Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Results
Discussion
Materials and Methods
Abbreviations
Supplementary Material
Acknowledgements
Conflicts of interest
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2018; 8(7):2044-2060. doi:10.7150/thno.23304 This issue Cite
Research Paper
3′-epi-12β-hydroxyfroside, a new cardenolide, induces cytoprotective autophagy via blocking the Hsp90/Akt/mTOR axis in lung cancer cells
Yan Sun1*, Yong-Hao Huang1*, Feng-Ying Huang1*, Wen-Li Mei2, Quan Liu3, Cai-Chun Wang1, Ying-Ying Lin1, Canhua Huang1,4, Yue-Nan Li1,5 ![]() , Hao-Fu Dai2
, Hao-Fu Dai2 ![]() , Guang-Hong Tan1
, Guang-Hong Tan1 ![]()
1. Key Laboratory of Tropical Diseases and Translational Medicine of the Ministry of Education & Hainan Provincial Key Laboratory of Tropical Medicine, Hainan Medical College, Haikou 571199, China
2. Institute of Tropical Bioscience and Biotechnology, Chinese Academy of Tropical Agricultural Sciences, Haikou 571199, China;
3. The Department of Medical Oncology, Affiliated Hospital of Jiangnan University and Fourth People's Hospital of Wuxi, Wuxi 214062, China;
4. State Key Laboratory of Biotherapy and Cancer Center, West China Hospital, Sichuan University, Chengdu 610041, China;
5. The Pathology Department of the First Affiliated Hospital, Hainan Medical College, Haikou 570103, China.
*Equal contribution.
Received 2017-10-12; Accepted 2018-1-19; Published 2018-2-15
Abstract

Rationale: Cardenolides have potential as anticancer drugs. 3′-epi-12β-hydroxyfroside (HyFS) is a new cardenolide structure isolated by our research group, but its molecular mechanisms remain poorly understood. This study investigates the relationship between its antitumor activities and autophagy in lung cancer cells.
Methods: Cell growth and proliferation were detected by MTT, lactate dehydrogenase (LDH) release, 5-ethynyl-20-deoxyuridine (EDU) and colony formation assays. Cell apoptosis was detected by flow cytometry. Autophagic and signal proteins were detected by Western blotting. Markers of autophagy and autophagy flux were also detected by immunofluorescence, transmission electron microscopy and acridine orange staining. Real time RT-PCR was used to analyze the gene expression of Hsp90. Hsp90 ubiquitination was detected by coimmunoprecipitation. The antitumore activities of HyFS were observed in nude mice.
Results: HyFS treatment inhibited cell proliferation and induced autophagy in A549 and H460 lung cancer cells, but stronger inhibition of cell proliferation and induction of cell apoptosis were shown when HyFS-mediated autophagy was blocked. The Hsp90/Akt/mTOR axis was found to be involved in the activation of HyFS-mediated autophagy. Evidence of direct interaction between Hsp90 and Akt was observed. HyFS treatment resulted in decreased levels of heat shock protein 90 (Hsp90) and phosphorylated Akt, overexpression of Hsp90 increased activation of autophagy, and inhibition of Hsp90 expression decreased autophagy. In addition, ubiquitin-mediated degradation of Hsp90 and subsequent dephosphorylation of its client protein Akt were also found in HyFS-treated lung cancer cells. Moreover, combination treatment with HyFS and chloroquine showed remarkably increased tumor inhibition in both A549- and H460-bearing mice.
Conclusion: Our results demonstrate that HyFS induced cytoprotective autophagy through ubiquitin-mediated degradation of Hsp90, which further blocked the Akt/mTOR pathway in lung cancer cells. Thus, a combination of a HyFS-like cardenolide and an autophagic inhibitor is a potential alternative approach for the treatment of lung cancer.
Keywords: 3′-epi-12β-hydroxyfroside, cardiac glycosides, autophagy, apoptosis, heat shock protein.
Introduction
Lung cancer is one of the leading causes of cancer-related death worldwide. There are more than 1.82 million people diagnosed with lung cancer annually, and over 1.59 million deaths each year [1]. Despite the increasing use of novel therapeutic strategies and anticancer agents for lung cancer treatment, their clinical efficacy remains unsatisfactory, with the high mortality and morbidity among lung cancer patients not improving as expected [2-4]. Thus, there is an urgent need for additional novel anticancer agents or more effective strategies.
3′-epi-12β-hydroxyfroside (HyFS) is a natural chemical component that was isolated from the roots of Calotropis gigantean by our research group [5]. Structural analysis by MS, NMR, and chemical evidence have shown that HyFS is a new cardiac glycoside with a special cardenolide structure (Fig. 1A) [5]. Traditionally, cardenolides have been widely used to treat congestive heart failure and arrhythmia [6-8]. However, recent research results have shown that certain cardenolides extracted from natural sources have anticancer activities, as they can block tumor cell proliferation and induce tumor cell apoptosis through the regulation of several cell signaling pathways [9-12] and sodium pump inhibition [13], and even through the induction of immunogenic cell death [14-16]. Recent studies have shown that some cardiac glycosides, such as digoxin and ouabain, can cause autophagy in lung cancer cells [17-19]. However, the molecular mechanism remains controversial.
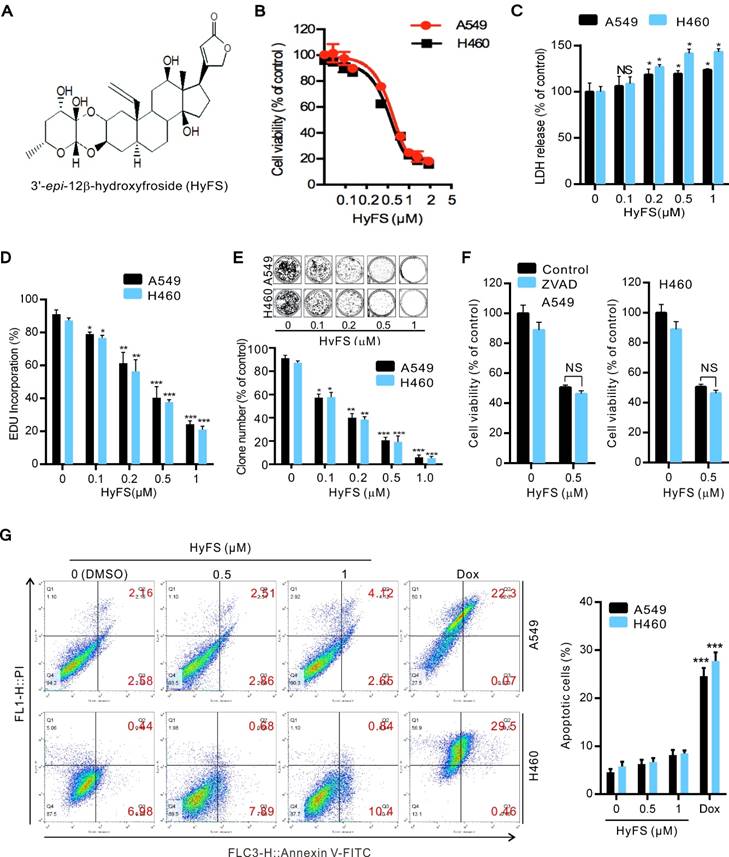
HyFS inhibits proliferation of lung cancer cells. (A) Structure of 3′-epi-12β-hydroxyfroside (HyFS). (B) A549 and H460 lung cancer cells were treated with indicated concentrations of HyFS for 24 h, and cell viability was measured by MTT assay. (C) Cells were treated for 24 h as in (B), and disrupted cell membranes were detected by LDH release assay. (D) Cells were treated for 24 h as in (B), and EDU-incorporating live cells were detected by EDU proliferation assay. (E) Cells were treated as in (B) for 7 days, and live cells were detected by colony formation assay. (F) Cells were treated with HyFS (0.5 μM) alone or in combination with ZVAD (20 μM) for 24 h, and cell viability was measured by MTT. (G) Cells were treated with HyFS as in (B) or doxorubicin (Dox, as positive control) for 24 h. Apoptotic cells were stained by PI/Annexin-V followed by flow cytometry. Data are expressed as mean ± SEM; * P < 0.05, ** P < 0.01, *** P < 0.001.
Autophagy, a conserved catabolic process, is important for the degradation of cell components via the lysosomal pathway [20]. During the autophagic process, protein aggregates and/or damaged organelles are encapsulated into a double-membraned vesicle known as an autophagosome [20-22]. Thereafter, the autophagosome further fuses with a lysosome to form a new structure known as an autolysosome in which the encapsulated protein aggregates and/or damaged organelles are degraded by hydrolytic enzymes and recycled [20-22]. Autophagy can be activated by various cellular stresses, such as nutrient stress, hypoxia, and cytotoxic chemotherapies [23]. At present, many cellular pathways have been reported to be involved in the initiation of autophagy, including the classic Akt/mTOR pathway by which autophagic processes are negatively regulated [24-28]. The role of autophagy in tumorigenesis is paradoxical, and both protective and suppressive roles have been observed in various types of cancer cells challenged with various cellular stresses, including anticancer chemotherapeutics [29]. Cancer cells can use energy or metabolites supplied by autophagy to survive stress, which thus plays a protective role in tumorigenesis [30]. However, autophagy can inhibit cancer cell proliferation and promote cancer cell death in certain contexts, which suggests a suppressive role in tumorigenesis. Accordingly, understanding the roles and the mechanisms of autophagy and related signaling pathways involved in cancer cells is important for targeting autophagy as an approach for cancer treatment.
Because HyFS is a cytotoxic chemical agent, it may cause some kinds of cellular stress just like other reported chemotherapeutics [29]. Therefore, it is reasonable to think that autophagic mechanisms may be related to the anticancer activities induced by HyFS. In this study, we found that HyFS induces inhibition of cell growth and autophagy, but not significant apoptosis, in several lung cancer cell lines. Inhibition of autophagy by autophagic inhibitors or RNA interference promoted anti-proliferation effects and led to lung cancer apoptosis, indicating a cytoprotective role for autophagy induced by HyFS in lung cancer cells. Furthermore, the cytoprotective autophagy is related to ubiquitin-mediated degradation of Hsp90 and subsequent dephosphorylation of its client protein Akt, leading to inactivation of the Akt/mTOR pathway and activation of the cytoprotective autophagy. Enhanced antitumor activities were observed in model mice with a combination treatment of HyFS and chloroquine (CQ). Our findings strongly indicate that the combination of HyFS and an autophagy inhibitor is a potential alternative approach for the treatment of lung cancer.
Results
3′-epi-12β-hydroxyfroside (HyFS) suppresses proliferation of lung cancer cells
We first investigated the anticancer activities induced by HyFS by observing cell proliferation in two human cancer cell lines, A549 and H460. The cells were treated with various concentrations of HyFS for 24 h followed by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT), lactate dehydrogenase (LDH), 5-ethynyl-20-deoxyuridine (EDU), and colony formation assays to observe whether HyFS suppressed cell proliferation. The results of MTT assays showed that cell viability decreased in a dose-dependent manner in both A549 and H460 cells (Fig. 1B). Consistently, similar cytotoxic effects induced by HyFS treatment for 24 h in lung cancer cells were revealed by LDH assays (Fig. 1C). In addition, the percentage of EDU-incorporating live cells was significantly decreased in HyFS-treated cells compared to the controls (Fig. 1D). Furthermore, colony formation assays also indicated that significant inhibition of cell proliferation occurred in HyFS-treated A549 and H460 cells (Fig. 1E). We also examined whether HyFS was toxic to normal human lung fibroblasts (MRC-5) and epithelial cells (BEAS-2B). We found that the tested doses of HyFs (< 0.5 μM) did not cause toxicity to normal lung cells, and the dose that caused significant toxicity was about 4 μM, 8-fold more than the dose used in our experiments (Fig. S1A and B). These results suggest that HyFS causes specific inhibition of cell growth and proliferation in lung cancer cells. As shown in Figure 1B, the IC50 of HyFS was about 0.5 μM in both A549 and H460 cells. Thus, a concentration of 0.5 μM was used in subsequent experiments.
We next investigated whether HyFS induced apoptosis in lung cancer cells. We found that disruption of apoptosis by ZVAD (a pan-caspase inhibitor) did not change the HyFS-induced cell death (Fig. 1F). This was further supported by the equivalent levels of cleaved PARP and cleaved caspase-3 in lung cancer cells with or without HyFS-treatment (Fig. S1C). Furthermore, lung cancer cells were treated with various concentrations of HyFS, doxorubicin (Dox), or DNase (as a positive control) for 24 h, and were then analyzed by flow cytometry (Fig. 1G) and TUNEL assays (Fig. S1D and E). Our results showed that there were few apoptotic cells observed in HyFS-treated lung cancer cells when compared to doxorubicin- or DNase-treated positive control cells (Fig. 1G, Fig. S1D and E). Taken together, these results suggest that HyFS profoundly inhibits cell proliferation, but does not obviously induce cell apoptosis in lung cancer cells in the first 24 h.
HyFS induces autophagy in lung cancer cells
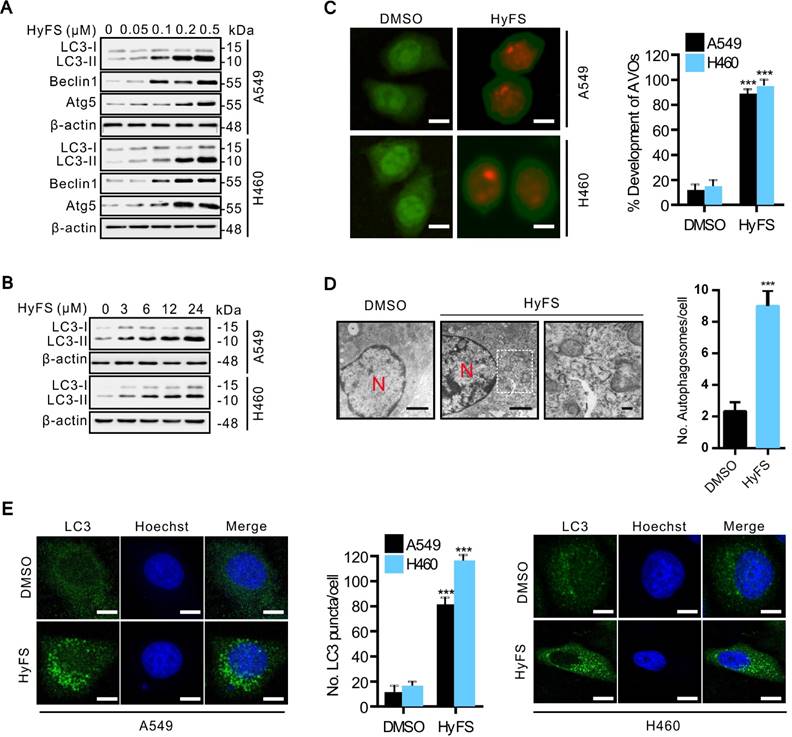
We next investigated whether HyFS induced autophagy in lung cancer cells. First, we measured the conversion of LC3-I to lipidated LC3-II, a classical marker of autophagosome formation. Our results showed that HyFS treatment resulted in increased autophagy as evidenced by increased LC3-II conversion, not only in A549 and H460 lung cancer cells (Fig. 2A and B), but also in various other cancer cell types, including DLD-1, HCT-116, H29, U87, U251, A172, HeLa, SiHa, C33A, HEP3B, BEC-7402, BEL-7404 and NCL-H1299 (Fig. S2A and B), indicating universal sensitivity of various cancer cells to HyFS. In addition, increased LC3-II conversion also occurred in a dose- and time-dependent manner in A549, H460, and H1299 lung cancer cells (Fig. 2A, B and Fig. S2B). Notably, the increased level of LC3-II in HyFS-treated lung cancer cells persisted to 48 h (Fig. S2C). To corroborate the evidence for formation of autophagosomes induced by HyFS, we analyzed the distribution of endogenous LC3 puncta, another classical marker of autophagosome formation. Our results showed that HyFS markedly increased endogenous LC3 puncta in HyFS-treated lung cancer cells compared to DMSO-treated control cells (Fig. 2E). Furthermore, we investigated the expression of beclin1 and Atg5, two autophagy-initiation related proteins. As shown in Fig. 2A, HyFS treatment promoted the expression of beclin1 and Atg5 in both A549 and H460 lung cancer cells. Moreover, since the formation of acidic vesicle organelles (AVO) is another characteristic of autophagy [25], we detected AVO by acridine orange staining. As predicted, abundant cytoplasmic AVO was readily observed in HyFS-treated lung cancer cells when compared to those treated with DMSO (Fig. 2C). To further test for HyFS-induced autophagy in lung cancer cells, the appearance of double-membraned autophagosomes was observed by transmission electron microscopy. As shown in Fig. 2D, there was significant accumulation of autophagosomes or autolysosomes in HyFS-treated cells but not in DMSO-treated cells. Collectively, these results demonstrated that HyFS induces autophagy in lung cancer cells.
HyFS stimulates autophagy in lung cancer cells. (A) A549 and H460 cells were treated with indicated concentrations of HyFS for 24 h, and LC3-I, LC3-II, Atg5, and beclin 1 were detected by western blot. (B) Cells were treated with HyFS (0.5 μM) for indicated hours, and LC3-I and LC3-II were detected by western blotting. (C) Cells were treated with DMSO or HyFS (0.5 μM) for 24 h and stained by acridine orange to observe the formation of acidic vesicle organelles (AVO). (D) A549 cells were treated with DMSO or HyFS (0.5 μM) for 24 h. Autophagosome formation was observed by a projection electron microscope. (E) Cells were treated with DMSO or HyFS (0.5 μM) for 24 h and stained with anti-LC3B antibody to detect the LC3 puncta and Hoechst to detect nuclei by immunofluorescence using a confocal microscope. The numbers of LC3-II positive cells in each group were counted from at least 100 random fields. Data are expressed as mean ± SEM; *** P < 0.001. Scale bar = 20 μm in immunofluorescence, bar = 0.1 μm in electron microscopy.
HyFS activates autophagy flux in lung cancer cells
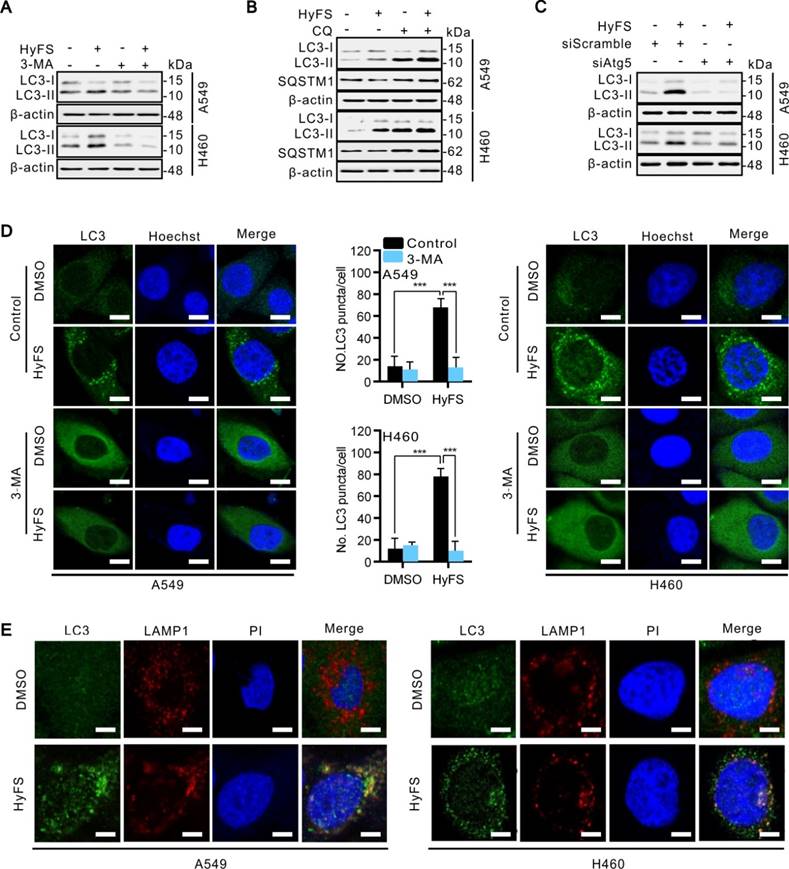
Since the inhibition of autophagosome turnover at the late stage also leads to LC3-II conversion and the accumulation of LC3 puncta [31], these phenomena do not mean that the autophagy progress is complete. Therefore, we clarified whether HyFS affected the complete autophagy progress by investigating the autophagic flux. Sequestosome-1 (SQSTM1) is a well-known autophagic substrate that is degraded through the autophagic pathway [32]. As shown in Fig. S3A, HyFS decreased SQSTM1 levels in a dose-dependent manner in HyFS-treated lung cancer cells. Combinatorial treatment with 3-methyladenine (3-MA, an autophagy early stage inhibitor) or wortmannin (Wort, a phosphoinositide 3-kinase inhibitor) decreased LC3-II conversion (Fig. 3A and Fig. S3B) and endogenous LC3 puncta (Fig. 3D and Fig. S3E). Conversely, combinatorial treatment with chloroquine (CQ, an autophagy late stage inhibitor that inhibits lysosome fusion with autophagosomes) or bafilomycin (Baf, an H+-ATPase inhibitor) resulted in the accumulation of LC3-II and SQSTM1 (Fig. 3B and Fig. S3C). In addition, silencing the expression of either beclin1 or Atg5 using siRNA partially blocked the LC3-II conversion and endogenous LC3 puncta accumulation in HyFS-treated cells (Fig. 3C and Fig. S3D), indicating that HyFS can trigger the initiation of autophagy in a PI3K-dependent manner. Moreover, in order to clarify the formation of autophagic lysosomes, we investigated whether there was colocalization of endogenous LC3 puncta and LAMP l (a lysosome marker) in HyFS-treated lung cancer cells [31]. Consistently, clear colocalization of endogenous LC3 puncta with LAMP1 was found in HyFS-treated lung cancer cells (Fig. 3E). These results strongly indicate that HyFS activates complete autophagic flux in lung cancer cells.
HyFS induces autophagy flux in lung cancer cells. (A) A549 and H460 cells were treated with or without HyFS (0.5 μM) and 3-MA as indicated for 24 h, and cell lysates were used for western blot analysis to detect LC3-I and LC3-II. (B) Cells were treated with or without HyFS (0.5 μM) and CQ as indicated for 24 h, and cell lysates were used to detect LC3-I, LC3-II, and SQSTM1 by western blot. (C) Cells were treated with HyFS (0.5 μM) alone or combined with siAtg5 as indicated for 24 h. Cell lysates were used for western blot to detect LC3-I and LC3-II. (D) Cells were treated with DMSO or HyFS (0.5 μM) and with or without 3-MA for 24 h. LC3 puncta and nuclei (Hoechst) were detected by confocal microscopy. (E) Cells were treated with or without HyFS (0.5 μM) for 24 h. LC3 puncta, LAMP1, and nuclei (PI) were detected by confocal microscopy and merged to observe the colocalization of LC3 puncta and LAMP1. Data are expressed as mean ± SEM; ** P < 0.01, *** P < 0.001. Scale bar = 20 μm.
Inhibition of HyFS-induced autophagy enhances antiproliferative activities and promotes cell apoptosis in lung cancer cells
Accumulated evidence indicates that cytoprotective, nonprotective, cytotoxic, and cytostatic autophagy are the four major forms of autophagy in the context of anticancer therapy [29]. Thus, we determined the relationship between HyFS-mediated autophagy and growth inhibition in lung cancer cells. We first inhibited HyFS-induced autophagy by autophagic inhibitors (3-MA or CQ) and observed the effects on cell proliferation and growth. As shown in Fig. 4A, MTT assays showed that inhibition of autophagy by 3-MA or CQ more significantly suppressed lung cell proliferation and enhanced HyFS-induced cell death. Similar results were found in EDU incorporation assays in which the inhibition of autophagy either by 3-MA or CQ sensitized lung cancer cells to HyFS (Fig. 4B). In addition, cell growth was determined by colony formation analysis, and the results showed that 3-MA or CQ treatment reversed the anticancer effects of HyFS (Fig. 4C and D). Moreover, inhibition of autophagy by beclin1 siRNA or Atg5 siRNA also markedly decreased cell viability and proliferation in HyFS-treated lung cancer cells (Fig. 4E and F). Collectively, these data suggest that inhibition of HyFS-induced autophagy can enhance HyFS-mediated antiproliferation activities.
Inhibition of HyFS-induced autophagy enhances antiproliferative activities and promotes cell apoptosis in lung cancer cells. (A) A549 and H460 cells were treated with HyFS (0.5 μM) alone or in combination with 3-MA (5 μM) or CQ (10 μM) for 24 h. Cell viability was measured by MTT assay. (B) Cells were treated for 24 h as in (A), and EDU-incorporating live cells were detected by EDU proliferation assay. (C and D) Cells were treated with HyFS (0.5 μM) alone or in combination with 3-MA (5 μM) or CQ (10 μM) for 7 days. Live cells were detected by colony formation assay. (E) Cells were treated with HyFS (0.5 μM) alone or combined with siAtg5 or siBeclin1 for 24 h. Cell viability was measured by MTT assay. (F) Cells were treated with HyFS (0.5 μM) alone or combined with siAtg5 or siBeclin1 for 24 h. EDU-incorporating live cells were detected by EDU proliferation assay. (G) Cells were treated with indicated concentrations of HyFS and in combination with CQ (10 μM) for 24 h. Apoptotic cells were stained by PI/Annexin-V followed by flow cytometry. Data are expressed as mean ± SEM; * P < 0.05, ** P < 0.01, *** P < 0.001.
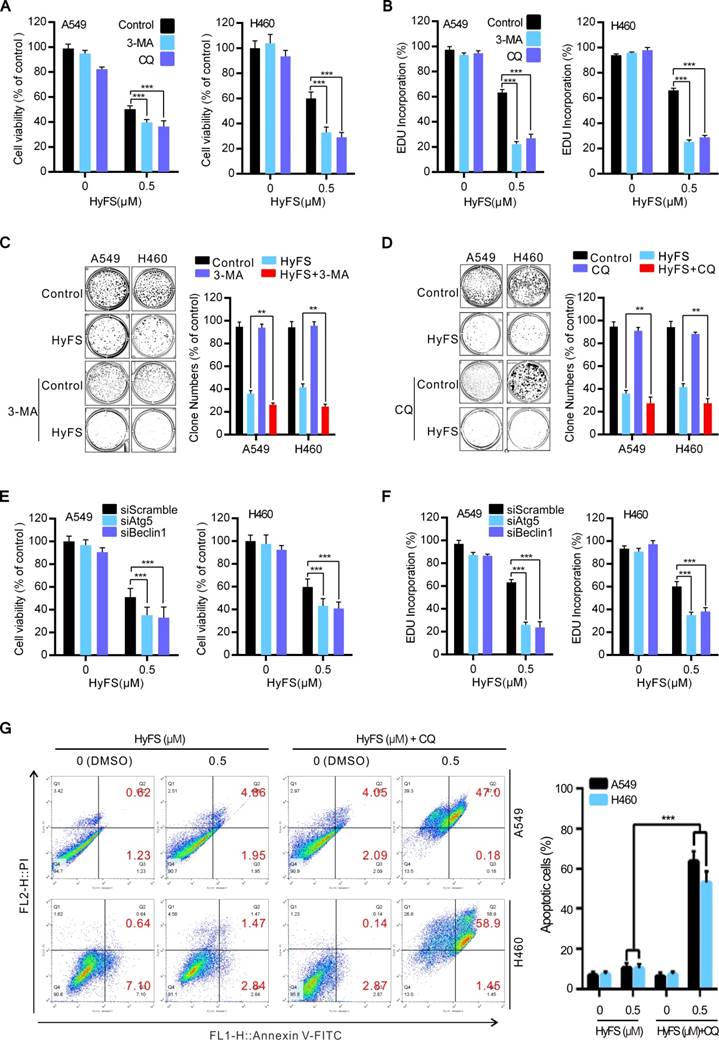
To investigate whether inhibition of HyFS-induced autophagy promotes cell apoptosis in lung cancer cells, we detected apoptotic cells using Annexin-V/PI staining followed by flow cytometry (Fig. 4G) and TUNEL assay (Fig. S4A and B). Our results showed that the number of apoptotic cells was significantly increased in lung cancer cells treated with both HyFS and CQ (Fig. 4G, Fig. S4A and B). Taken together, these results suggest that HyFS-induced autophagy plays a cytoprotective role against HyFS-induced cell apoptosis and anti-proliferation in lung cancer cells, and that combination therapy with HyFS and an autophagic inhibitor such as CQ or 3-MA is a potential alternative approach for treatment of lung cancer.
HyFS-induced autophagy is associated with inactivation of the Akt/mTOR pathway
Previous autophagy-related studies have shown that the Akt/mTOR pathway is a major negative regulatory pathway of autophagy [24-26]. Therefore, we investigated whether the Akt/mTOR pathway was inhibited in HyFS-treated lung cancer cells. First, we examined the phosphorylation status of Akt, mTOR, and p70S6K (a characterized target of the mTOR1 complex) by western blot. As shown in Fig. 5A, HyFS-mediated blockage of the Akt/mTOR pathway in A549 and H460 lung cancer cells was evidenced by decreasing phosphorylation levels of Akt, mTOR, and p70S6K. Moreover, we rescued HyFS-induced Akt/mTOR inhibition by overexpressing CA-Akt (the active form of Akt) and observed the classical marker of autophagy (LC3-II conversion and LC3 puncta accumulation) in HyFS-treated lung cancer cells. We found significantly decreased levels of LC3-II conversion and LC3 puncta accumulation in HyFS-treated cells (Fig. 5B and D). In contrast, combinatorial treatment of lung cancer cells with MK-2206 (an Akt inhibitor) and HyFS significantly increased the levels of LC3-II conversion in HyFS-treated cells (Fig. 5C). These results strongly indicate that HyFS-induced autophagy is associated with inactivation of the Akt/mTOR pathway in lung cancer cells.
HyFS induces autophagy through inhibition of the AKT/mTOR pathway. (A) A549 and H460 cells were treated with indicated concentrations of HyFS for 24 h. Cell lysates were used to detect Akt (S473), mTOR (S2448), P70S6K (S424/T421), and their phosphorylated counterparts by western blot. (B) Cells were treated with or without HyFS (0.5 μM) in combination with transfection by pCA-Akt plasmid or control vector for 24 h. Phosphorylated Akt (P-Akt), LC3-1, and LC3-II were detected by western blot. (C) Cells were treated with or without HyFS (0.5 μM) in combination with MK-2206 (an Akt inhibitor) for 24 h. Phosphorylated Akt (P-Akt), LC3-1, and LC3-II were detected by western blot. (D) Cells were treated with DMSO or HyFS (0.5 μM) in combination with or without transfection by pCA-Akt plasmid for 24 h. LC3 puncta and nuclei were observed by confocal microscopy. Data are expressed as mean ± SEM; * P < 0.05, ** P < 0.01, *** P < 0.001.
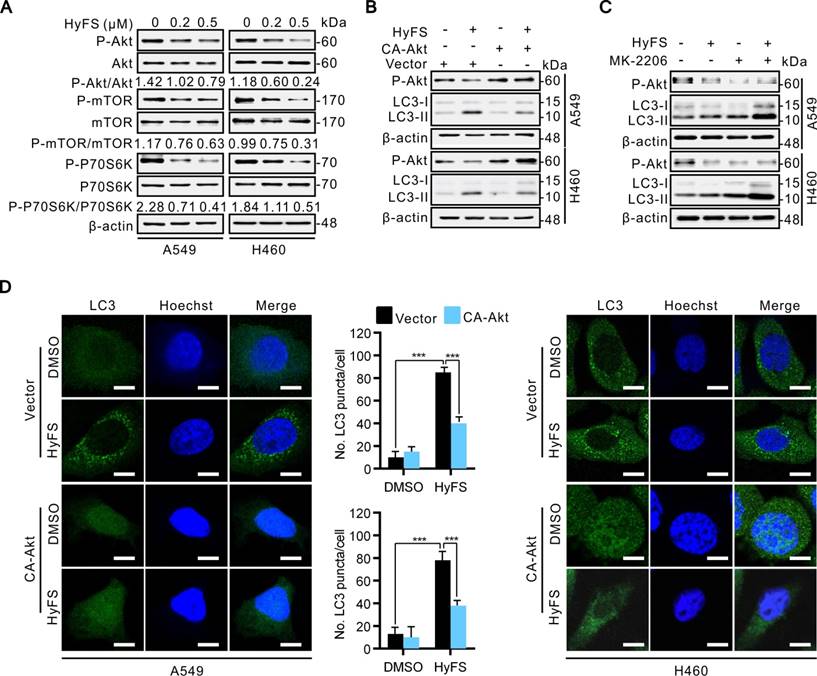
HyFS induces autophagy through blockage of the Hsp90/Akt/mTOR axis in lung cancer cells
Several lines of evidence show that some heat shock protein 90 (Hsp90) inhibitors can induce autophagy through inactivation of the Akt/mTOR pathway [33-36]. In addition, previous studies have also demonstrated that Hsp90 is overexpressed in many cancer cells and clinical cancer samples [36-39]. Hsp90 is a molecular chaperone responsible for folding and activating certain denatured proteins under stress conditions, which are generally called “client proteins” [40-42]. Considering that Akt is an important Hsp90 client protein [41], we thus detected whether Hsp90 was involved in HyFS-induced autophagy through inactivation of the Akt/mTOR pathway. Therefore, we first examined whether Hsp90 expression was involved in HyFS-induced autophagy in A549 and H460 cells by western blot. Our results showed that Hsp90 was markedly expressed in control lung cancer cells, but HyFS-treated cells showed significantly decreased levels of Hsp90 expression (Fig. 6A and B), and the decreased level of Hsp90 led to activation of autophagy, which was evidenced by significantly increased LC3-II conversion (Fig. 6A and B) and LC3 puncta accumulation (Fig. 6G and H) in HyFS-treated lung cancer cells. Downregulation of Hsp90 expression and activation of autophagy by HyFS treatment in A549 and H460 cells were further demonstrated through with HA-HSP90 plasmid transfection (high expression), Hsp90 RNA interference (siHsp90), and treatment with an inhibitor (17-AAG), respectively. As shown in Fig. 6B and G, overexpression of Hsp90 resulted in decreased LC3-II conversion (Fig. 6B) and LC3 puncta accumulation (Fig. 6G) in both A549 and H460 cells transfected with the HA-HSP90 plasmid. In contrast, inhibition of Hsp90 expression in A549 and H460 cells by siRNA against Hsp90 or treatment with 17-AAG (an Hsp90 inhibitor) significantly increased LC3 puncta accumulation (Fig. 6H) and LC3-II conversion (Fig. 6C) in HyFS-treated cells. These data strongly suggest that HyFS induces autophagy in lung cancer cells partly by suppressing Hsp90 expression.
HyFS induces autophagy through blockage of the Hsp90/Akt/mTOR axis in lung cancer cells. (A) A549 and H460 cells were treated with or without HyFS (0.5 μM) for 24 h. Hsp90, LC3-I, and LC3-II were detected by western blot. (B) Cells were treated with or without HyFS (0.5 μM) in combination with transfection of HA-HSP90 plasmid or vector for 24 h. Hsp90, phosphorylated Akt (P-Akt), LC3-1, and LC3-II were detected by western blot. (C) Cells were treated with or without HyFS (0.5 μM) in combination with or without 17-AAG (an Hsp90 inhibitor) for 24 h. Hsp90, LC3-1, and LC3-II were detected by western blot. (D) Cells were treated with or without HyFS (0.5 μM) in combination with transfection with HA-HSP90 and GFP-Akt plasmids for 24 h. Immunoprecipitation (IP) was performed with GFP and HA antibodies. (E) Cells were treated with or without HyFS (0.5 μM) in combination with transfection with HA-HSP90 plasmid or control vector. Hsp90 and phosphorylated Akt (P-Akt) were detected by western blot. (F) Cells were treated with siHSP90 or control siRNA (siScamble) for 24 h. Hsp90 and phosphorylated Akt (P-Akt) were detected by western blot. (G) Cells were treated with DMSO or HyFS (0.5 μM) in combination with transfection of HA-HSP90 plasmid or control vector for 24 h. LC3 puncta and nuclei were observed by confocal microscopy. (H) Cells were treated with siHsp90 and control siRNA (siScramble) for 24 h. LC3 puncta and nuclei were observed by confocal microscopy. Data are expressed as mean ± SEM; * P < 0.05, ** P < 0.01, *** P < 0.001.
To further unveil the mechanisms underlying HyFS-induced autophagy, we investigated the relationship between Hsp90 and Akt. We used a global protein-protein interaction network to identify the Akt-interacting proteins [43], and we found that both Hsp90 and mTOR can interact with Akt (Fig. S5). Distinctly decreased levels of both Hsp90 expression (Fig. 6A) and phosphorylated Akt (Fig. 6B) were observed in HyFS-treated lung cancer cells. To further investigate whether Hsp90 was involved in the phosphorylation of Akt in HyFS-treated lung cancer cells, we overexpressed Hsp90 with the HA-HSP90 plasmid. As shown in Fig. 6B and E, overexpressed Hsp90 increased the level of Akt phosphorylation in HyFS-treated A549 and H460 cells. Conversely, knockdown of Hsp90 by siRNA markedly decreased the level of phosphorylated Akt in the HyFS-treated lung cancer cells (Fig. 6F). Moreover, coimmunoprecipitation assays further confirmed that high levels of Hsp90 and phosphorylated Akt were found in lung cancer cells not treated with HyFS, but were significantly lower in HyFS-treated cells (Fig, 6D), indicating that Hsp90 interacts with Akt by direct binding, and that HyFS treatment interferes with the interaction between Hsp90 and phosphorylated Akt. Taken together, these data suggest that the Hsp90/Akt/mTOR axis is an important signaling pathway of HyFS-induced autophagy in lung cancer cells.
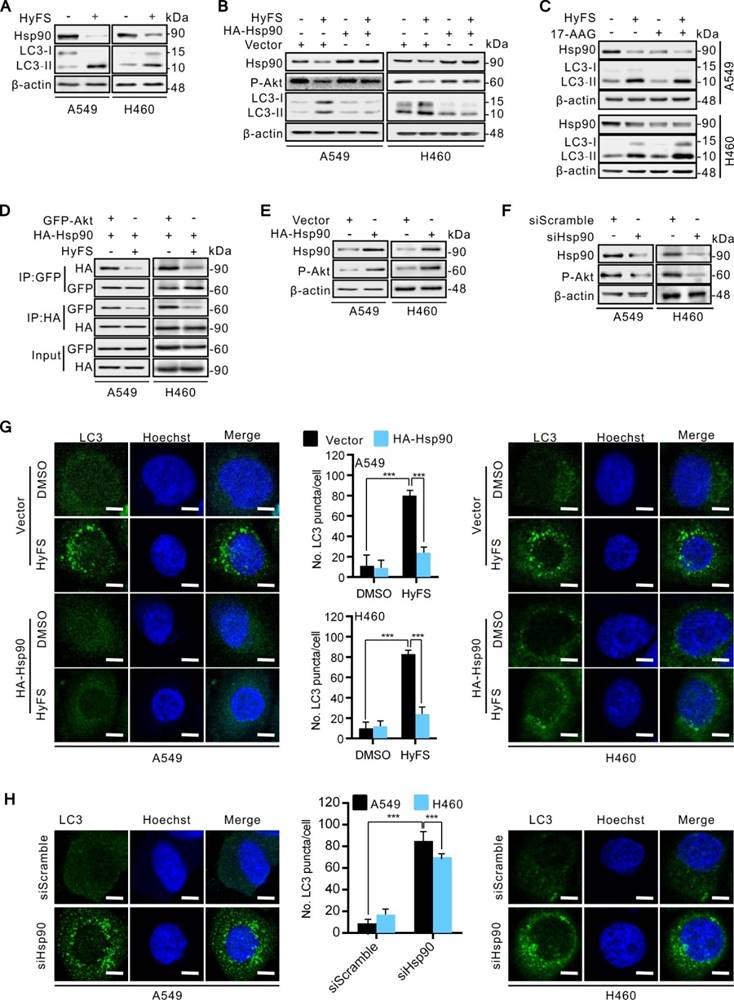
Ubiquitination-mediated degradation of Hsp90 leads to autophagy activation in lung cancer cells
To investigate how Hsp90 is downregulated by HyFS, we used reverse transcription PCR (RT-PCR) to detect the expression levels of Hsp90 mRNA. As shown in Fig. 7A, HyFS treatment showed no obvious effect on Hsp90 mRNA expression, indicating that transcription regulation of Hsp90 cannot account for the decreased Hsp90 expression following HyFS treatment. Therefore, we next examined whether Hsp90 was degraded through the proteasome/ubiquitination pathway. We pretreated A549 and H460 lung cancer cells with HyFS followed by treatment with cycloheximide (CHX, a protein synthesis inhibitor) or MG132 (a proteasome inhibitor). Our data showed that the level of Hsp90 clearly decreased in lung cancer cells treated with both HyFS and CHX (Fig. 7B). In contrast, the level of Hsp90 was kept stable in cells treated with HyFS and MG132 (Fig. 7C). These data suggest that Hsp90 may be degraded by the proteasome/ubiquitination pathway. To further determine whether the HyFS-induced Hsp90 decrease was due to proteasome-mediated degradation, we measured the effect of HyFS on Hsp90 ubiquitination by cotransfecting HA-HSP90 and Flag-ubiquitin expression vectors in A549 and H460 cells with or without MG132 treatment followed by coimmunoprecipitation assays. As shown in Fig. 7D, HyFS treatment markedly induced ubiquitin (Ub)-HyFS conjugation, which was further enhanced by MG132 treatment. Taken together, our results show that HyFS downregulates the expression of Hsp90 by ubiquitin-mediated degradation in lung cancer cells.
Ubiquitin-mediated degradation of Hsp90 leads to autophagy activation in lung cancer cells. (A) A549 and H460 cells were treated with indicated concentrations of HyFS for 24 h. Total RNA was extracted and Hsp90 cDNA was amplified by real time RT-PCR analysis. The mRNA level of the DMSO (0 μM) group was counted as 100%, and those of other groups were relative to that. (B) Cells were treated with or without HyFS (0.5 μM) in combination with or without cycloheximide (CHX, a protein synthesis inhibitor) for 24 h. Hsp90 was detected by western blot. (C) Cells were treated with or without HyFS (0.5 μM) in combination with or without MG132 (a proteasome inhibitor) for 24 h. Hsp90 was detected by western blot. (D) Cells were cultured in complete medium and transfected with both Flag-UB and HA-HSP90 plasmids in combination with without MG132 or HyFS (0.5 μM) for 24 h. Immunoprecipitation (IP) was performed with Flag and HA antibodies. Data are expressed as mean ± SEM; * P < 0.05, ** P < 0.01, *** P < 0.001.
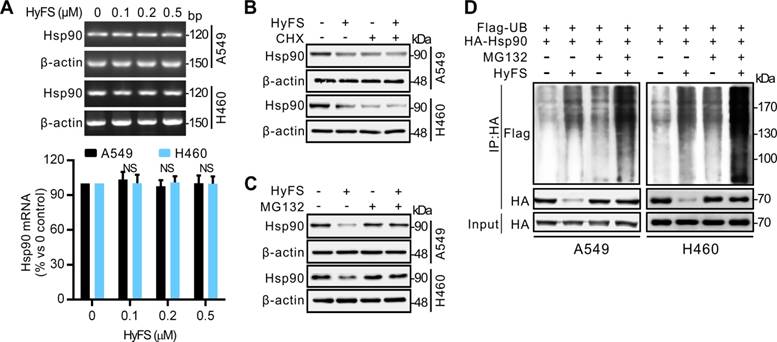
Combination therapy with HyFS and chloroquine enhances antitumor activities in vivo
A549 and H460 cells were subcutaneously implanted into immunodeficient (BALB/c Nu/Nu) mice. When tumor masses were palpable (about 50 mm3), the mice were treated without HyFS (DMSO control), with HyFS, or with a combination of HyFS and CQ in intervals of 2 days for 6 total treatments and observed to 21 days. As shown in Fig. 8A, compared to the DMSO group, the tumors in HyFS and HyFS+CQ groups grew significantly slower, and the HyFS+CQ group grew the slowest, respectively. Figure 8B shows images of tumor masses (10 in each group) that were collected at day 21. These data strongly indicate that disruption of HyFS-induced autophagy by CQ enhances the anti-tumor activities of HyFS.
Combination therapy with HyFS and chloroquine enhances antitumor activities in vivo. Human A549 and H460 lung tumor cells were injected into the right flanks of Nu/Nu mice (10 mice/group). When tumor volumes reached 50 mm3, mice were treated with indicated formulations every 2 days for 6 total treatments. (A) Tumor volumes at different time points. (B) Tumor mass images of both A549 and H460 model mice at day 21. Data are expressed as mean ± SEM; * P < 0.05, ** P < 0.001. Scale bar = 4.5 mm
Discussion
Recent studies have indicated that many anticancer agents can induce autophagy in cancer cells, but the role of autophagy in cancer therapy is still controversial [44-46]. Up to now, four different functional forms of autophagy—cytoprotective, nonprotective, cytotoxic, and cytostatic autophagy—have been recognized as being involved in anticancer treatment [29]. Cytoprotective autophagy works as a drug-resistant mechanism that endows cancer cells with the capability of compromising anticancer activities in chemotherapy. Thus, intervening in cytoprotective autophagy to enhance drug sensitivity is important to enable rational manipulation of autophagy for cancer treatment. In the current study, we tested whether HyFS could inhibit cancer cell growth (proliferation) and induction of cell apoptosis and autophagy, and evaluated their relationship. We found that HyFS mediated cell growth inhibition and autophagy, but did not induce obvious cell apoptosis in A549 and H460 lung cancer cells in the first 24 h. However, many previous studies have demonstrated that the induction of apoptosis is the major form of cell death by which cardenolides derive their anticancer activities [47-49]. Therefore, to answer why apoptosis is not observed in HyFS-treated lung cancer cells, we blocked HyFS-mediated autophagy either by inhibitors or siRNA against Atg5 or beclin-1 (two target proteins necessary for autophagy formation), and observed whether apoptosis could be restored. As expected, our data showed that stronger inhibition of cell growth and definite cell apoptosis were found in HyFS-treated lung cancer cells when HyFS-mediated autophagy was interrupted by autophagic inhibitors or siRNA. These data strongly suggest that HyFS induces cytoprotective autophagy in A549 and H460 lung cancer cells. In addition, our data also indicate that combination treatment with HyFS and an autophagic inhibitor may be a feasible strategy for the treatment of lung cancer, if HyFS is used as an anticancer drug in the future. However, the complicated intrinsic relationship between autophagy and apoptosis induced by HyFS is unknown, and thus it requires further investigation.
Natural products derived from plants such as cardenolides, flavonoids, and terpenes have received considerable attention in recent years due to their diverse pharmacological properties, especially anticancer activities [50-52]. C. gigantea is a common wild weed of the Asclepiadaceae family. C. gigantea has tremendous potential for medicinal purposes, because its extracts and preparations have various benefits such as anti-abdominal tumor, anti-inflammatory, and antimicrobial effects [52-55]. 3′-epi-12β-Hydroxyfroside (HyFS) is a natural product with a special cardenolide chemical structure (Fig. 1A) isolated from the roots of C. gigantean by our collaborating laboratory, and has been demonstrated to have anticancer activities in several cancer cell lines [5]. Recent studies have demonstrated that cardenolides derive their anticancer activities through various mechanisms, among which apoptosis is still the major theme [9-11]. However, some studies have shown that autophagy also plays an important role in cardenolide-induced anticancer activities. Wang and colleagues found that two traditional cardenolides, digoxin and ouabain, induce cytotoxic autophagy through mTOR and ERK1/2 pathways in A549 and H460 lung cancer cells [17, 18]. Similar cytotoxic autophagy in A549 and H1975 lung cancer cells was reported by Trenti and colleagues [19]. HyFS is a new cardenolide structure, and its cytotoxic activities have only been established by MTT assays [5]. Thus, the mechanisms underlying HyFS-mediated anticancer activities have remained largely unknown.
In this study, our data showed that HyFS inhibited cell growth and induced autophagy, but did not clearly induce cell apoptosis in A549 and H460 lung cancer cells in the first 24 h. Inhibition of HyFS-mediated autophagy by inhibitors or RNA interference led to increased antiproliferation effects and a dramatic induction of cancer cell apoptosis, indicating that HyFS activates protective autophagy in lung cancer cells, which is different from previous studies [17-19]. In addition, a few lines of evidence have shown that drug-induced autophagy may precede apoptosis in some cancer cells [21, 56]. Thus, it is reasonable to manipulate autophagy for cancer treatment in combination with traditional apoptosis-inducing drugs. Because of the complex nature between apoptosis and autophagy in cell fate determination [21], further studies are needed to unveil the crosstalk between these tightly regulated biological processes.
Recent studies have shown that some heat shock protein 90 (Hsp90) inhibitors can induce autophagy through inactivation of the Akt/mTOR pathway [33-36]. In addition, a study has also indicated that the Akt/mTOR pathway is involved in regulating the proliferation and apoptosis of cancer cells [57]. Since Akt is an Hsp90 client protein [40, 41], we investigated whether the Hsp90/Akt/mTOR axis is involved in the HyFS activation of autophagy in lung cancer cells. Our data show that decreased phosphorylation levels of Akt, mTOR, and p70S6K were observed in HyFS-treated lung cancer cells. Consistently, overexpressing CA-Akt (the active form of Akt) also leads to decreased levels of LC3-II conversion and LC3 puncta accumulation in HyFS-treated lung cancer cells. In contrast, combination treatment of lung cancer cells with MK-2206 (an Akt inhibitor) and HyFS significantly increased the levels of LC3-II conversion in HyFS-treated cells. These data strongly suggest that the Akt/mTOR signaling pathway is involved in HyFS-mediated autophagy. Furthermore, our data showed that HyFS markedly decreased the expression of Hsp90 and increased the levels of LC3-II conversion and LC3 puncta accumulation in HyFS-treated lung cancer cells. Moreover, our results also showed that HyFS downregulated Hsp90 and its client protein Akt by ubiquitin-mediated degradation of Hsp90. These results indicate that the Hsp90/Akt/mTOR axis is a novel pathway in HyFS-induced autophagy, suggesting that HyFS may be a potent agent in the induction of ubiquitin-mediated degradation of Hsp90. HyFS-mediated inhibition of the Hsp90/Akt/mTOR signaling pathway merits exploration as a therapeutic strategy for lung cancer treatment.
In our current study, although we have solidly confirmed the Hsp90/Akt/mTOR axis involved in the HyFS-induced protective autophagy, the relevant target protein of HyFS still remains unknown. Before and during this study, we have hypothesized that the physical binding between HyFS and Hsp90 is the determining factor that causes the ubiquitin-mediated degradation of Hsp90. Thus, we have tried to quantitatively measure the HyFS-Hsp90 binding using the cellular thermal shift assay (CETSA), a recently described method that permits rapid and simple assessment of target engagement of drugs in a cellular context [58-60]. However, we failed to find any evidence of HyFS-Hsp90 direct binding. The possible reason may be due to technical inexperience or really not existence of HyFS-Hsp90 direct binding. It still need further study to address this issue.
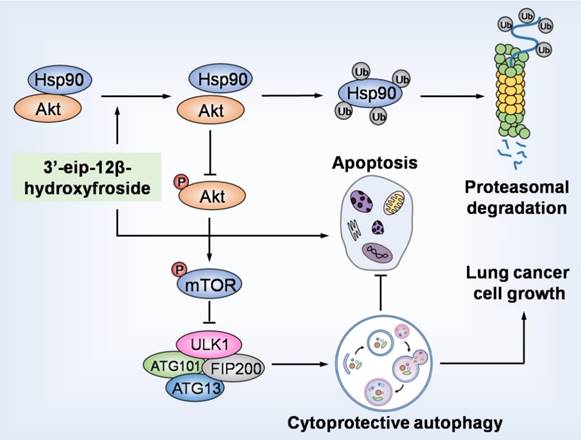
In summary, our study demonstrated that HyFS is a potential agent against lung cancer. HyFS induces cytoprotective autophagy through ubiquitin-mediated degradation of Hsp90 and further blockage of the Hsp90/Akt/mTOR axis. Our findings unveil the molecular mechanism underlying HyFS anticancer activity (summarized in Fig. 9), and strongly indicate that a combination therapy with HyFS and an autophagic inhibitor is a potential alternative approach for the treatment of lung cancer.
HyFS induces cytoprotective autophagy by blocking the Hsp90/Akt/mTOR axis in lung cancer cells. HyFS treatment promotes ubiquitin-mediated degradation of Hsp90, leading to inactivation of its client protein Akt, which subsequently induces blockage of the Akt/mTOR signaling pathway. The inhibition of Akt/mTOR signaling ultimately results in elevated autophagic flux, which promotes cell growth and suppresses apoptosis of lung cancer cells, indicating that HyFS induces cytoprotective autophagy. Ub: ubiquitin.
Materials and Methods
Reagents and antibodies
3′-epi-12β-hydroxyfroside (HyFS) was isolated and purified (> 95%) by our collaborating group [5]. Other reagents used in this study were purchased from Chinese suppliers indicated in the brackets. 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT, Sigma, M2128), dimethylsulfoxide (DMSO, Sigma, D2650), Z-VAD-fmk (Sigma, V116), wortmannin (Sigma, W1628), bafilomycin A1 (Sigma, B1793), CQ (Sigma, C6628), 3-MA (MCE, HY-13259), MK-2206 (MCE, HY-10358), 17-AAG (MCE, HY-10211), MG-132 (Sigma, M7449), CHX (MCE, HY-12320). The following antibodies were also purchased from Chinese suppliers indicated in the brackets. β-actin (Santa Cruz, sc-1616), caspase 3 (Millipore, MAB4703), PARP (Millipore, 04-575), LC3B (MBL, PM036), BECLIN1 (Santa Cruz, sc-11427), ATG5 (Cell Signaling Technology, 2630), SQSTM1 (MBL, M162-3), Akt (Cell Signaling, 4685), phospho-Akt (Cell Signaling, 4051), p70S6K (Santa Cruz, sc-9027), phosphop70S6K (Santa Cruz, sc-7984-R), mTOR (Millipore, 04-385), p-mTOR (Millipore, 09-213), Hsp90 (Santa Cruz, sc-27987), HA-tag (Santa Cruz, sc-53516).
Cell lines and cell culture
Human glioblastoma cell line U251, human hepatocellular carcinoma cell lines Bel-7402 and Bel-7404, and lung normal cell lines MRC-5 and BEAS-2B were purchased from Shanghai Institute of Biochemistry and Cell biology, Chinese Academy of Science. The following human cancer cell lines were obtained from ATCC and kept by our three collaborating laboratories: human lung cancer cell lines NCL-H1299 CRL-5803), A549 (CRM-CCL-185), and H460 (HTB-177); human colorectal cancer cell lines DLD-1 (CCL-221), HCT116 (CCL-247), and HT29 (HTB-38); human glioblastoma cell lines U87(HTB-14) and A-172(CRL-1620); human cervical cancer cell lines HeLa (CCL-2), SiHa (HTB-35), and C33A (HTB-31); and human hepatocellular carcinoma cell line Hep3B (HB-8064). All cells were cultured in RPMI 1640 or DMEM (GIBCO) supplemented with 10% fetal bovine serum (GIBCO), 100 U/mL penicillin (Sigma), and 100 μg/mL streptomycin (Sigma, P0781) at 37 °C in an atmosphere containing 5% CO2.
Cell viability assays
Cells were plated in 96-well plates and incubated overnight, and then cells were exposed to test compounds for 24 h. Subsequently, cells were evaluated by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) assay. Briefly, 10 μL of MTT was added to each well and incubated for 4 h. The media was removed, and 150 μL of DMSO was added to each well to dissolve the crystal formazan dye. The absorbance was measured at 570 nm on an enzyme-linked immunosorbent assay reader (ELX808IU, Bio-Tek).
LDH assay
Lactate dehydrogenase (LDH) activity was determined by a LDH release kit (Beyotime, C0016). The assay was performed according to the manufacturer's instructions. In brief, LDH converts pyruvate into lactate that reduces the developer to a colored product with absorbance at 450 nm as measured by an enzyme-linked immunosorbent assay reader (ELX808IU, Bio-Tek).
EDU proliferation assay
To assess cell proliferation, cells were seeded in 96-well plates and exposed to the test compound under standard conditions in complete media for 24 h. Cell proliferation was detected by the incorporation of 5-ethynyl-20-deoxyuridine (EDU) with a EDU Cell Proliferation Assay Kit (Ribobio, Guangzhou, China). Briefly, cells were incubated with 50 mM EDU for 6 h before fixation, permeabilization, and EDU staining, which were performed according to the manufacturer's protocol. Cell nuclei were stained with DAPI (Sigma) at a concentration of 1 mg/mL for 20 min. The proportion of cells incorporating EDU was determined by fluorescence microscopy (Nikon, 80i, Japan).
Colony formation assay
Cells were seeded at a density of 300 cells/well in 24-well plates and treated with the indicated concentration of HyFS or vehicle control. The medium was changed every 3 days. After 14 days, the colonies were stained with Giemsa for 15 min and washed three times. Visible colonies were photographed by a Molecular Imager Gel Do XR+ System (Bio-Rad) and counted using Image J software (NIH).
TUNEL assay
Terminal-deoxynucleoitidyl transferase mediated nick end labeling (TUNEL) was performed using the Dead End Fluorometric TUNEL system (Promega, G3250). Briefly, cells were cultured in 24-well plates and exposed to test compounds for 24 h, and then fixed in 4% paraformaldehyde followed by permeabilization with 0.2% Triton X-100. TUNEL staining was performed according to the manufacturer's instructions and observed under a fluorescence microscope (Nikon, 80i, Japan).
Flow cytometry
Cells were cultured and treated for 24 h, harvested by trypsinization, washed with PBS, then resuspended and incubated in PI/Annexin-V solution (KeyGEN Biotech, KGA106) for apoptosis analysis. At least 10,000 live cells were analyzed on a BD FACSCalibur Flow cytometer. Data were analyzed by FlowJo software (BD Biosciences).
Western blotting
Cells were lysed with RIPA buffer (50 mM Tris, 1.0 mM EDTA, 150 mM NaCl, 0.1% Triton X-100, 1% sodium deoxycholate, 1 mM PMSF). Protein concentrations were quantified by the BCA protein assay kit (Pierce, 23225). Proteins were resolved on 12% SDS-PAGE, then transferred to PVDF (Bio-Rad) membranes. After blocking, the membranes were incubated with primary antibodies at 4 °C overnight, then incubated with secondary antibodies at room temperature for 1 h. Target proteins were examined using Enhanced Chemiluminescence reagents (Millipore, WBKLS0100).
Immunofluorescence
Cells were fixed with 4% paraformaldehyde for 30 min, washed with PBS, and then incubated with 0.1% Triton X-100 for permeabilization. Cells were stained with anti-LC3B polyclonal antibody at 4 °C overnight, and subsequently with Alexa Fluor 488-conjugated goat anti-rabbit LgG (Abcam, ab150077) at 37 °C for 1 h. Nuclei were stained with DAPI for 5 min. Images were captured using a confocal laser scanning microscope (Olympus, FV3000).
Transmission electron microscopy
Transmission electron microscopy was performed as described previously [61]. Briefly, A549 and H460 cells were fixed in 4% glutaraldehyde (Sigma). A sorvall MT5000 microtome (DuPont Instruments, MT5000) was used to prepare ultrathin sections after dehydration. Lead citrate and/or 1% uranyl acetate were used to stain the sections, and the autophagic vacuoles in the cytoplasmic area were calculated using Image Pro Plus version 3 software.
Acridine orange staining
Cells were treated with HyFS at a given concentration for 24 h, then cultured cells were fixed by 4% paraformaldehyde, washed three times with PBS, and stained with 1 mM acridine orange (Sigma) at room temperature in PBS containing 5% FBS for 15 min. The cells were washed again and then observed under a fluorescence microscope (Nikon, 80i, Japan).
Real time RT-PCR analysis
Hsp90 and control β-actin RNA and cDNA were prepared by the manufacturer's recommended protocol using reverse transcriptase and random hexamers from a RevertAid First Strand cDNA Synthesis Kit (Fermentas). The previously reported primers used for quantifying Hsp90 mRNA expression were synthesized by TaKaRa (Dalian, China) [38]. The PCR reaction was also performed with rTaq (TaKaRa) in a DNA thermal cycler (Maxygen) according to a standard protocol as reported in a previous study [38].
Small RNA interference
Atg5, beclin1, Hsp90, and negative control small interfering RNA (siRNA) were synthesized by Genephama. The sequences of siRNAs were as follows. For human Atg5, siAtg5: 5'- GGAUGAGAUAACUGAAAGGTT-3'; for human beclin1, siBeclin1: 5'-CAGUUUGGCACAAUCAAUATT-3'; for human Hsp90, siHSP90: 5'-GGAAAGAGCUG-CAUAUUA-3'; for negative control of Atg5, siScramble (1): 5'-GGAAAGAGCUGCAUAUUAATT-3'; for negative control of beclin1, siScramble (2): 5'-UUCUCCGAACGUGUCACGUTT-3'. Human lung cancer A549 and H460 cell lines were transfected with siRNA by using Lipofectamine 2000 reagent (Invitrogen, 11668027) and cultured according to the manufacturer's protocol.
Plasmids and transfection of cells
The HA-HSP90 expression plasmid was kindly provided by Dr. Lu Zhang (Sichuan University, China). CA-Akt (myrAkt delta4-129) was purchased from Addgene (Chendu, China). The plasmids were introduced into cells using Lipofectamine 2000 according to the manufacturer's instructions.
Coimmunoprecipitation
Cells were lysed with RIPA buffer supplemented with protease inhibitor cocktail. Whole cell lysates obtained by centrifugation were incubated with 1-4 μg of the indicated antibodies overnight at 4 °C, followed by incubation for 2 h at 4 °C with protein G-Sepharose beads (40 μL, GE Healthcare). The immune-complexes were washed 3 times with RIPA buffer and then analyzed by immunoblotting with the indicated antibodies.
Observation of anti-tumor effect in vivo
The animal protocols in this study were approved by the Animal Care and Use Committee of Hainan Medical College. A549 and H460 tumor models were established in BALB/c immunodeficient nude (Nu/Nu) mice at 6-8 weeks of age. The mice were injected with 1 × 106 corresponding tumor cells (10 mice in each group). When the tumor volume was palpable (~50 mm3 at day 6), mice were randomly divided into three groups in each tumor model and injected with DMSO (HyFS 0 mg/kg), HyFS (50 mg/kg), and HyFS (50 mg/kg) + CQ (80 mg/kg), respectively, through the tail vein once every 2 days. Treatment was continued for 6 total times (12 days), and mice were observed to 21 days. The tumor volumes were monitored every 3 days using a handheld imaging device (TM900, Peira, Belgium).
Statistical analysis
Statistical analysis was performed using Prism 6. Statistical differences were determined by one-way ANOVA followed by the post hoc test or combined with two-sample equal variance Student's t test. Data are expressed as mean ± SEM and deemed to be statistically significant if P < 0.05. Error bars indicate SEM unless otherwise indicated.
Abbreviations
3-MA: 3-methyladenine; Akt: protein kinase B; Atg: aAutophagy-related gene; AVO: acidic vesicular organelles; CQ: choroquine; DMSO: dimethyl sulfoxside; EDU: 5-ethynyl-20-deoxyuridine; Hsp90: heat shock protein 90; HyFS: 3'-epi-12β-hydroxyfroside; LC3: Microtubule-associated protein 1 light chain 3; LDH: lactate dehydrogenase; mTOR: Mammalian Target of Rapamycin; MTT: 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide; SiRNA: Small interfering RNA; Ub: Ubiquitin.
Supplementary Material
Supplementary figures.
Acknowledgements
The authors are grateful to PhD. Lu Zhang (State Key Laboratory of Biotherapy, Sichuan University, China) for offering the HA-HSP90 plasmid. Most of the experiments were performed in State Key Laboratory of Biotherapy, China. This work was funded by the National Natural Science Foundation of China (81560599, 81460557, 81460020, 81360482 and 81560572) and Jiangsu Province Science Foundation for Youths, China (BK2012101).
Author contributions
The study was conceived and designed by G.-H.T., C.H. and Y.-N.L. Most of the agents and materials were offered by C.H. Most of the experiments were supervised by C.H. The statistical analysis was performed by G.-H.T., Y.S., Y.-H.H., and Y.-N.L. Partial Western blot analysis was performed by Q.L. and W.Z. Isolation and purification of 3′-epi-12β-hydroxyfroside and partial cytotoxic assays were performed by W.-L.M. and H.-F.D. All other experiments were performed by Y.S., Y.-H.H., F.-Y.H., C.-C.W. and Y.-Y.L. The manuscript was written by G.-H.T., Y.S. and Y.-H.H.
Conflicts of interest
No potential conflicts of interest were disclosed.
References
1. Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M. et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015;136:E359-86
2. Thomas A, Liu SV, Subramaniam DS, Giaccone G. Refining the treatment of NSCLC according to histological and molecular subtypes. Nat Rev Clin Oncol. 2015;12:511-26
3. Iacono D, Chiari R, Metro G, Bennati C, Bellezza G, Cenci M. et al. Future options for ALK-positive non-small cell lung cancer. Lung Cancer. 2015;87:211-9
4. Qiu YF, Liu ZG, Yang WJ, Zhao Y, Tang J, Tang WZ. et al. Research progress in the treatment of small cell lung cancer. J Cancer. 2017;8:29-38
5. Maoyuan W, Wenli M, Yuanyuan D, Shenglan L, Zhunian W, Haofu D. Cytotoxic cardenolides from the roots of Calotropis gigantea. Modern Pharm Res. 2008;1:4-9
6. Hamad E, Mather PJ, Srinivasan S, Rubin S, Whellan DJ, Feldman AM. Pharmacologic therapy of chronic heart failure. Am J Cardiovasc Drugs. 2007;7:235-48
7. Abarquez RF Jr, Reganit PF, Chungunco CN, Alcover J, Punzalan FE, Reyes EB. et al. Chronic Heart Failure Clinical Practice Guidelines' Class 1-A Pharmacologic Recommendations: Start-to-End Synergistic Drug Therapy? ASEAN Heart J. 2016;24:4
8. Gheorghiade M, van Veldhuisen DJ, Colucci WS. Contemporary use of digoxin in the management of cardiovascular disorders. Circulation. 2006;113:2556-64
9. Roberts DM, Gallapatthy G, Dunuwille A, Chan BS. Pharmacological treatment of cardiac glycoside poisoning. Br J Clin Pharmacol. 2016;81:488-95
10. Kaushik V, Azad N, Yakisich JS, Iyer AK. Antitumor effects of naturally occurring cardiac glycosides convallatoxin and peruvoside on human ER+ and triple-negative breast cancers. Cell Death Discov. 2017;3:17009
11. Karasneh RA, Murray LJ, Cardwell CR. Cardiac glycosides and breast cancer risk: A systematic review and meta-analysis of observational studies. Int J Cancer. 2017;140:1035-41
12. Kulkarni YM, Kaushik V, Azad N, Wright C, Rojanasakul Y, O'Doherty G. et al. Autophagy-Induced Apoptosis in Lung Cancer Cells by a Novel Digitoxin Analog. J Cell Physiol. 2016;231:817-28
13. Dietze R, Konrad L, Shihan M, Kirch U, Scheiner-Bobis G. Cardiac glycoside ouabain induces activation of ATF-1 and StAR expression by interacting with the alpha4 isoform of the sodium pump in Sertoli cells. Biochim Biophys Acta. 2013;1833:511-9
14. Diederich M, Muller F, Cerella C. Cardiac glycosides: From molecular targets to immunogenic cell death. Biochem Pharmacol. 2017;125:1-11
15. Menger L, Vacchelli E, Kepp O, Eggermont A, Tartour E, Zitvogel L. et al. Trial watch: Cardiac glycosides and cancer therapy. Oncoimmunology. 2013;2:e23082
16. Menger L, Vacchelli E, Adjemian S, Martins I, Ma Y, Shen S. et al. Cardiac glycosides exert anticancer effects by inducing immunogenic cell death. Sci Transl Med. 2012;4:143ra99
17. Wang Y, Qiu Q, Shen JJ, Li DD, Jiang XJ, Si SY. et al. Cardiac glycosides induce autophagy in human non-small cell lung cancer cells through regulation of dual signaling pathways. Int J Biochem Cell Biol. 2012;44:1813-24
18. Wang Y, Zhan Y, Xu R, Shao R, Jiang J, Wang Z. Src mediates extracellular signal-regulated kinase 1/2 activation and autophagic cell death induced by cardiac glycosides in human non-small cell lung cancer cell lines. Mol Carcinog. 2015;54(Suppl 1):E26-34
19. Trenti A, Grumati P, Cusinato F, Orso G, Bonaldo P, Trevisi L. Cardiac glycoside ouabain induces autophagic cell death in non-small cell lung cancer cells via a JNK-dependent decrease of Bcl-2. Biochem Pharmacol. 2014;89:197-209
20. Boya P, Reggiori F, Codogno P. Emerging regulation and functions of autophagy. Nat Cell Biol. 2013;15:713-20
21. Marino G, Niso-Santano M, Baehrecke EH, Kroemer G. Self-consumption: the interplay of autophagy and apoptosis. Nat Rev Mol Cell Biol. 2014;15:81-94
22. Fulda S, Kogel D. Cell death by autophagy: emerging molecular mechanisms and implications for cancer therapy. Oncogene. 2015;34:5105-13
23. Kroemer G, Marino G, Levine B. Autophagy and the integrated stress response. Mol Cell. 2010;40:280-93
24. Jung CH, Ro SH, Cao J, Otto NM, Kim DH. mTOR regulation of autophagy. FEBS Lett. 2010;584:1287-95
25. Schmelzle T, Hall MN. TOR, a central controller of cell growth. Cell. 2000;103:253-62
26. Moore J, Megaly M, MacNeil AJ, Klentrou P, Tsiani E. Rosemary extract reduces Akt/mTOR/p70S6K activation and inhibits proliferation and survival of A549 human lung cancer cells. Biomed Pharmacother. 2016;83:725-32
27. He C, Klionsky DJ. Regulation mechanisms and signaling pathways of autophagy. Annu Rev Genet. 2009;43:67-93
28. Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132:27-42
29. Gewirtz DA. The four faces of autophagy: implications for cancer therapy. Cancer Res. 2014;74:647-51
30. Efeyan A, Comb WC, Sabatini DM. Nutrient-sensing mechanisms and pathways. Nature. 2015;517:302-10
31. Klionsky DJ, Abdelmohsen K, Abe A, Abedin MJ, Abeliovich H, Acevedo Arozena A. et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy. 2016;12:1-222
32. Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen H. et al. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem. 2007;282:24131-45
33. Mori M, Hitora T, Nakamura O, Yamagami Y, Horie R, Nishimura H. et al. Hsp90 inhibitor induces autophagy and apoptosis in osteosarcoma cells. Int J Oncol. 2015;46:47-54
34. Liu KS, Liu H, Qi JH, Liu QY, Liu Z, Xia M. et al. SNX-2112, an Hsp90 inhibitor, induces apoptosis and autophagy via degradation of Hsp90 client proteins in human melanoma A-375 cells. Cancer Lett. 2012;318:180-8
35. Chen MH, Chiang KC, Cheng CT, Huang SC, Chen YY, Chen TW. et al. Antitumor activity of the combination of an HSP90 inhibitor and a PI3K/mTOR dual inhibitor against cholangiocarcinoma. Oncotarget. 2014;5:2372-89
36. Bekki H, Kohashi K, Maekawa A, Yamada Y, Yamamoto H, Harimaya K. et al. Elevated expression of HSP90 and the antitumor effect of an HSP90 inhibitor via inactivation of the Akt/mTOR pathway in undifferentiated pleomorphic sarcoma. BMC Cancer. 2015;15:804
37. Zhang H, Burrows F. Targeting multiple signal transduction pathways through inhibition of Hsp90. J Mol Med (Berl). 2004;82:488-99
38. Soudry E, Stern Shavit S, Hardy B, Morgenstern S, Hadar T, Feinmesser R. Heat shock proteins HSP90, HSP70 and GRP78 expression in medullary thyroid carcinoma. Ann Diagn Pathol. 2017;26:52-6
39. Kang GH, Lee EJ, Jang KT, Kim KM, Park CK, Lee CS. et al. Expression of HSP90 in gastrointestinal stromal tumours and mesenchymal tumours. Histopathology. 2010;56:694-701
40. Beck JT, Ismail A, Tolomeo C. Targeting the phosphatidylinositol 3-kinase (PI3K)/AKT/mammalian target of rapamycin (mTOR) pathway: an emerging treatment strategy for squamous cell lung carcinoma. Cancer Treat Rev. 2014;40:980-9
41. Sato S, Fujita N, Tsuruo T. Modulation of Akt kinase activity by binding to Hsp90. Proc Natl Acad Sci U S A. 2000;97:10832-7
42. Lorenz OR, Freiburger L, Rutz DA, Krause M, Zierer BK, Alvira S. et al. Modulation of the Hsp90 chaperone cycle by a stringent client protein. Mol Cell. 2014;53:941-53
43. Fu L, Zhang S, Zhang L, Tong X, Zhang J, Zhang Y. et al. Systems biology network-based discovery of a small molecule activator BL-AD008 targeting AMPK/ZIPK and inducing apoptosis in cervical cancer. Oncotarget. 2015;6:8071-88
44. Galluzzi L, Bravo-San Pedro JM, Demaria S, Formenti SC, Kroemer G. Activating autophagy to potentiate immunogenic chemotherapy and radiation therapy. Nat Rev Clin Oncol. 2016
45. Maycotte P, Aryal S, Cummings CT, Thorburn J, Morgan MJ, Thorburn A. Chloroquine sensitizes breast cancer cells to chemotherapy independent of autophagy. Autophagy. 2012;8:200-12
46. Li H, Wang P, Yu J, Zhang L. Cleaving Beclin 1 to suppress autophagy in chemotherapy-induced apoptosis. Autophagy. 2011;7:1239-41
47. Cerella C, Muller F, Gaigneaux A, Radogna F, Viry E, Chateauvieux S. et al. Early downregulation of Mcl-1 regulates apoptosis triggered by cardiac glycoside UNBS1450. Cell Death Dis. 2015;6:e1782
48. Frese S, Frese-Schaper M, Andres AC, Miescher D, Zumkehr B, Schmid RA. Cardiac glycosides initiate Apo2L/TRAIL-induced apoptosis in non-small cell lung cancer cells by up-regulation of death receptors 4 and 5. Cancer Res. 2006;66:5867-74
49. McConkey DJ, Lin Y, Nutt LK, Ozel HZ, Newman RA. Cardiac glycosides stimulate Ca2+ increases and apoptosis in androgen-independent, metastatic human prostate adenocarcinoma cells. Cancer Res. 2000;60:3807-12
50. Chew AL, Jessica JJ, Sasidharan S. Antioxidant and antibacterial activity of different parts of Leucas aspera. Asian Pac J Trop Biomed. 2012;2:176-80
51. Mann CD, Neal CP, Garcea G, Manson MM, Dennison AR, Berry DP. Phytochemicals as potential chemopreventive and chemotherapeutic agents in hepatocarcinogenesis. Eur J Cancer Prev. 2009;18:13-25
52. Seeka C, Sutthivaiyakit S. Cytotoxic cardenolides from the leaves of Calotropis gigantea. Chem Pharm Bull (Tokyo). 2010;58:725-8
53. Adak M, Gupta JK. Evaluation of anti-inflammatory activity of Calotropis gigantea (AKANDA) in various biological system. Nepal Med Coll J. 2006;8:156-61
54. Deshmukh PT, Fernandes J, Atul A, Toppo E. Wound healing activity of Calotropis gigantea root bark in rats. J Ethnopharmacol. 2009;125:178-81
55. Wang ZN, Wang MY, Mei WL, Han Z, Dai HF. A new cytotoxic pregnanone from Calotropis gigantea. Molecules. 2008;13:3033-9
56. Sy LK, Yan SC, Lok CN, Man RY, Che CM. Timosaponin A-III induces autophagy preceding mitochondria-mediated apoptosis in HeLa cancer cells. Cancer Res. 2008;68:10229-37
57. Shinojima N, Yokoyama T, Kondo Y, Kondo S. Roles of the Akt/mTOR/p70S6K and ERK1/2 signaling pathways in curcumin-induced autophagy. Autophagy. 2007;3:635-7
58. Martinez Molina D, Jafari R, Ignatushchenko M, Seki T, Larsson EA, Dan C. et al. Monitoring drug target engagement in cells and tissues using the cellular thermal shift assay. Science. 2013;341:84-7
59. Jafari R, Almqvist H, Axelsson H, Ignatushchenko M, Lundback T, Nordlund P. et al. The cellular thermal shift assay for evaluating drug target interactions in cells. Nat Protoc. 2014;9:2100-22
60. Jensen AJ, Martinez Molina D, Lundback T. CETSA: a target engagement assay with potential to transform drug discovery. Future Med Chem. 2015;7:975-8
61. Dou Q, Chen HN, Wang K, Yuan K, Lei Y, Li K. et al. Ivermectin induces cytostatic autophagy by blocking the PAK1/Akt axis in breast cancer. Cancer Res. 2016;76:4457-69
Author contact
![]() Corresponding author: Guang-Hong Tan (email: tanhohocom) or Hao-Fu Dai (email: daihaofuorg.cn) or Yue-Nan Li (email: liyuenan80com)
Corresponding author: Guang-Hong Tan (email: tanhohocom) or Hao-Fu Dai (email: daihaofuorg.cn) or Yue-Nan Li (email: liyuenan80com)