Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2019; 9(10):2882-2896. doi:10.7150/thno.32693 This issue Cite
Research Paper
The cell-penetrating FOXM1 N-terminus (M1-138) demonstrates potent inhibitory effects on cancer cells by targeting FOXM1 and FOXM1-interacting factor SMAD3
Zhenwang Zhang, Huitong Bu, Jingwei Yu, Yan Chen, Chaozhu Pei, Li Yu, Xiaoqin Huang, Guixiang Tan ![]() , Yongjun Tan
, Yongjun Tan ![]()
State Key Laboratory of Chemo/Biosensing and Chemometrics, College of Biology, Hunan Engineering Research Center for Anticancer Targeted Protein Pharmaceuticals, Hunan University, Changsha, Hunan 410082, China.
Received 2018-12-31; Accepted 2019-3-29; Published 2019-4-25
Abstract

Transcription factor FOXM1 is involved in stimulating cell proliferation, enhancing DNA damage repair, promoting metastasis of cancer cells, and the inhibition of FOXM1 has been shown to prevent the initiation and progression of multiple cancers and FOXM1 is considered to be an effective target for tumor therapeutic drug development. The N-terminus of FOXM1 has been found to prevent transcriptional activities of FOXM1 and to mediate the interaction between FOXM1 and SMAD3.
Methods: A recombinant FOXM1 N-terminal domain (1-138aa) fused with a nine arginine cell-penetrating peptide is produced with an E. coli expression system and named as M1-138. The effects of M1-138 on the proliferation, migration, and tumorigenic ability of cancer cells are analyzed in vitro with cell counting, transwell assays, and colony formation assays. Electrophoretic mobility shift assays (EMSAs) and Luciferase activity assays are used to test the DNA binding ability and transcriptional activity of transcription factors. The levels of mRNAs and proteins are measured by quantitative-PCR, Western blotting or Immunohistochemistry. The interactions among proteins are analyzed with Pull-down and Co-immunoprecipitation (Co-IP) assays. The nude mouse engrafted tumor models are used to test the inhibitory effects of M1-138 in vivo.
Results: M1-138 diminishes the proliferation and migration abilities of cancer cells through binding to FOXM1 and FOXM1-interacting factor SMAD3, and consequently attenuating FOXM1 transcriptional activities from both direct and indirect FOXM1-promoter binding mechanisms and interfering with the interaction between FOXM1 and SMAD3. Treatment of M1-138 prevents tumorigenicity of cancer cells and inhibits tumor growth in nude mouse xenograft models with no obvious signs of toxicity.
Conclusion: M1-138 is a promising drug candidate for the development of anti-cancer therapeutics targeting FOXM1 and SMAD3.
Keywords: Anticancer protein therapeutics, Transcription factor FOXM1, Targets for tumor suppression, Cell-penetrating peptide
Introduction
FOXM1 is a typical proliferation-associated transcription factor that belongs to the Forkhead box (FOX) transcription factor family [1]. From the perspective of gene function, FOXM1 is first identified as a protein that regulates cell cycle and cell proliferation [2]. FOXM1 is involved in the regulation of cell cycle-associated transcription of multiple genes, thereby controlling DNA replication and mitosis in cells, and the inhibition of FOXM1 effectively terminates the cell proliferation [3]. FOXM1 also participates in stimulating the expression of DNA damage repair genes [4]. In addition, FOXM1 is involved in cell stemness maintenance [5] and inhibition of FOXM1 disturbs the process of inducible pluripotent stem cell reprogramming [6]. Furthermore, FOXM1 is a key molecule that stimulates epithelial-mesenchymal transition in tumor cells and the inhibition of FOXM1 prevents cancer cell metastasis [7, 8].
The expression of FOXM1 is found to elevate in cancer cells, based on the gene expression analysis of various clinical tumor samples and its levels have been proposed as indicators for the diagnosis and prognosis of cancers [9]. The conditional knockout of FOXM1 in different mouse organs inhibits the development and progression of solid tumors, such as liver cancer, lung cancer, and colorectal cancer [10-12]. Since its multiple roles in regulating the operations of cancer cells, FOXM1 is considered to be an effective target for tumor therapeutic drug development [13]. This concept has been confirmed by our cancer gene therapy studies for liver cancer, breast cancer, and nasopharyngeal carcinoma, with an adenovirus-mediated specific interference of FOXM1 expression [14-16]. Small molecule drug screening for inhibiting FOXM1 has also progressed and small molecule compounds, such as Thiostrepton, antibiotic Siomycin A, and FDI-6, are selected to prevent the transcriptional activities of FOXM1 and consequently abolish the proliferation of cancer cells [17-19]. A single-strand DNA aptamer specific to the DNA binding domain (DBD) of FOXM1 has been obtained recently to act as a probe for FOXM1 detection and an inhibitor of FOXM1 transcriptional functions in cancer cells [20]. Besides, the cell-penetrating ARF26-44 peptide derived from the tumor suppressor protein p19ARF and the 9R-P201 peptide selected from a random dodecapeptide library against the FOXM1 DBD can inhibit the transcriptional activities of FOXM1 in liver cancer cells [21, 22]. However, there are no commercialized anti-cancer therapeutics developed directly against FOXM1 so far.
In terms of the mechanisms of FOXM1 stimulating transcription, FOXM1 has been found to activate gene expression by directly binding to the classic forkhead binding sequence RYAAAYA (FKH motif) that exists in the upstream of promoters of multiple cancer-related genes [23]. On the other hand, there is growing evidence that FOXM1 can be recruited to the promoters of proliferation-related genes by so called MuvB complex, especially during G2/M phase of cell cycle [24, 25]. The human MuvB complex consists of five proteins, LIN9, LIN37, LIN52, LIN54 and RBBP4; binds to the CHR (cell cycle genes homology region) motif (TTTGAA or TTTAAA) typically located within proximal promoters of target genes through LIN54; and acts as the core to control precisely timed transcription of cell cycle [26]. FOXM1 interacts with LIN9 in MuvB complex through the FOXM1 N-terminal domain (1-367 aa) and FOXM1 ChIP-seq data reveal that CHR sites and LIN9 binding sites are highly enriched at >60% of FOXM1-bound regions during the late cell cycle [25]. Obviously, FOXM1 can stimulate the transcription of its G2/M phase-specific targets not by binding directly to the typical FKH motifs, but through an indirect DNA binding mechanism mediated by MuvB complex [24, 25, 27].
FOXM1 promotes phenotypes of cancer cells by not only activating cancer-related gene transcription alone but also in concert with other transcription factors under certain circumstances. For example, during the tumorigenesis mediated by Wnt/β-catenin signaling, FOXM1 binds directly to β-catenin and enhances β-catenin nuclear localization and transcriptional activity. The disruption of the FOXM1-β-catenin interaction prevents β-catenin nuclear accumulation and abolishes Wnt target-gene expression in cancer cells [28]. During the TGF-β signaling activation in cancer cells, FOXM1 interacts with SMAD3 to sustain activation of the SMAD3/SMAD4 complex in the nucleus. The FOXM1-SMAD3 interaction is required for TGF-β-induced breast cancer invasion through promoting TGF-β/SMAD3-mediated transcriptional activity and target gene expression [29]. Therefore, the inhibition of FOXM1 transcriptional activity or the disruption of FOXM1 interaction with other cancer-related transcription factors or both may become viable strategies for the development of anti-tumor drugs.
Based on the facts that the N-terminus of FOXM1 protein is capable of inhibiting the transcriptional activity of FOXM1 [30] and also mediates the interaction between FOXM1 and other proteins such as LIN9 of MuvB complex [25] and SMAD3 [29], we have intended to develop a protein/peptide anti-cancer drug to abolish the activities and functions of FOXM1 by fusing FOXM1 N-terminal region (1-138aa) with a nine arginine cell-penetrating peptide. This purified recombinant protein (named as M1-138) demonstrates potent inhibitory effects on the proliferation and migration of cancer cells by attenuating FOXM1 transcriptional activities from both direct and indirect FOXM1-promoter binding mechanisms and abolishing the interaction between FOXM1 and SMAD3. M1-138 can inhibit the tumorigenic ability of cancer cells and tumor growth in nude mouse xenograft models with no obvious signs of toxicity. This work suggests that M1-138 is a promising drug candidate for the development of anti-cancer therapeutics.
Materials and Methods
Cell lines and cell culture
HEK 293T, MDA-MB-231, MCF-7, HepG2, A549, and Hela cells were obtained from ATCC (Manassas, USA). FOXM1-expressing MCF-7 clone cell line (M1 clone) was described previously [7]. All cells were grown in Dulbecco's Modified Eagle Media (Gibco) supplemented with 10% fetal bovine serum (Gibco). All cells were cultured in a 5% CO2-humidified atmosphere at 37 °C.
Construction of plasmids
The construction procedure of pHis-M1-138-R9, pHis-GFP-R9, pCMV-AVI-FOXM1, pCMV-M1-138, pCMV-M1-139-748, pCMV-SMAD3-RFP, pPLK1pro(-1.4 kb)-Luc, and pCDC25Bpro(-1.8 kb)-Luc was detailed in Supplementary Materials and Methods.
Expression and purification of recombinant proteins
Certain plasmids were transformed into Rosetta/DE3 E. Coli cells and positive colonies were identified by PCR of selected colonies. Cells were grown at 37 °C in LB medium until an optical density of 0.8 (OD600) was reached, and protein expression was induced by further adding 1 mM IPTG for additional 24 h culture at 28 °C. M1-138, R9-GFP, or FOXM1 recombinant protein was purified by Ni-SepharoseTM 6 Fast Flow (GE) following the manufacturer's instructions. The GST or GST-FOXM1 (688-748aa) recombinant protein was purified by Glutathione SepharoseTM 4B (GE) following the manufacturer's instructions. For large scale purification of M1-138 from E. coli cell lysates, an AKTA Protein Purifier with a Ni-NTA agarose affinity chromatography column (GE) was used according to the standard protocol of the manufacturer. The absorbance at 280 nm of the purification process was recorded during gradient washing and elution of samples.
Cell viability, Colony formation, and Transwell assays
The cell viability, in vitro tumorigenesis, and migration abilities of selected cancer cells were analyzed by standard CCK-8 activity, colony formation, or transwell assays, respectively. The detailed procedure was described in Supplementary Materials and Methods.
Quantitative real‑time PCR (qPCR)
Total RNA was extracted using Trizol reagent (Omega) according to the manufacturer's instructions. Total RNA (2.0 μg) was reverse transcribed into 20 μl cDNA by RevertAid First Strand Kit (Promega). The qPCR was performed with SYBR Green (Toyobo) with certain sense (S) and antisense (AS) primers. The detail information of primers was described in Supplementary Materials and Methods. The qPCR was performed in the realplex2 qPCR system (Eppendorf).
RNA interference
Transfection of LIN9 siRNA (SantaCruz sc-88786) was performed using Lipofectamine 2000 (Invitrogen) according to the manufacturer's protocols.
Protein extraction and Western blotting
The preparation procedure of total protein lysates and cytoplasmic/nuclear extracts was described in Supplementary Materials and Methods. Lysates were separated by SDS-PAGE and transferred onto PVDF membrane for western blotting. The detail information of antibodies was described in Supplementary Materials and Methods. The bands of Western blotting were quantified by ImageJ software.
Pull-down and Co-immunoprecipitation (Co-IP) assays
Cells were harvested and lysed with IP buffer (50 mM Tris-Cl, 100 mM NaCl, 2.5 mM EDTA, 2.5 mM EGTA, 1% NP-40, and 5% Glycerol) on ice for 30 min. The lysates were obtained by centrifuge at 12,000 rpm for 15 min at 4 °C. Usually 500 µg protein lysates were incubated with 20 µL of selected beads (Ni-beads or Streptavidin agarose beads, GE) at 4 °C for 2 h. At certain circumstances, M1-138 was added to the reactions at increased concentration (2, 4, 8 µM) to act as the competitor of FOXM1-SMAD3 interactions. The beads were washed three times in IP buffer and subjected to Western blotting.
For Co-IP, 500 µg protein lysates were incubated with 20 µL of Protein A/G PLUS-Agarose beads (SantaCruz sc-2003) and 2 µg anti-FOXM1 antibody (C-20) (recognizing FOXM1 C-terminus) or 2 µg anti-IgG antibody (CST # 2729S) at 4 °C for 4 h. At certain circumstances, M1-138 was added to the reactions at increased concentration (4 or 8 µM) to act as the competitor of FOXM1-mediated interactions. The beads were washed five times in IP buffer and subjected to Western blotting.
Electrophoretic mobility shift assays (EMSAs) and Luciferase activity assays
The DNA binding ability and transcriptional activity of FOXM1 and SMAD3 were analyzed by EMSAs or luciferase activity assays, respectively. The detailed procedure is described in Supplementary Materials and Methods.
Tumorigenesis assays in nude mice
All animal experiments were conducted in accordance with institutional animal care and use guidelines, following approval by the Laboratory Animal Center of Hunan, China (Protocol No. SYXK [Xiang] 2013-0001). BALB/c nude mice (female, 4-week old) were purchased from Hunan SJA Laboratory Animal Co., Ltd (Changsha, China). To study the effect of M1-138 on tumor formation and growth in vivo, three separate experiments were performed. In the first experiment to test whether M1-138 affected the tumorigenicity of cancer cells, mice were randomized into two groups (three mice per group) and right armpit injected with 1×107 MDA-MB-231 cells, which were treated by R9-GFP (8 μM) and M1-138 (8 μM) for 12 h, respectively. The mice were observed for tumor formation routinely and the volumes of tumors were measured at Day 35 day post the treatment. In the second experiment to further confirm the inhibitory ability of M1-138 on the tumorigenicity of cancer cells, mice were randomized into two groups (four mice per group) and right armpit injected with 1×106 MDA-MB-231 cells. One day later, R9-GFP (4 mg/Kg) or M1-138 (4 mg/Kg) was intraperitoneally injected the two groups respectively. The engrafted tumors were observed for 35 days. In the third experiment to test the inhibitory effects of M1-138 to engrafted tumors, mice (n=10) were subcutaneously (S.C.) injected with MDA-MB-231 cells (106 cells/mouse) into the right axilla. When the tumor volumes reached 50-100 mm3, the mice were randomly divided into M1-138 group (n=6) or R9-GFP group (n=4). R9-GFP or M1-138 (4 mg/Kg) was injected directly into tumors (orthotopic injection) once a day for 14 days (total 14 times). The size of engrafted tumors was measured at the interval of two days and tumor samples were collected at Day 35 post the injection of cancer cells. The weight of each mouse was monitored simultaneously. The tumors were measured by a vernier caliper and the tumor volumes (V) were calculated by: V = length × diameter2 × 1/2. Immunohistochemistry analysis of selected sample sections was performed according to a standard procedure detailed in Supplementary Materials and Methods.
Statistical analysis
We used Microsoft Excel Program or Prism 7 to calculate the mean ± standard deviation among samples. Briefly, we calculated the mean value ± SD (standard deviation) of repeated samples for both the control group and the experimental group. Two-way ANOVA or T-test was conducted with each sample values between the control group and the experimental group. *, P≤0.05; **, P≤0.01; and ***, P≤0.001. P≤0.05 was considered to indicate a statistically significant difference.
Results
M1-138 possessed an inhibitory effect to cancer cells
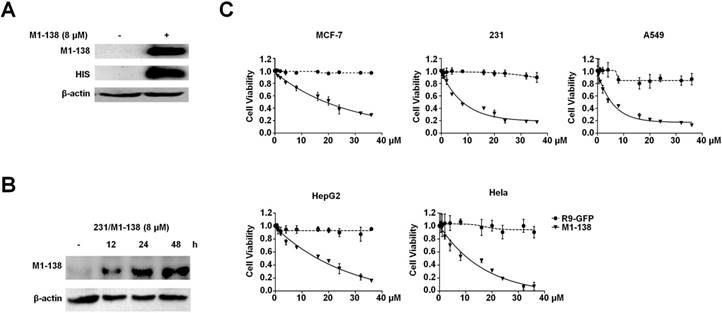
The N-terminus of FOXM1 protein was found to act as an inhibitory domain to FOXM1 transcriptional activities [30]. Before producing the FOXM1 N-terminus as an anti-cancer reagent, we first confirmed the published findings with co-transfection experiments, in which the N-terminus-deleted FOXM1 protein (139-748aa) possessed stronger activities than FOXM1 full length protein in stimulating the FOXM1-responded promoter (Figure S1A). The expression of FOXM1 N-terminus (1-138aa) in cancer cells abolished the transcriptional activities of FOXM1 and consequently down-regulated the expression of FOXM1's target genes such as CDC25B (Figure S1B-C). To determine the appropriate cell-penetrating peptide to mediate the entry of a recombinant protein into cancer cells, we tested the cell-penetrating efficiency of the polyarginine peptide (R9) [31], which resulted in the effective cell entry of the Green Fluerecent Protein (GFP) when fused with the R9 peptide (Figure S2). We then constructed a prokaryotic expression vector pHis-M1-138-R9 that contained a 6xHis tag, FOXM1 N-terminus (1-138aa), and the R9 peptide (Figure S3A). The recombinant protein M1-138 was produced by the E. coli expression system and purified with Ni-Sepharose (Figure S3B). The obtained M1-138 effectively entered into cancer cells at a reasonable concentration (8 μM) and remained stable for at least 48 h in MDA-MB-231 cells treated with M1-138 (Figure 1A-B). To test the effects of M1-138 on the growth of cancer cells, M1-138 at various concentrations (0.5, 1, 2, 4, 8, 16, 20, 24, 32, 36 µM) was used to treat multiple cell lines from different types of cancers, including breast cancer MCF-7 and MDA-MB-231 cells, lung carcinoma A549 cells, hepatocellular carcinoma HepG2 cells, and cervical adenocarcinoma Hela cells. The growth of all these cells was significantly inhibited by a 12 h treatment of M1-138 at a dose-dependent manner (Figure 1C). The half maximal inhibitory concentration (IC50) of M1-138 post the 12 h treatment was calculated for the five tested cell lines (17.9 µM for MCF-7, 7.8 µM for MBA-MD-231, 5.3 µM for A549, 13.8 µM for HepG2, and 8.9 µM for Hela, respectively), suggesting that the M1-138 sensitivity varied in different cancer cells (Figure S4).
M1-138 possessed an inhibitory effect on cancer cells. A, MDA-MB-231 cells (2×105 cells/well) were seeded in 6-well plates for 24 h and treated with M1-138 (8 µM) or untreated. 12 h later, the cells were harvested for preparation of total proteins. M1-138 was detected in the each lysate sample (40 µg) by Western blotting with anti-FOXM1 antibody (K-19, recognizing FOXM1 N-terminus) and anti-His-tag antibody respectively. The detection of β-actin was performed as the loading control. B, MDA-MB-231 cells were treated with M1-138 (8 µM) and the lysates were prepared at 0, 12, 24, 48 h post the treatment. M1-138 was detected in the each sample by Western blotting with anti-FOXM1 (K-19) antibody. C, MCF-7, MBA-MD-231, A549, HepG2, or Hela cells (4×103 cells/well) were seeded in 96-well plates for 12 h and treated with different concentrations of R9-GFP and M1-138 (0.5, 1, 2, 4, 8, 16, 20, 24, 32, 36 µM). 12 h later, CCK-8 solution (10 µL) was added to each well and incubated for another 2 h. The absorbance at 450 nm was measured and the relative cell viability in each well was calculated. The percentage of cell activity versus the concentration of M1-138 was plotted. Values represented the mean ± SD of four replicates.
The M1-138 sensitivity corresponded to the expression levels of FOXM1 in cancer cells
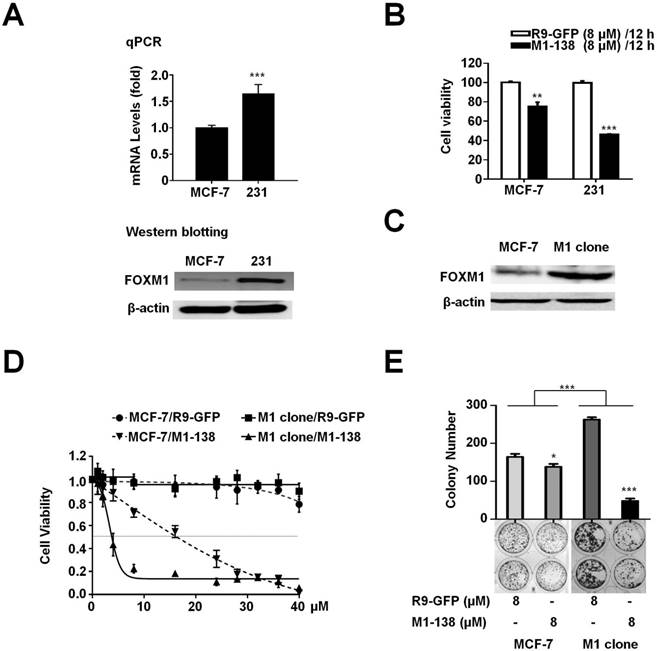
The M1-138 IC50 value of MCF-7 cells was 2 fold higher than that of MBA-MD-231 cells (17.9 µM of MCF-7 versus 7.8 µM of MBA-MD-231, post 12 h treatment). To confirm whether this variation was caused by the different levels of FOXM1 in cells, we first detected that the mRNA and protein levels of FOXM1 in MBA-MD-231 cells were significantly higher than those in MCF-7 cells (Figure 2A). We also compared the FOXM1 mRNA and protein levels of HepG2, Hela, and A549 cells to that of MBA-MD-231 cells, whose mRNA levels of FOXM1 were set to one (Figure S5). Consequently, MBA-MD-231 cells were more sensitive to M1-138 than MCF-7 cells, evidenced by the lower cell viability of MBA-MD-231 cells post M1-138 (8 μM) 12 h treatment (Figure 2B). Furthermore, we established a FOXM1-overexpressed MCF-7 cell clone (M1 clone) [7] (Figure 2C) and found that M1 clone cells were much more sensitive to M1-138 than parent MCF-7 cells (IC50 of M1 clone was 4.5 μM post 12 h treatment) (Figure 2D, Figure S6), providing a direct evidence that the sensitivity to M1-138 corresponded to the expression levels of FOXM1 in cancer cells. This view was further supported by the observation that the M1-138 treatment resulted in higher inhibitory rate of colony formation in M1 clone cells than that in MCF-7 cells (Figure 2E).
M1-138 sensitivity corresponded to the expression levels of FOXM1 in cancer cells. A, MCF-7 and MBA-MD-231 (231) cells were harvested to prepare total RNAs and protein lysates. The mRNA levels of FOXM1 were measured by qPCR and the protein levels of FOXM1 were measured by Western blotting. The detection of GAPDH mRNA and β-actin protein was performed as loading controls for RNAs and protein lysates respectively. Relative mRNA levels were normalized to GAPDH. The levels of MCF-7 cells were referred as One. B, MCF-7 and MDA-MB-231 cells (4×103 cells/well) were seeded in 96-well plates for 12 h and treated with R9-GFP and M1-138 (8 µM) for 12 h. 12 h later, CCK-8 solution (10 µL) was added to each well and incubated for another 2 h. The absorbance at 450 nm was measured and the relative cell viability in each well was calculated. Values represented the mean ± SD of four replicates. C, FOXM1-overexpressed MCF-7 cell clone (M1 clone) was generated by the infection of a lentivirus encoding human FOXM1 to MCF-7 cells. The protein levels of FOXM1 in MCF-7 and M1 clone cells were measured by Western blotting with anti-FOXM1 antibody (C-20, recognizing FOXM1 C-terminus). D, MCF-7 and M1 clone cells were treated with different concentrations of R9-GFP and M1-138 (1, 2, 4, 8, 16, 20, 24, 28, 32, 36, 40 µM). 12 h later, the measurement of cell viability with CCK-8 solution was performed. Values represented the mean ± SD of four replicates. E, MCF-7 and M1 clone cells (400 cells/well) were seeded in 6-well plates and treated with R9-GFP or M1-138 (8 µM) for 14 days. The cells were fixed and stained with crystal violet. The colonies were imaged and the graph represented the number ± SD of colonies/well. (*, P≤0.05; **, P≤0.01, ***, P≤0.001).
M1-138 inhibited the proliferation, migration, and tumorigenic ability of cancer cells
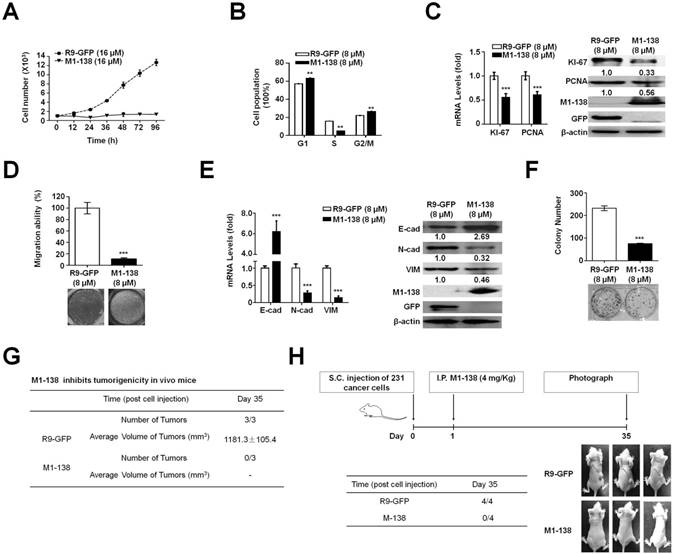
Next, we focused on MBA-MD-231 cells to analyze the effects and mechanisms of M1-138 on cancer cells. MBA-MD-231 cells were treated with M1-138 (16 µM, 2 fold of IC50 dosage) or control R9-GFP (16 µM) and counted at different time points (12h, 24h, 36h, 48h, 72h, and 96h) post the treatment. The growth curves showed that the proliferation of the cells was inhibited completely by M1-138 at this dosage (16 µM) (Figure 3A). We then treated MDA-MB-231 cells with M1-138 at the dosage of IC50 (8 μM) and collected the cells at 24 h post the treatment. Flow cytometry was performed to analyze the cell cycle of the samples, which showed an abolished proliferation in the M1-138-treated cells with the increase of G1 and G2/M phase population and the decrease of S phase population (Figure 3B). These phenomena correlated with the decreased expression of cell proliferative markers such as KI-67 and PCNA in the M1-138-treated MBA-MD-231 cells (Figure 3C). The inhibitory effects of M1-138 (8 μM) on the migration of MBA-MD-231 cells were demonstrated by Transwell experiments (Figure 3D) and the expression of migration-related genes (E-cadherin, N-cadherin, and Vimentin) was measured by qPCR and Western blotting, which showed a significant up-regulation of E-cadherin levels and a down-regulation of N-cadherin and Vimentin levels in the M1-138 treatment group (Figure 3E), supporting that M1-138 could inhibit the mobility of cancer cells. To test whether M1-138 affected the tumorigenicity of cancer cells, the colony formation in vitro and the tumor formation in vivo were performed for MDA-MB-231 cells treated with M1-138 (8 μM) or control R9-GFP (8 μM). The colony number of M1-138-treated cells was 3 fold less than that of control cells after the 14-day colony formation process (Figure 3F). The M1-138-treated cells generated no engrafted tumors in nude mice (n=3) after 35 days post the subcutaneous injection of the cells, when the tumors of average size>1000 mm3 were observed in the control group (n=3) (Figure 3G). Furthermore, we implanted MDA-MB-231 cells in nude mice (n=8) and one day later the mice were separated into two groups and treated with M1-138 (n=4) or R9-GFP (n=4) at dosage of 4 mg/Kg via intraperitoneal (I.P.) injection. The tumor formation was observed in the control group but no obvious tumors were found among the M1-138 group at Day 35 (Figure 3H), further confirming the inhibitory ability of M1-138 on the tumorigenicity of cancer cells.
M1-138 inhibited the proliferation, migration, and tumorigenic ability of cancer cells. A, MDA-MB-231 cells (1×103 cells/well) were seeded in 24-well plates for 12 h and treated with R9-GFP or M1-138 (16 µM). The cell numbers in each well were counted at different time points (12, 24, 36, 48, 72, and 96 h) post the treatment. B, MDA-MB-231 cells were treated R9-GFP or M1-138 (8 μM) and 24 h later the cells were collected, stained with propidium iodide, and analyzed for DNA content on a flow cytometer. Values represented the mean ± SD of three independent experiments. C, MDA-MB-231 cells were treated with R9-GFP or M1-138 (8 μM) and 24 h later the cell samples were harvested for the preparation of total RNA and total proteins. Levels of KI-67 and PCNA in the cells were examined by qPCR for mRNA levels and by Western blotting for protein levels. Relative mRNA levels were normalized to GAPDH. The levels of R9-GFP-treated cells were referred as One. Western blotting was performed for the detection of M1-138 and GFP with anti-FOXM1 (K-19) antibody or anti-GFP antibody respectively. D, MBA-MD-231 cells were treated with R9-GFP and M1-138 (8 μM) for 12 h. Subsequently, 1×105 cells were seeded in a top chamber of 200 μl of serum-free DMEM and 600 μl of DMEM containing 10% FBS was applied to the lower chamber. After incubation for 24 h, the cells on the upper membrane were gently removed and the cells on the interior membrane were fixed with 4% paraformaldehyde and stained with 0.1% crystal violet. Cells passing through the membrane were captured by a light microscope. The average cell counts were calculated from five random fields. E, MDA-MB-231 cells were treated with R9-GFP or M1-138 (8 μM) and 24 h later the cell samples were harvested for the preparation of total RNA and total proteins. The levels of E-cadherin (E-cad), N-cadherin (N-cad), and Vimentin (VIM) in the cells were examined by qPCR for mRNA levels and by Western blotting for protein levels. Relative mRNA levels were normalized to GAPDH. The levels of R9-GFP-treated cells were referred as One. The detection of M1-138 and GFP proteins was also performed with the protein lysates. F, MBA-MD-231 cells were treated with R9-GFP and M1-138 (8 µM) for 14 days. The cells were fixed and stained with crystal violet. The colonies were imaged and the graph represented the number ± SD of colonies/well. G. MDA-MB-231 cells were treated with R9-GFP or M1-138 (8 μM) for 12 h. Mice (female, 4-week old) were randomized into two groups (three mice per group) and subcutaneously injected with R9-GFP-treated or M1-138-treated cells (1×107 cells /mouse) respectively. The mice were observed for tumor formation routinely and the volumes of tumors were measured at Day 35 day post the treatment. The tumor volume (V) was calculated by: V = length × diameter2 × 1/2. H. Mice (female, 4-week old) were subcutaneously (S.C.) injected with MDA-MB-231 cells (1×106 cells /mouse). One day later, the mice were randomized into two groups (four mice per group) and intraperitoneally (I.P.) injected with R9-GFP or M1-138 (4 mg/Kg) respectively. The engrafted tumors were observed and the representative animals were photographed at Day 35 post the treatments. (**, P≤0.01, ***, P≤0.001).
M1-138 inhibited FOXM1 transcriptional activities
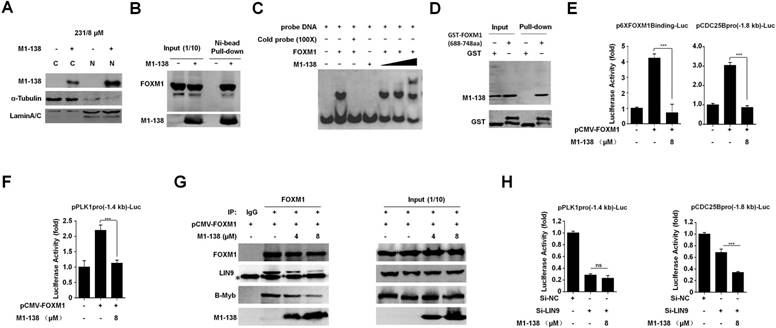
M1-138 was developed based on the N-terminus of FOXM1 protein as an inhibitory domain to FOXM1 transcriptional activities. We then tested whether M1-138 targeted FOXM1 and inhibited its activities. Even though M1-138 was found to distribute in both cytoplasm and nucleus of MBA-MD-231 cells treated with M1-138 (8 μM for 12 h), the majority of the protein could stay at the nuclei where FOXM1 functioned as a transcription factor (Figure 4A, Figure S7). The direct interaction between M1-138 and FOXM1 was confirmed by Pull-down assays in that Ni-agarose beads could pull down FOXM1 proteins through the His-tag of M1-138 from the M1-138-treated MBA-MD-231 cell lysates (Figure 4B), suggesting that M1-138 recognized FOXM1 specifically from a mixture of cellular proteins. The M1-138-FOXM1 interaction was further determined by Electrophoretic Mobility Shift Assays (EMSAs). A purified recombinant FOXM1 full-length protein (2 µM) and a FAM-labeled DNA probe containing FOXM1 putative binding sequence (50 nM) were used for the basic setting of EMSAs. M1-138 of increased dosage (0.5, 1, and 2 µM) was added to the reaction mixture and a super-shift band became obvious when the molecule ratio of M1-138 to FOXM1 was 1:1 (Figure 4C), implicating that the binding of M1-138 to FOXM1 did not disturb the DNA binding ability of FOXM1. To identify the region of FOXM1 proteins was recognized by M1-138, we purified a recombinant protein of FOXM1 C-terminus (688-748aa) fused with the GST tag (Figure S8). The fusion protein GST-FOXM1(688-748aa) but not the pure GST control was able to pull down M1-138 from the M1-138-treated MBA-MD-231 cell lysates (Figure 4D), demonstrating that M1-138 bound to the C-terminus of FOXM1. This result was also consistent with the published finding that the interaction existed between N-terminus and C-terminus of FOXM1 [30]. The C-terminus of FOXM1 acted as the transcriptional activation domain for FOXM1 [32] so we tested whether M1-138 could abolish FOXM1 transcriptional activities in cotransfection experiments. FOXM1-mediated stimulation on the FOXM1-binding promoters (an artificial 6xFOXM1 binding sequence-containing promoter or an endogenous -1.8 kb promoter of CDC25B) was significantly inhibited by the treatment of M1-138 (Figure 4E).
M1-138 inhibited FOXM1 transcriptional activities. A, MDA-MB-231 cells (2×105 cells/well) were seeded in 6-well plates for 24 h and then treated with M1-138 (8 µM). 12 h later, the cells were harvested for preparation of cytoplasmic and nuclear proteins. The levels of M1-138 in the cytoplasm (C) or the nucleus (N) were examined by Western blotting. α-Tubulin or LaminA/C was also used as a cytoplasmic or nuclear marker, respectively. B, The lysates (500 μg) of M1-138-treated MDA-MB-231 cells were incubated with Ni-NTA agarose beads to pull down His-tagged M1-138/protein complexes. FOXM1 proteins and M1-138 in the pull-down sample were detected by Western Blotting with anti-FOXM1 C-20 or K-19 antibodies respectively. Fifty μg lysates (10% of input) was also used for Western Blotting as input controls. C, The FAM-labeled DNA probe (50 nM) was mixed with recombinant FOXM1 proteins (2 µM) for EMSAs. M1-138 was added to the reactions at increased concentration (0.5, 1, and 2 µM). The 100×unlabeled DNA probe (Cold probe, 5000 nM) was used to show specificity of FOXM1/DNA complex formation. The reactions were resolved in 4% native polyacrylamide gel electrophoresis in 0.5×TBE and visualized with Kodak 4000 MM Imaging System (EX: 465 nm, EM: 535 nm for FAM). D, The lysates (500 μg) of M1-138-treated MDA-MB-231 cells were incubated with recombinant GST-FOXM1(688-748aa) or GST control proteins (8 μg). Glutathione SepharoseTM 4B beads were added to the reactions and the pull-down samples were detected by Western blotting with anti-FOXM1 K-19 antibody or anti-GST antibody. 10% of the reactions (50 μg) were also used as input controls. E-F, The FOXM1 expression vector (pCMV-FOXM1, 1 μg) was transfected with a luciferase reporter plasmid containing 6xFOXM1 binding sequences in its promoter (p6xFOXM1 Binding-Luc, 1 μg), a -1.8 kb CDC25B promoter-luciferase reporter plasmid (pCDC25Bpro(-1.8 kb)-Luc, 1 μg), or a -1.4 kb PLK1 promoter-luciferase reporter plasmid (pPLK1pro(-1.4 kb)-Luc, 1 μg) plus loading control pRL-CMV luciferase reporter plasmid (20 ng) into HEK293T cells. M1-138 (8 µM) was added to the selected transfections. Protein lysates were prepared at 48 h later and used for the measurement of dual Luciferase activities. G, Hela cells were transfected with pCMV-FOXM1 and 48 h later the cell lysates were prepared. The lysates (500 μg) were used for Co-IP experiments. M1-138 was added to the reactions at increased concentrations (4, 8 µM). LIN9, B-MYB, M1-138, and FOXM1 proteins in Co-IP samples were detected by Western Blotting with certain antibodies. 10% of lysates (50 μg) were used as input controls. (*) IgG bands. H, The FOXM1 expression vector (pCMV-FOXM1, 1 μg) was transfected with pPLK1pro(-1.4 kb)-Luc or pCDC25Bpro(-1.8 kb)-Luc (1 μg) plus pRL-CMV plasmid (20 ng) into Hela cells. Si-NC, si-LIN9, and M1-138 (8 µM) was added to the selected transfections. Protein lysates were prepared at 48 h later and used for the measurement of dual Luciferase activities. Values represented the mean ± SD (**, P≤0.01, ***, P≤0.001).
Besides it activates gene expression by directly binding to FKH motif (RYAAAYA), FOXM1 can stimulate the transcription of its G2/M phase-specific targets by an indirect DNA binding mechanism mediated by MuvB complex through CHR motif (TTTGAA or TTTAAA) [26]. The -1.8 kb CDC25B promoter did contain FKH sites (at -652 to -646, -952 to -839, -1105 to -1099 bp) but no any CHR site, providing a negative control for testing FOXM1-MuvB-dependent gene activation. We also cloned a -1.4 kb PLK1 promoter that contained CHR sites (at -30 to -25, -89 to -84, -149 to -144 bp) but no perfect FKH site. We found that M1-138 inhibited FOXM1 transcriptional activities on both FKH-containing -1.8 kb CDC25B promoter (Figure 4E) and CHR-containing -1.4 kb PLK1 promoter (Figure 4F), suggesting that M1-138 could affect both the direct FOXM1-promoter binding mechanism and the indirect FOXM1-promoter binding mechanism (FOXM1-MuvB-dependent). Next, we performed Co-IP experiments to confirm that M1-138 disrupted the interaction of FOXM1 with MuvB complex, evidenced by the decreased FOXM1-LIN9 interaction after the addition of M1-138 in a dose-dependent manner (Figure 4G). Furthermore, the addition of M1-138 also disrupted the interaction of FOXM1with B-MYB (Figure 4G). Due to that B-MYB was required to recruit FOXM1 to MuvB complex [24], the disruption of FOXM1-B-MYB interaction by M1-138 further explained how M1-138 prevented FOXM1-MuvB-dependent gene activation. Because LIN9 mediates the interaction between FOXM1 and MuvB complex [24, 25], we used LIN9 siRNA to abolish the LIN9 expression (Figure S9) and consequently to disrupt FOXM1 interacting with MuvB and indirect binding to CHR sites. We found that LIN9 siRNA could abolish FOXM1 transcriptional activities on either -1.4 kb PLK1 promoter or -1.8 kb CDC25B promoter but more dramatic decrease (~70%) was observed at -1.4 kb PLK1 promoter compared to -1.8 kb CDC25B promoter (~30%) (Figure 4H). Moreover, the addition of M1-138 resulted in further decrease of FOXM1 transcriptional activities only on -1.8 kb CDC25B promoter but not on -1.4 kb PLK1 promoter (Figure 4H). This data implicated that, after the disruption of FOXM1-MuvB interaction by LIN9 siRNA and the indirect FOXM1-MuvB-dependent mechanism no long existed, M1-138 would not affect the FOXM1 transcriptional activities on -1.4 kb PLK1 promoter anymore. As a negative control of FOXM1-MuvB-dependent mechanism, the -1.8 kb CDC25B promoter was still affected by M1-138 because of the existence of the direct FOXM1-promoter binding mechanism. Combined the data presented in Figure 4F, 4G, and 4H, we concluded that M1-138 could also affect FOXM1-MuvB-dependent gene activation.
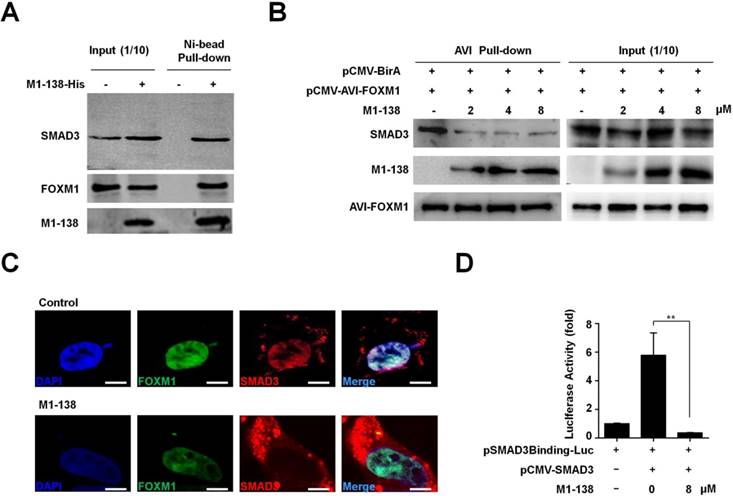
M1-138 prevented the interaction between FOXM1 and SMAD3 and inhibited the transcriptional activities of SMAD3
The N-terminus of FOXM1 interacted with SMAD3 to keep the SMAD3/SMAD4 complex in nuclei for promoting TGF-β/SMAD3 transcriptional activities. Among various types of human malignancies, the cancer-related transcription of SMAD3 target genes required FOXM1-mediated SMAD3 binding to its target gene promoters [29]. So, we performed Pull-down assays with the M1-138-treated MBA-MD-231 cell lysates to confirm the interaction of M1-138 with SMAD3 and FOXM1. Ni-agarose beads were able to pull down SMAD3 and FOXM1 proteins through the His-tag of M1-138 from the mixture of cellular proteins (Figure 5A), demonstrating that M1-138 bound to both SMAD3 and FOXM1 specifically. The similar results were obtained from Hela cells (Figure S10). To study whether M1-138 disrupted the interaction between FOXM1 and SMAD3 in cells, we generated an intracellular biotinylation system for FOXM1 by fusing an AviTag [33] with FOXM1. The AviTag could be recognized and biotinylated by the E. coli biotin ligase BirA when AviTag-fused FOXM1 and BirA were co-expressed by pCMV-AVI-FOXM1 and pCMV-BirA vectors in cells. Due to the strong affinity between biotin and streptavidin, the biotinylated FOXM1 had a huge advantage to test interactions between FOXM1 and FOXM1-interacting proteins such as SMAD3 (Figure S11). We transfected cells with pCMV-AVI-FOXM1 and pCMV-BirA and treated the cells with M1-138 at different dosages (2, 4, or 8 µM). One day later, cell lysates were prepared and streptavidin-agarose beads were used to pull down the biotinylated FOXM1. SMAD3 was pulled down at the same time (Figure 5B), proving the published interaction between FOXM1 and SMAD3 [29]. Moreover, the addition of M1-138 prevented the FOXM1-SMAD3 interaction in a dose-dependent manner, evidenced by the dramatic decrease of SMAD3 levels but no obvious changes of FOXM1 levels in the M1-138-treated pull-down samples (Figure 5B). To test the effects of M1-138 on the cellular distribution of FOXM1 and SMAD3 proteins in cells, we expressed FOXM1-GFP and SMAD3-RFP fusion proteins in cells, followed by the treatment of M1-138 (8 µM). In the control samples, the GFP signals of FOXM1-GFP mainly located in the nucleus, while the RFP signals of SMAD3-RFP were also dominant in the nucleus (Figure 5C), proving the published finding that FOXM1 promoted the nuclear localization of SMAD3 [29]. Under the M1-138-treated condition, the nuclear localization of FOXM1-GFP was maintained, implicating that M1-138 did not affect the nuclear localization of FOXM1 in M1-138-treated cells. Importantly, FOXM1-mediated SMAD3 nuclear localization was interrupted by M1-138, evidenced by that the RFP signals of SMAD3-RFP distributed mainly in the cytoplasm of FOXM1-GFP-expressed cells (Figure 5C). The decrease of SMAD3 nuclear localization was further confirmed by Western blotting of the cytosol and nuclear lysate samples from M1-138-treated cells (Figure S12). The results suggested that M1-138 could prevent the nuclear translocation of SMAD3 by disturbing the FOXM1-SMAD3 interaction. Therefore, it was reasonable to test whether M1-138 inhibited SMAD3 transcriptional activities. The SMAD3-mediated stimulation on the SMAD3-responded promoter (an artificial SMAD3 binding sequence-containing promoter) was significantly inhibited by the treatment of M1-138 (8 μM) (Figure 5D), demonstrating that M1-138 abolished SMAD3 transcriptional activities in cells.
M1-138 prevented the interaction between FOXM1 and SMAD3 and inhibited the transcriptional activities of SMAD3. A, The lysates (500 μg) of M1-138-treated MDA-MB-231 cells were incubated with Ni-NTA agarose beads to pull down His-tagged M1-138/protein complexes. SMAD3, FOXM1, and M1-138 in the pull-down sample were detected by Western blotting with anti-SMAD3 antibody or anti-FOXM1 antibodies respectively. Fifty μg lysates (10% of input) was also used for Western blotting as positive loading controls. B, HEK293T cells were transfected with pCMV-AVI-FOXM1 and pCMV-BirA and 48 h later the cell lysates were prepared. The lysates (500 μg) were incubated with Streptavidin agarose beads to pull down AVI-FOXM1/protein complexes. M1-138 was added to the reactions at increased concentrations (2, 4, 8 µM). SMAD3, M1-138, and AVI-FOXM1 proteins in the pull-down sample were detected by Western blotting with anti-SMAD3 antibody, anti-FOXM1 K-19 antibody, or HRP-labeled Streptavidin. 10% of the reactions (50 μg) was also used for Western blotting as input controls. C, HEK293T cell was transfected with pCMV-FOXM1-GFP and pCMV-SMAD3-RFP (5 μg) and 24 h later were treated with M1-138 (8 μM) or untreated. The localization of FOXM1-GFP and SMAD3-RFP was imaged with the fluorescent Con-focal microscope (Olympus FluoView FV1000). DAPI staining showed the location of nuclei of cells. (Scale bars: 5 μm). D, The SMAD3 expression vector (pCMV-SMAD3, 1 μg) was cotransfected with a luciferase reporter plasmid containing SMAD3 binding sequences in its promoter (pSMAD3Binding-Luc, 1 μg) plus loading control pRL-CMV luciferase reporter plasmid (20 ng) into HEK293T cells. M1-138 (8 µM) was added to the selected transfections. Protein lysates were prepared at 48 h later and used for the measurement of dual Luciferase activities. Values represented the mean ± SD (**, P≤0.01).
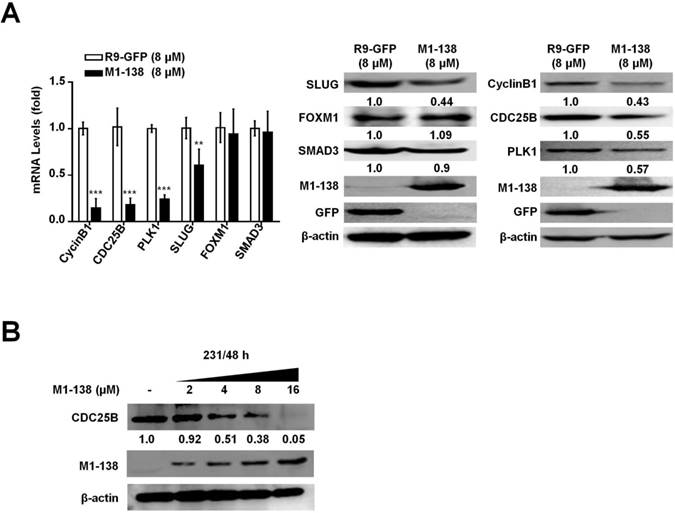
M1-138 inhibited the expression of FOXM1/SMAD3-downstream target genes involved in the stimulation of proliferation and migration of cancer cells
The results presented above demonstrated that M1-138 was able to directly bind to FOXM1 and SMAD3 and consequently prevented the transcriptional activities of these two transcription factors. To test whether M1-138 affected the expression of target genes of FOXM1 and SMAD3 in cancer cells, we chose four genes as typical examples from the list of multiple targets regulated by these two transcription factors: proliferation-related CyclinB1 [34], CDC25B [35], and PLK1 [25] as typical FOXM1 targets, and migration-related SLUG [29] as a typical SMAD3 target. MBA-MD-231 cells were treated with M1-138 (8 μM) or the control R9-GFP (8 μM) for 24 h and then harvested for RNA and cell lysate preparation. The mRNA and protein levels of CyclinB1, CDC25B, PLK1, and SLUG were measured by qPCR and Western blotting, which showed a significant down-regulation of levels of these four genes in the M1-138-treated group (Figure 6A), confirming that M1-138 could prevent the transcription of FOXM1/SMAD3-stimulated target genes in cancer cells. Next, MBA-MD-231 cells were treated with M1-138 at different dosages (2, 4, 8, and 16 μM) for 48 h and the protein levels of CDC25B were measured by Western blotting, which demonstrated a dose-dependent inhibition of target gene expression by M1-138 (Figure 6B).
M1-138 inhibited the expression of FOXM1/SMAD3-downstream target genes. A, MDA-MB-231 cells (2×105 cells/well) were plated on 6-well plates and treated with R9-GFP or M1-138 (8 µM) and 24 h later the cell samples were harvested for the preparation of total RNA and total proteins. The levels of CyclinB1, CDC25B, PLK1, and SLUG were examined by qPCR for mRNA levels and the levels of CyclinB1, CDC25B, PLK1, SLUG, FOXM1, and SMAD3 were detected by Western blotting for protein levels. Relative mRNA levels were normalized to GAPDH. The levels of R9-GFP-treated cells were referred as One. The detection of M1-138 and GFP proteins was also performed with the protein lysates. B, MDA-MB-231 cells (2×105 cells/well) were plated on 6-well plates and treated with M1-138 (2, 4, 8, and 16 µM) and 48 h later the cell samples were harvested for the preparation of total proteins. The levels of CDC25B in the cells were examined by Western blotting. (**, P≤0.01, ***, P≤0.001).
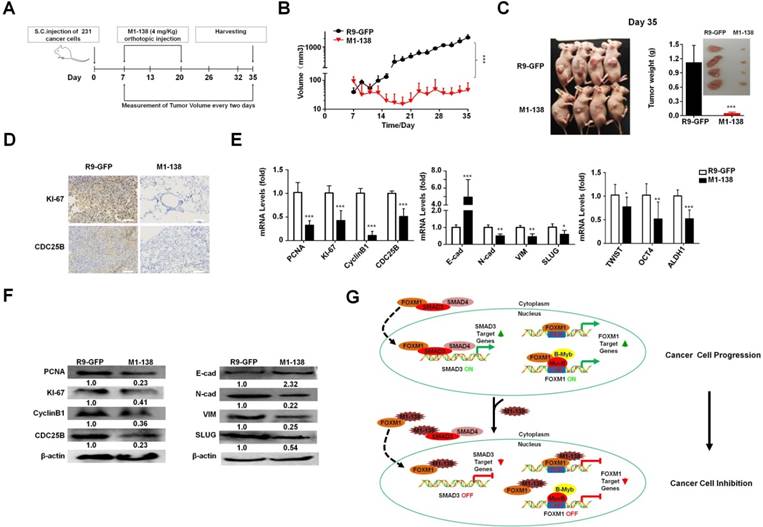
M1-138 suppressed tumor growth in vivo
To test the inhibitory effects of M1-138 to engrafted tumors, we implanted MDA-MB-231 cells (1x106 cells/injection) via subcutaneous (S.C.) injection in female nude mice (n=10) and seven days later the mice were separated into two groups and treated with M1-138 (n=6) or R9-GFP (n=4) at dosage of 4 mg/Kg via orthotopic injection, once a day for 14 days (total 14 times). The size of engrafted tumors was measured at the interval of two days and tumor samples were collected at Day 35 post the cancer cell implantation (Figure 7A). The continuous tumor growth was observed in the control group, but no obvious tumor growth occurred among the mice of the M1-138 group (Figure 7B). The dramatic difference of the tumor size between the M1-138-treated group and the control group was presented in Figure 7C. The levels of KI-67 and CDC25B proteins in the tumors were measured by immunostaining with the corresponding antibodies and typical positive staining of KI-67 and CDC25B was detected in the tumor tissue sections of the control group but not in that of the M1-138-treated group (Figure 7D). Tissue sections of the control group also showed a condensed morphology compared with that of the M1-138-treated group (Figure 7D). In addition, the pools of combined RNA samples from each group were tested with qPCR to measure the mRNA levels of cancer-related genes from three phenotype categories, including proliferation-related genes (PCNA, KI-67, CyclinB1, CDC25B), migration-related genes (E-cadherin, N-cadherin, Vimentin, SLUG), and stemness-related genes (TWIST, OCT4, and ALDH1). As predicted, in the M1-138-treated group, the mRNA levels of the cancer phenotype-promoting genes were significantly down-regulated except the migration-blocking E-cadherin mRNA levels were up-regulated (Figure 7E). Furthermore, Western blotting was performed to confirm the expression changes of most of the tested genes at protein levels (Figure 7F) and down-regulation of FOXM1 and SMAD3 protein levels in M1-138-treated group (Figure S13). Together, these results indicated that M1-138 elicited a strong antitumor effect in vivo.
M1-138 suppressed tumor growth in vivo. A, The protocol of M1-138 treatment in vivo. BALB/c nude mice (n=10) were subcutaneously (S.C.) injected with MDA-MB-231 cells (106 cells/mouse) into the right axilla. When the tumor volumes reached 50-100 mm3, the mice were randomly divided into M1-138 group (n=6) or R9-GFP group (n=4). R9-GFP or M1-138 (4 mg/Kg) was injected directly into tumors (orthotopic injection) once a day for 14 days. The size of engrafted tumors was measured at the interval of two days and tumor samples were collected at Day 35 post the injection of cancer cells. B, Tumor volume was measured from the first day to the end of experiments after grouping. The tumor volume (V) was calculated by: V = length × diameter2 × 1/2. Each data point represented the mean tumor volume in mm3 ±SD. C, Representative mice at Day 35 post the injection of cancer cells were imaged. The tumors were harvested and the weight of the tumors were compared between the R9-GFP group (n=4) and the M1-138 group (n=6). Inset showed the representative collected tumors. D, The tumor tissue sections of the two groups were immunostained with anti-KI-67 or anti-CDC25B antibodies. Photos were taken by the inverted microscope (200×, scale bar 50 µm). E, Total RNAs were prepared with the harvested tumors and the pools of combined RNA samples for the two groups were obtained. The mRNA levels of PCNA, KI-67, CyclinB1, CDC25B, E-cadherin (E-cad), N-cadherin (N-cad), Vimentin (VIM), SLUG, TWIST, OCT4, and ALDH1 were examined by qPCR. Relative mRNA levels were normalized to GAPDH. The levels of R9-GFP-treated cells were referred as One. F, Total proteins were prepared with the harvested tumors and the pools of combined protein lysates for the two groups were obtained. The protein levels of PCNA, KI-67, CyclinB1, CDC25B, E-cadherin (E-cad), N-cadherin (N-cad), Vimentin (VIM), and SLUG were measured by Western blotting. The detection of β-actin was performed as the loading control. (*, P≤0.05; **, P≤0.01; ***, P≤0.001). G. Schematic depicting the mechanisms of M1-138 to prevent cancer progression. In cancer cells, nucleus-localized FOXM1 promoted the transcription of its target genes that are involved in cancer progression. At the same time, FOXM1 interacts with SMAD3 and promotes the nuclear translocation of SMAD3. Consequently nucleus-localized SMAD3 promotes the transcription of its target genes and activates cancer progression. During the treatment of M1-138 to cancer cells, M1-138 binds to the C-terminal transcriptional activation domain of FOXM1 without disturbing its direct FKH site-binding abilities. It also affected the indirect DNA binding FOXM1-mediated gene activation by disrupting FOXM1 interacting with MuvB complex that binds to CHR sites. M1-138 can prevent the nuclear translocation of SMAD3 through competing away the interaction between FOXM1 and SMAD3, therefore abolishing SMAD3 transcriptional activities. Under the effects of M1-138, the stimulation of FOXM1 and SMAD3 on their target genes is prevented and the inhibition of cancer cell proliferation and migration is achieved.
Discussion
The global burden of cancer continues to increase, mainly due to the aging and growth of the world's population and the adoption of more and more cancer-causing behaviors [36]. Some specific transcription factor groups that are overactive in cancers have been recommended as the most suitable targets, including STATs, NF-κB, β-catenin, SMADs, and FOXM1 [37-41]. Different types of reagents, including small molecule compounds, peptides/proteins, and even antibodies, have been developed for effective interventions of these transcription factors in cancer cells. These reagents may act as potential therapeutics of cancers through inhibiting the expression of the transcription factors at the levels of transcription, translation, post-translation, and/or abolishing their interactions with target promoter binding sites, nuclear localization, and protein-protein interactions in cancer cells. Although few therapeutic examples are clinically successful so far, targeting transcription factors has been considered a promising and potentially successful approach to develop drugs for the treatment of cancer [42]. Among the choices of targeting transcription factors, the development of cell-penetrating peptide-involved peptide/protein drugs has attracted a great attention. For examples, cell-penetrating peptides selectively targeting SMAD3 was produced to inhibit TGF-β signaling [43]; a modified FOXO4-p53 interfering peptide could result in p53 nuclear exclusion and induce targeted apoptosis of senescent cells [44], and a cell-penetrating peptide was developed to suppress breast tumorigenesis by inhibiting β-catenin/LEF-1 signaling [45].
In this study, we showed a new cell-penetrating protein reagent, M1-138, which possessed potent inhibitory effects to cancer cells and targeted transcription factors FOXM1 and SMAD3 at the same time. With three different in vivo nude mouse models, we confirmed that M1-138 prevented the tumorgenicity of cancer cells (Figure 3G-H) and the progression of engrafted tumors (Figure 7). From the perspective of inhibitory mechanisms to cancer cells by M1-138, we proved that M1-138 bound to the C-terminal transcriptional activation domain of FOXM1 without disturbing its direct FKH site-binding abilities. It also affected the indirect DNA binding FOXM1-mediated gene activation by disrupting FOXM1 interacting with MuvB complex that binds to CHR sites. Therefore, M1-138 attenuated FOXM1 transcriptional activities from both direct and indirect FOXM1-promoter binding mechanisms. At the same time, M1-138 could prevent the nuclear translocation of SMAD3 through competing away the interaction between FOXM1 and SMAD3, therefore abolishing SMAD3 transcriptional activities. Under the effects of M1-138, the stimulation of FOXM1 and SMAD3 on their target genes was prevented and the inhibition of cancer cell proliferation and migration was achieved (Figure 7G). In addition, M1-138 might result in cancer suppression through other mechanisms. For an example, FOXM1 proteins could be phosphorylated by cell cycle-dependent kinases (CDKs) as substrates. A systematic screen for substrates of CDK4/6 identified FOXM1 as a common critical phosphorylation target of the kinases, which stabilized and activated FOXM1 and thereby maintained expression of G1/S phase genes [46]. The result supported the CDK4/6 inhibitor Palbociclib as a new First-in-Class drug in breast cancer therapy [47]. Moreover, both CDK2 and CDK1 activated FOXM1 by strictly phosphorylating N-terminus of FOXM1 and then releasing its transactivation domain from the repression of the N-terminus [48]. Theoretically, M1-138 might be able to act as a competitive substrate to block the CDK-mediated phosphorylation of cell cycle-promoting proteins (such as FOXM1) in cancer cells and to prevent cancer cell progression. This plausible mechanism of M1-138 actions will be elucidated in future studies.
Based on the inhibitory effects of M1-138 to cancer cells, the drug tolerance in animals is an issue worth considering before conducting clinical trials in the next step. During the period of testing M1-138 effects on tumor growth in vivo (Figure 7), we had observed no apparent side effects from the tested animals, evidenced by no difference in the body weight between the M1-138-treated group (4 mg/Kg/day×14 days) and the control group at the end of the experiments (Figure S14A). From the hemolytic test of M1-138 to blood cells, we found that a relative high concentration of M1-138 treatment (200 µg/mL, 3 h) resulted in very mild hemolysis to erythrocytes (Hemolytic Rate < 2%) (Figure S14B), proving that M1-138 was well tolerated by normal cells. We also found that wild type ICR/JCL mice could tolerate the dosage of M1-138 as high as 180 mg/Kg body weight by intraperitoneal (I.P.) injection with no observed toxicity (Figure S14C). Another issue worth concerning is the potential immunogenicity of M1-138. From the serum of wild type ICR/JCL mice treated with M1-138 (4 mg/Kg/week×4 weeks, IP injection), we detected the generation of an antibody specific to M1-138 (data not shown). If multiple dosing of M1-138 is needed in the future cancer therapy, the immunogenicity of M1-138 is worthy of attention but can be controlled according to current clinical practice rules for drugs with immunogenicity [49]. Interestingly, it has been found that certain peptides from different regions of FOXM1 protein can prime HLA-A2-restricted cytotoxic T lymphocytes [50] and a phase I clinical trial of FOXM1 peptide to vaccinate cervical cancer patients is finished with a promising conclusion [51], suggesting that FOXM1 protein-induced immune response can be utilized for immunotherapy against cancers. It is an ongoing study in our lab to test whether M1-138-induced immune response prevents the primary tumor generation in the mouse model and whether M1-138 can be considered as a vaccine against cancers, hopefully providing an additional advantage for the cancer treatment of M1-138. Though a more thorough analysis is required, at least as far as tested here M1-138 appears to be well tolerated, which is an absolutely critical milestone to pass when aiming to perform the next step for clinical trials.
M1-138 was designed and produced as a cell-penetrating protein. There are more than 100 peptide sequences capable of penetrating the plasma membrane being identified [52]. Among them, polyarginine peptide (such as R9) is a purely synthetic penetrating peptide and can be modified to improve stability and transmembrane efficiency [31]. It is generally believed that penetrating peptides enter into cells by an energy-dependent endocytosis or by physical endocytosis and direct cell membrane translocation [53]. R9 is found to enter into cells at a low concentration (≤5 µM) through endocytosis but directly cross the plasma membrane at high concentrations (≥10 µM) [31]. In this study, R9 was confirmed again to penetrate the plasma membrane of cells and nuclei effectively because of the distribution of M1-138 in both cytoplasm and nucleus of cancer cells treated at 8 μM, while the majority of the protein staying at nuclei. Furthermore, cell type-specific penetrating peptides that enter only certain types of cancer cells are also available to mediate bioactive substances into particular cancer cells without entering normal or other types of cancer cells [54]. It will be worth studying in the future whether modifying M1-138 with cell type-specific penetrating peptides can improve its targeting and reduce its cytotoxicity for the treatment of certain type of cancers.
Compared to broad-range inhibitors that disrupt the activity of single domains in target proteins, M1-138 might have advantages to reduce the possibility of drug resistance in clinical applications. It is well known that continuous dosing of the inhibitors targeting single domains of proteins eventually induces the emergence of drug resistance, leading to limited therapeutic durability [55]. In contrast, M1-138 was selected from the natural sequence of the N-terminus of FOXM1 protein and possessed potentials to block multiple FOXM1 N-terminus-involved protein-protein interactions. In so doing, M1-138 can selectively modulate multiple specific downstream signaling events in cancer cells. In this study, we confirmed that M1-138 jeopardized the functions of both FOXM1 and SMAD3, leading to a variety of phenotypic inhibition of cancer cells. Currently we are performing Mass Spectrometry Analysis with M1-138-pull down lysate samples to identify M1-138-interacting proteins, which may provide a whole picture of M1-138 potential impact on multiple cancer-related signaling pathways. In theory, it will also provide evidence that the effects of M1-138 may be better than that of FOXM1 small molecule inhibitors and FOXM1 shRNAs in cancer therapy.
Supplementary Material
Supplementary methods, figures and tables.
Acknowledgements
The authors thank Dr. Xiaohua Shen of Tsinghua University, China for sharing the pCMV-BirA vector.
Authors' Contributions
Conception and design: Z. Zhang, J. Yu, Y. Tan
Development of methodology: Z. Zhang, Y. Chen, L. Yu, G. Tan
Acquisition of data (provided animals, acquired and managed patients, provided facilities, etc.): Y. Chen, C. Pei, H. Bu
Analysis and interpretation of data (e.g., statistical analysis, biostatistics, computational analysis): Z. Zhang, J. Yu, Y. Tan
Writing, review, and/or revision of the manuscript: Z. Zhang, Y. Tan
Administrative, technical, or material support (i.e., reporting or organizing data, constructing databases): Y. Chen, L. Yu, X. Huang, G. Tan
Study supervision: G. Tan, Y. Tan
Grant Support
This work was supported by the National Natural Science Foundation of China (grant number 81773169 to Y.T., 81472718 to Y.T., 31701245 to G.T.), China Changsha Development and Reform Commission “Mass entrepreneurship and innovation program” (2018-68) and “Innovation platform construction program” (2018-216).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Kaestner KH, Knochel W, Martinez DE. Unified nomenclature for the winged helix/forkhead transcription factors. Genes Dev. 2000;14:142-6
2. Ye H, Kelly TF, Samadani U, Lim L, Rubio S, Overdier DG. et al. Hepatocyte nuclear factor 3/fork head homolog 11 is expressed in proliferating epithelial and mesenchymal cells of embryonic and adult tissues. Mol Cell Biol. 1997;17:1626-41
3. Laoukili J, Stahl M, Medema RH. FoxM1: at the crossroads of ageing and cancer. Biochim Biophys Acta. 2007;1775:92-102
4. Tan Y, Raychaudhuri P, Costa RH. Chk2 mediates stabilization of the FoxM1 transcription factor to stimulate expression of DNA repair genes. Mol Cell Biol. 2007;27:1007-16
5. Xie Z, Tan G, Ding M, Dong D, Chen T, Meng X. et al. Foxm1 transcription factor is required for maintenance of pluripotency of P19 embryonal carcinoma cells. Nucleic Acids Res. 2010;38:8027-38
6. Tan G, Cheng L, Chen T, Yu L, Tan Y. Foxm1 Mediates LIF/Stat3-Dependent Self-Renewal in Mouse Embryonic Stem Cells and Is Essential for the Generation of Induced Pluripotent Stem Cells. PLoS One. 2014;9:e92304
7. Yang C, Chen H, Tan G, Gao W, Cheng L, Jiang X. et al. FOXM1 promotes the epithelial to mesenchymal transition by stimulating the transcription of Slug in human breast cancer. Cancer Lett. 2013;340:104-12
8. Huang C, Xie D, Cui J, Li Q, Gao Y, Xie K. FOXM1c promotes pancreatic cancer epithelial-to-mesenchymal transition and metastasis via upregulation of expression of the urokinase plasminogen activator system. Clin Cancer Res. 2014;20:1477-88
9. Koo CY, Muir KW, Lam EW. FOXM1: From cancer initiation to progression and treatment. Biochim Biophys Acta. 2011;1819:28-37
10. Kalinichenko VV, Major ML, Wang X, Petrovic V, Kuechle J, Yoder HM. et al. Foxm1b transcription factor is essential for development of hepatocellular carcinomas and is negatively regulated by the p19ARF tumor suppressor. Genes Dev. 2004;18:830-50
11. Kim IM, Ackerson T, Ramakrishna S, Tretiakova M, Wang IC, Kalin TV. et al. The Forkhead Box m1 transcription factor stimulates the proliferation of tumor cells during development of lung cancer. Cancer Res. 2006;66:2153-61
12. Yoshida Y, Wang IC, Yoder HM, Davidson NO, Costa RH. The forkhead box M1 transcription factor contributes to the development and growth of mouse colorectal cancer. Gastroenterology. 2007;132:1420-31
13. Halasi M, Gartel AL. Targeting FOXM1 in cancer. Biochem Pharmacol. 2013;85:644-52
14. Chen H, Yang C, Yu L, Xie L, Hu J, Zeng L. et al. Adenovirus-mediated RNA interference targeting FOXM1 transcription factor suppresses cell proliferation and tumor growth of nasopharyngeal carcinoma. J Gene Med. 2012;14:231-40
15. Chen T, Xiong J, Yang C, Shan L, Tan G, Yu L. et al. Silencing of FOXM1 transcription factor expression by adenovirus-mediated RNA interference inhibits human hepatocellular carcinoma growth. Cancer Gene Ther. 2014;21:133-8
16. Yang C, Chen H, Yu L, Shan L, Xie L, Hu J. et al. Inhibition of FOXM1 transcription factor suppresses cell proliferation and tumor growth of breast cancer. Cancer Gene Ther. 2013;20:117-24
17. Gormally MV, Dexheimer TS, Marsico G, Sanders DA, Lowe C, Matak-Vinkovic D. et al. Suppression of the FOXM1 transcriptional programme via novel small molecule inhibition. Nat Commun. 2014;5:5165
18. Kwok JM, Myatt SS, Marson CM, Coombes RC, Constantinidou D, Lam EW. Thiostrepton selectively targets breast cancer cells through inhibition of forkhead box M1 expression. Mol Cancer Ther. 2008;7:2022-32
19. Radhakrishnan SK, Bhat UG, Hughes DE, Wang IC, Costa RH, Gartel AL. Identification of a chemical inhibitor of the oncogenic transcription factor forkhead box M1. Cancer Res. 2006;66:9731-5
20. Xiang Q, Tan G, Jiang X, Wu K, Tan W, Tan Y. Suppression of FOXM1 Transcriptional Activities via a Single-Stranded DNA Aptamer Generated by SELEX. Sci Rep. 2017;7:45377
21. Gusarova GA, Wang IC, Major ML, Kalinichenko VV, Ackerson T, Petrovic V. et al. A cell-penetrating ARF peptide inhibitor of FoxM1 in mouse hepatocellular carcinoma treatment. J Clin Invest. 2007;117:99-111
22. Bi Z, Liu W, Ding R, Wu Y, Dou R, Zhang W. et al. A novel peptide, 9R-P201, strongly inhibits the viability, proliferation and migration of liver cancer HepG2 cells and induces apoptosis by down-regulation of FoxM1 expression. Eur J Pharmacol. 2017;796:175-89
23. Wang IC, Chen YJ, Hughes D, Petrovic V, Major ML, Park HJ. et al. Forkhead box M1 regulates the transcriptional network of genes essential for mitotic progression and genes encoding the SCF (Skp2-Cks1) ubiquitin ligase. Mol Cell Biol. 2005;25:10875-94
24. Sadasivam S, Duan S, DeCaprio JA. The MuvB complex sequentially recruits B-Myb and FoxM1 to promote mitotic gene expression. Genes Dev. 2012;26:474-89
25. Chen X, Muller GA, Quaas M, Fischer M, Han N, Stutchbury B. et al. The forkhead transcription factor FOXM1 controls cell cycle-dependent gene expression through an atypical chromatin binding mechanism. Mol Cell Biol. 2013;33:227-36
26. Fischer M, Muller GA. Cell cycle transcription control: DREAM/MuvB and RB-E2F complexes. Crit Rev Biochem Mol Biol. 2017;52:638-62
27. Sanders DA, Gormally MV, Marsico G, Beraldi D, Tannahill D, Balasubramanian S. FOXM1 binds directly to non-consensus sequences in the human genome. Genome Biol. 2015;16:130
28. Zhang N, Wei P, Gong A, Chiu WT, Lee HT, Colman H. et al. FoxM1 promotes beta-catenin nuclear localization and controls Wnt target-gene expression and glioma tumorigenesis. Cancer Cell. 2011;20:427-42
29. Xue J, Lin X, Chiu WT, Chen YH, Yu G, Liu M. et al. Sustained activation of SMAD3/SMAD4 by FOXM1 promotes TGF-beta-dependent cancer metastasis. J Clin Invest. 2014;124:564-79
30. Wierstra I, Alves J. Despite its strong transactivation domain, transcription factor FOXM1c is kept almost inactive by two different inhibitory domains. Biol Chem. 2006;387:963-76
31. Mitchell DJ, Kim DT, Steinman L, Fathman CG, Rothbard JB. Polyarginine enters cells more efficiently than other polycationic homopolymers. J Pept Res. 2000;56:318-25
32. Laoukili J, Alvarez M, Meijer LA, Stahl M, Mohammed S, Kleij L. et al. Activation of FoxM1 during G2 requires cyclin A/Cdk-dependent relief of autorepression by the FoxM1 N-terminal domain. Mol Cell Biol. 2008;28:3076-87
33. de Boer E, Rodriguez P, Bonte E, Krijgsveld J, Katsantoni E, Heck A. et al. Efficient biotinylation and single-step purification of tagged transcription factors in mammalian cells and transgenic mice. Proc Natl Acad Sci U S A. 2003;100:7480-5
34. Zhu G, Spellman PT, Volpe T, Brown PO, Botstein D, Davis TN. et al. Two yeast forkhead genes regulate the cell cycle and pseudohyphal growth. Nature. 2000;406:90-4
35. Ye H, Holterman AX, Yoo KW, Franks RR, Costa RH. Premature expression of the winged helix transcription factor HFH-11B in regenerating mouse liver accelerates hepatocyte entry into S phase. Mol Cell Biol. 1999;19:8570-80
36. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68:394-424
37. Chai EZ, Shanmugam MK, Arfuso F, Dharmarajan A, Wang C, Kumar AP. et al. Targeting transcription factor STAT3 for cancer prevention and therapy. Pharmacol Ther. 2016;162:86-97
38. Li F, Sethi G. Targeting transcription factor NF-kappaB to overcome chemoresistance and radioresistance in cancer therapy. Biochim Biophys Acta. 2010;1805:167-80
39. Luu HH, Zhang R, Haydon RC, Rayburn E, Kang Q, Si W. et al. Wnt/beta-catenin signaling pathway as a novel cancer drug target. Curr Cancer Drug Targets. 2004;4:653-71
40. Javelaud D, Alexaki VI, Dennler S, Mohammad KS, Guise TA, Mauviel A. TGF-beta/SMAD/GLI2 signaling axis in cancer progression and metastasis. Cancer Res. 2011;71:5606-10
41. Adami GR, Ye H. Future roles for FoxM1 inhibitors in cancer treatments. Future Oncol. 2007;3:1-3
42. Darnell JE Jr. Transcription factors as targets for cancer therapy. Nat Rev Cancer. 2002;2:740-9
43. Kang JH, Jung MY, Yin X, Andrianifahanana M, Hernandez DM, Leof EB. Cell-penetrating peptides selectively targeting SMAD3 inhibit profibrotic TGF-beta signaling. J Clin Invest. 2017;127:2541-54
44. Baar MP, Brandt RMC, Putavet DA, Klein JDD, Derks KWJ, Bourgeois BRM. et al. Targeted Apoptosis of Senescent Cells Restores Tissue Homeostasis in Response to Chemotoxicity and Aging. Cell. 2017;169:132-47 e16
45. Hsieh TH, Hsu CY, Tsai CF, Chiu CC, Liang SS, Wang TN. et al. A novel cell-penetrating peptide suppresses breast tumorigenesis by inhibiting beta-catenin/LEF-1 signaling. Sci Rep. 2016;6:19156
46. Anders L, Ke N, Hydbring P, Choi YJ, Widlund HR, Chick JM. et al. A systematic screen for CDK4/6 substrates links FOXM1 phosphorylation to senescence suppression in cancer cells. Cancer Cell. 2011;20:620-34
47. McCain J. First-in-Class CDK4/6 Inhibitor Palbociclib Could Usher in a New Wave of Combination Therapies for HR+, HER2- Breast Cancer. P T. 2015;40:511-20
48. Wierstra I, Alves J. FOXM1c is activated by cyclin E/Cdk2, cyclin A/Cdk2, and cyclin A/Cdk1, but repressed by GSK-3alpha. Biochem Biophys Res Commun. 2006;348:99-108
49. De Groot AS, Martin W. Reducing risk, improving outcomes: bioengineering less immunogenic protein therapeutics. Clin Immunol. 2009;131:189-201
50. Yokomine K, Senju S, Nakatsura T, Irie A, Hayashida Y, Ikuta Y. et al. The forkhead box M1 transcription factor as a candidate of target for anti-cancer immunotherapy. Int J Cancer. 2010;126:2153-63
51. Hasegawa K, Ikeda Y, Kunugi Y, Kurosaki A, Imai Y, Kohyama S. et al. Phase I Study of Multiple Epitope Peptide Vaccination in Patients With Recurrent or Persistent Cervical Cancer. J Immunother. 2018;41:201-7
52. Koren E, Torchilin VP. Cell-penetrating peptides: breaking through to the other side. Trends Mol Med. 2012;18:385-93
53. Jiao CY, Delaroche D, Burlina F, Alves ID, Chassaing G, Sagan S. Translocation and endocytosis for cell-penetrating peptide internalization. J Biol Chem. 2009;284:33957-65
54. Kondo E, Saito K, Tashiro Y, Kamide K, Uno S, Furuya T. et al. Tumour lineage-homing cell-penetrating peptides as anticancer molecular delivery systems. Nat Commun. 2012;3:951
55. Gottesman MM. Mechanisms of cancer drug resistance. Annu Rev Med. 2002;53:615-27
Author contact
![]() Corresponding author: Guixiang Tan and Yongjun Tan State Key Laboratory of Chemo/Biosensing and Chemometrics, College of Biology, Hunan Engineering Research Center for Anticancer Targeted Protein Pharmaceuticals, Hunan University, Changsha, Hunan 410082, China. Email: yjtanedu.cn and TGX2222com
Corresponding author: Guixiang Tan and Yongjun Tan State Key Laboratory of Chemo/Biosensing and Chemometrics, College of Biology, Hunan Engineering Research Center for Anticancer Targeted Protein Pharmaceuticals, Hunan University, Changsha, Hunan 410082, China. Email: yjtanedu.cn and TGX2222com