Impact Factor
 Global reach, higher impact
Global reach, higher impact
Theranostics 2016; 6(11):1740-1752. doi:10.7150/thno.15169 This issue Cite
Research Paper
Ultrasound Molecular Imaging of the Breast Cancer Neovasculature using Engineered Fibronectin Scaffold Ligands: A Novel Class of Targeted Contrast Ultrasound Agent
Lotfi Abou-Elkacem1, Katheryne E. Wilson1, Sadie M. Johnson2, Sayan M. Chowdhury1, Sunitha Bachawal1, Benjamin J. Hackel2, Lu Tian3, Jürgen K. Willmann1 ![]()
1. Department of Radiology, Molecular Imaging Program at Stanford, Stanford University, School of Medicine, Stanford, California, USA;
2. Department of Chemical Engineering and Materials Science, University of Minnesota, Minnesota, USA;
3. Department of Health, Research & Policy, Stanford University, Stanford, California, USA.
Received 2016-2-1; Accepted 2016-5-19; Published 2016-6-28
Abstract

Molecularly-targeted microbubbles (MBs) are increasingly being recognized as promising contrast agents for oncological molecular imaging with ultrasound. With the detection and validation of new molecular imaging targets, novel binding ligands are needed that bind to molecular imaging targets with high affinity and specificity. In this study we assessed a novel class of potentially clinically translatable MBs using an engineered 10th type III domain of human-fibronectin (MB-FN3VEGFR2) scaffold-ligand to image VEGFR2 on the neovasculature of cancer. The in vitro binding of MB-FN3VEGFR2 to a soluble VEGFR2 was assessed by flow-cytometry (FACS) and binding to VEGFR2-expressing cells was assessed by flow-chamber cell attachment studies under flow shear stress conditions. In vivo binding of MB-FN3VEGFR2 was tested in a transgenic mouse model (FVB/N Tg(MMTV/PyMT634Mul) of breast cancer and control litter mates with normal mammary glands. In vitro FACS and flow-chamber cell attachment studies showed significantly (P<0.01) higher binding to VEGFR2 using MB-FN3VEGFR2 than control agents. In vivo ultrasound molecular imaging (USMI) studies using MB-FN3VEGFR2 demonstrated specific binding to VEGFR2 and was significantly higher (P<0.01) in breast cancer compared to normal breast tissue. Ex vivo immunofluorescence-analysis showed significantly (P<0.01) increased VEGFR2-expression in breast cancer compared to normal mammary tissue. Our results suggest that MBs coupled to FN3-scaffolds can be designed and used for USMI of breast cancer neoangiogenesis. Due to their small size, stability, solubility, the lack of glycosylation and disulfide bonds, FN3-scaffolds can be recombinantly produced with the advantage of generating small, high affinity ligands in a cost efficient way for USMI.
Keywords: Ultrasound molecular imaging, breast cancer, vascular endothelial growth factor receptor type 2, fibronectin scaffold, protein engineering, yeast display.
Introduction
Ultrasound molecular imaging (USMI) using molecularly-targeted gas-filled echogenic microbubbles (MBs) as contrast agents is emerging as a promising tool in oncological imaging [1-3]. Molecularly-targeted MBs consist of a gas-filled core, stabilized by a shell. The shell is only a few micrometers in diameter and can be functionalized using various types of ligands to make MBs attach to receptors that are differentially expressed on the neovasculature of different cancer types including breast cancer. Among those receptors differentially expressed in breast cancer is vascular endothelial growth factor receptor type 2 (VEGFR2) [4-6]. VEGFR2 is one of the key regulators of tumor angiogenesis and several studies have shown that targeting VEGFR2 allows detection and monitoring of breast cancer using USMI [7, 8].
With the deeper understanding of cancer biology and the discovery and validation of new receptors differentially expressed on the cancer neovasculature [3, 9, 10], there is a need for a robust platform to allow rapid production of next generation MBs using bio-compatible ligands binding to those receptors with high affinity and specificity. Traditional binding ligands that have been used to functionalize MBs are monoclonal antibodies generated by the hybridoma technology [11]. However, the use of antibodies as binding ligands on MBs can be problematic because of potential immune reactions as well as the time-consuming and high-cost production of antibodies [12]. A potential alternative to antibodies are engineered proteins that could be produced more readily in recombinant bacterial culture with the advantage of generating small, high affinity ligands for molecular imaging in a cost efficient way. One evolving technique is the engineered protein scaffold (EPS) technique that uses a scaffold that can be modified to bind to a specific receptor [13]. Several EPSs have been explored including knottins [14], single-chain variable fragments [15], or nanobodies [16]. Recently, the 10th type III domain of human fibronectin (FN3), has been shown to be a promising platform for designing binding ligands for molecular imaging [17, 18]. The FN3 is a small (~10 kDa), cysteine-free protein scaffold containing a β-sandwich and loop structures, termed BC, DE, and FG, which are structurally similar to antibody complementarity-determining regions. In contrast to antibodies, they are inexpensive to produce in large quantities, because of their relatively small size, solubility, and the lack of glycosylation and disulfide bonds [18-24]. A VEGFR2-targeted FN3-scaffold (FN3VEGFR2) for therapeutic purposes has been shown to be safe in patients in early clinical trials [25-27].
The purpose of our study was to develop and test a molecularly-targeted MB using FN3VEGFR2 for ultrasound imaging of the neovasculature of breast cancer at the molecular level as a proof-of-principle for the future design of next generation MBs targeted at molecular markers beyond VEGFR2.
Materials and Methods
Recombinant production of engineered VEGFR2-binding ligand (FN3VEGFR2)
The engineered FN3VEGFR2 scaffold FN3VEGFR2 containing the specific loop sequences for BC, DE, and FG with high affinity to human (KD: 1.6 ± 0.4 nM) and murine VEGFR2 (KD: 1.3 ± 0.2 nM) was recombinantly expressed in E. coli [25]. In brief, a plasmid was generated from a derivative of pET24-FN3 [18] to encode for FN3VEGFR2 with a C-terminal 6xHis-tag (Fig. 1 A) [25]. An analogous plasmid encoding for a scrambled wild-type FN3 (FN3Scrambled) [19] was used as a non-binding control. The proteins were recombinantly expressed in BL21 E. coli in lysogeny broth upon induction with 0.5 mM isopropyl β-D-thiogalactopyranoside. Cells were lysed by freeze/thaw in 50 mM sodium phosphate, pH 8.0, 5% glycerol, 500 mM sodium chloride, 5 mM 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate (CHAPS), and 25 mM imidazole. After centrifugation at 12,000g for 10 minutes to remove the insoluble fraction, FN3VEGFR2 was purified by metal affinity chromatography using a HisPur™ cobalt resin gravity-flow column (Thermo Scientific, USA) at room temperature. Protein was further purified by reversed-phase high-performance liquid chromatography on a C18 column using a 9-81% gradient of acetonitrile in water with 0.1% trifluoroacetic acid. Protein was lyophilized and resuspended.
A, structure of the FN3-scaffold with the corresponding amino acid sequence of FN3VEGFR2 binder backbone and loop sequences BC (red), DE (green), and FG (blue) with a C-terminal 6xHis-tag (purple). Source: protein data bank (http://www.rcsb.org/pdb/home/home.do); search option with ID: 1TTG. B-D, overview of the overall study design. MB-FN3VEGFR2 was generated by covalently attaching the FN3-scaffold on the surface of MB. C, molecularly-targeted MBs were tested both in vitro and D, in vivo in a transgenic mouse model of breast cancer followed by ex vivo quantitative immunofluorescence of VEGFR2 expression on the tumor neovasculature. USMI signal was measured using the destruction-replenishment technique.
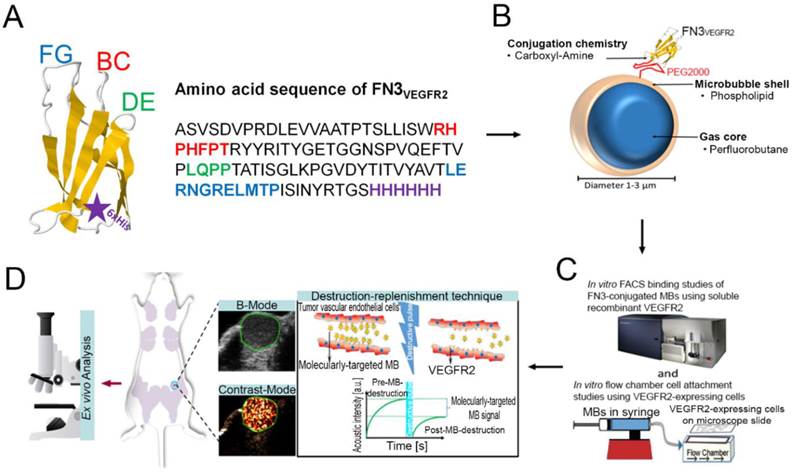
Preparation of VEGFR2-targeted MBs using FN3VEGFR2
Molecularly-targeted MBs were designed as previously described [28-32], using the following compounds: 2-Distearoyl-sn-glycero-3-phosphocholine (DSPC; Avanti Polar Lipids, USA), an active functionalized N-Hydroxysulfosuccinimide-PEG2000-DSPE (DSPE-020GS-NHS, Sunbright NOF America Corporation, USA), and polyoxyethylene-40 stearate (PEG40S; Sigma-Aldrich, USA) at a molar ratio of 8:1:1. 1 mg DSPC was evaporated and the dried lipid film was then hydrated with sterile phosphate-buffered serum (PBS) and mixed with DSPE-020GS-NHS and PEG40S (Sigma-Aldrich, USA) to a final concentration of 1 mg/ml. The lipid mixture was first preheated (55 ºC) and perfluorobutane gas (FluoroMed, L.P., USA) was introduced into the lipid suspension, and subsequently sonicated with a high-frequency, high-power, probe sonicator at 500W for 45 s (QSonica, USA) to generate MBs. Subsequently, the lysine group of the ligand FN3VEGFR2 was conjugated to the active N-hydroxysulfosuccinimide ester of the DSPE-020GS-NHS using a direct one-step carboxyl-amine-conjugation chemistry and attached to the shell of synthesized perfluorobutane-filled phospholipid MBs (Fig. 1 B). All ligands were added in excess amounts (10-fold molar excess) to occupy all binding sites on the MB surface. Unbound FN3VEGFR2 ligand was removed by centrifugation at 300g for 2 min, and MB-FN3VEGFR2-containing supernatant were collected and reconstituted in sterile saline (0.9% sodium chloride). As a negative control MB, FN3Scrambled was coupled to MBs (MB-FN3Scrambled). A second type of control non-targeted MB (MBNon-targeted) was synthesized using the same techniques but without attaching a binding ligand to the MB shell. The mean diameter, concentration, and total particle surface area of all MBs were analyzed using a cell counter and sizer (Multisizer III Coulter Counter; Beckman Coulter). Approximately 90-95% of MBs could be recovered after washing steps.
For comparison purposes, commercially available streptavidin-containing MBs with a mean diameter of 1.5 μm (range, 1-2 μm) [44] (Target-Ready MicroMarker Contrast Agents; VisualSonics, Canada) coupled to a biotinylated anti-VEGFR2 monoclonal antibody (MBVEGFR2) were prepared according to manufacturer's instructions. In brief, lyophilized streptavidin coated MBs were suspended in 1 mL of sterile saline (0.9% sodium chloride) and 6 μg of biotinylated anti-mouse VEGFR2 monoclonal antibody (eBioscience, USA) were incubated with 5 × 107 MBs for 10 minutes at room temperature to allow attachment of the antibodies to the MB shell via biotin-streptavidin interactions. Non-bound antibodies were removed by centrifugation at 300g for 2 min.
Validation of FN3 coupling to the MB surface
Assessment of FN3 ligand conjugation on MB surface by using flow cytometry and microscopy
The successful coupling of both FN3VEGFR2 and FN3Scrambled on the MB shell was confirmed by fluorescence-activated cell sorting flow cytometry (FACS, Becton-Dickinson Biosciences, USA) and microscopy. Synthesized targeted MBs (1 x 105 each) coupled with either FN3VEGFR2 and FN3Scrambled were incubated with an anti-His antibody-AF488 (Thermo Fisher, USA) for 1h. The labeled molecularly-targeted MBs were washed three times by centrifugation at 300g for 2 min and analyzed by FACS. FACS was used to confirm MB ligand coating by fluorescence intensity. Voltage, forward and side light scattering (FSC and SSC) settings were adjusted to detect solely MB populations. 50 μL freshly synthesized MB solutions were diluted with 200 µL PBS prior to each measurement. Subsequent data analysis was done using FlowJo software (Stanford University, CA, USA).
Furthermore, direct visual confirmation of MB size was performed after the samples were prepared using a Zeiss inverted microscope (Axio Imager.M2 Zeiss, Germany). The MB samples were taken directly from the vials and imaged at room temperature. Images were captured in bright-field mode. The FN3 ligand conjugation on the MB surface was confirmed by fluorescence microscopy (Axio Imager.M2 Zeiss, Germany) using anti-His antibody-AF488-labeled molecularly-targeted MBs.
Assessment of ligand purity and conjugation on MB surface
To confirm purity and stable ligand conjugation of FN3 ligands on the MB shell, SDS-PAGE analysis was performed [15]. SDS-PAGE was performed according to standard protocols with a Novex ExCell Sure lock SDS-PAGE Electrophoresis System (Life Technologies, USA). Three hours after MB synthesis, MBVEGFR2, MB-FN3VEGFR2, MB-FN3Scrambled, and MBNon-targeted were washed four times by centrifugation at 300g for 2 min and were dissolved in SDS-loading buffer (Life Technologies, USA) at 94ºC for 1 min in order to release ligands from the surface of MBs. The MB solution was centrifuged and the supernatant was loaded on a gradient gel and then electrophoresed on a 7.5 to 10% Tris gel with Tris-running buffer. After migration, gels were stained with Coomassie stain (Thermo Fisher Scientific, USA) to reveal protein.
In vitro flow cytometry VEGFR2-binding studies of FN3-conjugated MBs using soluble recombinant VEGFR2
FACS analysis was performed in order to assess the binding specificity of molecularly-targeted MBs to a soluble human and mouse VEGFR2 protein (R&D Systems, USA). To confirm successful ligand conjugation of FN3 ligands including His-tag to the MB shell, 1 x 105 MBVEGFR2 (no His-tag), MB-FN3VEGFR2, MB-FN3Scrambled, and MBNon-targeted (no His-tag) were pre-labeled with anti-His antibody-AF488 for 30 minutes at room temperature. The MBs were washed twice by centrifugation at 300g for 2 min. Subsequently, pre-labeled MBs were incubated with 11 nM soluble human and mouse IgG-Fc-conjugated VEGFR2 with 900 μL mouse serum (Sigma-Aldrich, USA) on a benchtop rotator for 40 minutes at room temperature. This was followed by washing by centrifugation at 300g for 2 min and incubation with anti-human IgG-Fc antibody-AF647 (eBioscience, USA) for 30 minutes on ice. This was followed by a final washing by centrifugation at 300g for 2 min. The measured median fluorescence intensity data was analyzed using FlowJo software. A strong correlation of anti-His antibody-AF488-labeled MBs and IgG-Fc antibody-AF647-labeled VEGFR2 indicates positive binding. All experiments were performed in quintuplicate.
In vitro flow chamber cell attachment studies of FN3-conjugated MBs
Binding specificity of MB-FN3VEGFR2 to the target VEGFR2 was also assessed in cell culture experiments under flow shear stress conditions simulating flow in blood capillaries by using a parallel flow chamber experimental set-up. Human umbilical vein endothelial cells (HUVEC) were obtained directly from ATCC (American Type Culture Collection, USA) that performs standard cell line characterizations of mycoplasma, yeast, bacterial and viral contamination. HUVEC were cultivated and activated in sterile vascular cell basal medium supplemented with endothelial cell growth kit-VEGF (ATCC), 10% fetal bovine serum, 100 U/ml penicillin, and 100 μg/ml streptomycin under standard culture conditions (37°C, 5% CO2) for fewer than 6 months after receipt. Cells between passages 2 and 4 were used in all experiments. HUVECs were cultivated in basal medium and were subsequently activated with endothelial cell growth kit-VEGF for 24h to stimulate VEGFR2 cell membrane expression. In addition, mouse VEGFR2-expressing angiosarcoma cells (SVR cells; ATCC) were used for in vitro flow chamber cell attachment studies. SVR cells were cultivated in Dulbecco's modified Eagle's medium with a high concentration of glucose (4.5 g/l) and L-glutamine (Invitrogen, USA), and supplemented with 10% fetal bovine serum and penicillin (100 U/ml) and streptomycin (100 μg/ml). Before flow chamber experiment, FACS analysis was performed in order to confirm VEGFR2 expression of activated HUVEC and SVR cells. The geometric mean fluorescence intensity was determined using FlowJo software (Fig. 3 B). VEGFR2-expressing HUVEC and SVR cells were grown on coated (Sigmacote; Sigma, St Louis, Mo) neutral-charged glass microscope slides (VWR, USA) for 24 hours and mounted on a parallel plate flow chamber (GlycoTech, Rockville, Md). A syringe infusion and withdrawal pump (Genie Plus; Kent Scientific, Torrington, Conn) was used to maintain the flow rate of 0.6 mL/min, corresponding to a wall shear stress rate of 100 sec-1, similar to that in tumor capillaries [33]. The flow chamber cell attachment study was performed as described [10, 34-36]. The glass microscope slides were inverted and positioned in the parallel flow chamber apparatus in order to allow MBs to float and then bind to VEGFR2-expressing cells. In brief, solutions were passed over cells in the following order: PBS for 2 minutes; 5 × 107 of either MBVEGFR2, MB-FN3VEGR2, MB-FN3Srambled, or MBNon-targeted in PBS for 4 minutes; and finally washing with PBS for 2 minutes. The adhered MB number on the HUVEC and SVR cells monolayer was quantified manually by counting attached MBs on HUVEC and SVR cells with a phase-contrast bright-field microscope (Axiovert 25; Carl Zeiss, Thornwood, NY; original magnification, x 100) to assess the number of attached MBs per cell. At least five random fields of view of these slides were immediately imaged. Note that MBs can be visualized as small, rounded particles and were considered to be attached to VEGFR2-expressing cells when there was direct contact with the cells without free floating. Blocking studies were performed to confirm binding specificity of MB-FN3VEGFR2 to VEGFR2. For this purpose, HUVEC and SVR cells were incubated with 80 μg/mL purified FN3VEGFR2 in PBS for 30 minutes at 37°C before flow chamber cell attachment study to block the VEGFR2 receptor. All flow chamber experiments were performed in triplicate.
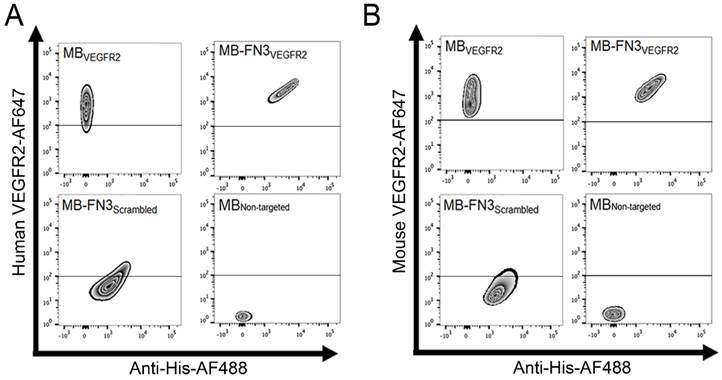
In vitro binding specificity assessment of MB-FN3VEGFR2. A, B binding specificity of MBVEGFR2, and MB-FN3VEGFR2 to a fluorescent soluble human (A) and mouse (B) VEGFR2 was analyzed using FACS. MB-FN3VEGFR2 and MB-FN3Scrambled showed enhanced geometric mean fluorescence intensity which indicates the presence of an anti-His-AF488-antibody binding to FN3-coated MBs (A and B). A correlation between MB-FN3VEGFR2 to the soluble human (A) and mouse (B) AF647-labeled VEGFR2 demonstrated specific binding of FN3VEGFR2 scaffold. As a reference standard, MBVEGFR2 shows enhanced geometric mean fluorescence intensity towards human (A) and mouse (B) AF647-labeled VEGFR2. As a negative control, MB-FN3Scrambled and MBNon-targeted showed no binding to human and mouse VEGFR2. The cut-off was defined based on the background geometric mean fluorescence intensity of MBNon-targeted and MB-FN3Scrambled.
Transgenic Breast Cancer Mouse Model
All experiments were approved by the Institutional Administrative Panel on Laboratory Animal Care. The transgenic mouse model of breast cancer development FVB/N-Tg(MMTV-PyMT)634Mul [8, 37-39] was used (Fig. 1 D). The mammary tissue of this transgenic mouse model progresses through four distinct histological stages from normal mammary tissues, through hyperplasia, to ductal carcinoma in situ (DCIS), and finally invasive breast carcinoma which highly recapitulates human breast cancer. For this study, inguinal mammary glands with invasive breast carcinoma were imaged by USMI (total of 12 female mice (mean age, 7 weeks; range, 4-10 weeks) with 12 mammary tumors). Four control litter mates with normal mammary glands were used as controls.
In vivo ultrasound molecular imaging
Mice were kept anesthetized with 2% isoflurane in oxygen at 2 L/min on a heated stage for 37ºC throughout the ultrasound imaging sessions. In an intra-animal comparison experiments, molecularly-targeted MBs (5x107 MBVEGFR2, MB-FN3VEGFR2, MB-FN3Scrambled, and MBNon-targeted) were injected intravenously through the tail vein of 8 mice in random order to minimize any bias. Between imaging sessions using the four different contrast agents, a waiting interval of 30 min was used to allow for clearance of MBs from the vasculature [40, 41]. Also, at 30 min it was confirmed by USMI that no remaining MBs were present from the previous injection.
In addition, control litter mates with normal mammary glands were scanned as tumor angiogenesis-negative models after the injection of all MB types to assess the contrast enhancement of non-angiogenic microvasculature. To further confirm binding specificity of molecularly-targeted MBs, an in vivo competition experiment was performed. In a subgroup of four breast cancer-bearing mice, in vivo blocking of VEGFR2 by injecting 125µg FN3VEGFR2 via the tail vein was performed in order to block binding of MB-FN3VEGFR2 to its target VEGFR2 [18, 42, 43].
All in vivo imaging studies were performed in contrast mode using a small-animal high resolution ultrasound imaging system (Vevo 2100; VisualSonics, Canada). Images were acquired with a 21-MHz high-resolution linear transducer (MS250, VisualSonics; lateral and axial resolution of 165 μm and 75 μm, respectively), and all imaging parameters (focal length, 8 mm; transmit power, 10%; mechanical index, 0.2; dynamic range, 40 dB and a center frequency of 18MHz) were kept constant during all imaging sessions. The transducer was fixed on a railing system to maintain the acoustic focus at the center of the mammary gland at the level of the largest transverse cross section.
To differentiate the acoustic signal owing to MBs adherent to VEGFR2 and the signal from freely circulating MBs in the bloodstream we used previously described principles of destruction-replenishment techniques [3, 44, 45]. In brief, the imaging signal in the field of view increases after intravenous injection of molecularly-targeted MBs and is composed of signal from attached and freely circulating MBs as well as tissue background signal. After 4 minutes, a high pressure destructive pulse (1-second continuous high-power destructive pulse of 3.7 MPa, transmit power, 100%; mechanical index, 0.63) was applied to destroy all bound and unbound MBs within the beam elevation. After ten seconds to allow freely circulation MBs to replenish into the field of view, an additional set of 200 frames was acquired to measure the signal intensity from the unbound circulating MBs. The difference in imaging signal pre- and post-destruction was calculated corresponding to the signal from attached MBs (see below).
Ultrasound molecular Imaging data analysis
The acoustic imaging signals were analyzed post image acquisition, averaged to compensate breathing motion artifacts by using commercially available analysis software (VevoCQ; VisualSonics). Data analysis was accomplished by selecting a frame with the plane of interest and manually drawing a region of interest (ROI) around the breast cancer-bearing mammary glands or placing ROI in the normal mammary glands of control litter mates. The magnitude of imaging signal (expressed in arbitrary units, a.u.) from attached microbubbles was assessed by calculating an average for pre- and post-destruction imaging signals and subtracting the average post-destruction signal from the average pre-destruction signal using Vevo2100 built-in software (VevoCQ; VisualSonics).
Ex vivo immunofluorescence
Immediately after USMI, all mice were sacrificed and 12 breast cancers and 4 normal mammary breast tissues were excised, fixed with ice-cold acetone, embedded in optimum cutting temperature compound (O.C.T.; Sakura Finetek), frozen on dry ice, and sectioned to 5-10 μm thickness. Slices were incubated for 1h at room temperature in 5% goat serum to block non-specific binding. Sections were co-incubated with a rabbit anti-mouse VEGFR2 antibody (Cell Signaling Technology Inc., Danvers, MA) and rat anti-mouse CD31 antibody (BD Biosciences, San Jose, CA) overnight at 4°C at a dilution of 1:200 and 1:100, respectively, and were visualized by using Cy3-conjugated anti-rabbit and FITC-conjugated anti-rat secondary antibodies, respectively (both at a dilution of 1:200; Jackson Immuno Research Laboratories, West Grove, Pa). Double staining for VEGFR2 and CD31 was performed to confirm co-localization of VEGFR2 on CD31-positive tumor vascular endothelial cells. Fluorescent images were acquired by microscopy at a magnification of 100X (Axiovert 25; Carl Zeiss, Germany). The total number of vessels was summed for at least 10 fields of view covering the whole tumor section for each tumor slice. Quantitative immunofluorescence analysis was performed using ImageQuant software (GE Healthcare, USA). The angiogenic activity was determined by VEGFR2 expression and the percentage area of blood vessels per field of view. At least 10 fields of view covering the whole tumor section for each tumor slice were quantified. CD31 was used to identify blood vessels, and the mean fluorescent intensity of VEGFR2 within the region of interest and percentage area of blood vessels were measured. The angiogenic activity was calculated by the ratio of overall mean expression of VEGFR2 and the mean percentage area of blood vessels per mm2 [46].
Statistical Analysis
Data were presented as mean ± standard deviation. An unpaired nonparametric one-sample Wilcoxon test for in vitro and a paired one-sample Wilcoxon test for in vivo experiments were used to analyze differences between the groups using commercially available software (IBM SPSS statistics software, version 20; IBM Corp, Chicago, IL). The significance level was set at 0.05.
Results
Engineered FN3VEGFR2 binding ligand covalently coupled to MB
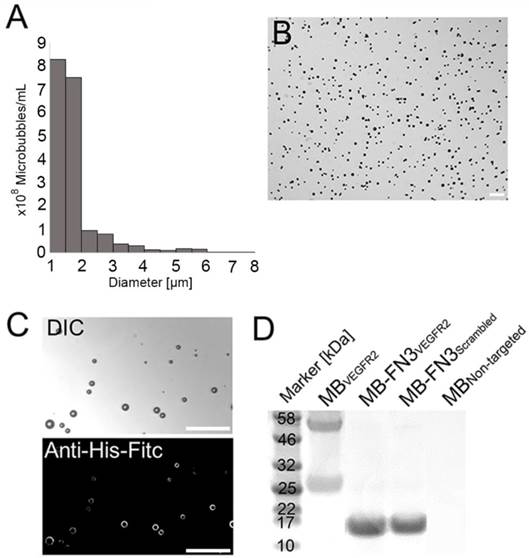
The mean diameter of synthesized MB-FN3VEGFR2 and MB-FN3Scrambled MBs was 1.8 μm (>95% range, 1-3 μm; Fig. 2 A and B). The mean concentration after reconstitution of a vial with 2 mL of saline was 8 x 108 MBs per mL. Successful FN3VEGFR2 conjugation to the surface of MBs was confirmed through fluorescence microscopy (Fig. 2 C) and SDS PAGE, showing ligand purity and conjugation in molecularly-targeted MBs compared to MBNon-targeted (Fig. 2 D).
Characterization of synthesized MB-FN3VEGFR2. A, size distribution analysis of synthesized molecularly-targeted MBs showed a mean diameter of 1.8 μm. B, representative micrograph of molecularly-targeted MBs showed no aggregation of particles in solution. Scale bar = 10 µm. C, successful conjugation of FN3VEGFR2 to the surface of MB was validated through fluorescence microscopy using anti-His-AF488-antibody binding to His-tagged FN3-coated MBs. DIC; Differential interference contrast. Scale bar = 10 µm. D, SDS PAGE showed the presence of successful stable ligand conjugation and purity of anti-VEGFR2 antibody, FN3VEGFR2, and FN3Scrambled scaffolds. Note that all synthesized molecularly-targeted MBs showed their corresponding expected molecular weight for each ligand type. As a negative control, no ligand was conjugated to the surface (MBNon-targeted).
In vitro binding study of MB-FN3VEGFR2 using FACS
FACS analysis was performed to analyze binding specificity of synthesized MB-FN3VEGFR2 to a soluble recombinant human and mouse VEGFR2 protein. Before FACS studies, all synthesized MBs were fluorescently pre-labeled by anti-His antibody-AF488 and were incubated with human and mouse VEGFR2 protein. Figure 3 A shows the fluorescence intensity values of MBs after incubation with AF647 post-labeled soluble recombinant human VEGFR2. Figure 3 B shows the fluorescence intensity values of MBs after incubation with AF647 post-labeled soluble recombinant mouse VEGFR2. In comparison to non-FN3 coated MBs (MBVEGFR2 and MBNon-targeted), solely His-tagged FN3-scaffolds conjugated to the surface of MBs (MB-FN3VEGFR2 and MB-FN3Scrambled) showed an enhanced fluorescence intensity signal using anti-His antibody-AF488, indicating successful conjugation of FN3-scaffolds to the surface of MBs (Fig. 3 A and B).
To confirm binding specificity of MB-FN3VEGFR2 to the recombinant VEGFR2 protein, a binding complex analysis between anti-His antibody-AF488 pre-labeled MB-FN3VEGFR2 and AF647 post-labeled soluble human and mouseVEGFR2 was performed and showed a significant (P < 0.01) and strong positive correlation (human VEGFR2 R2=0.97 ± 0.02; mouse VEGFR2 R2=0.96 ± 0.04), whereas, MB-FN3Scrambled (R2=0.24 ± 0.09; R2=0.19 ± 0.03), and MBNon-targeted (R2=0.07 ± 0.03; R2=0.05 ± 0.0.04) showed no correlation, indicating no binding to soluble AF647-labeled VEGFR2 (Fig. 3 A and B). As a positive control, a binding complex between pre-labeled VEGFR2 and MBVEGFR2 (no His-tag) depicted an enhanced AF647 fluorescence intensity signal (Fig. 3A and B).
Flow chamber cell attachment studies
FACS analysis of VEGFR2 expression on activated HUVEC and SVR cells confirmed overexpression of VEGFR2 on cells (Fig. 4 A and D). Activated HUVEC as well as SVR cells showed significantly (P < 0.01) increased VEGFR2 expression (HUVEC: 2921.17 ± 201.73 a.u.; SVR: 3332.28 ± 332.67 a.u.) compared to non-activated HUVEC cells (448.83 ± 57.65 a.u.) and non-stained SVR (negative control incubated solely with secondary antibody) cells (515.5 ± 126.39a.u.). Figure 4 B and E illustrates binding of MBVEGFR2, MB-FN3VEGFR2, MB-FN3Scrambled, and MBNon-targeted to VEGFR2-expressing HUVEC and SVR cells. The average number of MBVEGFR2 (2.1± 1.1 MBs) and MB-FN3VEGFR2 attached per HUVEC cell (2.07 ± 0.88 MBs) were significantly higher (P < 0.01) than MB-FN3Scrambled (0.67 ± 0.72 MBs). Blocking of VEGFR2 receptors with free FN3VEGFR2 resulted in significantly (P < 0.01) decreased MB-FN3VEGFR2 attachment (0.53 ± 0.64 MBs), confirming binding specificity of MB-FN3VEGFR2 to VEGFR2-expressing HUVEC cells. There was only minimal non-specific attachment of MBNon-targeted (0.47 ± 0.51 MBs) to VEGFR2-expressing HUVEC cells compared with MB-FN3VEGFR2 and MBVEGFR2, respectively (P < 0.01).
A, activated HUVEC cells showed significantly increased fluorescence intensity on FACS, indicating strong VEGFR2 expression (**, P < 0.01) compared to non-activated HUVEC cells. B, C, representative photomicrographs from cell culture experiments using a parallel plate flow chamber setting with VEGFR2-expressing HUVEC cells exposed to MBVEGFR2, MB-FN3VEGFR2, MB-FN3Scrambled, and MBNon-targeted. There was significantly lower accumulation of MB-FN3Scrambled, MBNon-targeted, and MB-FN3VEGFR2 after blocking the receptors with the free FN3VEGFR2 compared with MB-FN3VEGFR2 (**, P < 0.01). D, mouse VEGFR2-expressing SVR cells showed significantly increased fluorescence intensity on FACS, indicating strong VEGFR2 expression (**, P < 0.01) compared to non-stained SVR cells. E, F, representative photomicrographs from cell culture experiments using a parallel plate flow chamber setting with VEGFR2-expressing SVR cells exposed to MBVEGFR2, MB-FN3VEGFR2, MB-FN3Scrambled, and MBNon-targeted. There was significantly lower accumulation of MB-FN3Scrambled, MBNon-targeted, and MB-FN3VEGFR2 after blocking the receptors with free FN3VEGFR2 compared with MB-FN3VEGFR2 (**, P < 0.01). MBs (arrows) are visualized as white spherical dots. Scale bar = 10 µm.
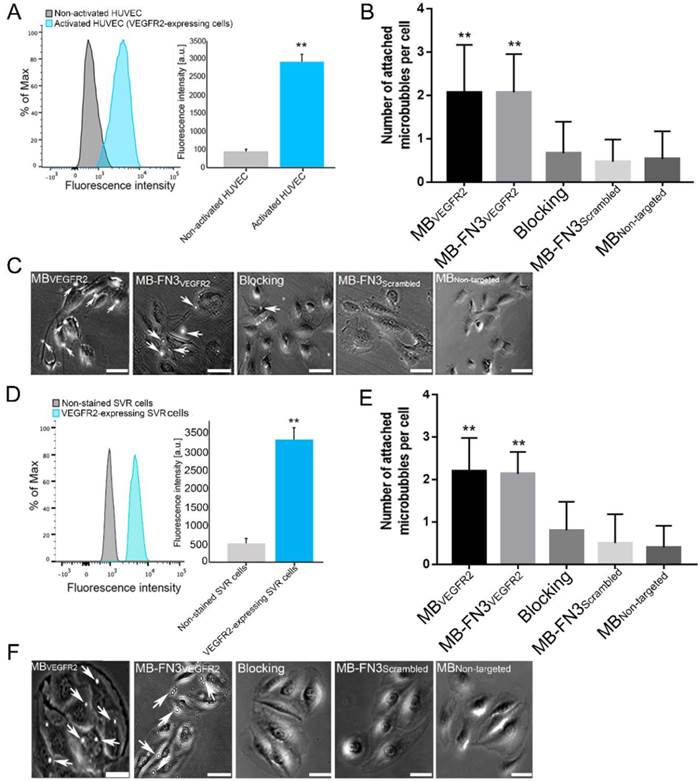
Similarly, the average number of MBVEGFR2 (2.2± 0.77 MBs) and MB-FN3VEGFR2 attached per SVR cell (2.13 ± 0.52 MBs) were significantly higher (P < 0.01) than MB-FN3Scrambled (0.50 ± 0.68 MBs), confirming attachment of both MB types to murine VEGFR2. Blocking of VEGFR2 receptors by free FN3VEGFR2 resulted in significantly (P < 0.01) decreased MB-FN3VEGFR2 attachment (0.8 ± 0.67 MBs), further confirming binding of MB-FN3VEGFR2 to murine VEGFR2 (Fig. 4 E). There was also only minimal non-specific attachment of MBNon-targeted (0.4 ± 0.51 MBs) to murine VEGFR2 compared with MB-FN3VEGFR2 and MBVEGFR2, respectively (P < 0.01).
In vivo ultrasound molecular imaging of tumor angiogenesis in breast cancer and ex vivo immunofluorescence analysis
In vivo USMI showed significantly higher signal following MB-FN3VEGFR2 (P < 0.01) and MBVEGFR2 (P < 0.01) administration compared to MBNon-targeted (Fig. 5 A). The imaging signals following administration of MB-FN3VEGFR2 (12.48 ± 3.41 a.u.) and following MBVEGFR2 (13.01 ± 3.08 a.u.) administration were not significantly different (P = 0.84, Fig. 5 B). Following injection of MB-FN3Scrambled (2.69 ± 1.51 a.u.), imaging signal was significantly (P < 0.01) lower compared to the signal after MB-FN3VEGFR2 and not significantly different (P = 0.62) compared to MBNon-targeted (2.5 ± 0.71 a.u.; Fig. 5 B). To further confirm VEGFR2-binding specificity of MB-FN3VEGFR2, in vivo blocking of VEGFR2 receptors with free FN3VEGFR2 was performed and showed significantly decreased (P < 0.05, 6.10 ± 3.12 a.u.) imaging signal by 60% compared to tumors without pre-administration of the blocking agent (Fig. 5 C, D). Finally, as a negative control for non-angiogenic vessels, normal mammary glands were scanned after intravenous administration of MB-FN3VEGFR2, MB-FN3Scrambled, and MBNon-targeted (Fig. 5 E). The imaging signal was significantly lower in normal mammary gland tissue (MB-FN3VEGFR2 1.35 ± 0.21 a.u.; MB-FN3Scrambled 1.27 ± 0.31 a.u., MBNon-targeted 1.18 ± 0.15 a.u.; P < 0.01) than in breast cancer following injection of MB-FN3VEGFR2.
In vivo USMI of breast cancer in transgenic mouse model. A, bar graph summarizes in vivo imaging signal using the various types of contrast MB. Error bar = standard deviation; **, P < 0.01. B, representative transverse contrast mode ultrasound images show strong signal in breast cancer (green ROI) after injection of MBVEGR2 and MB-FN3VEGFR2 but only background signal following injection of the two types of control MBs (MB-FN3Scrambled and MBNon-targeted). C and D, bar graph and representative in vivo USMI example show substantial decrease of imaging signal after intravenous administration of free blocking FN3VEGFR2 ligand. Error bar = standard deviation; *, P < 0.05. E, representative transverse contrast mode ultrasound images show a strong signal in breast cancer but only background signal in control normal mammary gland tissue after injection of MB-FN3VEGFR2, MB-FN3Scrambled, and MBNon-targeted. F, representative immunofluorescence staining of breast cancer and normal mammary tissue shows strong expression of VEGFR2 on the neovasculature of cancer (co-localization of VEGFR2 (red) on CD31-stained (green) tumor vessels) compared to normal control tissue. Scale bar = 50 µm.
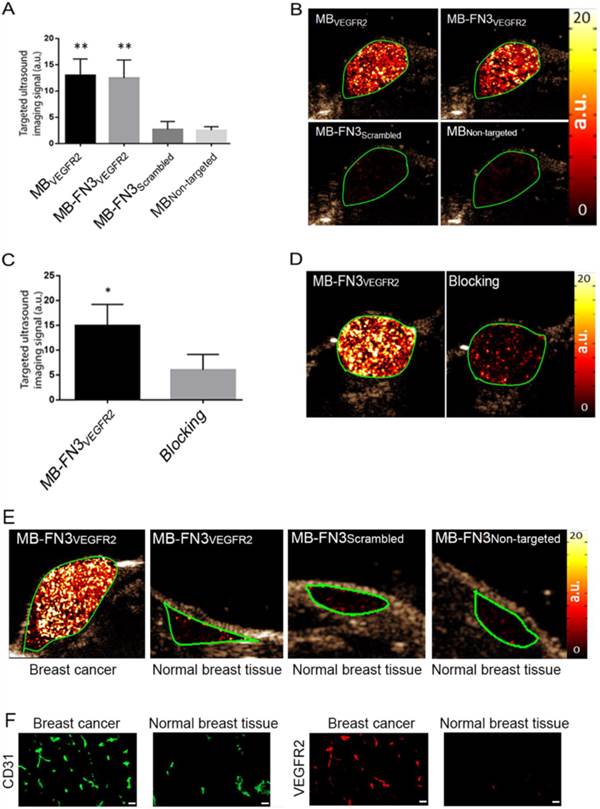
Similar to in vivo USMI parameters, quantitative immunofluorescence showed significantly (P < 0.01) increased angiogenic activity (co-localization of VEGFR2 (red) on CD31-stained (green) tumor vessels) in breast cancer (0.64 ± 0.08 a.u.) compared to normal breast tissue (0.12 ± 0.05 a.u.) (Fig. 5 F).
Discussion
This study showed that breast cancer neovasculature can be visualized by ultrasound using molecularly-targeted MBs coupled with a new class of targeting ligands, small FN3VEGFR2 scaffolds. Specific binding of MB-FN3VEGFR2 to both soluble human and mouse VEGFR2 as well as VEGFR2 expressed on live cells was demonstrated, and VEGFR2 specific ultrasonic molecular imaging signal using MB-FN3VEGFR2 was shown in transgenic mice with breast cancer in vivo.
USMI using molecularly-targeted MBs is emerging as a promising new modality for oncological imaging. In particular for earlier cancer detection where repetitive imaging exams are warranted, such as in a screening setting of women with dense breast tissue where mammography has limited accuracy in detecting breast cancer [47-51], USMI is advantageous as it has the potential to detect small foci of cancer with high sensitivity and specificity without the use of potentially harmful ionizing radiation [8, 10]. Also, USMI comes at relatively low cost compared to other imaging techniques. While novel molecular imaging targets are being discovered and validated that may further improve diagnostic accuracy of USMI for better detection of various cancer types including breast cancer [9, 10, 52, 53], there is a critical need for a platform for quickly generating biocompatible high affinity binding ligands that are smaller and cheaper to produce than conventional full-length antibodies [54, 55].
Engineered protein scaffold (EPS) techniques are promising approaches to enhance binding to a specific molecular imaging target by using antibody-like molecules with exposed loops or surfaces that can be randomized, modified, and screened using selective engineered libraries [55]. Several EPSs have been explored including knottins [14], single-chain variable fragments [15], or nanobodies [16]. Recently, engineered FN3-scaffolds have been introduced as an alternative platform for designing binding ligands for molecular imaging with several advantages [17, 18]. FN3 domains are small (~10 kDa) immunoglobulin (Ig)-like domains. Their hydrophobic core of the β-sandwich domain provides a stable framework structure, conveying high thermo-stability (84 ºC) [56]. Hence, wild-type FN3 and most mutants are stable monomers lacking cysteine residues and post-translational modifications, which facilitates high-level expression in bacteria. Also, fibronectin is already present at high levels (300 to 400 μg/mL) in normal human plasma [57], thereby minimizing the risk for immunogenicity to the FN3-scaffold. FN3 loops which are structurally similar to antibody complementarity-determining regions provide a platform to engineer novel binders with antibody-like affinity and specificity. FN3 libraries using phage, mRNA, and yeast display with randomized BC, DE and FG loops, or loop/sheet combinations, have been successfully engineered for screening novel binding ligands to various molecular targets [25, 42]. FN3VEGFR2 is a scaffold with high affinity to human VEGFR2 with simultaneous introduction of cross-reactivity to murine VEGFR2 [25]. It has shown good safety profile in phase I and II clinical trials in patients used as an anti-angiogenic drug [26, 27]. The absence of cysteine and lysine residues near the engineered binding loops of FN3-scaffolds allows site-specific coupling of the ligand to the MB shell without compromising binding affinity. We demonstrated that FN3VEGFR2 can also be used as binding ligand for designing a new class of molecularly-targeted MBs using derivatives of the biocompatible human fibronectin.
Binding of MB-FN3VEGFR2 was significantly higher compared to the two types of control MBs (MBNon-targeted and MB-FN3Scrambled) in both in vitro and in vivo experiments and could be blocked by free FN3VEGFR2, further confirming binding specificity of MB-FN3VEGFR2 to VEGFR2. Also, MB-FN3VEGFR2 resulted in similar in vivo imaging signal in breast cancer compared to MBVEGFR2, the most commonly used targeted contrast MB in preclinical ultrasound imaging experiments [44]. MBVEGFR2 contains streptavidin in the MB shell and biotinylated anti-VEGFR2-antibody were attached to the MB shell via the strong biotin/streptavidin interaction [1]. Since streptavidin is immunogenic and can cause severe allergic reactions in patients [58, 59], MBVEGFR2 can only be used for preclinical experiments. In contrast, we covalently bound FN3VEGFR2 to the MB shell using carboxyl-amine-conjugation chemistry in our study to generate a potentially clinically translatable MB platform that does not use biotin/streptavidin interactions for ligand attachment.
Recently, another VEGFR2-targeted ultrasound contrast agent (BR55), using an engineered heterodimeric peptide identified by phage display and covalently bound to the MB shell, has been designed for better cancer visualization [4, 8]. BR55 is currently the only clinical grade molecularly-targeted ultrasound contrast agent and has been moved into early phase clinical trials in Europe and the USA for cancer imaging (ClinicalTrials.gov identifiers: NCT01253213, NCT02142608, 2012-000699-40) [60, 61]. As an alternative to engineered peptides, versatile FN3-scaffolds are small, stable, and efficiently conjugated immunoglobulin (Ig)-like domains that can be readily modified by direct evolution and produced in bacteria. As such, FN3-scaffolds are excellent candidates for molecular recognition with unique advantages in several applications and can be further explored for the design of next generation ultrasound contrast agents targeted at novel molecular imaging targets beyond VEGFR2.
A limitation of our study was that USMI was limited to two-dimensional planes. Efforts are under way for three-dimensional USMI which better captures the spatial heterogeneity of the neovasculature of cancer and of the expression levels of molecular markers such as VEGFR2 [62]. Automated whole breast ultrasound scanners have been clinically introduced [63] that are currently being further developed to allow automated whole breast USMI of breast cancer in an operator-independent manner. Furthermore, additional characterization and refinement of FN3-conjugated MBs are needed to assess its stability and functionality over longer periods of time. Also, future experiments are warranted to assess the relationship between the number of targeting FN3VEGFR2 ligands on the MB surface and the binding properties of the MB to VEGFR2.
In conclusion, our results suggest that potentially clinically translatable molecularly-targeted MBs coupled to FN3-scaffolds can be designed and used for USMI of breast cancer neoangiogenesis. Due to their small size and stability, FN3-scaffolds can be recombinantly produced with the advantage of generating small, high affinity ligands in a cost efficient way for USMI.
Abbreviations
MBs: Molecularly-targeted microbubbles; USMI: ultrasound molecular imaging; FN3: 10th type III domain of human-fibronectin; EPS: engineered protein scaffold; VEGFR2: Vascular endothelial growth factor receptor type 2; HUVEC: Human umbilical vein endothelial cells; SVR: Mouse angiosarcoma cells; KD: Dissociation constant; ROI: Region of interest.
Acknowledgements
We would like to acknowledge the SCi3 Core imaging facilities and the Canary Center at Stanford for the technical support of this study.
Funding
This research was supported by the NIH R01 CA155289-01 grant (JKW), the R01DK092509-01 grant (JKW), and by the Canary Foundation. The Mildred-Scheel Cancer Foundation from Germany provided funding for Dr. Lotfi Abou-Elkacem to study abroad at Stanford University.
Conflict of Interest
The authors report no potential conflicts of interest.
References
1. Kiessling F, Fokong S, Bzyl J, Lederle W, Palmowski M, Lammers T. Recent advances in molecular, multimodal and theranostic ultrasound imaging. Adv Drug Deliv Rev. 2014;72:15-27
2. Klibanov AL, Hossack JA. Ultrasound in Radiology: From Anatomic, Functional, Molecular Imaging to Drug Delivery and Image-Guided Therapy. Invest Radiol. 2015;50:657-70
3. Abou-Elkacem L, Bachawal SV, Willmann JK. Ultrasound molecular imaging: Moving toward clinical translation. Eur J Radiol. 2015;84:1685-93
4. Pochon S, Tardy I, Bussat P, Bettinger T, Brochot J, von Wronski M. et al. BR55: a lipopeptide-based VEGFR2-targeted ultrasound contrast agent for molecular imaging of angiogenesis. Invest Radiol. 2010;45:89-95
5. Lyshchik A, Fleischer AC, Huamani J, Hallahan DE, Brissova M, Gore JC. Molecular imaging of vascular endothelial growth factor receptor 2 expression using targeted contrast-enhanced high-frequency ultrasonography. J Ultrasound Med. 2007;26:1575-86
6. Monsky WL, Mouta Carreira C, Tsuzuki Y, Gohongi T, Fukumura D, Jain RK. Role of host microenvironment in angiogenesis and microvascular functions in human breast cancer xenografts: mammary fat pad versus cranial tumors. Clin Cancer Res. 2002;8:1008-13
7. Bzyl J, Lederle W, Palmowski M, Kiessling F. Molecular and functional ultrasound imaging of breast tumors. Eur J Radiol. 2012;81(Suppl 1):S11-2
8. Bachawal SV, Jensen KC, Lutz AM, Gambhir SS, Tranquart F, Tian L. et al. Earlier detection of breast cancer with ultrasound molecular imaging in a transgenic mouse model. Cancer Res. 2013;73:1689-98
9. Foygel K, Wang H, Machtaler S, Lutz AM, Chen R, Pysz M. et al. Detection of pancreatic ductal adenocarcinoma in mice by ultrasound imaging of thymocyte differentiation antigen 1. Gastroenterology. 2013;145:885-94 e3
10. Bachawal SV, Jensen KC, Wilson KE, Tian L, Lutz AM, Willmann JK. Breast Cancer Detection by B7-H3-Targeted Ultrasound Molecular Imaging. Cancer Res. 2015;75:2501-9
11. Tami JA, Parr MD, Brown SA, Thompson JS. Monoclonal antibody technology. Am J Hosp Pharm. 1986;43:2816-25
12. Shukla AA, Thommes J. Recent advances in large-scale production of monoclonal antibodies and related proteins. Trends Biotechnol. 2010;28:253-61
13. Skerra A. Alternative non-antibody scaffolds for molecular recognition. Curr Opin Biotechnol. 2007;18:295-304
14. Willmann JK, Kimura RH, Deshpande N, Lutz AM, Cochran JR, Gambhir SS. Targeted contrast-enhanced ultrasound imaging of tumor angiogenesis with contrast microbubbles conjugated to integrin-binding knottin peptides. J Nucl Med. 2010;51:433-40
15. Wang X, Hagemeyer CE, Hohmann JD, Leitner E, Armstrong PC, Jia F. et al. Novel single-chain antibody-targeted microbubbles for molecular ultrasound imaging of thrombosis: validation of a unique noninvasive method for rapid and sensitive detection of thrombi and monitoring of success or failure of thrombolysis in mice. Circulation. 2012;125:3117-26
16. Hernot S, Unnikrishnan S, Du Z, Shevchenko T, Cosyns B, Broisat A. et al. Nanobody-coupled microbubbles as novel molecular tracer. J Control Release. 2012;158:346-53
17. Koide S, Koide A, Lipovšek D. Target-binding proteins based on the 10th human fibronectin type III domain (¹⁰Fn3). Methods Enzymol. 2012;503:135-56
18. Hackel BJ, Kimura RH, Gambhir SS. Use of (64)Cu-labeled fibronectin domain with EGFR-overexpressing tumor xenograft: molecular imaging. Radiology. 2012;263:179-88
19. Garcia-Ibilcieta D, Bokov M, Cherkasov V, Sveshnikov P, Hanson SF. Simple method for production of randomized human tenth fibronectin domain III libraries for use in combinatorial screening procedures. Biotechniques. 2008;44:559-62
20. Hosse RJ, Rothe A, Power BE. A new generation of protein display scaffolds for molecular recognition. Protein Sci. 2006;15:14-27
21. Koide A, Bailey CW, Huang X, Koide S. The fibronectin type III domain as a scaffold for novel binding proteins. J Mol Biol. 1998;284:1141-51
22. Ruigrok VJB, Levisson M, Eppink MHM, Smidt H, van der Oost J. Alternative affinity tools: more attractive than antibodies? Biochem J. 2011;436:1-13
23. Taussig MJ, Stoevesandt O, Borrebaeck CAK, Bradbury AR, Cahill D, Cambillau C. et al. ProteomeBinders: planning a European resource of affinity reagents for analysis of the human proteome. Nat Methods. 2007;4:13-7
24. Wurch T, Pierré A, Depil S. Novel protein scaffolds as emerging therapeutic proteins: from discovery to clinical proof-of-concept. Trends Biotechnol. 2012;30:575-82
25. Getmanova EV, Chen Y, Bloom L, Gokemeijer J, Shamah S, Warikoo V. et al. Antagonists to human and mouse vascular endothelial growth factor receptor 2 generated by directed protein evolution in vitro. Chem Biol. 2006;13:549-56
26. Tolcher AW, Sweeney CJ, Papadopoulos K, Patnaik A, Chiorean EG, Mita AC. et al. Phase I and pharmacokinetic study of CT-322 (BMS-844203), a targeted Adnectin inhibitor of VEGFR-2 based on a domain of human fibronectin. Clin Cancer Res. 2011;17:363-71
27. Schiff D, Kesari S, de Groot J, Mikkelsen T, Drappatz J, Coyle T. et al. Phase 2 study of CT-322, a targeted biologic inhibitor of VEGFR-2 based on a domain of human fibronectin, in recurrent glioblastoma. Invest New Drugs. 2015;33:247-53
28. Feshitan JA, Chen CC, Kwan JJ, Borden MA. Microbubble size isolation by differential centrifugation. J Colloid Interface Sci. 2009;329:316-24
29. Weller GER, Wong MKK, Modzelewski RA, Lu E, Klibanov AL, Wagner WR. et al. Ultrasonic imaging of tumor angiogenesis using contrast microbubbles targeted via the tumor-binding peptide arginine-arginine-leucine. Cancer Res. 2005;65:533-9
30. Anderson CR, Rychak JJ, Backer M, Backer J, Ley K, Klibanov AL. scVEGF microbubble ultrasound contrast agents: a novel probe for ultrasound molecular imaging of tumor angiogenesis. Invest Radiol. 2010;45:579-85
31. Klibanov AL, Rychak JJ, Yang WC, Alikhani S, Li B, Acton S. et al. Targeted ultrasound contrast agent for molecular imaging of inflammation in high-shear flow. Contrast Media Mol Imaging. 2006;1:259-66
32. Klibanov AL, Hughes MS, Villanueva FS, Jankowski RJ, Wagner WR, Wojdyla JK. et al. Targeting and ultrasound imaging of microbubble-based contrast agents. MAGMA. 1999;8:177-84
33. Jain RK. Determinants of tumor blood flow: a review. Cancer Res. 1988;48:2641-58
34. Bachmann C, Klibanov AL, Olson TS, Sonnenschein JR, Rivera-Nieves J, Cominelli F. et al. Targeting mucosal addressin cellular adhesion molecule (MAdCAM)-1 to noninvasively image experimental Crohn's disease. Gastroenterology. 2006;130:8-16
35. Guenther F, von zur Muhlen C, Ferrante EA, Grundmann S, Bode C, Klibanov AL. An ultrasound contrast agent targeted to P-selectin detects activated platelets at supra-arterial shear flow conditions. Invest Radiol. 2010;45:586-91
36. Fokong S, Fragoso A, Rix A, Curaj A, Wu Z, Lederle W. et al. Ultrasound molecular imaging of E-selectin in tumor vessels using poly n-butyl cyanoacrylate microbubbles covalently coupled to a short targeting peptide. Invest Radiol. 2013;48:843-50
37. Hutchinson JN, Muller WJ. Transgenic mouse models of human breast cancer. Oncogene. 2000;19:6130-7
38. Guy CT, Cardiff RD, Muller WJ. Induction of mammary tumors by expression of polyomavirus middle T oncogene: a transgenic mouse model for metastatic disease. Mol Cell Biol. 1992;12:954-61
39. Lin EY, Jones JG, Li P, Zhu L, Whitney KD, Muller WJ. et al. Progression to malignancy in the polyoma middle T oncoprotein mouse breast cancer model provides a reliable model for human diseases. Am J Pathol. 2003;163:2113-26
40. Willmann JK, Cheng Z, Davis C, Lutz AM, Schipper ML, Nielsen CH. et al. Targeted microbubbles for imaging tumor angiogenesis: assessment of whole-body biodistribution with dynamic micro-PET in mice. Radiology. 2008;249:212-9
41. Palmowski M, Morgenstern B, Hauff P, Reinhardt M, Huppert J, Maurer M. et al. Pharmacodynamics of streptavidin-coated cyanoacrylate microbubbles designed for molecular ultrasound imaging. Invest Radiol. 2008;43:162-9
42. Natarajan A, Hackel BJ, Gambhir SS. A novel engineered anti-CD20 tracer enables early time PET imaging in a humanized transgenic mouse model of B-cell non-Hodgkins lymphoma. Clin Cancer Res. 2013;19:6820-9
43. Park S-H, Park S, Kim D-Y, Pyo A, Kimura RH, Sathirachinda A. et al. Isolation and Characterization of a Monobody with a Fibronectin Domain III Scaffold That Specifically Binds EphA2. PLoS One. 2015;10:e0132976
44. Willmann JK, Paulmurugan R, Chen K, Gheysens O, Rodriguez-Porcel M, Lutz AM. et al. US imaging of tumor angiogenesis with microbubbles targeted to vascular endothelial growth factor receptor type 2 in mice. Radiology. 2008;246:508-18
45. Baetke SC, Rix A, Tranquart F, Schneider R, Lammers T, Kiessling F. et al. Squamous Cell Carcinoma Xenografts: Use of VEGFR2-targeted Microbubbles for Combined Functional and Molecular US to Monitor Antiangiogenic Therapy Effects. Radiology. 2015:142899
46. Weidner N. Current pathologic methods for measuring intratumoral microvessel density within breast carcinoma and other solid tumors. Breast Cancer Res Treat. 1995;36:169-80
47. Stomper PC, D'Souza DJ, DiNitto PA, Arredondo MA. Analysis of parenchymal density on mammograms in 1353 women 25-79 years old. AJR Am J Roentgenol. 1996;167:1261-5
48. Kolb TM, Lichy J, Newhouse JH. Comparison of the performance of screening mammography, physical examination, and breast US and evaluation of factors that influence them: an analysis of 27,825 patient evaluations. Radiology. 2002;225:165-75
49. Mandelson MT, Oestreicher N, Porter PL, White D, Finder CA, Taplin SH. et al. Breast density as a predictor of mammographic detection: comparison of interval- and screen-detected cancers. J Natl Cancer Inst. 2000;92:1081-7
50. Boyd NF, Guo H, Martin LJ, Sun L, Stone J, Fishell E. et al. Mammographic density and the risk and detection of breast cancer. N Engl J Med. 2007;356:227-36
51. Berg WA, Blume JD, Cormack JB, Mendelson EB, Lehrer D, Böhm-Vélez M. et al. Combined screening with ultrasound and mammography vs mammography alone in women at elevated risk of breast cancer. JAMA. 2008;299:2151-63
52. Lutz AM, Bachawal SV, Drescher CW, Pysz MA, Willmann JK, Gambhir SS. Ultrasound molecular imaging in a human CD276 expression-modulated murine ovarian cancer model. Clin Cancer Res. 2014;20:1313-22
53. Olopade OI, Grushko TA, Nanda R, Huo D. Advances in breast cancer: pathways to personalized medicine. Clin Cancer Res. 2008;14:7988-99
54. Gebauer M, Skerra A. Engineered protein scaffolds as next-generation antibody therapeutics. Curr Opin Chem Biol. 2009;13:245-55
55. Stern LA, Case BA, Hackel BJ. Alternative Non-Antibody Protein Scaffolds for Molecular Imaging of Cancer. Curr Opin Chem Eng. 2013:2
56. Hackel BJ, Kapila A, Wittrup KD. Picomolar affinity fibronectin domains engineered utilizing loop length diversity, recursive mutagenesis, and loop shuffling. J Mol Biol. 2008;381:1238-52
57. Mosher DF. Plasma fibronectin concentration: a risk factor for arterial thrombosis? Arterioscler Thromb Vasc Biol. 2006;26:1193-5
58. Marshall D, Pedley RB, Boden JA, Boden R, Melton RG, Begent RH. Polyethylene glycol modification of a galactosylated streptavidin clearing agent: effects on immunogenicity and clearance of a biotinylated anti-tumour antibody. Br J Cancer. 1996;73:565-72
59. Meyer DL, Schultz J, Lin Y, Henry A, Sanderson J, Jackson JM. et al. Reduced antibody response to streptavidin through site-directed mutagenesis. Protein Sci. 2001;10:491-503
60. Tranquart F, Arditi M, Bettinger T, Frinking P, Hyvelin JM, Nunn A. et al. Ultrasound Contrast Agents For Ultrasound Molecular Imaging. Z Gastroenterol. 2014;52:1268-76
61. Abou-Elkacem L, Bachawal SV, Willmann JK. Ultrasound molecular imaging: Moving toward clinical translation. Eur J Radiol. 2015
62. Wang H, Kaneko OF, Tian L, Hristov D, Willmann JK. Three-Dimensional Ultrasound Molecular Imaging of Angiogenesis in Colon Cancer Using a Clinical Matrix Array Ultrasound Transducer. Invest Radiol. 2015
63. Giuliano V, Giuliano C. Improved breast cancer detection in asymptomatic women using 3D-automated breast ultrasound in mammographically dense breasts. Clin Imaging. 2013;37:480-6
Author contact
![]() Corresponding author: Jürgen K. Willmann, M.D. Department of Radiology and Molecular Imaging Program at Stanford, School of Medicine, Stanford University, 300 Pasteur Drive, Room H1307, Stanford, CA 94305-5621 P: 650-723-5424 Fax: 650-723-1909 Email: willmannedu.
Corresponding author: Jürgen K. Willmann, M.D. Department of Radiology and Molecular Imaging Program at Stanford, School of Medicine, Stanford University, 300 Pasteur Drive, Room H1307, Stanford, CA 94305-5621 P: 650-723-5424 Fax: 650-723-1909 Email: willmannedu.