Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Results
Discussion
Materials and Methods
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2017; 7(16):4001-4012. doi:10.7150/thno.18788 This issue Cite
Research Paper
Ginsenoside Rg1 Inhibits Glucagon-Induced Hepatic Gluconeogenesis through Akt-FoxO1 Interaction
Qun Liu1, 2*, Fei-Ge Zhang1*, Wen-Song Zhang1, An Pan1, Yi-Lin Yang1, Jin-Feng Liu1, Ping Li1, Bao-Lin Liu1, Lian-Wen Qi1, 2 ![]()
1. State Key Laboratory of Natural Medicines, China Pharmaceutical University, Nanjing 210009, China;
2. Clinical Metabolomics Center, China Pharmaceutical University, Nanjing 211198, China.
* The authors contributed equally to the work.
Received 2016-12-16; Accepted 2017-8-14; Published 2017-9-20
Abstract

Rationale: Glucagon is involved in hepatic gluconeogenesis, playing a key role in type 2 diabetes. Ginsenosides are reported to have antidiabetic activities. Ginsenoside Rg1 is a major propanaxatriol-type saponin in ginseng. This study aims to investigate the regulatory effects of Rg1 on glucagon-induced hepatic glucose production.
Methods: The effects of Rg1 were investigated in high-fat-diet (HFD)-fed mice and glucagon-challenged C57BL/6J mice. Glucose metabolism was evaluated by oral glucose tolerance test and pyruvate tolerance test. The role of Rg1 on the regulation of Akt-FoxO1 interaction was performed using immunofluorescence, immunoprecipitation, siRNA silencing, pharmacological inhibitor and active-site mutant in primary hepatocytes or HepG2 cells.
Results: Abnormally elevated fasting glucagon levels were observed in HFD-fed mice, contributing significantly to increased fasting plasma glucose levels. Inappropriate fasting glucagon secretion inactivated Akt and promoted hepatic glucose production via upregulation of FoxO1 activity. Rg1 preserved glucagon-impaired Akt activation partly by binding to Akt at Ser473 site. Rg1 also promoted Akt binding to FoxO1 and inactivated FoxO1 by phosphorylation. Consequently, Rg1 decreased the hepatic glucose production through a decrease in transcription of phosphoenolpyruvate carboxykinase (PEPCK) and glucose 6-phosphatase (G6Pase). Both siRNA silencing of Akt and Akt inhibitor triciribine attenuated the effects of Rg1 in response to fasting hormone glucagon.
Conclusion: Akt phosphorylation at Ser473 by ginsenoside Rg1 is critical for its gluconeogenesis-lowering effect, suggesting a potential for pharmaceutical intervention in response to fasting hormone glucagon.
Keywords: ginsenoside Rg1, glucagon, gluconeogenesis, Akt, FoxO1.
Introduction
Glucose homeostasis is regulated by the balance between insulin and glucagon. Insulin prevents postprandial hyperglycemia by promoting glucose disposal, while glucagon contributes to the maintenance of fasting euglycemia through hepatic glucose production [1]. Although diabetes has been defined as a metabolic disorder owing to insulin deficiency or insulin resistance, accumulating evidence demonstrates that endogenous glucagon plays an essential role in the pathogenesis of diabetes, especially for hyperglycemia during fasting [2]. In addition to the insulin abnormality, hyperglucagonemia is often observed in diabetic patients [3]. In diabetic and obesity animals, enhanced hepatic glucagon response is associated with cellular stress and metabolic disorders [4, 5, 6]. Gene knockout of glucagon receptor in diabetic mice prevents metabolic consequences, further confirming that glucagon is indispensable in the development of diabetes [7].
Glucagon increases hepatic glucose output through gluconeogenesis, while insulin counteracts the action of glucagon on hepatic gluconeogenesis mainly through PI3K/ Akt signaling [8]. FoxO1 is a transcriptional factor, and in the liver, it collaborates with peroxisome proliferator-activated receptor gamma co-activator 1α (PGC-1α) to increase coordinately the expression of the genes encoding in gluconeogenesis, including phosphoenolpyruvate carboxykinase (PEPCK) and glucose 6-phosphatase (G6Pase) [9]. In response to insulin, Akt activation promotes nuclear exclusion of FoxO1 by phosphorylation, thereby blocking transcriptional regulation of gluconeogenic genes. However, the canonical Akt/FoxO1 regulation in insulin signaling cascades has been challenged by recent studies which show that Akt/FoxO1 is dispensable in the suppression of hepatic gluconeogenesis by insulin [10, 11]. These findings raise the question of whether or not Akt could suppress hepatic glucose production through regulation of FoxO1 in a manner independent of insulin.
Hepatic gluconeogenesis is mainly mediated through glucagon response. In response to glucagon, cAMP accumulates and induces PKA activation, which in turn phosphorylates cAMP response element binding (CREB) protein. Phosphorylated CREB, together with different co-activators, increases transcription of gluconeogenic genes. A recent study demonstrated that glucagon activates FoxO1 through the regulation of calcium-sensing enzyme CaMKII to promote excessive hepatic glucose production in obesity, indicating the special role of FoxO1 in enhanced hepatic glucagon response [12]. Given the inactivation of FoxO1 by Akt, it is reasonable to hypothesize that pharmacological activation of Akt could restrain hepatic glucagon response via suppressing FoxO1 activation.
Integrative medicine plays an important role in metabolic syndrome treatment [13]. Traditional Chinese Medicine has been widely used in the prevention and treatment of metabolic syndrome [14, 15, 16]. Ginseng is one of the world's most widely used natural products. Due to its diverse pharmacological activities, ginseng has always been an attractive herbal product to study and understand. Ginsenosides are the major bioactive constituents of ginseng. Ginsenoside Rg1 (Rg1) is a protopanaxtriol type saponin abundant in ginseng products [17]. The antidiabetic activities of ginsenosides have been widely investigated, and most of these studies focus on the regulation of insulin signaling pathway [18, 19, 20]. It remains largely unknown whether ginsenosides could regulate glucagon-induced hepatic glucose production. Metformin is an antidiabetic agent widely used as a first line treatment for diabetes, and recent studies demonstrate that its action to reduce hyperglycemia is mainly mediated by combating hepatic glucagon response [21, 22]. This work was designed to investigate the effects of Rg1 on glucagon-induced hepatic gluconeogenesis during fasting in the setting of metabolic disorders.
Results
Rg1 reduced endogenous glucose production in mice
High-fat-diet (HFD) feeding elevated fasting blood glucose levels and impaired glucose tolerance in mice (Figure 1A, 1B). Oral administration of 50 mg/kg Rg1 lowered fasting blood glucose and improved glucose tolerance in HFD mice but not in chow-fed mice, demonstrating its beneficial effect on glucose homeostasis (Figure 1A, 1B). HFD feeding increased glucagon content in the blood, whereas the increased blood glucagon was reduced by Rg1 treatment (Figure 1C). Rg1 showed no significant effects on glucagon levels in chow-fed mice. Because fasting blood glucose is mainly regulated by endogenous glucose production, we performed pyruvate tolerance, a test for hepatic glucose output, and found that HFD-fed mice showed higher blood glucose in response to pyruvate load (Figure 1D). Rg1 administration attenuated blood glucose rise in the pyruvate tolerance test in HFD-fed mice but not in chow-fed mice (Figure 1D). In glucagon-challenged (Figure 1E) and pyruvate-challenged (Figure 1F) normal mice, Rg1 treatment also reduced blood glucose concentrations, suggesting that Rg1 lowered blood glucose by suppressing endogenous glucose production. Besides, we observed that Rg1 reduced elevated levels of blood triglyceride (TG), total cholesterol (T-CHO) and free fatty acids (FFAs) in HFD-fed mice. Rg1 reduced body gains without significant influence on food intake during HFD feeding course (data not shown). Metformin, an anti-diabetic agent that lowers blood glucose by blocking hepatic gluconeogenesis [23], demonstrated a similar regulation as Rg1 in mice (Figure 1).
Ginsenoside Rg1 reduced endogenous glucose production in mice. (A) Fasting blood glucose in chow or HFD-fed C57BL/6J mice after 8 weeks; (B) Oral glucose tolerance test (OGTT, 2 g/kg body weight, p.o.) in mice fed the indicated diet after 8 weeks; (C) Blood glucagon content in mice fed the indicated diet; (D) Pyruvate tolerance test (PTT, pyruvate 2.0 g/kg, i.p.) in mice fed the indicated diet after 8 weeks of feeding; (E) Glucagon tolerance test in chow-fed mice (glucagon 2 mg/kg, i.p.). (F) PTT in chow-fed mice (pyruvate 2.0 g/kg, i.p.). (G) Plasma concentration versus time profiles of Rg1 in fasting mice after oral administration of Rg1. Rg1, 50 mg/kg; Met: metformin, 200 mg/kg; AUC: area under the curve. Data are shown as the mean ± SEM (n=8-10). *p < 0.05 vs. vehicle; #p < 0.05 vs. chow or control. The difference was determined using Wilcoxon rank test.
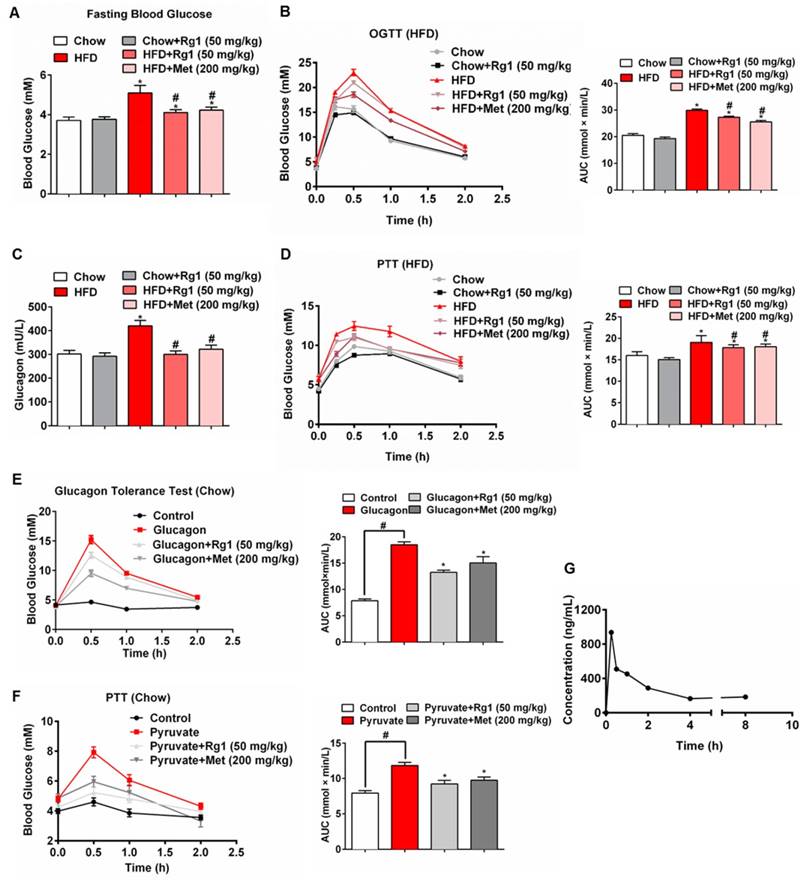
We investigated the pharmacokinetic properties of Rg1 in mice plasma using a liquid chromatography-tandem mass spectrometry method. In the fasting state, Rg1 reached Cmax at 15 min after oral administration, and returned to stable low levels after 4 h (Figure 1G). Consistent with the rise in blood concentration, Rg1 reduced fasting blood glucose in normal mice at 1 h after oral administration (Figure S1).
Rg1 suppressed glucagon-induced hepatic gluconeogenesis in fasting mice
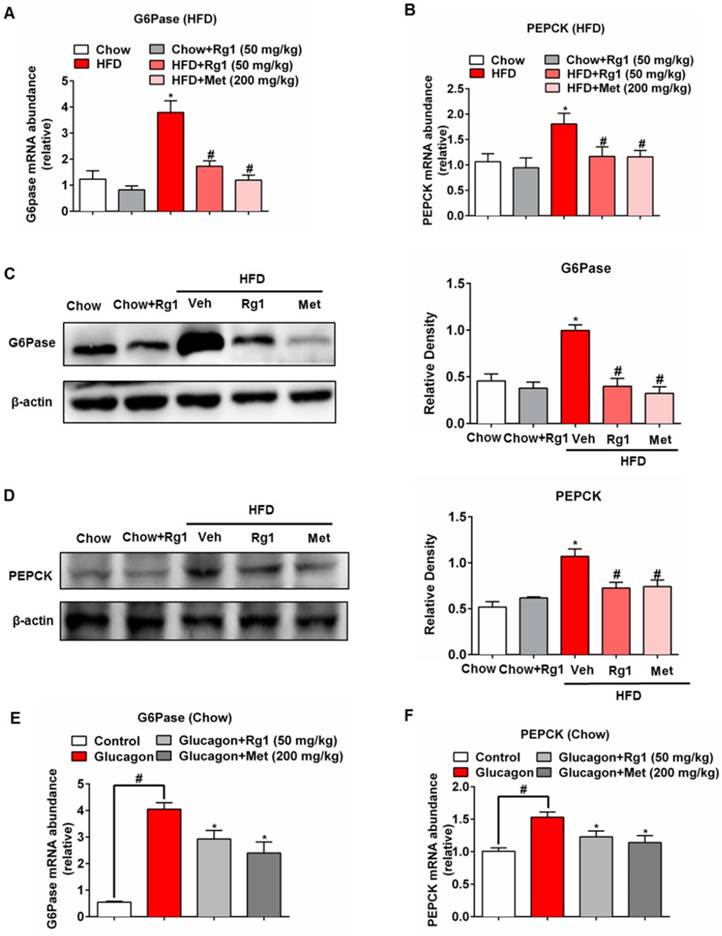
Next, we examined whether Rg1 could modulate hepatic gluconeogenesis in HFD-fed mice. PEPCK and G6Pase are the two rate-limiting enzymes controlling gluconeogenesis in the liver. HFD feeding increased gene expressions for G6Pase and PEPCK in the liver, but the transcriptional regulation was inhibited by oral administration of Rg1 and metformin (Figure 2A, 2B). Consistently, increased protein expressions for G6Pase and PEPCK were also attenuated in HFD-fed mice by Rg1 and metformin (Figure 2C, 2D). Rg1 did not affect gene and protein expressions for G6Pase and PEPCK (Figure 2A-2D) in chow-fed mice. Rg1 also reduced glucagon-induced G6Pase and PEPCK mRNA expression (Figure 2E, 2F). In normal mice during fasting, Rg1 inhibited G6Pase and PEPCK gene expression in the liver at 1 h after oral administration (Figure S2), providing direct evidence that Rg1 inhibited endogenous glucose production via blocking hepatic glucagon response.
Ginsenoside Rg1 inhibited hepatic G6Pase and PEPCK expression. (A, B) mice fed the indicated diet for 8 weeks (n=10/group). Liver mRNA levels of G6Pase and PEPCK were determined by quantitative RT-PCR; (C, D) G6Pase and PEPCK protein expressions in the liver of chow or HFD-fed mice were determined by western blot; (E, F) Chow-fed mice were orally administered Rg1 or Met, and then intraperitoneally injected with glucagon (2 mg/kg). G6Pase and PEPCK mRNA expressions in liver were determined by qPCR. Rg1, 50 mg/kg; Met: metformin, 200 mg/kg. Data are shown as the mean ± SEM after normalization to the control (n=6/group). *p < 0.05, vs. chow or control; #p < 0.05 vs. HFD or control. The difference was determined using Wilcoxon rank test.
Rg1 suppressed glucagon-mediated FoxO1 activation in hepatocytes
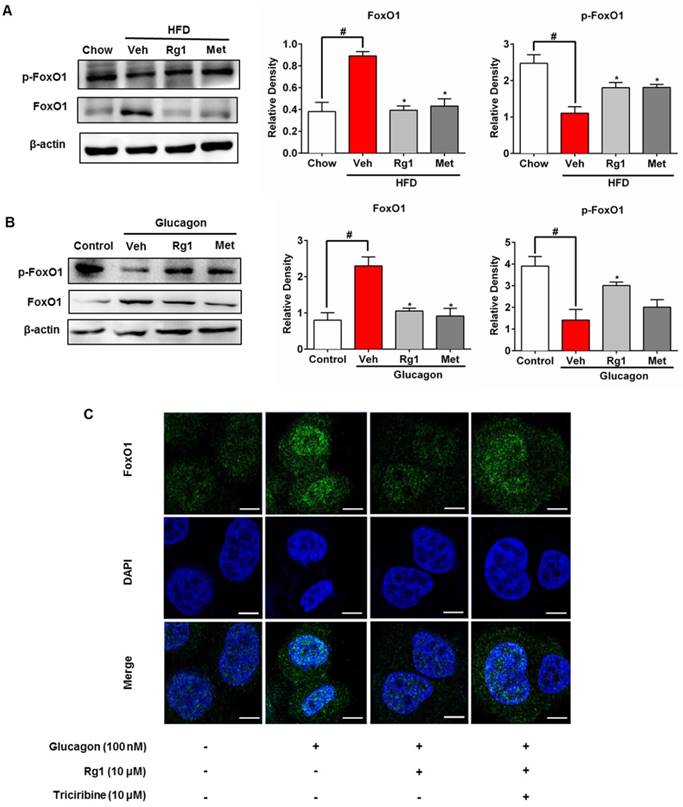
FoxO1 is a transcriptional factor encoding genes for gluconeogenesis [12], and its nuclear localization is modulated by phosphorylation. HFD feeding increased FoxO1 protein induction and activated FoxO1 by dephosphorylation in the liver, whereas these alternations were reversed by Rg1 treatment (Figure 3A). We treated C57BL/6J mice with glucagon to induce FoxO1 activation and found that Rg1 also effectively attenuated FoxO1 protein expression and inactivated FoxO1 by phosphorylation (Figure 3B). The view of confocal microscope showed that Rg1 effectively blocked nuclear translocation of FoxO1 in response to glucagon stimulation (Figure 3C). Akt inhibitor triciribine diminished the effect of Rg1 on nuclear exclusion of FoxO1 in HepG2 cells, indicative of the potential role of Akt in the action of Rg1 (Figure 3C).
Ginsenoside Rg1 suppressed FoxO1 activation. (A) Total FoxO1 protein and FoxO1 phosphorylation (Ser256) in liver of HFD-fed mice were assayed by western blot; (B) Chow-fed mice were orally administered Rg1 or Met, and then intraperitoneally injected with glucagon (2 mg/kg). Total FoxO1 protein and FoxO1 phosphorylation (Ser256) in liver were determined using western blot; (C) Primary mouse hepatocytes were pretreated with Rg1 in the absence or presence of triciribine, followed by incubation with glucagon for 2 h. The image of FoxO1 nucleus translocation was viewed with confocal scanning microscope (Green: FoxO1; Blue: DAPI. Scale bars: 10 μm). Scale bar, 10 μm. Data are shown as the mean ± SEM after normalization to the control (n=4). *p < 0.05 vs. vehicle; # p < 0.05 vs. chow or control. The difference was determined using Wilcoxon rank test.
Rg1 regulated Akt activity in hepatocytes
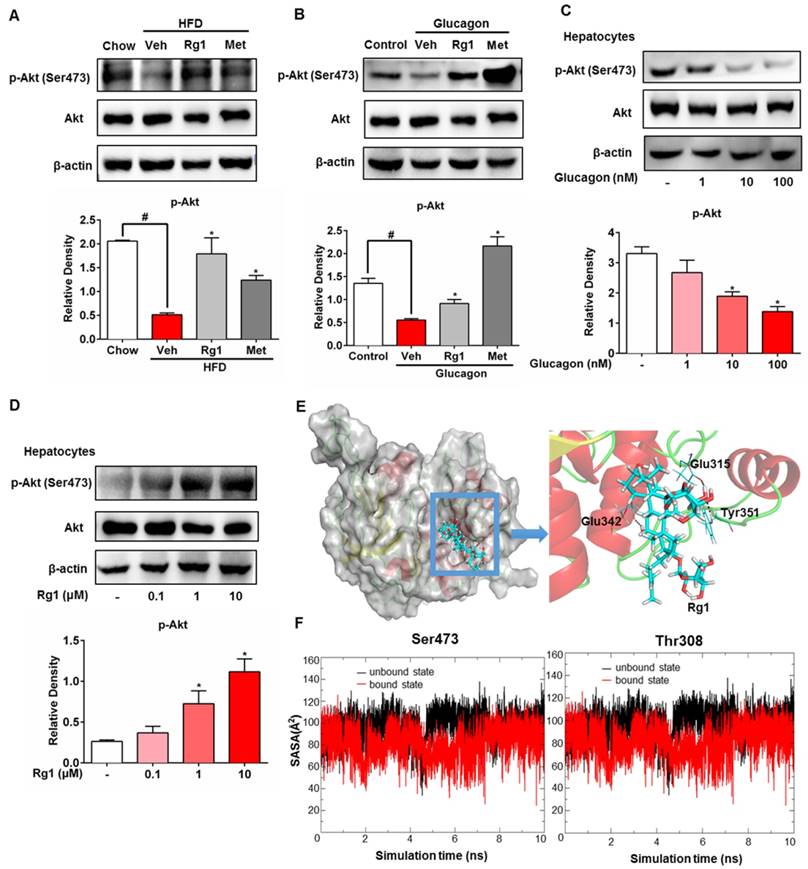
HFD feeding impaired Akt activity in the liver, whereas Rg1 administration restored Akt activity by preserving phosphorylation (Figure 4A). Consistently, Rg1 increased hepatic Akt phosphorylation in fasting mice at 1 h after oral administration (Figure S3). Interestingly, normal mice injected with glucagon displayed Akt phosphorylation inhibition at Ser473 in the liver, while Rg1 could counteract the effect of glucagon on Akt inactivation (Figure 4B). In hepatocytes, glucagon decreased Akt phosphorylation (Ser473) at concentrations ranging from 1-100 nM, indicative of Akt inactivation (Figure 4C). Rg1 increased Akt phosphorylation (Ser473) at concentrations from 0.1-10 μM, demonstrating its ability to activate Akt (Figure 4D). To explore the interaction between Rg1 and Akt, an in silico molecular docking study was performed. The binding modes of Rg1 to Akt were predicted by Autodock. As shown in Figure 4E, Rg1 formed three hydrogen-bonds with Akt by interacting with Glu315, Glu342 and Tyr351. The binding energy of Rg1 to Akt is -5.08 kcal/mol. We then investigated the protein conformational changes by performing molecular dynamics (MD) simulations to reveal the connections between Rg1 binding and protein phosphorylation. Figure 4F shows the solvent accessible surface areas (SASA) of Akt Ser473 and Thr308 evolution during the last 10 ns of MD simulations. For the Akt Ser473 site, the Rg1-bound state had a significantly larger SASA value (89.7 Å2) than the unbound state (38.9 Å2). The Rg1 binding did not cause significant changes at the Akt Thr308 site compared with the unbound state. These simulation results indicated that Rg1 binding to Akt could cause Ser473 to become more apt to phosphorylation, but has less impact on the Thr308 site.
Ginsenoside Rg1 regulated Akt activity in hepatocytes. (A) Akt phosphorylation (Ser473) in the liver of HFD-fed mice; (B) Akt phosphorylation (Ser473) and total Akt expression in the liver of normal mice injected with glucagon (2 mg/kg); (C) Akt phosphorylation (Ser473) in primary mouse hepatocytes treated with glucagon for 2 h; (D) Akt phosphorylation (Ser473) in primary mouse hepatocytes treated with Rg1 for 2 h; (E) Molecular docking analysis of the interaction between Rg1 and Akt; (F) The SASA evolution of Ser473 and Thr308 for the bound and unbound states of Akt during the MD simulation, along with the corresponding accumulative averages. Rg1, 50 mg/kg; Met: metformin, 200 mg/kg. Data are shown as the mean ± SEM after normalization to the control. *p < 0.05 vs. vehicle; # p < 0.05 vs. chow or control. The difference was determined using Wilcoxon rank test.
Rg1 suppressed hepatic glucagon response via regulation of Akt/FoxO1
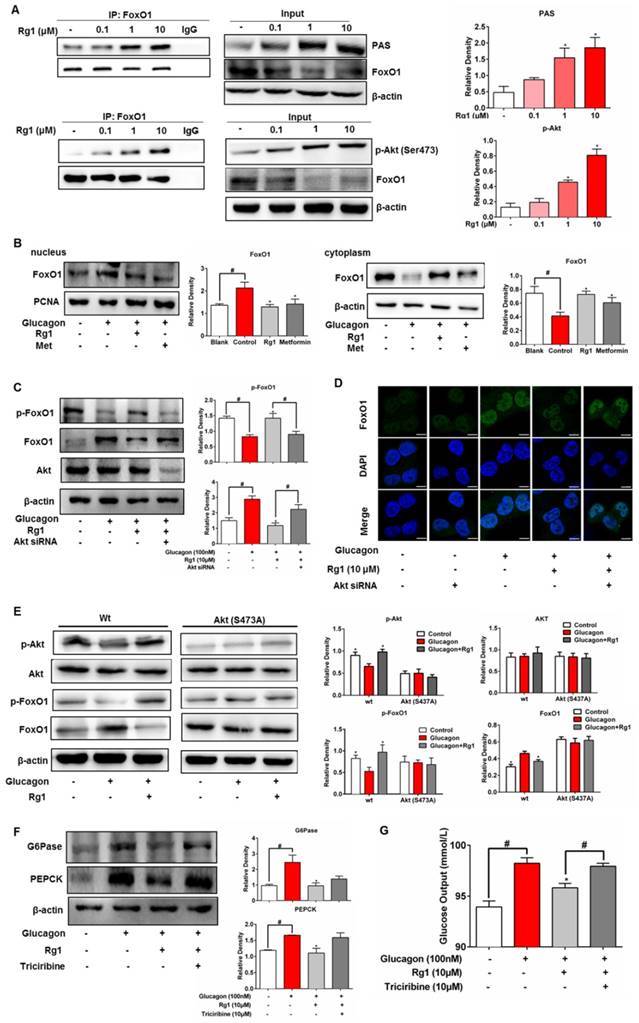
The specific bioactivity of Akt is dependent on the location of subcellular compartments. In HepG2 cells, when FoxO1 protein was immunoprecipitated and then blotted using p-Akt (Ser473) antibody, Rg1 increased Akt expression in FoxO1 in a concentration-dependent manner (Figure 5A). Immunoprecipitation examination showed that Akt consensus sequence (PAS) presented in FoxO1, and Rg1 treatment induced PAS expression (Figure 5A). These results showed that Rg1 promoted Akt binding to FoxO1 and induced Akt activation in FoxO1 (Figure 5A). Similarly, Rg1 could also promote Akt binding to FoxO1 in the presence of glucagon (Figure S4). We separated the cytoplasmic and nuclear fractions of HepG2 cells to determine the nuclear translocation of FoxO1. Western blotting was performed to test the effect of Rg1 on subcellular localization of FoxO1 in response to glucagon stimulation. As shown in Figure 5B, glucagon induced the nuclear translocation of FoxO1. When the cells were treated with Rg1, FoxO1 nuclear translocation was remarkably attenuated. We transfected HepG2 cell with siRNA to silence Akt, and found that the action of Rg1 to attenuate glucagon-induced FoxO1 induction was abrogated (Figure 5C). Likewise, the enhanced effect of Rg1 on FoxO1 phosphorylation was also weakened by knockdown of Akt (Figure 5C). Consistently, knockdown of Akt attenuated the effect of Rg1 on nuclear exclusion of FoxO1 (Figure 5D). Rg1 could inhibit the expression of FoxO1 in HepG2, but when the cells were transfected with Akt/S473A mutant, the effect of Rg1 on FoxO1 was diminished (Figure 5E), providing direct evidence that Rg1 regulated FoxO1 depending on Akt activation by phosphorylation at Ser473. In primary hepatocytes, Akt inhibitor triciribine diminished the inhibitory effects of Rg1 on glucagon-induced G6Pase, PEPCK expression and glucose production, further confirming the role of Akt in Rg1 action (Figure 5F, 5G). Together, these results demonstrated that Rg1 restrained FoxO1 activation via Akt activation, and thereby suppressed hepatic glucagon response (Figure 6).
Ginsenoside Rg1 inhibited FoxO1 activation dependent on Akt. (A) After treating HepG2 cells with Rg1 for 2 h, Akt consensus sequence (PAS) and Akt phosphorylation in FoxO1 protein were determined by immunoprecipitation and western blot; (B) Cytoplasmic and nuclear proteins were collected, and expression of FoxO1 in cytoplasmic and nuclear fraction determined by western blot. Cytoplasmic FoxO1 and nuclear FoxO1 band intensities were normalized to β-Actin or PCNA intensity; (C, D) HepG2 cells were transfected with Akt or control scrambled siRNA and then incubated with Rg1 in the presence of glucagon for 2 h. p-FoxO1 and FoxO1 expressions were determined using western blot and FoxO1 translocation into the nucleus was viewed with confocal scanning microscope (Green: FoxO1, Blue: DAPI). Scale bar, 10 μm; (E) HepG2 cells transiently transfected with vector or Akt/S473A mutant and probed with p-Akt, Akt, p-FoxO1 and FoxO1 antibodies; (F) Primary mouse hepatocytes were pretreated with Rg1 in the absence or presence of triciribine and then stimulated with glucagon for 2 h. Total FoxO1 and FoxO1 phosphorylation (Ser256) were determined by western blot; (G) Primary mouse hepatocytes were pretreated with Rg1 in the absence or presence of triciribine, and glucose output was induced by 2 h glucagon stimulation. Data are shown as the mean ± SEM after normalization to the control. *p < 0.05 vs. vehicle; #p < 0.05 vs. indicated treatment. The difference was determined using Wilcoxon rank test.
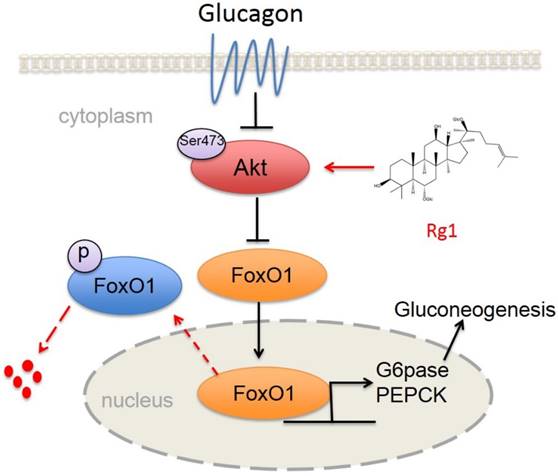
The proposed mechanism for ginsenoside Rg1 to restrain hepatic glucagon response. Glucagon inactivated Akt to inhibit FoxO1 phosphorylation. Rg1 preserved glucagon-impaired Akt activation by binding to Akt at Ser473 site and promoted Akt binding to FoxO1. Subsequently, Rg1 increased Akt-dependent FoxO1 phosphorylation and promoted FoxO1 nuclear exclusion. Finally, the hepatic glucose production was reduced through a decrease in transcription of PEPCK and G6Pase.
Discussion
Insulin inhibits hepatic gluconeogenesis through insulin receptor substrate 1/2 and PI3K signaling. Therefore, the suppression of gluconeogenesis by Akt/FoxO1 is usually considered to result from upstream insulin signaling. In this study, we showed that Akt/FoxO1 signaling pathway could combat hepatic glucagon response in a way independent of insulin. Rg1 inhibited FoxO1 translocation into the nucleus via direct activation of Akt, resultantly reducing excessive hepatic glucose output in response to fasting glucagon.
Glucagon action in the liver is to promote glucose output through gluconeogenesis and the basal glucagon level in fasting accounts for up to 70% of glucose production [24]. The mechanism by which glucagon promotes gluconeogenesis is not well established. FoxO transcription factors are major targets of insulin action and contribute to the regulation of gluconeogenesis in the liver. FoxO1 is active during fasting and inactivated after feeding [25]. It is generally accepted that FoxO1 action is controlled by insulin PI3K/Akt signaling. Although it is generally accepted that glucagon activates FoxO1 though PKA and CREB pathway, a previous study revealed that glucagon could induce FoxO1 activation through calcium/CaMKII pathway in the liver [12], indicating the special role of FoxO1 in hepatic glucagon response. In line with this observation, our work found that glucagon promoted FoxO1 activation in hepatocytes in the absence of insulin. Interestingly, we found that glucagon inactivated Akt by dephosphorylation and prevented activated Akt binding to FoxO1 (Figure S4), thereby releasing FoxO1 to regulate genes encoding to gluconeogenesis. This finding indicated that preserving the association of Akt with FoxO1 could block hepatic glucagon response. Taken together, Akt/FoxO1 signaling pathway is involved in the regulation of hepatic glucagon response in an insulin-independent manner.
Ginseng root has been used for thousands of years in Traditional Chinese Medicine to treat type 2 diabetes. Results of in vitro and in vivo animal studies as well as clinical trials support the claim that ginseng possesses hyperglycemia-lowering activity [26-28]. The mechanisms underlying ginseng and its active components ginsenosides remain largely unknown and most studies focused on the regulation of insulin signaling pathway [16-18]. In this work, we investigated whether ginsenosides regulate glucagon-induced hepatic glucose production. We showed that Rg1 reduced hyperglycemia both in HFD-fed mice and in glucagon-challenged mice by the inhibition of endogenous glucose production. Different from the regulation in HFD-fed mice, Rg1 produced no significant effects in glucagon and fasting blood glucose levels and had no significant effect on metabolic parameters in chow-fed mice. Because these results were derived from the regulation under physiological conditions, it is reasonable to speculate that Rg1 administration is relatively safe. To verify the effect of Rg1 in vivo in short time, we examined the effect of Rg1 in normal mice during fasting 1 h after oral administration, and found that fasting blood glucose and hepatic gene expressions of G6Pase and PEPCK were downregulated, while hepatic Akt phosphorylation increased (Figure S1, S2 and S3). Given the enhanced hepatic gluconeogenesis during fasting, these results indicated the special role of Rg1 in the suppression of hepatic glucose production.
The mechanisms by which Rg1 regulated glucagon response were elucidated, at least in part. Specifically, Rg1 activated Akt, promoting Akt to interact with FoxO1. Consequently, Rg1 reduced glucagon response by blocking transcriptional regulation of gluconeogenic genes. These results provided evidence for our hypothesis that Rg1 prevented hyperglycemia in HFD-fed mice by restraining hepatic glucagon response. The proposed pathway is shown in Figure 6.
In conclusion, this work not only identified an unrecognized role for ginsenosides in the regulation of glucagon response, but also suggested that pharmacological regulation of Akt/FoxO1 might be effective in reducing fasting hyperglycemia in diabetes.
Materials and Methods
Reagents and mice
Ginsenoside Rg1 (purity=95%) was a product of Jilin University. Metformin was purchased from Sigma-Aldrich, glucagon was from Novo Nordisk and triciribine was purchased from Apex Bio (Houston, USA).
Male C57BL/6J mice (18-22 g) were offered from Yangzhou University and housed in colony cages with 12 h dark-light cycles. For HFD feeding, the mice were fed a regular chow diet (SLRC, Shanghai, People's Republic of China) or a HFD (60% fat, D12492; Research Diets, New Brunswick, NJ) ad libitum for 8 weeks simultaneously with administration of Rg1 (50 mg/kg) or metformin (200 mg/kg) daily by oral gavage. All experiments and animal care were approved by Animal Ethics Committee of China Pharmaceutical University. At the end of HFD feeding, the mice were fasted for 15 h before blood collection. Glucagon in the blood was determined using ELISA kit (Biocalvin, Suzhou, China). Blood glucose, TG, T-CHO and FFAs, were assayed with commercial kits according to the manufacturers' instructions.
Glucose, pyruvate and glucagon tolerance tests
Mice were fasted overnight and orally gavaged with glucose (2.0 g/kg) for glucose tolerance test in HFD-fed mice. For pyruvate tolerance test in HFD-fed mice, fasted mice were intraperitoneally injected with pyruvate (2.0 g/kg). For glucagon tolerance test and pyruvate tolerance test in normal mice, fasting blood (15 h) was first determined, and then ginsenoside Rg1 (50 mg/kg) or metformin (200 mg/kg) were administrated 1 h before glucagon (2 mg/kg) and pyruvate (2.0 g/kg) challenge via intraperitoneal injection. Blood was collected at regular intervals for the assay of glucose contents.
Pharmacokinetic study
Twenty clean grade C57BL/6J male mice were obtained from the Sino-British SIPPR/BK Lab Animal Ltd. (Shanghai, China). The mice were maintained on a 12 h light-dark cycle with free access to food and water for 1 week. Prior to the experiments, the mice were allowed to fast for 12 h but with access to water. The mice received a single dose of Rg1 (prepared in physiological saline) of 50 mg/kg by oral administration. Blood samples (~200 mL) were collected from the retro orbital vein of ether anesthetized mice into heparinized centrifuge tubes at 0.25, 0.5, 1, 1.5, 2, 4, 6, and 8 h after oral administration. Plasma was separated by centrifugation of the whole blood samples at 1500 × g for 10 min and stored at -80°C until subsequent analysis. Chromatographic conditions: Chromatographic separation was performed on LC-30AD high performance liquid chromatography system. All samples were analyzed using an Agilent Zorbax Eclipse Plus C18 (2.1 mm × 100 mm, 1.8 μM) column with a column temperature of 55°C and a sample temperature of 4°C, sample volume of 1 μL, and mobile phase flow rate of 0.45 mL/min. Mode Flow Phase Composition: A is a solution of acetonitrile / water (v/v = 2:3) containing 10 mM ammonium acetate, and B is a solution of acetonitrile / isopropanol (v/v =1:1) containing 10 mM ammonium acetate. The elution conditions were as follows: 0-2 min was 30% B phase, B phase increased to 100% in 2.10-3 min, and the effluent was directly introduced into the mass spectrometry system without shunt. Mass spectrometry conditions: Mass spectrometry was performed using a triple quadrupole mass spectrometer (Shimadzu LC-8050 type QQQ-MS, Japan) using an electrospray ionization source in negative ion mode. MRM transitions were performed at m/z 845.5 → 637.4 for Rg1, and m/z 829.4 → 783.2 for IS (Astragaloside). Optimized values for Q1 Pre Bias, collision energy, and Q3 Pre Bias were 30 V, 31 V, and 15 V for ginsenoside Rg1, and 30 V, 25 V, and 38 V for IS, respectively. The MS parameters were as follows: Nebulizing Gas Flow of 3 L/min; Heating Gas Flow of 10 L/min; Interface Temperature of 300°C; DL Temperature of 250°C; Heat Block Temperature of 400°C; Drying Flow of 10 L/min.
Cell culture
The primary hepatocytes were prepared as described elsewhere [29]. In brief, the liver was perfused with Krebs-HEPES and digested with collagenase IV by perfusion through the inferior vena cava at a 3 mL/min. Hepatocytes were collected and cultured in Dulbecco's Modified Eagle's Medium (DMEM) supplemented with 10% fetal bovine serum (FBS). The human hepatoma cell line HepG2 was obtained from the American Type Culture Collection (HB-8065, Manassas, VA 20110) and cultured in DMEM for transfection treatment.
mRNA quantification by RT-PCR
Total RNA was isolated from cultured cells or liver using Trizol reagent (Invitrogen, Carlsbad, USA). RNA was reverse-transcribed to cDNA using HiScript Synthesis kit (Vazyme, Nanjing, China). Quantitative PCR was performed on the Roche LightCycler 96 System using the Fast SYBR Green Master Mix (Roche, Germany). The mRNA level of individual genes was normalized and calculated using the ΔΔCT method. The primers used for each target gene were as follows, G6pase (forward-5'-CGA CTC GCT ATC TCC AAG TGA-3'; reverse-5'-GTT GAA CCA GT C TCC GAC CA-3'), PEPCK (forward-5'-AAG CAT TCA ACG CCA GGT TC-3'; reverse-5'- GGG CGA GTC TGT CAG TTC AAT-3'), β-actin (forward-5'- AGT GTG ACG TTG ACA TCC GTA-3'; reverse-5'-GCC AGA GCA GTA ATC TCC TTC T-3').
Western Blotting
Proteins from total cell lysates were separated by SDS-PAGE and probed with different primary antibodies against p-Akt (Ser473), Akt, p-FoxO1 (Ser256), FoxO1 (Cell Signaling, Danvers, MA), G6Pase and PEPCK (Santa Cruz Biotechnology, Santa Cruz, CA); β-actin (Sigma-Aldrich, St. Louis, MO).
Immunoprecipitation
Cells were treated with Rg1 (0.1, 1 or 10 μM) for 2 h. After treatment, the cells were washed once with PBS and lysed for 15 min on ice. Cell lysates were then centrifuged at 12 000 × g for 20 min and the soluble fraction was collected. Next, anti-FoxO1 antibody was immunoprecipitated overnight at 4°C and then with protein A+G agarose beads (Beyotime Institute of Biotechnology, Shanghai, China) for another 2 h. After that, the protein A+G agarose beads were washed four times with the lysis buffer. The beads were then boiled in 1% SDS loading buffer for western blotting with the indicated antibodies.
Immunofluorescence
Cells were washed with cold PBS, then fixed with 4% paraformaldehyde for 20 min. After that, cells were permeabilized with 0.2% Triton X-100 and incubated with 5% BSA to block non-specific staining, and then incubated with specific primary antibodies staining (anti-FoxO1) overnight at 4°C in a humidified chamber. After several washings, the cells were incubated with Alexa Fluor 488-labeled goat anti-rabbit IgG (H+L) antibody for 1 h at 37°C. They were then washed in PBS twice and incubated in DAPI for 15 min at 37°C. The cells were mounted on a medium and visualized under a confocal scanning microscope.
Molecular docking and modeling
The initial protein structures used in this study were downloaded from the protein data bank (PDB). The PDB ID for Akt is 3doe. The Autodock program (version 4.2.6) [30] was employed to generate an ensemble of docked conformations for Rg1 bound to its targets. We used the genetic algorithm (GA) for conformational search, and performed 100 individual GA runs to generate 100 docked conformations for each ligand. For each complex, the top ranked docking pose was optimized in the binding pocket, and used as the initial geometry in the following MD. We performed 20 ns MD simulation for Rg1 bound and unbound states, respectively. All of the MD simulations were carried out with Amber14 program [31]. Then, the SASA of Thr308 and Ser473 were evaluated to investigate whether Rg1 binding could facilitate the phosphorylation of both sites. The SASA values were determined with probe radii of 1.4 Å.
Transfection and RNA Interference
HepG2 cells were seeded onto 6-well plates and transfected with Akt siRNA (sc-108063, Santa Cruz, CA, USA) or control siRNA (sc-37007, Santa Cruz, CA, USA) at 80% confluence using siRNA transfection reagent (Santa Cruz, CA, USA) or Lipofectamine® 2000 DNA transfection reagent (Invitrogen, Oregon, USA), respectively. At 24 h post-transfection, the cells with Akt siRNA transfection were treated with Rg1 for 2 h. Similarly, for the cells with control siRNA transfection, 100 nM glucagon was added for 2 h.
Cytoplasmic and Nuclear Protein Extraction
The nuclear extraction was prepared using an NE-PER Nuclear Cytoplasmic Extraction Reagent kit (Pierce, Rockford, IL, USA) according to the manufacturer's instruction. Briefly, the treated HepG2 cells were washed twice with cold PBS and centrifuged at 500 × g for 3 min. The cell pellet was suspended in cytoplasmic extraction reagent I by vortexing. The suspension was incubated on ice for 10 min, 11 μL of a second cytoplasmic extraction reagent II was added, then the sample was vortexed for 5 s, incubated on ice for 1 min and centrifuged for 5 min at 16 000 × g. The supernatant fraction (cytoplasmic extract) was transferred to a pre-chilled tube. The insoluble pellet fraction, which contains crude nuclei, was resuspended in 100 μL of nuclear extraction reagent by vortexing for 15 s and incubated on ice for 10 min, then centrifuged for 10 min at 16 000 × g. The resulting supernatant, constituting the nuclear extract, was used for subsequent experiments.
Akt mutant assay
Akt/S473A protein was expressed and purified from baculovirus-infected HepG2 cells. Briefly, HepG2 cells were maintained in Trypsin-EDTA media (Invitrogen) and infected with baculovirus encoding pEX-3-Akt for 24 h. The infected cells were treated with 10% FBS for 15 min prior to lysis. The infected cells were lysed in PBS containing 1% Triton X-100 and 10 mM imidazole. Transient transfection of HepG2 cell was carried out by using Effectene reagents (Qiagen). Lipofectamine 2000 (Invitrogen) was used to transfect Akt mut into HepG2 cells.
Hepatic glucose output
Primary mouse hepatocytes were maintained in DMEM medium with 10% FBS. After planting, the media was replaced with serum-free DMEM for 18 h. After wash, the cells were incubated in glucose-free media supplemented with 10 mM pyruvate, 100 nM glucagon or with Rg1 at given concentrations for 4 h. Then the cell supernatant was collected for glucose analysis.
Statistical analysis
All results are expressed as the mean ± SEM. All two group comparisons were deemed statistically significant by Wilcoxon rank test if p<0.05 (GraphPad Prism 5 software). Number of mice used in each in vivo condition is indicated in the figure legends.
Abbreviations
PGC-1α: peroxisome proliferator-activated receptor gamma co-activator 1α; PEPCK: phosphoenolpyruvate carboxykinase; G6Pase: glucose 6-phosphatase; CREB: cAMP response element binding; HFD: high-fat-diet; TG: triglyceride; T-CHO: total cholesterol; FFAs: free fatty acids; MD: molecular dynamics; SASA: solvent accessible surface areas; DMEM: Dulbecco's Modified Eagle's Medium; FBS: fetal bovine serum; PDB: protein data bank; GA: genetic algorithm; OGTT: oral glucose tolerance test; PTT: pyruvate tolerance test; AUC: area under the curve.
Supplementary Material
Supplementary figures. Ginsenoside Rg1 reduced fasting glucose, inhibited hepatic G6Pase and PEPCK expression, and regulated Akt activity in normal mice. Ginsenoside Rg1 inhibited FoxO1 activation dependent on Akt under the glucagon stimulation.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant Nos. 81603353, 91639115, and 81421005) and Natural Science Foundation of Jiangsu Province (BK20160762). The authors thank Dr. Raphael N. Alolga, a native English teacher for editing our manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Ikeda T, Hoshino T, Honda M. et al. Effect of glucagon on glucose output from bivascularly perfused rat liver. Exp Clin Endocrinol. 1989;94:383-6
2. Unger RH, Cherrington AD. Glucagonocentric restructuring of diabetes: a pathophysiologic and therapeutic makeover. J Clin Invest. 2012;122:4-12
3. Unger RH. Role of glucagon in the pathogenesis of diabetes: the status of the controversy. Metabolism. 1978;27:1691-1709
4. Wang Y, Vera L, Fischer WH. et al. The CREB coactivator CRTC2 links hepatic ER stress and fasting gluconeogenesis. Nature. 2009;460:534-7
5. Ozcan L, Wong CC, Li G. et al. Calcium signaling through CaMKII regulates hepatic glucose production in fasting and obesity. Cell Metab. 2012;15:739-51
6. Song WJ, Mondal P, Wolfe A. et al. Glucagon Regulates Hepatic Kisspeptin1 to Impair Insulin Secretion. Cell Metab. 2014;19:667-81
7. Lee Y, Wang MY, Du XQ. et al. Glucagon receptor knockout prevents insulin-deficient type 1 diabetes in mice. Diabetes. 2011;60:391-7
8. Saltiel AR, Kahn CR. Insulin signalling and the regulation of glucose and lipid metabolism. Nature. 2001;414:799-806
9. Puigserver P, Rhee J, Donovan J. et al. Insulin-regulated hepatic gluconeogenesis through FOXO1-PGC-1alpha interaction. Nature. 2003;423:550-5
10. Lu M, Wan M, Leavens KF. et al. Insulin regulates liver metabolism in vivo in the absence of hepatic Akt and Foxo1. Nat Med. 2012;18:388-95
11. Titchenell PM, Chu Q, Monks BR. et al. Hepatic insulin signaling is dispensable for suppression of glucose output by insulin in vivo. Nat Commun. 2015;6:7078
12. Ozcan L, Wong CC, Li G. et al. Calcium signaling through CaMKII regulates hepatic glucose production in fasting and obesity. Cell Metab. 2012;15:739-51
13. Wang WJ. Enhancing the treatment of metabolic syndrome with integrative medicine. J Integr Med. 2013;11:153-6
14. Zhang Q, Xiao XH, Li M. et al. Chromium-containing traditional Chinese medicine, Tianmai Xiaoke Tablet improves blood glucose through activating insulin-signaling pathway and inhibiting PTP1B and PCK2 in diabetic rats. J Integr Med. 2014;12:162-70
15. Yao Z, Zhang L, Ji G. Efficacy of polyphenolic ingredients of Chinese herbs in treating dyslipidemia of metabolic syndromes. J Integr Med. 2014;12:135-46
16. Perry B, Zhang J, Saleh T. et al. Liuwei Dihuang, a traditional Chinese herbal formula, suppresses chronic inflammation and oxidative stress in obese rats. J Integr Med. 2014;12:447-54
17. Qi LW, Wang CZ, Yuan CS. Isolation and analysis of ginseng: advances and challenges. Nat Prod Rep. 2011;28:467-95
18. Chen W, Wang J, Luo Y. et al. Ginsenoside Rb1 and compound K improve insulin signaling and inhibit ER stress-associated NLRP3 inflammasome activation in adipose tissue. J Ginseng Res. 2016;40:351-8
19. Shen L, Haas M, Wang DQ. et al. Ginsenoside Rb1 increases insulin sensitivity by activating AMP-activated protein kinase in male rats. Physiol Rep. 2015:3 pii: e12543
20. Chu JM, Lee DK, Wong DP. et al. Ginsenosides attenuate methylglyoxal-induced impairment of insulin signaling and subsequent apoptosis in primary astrocytes. Drug Metab Dispos. 2015;43:1702-10
21. He L, Sabet A, Djedjos S. et al. Metformin and insulin suppress hepatic gluconeogenesis through phosphorylation of CREB binding protein. Cell. 2009;137:635-46
22. Miller RA, Chu Q, Xie J. et al. Biguanides suppress hepatic glucagon signalling by decreasing production of cyclic AMP. Nature. 2013;494:256-60
23. Foretz M, Guigas B, Bertrand L. et al. Metformin: from mechanisms of action to therapies. Cell Metab. 2014;20:953-66
24. Cherrington AD, Liljenquist JE, Shulman GI. et al. Importance of hypoglycemia-induced glucose production during isolated glucagon deficiency. Am J Physiol. 1979;236:E263-71
25. Zhang W, Patil S, Chauhan B. et al. FoxO1 regulates multiple metabolic pathways in the liver: effects on gluconeogenic, glycolytic, and lipogenic gene expression. J Biol Chem. 2006;281:10105-17
26. Luo J Z, Kim J W, Luo L G. Effects of Ginseng and its four purified ginsenosides (Rb2, Re, Rg1, Rd) on human pancreatic islet β cell in vitro. Eur J Pharm Med Res. 2016;3:110
27. Oh M J, Kim H J, Park E Y. et al. The effect of Korean Red Ginseng extract on rosiglitazone-induced improvement of glucose regulation in diet-induced obese mice. J Ginseng Res. 2017;41:52-9
28. Vuksan V, Sung M K, Sievenpiper J L. et al. Korean red ginseng (Panax ginseng) improves glucose and insulin regulation in well-controlled, type 2 diabetes: results of a randomized, double-blind, placebocontrolled study of efficacy and safety. Nutr Metab Cardiovasc Dis. 2008;18:46-56
29. Ke B, Zhao Z, Ye X. et al. Inactivation of NF-κB p65 (RelA) in liver improves insulin sensitivity and inhibits cAMP/PKA pathway. Diabetes. 2015;64:3355-62
30. Morris G M, Goodsell D S, Halliday R S. et al. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J Comput Chem. 1998;19:1639-62
31. Case D A, Cheatham T E, Darden T. et al. The Amber biomolecular simulation programs. J Comput Chem. 2005;26:1668-88
Author contact
![]() Corresponding author: Dr. Lian-Wen Qi, Clinical Metabolomics Center, China Pharmaceutical University, No. 639 Longmian Road, Nanjing 211198, China, Email: Qilwedu.cn, Tel.:/Fax: +86 25 86185559.
Corresponding author: Dr. Lian-Wen Qi, Clinical Metabolomics Center, China Pharmaceutical University, No. 639 Longmian Road, Nanjing 211198, China, Email: Qilwedu.cn, Tel.:/Fax: +86 25 86185559.