Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Theranostic targets for ischemic...
Theranostic nanoplatforms
Molecular recognition of...
Diagnostic function of...
Therapeutic function of...
Conclusions
Methods
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2014; 4(1):90-105. doi:10.7150/thno.7088 This issue Cite
Research Paper
In Vivo Theranostics at the Peri-Infarct Region in Cerebral Ischemia
Jesús Agulla1,2*, David Brea1,3*, Francisco Campos1, Tomás Sobrino1, Bárbara Argibay1, Wajih Al-Soufi4, Miguel Blanco1, José Castillo1, Pedro Ramos-Cabrer1 ![]()
1. Department of Neurology, Neurovascular Area, Clinical Neurosciences Research Laboratory, Hospital Clínico Universitario, Health Research Institute of Santiago de Compostela (IDIS), University of Santiago de Compostela, E-15706 Santiago de Compostela, Spain.
2. Research Unit, University Hospital of Salamanca and Institute of Health Sciences of Castilla and Leon, E-37007 Salamanca, Spain.
3. Cellular and Molecular Neurobiology Research Group and Grup de Recerça en Neurociencies del IGTP, Department of Neurosciences, Fundació Institut d'Investigació en Ciències de la Salut Germans Trias I Pujol-Universitat Autónoma de Barcelona, E-08916 Badalona, Spain.
4. Single Molecule Fluorescence Research Unit, Department of Physical Chemistry, Faculty of Science, University of Santiago de Compostela, E-27002 Lugo, Spain.
* These two authors have contributed equally to this work.
Received 2013-7-4; Accepted 2013-9-12; Published 2013-12-12
Abstract

The use of theranostics in neurosciences has been rare to date because of the limitations imposed on the free delivery of substances to the brain by the blood-brain barrier. Here we report the development of a theranostic system for the treatment of stroke, a leading cause of death and disability in developed countries. We first performed a series of proteomic, immunoblotting and immunohistological studies to characterize the expression of molecular biomarkers for the so-called peri-infarct tissue, a key region of the brain for stroke treatment. We confirmed that the HSP72 protein is a suitable biomarker for the peri-infarct region, as it is selectively expressed by at-risk tissue for up to 7 days following cerebral ischemia. We also describe the development of anti-HSP72 vectorized stealth immunoliposomes containing imaging probes to make them traceable by conventional imaging techniques (fluorescence and MRI) that were used to encapsulate a therapeutic agent (citicoline) for the treatment of cerebral ischemia. We tested the molecular recognition capabilities of these nano-platforms in vitro together with their diagnostic and therapeutic properties in vivo, in an animal model of cerebral ischemia. Using MRI, we found that 80% of vectorized liposomes were located on the periphery of the ischemic lesion, and animals treated with citicoline encapsulated on these liposomes presented lesion volumes up to 30% smaller than animals treated with free (non-encapsulated) drugs. Our results show the potential of nanotechnology for the development of effective tools for the treatment of neurological diseases.
Keywords: cerebral ischemia, peri-infarct region, MRI, Theranostics, Drug delivery.
Introduction
Nanomedicine is a discipline that offers new and exciting tools for the medical field, such as theranostics, that enable the development of the concept of personalized medicine. Theranostics consist of the design, construction and use of molecular platforms with dual diagnostic and therapeutic roles. Such nano-platforms include imaging probes for the detection of molecular targets in order to identify specific cell populations or cellular functional states in vivo and, at the same time, promoting the stabilization, transport and controlled release of these drugs at targeted sites by virtue of including therapeutics in their structure [1]. In this way, theranostics represents a perfect marriage between molecular imaging and targeted drug delivery technologies in a single molecular platform. Exciting examples of theranostic applications have been reported for cancer [2], atherosclerosis [3], and gene therapy [4], among others.
The incidence of neurological diseases is rapidly increasing in developed countries due to the progressive aging of their population, representing a huge burden for society [5]. In this sense, theranostics may offer new and stimulating opportunities for the development of novel treatments in this field. However, most of the reported applications for theranostic agents imply the action of these agents at a vascular level (e.g., atherosclerosis) or in tissues with high vascular density and permeability (e.g., tumors). Theranostic approaches for targets located inside the brain are rare due to the difficulties experienced by systemically administered substances in crossing the blood-brain barrier (BBB), the functional structure that tightly controls the efflux of substances from and to the brain [6].
In terms of stroke, one of the leading causes of mortality and morbidity in developed countries, it is noteworthy that treatment by recanalization therapies is currently limited to less than 5% of patients admitted to hospitals (though this figure may be higher in centers with specialized stroke units) [7]. We believe that, despite the challenge represented by crossing the BBB, theranostics may help us to provide a better understanding of the underlying molecular mechanisms of this devastating disease, enabling the development of future treatments at the clinical level.
Ischemic stroke consists of a rapid loss of cerebral function as consequence of the obstruction of a brain vessel, followed by subsequent damage to neighboring tissue. Cerebral tissue quickly becomes irreversibly damaged in the vicinity of the closed vessel (this region is known as the infarct core), but tissue is potentially salvageable towards the periphery of the lesion (in an area known as the peri-infarct region). Tissue at the peri-infarct region represents a key target for the development of new treatments against stroke, especially at sub-acute and chronic phases of this disease, for which no effective therapies are currently available [8, 9].
Here we report the development of a novel theranostic nanocarrier that specifically targets the peri-infarct tissue in cerebral ischemia. This is one of the few reports that includes a complete set of in vitro, in vivo and ex vivo studies of the design and testing of the diagnostic and therapeutic capacities of a novel theranostic nano-platform for cerebral ischemia. This study demonstrates that theranostics beyond the blood-brain barrier is feasible and represents an example of the potential that nanotechnology offers for the treatment of neurological diseases.
Theranostic targets for ischemic stroke
The peri-infarct region is a complex and continuously evolving portion of the ischemic brain that is difficult to delineate with conventional imaging techniques. Theranostics may provide an effective way to define this region in detail through molecular recognition processes [6, 8, 9].
The selection of a biomarker that unequivocally characterizes the targeted tissue is a key step in the design of a theranostic agent [6]. A handful of published literature has reported the upregulation of particular proteins in the periphery of an ischemic lesion [10-16], but comparisons of their appropriateness as biomarkers cannot be extracted from individualized analyses. Thus, we conducted a complete proteomic, immunoblot and immunohistological study (schematically described in figure 1), in order to achieve a full characterization of protein expression at the peri-infarct region in an animal model of cerebral ischemia. Once a suitable target was selected, we performed a spatiotemporal description of its expression for a period of 14 days following the induction of the lesion.
a) Infarct core of a section of an ischemic rat brain stained (in beige) by tetrazole chloride. A 2 mm strip around this area (dotted line) and a mirrored contralateral section were selected as peri-infarct and control tissues, respectively. Following tissue disaggregation, proteins were isolated in 3 fractions (insoluble, soluble and membrane proteins). 2D-PAGE analysis was performed, and proteins over-expressed in the peri-infarct tissue were evaluated by Western blot (WB) and immunohistochemistry (IHC). b) Proteomic gels showing the expression of peri-infarct and contralateral proteins (spots) with magnification (red frames) of the region where HSP70 proteins is located. c) 2D-WB results showing (top row) that HSP72 (circled in red) is the only member of the HSP70 family specifically expressed at the peri-infarct tissue, while other members of the family are also expressed in contralateral tissues (actin bands used for protein load control are also shown). d) Immunohistochemical (IHC) study of consecutive brain slices (10 µm) stained for HSP72 (top-row), HSC70 (middle-row) and GFAP (bottom-row). Left-to-right columns: complete brain slices, 4X magnifications of the ischemic and contralateral hemispheres, and a 10X magnification of the peri-infarct region. e) Double fluorescence IHC of peri-infarct tissue. HSP72 (red) is mostly expressed by neurons (NeuN green, top) but not by astrocytes (GFAP green, bottom), 48 h after ischemia. f) Temporal profile of HSP72 expression in the ischemic brain. Regions selected for cell counting analysis are indicated in the MR image (where infarct core appears hyper-intense). g) Color-coded density maps (overlaid on MR images) presenting the spatiotemporal expression profile of HSP72 in the ischemic brain.
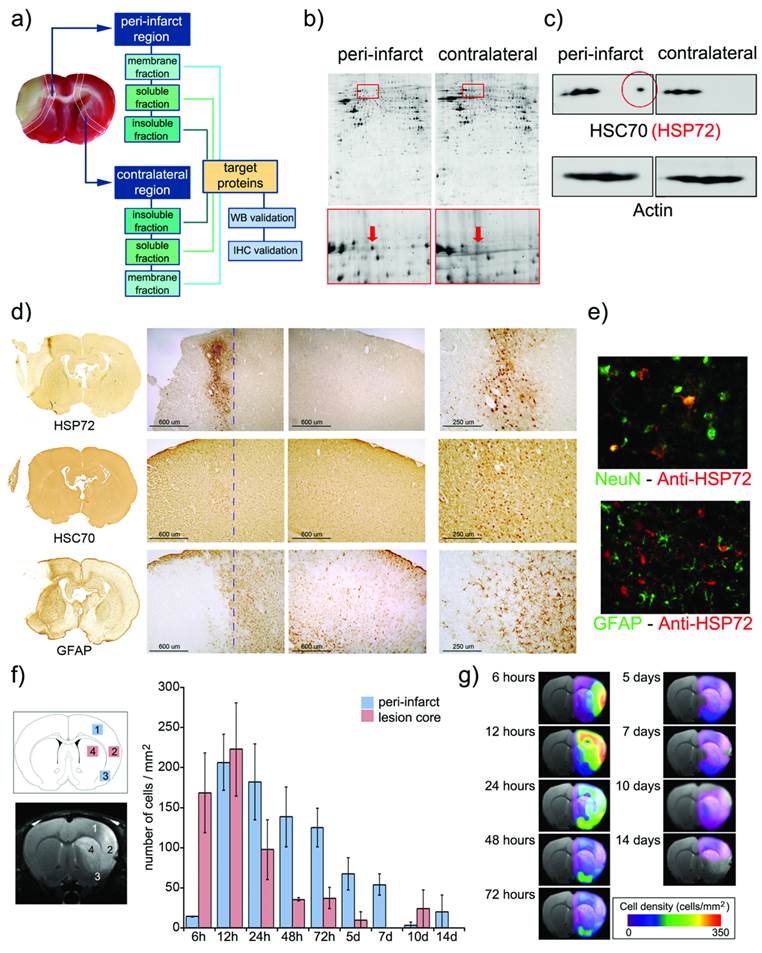
For this purpose, a permanent focal cerebral ischemia was induced in 6 rats (see methods), extracting and processing their brains (t= 48 h port surgery) to identify the ischemic core by staining necrotic tissue with 2,3,5-triphenyl-tetrazolium chloride (TTC, white staining in figure 1a). Strips of tissue (2 mm thick) were dissected around the infarct core (considered peri-infarct tissue) and in the contralateral brain hemisphere (considered control tissue). Proteins from both tissue sections were extracted and separated into membrane, soluble and insoluble protein fractions (see methods below). 2D-Gel electrophoresis was performed on 2 sets of polyacrylamide gels (peri-infarct vs. healthy tissue), separating proteins by their isoelectric points and molecular weights (figure 1b). Paired comparisons (peri-infarct vs. control) of stained spots in gels (n=962, 1632 and 814 spots found for membrane, insoluble and soluble protein fractions, respectively) were analyzed, revealing 11 significant differences in protein expression at the peri-infarct tissue with respect to control tissue. The most robust difference (peri-infarct/control tissue ratio of expression > 20) corresponded to the 70 kDa family of Heat Shock Proteins (arrows in figure 1b).
Following 2D Western-blotting (figure 1c), isolated HSP70 protein bands were stained with a monoclonal anti-HSP70 antibody (using β-actin as the protein load control). A fraction of this family, known as the constitutive form of HSP70 (or HSC70), was equally expressed in both peri-infarct and healthy tissues, so it was not suitable as targeting vector. However, a small fraction of this family, identified as the HSP72 protein (or inducible form of HSP70), was specifically expressed at the peri-infarct tissue (circled spot in figure 1c) and selected as a specific target for the peri-infarct tissue.
These results were confirmed by immunohistochemical studies (figure 1d) that revealed the specific expression of HSP72 in the peri-infarct region (top row in figure 1d), while HSC70 is constitutively expressed in the whole brain, although higher expression of this protein can be found in the peri-infarct tissue (middle row in figure 1d). The infarct core was delineated as the area with a very low density of cells, surrounded by an accumulation of glial cells in the periphery (GFAP staining, bottom row in figure 1d) as previously described [17].
Double-fluorescence immunohistochemistry studies (figure 1e) confirmed that HSP72 (red fluorescence) is mainly expressed by neurons in the ischemic brain, 48 hours after surgery (Green NeuN fluorescence in the top image). Astrocytes, in contrast, did not significantly express the protein at this time point (Green GFAP fluorescence in the bottom image). We analyzed these two cell types because they represent the major fraction of brain cell populations. However, few other scattered cells were also positive for HSP72, and we cannot rule out the possibility that microglia, endothelial cells or other cell types also exhibit some degree of HSP72 expression using current ICH material. It should be mentioned that the type of brain cells expressing HSP72 is dependent on the time after ischemia [13].
The fact that HSP72 is mostly a cytosolic protein may be a concern with respect to its suitability as a target for molecular recognition processes with theranostic molecules. However, under stress conditions, such as hyperthermia or ischemia, HSP72 has been shown to translocate to the membrane and is even released into the extracellular space [18, 19]. Thus, we believe that the HSP72 protein can still be considered a valid target for theranostic molecules (as we demonstrate later in our in vitro studies).
Brains sections obtained at 6 h, 12 h and 1, 2, 3, 5, 7, 10 and 14 days after surgery were processed to establish a spatiotemporal profile of expression of HSP72 in the ischemic brain. The number of HSP72-positive cells was quantified in 2 square regions located in the infarct core (as delineated by diffusion-weighted or T2-weighted MR images) and in another 2 regions in its periphery (figure 1f). During the first hours of lesion evolution, MR images show a hyperintense region on the diffusion-weighted images, which is generally accepted as the lesion core. However, this hyperintense area includes necrotic cells as well as surviving cells that usually die after a period of time by different molecular mechanisms (such as apoptosis). Additionally, it has been reported that HSP72 is absent in dead neurons in necrotic tissue, but the protein can be found in surviving glia and in the microvasculature [13]. Thus, high expression of HSP72 was found in the core during the first 12 h of the evolution of the lesion, followed by a rapidly drop after this period, when the whole region becomes necrotic. Damage to the surrounding tissue, the peri-infarct region, progresses in a slower fashion, reaching maximum HSP72 expression at 24-72 h post ischemia (figure 1f) and lasting for a period of one week. The number of positive cells beyond this time point presented large variability, and observed values are not statistically significant.
Using the numeric values presented in figure 1f, we were able to construct density maps of HSP72 expression in the ischemic brain over a period of 2 weeks (figure 1g). The high and specific upregulation of HSP72 in the peri-infarct tissue during a period of 24 h to 7 days qualifies this protein as a suitable target for theranostic molecules in the treatment of stroke at the subacute phase of the disease.
Theranostic nanoplatforms
Several types of nanosystems (e.g., peptides, dendrimers or liposomes) have the potential to be used as theranostic agents [20-27]. We used liposomes as the synthetic basis for our agent because they are very well-known biocompatible drug carriers, with the capacity to effectively cross the BBB when their size is controlled [28, 29]. In addition, liposomes are useful for proof-of-principle purposes, and the preparation of these self-assembling systems is affordable in research laboratories with limited synthetic capacities, which is an important issue, considering a potential translationality of our methodology to clinical practice.
Thus, stealth cholesterol/DSPC liposomes were prepared as reported elsewhere [30] (see methods for further details), including a fraction of PEG-conjugated lipids to avoid in vivo aggregation, opsonization and rapid clearance from blood by the reticuloendothelial system [20, 31-34]. The size of liposomes was limited to circa 100 nm by extrusion. This size is large enough to allow the entrapment of therapeutic doses of a drug, while still allowing passage across the BBB by different molecular mechanisms, as has been reported in the literature [6, 29, 35]. The transient opening of the BBB that takes place after cerebral ischemia may also facilitate the access of liposomes to the brain parenchyma.
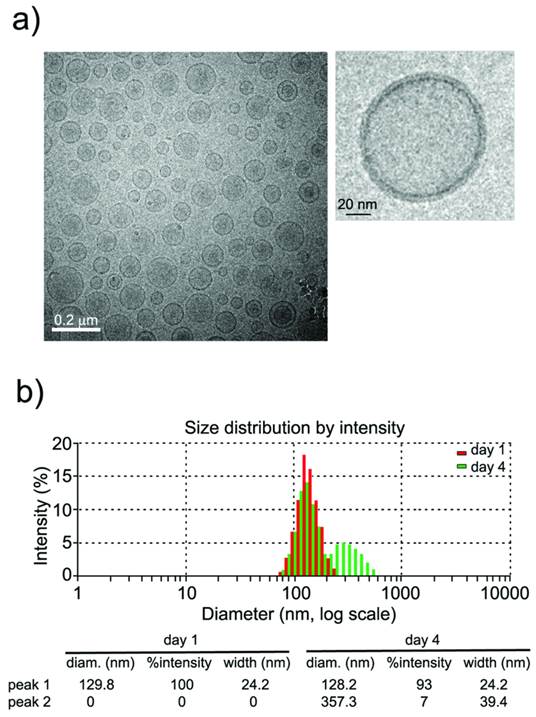
Characterization of liposomes by dynamic light scattering (DLS) and transmission electronic microscopy (TEM) techniques (see methods) indicated the formation of small unilamellar vesicles (SUVs) with a narrow distribution of hydrodynamic radii around 100 nm (figure 2). Liposomes were stored at 4°C and remained stable for at least 4 days (figure 2b, <7% aggregation), which is an important factor in the performance of longitudinal in vivo studies.
a) CryoTEM micrograph of liposomes showing the formation of (mostly) unilamellar vesicles, and magnification of a single liposome. b) Liposome sizes (measured by Dynamic Light Scattering) are homogeneous (~100 nm) and remain stable (at 4˚C) for at least 4 days (<7% of large aggregates at this time point).
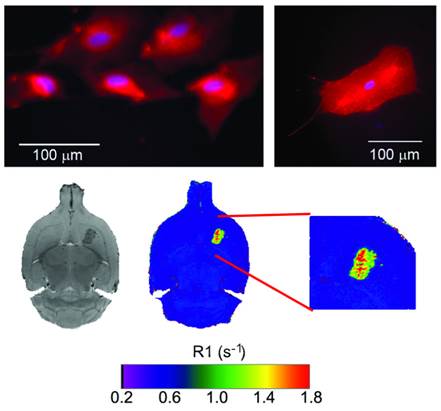
Following a multimodal approach [36, 37], phospholipids containing rhodamine or gadolinium were used for the preparation of liposomes to make them traceable by fluorescence and magnetic resonance imaging (MRI) techniques, enabling their use as diagnostic tools. Detectability by fluorescence was tested with cultured astrocytes transfected with (red) rhodamine-loaded liposomes (Figure 3). Liposomes were also detectable in vivo by MRI as hyperintense areas on R1 parametric maps (R1 = 1/T1) or hypointense areas on T2-weighted MR images, as a consequence of a reduction of longitudinal (T1) and transversal (T2) relaxation times induced by the gadolinium carried by liposomes (Figure 3).
Top: fluorescence images of cultured astrocytes transfected with liposomes, showing red fluorescence due the rhodamine content of liposomes. Liposomes do not penetrate cell nuclei (blue Hoechst staining). Bottom: MR T2-weighted images (left) and R1 (=1/T1) parametric maps of a rat brain 24 hours after intra-parenchymal injection of liposomes. The presence of gadolinium in liposomes enables their detection as hypointense areas on T2w images and hyperintense regions on R1 maps.
Furthermore, citicoline (CDP-Choline), a drug used in the treatment of stroke [30, 38], was encapsulated in the liposomes for therapeutic purposes. We previously optimized citicoline encapsulation for our particular liposomal composition and size [30], achieving a drug content of 8% in weight with an encapsulation efficiency of 1.5 ml/mmol.
Synthesis of the theranostic nano-platform ended with the vectorization of liposomes against HSP72, a specific molecular biomarker of the peri-infarct region. The covalent union of HSP72 antibodies to liposomes was achieved by the formation of thioether bonds between the maleimide groups present in PEGylated phospholipids and thiol groups from SATA-modified amino acids in HSP72 antibodies, as described elsewhere [37]. The efficiency achieved in this step was 64% (as determined by the Lowry method) [39]. Finally, we used the method described by Hutchinson et al. [40] to estimate that each liposome contained 12 anti-HSP72 units attached to its surface. For the calculations, we used a molar ratio of anti-HSP72/lipids of 3.13 10-4/1 (used in the synthesis of liposomes), a protein molecular weight of 160 kDa, a liposome radius of 50 nm, an area per lipid molecule in the liposomal bilayer of 0.50 nm2 [40], and efficiency of protein coupling to liposomes of 64%.
Molecular recognition of targeted cells
Molecular recognition of HSP72-vectorized liposomes was tested using an in vitro model. At this point it is not particularly relevant which cell type or culture conditions are used, because our sole intention was to demonstrate that synthetized HSP72-targeting liposomes can effectively participate in molecular recognition processes with cells that express that protein. Following a model previously described in literature, we found that cultures of rat astrocytes subjected to stress by hyperthermia showed a noticeable expression of HSP72 protein [41], making them a good model for our purposes.
Primary cultures of astrocytes were obtained from the brains of 1-day-old rats (see methods), subjecting one set of cells to a thermal shock (42 °C, 30 min), followed by a resting period of 6 hours under regular conditions (37 °C, 5% CO2) to induce the expression of HSP72 [41]. A set of cells was maintained in parallel under normal conditions as controls. Both sets of cells were incubated for 30 min with regular (non-targeted) and anti-HSP72 vectorized liposomes in all four possible combinations as follows: 1) control cells + control liposomes, 2) control cells + vectorized liposomes, 3) HSP72 expressing cells + control liposomes, and 4) HSP72 expressing cells + vectorized liposomes (figure 4a).
a) Fluorescence images of cell cultures of HSP72-positive and negative cells incubated for 30 min with anti-HSP72 vectorized or with regular (non-targeting) fluorescent liposomes. Only the incubation of HSP72-positive cells with anti-HSP72 vectorized liposomes led to the observation of significant fluorescence (molecular recognition). b) T1 parametric maps (and quantification of T1 relaxation times) of cell pellets obtained from the previously described cell cultures using a 9.4 Tesla MRI system. Only the incubation of HSP72-positive cells with anti-HSP72 vectorized liposomes led to the observation of a significant reduction of T1 relaxation times.
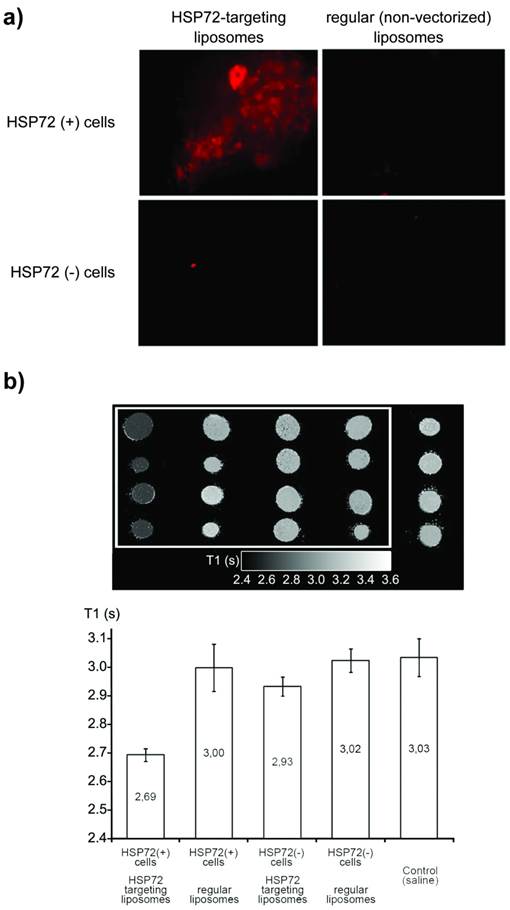
Cells tagged with the rhodamine contained in liposomes were only observed when HSP72 positive cells were combined with anti-HSP72 vectorized liposomes. No other liposomes-cell combinations presented fluorescence under the microscope (figure 4a).
Cell pellets were collected from all cultures by centrifugation and, after removing the supernatant fluid, the corresponding parametric maps of the longitudinal relaxation times (T1) of the pellets were obtained on a MR scanner at 9.4 T (figure 4b). Combinations of anti-HSP72 vectorized liposomes with HSP72-positive cells presented a significantly reduced T1 value (T1=2.69 s) with respect to all the other studied cell-liposome combinations, including the culture medium used as a control (ANOVA, p<0.05).
Fluorescence and MRI studies confirmed the effective molecular recognition of anti-HSP72 vectorized liposomes by HSP72-expressing cells.
Diagnostic function of theranostic nanoplatforms
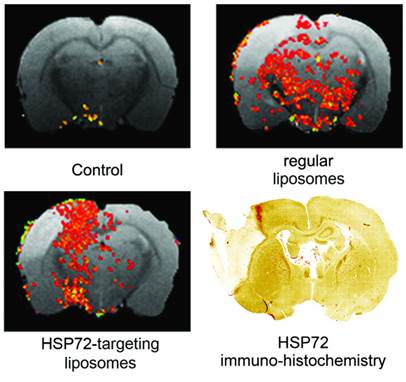
The capacity of the theranostic agent to delineate the peri-infarct tissue in ischemic animals in vivo (diagnostic function) was assessed in 12 ischemic rats, randomly divided in 3 groups receiving 6 intravenous injections of v=1 ml (at t=45 min and 6, 12, 18, 24, 30 h after surgery) of one of the following: (1) saline (control group), (2) regular (non-vectorized) liposomes, or (3) HSP72-vectorized liposomes. In those groups of animals treated with liposomes, a total concentration of 24 μmol/ml of lipids was used. MR longitudinal relaxation (T1) maps of their brains were obtained 1 day before and 1, 3 and 7 days post-surgery (see methods). Images were processed (see methods and figure 5 for a detailed description of image analysis) to display regions of abnormally reduced T1 relaxation times assigned to areas where liposomes doped with gadolinium are present. It is important to remember that liposomes reduce T1 relaxation times (as shown in figure 4b and in the histogram presented in figure 5a). Representative images showing the distribution of liposomes in the brain of ischemic rats 24 h after systemic injection of liposomes are presented in figure 6. Saline-treated animals showed no R1-enhanced areas (except for some scattered pixels that may correspond to fitting noise), while anti-HSP72 vectorized liposomes show highlighted areas in the periphery of the ischemic lesion. Interestingly, regular (non-targeted) liposomes appear to be randomly distributed along both brain hemispheres (figure 6). An immunohistological image of HSP72 expression in the ischemic brain (figure 6) shows that the in vivo distribution of liposomes (as seen on MR images) presents a considerable degree of correlation with the ex vivo expression of the protein, corroborating the molecular recognition capacity of the theranostic agent and its diagnostic capabilities. In Figure 6, although there is a high expression of HSP72 in a thin strip around the lesion upper corner, a closer look at the histological image suggests the overexpression of the protein in a wider region around the ischemic lesion, as seen on MRI images. However, the sensitivity of both techniques is very different, and a direct correlation between the expression of protein and liposome distribution in MR images may be difficult to establish.
a) Histogram of T1 values corresponding to healthy tissue (pink), ischemic tissue (blue) and tissue containing Gd-doped liposomes (green) (the last obtained after an intra-parenchymal injection of liposomes into healthy animals). b) Image processing to obtain the following distribution maps of liposomes: (1) Manually segmented T1 maps obtained from saturation-recovery MRI studies. (2) Color encoding of maps. (3) Image smoothing (r=0.6 Gaussian filter). (4) Clipping process to remove pixels with T1>2.25 s (thresholding). (5) Color re-scaling. (6) Overlaying of maps on corresponding proton density MR images. (7) Clustering process to eliminate groups of n< 3 pixels (considered noise).
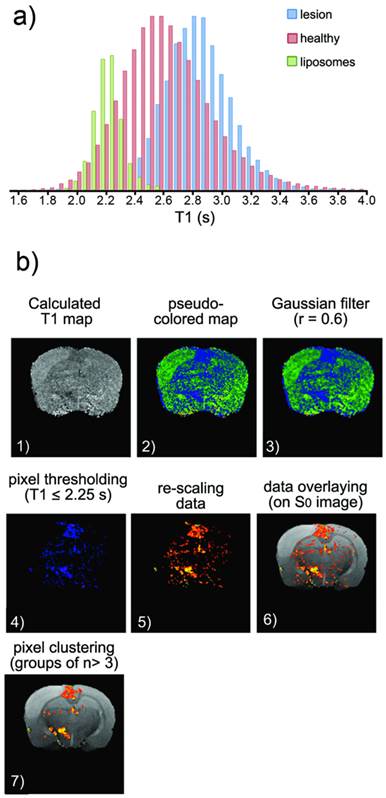
Representative MR images of ischemic brains from rats treated (i.v.) with 1 ml of saline (top left), regular (non-vectorized) liposomes (top right) or anti-HSP72 vectorized liposomes (bottom left) showing the in vivo distribution of liposomes 24 h after treatment. An immunohistological image of HSP72 protein expression (bottom right) shows good agreement between the in vivo locations of liposomes with the areas of expression of HSP72 observed ex vivo.
Furthermore, in an attempt to quantify the tendency of HSP72-targeting liposomes to accumulate in the peri-infarct region, we measured the number of highlighted pixels in each brain hemisphere (the number of pixels w
ith T1 values lower than 2.25 s, see methods and figure 5a). Figure 7a shows the percentage of all counted pixels that are located in the left (ischemic) hemisphere with respect to the total number of pixels found in the whole brain of the animals. For anti-HSP72 liposomes, the percentage of pixels with reduced T1 times (attributed to the presence of liposomes) in the affected hemisphere ranged between 70-80%, at day 1, 3, and 7, while regular liposomes appear to be equally distributed between both brain hemispheres (figure 7a).
a) Plot showing the percentage of the total amount of liposomes (image pixels of reduced T1 values on parametric maps) found in the ipsilesional hemisphere with respect to the total amount of liposomes found in the entire brain. Differences between regular liposomes (evenly distributed on both hemispheres) and HSP72-targeting liposomes (mostly located at the infarcted hemisphere) are significant between both groups at all studied time points (1, 3 and 7 days post injection), but no significant differences were found within groups. (*, $ and # represent p<0.05). b) Plot showing the percentage of pixels that contain liposomes (image pixels of reduced T1 values) with respect to the total amount of pixels of the brain MRI. Liposomes levels were relatively higher for regular liposomes on day 1 and for targeting liposomes on day 7. No significant differences were found between groups on day 1. ($ and # represent p<0.05).
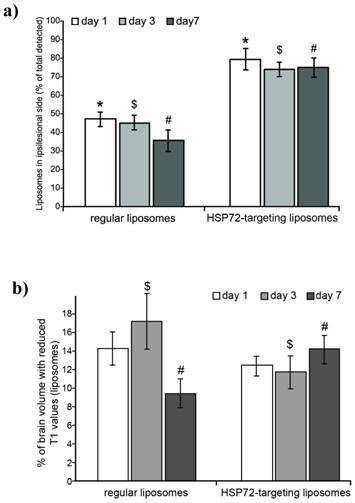
On the other hand, the percentage of the total number of pixels of the brain that presented a reduced T1 value (a rough estimation of the amount of liposomes that reach the brain) was similar for both groups and ranged between 9 to 17% (figure 7b). On day 1, there was no significant difference between vectorized and non-vectorized liposomes (14.32±1.9 vs. 12.58±0.81), while the percentage of brain pixels that contain liposomes was higher for control liposomes on day 3 (17.43±1.62 vs. 11.89±1.76), and higher for targeted liposomes on day 7 (9.46±1.58 vs. 15.67±1.35).
These studies demonstrate the capacity of HSP72-targeted liposomes to delineate and follow the evolution of the peri-infarct tissue in vivo, confirming the diagnostic functionality of theranostic liposomes.
Therapeutic function of theranostic nanoplatforms
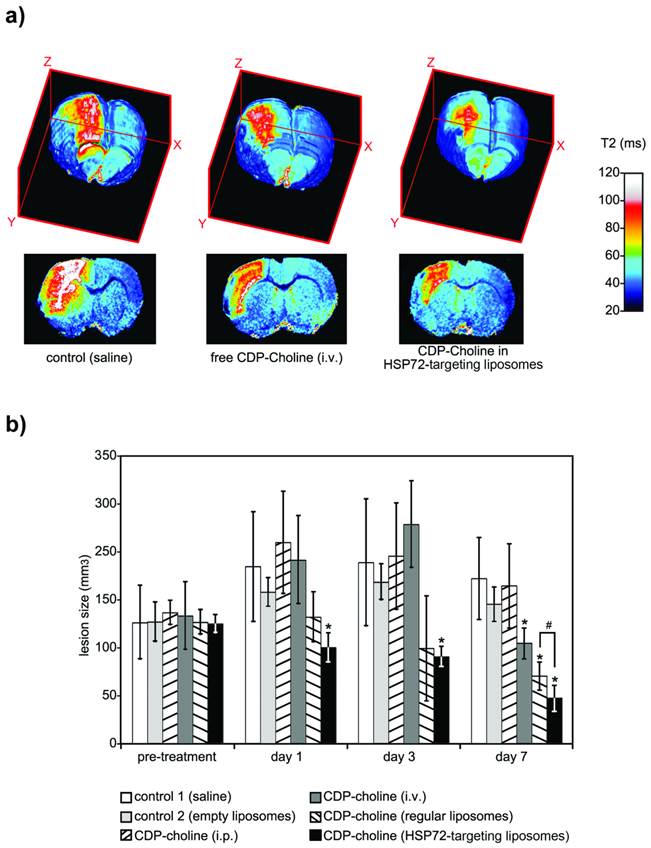
The therapeutic capacity of the theranostic agent was demonstrated with a second in vivo study. Qualitatively speaking, the therapeutic effects of citicoline (CDP-Choline, a therapeutic agent that reduces lesion volumes in this animal model) [30, 38, 42] were highly increased by encapsulation of the drug in HSP72-targeted liposomes. Animals treated with citicoline-loaded HSP72 targeting liposomes presented reduced lesion volumes on T2 parametric maps compared to untreated animals or animals treated with the free (non-encapsulated) drug (figure 8a).
a) 3D color-coded MR parametric maps of transverse relaxation times (T2) of the brain of 3 ischemic rats, 7 days after treatment (i.v.) with saline (left), free CDP-Choline (center), or CDP-Choline encapsulated in HSP72-targeting liposomes (right). b) Plot showing the evolution of the lesion sizes of ischemic animals previously to and during the 7 day post-treatment period with intravenous injections of saline (white), empty liposomes (light grey), an intraperitoneal injection of CDP-choline (left-tilted bars), or intravenous injections of free CDP-choline (dark grey), CDP-choline encapsulated in regular liposomes (right-tilted bars) or CDP-choline encapsulated in HSP72-targeted liposomes (black). In this plot, (*) represents significant differences (p<0.05) found by ANOVA for all groups of animals, while (#) represents significant differences (p<0.05) based on Student's t-test between selected pairs of groups.
Quantitation of the superior therapeutic effect offered by the theranostic agent was performed via a study in which 24 Sprague-Dawley rats were given to a permanent focal cerebral ischemia and randomized into 6 groups of n=4 rats. One group (group 3 in figure 8b) was treated with a single intraperitoneal injection of 500 mg/kg body weight of citicoline (described as an effective neuroprotective dose for ischemic rats) [30]. The other 5 treatment groups received 6 (t= 45 min, 6, 12, 24 and 30 h post-surgery) intravenous injections (jugular vein) of v=1 ml of saline (group 1), empty liposomes (group 2), a dose of 8 mg/kg body weight of citicoline dissolved in saline (group 4), and the same amount of citicoline encapsulated in regular liposomes (group 5) or encapsulated in HSP72-targeted liposomes (group 6). Diffusion-weighted imaging (pre-treatment) or T2-weighted MR imaging (days 1, 3 and 7 post-treatment) were used to quantify the lesion volumes for all animals (see methods).
Before treatment, measured lesion volumes (figure 8b) were comparable for all groups (Vt=0= 147±45, 148±24, 159±15, 155±41, 147±15 and 146±11 mm3 for groups 1, 2, 3, 4, 5 and 6, respectively). As expected for this animal model [43, 44], the lesion volumes of control animals increased, reaching their maximum values at day 3 post surgery and dropping to intermediate values at day 7 (Vgroup 1= 215±67, 220±77 and 201±50 mm3 at days 1, 3 and 7). The results were not significantly different for animals treated with empty liposomes or an i.p. injection of citicoline (ANOVA test, p<0.05). However, intravenous injections of free citicoline (group 4) or citicoline encapsulated in regular liposomes significantly reduced lesion volumes on day 7 (Vgroup4= 122±19 and Vgroup5= 82±17 mm3, respectively, vs. Vgroup1= 201±50 mm3 for controls; p<0.05). The highest therapeutic effect was achieved when citicoline was encapsulated in HSP72-targeting liposomes, which resulted in significantly smaller lesion volumes compared to controls at day 1 (Vgroup6= 116±18 vs. Vgroup1= 215±67 mm3), day 3 (Vgroup6= 106±12 vs. Vgroup1=220±77 mm3) and day 7 (Vgroup6=55±16 vs. Vgroup1=201±50 mm3) and compared to citicoline encapsulated in regular liposomes at day 7 (Vgroup6= 55±16 vs. Vgroup5= 82±17 mm3).
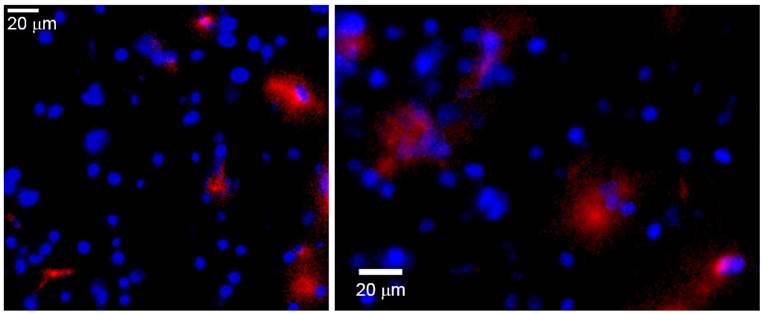
The presence of HSP72-targeted liposomes in the peri-infarct tissue in the brain was confirmed by ex vivo fluorescence microscopy of tissue sections obtained on day 7 (figure 9). In this figure, red rhodamine (from liposomes) was detected in a few cells of peri-infarct tissue (nuclei stained in blue), although the current data cannot provide information about which particular type of cell is positive for rhodamine. In any case, those pictures show that it is feasible for a load of treatment to reach the brain parenchyma after an intravenous injection of liposomes.
Ex vivo fluorescence microscopy images of the peri-infarct region of animals obtained 7 days after intravenous injections of CDP-choline encapsulated in anti-HSP72 vectorized liposomes. Rhodamine fluorescence (red) confirms the presence of the theranostic agent in the brain parenchyma (cell nuclei stained in blue with Hoescht).
Conclusions
In this study we report a new theranostic nano-platform vectorized towards peri-infarct tissue, a key target for the treatment of cerebral ischemia. The selection of the HSP72 protein as a selective targeting vector was made after a complete characterization of protein expression in the ischemic brain. Anti-HSP72 stealth immunoliposomes containing a load of citicoline and fluorescence and MR imaging probes were used to delineate the peri-infarct region in vivo (diagnostic function) and to achieve a superior therapeutic effect in comparison to other non-targeted drug delivery means. Thus, despite the challenge of crossing the blood-brain barrier, this study demonstrates that theranostics inside the brain parenchyma is feasible and represents a good example of the potential that nanotechnology offers for the treatment of neurological disorders such as stroke.
Methods
Animal studies
All experimental protocols involving the use of research animals were performed according to the guidelines of the Animal Welfare Committee of our host institution and were in accordance with applicable legislation of the European Union (DC 86/609/EEC, 2003/65/EC, 2010/63/EU). Studies were designed and conducted according to the following Stroke Therapy Academic Industry Roundtable group guidelines (STAIR) criteria: 1) Monitoring: the temperature of the animals was continuously monitored and corrected during surgery. Blood glucose levels were recorded prior to surgery. A laser Doppler blood flow-meter was used to measure cerebral blood flow during surgery. 2) Randomization: all animals were randomly assigned to treatment groups. 3) Allocation concealment: the scientist performing the surgery or treatment application was blinded to the procedures. Scientists acquiring images on MR were also blinded to procedures. 4) Blinded assessment of outcome: scientists performing the image analysis and outcome assessment were blinded to all surgical procedures and treatment applications. 5) Conflict of interest: all potential conflicts of interest and study funding have been reported on this manuscript.
A total of 72 male Sprague-Dawley rats (Harlan Laboratories, Barcelona, Spain) weighing 348±63 g were used in this study. Animals were kept in a controlled environment at 22±1 °C and 60±5% humidity with 12/12 h light/darkness cycles, and were fed ad libitum with standard diet pellets and tap water. All surgical procedures and MRI studies were conducted under Sevofluorane (Abbott Laboratories, IL, USA) anesthesia (3-4% carried by a 65:35 mixture of N20:02). Permanent focal cerebral ischemia was induced by an intracranial occlusion of the left middle cerebral artery (MCA) following the method of Shigeno et al. [45] as described elsewhere [30]. When required, intravenous injections of typically v=1 ml were administered in the jugular vein (alternating left and right jugular veins for treatments requiring multiple injections) of anesthetized animals with a 30 gauge needle. Animals were sacrificed at the end of the studies by exposure to a lethal dose of Sevofluorane (8%).
Synthesis and Characterization of liposomes
Liposomes were prepared by the lipid film rehydration method [30, 46]. Mixtures of lipids (typically 24 µmol) were prepared in a 6:1 v/v mixture of chloroform: methanol (Panreac Química, Barcelona, Spain). The mixtures contained cholesterol (molar fraction: x=0.33), 1,2-dioctadecanoyl-sn-glycero-3-phosphocholine (DSPC: x=0.57), diethylenetriamine pentaacetic acid)-bis(stearylamide) gadolinium salt (Gd-DTPA-BSA: x=0.03), 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[amino(polyethyleneglycol)-2000] (ammonium salt) (PEG-DSPE: x=.05 in regular and x=.025 in vectorized liposomes), 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[maleimide(polyethylene glycol)-2000] (ammonium salt) (Mal-PEG-DSPE: x=0.025, only in vectorized liposomes), and 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-(lissamine rhodamine B sulfonyl) ammonium salt (Liss Rhodamine PE, 0.1% w/w). All products were obtained from Avanti Polar Lipids, AL, USA.
After evaporation of the organic solvent on a rotavapor operated under vacuum at 30°C and overnight drying under a nitrogen flow, the lipid films formed were hydrated in HEPES-buffered saline (HBS, 10 mM HEPES, 135 mM NaCl, pH 7.4) at 65°C. Liposomes were then extruded in a Lipex Extruder (Northern Lipids, B.C., Canada), keeping the mixture at 65°C at all times. Liposomes were consecutively extruded 2, 4, 6 and 8 times through 400-, 200-, 100- and 80-nm polycarbonate membranes, respectively (Whatman, GE Healthcare, WI, USA). The final volumes were adjusted with HBS to achieve the desired total concentrations of lipids.
The total lipid concentrations in liposomes were determined based on phosphate quantification according to Rouser et al [47]. The liposome protein content (vectorized liposomes) was determined using the Lowry protocol [39] as described elsewhere [46]. The distribution of liposome sizes in solution (hydrodynamic diameters) was determined by dynamic light scattering (DLS, ZetaSizer NanoS, Malvern Instruments, Worcestershire, UK) at 23°C.
Liposomal solutions were vitrified on carbon-coated grids using a Vitrobot Mark III (FEI, OR, USA) for cryogenic transmission electron microscopy (cryo-TEM), and 25000X images were acquired on a Tecnai 20 Sphera TEM instrument (FEI, OR, USA) equipped with a LaB6 filament (200 kV) and Gatan cryoholder (-170°C).
Protein conjugation to liposomes
Regular liposomes were prepared as described above using a molar fraction of x=0.025 of maleimide containing PEGylated phospholipids (Mal-PEG-DSPE). In parallel, azide-free antibodies were obtained by three successive centrifugations of monoclonal antibodies against HSP72 (clone C92F3A-5, Enzo Life Sciences, NY, USA) on Vivaspin concentrator filters with a molecular weight cut-off (MWCO) threshold of 30 kDa (GE Healthcare, WI, USA) in the presence of 2 ml of HBS (pH=6.7). The resulting solutions were incubated for 45 min with N-succinimidyl S-acetylthioacetate (SATA, Sigma-Aldrich, MO, USA) using an antibody:SATA ratio of 1:80. Free SATA was removed by three consecutive centrifugations in Vivaspin filters with a 30 kDa MWCO in the presence of 4 ml of HBS (pH=6.7), and SATA-treated antibodies were incubated for 60 min in a hydroxylamine solution (50 mM; 1:10 H3NO:antibody ratio, Sigma-Aldrich, MO, USA). This solution was added to maleimide liposomes (50 µg of protein per µmol of lipid) and incubated overnight at 4°C. Uncoupled antibody units were removed by ultra-centrifugation at 65000 rpm for 45 min, suspending the resulting HSP72-coupled liposomes in HBS (pH=7.4) and storing them at 4 °C until use.
Tissue samples
Rat brains were quickly extracted after sacrifice for proteomic and Western-blotting studies and sliced into 5x 2-mm-thick sections (rostro-caudal direction, starting at 1 mm from the rhinal fissure) and immersed for 5-10 min in a saturated solution of Triphenyl Tetrazolium Chloride (Parnreac Química, Barcelona, Spain) in 1x phosphate-buffered saline (Gibco Life Technologies, CA, USA), to stain dead cells (infarct core) in white (figure 1a). For immunohistological studies, animals were transcardially perfused with a lethal dose of anesthetic (Sevofluorane at 8%) in 100 ml of saline followed by 300 ml of 4% formaldehyde (BDH Prolabo, VWR, PA, USA). Brains were then extracted and sliced in 4x 3-mm-thick blocks (rostro-caudal direction, starting at 1 mm from the rhinal fissure). Tissue blocks were immersed in 4% formaldehyde (overnight), 20% sucrose (24 h), and finally in liquid N2-cooled pentane. Frozen brain blocks were preserved at -80 °C until further use.
Proteomic studies
Proteins from peri-infarct and contralateral (control) tissue strips (manually dissected from TTC-stained brain sections, figure 1a) were extracted using the ProteoExtract® Native Membrane Protein Extraction Kit (Calbiochem Merck KGaA, Darmstadt, Germany) following the manufacturer's instructions. Protein contents were quantified with the modified Bradford method [48]. A series of 72 proteomic gels were used for 2D electrophoresis (2 replicas x 6 animals x 2 tissue samples x 3 protein fractions), each containing 40 µg of protein. The proteins were separated by isoelectric point (1st dimension) and molecular weight (2nd dimension) with 17 cm 3-10 NL IPG strips (BioRad, CA, USA) and 12.5% polyacrylamide gels on a ETTAN-DALTSIX system (GE Healthcare, WI, USA), respectively. Procedures used for 2D electrophoresis and data processing were performed as described by Brea et al. [49].
Immunoblotting studies
Twelve additional 2D electrophoresis gels (2 replicas x 2 animals x 3 protein fractions) were prepared as described above. The separated proteins were transferred from those gels to low-fluorescence PVDF membranes (Millipore, MA, USA) in semi-dry conditions at 15 V for 45 minutes, blocked in 5% non-fat dry milk (2 h, room temperature) and incubated overnight with mouse monoclonal anti-rat HSP70 antibody (1:1000, Abcam, MA, USA). Protein loads were verified by simultaneous incubation with rabbit polyclonal anti-rat β-actin (1:3000, Abcam, MA, USA). Detection was achieved by Cy3-labelled goat anti-mouse (1:3000, GE, Barcelona, Spain) or Cy5-labelled goat anti-rabbit (1:3000, GE, Barcelona, Spain) secondary antibodies. Gels were scanned on a Molecular Imager FX Pro-plus (BioRad, CA, USA) for data analysis with PDQuest software package (BioRad, CA, USA).
Immunohistochemistry studies
Frozen brain blocks were sliced in 10 µm coronal sections with a Tissue-Tek Cryo3 Cryostat (Sakura Finetek, Alphen aan dem Rijn, The Netherlands) and incubated with 3% H2O2 and 10% methanol in PBS to block endogenous peroxidases, and with 3% normal serum and 0,2% Triton X-100 in PBS to block unspecific binding sites.
For light microscopy, the inducible form of HSP70 was revealed by a 1 h incubation with a 1:50 dilution of HSP70/HSP72 mAb (C92F3A-5) primary antibody (Enzo life sciences, NY, USA), while nonspecific inducible and constitutive HSP70 protein mixtures were identified by a 1 h incubation with a 1:50 dilution of anti-HSP70 antibody (ab5439, 1:50 dilution Abcam, Cambridge, MA). Samples were further processed with biotin-conjugated secondary anti-rabbit antibody (1:200; Vector Laboratories, CA, USA) and streptavidin-conjugated peroxidase (Vecstatin Abc kit, Vector Laboratories, CA, USA). Color was developed by the addition of DAB (Dako, Glostrup, Denmark).
For in vitro fluorescence studies, HSP72 was labeled with HSP70/HSP72 mAb (C92F3A-5; 1:50 dilution; Enzo Life Sciences, NY, USA) and biotin-conjugated anti-mouse secondary antibody (BA-2001; 1:200; Vector Laboratories CA, USA) attached to DyLight 549 Streptavidin (SA-5549-1; 1:200; Vector Laboratories, CA, USA) as the secondary antibody. Neurons and astrocytes were labeled with anti-FOX3 (ab104225; 1:50 dilution; Abcam, MA, USA) or polyclonal anti-GFAP (G4546; 1:100: Sigma-Aldrich, MO, USA), respectively, as primary antibodies, directly attached to DyLight 488 horse anti-rabbit IgG antibody (DI-1488; 1:200; Vector Laboratories, CA, USA).
Cell cultures
Primary cultures of rat astrocytes were obtained from 1-day-old rats by disaggregation and digestion of their freshly extracted brains following the method of McCarthy et al [50]. Cultures were maintained in flasks or dishes (BD Biosciences, CA, USA) in a humidified (97%) incubator at 37 °C and 95:5 air:CO2 atmosphere. Confluence levels were typically maintained at 70-80%. Upregulation of HSP72 proteins in the membrane of cultured astrocytes (in vitro model for peri-infarct tissue) was achieved following the process described by de Freitas et al [41].
Microscopy
Fluorescence and light microscopy images were acquired on a IX-51 microscope (Olympus GMBH, Hamburg, Germany) attached to a DS-U2 LCD camera (Nikon Instruments, NY, USA).
MR imaging
Magnetic resonance imaging (MRI) studies were conducted on a 9.4 T MR system (Bruker Biospin, Ettlingen, Germany) with 440 mT/m gradients, using a combination of a linear birdcage resonator of 7 cm of diameter for signal transmission and a 2x2 arrayed surface coil for signal detection.
T1-weighted images (T1w) were typically acquired using a RARE sequence (rare factor=4, averages=2) with an effective echo time of TE=22 ms and a repetition time of TR=900 ms (flip angle = 90°). The whole brain was covered with 14 consecutive axial slices of 1 mm thickness with a field-of-view (FOV) of 19.2x19.2 mm (with saturation bands to suppress the signal outside the brain) and an in-plane resolution of 100 microns (192x192 points matrix). When required, coronal images where acquired using a FOV of 32x19.2 mm, keeping the same spatial resolution. The FOV was adapted to the required size for MR imaging of solutions (placed in 1.5 ml Eppendorf tubes) in in vitro studies. To obtain T1 maps, we used the same RARE sequence following a saturation-recovery approach [51] to acquire a series of 12 images with repetition times ranging from 250 ms to 12 s (exponentially distributed).
T2-weighted images (and maps) were typically acquired using a multi-slice multi-echo (MSME) sequence (averages=2) with a train of 16 echoes (TE=9.5 ms) and a repetition time of TR=2.5 s (flip angle = 90°). The spatial orientations and resolutions were the same as described above.
Apparent diffusion coefficient (ADC) maps were obtained from diffusion-weighted images (DWI) by pixel-by-pixel mono-exponential fitting of a series of 7 images acquired using an echo-planar sequence (DWI-EPI) with the following acquisition parameters: field-of-view 19.2x19.2 mm2 (saturation bands out of this FOV), an image matrix of 128x128 point (in-plane resolution 0.15 mm/pixel), 14 consecutive slices of 1 mm thickness, repetition time = 4 s, echo time = 30 ms, and diffusion b values of 0, 100, 300, 600, 800, 1000, and 1400 s/mm2.
ADC maps were constructed using ParaVision 5.1 software from Bruker Biospin (Ettlingen, Germany), while proton density images and T1 and T2 relaxation maps were calculated by a pixel-by-pixel mono-exponential fitting of the data with self-developed applications for Image-J (Rasband, W.S., ImageJ, U. S. National Institutes of Health, Bethesda, Maryland, USA, http://imagej.nih.gov/ij/, 1997-2012).
Lesion sizes from all animals were calculated from ADC maps (pre-treatment) or from T2 parametric maps (days 1, 3 and 7 post-treatment) by manual selection of the (hyperintense) lesion area on each of the 14 acquired brain slices per brain, and the areas calculated for the 14 measured 1-mm-thick brain slices were added. Considering the large increase in T2 values induced by an ischemic lesion and the relatively low transversal relaxivity of our liposomes (r2=8.38 mM-1s-1), the potential presence of Gd-liposomes in the brain did not significantly affect the measure of lesion volumes.
MRI maps of the distribution of liposomes
T1 parametric maps of control (saline-treated) animals were used to obtain histograms of T1 values for healthy tissue (pink bars in figure 5a, distribution centered at T1=2.47 s with FWHM=0.48 s) and ischemic tissue (blue bars in figure 5a, distribution centered at T1=2.80 s with a FWHM of 0.54 s).
The distribution of T1 values for brain tissue regions that contain Gd-doped liposomes in vivo cannot be directly calculated because the exact concentration of liposomes in each region of the brain remains unknown. Thus, an approximation has to be made to estimate the distribution. We followed an approach based on obtaining the T1 maps of animals receiving intraparenchymal injections of liposomes (study presented in figure 3). After the injection of liposomes, a gradient of concentrations was established with the distance from the injection point. The selection of a reasonable proportion of tissue around the injection site allowed the generation of a histogram of pixels that contain a wide range of T1 values based on the concentration of liposomes on each pixel of the tissue that dilutes to zero with the distance to the injection point (green bars in figure 5a, centered at T1=2.25 s with FWHM=0.32 s). This is a limitation of the present study and the most critical part of the image analysis described here (and graphically summarized in figure 5b). Future studies will include the preparation of liposomes with higher r1 relaxivity to allow better characterization of liposome-loaded tissue (resulting in fewer overlapping of populations in the histogram).
For image analysis, we first obtained T1 parametric maps of each brain slice (pixel-by-pixel mono-exponential fitting) for all animals. Then, a colored look-up table (LUT) was applied to emphasize differences in T1 values. For the third step, noisy maps were smoothed using a Gaussian filter (r=0.6). Subsequently, a clipping filter was used to eliminate pixels with T1 ≥ 2.25 s, which corresponds to the peak T1 distribution of liposome-loaded tissues (green bars in figure 5a). The selection of this threshold is the most critical step of our analysis and represents a limitation to this work because the tails of the healthy tissue and liposome-loaded tissue distributions of T1 values overlap. Thus, pixels corresponding to the left tail of the distribution of T1 values for healthy tissue may be counted as liposomes-containing tissue (false positives) while pixels of the right tail of the distribution of liposomes-containing tissue are obviated (false negatives). Our work must be improved in the future through more complex models for image analysis, which may not be necessary if optimized liposomal designs are used to achieve higher relaxometries (lower T1 values), reducing the overlap of the two aforementioned histograms.
Assuming that the results of this last step are acceptable (at least for proof-of-principle purposes), we proceeded to enhance the contrast of the remaining pixels against the background by rescaling the images. Finally, images of “liposomes” (low T1 pixels) were overlaid with the corresponding proton density images of the brain, and a clustering process was performed to remove groups of n<3 pixels (considered random noise).
Statistical analysis
Quantitative data are always presented as the mean ± standard deviation in this work. Differences between pairs were compared with Student's t-test. Differences among multiple groups (in vivo studies) were compared with ANOVA tests. Differences were considered statistically significant when a level of p<0.05 was achieved.
Acknowledgements
Financial support from the Instituto de Salud Carlos III of the Spanish Ministry of Health (projects PI11/02161 and CP09/00074), the Spanish Ministry of Economy and Competence (projects SAF2008-02190, SAF2011-30517 and RETICS-INVICTUS R012/0014), and the European Union (FEDER program) is gratefully acknowledged. Research contracts from the Miguel Servet program (PRC and TS) and the Sara Borrell program (JA and DB) of the Instituto de Salud Carlos III are acknowledged. Authors thank Prof. Dr. Klaas Nicolay and Dr. Gustav Strijkers, from the Technical University of Eindhoven (The Netherlands) for sharing their knowledge on the synthesis and characterization of liposomes.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Picard FJ, Bergeron MG. Rapid molecular theranostics in infectious diseases. Drug Discov Today. 2002;7:1092-101
2. Fernandez-Fernandez A, Manchanda R, McGoron A. Theranostic Applications of Nanomaterials in Cancer: Drug Delivery, Image-Guided Therapy, and Multifunctional Platforms. Applied Biochemistry and Biotechnology. 2011;165:1628-51
3. Lobatto ME, Fuster V, Fayad ZA, Mulder WJM. Perspectives and opportunities for nanomedicine in the management of atherosclerosis. Nat Rev Drug Discov. 2011;10:835-52
4. Shim MS, Kwon YJ. Stimuli-responsive polymers and nanomaterials for gene delivery and imaging applications. Advanced Drug Delivery Reviews. 2012;64:1046-59
5. Kinlay S. Changes in Stroke Epidemiology, Prevention, and Treatment. Circulation. 2011;124:e494-e6
6. Ramos-Cabrer P, Campos F. Liposomes and nanotechnology in drug development: focus on neurological targets. Int J Nanomed. 2013;8:951-60
7. Adeoye O, Hornung R, Khatri P, Kleindorfer D. Recombinant Tissue-Type Plasminogen Activator Use for Ischemic Stroke in the United States. Stroke. 2011;42:1952-5
8. Lo EH. A new penumbra: transitioning from injury into repair after stroke. Nat Med. 2008;14:497-500
9. Ramos-Cabrer P, Campos F, Sobrino T, Castillo J. Targeting the Ischemic Penumbra. Stroke. 2011;42:S7-S11
10. Hata R, Maeda K, Hermann D, Mies G, Hossmann KA. Evolution of brain infarction after transient focal cerebral ischemia in mice. J Cereb Blood Flow Metab. 2000;20:937-46
11. Hill WD, Hess DC, Martin-Studdard A, Carothers JJ, Zheng J, Hale D. et al. SDF-1 (CXCL12) is upregulated in the ischemic penumbra following stroke: association with bone marrow cell homing to injury. J Neuropathol Exp Neurol. 2004;63:84-96
12. Kokubo Y, Matson GB, Liu J, Mancuso A, Kayama T, Sharp FR. et al. Correlation between changes in apparent diffusion coefficient and induction of heat shock protein, cell-specific injury marker expression, and protein synthesis reduction on diffusion-weighted magnetic resonance images after temporary focal cerebral ischemia in rats. J Neurosurg. 2002;96:1084-93
13. Li Y, Chopp M, Garcia JH, Yoshida Y, Zhang ZG, Levine SR. Distribution of the 72-kd heat-shock protein as a function of transient focal cerebral ischemia in rats. Stroke. 1992;23:1292-8
14. Rami A. Upregulation of Beclin 1 in the ischemic penumbra. Autophagy. 2008;4:227-9
15. Sanz O, Estrada A, Ferrer I, Planas AM. Differential cellular distribution and dynamics of HSP70, cyclooxygenase-2, and c-Fos in the rat brain after transient focal ischemia or kainic acid. Neuroscience. 1997;80:221-32
16. Soriano MA, Ferrer I, Rodriguez-Farre E, Planas AM. Expression of c-fos and inducible hsp-70 mRNA following a transient episode of focal ischemia that had non-lethal effects on the rat brain. Brain Res. 1995;670:317-20
17. Shimada IS, Borders A, Aronshtam A, Spees JL. Proliferating reactive astrocytes are regulated by Notch-1 in the peri-infarct area after stroke. Stroke. 2011;42:3231-7
18. Vega VL, Rodríguez-Silva M, Frey T, Gehrmann M, Diaz JC, Steinem C. et al. Hsp70 Translocates into the Plasma Membrane after Stress and Is Released into the Extracellular Environment in a Membrane-Associated Form that Activates Macrophages. The Journal of Immunology. 2008;180:4299-307
19. Gupta A, Cooper ZA, Tulapurkar ME, Potla R, Maity T, Hasday JD. et al. Toll-like receptor agonists and febrile range hyperthermia synergize to induce heat shock protein 70 expression and extracellular release. J Biol Chem. 2013;288:2756-66
20. Allen TM. Long-circulating (sterically stabilized) liposomes for targeted drug delivery. Trends Pharmacol Sci. 1994;15:215-20
21. Drummond DC, Meyer O, Hong K, Kirpotin DB, Papahadjopoulos D. Optimizing liposomes for delivery of chemotherapeutic agents to solid tumors. Pharmacol Rev. 1999;51:691-743
22. Kabanov AV. Polymer genomics: an insight into pharmacology and toxicology of nanomedicines. Adv Drug Deliv Rev. 2006;58:1597-621
23. Kataoka K, Harada A, Nagasaki Y. Block copolymer micelles for drug delivery: design, characterization and biological significance. Adv Drug Deliv Rev. 2001;47:113-31
24. Lee CC, MacKay JA, Frechet JM, Szoka FC. Designing dendrimers for biological applications. Nat Biotechnol. 2005;23:1517-26
25. Lu Y, Low PS. Folate-mediated delivery of macromolecular anticancer therapeutic agents. Adv Drug Deliv Rev. 2002;54:675-93
26. Nori A, Kopecek J. Intracellular targeting of polymer-bound drugs for cancer chemotherapy. Adv Drug Deliv Rev. 2005;57:609-36
27. Widera A, Norouziyan F, Shen WC. Mechanisms of TfR-mediated transcytosis and sorting in epithelial cells and applications toward drug delivery. Adv Drug Deliv Rev. 2003;55:1439-66
28. Denora N, Trapani A, Laquintana V, Lopedota A, Trapani G. Recent advances in medicinal chemistry and pharmaceutical technology--strategies for drug delivery to the brain. Curr Top Med Chem. 2009;9:182-96
29. Vlieghe P, Khrestchatisky M. Medicinal Chemistry Based Approaches and Nanotechnology-Based Systems to Improve CNS Drug Targeting and Delivery. Med Res Rev. 2013;33:457-516
30. Ramos-Cabrer P, Agulla J, Argibay B, Perez-Mato M, Castillo J. Serial MRI study of the enhanced therapeutic effects of liposome-encapsulated citicoline in cerebral ischemia. Int J Pharm. 2011;405:228-33
31. Bulte JW, de Cuyper M, Despres D, Frank JA. Short- vs. long-circulating magnetoliposomes as bone marrow-seeking MR contrast agents. J Magn Reson Imaging. 1999;9:329-35
32. Aragnol D, Leserman LD. Immune clearance of liposomes inhibited by an anti-Fc receptor antibody in vivo. Proc Natl Acad Sci U S A. 1986;83:2699-703
33. Gabizon A, Papahadjopoulos D. The role of surface charge and hydrophilic groups on liposome clearance in vivo. Biochim Biophys Acta. 1992;1103:94-100
34. Torchilin VP, Omelyanenko VG, Papisov MI, Bogdanov AA Jr, Trubetskoy VS, Herron JN. et al. Poly(ethylene glycol) on the liposome surface: on the mechanism of polymer-coated liposome longevity. Biochim Biophys Acta. 1994;1195:11-20
35. van Rooy I, Cakir-Tascioglu S, Hennink WE, Storm G, Schiffelers RM, Mastrobattista E. In vivo methods to study uptake of nanoparticles into the brain. Pharmaceutical research. 2011;28:456-71
36. Mulder WJ, Griffioen AW, Strijkers GJ, Cormode DP, Nicolay K, Fayad ZA. Magnetic and fluorescent nanoparticles for multimodality imaging. Nanomedicine (Lond). 2007;2:307-24
37. Mulder WJ, Strijkers GJ, van Tilborg GA, Griffioen AW, Nicolay K. Lipid-based nanoparticles for contrast-enhanced MRI and molecular imaging. NMR Biomed. 2006;19:142-64
38. Adibhatla RM. Citicoline in stroke and TBI clinical trials. Nat Rev Neurol. 2013;9:173
39. Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. Protein measurement with the Folin phenol reagent. J Biol Chem. 1951;193:265-75
40. Hutchinson FJ, Francis SE, Lyle IG, Jones MN. The characterisation of liposomes with covalently attached proteins. Biochim Biophys Acta. 1989;978:17-24
41. de Freitas MS, Spohr TC, Benedito AB, Caetano MS, Margulis B, Lopes UG. et al. Neurite outgrowth is impaired on HSP70-positive astrocytes through a mechanism that requires NF-kappaB activation. Brain Res. 2002;958:359-70
42. Hurtado O, Moro MA, Cárdenas A, Sánchez V, Fernández-Tomé P, Leza JC. et al. Neuroprotection afforded by prior citicoline administration in experimental brain ischemia: effects on glutamate transport. Neurobiology of Disease. 2005;18:336-45
43. Agulla J, Argibay B, Perez-Mato M, Brea D, Ramos-Cabrer P, Castillo J. [Comparison of the lesion produced by permanent focal cerebral ischaemia in three animal models using magnetic resonance imaging]. Rev Neurol. 2011;53:265-74
44. Wegener S, Weber R, Ramos-Cabrer P, Uhlenkueken U, Sprenger C, Wiedermann D. et al. Temporal profile of T2-weighted MRI distinguishes between pannecrosis and selective neuronal death after transient focal cerebral ischemia in the rat. J Cereb Blood Flow Metab. 2006;26:38-47
45. Shigeno T, Teasdale GM, McCulloch J, Graham DI. Recirculation model following MCA occlusion in rats. Cerebral blood flow, cerebrovascular permeability, and brain edema. J Neurosurg. 1985;63:272-7
46. Hak S, Sanders HM, Agrawal P, Langereis S, Grull H, Keizer HM. et al. A high relaxivity Gd(III)DOTA-DSPE-based liposomal contrast agent for magnetic resonance imaging. Eur J Pharm Biopharm. 2009;72:397-404
47. Rouser G, Fkeischer S, Yamamoto A. Two dimensional then layer chromatographic separation of polar lipids and determination of phospholipids by phosphorus analysis of spots. Lipids. 1970;5:494-6
48. Ramagli LS, Capetillo S, Becker FF, Rodriguez LV. Alterations in nonhistone chromatin proteins during hepatocarcinogenesis induced by diverse acting carcinogens. Carcinogenesis. 1985;6:367-75
49. Brea D, Rodriguez-Gonzalez R, Sobrino T, Rodriguez-Yanez M, Blanco M, Castillo J. Proteomic analysis shows differential protein expression in endothelial progenitor cells between healthy subjects and ischemic stroke patients. Neurol Res. 2011;33:1057-63
50. McCarthy KD, de Vellis J. Preparation of separate astroglial and oligodendroglial cell cultures from rat cerebral tissue. J Cell Biol. 1980;85:890-902
51. Haacke EM, Brown RW, Thompson MR, Venkatesan R. Magnetic resonance imaging: physical principles and sequence design. New York; CHichester: J. Wiley & Sons. 1999
Author contact
![]() Corresponding author: Dr. Pedro Ramos Cabrer. Laboratorio de Neurociencias Clínicas. Hospital Clínico Universitario. Travesa da Choupana s/n. 15706 Santiago de Compostela. Spain. Phone: +34981951097, Fax: +34981951098, e-mail: pedro.ramos.cabreres.
Corresponding author: Dr. Pedro Ramos Cabrer. Laboratorio de Neurociencias Clínicas. Hospital Clínico Universitario. Travesa da Choupana s/n. 15706 Santiago de Compostela. Spain. Phone: +34981951097, Fax: +34981951098, e-mail: pedro.ramos.cabreres.