Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and methods
Results and discussion
Conclusions
Supplementary Material
Abbreviations
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2015; 5(10):1154-1165. doi:10.7150/thno.12398 This issue Cite
Research Paper
Structure-based Design of Peptides with High Affinity and Specificity to HER2 Positive Tumors
Lingling Geng, Zihua Wang, Xiaoliang Yang, Dan Li, Wenxi Lian, Zhichu Xiang, Weizhi Wang, Xiangli Bu, Wenjia Lai, Zhiyuan Hu ![]() , Qiaojun Fang
, Qiaojun Fang ![]()
CAS Key Laboratory for Biomedical Effects of Nanomaterials & Nanosafety, National Center for Nanoscience and Technology, Beijing 100190, China
Received 2015-4-14; Accepted 2015-7-5; Published 2015-8-1
Abstract

To identify peptides with high affinity and specificity against human epidermal growth factor receptor 2 (HER2), a series of peptides were designed based on the structure of HER2 and its Z(HER2:342) affibody. By using a combination protocol of molecular dynamics modeling, MM/GBSA binding free energy calculations, and binding free energy decomposition analysis, two novel peptides with 27 residues, pep27 and pep27-24M, were successfully obtained. Immunocytochemistry and flow cytometry analysis verified that both peptides can specifically bind to the extracellular domain of HER2 protein at cellular level. The Surface Plasmon Resonance imaging (SPRi) analysis showed that dissociation constants (KD) of these two peptides were around 300 nmol/L. Furthermore, fluorescence imaging of peptides against nude mice xenografted with SKBR3 cells indicated that both peptides have strong affinity and high specificity to HER2 positive tumors.
Keywords: structure-based design, HER2 targeted peptide, breast cancer, MD simulation
Introduction
The human epidermal growth factor receptor (EGFR/ERBB) family of receptor tyrosine kinases (RTKs) consists of four members: HER1 (EGFR, ErbB1), HER2 (neu, ErbB2), HER3 (ErbB3), and HER4 (ErbB4). Overexpression or mutations of these proteins are associated with many types of human cancers. Except HER2, the other three receptors can be activated by EGF, EGF-like, or neuregulin ligands. Although it lacks a ligand binding domain, HER2 can form homodimers or heterodimers with the other three HER (1, 3 and 4) family members. Upon dimerisation, the cytoplasmic tyrosine kinase domain activates several important oncogenes and drives downstream signaling cascade, leading to the stimulation of cell proliferation, invasion, and anti-apoptosis [1-4]. Amplification and/or high expression of the HER2 occurs in approximately 20‑25% of invasive breast cancers, which is classified as HER2 positive breast cancer [5, 6]. Compared with HER2-normal breast cancer, HER2-positive breast cancer is associated with worse prognosis, including higher mortality in early-stage disease, increased incidence of metastasis, and reduced time to relapse [5, 7, 8]. Therefore, HER2 has become a validated therapeutic target in breast cancer. Trastuzumab (Herceptin), a recombinant humanized monoclonal antibody directed against the extracellular domain of HER2, is mainly used in treating patients with HER2-positive breast cancer [9]. However, approximately 15% of patients with HER2-positive metastatic breast cancer eventually relapse after therapy due to de novo or acquired resistance [10]. To solve this problem, other anti-HER2 agents, including monoclonal antibody pertuzumab, small molecule kinase inhibitors like lapatinib, and antibody-drug conjugate such as trastuzumab emtansine have been developed. However, the therapeutic potential of these new agents is still unproven and there remain needs for a novel therapeutic agent that can have a long term effect on the treatment of breast cancer and ensure disease free survival [11, 12].
Affibody is a new class of affinity ligands, developed as a stabilized variant of the “B domain” of the IgG-binding staphylococcal protein A, and contains 58 amino acids that folds into a stable three-helix bundle [13]. HER2 targeted affibody molecule ZHER2 displays alternative properties compared to HER2 antibodies. It does not compete with trastuzumab [14] or pertuzumab [15] because the binding site of ZHER2 as shown in the crystal structure of HER2/ZHER2 complex is different from the epitopes for trastuzumab and pertuzumab determined previously [16-18]. ZHER2 does not appear to mediate detectable biological effects by itself [19]. However, its different binding site makes it suitable for molecular imaging as a tracer [15, 20], even in the presence of therapeutic monoclonal antibodies. Therefore, ZHER2 has the potential for diagnostic and may be used as a carrier to direct therapeutic agents to their HER2 target. Up to now, a range of modified HER2-binding affibody molecules have been thoroughly investigated, such as ZHER2:4 [21], dimeric clone (ZHER2:4)2 [22] and an high affinity monomeric version Z(HER2:342) with KD of 22 pM [23]. Although these affibody molecules are promising for development into imaging agents, with a length of 58 amino acids, it is difficult to synthesize using solid phase peptide synthesis method.
Small peptides combine both the advantages of small molecule drugs which are cost effective, good tissue and membrane permeability and those of antibodies which are high target specificity and low toxicity. To identify peptides with high affinity and specific to HER2 for imaging and therapeutic application, we designed a series of novel peptides with shorter lengths than Z(HER2:342) based on the interactions between Z(HER2:342) and HER2. Basically, we kept important binding residues and linked them together with suitable number of glycines according to distances between them. A combination protocol of molecular dynamics modeling, MM/GBSA binding free energy calculations [24-26], and binding free energy decomposition analysis [27-29] were then applied to study interactions of HER2 and designed peptides. After several rounds of mutations and simulations, those with lowest binding free energy were selected for experimental validation.
As a result, two peptides with 27 residues named pep27 and pep27-24M were successfully obtained by computational simulation. Both peptides were demonstrated to bind specifically to the extracellular domain of HER2 protein with KD values of 346 and 293 nmol/L, respectively. Ex vivo and in vivo experiments verified that both novel peptides have strong affinity and high specificity for HER2 using mice xenografted with HER2 positive tumors. We expect that both peptides may be used as new probes in diagnosis and treatment of HER2 positive breast cancer.
Materials and methods
Prepare the initial structure
Primary sequences of Z(HER2:342), pep32, pep27, pep26 and pep23 were aligned by using ClustalW program available on the web of EMBnet [30].
The model of HER2/Z(HER2:342) complex comes from crystal structure of HER2 extracellular region and 3-helix affibody Z(HER2:342) (PDB entry: 3MZW) [16] in the RCSB Brookhaven Protein Data Bank (PDB) [31]. The model of HER2/pep32 was constructed based on the crystal structure of HER2/Z(HER2:342) by deleting and adding suitable number of amino acids in affibody Z(HER2:342). Other models like HER2/pep27, HER2/pep26 and HER2/pep23 complexes were constructed based on HER2/pep32. The mutant complexes of HER2/pep27 were acquired by mutating the amino acids in the original model.
MD Simulations
The AMBER03 force field was used to establish the potentials of the proteins in the following molecular mechanics (MM) minimizations and molecular dynamics (MD) simulations [32]. The whole system was solvated in a truncated octahedron box filled with 8 Å TIP3P water molecules, with a minimum solute-wall distance of 12 Å [33], and the missing hydrogen atoms of each model were added using the tleap program. Then, counter-ions Na+ were placed on the grids with the largest negative coulombic potentials around the protein.
The sander program was used before the MD simulations to minimize the structure via three steps: firstly, the whole protein was fixed and the water molecules and counter-ions were minimized with 5000 cycles of steepest descent and 2500 cycles of conjugate gradient minimizations; secondly, the backbone atoms of the protein were fixed and the side chains were minimized using the same settings as above (5000 cycles of steepest descent and 2500 cycles of conjugate gradient minimizations); thirdly, the whole system was minimized without any constrain with 10000 cycles of steepest descent and 5000 cycles of conjugate gradient minimizations. The SHAKE procedure was applied, and the time step was set to 2.0 fs [34]. Particle Mesh Ewald (PME) was employed to deal with the long-range electrostatic interactions in the MD simulations [35]. Followed by minimization, the entire system was gradually heated from 0 to 310 K via seven steps in the NVT (canonical ensemble). Finally, 10 ns MD simulations were carried out under the constant temperature of 310 K. During the sampling process, the coordinates were saved every 0.2 ps, and the conformations generated from the simulations were used for further binding free energy calculations and decomposition analysis.
Binding free energy calculations
The binding free energy of each system was calculated using MM/GBSA technique according to the following equation [25].
∆Gbind = Gcomplex-Gprotein-Gligand
= ∆EMM +∆GGB+∆GSA-T∆S (1)
= ∆EvdW +∆Eele+∆GGB+∆GSA-T∆S
where ∆EMM is the interaction energy between protein and ligand in gas-phase, including the parts: the van der Waals energies (∆EvdW) and the electrostatic (∆Eele); ∆GGB and ∆GSA are the relative polar and nonpolar contributions to desolvation free energy, respectively, and -T∆S represents the conformational entropic contribution at temperature T. In this study, the polar solvation free energy was calculated by the generalized born (GB) model [36]. In the GB calculations, the solvent and the solute dielectric constants were set to 80 and 4, respectively. The nonpolar solvation term was estimated based on the solvent accessible surface area (SASA) model by the LCPO method with a solvent-probe radius of 1.4 Å: ∆GSA =0.0072×∆SASA [37]. The binding free energy of each system was calculated based on 500 snapshots from 7 to 10 ns MD simulation trajectories of each complex by using the mm_pbsa program in AMBER12 [38]. The conformational entropy (translation, rotation and vibration) upon the ligand binding (-T∆S) was calculated using normal-mode analysis via the nmode program in AMBER12 [38]. Due to the high computational demand and low prediction accuracy, 50 snapshots evenly extracted from the last 3 ns were used to estimate the entropic contribution [39-43].
Free energy decomposition analysis
To have a clear understanding of key residues in the interaction between HER2 and Z(HER2:342), the MM/GBSA free energy decomposition process was applied by the mm_pbsa program in AMBER12 [44]. During the decomposition process, the polar contribution of desolvation free energy (∆GGB) was calculated using the generalized Born (GB) approximation model developed by Onufriev et al. [45], and the nonpolar solvation contribution (∆GSA) part was computed based on the SASA determined with the ICOSA method [37]. All the energy components were calculated using 500 snapshots extracted from the MD trajectory from 7 to 10 ns. After the decomposition process, the free energy contribution can be allocated to each residue from the association of the receptor and the ligand.
Peptides synthesis
All peptides were synthesized using Fmoc strategy SPPS (solid phase peptide synthesis). The synthesized peptides were purified by using a Hitachi HPLC system (L-7100, Japan) on a TSK gel ODS-100V column (150 mm×4.6 mm) at a flow rate of 2 mL min-1. Gradient: 0-25 min, 5-80% acetonitrile containing 0.1% Trifluoroacetic acid (TFA). After purification, peptides were characterized by MALDI-TOF MS (Bruker Daltonics).
9-Fluorenylmethoxycarbonyl (Fmoc)-protected amino acids and 2-(1H-benzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HBTU) were purchased from GL Biochem (China). Trifluoroacetic acid (TFA), and fluorescein 5-isothiocyanate were from Sigma-Aldrich (USA). N-Methyl morpholine (NMM) and N,N-dimethylformamide (DMF) were from Beijing chemical plant (China).
SPRi assay
Surface Plasmon Resonance imaging (SPRi) chip used in this work is a gold layer of 47.5 nm thickness bare gold Plexera® Nanocapture® Chip. For detection, all peptides were linked to a cysteine residue in the amino terminal for interacting with bare gold. First, 1mg/mL peptides were added to the gold surface of the chip and incubated at 4oC overnight in water box. Then, SPRi chip was washed with phosphate buffer saline (PBS) and water for 4 times before 5% non-fat milk was applied to block overnight at 4oC. After the chip was washed again with PBS and water, it was dried with nitrogen for later usage. The purified HER2 protein purchased from Sino Biological Inc (China) was used as the mobile phase. HER2 protein was dissolved in PBST (PBS with 0.1% Tween-20) to a stock concentration of 20 μg/mL and further diluted to 10, 5, 2.5, 1.25 and 0.625 μg/mL. The SPRi analysis procedure was carried out on the prepared SPRi chip by injection of PBST running buffer for baseline stabilization, followed by protein sample (one of the five concentration of protein, binding), then PBST running buffer for washing and finally 0.5% (vol/vol) H3PO4 in deionized water for regeneration. The above cycle was repeated for each concentration of the HER2 protein at 20, 10, 5, 2.5, 1.25 and 0.625 μg/mL. Real-time binding signals were recorded and analyzed by PlexArray HT system (Plexera LLC, Bothell, WA, USA). The dissociation constant was calculated by fitting the association-dissociation curves.
Flow cytometry, immunocytochemistry and MTT assay
Human breast cancer cell lines SKBR3 (HER2 high expression), MCF-7 (HER2 medium expression), MDA-MB-231 (HER2 medium expression), 468 (HER2 low expression) and human embryonic kidney cell line 293A (HER2 low expression) were used in this study [46, 47]. SKBR3 cells were cultured in RPMI 1640 medium (Hyclone) supplemented with 10% fetal bovine serum (Gibco). MCF-7, MDA-MB-231, 468 and 293A cells were cultured in DMEM/High glucose (Hyclone) medium, with 10% fetal bovine serum, respectively. All of the following flow cytometry, immunocytochemistry and MTT assay were repeated three times.
For flow cytometry analysis, all cells were cultured overnight, and approximately 1×106 mL-1 cells (100 μL) were collected. FITC-labeled peptides were dissolved to a concentration of 50 μM using cell culture mediums and the FITC-labeled anti-HER2 antibody (eBioscience) was used with the recommended dilution ratio (1:20). The collected cells were incubated with FITC-labeled peptides on ice for 20 minutes. The non-bound peptides were rinsed with PBS for three times. The whole assay was performed on ice with minimal light exposure. A Beckman Quanta SC flow cytometry was used to sort cells. Cell culture medium was used as the control. Each flow cytometry analysis was repeated for three times.
For immunocytochemistry (ICC), approximately 1×105 mL-1 cells (1 mL) were seeded into culture dishes and cultured overnight. FITC-labeled peptide (50 μM) with 1 mM Hoechst 33342 was dissolved in cell culture medium, and added to the culture dishes. Then cells were incubated in the solution for 20 minutes at 4℃. Finally, cells were washed three times with cold PBS for observation. For the colocalization analysis, cells were incubated with phycoerythrin (PE) conjugated anti-human HER2 antibody (LS-C213848, 1:20 dilution) as well as FITC-labeled peptide (50 μM) and 1 mM Hoechst 33342 simultaneously. An Olympus FV1000-IX81 confocal-laser scanning microscope was used for confocal fluorescence imaging. For FITC, A FV5-LAMAR 488 nm laser was used as the excitation source, and emission was collected between 520 and 620 nm. Hoechst 33342 was excited by a FV5- LD405-2 405 nm laser and collected within the range of 422 to 472 nm. PE was excited by a FV10-LD559 559 nm laser with an emission maximum of 573 nm. All parameters of the microscope were set to be the same for observations of different samples to allow comparisons of the binding ability of different peptides.
The toxicity of pep27 and pep27-24M against SKBR3 breast cancer cells and 3T3 fibroblast cells was investigated by MTT assay. Cells resuspended at a concentration of 3 × 104 cells/mL in fresh cell culture medium were seeded into 96-well plates at 3 × 103 cells/well and cultured for 24 h with 5% CO2 at 37 °C. Then, the medium was discarded and replaced with the fresh one containing pep27 or pep27-24M at the final concentration from 1 nM to 100 μM and cultured for another 24 hours. The MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide, obtained from Sigma USA) solution was added to each well and the plates were incubated at 37°C for 4 hours. Afterwards, the MTT solution was removed and 200 µL of dimethyl sulfoxide (DMSO) was added to each well. The optical density (OD) of the cells treated with and without (control) peptides was measured by using an ELISA reader at 570 nm after 10 minutes of vibration mixing.
In Vivo and Ex Vivo Fluorescence Imaging
All animal experiments were conducted in compliance with the guide for the care and use of laboratory animals of Beijing University Animal Study Committee's requirements. The Beijing University Animal Study Committee approved the experiments. 1 × 107 SKBR3 cells were injected by subcutaneously (s.c.) into the right flank of the 6-7 week-old female BALB/c nude mice to establish xenografted tumors. After tumors were established, tumor size was measured periodically using calipers, and mice with tumors of 6-8 mm in diameter were selected for the following small animal experiments. Cy5.5-NHS purchased from Lumiprobe was used to label peptides. The conjugation of peptides and Cy5.5-NHS was carried out following the protocol provided by Lumiprobe and the crude product was purified by semi-preparative reverse phase HPLC. Either Cy5.5-peptide (1 μM, 200 μL) or the control Cy5.5 (1 μM, 200 μL) was intravenously administered into tumor-bearing nude mice via the tail vein. Half an hour after the injection, the mice were anesthetized and placed into the small animal in vivo imaging system (CRI Maestro 2) and scanned for the Cy5.5 signals. For each peptide and control, at least three mice were used. Near-infrared (NIR) fluorescence imaging of nude mice bearing subcutaneous tumor were taken with an exposure time of 50 ms using the Cy5.5 filter sets (excitation: 673 nm, emission: 707 nm) and the intensities were quantified using the same software. Then the nude mice were sacrificed and the main organs as well as tumors were harvested. The fluorescence images of these organs were individually taken as above.
Results and discussion
Structure analysis of HER2/Z(HER2:342) complex revealed key residues on the interface
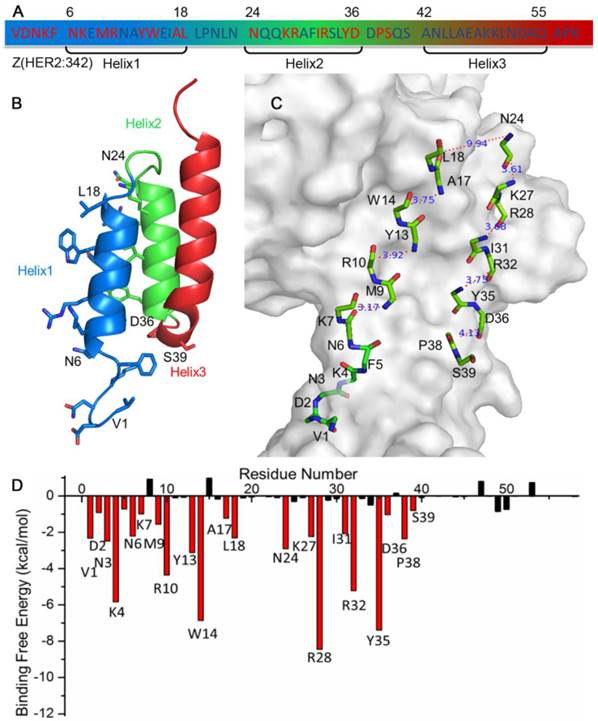
Z(HER2:342), a 58 amino acids affibody, has three helixes: helix1 (Asn6-Leu18), helix2 (Asn24-Asp36) and helix3 (Ala42-Gln55) (Figure 1A,B). In order to find the key residues for HER2 and Z(HER2:342) interaction, molecular dynamics (MD) simulation was carried out based on the crystal structure of Z(HER2:342) binding to HER2 (PDB ID: 3MZW) reported by Eigenbrot et al. [16]. MM/GBSA free energy was then calculated based on 500 snapshots from 7 to 10 ns MD simulation trajectories of each complex by using the mm_pbsa program in AMBER12 [38]. Details of MD simulation and MM/GBSA free energy calculation were described in Materials and Methods. The results showed that the predicted binding free energy between Z(HER2:342) and HER2 is -95.63 kcal/mol. Although, the electrostatic (ΔEele) contribution is the most strongest (-171.57 kcal/mol), the net electrostatic (ΔGGB +ΔEele) contribution is unfavorable for the binding. Therefore, the van der Waals (ΔEvdw) contribution is the main component (Table 1).
Sequence (A), structure (B, C) and binding free energy decomposition analysis (D) of affibody Z(HER2:342) in the HER2/Z(HER2:342) complex. Residues highlighted in red (A, D) are 22 key residues for HER2 binding, while the less contribution and unfavorable residues were colored in black (A, D). Side chain (B) and backbone (C) of these 22 key residues were shown in stick in the structure, and the distances between the backbone carboxyl carbon and amino nitrogen of each gap were calculated and shown in panel C.
Binding free energies and individual energy terms of complexes of HER2 and Z(HER2:342) or the designed peptides calculated by MM/GBSA (kcal/mol)
| ΔEvdw | ΔEele | ΔGGB | ΔGSA | -TΔS | ΔGpred | |
|---|---|---|---|---|---|---|
| HER2/Z(HER2:342) | -126.77±6.63 | -171.57±11.86 | 183.40±11.04 | -19.16±0.64 | 38.47±3.55 | -95.63±5.01 |
| HER2/pep32 | -88.38±10.07 | -238.11±10.85 | 238.91±10.01 | -14.32±1.19 | 41.52±4.21 | -60.37±6.80 |
| HER2/pep27 | -100.49±6.63 | -275.80±14.03 | 274.42±13.08 | -15.08±0.75 | 40.93±5.01 | -76.02±5.79 |
| HER2/pep26 | -86.34±5.03 | -304.28±10.56 | 300.90±9.57 | -13.13±0.57 | 39.24±3.99 | -63.61±4.44 |
| HER2/pep23 | -62.15±5.61 | -259.91±15.98 | 256.15±14.63 | -10.71±0.71 | 34.11±4.18 | -42.50±4.66 |
ΔEvdw, van der Waals contribution; ΔEele, electrostatic contribution; ΔGGB, the polar contribution of desolvation; ΔGSA, nonpolar contribution of desolvation; -TΔS, the conformational entropy at temperature T; ΔGpred, the total binding free energy.
To test whether MD simulation and MM/GBSA free energy calculation used in this study can reflect actual binding affinity, two similar HER2 affibody complexes: HER2/Zwt and HER2/ZHER2:4 were also simulated using same parameters (see Supplementary Material). HER2/Zwt was constructed by replacing Z(HER2:342) in the HER2/Z(HER2:342) complex with Zwt (PDB ID: 1Q2N [48]). HER2/ZHER2:4 was constructed based on HER2/Z(HER2:342) by changing to different residues according to the sequence. Both constructions were carried out by PyMOL [49]. For all three complexes, Root-Mean-Square Deviation (RMSD) between the initial structure and simulated ones with time for three HER2 and ligands complexes converged after ∼500 ps (Figure S1). Our calculation of the binding free energies of HER2/Zwt, HER2/ZHER2:4 and HER2/Z(HER2:342) are -42.33, -82.84 and -95.63 kcal/mol, respectively. The trend of free energy change is consistent with the experimentally measured dissociation constant (KD) (Table S1) that lower free energy corresponds to lower KD. This indicates that when the receptor remains the same and the structure of ligands are very close, MD simulation and MM/GBSA free energy calculation can reliably reflect the relative binding affinity of the ligand. Based on this, we set out to design peptides and select peptides based on the binding free energy calculated from MD simulations.
The binding free energy was then decomposed to study the contribution of each residue in the receptor and ligand interactions (see Materials and Methods). The analysis indicated that residues Val1-Lys7, Met9-Arg10, Tyr13-Trp14, Ala17-Leu18, Asn24, Lys27-Arg28, Ile31-Arg32, Tyr35-Asp36 and Pro38-Ser39 contribute most to the binding between Z(HER2:342) and HER2 (Figure 1D). These 22 key residues are located at helix1 and helix2, but none in helix3. We can see that these key residues are made up by residues with an interval of 3-4 of the sequence, locating on the side of both helices facing HER2.
Pep27 has the lowest binding energy to HER2 among all designed peptides
To design a short peptide with high affinity based on Z(HER2:342), we decided to eliminate residues that are not interacting with HER2, and replace them with proper number of glycines, so that interactions between the key residues and HER2 remained. The numbers of glycine residue needed to link the gaps between key residues were estimated by calculating the distance between the backbone carboxyl carbon of a previous residue and amino nitrogen of the next residue for each gap. As shown in Figure 1C, most of these distances were between 3Å and 4Å which can be linked by one glycine residue. The only exception is that Leu18 is 9.94 Å away from Asn24, and therefore three glycine residues were required. Based on these calculations, a 32-mer peptide named pep32 (Figure 2A) was derived and HER2/pep32 complex was constructed based on the crystal structure of HER2/Z(HER2:342) complex (PDB code: 3MZW) by PyMOL [49], by keeping the original receptor-ligand interactions.
Alignment of sequences of pep23, pep26, pep27, pep32 and Z(HER2:342), all glycines that linked the gaps among key residues were colored in brown (A); Backbone RMSDs as a function of time for the initial and successive structures of HER2/peptides complexes in MD trajectories (B-E). (B) HER2/pep32; (C) HER2/pep27; (D) HER2/pep26; (E) HER2/pep23. MD simulation for each complex was performed three times and shown in each panel.
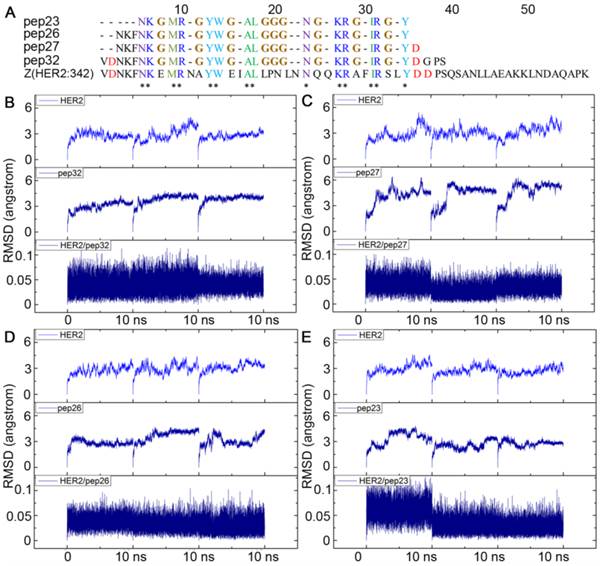
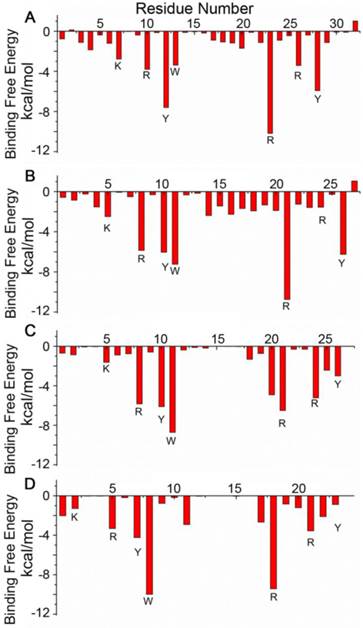
The designed pep32 was more extended without the original helical turns in Z(HER2:342). This might raise the concern of peptide structure stability. To check this, MD simulations were carried out three times. Figure 2B showed the RMSD values of the backbone atoms calculated from the initial structure and successive simulated structures became stable after about 500 ps in all three MD trajectories, indicating convergence of peptide structure toward an equilibrium state. The binding free energy between pep32 and HER2 is -60.37 kcal/mol which is higher than that of Z(HER2:342)/HER2 system (Table 1). The free energy decomposition analysis showed the contributions of residues Val1-Asp2pep32 and Gly30-Pro31-Ser32pep32 are very small, even unfavorable for HER2 binding (Figure 3A). With the purpose to get a shorter peptide, these five unfavorable residues were deleted from pep32, resulting a 27-mer named pep27. Similarly, MD simulation, MM/GBSA binding free energy calculation and decomposition analysis was applied to pep27/HER2 complex. HER2/pep27 has a binding free energy of -76.02 kcal/mol (Table 1) which is lower than HER2/pep32, indicating a higher affinity of pep27 binding to HER2. Figure 2C shows convergence of peptide structure, suggesting that the peptide is stable in the complex. Key residues shown in Figure 3B indicated that the last residue Asp27pep27 was unfavorable for HER2 binding. Similarly, residue Asp27pep27 was deleted from pep27 and pep26 was obtained. However, the binding free energy of HER2/pep26 was -63.61 kcal/mol (Table 1), higher than HER2/pep27 but lower than HER2/pep32. Finally, the pep23 was designed by deleting the first three residues Asn1-Lys2-Phe3pep26 which showed less contribution in pep26 (Figure 3C), but the binding free energy increased to -42.50 kcal/mol (Table 1 and Figure 3D). We proposed that although these terminal residues show less contribution to receptor-ligand interactions, they might function as stabilizing the peptide and thus facilitate binding. Although we would like to get a peptide shorter than 27 residues, there is a trade-off of rather big increase of free binding energy after deletion of residues. As shown above, a significant increase of free binding energy indicated low affinity of the ligand, therefore we decided to select pep27 for further analysis.
Binding free energy decomposition for each residue of the ligands in the four complexes: HER2/pep32 (A), HER2/pep27 (B), HER2/pep26 (C) and HER2/pep23 (D).
Mutation based on pep27
To improve affinity of pep27 against HER2, we examined the structure of the complex to design single mutations. The properties of amino acids in both HER2 and peptide, as well as the space among the interactions were considered. Basic rules were that mutations should favor electrostatic and van der Waals interactions, without causing steric overlap. Finally a list of residues of the pep27 and their mutations were made, including residues with less contributions to free energies (Phe3Glu, Gly22Met and Arg24Met), and unfavorable residues (Asp27Met, Asp27Asn and Asp27Arg). Residues with more contribution were also tried with amino acids of the same property: such as Tyr10Trp and Trp11Tyr to see whether there is improvement of binding. The single mutated sequences and binding free energies were shown in the Supplementary Material (Figure S2 and Table S2). Among the eight mutants, the binding free energy of pep27-24M is the lowest and is expected to have a tight binding to HER2. Two other mutants, pep27-27N and pep27-27R, were almost the same with pep27. From the results of computational simulation and analysis, pep27 and pep27-24M were selected for experimental validation using cells and animal models.
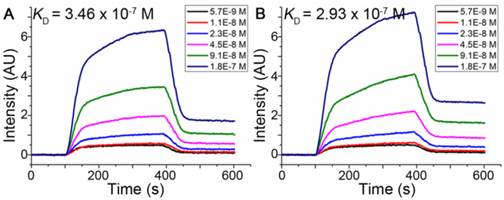
Dissociation constants (KD) of pep27 and pep27-24M measured by SPRi
Surface Plasmon Resonance imaging (SPRi), an acceptable method to estimate interactions of macromolecules for disease diagnosis, drug discovery and peptide screening [50-54], was used to estimate dissociation constants of peptides binding to HER2 as described in Methods. The dissociation constant was calculated from kinetic constants obtained by curve-fitting association and dissociation rates to real-time binding and washing data. The results shown in Figure 4 indicated that the dissociation constants (KD) of the pep27 and pep27-24M with HER2 protein were 346 nmol/L and 293 nmol/L, respectively. The SPRi results were consistent with the computational simulation results that peptide pep27-24M has a lower binding free energy. Compared to ZHER2:4 [55] which has a dissociation rate constant (kd) of around 9.9 × 10-3 s-1, kd of pep27 and pep27-24M are 10.7 × 10-3 and 12.1 × 10-3 s-1, which are close. On the contrary, the association rate showed much difference between ZHER2:4 and peptides (Table S3).
SPRi detection of the binding affinity of pep27 (A) and pep27-24M (B) toward HER2.
Validation of affinity and specificity of peptides to HER2 high expression cells
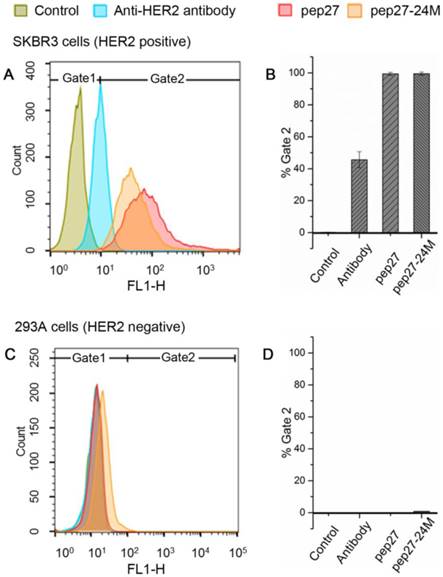
Human breast cancer cell line SKBR3 has a high expression of HER2 proteins and adenocarcinoma cell and human embryonic kidney cell line 293A has a low expression of HER2 proteins. We performed flow cytometry analysis for pep27 and pep27-24M on both cells as described in Methods. As shown in Figure 5, for SKBR3 cells, 99.4% and 99.5% of the cells incubated with FITC-labeled pep27 and pep27-24M respectively have higher intensity than the control. However, for 293A cells, less than 1% cells have higher fluorescence intensity than the control background. This result suggests high binding affinity and specificity of pep27 and pep27-24M to HER2 high expression cells. It is surprising to see that both peptides bind better than anti-HER2 antibody used in this experiment. Unfortunately, the KD value of this antibody is not available for us to make a comparison.
Flow cytometry analysis showed pep27 and pep27-24M have high affinity and specificity for HER2 high expression cells. The fluorescence intensities of cells bound with peptides or antibody for SKBR3 and 293A cell lines are shown in A and C, respectively, and the binding percentages in two cell lines are plotted as bars in panel B and D, respectively (n = 3).
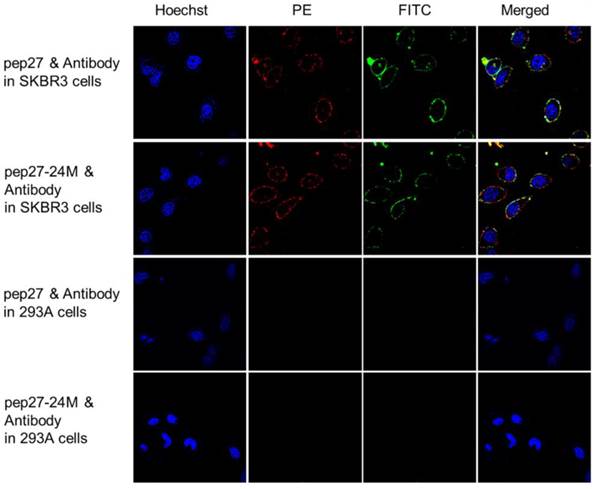
Immunocytochemistry analyses were then performed using 6 cell lines expressing different levels of HER2: SKBR3 has high expression, MCF-7 and MDA-MB-231 medium, and 468 and 293A has low expression [46, 47]. Figure S3 showed that both peptides labeled with FITC bound strongly to SKBR3 cells; weakly to MCF-7 and MDA-MB-231 cells and no signals in 468 and 293A cells. PE labeled anti-HER2 antibody raised against sequence 30-59 of the N-terminal region of human HER2 (LS-C213848) together with FITC labeled peptides were used in colocalization analysis using immunocytochemistry staining. Figure 6 indicated that both peptides (labeled with FITC in green color) and anti-HER2 antibody (labeled with PE in red color) have strong fluorescence signals on SKBR3 cells. The fluorescence signals overlapped after merging and colocalized on cell surface. All these results confirmed that pep27 and pep27-24M can bind to the extracellular domain of HER2 protein at the cellular level.
Colocalization analysis of peptides (labeled with FITC, green) and anti-HER2 antibody (labeled with PE, red) in high (SKBR3) and low (293A) expression of HER2 cell lines. Both peptides showed significant fluorescence signals and overlap in SKBR3 cell surface, but no signals were detected in 293A cells.
Confirmation of affinity and specificity of peptides to HER2 positive tumors
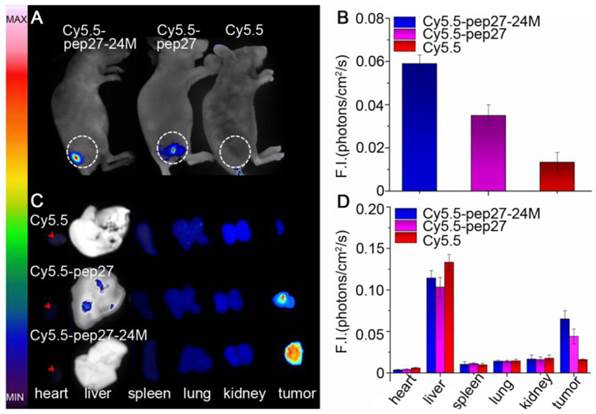
Subsequently, in vivo and ex vivo studies were conducted to investigate the affinity and specificity of peptides to HER2 positive tumors. Nude mice were inoculated with SKBR3 human breast cancer cells to generate xenografted tumors of 6-8 mm in diameters. Cy5.5 labeled peptides (Cy5.5-pep27, Cy5.5-pep27-24M) and control Cy5.5 were then injected into the tail vein of mice (see Methods for details) and in vivo tumor detection and imaging were carried out half an hour after the injection. Fluorescence images of nude mice were acquired using the small animal in vivo imaging system (CRI Maestro 2) as described in Methods. Figure 7A showed clear differences of the tumor images of mice with Cy5.5-pep27-24M or Cy5.5-pep27 injected with those of the control mice. With data from three mice in each group, Figure 7B indicated that the intensity of signals increased 4.43-fold for pep27-24M and 2.63-fold for pep27 as compared to the control. After in vivo imaging, the nude mice were sacrificed and the main organs as well as tumors were harvested and the fluorescence images were individually taken. We saw that tumors targeted with pep27-24M and pep27 have higher fluorescence signals than controls and other normal organs except liver. (Figure 7C and 7D). This is consistent with the in vivo results that pep27 and pep27-24M have high specificity and affinity for HER2 positive tumors. Livers have much high signals for all the three labels, with the control Cy5.5 as much intense as those peptide linked Cy5.5. This is consistent with the previous report by Hue et. al that there is a high distribution of free Cy5.5 dye in the liver within day one post injection [56].
In vivo and ex vivo imaging of tumor targeting by pep27 and pep27-24M: (A) in vivo fluorescence imaging of pep27 and pep27-24M to tumor; (C) ex vivo fluorescence imaging of tumor accumulation and biodistribution; and (B, D) quantification of the fluorescence signals in vivo and ex vivo. Fluorescence intensity was measured in terms of counts/energy/area and is presented as an average (n = 3).
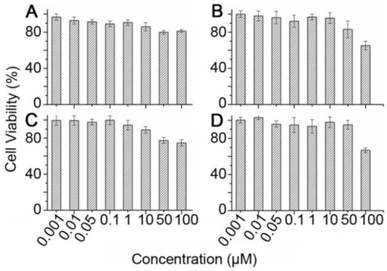
The high accumulation of Cy5.5-pep27-24M and Cy5.5-pep27 in the liver raised the concern of the toxicity of these peptides. To test this, in vitro cytotoxicity analyses of pep27 and pep27-24M against SKBR3 and 3T3 cells were measured by MTT assay. Cell viability at different concentrations of peptides in Figure 8 suggested that both peptides have no significant cytotoxicity at low concentrations (1 nM-10 μM) and low cytotoxicity at high concentration (50 μM-100 μM) in both cell lines. Compared to 1μM concentration used in the above imaging experiments, both peptides should be rather safe to be used as imaging probe.
The in vitro cytotoxicity assay of pep27 (A) and pep27-24M (C) against SKBR3 cells, pep27 (B) and pep27-24M (D) against 3T3 cells tested by MTT assay (n = 3).
Conclusions
Based on affibody Z(HER2:342), we designed peptides at half size of Z(HER2:342) to targeted at HER2 protein with a combination protocol of molecular dynamics modeling, MM/GBSA binding free energy calculations, and binding free energy decomposition analysis. By this method, when the receptor-ligands interactions are similar with only a few residue differences, MM/GBSA free energy can be used to reflect the relative affinity of peptides binding, that is, lower free energy corresponds to tighter binding. We obtained two 27-mer peptides named pep27 and pep27-24M with dissociation constants (KD) of 346 nmol/L and 293 nmol/L, respectively. Both peptides showed high affinity and specificity of binding to the extracellular domain of HER2 proteins by immunocytochemistry and flow cytometry analyses. Furthermore, in vivo imaging showed both peptides have strong affinity and high specificity for HER2 positive tumors. In the meantime, these peptides showed low cytotoxicity to SKBR3 cells even at the concentration 50 times higher than the one used for imaging experiments.
Because most residues responsible for HER2/Z(HER2:342) interactions were unchanged and have low binding free energy, both peptides should dock to HER2 in a much similar way as Z(HER2:342). These novel designed peptides with a smaller size than Z(HER2:342) may provide alternative probes to improve HER2 positive breast cancer early detection, diagnosis, and targeted therapy. In addition, our study proved that MM/GBSA binding free energy calculations based on MD simulation can be used to design and guide the selection of high affinity peptides, saving laborious wet-lab work.
Supplementary Material
Supplementary Tables S1-S3. Supplementary Figures S1-S3.
Abbreviations
HER2: human epidermal growth factor receptor 2; MD: molecular dynamics; SPRi: Surface Plasmon Resonance Imaging; KD: dissociation constants; EGFR/ERBB: epidermal growth factor receptor; RTK: receptor tyrosine kinases; MM: molecular mechanics; PME: Particle Mesh Ewald; ∆EvdW: van der Waals energy; ∆Eele: electrostatic energy; GB: generalized born; SASA: solvent accessible surface area; ∆GGB: polar contribution of desolvation free energy; ∆GSA: nonpolar solvation contribution; SPPS: solid phase peptide synthesis; HBTU: 2-(1H-benzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate; TFA: Trifluoroacetic acid; FITC: fluorescein 5-isothiocyanate; NMM: N-Methyl morpholine; DMF: N,N-dimethylformamide; PBS: phosphate buffer saline; ICC: immunocytochemistry; PE: phycoerythrin; MTT: 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide; DMSO: dimethyl sulfoxide; OD: optical density; s.c.: subcutaneously; NIR: Near-infrared.
Acknowledgements
This work is supported by Chinese Academy of Science (100 Talents Program of the Chinese Academy of Sciences: Y4362911ZX), Beijing Natural Science Foundation Beijing Natural Science Foundation (5142021) and National Natural Science Foundation of China (No. 31270875, 31470049). We express our gratitude to Dr. Chen Wang and Dr. Yanlian Yang (National Center for Nanoscience and Technology of China) for providing SKBR3 cell line.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Yarden Y, Sliwkowski MX. Untangling the ErbB signalling network. Nat Rev Mol Cell Biol. 2001;2:127-37
2. Burgess AW, Cho H-S, Eigenbrot C, Ferguson KM, Garrett TPJ, Leahy DJ. et al. An Open-and-Shut Case? Recent Insights into the Activation of EGF/ErbB Receptors. Mol Cell. 2003;12:541-52
3. Holbro T, Civenni G, Hynes NE. The ErbB receptors and their role in cancer progression. Exp Cell Res. 2003;284:99-110
4. Yarden Y. The EGFR family and its ligands in human cancer: signalling mechanisms and therapeutic opportunities. Eur J Cancer. 2001;37(Suppl 4):S3-S8
5. Slamon DJ, Clark GM, Wong SG, Levin WJ, Ullrich A, McGuire WL. Human breast cancer: correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science. 1987;235:177-82
6. Slamon DJ, Godolphin W, Jones LA, Holt JA, Wong SG, Keith DE. et al. Studies of the HER-2/neu proto-oncogene in human breast and ovarian cancer. Science. 1989;244:707-12
7. Seshadri R, Firgaira FA, Horsfall DJ, McCaul K, Setlur V, Kitchen P. Clinical significance of HER-2/neu oncogene amplification in primary breast cancer. The South Australian Breast Cancer Study Group. J Clin Oncol. 1993;11:1936-42
8. Gabos Z, Sinha R, Hanson J, Chauhan N, Hugh J, Mackey JR. et al. Prognostic Significance of Human Epidermal Growth Factor Receptor Positivity for the Development of Brain Metastasis After Newly Diagnosed Breast Cancer. J Clin Oncol. 2006;24:5658-63
9. Carter P, Presta L, Gorman CM, Ridgway JB, Henner D, Wong WL. et al. Humanization of an anti-p185HER2 antibody for human cancer therapy. Proc Natl Acad Sci U S A. 1992;89:4285-9
10. Nahta R, Esteva FJ. Herceptin: mechanisms of action and resistance. Cancer Lett. 2006;232:123-38
11. Tolaney S. New HER2-positive targeting agents in clinical practice. Curr Oncol Rep. 2014;16:013-0359
12. Schroeder RL, Stevens CL, Sridhar J. Small molecule tyrosine kinase inhibitors of ErbB2/HER2/Neu in the treatment of aggressive breast cancer. Molecules. 2014;19:15196-212
13. Nilsson B, Moks T, Jansson B, Abrahmsen L, Elmblad A, Holmgren E. et al. A synthetic IgG-binding domain based on staphylococcal protein A. Protein Eng. 1987;1:107-13
14. Lee SB, Hassan M, Fisher R, Chertov O, Chernomordik V, Kramer-Marek G. et al. Affibody molecules for in vivo characterization of HER2-positive tumors by near-infrared imaging. Clin Cancer Res. 2008;14:3840-9
15. Tolmachev V, Orlova A, Pehrson R, Galli J, Baastrup B, Andersson K. et al. Radionuclide therapy of HER2-positive microxenografts using a 177Lu-labeled HER2-specific Affibody molecule. Cancer Res. 2007;67:2773-82
16. Eigenbrot C, Ultsch M, Dubnovitsky A, Abrahmsen L, Hard T. Structural basis for high-affinity HER2 receptor binding by an engineered protein. Proc Natl Acad Sci U S A. 2010;107:15039-44
17. Cho HS, Mason K, Ramyar KX, Stanley AM, Gabelli SB, Denney DW Jr. et al. Structure of the extracellular region of HER2 alone and in complex with the Herceptin Fab. Nature. 2003;421:756-60
18. Franklin MC, Carey KD, Vajdos FF, Leahy DJ, de Vos AM, Sliwkowski MX. Insights into ErbB signaling from the structure of the ErbB2-pertuzumab complex. Cancer Cell. 2004;5:317-28
19. Ekerljung L, Lindborg M, Gedda L, Frejd FY, Carlsson J, Lennartsson J. Dimeric HER2-specific affibody molecules inhibit proliferation of the SKBR-3 breast cancer cell line. Biochem Biophys Res Commun. 2008;377:489-94
20. Baum RP, Prasad V, Muller D, Schuchardt C, Orlova A, Wennborg A. et al. Molecular imaging of HER2-expressing malignant tumors in breast cancer patients using synthetic 111In- or 68Ga-labeled affibody molecules. J Nucl Med. 2010;51:892-7
21. Wikman M, Steffen AC, Gunneriusson E, Tolmachev V, Adams GP, Carlsson J. et al. Selection and characterization of HER2/neu-binding affibody ligands. Protein Eng Des Sel. 2004;17:455-62
22. Steffen AC, Orlova A, Wikman M, Nilsson FY, Stahl S, Adams GP. et al. Affibody-mediated tumour targeting of HER-2 expressing xenografts in mice. Eur J Nucl Med Mol Imaging. 2006;33:631-8
23. Orlova A, Magnusson M, Eriksson TL, Nilsson M, Larsson B, Hoiden-Guthenberg I. et al. Tumor imaging using a picomolar affinity HER2 binding affibody molecule. Cancer Res. 2006;66:4339-48
24. Wang J, Hou T, Xu X. Recent Advances in Free Energy Calculations with a Combination of Molecular Mechanics and Continuum Models. Curr Comput Aided Drug Des. 2006;2:287-306
25. Kollman PA, Massova I, Reyes C, Kuhn B, Huo S, Chong L. et al. Calculating Structures and Free Energies of Complex Molecules: Combining Molecular Mechanics and Continuum Models. Acc Chem Res. 2000;33:889-97
26. Homeyer N, Gohlke H. Free Energy Calculations by the Molecular Mechanics Poisson-Boltzmann Surface Area Method. Mol Inf. 2012;31:114-22
27. Gohlke H, Kiel C, Case DA. Insights into protein-protein binding by binding free energy calculation and free energy decomposition for the Ras-Raf and Ras-RalGDS complexes. J Mol Biol. 2003;330:891-913
28. Hou T, Xu Z, Zhang W, McLaughlin WA, Case DA, Xu Y. et al. Characterization of domain-peptide interaction interface: a generic structure-based model to decipher the binding specificity of SH3 domains. Mol Cell Proteomics. 2009;8:639-49
29. Hou T, Zhang W, Case DA, Wang W. Characterization of domain-peptide interaction interface: a case study on the amphiphysin-1 SH3 domain. J Mol Biol. 2008;376:1201-14
30. Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H. et al. Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23:2947-8
31. Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H. et al. The Protein Data Bank. Nucleic Acids Res. 2000;28:235-42
32. Duan Y, Wu C, Chowdhury S, Lee MC, Xiong G, Zhang W. et al. A point-charge force field for molecular mechanics simulations of proteins based on condensed-phase quantum mechanical calculations. J Comput Chem. 2003;24:1999-2012
33. Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. Comparison of simple potential functions for simulating liquid water. J Chem Phys. 1983;79:926-35
34. Ryckaert J-P, Ciccotti G, Berendsen HJC. Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J Comput Phys. 1977;23:327-41
35. Darden T, York D, Pedersen L. Particle mesh Ewald: An N [center-dot] log(N) method for Ewald sums in large systems. J Chem Phys. 1993;98:10089-92
36. Tsui V, Case DA. Theory and applications of the generalized Born solvation model in macromolecular simulations. Biopolymers. 2000;56:275-91
37. Weiser Jr, Shenkin PS, Still WC. Approximate atomic surfaces from linear combinations of pairwise overlaps (LCPO). J Comput Chem. 1999:217-30
38. Case DA, Cheatham TE 3rd, Darden T, Gohlke H, Luo R, Merz KM Jr. et al. The Amber biomolecular simulation programs. J Comput Chem. 2005;26:1668-88
39. Sun H, Li Y, Shen M, Tian S, Xu L, Pan P. et al. Assessing the performance of MM/PBSA and MM/GBSA methods. 5. Improved docking performance using high solute dielectric constant MM/GBSA and MM/PBSA rescoring. Phys Chem Chem Phys. 2014;16:22035-45
40. Hou T, Wang J, Li Y, Wang W. Assessing the performance of the MM/PBSA and MM/GBSA methods. 1. The accuracy of binding free energy calculations based on molecular dynamics simulations. J Chem Inf Model. 2011;51:69-82
41. Cui W, Cheng YH, Geng LL, Liang DS, Hou TJ, Ji MJ. Unraveling the allosteric inhibition mechanism of PTP1B by free energy calculation based on umbrella sampling. J Chem Inf Model. 2013;53:1157-67
42. Hou T, Yu R. Molecular dynamics and free energy studies on the wild-type and double mutant HIV-1 protease complexed with amprenavir and two amprenavir-related inhibitors: mechanism for binding and drug resistance. J Med Chem. 2007;50:1177-88
43. Geng L, Gao J, Cui W, Tang Y, Ji M, Chen B. Computational insights into the selectivity mechanism of APP-IP over matrix metalloproteinases. J Comput Aided Mol Des. 2012;26:1327-42
44. Gohlke H, Kiel C, Case DA. Insights into Protein-Protein Binding by Binding Free Energy Calculation and Free Energy Decomposition for the Ras-Raf and Ras-RalGDS Complexes. J Mol Biol. 2003;330:891-913
45. Onufriev A, Bashford D, Case DA. Exploring protein native states and large-scale conformational changes with a modified generalized born model. Proteins. 2004;55:383-94
46. Neve RM, Chin K, Fridlyand J, Yeh J, Baehner FL, Fevr T. et al. A collection of breast cancer cell lines for the study of functionally distinct cancer subtypes. Cancer Cell. 2006;10:515-27
47. Subik K, Lee JF, Baxter L, Strzepek T, Costello D, Crowley P. et al. The Expression Patterns of ER, PR, HER2, CK5/6, EGFR, Ki-67 and AR by Immunohistochemical Analysis in Breast Cancer Cell Lines. Breast Cancer. 2010;4:35-41
48. Zheng D, Aramini JM, Montelione GT. Validation of helical tilt angles in the solution NMR structure of the Z domain of Staphylococcal protein A by combined analysis of residual dipolar coupling and NOE data. Protein Sci. 2004;13:549-54
49. Schrodinger LLC. The PyMOL Molecular Graphics System, Version 1.3r1. 2010.
50. Zhu L, Wang K, Cui J, Liu H, Bu X, Ma H. et al. Label-free quantitative detection of tumor-derived exosomes through surface plasmon resonance imaging. Anal Chem. 2014;86:8857-64
51. Gorodkiewicz E, Guszcz T, Roszkowska-Jakimiec W, Kozlowski R. Cathepsin D serum and urine concentration in superficial and invasive transitional bladder cancer as determined by surface plasmon resonance imaging. Oncol Lett. 2014;8:1323-7
52. Villiers MB, Cortay JC, Cortes S, Bloquel B, Brichler S, Brakha C. et al. Protein-peptide arrays for detection of specific anti-hepatitis D virus (HDV) genotype 1, 6, and 8 antibodies among HDV-infected patients by surface plasmon resonance imaging. J Clin Microbiol. 2015;53:1164-71
53. Wang W, Zhang D, Wei Z, Wang Z, Bu X, Yang S. et al. Label-free detection microarray for novel peptide ligands screening base on MS-SPRi combination. Talanta. 2015;134:705-11
54. Wang W, Wei Z, Zhang D, Ma H, Wang Z, Bu X. et al. Rapid screening of peptide probes through in situ single-bead sequencing microarray. Anal Chem. 2014;86:11854-9
55. Steffen AC, Wikman M, Tolmachev V, Adams GP, Nilsson FY, Stahl S. et al. In vitro characterization of a bivalent anti-HER-2 affibody with potential for radionuclide-based diagnostics. Cancer Biother Radiopharm. 2005;20:239-48
56. Hue JJ, Lee HJ, Jon S, Nam SY, Yun YW, Kim JS. et al. Distribution and accumulation of Cy5.5-labeled thermally cross-linked superparamagnetic iron oxide nanoparticles in the tissues of ICR mice. J Vet Sci. 2013;14:473-9
Author contact
![]() Corresponding authors: Qiaojun Fang, National Center for Nanoscience and Technology, Beijing 100190, China. Phone: +86-10-82545562; Fax: +86-10-82545643; E-mail: fangqjcn. Zhiyuan Hu, National Center for Nanoscience and Technology, Beijing 100190, China. Phone: +86-10-82545643; Fax: +86-10-82545643; E-mail: huzycn.
Corresponding authors: Qiaojun Fang, National Center for Nanoscience and Technology, Beijing 100190, China. Phone: +86-10-82545562; Fax: +86-10-82545643; E-mail: fangqjcn. Zhiyuan Hu, National Center for Nanoscience and Technology, Beijing 100190, China. Phone: +86-10-82545643; Fax: +86-10-82545643; E-mail: huzycn.