Impact Factor
- Issue 9; 2026
- Issue 8; 2026
- Issue 7; 2026
- Issue 6; 2026
- Issue 5; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Conclusions
Supplementary Material
Abbreviations
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2017; 7(18):4383-4398. doi:10.7150/thno.20745 This issue Cite
Research Paper
Rational design of Polymeric Hybrid Micelles to Overcome Lymphatic and Intracellular Delivery Barriers in Cancer Immunotherapy
Hanmei Li1, 2*, Yanping Li1*, Xue Wang1, Yingying Hou1, Xiaoyu Hong1, Tao Gong1, Zhirong Zhang1, Xun Sun1 ![]()
1. Key Laboratory of Drug Targeting and Drug Delivery Systems, Ministry of Education, West China School of Pharmacy, Sichuan University, Chengdu 610041, China;
2. College of Pharmacy and Biological Engineering, Chengdu University, Chengdu 610106, China.
* Contributed equally to this work.
Received 2017-4-26; Accepted 2017-8-21; Published 2017-9-26
Abstract

Poor distribution of antigen/adjuvant to target sites and inadequate induction of T cell responses remain major challenges in cancer immunotherapy because of the lack of appropriate delivery systems. Nanocarrier-based antigen delivery systems have emerged as an innovative strategy to improve vaccine efficacy. Here we present polymeric hybrid micelles (PHMs) as a simple and potent antigen/adjuvant co-delivery system with highly tunable properties. PHMs consist of two amphiphilic diblock copolymers, polycaprolactone-polyethylenimine (PCL-PEI) and polycaprolactone-polyethyleneglycol (PCL-PEG). PHMs with different proportions of cationic PCL-PEI were prepared and loaded with tyrosinase-related protein 2 (Trp2) peptide and adjuvant CpG oligodeoxynucleotide to generate the Trp2/PHM/CpG co-delivery system. Lymphatic and intracellular antigen delivery as a function of PCL-PEI proportion was evaluated in vitro and in vivo. PHMs containing 10% (w/w) PCL-PEI (Trp2/PHM10/CpG) showed the optimal balance of good distribution to lymph nodes, strong immunization effect after subcutaneous administration, and low toxicity to dendritic cells. In a mouse model of B16F10 melanoma, Trp2/PHM10/CpG showed significantly higher antigen-specific cytotoxic T lymphocyte activity and greater anticancer efficacy than Trp2/PHM0/CpG without PCL-PEI or a mixture of free Trp2 and CpG. These results provide new insights into how cationic segments affect the efficiency of antigen delivery by cationic nanocarriers. They also suggest that PHMs can serve as a structurally simple and highly tunable platform for co-delivery of antigen and adjuvant in cancer immunotherapy.
Keywords: cancer immunotherapy, co-delivery system, amphiphilic diblock copolymers, tyrosinase-related protein 2 peptide, subcutaneous administration.
Introduction
In recent years, active immunotherapy has shown great promise in cancer treatment, especially for aggressive and metastatic cancers. Not only can the immune system be harnessed to specifically target existing tumor cells, but it may also provide long-term memory against recurrence [1-3]. Antigen-presenting cells, especially dendritic cells (DCs), play a central role in cancer immunotherapy. DCs process and present tumor antigens on their surface via MHC-I molecules [4, 5]. Upon activation and maturation, antigen-presenting DCs activate an antigen-specific cytotoxic T lymphocyte (CTL) response, which is important for tumor eradication [6]. An efficient CTL response depends on optimal antigen loading and activation of DCs [7].
DC activation in immunotherapy is challenging for several reasons [8, 9]. One is the instability of antigen and adjuvant: most antigens and adjuvants are proteins, peptides or nucleic acids, which degrade easily in the body before reaching the target site. Another problem is that antigen and adjuvant do not distribute efficiently into secondary lymphoid organs. Instead, antigens and adjuvants distribute to irrelevant organs and tissues, resulting in low abundance at the target site. A third problem is that antigen and adjuvant inefficiently enter DCs, limiting their ability to trigger an immune response. A fourth challenge is that antigens and adjuvants usually do not reach the same DCs at the same time. This results in weaker induction of antigen-specific humoral and cellular immune responses than when antigen is co-presented simultaneously with immunoregulatory adjuvants.
Co-delivering antigen and adjuvant to antigen-presenting cells may lead to more potent immune responses, but the two molecules usually have quite distinct biodistribution properties. This highlights the need for appropriate nanocarriers that can stably accommodate both antigen and adjuvant, protecting them from degradation in vivo and ensuring synchronized delivery to antigen-presenting cells.
Polymer hybrid micelles (PHMs), consisting of different amphiphilic diblock copolymers, have become promising carriers for co-delivery of nucleic acids and small-molecule drugs [10]. In general, nanoparticle-based drug delivery systems can efficiently co-deliver hydrophobic drugs and nucleic acids to tumors [11-13]. Hydrophobic agents can be encapsulated within the hydrophobic core of PHMs via hydrophobic interaction, while cationic hydrophilic segments can encapsulate nucleic acids. The physicochemical properties of PHMs, including the proportion of cationic segments, can be optimized simply by adjusting the ratio of the two diblock copolymers, which is much easier than altering the copolymer architecture as in conventional cationic nanocarriers.
Our laboratory has developed PHMs as a new intracellular delivery system for co-delivery of microRNA and small-molecule drugs for cancer therapy [10]. Those studies indicated that PHMs could efficiently co-deliver different therapeutics simultaneously into the cytoplasm of target cells. Here we aimed to explore the potential of PHMs as a system for co-delivering peptide antigen and nucleic acid adjuvant for cancer immunotherapy. As model antigen, we selected the melanoma antigen tyrosinase-related protein 2 (Trp2) peptide, comprising residues 180-188 of the Trp2 protein (SVYDFFVWL) [14]. Meanwhile, we selected CpG oligodeoxynucleotide (CpG ODN) as the adjuvant, which activates immature DCs via Toll-like receptor (TLR) 9 to initiate the immune response [15]. We encapsulated the hydrophobic Trp2 peptide within the hydrophobic core of PHMs via hydrophobic interactions and encapsulated CpG via interactions with cationic hydrophilic segments in the PHMs.
With these PHM preparations, we investigated relationships between their physicochemical properties and their immunization effect after subcutaneous administration. These insights may help guide and improve the synthesis of novel materials and rational design of PHMs for cancer immunotherapy. To this end, we formulated PHMs with different ratios of the two diblock copolymers, loaded them with Trp2 peptide and CpG, and compared them in terms of in vitro stability, cytotoxicity, intracellular delivery, as well as in vivo tissue biodistribution and ability to elicit CTL activity (Scheme 1). The preparations were also compared in terms of their immunoregulatory effects and therapeutic efficacy against the metastatic melanoma cell line B16F10 in culture and a mouse model.
Materials and Methods
Materials
PCL-PEI and PCL-PEG were synthesized in our laboratory as previously reported [16], using branched polyethylenimines (PEI, Mw=2000, Sigma-Aldrich, USA) or methoxy-poly(ethylene glycol) (mPEG-NH2, Mw=5000, JianKai, China) and PCL-NPC. Structures were confirmed using 1H NMR [16]. Trp2180-188 peptide (SVYDFFVWL, Mw=1175) and FITC-labeled Trp2180-188 were purchased from Kaijie Peptide Company (Chengdu, China). All organic solvents were of analytical grade. 5'-SPO3-CpG ODN1826 (50'-TCCATGACGTTCCTGACGTT-30') and FAM/Cy5-labeled CpG ODN1826 were synthesized by ShengGong (Shanghai, China).
Cell lines and mice
Murine melanoma cell line B16F10 was obtained from the American Type Culture Collection (ATCC, USA), and dendritic cell line DC2.4 was kindly donated by the Third Military Medical University. B16F10 and DC2.4 cells were cultured in RPMI-1640 supplemented with 10% fetal bovine serum, 100 μg/mL streptomycin and 100 U/mL penicillin (Invitrogen, Carlsbad, CA). Male C57BL/6 mice 6-8 weeks old were obtained from the Laboratory Animal Center of Sichuan University (Chengdu, China). All animal experiments were carried out under protocols approved by the Institutional Animal Care and Use Committee of Sichuan University.
Schematic illustration of Trp2/PHM/CpG vaccine preparation and injection into mice. Cationic PCL-PEI is mixed with neutral PCL-PEG, then loaded with Trp2 by solvent injection and loaded with CpG by electrostatic adsorption. The resulting Trp2/PHM/CpG activates immature DCs, which in turn activate cytotoxic T cells by presenting the antigen-MHC I complex to T cell receptors and facilitating co-receptor binding between T cells and DCs.
Preparation and characterization of Trp2/PHM/CpG nanoparticles
Trp2 was encapsulated into PHM to obtain Trp2/PHM, which was incubated with CpG to yield Trp2/PHM/CpG. Trp2 powder (1 mg) and a mixture of PCL-PEG and PCL-PEI (12.5 mg), composed of PCL-PEG and PCL-PEI in mass ratios of 100:0, 95:5, 90:10, 75:25 or 50:50, were dissolved in 1 mL of methanol. This solution was slowly added to 10 mL of pure water and stirred with moderate speed for 10-15 min at room temperature. Then organic solvents were removed by rotary vacuum evaporation at 37 °C, yielding 1 mL of Trp2/PHM. A solution of 100 μg/mL CpG in sterile water was mixed with an equal volume of Trp2/PHM by gentle pipetting, then the mixture was incubated at room temperature for 30 min to obtain Trp2/PHM/CpG. Dynamic light scattering was used to measure Trp2/PHM/CpG size and zeta potential (Zetasizer Nano ZS90, Malvern, UK). Encapsulation efficiency (EE%) of Trp2 and CpG in Trp2/PHM/CpG was determined using ultrafiltration (300 kDa, 3000 × g, 30 min). FITC-Trp2 and FAM-labeled CpG were used in order to allow fluorometric measurement of Trp2 and CpG concentration. The total amount of FITC-Trp2 or FAM-labeled CpG was determined by fluorescence spectrophotometry. Briefly, FITC-Trp2/PHM/CpG or Trp2/PHM/FAM-CpG was ultrafiltered, and the filtrate was collected to analyze the amount of free FITC-Trp2 or FAM-CpG by fluorescence spectrophotometry. EE% was calculated according to the following equation:
EE% = (Weight of drug in PHM) / (Weight of drug added) × 100%.
To investigate the release kinetics of FITC-labeled Trp2 and Cy5-CpG from Trp2/PHM/CpG, 1 mL of FITC-Trp2/PHM/Cy5-CpG (0.5 mg of FITC-Trp2 and 0.05 mg of Cy5-CpG) was dialyzed using a molecular weight cut-off of 8-12 kDa (Millipore, USA), against 50 mL PBS (pH 7.4). At designated time intervals, aliquots were removed from the dialysate and replaced with 1 mL of PBS. The amount of FITC-Trp2 and Cy5-CpG in the dialysate was determined by fluorescence spectrophotometry.
Transmission electron microscopy of blank PHM10 and Trp2/PHM10/CpG
The morphology of blank PHM10 and Trp2/PHM10/CpG was examined by transmission electron microscopy (TEM; H-600, Hitachi, Japan) using a negative stain technique. Blank PHM10 and Trp2/PHM10/CpG (containing 1 mg/mL of PCL-PEG) were dispersed onto a copper grid for 60 s, then the bulk solution was removed and stained with phosphotungstic acid (1%) for 20 s. After removal of the dye, the sample was observed by TEM.
Gel retardation assay
Condensation of CpG into PHMs was monitored by gel electrophoresis. Different Trp2/PHM/CpG formulations were prepared as described above. Trp2/PHM/CpG (20 μL) was mixed with 6× loading buffer (4 μL), then the mixture was loaded onto a 1% agarose gel containing 500 ng/mL ethidium bromide. Electrophoresis was carried out at 120 mV for 15 min in 1× TAE running buffer. Finally, the results were recorded with a UV illuminator. This allowed detection of CpG that was entirely uncomplexed or exposed on the micelle surface.
Stability of Trp2/PHM/CpG
Trp2/PHM/CpG complexes were stored at 4 °C for 1 week, and changes in particle size were assessed by dynamic light scattering (Zetasizer Nano ZS90, Malvern, UK). Samples were measured in triplicate.
Uptake of PHMs by DCs
DC2.4 cells were seeded into 12-well culture plates, grown for 24 h, then incubated for 2 h with different Trp2/PHM/Cy5-CpG formulations (Cy5-CpG, 800 ng/mL). Cells were washed with cold PBS, resuspended in 1 mL of PBS, and analyzed by flow cytometry (CytomicsTM FC500, Beckman).
To evaluate the co-delivery efficiency of Trp2/PHM/CpG, PHMs loaded with FITC-Trp2 and Cy5-CpG were prepared and incubated with DC2.4 cells for 2 h. The percentage of double-positive cells was determined by flow cytometry. Confocal laser scanning microscopy (Leica DM6000, Solms, Germany) was used to observe the co-localization of FITC-Trp2 and Cy5-CpG.
Cytotoxicity of PHMs
DC2.4 cells were seeded in 96-well plates, incubated for 24 h, then exposed for 24 h to various Trp2/PHM/CpG formulations (CpG, 800 ng/mL). The medium was removed and 100 μL of fresh serum-free RPMI-1640 medium containing 10% (v/v) Cell Counting Kit-8 (CCK-8) reagent was added. Plates were incubated at 37 °C for 2 h, and absorbance at 450 nm was measured using a Varioskan Flash microplate reader (Thermo Scientific). Cell viability (%) was calculated according to the following equation, where Atreat and Acontrol represent the absorbance of Trp2/PHM/CpG-treated cells and untreated cells, respectively:
Cell viability % = (Atreat/Acontrol) × 100%
Induction of BMDC maturation and proliferation by Trp2/PHM/CpG
Different Trp2/PHM/CpG formulations were incubated with immature BMDCs for 24 h at 37 °C. As a positive control, some cultures were incubated with LPS (1 μg/mL). After the treatment, cells were harvested and marked with PE-Cy5-labeled anti-mouse CD86 monoclonal antibody as well as FITC-labeled anti-mouse CD80 monoclonal antibody (eBioscience, USA). Then the labeling of cells with each antibody was analyzed by flow cytometry (Cytomics™ FC500). In addition, the level of pro-inflammatory cytokines IFN-γ and IL-12 secreted from BMDCs after treatment by various Trp2/PHM/CpG formulations were assayed by using enzyme-linked immunosorbent assay (ELISA) kits (R&D Systems, Minneapolis, MN, USA).
To assay BMDC proliferation, immature BMDCs were seeded in 96-well plates and incubated with PBS, LPS (1 μg/mL) or various Trp2/PHM/CpG formulations for 24 h at 37 °C. Numbers of viable cells (Cell viability %) were quantified as described in previous section.
Biodistribution of Trp2/PHM/CpG
C57BL/6 mice were injected in the hind foot pad with saline, DiD/CpG, DiD/PHM0/CpG, DiD/PHM5/CpG, DiD/PHM10/CpG, DiD/PHM25/CpG or DiD/PHM25/CpG (1 µg DiD in all cases). Mice were imaged at 5 min, 3 h, 24 h and 48 h after injection using an IVIS® Spectrum system (Caliper, Hopkington, MA, USA). At 24 h after injection, mice were sacrificed and the specific organ distribution of each formulation was assessed by measuring fluorescence in heart, liver, spleen, lung, kidney, and lymph nodes closer or farther from the injection site. Lymph nodes were homogenized using a cell strainer to obtain single-cell suspensions, which were stained with FITC-labeled monoclonal antibody against CD11c+ to mark dendritic cells. Percentages of cells positive for DiD and FITC were measured using flow cytometry.
PHMs were co-loaded with FITC-Trp2 and Cy5-CpG, then injected into the right hind foot pad of mice. Control animals were injected with saline or with a mixture of free FITC-Trp2 and Cy5-CpG. At 6 h after injection, mice were sacrificed and lymph nodes were excised and digested in 1 mg/mL of collagenase D for 30 min at 37 °C. Then the digested lymph nodes were homogenized with a cell strainer to obtain single-cell suspensions. Percentages of cells positive for both FITC-Trp2 and Cy5-CpG were determined using flow cytometry. Distribution of FITC-Trp2 and Cy5-CpG was also analyzed in distal lymph nodes by freezing the excised nodes in tissue freezing medium (Leica CM 1950), then cutting them into sections 10 mm thick, fixing with 4% paraformaldehyde and staining with DAPI. Signals from DAPI, FITC and Cy5 were observed in the stained sections using fluorescence microscopy (TCS SP5, Leica, Germany).
Cytotoxic T lymphocyte (CTL) assay in vivo
C57BL/6 mice aged 6-8 weeks were subcutaneously immunized with saline, Trp2/CpG, Trp2/PHM0/CpG, Trp2/PHM5/CpG, Trp2/PHM10/CpG, Trp2/PHM25/CpG or Trp2/PHM50/CpG (16 μg Trp2, 1.6 μg CpG) on days 0, 7 and 14. On day 21, naïve C57BL/6 mice were sacrificed and splenocytes were harvested. The harvested splenocytes were divided into two aliquots and pulsed with either 10 μM Trp2 peptide or complete medium for 2 h at 37 °C. The Trp2 peptide-pulsed cells were stained with 4 μM CFSE (CFSEhigh), while the medium-treated cells were stained with 0.4 μM CFSE (CFSElow). Then the two aliquots, each containing 5 × 107/mL cells, were mixed together and administered intravenously into control or immunized mice. After 18 h, the spleen was isolated from the treated mice, then splenocytes were collected and analyzed by flow cytometry. The numbers of CFSEhigh and CFSElow cells were determined. The percentage of Trp2-specific lysis was calculated according to the following equation [17], where X is the ratio CFSEhigh/CFSElow in naive mice:
Specific lysis % = [1 - CFSEhigh/(CFSElow × X)] × 100%
Tumor growth inhibition
On day 0, male C57BL/6 mice aged 6-8 weeks were inoculated subcutaneously with B16F10 cells (1×105) on the back and randomly allocated into 7 groups (n=10 per treatment). On days 4, 11 and 18, the animals were subcutaneously injected with various Trp2/PHM/CpG (16 μg Trp2, 1.6 μg CpG) formulations on the hind foot pad. Tumor growth, dates of the animal death and body weight were monitored every 2-3 days, and tumor volume was calculated using the formula (L × W2 / 2), where W refers to the smaller diameter and L to the larger. Mice were sacrificed on day 20.
Ability of Trp2/PHM/CpG to stimulate the production of IFN-γ and IL-4
To determine the ability of various formulations to stimulate production of IFN-γ and IL-4 by CD8+ and CD4+ T cells in splenocytes. C57BL/6 mice aged 6-8 weeks were subcutaneously immunized with saline, Trp2/CpG, Trp2/PHM0/CpG, Trp2/PHM5/CpG, Trp2/PHM10/CpG, Trp2/PHM25/CpG or Trp2/PHM50/CpG (16 μg Trp2, 1.6 μg CpG) on day 0, 7 and 14. On day 21, splenocyte suspensions were prepared from immunized mice and incubated with 0.2 μM Trp2 peptide for 1 h at 37 °C. Then brefeldin A was added and incubated for further 5 h to prevent extracellular secretion. After washing, splenocytes were stained with FITC-labeled anti-mouse antibody FITC-anti-CD8 or FITC-anti-CD4 (eBioscience). Then cells were mixed with IC fixation buffer, followed by permeabilization buffer. Finally, splenocytes were incubated with PE-labeled anti-mouse antibody PE-anti-IFN-γ or PE-anti-IL-4 (eBioscence). Flow cytometry was used to determine the proportions of CD8+/IFN-γ+ cells and CD4+/IL-4+ cells in the splenocytes. In addition, to determine the concentration of IFN-γ or IL-4 produced by splenocytes after stimulation by various formulations, splenocytes prepared from immunized mice were incubated with 10 μM Trp2 peptide for 60 h at 37 °C. Then the concentrations of the cytokines IFN-γ and IL-4 in the culture supernatant were determined by ELISA kit (R&D Systems).
Statistical Analysis
All quantitative data is expressed as mean ± SD from triplicate measurements, unless otherwise noted. Differences between treatment groups were assessed for significance using single factor analysis of variance, followed by Student's t-test. The threshold for significance was p < 0.05.
Results
Preparation and characterization of various PHMs containing Trp2 peptide and CpG
Co-delivery of Trp2 peptide antigen and the potent adjuvant CpG to antigen-presenting cells aimed to improve the efficiency of cancer vaccines. PHMs are stable micelles prepared from the self-assembly of amphiphilic diblock copolymers PCL-PEI and PCL-PEG. They both possess the same hydrophobic poly-(caprolactone) (PCL), which forms the inner core of PHMs to encapsulate hydrophobic drugs. PCL-PEI and PCL-PEG differ in their hydrophilic block: PCL-PEI contains PEI, which possesses partially protonated amine groups and could complex with negatively charged nucleic acid drugs and facilitate their uptake by cells [12]; PCL-PEG contains uncharged PEG, which gives the micelles colloidal stability.
We prepared PHMs by synthesizing PCL-PEI and PCL-PEG as we described previously [10]. PCL-PEI was synthesized by conjugating branched poly-(ethyleneimine) (MW 2000 Da, PEI 2k) with 4-nitrophenyl chloroformate-activated PCL (PCL-NPC). PCL-PEG was synthesized by conjugating primary amine-terminated polyethylene glycol (MW 5000 Da, mPEG5k-NH2) with PCL-NPC. The synthesized PCL-PEG and PCL-PEI had respective molecular weights of 8400 Da and 4900 Da based on gel permeation chromatography.
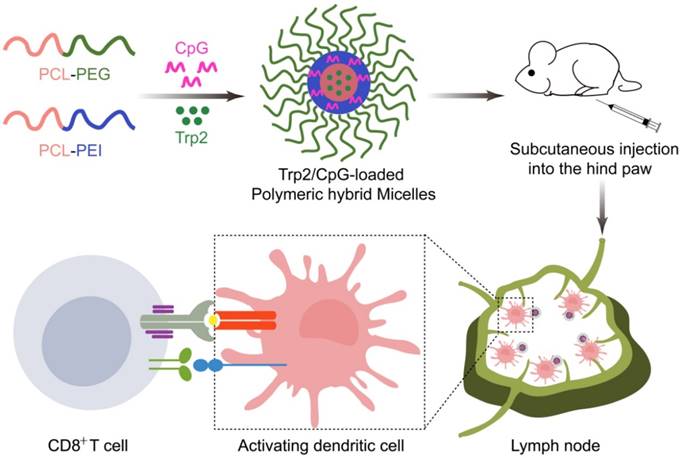
To investigate how the proportion of PCL-PEI affected the properties of Trp2/PHM/CpG, we prepared Trp2/PHM/CpG with different weight ratios of PCL-PEI and PCL-PEG as described in Table S1. Increasing the weight ratio of PCL-PEI did not affect the size of Trp2/PHM/CpG, but it did increase the zeta potential from -10.21 mV to +18.23 mV (Table 1). All Trp2/PHM/CpG preparations showed sizes around 80 nm, and they showed uniform spherical morphology based on transmission electron microscopy, similar to blank PHMs (Figure 1A, B). Encapsulation efficiency was determined by tracking the amount of FITC-labeled Trp2 peptide (FITC-Trp2) and FAM-labeled CpG (FAM-CpG). Trp2 was incorporated into all PHMs with encapsulation efficiencies >97%, as was CpG (>98%) with the exception of a formulation containing 100% PCL-PEG and 0% PCL-PEI (PHM0). This result indicated that PCL-PEI was essential for CpG encapsulation.
Efficient CpG delivery depended on the condensation of CpG in the nanocarriers, so we assessed this in various Trp2/PHM/CpG formulations. Retardation assays showed no detectable free CpG in Trp2/PHM5/CpG, Trp2/PHM10/CpG, Trp2/PHM25/CpG or Trp2/PHM50/CpG (Figure 1C), indicating efficient encapsulation of CpG in these PHMs. In contrast, the Trp2/PHM0/CpG group without PCL-PEI showed an obvious free CpG band, suggesting that the cationic charges of PCL-PEI played a critical role in condensing negatively charged nucleic acid drugs. All Trp2/PHM/CpG preparations remained stable, retained their diameters of approximately 80 nm and encapsulation efficiencies above 97% after storage at 4 °C for 72 h (Figure S1, S2). In addition, the release kinetics of FITC-labeled Trp2 and Cy5-labeled CpG from various Trp2/PHM/CpG formulations in PBS (pH=7.4) are shown in Figure S3. As shown in Figure S3A, PHM0 showed a sustained drug release of Trp2 and the cumulative release amount of Trp2 reached 100% in 48 h. The cumulative release amounts of Trp2 from PHM5, PHM10, PHM25 and PHM50 in 48 h were less than 5%, which were much less than free Trp2. PHM5, PHM10, PHM25 and PHM50 possess positively charged PCL-PEI, which could interact with negatively charged CpG and form a protective shell around the nanoparticles. This protective shell could improve the stability of nanoparticles and inhibit the release of Trp2 from PHM5, PHM10, PHM25 and PHM50. As shown in figure S3B, the cumulative release amounts of CpG from PHM5, PHM10, PHM25 and PHM50 in 48 h were less than 10%, indicating that CpG could form stable complexes with them. These data suggested that PHM5, PHM10, PHM25 and PHM50 were quite stable before they were uptaken by DCs, which was beneficial to the co-delivery of Trp2 and CpG to DCs in lymph nodes. After PHM5, PHM10, PHM25 and PHM50 entered DCs, they could release Trp2 and CpG under the acidic microenvironment of endosomes and lysosomes, which was followed by antigen presentation and activation of TLR9 receptor.
Size, zeta potential and encapsulation efficiency of various Trp2/PHM/CpG.
| PHM0 | PHM5 | PHM10 | PHM25 | PHM50 | |
|---|---|---|---|---|---|
| Size (nm) | 75.24±2.23 | 78.42±1.57 | 80.12±2.62 | 82.45±3.45 | 79.04±3.08 |
| PDI | 0.237±0.013 | 0.259±0.034 | 0.219±0.007 | 0.229±0.016 | 0.289±0.045 |
| Zata (mV) | -10.21±0.67 | +10.29±0.56 | +17.42±0.98 | +17.00±0.89 | +18.23±0.43 |
| EE% of Trp2 | 98.32±1.22 | 97.34±0.91 | 99.43±0.87 | 98.21±0.37 | 98.95±0.59 |
| EE% of CpG | 0.39±0.02 | 98.84±0.89 | 97.89±0.59 | 98.30±0.94 | 98.59±1.23 |
In vitro characterization of Trp2/PHM/CpG. (A) Transmission electron micrographs of blank PHM10. (B) Transmission electron micrographs of Trp2/PHM10/CpG. Scale bar, 100 nm. (C) Electrophoretic mobility assays to detect unencapsulated CpG after loading in various Trp2/PHM/CpG formulations.
Uptake of Trp2/PHM/CpG by DC2.4 cells and efficiency of co-delivery
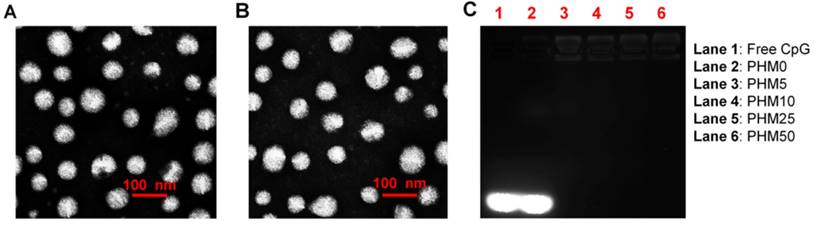
Cy5-labeled CpG (Cy5-CpG) was encapsulated in various PHMs and incubated with DC2.4 cultures in the absence of serum for 2 h. Flow cytometry analysis showed that the cellular uptake of the Trp2/PHM0/CpG group was quite low with a mean fluorescence intensity (MFI) of 2.23, similar to that of free CpG (Figure 2A). Cellular uptake increased with increasing PCL-PEI ratio up to 10%, when cellular uptake was most efficient (MFI 17.8). At PCL-PEI ratios of 25% and above, cellular uptake gradually decreased, such that the MFI of Trp2/PHM50/CpG was only 9.43. The lower cellular uptake of Trp2/PHM25/CpG and Trp2/PHM50/CpG may reflect their cytotoxicity to DC2.4 cells; those formulations were more cytotoxic than Trp2/PHM0/CpG, Trp2/PHM5/CpG, or Trp2/PHM10/CpG (Figure 2B). Since PCL-PEI is positively charged, excessive PCL-PEI may interact with the cell membrane and lead to cytotoxicity [18], which may help explain why the ratio of PCL-PEI in our formulations strongly affected cellular uptake of Trp2/PHM/CpG. In other words, our results suggest the need to strike a balance between having sufficient PCL-PEI to condense CpG in Trp2/PHM/CpG and enhance cellular uptake, but not so much PCL-PEI that cytotoxicity arises and reduces uptake.
The ability of our Trp2 peptide vaccine to induce potent immune responses depends on how well Trp2/PHM/CpG co-delivers antigen and adjuvant to antigen-presenting cells. We investigated this by studying the uptake of FITC-Trp2/PHM/Cy5-CpG into DC2.4 cells using flow cytometry and confocal microscopy. DC2.4 cells were incubated with these nanoparticles for 2 h, then the proportion of double-positive FITC-Trp2+/Cy5-CpG+ cells was measured (Figure 2C). This proportion was similarly low for Trp2/PHM0/CpG and for free Trp2/CpG. Consistent with these results, flow cytometry showed that the proportion of double-positive FITC-Trp2+/Cy5-CpG+ cells increased with PCL-PEI ratio up to 10%, after which it decreased. This provides further evidence that an appropriate amount of PCL-PEI is needed for co-delivery of FITC-Trp2 and Cy5-CpG. The proportion of double-positive FITC-Trp2+/Cy5-CpG+ cells was highest for Trp2/PHM10/CpG, containing 10% PCL-PEI. Confocal microscopy of DC2.4 cells incubated for 2 h with Trp2/PHM10/Cy5-CpG showed co-localization (yellow) of FITC-Trp2 and Cy5-CpG in the cytoplasm (Figure 2D). These results indicate that PCL-PEI is necessary for Trp2/PHM/CpG to co-deliver antigen and adjuvant into the cytosol of DC2.4 cells, and that the optimal PCL-PEI proportion is 10% under our experimental conditions.
Uptake of Trp2/PHM/CpG formulations by DC2.4 cells and associated cytotoxicity. (A) DC2.4 cells were incubated for 2 h with various Trp2/PHM/Cy5-CpG formulations, then intracellular fluorescence was analyzed by flow cytometry. Data are shown as mean ± SD (n=3). (B) Viability of DC2.4 cells treated for 24 h with different Trp2/PHM/CpG formulations. The percentage of viable cells was calculated relative to untreated cells and is shown as mean ± SD (n=5). *, p < 0.05. (C) Efficiency of Trp2 and CpG co-delivery by Trp2/PHM/CpG into DC2.4 cells in vitro. DC2.4 cells were incubated with FITC-Trp2/PHM/Cy5-CpG for 2 h at 37 °C, and the percentage of double-positive FITC-Trp2+/Cy5-CpG+ cells was measured using flow cytometry. (D) Confocal micrographs of DC2.4 cells incubated for 2 h at 37 °C with FITC-Trp2/PHM10/Cy5-CpG. Scale bar, 50 μm.
Maturation and proliferation of bone marrow-derived dendritic cells induced by Trp2/PHM/CpG
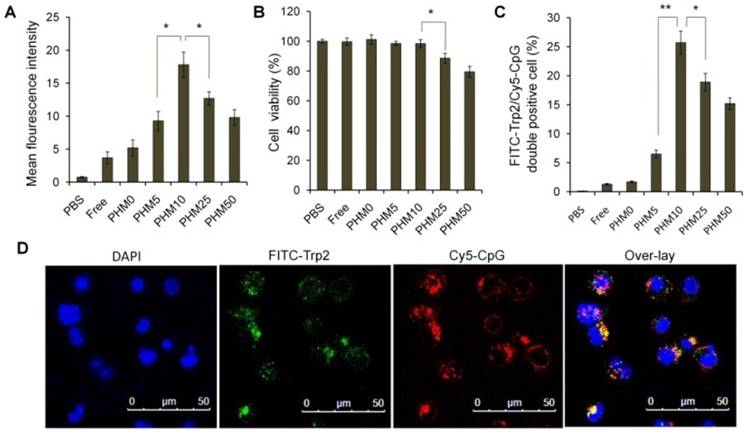
Various Trp2/PHM/CpG formulations were incubated with bone marrow-derived dendritic cells (BMDCs) for 24 h, and then the expressions of the co-stimulators CD86 and CD80 were measured as an index of BMDC maturation. Levels of CD86 and CD80 were similar for Trp2/PHM0/CpG as for free Trp2/CpG and slightly higher than for PBS (Figure 3A, B). These results indicate that free CpG can promote BMDC maturation to some extent. Levels of CD86 and CD80 were substantially higher for Trp2/PHM5/CpG, Trp2/PHM10/CpG, Trp2/PHM25/CpG and Trp2/PHM50/CpG. Levels for Trp2/PHM10/CpG were highest, even higher than the levels after lipopolysaccharide (LPS) treatment. In addition, the secretions of IL-12 and IFN-γ by BMDCs treated with various Trp2/PHM/CpG formulations were measured. The result showed PHM10 can increase the secretions of cytokine IL-12 and IFN-γ by BMDCs significantly compared with other groups (Figure S4).
Next, we investigated the effect of Trp2/PHM/CpG on BMDC proliferation. Cells were incubated with Trp2/PHM/CpG, LPS or PBS for 24 h. Proliferation in the presence of Trp2/PHM10/CpG was 6.5-fold more than that of cells treated with PBS and 2.13-fold more than that of cells treated with LPS (Figure 3C). These results indicate that PCL-PEI is necessary for promoting BMDC proliferation and maturation. It makes sense that Trp2/PHM10/CpG is the PHM formulation that was taken up most efficiently by DC 2.4 cells and that promoted BMDC proliferation and maturation to the greatest extent. Higher uptake of CpG translates to stronger stimulation of TLR9, which in turn means greater activation of innate and acquired immune responses [15].
Induction of BMDC maturation and proliferation by various Trp2/PHM/CpG formulations. Cells were treated with PHM formulations at 37 °C for 24 h. (A, B) BMDC maturation was measured by labeling cells with anti-mouse monoclonal antibodies against the co-stimulators (A) CD86 and (B) CD80. Labeled cells were analyzed by flow cytometry. Results are expressed as overall mean fluorescence intensity. (C) BMDC proliferation after treatment with various formulations. Data shown are mean ± SD. Statistical comparisons were made relative to PHM10. *p < 0.05, **p < 0.01 (n=5).
Accumulation of Trp2/PHM/CpG in lymph nodes and uptake by DCs in vivo
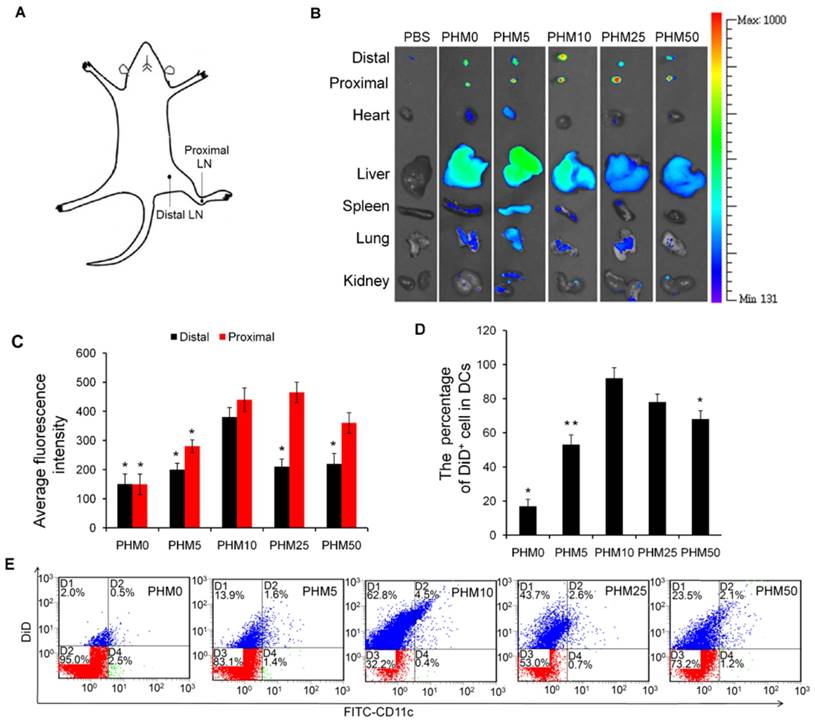
Since antigen-presenting cells are activated in the lymph nodes and immune responses begin there, we wanted to test whether our PHM formulations would cause accumulation of antigen and adjuvant in lymph nodes [19]. PHMs with different proportions of PCL-PEI and loaded with the fluorescent molecule 1,1-dioctadecyl-3,3,3′,3′-tetramethyl indodicarbocyanine,4-chlorobenzenesulfonate salt (DiD) as well as CpG were injected subcutaneously into the foot pad of mice. Fluorescence imaging showed that DiD/PHM0/CpG dispersed throughout the body within just 24 h, while DiD/PHM5/CpG dispersed throughout the body within 48 h (Figure S5). In contrast, DiD/PHM10/CpG, DiD/PHM25/CpG and DiD/PHM50/CpG did not disperse throughout the body even after 48 h, remaining mostly at the site of injection. Migration of PHMs away from the injection site decreased with increasing PCL-PEI ratio, indicating that PCL-PEI can hinder the migration of DiD/PHM/CpG.
Next, we examined the biodistribution of DiD/PHM/CpG in greater detail. Lymph nodes and other organs were harvested from mice at 24 h after injection with DiD/PHM/CpG, and subjected to fluorescence imaging. DiD/PHM0/CpG showed good dispersion throughout the body, with low signal in the lymph nodes (Figure 4B). Initially as the PCL-PEI proportion increased, fluorescence signal in the lymph nodes also increased, because PCL-PEI can prevent DiD/PHM/CpG from dispersing throughout the body too quickly. DiD/PHM10/CpG showed the strongest fluorescence signal in lymph nodes, and much lower fluorescence signal in other organs. As the PCL-PEI proportion continued to increase, DiD/PHM/CpG migrated less from the site of injection (Figure S6). DiD/PHM50/CpG accumulated primarily in lymph nodes near the injection site but not in distant lymph nodes or other organs (Figure 4A, B, C). These results indicate that DiD/PHM50/CpG, which has a relatively high proportion of PCL-PEI, does not migrate efficiently away from the site of injection after subcutaneous administration.
To initiate a CD8+ T cell immune response, antigen needs to be captured by DCs after arriving at lymph nodes. Therefore, we used flow cytometry to measure uptake of DiD/PHM/CpG by DCs in mice. Including even small proportions of PCL-PEI increased uptake by DCs (Figure 4D, E); more than 90% of DCs in lymph nodes took up DiD/PHM10/CpG. DCs in lymph nodes took up DiD/PHM50/CpG less efficiently. These results suggest that the appropriate PCL-PEI proportion can enhance accumulation of the antigen-adjuvant delivery system in the lymph nodes as well as uptake by DCs. This should translate to greater potential for inducing effective immune responses.
Accumulation of various DiD/PHM/CpG formulations in lymph nodes after subcutaneous injection in mice, and intracellular trafficking of the formulations within DCs. (A) Illustration of the location of proximal LNs and distal LNs. (B) Ex vivo imaging of lymph nodes, heart, liver, spleen, lung and kidney from mice at 24 h after injection. Distal refers to lymph nodes farther from the injection site; proximal, to lymph nodes closer to the injection site. (C) Mean fluorescence intensity of DiD in various formulations in distal and proximal lymph nodes at 24 h after injection. (D, E) Percentages of DiD+ cell in DCs (CD11c+) in lymph nodes at 24 h after injection. (D) Percentage of DiD+ cells in DCs. (E) Representative dot plot for each sample. Statistical comparisons were made relative to PHM10. *p < 0.05, **p < 0.01, n=3.
Co-delivery of Trp2 and CpG to lymph nodes in vivo by PHMs
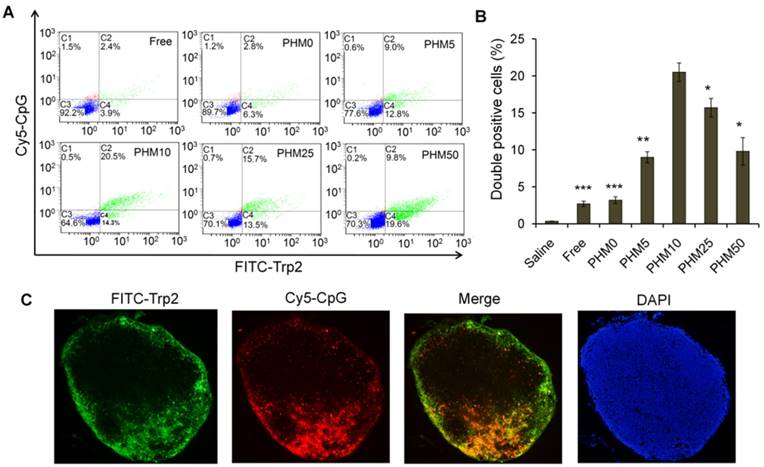
Since co-delivering antigen and adjuvant into target tissue is important for achieving an immune response [20-22], we examined the ability of PHMs to co-deliver Trp-2 and CpG to lymph nodes in vivo. Various FITC-Trp2/PHM/Cy5-CpG formulations were injected into the right hind foot pad of mice and 24 h later, the percentage of double-positive FITC+/Cy5+ cells in lymph nodes was measured by flow cytometry. The percentage of double-positive FITC+/Cy5+ cells in lymph nodes correlated closely with the proportion of PCL-PEI in the formulation (Figure 5A, B). The lowest percentage (about 2.8%) was observed in cells treated with FITC-Trp2/PHM0/Cy5-CpG, which contained no PCL-PEI. As the PCL-PEI proportion increased, the percentage of double-positive FITC+/Cy5+ cells increased at first and then decreased: FITC-Trp2/PHM5/Cy5-CpG, 9%; FITC-Trp2/PHM10/Cy5-CpG, 20.5%; FITC-Trp2/PHM25/Cy5-CpG, 15.7%; FITC-Trp2/PHM50/Cy5-CpG, 9.8%. The highest percentage was observed with FITC-Trp2/PHM10/Cy5-CpG, which therefore provided the best co-delivery of FITC-Trp2 and Cy5-CpG to lymph nodes.
To gain higher-resolution insights into the biodistribution of co-delivered FITC-Trp2 and Cy5-CpG, FITC-Trp2/PHM10/Cy5-CpG was injected into the right hind footpad of mice, and the biodistribution of FITC-Trp2 and Cy5-CpG in frozen distal lymph node sections was observed under the fluorescence microscope. Abundant signal from FITC-Trp2 and Cy5-CpG appeared at the same position in the frozen distal lymph node sections (Figure 5C), suggesting efficient co-delivery of both molecules. These results suggest that an appropriate amount of PCL-PEI is necessary for co-delivering Trp2 and CpG to lymph nodes, and that the optimal PCL-PEI ratio is 10% under our experimental conditions.
These results are consistent with the ability of PEI to potentiate the effects of Trp2 antigen and CpG adjuvant in vivo [23-25]. PCL-PEI protects CpG from degradation by DNases, enhances its uptake by immune cells, helps it bind the TLR-9 receptor in the endosome, prolongs its retention in the lymph nodes and enables persistent stimulation of immune cells [26, 27]. However, our experiments show that these positive effects of PCL-PEI have their limits: excessive levels of the cationic copolymer can lead to drug retention at the injection site and toxicity to cells, reducing drug uptake.
Co-delivery of FITC-Trp2 and Cy5-CpG into lymph nodes by PHMs. Mice were injected with PHMs loaded with FITC-Trp2/Cy5-CpG or with a simple mixture of free FITC-Trp2 and Cy5-CpG. (A, B) At 6 h after injection of FITC-Trp2/PHM/Cy5-CpG, the percentage of cells that internalized FITC-Trp2 and Cy5-CpG simultaneously was detected by flow cytometry. (A) Representative dot plot for each sample. (B) The percentage of double-positive FITC-Trp2+/Cy5-CpG+ cells was measured in each sample (n=3, mean ± SD). Statistical comparisons were made relative to PHM10. *p < 0.05, **p < 0.01, ***p < 0.001. (C) Fluorescence micrographs of distal lymph node sections from mice at 6 h after injection with FITC-Trp2/PHM10/Cy5-CpG (16 μg FITC-Trp2 and 1.6 μg Cy5-CpG per mouse). Green shows the location of FITC-Trp2; red, the location of Cy5-CpG.
Induction of antigen-specific CTL activity by Trp2/PHM/CpG
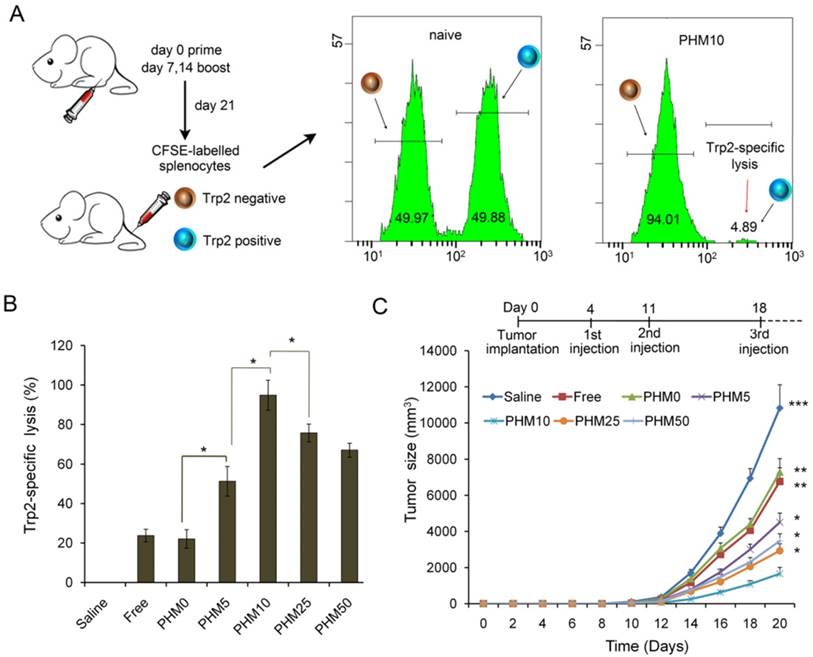
A primary CTL response is important for stopping tumor growth and preventing recurrence [28, 29], so the ability of Trp2/PHM/CpG to elicit an antigen-specific CTL response in vivo was analyzed by carboxyfluorescein succinimidyl ester (CFSE) method. The schematic of the CFSE method to quantify the Trp2-specific CTL response is shown in Figure 6A. First, splenocytes were isolated from naive mice and pulsed with or without Trp2 peptide. Then splenocytes were labeled with a low or high concentration of CFSE solution, creating two populations: non-Trp2-pulsed CFSElow and Trp2-pulsed CFSEhigh. Equal amounts of the two populations were mixed and injected intravenously into mice previously vaccinated with various Trp2/PHM/CpG formulations. To evaluate whether Trp2/PHM/CpG can stimulate strong, antigen-specific CD8+ T cell immunity, the ability of Trp2-specific CD8+ T cells from vaccinated mice to lyse Trp2-pulsed CFSEhigh splenocytes was analyzed using flow cytometry at 18 h after the adoptive transfer. Specific lysis was greater for mice immunized with Trp2/PHM5/CpG (51.3%), Trp2/PHM10/CpG (94.8%), Trp2/PHM25/CpG (79.7%) or Trp2/PHM50/CpG (71.0%) than for mice immunized with Trp2/PHM0/CpG (22.1%) or a mixture of free antigen and adjuvant (17.3%) (Figure 6B). Specific lysis initially increased with increasing proportion of PCL-PEI (peaking at PHM10), then later decreased.
In vivo CTL response and anti-tumor activity of Trp2/PHM5/CpG. (A) The schematic of carboxyfluorescein succinimidyl ester (CFSE) method to quantify the Trp2-specific CTL response. (B) In vivo CTL response after vaccination. Target cells pulsed with or without Trp2 were stained with high or low concentrations of CFSE and injected intravenously into vaccinated mice. After 18 h, spleens were harvested and the splenocytes were analyzed by flow cytometry to determine the proportion of Trp2-specific lysis (n=5, mean ± SD). (C) Anti-tumor activity of vaccine formulations in a subcutaneous B16F10 tumor model. C57BL/6 mice were inoculated with B16F10 cells (1 × 105 ) subcutaneously on day 0. Animals were vaccinated with saline, Trp2/CpG, Trp2/PHM0/CpG, Trp2/PHM5/CpG, Trp2/PHM10/CpG, Trp2/PHM25/CpG or Trp2/PHM50/CpG (16 μg Trp2, 1.6 μg CpG) on days 4, 11 and 18. Tumor growth was measured every 2 days (n=10). Statistical comparisons were made relative to PHM10. *p < 0.05, **p < 0.01, ***p < 0.001.
These results are consistent with previous reports that co-delivering antigen and adjuvant elicited stronger CD8+ T cell immunity than delivering a simple mixture of the two. Other co-delivery systems have been described, such as multifunctional nanoparticles [12], micelle nanoparticles assembled from a pH-responsive polymers [30], and gold nanoparticles [31]. These systems involve chemically conjugating antigen or adjuvant to the carrier, which makes the nanoparticles difficult to prepare and impairs the activity of antigen or adjuvant. In contrast, Trp2/PHM10/CpG self-assembles and encapsulates peptide antigen by hydrophobic interaction and CpG adjuvant by electrostatic adsorption.
Inhibition of B16F10 mouse melanoma tumor growth by Trp2/PHM/CpG
C57BL/6 mice were inoculated with B16F10 melanoma subcutaneously on day 0, and vaccine formulations were injected subcutaneously on the right hind foot pad on days 4, 11 and 18. Tumor size and body weight were monitored every 2 days for 20 days. Trp2/CpG and Trp2/PHM0/CpG inhibited tumor growth to a limited extent, while Trp2/PHM5/CpG, Trp2/PHM10/CpG, Trp2/PHM25/CpG and Trp2/PHM50/CpG inhibited tumor growth to a greater extent that was proportional to the PCL-PEI proportion (Figure 6C). Trp2/PHM10/CpG inhibited growth significantly more than Trp2/CpG and Trp2/PHM0/CpG (p < 0.01). The mice of group Trp2/PHM10/CpG also lived substantially longer than the other groups (Figure S7). In addition, body weight increased slightly in the saline, Trp2/CpG and Trp2/PHM0/CpG groups, which may reflect tumor growth. None of the groups showed loss of body weight, suggesting that the treatments did not cause substantial toxicity (Figure S8). The observed tumor inhibition was due primarily to the direct eradication of tumor cells by antigen-specific CTLs. These results suggest the promise of triggering a therapeutic immune response to B16F10 mouse melanoma by using PHM10 to co-deliver Trp2 and CpG.
Ability of Trp2/PHM/CpG to elicit Trp2 responsiveness in vaccinated spleen
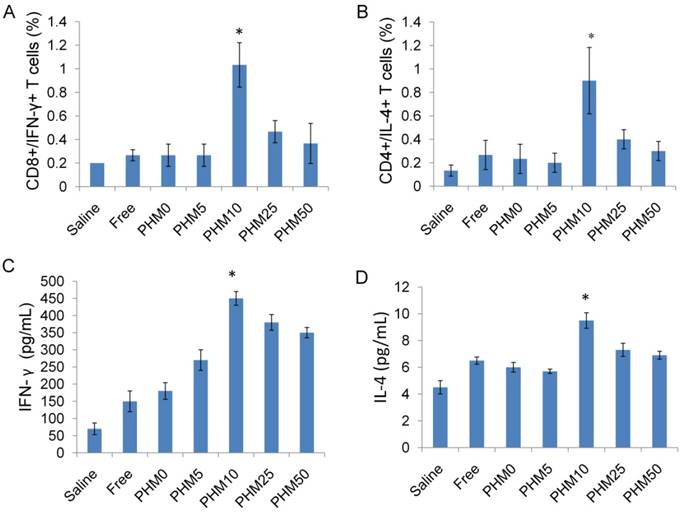
Among all the lymphocytes, CD8+/IFN-γ+ cells seem to be the most important T cell population for mediating tumor rejection upon adoptive transfer. CD4+/IL-4+ cells are also important because they could modulate and prolong the CD8+ T cell immune response [32, 33]. So, the ability of vaccinated splenocytes to produce CD8+/IFN-γ+ T cells and CD4+/IL-4+ T cells after being stimulated with Trp2 was determined. Mice were vaccinated with saline and various Trp2/PHM/CpG formulations on days 0, 7 and 14. Subsequently, mice were sacrificed on day 21, and single-cell suspensions of splenocytes were prepared and stimulated with Trp2 peptide for 1 h at 37 °C. Proportions of CD4+ T cells producing IL-4 and CD8+ T cells producing IFN-γ in the spleen were determined by flow cytometry. As shown in Figure 7 (A,B), the proportions of CD8+/IFN-γ+ cells in splenocytes from mice treated with saline and free Trp2 peptide and CpG were 0.20% and 0.27%, respectively. The corresponding proportions of CD4+/IL-4+ T cells were 0.13% and 0.27%, respectively. The proportions of CD8+/IFN-γ+ T cells and CD4+/IL-4+ T cells were much higher in spleens from mice vaccinated with PHM10, in which the proportion of CD8+/IFN-γ+ cells was 1.03%, while that of CD4+/IL-4+ T cells was 0.90%. In a separate experiment, single-cell suspensions of splenocytes prepared from immunized mice were incubated with Trp2 peptide for 60 h in the cell culture medium. Then the concentration of the cytokines IFN-γ and IL-4 in the culture supernatant was determined by ELISA kit. As shown in Figure 7 (C,D), consistent with the results from antigen-specific CTL response in vivo, Trp2/PHM10/CpG exhibited the highest levels of IFN-γ and IL-4 compared with other groups.
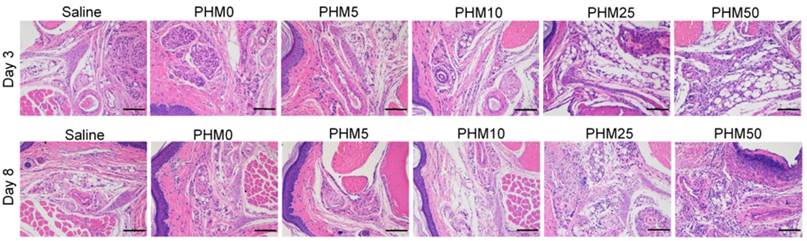
Side effects of Trp2/PHM/CpG at the injection site
As PCL-PEI is a cationic polymer, it may cause local dermal irritation and lead to an inflammatory response [23, 25]. So, we examined the pathology change at the injection site after immunization with various formulations. Briefly, C57BL/6 mice were subcutaneously injected with various formulations in the hind foot pad. After 3 days and 8 days, the injected foot pad were excised and examined for histopathology using hematoxylin-eosin staining. As shown in the figure 8, the foot pad treated with PHM0, PHM5 and PHM10 didn't show obvious inflammation compared with saline. In contrast, severe inflammation was observed from PHM25 and PHM50. Furthermore, the inflammation pathology of PHM25 and PHM50 was not eliminated at 8 days post-injection. These results indicated that PHM10 caused much less irritation at the injection site than PHM25 and PHM50, suggesting that PHM10 is a biocompatible and safe formulation for co-delivering CpG and Trp2.
Ability of Trp2/PHM/CpG to elicit Trp2 responsiveness in vaccinated spleen. (A, B). Proportion of CD8+/IFN-γ+ (A) or CD4+/IL-4+ (B) T cells. Splenocytes were prepared from immunized mice and incubated with 0.2 μM Trp2 peptide for 1 h at 37 °C. After washing, the cells were stained with anti-mouse monoclonal antibodies against CD8+/IFN-γ+ or CD4+/IL-4+. The proportions of CD8+/IFN-γ+ and CD4+/IL-4+ T cells were determined by flow cytometry. (C, D) IFN-γ (C) and IL-4 (D) production from vaccinated mice. Splenocytes prepared from immunized mice were stimulated with Trp2 for 60 h. IFN-γ or IL-4 production was measured by ELISA. Results are shown as mean ± SD (n = 5). Statistical comparisons were made relative to saline. *P <0.05.
Irritation at the injection site. C57BL/6 mice were given an injection of various formulations subcutaneously in the hind foot pad. After 3 days and 8 days, the injected foot pad were excised and examined for histopathology using hematoxylin-eosin staining. Inflammation at the injection site was assessed using hematoxylin-eosin staining. Scale bar, 100 µm.
Discussion
A major challenge to developing antitumor vaccines is efficiently delivering antigen and adjuvant to lymph nodes, where they can be taken up by DCs, the antigen can be presented and T cell-mediated immune responses can be initiated [34, 35]. Various nanocarriers have been developed to protect antigen and adjuvant in the circulation and improve their co-delivery to lymph nodes [36]. Here we describe a novel PHM co-delivery system that is highly tunable simply through manipulation of the ratio of two constitutive block copolymers, and we show that a small amount of cationic polymer (approximately 10% under our conditions) is optimal for stimulating efficient uptake by DCs without causing cytotoxicity, as well as for eliciting immune responses in vitro and in vivo. PHMs are mainly composed of hydrophobic segments (PCL), cationic hydrophilic segments (PEI) and PEG segments. The components and fundamental structure of PHMs are similar with other cationic polymer nanoparticles. The lymphatic and intracellular antigen delivery ability of PHMs as a function of their physicochemical properties was investigated in the study. The results in the study provide new insights into how cationic segments affect the efficiency of antigen delivery by cationic nanocarriers and the findings in the study are also applicable to other cationic polymer nanoparticles. The model antigen in our study, Trp2 peptide, presents several drawbacks, including hydrophobicity and poor immunogenicity, yet encapsulating it into our PHM system strongly enhanced its immunization efficacy. This suggests the potential of our co-delivery system for potentiating this and other peptide vaccines, which show great promise against various cancers, such as nasopharyngeal tumors, breast tumors and melanomas [37, 38], but which show limited immunogenicity because of poor pharmacokinetics, including rapid clearance before reaching DCs.
Our highly tunable PHM system works differently from other systems that have been described for improving the immunotherapeutic efficacy of Trp-2, including PLGA nanoparticles, AVE3 liposomes and cationic lipids [39, 40]. While Trp2 delivered with those previous systems also inhibits tumor growth, our system can be optimized by titrating the amount of cationic PCL-PEI segments, which protect the CpG adjuvant from nuclease degradation, facilitate micelle interaction with the cell membrane, and promote nanocarrier distribution to lymph nodes.
Screening Trp2/PHM/CpG formulations containing 0% (PHM0) to 50% (PHM50) of PCL-PEI showed that a minimum amount of PCL-PEI is necessary to allow CpG to condense within the micelles. The PHM formulations were able to efficiently encapsulate Trp2, and the complete particles remained stable at 4 °C for at least one week. It should be possible to encapsulate nearly any hydrophobic peptide into these PHMs together with CpG adjuvant. This may make our system a powerful, highly flexible platform for delivering peptide vaccines.
Cationic components in polymeric nanocarriers are a double-edged sword. On the one hand, they are needed to ensure condensation, protection and delivery of CpG. On the other hand, they can be toxic because they disrupt the cell membrane, leading to cell death [10]. Therefore, the amount of cationic PCL-PEI in our PHMs had to be optimized in order to strike a balance between efficacy and safety. Under our experimental conditions, uptake of PHMs by DC2.4 cells increased with increasing PCL-PEI proportion up to 10%, decreasing thereafter. This decrease in uptake correlated with increasing cytotoxicity to DC2.4 cells in standard viability assays. It is likely that the PCL-PEI proportion will need to be optimized individually for different antigens and adjuvants.
Our fluorescence studies of the biodistribution of PHMs in mice following foot pad injection indicate that toxicity is not the only problem that can occur when the PCL-PEI proportion is suboptimal. DiD/PHM0/CpG dispersed throughout the body within just 24 h, and DiD/PHM5/CpG dispersed throughout the body within 48 h. In contrast, DiD/PHM10/CpG, DiD/PHM25/CpG and DiD/PHM50/CpG remained primarily at the site of injection even after 48 h. These results suggest that PCL-PEI can hinder the migration of DiD/PHM/CpG, while also helping to ensure accumulation in lymph nodes: DiD/PHM0/CpG and DiD/PHM5/CpG accumulated primarily in the liver and other organs. This may decrease their distribution to lymph nodes and increase systemic CpG toxicity. Under our experimental conditions, 10% PCL-PEI seemed the best solution: DiD/PHM10/CpG showed the highest distribution to lymph nodes, where it was internalized by >90% of DCs. These results illustrate the need to optimize the cationic component of PHMs to ensure lymph node accumulation and delivery to DCs.
Indeed, our in vivo studies with a mouse model of melanoma showed that Trp2/PHM10/CpG significantly inhibited tumor growth, while Trp2/PHM0/CpG or a mixture of free Trp2 and CpG did not. This tumor inhibition correlates with the finding that Trp2/PHM10/CpG elicited much stronger CTL activity (94.8% specific lysis of Trp2-pulsed targets) than either Trp2/PHM0/CpG (22.1%) or the mixture of free Trp2 and CpG (17.3%). These results suggest that the PCL-PEI proportion must be optimized to ensure a strong therapeutic immune response against melanoma.
Conclusions
Here we report a novel PHM as a multifunctional carrier for co-delivering tumor peptide antigen and human adjuvant for treating melanomas. PHMs with different proportions of cationic PCL-PEI segments were rationally designed and used to co-encapsulate Trp2 antigen and CpG adjuvant. The proportion of PCL-PEI in the micelle formulations strongly influenced uptake of PHMs by DCs, viability of DCs, migration of PHMs from the site of injection in vivo, and ability of PHMs to inhibit tumor growth in vivo. These results highlight the potential of our PHM system for immunotherapy against melanoma. It should also be adaptable to other tumors, since the PHMs should be able to encapsulate a range of peptides, and the micelles can tolerate a broad range of PCL-PEI proportions, allowing fine-tuning of nanoparticle properties.
Supplementary Material
Supplementary figures and tables.
Abbreviations
PHMs: polymeric hybrid micelles; PCL-PEI: polycaprolactone-polyethylenimine; PCL-PEG: polycaprolactone-polyethyleneglycol; Trp2: tyrosinase-related protein 2; DCs: dendritic cells; TLR: Toll-like receptor; ELISA: enzyme-linked immunosorbent assay; CTL: cytotoxic T lymphocyte; MFI: mean fluorescence intensity; BMDCs: bone marrow-derived dendritic cells; LPS: lipopolysaccharide; DiD: 1,1-dioctadecyl-3,3,3′,3′-tetramethyl indodicarbocyanine, 4-chlorobenzenesulfo- nate salt; CFSE: carboxy fluorescein succinimidyl ester.
Acknowledgements
This research was financially supported by the National Natural Science Foundation of China (No. 81690261 & 81673362).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Yamshchikov G, Thompson L, Ross WG, Galavotti H, Aquila W, Deacon D, Caldwell J, Patterson JW, Hunt DF, Slingluff CL Jr. Analysis of a natural immune response against tumor antigens in a melanoma survivor: lessons applicable to clinical trial evaluations. Clin Cancer Res. 2001;7:909s-16s
2. Finn OJ. Immuno-oncology: understanding the function and dysfunction of the immune system in cancer. Ann Oncol. 2012;23(Suppl 8):viii6-9
3. Jackaman C, Lew AM, Zhan Y, Allan JE, Koloska B, Graham PT, Robinson BW, Nelson DJ. Deliberately provoking local inflammation drives tumors to become their own protective vaccine site. Int Immunol. 2008;20:1467-79
4. Harimoto H, Shimizu M, Nakagawa Y, Nakatsuka K, Wakabayashi A, Sakamoto C, Takahashi H. Inactivation of tumor-specific CD8(+) CTLs by tumor-infiltrating tolerogenic dendritic cells. Immunol Cell Biol. 2013;91:545-55
5. Endo S, Sakamoto Y, Kobayashi E, Nakamura A, Takai T. Regulation of cytotoxic T lymphocyte triggering by PIR-B on dendritic cells. Proc Natl Acad Sci U S A. 2008;105:14515-20
6. Hu S, Li B, Shen X, Zhang R, Gao D, Guo Q, Jin Y, Fei Z. Induction of antigen-specific cytotoxic T-cell response by dendritic cells generated from ecto-mesenchymal stem cells infected with an adenovirus containing the MAGE-D4a gene. Oncol Lett. 2016;11:2886-92
7. van den Ancker W, van Luijn MM, Westers TM, Bontkes HJ, Ruben JM, de Gruijl TD, Ossenkoppele GJ, van de Loosdrecht AA. Recent advances in antigen-loaded dendritic cell-based strategies for treatment of minimal residual disease in acute myeloid leukemia. Immunotherapy. 2010;2:69-83
8. Choi B, Moon H, Hong SJ, Shin C, Do Y, Ryu S, Kang S. Effective Delivery of Antigen-Encapsulin Nanoparticle Fusions to Dendritic Cells Leads to Antigen-Specific Cytotoxic T Cell Activation and Tumor Rejection. ACS Nano. 2016;10:7339-50
9. Kranz LM, Diken M, Haas H, Kreiter S, Loquai C, Reuter KC, Meng M, Fritz D, Vascotto F, Hefesha H, Grunwitz C, Vormehr M, Husemann Y, Selmi A, Kuhn AN, Buck J, Derhovanessian E, Rae R, Attig S, Diekmann J, Jabulowsky RA, Heesch S, Hassel J, Langguth P, Grabbe S, Huber C, Tureci O, Sahin U. Systemic RNA delivery to dendritic cells exploits antiviral defence for cancer immunotherapy. Nature. 2016;534:396-401
10. Li H, Fu Y, Zhang T, Li Y, Hong X, Jiang J, Gong T, Zhang Z, Sun X. Rational Design of Polymeric Hybrid Micelles with Highly Tunable Properties to Co-Deliver MicroRNA-34a and Vismodegib for Melanoma Therapy. Advanced Functional Materials. 2015;25:7457-69
11. Thomas SN, Vokali E, Lund AW, Hubbell JA, Swartz MA. Targeting the tumor-draining lymph node with adjuvanted nanoparticles reshapes the anti-tumor immune response. Biomaterials. 2014;35:814-24
12. Xu Z, Ramishetti S, Tseng YC, Guo S, Wang Y, Huang L. Multifunctional nanoparticles co-delivering Trp2 peptide and CpG adjuvant induce potent cytotoxic T-lymphocyte response against melanoma and its lung metastasis. J Control Release. 2013;172:259-65
13. Zhao Y, Huo M, Xu Z, Wang Y, Huang L. Nanoparticle delivery of CDDO-Me remodels the tumor microenvironment and enhances vaccine therapy for melanoma. Biomaterials. 2015;68:54-66
14. Miconnet I, Koenig S, Speiser D, Krieg A, Guillaume P, Cerottini JC, Romero P. CpG are efficient adjuvants for specific CTL induction against tumor antigen-derived peptide. J Immunol. 2002;168:1212-8
15. Krieg AM. Therapeutic potential of Toll-like receptor 9 activation. Nat Rev Drug Discov. 2006;5:471-84
16. Li H, Jiang H, Zhao M, Fu Y, Sun X. Intracellular redox potential-responsive micelles based on polyethylenimine-cystamine-poly(ε-caprolactone) block copolymer for enhanced miR-34a delivery. Polym Chem. 2015;6:1952-60
17. Byers AM, Kemball CC, Moser JM, Lukacher AE. Cutting edge: rapid in vivo CTL activity by polyoma virus-specific effector and memory CD8+ T cells. J Immunol. 2003;171:17-21
18. Zhang Y, Mao L, Liu J, Liu T. Self-fluorescent drug delivery vector based on genipin-crosslinked polyethylenimine conjugated globin nanoparticle. Mater Sci Eng C Mater Biol Appl. 2017;71:17-24
19. Trevaskis NL, Kaminskas LM, Porter CJ. From sewer to saviour - targeting the lymphatic system to promote drug exposure and activity. Nat Rev Drug Discov. 2015;14:781-803
20. Wang C, Sun W, Wright G, Wang AZ, Gu Z. Inflammation-Triggered Cancer Immunotherapy by Programmed Delivery of CpG and Anti-PD1 Antibody. Adv Mater. 2016;28:8912-20
21. Duan F, Feng X, Yang X, Sun W, Jin Y, Liu H, Ge K, Li Z, Zhang J. A simple and powerful co-delivery system based on pH-responsive metal-organic frameworks for enhanced cancer immunotherapy. Biomaterials. 2017;122:23-33
22. Li Z, Dong K, Zhang Y, Ju E, Chen Z, Ren J, Qu X. Biomimetic nanoassembly for targeted antigen delivery and enhanced Th1-type immune response. Chem Commun (Camb). 2015;51:15975-8
23. Zeng Q, Jiang H, Wang T, Zhang Z, Gong T, Sun X. Cationic micelle delivery of Trp2 peptide for efficient lymphatic draining and enhanced cytotoxic T-lymphocyte responses. J Control Release. 2015;200:1-12
24. Li M, Zhao M, Fu Y, Li Y, Gong T, Zhang Z, Sun X. Enhanced intranasal delivery of mRNA vaccine by overcoming the nasal epithelial barrier via intra- and paracellular pathways. J Control Release. 2016;228:9-19
25. Zhang H, Feng S, Yan T, Zhi C, Gao XD, Hanagata N. Polyethyleneimine-functionalized boron nitride nanospheres as efficient carriers for enhancing the immunostimulatory effect of CpG oligodeoxynucleotides. Int J Nanomedicine. 2015;10:5343-53
26. Qin T, Yin Y, Huang L, Yu Q, Yang Q. H9N2 influenza whole inactivated virus combined with polyethyleneimine strongly enhances mucosal and systemic immunity after intranasal immunization in mice. Clin Vaccine Immunol. 2015;22:421-9
27. Yim H, Park W, Kim D, Fahmy TM, Na K. A self-assembled polymeric micellar immunomodulator for cancer treatment based on cationic amphiphilic polymers. Biomaterials. 2014;35:9912-9
28. Melief CJ, Van Der Burg SH, Toes RE, Ossendorp F, Offringa R. Effective therapeutic anticancer vaccines based on precision guiding of cytolytic T lymphocytes. Immunol Rev. 2002;188:177-82
29. Melief CJ. Tumor eradication by adoptive transfer of cytotoxic T lymphocytes. Adv Cancer Res. 1992;58:143-75
30. Wilson JT, Keller S, Manganiello MJ, Cheng C, Lee CC, Opara C, Convertine A, Stayton PS. pH-Responsive nanoparticle vaccines for dual-delivery of antigens and immunostimulatory oligonucleotides. ACS Nano. 2013;7:3912-25
31. Tao Y, Zhang Y, Ju E, Ren H, Ren J. Gold nanocluster-based vaccines for dual-delivery of antigens and immunostimulatory oligonucleotides. Nanoscale. 2015;7:12419-26
32. Becker C, Pohla H, Frankenberger B, Schuler T, Assenmacher M, Schendel DJ, Blankenstein T. Adoptive tumor therapy with T lymphocytes enriched through an IFN-gamma capture assay. Nat Med. 2001;7:1159-62
33. Barth RJ Jr, Mule JJ, Spiess PJ, Rosenberg SA. Interferon gamma and tumor necrosis factor have a role in tumor regressions mediated by murine CD8+ tumor-infiltrating lymphocytes. J Exp Med. 1991;173:647-58
34. Qian Y, Jin H, Qiao S, Dai Y, Huang C, Lu L, Luo Q, Zhang Z. Targeting dendritic cells in lymph node with an antigen peptide-based nanovaccine for cancer immunotherapy. Biomaterials. 2016;98:171-83
35. Schuler G. Dendritic cells in cancer immunotherapy. Eur J Immunol. 2010;40:2123-30
36. Hamdy S, Molavi O, Ma Z, Haddadi A, Alshamsan A, Gobti Z, Elhasi S, Samuel J, Lavasanifar A. Co-delivery of cancer-associated antigen and Toll-like receptor 4 ligand in PLGA nanoparticles induces potent CD8+ T cell-mediated anti-tumor immunity. Vaccine. 2008;26:5046-57
37. Frazer IH, Quinn M, Nicklin JL, Tan J, Perrin LC, Ng P, O'Connor VM, White O, Wendt N, Martin J, Crowley JM, Edwards SJ, McKenzie AW, Mitchell SV, Maher DW, Pearse MJ, Basser RL. Phase 1 study of HPV16-specific immunotherapy with E6E7 fusion protein and ISCOMATRIX adjuvant in women with cervical intraepithelial neoplasia. Vaccine. 2004;23:172-81
38. Ressing ME, van Driel WJ, Brandt RM, Kenter GG, de Jong JH, Bauknecht T, Fleuren GJ, Hoogerhout P, Offringa R, Sette A, Celis E, Grey H, Trimbos BJ, Kast WM, Melief CJ. Detection of T helper responses, but not of human papillomavirus-specific cytotoxic T lymphocyte responses, after peptide vaccination of patients with cervical carcinoma. J Immunother. 2000;23:255-66
39. Fourcade J, Kudela P, Andrade Filho PA, Janjic B, Land SR, Sander C, Krieg A, Donnenberg A, Shen H, Kirkwood JM, Zarour HM. Immunization with analog peptide in combination with CpG and montanide expands tumor antigen-specific CD8+ T cells in melanoma patients. J Immunother. 2008;31:781-91
40. Zhang Z, Tongchusak S, Mizukami Y, Kang YJ, Ioji T, Touma M, Reinhold B, Keskin DB, Reinherz EL, Sasada T. Induction of anti-tumor cytotoxic T cell responses through PLGA-nanoparticle mediated antigen delivery. Biomaterials. 2011;32:3666-78
Author contact
![]() Corresponding author: E-mail: sunxunedu.cn
Corresponding author: E-mail: sunxunedu.cn