Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Material and methods
Results
Discussion
Supplementary Material
Abbreviations
Acknowledgements
References
Author biography
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2017; 7(6):1749-1769. doi:10.7150/thno.18415 This issue Cite
Research Paper
SAK-HV Triggered a Short-period Lipid-lowering Biotherapy Based on the Energy Model of Liver Proliferation via a Novel Pathway
Chao Zhang, Zhiguang Huang, Haoran Jing, Wenliang Fu, Min Yuan, Wenrong Xia, Ling Cai, Xiangdong Gan, Yao Chen, Minji Zou, Minhui Long, Jiaxi Wang, Min Wang* ![]() , Donggang Xu*
, Donggang Xu* ![]()
Laboratory of Genome Engineering, Beijing Institute of Basic Medical Sciences, Beijing, PR China.
*These authors share corresponding authorship.
Received 2016-11-19; Accepted 2017-3-2; Published 2017-4-10
Abstract

The accumulations of excess lipids within liver and serum are defined as non-alcoholic fatty liver disease (NAFLD) and hyperlipemia respectively. Both of them are components of metabolic syndrome that greatly threaten human health. Here, a recombinant fusion protein (SAK-HV) effectively treated NAFLD and hyperlipemia in high-fat-fed ApoE-/- mice, quails and rats within just 14 days. Its triglyceride and cholesterol-lowering effects were significantly better than that of atorvastatin during the observation period. We explored the lipid-lowering mechanism of SAK-HV by the hepatic transcriptome analysis and serials of experiments both in vivo and in vitro. Unexpectedly, SAK-HV triggered a moderate energy and material-consuming liver proliferation to dramatically decrease the lipids from both serum and liver. We provided the first evidence that PGC-1α mediated the hepatic synthesis of female hormones during liver proliferation, and proposed the complement system-induced PGC-1α-estrogen axis via the novel STAT3-C/EBPβ-PGC-1α pathway in liver as a new energy model for liver proliferation. In this model, PGC-1α ignited and fueled hepatocyte activation as an “igniter”; PGC-1α-induced estrogen augmented the energy supply of PGC-1α as an “ignition amplifier”, then triggered the hepatocyte state transition from activation to proliferation as a “starter”, causing triglyceride and cholesterol-lowering effects via PPARα-mediated fatty acid oxidation and LDLr-mediated cholesterol uptake, respectively. Collectively, the SAK-HV-triggered distinctive lipid-lowering strategy based on the new energy model of liver proliferation has potential as a novel short-period biotherapy against NAFLD and hyperlipemia.
Keywords: non-alcoholic fatty liver disease (NAFLD), hyperlipemia, liver proliferation, PGC-1α, estrogen, biotherapy.
Introduction
With the tremendous rise of prevalence in obesity, the morbidity of metabolic syndrome has been increased each year, greatly threatening human health. The accumulations of excess lipids within liver and serum are defined as non-alcoholic fatty liver disease (NAFLD) and hyperlipemia respectively. Both of them are components of metabolic syndrome, thus are closely associated with each other (1). Epidemiological and prospective studies have established the benefit of reducing LDL-cholesterol (2). Meanwhile, high triglycerides (TGs) are also closely related to increased CVD risk through their effects on HDL and LDL particle sizes that increase the atherogenicity of LDL (3, 4). Although there are many different kinds of lipid-lowering medications available, these drugs take effect on their specialized indications and are limited by their side effects (5). Even for the statins, the first choice lipid-lowering drugs (6), only 50% of the high-risk patients attain LDL-cholesterol targets with statins (7). Besides, the treatment cycles of these drugs are very long, which usually last for several months. Therefore, the exploration of novel lipid-lowering strategies may provide more choices for patients who cannot achieve current targets or who are intolerant to the existing therapies (5). Especially, the efficient lipid-lowering drugs with short treatment cycles are still needed to be developed.
The immune system plays an important role in metabolic regulation (8), and the complement system, as a central part of the innate immune system, is inextricably linked to metabolic alterations. Persson et al. found that intravenous immunoglobulin reduced the atherosclerotic lesions in the apolipoprotein E-deficient (ApoE-/-) and LDL receptor-deficient (Ldlr-/-) mice via the complement system, however its mechanism remains unclear (9). They also found that the complement component 3 (C3) knockout resulted in the deterioration of hyperlipidemia and atherosclerosis in ApoE-/- Ldlr-/- mice (10). In addition, the complement activation is critically involved in regulating liver proliferation (11-13). Strey et al. demonstrated that IL-6-STAT3 pathway played key roles in regulating liver proliferation mediated by C3a and C5a (14). Liver proliferation is an energy and material-consuming process that causes a dramatic consuming of both TG and cholesterol from serum and liver in a short time (15). Although the detailed lipid regulatory pathway is still unclear, lots of studies have reached a consensus on the lipid metabolic characteristics during this process (15-19). It enhanced fatty acid oxidation in liver for energy production, and upregulated both hepatic cholesterol synthesis and uptake for cell membrane synthesis (15, 16). The increased fatty acid oxidation in liver ameliorates the hepatic steatosis, and decreases the VLDL secretion to lower serum TG (17). Meanwhile, the upregulated hepatic cholesterol uptake dramatically lowers serum total cholesterol (TC) (18). Especially when serum cholesterol is high, the cholesterol uptake from serum may become a more important source in comparison with the hepatic cholesterol synthesis (18), which is not induced until preexisting cholesterol has become insufficient to meet the cellular demand (16). Collectively, these studies suggested that complement system might be involved in lipid metabolism regulation via liver proliferation, and liver proliferation might be a potential high-efficiency treatment strategy with a very short treatment cycle against NAFLD, hypertriglyceridemia and hypercholesteremia.
SAK-HV composed of a staphylokinase variant, tripeptide of arg-gly-asp (RGD), and a segment of 12 amino acid residues at the C-terminus of hirudin, is a fusion protein with the combined functions of thrombolysis, anticoagulation, and inhibition of platelet aggregation (20). We found surprisingly that SAK-HV significantly decreased the serum total TC and TG in ApoE-/- mice, and also markedly ameliorated the hepatic steatosis. During the observation period, the lipid-lowering effects of SAK-HV were even significantly better than that of atorvastatin.
The outstanding lipid-lowering effect of SAK-HV implied its distinctive mechanism of drug action. The liver is a key organ of lipid metabolism. PPARγ coactivator-1 α (PGC-1α), PPARα, and their target genes, regulating lipid oxidation in liver, not only ameliorate hepatic steatosis, but also decrease blood triglyceride. Besides, liver is the main site for lowering serum TC. Steroid hormones, such as estrogen that is closely related to cell cycle alteration in liver (21), also play important roles in hepatic lipid metabolism (22, 23). Therefore, we hypothesized that liver may be a target organ of SAK-HV.
Briefly speaking, we explored the lipid-lowering mechanism of SAK-HV and proposed a novel energy model for liver proliferation. Our study suggested that the SAK-HV-triggered distinctive lipid-lowering strategy based on the new energy model of liver proliferation had potential as a novel short-period biotherapy against NAFLD and hyperlipemia.
Material and methods
Animal and experimental protocol
Male ApoE-/- mice with a C57BL/6 background and wild-type mice were purchased from Beijing Huafukang Biotechnology Co. Ltd (Beijing Huafukang Biotechnology Co. Ltd, Beijing, China). The male C3-/- mice with a C57BL/6 background and wild-type C3+/+ mice were kindly provided by Dr. Chen Guojiang, Beijing Institute of Basic Medical Sciences. Dr. Chen purchased both male and female C3 homozygous knockout mice from Jackson Laboratory (Bar Harbor, ME, USA), and provided us with their male offspring.
Experimental animal feeding
Male mice aged 14 weeks were used as experiments mice. After adaptive feeding for one week, these ApoE-/- mice with a C57BL/6 background were fed on a high-fat-diet (cholesterol 2%, lard 20% the basic feed 78%) for one week. The male wild-type C57BL/6J mice with the same genetic background fed on a normal diet were used as blank control. For the PGC-1α knockdown experiment, ApoE-/- male mice aged 8 weeks were used as experiment mice. After adaptive feeding for one week, these mice were injected via tail veil with lentivirus at 100 μL/mouse, and after 3 days the PGC-1α knockdown model was established. Additionally, these C3-/- mice with a C57BL/6 background and wild-type animals C3+/+ were fed on a normal diet. All the mice were fasted for 12 h before sacrifice, and then the liver tissues and the blood samples drawn from the orbits were stored at -80℃.
RNA isolation and microarray analysis
The total RNA was extracted from mouse tissues by the Trizol reagent (Invitrogen, California, USA). The mRNA samples of mice liver (7 from SAK-HV 0.125mg/kg group and 5 from control group) were analyzed for gene expression profiling on the illumina WG-6V2 transcriptome chips (Illumina, San Diego, CA) using the fluorogenic dyes Cy3 and Cy5 for control and protease-treated conditions, respectively. The raw data are available from the Gene Expression Omnibus (GEO) repository, accession number GSE88964.
Differential expressed gene analysis
An expression matrix was processed for differential expression by employing the Rank Product method (RankProd) with the RankProd package in R. The differentially expressed genes were analyzed by WEB-based GEne SeT AnaLysis Toolkit (WebGestalt) for Gene Ontology (GO) enrichment, Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment, etc (24).
Hepatic weighted gene co-expression network analysis
The source code, the R packet of weighted gene co-expression network analysis (WGCNA), and the additional materials are freely available at http://www.genetics.ucla.edu/labs/horvath/CoexpressionNetwork/Rpackages/WGCNA (25). The lipid metabolism-related modules for SAK-HV group were obtained by identifying the modules that were significantly associated with the serum lipid levels through employing WGCNA packet with the correlation coefficient of 0.7 as the cutoff.
Blood biochemical tests
Serum levels of total cholesterol and triglyceride were measured by total cholesterol assay kit and triglyceride assay kit (Sekisui Medical Technology (China) Ltd., Beijing, China) respectively. The serum MDA level and SOD activity of mice were determined by Maleic Dialdehyde (MDA) Assay Kit (TBA method) and Superoxide Dismutase (SOD) assay Kit (WST-1 method) (Nanjing Jiancheng Bioengineering Co., Ltd., Nanjing, China) respectively. The tests of both liver function and coagulation parameters were performed by Beijing CIC Clinical Laboratory.
ELISA assay
FUT-175 (Becton, Dickinson and Company, Franklin Lakes, NJ) was immediately added to the blood samples for determining the serum level of both C3a and C5a before centrifugation. The serum levels of IL-6, C3a and C5a were determined respectively by following ELISA kits: IL-6 (eBioscience, San Diego, CA), C3a (Abgent, San Diego, CA), and C5a ELISA Kits (Abgent, San Diego, CA).
Histology and immunohistochemistry
Liver sections were fixed in formalin, and embedded in paraffin or OCT, then, were stained with Oil Red O (Sigma Chemical Co., St. Louis, MO), PCNA (Zhong-Shan Golden Bridge Biotechnology, Beijing, China), Masson trichrome staining (Soonbio, Beijing, China), TUNEL (Roche, Shanghai, China), and Hematoxylin and Eosin for analysis of hepatic fat accumulation, liver proliferation, liver apoptosis, liver fibrosis and pathological changes, separatively.
Western blot
The protein concentration was determined in the supernatant of liver tissue. The samples with equal amounts of protein were analyzed for Phospho-Tyr705-STAT3 (Cell signaling Technology, Boston, MA), Phospho-217-C/EBPβ (Santa Cruz Biotechnologies, CA, USA), PGC-1α (Abcam plc, Cambridge, UK), Phospho-Ser32-IқBα (Cell signaling Technology, Boston, MA), PPARα (Santa Cruz Biotechnologies, CA, USA), ABCG5 (Santa Cruz Biotechnologies, CA, USA), ABCG8 (Novus Biological, Littleton, USA), CYP7A1 (Abcam plc, Cambridge, UK), CYP19A1 (Abcam plc, Cambridge, UK), β-actin (Abgent, San Diego, CA, USA) and GAPDH (CWBIO, Beijing, China) by SDS-PAGE and Western blotting.
Assaying the levels of steroid hormones, acetyl-CoA, and lipids in the liver tissue
The liver tissue was homogenized on ice in normal saline. Then, the hepatic E1,E2,E3 and progesterone were measured in the liver tissue supernatant according to the manufacturer's instructions using enzyme radioimmunoassay kits (Beijing Zhichengkewei Biotechnology Science and Technology Co., Ltd., Beijing, China) by a radioimmunoanalyzer (model: XH6080) (Xi'an nuclear instrument factory, Shanxi, China). The hepatic acetyl-Coa level was measured according to the manufacturer's instructions using spectrophotometry kits (Beijing Zhichengkewei Biotechnology Science and Technology Co., Ltd., Beijing, China). In addition, the hepatic total cholesterol was measured using ELISA kits (Applygen Technologies Inc., Beijing, China) according to the manufacturer's instructions. The hepatic triglyceride was measured using ELISA kits (KeyGEN BioTECH, Co., Ltd., Nanjing, China) according to the manufacturer's instructions.
MTT assay
20μL was taken from 5mg/mL MTT solution and added to each well of plate, then the plate was further incubated at 37°C for 4 hours. Thereafter, the medium was pipetted out, and 150μL DMSO was added to each well. The microtiter plate was shaked on a shaker, so as to dissolve the dye. After the formazan was fully dissolved, the absorbance at 490 nm was measured on a microplate reader.
Flow cytometry
The samples were processed according to the manufacturer's instructions using cell cycle detection kit (KeyGEN BioTECH, Co., Ltd., Nanjing, China), then examined by flow cytometry (model: FACSCalibur) (Becton, Dickinson and Company, Franklin Lakes, NJ).
Real-time quantitative polymerase chain reaction (qPCR)
The gene expressions were analyzed by qPCR. The primers for qPCR are listed in Table S1.
Construction of cDNA expression plasmids, shRNA , and lentiviruses
The shPGC-1 lentivirus (5'-GGTGGATTGAAGTGGTGTAGA-3') (26) was bought from GenePharma (Shanghai GenePharma Co., Ltd, Shanghai, China). For construction of shRNA lentiviruses, each short hairpin preceded by the human U6 promoter was inserted into LV3 (H1/GFP&Puro). Recombinant lentivirus (0.5×109 TU) was delivered by tail-vein injection to each mouse 4 days before SAK-HV or PBS injection to construct the PGC-1α knockdown mice model.
Chromatin immunoprecipitation (ChiP) Assay
The ChiP kit was purchased from Merk Milipore (Merk Milipore, Shanghai, Beijing). The fresh liver tissues were minced in cold PBS, and was crosslinked in 1% formaldehyde at room temperature for 15 mins. Following PBS washes, formaldehyde was quenched by the glycine. Then these liver tissues were grinded with a homogenizer and resuspend in SDS Lysis Buffer containing Protease Inhibitor Cocktail II. Then these tissues were sonicated on ice for DNA shearing, and centrifuged at 12,000×g for 10 min at 4℃ to get chromatin DNA samples. After preclearing of these chromatin DNA samples, 2μg of anti-C/EBPβ (Abcam plc, Cambridge, UK)/anti-STAT3 (Cell signaling Technology, Boston, MA) or anti-rat IgG antibody (Thermo Scientific, Fremont, CA), and 60μL of Protein G Agarose (Biowest, Vieux Bourg, France) were added to carry out chromatin immunoprecipitation. Then the Protein G Agarose-antibody/chromatin complex was washed by various buffers. Following elution, reverse crosslinking, and purification of these complex, the immunoprecipitated DNA were prepared. Then qPCR analysis was performed using both immunoprecipitated DNA of each sample and input DNA as templates, in which the input DNA was adopted as control. The following primers were used to amplify C/EBPβ and PGC-1α promoters respectively. C/EBPβ promoter: Forward, 5' -CACACCAGGCACACCAAGCACAC 3', and Reverse, 5' CCACGGGGAGGCCAGAGGAT 3' (27). PGC-1α promoter: Forward, 5' CAAAGGCCAAGTGTTTCCTT 3', and Reverse, 5' TTGCTGCACAAACTCCTGA 3' (28); All reactions were carried out with three to five independent biological replicates and performed with reference dye normalization. The median cycle threshold (Ct) value was used for analysis. ChIP qPCRs were normalized to total input by calculating the fold enrichment. Before qPCR analysis, semi-quantitative PCR analysis was performed to verify both the binding of C/EBPβ to the PGC-1α promoter and the binding of STAT3 to the C/EBPβ promoter.
Detailed Animal experiments designs are described in Supplemental Methods.
Statistics
Data in the figures were presented as the mean ± SEM of one representative experiment. Statistical analysis was performed in R using one-way analysis of variance (ANOVA) or two-way ANOVA, both with Tukey's multiple comparisons test, and unpaired 2-tailed Student's t test according to the experiment grouping designs. The differences at 5% level were considered statistically significant.
Ethics Committee Approval
All animal experiments were approved by the local government authorities and were carried out according to the guidelines of the Institutional Animal Care and Use Committee of the Academy of Military Medical Sciences (Approval number: IACUC of AMMS-13-2015-008).
Results
Pharmacodynamic evaluation of SAK-HV
The effective dosages of SAK-HV for ApoE-/- mice ranged from 0.0625 to 0.5 mg/kg. Thus, the doses of 0.0625, 0.125, 0.25, and 0.5 mg/kg were chosen to explore the optimal dosage to achieve a lipid-reducing effect. The serum TC and/or TG were decreased in all SAK-HV-treated groups on the 14th day, and the lipid-lowering effect of SAK-HV in 0.125 mg/kg group was optimal (Figure 1A). Hepatic steatosis in the 0.125 mg/kg group was ameliorated (Figure 1B), and the hepatocytic morphology was mildly improved without necrosis (Figure S1A). The inflammatory levels in both liver and serum also decreased to be similar to that of the normal group (Figure S1, B-E). The TUNEL detection and Masson trichrome staining indicated that neither apoptosis nor fibrosis in liver was caused by SAK-HV treatment (Figure S2, A and B). Besides that, although SAK-HV caused no significant changes of coagulation parameters in ApoE-/- mice, it mildly improved these indexes to be similar to these of the normal group (Figure S2, C-F), demonstrating that SAK-HV administration at therapeutic dose did not aggravate the bleeding tendency.
Pharmacodynamics evaluation of SAK-HV in ApoE-/- mice. (A) The dosage-response relationship of SAK-HV was in a U-shape, and the lipid-lowering effect of 0.125 mg/kg group was optimal (n=8). (B) The curative effect of SAK-HV (0.125 mg/kg) on hepatic steatosis (magnification, 20×; n=5). (C) The time-effect relationship of SAK-HV (0.125 mg/kg). No significant lipid-lowering effects were observed at the 7th and 10th days; however, they were significantly decreased at the 14th day (n=10). (D) SAK-HV (0.125mg/kg) showed better lipid-lowering effects than atorvastatin (100mg/kg) at the 14th day (n=8). ApoE-/- mice were injected via tail vein with SAK-HV and PBS for SAK-HV groups and the model group respectively. An oral dosage of atorvastatin (100mg/kg) was given to the atorvastatin group. The C57BL/6 mice were used as the normal control. Abbreviations: M, model; NC, normal control; S, SAK-HV; TG, triglyceride; TC, total cholesterol, NS, no significance. One-way ANOVA with Tukey's multiple comparisons test for A, C and D. *P < 0.05; **P < 0.01; ***P < 0.001.
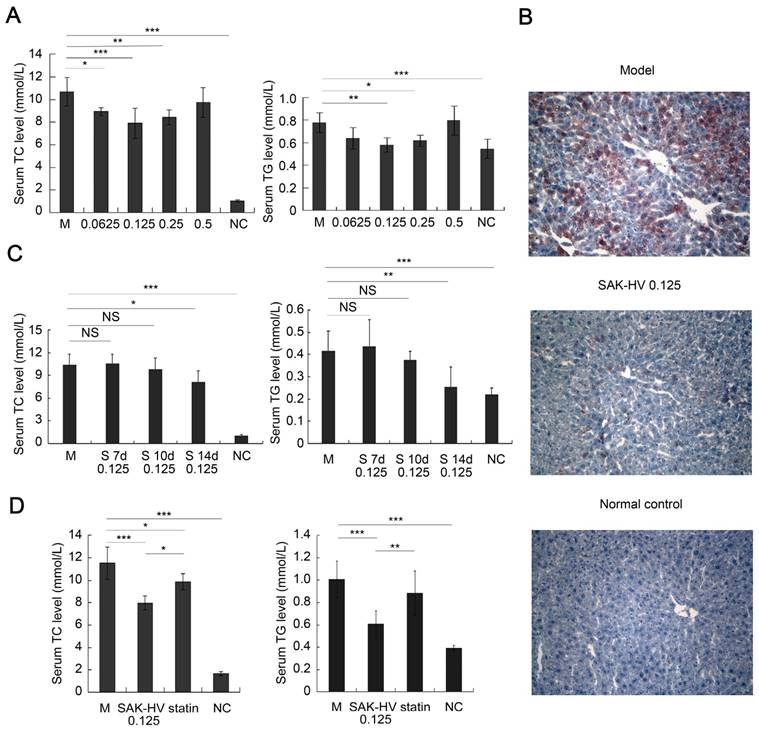
Then we treated the mice with 0.125 mg/kg of SAK-HV and evaluated its time-effect relationship. Unexpectedly, no significant changes of the serum lipid levels were found on the 7th and 10th day; however, both serum TC and TG were significantly decreased on the 14th day (Figure 1C). These results indicated that the lipid-lowering process of SAK-HV was swift and violent, and broke out suddenly with an about 10 days' delay, showing that the time-window of lipid-lowering was just about 4 days. Additionally, its dosage-response relationship was in a U-shape, but not in a dose-dependent manner.
Furthermore, SAK-HV displayed better lipid-lowering effects than atorvastatin (100mg/kg (29)) during the 14-day observation period (Figure 1D). Its effect of anti-oxidative stress was similar to that of atorvastatin (Figure S3). SAK-HV treatment also improved the liver function to be similar to that of normal group, except for the serum cholinesterase, which was still significantly better than that of atorvastatin group (Figure S4).
Besides, SAK-HV exerted effective lipid-lowering effects on high-fat-fed rats (Figure S5) and quails (Figure S6), and significantly decreased their inflammation and oxidative stress in serum as well (data not shown), both of which indicated its general applicability.
Differential gene expression analysis suggested that complement activation triggered hepatocytic proliferation to lower lipids
513 differentially expressed genes were identified from the liver transcriptome of ApoE-/- mice, including 418 up-regulated genes and 95 downregulated genes (Table S2). Next, we focused on the upregulated genes, which were the main differentially expressed genes, for the following analysis. The main biological activities of the upregulated genes included 3 categories: 1) the mainly exogenous stimulus-based immune response; 2) the cell activation and proliferation-related biological activities; and 3) the biological process of metabolic process (Table S3). KEGG analysis of these upregulated genes identified complement and coagulation cascades (p = 1.00E-04), antigen processing and presentation (p = 1.67E-05), steroid biosynthesis (p = 3.21E-06), steroid hormone biosynthesis (p = 5.40E-03), and PPAR signaling pathway (p = 2.00E-04) (Table S4). These results indicated that the SAK-HV-induced biological function disturbances involved in complement activation, cell activation and proliferation, and both cholesterol and triglyceride metabolisms, suggesting that the foreign SAK-HV may induce the complement activation-mediated hepatocytic proliferation, and therefore cause an increased demand of energy and materials to alter lipid metabolism. During this process, the serum TG might be reduced by the PPAR signaling pathway. Besides, cholesterol-related biological disturbances included steroid biosynthesis and steroid hormone biosynthesis. Steroid biosynthesis during liver proliferation is necessary for the cell division (30), and may not cause cholesterol-lowering effects. Thus, our results suggested a potential correlation between the declined serum TC and the enhanced steroid hormone biosynthesis in liver.
STAT3-C/EBPβ-PGC-1α was screened out by weighted gene co-expression network analysis (WGCNA) as a potential novel lipid-reducing pathway of SAK-HV
The two most relevant modules of cholesterol metabolism, cyan and magenta (r = -0.93, p < 0.01; r = -0.75, p < 0.05; respectively), and the most relevant module of triglyceride metabolism, turquoise (r = -0.71, p < 0.05), were identified by WGCNA from liver transcriptome of SAK-HV group (Table S5). Analysis for the cyan module identified the cell proliferation-related metabolic process, including DNA metabolic process (p = 1.65E-02), purine nucleoside metabolic process (p = 3.06E-02), and some anabolism-related biological process, such as macromolecule biosynthetic process (p = 2.51E-02), cellular component biogenesis (p = 1.32E-02) and ribosome biogenesis (p = 6.20E-03) (Table S6), suggesting that the decreased serum TC was closely related to the proliferation-related anabolism. In general, the important hub genes are usually the differentially expressed genes. Based on this hypothesis, Pgc1α and C6 were screened out by determining the intersection of the upregulated genes and hub genes with high intramodular connectivity of the cyan module. C6 again suggested that the decreased serum TC was closely related to the complement system. Pgc1α is involved in regulating steroid hormone biosynthesis (31-33). Thus, the complement system and PGC-1α may be effectors in the SAK-HV-induced cholesterol metabolism disturbance. Cyclin D (Ccnd) 2 and Nuclear receptor 5a (Nr5a)2 were also identified from the magenta module in this manner. Ccnd2 further suggested that the decreased TC was closely related to cell proliferation. Additionally, Nr5a2 and its target hydroxy-delta-5-steroid dehydrogenase, 3 beta- and steroid delta-isomerase 2 (Hsd3b2) are the critical genes in steroid hormone biosynthesis (34), and Hsd3b2 was also up-regulated (Table S2). PGC-1α up-regulates the transcription levels of both Nr5a2 and Hsd3b2 (35, 36), suggesting that PGC-1α-mediated steroid hormone biosynthesis may play important roles in the cholesterol metabolism. These results further supported the conclusions of differential gene expression analysis, and suggested the central role of PGC-1α in cholesterol metabolism in liver during SAK-HV treatment.
KEGG analysis for the turquoise module identified peroxisome (p = 4.00E-04), PPAR signaling pathway (p = 3.88E-02), and fatty acid metabolism (p = 1.21E-02), suggesting that PPARα-mediated fatty acid oxidation pathway played important roles in SAK-HV-regulated triglyceride metabolism (Table S7). PPARα is coactivated by PGC-1α to enhance the fatty acid oxidation pathway (37, 38). Thus, PGC-1α may be also the effector of triglyceride metabolism during SAK-HV treatment. 136 genes were screened out by determining the intersection of the upregulated genes and the genes of turquoise (Table S8). Wikipathways analysis for these genes identified the IL-6 signaling pathway (IL-6-STAT3 pathway) (p = 3.84E-02) (Table S9). Complement activation activates IL-6 signaling pathway, which is closely related to the liver proliferation rate (39). Additionally, as a member of IL-6 signaling pathway, C/EBPβ activates PGC-1α during liver proliferation with an amplitude of induction that exceeds the fasting response (28). These data suggested that SAK-HV triggered complement activation to induce the IL-6-STAT3 pathway, leading to the PGC-1α-mediated lipid regulation. Therefore, the complement system-mediated STAT3-C/EBPβ-PGC-1α may constituted a novel lipid-reducing pathway of SAK-HV.
SAK-HV activated the classical complement pathway and the potential downstream STAT3-C/EBPβ-PGC-1α pathway, and triggered the moderate liver proliferation in ApoE-/- mice
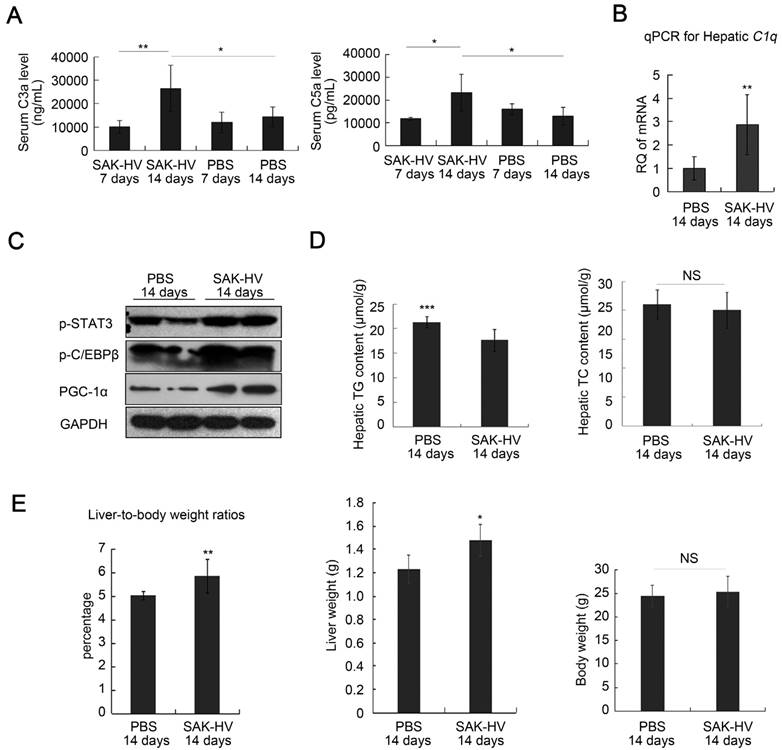
The specific IgG against SAK-HV in serum was not generated on the 7th day, but its antibody titer reached 102.88 on the 14th day (Figure S7A). The consensus result was that the serum C3a and C5a did not changed significantly on the 7th day, but up-regulated on the 14th day, accompanied with raised hepatic transcriptional level of C1q (Figure 2, A and B). The phosphorylation levels of STAT3 and C/EBPβ, and the expression of PGC-1α were also upregulated in liver on the 14th day (Figure 2C). These data supported that SAK-HV triggered the classical complement activation and induced its potential downstream STAT3-C/EBPβ-PGC-1α pathway. While there were no significant changes in the hepatic TC content after SAK-HV treatment, the declined hepatic TG level confirmed the ameliorated hepatic steatosis (Figure 2D). Given that SAK-HV effectively ameliorated hepatic steatosis, the elevated liver-to-body weight ratios in SAK-HV group, as well as the corresponding liver weights and body weights, suggested that SAK-HV triggered a moderate liver proliferation (Figure 2E).
SAK-HV-triggered pharmacodynamic effects in ApoE-/- mice I. (A and B) SAK-HV triggered the classical complement activation on the 14th day but not on the 7th day (n=6). (C) SAK-HV activated the potential lipid-lowering pathway STAT3-C/EBPβ-PGC-1α in liver (n=4). (D) SAK-HV significantly ameliorated the hepatic steatosis (n=8). (E) Given the significantly ameliorated hepatic steatosis by SAK-HV (Figure 2D), the increased liver-to-body weight ratios, as well as the corresponding liver weights and body weights suggested that SAK-HV triggered a moderate liver proliferation on the 14th day. (n=8). ApoE-/- mice were divided into four groups, and were injected with SAK-HV or PBS via tail vein for 7 or 14 days, separatively. Abbreviation: C, complement component. PGC-1α, PPARγ coactivator-1 α. Two-way ANOVA with Tukey's multiple comparisons test for A; Student's t test for B, D and E.
SAK-HV-triggered lipid and steroid metabolism alterations in liver of ApoE-/- mice
The up-regulated transcription levels of both Ppara and carnitine palmitoyltransferase I α (Cpt1a), and the increased acetyl-CoA level suggested that fatty acid oxidation in liver was enhanced (Figure 3A). Additionally, the up-regulated transcription levels of Ldlr in liver after SAK-HV treatment suggested an enhanced LDLr-mediated cholesterol uptake (Figure 3B). While both the transcriptional and protein levels of liver X receptor (LXR) transcriptome in liver involved in cholesterol catabolism secretion were not significantly changed after SAK-HV treatment (Figure 3, C and D).
SAK-HV-triggered pharmacodynamic effects ApoE-/- mice II. (A) The up-regulated transcription levels of Ppara and its target gene Cpt1a (Left, n=6), and the increased acetyl CoA level in liver (Right, n=8) suggested that SAK-HV enhanced PPARα-mediated fatty acid oxidation in liver. (B) The up-regulated transcription level of Ldlr in liver suggested an enhanced LDLr-mediated hepatic cholesterol uptake (n=6). (C) SAK-HV treatment caused no significant changes in the transcription levels of LXR transcriptome involved in cholesterol catabolism secretion in liver (n=6). (D) SAK-HV treatment caused no significant changes in the protein levels of LXR transcriptome, but upregulated CYP19A1, the estrogen synthase (n=4); (E) The genes involved in female hormone synthesis were transcriptionally up-regulated in liver after SAK-HV treatment. (n=6). (F) The female hormones, estrogen (Right) and progesterone (Left), were significantly up-regulated in liver (n=8); (G) The serum levels of estrogen (Right) and progesterone (Left) were not statistically significant upregulated (n=8). Abbreviation: carnitine palmitoyltransferase I α (Cpt1a), Ldlr, ldl receptor; LXR, liver X receptor; Cyp7a1, cholesterol 7-alpha-monooxygenase or cytochrome p450 7a1; Abcg, ATP-cassette binding proteins G; Nr5a, nuclear receptor 5a 2; Cyp19a1, cytochrome P450 family 19 subfamily a member 1; Hsd3b2, hydroxy-delta-5-steroid dehydrogenase, 3 beta- and steroid delta-isomerase 2. Student's t test for A-F.
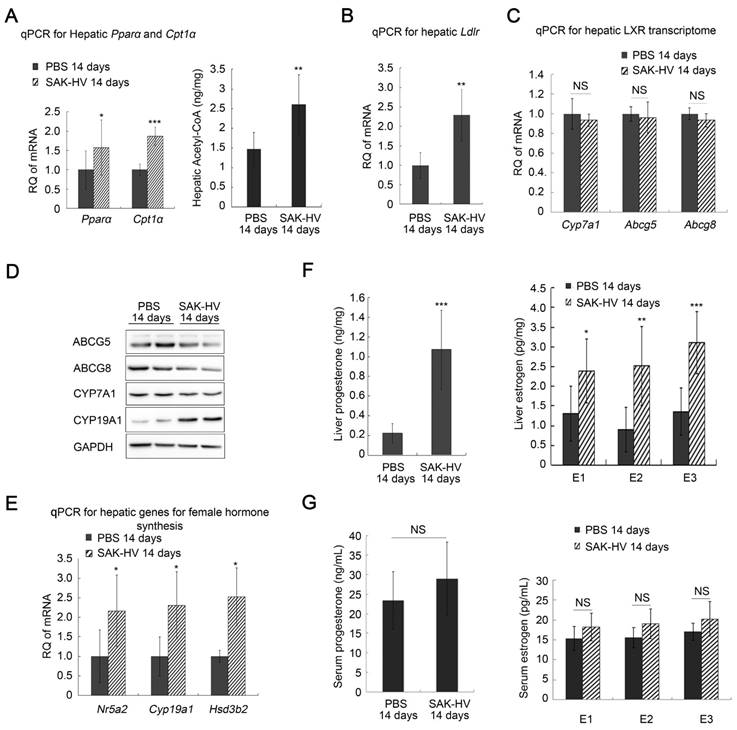
Meanwhile, the transcriptional levels of Nr5a2, cytochrome P450 family 19 subfamily a member 1 (Cyp19a1), and Hsd3b2 were all up-regulated in liver by SAK-HV, as well as the protein level of CYP19A1, namely estrogen synthase (Figure 3,D and E). Their products, estrogen and progesterone, were also significantly up-regulated in liver (Figure 3F). These results indicated that SAK-HV enhanced the synthesis of female hormones in liver. However, neither of the serum levels of these two hormones was statistically significantly increased (Figure 3G).
Among these female hormones, estrogen is a potent TC and TG-lowering hormone (40), and promotes liver proliferation (41). So we focused our attentions on the estrogen metabolism induced by SAK-HV. Liver is the major organ for estrogen metabolism and excretion. We further checked the transcriptional levels of several genes involved in this process, including cytochrome P450 family 1 subfamily A member 2 (Cyp1a2) and cytochrome P450 family 3 subfamily A member 11 (Cyp3a11) for phase I elimination of estrogen, and UDP Glucuronosyltransferase Family 1 Member A1 (Ugt1a1), sulfotransferase family 1E member 1 (Sult1e1) and catechol-O-methyltransferase (Comt) for phase II elimination of estrogen, and ATP Binding Cassette Subfamily C Member 2 (Abcc2) for biliary excretion of estrogen. Among them, the transcription levels of Cyp1a2 and Sult1e1 were significantly decreased after SAK-HV treatment, although the others were not significantly changed (figure S7B). These results suggested that the SAK-HV-induced upregulation of estrogen in liver might be attributed to both increased synthesis and decreased inactivation of estrogen in liver. The elevated estrogen levels in liver may explain the potential correlation of the decreased serum TC with the enhanced synthesis of steroid hormone.
Based on the hepatic transcriptome analysis and all these investigations, we hypothesized that SAK-HV decreased the lipid levels via an energy-consuming hepatocytic proliferation, and the complement system-mediated STAT3-C/EBPβ-PGC-1α in liver constituted the novel lipid-lowering pathway, in which PGC-1α-triggered hepatic intracrine estrogen may play important roles.
Estrogen inhibition blocked liver proliferation and weakened lipid-lowering effects of SAK-HV
Letrozole, the aromatase inhibitor, effectively inhibited the SAK-HV-induced estrogen in liver (Figure 4A), suggesting that estrogen synthesis played a more important role than its decreased inactivation in increasing estrogen in liver by SAK-HV. As a result, the upregulation of proliferating cell nuclear antigen (PCNA) induced by SAK-HV was significantly inhibited by letrozole (Figure 4B), indicating the important role of estrogen in triggering liver proliferation.
Estrogen inhibition broke the SAK-HV-triggered liver proliferation, and blocked the SAK-HV-induced lipid metabolism alteration. (A) Letrozole effectively inhibited the upregulation effect of SAK-HV on hepatic synthesis of estrogen (n=10). (B) The immunohistochemical detection of PCNA indicated that letrozole remarkably inhibited the SAK-HV-triggered liver proliferation (magnification, 200×; n=5). (C) Letrozole partially inhibited the SAK-HV-induced upregulation trends of both the transcription level of Ppara and its target genes (Left, n=6), as well as protein level of PPARα (Right, n=4) and acetyl CoA levels in liver (Right, n=10). (D) Letrozole inhibited the SAK-HV-induced transcriptional upregulation of Ldlr in liver (n=6). The aromatase inhibitor letrozole or its PBS solution was injected subcutaneously to the ApoE-/- mice to explore the role of estrogen in the drug action of SAK-HV, and these ApoE-/- mice were divided into four groups, model, SAK-HV, Letrozole, and SAK-HV+letrozole group. Abbreviation: Acot, Acyl-CoA thioesterase. Two-way ANOVA with Tukey's multiple comparisons test for A, C and D.
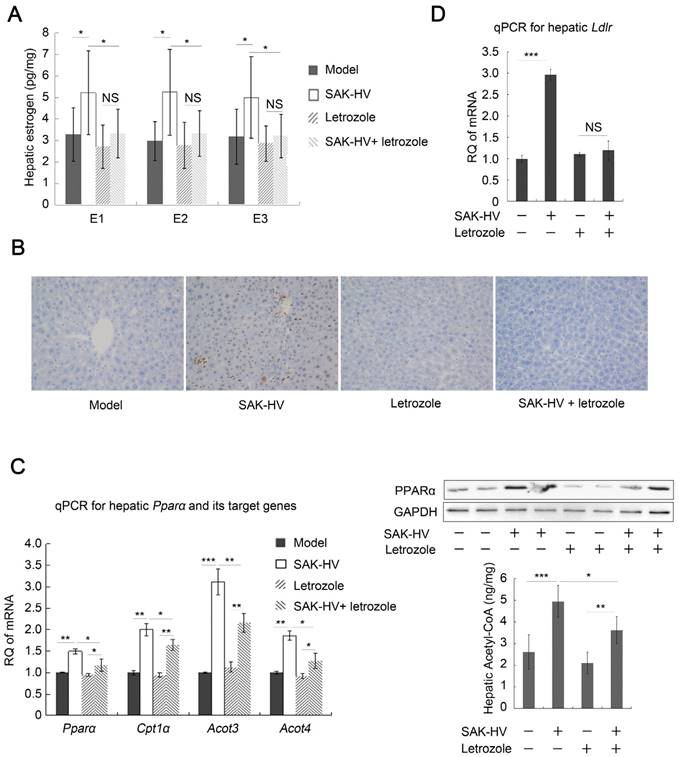
Furthermore, it remarkably weakened, but not completely inhibited the SAK-HV-induced upregulation trends of Ppara and its target genes in liver, as well as hepatic protein level of PPARα and acetyl-CoA level (Figure 4C), indicating that estrogen played partial roles in SAK-HV-induced hepatic fatty acid oxidation. The Ldlr was transcriptionally upregulated in SAK-HV group, but not in SAK-HV+letrozole group (Figure 4D), suggesting an enhanced LDLr-mediated cholesterol uptake induced by estrogen after SAK-HV treatment. Consequently, letrozole impaired the serum TC and TG-lowering effect of SAK-HV in ApoE-/- mice (Figure 5A). Besides, it also significantly compromised the hepatic TG content-lowering effect of SAK-HV, although no significant changes of hepatic TC content were found among all groups (Figure 5B), indicating that estrogen inhibition weakened the curative effect of SAK-HV on hepatic steatosis. These results were further intensified by liver sections stained with oil red (Figure 5C).
Estrogen inhibition weakened the lipid-lowering effect of SAK-HV. (A) Estrogen inhibition compromised the serum lipid-reducing effects of SAK-HV (n=10). (B) Although no significant changes were found in hepatic TC content in liver, letrozole significantly inhibited the hepatic TG contend-reducing effect of SAK-HV (n=10). (C) Estrogen inhibition weaken the curative effect of SAK-HV on hepatic steatosis (magnification, 20×; n=5). Two-way ANOVA with Tukey's multiple comparisons test for A and B.
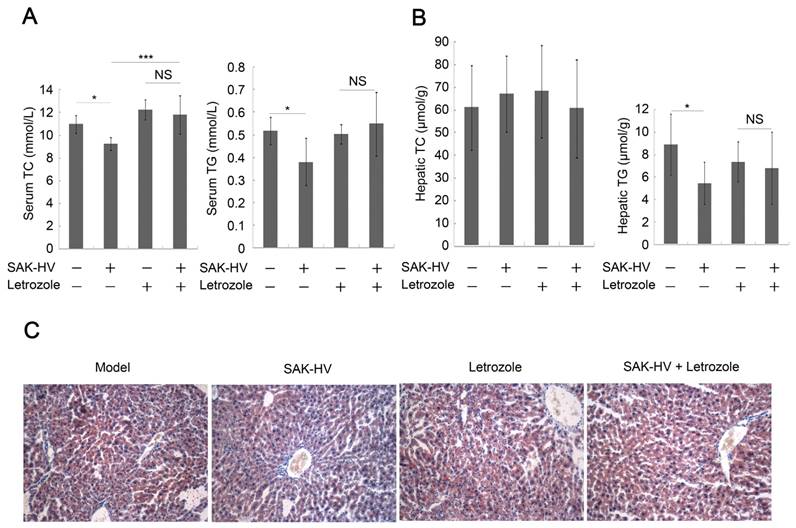
In conclusion, these results verified that the estrogen played important roles in triggering liver proliferation and lowering serum TC and TG, as well as ameliorating hepatic steatosis.
PGC-1α knockdown downregulated female hormone synthesis in liver, inhibited the lipid-reducing effect of SAK-HV, and blocked the liver proliferation
PGC-1α knockdown significantly down-regulated its expression in liver and remarkably inhibited its up-regulation induced by SAK-HV (Figure 6A). It also blocked the SAK-HV-triggered up-regulation of the hepatic Nr5a2, Cyp19a1 and Hsd3b2 (Figure 6B), and inhibited the hepatic levels of estrogen and progesterone as their products (Figure 6C). The alterations of protein levels of CYP19A1 by PGC-1α knockdown further confirmed that SAK-HV enhanced estrogen synthesis via PGC-1α in liver (Figure 6D). These results provided the first evidence that PGC-1α mediated the synthesis of female hormones in liver.
PGC-1α knockdown inhibited the hepatic synthesis of female hormones and broke the SAK-HV-induced liver proliferation. (A) The expression of PGC-1α in liver was significantly knocked down, and its up-regulation induced by SAK-HV was also remarkably inhibited (n=4). (B) PGC-1α knockdown blocked the SAK-HV-triggered up-regulation of the hepatic transcription levels of genes involved in female hormones synthesis (n=4). (C) PGC-1α knockdown effectively inhibited the upregulation effect of SAK-HV on estrogen and progesterone levels in liver (n=7). (D) PGC-1α knockdown blocked the SAK-HV-triggered up-regulation of CYP19A1 (n=4). (E) The immunohistochemical detection of PCNA indicated that PGC-1α knockdown remarkably inhibited the SAK-HV-triggered liver proliferation (magnification, 200×; n=4). ApoE-/- mice were injected with either PGC-1α RNAi lentivirus or control virus 4 days before they were treated with SAK-HV or PBS, and these ApoE-/- mice were divided into four groups, model, SAK-HV, model PGC-1α KD and SAK-HV PGC-1α KD group. Abbreviation: KD, knockdown. Two-way ANOVA with Tukey's multiple comparisons test for B and C.
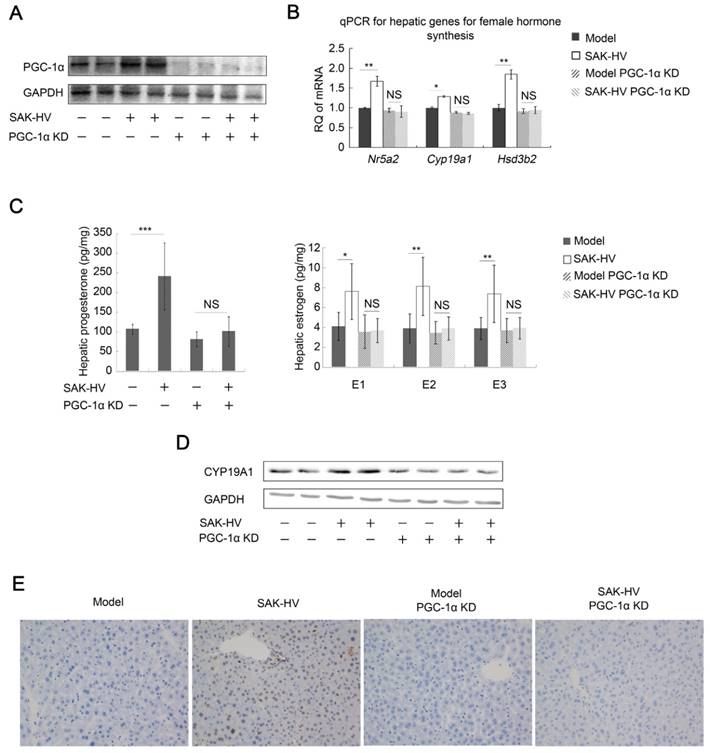
In line with estrogen inhibition, the upregulation of PCNA by SAK-HV was inhibited by PGC-1α knockdown (Figure 6E). The serum TG and TC-lowering effects of SAK-HV were also inhibited in PGC-1α knockdown ApoE-/- mice (Figure 7A), as well as its curative effect on hepatic steatosis (Figure 7B). Indeed, the hepatic TG level was remarkably decreased in ApoE-/- mice but elevated in PGC-1α knockdown ApoE-/- mice after SAK-HV treatment, although the hepatic TC levels were not statistically different among all groups (Figure 7C). This elevated hepatic TG content may be caused by the transient TG accumulation during early liver proliferation, but it was not observed in SAK-HV-treated ApoE-/- mice after estrogen inhibition (Figure 5B), indicating that PGC-1α partially lowered the hepatic TG level without the effect of estrogen. Similarly, PGC-1α knockdown caused a nearly complete inhibition of Ppara and its target genes in liver of ApoE-/- mice after SAK-HV treatment, as well as hepatic protein level of PPARα and acetyl-CoA level (Figure 7D), but not a partial inhibition of them as that caused by estrogen inhibition (Figure 4C). These results indicated that the PGC-1α-triggered TG-lowering effect by inducing PPARα-mediated fatty acid oxidation was further strengthened by PGC-1α-induced estrogen in liver.
PGC-1α KD blocked the lipid-lowering effect of SAK-HV. (A) PGC-1α KD broken the lipid-reducing effect of SAK-HV on PGC-1α KD ApoE-/- mice. It also caused decreased serum TC and TG, which might be caused by PGC-1α KD-induced CNS-linked hyperactivity (n=7). (B) PGC-1α KD broke the curative effect of SAK-HV on hepatic steatosis (magnification, 20×; n=4). (C) The hepatic TC levels were not statistically different among all groups. The hepatic TG level was decreased in ApoE-/- mice but elevated in PGC-1α KD ApoE-/- mice after SAK-HV treatment (n=7). The elevated hepatic TG may be caused by the transient TG accumulation during early liver proliferation, but it was not observed in SAK-HV-treated mice after estrogen inhibition (Figure 6B), indicating that PGC-1α partially lowered the hepatic TG level without the effect of estrogen. (D) PGC-1α KD caused a complete inhibition of the SAK-HV-induced Ppara and its target genes (Left, n=4), as well as the protein level of PPARα (Right, n=4) and acetyl CoA levels in liver (Right, n=7), but not a partial inhibition as that caused by letrozole (Figure 5C). Two-way ANOVA with Tukey's multiple comparisons test for A, C and D.
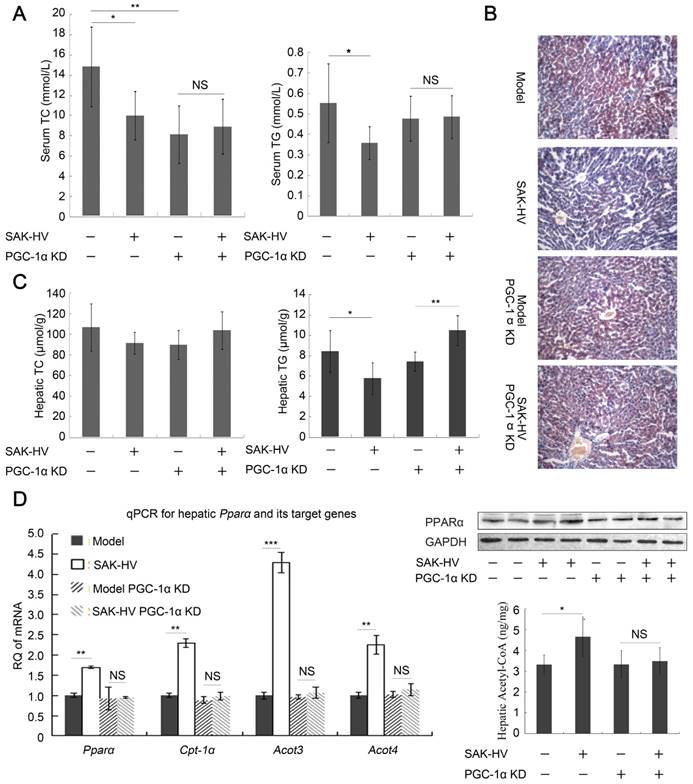
Unexpectedly, the serum TC was significantly decreased after PGC-1α knockdown, and the serum TG also displayed a decreasing trend, but without statistical significance (Figure 7A). It was reported that PGC-1α knockout caused CNS-linked hyperactivity to decrease serum TC and TG in ApoE-/- Pgc1a-/- mice (42, 43). We actually found that the PGC-1α knockdown ApoE-/- mice performed a profound hyperactivity with stimulus-induced myoclonus, which may explain their decreased serum lipids.
Collectively, we concluded that PGC-1α induced PPARα-mediated fatty acid oxidation in liver to decrease serum TG and ameliorate the hepatic steatosis, and triggered hepatic estrogen synthesis to induce hepatocytic proliferation and lower serum TC; its TG-lowering effect was also amplified by estrogen. Most importantly, this conclusion indicated the important role of the PGC-1α-estrogen axis in the liver proliferation-dependent lipid-lowering process (Please refer to Discussion section for details).
The STAT3-C/EBPβ-PGC-1α in liver constituted a novel lipid-lowering pathway of SAK-HV
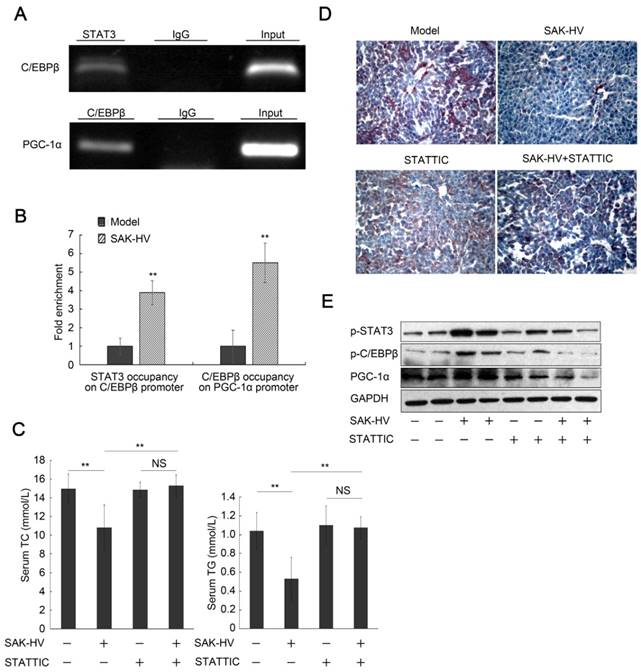
C/EBPβ, the downstream protein of STAT3 (27), is the transcriptional regulator of PGC-1α during liver proliferation (28). The ChiP assays verified both STAT3 occupancy on C/EBPβ promoter and C/EBPβ occupancy on PGC-1α promoter after SAK-HV treatment (Figure 8A), and further demonstrated that both of them were increased by SAK-HV treatment in vivo (Figure 8B). STATTIC, a STAT3 phosphorylation inhibitor, significantly blocked the serum lipid-lowering effect of SAK-HV (Figure 8C), and remarkably counteracted its therapeutic efficacy on hepatic steatosis (Figure 8D). Both the phosphorylation level of C/EBPβ and the expression of PGC-1α were markedly downregulated by STAT3 inhibition (Figure 8E). These results demonstrated that the STAT3-C/EBPβ-PGC-1α in liver constituted a novel lipid-lowering pathway of SAK-HV.
The blockage of STAT3 phosphorylation inhibited the pharmacodynamic effect of SAK-HV. (A) The semi-quantitative PCR analysis of ChiP assay verified both STAT3 occupancy on C/EBPβ promoter and C/EBPβ occupancy on PGC-1α promoter after SAK-HV treatment in vivo. (B) The qPCR analysis of ChiP assay demonstrated that both STAT3 occupancy on C/EBPβ promoter and C/EBPβ occupancy on PGC-1α promoter were increased by SAK-HV treatment in vivo. (C) The inhibition of STAT3 phosphorylation blocked the lipid-reducing effect of SAK-HV (n=8). (D) The inhibition of STAT3 phosphorylation broke the therapeutic effect of SAK-HV on hepatic steatosis (magnification, 20×; n=5). (E) The inhibition of STAT3 phosphorylation significantly down-regulated both the phosphorylation levels of C/EBPβ and the expression level of PGC-1α, the downstream protein of C/EBPβ during liver proliferation (n=5). ApoE-/- mice were injected intraperitoneally with either STAT3 phosphorylation inhibitor STATTIC or PBS, and these ApoE-/- mice were divided into model, SAK-HV, STATTIC and SAK-HV+STATTIC group. Two-way ANOVA with Tukey's multiple comparisons test for A. Abbreviation: ChiP, Chromatin immunoprecipitation.
Complement inhibitor blocked STAT3-C/EBPβ-PGC-1α pathway and liver proliferation, and ameliorated the lipid-lowering effect of SAK-HV
We checked the role of complement system in the lipid-lowering effect of SAK-HV by the complement inhibitor FUT-175. The titers of SAK-HV-specific IgG in serum reached 102.63 and 102.57 on the 14th day in the FUT-175+SAK-HV and SAK-HV groups, respectively (Figure S7C). IL-6-STAT3 pathway is activated by C3a and C5a during liver proliferation (14). FUT-175 effectively blocked the upregulations of serum C5a and IL-6 by SAK-HV (Figure 9, A and B), and further inhibited the STAT3-C/EBPβ-PGC-1α pathway (Figure 9C). These results suggested that the complement system upregulated STAT3-C/EBPβ-PGC-1α pathway through IL-6. Consequently, FUT-175 effectively compromised the lipid-lowering effect of SAK-HV (Figure 9D) and its therapeutic effect on hepatic steatosis (Figure 9E). Furthermore, the upregulation of PCNA by SAK-HV was also inhibited by FUT-175 (Figure 9F). Collectively, these data revealed the causal relationships among SAK-HV-triggered complement activation, hepatocytic proliferation, activation of the STAT3-C/EBPβ-PGC-1α pathway, and its lipid-lowering effects.
The complement inhibition blocked the pharmacodynamic effect of SAK-HV. (A and B) The complement inhibition effectively compromised the increasing trends of C5a and its downstream protein IL-6 in serum (n=8). (C) The complement inhibition compromised the STAT3-C/EBPβ-PGC-1α pathway in liver (n=4). (D) The complement inhibition compromised the lipid-reducing effects of SAK-HV in serum (n=8). (E) The complement inhibition weakened the therapeutic effect of SAK-HV on hepatic steatosis (magnification, 20×; n=5). (F) The immunohistochemical detection of PCNA indicated that the complement inhibition blocked the SAK-HV-triggered liver proliferation (magnification, 200×; n=5). The complement inhibitor FUT-175 or its PBS solution was injected intraperitoneally to the ApoE-/- mice, and these ApoE-/- mice were divided into four groups, model, SAK-HV, FUT-175, and SAK-HV+FUT-175 group. Two-way ANOVA with Tukey's multiple comparisons test for A, B and D.
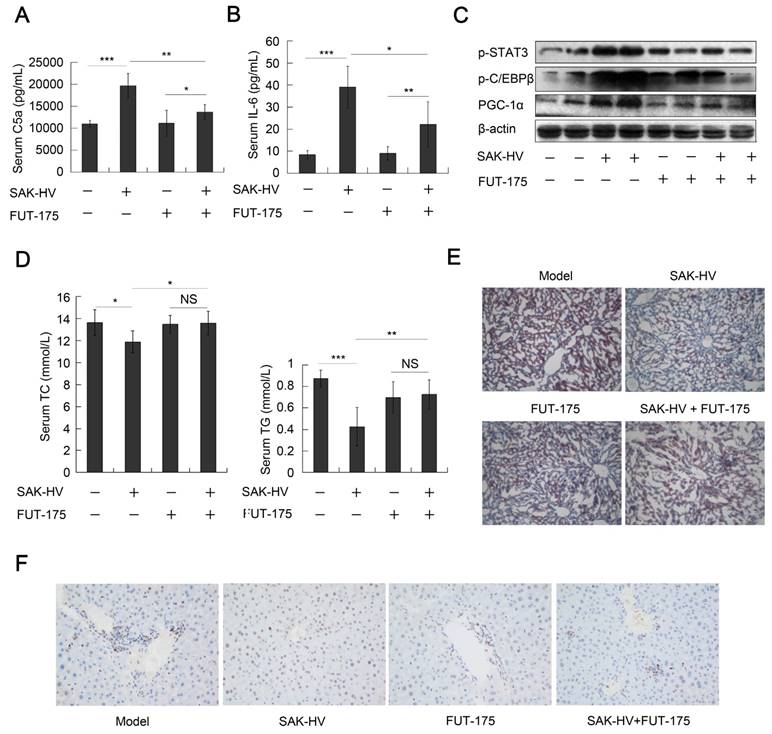
STAT3-C/EBPβ-PGC-1α pathway and liver proliferation were mediated by complement activation
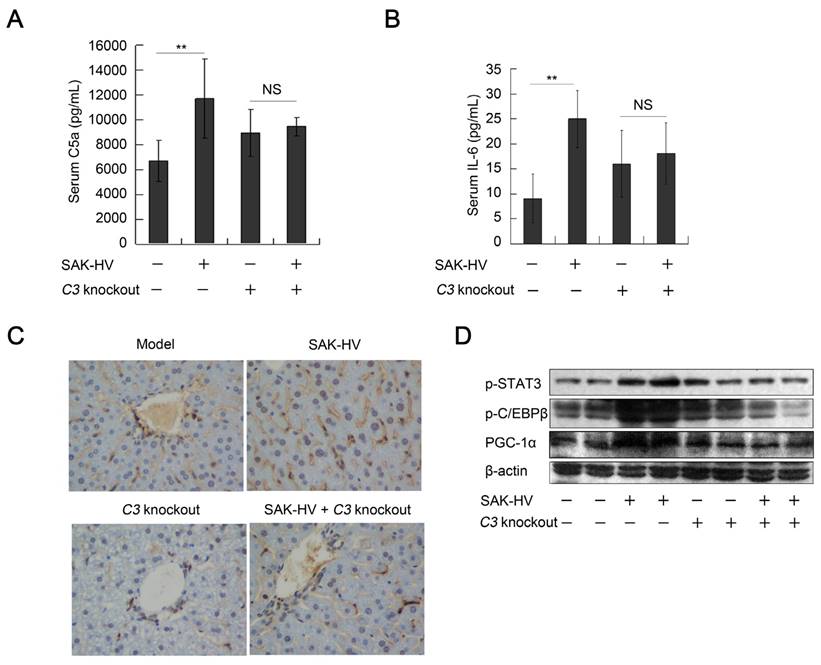
The FUT-175 is not a specific inhibitor of complement system. Thus, C3-/- mice were used to further verify the regulation of complement activation to STAT3-C/EBPβ-PGC-1α pathway. The titers of SAK-HV-specific IgG in serum reached 102.6 and 102.43 on the 14th day in the SAK-HV C3-/- and SAK-HV C3+/+ group, respectively (Figure S7D). Both serum C5a and IL-6 were effectively upregulated in C3+/+ mice but not in C3-/- mice after SAK-HV treatment (Figure 10, A and B). Furthermore, STAT3-C/EBPβ-PGC-1α pathway was activated in SAK-HV C3+/+ group but not in SAK-HV C3-/- group (Figure 10D). The upregulation of PCNA by SAK-HV was also inhibited in C3-/- mice (Figure 10C). These results again strongly supported that the activation of STAT3-C/EBPβ-PGC-1α pathway and liver proliferation were mediated by complement activation via IL-6.
The regulation of complement activation on STAT3-C/EBPβ-PGC-1α pathway and liver proliferation. (A and B) C3 knockout significantly down-regulated the serum C5a and its downstream IL-6 (n=6). (C) The immunohistochemical detection of PCNA indicated that C3 knockout blocked the SAK-HV-triggered liver proliferation (magnification, 400×; n=4). (D) C3 knockout inhibited the SAK-HV-induced activation of STAT3-C/EBPβ-PGC-1α pathway in liver (n=4). Male C3-/- mice and their wild type C3+/+ mice were divided into 4 groups to be injected with SAK-HV or PBS separatively: C3-/- SAK-HV , C3-/- PBS , C3+/+ SAK-HV and C3+/+ PBS groups. Two-way ANOVA with Tukey's multiple comparisons test for A, B.
The SAK-HV stimulus to BNL-Cl2 in vitro could not trigger STAT3-C/EBPβ-PGC-1α pathway or proliferated effect
The RGD sequence activates integrin-linked kinase through integrin (44), promoting NF-қB p65 to activate the IL-6-STAT3 pathway (45). Based on the dosage administered in vivo (0.125 mg/kg), we used a concentration gradient of SAK-HV (1, 2.5, and 5 μg/mL) to stimulate BNL-Cl2 cells. The SAK-HV stimulus to BNL-Cl2 cells for 24 h at all concentrations caused no proliferated effects, and led to no changes in the cell cycle (Figure S8, A and B). The activation levels of IқBα and STAT3-C/EBPβ-PGC-1α pathway were not obviously altered either (Figure S8C). At the maximum concentration of 5μg/mL, the activation levels of the IқBα and STAT3-C/EBPβ-PGC-1α pathway were not significantly changed at any time point (Figure S8D). It was possible that the function domain of RGD was hidden or altered in the new structure of SAK-HV. The results in vitro indicated that STAT3-C/EBPβ-PGC-1α pathway and liver proliferation was activated by SAK-HV via the complement system, but not by its direct stimulus to liver.
PPARα may not be involved in the SAK-HV-activated complement system
We have demonstrated that SAK-HV induced the generation of SAK-HV-specific IgG and triggered classical complement activation after SAK-HV treatment. Recently it was established that C3 expression is regulated by nuclear receptors (46-48). Especially, PPARα was reported to regulate complement system through regulation of C3 transcription in HepG2 cells (46). So it was reasonable to postulate the involvement of PPARα in SAK-HV-triggered complement activation. We thus determined whether the inhibition of PPARα by PGC-1α knockdown reduced the level of SAK-HV-induced complement activation. The results indicated that PPARα inhibition caused no significant changes in serum levels of either C3a or C5a (Figure S9), suggesting that PPARα may not be involved in the SAK-HV-promoted complement activation.
Discussion
The complement system is involved in hepatic cell cycle alterations, and is closely related to lipid metabolism (49). Fatty acid oxidation consumes blood triglycerides and fatty acids to fuel hepatocytic proliferation (50). Meanwhile, cholesterol is largely consumed by proliferating cells for cell membrane synthesis (16). Thus, our study verified a distinctive short-period treatment strategy against NAFLD and hyperlipidemia, lowering lipids via liver proliferation.
First, SAK-HV triggers a distinctive lipid-lowering biotherapy via liver proliferation. The time-window of lipid-lowering of SAK-HV was just about 4 days (Figure 1C). This dramatical lipid-lowering hallmark was consistent with the rapid energy-consuming feature of liver proliferation. Indeed, SAK-HV treatment caused moderate liver proliferation (Figure 2, D and E). Furthermore, estrogen increased the hepatic cholesterol uptake from serum without upregulation of hepatic cholesterol content (Figure 2D; Figure 5B; Figure 7C). The general way in liver to decrease cholesterol is to output them by their catabolism to bile acids and biliary cholesterol secretion. However, SAK-HV caused no significant alterations of hepatic LXR transcriptome that promoted cholesterol catabolism (Figure 3, C and D), and decreased the serum total bile acids (Figure S4F). Indeed, the increased cholesterol transfer from serum was consumed by the liver growth, but not output by cholesterol catabolism during liver proliferation (16). The blocked liver proliferation and the inhibited lipid-lowering effect caused by estrogen inhibition (Figure 4B; Figure 5), PGC-1α knockdown (Figure 6E; Figure 7, A-C) or complement inhibition (Figure 9) highly supported the proliferation-dependent lipid-lowering effect of SAK-HV.
Second, the complement system-induced STAT3-C/EBPβ-PGC-1α constituted a novel lipid-lowering pathway for SAK-HV biotherapy. We carried out PGC-1α knockdown, ChiP assays for both C/EBPβ occupancy on PGC-1α promoter and STAT3 occupancy on C/EBPβ promoter, and STAT3 inhibition in ApoE-/- mice to verify this novel lipid-lowering pathway STAT3-C/EBPβ-PGC-1α during liver proliferation (Figure 7; Figure 8).
Complement system is critically involved in liver proliferation (11-14). SAK-HV triggered classical complement activation (Figure S7, A,C,D; Figure 2B), which was not induced until more than one week after SAK-HV administration (Figure S7A and Figure 2A), implying the causal relationship between complement activation and the delayed lipid-lowering effect. Indeed, SAK-HV lowered lipids through this proliferation-dependent lipid-lowering pathway via the complement system, rather than its direct stimulus to hepatocytes, which was demonstrated by complement inhibition (Figure 9) and C3 knockout (Figure 10) in vivo, and was further demonstrated by the SAK-HV stimulus to BNL-Cl2 cells in vitro without complement activation (Figure S8). Neither C3a nor C5a in serum was significantly affected by PGC-1α knockdown in ApoE-/- mice (Figure S9), suggesting that SAK-HV-upregulated PPARα coactivated by PGC-1α may not be involved in the SAK-HV-activated complement system, and further supporting that the complement activation is a cause but not an effect of lipid-lowering process. Interestingly, a recent study of liver proliferation implied a potential regulation of PGC-1α and cholesterol metabolism by complement system, however the mechanism is unclear (49). The complement system-induced STAT3-C/EBPβ-PGC-1α pathway may provide a possible mechanism for this study. Besides, the unusual manner of SAK-HV to take effect via complement system may cause the distinctive does-independent, U-shape dose-response relationship of SAK-HV (Figure 1A).
Third, we first demonstrated that PGC-1α mediated the hepatic synthesis of female hormones during liver proliferation. Female hormone synthesis is involved in liver proliferation (21). SAK-HV promoted the synthesis of progesterone and estrogen in liver (Figure 3, D-F), which was significantly blocked by PGC-1α knockdown (Figure 6, B-D). Indeed, PGC-1α was reported to trigger the female hormones synthesis in breast adipose tissue and ovarian granulosa cells (35, 36). While it was also reported that fasting-induced PGC-1α upregulation mediated the hepatic synthesis of female hormone precursors without significant changes in these hormones themselves in vitro (32). C/EBPβ activates PGC-1α during liver proliferation with an amplitude of induction that exceeds the fasting response (28), which may lead to the regulation of PGC-1α to these female hormones during liver proliferation. Noteworthy is the fact that the hepatic intracrine production of these hormones without biologically significant release in serum (Figure 3G) effectively avoided the systemic effects that may cause sex-specific responses (51).
Fourth, our results indicated a potential PGC-1α-estrogen axis as an interesting energy model for the liver proliferation, which requires two steps: “ignition” by PGC-1α (Step 1) and both “ignition amplifier” and “starter” by PGC-1α-induced estrogen in liver (Step 2). The blocked liver proliferation caused by PGC-1α knockdown (Figure 6E) or estrogen inhibition (Figure 4B) strongly supported this energy model.
During the Step 1, PGC-1α enhanced fatty acid oxidation as an “ignition” to ignite and fuel the hepatocyte activation. It was reported to bind to PPARα and upregulate LIPIN 1, and these 3 proteins together strengthened the fatty acid oxidation in liver to rapidly decrease serum TG (38, 52). Besides the upregulated expression of PGC-1α (Figure 2C) and transcriptional level of Ppara (Figure 3A) in liver, Lipin 1 was also upregulated (Table S2), suggesting that SAK-HV may activate PGC-1α-LIPIN 1-PPARα pathway to trigger a rapid TG-lowering effect.
During the Step 2, PGC-1α-induced estrogen triggered the liver proliferation as a “starter” to lower serum TC, and strengthened the TG-lowering effect of PGC-1α as an “ignition amplifier” to fuel the liver proliferation. The liver transcriptome analysis suggested that the decreased serum TC might be caused by the steroid hormones synthesis. By estrogen inhibiting, we verified that PGC-1α-induced estrogen in liver not only enhanced the TG-lowering effects of PGC-1α as an “ignition amplifier” (Figure 4C; Figure 5B; Figure 7, C and D), but also lowered serum TC (Figure 5A) partially by inducing the hepatic LDLr-mediated cholesterol uptake (Figure 4D). Indeed, estrogen is not only a potent serum TG-lowering hormone enhancing PPARα-mediated fatty acid oxidation (53-55), but also a potent serum TC-lowering hormone promoting hepatic cholesterol uptake from serum (56, 57). Most importantly, the estrogen stimulus to activated hepatocytes might cause a stronger serum TC-lowering effect than its stimulus to quiescent hepatocytes by inducing hepatocytic proliferation. The liver transcriptome analysis suggested that the decreased serum TC was closely associated with the proliferation-related anabolism and cell cycle alteration. Estrogen promotes liver proliferation, and is closely related to the cell cycle alteration in liver (41, 21), which establishes the closely association among them. As already reported (41), its inhibition broke the liver proliferation (Figure 4B), further explaining why letrozole compromised the lipid-lowering effect of SAK-HV (Figure 5), and verifying its role as a “starter” for liver proliferation. Although we detected the upregulation of Ldlr in liver (Figure 3B; Figure 4D), the dramatical serum TC-lowering process by estrogen-induced liver proliferation might involve a complex regulatory network, which still needs further research. Interestingly, a recent study indicates that when PGC-1α is highly expressed in liver, the physiological level of estrogen synergizes with PGC-1α activity and augments the expression of PGC-1α to protect against oxidative damage (58). Both this study and our study demonstrated that there was a synergy effect between PGC-1α and estrogen in liver: the prior one focused on the antioxidant response, and the latter one focused on both the fatty acid oxidation and the hepatocyte state transition from activation to proliferation. Furthermore, the prior study proposed an estrogen-induced upregulation of PGC-1α in liver, while our study put forward a PGC-1α-mediated hepatic estrogen synthesis during liver proliferation. Both scenarios may require a precondition, the high expression of PGC-1α. Both studies strongly suggested the potential positive feedback interaction between PGC-1α and estrogen in liver, and highly supported the “ignition amplifier” role of estrogen in PGC-1α-estrogen axis during liver proliferation. Moreover, it also suggested the synergistic protective effect of PGC-1α-estrogen axis on the potential enhanced oxidative stress during liver proliferation, which may provide a possible explanation for the anti-oxidative stress effect of SAK-HV (Figure S3). Taken together, the novel complement system-induced PGC-1α-estrogen axis via the STAT3-C/EBPβ-PGC-1α pathway may provide new perspectives on the lipid metabolism during liver proliferation (Figure 11).
The potential lipid-lowering mechanism of SAK-HV in liver. SAK-HV activated the complement system to induce the PGC-1α-estrogen axis via STAT3-C/EBPβ-PGC-1α pathway, triggering the hepatocyte state transition from activation to proliferation. During this energy-consuming process, PGC-1α as an “igniter” ignited and fueled the hepatocyte activation by enhancing PPARα-mediated fatty acid oxidation. PGC-1α-induced estrogen in liver further strengthened the PPARα-mediated fatty acid oxidation in liver as an “ignition amplifier”, then initiated and fueled the liver proliferation as a “starter”, causing the dramatically decreased serum TC partially by inducing the hepatic LDLr-mediated cholesterol uptake. Meanwhile, the upregulated PPARα-mediated fatty acid oxidation by the synergy effect of PGC-1α-estrogen axis led to the swift and violent decrease of serum TG, and significantly ameliorated the hepatic steatosis as well.
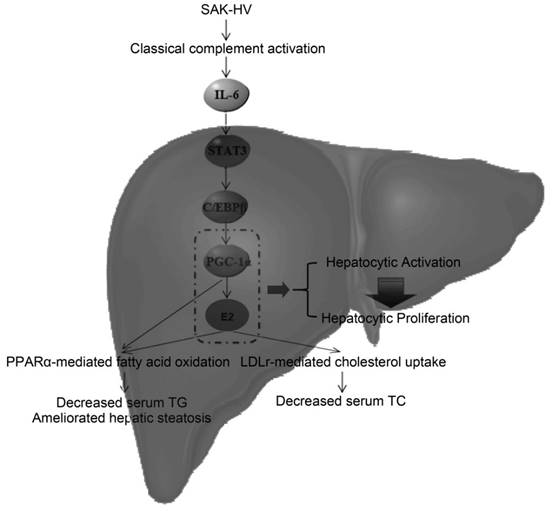
Fifth, although the mechanism of SAK-HV to induce mild liver proliferation is unclear, this might be adaptive and non-adverse liver enlargement caused by xenobiotics metabolism, which may benefit the organism by increasing metabolic capability (59, 60). Liver enlargement without histologic or clinical pathology alterations indicative of liver toxicity can be considered to be non-adverse (60). SAK-HV caused neither apoptosis nor fibrosis in liver (Figure S2, A and B). It also mildly improved the hepatocytic morphology without necrosis, and ameliorated both liver and serum inflammation (Figure S1), as well as both liver and coagulation functions in ApoE-/- mice (Figure S4; Figure S2, C-F). Its effect of anti-oxidative stress was similar to that of atorvastatin (Figure S3). The acute toxicity on mice and rats further supported its safety (Supplementary material). Besides that, the short treatment cycle may avoid the potential liver damage caused by long-term exposure of SAK-HV, although it required further research.
Finally, SAK-HV exerted rapid lipid-lowering effects on high-fat-fed rats and quails as well (Figure S5, Figure S6), indicating its general applicability. Most importantly, it triggers a much better curative effect than atorvastatin, the best lipid-lowering drugs available (6), within just 14 days (Figure 1D). These data showed that SAK-HV may be a promising lipid-lowering drug candidate with clinical potential.
In conclusion, our study suggests that the SAK-HV-triggered distinctive lipid-lowering strategy based on the new energy model of liver proliferation has potential as a novel short-period biotherapy against NAFLD and hyperlipemia.
Supplementary Material
Additional File 1Supplementary figures and tables (except Tables S2, S5, S8).
Table S2.
Table S5.
Table S8.
Abbreviations
NAFLD: non-alcoholic fatty liver disease;
TG: high triglyceride;
ApoE-/-: apolipoprotein E-deficient;
Ldlr-/-: LDL receptor-deficient;
C3: complement component 3;
RGD: tripeptide of arg-gly-asp;
TC: total cholesterol;
PGC-1α: peroxisome proliferator-activated receptor coactivator-1 α;
Pparα: peroxisome proliferator-activated receptor alpha;
WGCNA: weighted gene co-expression network analysis;
KEGG: Kyoto Encyclopedia of Genes and Genomes;
ANOVA: one-way analysis of variance;
Ccnd: Cyclin D;
Nr5a: nuclear receptor 5A;
C/EBPβ: CCAAT enhancer binding protein beta;
IgG: immunoglobulin G;
Hsd3b2: hydroxy-delta-5-steroid dehydrogenase, 3 beta- and steroid delta-isomerase 2; LXR: liver X receptor;
Cyp19a1: cytochrome P450 family 19 subfamily a member 1;
Cyp1a2: cytochrome P450 family 1 subfamily A member 2;
Cyp3a11: cytochrome P450 family 3 subfamily A member 11;
Ugt1a1: UDP Glucuronosyltransferase Family 1 Member A1;
Sult1e1: sulfotransferase family 1E member 1;
Comt: catechol-O-methyltransferase;
Abcc2: ATP Binding Cassette Subfamily C Member 2;
RankProd: Rank Product method;
WebGestalt: WEB-based GEne SeT AnaLysis Toolkit;
GO: Gene Ontology;
IL: interleukin;
STAT3: signal transducer and activator of transcription 3;
GEO: Gene Expression Omnibus;
qPCR: Real-time quantitative polymerase chain reaction;
PCNA: proliferating cell nuclear antigen;
Cpt1a: carnitine palmitoyltransferase I α;
Acot: Acyl-CoA thioesterase;
Acknowledgements
This work was supported by the National Science Foundation of China (grant no.: 81170255), and the National Science and Technology Major Project (grant no.: 2012ZX09102301-017). We thank Dr. Chen Guojiang (Beijing Institute of Basic Medical Sciences, Beijing, China) for providing the male C3-/- with a C57BL/6 background and wild-type animals (C3+/+) and Dr. Zhang Yi and Dr. Guo Ning (Beijing Institute of Basic Medical Sciences, Beijing, China) for their critical review of this article. We also thank Mr. Zhang Dongdong (Beijing Institute of Basic Medical Sciences, Beijing, China) for providing technical support for the ChiP assay.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Sapp V, Gaffney L, EauClaire SF. et al. Fructose leads to hepatic steatosis in zebrafish that is reversed by mechanistic target of rapamycin (mTOR) inhibition. Hepatology. 2014;60(5):1581-92
2. Executive Summary of the Third Report of the National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III). JAMA. 2001;285(19):2486-2497
3. Chapman MJ, Ginsberg HN, Amarenco P. et al. Triglyceride-rich lipoproteins and high-density lipoprotein cholesterol in patients at high risk of cardiovascular disease: evidence and guidance for management. Eur Heart J. 2011;32(11):1345-1361
4. Mikhailidis DP, Elisaf M, Rizzo M. et al. “European panel on low density lipoprotein (LDL) subclasses”: a statement on the pathophysiology, atherogenicity and clinical significance of LDL subclasses. Curr Vasc Pharmacol. 2011;9(5):533-571
5. Ewang-Emukowhate M, Wierzbicki AS. Lipid-lowering agents. J Cardiovasc Pharmacol Ther. 2013;18(5):401-11
6. Cholesterol Treatment Trialists' (CTT) Collaboration, Baigent C, Blackwell L. et al. Efficacy and safety of more intensive lowering of LDL cholesterol: a meta-analysis of data from 170,000 participants in 26 randomised trials. Lancet. 2010;376(9753):1670-81
7. Santos RD, Waters DD, Tarasenko L. et al. A comparison of non- HDL and LDL cholesterol goal attainment in a large, multinational patient population: the Lipid Treatment Assessment Project 2. Atherosclerosis. 2012;224(1):150-3
8. Hotamisligil GS. Inflammation and metabolic disorders. Nature. 2006;444(7121):860-7
9. Persson L, Borén J, Nicoletti A. et al. Immunoglobulin treatment reduces atherosclerosis in apolipoprotein E-/- low-density lipoprotein receptor-/- mice via the complement system. Clin Exp Immunol. 2005;142(3):441-5
10. Persson L, Borén J, Robertson AK. et al. Lack of complement factor C3, but not factor B, increases hyperlipidemia and atherosclerosis in apolipoprotein E-/- low-density lipoprotein receptor-/- mice. Arterioscler Thromb Vasc Biol. 2004;24(6):1062-7
11. DeAngelis RA, Markiewski MM, Lambris JD. Liver regeneration: a link to inflammation through complement. Adv Exp Med Biol. 2006;586:17-34
12. DeAngelis RA1, Markiewski MM, Kourtzelis I. A complement-IL-4 regulatory circuit controls liver regeneration. J Immunol. 2012;188(2):641-8
13. Mastellos D, Papadimitriou JC, Franchini S. et al. A novel role of complement: mice deficient in the fifth component of complement (C5) exhibit impaired liver regeneration. J Immunol. 2001;166(4):2479-86
14. Strey CW, Markiewski M, Mastellos D. et al. The proinflammatory mediators C3a and C5a are essential for liver regeneration. J Exp Med. 2003;198(6):913-23
15. Rudnick DA, Davidson NO. Functional Relationships between Lipid Metabolism and Liver Regeneration. Int J Hepatol. 2012;2012:549241
16. Lo Sasso G, Celli N, Caboni M. et al. Down-regulation of the LXR transcriptome provides the requisite cholesterol levels to proliferating hepatocytes. Hepatology. 2010;51(4):1334-44
17. Newberry EP, Kennedy SM, Xie Y. et al. Altered hepatic triglyceride content after partial hepatectomy without impaired liver regeneration in multiple murine genetic models. Hepatology. 2008;48(4):1097-105
18. Takeuchi N, Katayama Y, Matsumiya K. et al. Feed-back control of cholesterol synthesis in partially hepatectomized rats. Biochim Biophys Acta. 1976;450(1):57-68
19. Huang J, Rudnick DA. Elucidating the Metabolic Regulation of Liver Regeneration. Am J Pathol. 2014;184(2):309-21
20. Wang M, Wang Y, Wang J. et al. Construction and characterization of a novel staphylokinase variant with thrombin-inhibitory activity. Biotechnol Lett. 2009;31(12):1923-7
21. Mullany LK, Hanse EA, Romano A. et al. Cyclin D1 regulates hepatic estrogen and androgen metabolism. Am J Physiol Gastrointest Liver Physiol. 2010;298(6):G884-95
22. Mauvais-Jarvis F, Clegg DJ, Hevener AL. et al. The role of estrogens in control of energy balance and glucose homeostasis. Endocr Rev. 2013;34(3):309-38
23. Zhang ZC, Liu Y, Xiao LL. et al. Upregulation of miR-125b by estrogen protects against non-alcoholic fatty liver in female mice. J Hepatol. 2015;63(6):1466-75
24. Wang J, Duncan D, Shi Z. et al. WEB-based GEne SeT AnaLysis Toolkit (WebGestalt): update 2013. Nucleic Acids Res. 2013;41(Web Server issue):W77-83
25. Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics. 2008;9:559
26. Estall JL, Ruas JL, Choi CS. et al. PGC-1 negatively regulates hepatic FGF21 expression by modulating the heme/Rev-Erb axis. Proc Natl Acad Sci U S A. 2009;106(52):22510-5
27. Zhang K, Guo W, Yang Y. et al. JAK2/STAT3 Pathway Is Involved in the Early Stage of Adipogenesis Through Regulating C/EBPβ Transcription. J Cell Biochem. 2011;112(2):488-97
28. Wang H, Peiris TH, Mowery A. et al. CCAAT/enhancer binding protein-beta is a transcriptional regulator of peroxisome-proliferator-activated receptor-gamma coactivator-1alpha in the regenerating liver. Mol Endocrinol. 2008;22(7):1596-605
29. Nachtigal P, Pospisilova N, Jamborova G. et al. Atorvastatin has hypolipidemic and anti-inflammatory effects in apoE/LDL receptor-double-knockout mice. Life Sci. 2008;82(13-14):708-17
30. Field FJ, Mathur SN, LaBrecque DR. Cholesterol metabolism in regenerating liver of the rat. Am J Physiol. 1985;249(6 Pt 1):G679-84
31. Chen YJ, Lee MT, Yao HC. et al. Crucial role of estrogen receptor-alpha interaction with transcription coregulators in follicle-stimulating hormone and transforming growth factor beta1 up-regulation of steroidogenesis in rat ovarian granulosa cells. Endocrinology. 2008;149(9):4658-68
32. Grasfeder LL, Gaillard S, Hammes SR. et al. Fasting-induced hepatic production of DHEA is regulated by PGC-1alpha, ERRalpha, and HNF4alpha. Mol Endocrinol. 2009;23(8):1171-82
33. Suganuma I, Mori T, Ito F. et al. Peroxisome proliferator-activated receptor gamma, coactivator 1α enhances local estrogen biosynthesis by stimulating aromatase activity in endometriosis. J Clin Endocrinol Metab. 2014;99(7):E1191-8
34. Fayard E, Auwerx J, Schoonjans K. LRH-1: an orphan nuclear receptor involved in development, metabolism and steroidogenesis. Trends Cell Biol. 2004;14(5):250-60
35. Safi R, Kovacic A, Gaillard S. et al. Coactivation of liver receptor homologue-1 by peroxisome proliferator-activated receptor gamma coactivator-1alpha on aromatase promoter II and its inhibition by activated retinoid X receptor suggest a novel target for breast-specific antiestrogen therapy. Cancer Res. 2005;65(24):11762-70
36. Yazawa T, Inaoka Y, Okada R. et al. PPAR-gamma coactivator-1alpha regulates progesterone production in ovarian granulosa cells with SF-1 and LRH-1. Mol Endocrinol. 2010;24(3):485-96
37. Montagner A, Polizzi A, Fouché E. et al. Liver PPARα is crucial for whole-body fatty acid homeostasis and is protective against NAFLD. Gut. 2016;65(7):1202-14
38. Finck BN, Gropler MC, Chen Z. et al. Lipin 1 is an inducible amplifier of the hepatic PGC-1alpha/PPARalpha regulatory pathway. Cell Metab. 2006;4(3):199-210
39. He S, Atkinson C, Qiao F. et al. A complement-dependent balance between hepatic ischemia/reperfusion injury and liver regeneration in mice. J Clin Invest. 2009;119(8):2304-16
40. Bourassa PA, Milos PM, Gaynor BJ. et al. Estrogen reduces atherosclerotic lesion development in apolipoprotein E- deficient mice. Proc Natl Acad Sci USA. 1996;93(19):10022-7
41. Francavilla A, Polimeno L, DiLeo A. et al. The Effect of Estrogen and Tamoxifen on Hepatocyte Proliferation in Vivo and in Vitro. Hepatology. 1989;9(4):614-620
42. Lin J, Wu PH, Tarr PT. et al. Defects in Adaptive Energy Metabolism with CNS-Linked Hyperactivity in PGC-1 Null Mice. Cell. 2004;119(1):121-35
43. Stein S, Lohmann C, Handschin C. et al. ApoE-/- PGC-1α-/- Mice Display Reduced IL-18 Levels and Do Not Develop Enhanced Atherosclerosis. PLoS One. 2010;5(10):e13539
44. Pinkse GG, Jiawan-Lalai R, Bruijn JA. et al. RGD peptides confer survival to hepatocytes via the beta1-integrin-ILK-pAkt pathway. J Hepatol. 2005;42(1):87-93
45. Wani AA, Jafarnejad SM, Zhou J. et al. Integrin-linked kinase regulates melanoma angiogenesis by activating NF-κB/interleukin-6 signaling pathway. Oncogene. 2011;30(24):2778-88
46. Mogilenko DA, Kudriavtsev IV, Shavva VS. et al. Peroxisome proliferator-activated receptor α positively regulates complement C3 expression but inhibits tumor necrosis factor α-mediated activation of C3 gene in mammalian hepatic-derived cells. J Biol Chem. 2013;288(3):1726-38
47. Mogilenko DA, Kudriavtsev IV, Trulioff AS. et al. Modified low density lipoprotein stimulates complement C3 expression and secretion via liver X receptor and Toll-like receptor 4 activation in human macrophages. J Biol Chem. 2012;287(8):5954-68
48. Shavva VS, Mogilenko DA, Dizhe EB. et al. Hepatic nuclear factor 4α positively regulates complement C3 expression and does not interfere with TNFα-mediated stimulation of C3 expression in HepG2 cells. Gene. 2013;524(2):187-92
49. Min JS, DeAngelis RA, Reis ES. et al. Systems Analysis of the Complement-Induced Priming Phase of Liver Regeneration. J Immunol. 2016;197(6):2500-8
50. Sydor S, Gu Y, Schlattjan M. et al. Steatosis does not impair liver regeneration after partial hepatectomy. Lab Invest. 2013;93(1):20-30
51. Simpson ER, Misso M, Hewitt KN. et al. Estrogen-the Good, the Bad, and the Unexpected. Endocr Rev. 2005;26(3):322-30
52. Barroso E, Rodríguez-Calvo R, Serrano-Marco L. et al. The PPARβ/δ activator GW501516 prevents the down-regulation of AMPK caused by a high-fat diet in liver and amplifies the PGC-1α-Lipin 1-PPARα pathway leading to increased fatty acid oxidation. Endocrinology. 2011;152(5):1848-59
53. Fernández-Pérez L, Santana-Farré R, de Mirecki-Garrido M. Lipid Profiling and Transcriptomic Analysis Reveals a Functional Interplay between Estradiol and Growth Hormone in Liver. PLoS One. 2014;9(5):e96305
54. Vehmas AP, Adam M, Laajala TD. et al. Liver lipid metabolism is altered by increased circulating estrogen to androgen ratio in male mouse. J Proteomics. 2016;133:66-75
55. Djouadi F, Weinheimer CJ, Saffitz JE. et al. A gender-related defect in lipid metabolism and glucose homeostasis in peroxisome proliferator- activated receptor alpha- deficient mice. J Clin Invest. 1998;102(6):1083-91
56. Croston GE, Milan LB, Marschke KB. et al. Androgen receptor-mediated antagonism of estrogen-dependent low density lipoprotein receptor transcription in cultured hepatocytes. Endocrinology. 1997;138(9):3779-86
57. Kovanen PT, Brown MS, Goldstein JL. Increased binding of low density lipoprotein to liver membranes from rats treated with 17 alpha-ethinyl estradiol. J Biol Chem. 1979;254(22):11367-73
58. Besse-Patin A, Léveillé M, Oropeza D. et al. Estrogen Signals Through Peroxisome Proliferator-Activated Receptor-γ Coactivator 1α to Reduce Oxidative Damage Associated With Diet-Induced Fatty Liver Disease. Gastroenterology. 2017;152(1):243-256
59. Schulte-Hermann R. Induction of Liver Growth by Xenobiotic Compounds and Other Stimul. CRC Crit Rev Toxicol. 1974;3(1):97-158
60. Hall AP, Elcombe CR, Foster JR. et al. Liver Hypertrophy: A Review of Adaptive (Adverse and Non-adverse) Changes-Conclusions from the 3rd International ESTP Expert Workshop. Toxicol Pathol. 2012;40(7):971-94
Author biography
 Dr. Donggang Xu is a professor and the director of Laboratory of Genome Engineering, Beijing Institute of Basic Medical Sciences. He is the project leader for the research and development of a thrombolytic drug, which is in phase III clinical trial in China. He has coauthored over 30 publications including Journal of Control Release, Plos One, Cytokine, Biochem Bioph Res Co, and Gene. The current research interests in Professor Xu's group include the development of recombinant protein drug and the study on the structure and function of cytokine and its receptor.
Dr. Donggang Xu is a professor and the director of Laboratory of Genome Engineering, Beijing Institute of Basic Medical Sciences. He is the project leader for the research and development of a thrombolytic drug, which is in phase III clinical trial in China. He has coauthored over 30 publications including Journal of Control Release, Plos One, Cytokine, Biochem Bioph Res Co, and Gene. The current research interests in Professor Xu's group include the development of recombinant protein drug and the study on the structure and function of cytokine and its receptor.
 Dr. Min Wang earned her Ph.D. degree in Laboratory of Genome Engineering, Beijing Institute of Basic Medical Sciences, where she worked on the research and development of recombinant SAK-HV with Prof. Donggang Xu. She is currently an Associate Research Assistant in Laboratory of Genome Engineering, Beijing Institute of Basic Medical, China. Her research interests involve fat metabolism and autophagy in atherosclerosis.
Dr. Min Wang earned her Ph.D. degree in Laboratory of Genome Engineering, Beijing Institute of Basic Medical Sciences, where she worked on the research and development of recombinant SAK-HV with Prof. Donggang Xu. She is currently an Associate Research Assistant in Laboratory of Genome Engineering, Beijing Institute of Basic Medical, China. Her research interests involve fat metabolism and autophagy in atherosclerosis.
 Chao Zhang obtained his bachelor's degree in Computer Science and Technology and master's degree in Pattem Recognition and Artificial Intelligence from the School of Computer and Information Technology, Beijing Jiaotong University of China. He is currently a Ph. D. student under the supervision of Prof. Donggang Xu. His research focuses on bioinformatics and lipid metabolism.
Chao Zhang obtained his bachelor's degree in Computer Science and Technology and master's degree in Pattem Recognition and Artificial Intelligence from the School of Computer and Information Technology, Beijing Jiaotong University of China. He is currently a Ph. D. student under the supervision of Prof. Donggang Xu. His research focuses on bioinformatics and lipid metabolism.
![]() Corresponding authors: Donggang Xu, Ph.D. Min Wang, Ph.D., Laboratory of Genome Engineering, Beijing Institute of Basic Medical Sciences, Beijing 100850, China. E-mail: xudgac.cn (D. Xu), wmmm12cn (M. Wang) Tel: +86 10 68213039
Corresponding authors: Donggang Xu, Ph.D. Min Wang, Ph.D., Laboratory of Genome Engineering, Beijing Institute of Basic Medical Sciences, Beijing 100850, China. E-mail: xudgac.cn (D. Xu), wmmm12cn (M. Wang) Tel: +86 10 68213039