAR (right, unpaired t test for both) were compared (n=7 for Map3k3fl/fl mice, n=6 for Map3k3CKO mice). (E) Representative fluorescence images of myeloid cells infiltrated in MI/RI tissue sections stained for DAPI (blue), Ly6G/Ly6C (green), MAP3K3 (red), *200, scale bar 50 μm; *400, scale bar 20 μm. All data were displayed as mean ± SEM. ns, P > 0.05; ** P < 0.01.
To explore the function of MAP3K3 in myeloid cells during MI/RI, we first conducted immunofluorescence staining of MI/RI tissue sections and found that deficiency of Map3k3 in myeloid cells led to a decrease of infiltrating myeloid cells in heart tissue (Figure 2E and Figure S2D). To further elucidate this diapedesis change caused by deficiency of Map3k3 in myeloid cells, we conducted flowcytometric dot plot analysis on MI/RI mice (Figure S3A). We found that Map3k3CKO mice showed a decreased proportion of both neutrophils and monocytes infiltrated in the heart (Figure 3A-B and Figure S1C). Tracing back along the diapedesis of myeloid cells, we found that neutrophils and monocytes also decreased in the blood of Map3k3CKO mice (Figure 3C-D and Figure S3B) but increased in the bone of Map3k3CKO mice (Figure 3E-F and Figure S3B). Given that myeloid cells didn't home to the spleen (Figure S3C-D), we concluded that MAP3K3 might regulate the diapedesis of myeloid cells from bone to blood and eventually to heart. Hematoxylin-eosin (HE) staining also showed less diapedesis and disorganization in the heart of Map3k3CKO mice (Figure S3E).
Myeloid-specific Map3k3 deficiency decreased diapedesis from bone after MI/RI and diapedesis in vivo. (A) Representative flowcytometric dot plot analysis of infiltrating myeloid cells in heart of Map3k3fl/fl mice and Map3k3CKO mice. (B) Proportion of leukocytes (top), monocytes (bottom left), and neutrophils (bottom right, one-way ANOVA test with Tukey's multiple comparisons test for all) in heart of control + Map3k3fl/fl mice (n=4), control + Map3k3CKO mice (n=4), MI/RI + Map3k3fl/fl mice (n=8), and MI/RI + Map3k3CKO mice (n=8). (C) Representative flowcytometric dot plot analysis of myeloid cells in blood of Map3k3fl/fl mice and Map3k3CKO mice. (D) Proportion of leukocytes (top, one-way ANOVA test with Tukey's multiple comparisons test), monocytes (bottom left, Kruskal-Wallis's test with Dunn's multiple comparisons test), and neutrophils (bottom right, one-way ANOVA test with Tukey's multiple comparisons test) in blood of control + Map3k3fl/fl mice (n=4), control + Map3k3CKO mice (n=4), MI/RI + Map3k3fl/fl mice (n=7), and MI/RI + Map3k3CKO mice (n=6). (E) Representative flowcytometric dot plot analysis of myeloid cells in bone of Map3k3fl/fl mice and Map3k3CKO mice. (F) Proportion of leukocytes (top, Kruskal-Wallis's test with Dunn's multiple comparisons test), monocytes (bottom left, one-way ANOVA test with Tukey's multiple comparisons test), and neutrophils (bottom right, one-way ANOVA test with Tukey's multiple comparisons test) in bone of control + Map3k3fl/fl mice (n=4), control + Map3k3CKO mice (n=4), MI/RI + Map3k3fl/fl mice (n=5), and MI/RI + Map3k3CKO mice (n=8). (G) Study design of myeloid cells diapedesis in vivo experiment. Mice were intraperitoneally injected (i.p.) with thioglycolate (TG) for 1.5 hours. Bone marrow myeloid cells were labeled with a CFSE or a far-red dye and tail vein injected (i.v.) or intraperitoneally injected (i.p.) to mice with peritonitis. Cells were collected 2.5 hours afterward from peripheral blood and peritoneum. (H) The ratios between CFSE and far-red-labeled cells collected from blood after i.v. (n=6 each, Kruskal-Wallis's test with Dunn's multiple comparisons test), from peritoneum after i.v. (n=6 each, one-way ANOVA test with Tukey's multiple comparisons test), and from peritoneum after i.p. (n=3 each, unpaired t test). All data were displayed as mean ± SEM. ns, P > 0.05; * P < 0.05; ** P < 0.01; *** P < 0.001; **** P < 0.0001.
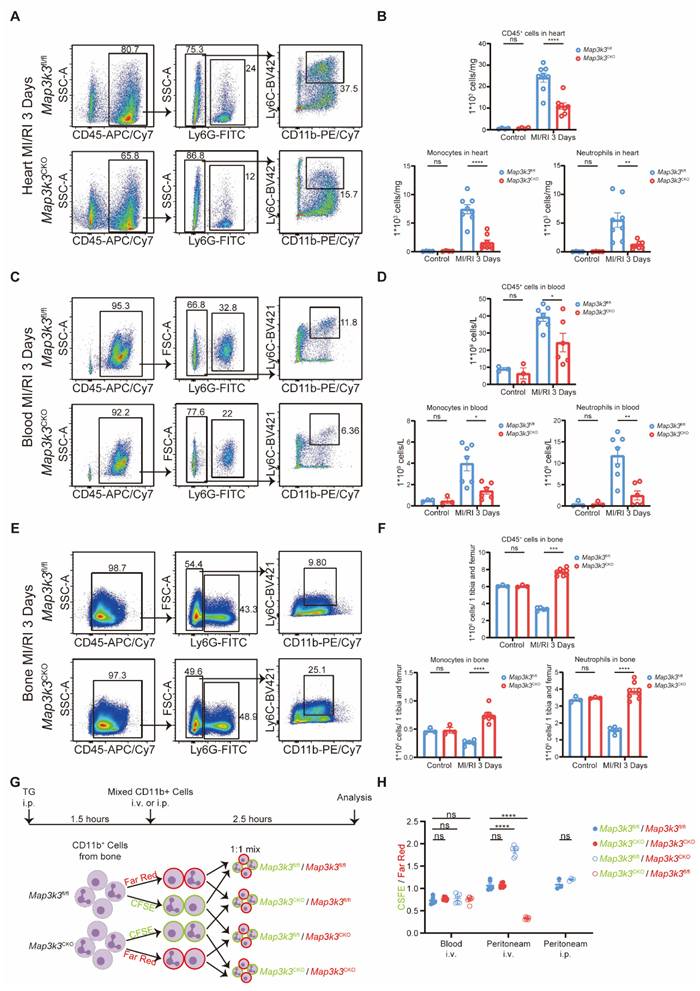
In addition, we conducted another inflammatory model, lipopolysaccharide (LPS)-induced sepsis, and examined diapedesis in targeted tissue lung and heart. As LPS-induced sepsis is an acute inflammation model, we focused on the diapedesis of neutrophils. As expected, deficiency of Map3k3 in myeloid cells decreased the diapedesis of neutrophils to the lung and heart from blood and bone (Figure S4).
We also performed another in vivo experiment as previously reported [38] to determine the function of MAP3K3 in diapedesis. After pre-treating mice with thioglycolate (TG) for 1.5 hours, a mixture of labeled myeloid cells from the bone of Map3k3CKO mice or Map3k3fl/fl mice was injected into circulation, with the ratio between the two myeloid cells populations assessed in the peritoneum and blood after 2.5 h (Figure 3G). All mixtures showed a mildly different label ratio in the blood, which confirmed the 1:1 mixture and no influence during the injection process. In the peritoneum, we observed a reduced proportion of myeloid cells of Map3k3CKO mice regardless of dye. While myeloid cells were injected directly into the peritoneum, we didn't observe significant changes in the proportion of myeloid cells (Figure 3H).
To this extent, we found that MAP3K3 played a vital role in the diapedesis of myeloid cells.
Myeloid-specific Map3k3 deficiency increased inhibitory transcription function of TAL1 on F11r by phosphorylation
To elucidate the downstream mechanism of MAP3K3 on diapedesis, we conducted RNA-seq comparing bone marrow myeloid cells from Map3k3CKO mice and Map3k3fl/fl mice. Sequencing results showed that many adhesion-related genes, such as Itgb3, Adgre4, and Itgax, were upregulated in Map3k3CKO mice, while the de-adhesion gene F11r was downregulated in Map3k3CKO mice (Figure 4A-B). Gene ontology (GO) analysis of the adhesion and migration pathway also showed increased enrichment of genes in adhesion-related terms and decreased enrichment of genes in migration-related genes from Map3k3CKO mice (Figure 4C). Genes of Kyoto Encyclopedia of Genes and Genomes (KEGG) term 'Cell adhesion molecules' (mmu04514) also showed upregulation of Itgb3 and Itgax and downregulation of F11r (Figure 4D). According to the function of JAM-A, we hypothesized that MAP3K3 regulated F11r expression, facilitated de-adhesion of myeloid cells to endothelial cells, and caused diapedesis.
Myeloid-Specific Map3k3 Deficiency increased inhibitory transcription function of TAL1 on F11r. (A) Heatmap of the DEGs in bone marrow myeloid cells from Map3k3fl/fl mice (n=3) and Map3k3CKO mice (n=3). Genes related to migration and adhesion were shown. (B) Volcano plot of the DEGs in bone marrow myeloid cells from Map3k3fl/fl mice (n=3) and Map3k3CKO mice (n=3). Genes related to migration and adhesion were shown. (C) GO analysis of DEGs. GO terms related to migration and adhesion were shown. (D) Heatmap of the DEGs from KEGG term 'Cell adhesion molecules' (mmu04514). (E) Four candidate transcription factors potentially involved in the regulation of F11r transcription predicted by RNA-seq. (F) Volcano plot of differentially expressed phosphorylated protein of phosphoproteomic analysis comparing bone marrow myeloid cells from Map3k3fl/fl mice (n=3) and Map3k3CKO mice (n=3). Phosphorylated protein related to MAP3K3 and candidate transcription factors were shown. (G) TAL1 and F11r promoter luciferase activity were detected by dual-luciferase assays (n=12 each, Brown-Forsythe test with Dunnett's T3 multiple comparisons test). (H) Western blots of MAP3K3, TAL1, and JAM-A proteins from blood myeloid cells of control mice and MI/RI mice. (I) Western blots of MAP3K3, TAL1, and JAM-A proteins from blood myeloid cells of Map3k3fl/fl mice and Map3k3CKO mice after MI/RI. (J) Western blots of MAP3K3, TAL1, and JAM-A proteins from bone marrow myeloid cells of Map3k3fl/fl mice and Map3k3CKO mice, treated with MCP-1 and Pazopanib or not. (K) Western blots of MAP3K3, TAL1, and JAM-A proteins from 293T, treated with MCP-1 and Pazopanib or not, transfected with MAP3K3 knockdown adenovirus or not. (L) Western blots of MAP3K3, TAL1, and JAM-A proteins from 293T, treated with MCP-1, transfected with TAL1 overexpression plasmid and MAP3K3 overexpression plasmid or not. (M) Western blots of MAP3K3, TAL1, and JAM-A proteins from 293T, treated with MCP-1, transfected with TAL1 siRNA and MAP3K3 knockdown adenovirus or not. (N) Western blots of MAP3K3, TAL1, and JAM-A proteins from 293T, treated with MCP-1, transfected with TAL1 siRNA, and Pazopanib or not. All data were displayed as mean ± SEM. ns, P > 0.05; **** P < 0.0001.
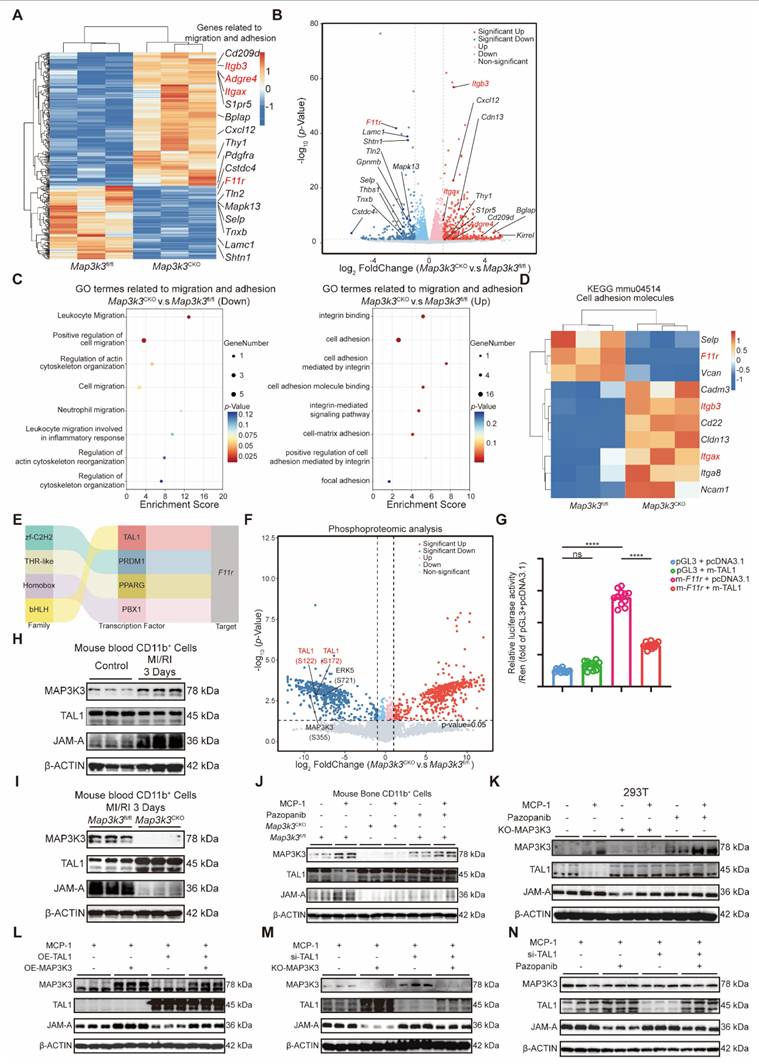
As the change of F11r was at the RNA levels, we traced back to its transcription factors and identified four candidate transcription factors potentially involved in the regulation of F11r transcription: TAL1, PRDM1, PPARG, and PBX1 (Figure 4E). To further elucidate the functional impact and downstream effects of MAP3K3, we performed phosphoproteomic analysis comparing bone marrow myeloid cells from Map3k3CKO mice and Map3k3fl/fl mice. The reduced phosphorylation at MAP3K3-S355 and its known downstream ERK5-S721 [39] in Map3k3CKO cells confirmed that MAP3K3 deletion impairs its phosphorylation-mediated signaling. Among the four transcription factors, only TAL1 showed reduced phosphorylation, specifically at S122 and S172, in Map3k3CKO cells (Figure 4F). Based on these findings, we hypothesized that MAP3K3 may regulate the transcription of F11r by modulating the phosphorylation of TAL1. We conducted dual-luciferase assays and found that TAL1 mainly suppressed the transcription of F11r (Figure 4G), as previously reported for the transcription inhibition function of TAL1 [40]. In myeloid cells from the blood of MI/RI mice (Figure S5A), we observed increasing expression levels of MAP3K3 and JAM-A, while TAL1 was inhibited (Figure 4H and Figure S5B). In Map3k3CKO mice, the above expression status was reversed (Figure 4I and Figure S5C). In vitro, bone marrow myeloid cells stimulated by monocyte chemokine MCP-1 can also cause an increase of MAP3K3 and JAM-A with a decrease of TAL1. Cells from Map3k3CKO mice or treated with pazopanib (a kinase inhibitor of MAP3K3 [16]) showed higher TAL1 expression and suppressed JAM-A (Figure 4J). To simplify the following experiments, the 293T cell line was used and we observed the same phenomenon. In 293T cells (Figure 4K), overexpressed TAL1 could reverse the up-regulation of JAM-A by overexpression of MAP3K3 (Figure 4L and Figure S5D), and the knockdown of TAL1 could reverse the down-regulation of JAM-A by MAP3K3 knockout or administration of pazopanib (Figure 4M-N and Figure S5E-F). The reverse experiments confirmed that MAP3K3 up-regulated F11r expression by downregulating its inhibitory transcription factor TAL1. This mechanism might be dependent on the kinase activity of MAP3K3 because the pazopanib showed a similar effect as that of MAP3K3 knockout.
Map3k3 knockout increased adhesion and decreased diapedesis by decreasing JAM-A expression and integrin internalization
Next, we focused on the function of MAP3K3 in the de-adhesion of myeloid cells in the manner of phosphorylation and regulation of JAM-A in vitro. We conducted a Transwell assay to evaluate the transmigration of myeloid cells through endothelial cells with different treatments. Compared to the myeloid cells from Map3k3fl/fl mice, myeloid cells from Map3k3CKO mice showed an impaired function of diapedesis during the time course (Figure 5A-D). Meanwhile, α-JAM-A antibody or pazopanib-treated cells also showed decreased diapedesis compared to the untreated or IgG1-treated cells (Figure 5A, E-F). However, in the adhesion assay, myeloid cells from Map3k3CKO mice showed an increased adhesion (Figure 5G-I).
Map3k3 Deficiency increased adhesion and decreased diapedesis through JAM-A in a phosphor-related manner. (A) Study design of Transwell assay for flowcytometric dot plot analysis on upper chamber and lower chamber myeloid cells. (B) Study design of Transwell assay for microscope photography. (C) Representative image of lower chamber cells from Map3k3fl/fl mice and Map3k3CKO mice after different time course during Transwell assay. Scale bar: 3000 μm. (D) Cell counts in the lower chamber after different time course during Transwell assay (n=6 each, Two-way ANOVA test). (E) Representative image of lower chamber cells from Map3k3fl/fl mice treated with IgG1, α-JAM-A, or Pazopanib after Transwell assay. Scale bar: 3000 μm. (F) Cell counts in the lower chamber after Transwell assay (n=6 each, Brown-Forsythe test with Dunnett's T3 multiple comparisons test). (G) Study design of Adhesion assay for microscope photography. (H) Representative image of adhered cells from Map3k3fl/fl mice and Map3k3CKO mice after Adhesion assay. Scale bar: 3000 μm. (I) Cell count adhesion to endothelial cells after Adhesion assay (n=6 each, unpaired t test). All data were displayed as mean ± SEM. ns, P > 0.05; * P < 0.05; ** P < 0.01; *** P < 0.001; **** P < 0.0001.
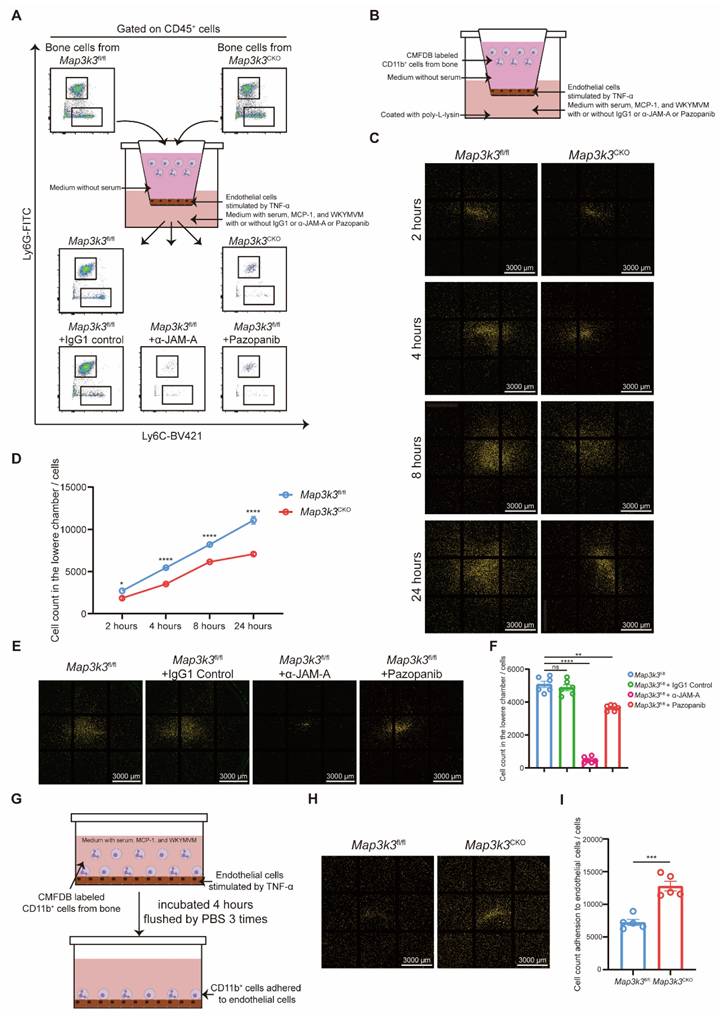
As JAM-A promotes diapedesis by controlling integrin internalization [25], we tested this function in vitro. Flowcytometric dot plot analysis without fixation and permeabilization mainly stained the surface protein, and we found that Map3k3 knockout, α-JAM-A, or pazopanib treatment showed higher integrin levels on plasma membrane after stimulated by chemokines, especially in neutrophils, but JAM-A on plasma membrane didn't change significantly (Figure S6A-D). Flowcytometric dot plot analysis after fixation and permeabilization showed the overall expression levels of protein in the cells. We found that Map3k3 knockout or pazopanib treatment showed higher integrin expression levels and lower JAM-A expression levels after being stimulated by chemokines, however, α-JAM-A treatment didn't influence the expression level of integrin and JAM-A significantly (Figure S6E-H). Immunofluorescence staining of chemokines stimulated cells showed that JAM-A was able to internalize integrins in normal conditions, while Map3k3 knockout or pazopanib treatment decreased JAM-A expression and kept integrin on the plasma membrane. α-JAM-A treatment kept JAM-A and integrin together on the plasma membrane as a complex without influencing the expression of JAM-A (Figure S6I). Plasma membrane-cytosol separation western blots also showed integrin internalization after MCP-1 stimulation which can be decreased by Map3k3 knockout or pazopanib treatment (Figure S6J).
Immunofluorescence staining of MI/RI tissue sections showed that after depletion of Map3k3, myeloid cells tended to adhere to the blood vessels instead of migrating through blood vessels, resulting in the decrease of infiltrating myeloid cells in heart tissue (Figure S7A).
MAP3K3 induced the proteasome-dependent degradation of TAL1 by phosphorylation on Ser-122
As we found above that the regulation of MAP3K3 on F11r was in a phosphorylation-dependent manner, we attempted to unveil whether TAL1 can be phosphorylated by MAP3K3. Previous studies on TAL1 all showed that TAL1 can be ubiquitinated after being phosphorylated on three highly conserved residues, Ser-122 [41], Ser-172 [42], or Thr-90 [43]. We first tested whether the decrease of TAL1 by MAP3K3 might be due to protein degradation. Proteasome inhibitor MG132 successfully decreased the degradation of TAL1 due to the overexpression of MAP3K3 (Figure 6A), while using cycloheximide (CHX) to suppress protein synthesis showed that the degradation of TAL1 protein was accelerated after overexpression of MAP3K3 and the stability of TAL1 protein increased after knockout of MAP3K3 (Figure 6B-C). These results supported that MAP3K3 reduces TAL1 by inducing its proteasome-dependent degradation.
MAP3K3 induced the proteasome-dependent degradation of TAL1 by phosphorylation on Ser-122. (A) Western blots of MAP3K3 and TAL1 proteins from 293T treated with MCP-1 and MG132 or not, transfected with MAP3K3 overexpression plasmid and MAP3K3 knockdown adenovirus or not. (B) Western blots of MAP3K3 and TAL1 proteins from 293T treated with MCP-1 and CHX time course, transfected with MAP3K3 overexpression plasmid and MAP3K3 knockdown adenovirus or not. (C) Relative protein level of TAL1 transfected with MAP3K3 overexpression plasmid, or MAP3K3 knockdown adenovirus along CHX time course. (D) Western blots of HA-TAL1 proteins from IP-myc and myc-UB, Flag-MAP3K3, and HA-TAL1 proteins from input. 293T was treated with MCP-1 and MG132, transfected with myc-UB plasmid, Flag-MAP3K3 plasmid or MAP3K3 knockdown adenovirus, and HA-TAL1 plasmid mutated at WT, T90A, S172A, S122A, S172E, or S122E. (E) In vitro kinase assay was performed using purified recombinant GST-MAPK3K3 kinase and purified SUMO-his-TAL1 protein (WT or S122A mutant). Protein expression levels of GST-MAP3K3, p-S/T, and SUMO-his-TAL1 were detected by western blots. (F) Western blots of HA-TAL1 proteins from 293T treated with MCP-1 and MG132, transfected with HA-TAL1 plasmid mutated at WT, S122A, or S122E. (G) Western blots of HA-TAL1 proteins from 293T treated with MCP-1 and CHX time course, transfected with HA-TAL1 plasmid mutated at WT, S122A, or S122E. (H) Relative protein level of HA-TAL1 transfected with HA-TAL1 plasmid mutated at WT, S122A, or S122E along CHX time course. (I) Western blots of MAP3K3, p-TAL1 (S122), and TAL1 proteins from bone marrow myeloid cells of Map3k3fl/fl mice and Map3k3CKO mice, treated with MCP-1, MG132, and Pazopanib or not. (J) Western blots of MAP3K3, p-TAL1 (S122), and TAL1 proteins from 293T treated with MCP-1, MG132, and Pazopanib or not, transfected with MAP3K3 overexpression plasmid and MAP3K3 knockdown adenovirus or not. (K) Western blots of p-TAL1 (S122) proteins from blood myeloid cells of control mice and MI/RI mice. (L) Western blots of p-TAL1 (S122) proteins from blood myeloid cells of Map3k3fl/fl mice and Map3k3CKO mice after MI/RI.
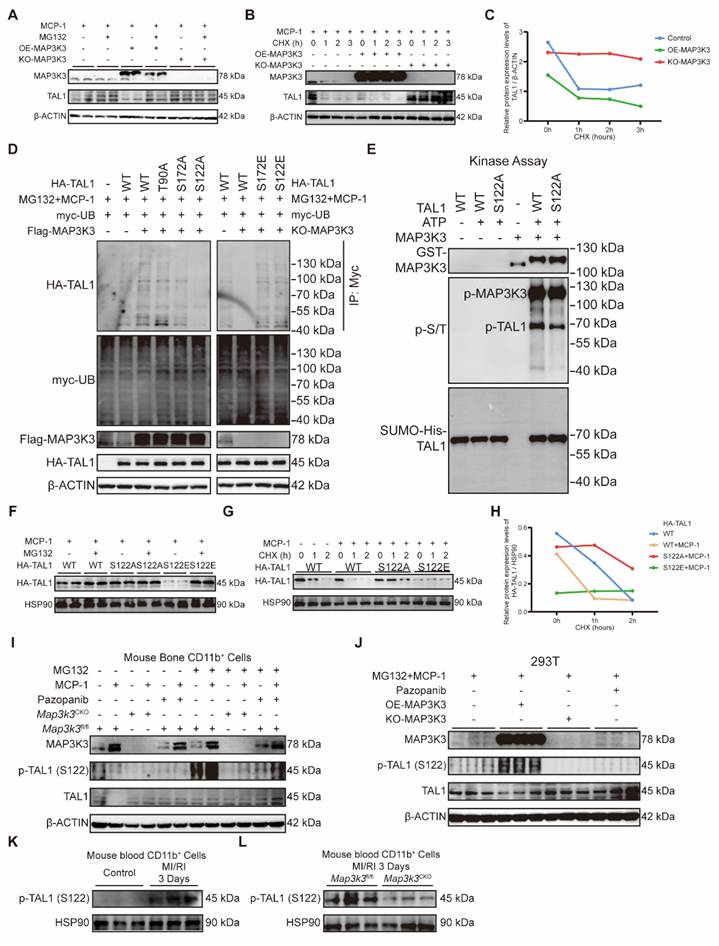
To determine which site on TAL1 was phosphorylated by MAP3K3, we predicted the kinase-specific phosphorylation sites in proteins on GPS 6.0 [44]. MAP3K3 and its downstream kinases were predicted to phosphorylate TAL1 on Ser-122 and Ser-172 at the highest score (Table S1). After comprehensive consideration of previous studies and our prediction results, we mutated Ser-122, Ser-172, or Thr-90 of TAL1 to Alanine (A). Overexpression of MAP3K3 increased the phosphorylation of TAL1 detected by pan phosphorylated antibody and increased the ubiquitination of TAL1. T90A mutation failed to decrease the phosphorylation and ubiquitination, while S172A mutation and S122A mutation decreased the phosphorylation and ubiquitination of TAL1 significantly. We further respectively mutated Ser-122 or Ser-172 to Glutamic acid (E) to mimic phosphorylation. Knockout of MAP3K3 reduced the phosphorylation and ubiquitination of TAL1 while S122E or S172E mutation maintained the ubiquitination of TAL1 (Figure 6D). To determine the direct phosphorylation function of MAP3K3 on TAL1, we performed an in vitro kinase assay and found that S122 could be phosphorylated directly by MAP3K3 (Figure 6E).
S122A mutation decreased the degradation of TAL1, while S122E mutation accelerated this process which can be reversed by MG132 (Figure 6F). Using CHX to suppress protein synthesis showed that the degradation of TAL1 protein was accelerated after S122E mutation and the stability of TAL1 protein increased after S122A mutation (Figure 6G-H). Bone marrow myeloid cells stimulated by monocyte chemokine MCP-1 can cause the increase of MAP3K3 and the degradation of TAL1 while using MG132 can help the observation of phosphorylation of TAL1 at Ser-122. Cells from Map3k3CKO mice or treated with pazopanib showed lower phosphorylation and higher stability of TAL1 (Figure 6I). The same phenomenon was also observed in 293T cell lines (Figure 6J). In myeloid cells from the blood of MI/RI mice, we also observed increasing expression levels of p-TAL1 (S122) (Figure 6K and Figure S7B), while in Map3k3CKO mice, this expression status was reversed (Figure 6L and Figure S7C).
Pazopanib ameliorated MI/RI by decreasing phosphorylation function of MAP3K3 and diapedesis from bone
The above findings suggested that MAP3K3 and its kinase activity would be a potential therapeutic target for treating MI/RI and other inflammatory diseases related to diapedesis. Pazopanib has shown curative effect in acute lung injury [16], and effective inhibition of diapedesis in vitro. Thus, we tested the effects of pazopanib in MI/RI. Pazopanib treatment led to amelioration of MI/RI reflected in elevated LVEF and LVFS (Figure 7A-B), reduced infarcted size (Figure 7C-D), and reduced apoptosis of cardiomyocyte stained by TUNEL (Figure S8A).
Pazopanib ameliorated MI/RI by decreasing phosphorylation function of MAP3K3 and diapedesis from bone. (A) Representative images of echocardiography of saline treated mice and Pazopanib treated mice undergoing MI/RI. (B) LVEF (left) and LVFS (right, unpaired t test for both) of MI/RI + Saline mice (n=4), and MI/RI + Pazopanib mice (n=4). (C) Representative image of left ventricular tissue sections stained with Evans blue and 2,3,5-triphenyl tetrazolium chloride at 3 days after MI/RI to delineate the area at risk (AAR, red) and the infarcted area (IR, white). (D) The ratios of AAR/LV (left) and IR/AAR (right, unpaired t test for both) were compared (n=7 for MI/RI + Saline mice, n=6 for MI/RI + Pazopanib mice). (E) Representative flowcytometric dot plot analysis of infiltrating myeloid cells in heart of saline treated mice and Pazopanib treated mice. (F) Proportion of leukocytes (left, unpaired t test), monocytes (middle, unpaired t test with Welch's correction), and neutrophils (right, unpaired t test) in heart of MI/RI + Saline mice (n=4), and MI/RI + Pazopanib mice (n=4). (G) Representative flowcytometric dot plot analysis of myeloid cells in blood of saline treated mice and Pazopanib treated mice. (H) Proportion of leukocytes (left, Mann-Whitney test), monocytes (middle, unpaired t test), and neutrophils (right, unpaired t test) in blood of MI/RI + Saline mice (n=4), and MI/RI + Pazopanib mice (n=4). (I) Representative flowcytometric dot plot analysis of myeloid cells in bone of saline treated mice and Pazopanib treated mice. (J) Proportion of leukocytes (left, unpaired t test with Welch's correction), monocytes (middle, unpaired t test), and neutrophils (right, unpaired t test) in bone of MI/RI + Saline mice (n=4), and MI/RI + Pazopanib mice (n=4). All data were displayed as mean ± SEM. ns, P > 0.05; * P < 0.05; ** P < 0.01; *** P < 0.001; **** P < 0.0001.
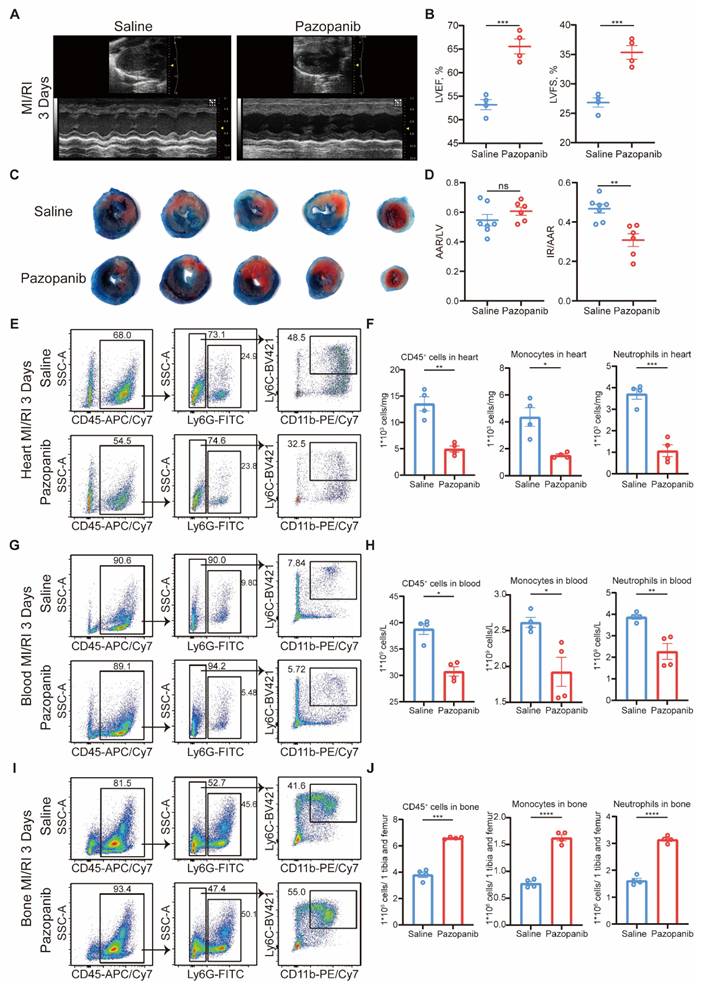
Flowcytometric dot plot analysis on MI/RI mice showed that pazopanib treatment decreased proportion of both neutrophils and monocytes diapedesis to heart (Figure 7E-F) and blood (Figure 7G-H) from bone (Figure 7I-J). The in vivo infiltration experiment also showed that myeloid cells treated with pazopanib decreased diapedesis to the peritoneum after TG injection (Figure S8B). HE staining also showed less diapedesis and disorganization in the heart after MI/RI with pazopanib treatment (Figure S8C). Immunofluorescence staining of MI/RI tissue sections also showed that after pazopanib treatment, myeloid cells tended to adhere to the blood vessels instead of diapedesis through blood vessels, resulting in the decrease of infiltrating myeloid cells in heart tissue (Figure S8D).
Thus, we found pazopanib as an excellent treatment to ameliorate MI/RI by decreasing the phosphorylation function of MAP3K3 and diapedesis.
Discussion
Leukocyte diapedesis plays a central role in inflammatory diseases. In the context of MI/RI, we disclosed the essential role of MAP3K3 in myeloid cell diapedesis. The high expression level of MAP3K3 in peripheral blood myeloid cells of MI/RI patients and its relationship with cardiac markers confirmed the important role of MAP3K3 in the MI/RI process. However, a larger clinical validation set is needed to further confirm this finding. Myeloid cell-specific deficiency of Map3k3 ameliorated MI/RI by inhibiting myeloid cell diapedesis from the bone. We expanded our research to a broader range of inflammatory models, such as LPS-induced sepsis, and found that MAP3K3 deficiency also led to impaired diapedesis. In vivo peritonitis experiments further confirmed the critical role of MAP3K3 in diapedesis.
Among the process of leukocyte diapedesis, the mechanisms of leukocyte chemotaxis [4] and leukocyte adhesion to vessels [6] have been studied extensively, while the processes governing leukocyte transmigration through vessels, particularly the upstream regulatory pathways, remain poorly understood. RNA-seq on monocytes found that depletion of MAP3K3 led to decreased F11r and increased adhesion-related genes Itgb3, Adgre4, and Itgax. As JAM-A showed an important function of integrin internalization and de-adhesion of leukocyte to endothelial cell, we considered MAP3K3 might influence myeloid cell transmigration through vessels by regulating F11r expression. Further exploring the potential transcription factors on F11r, we focused on an inhibitory transcription factor, TAL1, and confirmed its inhibitory effect on F11r transcription. JAM-A expression changed in the same direction as MAP3K3 while altering TAL1 expression in the opposite direction was able to reverse changes in JAM-A. To determine whether the phenotypes that we observed here are dependent on the kinase activity of MAP3K3, we utilized pazopanib, a known MAP3K3 kinase inhibitor, and observed similar changes in TAL1 and JAM-A expression. In vitro transwell assay and adhesion assay confirmed the role of MAP3K3/JAM-A in the de-adhesion and transmigration of myeloid cells, and the regulation of integrin internalization by MAP3K3/JAM-A was also verified. For the first time, we identified the MAP3K3/TAL1/JAM-A regulatory pathway as a key mechanism controlling myeloid cell transmigration through blood vessels. Due to the important role of JAM-A on platelets and the discovery of JAM-A on myeloid cells in our study, the interaction of JAM-A between different cell types is a topic worth exploring in the future. To be noticed, although decreasing myeloid cell transmigration could decrease inflammation in target tissue, inflammation on the blood vessels, such as atherosclerosis, might aggravate [29]. Therefore, more precise tissue- and time- treatments are required for complex diseases such as MI/RI with atherosclerosis. For instance, a short-term intervention immediately post-MI. Alternatively, developing delivery systems (e.g., nanoparticles activated by myeloid cell-specific enzymes) that preferentially target the infarct zone could spare the systemic vasculature. Meanwhile, combined adjunctive plaque-stabilizing therapy, such as high-intensity statins or novel anti-inflammatory drugs like colchicine and Firsekibart, could counteract any potential pro-inflammatory effects on the vasculature, resulting in a synergistic therapeutic outcome.
Since the phenotypes are dependent on the kinase activity of MAP3K3, we further explored the phosphorylation and ubiquitination of TAL1. Based on a combined analysis of previous research and our GPS6.0 prediction results, we identified Ser-122, Ser-172, and Thr-90 as potential phosphorylated sites on TAL1. In vitro kinase assays confirmed that MAP3K3 can directly phosphorylate TAL1 at Ser-122, which is a new substrate of MAP3K3 kinase. In addition, mutation at Ser-122 resulted in the most pronounced change in ubiquitination.
Given that MAP3K3 showed an important regulatory function in MI/RI through myeloid diapedesis in a phosphorylation-dependent manner, we administrated pazopanib to treat MI/RI for potential future clinical transformation. As an anti-tumor drug, the cardiotoxicity of pazopanib (potential QT prolongation and heart failure) has been the focus of previous research [45, 46]. However, our study found that strictly controlled use of pazopanib (low-dose, 1.5 mg/kg vs. >30 mg/kg, and short-term administration, 3 days vs. long-term) improved MI/RI by decreasing myeloid diapedesis, and weakening the inflammatory response. Future basic and clinical studies may help to determine the optimal dosage and timing of pazopanib administration more precisely, as well as possible remedial measures [45] (such as Bisoprolol fumarate, spironolactone, furosemide, and ramipril, which are commonly used treatments after PCI), to better exert pazopanib 's anti-inflammatory effects and reduce its cardiac toxicity. In addition, XJ-8, a natural compound isolated from Sanguis draxonis, is reported to inhibit MAP3K3 function in platelets [18], and whether it could inhibit diapedesis is worth further research. MicroRNA-145 [47] and miR-124-3p [48] are also reported to regulate MAP3K3 function, which might also be potential targets to treat MI/RI.
Conclusion
In conclusion, we revealed the MAP3K3/TAL1/JAM-A pathway as an important regulator of myeloid cell diapedesis. Inhibiting this pathway can decrease infiltrating myeloid cells in inflammatory tissue and ameliorate injuries, such as MI/RI and sepsis. The following in-depth mechanism study showed that MAP3K3 could phosphorylate TAL1 at Ser-122, which triggered the ubiquitination of TAL1 and decreased its inhibitory transcription function on F11r. Through JAM-A, MAP3K3 regulated integrin internalization and facilitated de-adhesion and transmigration of myeloid cells. As a kinase inhibitor of MAP3K3, pazopanib inhibited JAM-A expression and myeloid cell diapedesis, thus ameliorating MI/RI. Pazopanib could be a potential treatment for excessive inflammation during MI/RI or other inflammatory diseases.
Abbreviations
MI: myocardial infarction; MI/RI: myocardial ischemia/reperfusion injury; ACS: acute coronary syndrome; STEMI: ST-elevated myocardial infarction; CAD: coronary artery disease; ICM: ischemic cardiomyopathy; MAP3K3: mitogen-activated protein kinase kinase kinase 3; TAL1: T cell acute lymphocytic leukemia; PBMCs: peripheral blood mononuclear cells; MCP-1: chemoattractant protein 1; JAM-A: junctional adhesion molecule-A; CK-MB: creatine kinase M-type; cTnT: cardiac troponin T; LDH: lactate dehydrogenase; pro-BNP: pro B-type natriuretic peptide; CRP: C-reactive protein; IL-6: interleukin-6; WBC: white blood cell; NLR: neutrophil-lymphocyte-ratio; LVEF: left ventricular ejection fraction; LVFS: left ventricular fraction shortening; TUNEL: TdT-mediated dUTP nick end labeling; LPS: Lipopolysaccharide; TG: thioglycolate; GO: gene ontology; KEGG: Kyoto Encyclopedia of Genes and Genomes; Ser: serine; Thr: threonine; CHX: cycloheximide.
Supplementary Material
Supplementary methods, figures and tables.
Acknowledgements
This work was supported by the National Key Research and Development Program of China grant 2024YFB4610100 (to Feng Zhang), 2023YFC3606500 (to Jiatian Cao); the National Natural Science Foundation of China grant numbers 82170340 (to Feng Zhang), 82470334 (to Feng Zhang), 82070320 (to Jiatian Cao), and T2288101 (to Junbo Ge); the Shanghai Science and Technology Committee (STCSM) Science and Technology Innovation Program (Shanghai, China) project No. 20JC140800 (to Juying Qian); The Clinical Research Plan of Shanghai Hospital Development Center (Shanghai, China) NO. SHDC2020CR5009 (to Junbo Ge); and the Shanghai Top Priority Research Center Construction Project (2022ZZ01010) (to Junbo Ge).
Author contributions
Conceptualization, Jiatian Cao, Shiyu Hu, Wenwen Tang, and Feng Zhang; Methodology, Shiyu Hu, Jiatian Cao, and Wenwen Tang; Investigation, Shiyu Hu, Jian Zhang, Jingpu Wang, Yiwen Wang, Jiayu Liang, Rong Huang, Ji'e Yang, Yang Gao, Yanan Qu, Juying Qian, Chenguang Li, and Hongbo Yang; Visualization, Shiyu Hu, Jiatian Cao, and Wenwen Tang; Funding acquisition, Feng Zhang, Jiatian Cao, Junbo Ge, Juying Qian, Chenguang Li, and Hongbo Yang; Supervision, Feng Zhang, Jiatian Cao, and Wenwen Tang; Writing - original draft, Shiyu Hu, Jiatian Cao, and Wenwen Tang; Writing - review & editing, Feng Zhang, Shiyu Hu, Jiatian Cao, and Wenwen Tang.
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Competing Interests
Patent applications are being filed based on the findings described here.
References
1. Niccoli G, Montone RA, Ibanez B, Thiele H, Crea F, Heusch G. et al. Optimized Treatment of ST-Elevation Myocardial Infarction. Circ Res. 2019;125:245-58
2. Hausenloy DJ, Yellon DM. Ischaemic conditioning and reperfusion injury. Nat Rev Cardiol. 2016;13:193-209
3. Filippi MD. Neutrophil transendothelial migration: updates and new perspectives. Blood. 2019;133:2149-58
4. Frangogiannis NG, Smith CW, Entman ML. The inflammatory response in myocardial infarction. Cardiovasc Res. 2002;53:31-47
5. Nourshargh S, Alon R. Leukocyte migration into inflamed tissues. Immunity. 2014;41:694-707
6. Gumina RJ, Newman PJ, Kenny D, Warltier DC, Gross GJ. The leukocyte cell adhesion cascade and its role in myocardial ischemia-reperfusion injury. Basic Res Cardiol. 1997;92:201-13
7. Blank JL, Gerwins P, Elliott EM, Sather S, Johnson GL. Molecular cloning of mitogen-activated protein/ERK kinase kinases (MEKK) 2 and 3. Regulation of sequential phosphorylation pathways involving mitogen-activated protein kinase and c-Jun kinase. J Biol Chem. 1996;271:5361-8
8. Deacon K, Blank JL. Characterization of the mitogen-activated protein kinase kinase 4 (MKK4)/c-Jun NH2-terminal kinase 1 and MKK3/p38 pathways regulated by MEK kinases 2 and 3. MEK kinase 3 activates MKK3 but does not cause activation of p38 kinase in vivo. J Biol Chem. 1997;272:14489-96
9. Ellinger-Ziegelbauer H, Brown K, Kelly K, Siebenlist U. Direct activation of the stress-activated protein kinase (SAPK) and extracellular signal-regulated protein kinase (ERK) pathways by an inducible mitogen-activated protein Kinase/ERK kinase kinase 3 (MEKK) derivative. J Biol Chem. 1997;272:2668-74
10. Zhang D, Facchinetti V, Wang X, Huang Q, Qin J, Su B. Identification of MEKK2/3 serine phosphorylation site targeted by the Toll-like receptor and stress pathways. EMBO J. 2006;25:97-107
11. Yang J, Lin Y, Guo Z, Cheng J, Huang J, Deng L. et al. The essential role of MEKK3 in TNF-induced NF-kappaB activation. Nat Immunol. 2001;2:620-4
12. Chang X, Liu F, Wang X, Lin A, Zhao H, Su B. The kinases MEKK2 and MEKK3 regulate transforming growth factor-beta-mediated helper T cell differentiation. Immunity. 2011;34:201-12
13. Yang J, Boerm M, McCarty M, Bucana C, Fidler IJ, Zhuang Y. et al. Mekk3 is essential for early embryonic cardiovascular development. Nat Genet. 2000;24:309-13
14. Choi JP, Wang R, Yang X, Wang X, Wang L, Ting KK. et al. Ponatinib (AP24534) inhibits MEKK3-KLF signaling and prevents formation and progression of cerebral cavernous malformations. Sci Adv. 2018;4:eaau0731
15. Fisher OS, Deng H, Liu D, Zhang Y, Wei R, Deng Y. et al. Structure and vascular function of MEKK3-cerebral cavernous malformations 2 complex. Nat Commun. 2015;6:7937
16. Yuan Q, Basit A, Liang W, Qu R, Luan Y, Ren C. et al. Pazopanib ameliorates acute lung injuries via inhibition of MAP3K2 and MAP3K3. Sci Transl Med. 2021 13
17. Fan X, Wang C, Shi P, Gao W, Gu J, Geng Y. et al. Platelet MEKK3 regulates arterial thrombosis and myocardial infarct expansion in mice. Blood Adv. 2018;2:1439-48
18. Zhu Z, Wang L, Guo R, Pang D, Wang W, Wu Y. et al. XJ-8, a natural compound isolated from Sanguis draxonis, inhibits platelet function and thrombosis by targeting MAP3K3. J Thromb Haemost. 2022;20:605-18
19. Lecuyer E, Hoang T. SCL: from the origin of hematopoiesis to stem cells and leukemia. Exp Hematol. 2004;32:11-24
20. Ferrando AA, Neuberg DS, Staunton J, Loh ML, Huard C, Raimondi SC. et al. Gene expression signatures define novel oncogenic pathways in T cell acute lymphoblastic leukemia. Cancer Cell. 2002;1:75-87
21. Martin TA, Mason MD, Jiang WG. Tight junctions in cancer metastasis. Front Biosci (Landmark Ed). 2011;16:898-936
22. Kamola P, Babinska A, Przygodzki T. F11R/JAM-A: why do platelets express a molecule which is also present in tight junctions? Platelets. 2023;34:2214618
23. Karshovska E, Zhao Z, Blanchet X, Schmitt MM, Bidzhekov K, Soehnlein O. et al. Hyperreactivity of junctional adhesion molecule A-deficient platelets accelerates atherosclerosis in hyperlipidemic mice. Circ Res. 2015;116:587-99
24. Rehder D, Iden S, Nasdala I, Wegener J, Brickwedde MK, Vestweber D. et al. Junctional adhesion molecule-a participates in the formation of apico-basal polarity through different domains. Exp Cell Res. 2006;312:3389-403
25. Cera MR, Fabbri M, Molendini C, Corada M, Orsenigo F, Rehberg M. et al. JAM-A promotes neutrophil chemotaxis by controlling integrin internalization and recycling. J Cell Sci. 2009;122:268-77
26. Khandoga A, Kessler JS, Meissner H, Hanschen M, Corada M, Motoike T. et al. Junctional adhesion molecule-A deficiency increases hepatic ischemia-reperfusion injury despite reduction of neutrophil transendothelial migration. Blood. 2005;106:725-33
27. Corada M, Chimenti S, Cera MR, Vinci M, Salio M, Fiordaliso F. et al. Junctional adhesion molecule-A-deficient polymorphonuclear cells show reduced diapedesis in peritonitis and heart ischemia-reperfusion injury. Proc Natl Acad Sci U S A. 2005;102:10634-9
28. Lakshmi SP, Reddy AT, Naik MU, Naik UP, Reddy RC. Effects of JAM-A deficiency or blocking antibodies on neutrophil migration and lung injury in a murine model of ALI. Am J Physiol Lung Cell Mol Physiol. 2012;303:L758-66
29. Schmitt MM, Megens RT, Zernecke A, Bidzhekov K, van den Akker NM, Rademakers T. et al. Endothelial junctional adhesion molecule-a guides monocytes into flow-dependent predilection sites of atherosclerosis. Circulation. 2014;129:66-76
30. Rao SV, O'Donoghue ML, Ruel M, Rab T, Tamis-Holland JE, Alexander JH. et al. 2025 ACC/AHA/ACEP/NAEMSP/SCAI Guideline for the Management of Patients With Acute Coronary Syndromes: A Report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines. Circulation. 2025;151:e771-e862
31. Shi H, Gao Y, Dong Z, Yang J, Gao R, Li X. et al. GSDMD-Mediated Cardiomyocyte Pyroptosis Promotes Myocardial I/R Injury. Circ Res. 2021;129:383-96
32. Kumar R, Knick VB, Rudolph SK, Johnson JH, Crosby RM, Crouthamel MC. et al. Pharmacokinetic-pharmacodynamic correlation from mouse to human with pazopanib, a multikinase angiogenesis inhibitor with potent antitumor and antiangiogenic activity. Mol Cancer Ther. 2007;6:2012-21
33. Hu S, Zhang F, Wang J, Zhang J, Li C, Lyu Y. et al. MMP9(High) Neutrophils are Critical Mediators of Neutrophil Extracellular Traps Formation and Myocardial Ischemia/Reperfusion Injury. Adv Sci (Weinh). 2025;12:e2415205
34. Vanhaverbeke M, Vausort M, Veltman D, Zhang L, Wu M, Laenen G. et al. Peripheral Blood RNA Levels of QSOX1 and PLBD1 Are New Independent Predictors of Left Ventricular Dysfunction After Acute Myocardial Infarction. Circ Genom Precis Med. 2019;12:e002656
35. Sweet ME, Cocciolo A, Slavov D, Jones KL, Sweet JR, Graw SL. et al. Transcriptome analysis of human heart failure reveals dysregulated cell adhesion in dilated cardiomyopathy and activated immune pathways in ischemic heart failure. BMC Genomics. 2018;19:812
36. Welsh P, Preiss D, Hayward C, Shah ASV, McAllister D, Briggs A. et al. Cardiac Troponin T and Troponin I in the General Population. Circulation. 2019;139:2754-64
37. Ammirati E, Cannistraci CV, Cristell NA, Vecchio V, Palini AG, Tornvall P. et al. Identification and predictive value of interleukin-6+ interleukin-10+ and interleukin-6- interleukin-10+ cytokine patterns in ST-elevation acute myocardial infarction. Circ Res. 2012;111:1336-48
38. Zhang N, Tang W, Torres L, Wang X, Ajaj Y, Zhu L. et al. Cell surface RNAs control neutrophil recruitment. Cell. 2024;187:846-60 e17
39. Tamargo IA, Baek KI, Xu C, Kang DW, Kim Y, Andueza A. et al. HEG1 Protects Against Atherosclerosis by Regulating Stable Flow-Induced KLF2/4 Expression in Endothelial Cells. Circulation. 2024;149:1183-201
40. Chagraoui H, Kristiansen MS, Ruiz JP, Serra-Barros A, Richter J, Hall-Ponsele E. et al. SCL/TAL1 cooperates with Polycomb RYBP-PRC1 to suppress alternative lineages in blood-fated cells. Nat Commun. 2018;9:5375
41. Tang T, Arbiser JL, Brandt SJ. Phosphorylation by mitogen-activated protein kinase mediates the hypoxia-induced turnover of the TAL1/SCL transcription factor in endothelial cells. J Biol Chem. 2002;277:18365-72
42. Li Y, Deng C, Hu X, Patel B, Fu X, Qiu Y. et al. Dynamic interaction between TAL1 oncoprotein and LSD1 regulates TAL1 function in hematopoiesis and leukemogenesis. Oncogene. 2012;31:5007-18
43. Terme JM, Lhermitte L, Asnafi V, Jalinot P. TGF-beta induces degradation of TAL1/SCL by the ubiquitin-proteasome pathway through AKT-mediated phosphorylation. Blood. 2009;113:6695-8
44. Chen M, Zhang W, Gou Y, Xu D, Wei Y, Liu D. et al. GPS 6.0: an updated server for prediction of kinase-specific phosphorylation sites in proteins. Nucleic Acids Res. 2023;51:W243-W50
45. Karaagac M, Eryilmaz MK. Pazopanib-induced fatal heart failure in a patient with unresectable soft tissue sarcoma and review of literature. J Oncol Pharm Pract. 2020;26:768-74
46. Justice CN, Derbala MH, Baich TM, Kempton AN, Guo AS, Ho TH. et al. The Impact of Pazopanib on the Cardiovascular System. J Cardiovasc Pharmacol Ther. 2018;23:387-98
47. Liu Y, Hu J, Wang W, Wang Q. MircroRNA-145 Attenuates Cardiac Fibrosis Via Regulating Mitogen-Activated Protein Kinase Kinase Kinase 3. Cardiovasc Drugs Ther. 2023;37:655-65
48. Zhai C, Cong H, Hou K, Hu Y, Zhang J, Zhang Y. et al. Effects of miR-124-3p regulation of the p38MAPK signaling pathway via MEKK3 on apoptosis and proliferation of macrophages in mice with coronary atherosclerosis. Adv Clin Exp Med. 2020;29:803-12
Author contact
![]() Corresponding authors: zhang.fengsh.cn (Zhang Feng); wenwen.tangedu (Wenwen Tang); cao.jiatiansh.cn (Jiatian Cao).
Corresponding authors: zhang.fengsh.cn (Zhang Feng); wenwen.tangedu (Wenwen Tang); cao.jiatiansh.cn (Jiatian Cao).