Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Material and Methods
Results
Discussion
Conclusion
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2016; 6(11):1821-1832. doi:10.7150/thno.15311 This issue Cite
Research Paper
Potentiation of Peptide Receptor Radionuclide Therapy by the PARP Inhibitor Olaparib
Julie Nonnekens1,2 ![]() , Melissa van Kranenburg1, Cecile E.M.T. Beerens3, Mustafa Suker4, Michael Doukas5, Casper H.J. van Eijck4, Marion de Jong2, Dik C. van Gent1
, Melissa van Kranenburg1, Cecile E.M.T. Beerens3, Mustafa Suker4, Michael Doukas5, Casper H.J. van Eijck4, Marion de Jong2, Dik C. van Gent1 ![]()
1. Department of Molecular Genetics, Erasmus MC, Rotterdam, The Netherlands
2. Department of Radiology & Nuclear Medicine, Erasmus MC, Rotterdam, The Netherlands
3. Department of Radiation Oncology, Erasmus MC, Rotterdam, The Netherlands
4. Department of Surgery, Erasmus MC, Rotterdam, The Netherlands
5. Department of Pathology, Erasmus MC, Rotterdam, The Netherlands
Received 2016-2-18; Accepted 2016-6-16; Published 2016-7-18
Abstract

Metastases expressing tumor-specific receptors can be targeted and treated by binding of radiolabeled peptides (peptide receptor radionuclide therapy or PRRT). For example, patients with metastasized somatostatin receptor-positive neuroendocrine tumors (NETs) can be treated with radiolabeled somatostatin analogues, resulting in strongly increased progression-free survival and quality of life. There is nevertheless still room for improvement, as very few patients can be cured at this stage of disease. We aimed to specifically sensitize replicating tumor cells without further damage to healthy tissues. Thereto we investigated the DNA damaging effects of PRRT with the purpose to enhance these effects through modulation of the DNA damage response. Although PRRT induces DNA double strand breaks (DSBs), a larger fraction of the induced lesions are single strand breaks (expected to be similar to those induced by external beam radiotherapy) that require poly-[ADP-ribose]-polymerase 1 (PARP-1) activity for repair. If these breaks cannot be repaired, they will cause replication fork arrest and DSB formation during replication. Therefore, we used the PARP-1 inhibitor Olaparib to increase the number of cytotoxic DSBs. Here we show that this new combination strategy synergistically sensitized somatostatin receptor expressing cells to PRRT. We observed increased cell death and reduced cellular proliferation compared to the PRRT alone. The enhanced cell death was caused by increased numbers of DSBs that are repaired with remarkably slow kinetics, leading to genome instability. Furthermore, we validated the increased DSB induction after PARP inhibitor addition in the clinically relevant model of living human NET slices. We expect that this combined regimen can thus augment current PRRT outcomes.
Keywords: Neuroendocrine tumors, somatostatin receptor, peptide receptor radionuclide therapy, 177Lu-DOTA-[Tyr3]octreotate, DNA damage response, combination treatment, PARP inhibitor
Introduction
Targeted anticancer therapies utilize specific molecular marks of the tumor to distinguish between tumor and normal healthy cells [1]. Neuroendocrine tumors (NET) overexpressing the somatostatin receptor subtype 2 (SSTR2) are target for treatment with radiolabeled peptides, called peptide receptor radionuclide therapy (PRRT) [2]. Clinical trials have successfully been finalized using the radionuclide Lutetium-177 (177Lu) coupled to somatostatin analogue DOTA-[Tyr3]octreotate (177Lu-DOTA-TATE) [3-5]. 177Lu-DOTA-TATE PRRT is currently applied as third line treatment and is the last hope for many patients suffering from metastasized neuroendocrine tumors. The results of clinical trials are favorable: high tumor response rates, low toxicity, extended progression-free survival and improved quality of life [3-5]. Unfortunately, the overall efficacy analysis shows that most tumors that initially respond well to the treatment, will eventually relapse. Simply administering a higher radionuclide dose might lead to healthy tissue damage, especially in the bone marrow and kidneys, due to circulating radioactivity and renal clearance and reabsorption of radioactivity, respectively [6].
The cell killing effect of 177Lu is likely to be due to DNA double strand breaks (DSBs) caused by the emitted ionizing beta particles. In recent years, increased knowledge of the DNA damage response (DDR) has been used to enhance anticancer therapies. A very attractive example is the use of poly-[ADP-ribose]-polymerase 1 (PARP-1) inhibitors to specifically kill cells with homologous recombination defects, e.g. Breast Cancer 1/2 (BRCA1/2)-deficient cells: inactivation of a DNA repair pathway is compatible with survival of normal cells, but not with survival of repair-deficient cancer cells [7, 8]. Drugs such as PARP inhibitors are not only being applied as single agents, but can also be used in new strategies to enhance the efficacy of radiation [9]. These strategies aim at increasing the therapeutic window by sensitizing tumors and not the normal tissue.
Many anticancer therapies are based on the well-described cytotoxicity of DSBs, produced by ionizing radiation or chemical agents, leading to tumor cell death. Next to DSBs, ionizing radiation produces less cytotoxic DNA lesions, such as base damage and DNA single-strand breaks (SSBs). 177Lu produces low energy transfer radiation and therefore the damage range is expected to be similar. PARP-1 facilitates SSB-repair by activating and engaging repair enzymes at the site of the lesion. If the SSBs are not repaired efficiently (for example by blocking PARP-1 functioning), they will cause replication fork arrest and DSB formation in replicating cells. Therefore, PARP inhibition provides an excellent opportunity for tumor-specific radiosensitization of proliferating tumor cells, while sparing non-proliferating normal tissue [10]. We show that SSTR2-positive human osteosarcoma cells and ex vivo cultured human NET slices are synergistically sensitized to PRRT using the PARP inhibitor Olaparib. This sensitization is caused by increased genome instability leading to cell death.
Material and Methods
Cell lines and treatment
Experiments were performed on human osteosarcoma cells (U2OS), U2OS cells stably expressing SSTR2 [11] and the SSTR positive rat pancreatic Ca20948 cells [12]. Cells were cultured in DMEM (Lonza), supplemented with 10% fetal bovine serum (Biowest), penicillin (50 units/mL) and streptomycin (50 µg/mL) (Sigma Aldrich), at 37°C and 5% CO2.
For PRRT experiments, cells were treated for 4 h with different activity quantities of 177Lu-DTPA (saturated with DTPA) or 177Lu-DOTA-TATE (specific activity 53 MBq/nmol, radiometal incorporation >95% and radiochemical purity >90%) (IDB Holland). This specific activity is the same as used during patient treatment [5, 13]. Activity concentrations are based on a previous study by Capello and collaborators [14]. Subsequently, the radioligands were removed, cells were washed with phosphate buffered saline (PBS) (Lonza) and incubated in non-radioactive medium with or without 1 μM Olaparib (AZD2281, Ku-0059436) (Selleckchem). The Olaparib concentration was based on previous screens (data not shown) and we have used 1 μM because it had minimal effect as monotreatment on our cells. For comparative external beam irradiation experiments, cells were pretreated with 1 μM Olaparib for 4 h and subsequently irradiated with a Cesium-137 source (0.6Gy/min, Gammacell 40, Theratronics).
All experiments were performed 2 or 3 times (with technical triplicates) and averages of experiments were plotted in the figures. In some figures, only 177Lu-DTPA and 177Lu-DOTA-TATE results are shown for simplicity. In these experiments, no difference was observed between non treated (NT) samples and 177Lu-DTPA treated samples. NT data can be found in the supplemental figures.
Colony survival assay
For measurement of cell killing, a colony survival was performed. U2OS, U2OS+SSTR2 or Ca20948 cells were seeded in 6-well plates (1×105 cells / well) in 2 mL medium and the next day adherent cells were incubated for 4 h at 37°C, 5% CO2 with 5×10-8 M / 5 MBq, 2×10-8 M / 2 MBq, 5×10-9 M / 0.5 MBq or 2×10-9 M / 0.2 MBq 177Lu-DOTA-TATE or with 5 MBq, 2 MBq, 0.5 MBq or 0.2 MBq 177Lu-DTPA in 2 mL medium. Cells were trypsinized and seeded in triplicate in 6 well plates (300 cells per well) in 2 mL normal medium or medium containing 1 μM Olaparib. Four days after treatment, medium was replaced for 2 mL medium without Olaparib for all conditions. Ten days after treatment, colonies were washed with PBS and stained with 0.1% Coomassie blue acetic acid staining solution for 15 min at room temperature (RT). Colonies were counted manually and normalized to untreated controls (with or without 1 μM Olaparib). The area under the curve was calculated using GraphPad Prism software.
Sulforhodamine beta assay
For measurement of cell number a sulforhodamine beta assay was performed. U2OS+SSTR2 cells were seeded in 6-well plates (5×105 cells / well) and the next day adherent cells were incubated for 4 h with 5×10-8 M / 5 MBq 177Lu-DOTA-TATE or with 5 MBq 177Lu-DTPA. Cells were trypsinized and seeded in triplicate in 12 well plates (1.5×104 cells per well) in 1 mL normal medium or medium containing 1 μM Olaparib and allowed to grow for one to six days. Subsequently, medium was removed and cells were fixed with 1 mL 10% trichloroacetic acid overnight at 4oC. Plates were washed five times with tap water and dried. Then cells were incubated in 500 μl 0.5% sulforhodamine beta (SRB) in 1% acetic acid for 20 minutes at RT. Plates were washed four times with 1% acetic acid and air-dried. SRB was dissolved in 500 μl 10 mM Tris solution and absorbance was measured at 560 nm using a GloMax®-Multi Detection System (Promega).
Immunofluorescent stainings cells
For analysis of DDR and apoptosis parameters, different immunofluorescent stainings were performed. U2OS+SSTR2 cells were grown on glass coverslips in 6-well plates (DSB and micronuclei) or 24-well plates (cytochrome c release) and were incubated for 4 h with 5×10-8 M / 5 MBq 177Lu-DOTA-TATE or 5 MBq 177Lu-DTPA in 2 mL medium (DSB and micronuclei) or 500 μL medium (cytochrome C release) for 4 h at 37°C. Subsequently, cells were washed twice with PBS and incubated in normal medium or medium containing 1 μM Olaparib and allowed to grow until different timepoints (2 mL or 500 μL, respectively). For cytochrome C release quantification, 20 μM Q-VD-OPH (QVD, MP Biomedicals) was added until fixation to block the cells in early apoptosis. Cells were fixed with 1 mL 2% paraformaldehyde (Sigma Aldrich) for 15 min at RT, permeabilized for 20 min at RT in PBS containing 0.1% Triton X-100 (Sigma Aldrich) and incubated in blocking buffer (PBS, 0.1% Triton X-100, 2% bovine serum albumin (Sigma Aldrich)) for 30 min at RT. Next, cells were incubated for 90 min at RT with the primary antibody, anti-53BP1 (NB100-304, Novus Biologicals, 1/1000) or anti-cytochrome C (556432, BD Biosciences 1/100) diluted in blocking buffer. Following incubation cells were washed with PBS 0.1% Triton X-100 and incubated with the secondary antibody (goat anti-rabbit Alexa Fluor 594 or goat anti-mouse Alexa Fluor 488, Life Technologies; 1/1000) in blocking buffer for 60 min at RT. Cells were washed with PBS and mounted with Vectashield containing DAPI (Vector Laboratories).
For replication analysis, U2OS+SSTR2 cells were grown on glass coverslips in 24-well plates in 500 μL medium and 10 μM 5-ethynyl-2'-deoxyuridine (EdU) (Thermo Fisher Scientific) was added 1 h before fixation. Staining was performed according to manufacturer protocol (with Alexa fluor 594).
Propidium iodide staining for cell cycle analysis
For cell cycle distribution measurement, cells were grown in 6 well plates and incubated for 4 h with 5×10-8 M / 5 MBq 177Lu-DOTA-TATE or 5 MBq 177Lu-DTPA in 2 mL medium. Subsequently, cells were washed twice with PBS and incubated in 2 mL normal medium or medium containing 1 μM Olaparib. Samples were harvested by trypsinization 3 or 6 days after treatment. Following, cells were washed with PBS and fixed in 5 mL ice cold 70% ethanol. Cells were stored at -20oC until radioactivity was decayed. Cells were washed with PBS and permeabilized in 0.2% Triton X-100 in PBS for 5 min at RT. Subsequently cells were resuspended in 500 μL propidium iodide (PI) mix (50 μg/mL RNase (Roche) and 10 μg/mL PI (Sigma Aldrich) in PBS) and transferred to Fluorescence-activated cell sorting (FACS) tubes. Tubes were stored on ice for ~4 h until analysis. Cell cycle distribution was measured using a LSRFORTESSA FACS machine (BD Bioscience). Gating using the forward scatter and side scatter was used to discard aggregates and dead cells. PI was detected using the 610/20 bandpass emission filter.
Ex vivo culturing of human NET slices and treatment
For ex vivo assessment of PRRT, fresh pancreatic neuroendocrine tissue was obtained from patients undergoing surgery. After macroscopic investigation by the pathologist, remaining tumor tissue was used for storage in the biobank which includes use for research purposes. Tissue was obtained according to the code of proper secondary use of human tissue in the Netherlands established by the Dutch Federation of Medical Scientific Societies and approved by the local Medical Ethical committees. Specimens were coded anonymously in a way that they were not traceable to the patient by lab workers. Tissue samples were kept at 4oC and transported in DMEM/F12 medium (Lonza), supplemented with 10% fetal bovine serum, penicillin (50 units/mL) and streptomycin (50 µg/mL). Tumor tissue was sliced in 300 μm slices using a Leica VT 1200S Vibratome (vibration amplitude 3.0 mm and slicing speed 0.3 mm/sec). Culturing was performed in Medium D (DMEM/F12 supplemented with 10% fetal bovine serum, penicillin (50 units/mL), streptomycin (50 µg/mL), 0.3 µg/mL hydrocortisone (Sigma), 4 µg/mL insulin (Sigma), 8 ng/mL epidermal growth factor (Sigma) and 7 ng/mL cholera toxin (Sigma) (adapted and optimized for NET slices from [15])), at 5% CO2 and 37oC. Slices were cultured in 6 well plates and subjected to rotation at 60 rpm using a Stuart SSM1 mini orbital shaker that was placed in the incubator. At different time point after treatment, slices were fixed in 10% neutral buffered formalin for 24 h at RT. Subsequently, tumor slices were embedded in paraffin and 4 μm sections from 2 different regions of the tumor slice were generated for staining and microscopy analysis.
For PRRT experiments, slices were treated for 4 h with 5×10-9 M / 0.5 MBq 177Lu-DOTA-TATE or 0.5 MBq 177Lu-DTPA in 2 mL medium D. Subsequently, the slices were removed from the medium, washed for 1 min in PBS and transferred to 3 mL non-radioactive medium D with or without 10 μM Olaparib in new 6 well plates. Slices were incubated for different timepoints (up to 4.5 days after treatment).
Hematoxylin and eosin staining NET slices
For histological analysis, paraffin sections were deparaffinized in xylene and subsequently rehydrated by incubation in decreasing concentrations of ethanol. Sections were incubated for 1 min at RT in hematoxylin (Sigma Aldrich) followed by washing with tap water. Sections were incubated in eosin (Applichem) for 1 min at RT and subsequently dehydrated in increasing concentrations of ethanol. Before mounting with Entellan (Merck) sections were incubated in xylene.
Immunofluorescent stainings NET slices
For immunofluorescent 53BP1 staining, paraffin sections were deparaffinized in xylene and subsequently rehydrated by incubation in decreasing concentrations of ethanol. Target antigen retrieval was performed using a Target Retrieval Solution pH6.1 (Dako) for 18 min in a microwave at 650 W. Cells were permeabilized for 2×5 min at RT in PBS containing 0.1% Triton X-100 (Sigma Aldrich) and incubated in blocking buffer (PBS, 0.1% Triton X-100, 2% bovine serum albumin (Sigma Aldrich)) for 30 min at RT. Sections were incubated with the primary antibody, anti-53BP1 (1/1000) in blocking buffer for 90 min at RT. Subsequently, sections were washed with PBS 0.1% Triton X-100 and incubated with the secondary antibody (goat anti-rabbit Alexa Fluor 594, 1/1000) in blocking buffer for 60 min at RT. Cells were washed with PBS and mounted with Vectashield containing DAPI.
Imaging and analysis
For DSB (cells and slices) and micronuclei quantification, Z-stack imaging was performed using a TCS SP5 confocal microscope (Leica) and foci were counted from at least 50 cells per condition per experiment (cells) or on average 650 cells per condition per slice (slices) using Image J software (settings: median blur 1.0, maximum projection and find maxima, noise tolerance 75 for cells and 100 for slices). Micronuclei of least 50 cells per condition per experiment were counted manually and expressed as fraction of cells with micronuclei. For cytochrome C release quantification, imaging was performed using a DM4000 fluorescent microscope (Leica). At least 50 cells per condition per experiment were counted manually and expressed as fraction of cells with released cytochrome C (cytoplasmic and total release). For EdU staining, imaging was performed using a DM4000 fluorescent microscope and at least 100 cells per condition per experiment were analyzed. H&E stainings are image using the Olympus BX40 phase contrast microscope. Quantification data is presented as mean with standard error.
Uptake assay
U2OS+SSTR2 cells were seeded in 12-well plates in 1 mL medium and the next day adherent cells (~1×105 cells/ well) were incubated with 5×10-9 M/ 0.25 MBq 177Lu-DOTA-TATE in 1 mL medium for 4 h at 37°C with 5% CO2. Subsequently, cells were washed with PBS. For day 0 measurements, the membrane-bound fraction was separated from the internalized fraction by incubating cells for 10 min in 1 mL 50 mM glycine and 100 mM NaCl, pH2.8. Then, cells were lysed in 1 mL 0.1 M NaOH to collect the internalized fraction. For day 1-6 measurements, medium was added to the cells and they were incubated at 37°C, 5% CO2. For every time point, medium, membrane-bound fractions and internalized fractions were collected as described for day 0. All fractions were counted in a 1480 WIZARD automatic gamma counter (PerkinElmer). Data is expressed as percentage of added dose (%AD). Experiments were performed in triplicate and gamma-counter measurements were corrected for decay.
Statistics
Significance was determined using a homoscedastic 2-tailed student t-test. Samples were considered statistically different if p<0.05. * p<0.05, ** p<0.01 and *** p<0.001.
Results
PARP inhibition reduced cell number and proliferation after 177Lu-DOTA-TATE treatment
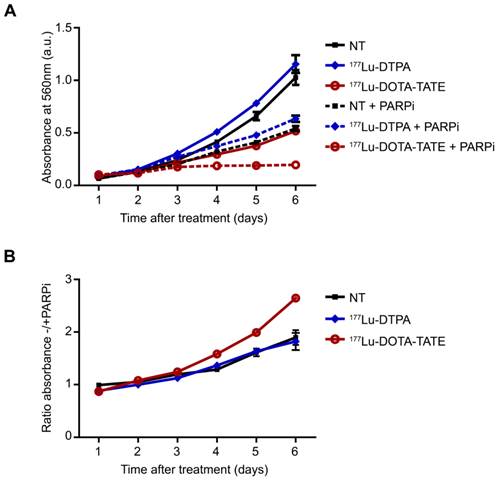
Cancer cells are characterized by their fast growing potential and anticancer treatments affect this via induction of apoptosis or inhibition of proliferation. Therefore, we examined whether inhibition of SSB-repair using the PARP inhibitor Olaparib affected cell number using a sulforhodamine beta (SRB) assay. Figure 1A shows that 177Lu-DOTA-TATE treatment significantly lowered the number of cells compared to 177Lu-DTPA treatment (DTPA is a chelator and does not bind SSTR2). Combination treatment of 177Lu-DOTA-TATE plus PARP inhibitor further reduced cell growth rate by 2.5 fold at day 6 after treatment (Figure 1B) and even completely blocked growth from day 3 at least until day 6 after treatment. PARP inhibition alone inhibited cell growth in non-treated or 177Lu-DTPA treated cells by 2 fold.
Effect of PARP inhibition on cell number after PRRT. (A) SRB assay; the absorbance in arbitrary units (a.u.) correlates with the relative number of cells. Cells were non-treated (NT) or treated with 177Lu-DTPA or 177Lu-DOTA-TATE and afterwards treated or not with PARP inhibitor (PARPi) and measured in triplicate at different time points after treatment. Error bars indicate the SEM. (B) Ratio of NT, 177Lu-DTPA or 177Lu-DOTA-TATE treated cells without/with PARPi. Error bars indicate the SEM.
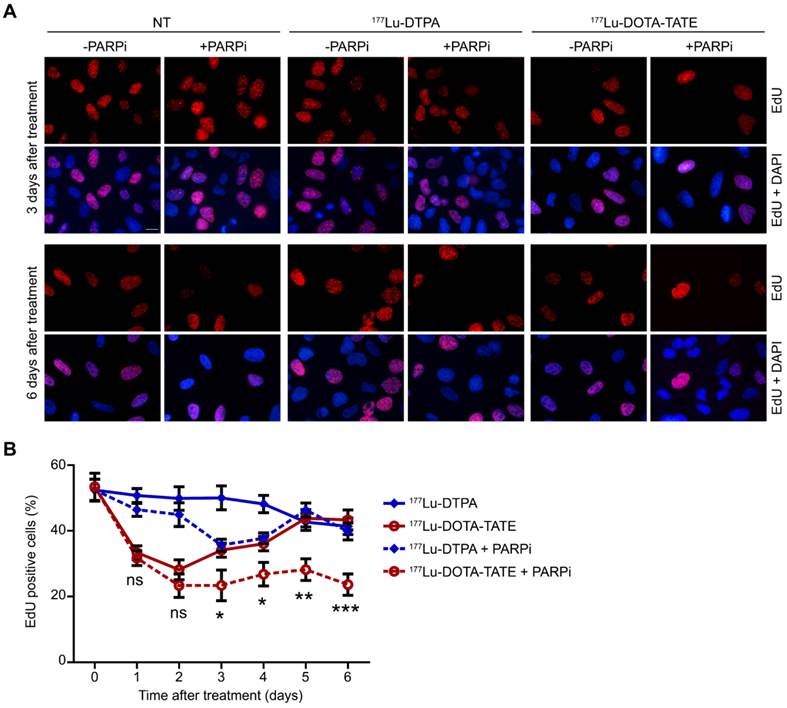
The total cell number is caused by a balance between cell death and proliferation. We therefore examined the effect of 177Lu-DOTA-TATE alone or combined with PARP inhibition on the cell cycle. EdU incorporation for 1 hour before fixation allowed quantification of S-phase cells (Figure 2A). Treatment with 177Lu-DOTA-TATE led to decreased replication from 1 to 4 days after exposure with 40-50%, which returned to normal levels at day 5. Combination treatment of 177Lu-DOTA-TATE with PARP inhibitor reduced the fraction of S-phase cells with 50% and this effect remained for at least 6 days after treatment. PARP inhibitor treatment alone did not significantly affect replication (Figure 2B and Figure S4A). The reduction of replication was caused by an increase of the number of cells in G2/M phase, as shown by propidium iodide (PI) FACS cell cycle analysis (Figure S1).
Effect of PARP inhibition on proliferation after PRRT.(A) Immunofluorescent EdU (red) and DAPI (blue) staining of a field of cells at T=3 days and T=6 days after treatment. Cells were non-treated (NT), treated with 177Lu-DTPA or with 177Lu-DOTA-TATE and afterwards treated or not with PARPi. Scale bar = 15 μm. (B) Quantification of the number of EdU positive cells at different time points after treatment. 8 microscope fields with on average 15 cells per field were quantified. Error bars represent the SEM and statistics represent the comparison between 177Lu-DOTA-TATE and 177Lu-DOTA-TATE + PARPi. Not significant (ns), * p<0.05, ** p<0.01 and *** p<0.001. NT data can be found in Figure S4A.
PARP inhibition enhanced 177Lu-DOTA-TATE induced cell death
The SRB assay showed a stabilization of the total cell population after the combination treatment of 177Lu-DOTA-TATE and PARP inhibitor. However, the EdU incorporation and PI assays showed a reduction, but not the complete absence of proliferation. This indicates that proliferation and cell death are in equilibrium. Therefore, we further investigated the effect of the combination treatment on cell death. Clonal survival experiments showed the specific cell killing by 177Lu-DOTA-TATE compared to 177Lu-DTPA treatment in both U2OS+SSTR (Figure 3A,B) and Ca20948 (Figure S2C,D) cells. U2OS cells without SSTR2 expression were not killed by 177Lu-DOTA-TATE treatment (Figure S2A,B), confirming the specificity of the treatment. Next, we investigated whether addition of the PARP inhibitor could specifically sensitize the SSTR2-positive U2OS cells. Combination treatment of 177Lu-DOTA-TATE and PARP inhibitor indeed significantly decreased the cellular survival compared to 177Lu-DOTA-TATE alone (reduction of 50% of the area under the curve), while PARP inhibition did not sensitize cells treated with 177Lu-DTPA (Figure 3A,B).
Effect of PARP inhibition on PRRT induced cell death.(A) Colony forming ability of cells after different activity quantities of PRRT (177Lu-DTPA or 177Lu-DOTA-TATE) and afterwards treated or not with PARP inhibitor (PARPi). The percentage of surviving cells was plotted against the applied PRRT activity, measured by counting surviving colonies of two independent experiments. Error bars indicate the SEM. (B) Area under the curve comparison from the data in panel 3A. Statistics: not significant (ns), ** p<0.01 and *** p<0.001. (C) Immunofluorescent cytochrome C (green) and DAPI (blue) staining of a field of cells 3 days after treatment. Cells with released cytochrome C were are marked with an asterisk. Cells were non-treated (NT), treated with 177Lu-DTPA or with 177Lu-DOTA-TATE and afterwards treated or not with PARPi. Scale bar = 15 μm. (C) Quantification of the number of cells with released cytochrome C at 1, 2 and 3 days after treatment. 8 microscope fields with on average 17 cells per field were quantified. Error bars represent the SEM. Statistics: not significant (ns), ** p<0.01 and *** p<0.001.
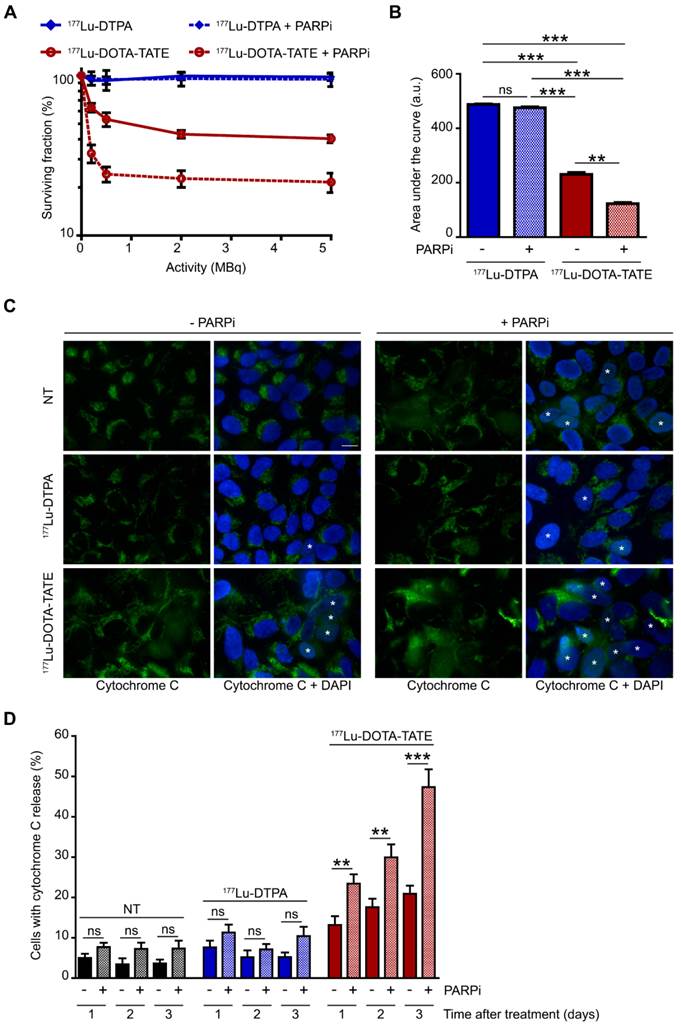
We then analyzed whether apoptosis was increased after combination treatment via measurement of cytochrome C release from the mitochondria into the cytoplasm (early step during apoptosis) (Figure 3C) [16]. The caspase inhibitor Q-VD-OPh was added to the cells directly after PRRT to block completion of apoptosis (step after cytochrome C release) and prevent cell detachment from the cell culture surface, enabling direct determination of accumulating apoptotic cells. As expected, 177Lu-DOTA-TATE treatment induced apoptosis over time. Consistent with the survival experiment, a significantly steeper increase in apoptosis was observed in cells treated with the combined treatment of 177Lu-DOTA-TATE and PARP inhibitor compared to 177Lu-DOTA-TATE alone (2.3 fold at 3 days after treatment) (Figure 3D).
Cytotoxicity is largely dependent on the absorbed dose, and thus on the quantity of radionuclide taken up by the cells. In order to exclude that the increased cell death might have been caused by increased radionuclide uptake after PARP inhibition, we determined membrane bound, internalized and medium (excreted plus released) radioactive fractions over time after PRRT in cells treated with and without PARP inhibitor (Figure S3). As we did not observe any difference between the two conditions, the increased cell death was not caused by the effect of the PARP inhibitor on radionuclide uptake/residence time, implying that a direct effect on the DDR is indeed the most likely explanation for its sensitizing effect.
PARP inhibition increased the number and persistence of DNA double strand breaks after 177Lu-DOTA-TATE treatment
The underlying cause of enhanced cell death after PARP inhibition is probably a difference in DNA damage induction and/or repair. Therefore, we determined the spatiotemporal distribution of DSBs after therapy. Accumulation of p53-binding protein 1 (53BP1) at DSB sites (so-called foci, Figure 4A) is a biomarker of radiation-induced DSBs, which has been used to monitor DNA damage and repair in a wide range of applications (reviewed in [17]). Figure 4 shows the timing and level of DSB induction and repair after PRRT by measuring the number of 53BP1 foci per nucleus. 177Lu-DOTA-TATE treatment of SSTR2-positive cells induced many DSBs which remained present for 3 days, whereas unbound 177Lu-DTPA only caused transient DSBs (Figure 4B and Figure S4B). Combination of 177Lu-DOTA-TATE treatment with PARP inhibition not only led to an increased number of DSBs at all time points, but surprisingly these DSBs were also present up to 6 days after 177Lu-DOTA-TATE treatment in the presence of PARP inhibitor, whereas DSBs were repaired at 4 days after treatment in the absence of PARP inhibition (Figure 4B). Cells treated with PARP inhibitor alone showed a small increase of DSBs, but this effect was minimal (Figure S4B).
Effect of PARP inhibition on double-strand break induction and genomic instability after PRRT. (A) Representative cells with immunofluorescent 53BP1 (red) and DAPI (blue) staining at 3 time points after treatment (0 hours, 3 days and 6 days). Cells were non-treated (NT), treated with 177Lu-DTPA or with 177Lu-DOTA-TATE and afterwards treated or not with PARP inhibitor (PARPi). Scale bar = 5 μm. (B) Quantification of 53BP1 foci of at least 50 cells per condition at different time points after treatment. Error bars represent the SEM. Statistics represent the comparison between 177Lu-DOTA-TATE and 177Lu-DOTA-TATE + PARPi. *** p<0.001. NT data can be found in Figure S4B. (C) Quantification of 53BP1 foci of at least 50 cells per condition of cells treated with 177Lu-DOTA-TATE and PARPi for 4 days. Following cells were treated or not with PARPi for different time points. Error bars represent the SEM. (D) Quantification of 53BP1 foci of at least 60 cells per condition of cells treated with or without PARPi combined or not with ionizing radiation exposure of 2 Gy (XRT). Cells were incubated with EdU directly after XRT and fixed 1 hour later to determine which cells were replicating at the time of radiation exposure. Error bars represent the SEM. Statistics: not significant (ns) and *** p<0.001. (E) Quantification of the number of cells with micronuclei. 10 microscope fields with on average 13 cells per field were quantified. Error bars represent the SEM and statistics represent the comparison between 177Lu-DOTA-TATE and 177Lu-DOTA-TATE + PARPi. ** p<0.01 and *** p<0.001. NT data can be found in Figure S4C and detailed quantifications can be found in Figure S5.
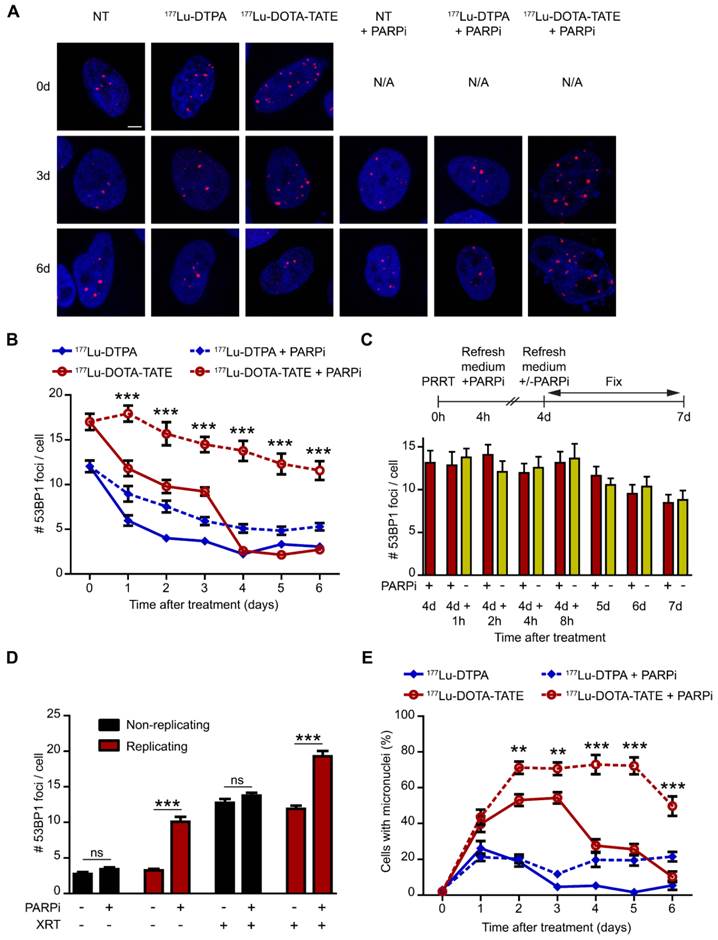
Ideally, one would prefer to minimize the time of patient PARP inhibitor treatment after PRRT. Therefore, we adapted the experiment such that the PARP inhibitor was removed at day 4 after treatment. Interestingly, cells treated with 177Lu-DOTA-TATE and PARP inhibitor for 4 days did not show a decrease in the number of DSBs when the PARP inhibitor was removed from the medium (Figure 4C), implying that a short interval of PARP inhibitor treatment may suffice to reach optimal tumor sensitization.
Theoretically, an increase of DSBs after PARP inhibition would only be expected in replicating cells, as they are expected to result from replication forks that encounter a single strand break. Therefore, cells were pretreated with PARP inhibitor and then exposed to an acute dose of ionizing radiation (external beam irradiation or XRT). Replicating cells were visualized by staining of the incorporated thymidine analogue 5-ethynyl-2'-deoxyuridine (EdU). XRT induced DSBs equally in replicating- and non-replicating cells. However, combination of XRT and PARP inhibition significantly increased the number of DSBs in replicating cells and not in non-replicating cells, indicating that a cell needs to go through replication for the PARP inhibitor to function as a sensitizer (Figure 4D).
Increased genomic instability after combination of 177Lu-DOTA-TATE treatment with PARP inhibition
Unrepaired DSBs may result in chromosome damage, leading to micronuclei formation. Micronuclei are extra-nuclear bodies that contain whole chromosomes or damaged chromosome fragments that were not incorporated into the nucleus after cell division (reviewed in [18]). In line with DSB induction, 177Lu-DOTA-TATE treatment led to an increased number of cells with micronuclei up to 4 days after treatment (Figure 4E and Figure S4C). As expected from transient DSB induction, 177Lu-DTPA treatment only caused a temporary increase in cells with micronuclei one day after treatment. Again, combination of 177Lu-DOTA-TATE treatment with PARP inhibition boosted the fraction of cells with micronuclei at later time points up to 6 days after treatment. Remarkably, not only the fraction of cells with micronuclei increased, but also the number of micronuclei per cell, especially at the later time points (Figure S5).
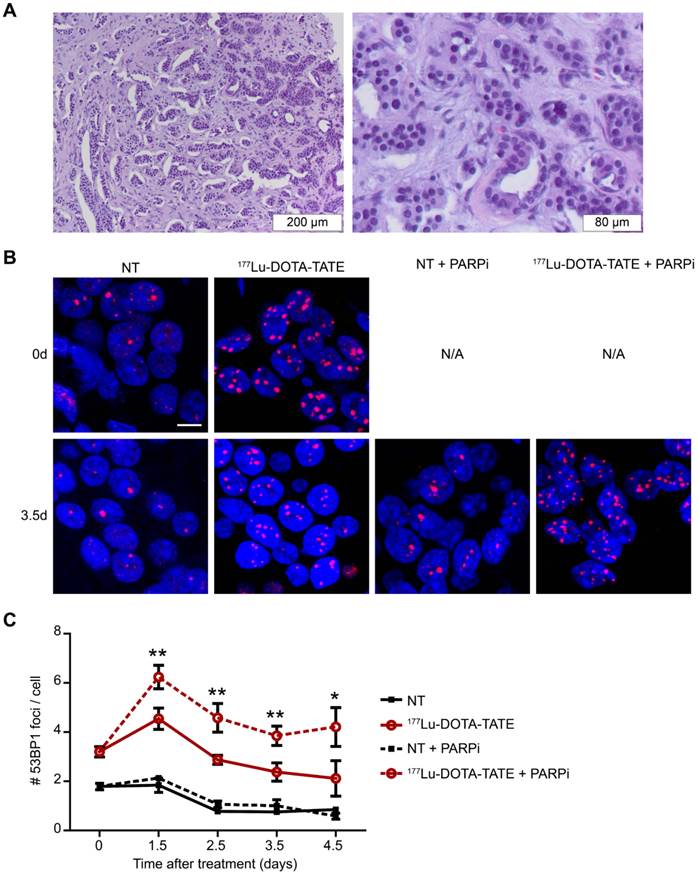
PARP inhibition increased the number of DNA double strand breaks after 177Lu-DOTA-TATE treatment in ex vivo cultured NET slices
The in vitro studies mentioned above have clearly shown that PARP inhibition can sensitize SSTR positive cells to PRRT. As several key parameters of tumors differ considerably from cells grown in 2D-culture (such as morphology and heterogeneity), we took the analysis of the DDR to the next level by using patient-derived NET-tissue (Figure 5A). As shown previously in cell cultures, tumor slices treated with 177Lu-DOTA-TATE showed an increase in the number of 53BP1 foci per nucleus, while treatment with 177Lu-DTPA did not induce DSBs (data not shown). Again, PARP inhibitor was able to further increase the number of 53BP1 foci induced by 177Lu-DOTA-TATE (Figure 5B,C). The tumor slices were examined up to 4.5 days after dissection (and 177DOTA-TATE treatment), but this timeframe was too short to observe effects on replication and apoptosis (data not shown).
Effect of PARP inhibition on double-strand break induction after PRRT in ex vivo cultured NET slices. (A) Hematoxylin and eosin staining of the fresh neuroendocrine pancreatic tumor; islands of NET cells are surrounded by stromal cells. (B) Immunofluorescent staining of 53BP1 (red) and DAPI (blue) of a field of cells at 2 time points after treatment (0 days and 3.5 days after treatment). Tissue slices were non-treated (NT) or treated with 177Lu-DOTA-TATE and afterwards treated or not with PARP inhibitor (PARPi). Scale bar = 5 μm. (C) Quantification of 53BP1 foci of different fields of cells at 2 locations per tumor slice of on average 660 cells per condition at different timepoints after treatment. Error bars represent the SEM. Statistics represent the comparison between 177Lu-DOTA-TATE and 177Lu-DOTA-TATE + PARPi. * p<0.05 and ** p<0.01.
Discussion
We explored the effect of combining PRRT with PARP inhibitors to sensitize tumor cells by increasing the number of cytotoxic DSBs. PARP inhibitors are associated with relatively mild side effects in the clinic when applied as a monotherapy [19] and are therefore ideal candidates for the use as combination agents. We showed that PARP inhibition synergistically sensitized SSTR2-expressing human tumor cells to 177Lu-DOTA-TATE treatment. This combination treatment reduced cell numbers and replication and induced cell death. The underlying mechanism is the inability to repair PRRT-induced SSBs that will be converted into DSBs during replication. Indeed we observed a vast increase in the number of DSBs that persisted over time, even when the PARP inhibitor was removed from the cells. As expected, this led to increased genome instability. An increase in the number of DSBs after combination treatment compared to 177Lu-DOTA-TATE treatment alone was also observed in ex vivo cultured NET slices. The innovating strategy of using PARP inhibitors to sensitize SSTR2-expressing tumor cells to PRRT has great potential to enhance the PRRT therapeutic index.
Several other studies have been performed to sensitize tumors to chemotherapeutics and radiotherapy using inhibitors of the DDR such as PARP inhibitors (reviewed in [9]). These approaches seem favorable in reducing tumor cells growth, but they are accompanied by significant damage to normal tissue. Several organs express (low levels) of the SSTR2, such as the pancreas, lung and cerebellum [20], which might be expected to acquire DNA damage by PRRT, as well. However, PARP inhibition only increases the number of DSBs in proliferating cells, as a result of replication through SSBs. Extra sensitization via PARP inhibition of the SSTR2-positive organs is therefore not to be expected, because little proliferation takes place in the adult organism. SSTR2 are also present on a small subset (<1%) of human bone marrow cells [20], however our experiments shows undetectable uptake of 177Lu-DOTA-TATE in the bone marrow (data not shown). Nonetheless, sensitization of the bone marrow by concomitant PARP inhibition might be expected, as they are proliferative, therefore close monitoring of bone marrow damage is essential. To minimize sensitization of bone marrow and non-targeted proliferative tissue, optimal timing for the combination treatment is essential. All unbound (circulating) 177Lu-DOTA-TATE needs to be cleared from the body before PARP inhibition treatment is initiated. As SSBs are generally repaired in the first few minutes after radionuclide exposure, one would expect that the sensitizing effect will be limited to the tumor as soon as the radionuclide has been cleared from circulation. It will be pivotal to monitor the effect of PARP inhibition in combination with PRRT closely in an in vivo model to optimize the timing of the combination regimen in order to reach maximum effect on the tumor and minimal normal tissue damage. Interestingly, triple combination treatment experiments in triple-negative breast cancer and pancreatic ductal adenocarcinoma xenograft models using radioimmunotherapy, chemotherapy and DDR inhibitors showed promising results in tumor eradication [21, 22].
We showed that the combination treatment increased the number of DSBs and, most interestingly, the breaks persisted for at least six days after treatment, whereas most DSBs were repaired within four days after 177Lu-DOTA-TATE monotherapy. Removal of the PARP inhibitor at day 4 after PRRT did not diminish the number of DSBs, indicating that they had been induced within the first 3 days after treatment and could not be repaired up to day 6 (even in the absence of the inhibitor). This implies that only a short treatment with the PARP inhibitor after PRRT is needed to obtain the favorable results. For patients this would mean that a brief treatment with the PARP inhibitor would be sufficient, minimizing the possibly enhanced side effects and reducing treatment costs.
Here we provided evidence of the sensitization of SSTR2-expressing human tumor cells and NET tissue slices to PRRT using the PARP inhibitor Olaparib. The knowledge gained in this study will also be applicable to other tumors that can be treated with PRRT, for example prostate tumors expressing the prostate specific membrane antigen (PSMA) [23]. Radionuclide treatment of PSMA-positive tumors is currently in clinical trials [24]. We expect that results of this project will also impact on this and similar radionuclide therapy applications. As Olaparib has been approved by the FDA and EMA, its application in PRRT in the clinic would be relatively easy and could be achieved in the near future.
Conclusion
In this study, we show that the innovative combination of sensitizing SSTR2-expressing human tumor cells to PRRT using the PARP inhibitor Olaparib; the combination treatment increased genotoxic stress and cell death and thereby has the potential to augment current PRRT outcomes without extra harm to healthy tissue.
Supplementary Material
Supplementary figures.
Acknowledgements
This study was supported by the Netherlands Organization for Scientific Research (ZON-MW grant 40-42600-98-018). Radionuclides were provided by IDB Holland. Fluorescent imaging was performed in collaboration with the optical imaging center (OIC) of the Erasmus MC. The authors thank Dr. E. de Blois and R. de Zanger for their technical assistance during radionuclide labeling procedures.
Conflict of interest
M. de Jong is shareholder in Advanced Accelerator Applications. The other authors declare that they have no competing interests.
References
1. Reubi JC. Peptide receptors as molecular targets for cancer diagnosis and therapy. Endocr Rev. 2003;24:389-427
2. Lamberts SWJ, Krenning EP, Reubi JC. The Role of Somatostatin and Its Analogs in the Diagnosis and Treatment of Tumors. Endocr Rev. 1991;12:450-82
3. de Jong M, Breeman WAP, Bakker WH, Kooij PPM, Bernard BF, Hofland LJ. et al. Comparison of In-111-labeled somatostatin analogues for tumor scintigraphy and radionuclide therapy. Cancer Res. 1998;58:437-41
4. de Jong M, Breeman WAP, Bernard BF, Bakker WH, Schaar M, van Gameren A. et al. [Lu-177-DOTA(0), Tyr(3)]octreotate for somatostatin receptor-targeted radionuclide therapy. Int J Cancer. 2001;92:628-33
5. Kwekkeboom DJ, de Herder WW, Kam BL, van Eijck CH, van Essen M, Kooij PP. et al. Treatment with the radiolabeled somatostatin analog [177 Lu-DOTA 0,Tyr3]octreotate: toxicity, efficacy, and survival. J Clin Oncol. 2008;26:2124-30
6. Vegt E, de Jong M, Wetzels JFM, Masereeuw R, Melis M, Oyen WJG. et al. Renal Toxicity of Radiolabeled Peptides and Antibody Fragments: Mechanisms, Impact on Radionuclide Therapy, and Strategies for Prevention. J Nucl Med. 2010;51:1049-58
7. Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E. et al. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature. 2005;434:913-7
8. Farmer H, McCabe N, Lord CJ, Tutt ANJ, Johnson DA, Richardson TB. et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434:917-21
9. O'Connor Mark J. Targeting the DNA Damage Response in Cancer. Mol Cell. 2015;60:547-60
10. Dungey FA, Loser DA, Chalmers AJ. Replication-Dependent Radiosensitization of Human Glioma Cells by Inhibition of Poly(Adp-Ribose) Polymerase: Mechanisms and Therapeutic Potential. Int J Radiat Oncol. 2008;72:1188-97
11. Dalm SU, Nonnekens J, Doeswijk GN, de Blois E, van Gent DC, Konijnenberg MW. et al. Comparison of the Therapeutic Response to Treatment with a 177Lu-Labeled Somatostatin Receptor Agonist and Antagonist in Preclinical Models. J Nucl Med. 2016;57:260-5
12. Bernard BF, Krenning E, Breeman WAP, Visser TJ, Bakker WH, Srinivasan A. et al. Use of the rat pancreatic CA20948 cell line for the comparison of radiolabelled peptides for receptor-targeted scintigraphy and radionuclide therapy. Nucl Med Commun. 2000;21:1079-85
13. Breeman WAP, Chan H, de Zanger RMS, Konijnenberg MK, de Blois E. Overview of Development and Formulation of 177Lu-DOTA-TATE for PRRT. Curr Radiopharm. 2016;9:8-18
14. Capello A, Krenning EP, Breeman WAP, Bernard BF, Konijnenberg MW, de Jong M. Tyr(3)-octreotide and Tyr(3)-octreotate radiolabeled with Lu-177 or Y-90: Peptide receptor radionuclide therapy results in vitro. Cancer Biother Radio. 2003;18:761-8
15. Naipal KAT, Verkaik NS, Sanchez H, van Deurzen CHM, den Bakker MA, Hoeijmakers JHJ. et al. Tumor slice culture system to assess drug response of primary breast cancer. Bmc Cancer. 2016:16
16. Goldstein JC, Rodier F, Garbe JC, Stampfer MR, Campisi J. Caspase-independent cytochrome c release is a sensitive measure of low-level apoptosis in cell culture models. Aging Cell. 2005;4:217-22
17. Panier S, Boulton SJ. Double-strand break repair: 53BP1 comes into focus. Nat Rev Mol Cell Biol. 2014Jan;15(1):7-18
18. Luzhna L, Kathiria P, Kovalchuk O. Micronuclei in genotoxicity assessment: from genetics to epigenetics and beyond. Frontiers in Genetics. 2013;4:131
19. Fong PC, Boss DS, Yap TA, Tutt A, Wu PJ, Mergui-Roelvink M. et al. Inhibition of Poly(ADP-Ribose) Polymerase in Tumors from BRCA Mutation Carriers. New Engl J Med. 2009;361:123-34
20. Unger N, Ueberberg B, Schulz S, Saeger W, Mann K, Petersenn S. Differential Expression of Somatostatin Receptor Subtype 1-5 Proteins in Numerous Human Normal Tissues. Exp Clin Endocr Diab. 2012;120:482-9
21. Al-Ejeh F, Shi W, Miranda M, Simpson PT, Vargas AC, Song S. et al. Treatment of Triple-Negative Breast Cancer Using Anti-EGFR-Directed Radioimmunotherapy Combined with Radiosensitizing Chemotherapy and PARP Inhibitor. Journal of Nuclear Medicine. 2013;54:913-21
22. Al-Ejeh F, Pajic M, Shi W, Kalimutho M, Miranda M, Nagrial AM. et al. Gemcitabine and CHK1 Inhibition Potentiate EGFR-Directed Radioimmunotherapy against Pancreatic Ductal Adenocarcinoma. Clinical Cancer Research. 2014;20:3187-97
23. Barve A, Jin W, Cheng K. Prostate cancer relevant antigens and enzymes for targeted drug delivery. J Control Release. 2014;187:118-32
24. Lutje S, Heskamp S, Cornelissen AS, Poeppel TD, van den Broek SAMW, Rosenbaum-Krumme S. et al. PSMA Ligands for Radionuclide Imaging and Therapy of Prostate Cancer: Clinical Status. Theranostics. 2015;5:1388-401
Author contact
![]() Corresponding authors: Erasmus MC, Department of Genetics, P.O. box 2040, 3000 CA Rotterdam, The Netherlands, Tel +31-107043449 (J.N.) and +31-107043932 (D.C.v.G), Fax +31-107044743, Email j.nonnekensnl (J.N.) and d.vangentnl (D.C.v.G).
Corresponding authors: Erasmus MC, Department of Genetics, P.O. box 2040, 3000 CA Rotterdam, The Netherlands, Tel +31-107043449 (J.N.) and +31-107043932 (D.C.v.G), Fax +31-107044743, Email j.nonnekensnl (J.N.) and d.vangentnl (D.C.v.G).