Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Methods
Results
Discussion
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2018; 8(16):4447-4461. doi:10.7150/thno.24284 This issue Cite
Research Paper
Aspirin cooperates with p300 to activate the acetylation of H3K9 and promote FasL-mediated apoptosis of cancer stem-like cells in colorectal cancer
Zhigang Chen1,2,*, Wenlu Li3,*, Fuming Qiu1,*, Qi Huang1, Zhou Jiang1, Jun Ye4, Pu Cheng1, Cho Low2, Yikun Guo2, Xinchi Yi2, Wenteng Chen5, Yongpin Yu5, YueHua Han4, Jun Wu1, Shenghang Jin1, Dong Kong2, ![]() , Jian Huang1,
, Jian Huang1, ![]()
1. Department of Surgical Oncology, Second Affiliated Hospital and Cancer Institute (Key Laboratory of Cancer Prevention & Intervention, National Ministry of Education, Provincial Key Laboratory of Molecular Biology in Medical Sciences), Zhejiang University School of Medicine, Hangzhou, China
2. Department of Neuroscience, Tufts University School of Medicine; Programs of Neuroscience and Cellular, Molecular and Development Biology, Tufts Sackler School of Graduate Biomedical Sciences, Boston, MA, USA
3. Department of Pharmacy, Second Affiliated Hospital, Zhejiang University School of Medicine, Hangzhou, China
4. Department of Gastroenterology, Second Affiliated Hospital, Zhejiang University School of Medicine, Hangzhou, China
5. College of Pharmaceutical Sciences, Zhejiang University, Hangzhou, China
*These authors contributed equally to this work.
Received 2017-12-10; Accepted 2018-6-24; Published 2018-8-7
Abstract

Cancer stem-like cells (CSCs) have been proposed as a key driving force of tumor growth and relapse in colorectal cancer (CRC), and therefore, they are promising targets for cancer therapy. Epidemiological evidence has suggested that the daily use of aspirin reduces overall mortality of CRC and the risk of distant metastasis. We investigated the effect and mechanism of aspirin on CSCs in CRC.
Methods: The ratio of CSCs was analyzed after aspirin treatment both in a cell model and patient samples. Chemically modified aspirin and immunoprecipitation were adopted to detect the target proteins of aspirin. A locus-specific light-inducible epigenetic modification system based on CRISPR technology was constructed to verify the causal relationship in these molecular events. In vivo characterization was performed in a xenograft model.
Results: We found that aspirin induces apoptosis in enriched colorectal CSCs, inhibits tumor progression, and enhances the anti-neoplastic effects of chemotherapeutic agents. Furthermore, aspirin directly interacts with p300 in the nucleus, promotes H3K9 acetylation, activates FasL expression, and induces apoptosis in colorectal CSCs. Notably, these effects of aspirin are absent in non-CSCs since H3K9 is hypermethylated in non-CSCs and the effects are not induced by other NSAIDs. In addition, aspirin can suppress oxaliplatin-enriched CSCs and serve as an adjuvant therapy.
Conclusions: Taken together, we revealed a unique epigenetic and cox-independent pathway (p300-AcH3K9-FasL axis) by which aspirin eliminates colorectal CSCs. These findings establish an innovative framework of the therapeutic significance of aspirin.
Keywords: Aspirin, FasL, apoptosis, cancer stem-like cells, colorectal cancer
Introduction
Colorectal cancer (CRC) is the third most common malignant disease, estimated to affect approximately 135,430 people and causes more than 50,260 deaths in the United States in 2017 [1]. The majority of deaths from colorectal cancer are associated with tumor cell metastasis and resistance to chemotherapy [2, 3]. Numerous studies have suggested that cancer stem-like cells (CSCs) mediate resistance to conventional therapies, drive tumor cell metastasis, and promote tumor recurrence in CRC [4-6]. Therefore, CSCs represent promising targets for effective prevention and treatment against colorectal cancer [7]. However, efficient and specific drugs for use in the clinic are still lacking.
Aspirin is a non-steroid anti-inflammatory drug (NSAID) with a number of indications including analgesia and cardiovascular prophylaxis. Epidemiological studies have suggested that the daily use of aspirin reduces the risk and overall mortality of CRC [8-10]. This favorable outcome has been reported to be related to aspirin therapy after diagnosis of colorectal cancer [11-13]. In addition, the daily use of aspirin (≥75 mg) was also observed to reduce the risk of distant metastasis in subsequent follow-ups of CRC patients without initial metastasis [14]. Thus, these studies indicate that aspirin can serve as a promising adjuvant therapy in CRC. Considering the major roles of CSCs in driving the incidence, mortality, and metastasis of CRC, it prompts us to know whether aspirin exerts an anti-cancer effect by targeting CSCs. It had been reported that aspirin inhibited colosphere formation in SW620 in CRC [15]. However, the evidence from patients with aspirin therapy is still lacking and the mechanism by which aspirin inhibits CSCs remains obscure.
Multiple models of action have been proposed to explain the anti-carcinogenic effects of aspirin in normal tumor cells [16]. The prostaglandin system and the cyclooxygenase enzyme COX-2 that oxidize arachidonic acid to prostaglandin, which are the central mechanisms of aspirin, are believed to be involved [17-20]. Other COX-2-independent mechanisms of the anti-neoplastic effects of aspirin have also been reported, such as inhibition of Wnt/β-catenin, and mTOR pathways [21-25] and activation of the AMPK pathway [25]. Finally, aspirin differs from other NSAIDs and can acetylate multiple cellular molecules including p53, histones, and immunoglobulins [26-30], which could also contribute to its anti-cancer mechanisms. Among these aforementioned mechanisms of action, how aspirin-induced anti-neoplastic effects are mediated in CSCs and its target still needs to be investigated.
In this study, we aimed to test the effect of aspirin on CSCs, decipher the mechanism behind these effects and demonstrate its potential to serve as an adjuvant therapy in CRC.
Methods
Reagents
Aspirin (Sigma) was dissolved in water using 10 N NaOH, and the pH was adjusted to 7.0. Secondary antibodies containing anti-rabbit IgG or anti-mouse conjugated to horseradish peroxidase, mammalian protein extraction reagent (MPER) lysis buffer, Halt protease inhibitor cocktail, and stripping buffer were obtained from Pierce (Rockford, IL, USA).
Primary colon cancer cell preparation and cell lines
The P1and P2 cells were primary cancer cells isolated from human colon adenocarcinoma specimens obtained during surgical procedures, which was approved by the Research Ethics Board at Second Affiliated Hospital of Zhejiang University School of Medicine. The preparation of primary tumor cells was performed as described previously [4, 31]. P1 cancer cells were cultured in RPMI-1640 (Invitrogen, Grand Island, NY) medium containing 10% FBS. P2 cancer cells were maintained in Dulbecco's modified Eagle's medium (Invitrogen, Grand Island, NY) with 10% FBS. Colon cancer cell line HT29 was purchased from the American Type Culture Collection and maintained in McCoy's 5A (Invitrogen, GrandIsland, NY) medium containing 10% FBS at 37 °C and 5% CO2.
Tumorsphere-forming assay
The tumorsphere-forming assay was performed as follows: cells were collected and washed to remove serum and then suspended in ultralow-attachment flasks in serum-free medium (SFM) (DMEM/F12 containing nonessential amino acids, antibiotic-antimycotic, N2 supplement (Invitrogen), B27 supplement (Invitrogen), heparin (4 μg/mL; Sigma), epidermal growth factor (20 ng/mL; Invitrogen), and basic fibroblast growth factor (10 ng/mL; Invitrogen)) at 37 ° C and 5% CO2.
Xenograft mouse model
Six-week-old female Balb/c athymic nude mice were purchased from and bred in defined conditions at the Laboratory Animal Research Center of Zhejiang Chinese Medicine University with the permission from the local animal care and ethical committee. Equal numbers (1×105) of CSCs were suspended in 100 μL Matrigel diluted to 1:1 in serum-free DMEM/F-12 and subcutaneously injected into the left rear flank of each mouse. 10 days after cell injection, the tumors reached a palpable size. A total of 16 mice were randomly assigned to receive either (1) control treatment or (2) aspirin treatment. Aspirin was injected once a day by intraperitoneal injection at a dosage of 20 mg/kg/body weight, which is equivalent to a daily human dosage of 80-110 mg. (3) Oxaliplatin treatment (once a week, 2 mg/kg/body weight) or (4) oxaliplatin combined aspirin treatment. Oxaliplatin was injected once a week by intraperitoneal injection at a dosage of 2 mg/kg/body weight. Aspirin was injected once a day by intraperitoneal injection at a dosage of 20 mg/kg/body weight. All groups received treatments for 4 weeks, tumors were measured by a slide gauge every 3 days and their volume was calculated as length×width×width/2. All mice were then euthanized by cervical dislocation.
Design and construction of plasmids in locus-specific histone acetylation
The SID4X-dCas9 and CIB1-dCas9 were constructed by replacing FokI in the FokI-dCas9 plasmid (Addgene52970) [32] with SID4X and CIB1 cDNA fragments amplified by PCR from LITE1.0_pAAV_hSyn_CRY2PHR-NLS-SID4X_2A_phiLOV2.1_WPRE_bGHpA plasmids (Addgene47454) and LITE2.0_pAAV_hSyn_TALE(Grm2)-GS-CIB1(NLS*,∆318-334)_WPRE_bGHpA plasmids (Addgene47456) [33]. The primers used for the above PCR amplifications were as follows: SID4X-forward: 5'-GACGATGACAAGATGGCCCCCAAGAAGAAGAGGAAGGTGGGCATTCACCGCGGGGTACCTGGAGGTTCTGGATCCATGAACATCCAGATGCTGCTG; SID4X-reverse: 5'-AGCTAAACCAATAGAATACTTTTTATCACTTTCGGGTGTGGCGGACTCTGAGGTCCCGGGAGTCTCGCTGCCGCTGGGCAGCATAGAGGCATAGCC; CIB1-forward: 5'-GACGATGACAAGATGGCCCCCAAGAAGAAGAGGAAGGTGGGCATTCACCGCGGGGTACCTGGAGGTTCTGGATCCAACGGCGCGATTGGTGGGGATTTG; CIB- reverse: 5'-AGCTAAACCAATAGAATACTTTTTATCACTTTCGGGTGTGGCGGACTCTGAGGTCCCGGGAGTCTCGCTGCCGCTACTGGAAGTATTCACCTGCTG.
gRNA-SID4X-dCas9 and gRNA-CIB1-dCas9 were then generated by introducing a chimeric gRNA cassette into the SID4X-dCas9 or CIB-dCas9 plasmids following PCR amplification of the px330 plasmid (Addgene42230). The gRNAs vectors were designed and further constructed according to a published protocol [34]. Briefly, a pair of oligos targeting the FasL promoter sequence were annealed and phosphorylated and then further cloned into the BbsI sites of px330. The primer sequences for gRNA cloning were as follows:
gRNA1 forward: CACCGCATAGCCTACTAACCTGTT, gRNA1 reverse: AAACAACAGGTTAGTAGGCTATGC; gRNA2 forward: CACCGTAGGCTATGCTCACCTTCC, gRNA2 reverse: AAACGGAAGGTGAGCATAGCCTAC; gRNA3 forward: CACCGACAGCAACTGAGGCCTTGA, gRNA 3 reverse: AAACTCAAGGCCTCAGTTGCTGTC; gRNA4 forward: CACCGGCTGTTATCAGAAAATTGT, gRNA4 reverse: AAACACAATTTTCTGATAACAGCC.
Light-inducible, locus-specific histone modification
Light-induced histone modification was performed similarly to a previous report [33, 35]. Briefly, cells were illuminated using a custom-built LED array aligned to a 6-well cell culture plate. LEDs were driven by a waveform generator (Rigol DG1022U) and powered by a DC power supply (Arksen 305D). Illumination was measured using a Thorlabs PM200 Power Meter and a S120C Power Sensor. The temperature inside the wells was measured using BMDS wireless temperature probes. Plates containing cells incubated in the dark were wrapped in aluminum foil. The following stimulation parameters were used for experiments: 466 nm, 5 mW/cm2 for 24 h. Pulses were delivered at 0.067 Hz with a duration of 7% corresponding to 1 s pulses.
Immunostaining
Tissue sections (5 μm) were deparaffinized, rehydrated, and treated with 3% hydrogen peroxide, followed by antigen retrieval in boiling 0.1 M citrate (pH 6.0) buffer for 10 min twice. The sections were then blocked with 20% goat/rabbit serum for 30 min. Immunostaining was performed as previously described [36]; antibodies against ALDH1 (1:100; Abcam,ab195255) or DLCK1(1:100; Abcam,ab31704 ) were used. The ratio of positively stained cells to tumor cells was scored and reported as the mean ± SEM.
Clinical samples
Paraffin specimens of CRCs from 18 patients who were taking aspirin (100 mg/day) and 20 patients who were not taking aspirin were obtained from the Pathology Department of the Second Affiliated Hospital, Zhejiang University School of Medicine. The pathologic type was adenoma and verified by the pathologist. All tissue samples and the experimental protocol were approved by the Review Board of the Second Affiliated Hospital of Zhejiang University, and written informed consent was obtained for each patient.
Statistics
Statistical analysis was performed using GraphPad Prism software.
Results
Aspirin eliminates colorectal cancer stem-like cells
To test the effects of aspirin treatment on colorectal CSCs in human patients, we recruited 18 patients with CRC who were taking 75-100 mg aspirin daily and 20 patients with CRC who were not taking aspirin. The clinical characteristics of the patients are shown in Table S1. We found that the percentage of stem cell marker-positive cells (ALDH1+, DLCK1+) in the patients taking aspirin decreased by almost 18-fold (from 18% to 1%) (Figure 1A) and 10-fold (from 10.6% to 1.3%) (Figure 1B). In addition, the ratios of lymph node metastasis (N1/N2) and advanced CRC (III/IV) were significantly decreased in the patients taking aspirin (p=0.041 and p=0.007, respectively) (Figure S1C-D). These results indicate that the CRC patients taking aspirin had a lower percentage of CSCs and a better prognosis.
Aspirin treatment eliminates colorectal CSCs. (A-B) Representative images (left) and quantification (right) of immunostaining assays used to detect the percentage of colorectal CSCs (ALDH1+, DLCK1+) in tumor cells in the paraffin specimens of patients. Scale bar indicates 100 μm. CRC: colorectal cancer. The results are presented as the mean ± SEM, n=3. *, p<0.01, unpaired t-test. (C-D) Tumorsphere-forming assay. Representative images (E) and quantification (F) of TCs (tumor spheres) formed from three colorectal cancer cell lines (HT29, P1, or P2) following 14-day treatment with aspirin (Asp) at the indicated concentrations. Tumor spheres formed from the adherent cells (ACs) of each cell line were also included. The results are presented as the mean ± SEM, n=3. *, p<0.01, one-way ANOVA. (E) The activity of ALDH1 analyzed using the ALDEFLUOR assay; diethylaminobenzaldehyde (DEAB) is a negative control, which was used to inhibit the reaction of ALDH with the ALDEFLUOR reagent. ALDH1-positive cells and their average percentages in the total cell counts are indicated in each panel (mean ± SEM, n=3). (F) Quantitative PCR (qPCR) results from HT29 and P1 ACs, TCs, or TCs with 2-day Asp treatment (5 mM) (mean ±SEM, n=3, normalized to Gapdh mRNA expression). *, p<0.01, one-way ANOVA compared to TCs without Asp treatment. (G) Tumor growth curve from xenograft assays following subcutaneous injection of 1×105 TCs of HT29 cells into 6-week-old female nude mice (mean ± SEM, n=6). In vivo effects of Asp analyzed by i.p. injection of 20 mg/kg Asp following the seeding of TCs. *, p<0.01, two-way ANOVA compared to untreated TCs at the indicated time points. (See also Figure S1, Table S1 and Table S2)
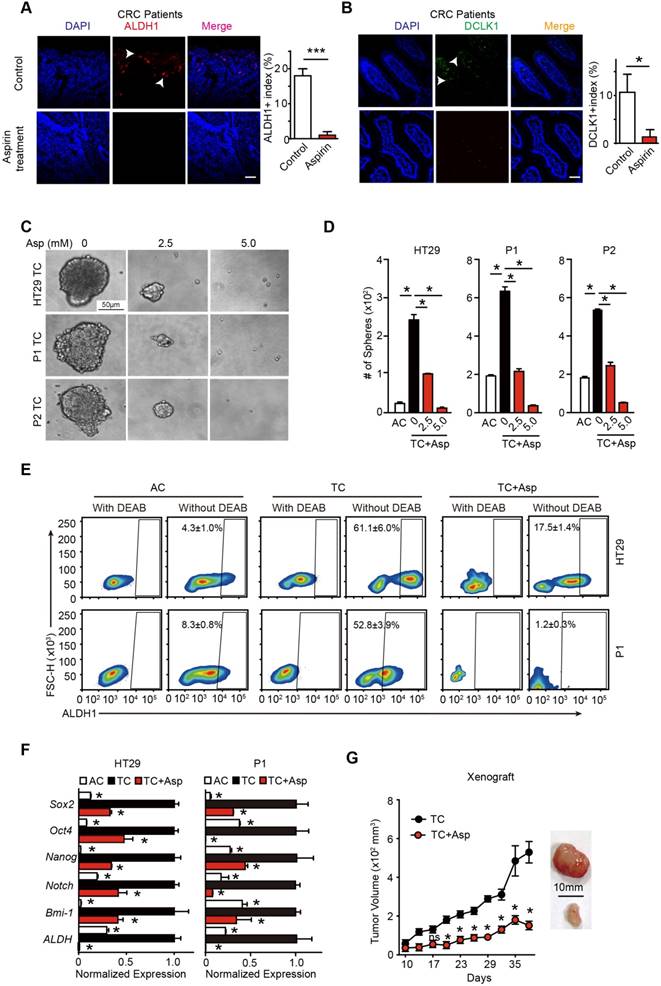
To determine the effects of aspirin treatment on colorectal CSCs, 3 different adherent cells (ACs) were incubated in serum-free medium. A small percentage of cells are able to form tumor spheres, and thus, are termed tumorsphere cells (TCs) (Figure 1C and Table S1). In accordance with our prior characterization [31], TCs exhibited enhanced tumor sphere formation ability (Figure 1D), highly expressed the major stem cell markers (Figure 1E and Figure S1A) and showed higher tumorigenicity in vivo compared with their respective ACs (Figure S1B). These results suggest that TCs fulfill the characteristics of CSCs. Then, we treated TCs with different concentrations of aspirin, and found that aspirin markedly reduced both the number and the diameter of the tumor spheres in a dose-dependent manner (Figure 1D). Secondly, aspirin decreased the expression of stem genes (Sox2, Oct4, Nanog, Notch1, Bmi-1, and ALDH1) (Figure 1F). Thirdly, we used the ALDEFLUOR assay to detect cells with ALDH activity in each group (Figure 1E) and found that aspirin significantly decreased the ratio of ALDH1 high activity CSCs from HT29 or P1 cell lines. In addition, the Lgr5+ and CD166+ expressing cell population was significantly enriched in TCs and greatly reduced following 14 days of incubation with 5 mM aspirin (Figure S1A). Limiting dilution assay also showed that aspirin treatment notably decreased the population of CSCs (Table S2). Lastly, we found that the tumor volumes significantly decreased in the groups administered aspirin (20 mg/kg/body weight, which is equivalent to a daily human dosage of 80-110 mg) (Figure 1G). Taken together, these results demonstrate that aspirin exerts inhibitory effects on colorectal CSCs.
Aspirin induces apoptosis in CSCs
We next compared the inhibitory effects of aspirin in ACs and TCs and found that aspirin treatment decreased cell viability of both ACs and TCs in time- and dose-dependent manners (Figure 2A). Following incubation with aspirin, there were significantly fewer viable TCs than ACs at the tested time points. In addition, high ALDH1 activity positive cells were markedly decreased following 5 mM aspirin treatment and were nearly completely depleted at 10 mM aspirin (Figure 2B). These results demonstrate that aspirin could inhibit both colorectal CSCs and non-CSCs, but colorectal CSCs are more vulnerable to aspirin.
Aspirin selectively induces apoptosis in CSCs. (A) Proliferation analysis of the survival of cells following 24 and 48 h exposure to Asp at the indicated concentrations. The results of Asp treatment of TCs from HT29 and P1 cell lines are presented (mean ±SEM, n=3). *, p<0.01, one-way ANOVA. (B) ALDH1 expression analyzed via FCM assays of ACs from HT29 and P1 cell lines, following 2-day treatment with Asp at the indicated concentrations. ALDH1 immunoreactive cells and their average percentages of the total cell counts are indicated in each panel (mean ± SEM, n=3). (C-D) TUNEL assay. Representative images (C) and quantification (D) of apoptotic cells of TCs from HT29 or P1 cell lines following 2-day treatment with Asp at the indicated concentrations. Scale bar in (C) indicates 50 μm. (D) The results are presented as the mean ± SEM, n=3. *, p<0.01, one-way ANOVA. (E) Cell cycle distributions analyzed via FCM assays of HT29 ACs or TCs following treatment with 5 mM Asp at the indicated time points. Sub-G1 was designated the apoptotic proportion and is listed in each panel (mean ± SEM, n=3). (See also Figure S2)
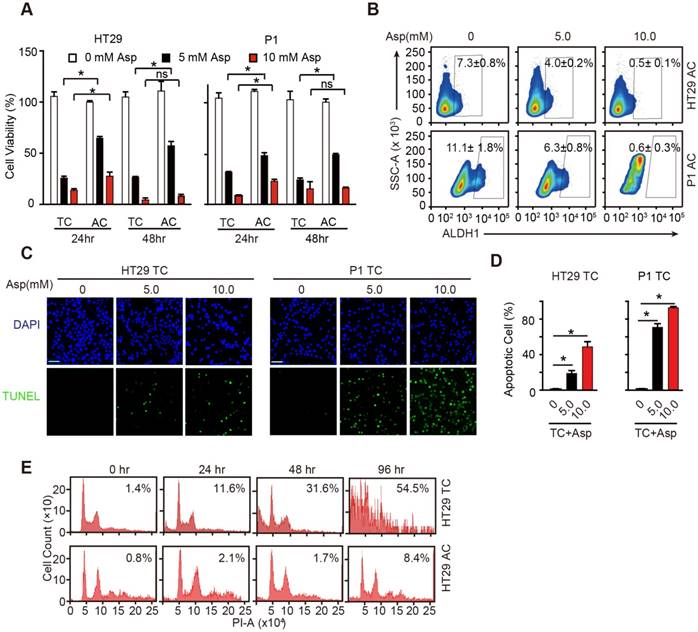
To better elucidate the inhibition of aspirin on CSCs, we performed TUNEL assays to investigate whether aspirin inhibits TCs by inducing apoptosis, which has been suggested to be responsible for aspirin-induced cell death [37]. TUNEL-positive cells were markedly increased in TCs following incubation with aspirin (Figure 2C-D). We next compared aspirin-induced apoptosis of TCs with that of ACs. Following 5 mM aspirin treatment, more apoptotic TCs (54.5%) were readily detected than ACs (8.4%) following 96 h treatment (Figure 2E). The results in Figure S2A-B also showed that less apoptosis was observed in ACs than TCs (Figure 2C-D) after 5 mM aspirin treatment for 48 h. These data demonstrate that aspirin inhibits TCs by inducing apoptosis, and TCs are more vulnerable to aspirin-induced apoptosis than regular ACs.
Aspirin-induced apoptosis in CSCs is mediated by FasL
To determine the apoptotic mechanisms, we screened a series of genes that have been reported to mediate aspirin-induced apoptosis, including Bcl-2, Bax, Fas, FasL, caspase-3, caspase-8 and caspase-9 [37-39]. Among these genes, FasL, Fas, caspase-3 and caspase-8 were significantly increased in TCs (Figure 3A). Interestingly, these apoptotic genes were largely suppressed in ACs (Figure 3B). In the flow cytometry assay, we also found FasL was greatly increased in TCs but not in ACs (Figure 3C), while Fas was not markedly affected (Figure 3D). We then assessed whether aspirin activates downstream molecules of the FasL pathway. As shown in Figure S3A, cleaved caspase-3 and caspase-8 proteins were markedly increased in TCs. Thus, downstream molecules of the FasL pathway were highly activated in colorectal CSCs following aspirin treatment. Next, we determined whether FasL is required for aspirin-induced apoptosis. TCs of HT29 cells were treated with 5 mM aspirin in the presence or absence of neutralizing antibody Nok-1. As indicated by cell viability assays (Figure 3E), flow cytometry (Figure 3F), and TUNEL assays (Figure S3B), aspirin-induced apoptosis in TCs was significantly attenuated by Nok-1, suggesting that aspirin-induced apoptosis in CSCs requires the activation of FasL.
Aspirin-induced apoptosis is mediated by FasL. (A-B) qPCR results of HT29 and P1 TCs (A) or ACs (B) following 2-day treatment with 5 mM Asp (mean ±SEM, n=3, normalized to Gapdh mRNA expression). *, p<0.01, one-way ANOVA. The control is set to 1 for every gene. (C-D) FasL and Fas expression in HT29 TCs (C) or ACs (D) analyzed via FCM assays. Immunoreactive cells and their average percentages of the total cell counts are indicated in the histogram and quantified (mean ± SEM, n=3). *, p<0.01, unpaired t-test. (E) Proliferation analysis of the survival of HT29 and P1 TCs following 48 h exposure to 5 mM Asp in the presence of an isotype-matched control antibody or FasL-blocking antibody (Nok-1) at the indicated concentrations (mean ±SEM, n=3). *, p<0.01, one-way ANOVA. (F) Representative results of HT29 TCs analyzed via FCM assays following 48 h exposure to 5 mM Asp with a control antibody or 2 μg/mL Nok-1. Sub-G1 was designated as the apoptotic proportion. Scale bar indicates 50 μm. (G-J) TUNEL assay (G, I) and FCM assay (H, J) for analyzing apoptotic cells and FasL expression, respectively, from HT29 TCs in the presence of Asp (5 mM), Asp (5 mM) and PGE2 (1 μM) co-treatment, and siRNA against COX-2 or NS-398 (75 μM). The results are presented as the mean ± SEM, n=3. *, p<0.01, one-way ANOVA. (See also Figure S3)
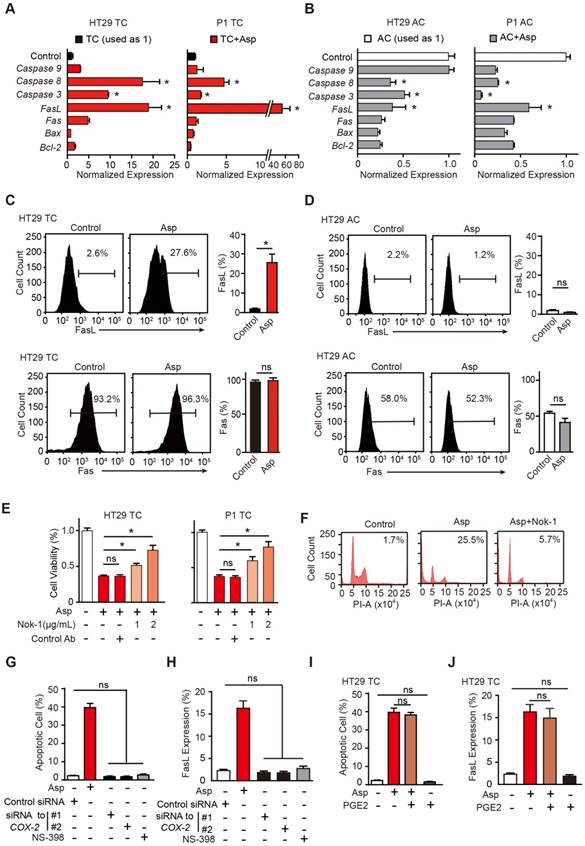
We next questioned whether aspirin stimulates the expression of FasL and induces apoptosis in a COX-2-dependent pathway (Figure S3C). COX-2 mRNA and enzyme activity were tested in both ACs and TCs, and no significant differences were found (Figure S3D-F). As shown in Figure S3C, siRNAs against COX-2, a selective inhibitor of COX-2, NS-398, or a substrate of the COX-2 pathway, PGE2, were used to assess both the sufficiency and necessity of COX-2 signaling in aspirin-induced apoptosis. The efficiencies of NS-398 and PGE2 were assessed in Figure S3G. Inhibition of COX-2 with either siRNAs or NS-398 failed to induce apoptosis or FasL expression (Figure 3G, H and Figure S3H-I). In addition, aspirin-induced apoptosis or FasL upregulation was not attenuated by the addition of PGE2 (Figure 3I-J and Figure S3H-I). These results suggest that aspirin induces apoptosis and FasL expression in colorectal CSCs independent of COX-2 inhibition.
Aspirin promotes the acetylation of histone H3
We compared the effects of aspirin on stimulating FasL expression in CSCs (Figure 4A) with other NSAIDs including salicylic acid, ibuprofen, sulindac, and indomethacin after 48 h treatment (Figure S4A). Interestingly, aspirin, but not any of the other NSAIDs, stimulated FasL expression. Compared to other NSAIDs, aspirin has a unique structural feature and possesses an acetyl moiety (Figure 4B and Figure S4B), which led us to investigate whether the acetylation of aspirin plays a role in aspirin-induced effects. Given that FasL is exclusively induced by aspirin, that acetylation of histone H3 in the FasL promoter region enhances its transcription in endothelial cells, and that aspirin acetylates histone proteins [16], we assessed whether aspirin induces FasL expression by acetylating histones in CSCs. We found histone deacetylase inhibitor trichostatin A (TSA, 1 μM) significantly increased FasL expression in HT29 TCs (Figure 4C-D) but not in ACs (Figure 4E-F), suggesting that histone acetylation in CSCs drives FasL expression. Furthermore, we observed that H3K9 was significantly acetylated following aspirin treatment (Figure 4G). Next, chromatin immunoprecipitation (ChIP) assay showed that aspirin treatment enriched the acetylated histone H3K9 (ac-H3K9) on the promoter of FasL, and this effect was only observed in HT29 TCs but not ACs (Figure 4H).
Aspirin promotes the acetylation of histone H3. (A) FCM assay of FasL expression in HT29 TCs in the presence of Asp (5 mM), salicylic acid (5 mM), ibuprofen (1 mM), sulindac (100 μM) or indomethacin (600 μM). The results are presented as the mean ± SEM, n=3. *, p<0.01, one-way ANOVA. (B) Chemical structure of aspirin. The red square indicates its acetyl moiety. (C-F) Expression of FasL in HT29 TCs (C-D) or ACs (E-F) in the presence of Asp (5 mM) or TSA (1 μM) analyzed via FCM assays. FasL immunoreactive cells and their average percentage of the total cell counts are indicated (C, E) and quantified (D, F) (mean ± SEM, n=3). *, p<0.01, one-way ANOVA. (G) Immunoblotting analysis with anti-acetylated antibodies against H2AK5, H2BK5, H3K9, or H4K8 of HT29 or P1 TCs after 48 h Asp treatment. (H) In vitro ChIP assay using antibodies against Ac-H3K9 followed by qPCR to detect a fragment on the promoter of the FasL gene of HT29 TCs or ACs treated with 5 mM Asp (mean ± SEM, n=3, normalized to the expression level of 10% input). *, p<0.01, unpaired t-tests. (I) Immunoblotting against Mono-Me-H3K9 of ACs or TCs from HT29 and P1 cell lines. Immunoblotting against Ac-H3K9. (J) FCM assay of FasL expression in HT29 ACs in the presence of Asp (5 mM), 5-AZA (5 μM), or both Asp (5 mM) and 5-AZA (5 μM). The average percentages of FasL immunoreactive cells in the total cell counts were quantified (mean ± SEM, n=3). *, p<0.01, one-way ANOVA. (See also Figure S4)
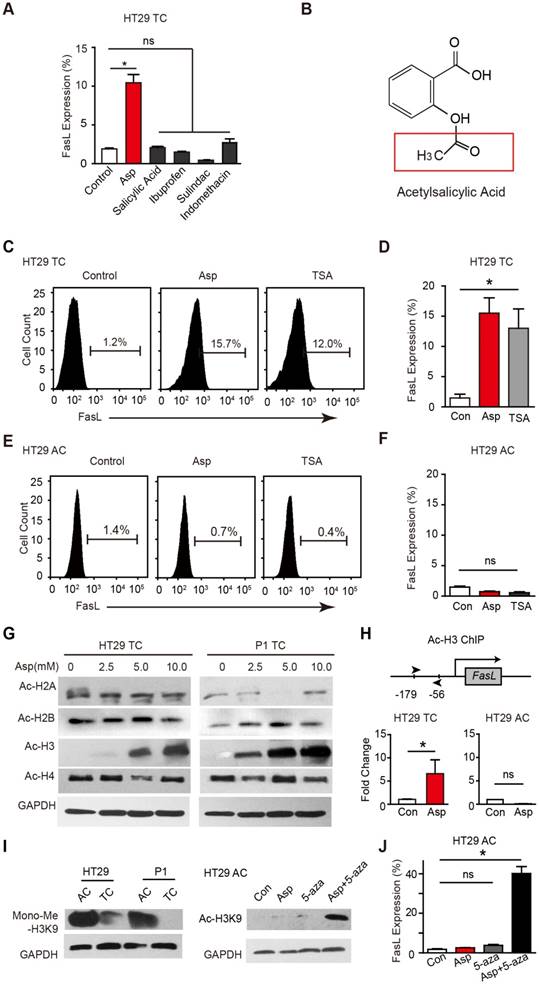
Of interest, it has been proposed that histone H3 is hypermethylated in ACs [40], raising the possibility that the hypermethylation of H3K9 in ACs prevents aspirin-induced FasL expression. Indeed, when monomethylation of H3K9 was evaluated (Figure 4I), the level of methylated H3K9 in ACs was found to be notably higher than that in TCs. In ACs, aspirin promoted the acetylation of H3K9 only when a methyltransferase inhibitor 5-aza-dC was added (Figure 4I). In accordance with the observed acetylation, FasL expression was also induced in ACs with aspirin and 5-aza-dC co-treatment (Figure 4J). These results demonstrate that aspirin induces FasL expression in TCs by promoting histone H3K9 acetylation and that this effect is occluded in ACs due to hypermethylation of the same residue.
Acetylation of histone H3 activates FasL transcription and promotes apoptosis
Aspirin is known to acetylate multiple substrates from nucleic acids to proteins [41]. To assess whether aspirin directly interacts with and acetylates histone H3, we chemically conjugated biotin to aspirin (Figure S5A-E) and synthesized biotinylated aspirin (Figure 5A, Biotin-Asp). The cell viability assays suggested that Biotin-Asp retained similar inhibitory effects as aspirin (Figure 5B). As shown in Figure 5C, co-localization of immunoreactive signals of biotin and DAPI demonstrated that aspirin is localized to nuclei. The co-IP these imaging data suggest an association of signals may be present between aspirin and histone H3 in the nucleus (Figure 5D).
Locus-specific analysis of aspirin-induced acetylation of histone H3K9. (A) Chemical structures of Biotin-Asp and Biotin-NH2. (B) Proliferation analysis of the survival of HT29 or P1 TCs following 48 h exposure to 5 mM Asp, Biotin-NH2, or Biotin-Asp (mean ±SEM, n=3). *, p<0.01, one-way ANOVA. (C) Immunocytochemical fluorescence staining with antibodies against biotin in HT29 TCs treated with 5 mM Biotin-NH2 or Biotin-Asp for 48 h. Scale bar indicates 10 μm. (D) In vitro IP assay. After 48 h treatment with 5 mM Biotin-NH2 or Biotin-Asp, HT29 TCs lysates were immunoprecipitated with anti-Biotin antibodies and immunoblotted with antibodies against Ac-H3K9 or GAPDH. (E) Schematics of locus-specific histone modification based on the CRISPR/dCas9 system (upper) and the associated gRNA-SID4X-dCas9 vector design (lower). (F) qPCR results of FasL mRNA from HT29 TCs transfected with the indicated plasmids (mean ±SEM, n=3, normalized to Gapdh mRNA expression) following 5 mM Asp treatment. *, p<0.01, one-way ANOVA. (G) Schematics of light-inducible locus-specific histone modification (upper) and the associated vectors (lower). (H) qPCR results of FasL mRNA from HT29 TCs transfected with the indicated plasmids (mean ± SEM, n=3, normalized to Gapdh mRNA expression); 5 mM Asp and 5 mW/cm2 466 nm blue light were applied as indicated. *, p<0.01, unpaired t-test. (See also Figure S5)
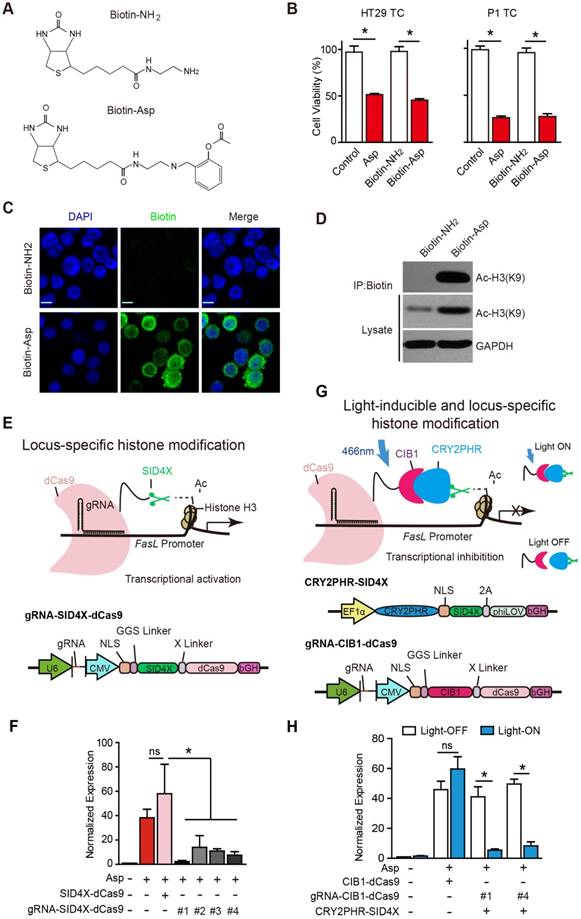
Although we observed that aspirin induces the acetylation of histone H3 on the promoter of FasL, the physiological significance of this specific modification is still undetermined given that traditionally used approaches, such as TSA, cause genome-wide alterations via histone modification. To assess the causal effects of aspirin-induced acetylation of histone H3, we modified recently developed programmable reagents based on transcription activator-like effectors (TALEs) [42] to achieve control of epigenetic modification. Briefly, we fused four concatenated mSin3 interaction domains (SID4X) that are able to reduce H3K9 acetylation with a catalytically dead Cas9 (dCas9) (Figure 5E). To allow SID4X-dCas9 protein to specifically bind to and remove histone acetylation on the promoter of FasL, four guide RNA (gRNA) sequences targeting different regions of the FasL promoter were designed and cloned into a plasmid containing SID4X-dCas9, driven by a U6 promoter (Figure 5E and Figure S5J). As shown in Figure 5F and Figure S5I, both aspirin-induced FasL expression and apoptosis were significantly attenuated by all four vectors expressing gRNAs.
To acutely modulate the acetylation of H3K9 on the FasL promoter, we further developed a light-inducible histone modification system by introducing a light-stimulated CRY2/CIB1 protein pair (Figure 5G). In this system, SID4X fused with CRY2PHR, a truncated light-sensitive cryptochrome 2 protein consisting of the photolyase homology region, was employed as previously described [33]. CIB1, which interacts with CRY2PHR upon light stimulation, was instead fused with dCas9. gRNA #1 and #4, which exhibited higher efficiency in guiding SID4X-dCas9 (Figure 5F), were cloned into the same plasmid and driven by a U6 promoter (Figure 5G). As shown in Figure 5H, transfection of the plasmids, i.e., CRY2PHR-SID4X, CIB1-dCas9, or gRNA-CIB1-dCas9, into HT29 TCs caused no significant changes in aspirin-induced expression of FasL when the cells were incubated in the dark (open bars). This system was stable under 24 h illumination, and 5 mW/cm2 466 nm blue light (Figure S5E-H). When the cells were illuminated for 24 h with 466 nm blue light, which induces the dimerization of CRY2PHR and CIB1 and, therefore, the anchoring of SID4X to the FasL promoter with the gRNA/dCas9 complex, aspirin-induced FasL expression was significantly attenuated (Figure 5H). Notably, similar light illumination of un-transfected HT29 TCs had no marked effects on FasL expression; light illumination also did not change aspirin-induced FasL expression in the absence of gRNA (Figure 5H). Taken together, these results demonstrate that specific acetylation of histone H3K9 on the promoter of FasL is directly required for the aspirin-induced effects in CSCs.
p300 is required for aspirin-induced acetylation of histone H3
The histone acetyltransferases (HATs) that are known to enhance H3K9 acetylation include p300, CREB-binding protein (CBP), p300/CBP-associated factor (PCAF), and GCN5L2. To determine which of the above HATs is required for aspirin-induced FasL, we designed siRNAs targeting p300, CBP, PCAF, and GCN5L2 and verified their efficiencies (Figure S6A-D) in HT29 TCs. As shown in Figure 6A-E, only the siRNA against p300 or a selective inhibitor against p300, C646, occluded aspirin-induced FasL expression. Similar results were also observed via flow cytometry (Figure S6F-G). These findings demonstrate that p300 but not PCAF, CBP or GCN5 is required for aspirin-induced FasL expression in CSCs.
p300 is required for aspirin-induced acetylation of histone H3. (A-D) FCM assay of FasL expression in HT29 TCs treated with 5 mM Asp in the presence of siRNA against p300 or its inhibitor C646 (10 μM) (A), or siRNAs against CBP (B), PCAF (C), or GCN5L2 (D) (mean ± SEM, n=3). *, p<0.01, one-way ANOVA. (E) In vitro ChIP assay with antibodies against p300 followed by qPCR to detect fragments on the promoter of the FasL gene in HT29 TCs treated with 5 mM Asp, siRNA against p300, or C646 (mean ± SEM, n=3, normalized to the expression level of 10% input). *, p<0.01, one-way ANOVA. (F) In vitro IP assay. After 48 h treatment with 5 mM Biotin-NH2 or Biotin-Asp, HT29 TC lysates were immunoprecipitated with anti-Biotin antibodies and immunoblotted with antibodies against p300 or GAPDH. (G) Diagram summarizing the difference between aspirin-induced effects in CSCs (upper) and non-CSCs (lower). (See also Figure S6)
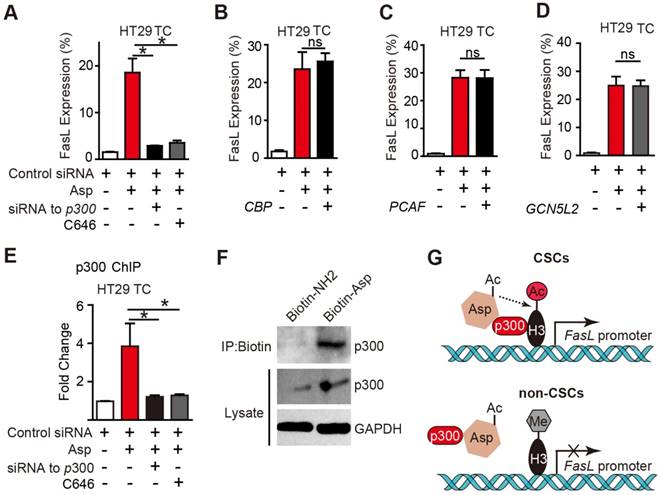
To determine whether p300 directly binds to the promoter region of FasL, a ChIP assay with antibodies against p300 was performed. As shown in Figure 6E, aspirin significantly increased the binding of p300 at the FasL promoter in HT29 TCs, and the binding was disrupted by either a siRNA against p300 or its inhibitor. A direct interaction between aspirin and p300 was revealed based on their co-IP (Figure 6F). Overall, these imaging data suggest an association of signals may be present between aspirin and p300, and p300 is recruited by aspirin in CSCs and is required for aspirin-induced acetylation of histone H3.
Aspirin suppresses oxaliplatin-enriched CSCs
To explore the potential of aspirin serving as an adjuvant therapy in CRC treatment, we treated the TCs and ACs with oxaliplatin, a first-line chemotherapeutic drug for colon cancer. In line with our prior findings, we found that TCs showed resistance to oxaliplatin compared to ACs, while 5 mM aspirin significantly sensitized oxaliplatin-induced cell death in TCs (Figure 7A-B), as evidenced by the reduced 50% inhibitory concentration (IC50) of oxaliplatin (Table S3). Given the different effects of aspirin on ACs and TCs, we expected that aspirin combined with oxaliplatin would exert a synergistic effect on inhibiting CRC. As shown in Figure 7C, both aspirin and oxaliplatin treatments decreased the number of viable cells, and the combination of aspirin and oxaliplatin induced a much greater reduction. The majority of tumor cells lose their CSC properties during tumor growth in vivo. It has been reported that oxaliplatin treatment can enrich the TC /CSC [6, 31]. We then employed oxaliplatin to enrich the CSCs and tested the effect of aspirin on them. We observed that the tumor volumes significantly decreased in the group of mice administered both aspirin and oxaliplatin compared with the group of mice treated with oxaliplatin only (Figure 7D). These results demonstrate that aspirin can inhibit oxaliplatin-enriched colorectal CSCs.
Aspirin suppresses oxaliplatin-enriched CSCs. (A-B) Proliferation analysis of the survival of cells following 48 h exposure to Oxa at the indicated concentrations. The results of ACs, TCs, or TCs with Asp and Oxa co-treatment (5 mM) from the HT29 (A) or P1 (B) cell lines are presented (mean ±SEM, n=3). (C) Proliferation analysis of the survived cells following 48 h treatment with saline (Control group), Oxa (20 μg/mL), Asp (5 mM), or both Oxa and Asp. ACs from both HT29 and P1 cell lines were used. The data are presented as the mean ±SEM. * p<0.01, one-way ANOVA. (D) Tumor growth curve from xenograft assays following subcutaneous injection of 1×105 HT29 TCs into 6-week-old female nude mice. The data are presented as the mean ± SEM and n=6 for each group. In vivo effects of Asp were analyzed by i.p. injection of both Asp (daily, 20 mg/kg body weight) and Oxa (once a week, 2 mg/kg/body weight) 10 days after seeding of TCs. The data are presented as the mean ± SEM. *, p<0.01 two-way ANOVA. (E) Representative images of TUNEL assays for detecting apoptotic cells from tumor tissues derived from HT29 TCs after Oxa treatment or both Oxa and Asp co-treatment. Scale bar indicates 20 μm. (F) Representative results of H&E staining and immunohistochemistry against ALDH1, FasL and ac-H3K9 on xenograft tumor sections from HT29 TCs mice treated with Oxa (once a week, 2 mg/kg/body weight) or Oxa combined with Asp (20 mg/kg body weight). Scale bar indicates 50 μm. (G) Quantification of immunohistochemistry on FasL or Ac-H3K9 positive cells in the tumors. Five visual fields were randomly selected and 100 cells in each visual field were counted. Data are shown as the percentage of the total number of positive cells. Values are presented as means ± SD, n=4. *P<0.05.
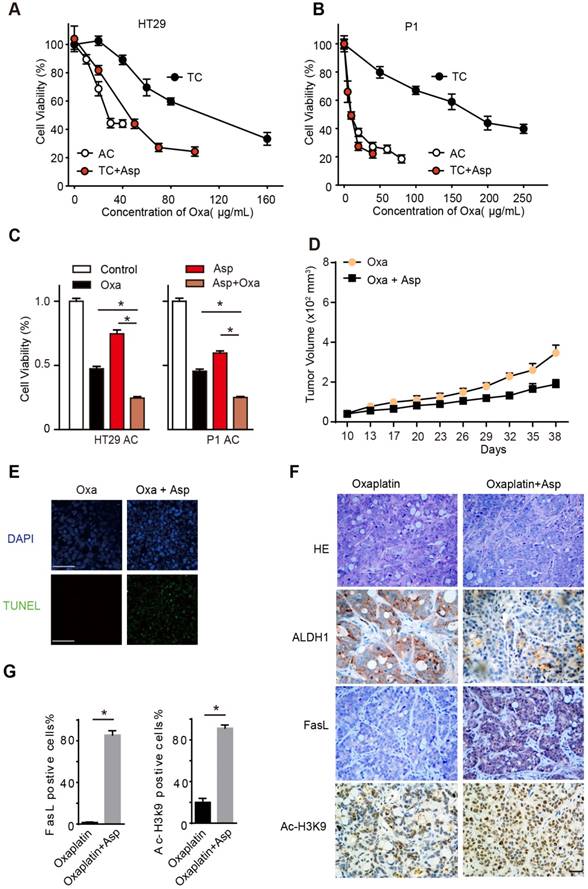
Then, we tested whether aspirin exerts these effects by activating Ac-H3K9-FasL signaling pathway in the tumor tissue. We observed that CSCs (ALDH-positive cells) were significantly enriched in the oxaliplatin treatment group, while the number of CSCs (ALDH-positive cells) decreased dramatically in the aspirin combined oxaliplatin treatment group (Figure 7F). In addition, aspirin combined oxaliplatin treatment induced significant apoptosis compared with the group of mice treated with oxaliplatin only (Figure 7E). The expression of FasL and Ac-H3K9 increased obviously in the group with aspirin combined oxaliplatin treatment (Figure 7F-G). Taken together, these results showed that aspirin suppresses oxaliplatin-enriched CSCs by activating Ac-H3K9-FasL signaling pathway; thus, it may serve as an adjuvant therapy in CRC treatment.
Discussion
CSCs are a small population of cells found within tumors that exhibit self-renewal, differentiation, and resistance to conventional therapies [7]. Their existence has been strongly implicated in carcinogenesis and tumor progression in CRC patients [43]. Many recent studies have focused on CSCs and have sought to develop strategies to selectively eliminate them, but efficient and specific drugs are still lacking. In the current study, we found that aspirin, a widely used NSAID, can serve as an efficient anti-CSC agent. We further demonstrated that aspirin inhibits CSCs by inducing FasL-mediated apoptosis. Importantly, we revealed a unique epigenetic pathway that underlies these effects, i.e., aspirin interacts directly with p300 and histone H3 in the nucleus, promotes the acetylation of H3K9 on the promoter of the FasL gene, and transcriptionally activates FasL expression. Notably, we found that the aforementioned effects of aspirin are absent in non-CSCs due to the hypermethylation of H3K9, which occludes their acetylation (Figure 6G). More importantly, aspirin suppresses oxaliplatin-enriched CSCs and sensitized CSCs to oxaliplatin. Our above findings have thus filled a major knowledge gap and identified aspirin as a unique agent that can target CSCs in CRC and serve as an adjuvant therapy besides traditional chemotherapy in CRC.
A few studies had suggested that aspirin decreases the number of CSCs in some types of cell lines [15], but in vivo evidence from patients is still lacking. In this study, we included CRC patients who were treated with or without aspirin at the second affiliated hospital of Zhejiang University School of Medicine from 2012 to 2016. Owing to the preventive effect of aspirin on CRC, we finally included 18 patients with CRC who were taking 75-100 mg aspirin daily. Thus, it is difficult to divide these patients into different subtypes according to sex, age and exposure time, or analyze the interindividual variability. We detected the ratio of CSCs in the CRC tissue from patients with or without aspirin treatment and found that patients taking aspirin had a lower percentage of CSCs and a better prognosis. However, more patients still need to be included in future studies to provide reliable evidence for the therapeutic effect of aspirin. Recently, an ongoing clinical trial (ADD-aspirin trial) has been initiated in patients with newly diagnosed cancers, including colorectal, gastroesophageal, breast, and prostate cancer, and is testing the efficacy of aspirin administration as an adjuvant for chemotherapy, which may offer us more evidence [44].
To test the effect of aspirin on CSCs in vitro, we treated TCs with different concentrations of aspirin (0-10 mM). It has been reported that low-dose aspirin (humans: 80-100 mg/day; mice: 20 mg/kg/day) can exert an antineoplastic effect. The concentration of aspirin used in this study is much higher than that in the human case. We chose the aspirin concentration range of 5-10 mM based on the following reasons. Firstly, aspirin was used daily for a long time in these clinical trials [10, 14, 45]. It was also reported that the ability of aspirin to reduce colorectal cancer incidence became evident only after regular use for more than 10 years, indicating that the effect was dependent upon both the dose and duration of aspirin intake. This suggested the importance of cumulative dosage as a determinant of aspirin efficacy in these settings [46]. Conversely, in the cell model, cancer cells were treated with aspirin once. Thus we adopted the concentration of aspirin which was widely used in previous studies [25, 47, 48]. Secondly, we treated the colorectal cancer stem-like cells with 1 mM daily for 5 days or 500 μM daily for 10 days, and found that lower concentration of aspirin exerted similar effects on colorectal cancer stem-like cells. Therefore, we used the most common concentration (5-10 mM) to test its effect in vitro. In the current study, we observed that aspirin can inhibit colorectal cancer stem-like cells and induce apoptosis. In addition, aspirin causes more reduction of TCs compared to ACs at the same time point. But, we did not offer more direct evidence that it selectively inhibits cancer stem cells in colorectal cancer, which needs more evidence in future studies. Inducing apoptosis is believed to be a superior strategy for eliminating CSCs. We found that the apoptosis induced by aspirin in CSCs requires the upregulation of FasL. These results demonstrate that FasL signaling is a promising route for eliminating CSCs and that the increased FasL expression following aspirin treatment could serve as a sensitive indicator of its efficacy. In addition, we found that Fas protein is significantly upregulated in CSCs. Fas is a prototypical death receptor that induces apoptosis upon ligand binding, and it is also a tumorigenic gene [49]. It has recently been observed that stimulation of Fas increases the number of CSCs in mouse colon cancer cell lines [50]. These findings suggest that Fas could contribute to the maintenance of CSCs and mediate their resistance to conventional therapy. Additionally, increased expression of Fas in CRC samples could also be a viable clinical marker for aspirin-sensitive therapies in CRC patients.
Whether aspirin suppresses tumor growth and progression by inhibiting COX-2 signaling has been extensively discussed. However, we revealed that aspirin inhibits colorectal CSCs in a COX-2-independent manner. Specifically, we found that 1) inhibition of COX-2 with either siRNAs or its selective inhibitor fails to reproduce the aspirin-induced effects; 2) the addition of PGE2, the functional product of COX-2 activation, fails to attenuate aspirin-induced effects; and 3) other NSAIDs that inhibit COX-2 activity fail to mimic aspirin's effects. These results strongly suggest that aspirin is engaged in a mechanism other than COX-2 inhibition to inhibit CSCs.
Compared to the chemical structures of other NSAIDs, aspirin exhibits a unique feature as it possesses an acetyl moiety, which makes it an efficient acetylating compound. Indeed, in past decades, a variety of bioactive molecules have been identified as substrates of aspirin-mediated acetylation. In addition to COX enzymes, aspirin can also acetylate DNA, RNA, metabolites, and proteins including p53, hemoglobin, transglutaminase, and histones [16, 26-30]. The biological consequences of aspirin-mediated acetylation, however, are largely undetermined. Especially, the acetylation of histone plays a vital role in epigenetic regulation of genes. In the current study, we found that aspirin could interact with acetyltransferase p300, and acetylate histone H3 at its H3K9 lysine residue. We further demonstrated that such acetylating modification of histone proteins is required for aspirin-induced, FasL-mediated apoptosis of CSCs, while it is prevented in non-CSCs due to hypermethylation of histone H3K9 residues. Importantly, to understand whether aspirin directly acetylates histones in the promoter region of FasL, we improved and established locus-specific histone modification (LSHM) and light-inducible LSHM systems based on CRIPSR/dCas9 technology. The results from these state-of-art approaches provide the critical evidence that histone acetylation mediates the anti-cancer effects of aspirin. Given the significance of histone acetylation in a variety of biological and cellular processes, the epigenetic modification of aspirin on histones could have broader meanings and provide a more universal basis for understanding the multifaceted effects of aspirin.
Finally, although aspirin has been reported to reduce the overall mortality of CRC and the risk of distant tumor metastasis [11, 12], treatments including aspirin as an adjuvant drug for CRC are still a topic of debate. The current study represents a critical advance toward clinical applications of aspirin in cancer therapy, and the molecular pathway revealed here could provide important guidelines regarding its clinical prescription. Our findings that aspirin exerts synergistic anti-neoplastic effects with oxaliplatin suggest that aspirin could be administered with traditional chemotherapy drugs in CRC treatments. Additional evidence from clinical studies is thus anticipated in the near future.
Abbreviations
CSCs: cancer stem-like cells; CRC: colorectal cancer; CRISPR: clustered regularly interspaced short palindromic repeats; NSAIDs: non-steroidal anti-inflammatory drugs; COX-2: cyclooxygenase-2; ACs: adherent cells; TCs: tumorsphere cells; ANOVA: one-way analysis of variance; TUNEL: terminal deoxynucleotidyltransferase dUTP nick end labelling; TALEs: transcription activator-like effectors; CBP: CREB-binding protein; PCAF: p300/CBP-associated factor; ChIP: chromatin immunoprecipitation; IC50: 50% inhibitory concentration; LSHM: locus-specific histone modification.
Supplementary Material
Supplementary experimental procedures, figures and tables.
Acknowledgements
The authors thank members in Drs. J.H and D.K.'s laboratories for helpful discussion. We thank Dr. X.W. He for his help with setup of the optic stimulation system.
Grant Support
J.H. is supported by the National Natural Science Foundation of China (NO.31471391, 81402440 and 81272672) and Zhejiang Provincial Program for the Cultivation of High-level Innovative Health Talents, Zhejiang provincial key subject of traditional Chinese medicine; D.K. is sponsored by NIH/NIDDK K01 DK094943, Charles Hood Foundation Grant, and Tufts CNR grant (P30 NS047243); Z.C. is supported by National Natural Science Foundation of China (NO.81502564 and NO.81402440), Natural Science Foundation of Zhejiang province (NO.LY16H160018, LY15H160037), the scholarship from China Scholarship Council (CSC) under the Grant CSC NO. 201706325003; F.Q. was sponsored by National Natural Science Foundation of China (NO.81472640).
Authors' Contributions
Z.C., D.K., and J.H. conceived the project, designed the experiments, analyzed the data and wrote the manuscript; Z.C. conducted experiments with assistance from W.L., F.Q., Q.H., Z.J., J.Y. and J.W.; W.C. and Y.Y. synthesized biotinylated Aspirin; Z.C. performed LSHM and light-inducible LSHM with help from C.L., Y.G. and X.Y.; Y.H., S.J., P.C. and W.C. provided advice and help in analyzing data.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2017. CA Cancer J Clin. 2017;67:7-30
2. Sonoshita M, Aoki M, Fuwa H, Aoki K, Hosogi H, Sakai Y. et al. Suppression of colon cancer metastasis by Aes through inhibition of Notch signaling. Cancer cell. 2011;19:125-37
3. Dallas NA, Xia L, Fan F, Gray MJ, Gaur P, van Buren G. et al. Chemoresistant colorectal cancer cells, the cancer stem cell phenotype, and increased sensitivity to insulin-like growth factor-I receptor inhibition. Cancer Res. 2009;69:1951-7
4. Wang YK, Zhu YL, Qiu FM, Zhang T, Chen ZG, Zheng S. et al. Activation of Akt and MAPK pathways enhances the tumorigenicity of CD133+ primary colon cancer cells. Carcinogenesis. 2010;31:1376-80
5. Li J, Yu B, Deng P, Cheng Y, Yu Y, Kevork K. et al. KDM3 epigenetically controls tumorigenic potentials of human colorectal cancer stem cells through Wnt/beta-catenin signalling. Nat Commun. 2017;8:15146
6. Wang K, Liu L, Zhang T, Zhu YL, Qiu F, Wu XG. et al. Oxaliplatin-incorporated micelles eliminate both cancer stem-like and bulk cell populations in colorectal cancer. Int J Nanomedicine. 2011;6:3207-18
7. Visvader JE, Lindeman GJ. Cancer stem cells: current status and evolving complexities. Cell stem cell. 2012;10:717-28
8. Flossmann E, Rothwell PM, British Doctors Aspirin T, the UKTIAAT. Effect of aspirin on long-term risk of colorectal cancer: consistent evidence from randomised and observational studies. Lancet. 2007;369:1603-13
9. Din FV, Theodoratou E, Farrington SM, Tenesa A, Barnetson RA, Cetnarskyj R. et al. Effect of aspirin and NSAIDs on risk and survival from colorectal cancer. Gut. 2010;59:1670-9
10. Rothwell PM, Wilson M, Elwin CE, Norrving B, Algra A, Warlow CP. et al. Long-term effect of aspirin on colorectal cancer incidence and mortality: 20-year follow-up of five randomised trials. Lancet. 2010;376:1741-50
11. McCowan C, Munro AJ, Donnan PT, Steele RJ. Use of aspirin post-diagnosis in a cohort of patients with colorectal cancer and its association with all-cause and colorectal cancer specific mortality. Eur J Cancer. 2013;49:1049-57
12. Chan AT, Ogino S, Fuchs CS. Aspirin use and survival after diagnosis of colorectal cancer. JAMA. 2009;302:649-58
13. Li P, Wu H, Zhang H, Shi Y, Xu J, Ye Y. et al. Aspirin use after diagnosis but not prediagnosis improves established colorectal cancer survival: a meta-analysis. Gut. 2015;64:1419-25
14. Rothwell PM, Wilson M, Price JF, Belch JF, Meade TW, Mehta Z. Effect of daily aspirin on risk of cancer metastasis: a study of incident cancers during randomised controlled trials. Lancet. 2012;379:1591-601
15. Moon CM, Kwon JH, Kim JS, Oh SH, Jin Lee K, Park JJ. et al. Nonsteroidal anti-inflammatory drugs suppress cancer stem cells via inhibiting PTGS2 (cyclooxygenase 2) and NOTCH/HES1 and activating PPARG in colorectal cancer. Int J Cancer. 2014;134:519-29
16. Alfonso L, Ai G, Spitale RC, Bhat GJ. Molecular targets of aspirin and cancer prevention. Br J Cancer. 2014;111:61-7
17. Thun MJ, Jacobs EJ, Patrono C. The role of aspirin in cancer prevention. Nat Rev Clin Oncol. 2012;9:259-67
18. Soumaoro LT, Uetake H, Higuchi T, Takagi Y, Enomoto M, Sugihara K. Cyclooxygenase-2 expression: a significant prognostic indicator for patients with colorectal cancer. Clin Cancer Res. 2004;10:8465-71
19. Baron JA, Sandler RS, Bresalier RS, Quan H, Riddell R, Lanas A. et al. A randomized trial of rofecoxib for the chemoprevention of colorectal adenomas. Gastroenterology. 2006;131:1674-82
20. Arber N, Eagle CJ, Spicak J, Racz I, Dite P, Hajer J. et al. Celecoxib for the prevention of colorectal adenomatous polyps. N Engl J Med. 2006;355:885-95
21. Bos CL, Kodach LL, van den Brink GR, Diks SH, van Santen MM, Richel DJ. et al. Effect of aspirin on the Wnt/beta-catenin pathway is mediated via protein phosphatase 2A. Oncogene. 2006;25:6447-56
22. He TC, Chan TA, Vogelstein B, Kinzler KW. PPARdelta is an APC-regulated target of nonsteroidal anti-inflammatory drugs. Cell. 1999;99:335-45
23. Yin MJ, Yamamoto Y, Gaynor RB. The anti-inflammatory agents aspirin and salicylate inhibit the activity of I(kappa)B kinase-beta. Nature. 1998;396:77-80
24. Stark LA, Reid K, Sansom OJ, Din FV, Guichard S, Mayer I. et al. Aspirin activates the NF-kappaB signalling pathway and induces apoptosis in intestinal neoplasia in two in vivo models of human colorectal cancer. Carcinogenesis. 2007;28:968-76
25. Din FV, Valanciute A, Houde VP, Zibrova D, Green KA, Sakamoto K. et al. Aspirin inhibits mTOR signaling, activates AMP-activated protein kinase, and induces autophagy in colorectal cancer cells. Gastroenterology. 2012;142:1504-15 e3
26. Jung SB, Kim CS, Naqvi A, Yamamori T, Mattagajasingh I, Hoffman TA. et al. Histone deacetylase 3 antagonizes aspirin-stimulated endothelial nitric oxide production by reversing aspirin-induced lysine acetylation of endothelial nitric oxide synthase. Circulation research. 2010;107:877-87
27. Marimuthu S, Chivukula RS, Alfonso LF, Moridani M, Hagen FK, Bhat GJ. Aspirin acetylates multiple cellular proteins in HCT-116 colon cancer cells: Identification of novel targets. Int J Oncol. 2011;39:1273-83
28. Sharma NP, Dong L, Yuan C, Noon KR, Smith WL. Asymmetric acetylation of the cyclooxygenase-2 homodimer by aspirin and its effects on the oxygenation of arachidonic, eicosapentaenoic, and docosahexaenoic acids. Mol Pharmacol. 2010;77:979-86
29. Bateman LA, Zaro BW, Miller SM, Pratt MR. An alkyne-aspirin chemical reporter for the detection of aspirin-dependent protein modification in living cells. J Am Chem Soc. 2013;135:14568-73
30. Alfonso LF, Srivenugopal KS, Bhat GJ. Does aspirin acetylate multiple cellular proteins? (Review). Mol Med Rep. 2009;2:533-7
31. Ni C, Wu P, Zhu X, Ye J, Zhang Z, Chen Z. et al. IFN-gamma selectively exerts pro-apoptotic effects on tumor-initiating label-retaining colon cancer cells. Cancer Lett. 2013;336:174-84
32. Guilinger JP, Thompson DB, Liu DR. Fusion of catalytically inactive Cas9 to FokI nuclease improves the specificity of genome modification. Nat Biotechnol. 2014;32:577-82
33. Konermann S, Brigham MD, Trevino AE, Hsu PD, Heidenreich M, Cong L. et al. Optical control of mammalian endogenous transcription and epigenetic states. Nature. 2013;500:472-6
34. Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F. Genome engineering using the CRISPR-Cas9 system. Nat Protoc. 2013;8:2281-308
35. Polstein LR, Gersbach CA. A light-inducible CRISPR-Cas9 system for control of endogenous gene activation. Nat Chem Biol. 2015;11:198-200
36. Huang EH, Hynes MJ, Zhang T, Ginestier C, Dontu G, Appelman H. et al. Aldehyde dehydrogenase 1 is a marker for normal and malignant human colonic stem cells (SC) and tracks SC overpopulation during colon tumorigenesis. Cancer Res. 2009;69:3382-9
37. Gao J, Liu X, Rigas B. Nitric oxide-donating aspirin induces apoptosis in human colon cancer cells through induction of oxidative stress. Proc Natl Acad Sci U S A. 2005;102:17207-12
38. Tian Y, Ye Y, Gao W, Chen H, Song T, Wang D. et al. Aspirin promotes apoptosis in a murine model of colorectal cancer by mechanisms involving downregulation of IL-6-STAT3 signaling pathway. Int J Colorectal Dis. 2011;26:13-22
39. Yan KH, Lee LM, Hsieh MC, Yan MD, Yao CJ, Chang PY. et al. Aspirin antagonizes the cytotoxic effect of methotrexate in lung cancer cells. Oncol Rep. 2013;30:1497-505
40. Tao H, Li H, Su Y, Feng D, Wang X, Zhang C. et al. Histone methyltransferase G9a and H3K9 dimethylation inhibit the self-renewal of glioma cancer stem cells. Mol Cell Biochem. 2014;394:23-30
41. Pinckard RN, Hawkins D, Farr RS. In vitro acetylation of plasma proteins, enzymes and DNA by aspirin. Nature. 1968;219:68-9
42. Maeder ML, Linder SJ, Cascio VM, Fu Y, Ho QH, Joung JK. CRISPR RNA-guided activation of endogenous human genes. Nat Methods. 2013;10:977-9
43. O'Brien CA, Kreso A, Ryan P, Hermans KG, Gibson L, Wang Y. et al. ID1 and ID3 regulate the self-renewal capacity of human colon cancer-initiating cells through p21. Cancer cell. 2012;21:777-92
44. Patrignani P, Patrono C. Aspirin and cancer. J Am Coll Cardiol. 2016;68:967-76
45. Fendrich V, Chen NM, Neef M, Waldmann J, Buchholz M, Feldmann G. et al. The angiotensin-I-converting enzyme inhibitor enalapril and aspirin delay progression of pancreatic intraepithelial neoplasia and cancer formation in a genetically engineered mouse model of pancreatic cancer. Gut. 2010;59:630-7
46. Chan AT, Ogino S, Fuchs CS. Aspirin and the risk of colorectal cancer in relation to the expression of COX-2. N Engl J Med. 2007;356:2131-42
47. Ying J, Zhou HY, Liu P, You Q, Kuang F, Shen YN. et al. Aspirin inhibited the metastasis of colon cancer cells by inhibiting the expression of toll-like receptor 4. Cell Biosci. 2018;8:1
48. Goel A, Chang DK, Ricciardiello L, Gasche C, Boland CR. A novel mechanism for aspirin-mediated growth inhibition of human colon cancer cells. Clin Cancer Res. 2003;9:383-90
49. Kleber S, Sancho-Martinez I, Wiestler B, Beisel A, Gieffers C, Hill O. et al. Yes and PI3K bind CD95 to signal invasion of glioblastoma. Cancer cell. 2008;13:235-48
50. Ceppi P, Hadji A, Kohlhapp FJ, Pattanayak A, Hau A, Liu X. et al. CD95 and CD95L promote and protect cancer stem cells. Nat Commun. 2014;5:5238
Author contact
![]() Corresponding authors: drhuangjianedu.cn (J.H.); dong.kongedu (D.K.).
Corresponding authors: drhuangjianedu.cn (J.H.); dong.kongedu (D.K.).