Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Abbreviations
Acknowledgements
Supplementary Material
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2017; 7(14):3446-3460. doi:10.7150/thno.20359 This issue Cite
Research Paper
rSj16 Protects against DSS-Induced Colitis by Inhibiting the PPAR-α Signaling Pathway
Lifu Wang1, 2, 3, Hui Xie1, 2, 3, Lian Xu1, 2, 3, Qi Liao4, Shuo Wan1, 2, 3, Zilong Yu1, 2, 3, Datao Lin 1, 2, 3, Beibei Zhang1, 2, 3, Zhiyue Lv1, 2, 3, Zhongdao Wu1, 2, 3 ![]() , Xi Sun1, 2, 3
, Xi Sun1, 2, 3 ![]()
1. Department of parasitology of Zhongshan School of Medicine, Sun Yat-sen University, Guangzhou, Guangdong 510080, China;
2. Key Laboratory of Tropical Disease Control (SYSU), Ministry of Education, Guangzhou, Guangdong 510080, China;
3. Provincial Engineering Technology Research Center for Biological Vector Control, Guangzhou, Guangdong 510080, China;
4. Department of Preventive Medicine, School of Medicine, Ningbo University, Zhejiang, Ningbo 315211, China.
Received 2017-3-31; Accepted 2017-6-17; Published 2017-8-15
Abstract

Background: Epidemiologic studies and animal model experiments have shown that parasites have significant modulatory effects on autoimmune disorders, including inflammatory bowel disease (IBD). Recombinant Sj16 (rSj16), a 16-kDa secreted protein of Schistosoma japonicum (S.japonicum) produced by Escherichia coli (E. coli), has been shown to have immunoregulatory effects in vivo and in vitro. In this study, we aimed to determine the effects of rSj16 on dextran sulfate sodium (DSS)-induced colitis.
Methods: DSS-induced colitis mice were treated with rSj16. Body weight loss, disease activity index (DAI), myeloperoxidase (MPO) activity levels, colon lengths, macroscopic scores, histopathology findings, inflammatory cytokine levels and regulatory T cell (Treg) subset levels were examined. Moreover, the differential genes expression after treated with rSj16 were sequenced, analyzed and identified.
Results: rSj16 attenuated clinical activity of DSS-induced colitis mice, diminished pro-inflammatory cytokine production, up-regulated immunoregulatory cytokine production and increased Treg percentages in DSS-induced colitis mice. Moreover, DSS-induced colitis mice treated with rSj16 displayed changes in the expression levels of specific genes in the colon and show the crucial role of peroxisome proliferator activated receptor α (PPAR-α) signaling pathway. PPAR-α activation diminished the therapeutic effects of rSj16 in DSS-induced colitis mice, indicating that the PPAR-α signaling pathway plays a crucial role in DSS-induced colitis development.
Conclusions: rSj16 has protective effects on DSS-induced colitis, effects mediated mainly by PPAR-α signaling pathway inhibition. The findings of this study suggest that rSj16 may be useful as a therapeutic agent and that PPAR-α may be a new therapeutic target in the treatment of IBD.
Keywords: parasites, rSj16, inflammatory bowel disease, protective effects, PPAR-α.
Introduction
Inflammatory bowel disease (IBD), which comprises Crohn's disease (CD) and ulcerative colitis (UC), is a chronic relapsing idiopathic disease characterized by epithelial barrier damage and inflammation homeostasis disruption in the intestinal tract [1, 2, 3]. The etiologies of both CD and UC remain unknown; however, it is generally believed that both diseases are associated with multiple pathogenic factors, including environmental changes, arrays of gene variants resulting in disease susceptibility, qualitative and quantitative gut microbiota abnormalities and broadly dysregulated immune responses [4]. Abdominal pain, diarrhea, rectal bleeding and weight loss are the most common symptoms in patients with IBD and affect millions of people worldwide [5, 6].
During the development of IBD, a number of proinflammatory molecules, such as tumor necrosis factor-alpha (TNF-α), interleukin-1beta (IL-1β), interleukin-6 (IL-6), interferon-gamma (IFN-γ) and interleukin-12 (IL-12), are expressed at higher levels, and have been shown to play important roles in mediating immune inflammatory responses [7, 8, 9]. Interleukin-12 (IL-12) production is increased in active CD lesions and in mice with dextran sulfate sodium (DSS)-induced disease [7]. Potent immunoregulatory cytokines, such as transforming growth factor (TGF)-β and interleukin-10 (IL-10), have been reported to be produced in equal or increased amounts in the gut of IBD compared with that of normal gut [10, 11]. Increases in the expression of interleukin-17 (IL-17), whose production is promoted by Chil1 and Chil3, have been noted in the mucosa and serum of most patients with IBD [12, 13]. The current treatments for IBD include mainly anti-inflammatory drugs, immunosuppressants and TNF blockers, such as 5-aminosalicylic acid (5-ASA)-based agents, as well as steroids, antimicrobials, and the monoclonal antibody (Ab) to TNF-α, infliximab (IFX) [14, 15]. However, as these drugs have adverse effects and limitations, studies aiming to devise novel therapeutic strategies for the treatment of IBD are urgently needed.
The “hygiene hypothesis” states that raising children in extremely hygienic environments in which exposure to parasitic infections is less common may negatively affect immune development and thus predispose children to developing autoimmune diseases, such as IBD, later in life [16]. This hypothesis is supported by the findings of epidemiological investigations and case-control experiments, which showed that positively deworming and early-childhood parasite infections negatively correlating with immunologic diseases [17]. Recent clinical studies have demonstrated that Trichuris of pig has shown considerable promise as a treatment for human autoimmune disorders, including IBD and multiple sclerosis [18], Ascaris lumbricoides attenuates wheezing in humans [19], and the human hookworm Necator americanus improves gluten tolerance in patients with celiac disease and attenuates disease in patients with chronic CD [20]. Additionally, Trichuris trichiura infections may reduce the risk of symptomatic colitis by promoting goblet cell hyperplasia and mucus production [21]. Furthermore, substantial evidence from studies involving rodents indicates that Heligmosomoides polygyrus [22], Hymenolepsis diminuta [23], Trichinella spiralis [24], Echinococcus granulosus [25], and the hookworms Ancylostoma ceylanicum and Ancylostoma caninum [26, 27] have protective effects against colitis.
Schistosoma japonicum (S. japonicum) and Schistosoma mansoni (S. mansoni) egg antigens have significant modulatory effect on DSS-induced colitis and can inhibit asthma development in murine models [28, 29], and S. japonicum ova can prevent epithelial damage and maintain epithelial barrier function in TNBS-induced colitis [30]. Helminths usually induce chronic infections that do not kill the host and have evolved a wide variety of approaches to suppress the immune response. Studies exploring the usefulness of helminths as new therapy have been conducted previously. However, therapies based on live helminths are associated with safety issues; thus, researchers have shifted their focus to helminth-derived molecules.
rSj16, a 16-kDa secreted protein of S.japonicum produced by E. coli, was initially described by our group. Our previous studies revealed that rSj16 has immunoregulatory effects in vivo and in vitro, as it dramatically suppressed the recruitment of thioglycolate-treated leukocytes to the peritoneal cavity in BALB/c mice [31], attenuated complete Freund's adjuvant-induced arthritis in a rat model [32] and decreased cytokine production by mouse immune cell lines to suppress T cell proliferation [33]. Moreover, rSj16 reduced lipopolysaccharide (LPS)-induced dendritic cell (DC) and RAW264.7 macrophage-mediated pro-inflammatory cytokine production [34]. Therefore, we hypothesized that rSj16 may be useful for the treatment of IBD. In this study, we confirmed that rSj16 can attenuate the development of DSS-induced colitis in mice by inhibiting the PPAR-α signaling pathway.
Materials and Methods
Animals
Male BALB/c mice (specific pathogen free, SPF) aged 6 weeks (18-20 g) were purchased from the Experimental Animal Center of Sun Yat-sen University. All animal experiments were approved by the Medical Research Ethics Committee of Sun Yat-sen University and conformed to the Chinese National Institute of Health Guide for the Care and Use of Laboratory Animals (NO. 2017-008). The mice were group-housed in ventilated cages in a temperature-control room (25 °C) and were fed standard mouse chow.
Recombinant protein (rSj16 and GST) expression and purification
Recombinant Sj16 and GST were produced and purified as previously described [32]. Briefly, the indicated plasmid (pGEX-4T-1/Sj16) was constructed and transformed into E. coli BL21 (DE3). rSj16 was expressed as a GST-Sj16 fusion protein via incubation with 1 mM isopropylthio-β-galactoside (IPTG, Sigma, USA) at 37°C. The GST-Sj16 fusion protein was purified using a GSTrap™ FFresin column (Amersham Pharmacia, USA) before being subjected to thrombin enzyme (Amersham, USA) digestion, resulting in the production of rSj16. GST was purified from the column by GST elution buffer and collected, after which the purified rSj16 and GST were excised from sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE), and identified by mass spectrometry and treated with AffinityPak™ Detoxi-Gel™ Endotoxin Removing Gel (Thermo, USA) to eliminate the endotoxin, which was detected using the Limulus amebocyte lysate test (sensitivity 0.25 EU/ml, Associates of Cape Cod, USA). The normal concentration of endotoxin in a 1 mg/ml gel was 0.01 EU/kg and has been found to be as high as 0.2 EU/kg, according to the endotoxin normative standard in the “American FDA Final Product Examination Guide”. The concentrations of rSj16 and GST were determined by Bicinchoninic Acid Protein Assay Kit (Beyotime Biotechnology, China).
DSS-induced colitis and treatment
Acute colitis was induced by replacing the drinking water with 5% (wt/vol) DSS (36-50 kDa, MP Biomedicals), which was administered to the mice for 6 days (day 1 to day 6). The DSS solution was changed every 2 days, and the control mice received water ad libitum. The mice were organized into the following groups: Water+PBS group, Water+rSj16 group, DSS+PBS group, DSS+rSj16 group, DSS+S. japonicum (Sj) group, DSS+GST group, and DSS+dexamethasone (DXM) group. Sj16, GST, and DXM were prepared with PBS and filtered using an ultrafiltration membrane (0.22 μm; Millipore, Germany). The Water+rSj16, DSS+GST, and DSS+rSj16 groups were intraperitoneally injected with 100 μg of rSj16 or GST daily from day 1 to day 6 of treatment, the DSS+DXM group was intraperitoneally injected with 20 μg of DXM daily from day 1 to day 6 of treatment, and the DSS+Sj group was infected with S. japonicum cercariae (20 per mouse) and then treated with DSS after 32 days. The Water+PBS, DSS+PBS and DSS+Sj groups were intraperitoneally injected with the same volume of PBS daily from day 1 to day 6 of treatment.
For the PPAR-α activation and inhibition study, DSS-induced colitis mice were administered either fenofibrate (a PPAR-α agonist; APExBIO, USA) at a dose of 20 mg/kg per day via intragastric gavage or GW6471 (a PPAR-α antagonist; APExBIO, USA) at a dose of 10 mg/kg per day via intraperitoneal injection for 6 days. To determine the role of PPAR-α activation and inhibition in colitis development, we divided the mice into the following 7 groups: Water+PBS group, DSS+PBS group, DSS+rSj16 group, DSS+PBS+PPAR-α(+) group, DSS+rSj16+PPAR-α(+) group, DSS+PBS+PPAR-α(-) group and DSS+rSj16+PPAR-α(-) group. These groups were intraperitoneally injected with 100 μg of rSj16 or the same volume PBS from day 1 to day 6 of treatment.
On day 6, the mice were sacrificed, after which their abdomens were opened, and their entire colons (from the caecum to the anus) were exposed and then removed. The length of each colon was recorded, after which each colon was washed in PBS, and 0.3-0.5-cm segments near the caecum and anus were removed for myeloperoxidase (MPO) activity measurements and quantitative real-time PCR (RT-PCR) analysis. The remaining colon tissues were fixed in 4% formaldehyde for histological and immunohistochemical analysis.
Clinical scoring of disease
During treatment, each group of mice was observed daily in the morning, and changes in body weight and diarrhea and bleeding were recorded. Scoring was performed according to the criteria described by Holger Sann [35]. Blood in the feces was identified using a Hemoccult assay kit (Nanjing Jiancheng Bio-engineering Institute, China). Body weight changes were calculated relative to day 1. The sum of the weight loss, diarrhea, and bloody stool scores served as the clinical disease score (disease activity index, DAI) (Table S1) [35].
Measurement of MPO activity
Inflammatory cell (polymorphonuclear neutrophils) infiltration into the colon was quantified by measuring tissue MPO activity using an MPO assay kit (Nanjing Jiancheng Bio-engineering Institute, China), according to the manufacturer's instructions. MPO activity in the colons were determined and expressed as a percentage relative to DSS+PBS group.
Macroscopic assessment and histologic evaluation
After the mice were sacrificed, colons length (as an indirect marker of inflammation) were measured and the macroscopic characteristics of their colons were assessed by an independent observer blinded to the treatment. The following parameters were assessed: hyperemia, wall thickening, ulceration, inflammation extension, and damage (Table S2) [36].
The colons were fixed in 4% formaldehyde and then processed according to standard procedures before being embedded in paraffin. Five-micrometer-thick paraffin-embedded colon sections were subsequently prepared and stained with hematoxylin and eosin (H&E). Histopathological scores were determined according to the criteria described by Holger Sann [35]. Briefly, the histopathological scores of the colonic lesions were determined based on extent of inflammation, neutrophil and lympho-histiocyte infiltration, crypt damage, crypt abscess formation, sub-mucosal edema, goblet cell loss, and reactive epithelial hyperplasia displayed by the sections (Table S3). The overall histopathological scores were determined by adding all the scores pertaining to the above parameters together.
RNA extraction and RT-PCR
mRNA expression levels were quantified using RT-PCR. Briefly, RNA extraction was performed by lysing 100 mg of mouse colon tissue samples with Trizol reagent (Invitrogen), according to the manufacturer's instructions. Then, the amount of extracted RNA was quantified by measuring the ratio of the absorbance at 260 to that at 280 nm using a NanoDrop ND-2000 spectrophotometer (Thermo Scientific, USA). Complementary DNA (cDNA) was synthesized from 1.0 μg of total RNA with oligo (dT) primers using a Thermo Scientific RevertAid First Strand cDNA Synthesis Kit (Thermo Scientific, USA), according to the manufacturer's protocol, and RT-PCR was performed using SYBR Green QPCR Master Mix (TaKaRa, Japan), according to manufacturer's instructions. The primers used for RT-PCR are listed in Table S4. PCR was performed with a reaction mixture with a total volume of 20 μl comprising the following: 10 μl of SYBR® Premix Ex TaqTM (2×), 0.8 μl of forward primer (10 μmol/l), 0.8 μl of reverse primer (10 μmol/l), 2 μl of template, and 6.4 μl of ddH2O. The reaction comprised the following steps: an initial denaturation at 95°C for 30 s, followed by amplification for 35 cycles at 95°C for 5 s and 60°C for 20 s. These steps were performed with a LightCycler® 480 instrument (Roche Diagnostics, Switzerland). The relative mRNA expression levels of the target genes were normalized to those of the indicated housekeeping gene (GAPDH) and were quantified using the comparative Ct method and the formula 2-ΔΔCT.
Immunohistochemistry
Immunohistochemistry was performed to evaluate IFN-γ, IL-10, IL-17A, TGF-β, TNF-α, Chil3 and PPAR-α expression levels in the abovementioned formalin-fixed paraffin-embedded colonic sections. The procedure comprised the following steps: the paraffin sections were deparaffinized via baking and dehydrated with xylene and ethanol. Antigen retrieval was subsequently performed by irradiating the samples in citrate buffer (pH 6.0) in a microwave for 20 minutes, after which the sections were incubated with 3% H2O2 for 15 minutes at room temperature to block endogenous peroxidase activity. After being blocked with 1% bovine serum albumin for 60 minutes, the sections were incubated with rabbit anti-mouse IFN-γ, IL-10, IL-17A, TGF-β, TNF-α, Chil3 and PPAR-α antibodies (1:100) overnight at 4℃. The sections were then incubated with the indicated secondary antibody (goat anti-rabbit IgG) for 30 min at room temperature. Horseradish peroxidase activity was detected by 3, 3′diaminobenzidine (DAB), which served as a chromogen (DAB; Dako, Copenhagen, Denmark), for 30 seconds. All the steps were washed 3 times in PBS for 5 minutes each. The sum of the integrated optical density (IOD) was analyzed by Image-Pro Plus 6.0.
Western blotting
PPAR-α protein expression in colon tissue was detected by western blotting. In briefly, colon tissues were lysed in a western blotting lysis buffer and run on SDS-PAGE according to standard methods. Then proteins were electrophoretically transferred to a PVDF membrane (Millipore, Germany) and blocked using 5 % skim milk. Then the membranes were incubated with anti-mouse PPAR-α (Abcam, UK) antibody overnight at 4 °C. Anti-rabbit IgG conjugated to horse-radish peroxidase (HRP) (Abcam, UK) were used as secondary antibodies and incubated for 2 h at room temperature. The membranes were visualized by ECL Western blotting detection system (Amersham, USA).
Flow cytometry
To determine Treg subset levels, we isolated cells from the spleens and mesenteric lymph nodes (MLNs) of BALB/C mice, washed with PBS and stained with fluorochrome-conjugated monoclonal antibodies (mAbs) against the following cell-surface markers: CD4, CD25, and Foxp3 (Mouse Regulatory T Cell Staining Kit #3, eBiosciences, San Diego, CA). The fluorescent monoclonal antibodies were used at concentrations recommended by the manufacturer and were applied according to the manufacturer's instructions. For Foxp3 staining, the cells were fixed and permeabilized using working fixation/permeabilization solution, according to the manufacturer's instructions. Images of all the samples were acquired on a COULTER EPICS XL (Beckman, USA). The numbers of Tregs in the MLNs and spleens were quantified and were expressed as percentages of the CD4 cell population. Postacquisition analyzes were performed using FlowJo software (TreeStar, Inc, Ashland, OR).
RNA and long noncoding RNA (lncRNA) sequencing and analysis
Total RNA was isolated from the colons of the Water+PBS (n=3), DSS+PBS (n=3) and DSS+rSj16 (n=3) groups and then subjected to quantitative and qualitative analyzes. Only samples with an RNA integrity number (RIN) greater than 8.0 and A class test results were considered for further analysis. A total of 3 μg of RNA from each sample was used as input material for preparation of the RNA library. Following cluster generation, the library samples were sequenced on an Illumina Hiseq 2500 platform, and 125-bp paired-end reads were generated. After sequencing, the data were subjected to the following preliminary analyzes and procedures, which were performed by the Novogene Corporation: quality control analysis, read mapping to the reference genome, transcriptome assembly, coding potential analysis, conservative analysis, target gene prediction, gene expression level quantification, differential expression analysis and Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analysis.
Small RNA sequencing and analysis
Total RNA was isolated from the colons of the Water+PBS, DSS+PBS and DSS+rSj16 groups and then subjected to quantitative and qualitative analyses. Only samples with an RIN greater than 8.0 and A class test results were considered for further analysis. A total of 3 μg RNA from each sample was used as input material for generation of the small RNA library. Following cluster generation, the libraries were sequenced on an Illumina Hiseq 2500/2000 platform, and 50-bp single-end reads were generated. After sequencing, the data were subjected to the following preliminary analyzes, which were performed by the Novogene Corporation: quality control analysis, read mapping to the reference sequence, known miRNA alignment, source tag removal, novel miRNA prediction, small RNA annotation summarization, miRNA editing analysis, miRNA family analysis, target gene prediction, miRNA quantification, mRNA differential gene expression analysis, and GO and KEGG enrichment analysis.
Statistical analysis
All data are expressed as the mean ± SEM. One-way ANOVA was used to analyze the significance of the differences between groups, and P <0.05 was considered statistically significant.
Results
rSj16 attenuates clinical activity of DSS-induced colitis mice
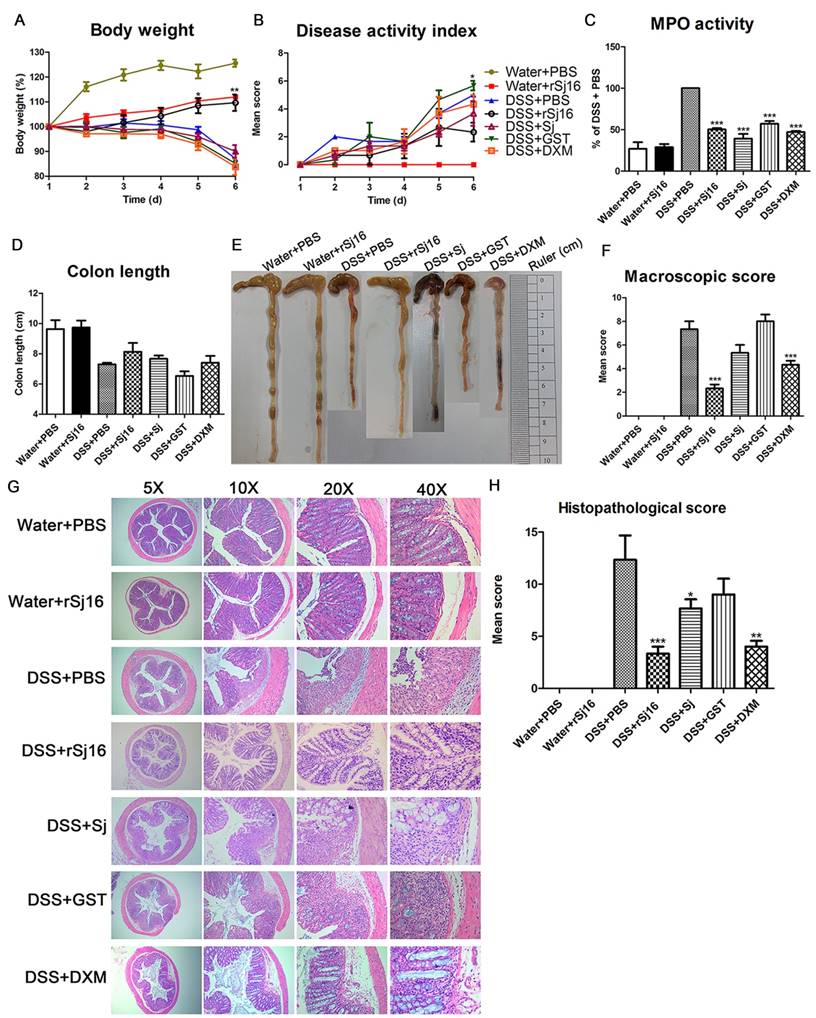
To assess the impact of rSj16 on acute colitis development, we established DSS-induced colitis model. The loss of body weight observed in DSS+PBS group was significantly aggravated from 5th day after DSS administration (Figure 1A) as well as DSS+Sj group, DSS+GST group and DSS+DXM group. However, after treated with rSj16, the loss of body weight in colitis model was significantly alleviated and close to Water +PBS group and Water+rSj16 group (Figure 1A). In addition, the DSS+rSj16 group exhibited a decreased cumulative DAI compared with the DSS+PBS, DSS+Sj, DSS+GST and DSS+DXM groups (Figure 1B). Moreover, our clinical and histological assessments of disease severity showed that MPO activity, an index of neutrophil accumulation, was significantly lower in the DSS+rSj16 group than in the DSS+PBS group (Figure 1C) and that colon length, a marker of intestinal inflammation, was significantly reduced by DSS treatment but was apparently not affected by Sj, GST or DXM treatment; however, colon length was improved by rSj16 treatment (Figure 1D and E). Additionally, mean colon macroscopic scores showed that treatment with rSj16 significantly suppressed DSS-induced increases in colon macroscopic scores compared with treatment with Sj, GST and DXM (Figure 1F). The results of our histological assessment showed that DSS induced a significant inflammatory response characterized by significant neutrophil and lympho-histiocyte infiltration, near-complete crypt loss, crypt abscess formation, sub-mucosal edema, goblet cell loss, and reactive epithelial hyperplasia. Representative H&E histopathology results indicated that treatment with rSj16 significantly reduced the inflammatory response compared with treatment with Sj, GST and DXM (Figure 1G). Blinded histological injury scores in the distal colon of the DSS+rSj16 group were significantly lower than those in the DSS+Sj, DSS+GST and DSS+DXM groups (Figure 1H). The body weight loss results, DAI, MPO activity levels, colon lengths, macroscopic scores, H&E staining results and histopathological scores of the Water+rSj16 group indicated that rSj16 has no side effects (Figure 1). Taken together, these results indicate that rSj16 attenuates clinical activity in DSS-induced colitis mice and that its efficacy is superior to that of DXM and S. japonicum infection.
rSj16 treatment attenuates clinical activity in DSS-induced colitis mice. The following groups of mice were used in the study: Water+PBS (n=5), Water+rSj16 (n=5), DSS+PBS (n=5), DSS+rSj16 (n=5), DSS+Sj (n=5), DSS+GST (n=5), DSS+DXM (n=5), and each group was used for three independent experiments. The mean values ± SEMs are represented by bars. (A) The daily mean weight change in each group (*P <0.05, DSS+rSj16 versus DSS+PBS). (B) The changes in DAI, scored from diarrhea, bleeding and body weight loss (*P <0.05, DSS+rSj16 versus DSS+PBS). (C) Colonic MPO activity. (D) On day 6, the mice were sacrificed, their colons were removed, and the lengths of their colons were measured and recorded. (E) Macroscopic appearance of the colon, as represented by the colon with the mean colon length and typical injury findings. (F) Mean colon macroscopic scores in each group. (G) The histopathological changes in the colon tissue samples were examined by H&E staining (5×, 10×, 20×, 40×). (H) Histopathological scores were determined for the colon tissue samples. Statistical analysis was performed with one-way ANOVA followed by Dunnett's multiple comparison test versus DSS+PBS. *P <0.05, **P <0.01, ***P <0.001.
rSj16 diminished pro-inflammatory cytokine production and up-regulated immunoregulatory cytokine production in the colons of DSS-induced colitis mice
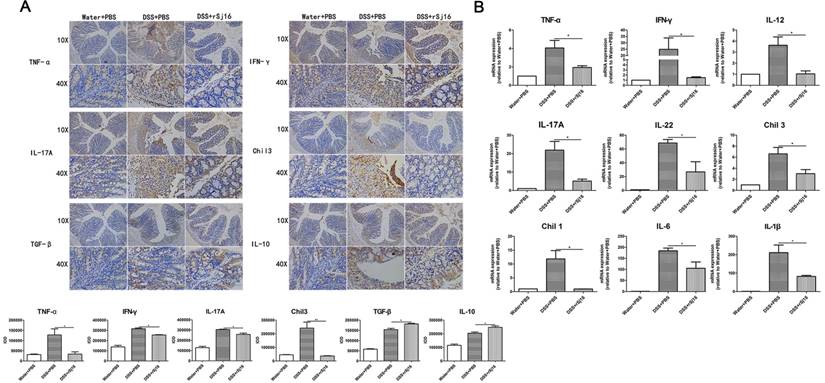
Pro-inflammatory cytokines play important roles in the pathogenesis of human IBD, and their expression levels are reportedly increased in DSS-induced colitis [37]. To determine whether the protective effects of rSj16 against DSS-induced colitis in mice are associated with reductions in pro-inflammatory mediator and cytokine production, we examined the protein expression levels of the following key pro-inflammatory cytokines in colon tissue by immunohistochemistry: TNF-α, IFN-γ, IL-17A, Chil3, TGF-β, and IL-10 (Figure 2A). Additionally, we examined the gene expression levels of the following pro-inflammatory cytokines by RT-PCR: TNF-α, IFN-γ, IL-12, IL-17A, IL-22, Chil3, Chil1, IL-6, and IL-1β (Figure 2B). As shown in Figure 2A, DSS-induced increases in the protein expression levels of the pro-inflammatory cytokines TNF-α, IFN-γ, IL-17A, and Chil3 were reduced by treatment with rSj16. In contrast, the protein expression levels of the anti-inflammatory cytokines TGF-β and IL-10 were increased by treatment with rSj16 compared with treatment with DSS. Consistent with the above results, the RT-PCR results showed that TNF-α, IFN-γ, IL-12, IL-17A, IL-22, Chil3, Chil1, IL-6, and IL-1β expression levels were significantly down-regulated by treatment with rSj16 (Figure 2B).
rSj16 regulates pro-inflammatory cytokine and immunoregulatory cytokine production in the colons of DSS-induced colitis mice. (A) TNF-α, IFN-γ, IL-17A, Chil3, TGF-β, and IL-10 protein expression in colon tissue was detected by immunohistochemistry. Positive immunoreactivity for TNF-α, IFN-γ, IL-17A, Chil3, TGF-β, and IL-10 protein expression is indicated by the red-brown color. The slides were counterstained with hematoxylin (blue). In addition, the sum of IOD was analyzed. (B) The relative mRNA expression levels of the inflammatory cytokines TNF-α, IFN-γ, IL-12, IL-17A, IL-22, Chil3, Chil1, IL-6, and IL-1β in colon tissue were determined by RT-PCR, and the housekeeping gene GAPDH was used as an internal reference. The bars indicate the fold changes in gene expression, which were determined by quantitative RT-PCR. The mean values ± SEMs are represented by bars. Statistical analysis was performed by one-way ANOVA followed by Dunnett's multiple comparison test versus the DSS+PBS. * P <0.05, ** P <0.01, *** P <0.001.
rSj16 increased Treg percentages in DSS-induced colitis mice
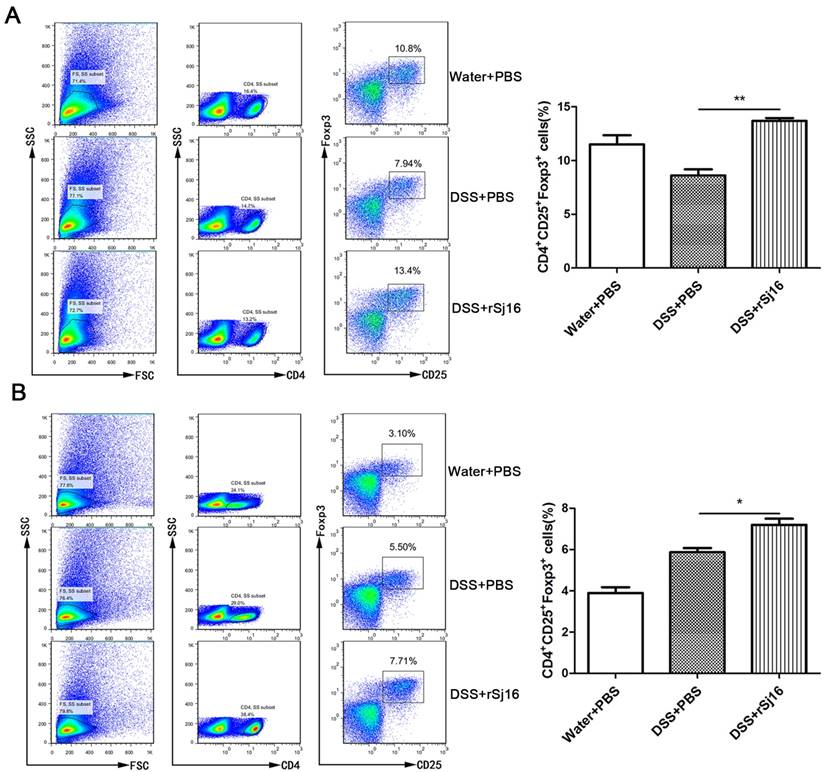
Treg subsets are key regulators of immune homeostasis and tolerance [38], and Treg function is linked to the development of inflammatory disorders, including IBD. To assess the changes in Treg subset levels induced by rSj16 in DSS-induced colitis mice, we isolated cells from the MLNs and spleens of BALB/C mice, washed them with PBS and stained with fluorochrome-conjugated mAbs against the following markers: CD4, CD25, and Foxp3. We then evaluated the changes in Treg subset levels by flow cytometry. We noted that the percentages of CD4+CD25+Foxp3+ cells in the spleens of rSj16-treated DSS-induced colitis mice was significantly increased (13.4%±1; P <0.01) compared with that in the spleens of vehicle-treated DSS-induced colitis mice (7.94%±2) (Figure 3A). In addition, we noted that the percentage of CD4+CD25+Foxp3+ cells in the MLNs of rSj16-treated DSS-induced colitis mice (7.71%±1; P <0.05) was significantly increased compared with that in the MLNs of vehicle-treated DSS-induced colitis mice (5.5%±1) (Figure 3B). Taken together, these findings indicate that rSj16 had protective effects against DSS-induced colitis partly mediated by increases in Treg percentages.
rSj16 expands the Treg population in experimental colitis in mice. Mice were sacrificed, and their spleens and MLNs were isolated. To detect Tregs, we stained the spleens (A) and MLNs (B) with anti-mouse CD4, CD25, and Foxp3 mAbs, as described in the Materials and Methods. Treg percentages were analyzed by FACS (left), and the results of the statistical analysis are shown (right). Data are shown as the means ± SEM (three independent experiments) of each group (n=5). Statistical analysis was performed by one-way ANOVA followed by Dunnett's multiple comparison test versus the DSS+PBS. * P <0.05, ** P <0.01, *** P <0.001
DSS-induced colitis mice treated with rSj16 display changes in the expression of specific genes in the colon, changes that are related to PPAR-α signaling pathway
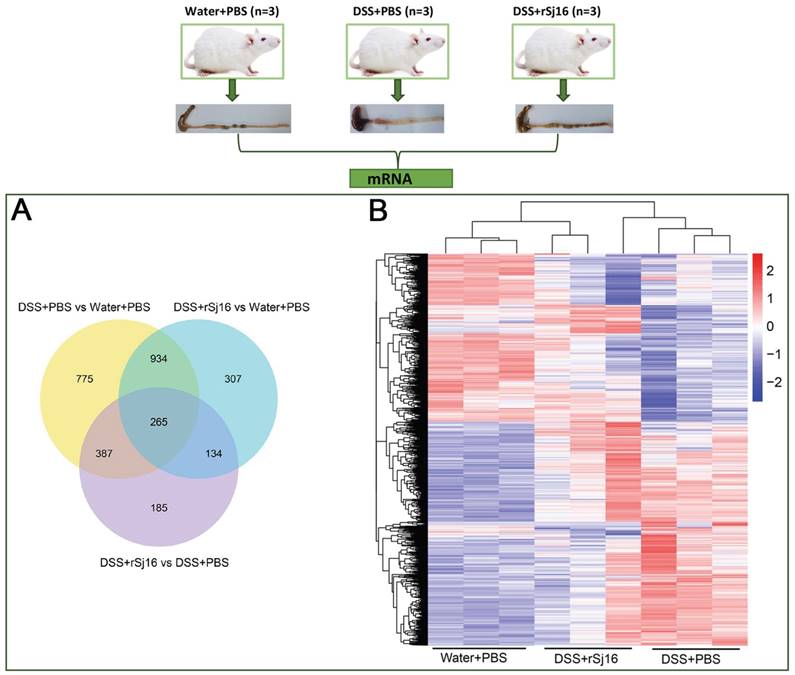
To better understand the effects of rSj16 in DSS-induced colitis mice, we sequenced and analyzed the RNA, lncRNA and small RNA of the Water+PBS (n=3), DSS+PBS (n=3) and DSS+rSj16 (n=3) groups. Our comparative assessments of the global mRNA expression profiles of various genes in the DSS+rSj16 and DSS+PBS groups revealed that 971 genes (545 up-regulated and 426 down-regulated) were significantly differentially expressed between these groups (Figure S1A). Moreover, analysis of global mRNA expression profiles of the DSS+rSj16 and Water+PBS groups revealed that 1640 genes (1051 up-regulated and 589 down-regulated) were significantly differentially expressed between these groups (Figure S1B), and analysis of the global mRNA expression profiles of the DSS+PBS and Water+PBS groups showed that 2361 genes (1424 up-regulated and 937 down-regulated) were differentially expressed between these groups (Figure S1C) (Figure 4A). The genes that were differentially expressed (based on fold changes) among the Water+PBS group, DSS+PBS and DSS+rSj16 group are shown as heatmaps in Figure 4B. GO analysis showed that the differentially expressed genes could be classified into the following three categories: biological process (ranked highest), molecular function and cellular component. Genes associated with immune system processes, the immune response, the innate immune response, the inflammatory response and immune system regulation ranked at the top of the biological process class (Figure S2-4). KEGG was used to identify the biological pathways that were enriched most significantly in the above groups. The pathway search results showed that the top 10 most significantly enriched pathways included those related to fat digestion and absorption, pancreatic secretion, cytokine-cytokine receptor interactions, protein digestion and absorption, and the PPAR signaling pathway (Figure S5).
The expression profiles of the Water+PBS (n=3), DSS+PBS (n=3) and DSS+rSj16 (n=3) groups. (A) Venn diagram showing the genes that were unique to each group and shared among the three groups. The cluster number for each component is listed. (B) Unsupervised hierarchical clustering was performed on the gene expression profiles of the three groups (Water+PBS, DSS+PBS and DSS+rSj16). A heat map of the differentially expressed genes, as determined by the clustering analysis, is shown in the figure. Each column represents a specimen, and each row represents a gene. The red and blue colors indicate the genes that were up-regulated and down-regulated, respectively.
We screened the top 100 genes that were up-regulated in the DSS+rSj16 group compared with the DSS+PBS group, as well as those that were down-regulated in the DSS+PBS group compared with the Water+PBS group. We also screened the top 100 genes that were down-regulated in the DSS+rSj16 group compared with the DSS+PBS group, as well as those that were up-regulated in the DSS+PBS group compared with the Water+PBS group. The 200 genes that were most significantly differentially expressed between the above sets of groups were subjected to additional GO analyzes and KEGG enrichment analyzes (Figure 5A and Figure S6). GO analysis showed that the genes that were most significantly differentially expressed between the DSS+rSj16 and DSS+PBS groups were those related to the defense response, the innate immune response, the stress response, the immune response, and immune system function (Figure S6). KEGG pathway analysis revealed that the genes that were most significantly differentially expressed between DSS+rSj16 and DSS+PBS groups were those associated with the PPAR-α signaling pathway (indicated by the arrows) (Figure 5A), namely, FATCD36, FABP, PPAR-α, HMGCS2, SCD1, LPL, ACSl1, aP2, and ADIPO (Figure 5B). The PPAR-α protein expression in colonic tissue was detected by immunohistochemistry and western blotting, the results showed that PPAR-α expression was decreased by rSj16 treatment compare with DSS+PBS group (Figure 5C and D). The change genes expression levels were confirmed by RT-PCR (Figure 5E).
The crucial role of the PPAR-α signaling pathway in DSS-induced colitis mice treated with rSj16, as determined by sequencing. We screened the top 100 genes that were up-regulated in DSS+rSj16 group compared with the DSS+PBS group, as well as the genes that were down-regulated in the DSS+PBS group compared with the Water+PBS group. We also screened the top 100 genes that were down-regulated in the DSS+rSj16 group compared with the DSS+PBS group, as well as those that were up-regulated in the DSS+PBS group compared with the Water+PBS group. (A) The 200 genes that were most significantly differentially expressed were analyzed by KEGG enrichment analysis, which showed that the genes in question were enriched in the PPAR signaling pathway. (B) The most significantly differentially expressed genes (DSS+rSj16 versus DSS+PBS) were those associated with the PPAR-α pathway. The down-regulated genes are boxed in red, and the up-regulated genes are boxed in blue. (C) PPAR-α protein expression in colon tissue was detected by immunohistochemistry and the sum of IOD was analyzed. (D) Western blotting analysis of PPAR-α protein expression in colon tissue. (E) The differentially expressed genes (DSS+rSj16 versus DSS+PBS) associated with the PPAR-α signaling pathway were identified by quantitative RT-PCR, and the housekeeping gene GAPDH was used as a reference. The bars indicate the fold changes in gene expression, as determined by quantitative RT-PCR results. The mean values ± SEMs are represented by bars. Statistical analysis was performed by one-way ANOVA followed by Dunnett's multiple comparison test against the DSS+PBS. *P <0.05, ** P <0.01, *** P <0.001.
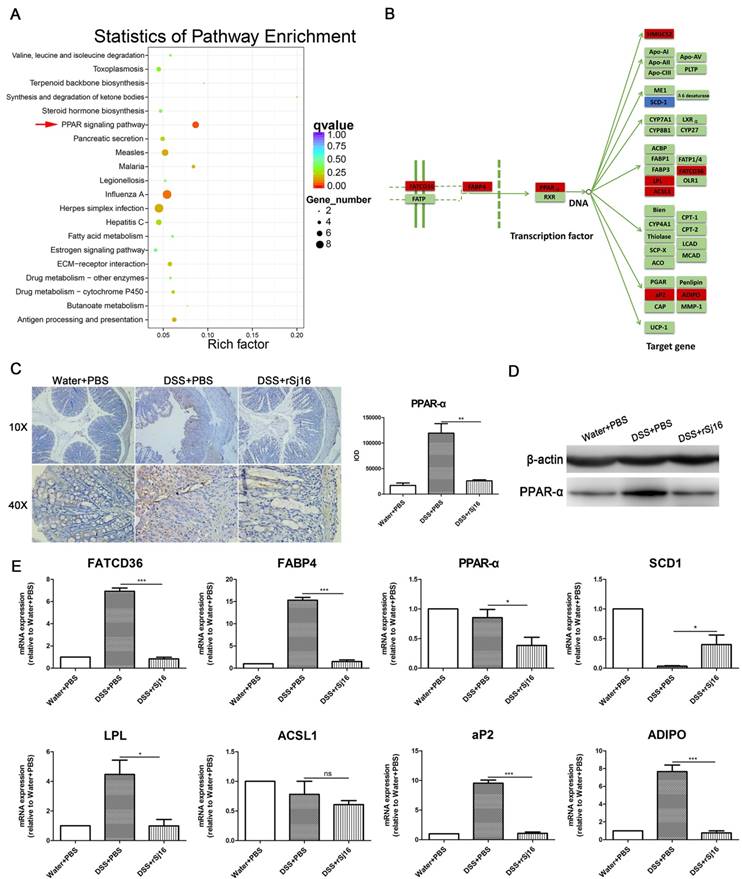
We also assessed the regulatory effects of lncRNA on the PPAR-a signaling pathway and found that rSj16 can reduce the expression of the lncRNAs 2410006H16Rik and Gm26883. We also found that ACSl1 was regulated by lncRNA 2410006H16Rik and that LPL was regulated by Gm26883 (Table S5). Furthermore, we determined that rSj16 can reduce the expression of mmu-miR-374c-3p and mmu-miR-335-3p and that the expression of ACSl1 and ADIPO was regulated by mmu-miR-374c-3p and mmu-miR-335-3p, respectively (Table S6).
Taken together, this data indicate that rSj16 protects against DSS-induced colitis by inhibiting the PPAR-α signaling pathway through specific lncRNA and miRNA.
PPAR-α activation diminished the therapeutic effects of rSj16 in DSS-induced colitis mice
Based on the above sequencing and analysis results, we sought to study the impact of PPAR-α agonist-mediated PPAR-α activation on the therapeutic effects of rSj16 in DSS-induced colitis mice. As expected, the results showed that DSS-induced colitis mice exhibited dramatic body weight loss, as did the mice treated with the PPAR-α agonist. However, the DSS+rSj16+PPAR-α(+) group displayed more significant body weight loss than the DSS+rSj16 group (Figure 6A). Additionally, the DAI was considerably increased in the DSS+rSj16+PPAR-α(+) group compared with the DSS+rSj16 group (Figure 6B). In addition, the DSS+rSj16+PPAR-α(+) group exhibited elevated MPO activity compared with the DSS+rSj16 group (Figure 6C). Moreover, PPAR-α activation resulted in a significant reduction in colon length in the DSS+rSj16+PPAR-α(+) group compared with the DSS+rSj16 group (Figure 6D and E). Mean colon macroscopic scores were significantly increased in the DSS+rSj16+PPAR-α(+) group compared with the DSS+rSj16 group (Figure 6F). We also evaluated the impact of PPAR-α signaling on the therapeutic effects of rSj16 in DSS-induced colitis mice by histopathological analysis using H&E staining (Figure 6G). We found that the DSS+rSj16+PPAR-α(+) group exhibited significant numbers of inflammatory lesions displaying ulceration, crypt damage, inflammatory cell infiltration, sub-mucosal edema, goblet cell loss and reactive epithelial hyperplasia compared with the DSS+rSj16 group. Histological scoring of the inflammatory lesions confirmed that the above differences between the two groups were significant (Figure 6H).
Activating PPAR-α diminished the therapeutic effects of rSj16 on DSS-induced colitis mice. The following groups of mice were used in the study: Water+PBS (n=5), DSS+PBS (n=5), DSS+rSj16 (n=5), DSS+PBS+PPAR-α(+) (n=5), DSS+rSj16+PPAR-α(+) (n=5), DSS+PBS+PPAR-α(-) (n=5), and DSS+rSj16+PPAR-α(-) (n=5). (A) Changes in body weight. (*P <0.05, DSS+rSj16 versus DSS+PBS; # P <0.05, DSS+rSj16+PPAR-α (+) versus DSS+PBS; &P <0.05, DSS+rSj16+PPAR-α (-) versus DSS+PBS) (B) DAI. (*P <0.05, DSS+rSj16 versus DSS+PBS; &P <0.05, DSS+rSj16+PPAR-α (-) versus DSS+PBS) (C) Colonic MPO activity. (D) Mean colon length. (E) Macroscopic appearance of the colon. (F) Mean colon macroscopic scores. (G) The histopathological changes in the colon tissue samples were examined by H&E staining. (H) Colon tissue histopathological scores. Mean values ± SEMs are represented by bars. Statistical analysis was performed by one-way ANOVA followed by Dunnett's multiple comparison test against the DSS+PBS. * P <0.05, ** P <0.01, *** P <0.001.
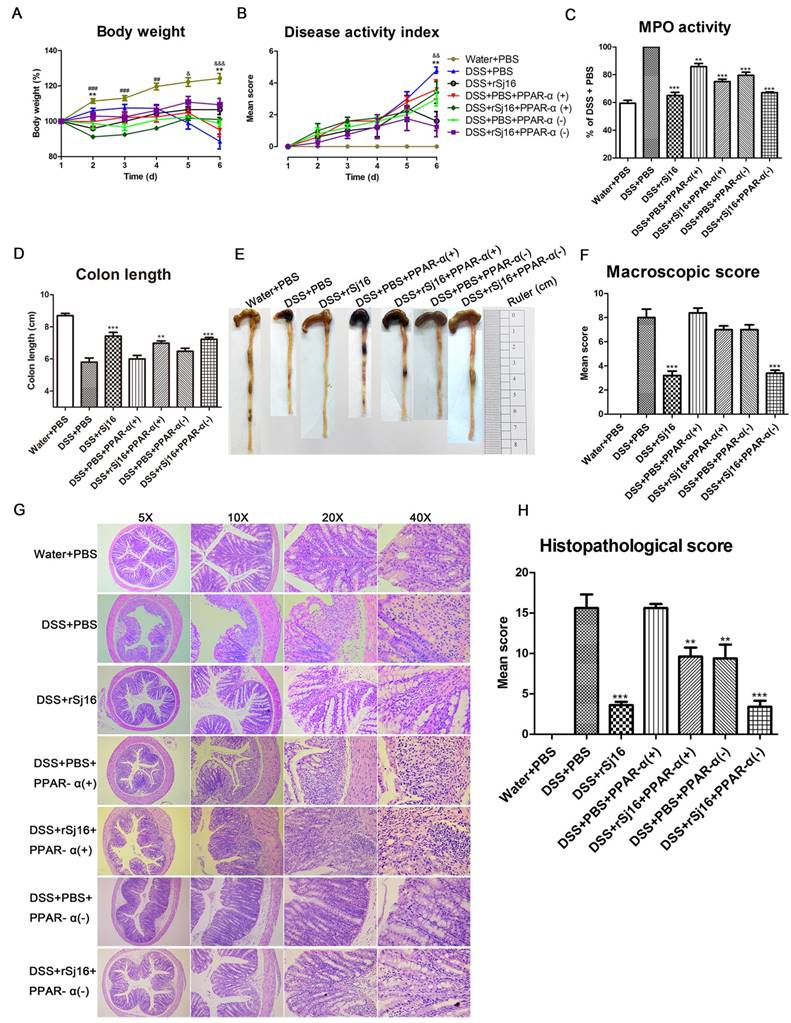
Compared with the DSS+PBS group, the DSS+PBS+PPAR-α(-) group exhibited ameliorated body weight loss, DAI, MPO, macroscopic scores and histopathological scores upon PPAR-α inhibition (Figure 6). Moreover, the body weight loss, DAI, MPO, macroscopic scores and histopathological scores of mice in the DSS+rSj16+PPAR-α(-) group were significantly decreased (Figure 6).
Discussion
In this study, we assessed whether rSj16 protects against the development of DSS-induced colitis. As expected, we found that rSj16 attenuates clinical disease activity in DSS-induced colitis mice. Specifically, rSj16 diminished pro-inflammatory cytokine production and up-regulated anti-inflammatory cytokine production in the colon of DSS-induced colitis mice. In addition, we showed that rSj16 increased Treg percentages in DSS-induced colitis mice. Moreover, we found that DSS-induced colitis mice treated with rSj16 displayed differences in the gene expression in colon through inhibits PPAR-α pathway. PPAR-α activation diminished the therapeutic effects of rSj16 in DSS-induced colitis mice.
IBD is a chronic relapsing idiopathic disease characterized by epithelial barrier damage and inflammation homeostasis disruption in the intestinal tract [1, 2, 39]. The pathogenesis of the disease remains poorly understood, and the available treatments for the disease are far from optimal. The results of epidemiologic studies support the hygiene hypothesis, which states that hyperhygienic environments may predispose individuals to developing IBD. Such environments may result from decreased exposure to parasitic infections [16]. Ongoing trials are testing the potential efficacy of helminth immunotherapy as a treatment for IBD. Heligmosomoides polygyrus [22], Hymenolepsis diminuta [23], Trichinella spiralis [24], Echinococcus granulosus [25], and the hookworms Ancylostoma ceylanicum and Ancylostoma caninum [26, 27] have been shown to protect against colitis in different animal models. In addition, S. japonicum and S. mansoni egg antigens have been shown to have significant modulatory effects on DSS-induced colitis [28, 29], and S. japonicum ova have been shown to prevent epithelial barrier damage and maintain epithelial barrier function in TNBS-induced colitis [30]. The results of our previous studies indicate that rSj16 has immunoregulatory effects in vivo and in vitro [31, 32, 33]. Therefore, to determine whether rSj16 can be used to treat IBD, we treated DSS-induced colitis mice with rSj16. The results revealed that body weight loss, DAI, and MPO activity levels decreased significantly in DSS-induced mice colitis treated with rSj16 (Figure 1A-C). Moreover, colon lengths improved, mean colons macroscopic scores decreased, and the inflammatory response was suppressed (Figure 1D-H). In addition, rSj16 treatment was a more effective treatment for DSS-induced colitis than S. japonicum. These results indicate that rSj16 attenuates clinical disease activity in DSS-induced colitis mice (Figure 1).
Studies have shown that in patients with CD, large amounts of IL-6 and TNF-α are produced by macrophages and DCs and contribute to IFN-γ production by mononuclear cells [40]. DCs also express higher levels of IL-12 in patients with CD than in patients with normal mucosa [8]. The levels of the pro-inflammatory cytokines IL-1β, IL-6 and TNF are predictably increased in mucosa affected by both CD and UC [41]. IL-22 production is increased in active Crohn's disease lesions and in mice with DSS [7]. Moreover, the expression level of the potent immunoregulatory cytokine IL-10 in guts affected by IBD has been reported to be equal to or greater than the expression level of IL-10 in normal gut [11]. TGF-β, which facilitates epithelial remodeling and fibrosis and has potent immunoregulatory effects, is expressed at high levels in human IBD tissue. Increases in IL-17 expression have been noted in the mucosa and serum of most patients with IBD. These increases can be induced by Chil1 and Chil3 [12, 13]. We found that the protective effects of rSj16 on DSS-induced colitis in mice were associated with the ability of rSj16 to regulate the production of inflammatory mediators and cytokines. Treatment with rSj16 down-regulated TNF-α, IFN-γ, IL-12, IL-17A, IL-22, Chil3, Chil1, IL-6, and IL-1β expression levels (Figure 2). Furthermore, the protein expression levels of the immunoregulatory cytokines TGF-β and IL-10 were increased (Figure 2). Treg subsets are key regulators of immune homeostasis and tolerance [38]. Tregs play an important role in controlling the immune response, and defects in their function underlie the development of autoimmune and chronic inflammatory conditions, including IBD [42]. Boosting Treg function may thus have beneficial effects in IBD [43]. Our results showed that the percentage of CD4+CD25+Foxp3+ cells in the spleens and MLNs were increased after treated with rSj16 in DSS-induced colitis mice (Figure 3), indicating that rSj16 had protective effects against the development of DSS-induced colitis that were partly mediated by increases in Treg percentages.
PPAR-α is a member of the nuclear receptor superfamily and plays a crucial role in regulating lipid homeostasis and metabolic diseases. Accumulating evidence indicates that PPAR-α influences multiple facets of inflammation and immunity and thus serves as an important link between metabolism and the immune system [44, 45]. However, the molecular and cellular mechanisms underlying the effects of PPAR-α on inflammation and immunity have yet to be fully elucidated. It has been proposed that PPAR-α plays an important role in the development of IBD, but studies regarding its specific effects have yielded contrasting results [46, 47, 48, 49]. The results of several previous reports suggest PPAR-α activation protects against the development of experimental colitis [46, 47, 48]. However, the results of the study by Xueyan Zhou et al. showed that treatment with PPAR-α agonists can significantly promote the development of DSS-induced colitis, whereas knocking out PPAR-α can significantly retard the development of DSS-induced colitis [49]. In addition, Fenofibrate (PPAR-α agonist) was found to exacerbate inflammation and tissue injury in experimental acute colitis mice [50]. Moreover, PPAR-α inhibitors reduced inflammation associated with endometriosis [51]. In this study, we sequenced and analyzed the mRNA of the Water+PBS (n=3), DSS+PBS (n=3) and DSS+rSj16 groups (n=3). We found that DSS-induced colitis mice treated with rSj16 display differences in the expression of specific genes in the colon compared with mice treated with vehicle (Figure 4). The genes that were differentially expressed between the two groups included those associated with the PPAR-α signaling pathway. The genes included FATCD36, FABP, PPAR-α, HMGCS2, LPL, ACS, aP2, ADIPO, and SCD1 (Figure 5). It has been suggested that lncRNAs and small RNA play important roles in biological functions and are thus associated with human diseases, including IBD [52, 53]. Analysis of the expression profiles of lncRNAs and small RNAs indicated that the lncRNAs 2410006H16Rik and Gm26883 and the small RNAs mmu-miR-374c-3p and mmu-miR-335-3p regulate the differentially expressed genes in the PPAR-α signaling pathway in DSS-induced colitis after treated with rSj16. We subsequently sought to study the impact of PPAR-α on the therapeutic effects of rSj16 in DSS-induced colitis mice by treating the mice with a PPAR-α agonist. The results indicated that the PPAR-α agonist significantly attenuated the therapeutic effects of rSj16 (Figure 6). However, PPAR-α antagonist can attenuates clinical activity of DSS-induced colitis mice (Figure 6). These data suggest that the therapeutic effects of rSj16 in DSS-induced colitis are mainly associated with PPAR-α signaling pathway inhibition.
In conclusion, our findings show that rSj16 attenuates clinical disease activity, diminishes pro-inflammatory cytokine production, up-regulates anti-inflammatory cytokine production and increases the percentages of Tregs in DSS-induced colitis mice and suggest that rSj16 has protective effects on DSS-induced colitis mediated mainly by PPAR-α inhibition. Taken together, these findings indicate that rSj16 may be useful as a therapeutic agent and that inhibiting PPAR-α may be a new strategy for treating IBD.
Abbreviations
IBD: inflammatory bowel disease; rSj16: Recombinant Sj16; S.japonicum: Schistosoma japonicum; E. coli: Escherichia coli; DSS: Dextran sulfate sodium; DAI: disease activity index; MPO: myeloperoxidase; Treg: regulatory T cell; PPAR-α: peroxisome proliferator activated receptorα; CD: Crohn's disease; UC: ulcerative colitis; TNF-α: tumor necrosis factor-alpha; IL-1β: interleukin-1beta; IL-6: interleukin-6; IFN-γ: interferon-gamma; IL-12: interleukin-12; IL-22: interleukin-22; TGF: transforming growth factor; IL-10: interleukin-10; IL-17: interleukin-17; 5-ASA: 5-aminosalicylic acid; IFX: infliximab; DC: dendritic cell; LPS: lipopolysaccharide; SPF: specific pathogen free; IPTG: isopropylthio-β-galactoside; SDS-PAGE: sodium dodecyl sulphate-polyacrylamide gel electrophoresis; DXM: dexamethasone; RT-PCR: real-time PCR; MLNs: mesenteric lymph nodes; GO: Gene Ontology; KEGG: Kyoto Encyclopedia of Genes and Genomes; Sj: S.japonicum; FATCD36: CD36 antigen; FABP: fatty acid-binding protein; HMGCS: hydroxymethylglutaryl-CoA synthase; SCD: stearoyl-CoA desaturase; LPL: lipoprotein lipase; ACS: long-chain acyl-CoA synthetase; aP2: fatty acid-binding protein; ADIPO: adiponectin.
Acknowledgements
These experiments were supported by grants from the National High Technology Research and Development Program of China (no. 2015AA020934), National Natural Science Foundation of China (grant no. 81201309 and 30972574), grant from the Doctoral Program of Higher Education of China (grant no. 20120171120049) and grant from the National Science Foundation of Guangdong Province (grant no. S2012040007256).
Authorship Contributions
XS and ZDW designed the study. LFW, HX, SW, ZLY, DTL and BBZ performed the study and the experiments. LX, ZYL and QL analyzed sequencing data. LFW, ZDW and XS wrote the manuscript.
Supplementary Material
Supplementary figures and tables.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Gupta RB, Harpaz N, Itzkowitz S. et al. Histologic inflammation is a risk factor for progression to colorectal neoplasia in ulcerative colitis: a cohort study. Gastroenterology. 2007;133:1099-105
2. Huang L-Y, He Q, Liang S-J. et al. ClC-3 chloride channel/antiporter defect contributes to inflammatory bowel disease in humans and mice. Gut. 2014;63:1587-95
3. Xiao B, Zhang Z, Viennois E. et al. Combination Therapy for Ulcerative Colitis: Orally Targeted Nanoparticles Prevent Mucosal Damage and Relieve Inflammation. Theranostics. 2016;6:2250-66
4. De Souza HS, Fiocchi C. Immunopathogenesis of IBD: current state of the art. Nat Rev Gastroenterol Hepatol. 2016;13:13-27
5. Boldeanu MV, Silosi I, Ghilusi M. et al. Investigation of inflammatory activity in ulcerative colitis. Rom J Morphol Embryol. 2014;55:1345-51
6. Machtaler S, Knieling F, Luong R. et al. Assessment of Inflammation in an Acute on Chronic Model of Inflammatory Bowel Disease with Ultrasound Molecular Imaging. Theranostics. 2015;5:1175-86
7. Brand S, Beigel F, Olszak T. et al. IL-22 is increased in active Crohn's disease and promotes proinflammatory gene expression and intestinal epithelial cell migration. Am J Physiol Gastrointest Liver Physiol. 2006;290:G827-38
8. Hart AL, Al-Hassi HO, Rigby RJ. et al. Characteristics of intestinal dendritic cells in inflammatory bowel diseases. Gastroenterology. 2005;129:50-65
9. Seo GS, Chae S-C. Biological therapy for ulcerative colitis: an update. World Journal of Gastroenterology. 2014;20:13234-8
10. Li MO, Flavell RA. TGF-β: a master of all T cell trades. Cell. 2008;134:392-404
11. Schreiber S, Heinig T, Thiele H-G. et al. Immunoregulatory role of interleukin 10 in patients with inflammatory bowel disease. Gastroenterology. 1995;108:1434-44
12. Fujino S, Andoh A, Bamba S. et al. Increased expression of interleukin 17 in inflammatory bowel disease. Gut. 2003;52:65-70
13. Sutherland TE, Logan N, Rückerl D. et al. Chitinase-like proteins promote IL-17-mediated neutrophilia in a tradeoff between nematode killing and host damage. Nat Immunology. 2014;15:1116-25
14. Baumgart DC, Sandborn WJ. Inflammatory bowel disease: clinical aspects and established and evolving therapies. The Lancet. 2007;369:1641-57
15. Steenholdt C, Brynskov J, Thomsen OØ. et al. Individualised therapy is more cost-effective than dose intensification in patients with Crohn's disease who lose response to anti-TNF treatment: a randomised, controlled trial. Gut. 2014;63:919-27
16. Moreels TG, Pelckmans PA. Gastrointestinal parasites. Potential therapy for refractory inflammatory bowel diseases. Inflamm Bowel Dis. 2005;11:178-84
17. Ramanan D, Bowcutt R, Lee SC. et al. Helminth infection promotes colonization resistance via type 2 immunity. Science. 2016;352:608-12
18. Jex AR, Nejsum P, Schwarz EM. et al. Genome and transcriptome of the porcine whipworm Trichuris suis. Nat Genet. 2014;46:701-6
19. Dagoye D, Bekele Z, Woldemichael K. et al. Wheezing, allergy, and parasite infection in children in urban and rural Ethiopia. Am J Respir Gut Care Med. 2003;167:1369-73
20. Croese J, O'neil J, Masson J. et al. A proof of concept study establishing Necator americanus in Crohn's patients and reservoir donors. Gut. 2006;55:136-7
21. Broadhurst MJ, Leung JM, Kashyap V. et al. IL-22+ CD4+ T cells are associated with therapeutic trichuris trichiura infection in an ulcerative colitis patient. Sci Transl Med. 2010;2:60ra88-60ra88
22. Elliott DE, Setiawan T, Metwali A. et al. Heligmosomoides polygyrus inhibits established colitis in IL-10 deficient mice. Eur J Immunol. 2004;34:2690-8
23. Hunter MM, Wang A, Hirota CL. et al. Neutralizing anti-IL-10 antibody blocks the protective effect of tapeworm infection in a murine model of chemically induced colitis. J Immunol. 2005;174:7368-75
24. Khan W, Blennerhasset P, Varghese A. et al. Intestinal nematode infection ameliorates experimental colitis in mice. Infect Immun. 2002;70:5931-7
25. Motomura Y, Wang H, Deng Y. et al. Helminth antigen-based strategy to ameliorate inflammation in an experimental model of colitis. Clin Exp Immunol. 2009;155:88-95
26. Cançado GGL, Fiuza JA, de Paiva NCN. et al. Hookworm products ameliorate dextran sodium sulfate-induced colitis in BALB/c mice. Inflamm Bowel Dis. 2011;17:2275-86
27. Soufli I, Toumi R, Rafa H. et al. Crude extract of hydatid laminated layer from Echinococcus granulosus cyst attenuates mucosal intestinal damage and inflammatory responses in Dextran Sulfate Sodium induced colitis in mice. J Inflamm(Lond). 2015;12:19
28. Hasby EA, Saad MAH, Shohieb Z. et al. FoxP3+ T regulatory cells and immunomodulation after Schistosoma mansoni egg antigen immunization in experimental model of inflammatory bowel disease. Cell Immunol. 2015;295:67-76
29. Yang J, Zhao J, Yang Y. et al. Schistosoma japonicum egg antigens stimulate CD4+ CD25+ T cells and modulate airway inflammation in a murine model of asthma. Immunology. 2007;120:8-18
30. Xia C-M, Zhao Y, Jiang L. et al. Schistosoma japonicum ova maintains epithelial barrier function during experimental colitis. World J Gastroenterol. 2011;17:4810-6
31. Hu S, Yang L, Wu Z. et al. Suppression of adaptive immunity to heterologous antigens by SJ16 of Schistosoma japonicum. J Parasitol. 2012;98:274-83
32. Sun X, Liu Y, Lv Z. et al. rSj16, a recombinant protein of Schistosoma japonicum-derived molecule, reduces severity of the complete Freund's adjuvant-induced adjuvant arthritis in rats' model. Parasite Immunol. 2010;32:739-48
33. Sun X, Li R, Sun X. et al. Unique roles of Schistosoma japonicum protein Sj16 to induce IFN-γ and IL-10 producing CD4+ CD25+ regulatory T cells in vitro and in vivo. Parasite Immunol. 2012;34:430-9
34. Yang F, Sun X, Shen J. et al. A recombined protein (rSj16) derived from Schistosoma japonicum induces cell cycle arrest and apoptosis of murine myeloid leukemia cells. Parasite Res. 2013;112:1261-72
35. Sann H, von Erichsen J, Hessmann M. et al. Efficacy of drugs used in the treatment of IBD and combinations thereof in acute DSS-induced colitis in mice. Life Sci. 2013;92:708-18
36. Yang Y, Yan H, Jing M. et al. Andrographolide derivative AL-1 ameliorates TNBS-induced colitis in mice: involvement of NF-кB and PPAR-γ signaling pathways. Sci Rep. 2016;6:29716
37. Bento AF, Leite DFP, Marcon R. et al. Evaluation of chemical mediators and cellular response during acute and chronic gut inflammatory response induced by dextran sodium sulfate in mice. Biochem pharmacol. 2012;84:1459-69
38. Sakaguchi S, Yamaguchi T, Nomura T. et al. Regulatory T cells and immune tolerance. Cell. 2008;133:775-87
39. Turker NS, Heidari P, Kucherlapati R. et al. An EGFR targeted PET imaging probe for the detection of colonic adenocarcinomas in the setting of colitis. Theranostics. 2014;4:893-903
40. Kamada N, Hisamatsu T, Okamoto S. et al. Unique CD14+ intestinal macrophages contribute to the pathogenesis of Crohn disease via IL-23/IFN-γ axis. J Clin Invest. 2008;118:2269-80
41. Youngman KR, Simon PL, West GA. et al. Localization of intestinal interleukin 1 activity and protein and gene expression to lamina propria cells. Gastroenterology. 1993;104:749-58
42. Mayne CG, Williams CB. Induced and natural regulatory T cells in the development of inflammatory bowel disease. Inflamm Bowel Dis. 2013;19:1772
43. Valencia X, Stephens G, Goldbach-Mansky R. et al. TNF downmodulates the function of human CD4+ CD25hi T-regulatory cells. Blood. 2006;108:253-61
44. Ghisletti S, Huang W, Ogawa S. et al. Parallel SUMOylation-dependent pathways mediate gene-and signal-specific transrepression by LXRs and PPARγ. Mol Cell. 2007;25:57-70
45. Kidani Y, Bensinger SJ. Liver X receptor and peroxisome proliferator-activated receptor as integrators of lipid homeostasis and immunity. Immunol Rev. 2012;249:72-83
46. Azuma Y-T, Nishiyama K, Matsuo Y. et al. PPARα contributes to colonic protection in mice with DSS-induced colitis. Int Immunopharmacol. 2010;10:1261-7
47. Lee JW, Bajwa PJ, Carson MJ. et al. Fenofibrate represses interleukin-17 and interferon-γ expression and improves colitis in interleukin-10 deficient mice. Gastroenterology. 2007;133:108-23
48. Tanaka T, Kohno H, Yoshitani S-i. et al. Ligands for peroxisome proliferator-activated receptors α and γ inhibit chemically induced colitis and formation of aberrant crypt foci in Rats. Cancer Res. 2001;61:2424-8
49. Zhou X, Cao L, Jiang C. et al. PPARα-UGT axis activation represses intestinal FXR-FGF15 feedback signalling and exacerbates experimental colitis. Nat Commun. 2014;5:4573
50. Gu X, Song Y, Chai Y. et al. GC-MS metabolomics on PPARα-dependent exacerbation of colitis. Mol Biosyst. 2015;11:1329-37
51. Hornung D, Waite LL, Ricke EA. et al. Nuclear peroxisome proliferator-activated receptors α and γ have opposing effects on monocyte chemotaxis in endometriosis. J Clin Endocrinol Metab. 2001;86:3108-14
52. Wu F, Huang Y, Dong F. et al. Ulcerative Colitis-Associated Long Noncoding RNA, BC012900, Regulates Intestinal Epithelial Cell Apoptosis. Inflamm Bowel Dis. 2016;22:782-95
53. Wang H, Chao K, Ng SC. et al. Pro-inflammatory miR-223 mediates the cross-talk between the IL23 pathway and the intestinal barrier in inflammatory bowel disease. Genome Biol. 2016;17:58
Author contact
![]() Corresponding authors: Xi Sun, Email: sunxi2sysu.edu.cn Zhongdao Wu, Email: wuzhdsysu.edu.cn
Corresponding authors: Xi Sun, Email: sunxi2sysu.edu.cn Zhongdao Wu, Email: wuzhdsysu.edu.cn