Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
RNA modifications: chemical...
Epitranscriptomic regulation of...
RNA modification-driven...
Challenges and perspectives
Conclusion
Abbreviations
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2026; 16(5):2576-2597. doi:10.7150/thno.124482 This issue Cite
Review
RNA Modifications in T cell Immunity: Mechanisms, Disease Relevance, and Therapeutic Potential
Xinyuan Zhao#, Meiyan Zou#, Weiyao Feng, Nina Li, Xu Chen, Pei Lin, Zihao Zhou, Yunfan Lin, Li Cui ![]()
Stomatological Hospital, School of Stomatology, Southern Medical University, Guangzhou, 510280, Guangdong, China.
# Equal contributors.
Received 2025-8-31; Accepted 2025-11-27; Published 2026-1-1
Abstract

RNA modifications constitute a versatile and dynamic layer of post-transcriptional regulation that enables T lymphocytes to fine-tune gene expression programs in response to developmental, environmental, and pathogenic cues. Chemical marks such as N6-methyladenosine (m6A), 5-methylcytosine (m5C), and pseudouridine (Ψ) shape transcript stability, splicing, localization, and translation through coordinated actions of writer, reader, and eraser proteins. Emerging evidence reveals that these pathways orchestrate T cell lineage specification, activation thresholds, effector-memory balance, and immune tolerance, while their dysregulation contributes to infection, autoimmunity, malignancy, and graft rejection. Integrating findings across m6A and other epitranscriptomic marks—including m5C, Ψ, N7-methylguanosine (m7G), N1-methyladenosine (m1A), N4-acetylcytidine (ac4C), and N6-2'-O-methyladenosine (m6Am) —this review delineates how distinct RNA modifications converge on shared molecular circuits controlling transcriptional, metabolic, and signaling networks in T cell immunity. Aberrant modification patterns reshape cytokine profiles, mitochondrial metabolism, and antigen-driven responses, thereby influencing disease trajectories across diverse pathological contexts. Collectively, these insights establish RNA modification as a central regulatory axis linking transcriptomic plasticity to immune function and therapeutic responsiveness. We further highlight unresolved challenges—such as defining spatiotemporal modification landscapes and achieving selective pharmacological modulation—and propose integrative multi-omics and in vivo perturbation approaches to translate epitranscriptomic mechanisms into targeted immunotherapies.
Keywords: RNA modifications, T cell immunity, therapeutic potential, epitranscriptomics, immune regulation
Introduction
T lymphocytes constitute the cornerstone of adaptive immunity, orchestrating antigen-specific recognition, clonal expansion, and effector responses that collectively ensure the elimination of infected or malignant cells while maintaining immune tolerance and tissue homeostasis [1-3]. Their capacity to mount precise and flexible immune responses relies on the coordinated interplay of multiple regulatory layers, encompassing transcriptional, epigenetic, post-transcriptional, and metabolic control [4-6]. These mechanisms operate in an integrated and temporally synchronized manner to align T cell activation, differentiation, and resolution of effector responses with immunological demands. Disruption of this multilayered regulation can result in either immune insufficiency or immunopathology, underscoring the necessity for precise control mechanisms that enable T cells to dynamically adapt to diverse environmental and pathogenic cues [7, 8].
Among these regulatory dimensions, post-transcriptional control is particularly crucial. Following antigen encounter, T cells must rapidly remodel their transcriptome and proteome to sustain proliferation, metabolic reprogramming, and effector molecule production [9-11]. Unlike transcriptional regulation, which requires new gene activation, post-transcriptional processes allow immediate adjustment of existing transcripts, conferring temporal precision and energetic efficiency. RNA modifications have recently emerged as a central post-transcriptional mechanism that adds a chemical layer of information to RNA molecules [12]. Through covalent modification of messenger RNAs, transfer RNAs, ribosomal RNAs, and non-coding RNAs, chemical marks such as N6-methyladenosine (m6A), 5-methylcytosine (m5C), pseudouridine (Ψ), N7-methylguanosine (m7G), N1-methyladenosine (m1A), and N4-acetylcytidine (ac4C) dynamically regulate RNA stability, splicing, export, localization, and translation [13]. These modifications are installed, recognized, and removed by dedicated “writer”, “reader”, and “eraser” proteins that collectively integrate upstream signaling events with downstream gene expression outcomes [14, 15].
Mounting evidence indicates that these RNA modification pathways act as key modulators of T cell fate and function [16]. By controlling the stability and translation of lineage-defining transcription factors, signaling mediators, and metabolic enzymes, RNA modifications influence T cell differentiation, activation thresholds, effector-memory transitions, and regulatory T cell stability [17-22]. They serve as molecular interfaces that couple cytokine and metabolic signals to transcriptional and functional reprogramming, ensuring the precise tuning of immune responses. Conversely, perturbations in RNA modification machinery—whether through altered enzyme expression, localization, or substrate availability—can rewire T cell programs, promoting immune dysregulation, chronic inflammation, or tumor immune evasion [10]. These findings position the epitranscriptome as a pivotal integrator that links transcriptional, metabolic, and signaling networks to T cell functionality and immune homeostasis.
Despite substantial advances, critical gaps remain in understanding how distinct RNA modifications are coordinated to regulate T cell biology. The temporal and spatial dynamics of modification deposition during T cell activation and differentiation remain incompletely mapped. Moreover, the functional interplay among different modification types—such as m6A, m5C, and Ψ—and their crosstalk with chromatin remodeling and non-coding RNA networks are only beginning to be explored. Technical limitations in detecting low-abundance modifications within small immune subsets, coupled with the challenge of causally linking modification events to specific functional outcomes in vivo, continue to constrain mechanistic insight. A comprehensive and integrative framework is therefore needed to elucidate how RNA modifications orchestrate the molecular circuitry underlying T cell immunity. Deciphering this interplay will deepen our understanding of immune regulation and open new therapeutic avenues. Selective targeting of RNA modification enzymes or pathways holds promise for reprogramming immune responses in autoimmunity, infection, cancer, and transplantation, representing an emerging frontier for precision immunotherapy.
In this work, we integrate current knowledge across three interrelated dimensions that collectively distinguish it from previous publications. First, we provide a comprehensive synthesis of multiple RNA modification types—including m6A, m5C, Ψ, m7G, m1A, ac4C, and N6-2'-O-methyladenosine (m6Am) —in the context of T cell biology, rather than focusing on a single modification or immune cell subset as in prior reviews. Second, we systematically examine how these epitranscriptomic mechanisms shape diverse disease landscapes, extending beyond cancer and autoimmunity to encompass viral and bacterial infections, cardiovascular disease, and transplant immunology. Third, we emphasize mechanistic depth, detailing how RNA modifications regulate signaling pathways, transcriptional networks, and metabolic programs underlying T cell activation, lineage specification, and immune tolerance. Finally, we outline future challenges and directions, such as spatiotemporal mapping and therapeutic translation, thereby establishing a conceptual and technological framework that advances the field beyond existing summaries of m6A-centric regulation.
RNA modifications: chemical principles, analytical strategies, and functional implications
Chemical basis and regulatory mechanisms of RNA modifications
RNA modifications constitute a dynamic post-transcriptional layer of gene regulation that fine-tunes RNA stability, translation, and localization [15]. More than 170 distinct chemical marks have been identified [23], among which m6A [24], m5C [25], Ψ [26], m7G [27], m1A [28], and ac4C [29] are the most functionally characterized. These modifications occur at defined sequence motifs or structural contexts and are catalyzed by dedicated enzymatic machineries commonly referred to as writers, erasers, and readers [30, 31].
The m6A writer complex, composed of METTL3, METTL14, and WTAP, acts co-transcriptionally on DRACH motifs to control mRNA turnover and translation [32-34]. Accessory factors such as VIRMA, RBM15/15B, and ZC3H13 regulate substrate selectivity and subcellular deposition [35-38]. In T cells, METTL3-mediated methylation promotes clonal expansion and effector differentiation, whereas its loss impairs memory maintenance and cytokine expression [17]. Reversibility is provided by erasers—FTO and ALKBH5—which demethylate m6A-marked transcripts to modulate apoptosis and metabolic adaptation [39, 40]. Reader proteins such as YTHDF1/2/3 and YTHDC1/2 interpret these marks to determine RNA stability, translation efficiency, and localization, thereby coupling post-transcriptional control to T cell activation thresholds [41-47].
Beyond m6A, other modification machineries participate in immune regulation. The NSUN family installs m5C marks that enhance translational fidelity and mitochondrial homeostasis [48-54], while DNMT2 methylates tRNAAsp to sustain protein synthesis under stress [55]. Ψ synthases (PUS10) and the acetyltransferase NAT10 introduce Ψ and ac4C modifications, respectively, to stabilize RNA conformation and improve decoding accuracy [56, 57]. The m7G cap is installed by the RNMT-RAM complex, which is essential for mRNA stability, nuclear export, and efficient translation initiation [58]. The TRMT6/61A complex catalyzes m1A modification primarily at position 58 of tRNAs, maintaining translational fidelity and tRNA stability [59]. Importantly, these enzymatic systems are not static; their abundance and activity are governed by metabolic and signaling pathways—such as mTOR, HIF-1α, and cytokine-driven JAK/STAT cascades—linking extracellular cues to post-transcriptional outputs that orchestrate T cell activation, differentiation, and exhaustion [60-62]. Together, these interdependent machineries establish a multilayered regulatory hierarchy through which diverse RNA marks confer transcriptional flexibility, metabolic adaptability, and immune precision upon T cells.
Technologies and analytical approaches for RNA modifications
Technological advances in biochemical and sequencing methodologies have revolutionized the detection and quantification of RNA modifications at transcriptome-wide and even single-molecule resolution. Conventional mapping strategies fall into three main categories: chemical or enzymatic probing, immunoprecipitation-based enrichment, and direct detection using nanopore or single-molecule real-time sequencing [63]. Among these, nanopore direct RNA sequencing (DRS) has emerged as a powerful approach that enables simultaneous identification of multiple modification types without chemical pretreatment, offering quantitative insights into modification stoichiometry and transcript distribution [64]. Computational frameworks complement these advances by integrating raw signal features, sequence context, and biological replicates to distinguish true modification events from background noise [65]. Deep learning-based algorithms such as modCNet further improve precision, allowing concurrent identification of multiple cytidine modifications, including m5C and ac4C, from a single sample [66]. These developments expand the analytical capacity to dissect complex modification networks that coordinate RNA stability, translation, and metabolism. Although direct single-cell mapping of RNA modifications remains technically challenging, integration of modification profiling with single-cell transcriptomic and spatial analysis has begun to reveal how RNA modification enzyme expression and activity vary across T cell states and tissue niches. Such combined approaches promise to elucidate how post-transcriptional regulation contributes to T cell activation, differentiation, and exhaustion within diverse immune microenvironments.
Biological significance and functional consequences of RNA modifications in T cells
RNA modifications form an essential post-transcriptional regulatory layer that enables T cells to fine-tune gene expression programs in response to environmental and metabolic cues [67, 68]. By introducing reversible chemical marks onto coding and non-coding RNAs, these pathways control transcript stability, splicing, export, and translation, allowing T cells to rapidly transition between quiescent, activated, and memory states [69]. This dynamic adaptability ensures that immune responses are both flexible and precisely timed without requiring complete transcriptional reprogramming. Functionally, the epitranscriptomic machinery establishes a coordinated network that balances activation, persistence, and exhaustion. Individual RNA modifications do not act in isolation but instead intersect to modulate shared molecular circuits governing transcriptional, metabolic, and signaling pathways [67, 70]. By integrating these post-transcriptional checkpoints, T cells dynamically adjust to antigenic load, nutrient fluctuations, and cytokine gradients, maintaining effector potential while preventing overstimulation or premature dysfunction.
Recent discoveries highlight that aberrant RNA modification programs can profoundly influence T cell fate and immune homeostasis. Elevated METTL16-mediated m6A deposition increases methylation of TCF-1 mRNA, limiting TCF-1 stability and constraining the pool of self-renewing precursors essential for sustained cytotoxic responses. Loss of METTL16 restores TCF-1-driven stem-like properties, improving chimeric antigen receptor (CAR)-T cell persistence and antitumor efficacy [71]. Conversely, excessive METTL3-dependent m6A methylation stabilizes inhibitory receptor transcripts and suppresses cytokine production, reinforcing T cell exhaustion under chronic stimulation. Inhibition of METTL3 reverses these effects and reactivates effector function [72]. Beyond m6A, additional RNA modification systems exert equally critical control over T cell biology. ac4C modification of mRNA by NAT10 enhances translational efficiency of MYC transcripts upon activation, driving cell cycle progression and clonal expansion. Loss of NAT10 impairs proliferation and antiviral immunity, and its age-associated decline contributes to reduced immune responsiveness, positioning RNA acetylation as a determinant of T cell vigor [73]. In parallel, NSUN4-mediated m5C modification promotes CD8+ T cell exhaustion in systemic lupus erythematosus (SLE) by elevating CD74 expression and activating CD44-mTOR-dependent mitophagy. Silencing NSUN4 with siRNA-loaded nanoparticles restores mitochondrial integrity and cytotoxic capacity, underscoring m5C methylation as a key post-transcriptional regulator of immune dysfunction [60]. Collectively, these insights define RNA modifications as integral mediators of T cell function that couple environmental sensing with metabolic and transcriptional programs. Their context-dependent and reversible nature endows T cells with functional precision yet also creates vulnerabilities that can be therapeutically exploited. Selective modulation of RNA-modifying enzymes therefore represents a promising approach to reprogram immune cell states and restore immune competence across cancer, infection, and autoimmune disease.
Epitranscriptomic regulation of T cell development and function
RNA modifications, particularly m6A, m5C, Ψ, and other chemical marks, have emerged as pivotal post-transcriptional regulators of T cell fate, effector function, and immune homeostasis [74-77]. These modifications influence mRNA stability, splicing, translation, and decay, thereby integrating environmental cues, metabolic status, and activation signals into precise gene-expression programs [78]. Accumulating evidence from distinct T cell subsets reveals that RNA modification enzymes and readers orchestrate immune responses through highly context-dependent mechanisms [79, 80].
m6A modification in CD8+ T cell differentiation and survival
In the cytotoxic T cell lineage, m6A methylation serves as a pivotal epitranscriptomic regulator that orchestrates both effector differentiation and memory maintenance [81, 82]. The methyltransferase METTL3 is essential for the expansion and lineage progression of CD8+ T cells during acute viral infection. Conditional Mettl3 deletion impairs effector proliferation and terminal differentiation, consequently compromising memory formation and secondary responses. Integrative RNA-seq and m6A-miCLIP-seq analyses demonstrate that METTL3 modulates a network of cell-cycle and transcriptional regulators, including stabilizing Tbx21 mRNA to sustain T-bet expression and effector programming. Ectopic T-bet expression partially restores differentiation defects, underscoring a direct mechanistic link between m6A-mediated mRNA stabilization and lineage-specifying transcriptional circuits [17]. In parallel, m6A demethylation provides a counter-regulatory mechanism that preserves cytotoxic T cell survival. The demethylase FTO fine-tunes activation-induced apoptosis by modulating m6A methylation on Fas transcripts. Loss of FTO leads to excessive methylation and IGF2BP3-dependent stabilization of Fas mRNA, resulting in upregulated Fas expression and enhanced apoptosis in activated CD8+ T cells. Disruption of the Fas m6A sites or IGF2BP3 knockdown restores normal Fas turnover and rescues T cell viability [79]. Collectively, these findings delineate an integrated post-transcriptional program in which m6A dynamically couples transcriptional reprogramming, cell-cycle progression, and apoptotic control to coordinate CD8+ T cell fate decisions. By adjusting the stability and translation of lineage-defining and pro-survival transcripts, m6A ensures the precise balance between effector expansion and memory persistence, providing a mechanistic framework for epitranscriptomic regulation of cytotoxic immunity.
m6A-dependent post-transcriptional regulation of CD4+ T cell activation and effector function
Building on the regulatory logic established in cytotoxic T cells, m6A methylation exerts multifaceted control over CD4+ T cell activation and lineage specialization by coordinating cytokine expression, co-stimulatory signaling, and transcriptional programming [83, 84]. In CD4+ T lymphocytes, m6A dynamically shapes effector responses through selective regulation of transcript stability. Upon activation, Tnf mRNA acquires increased m6A deposition within its 3′ untranslated region, enabling recognition by the reader protein YTHDF2, which accelerates mRNA decay and constrains TNF production. This post-transcriptional mechanism links RNA methylation to the resolution of inflammatory signaling, preventing excessive cytokine output while sustaining appropriate effector activity [80]. m6A methylation also governs the expression of CD40L, a co-stimulatory molecule essential for CD4+ T cell activation and immune coordination. The antagonistic actions of METTL3 and FTO modulate the methylation status of Cd40l transcripts, whereas YTHDF2 promotes degradation of methylated mRNAs. This regulatory circuit fine-tunes CD40L expression and downstream cytokine programs, thereby determining the amplitude and qualitative polarization of CD4+ T cell responses [85]. In specialized helper subsets, METTL3-catalyzed m6A deposition preserves the stability of lineage-defining transcripts and maintains the transcriptional network of T follicular helper (Tfh) cells. Loss of METTL3 in CD4+ T cells impairs germinal-center formation and reduces expression of Tcf7, Bcl6, Icos, and Cxcr5 in a methyltransferase activity-dependent manner. m6A deposition within the Tcf7 3′UTR stabilizes the transcript, whereas its removal accelerates decay and disrupts TCF-1-driven gene programs. Restoration of TCF-1 rescues these defects, directly linking m6A-dependent RNA stabilization to the maintenance of helper-cell identity and humoral immunity [86]. Within the regulatory lineage, METTL14 expression increases during the induction of induced regulatory T cells (iTregs) from naïve CD4+ precursors, where it sustains FOXP3 expression and suppressive function. Silencing METTL14 destabilizes Foxp3 mRNA, impairs iTreg differentiation, and enhances expression of pro-inflammatory cytokines such as IFN-γ and IL-17A, leading to compromised immunosuppression in vitro and in colitis models. Mechanistically, METTL14 deficiency activates the mTOR-p70S6K signaling cascade, a pathway known to undermine iTreg stability and function [87]. Collectively, these findings reveal m6A methylation as a key post-transcriptional mechanism that integrates signaling intensity, transcriptional programming, and metabolic state to coordinate CD4+ T cell activation and lineage differentiation. Through the concerted actions of methyltransferases, demethylases, and reader proteins, m6A dynamically governs the stability and translation of transcripts that define effector, helper, and regulatory fates. This integrated framework underscores the central role of RNA methylation in maintaining CD4+ T cell functional balance and highlights its pathological relevance in autoimmunity, chronic inflammation, and tumor immunity.
m6A-mediated specification of γδ T and iNKT cell lineages
Extending beyond conventional αβ T cells, m6A methylation also governs unconventional T cell subsets that bridge innate and adaptive immunity [88]. γδ T cells rapidly respond to stress signals and maintain epithelial integrity, whereas invariant natural killer T (iNKT) cells recognize lipid antigens presented by CD1d and exert potent immunoregulatory and antitumour functions through immediate cytokine secretion [89]. These lineages rely on distinct transcriptional programs and thymic developmental cues, both of which are subject to post-transcriptional regulation by m6A.
Loss of the RNA demethylase ALKBH5 in lymphocytes results in a pronounced expansion of γδ T cells, enhancing mucosal defence against Salmonella typhimurium. In thymocytes, ALKBH5 deficiency elevates global m6A levels and represses critical Notch pathway components—including Jagged1 and Notch2—thereby attenuating Notch signalling. The resulting transcriptional shift promotes γδ T cell precursor proliferation and differentiation, expanding the mature γδ T cell compartment [90]. In addition, METTL3-dependent methylation fine-tunes γδ T cell subset balance by regulating mRNA stability and endogenous double-stranded RNA (dsRNA) levels. By promoting Stat1 mRNA degradation and preventing dsRNA accumulation, m6A methylation limits aberrant STAT1 activation, thereby sustaining γδ T17 differentiation and IL-17 production [91].
In the iNKT lineage, METTL14-dependent m6A modification is indispensable for developmental programming and survival. Deletion of METTL14 increases apoptosis in double-positive thymocytes, impairs Vα14-Jα18 rearrangement, and markedly reduces iNKT cell numbers. Residual iNKT cells exhibit defective maturation, enhanced apoptosis, and impaired cytokine production due to compromised IL-2/IL-15 responsiveness, diminished TCR signalling, and elevated Cish expression [92]. Similarly, METTL3-dependent methylation intrinsically maintains iNKT cell homeostasis and effector specification by stabilizing Creb1 mRNA. Conditional ablation of METTL3 in CD4+CD8+ double-positive thymocytes disrupts m6A-dependent CREB1 translation and phosphorylation, impairing iNKT cell proliferation, lineage differentiation, and cytokine production, ultimately compromising antitumour immunity against melanoma. Restoration of Creb1 expression rescues these defects, defining a critical m6A-CREB1 regulatory axis that orchestrates transcriptional networks controlling iNKT cell development and effector programming [93] (Figure 1). Together, these findings position m6A as a key post-transcriptional regulator that integrates thymic developmental signalling, transcription factor networks, and cytokine responsiveness to shape unconventional T cell lineages. Through coordinated actions of its writer and eraser enzymes, m6A establishes transcriptional thresholds governing γδ T cell subset composition and iNKT cell maturation, thereby aligning innate-like T cell plasticity with tissue immunity and antitumour defence.
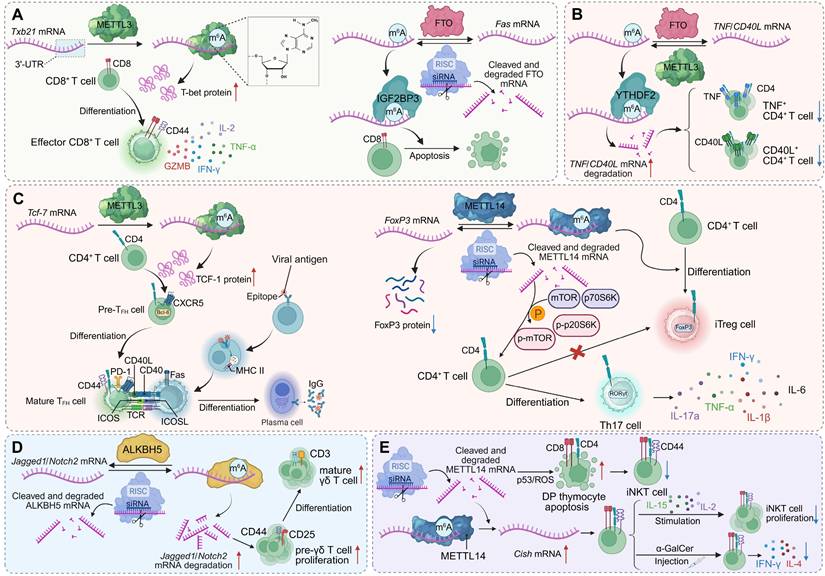
m6A modification regulates T cell lineage commitment and effector functions. (A) METTL3-mediated m6A modification of Tbx21 mRNA enhances T-bet expression, promoting CD8+ T cell differentiation and effector cytokine production. FTO demethylation of Fas mRNA limits apoptosis, while IGF2BP3 stabilizes m6A-modified Fas mRNA to promote it. (B) Regulation of TNF/CD40L transcripts in CD4+ T cells depends on the balance between FTO and YTHDF2. METTL3-mediated m6A modification stabilizes these mRNAs, supporting CD4+ T cell activation and cytokine secretion. (C) m6A modifications orchestrate CD4+ T cell lineage fate. METTL3 stabilizes Tcf-7 mRNA (encoding TCF-1) to drive Tfh cell differentiation and B cell help, whereas METTL14 stabilizes Foxp3 mRNA to promote iTreg development. Loss of METTL14 disrupts mTOR signaling, favoring Th17 differentiation. (D) ALKBH5 demethylation of Jagged1/Notch2 mRNA prevents transcript degradation, sustaining Notch signaling. This promotes pre-γδ T cell proliferation and the differentiation of mature γδ T cells. (E) METTL14-mediated m6A modification of Cish mRNA preserves thymocyte viability by limiting p53/ROS-driven apoptosis. Invariant NKT cells expand upon IL-15 stimulation and α-GalCer injection, producing cytokines including IFN-γ and IL-4.
RNA modifications beyond m6A in T cell immunity
Beyond m6A, a diverse repertoire of RNA modifications fine-tunes T cell activation, effector programming, and immune tolerance through distinct molecular pathways. The cytosine-5 methyltransferase NSUN2 enhances IL-17A expression by methylating cytosine C466 within Il17a mRNA, promoting translational efficiency without altering transcript abundance. Deletion of Nsun2 abolishes homocysteine-induced IL-17A upregulation, establishing a mechanistic link between metabolic stress and T cell-driven inflammation [94].
At the level of Ψ modification, comparative Ψ-seq profiling reveals that Ψ landscapes in primary and immortalized human T cells are highly conserved, with 87% of Ψ sites shared and enriched in transcripts encoding core cellular machinery, including RNA-processing enzymes. Divergent Ψ marks correspond primarily to transcript availability rather than enzymatic expression: Jurkat-specific Ψ sites associate with immune activation and oncogenic pathways, whereas primary T cells display Ψ-modified transcripts related to calcium signalling and vesicular trafficking. Despite similar expression of Ψ synthases, the differential Ψ distribution reflects trans-acting regulatory control that dynamically shapes T cell states across physiological and transformed contexts [95]. In innate-immune crosstalk, the Ψ synthase PUS10 modulates tRNA pseudouridylation and fragmentation, thereby maintaining RNA homeostasis. PUS10 deficiency increases tRNA-derived small RNAs and retroelement transcription, generating RNA-DNA hybrids that activate the cGAS-STING pathway. This intrinsic inflammatory loop heightens interferon responses and perturbs immune regulation, altering T cell activation thresholds and promoting autoimmune susceptibility. Supplementation with defined tRNA-derived fragments restores immune balance, identifying PUS10 as a pivotal regulator of RNA-driven inflammatory tone within T cell-relevant networks [96]. Chemical modifications of uridine residues can also be synthetically leveraged to modulate T cell activity. Unmodified in-vitro-transcribed mRNA triggers robust chemokine secretion and lymphocyte recruitment, whereas uridine modifications—such as Ψ, m1Ψ, and 5moU—progressively attenuate this immunogenicity. These synthetic nucleoside chemistries allow precise tuning of mRNA-induced immune activation, enhancing the immunosuppressive potential of stromal cells and augmenting IL-10 mRNA anti-inflammatory efficacy, thereby influencing T cell proliferation and functional polarization [97].
tRNA and mRNA cap modifications further integrate transcriptional activation with translational output during early T cell responses. Antigenic stimulation induces TRMT61A and TRMT6, which catalyse m1A58 modification on early-expressed tRNAs. These modified tRNAs boost translation of proteins such as MYC, supporting the metabolic and biosynthetic reprogramming required for clonal expansion. Loss of Trmt61a in CD4+ T cells reduces MYC synthesis, induces cell-cycle arrest, and limits T cell-mediated pathology in colitis, revealing a tRNA-modification-driven translational program essential for effector activation [10]. In parallel, the RNA-cap methyltransferase RNMT promotes ribosome biogenesis via m7G capping of transcripts encoding TOP mRNAs and snoRNAs. RNMT upregulation ensures coordinated rRNA and mRNA production for ribosomal assembly, whereas RNMT loss disrupts LARP1-dependent expression, impairs ribosome synthesis, and restricts CD4+ T cell proliferation and effector differentiation [98].
Collectively, these findings delineate a multilayered epitranscriptomic network in which diverse RNA modifications cooperate to orchestrate T cell development, activation, and functional plasticity. Acting at multiple hierarchical levels—from tRNA charging and mRNA translation to cytokine secretion and innate-immune feedback—these chemical marks integrate metabolic cues, stress signals, and immune pathways into coherent post-transcriptional programs. This integrated framework endows T cells with the capacity to rapidly reconfigure gene expression in response to environmental and immunological perturbations, sustaining immune homeostasis while preventing uncontrolled activation or tolerance breakdown [99] (Figure 2, Table 1).
Mechanisms of T Cell Regulation by RNA Modifications
| T cell subset | Regulator | Modification | Target | Mechanism | Functional Outcome | Ref. |
|---|---|---|---|---|---|---|
| CD8+ T cells | METTL3 | m6A writer | Tbx21 (T-bet) mRNA | Enhancing mRNA stability and translation | Promoting effector differentiation and cytokine production | [17] |
| FTO/IGF2BP3 | m6A eraser/reader | Fas mRNA | FTO: Preventing excessive stabilization and translation of Fas mRNA; IGF2BP3: Stabilizing Fas mRNA and promoting its translation upon FTO loss | Preventing excessive apoptosis, maintaining CD8+ T cell survival and effector function | [79] | |
| CD4+ T cells | YTHDF2 | m6A reader | TNF mRNA | Binding m6A-modified transcripts and promoting their decay | Limiting TNF production to fine-tune inflammatory responses | [80] |
| METTL3/FTO/YTHDF2 | m6A writer/eraser/reader | CD40L mRNA | METTL3: Dynamically regulating mRNA stability; FTO/YTHDF2: Fine-tuning mRNA stability | Controlling T cell activation thresholds and cytokine production | [85] | |
| METTL3 | m6A writer | Tcf7 (TCF-1) mRNA | Stabilizing Tcf7 mRNA and defining Tfh identity | Promoting Tfh cell differentiation and germinal center formation | [86] | |
| METTL14 | m6A writer | Foxp3 mRNA | Maintaining Foxp3 mRNA stability and restraining mTOR-p70S6K signaling | Preserving iTreg suppressive function and preventing inflammation | [87] | |
| γδ T cells | ALKBH5 | m6A eraser | Jagged1/Notch2 mRNA | Suppressing Notch signaling | Expanding the γδ T cell pool and enhancing mucosal defense | [90] |
| METTL3 | m6A writer | Stat1 mRNA | Promoting Stat1 mRNA decay and limiting dsRNA accumulation | Preserving γδT17 cell differentiation and IL-17 production | [91] | |
| iNKT cells | METTL14 | m6A writer | Cish mRNA | Regulating IL-2/IL-15 responsiveness and Vα14-Jα18 rearrangement | Supporting iNKT cell survival, maturation, and cytokine production | [92] |
| METTL3 | m6A writer | Creb1 transcripts | Ensuring sufficient CREB1 protein abundance and phosphorylation | Maintaining iNKT lineage homeostasis, functional specialization and antitumor immunity | [93] | |
| CD4+ T cells | NSUN2 | m5C writer | IL-17A mRNA (C466 site) | Enhancing translational efficiency without altering mRNA abundance | Promoting IL-17A expression in Th17 cells | [94] |
| T cells | PUS10 | Ψ modification | tRNA | Preventing tRNA fragmentation and inhibiting retrotransposon expression to avoid cGAS-STING pathway activation | Maintaining innate immune homeostasis and preventing autoimmunity | [96] |
| CD4+ T cells | TRMT61A/ TRMT6 | m1A58 writer | Early-expressed tRNA subset | Enhancing translation of MYC and key effector proteins | Promoting cell cycle entry and clonal expansion | [10] |
| RNMT | m7G writer | TOP mRNAs and snoRNAs | Promoting translation via ribosome biogenesis | Enabling proliferation and effector differentiation | [98] |
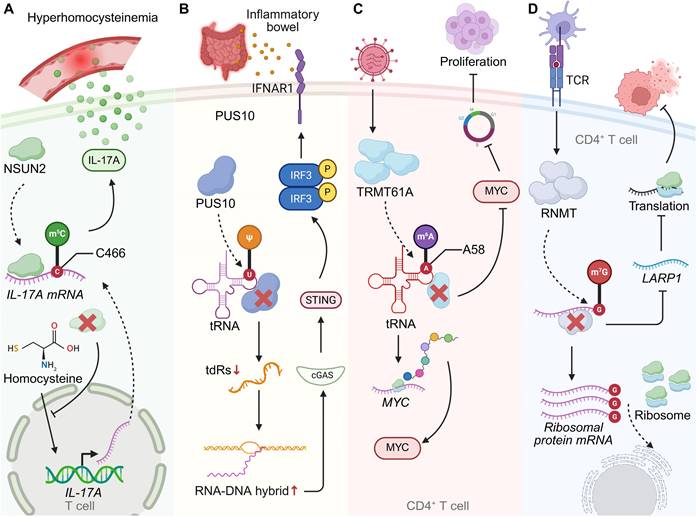
RNA modifications shape T cell function and immune regulation under pathological conditions. (A) In hyperhomocysteinemia, NSUN2 catalyzes m5C modification of IL-17A mRNA at cytosine 466, enhancing its translational efficiency and promoting IL-17A production in T cells. NSUN2 deficiency blocks the homocysteine-driven increase in IL-17A, highlighting a link between metabolic stress and T cell-mediated inflammatory responses. (B) PUS10-mediated pseudouridylation of tRNA regulates innate immune sensing. Loss of PUS10 reduces tRNA stability, increases RNA-DNA hybrid formation, and activates the cGAS-STING pathway, leading to inflammatory signaling. This mechanism contributes to intestinal immune dysregulation in inflammatory bowel disease (IBD). (C) TRMT61A installs m1A at position A58 of tRNA, thereby sustaining MYC expression in CD4+ T cells. MYC-driven transcriptional programs promote cell proliferation and clonal expansion, coupling RNA modification to T cell metabolic and proliferative fitness. (D) RNMT-dependent m7G modification of ribosomal protein mRNAs regulates CD4+ T cell activation. In response to TCR stimulation, RNMT and LARP1 coordinate ribosome biogenesis and translation, thereby enabling efficient protein synthesis during T cell responses.
Potential epitranscriptomic crosstalk integrates RNA modification networks in T cell immunity
Epitranscriptomic modifications have emerged as critical regulators of immune homeostasis, adding a dynamic layer of control that complements transcriptional and translational programs. Among the diverse RNA modifications identified to date, examples such as m6A and m5C illustrate how chemical marks on RNA can fine-tune transcript fate and coordinate cellular adaptation to stress. Once viewed as discrete regulatory systems, these modification pathways are now recognized to engage in extensive cross-regulatory interactions. Reciprocal modification of their effector transcripts forms intricates post-transcriptional feedback loops, while coordinated expression of regulators such as the m6A eraser ALKBH5 and the m5C writer NSUN4 links RNA methylation to proteasomal turnover, mitochondrial function, and post-translational signaling networks [100]. This interplay reveals a hierarchical control system in which distinct RNA marks converge to modulate gene expression in a context-dependent manner.
Within the immune system, such crosstalk provides a conceptual basis for understanding how metabolic and environmental cues are integrated at the RNA level to calibrate T cell activation, differentiation, and effector function. The coordination between different RNA modification machineries may enable T cells to dynamically balance biosynthetic demands, redox homeostasis, and cytokine production during immune activation. Supporting this notion, the cooperative m6A and m5C modification of GPX4 by RBM15B, IGFBP2, and NSUN5 sustains redox equilibrium and activates the cGAS-STING pathway, thereby enhancing antitumor immune responses [101]. By analogy, similar dual-modification networks may operate in T cells to link mitochondrial metabolism and RNA stability with immune signaling precision. These insights collectively point to potential RNA modification crosstalk as an emerging axis of immune regulation—one that integrates metabolic resilience with epitranscriptomic plasticity to fine-tune T cell-mediated immunity and therapeutic responsiveness.
RNA modification-driven remodeling of T cell immunity across disease contexts
RNA modification-mediated regulation of CD8+ T cell function in cancer
In the tumor microenvironment, RNA modifications act as critical determinants of CD8+ T cell persistence, metabolic fitness, and responsiveness to immunotherapy, with distinct regulatory enzymes exerting either supportive or suppressive effects on antitumor immunity [102-104]. YTHDF2 exemplifies a positive regulator by sustaining polyfunctionality in early effector-like CD8+ T cells through facilitation of nascent RNA synthesis and modulation of chromatin architecture. High YTHDF2 expression is indispensable for preserving mitochondrial fitness and maintaining cytotoxic potential, whereas its loss accelerates tumor progression, compromises effector responses, and diminishes the efficacy of PD-1 blockade. Mechanistically, YTHDF2 interacts with IKZF1/3 to maintain transcription of key target genes, and pharmacological intervention with lenalidomide can rescue these defects, underscoring the therapeutic relevance of targeting RNA modification pathways [18]. In contrast, the m6Am methyltransferase PCIF1 functions as a negative regulator of CD8+ T cell antitumor activity. Deletion of Pcif1, either systemically or specifically in T cells, not only reduces tumor burden but also increases the infiltration of activated cytotoxic CD8+ T cells. This effect is mediated by the upregulation of m6Am -modified ferroptosis suppressor genes such as Fth1 and Slc3a2, along with the activation marker Cd69, thereby enhancing resistance to ferroptosis and augmenting antitumor function. Pcif1 deficiency also potentiates the efficacy of anti-PD-1 therapy and improves tumor control in CAR-T cell models, with clinical data linking low PCIF1 expression to superior immunotherapy outcomes [105]. Complementing these findings, the m6A methyltransferase adaptor WTAP contributes to the onset of CD8+ T cell exhaustion in hepatocellular carcinoma (HCC) by increasing PD-1 mRNA translation in a YTHDF1-dependent manner. Elevated WTAP expression in tumor-infiltrating CD8+ T cells enhances PD1 levels, suppressing proliferation and effector activity, whereas WTAP silencing—especially in combination with PD1 knockdown—restores immune function, inhibits tumor growth, and improves responses to PD-1 blockade. Together, these examples illustrate how distinct m6A and m6Am regulators orchestrate the balance between functional resilience and exhaustion in CD8+ T cells, ultimately shaping antitumor immunity and therapeutic responsiveness. Notably, the tRNA m1A methyltransferase TRMT61A enhances CD8+ T cell antitumor immunity by promoting cholesterol biosynthesis. TRMT61A-driven m1A modification facilitates translation of ATP citrate lyase, a key enzyme in the cholesterol synthesis pathway, supporting T cell proliferation and cytotoxic function. Loss of TRMT61A impairs cholesterol production, leading to diminished effector responses and tumor control, which can be restored by cholesterol supplementation [106]. These findings position tRNA m1A as a crucial metabolic regulator linking translational control to the functional competence of CD8+ T cells in the tumor microenvironment. Single-cell transcriptomic profiling of nasopharyngeal carcinoma (NPC) reveals that m7G RNA modification is intricately linked to immune dysregulation within the tumor microenvironment. CD4+ and CD8+ T cells in NPC exhibit reduced m7G scores compared to nonmalignant tissues, alongside altered expression of immune co-stimulatory and co-inhibitory molecules, suggesting impaired antitumor function. m7G-associated interactions between T cells and other stromal and immune populations, including macrophages and fibroblasts, further reshape the immunosuppressive milieu [107] (Figure 3).
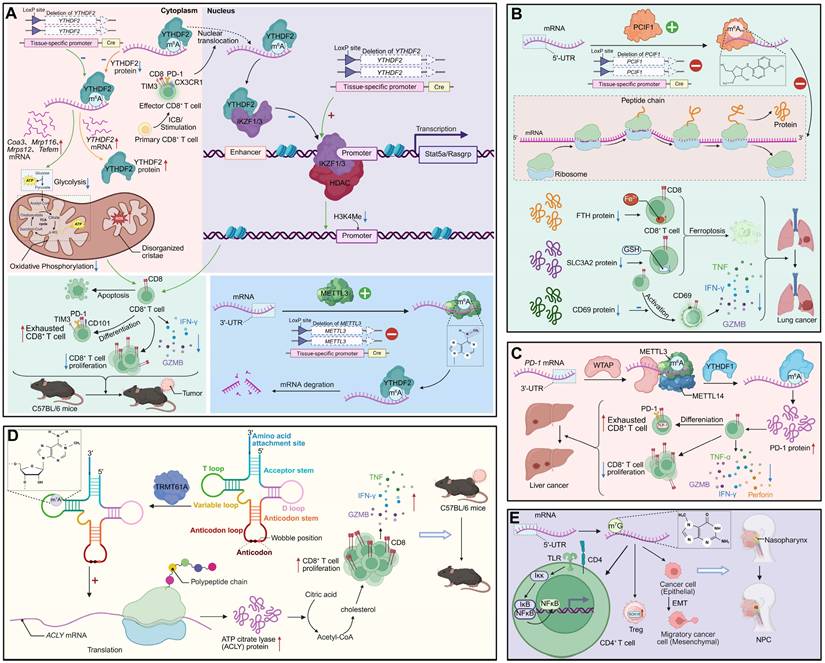
RNA modifications regulate CD8+ T cell-mediated antitumor immunity and shape the immunosuppressive tumor microenvironment. (A) YTHDF2 controls mitochondrial metabolism and epigenetic programming of CD8+ T cells. Conditional deletion of Ythdf2 impairs oxidative phosphorylation, disrupts cristae structure, and upregulates genes such as Cox3, Mtp16, and Tefm. Nuclear YTHDF2 stabilizes IKZF1/3 transcriptional activity and represses HDAC-mediated H3K4me modifications, thereby sustaining effector function. Loss of YTHDF2 accelerates CD8+ T cell exhaustion, with increased PD-1, TIM3, and CD101 expression, impaired proliferation and tumor progression. METTL3-dependent m6A modification further stabilizes transcripts critical for effector CD8+ T cell activity, whereas YTHDF2 deletion promotes mRNA decay. (B) PCIF1 regulates m6Am modification and translation in CD8+ T cells. PCIF1-mediated m6Am modification diminishes ribosome-associated peptide synthesis, lowering proteins such as FTH, SLC3A2 and CD69. This triggers ferroptosis and compromises CD8+ T cell cytotoxicity, reducing TNF, IFN-γ, and GZMB production, and accelerating lung cancer progression. (C) METTL3-METTL14-YTHDF1 complex maintains effector function in liver cancer. m6A modification of Pdcd1 (PD-1) transcripts enhances PD-1 protein expression, promoting CD8+ T cell exhaustion. YTHDF1 stabilizes effector transcripts, sustaining cytotoxicity (TNF, IFN-γ, perforin, GZMB), while WTAP regulates m6A deposition. Dysregulation of this axis reduces CD8+ T cell proliferation and accelerates immune dysfunction. (D) TRMT61A installs m1A modification on tRNA, regulating translation of ACLY and metabolic reprogramming in CD8+ T cells. m1A at wobble positions enhances ACLY-driven acetyl-CoA production, fueling cholesterol and citric acid metabolism. This sustains effector function and cytokine release (TNF, IFN-γ, GZMB), enabling tumor control in C57BL/6 mice. (E) m7G modification shapes regulatory T cell (Treg) stability and epithelial-mesenchymal transition (EMT) in the tumor microenvironment of NPC. m7G-modified transcripts activate the IKK-NFκB pathway in CD4+ T cells, promoting Treg differentiation. In parallel, RNA modification stabilizes EMT-associated transcripts, driving cancer cell migration and NPC progression.
RNA modification-mediated regulation of CD4+ T cell function in diseases
Autoimmune diseases
SLE
In SLE, CD4+ T cells undergo extensive remodeling of RNA modifications, encompassing m6A, m5C, and ac4C marks on both nuclear- and mitochondrial-encoded transcripts. These modifications collectively integrate transcriptional, metabolic, and post-transcriptional networks that shape lineage commitment, effector activity, and pathogenic potential [108].
METTL3 functions as a pivotal regulator of CD4+ T cell activation and lineage commitment in SLE. Reduced METTL3 expression correlates with aberrant activation dynamics and skewed effector differentiation. Pharmacological inhibition of METTL3 destabilizes Foxp3 mRNA by impairing m6A-dependent protection, thereby limiting regulatory T cell development and favoring pro-inflammatory responses. In vivo, METTL3 blockade augments CD4+ T cell activation, diminishes Treg differentiation, enhances antibody production, and exacerbates lupus-like pathology [109]. Mitochondrial dysfunction further links m6A dysregulation to pathogenic activation. The mitochondrial transcript MT-ND6 exhibits hypermethylation and reduced expression in SLE CD4+ T cells, correlating with disease activity and autoantibody titres. MT-ND6 deficiency impairs ATP production, increases total and mitochondrial ROS, and activates inflammatory transcriptional programs—defects reversible by mitochondrial antioxidant treatment—demonstrating that m6A safeguards mitochondrial integrity to restrain pathogenic activation [110].
Beyond m6A, SLE CD4+ T cells display widespread perturbation of m5C and ac4C marks. Global m5C levels are reduced despite an increased number of m5C-modified transcripts, particularly near translation initiation sites, implicating translational dysregulation in aberrant immune activation. Downregulation of NSUN2 contributes to this disrupted methylation pattern and altered mRNA metabolism [111]. In parallel, reduced ac4C and its writer NAT10 coincide with altered acetylation of coding sequences enriched in metabolic and NF-κB signalling genes. Dysregulated ac4C distribution on immune-regulatory transcripts such as USP18, GPX1, and RGL1 affects mRNA stability and translation, linking ac4C remodeling to oxidative stress and hyperinflammatory states [112]. Together, these findings define a multilayered epitranscriptomic landscape in which m6A, m5C, and ac4C cooperatively modulate transcriptional fidelity, mitochondrial function, and redox homeostasis to shape autoimmune pathology in SLE.
Psoriasis
Psoriasis is a chronic inflammatory skin disorder driven by Th17-mediated pathology. In CD4+ T cells, enhanced m6A deposition on Il17a transcripts increases their stability and abundance, amplifying IL-17A production and psoriatic inflammation. Loss of the demethylase ALKBH5 exacerbates disease severity, whereas METTL3 ablation alleviates pathology, underscoring that m6A-dependent stabilization of Il17a mRNA drives Th17 expansion and disease progression [113].
Ocular immune-mediated disorders
In ocular autoimmunity, including Graves' ophthalmopathy (GO) and autoimmune uveitis, aberrant m6A modification reinforces dysregulated CD4+ T cell metabolism and inflammatory programming. In GO, WTAP expression is markedly upregulated in CD4+ T cells, promoting m6A deposition on THBS1 transcripts. This hypermethylation stabilizes THBS1, driving glycolytic flux—reflected by increased glucose uptake and lactate production—thereby enhancing Th17 differentiation and suppressing Treg induction. Silencing WTAP reduces THBS1 methylation, normalizes metabolism, and restores T cell balance, defining a WTAP-THBS1-glycolysis axis as a central driver of GO pathology [114]. Conversely, in experimental autoimmune uveitis, METTL3 plays a protective role. Reduced METTL3 expression and global m6A levels correlate with heightened inflammation, whereas enforced METTL3 expression mitigates disease. Mechanistically, METTL3 promotes YTHDC2-dependent m6A modification of ASH1L transcripts, stabilizing them and suppressing IL17 and IL23R expression, thereby limiting Th17 effector activity and ocular inflammation [115]. These dual mechanisms illustrate how m6A-dependent reprogramming can either amplify or restrain CD4+ T cell pathogenicity, depending on the molecular context.
IBD (ulcerative colitis)
In ulcerative colitis, sustained T cell-driven inflammation is maintained by cooperative actions of ac4C and m5C modifications. The acetyltransferase NAT10 catalyzes ac4C modification on Bag3 mRNA, stabilizing this anti-apoptotic transcript and supporting pathogenic CD4+ T cell survival. NAT10 deletion accelerates apoptosis and diminishes T cell persistence, thereby reducing colitogenic potential [116]. Concurrently, the methyltransferase NSUN2 promotes Th17 differentiation by forming a complex with RORγt to methylate Il17a and Il17f transcripts, enhancing their stability and cytokine production. NSUN2 deficiency impairs Th17 polarization and ameliorates colitis, revealing a RORγt-NSUN2 axis that sustains Th17 effector function [117]. Collectively, ac4C- and m5C-mediated programs integrate metabolic, survival, and cytokine circuits to maintain chronic inflammation in ulcerative colitis, exemplifying how diverse RNA modifications converge to stabilize pro-inflammatory states (Figure 4).
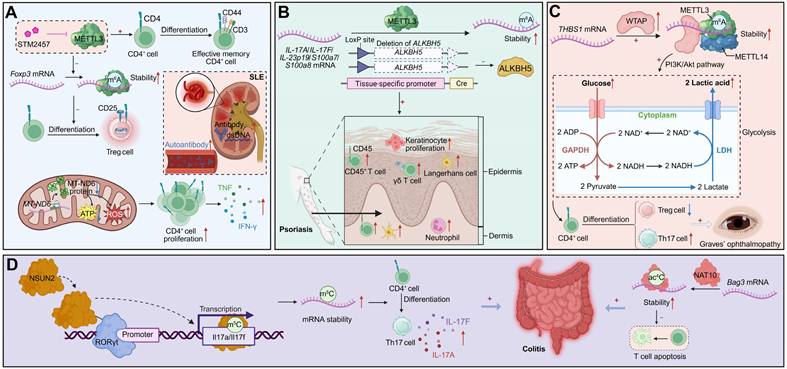
RNA modifications regulate CD4+ T cell differentiation and autoimmune pathogenesis. (A) METTL3-mediated m6A modification of Foxp3 mRNA promotes its stability and supports Treg differentiation, a process that can be pharmacologically inhibited by STM2457. Concurrently, METTL3-mediated m6A modification of MT-ND6 mRNA reduces its expression, leading to mitochondrial dysfunction (ATP loss, ROS accumulation), aberrant CD4+ T cell proliferation, and cytokine production. In SLE, impaired Treg function and mitochondrial defects contribute to enhanced autoantibody production, and systemic inflammation. (B) Deletion of ALKBH5 enhances METTL3-mediated m6A methylation on pro-inflammatory transcripts (IL-17A, IL-17F, IL-23p19, S100a7/8), increasing their stability. This drives keratinocyte hyperproliferation, epidermal thickening, and infiltration of inflammatory cells (CD45+ T cells, γδ T cells, neutrophils, Langerhans cells) in psoriasis. (C) WTAP-METTL3-METTL14 complex stabilizes THBS1 mRNA via m6A modification, activating the PI3K-Akt pathway and reinforcing glycolysis. This metabolic rewiring skews CD4+ T cell differentiation toward Th17 cells at the expense of Tregs, exacerbating disease manifestations such as GO. (D) NSUN2-mediated m5C modification of Il17a/Il17f transcripts enhances their stability, driving Th17 differentiation and excessive IL-17A/F secretion, which exacerbates colitis. Additionally, NAT10-catalyzed ac4C modification stabilizes Bag3 mRNA, preventing T cell apoptosis and sustaining inflammation.
Transplantation and graft rejection
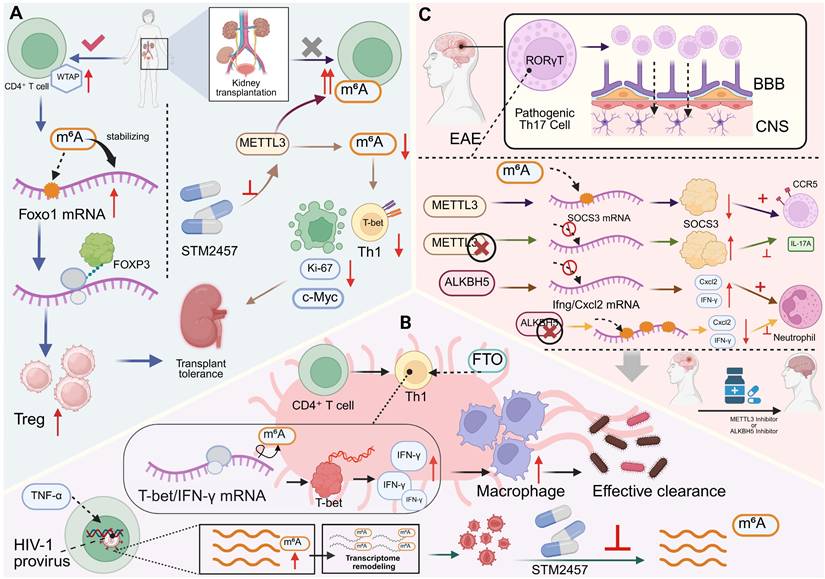
In kidney transplantation, epitranscriptomic remodeling of CD4+ T cells critically determines the equilibrium between immune tolerance and graft rejection. In states of operational tolerance, the m6A methyltransferase component WTAP is upregulated in CD4+ T cells and correlates with expansion of the Treg compartment. Mechanistically, WTAP promotes m6A deposition within the coding region of Foxo1 mRNA, stabilizing its transcript and sustaining FOXO1 expression. This enhances Treg differentiation and suppressive activity, thereby maintaining peripheral tolerance. In vivo, WTAP overexpression prolongs allograft survival by augmenting Treg-mediated immunoregulation and dampening inflammatory activation, establishing a WTAP-Foxo1-m6A axis as a central safeguard of transplant tolerance [118]. Conversely, during allograft rejection, alloreactive CD4+ T cells exhibit globally elevated m6A modification, partly driven by METTL3. Pharmacological inhibition of METTL3 using STM2457 reduces overall m6A levels, inducing G₀ cell-cycle arrest, increasing apoptosis, and impairing both proliferation and effector differentiation of polyclonal and alloantigen-specific CD4+ T cells. These effects are accompanied by attenuated Th1 polarization and downregulation of key transcriptional drivers, including Ki-67, c-Myc, and T-bet [119]. Collectively, these findings delineate a dual role for m6A-dependent regulation in transplantation: promoting tolerance through Treg stabilization while sustaining rejection by supporting effector T cell fitness and persistence. This functional dichotomy positions the RNA methylation machinery as a tunable checkpoint in graft-specific immunity—one that could be therapeutically manipulated to tip the balance between durable tolerance and immune rejection.
Infectious diseases
Epitranscriptomic regulation has emerged as a pivotal determinant of CD4+ T cell fate and function during infection [120, 121]. In acute bacterial challenge, the m6A demethylase FTO safeguards the Th1 differentiation program by maintaining Tbx21 (T-bet) expression and sustaining IFN-γ production. Genetic ablation of FTO leads to defective antigen-specific Th1 expansion, resulting in impaired pathogen clearance in both in vitro and in vivo models. Mechanistically, FTO dynamically modulates the stability and translational efficiency of lineage-defining transcripts, thereby coordinating transcriptional and metabolic programs that underpin effective antibacterial immunity [122]. A comparable dependence on m6A regulation is observed in chronic viral infection, where reactivation of latent HIV-1 in CD4+ T cells induces a transient but pronounced increase in global m6A RNA methylation, independent of changes in writer or eraser abundance. Silencing m6A methyltransferases or pharmacologically inhibiting methylation markedly suppresses HIV-1 reactivation, demonstrating that m6A facilitates viral transcriptional reawakening. m6A-seq profiling reveals widespread remodeling of both host and viral transcript methylation during latency reversal, suggesting that RNA methylation reprograms the T cell transcriptome to favor viral gene expression and reservoir destabilization [123]. In acute viral infections, particularly COVID-19, altered m6A landscapes in peripheral blood leukocytes correlate with CD4+ T cell activation states and clinical outcomes. Unsupervised clustering of patient transcriptomes reveals two distinct m6A-modification patterns: an activation-enriched cluster characterized by enhanced T cell effector signatures and improved recovery, and a metabolically reprogrammed, checkpoint-suppressed cluster marked by reduced m6A scores and poor prognosis. A nine-gene m6A-related transcriptional signature derived from these datasets reliably predicts disease trajectory, linking the intensity and quality of antiviral responses to the m6A regulatory landscape [124]. Collectively, these studies identify m6A-dependent post-transcriptional regulation as a unifying mechanism that enables CD4+ T cells to tailor their effector programs across diverse infectious contexts. By coupling transcriptional activation with translational control, m6A modification integrates metabolic readiness, cytokine production, and viral sensing into a coherent immune response, thereby shaping both pathogen clearance and the persistence of infection.
Neuroinflammatory and CNS autoimmune disorders
Neuroinflammatory and central nervous system (CNS) autoimmune diseases—exemplified by experimental autoimmune encephalomyelitis (EAE)—are critically governed by m6A-dependent regulation of pathogenic CD4+ T cell subsets. m6A RNA methylation reinforces Th17 lineage integrity and effector persistence in CNS autoimmunity: deletion of the methyltransferase METTL3 in total T cells or specifically in Th17 cells markedly reduces CNS infiltration and ameliorates disease severity. Mechanistically, loss of METTL3 disrupts m6A-dependent decay of Socs3 mRNA, leading to its stabilization and the suppression of Il17a and Ccr5 expression, thereby dismantling the Th17 transcriptional circuit essential for pathogenic activity [125]. Conversely, the m6A demethylase ALKBH5 sustains inflammatory effector programs by preventing methylation-dependent degradation of proinflammatory transcripts such as Ifng and Cxcl2. ALKBH5 ensures their stability, maintaining cytokine output and promoting neutrophil recruitment within inflamed CNS tissues. In its absence, hypermethylation of these transcripts accelerates decay, dampens effector cytokine production, and mitigates autoimmune pathology [126]. Together, these findings delineate a bidirectional m6A-regulatory circuit that dynamically balances methylation-driven transcript turnover and demethylation-mediated transcript stabilization. This interplay fine-tunes cytokine networks and effector persistence, preserving the identity and inflammatory tone of CNS-infiltrating CD4+ T cells. Such dynamic epitranscriptomic regulation integrates transcriptional feedback, metabolic adaptation, and immune signaling to orchestrate neuroinflammatory responses, highlighting RNA methylation machinery as a potential therapeutic target in CNS autoimmune disease (Figure 5).
m6A RNA modifications orchestrate CD4+ T cell fate and immune-mediated disease. (A) The dual role of m6A RNA modification in transplantation. The WTAP-m6A-FOXO1 axis promotes Treg-mediated tolerance and graft survival. In contrast, METTL3-mediated m6A modification enhances effector T cell proliferation, differentiation, and function, driving rejection. Targeting METTL3 can inhibit its activity through drugs such as STM2457, reduce T-bet, Ki-67, and c-Myc to suppress Th1 polarization, while enhancing Treg responses, thereby inhibiting this alloreactive response. (B) RNA modification fine-tunes CD4+ T cell responses to infections. (Top) FTO-mediated demethylation regulates Th1-driven macrophage activation. By controlling m6A on T-bet/IFN-γ transcripts, FTO enhances IFN-γ secretion, which augments macrophage effector functions and pathogen clearance. (Bottom) In HIV-1 latency, reactivation signals induce m6A methylation on viral and host transcripts, promoting viral gene expression and reactivation, which can be blocked by METTL3 inhibition. (C) m6A modifications influence Th17 pathogenicity in autoimmune disease. In EAE, the presence of METTL3 reduces the stability of SOCS3 mRNA, which in turn leads to increased expression of IL-17A and CCR5, the m6A demethylase ALKBH5 preserves the stability of pro-inflammatory transcripts such as Ifng and Cxcl2, thereby maintaining CD4+ T cell effector potential and promoting neutrophil recruitment during neuroinflammation. Pharmacological inhibition of METTL3 or ALKBH5 impairs Th1 responses and reduces immune clearance, underscoring therapeutic potential of m6A modulators.
Other diseases
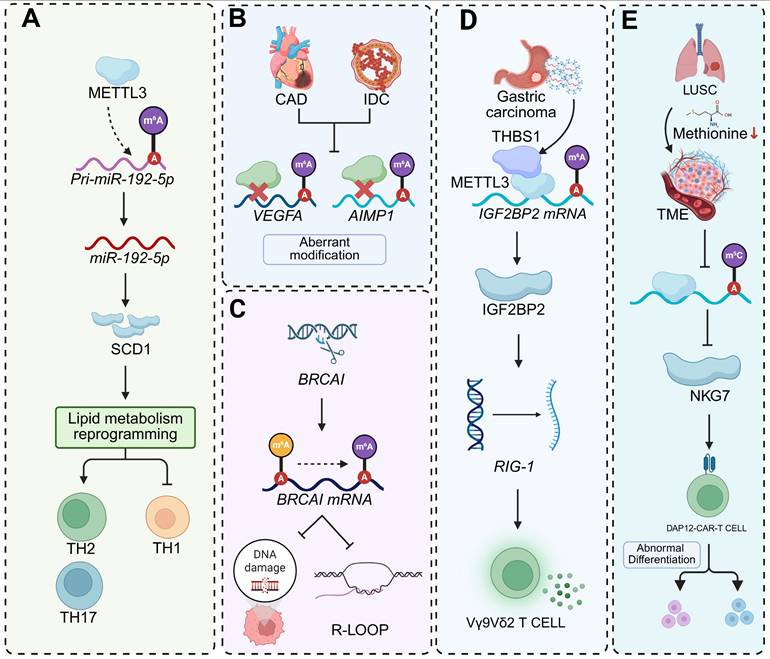
Beyond classical autoimmune, infectious, and neuroinflammatory disorders, RNA modifications in CD4+ T cells contribute to the pathogenesis of diverse immune-related conditions. In asthma, skewed differentiation toward Th2 and Th17 lineages is coupled to metabolic reprogramming through m6A-dependent control. METTL3-mediated m6A methylation inhibits processing of pri-miR-192-5p, thereby relieving repression of stearoyl-CoA desaturase 1 (SCD1). Elevated SCD1 promotes lipid metabolic adaptation that increases Th2 and Th17 frequencies while limiting Th1 cells. Restoration of miR-192-5p or pharmacological inhibition of SCD1 reverses this lineage bias and mitigates airway inflammation, whereas oleic-acid supplementation intensifies the inflammatory phenotype [127]. In parallel, METTL3-driven m6A deposition within the 3′ UTR of Foxp3 mRNA enables YTHDF2-mediated degradation, reducing Treg proportions and shifting cytokine secretion toward a pro-inflammatory profile [128]. Aberrant m6A methylation in CD4+ T cells also forms a shared epitranscriptomic signature linking coronary artery disease (CAD) and invasive ductal carcinoma (IDC). Comparative profiling identifies dysregulated m6A marks on inflammation-associated transcripts, including VEGFA and AIMP1, in both conditions. CRISPR-Cas9-mediated modulation of BRCA1 alters m6A distribution, influences R-loop formation and DNA-damage susceptibility, and modulates p53 activity, thereby affecting immune-tumour interactions and enhancing cytotoxic T lymphocyte-mediated killing of breast-cancer cells [129]. Together, these findings underscore that RNA modification-dependent control of T cell fate and function extends beyond canonical autoimmune and infectious diseases, bridging metabolic, inflammatory, and oncogenic pathways across distinct pathological contexts.
RNA modification-mediated regulation of unconventional T cell function in diseases
Unconventional T cell subsets, such as Vγ9Vδ2 T cells and CAR-T cells, are emerging as crucial mediators of antitumor immunity, with their activation and persistence tightly governed by RNA modifications and metabolic cues. In gastric cancer, tumor-derived exosomal THBS1 enhances the cytotoxicity and effector cytokine production of Vγ9Vδ2 T cells by remodeling the m6A RNA methylation landscape. Mechanistically, THBS1 directly interacts with the methyltransferase METTL3 and induces expression of the m6A reader IGF2BP2, leading to stabilization of transcripts encoding RIG-I-like receptor signaling components, including IRF7, ISG15, and PKR. This enhanced transcript stability sustains antiviral signaling and effector activation, whereas pharmacological inhibition of the m6A machinery abolishes THBS1-driven functional gains [130]. In parallel, B7H3-targeted DAP12-CAR-T cells exhibit strong cytotoxicity against lung squamous cell carcinoma (LUSC) but are metabolically constrained within methionine-depleted tumor microenvironments. Nutrient restriction reduces m5C RNA modification and downregulates NKG7, a key cytolytic effector gene, thereby driving CAR-T cells toward functional exhaustion. Restoration of NKG7 expression or blockade of tumor SLC7A5-mediated methionine uptake rescues CAR-T cell cytotoxicity under nutrient stress, linking methionine availability and RNA methylation to effector competency [131]. Together, these findings define RNA modification as a dynamic regulatory layer that integrates metabolic inputs and tumor-derived signals to fine-tune the effector programs of unconventional T cells. By coupling post-transcriptional control with metabolic adaptation, m6A and m5C modifications determine whether γδ T cells and CAR-T cells sustain cytotoxic persistence or enter functional exhaustion, underscoring the therapeutic potential of targeting epitranscriptomic-metabolic crosstalk to enhance T cell-based immunotherapies (Figure 6, Table 2).
RNA modification-driven remodeling of T cell immunity in disease
| Disease | RNA modifier | Molecular target | Immunological outcome | Implication | Ref. |
|---|---|---|---|---|---|
| Lung cancer | YTHDF2 | Epi-transcriptional and transcriptional networks | Promoting T cell polyfunctionality | Potentiating T cell immunity | [18] |
| Oral squamous cell carcinoma | PCIF1 | Ferroptosis-inhibitory gene | CD8+ T cell activation↑ | Boosting anti-PD-1 immunotherapy | [105] |
| HCC | WTAP | Promoting PD1 expression | Inhibiting CD8+ T cells proliferation | Promoting HCC progression | [132] |
| NPC | / | / | CD8+ T cells and CD4+ T cells↑ | Immune dysregulation | [107] |
| SLE | METTL3 | Stabilizing Foxp3 mRNA | CD4+ T cell activation↓ | Suppressing Treg cell differentiation | [109] |
| SLE | MT-ND6 | Increasing ROS and ATP | Inflammatory CD4+ T cells↓ | Exhibiting mitochondrial dysfunction | [110] |
| SLE | NSUN2 | / | Altering mRNA methylation patterns | Immune and inflammatory dysregulation | [111] |
| Colitis | NSUN2 | Chromatin regions | Inhibiting Th17 cell differentiation | Enhancing mRNA stability | [117] |
| SLE | NAT10 | / | Reducing ac4C levels | Modulating mRNA stability and translational initiation | [112] |
| Psoriasis | ALKBH5 | Enhancing stability of IL-17A mRNA | Increasing IL-17A expression in CD4+ T cells | Promoting psoriasis-like phenotype and inflammation | [113] |
| GO | WTAP | promoting m6A deposition on THBS1 transcripts | Promoting glycolysis of CD4+ T cells | Restoring T cell balance | [114] |
| Uveitis | METTL3 | Enhancing stability in YTHDC2-dependent manner | Decreasing IL-17 and IL-23 receptor expression | Reducing pathogenic Th17 responses | [115] |
| IBD | NAT10 | Diminishing Bag3 stability | Accelerating T cell apoptosis | Preserving T cell balance | [116] |
| Kidney transplantation | WTAP | Upregulation of Foxo1 | Naïve T cells↓ | Promoting Treg differentiation and function | [118] |
| Allograft rejection | METTL3 | Decreasing in m6A levels | Reduction TEa cell proliferation | Suppressing alloreactive CD4+ T cell effector function and differentiation | [119] |
| Bacterial infection | FTO | / | CD4+ T cell differentiation↑ | Decreasing T-bet and IFN-γ expression | [122] |
| EAE | METTL3 | Stabilizing SOCS3 mRNA | Disrupting Th17 pathogenic programs | Repressing IL-17A and CCR5 expression | [125] |
| Asthma | METTL3 | Suppressing pri-miR-192-5p processing | Th1 cells↓ | Driving to lipid metabolic reprogramming | [127] |
| IDC CAD | BRCA1 | m6A RNA methylation alterations | CTL mediated cytotoxicity↑ | Regulating tumor suppressor gene P53 | [129] |
Schematic representations of RNA modification (m6A, m5C)-mediated regulatory mechanisms in diverse pathological contexts. (A) METTL3-catalyzed m6A modification of pri-miR-192-5p promotes its processing into mature miR-192-5p. This miR-192-5p then targets SCD1, triggering lipid metabolism reprogramming and shaping the functional phenotypes of Th1, Th2, and Th17 cells. (B) In CAD and IDC, abnormal m6A modifications occur on the mRNAs of VEGFA and AIMP1, implying dysregulated post-transcriptional regulation in these cardiovascular disorders. (C) During BRCA1 transcription, m6A modification on BRCA1 mRNA modulates R-loop formation, thereby influencing the cellular response to DNA damage, a process crucial for maintaining genomic stability. (D) In gastric carcinoma, THBS1-associated METTL3 mediates m6A modification of IGF2BP2 mRNA, enhancing IGF2BP2 expression. Subsequently, IGF2BP2 regulates RIG-1, which in turn impacts the functional activity of Vγ9Vδ2 T cells in anti-tumor immunity. (E) In LUSC, reduced methionine levels affect m5C modifications. This alteration regulates NKG7 expression and leads to the abnormal differentiation of DAP12-CAR-T cells within the tumor microenvironment (TME).
Challenges and perspectives
Despite remarkable advances in uncovering the epitranscriptomic regulation of T cell biology, a mechanistic and clinically actionable understanding remains incomplete. Knowledge gaps persist across multiple layers—from modification dynamics and functional hierarchies to clinical translation—largely because existing data are context-specific, technically fragmented, and lack longitudinal resolution. Bridging these gaps will require the convergence of technological innovation, mechanistic dissection, and translational standardization.
Defining the spatiotemporal landscape of RNA modifications in T cells
Although transcriptional and epigenetic trajectories of T cell differentiation have been resolved at single-cell resolution, an equivalent atlas for RNA modifications is still lacking. Most current studies examine individual marks within limited subsets, without capturing developmental continuity or microenvironmental influences. This piecemeal approach obscures stage-specific dependencies, as the same modification may promote proliferation during activation but suppress memory maintenance later.
To overcome these limitations, single-cell and direct RNA sequencing technologies should be systematically integrated. Nanopore DRS now enables single-molecule, single-nucleotide identification of multiple RNA modifications, offering a unified framework for quantitative profiling [133]. Deep-learning frameworks such as modCnet further improve precision by distinguishing diverse cytidine modifications (e.g., ac4C and m5C) from a single sample, thus revealing combinatorial modification patterns [66]. For immunological applications, clinical standardization of single-cell epitranscriptomics will require three interdependent components: (1) reference cell lines and synthetic spike-in controls to calibrate detection sensitivity and ensure reproducibility across laboratories [134]; (2) automated, low-input library preparation workflows that integrate DRS with optimized barcoding and multiplexing for small patient samples [135]; and (3) harmonized computational pipelines and annotation standards for basecalling, signal normalization, and modification calling [136]. Together, these developments will enable construction of a spatiotemporal “T cell epitranscriptomic atlas,” facilitating causal analysis of stage-specific RNA-modification events through inducible perturbation models.
Capturing early epitranscriptomic signatures of T cell exhaustion
T cell exhaustion in chronic infection and cancer reflects progressive functional decline accompanied by stable epigenetic remodeling. Current markers—based on inhibitory receptors or transcriptional states—identify exhaustion only after irreversible dysfunction has occurred [137, 138]. Whether changes in RNA modifications precede these fixed states, acting as early molecular sentinels of exhaustion, remains unresolved. To clarify this, longitudinal single-cell epitranscriptomic profiling should be employed to trace modification dynamics across the exhaustion continuum in vivo [139, 140]. Integration of these datasets with transcriptomic, metabolic, and chromatin-accessibility profiles would identify reversible nodes that define pre-exhaustion states [141]. Transient perturbation of RNA-modifying enzymes at these early time points—via CRISPR interference or programmable RNA editors—can establish causality and identify modifiable checkpoints [142, 143]. Mapping such temporal dynamics will yield predictive biomarkers for immune decline and guide the timing of therapeutic interventions to sustain durable effector competence.
Translating RNA-modification biology into clinical and therapeutic contexts
The translation of RNA-modification biology into clinical applications is still in its infancy, constrained by technical, mechanistic, and safety challenges. Two major barriers hinder its progress: the lack of standardized, clinically compatible assays for limited immune samples, and the difficulty of safely modulating RNA-modifying enzymes whose pleiotropic functions extend across multiple cell types.
To address the first challenge, low-input, high-sensitivity detection platforms must be adapted for clinical workflows. Nanopore-based DRS enables single-molecule, single-nucleotide identification of multiple RNA modifications without chemical pretreatment, providing an efficient framework for clinical translation [143]. Integration of machine-learning-driven basecalling algorithms and harmonized cloud-based analysis pipelines will allow reproducible quantification and regulatory standardization [144]. Establishing clinical SOPs for RNA isolation, library preparation, and data interpretation will further facilitate the incorporation of RNA-modification biomarkers into patient monitoring and immune stratification programs.
Therapeutically, pharmacologic targeting of RNA-modifying enzymes is emerging as a promising approach for cancer immunotherapy. The oral METTL3 inhibitor STC-15 exemplifies this strategy: METTL3 inhibition enhances interferon signalling and dsRNA accumulation, leading to upregulation of interferon-stimulated genes and increased T cell cytotoxicity. In syngeneic tumour models, STC-15 induces durable antitumour immunity and synergizes with PD-1 blockade to achieve complete tumour regression. The completed Phase 1 trial (NCT05584111) established its safety and pharmacologic activity, while an ongoing Phase 1b/2 trial (NCT06975293) is evaluating STC-15 in combination with the PD-1 antibody toripalimab across advanced cancers—including non-small-cell lung carcinoma, melanoma, endometrial carcinoma, and head and neck squamous cell carcinoma—to define optimal dosing, safety, and T cell-modulating efficacy of METTL3 blockade. These findings underscore that manipulating RNA methylation can remodel the tumour-immune interface by enhancing antigen presentation, T cell infiltration, and effector persistence, positioning epitranscriptomic modulation as a viable adjunct to checkpoint therapy.
Beyond METTL3 inhibition, the functional tuning of m6A reader proteins also offers therapeutic promise. Loss of YTHDF2 enhances Th9 differentiation by stabilizing Gata3 and Smad3 mRNAs under IL-4 and TGF-β signalling, promoting IL-9 and IL-21 production, increased CD8+ T cell and NK cell infiltration, and improved antitumour efficacy. In CAR-Th9 cells, YTHDF2 depletion sustains immune activation and augments cytotoxic potential, identifying YTHDF2 as a negative epitranscriptomic checkpoint restraining T cell-mediated immunity [145].
Nevertheless, the dual identity of RNA-modifying enzymes complicates their therapeutic targeting. The same factor can exert oncogenic or tumour-suppressive effects depending on cellular context—for example, METTL3 deletion in melanoma cells suppresses tumour progression and enhances CD8+ T cell infiltration [20], whereas METTL3 loss in macrophages promotes tumour growth and impairs PD-1 blockade efficacy [146]. Such context-dependent outcomes highlight the necessity for cell-type-specific delivery systems (e.g., lipid nanoparticles or virus-like particles) and temporal control mechanisms, including drug-inducible degrons or switchable RNA editors, to achieve precise and reversible modulation during T cell manufacturing or post-infusion phases.
Because RNA modifications are broadly involved in essential physiological processes, selective targeting within the tumour microenvironment without perturbing normal homeostasis remains a major challenge. Although advances in structure-guided drug design have yielded molecules with improved specificity, complete separation of therapeutic and physiological effects has not yet been achieved. The integration of standardized detection frameworks, cell-type-restricted delivery, and temporal control strategies will be key to transforming RNA modifications from static biomarkers into actionable determinants of immune behavior. Collectively, these advances establish a conceptual foundation for precision epitranscriptomic immunotherapy, in which RNA-modification pathways are harnessed to reprogram T cell metabolism, activation, and persistence in cancer and beyond.
Conclusion
RNA modifications have emerged as a fundamental epitranscriptomic mechanism that integrates transcriptional, metabolic, and post-transcriptional regulation to orchestrate T cell activation, differentiation, and functional plasticity. Through the coordinated interplay of methyltransferases, demethylases, and reader proteins, these chemical modifications remodel the transcriptomic landscape and control RNA metabolism at multiple levels, coupling extracellular cues and intracellular metabolic states to precise gene-expression programs. This multilayered regulatory system enables T cells to balance effector expansion with immune tolerance, thereby maintaining immune homeostasis while preserving responsiveness to antigenic stimulation.
Across distinct T cell lineages, RNA modifications act as key determinants of lineage commitment, effector specification, and memory persistence. By modulating transcript stability, translational efficiency, and RNA decay, they synchronize transcriptional output with the biosynthetic and energetic demands of immune activation. Functioning as molecular rheostats, these modifications translate transient signaling inputs into durable transcriptional states that define activation thresholds, clonal fitness, and survival potential. Such epitranscriptomic regulation operates in concert with transcriptional and epigenetic programs, establishing a hierarchical control network that safeguards immune precision and adaptability.
The primary objective of this review is to delineate the conceptual and mechanistic framework through which RNA modifications govern T cell fate and function. By synthesizing recent advances across molecular, cellular, and pathological contexts, this work consolidates current understanding of how distinct RNA modifications converge to regulate transcript stability, translational control, and metabolic reprogramming, thereby shaping T cell behavior in immunity and disease. The rationale underlying this synthesis is to interpret the epitranscriptome as a dynamic and programmable interface that integrates signaling intensity, metabolic flux, and chromatin architecture into coherent immune outcomes. Through this perspective, RNA modification machinery emerges as a set of tunable molecular nodes with substantial therapeutic potential for the modulation of immune responses.
Despite these advances, several conceptual and technical challenges constrain current progress. Most available studies examine individual RNA modifications or single regulatory enzymes in isolation, providing limited insight into their combinatorial interactions or site-specific cross-regulation within the same transcript. The temporal and spatial dynamics of RNA modifications throughout T cell activation, differentiation, and memory maintenance remain insufficiently resolved. Furthermore, the interplay between RNA modifications and other epigenetic layers—such as DNA methylation, histone modifications, and chromatin accessibility—remains largely unexplored, despite their likely role in establishing integrated regulatory feedback. Methodological limitations in quantitative mapping, stoichiometric resolution, and single-cell detection further restrict mechanistic dissection. Translational application also requires the development of selective, reversible modulators of RNA-modifying enzymes and effective strategies for in vivo delivery, which remain in early stages of development.
Future research should aim to construct an integrative and spatiotemporally resolved map of the T cell epitranscriptome. The integration of single-cell multi-omics, spatial transcriptomics, and metabolic tracing will be instrumental in defining the dynamic coordination between RNA modifications, transcriptional programs, and metabolic adaptation. Advances in programmable RNA-modification editing and synthetic epitranscriptomic technologies may allow precise reconfiguration of T cell states, enabling targeted modulation of activation, persistence, and exhaustion. Ultimately, elucidating how RNA modifications encode, stabilize, and reprogram immune identity will provide the conceptual foundation for next-generation immunotherapeutic strategies based on epitranscriptomic modulation.
In summary, RNA modifications constitute a central regulatory axis that confers flexibility, precision, and endurance upon T cell immunity. They form the molecular syntax through which immune cells integrate signaling and metabolic information into defined transcriptional and functional states. Deciphering this epitranscriptomic code will not only advance our fundamental understanding of T cell biology but also open translational avenues for rationally engineering immune responses in infection, autoimmunity, and cancer immunotherapy.
Abbreviations
ac4C: N4-acetylcytidine; CAD: coronary artery disease; CAR: chimeric antigen receptor; CNS: central nervous system; dsRNA: double-stranded RNA; EAE: experimental autoimmune encephalomyelitis; EMT: epithelial-mesenchymal transition; GO: Graves' ophthalmopathy; HCC: hepatocellular carcinoma; IBD: inflammatory bowel disease; IDC: invasive ductal carcinoma; iNKT: invariant natural killer T; iTregs: induced regulatory T cells; LUSC: lung squamous cell carcinoma; m1A: N1-methyladenosine; m5C: 5-methylcytosine; m6A: N6-methyladenosine; m7G: N7-methylguanosine; MT-ND6: mitochondrial-encoded NADH dehydrogenase 6; NPC: nasopharyngeal carcinoma; SCD1: stearoyl-CoA desaturase 1; SLE: Systemic lupus erythematosus; Tfh: T follicular helper; THBS1: thrombospondin 1; Treg: regulatory T cell; Ψ: pseudouridine.
Acknowledgements
Funding
This work was supported by National Natural Science Foundation of China (82372905, 81901006), Young Top-notch Talent of Pearl River Talent Plan (0920220228), Guangdong Provincial Science and Technology Project Foundation (2022A0505050038), and Science Research Cultivation Program of Stomatological Hospital, Southern Medical University (PY2020002, PY2022019).
Author contributions
LC conceived the idea and supervised the review. XYZ drafted the manuscript. MYZ edited the text and designed the figures and tables. WYF, NNL and XC developed the visualizations and graphs. PL, ZHZ and YFL participated in writing and revision. All authors reviewed and approved the final manuscript.
Competing interests
The authors have declared that no competing interest exists.
References
1. Pishesha N, Harmand TJ, Ploegh HL. A guide to antigen processing and presentation. Nat Rev Immunol. 2022;22:751-64
2. Jensen PE. Mechanisms of antigen presentation. Clin Chem Lab Med. 1999;37:179-86
3. Kaech SM, Wherry EJ, Ahmed R. Effector and memory T-cell differentiation: implications for vaccine development. Nat Rev Immunol. 2002;2:251-62
4. Rossi F, Turner M. TEAMwork: Interplay of Post-Transcriptional Mechanisms, Epigenetics and Metabolism in (Auto-)Immunity. Eur J Immunol. 2025;55:e70064
5. Kanno Y, Vahedi G, Hirahara K, Singleton K, O'Shea JJ. Transcriptional and epigenetic control of T helper cell specification: molecular mechanisms underlying commitment and plasticity. Annu Rev Immunol. 2012;30:707-31
6. Zhang J, Bi G, Xia Y, Li P, Deng X, Wei Z. et al. Transcriptional Regulation of T Cell Metabolism Reprograming. Adv Exp Med Biol. 2017;1011:131-52
7. Chapman NM, Boothby MR, Chi H. Metabolic coordination of T cell quiescence and activation. Nat Rev Immunol. 2020;20:55-70
8. Basu A, Ramamoorthi G, Albert G, Gallen C, Beyer A, Snyder C. et al. Differentiation and Regulation of T(H) Cells: A Balancing Act for Cancer Immunotherapy. Front Immunol. 2021;12:669474
9. Jurgens AP, Popović B, Wolkers MC. T cells at work: How post-transcriptional mechanisms control T cell homeostasis and activation. Eur J Immunol. 2021;51:2178-87
10. Liu Y, Zhou J, Li X, Zhang X, Shi J, Wang X. et al. tRNA-m(1)A modification promotes T cell expansion via efficient MYC protein synthesis. Nat Immunol. 2022;23:1433-44
11. Karginov TA, Ménoret A, Leclair NK, Harrison AG, Chandiran K, Suarez-Ramirez JE. et al. Autoregulated splicing of TRA2β programs T cell fate in response to antigen-receptor stimulation. Science. 2024;385:eadj1979
12. Song J, Yi C. Chemical Modifications to RNA: A New Layer of Gene Expression Regulation. ACS Chem Biol. 2017;12:316-25
13. Yang W, Zhao Y, Yang Y. Dynamic RNA methylation modifications and their regulatory role in mammalian development and diseases. Sci China Life Sci. 2024;67:2084-104
14. Flamand MN, Tegowski M, Meyer KD. The Proteins of mRNA Modification: Writers, Readers, and Erasers. Annu Rev Biochem. 2023;92:145-73
15. Shi H, Wei J, He C. Where, When, and How: Context-Dependent Functions of RNA Methylation Writers, Readers, and Erasers. Mol Cell. 2019;74:640-50
16. Li X, Xiong X, Yi C. Epitranscriptome sequencing technologies: decoding RNA modifications. Nat Methods. 2016;14:23-31
17. Guo W, Wang Z, Zhang Y, Li Y, Du Q, Zhang T. et al. Mettl3-dependent m(6)A modification is essential for effector differentiation and memory formation of CD8(+) T cells. Sci Bull (Beijing). 2024;69:82-96
18. Zhang H, Luo X, Yang W, Wu Z, Zhao Z, Pei X. et al. YTHDF2 upregulation and subcellular localization dictate CD8 T cell polyfunctionality in anti-tumor immunity. Nat Commun. 2024;15:9559
19. Meister H, Look T, Roth P, Pascolo S, Sahin U, Lee S. et al. Multifunctional mRNA-Based CAR T Cells Display Promising Antitumor Activity Against Glioblastoma. Clin Cancer Res. 2022;28:4747-56
20. Wang L, Hui H, Agrawal K, Kang Y, Li N, Tang R. et al. m(6) A RNA methyltransferases METTL3/14 regulate immune responses to anti-PD-1 therapy. Embo j. 2020;39:e104514
21. Li HB, Tong J, Zhu S, Batista PJ, Duffy EE, Zhao J. et al. m(6)A mRNA methylation controls T cell homeostasis by targeting the IL-7/STAT5/SOCS pathways. Nature. 2017;548:338-42
22. Cui L, Ma R, Cai J, Guo C, Chen Z, Yao L. et al. RNA modifications: importance in immune cell biology and related diseases. Signal Transduct Target Ther. 2022;7:334
23. Cappannini A, Ray A, Purta E, Mukherjee S, Boccaletto P, Moafinejad SN. et al. MODOMICS: a database of RNA modifications and related information. 2023 update. Nucleic Acids Res. 2024;52:D239-d44
24. Yang Y, Hsu PJ, Chen YS, Yang YG. Dynamic transcriptomic m(6)A decoration: writers, erasers, readers and functions in RNA metabolism. Cell Res. 2018;28:616-24
25. Chen YS, Yang WL, Zhao YL, Yang YG. Dynamic transcriptomic m(5) C and its regulatory role in RNA processing. Wiley Interdiscip Rev RNA. 2021;12:e1639
26. Karijolich J, Yi C, Yu YT. Transcriptome-wide dynamics of RNA pseudouridylation. Nat Rev Mol Cell Biol. 2015;16:581-5
27. Cowling VH. Regulation of mRNA cap methylation. Biochem J. 2009;425:295-302
28. Dominissini D, Nachtergaele S, Moshitch-Moshkovitz S, Peer E, Kol N, Ben-Haim MS. et al. The dynamic N(1)-methyladenosine methylome in eukaryotic messenger RNA. Nature. 2016;530:441-6
29. Jin G, Xu M, Zou M, Duan S. The Processing, Gene Regulation, Biological Functions, and Clinical Relevance of N4-Acetylcytidine on RNA: A Systematic Review. Mol Ther Nucleic Acids. 2020;20:13-24
30. Dominguez D, Freese P, Alexis MS, Su A, Hochman M, Palden T. et al. Sequence, Structure, and Context Preferences of Human RNA Binding Proteins. Mol Cell. 2018;70:854-67.e9
31. Höfler S, Duss O. Interconnections between m(6)A RNA modification, RNA structure, and protein-RNA complex assembly. Life Sci Alliance. 2024 7
32. Schöller E, Weichmann F, Treiber T, Ringle S, Treiber N, Flatley A. et al. Interactions, localization, and phosphorylation of the m(6)A generating METTL3-METTL14-WTAP complex. Rna. 2018;24:499-512
33. Liu J, Yue Y, Han D, Wang X, Fu Y, Zhang L. et al. A METTL3-METTL14 complex mediates mammalian nuclear RNA N6-adenosine methylation. Nat Chem Biol. 2014;10:93-5
34. He PC, He C. m(6) A RNA methylation: from mechanisms to therapeutic potential. Embo j. 2021;40:e105977
35. Yue Y, Liu J, Cui X, Cao J, Luo G, Zhang Z. et al. VIRMA mediates preferential m(6)A mRNA methylation in 3'UTR and near stop codon and associates with alternative polyadenylation. Cell Discov. 2018;4:10
36. Wen J, Lv R, Ma H, Shen H, He C, Wang J. et al. Zc3h13 Regulates Nuclear RNA m(6)A Methylation and Mouse Embryonic Stem Cell Self-Renewal. Mol Cell. 2018;69:1028-38.e6
37. Deng T, Ma J. Structures and mechanisms of the RNA m (6)A writer. Acta Biochim Biophys Sin (Shanghai). 2024;57:59-72
38. Patil DP, Chen CK, Pickering BF, Chow A, Jackson C, Guttman M. et al. m(6)A RNA methylation promotes XIST-mediated transcriptional repression. Nature. 2016;537:369-73
39. Zheng G, Dahl JA, Niu Y, Fedorcsak P, Huang CM, Li CJ. et al. ALKBH5 is a mammalian RNA demethylase that impacts RNA metabolism and mouse fertility. Mol Cell. 2013;49:18-29
40. Jia G, Fu Y, Zhao X, Dai Q, Zheng G, Yang Y. et al. N6-methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat Chem Biol. 2011;7:885-7
41. Zaccara S, Jaffrey SR. A Unified Model for the Function of YTHDF Proteins in Regulating m(6)A-Modified mRNA. Cell. 2020;181:1582-95.e18
42. Wang X, Zhao BS, Roundtree IA, Lu Z, Han D, Ma H. et al. N(6)-methyladenosine Modulates Messenger RNA Translation Efficiency. Cell. 2015;161:1388-99
43. Zhou J, Wan J, Gao X, Zhang X, Jaffrey SR, Qian SB. Dynamic m(6)A mRNA methylation directs translational control of heat shock response. Nature. 2015;526:591-4
44. Wang X, Lu Z, Gomez A, Hon GC, Yue Y, Han D. et al. N6-methyladenosine-dependent regulation of messenger RNA stability. Nature. 2014;505:117-20
45. Xiao W, Adhikari S, Dahal U, Chen YS, Hao YJ, Sun BF. et al. Nuclear m(6)A Reader YTHDC1 Regulates mRNA Splicing. Mol Cell. 2016;61:507-19
46. Roundtree IA, Luo GZ, Zhang Z, Wang X, Zhou T, Cui Y. et al. YTHDC1 mediates nuclear export of N(6)-methyladenosine methylated mRNAs. Elife. 2017 6
47. Hsu PJ, Zhu Y, Ma H, Guo Y, Shi X, Liu Y. et al. Ythdc2 is an N(6)-methyladenosine binding protein that regulates mammalian spermatogenesis. Cell Res. 2017;27:1115-27
48. Liao H, Gaur A, McConie H, Shekar A, Wang K, Chang JT. et al. Human NOP2/NSUN1 regulates ribosome biogenesis through non-catalytic complex formation with box C/D snoRNPs. Nucleic Acids Res. 2022;50:10695-716
49. Yang X, Yang Y, Sun BF, Chen YS, Xu JW, Lai WY. et al. 5-methylcytosine promotes mRNA export - NSUN2 as the methyltransferase and ALYREF as an m(5)C reader. Cell Res. 2017;27:606-25
50. Nakano S, Suzuki T, Kawarada L, Iwata H, Asano K, Suzuki T. NSUN3 methylase initiates 5-formylcytidine biogenesis in human mitochondrial tRNA(Met). Nat Chem Biol. 2016;12:546-51
51. Metodiev MD, Spåhr H, Loguercio Polosa P, Meharg C, Becker C, Altmueller J. et al. NSUN4 is a dual function mitochondrial protein required for both methylation of 12S rRNA and coordination of mitoribosomal assembly. PLoS Genet. 2014;10:e1004110
52. Heissenberger C, Liendl L, Nagelreiter F, Gonskikh Y, Yang G, Stelzer EM. et al. Loss of the ribosomal RNA methyltransferase NSUN5 impairs global protein synthesis and normal growth. Nucleic Acids Res. 2019;47:11807-25
53. Haag S, Warda AS, Kretschmer J, Günnigmann MA, Höbartner C, Bohnsack MT. NSUN6 is a human RNA methyltransferase that catalyzes formation of m5C72 in specific tRNAs. Rna. 2015;21:1532-43
54. Harris T, Marquez B, Suarez S, Schimenti J. Sperm motility defects and infertility in male mice with a mutation in Nsun7, a member of the Sun domain-containing family of putative RNA methyltransferases. Biol Reprod. 2007;77:376-82
55. Huang ZX, Li J, Xiong QP, Li H, Wang ED, Liu RJ. Position 34 of tRNA is a discriminative element for m5C38 modification by human DNMT2. Nucleic Acids Res. 2021;49:13045-61
56. McCleverty CJ, Hornsby M, Spraggon G, Kreusch A. Crystal structure of human Pus10, a novel pseudouridine synthase. J Mol Biol. 2007;373:1243-54
57. Ito S, Horikawa S, Suzuki T, Kawauchi H, Tanaka Y, Suzuki T. et al. Human NAT10 is an ATP-dependent RNA acetyltransferase responsible for N4-acetylcytidine formation in 18 S ribosomal RNA (rRNA). J Biol Chem. 2014;289:35724-30
58. Varshney D, Lombardi O, Schweikert G, Dunn S, Suska O, Cowling VH. mRNA Cap Methyltransferase, RNMT-RAM, Promotes RNA Pol II-Dependent Transcription. Cell Rep. 2018;23:1530-42
59. Safra M, Sas-Chen A, Nir R, Winkler R, Nachshon A, Bar-Yaacov D. et al. The m1A landscape on cytosolic and mitochondrial mRNA at single-base resolution. Nature. 2017;551:251-5
60. Ren B, He K, Wei N, Liu S, Cui X, Yang X. et al. Nanoparticle-Delivered siRNA Targeting NSUN4 Relieves Systemic Lupus Erythematosus through Declining Mitophagy-Mediated CD8+T Cell Exhaustion. MedComm (2020). 2025;6:e70311
61. Zhu Y, Zhao Y, Zou L, Zhang D, Aki D, Liu YC. The E3 ligase VHL promotes follicular helper T cell differentiation via glycolytic-epigenetic control. J Exp Med. 2019;216:1664-81
62. Xu H, Shen K, Jiang B, Xu Q, Li B, Ke Z. et al. VISTA expressed on tumor cells is regulated by m6A and influences immune microenvironment through STAT3/CCL22 in NSCLC. J Transl Med. 2025;23:1043
63. Zhang Y, Lu L, Li X. Detection technologies for RNA modifications. Exp Mol Med. 2022;54:1601-16
64. Zhao X, Zhang Y, Hang D, Meng J, Wei Z. Detecting RNA modification using direct RNA sequencing: A systematic review. Comput Struct Biotechnol J. 2022;20:5740-9
65. Zhang Y, Huang D, Wei Z, Chen K. Primary sequence-assisted prediction of m(6)A RNA methylation sites from Oxford nanopore direct RNA sequencing data. Methods. 2022;203:62-9
66. Wu Y, Shao W, Liu S, Wang L, Xu P, Zhang X. et al. Simultaneous profiling of ac(4)C and m(5)C modifications from nanopore direct RNA sequencing. Int J Biol Macromol. 2025;305:140863
67. Kan RL, Chen J, Sallam T. Crosstalk between epitranscriptomic and epigenetic mechanisms in gene regulation. Trends Genet. 2022;38:182-93
68. Chen Y, Wan R, Zou Z, Lao L, Shao G, Zheng Y. et al. O-GlcNAcylation determines the translational regulation and phase separation of YTHDF proteins. Nat Cell Biol. 2023;25:1676-90
69. Reichle VF, Kaiser S, Heiss M, Hagelskamp F, Borland K, Kellner S. Surpassing limits of static RNA modification analysis with dynamic NAIL-MS. Methods. 2019;156:91-101
70. Xu W, He C, Kaye EG, Li J, Mu M, Nelson GM. et al. Dynamic control of chromatin-associated m(6)A methylation regulates nascent RNA synthesis. Mol Cell. 2022;82:1156-68.e7
71. Li J, Kang H. m6A hypermethylation of TCF-1 regulated by METTL16 promotes acute myeloid leukemia. Clin Exp Med. 2025;25:129
72. Wu K, Li S, Hong G, Dong H, Tang T, Liu H. et al. Targeting METTL3 as a checkpoint to enhance T cells for tumour immunotherapy. Clin Transl Med. 2024;14:e70089
73. Sun L, Li X, Xu F, Chen Y, Li X, Yang Z. et al. A critical role of N(4)-acetylation of cytidine in mRNA by NAT10 in T cell expansion and antiviral immunity. Nat Immunol. 2025;26:619-34
74. Wang A, Huang H, Shi JH, Yu X, Ding R, Zhang Y. et al. USP47 inhibits m6A-dependent c-Myc translation to maintain regulatory T cell metabolic and functional homeostasis. J Clin Invest. 2023 133
75. Liu S, Liu Y, Zhou Y, Xia G, Liu H, Zeng Y. et al. NSUN5 promotes tumorigenic phenotypes through the WNT signaling pathway and immunosuppression of CD8+ T cells in gastric cancer. Cell Signal. 2024;124:111475
76. Knudson CJ, Alves-Peixoto P, Muramatsu H, Stotesbury C, Tang L, Lin PJC. et al. Lipid-nanoparticle-encapsulated mRNA vaccines induce protective memory CD8 T cells against a lethal viral infection. Mol Ther. 2021;29:2769-81
77. m(1)A tRNA modification facilitates rapid T cell proliferation. Nat Immunol. 2022; 23: 1408-9.
78. Roundtree IA, Evans ME, Pan T, He C. Dynamic RNA Modifications in Gene Expression Regulation. Cell. 2017;169:1187-200
79. Sun L, Zhang T, Ge Y, Yao Z, Su Y, Wang Q. et al. FTO controls CD8(+) T cell survival and effector response by modulating m(6)A methylation of Fas. Cell Death Dis. 2025;16:301
80. van Vroonhoven ECN, Picavet LW, Scholman RC, Sijbers L, Kievit CRE, van den Dungen NAM. et al. N6-methyladenosine promotes TNF mRNA degradation in CD4+ T lymphocytes. J Leukoc Biol. 2024;116:807-15
81. Fang M, Li Y, Wang P, Wang Y, Wang X, Wa X. et al. METTL3 Inhibition Restores PD-L1 Expression and CD8+ T-cell Cytotoxic Function in Immunotherapy-Treated Gastric Cancer. Cancer Immunol Res. 2025;13:1037-52
82. Bao Y, Zhai J, Chen H, Wong CC, Liang C, Ding Y. et al. Targeting m(6)A reader YTHDF1 augments antitumour immunity and boosts anti-PD-1 efficacy in colorectal cancer. Gut. 2023;72:1497-509
83. Chen X, Tong X, Zhou L, Huang J, Gao L, Zeng J. et al. Critical role of m(6)A modification in T-helper cell disorders. Mol Immunol. 2022;151:1-10
84. Wang K, Liu F, Muchu B, Deng J, Peng J, Xu Y. et al. E3 ubiquitin ligase RNF180 mediates the ALKBH5/SMARCA5 axis to promote colon inflammation and Th17/Treg imbalance in ulcerative colitis mice. Arch Pharm Res. 2024;47:645-58
85. van Vroonhoven ECN, Picavet LW, Scholman RC, van den Dungen NAM, Mokry M, Evers A. et al. N(6)-Methyladenosine Directly Regulates CD40L Expression in CD4(+) T Lymphocytes. Biology (Basel). 2023 12
86. Yao Y, Yang Y, Guo W, Xu L, You M, Zhang YC. et al. METTL3-dependent m(6)A modification programs T follicular helper cell differentiation. Nat Commun. 2021;12:1333
87. Liu Y, Yuan Y, Zhou Z, Jiang X, He S, Wei F. et al. Mettl14 sustains FOXP3 expression to promote the differentiation and functions of induced-regulatory T cells via the mTOR signaling pathway. Immunol Lett. 2023;258:35-44
88. Zhao J, Ding C, Li HB. N(6) - Methyladenosine defines a new checkpoint in γδ T cell development. Bioessays. 2023;45:e2300002
89. Pellicci DG, Koay HF, Berzins SP. Thymic development of unconventional T cells: how NKT cells, MAIT cells and γδ T cells emerge. Nat Rev Immunol. 2020;20:756-70
90. Ding C, Xu H, Yu Z, Roulis M, Qu R, Zhou J. et al. RNA m(6)A demethylase ALKBH5 regulates the development of γδ T cells. Proc Natl Acad Sci U S A. 2022;119:e2203318119
91. Xiao Z, Wang S, Tian Y, Lv W, Sheng H, Zhan M. et al. METTL3-mediated m6A methylation orchestrates mRNA stability and dsRNA contents to equilibrate γδ T1 and γδ T17 cells. Cell Rep. 2023;42:112684
92. Cao L, Morgun E, Genardi S, Visvabharathy L, Cui Y, Huang H. et al. METTL14-dependent m(6)A modification controls iNKT cell development and function. Cell Rep. 2022;40:111156
93. You M, Liu J, Li J, Ji C, Ni H, Guo W. et al. Mettl3-m(6)A-Creb1 forms an intrinsic regulatory axis in maintaining iNKT cell pool and functional differentiation. Cell Rep. 2023;42:112584
94. Wang N, Tang H, Wang X, Wang W, Feng J. Homocysteine upregulates interleukin-17A expression via NSun2-mediated RNA methylation in T lymphocytes. Biochem Biophys Res Commun. 2017;493:94-9
95. Fanari O, Bloch D, Qiu Y, Meseonznik M, Boyko D, Makhamreh A. et al. Pseudouridine reprogramming in the human T-cell epitranscriptome: from primary to immortalized states. Rna. 2025;31:1320-34
96. Madej M, Ngoc PCT, Muthukumar S, Konturek-Cieśla A, Tucciarone S, Germanos A. et al. PUS10-induced tRNA fragmentation impacts retrotransposon-driven inflammation. Cell Rep. 2025;44:115735
97. Drzeniek NM, Kahwaji N, Picht S, Dimitriou IM, Schlickeiser S, Moradian H. et al. In vitro Transcribed mRNA Immunogenicity Induces Chemokine-Mediated Lymphocyte Recruitment and Can Be Gradually Tailored by Uridine Modification. Adv Sci (Weinh). 2024;11:e2308447
98. Galloway A, Kaskar A, Ditsova D, Atrih A, Yoshikawa H, Gomez-Moreira C. et al. Upregulation of RNA cap methyltransferase RNMT drives ribosome biogenesis during T cell activation. Nucleic Acids Res. 2021;49:6722-38
99. Zha LF, Wang JL, Cheng X. The effects of RNA methylation on immune cells development and function. Faseb j. 2022;36:e22552
100. Orji OC, Stones J, Rajani S, Markus R, Öz MD, Knight HM. Global Co-regulatory Cross Talk Between m(6)A and m(5)C RNA Methylation Systems Coordinate Cellular Responses and Brain Disease Pathways. Mol Neurobiol. 2025;62:5006-21
101. Chen B, Hong Y, Zhai X, Deng Y, Hu H, Tian S. et al. m6A and m5C modification of GPX4 facilitates anticancer immunity via STING activation. Cell Death Dis. 2023;14:809
102. Liu Z, Wang T, She Y, Wu K, Gu S, Li L. et al. N(6)-methyladenosine-modified circIGF2BP3 inhibits CD8(+) T-cell responses to facilitate tumor immune evasion by promoting the deubiquitination of PD-L1 in non-small cell lung cancer. Mol Cancer. 2021;20:105
103. Zhai J, Chen H, Wong CC, Peng Y, Gou H, Zhang J. et al. ALKBH5 Drives Immune Suppression Via Targeting AXIN2 to Promote Colorectal Cancer and Is a Target for Boosting Immunotherapy. Gastroenterology. 2023;165:445-62
104. Tan Z, Hei F, Ma K, Lv Z, Zhang H, Sun N. et al. m(6)A reader YTHDF2 orchestrates CD8(+) T cell infiltration to promote pancreatic cancer progression and predicts clinical outcome. Int Immunopharmacol. 2024;142:113079
105. Xiang B, Zhang M, Li K, Zhang Z, Liu Y, Gao M. et al. The epitranscriptional factor PCIF1 orchestrates CD8(+) T cell ferroptosis and activation to control antitumor immunity. Nat Immunol. 2025;26:252-64
106. Miao S, Li H, Song X, Liu Y, Wang G, Kan C. et al. tRNA m1A modification regulates cholesterol biosynthesis to promote antitumor immunity of CD8+ T cells. J Exp Med. 2025 222
107. Long Z, Li X, Deng W, Tan Y, Liu J. Tumor-associated characteristics and immune dysregulation in nasopharyngeal carcinoma under the regulation of m7G-related tumor microenvironment cells. World J Surg Oncol. 2024;22:166
108. Lv X, Liu X, Zhao M, Wu H, Zhang W, Lu Q. et al. RNA Methylation in Systemic Lupus Erythematosus. Front Cell Dev Biol. 2021;9:696559
109. Lu S, Wei X, Zhu H, Hu Z, Zheng M, Wu J. et al. m(6)A methyltransferase METTL3 programs CD4(+) T-cell activation and effector T-cell differentiation in systemic lupus erythematosus. Mol Med. 2023;29:46
110. Abdukiyum M, Tang X, Zhao N, Cui Y, Zhang J, Alim T. et al. Reduced mitochondrial-encoded NADH dehydrogenase 6 gene expression drives inflammatory CD4(+)T cells in patients with systemic lupus erythematosus. Free Radic Biol Med. 2024;213:79-89
111. Guo G, Wang H, Shi X, Ye L, Yan K, Chen Z. et al. Disease Activity-Associated Alteration of mRNA m(5) C Methylation in CD4(+) T Cells of Systemic Lupus Erythematosus. Front Cell Dev Biol. 2020;8:430
112. Guo G, Shi X, Wang H, Ye L, Tong X, Yan K. et al. Epitranscriptomic N4-Acetylcytidine Profiling in CD4(+) T Cells of Systemic Lupus Erythematosus. Front Cell Dev Biol. 2020;8:842
113. Yuan L, Chen S, Ding K, Wang X, Lv W, Liu Y. et al. The m(6) A modification of Il17a in CD4(+) T cells promotes inflammation in psoriasis. Exp Dermatol. 2024;33:e14879
114. Li LN, Wu JM, Zheng ZJ, Li SX, Cai MY, Zou MC. N6-methyladenosine modification of THBS1 induced by affluent WTAP promotes Graves' ophthalmopathy progression through glycolysis to affect Th17/Treg balance. Autoimmunity. 2025;58:2433628
115. Zhao L, Liu Y, Ma B, Liu X, Wei R, Nian H. METTL3 inhibits autoreactive Th17 cell responses in experimental autoimmune uveitis via stabilizing ASH1L mRNA. Faseb j. 2023;37:e22803
116. Li H, Cai X, Xu C, Yang X, Song X, Kong Y. et al. RNA cytidine acetyltransferase NAT10 maintains T cell pathogenicity in inflammatory bowel disease. Cell Discov. 2025;11:19
117. Yang WL, Qiu W, Zhang T, Xu K, Gu ZJ, Zhou Y. et al. Nsun2 coupling with RoRγt shapes the fate of Th17 cells and promotes colitis. Nat Commun. 2023;14:863
118. Wang Z, Qi Y, Feng Y, Xu H, Wang J, Zhang L. et al. The N6-methyladenosine writer WTAP contributes to the induction of immune tolerance post kidney transplantation by targeting regulatory T cells. Lab Invest. 2022;102:1268-79
119. Li S, Zou D, Chen W, Britz GW, Liu Z, Weng YL. METTL3 inhibition reduces N(6) -methyladenosine levels and prevents allogeneic CD4(+) T-cell responses. Immunol Cell Biol. 2022;100:718-30
120. Huang M, Mark A, Pham J, Vera K, Saravia-Butler AM, Beheshti A. et al. RNA editing regulates host immune response and T cell homeostasis in SARS-CoV-2 infection. PLoS One. 2024;19:e0307450
121. Tirumuru N, Zhao BS, Lu W, Lu Z, He C, Wu L. N(6)-methyladenosine of HIV-1 RNA regulates viral infection and HIV-1 Gag protein expression. Elife. 2016 5
122. Yao Z, Sun L, Gao Y, Su Y, He B, Ge Y. et al. The m(6)A demethylase FTO controls Th1 differentiation and immunity against infections. Mol Immunol. 2025;183:172-81
123. Mishra T, Phillips S, Zhao Y, Wilms B, He C, Wu L. Epitranscriptomic m(6)A modifications during reactivation of HIV-1 latency in CD4(+) T cells. mBio. 2024;15:e0221424
124. Qiu X, Hua X, Li Q, Zhou Q, Chen J. m(6)A Regulator-Mediated Methylation Modification Patterns and Characteristics of Immunity in Blood Leukocytes of COVID-19 Patients. Front Immunol. 2021;12:774776
125. Wang X, Chen C, Sun H, Mao K, Yao J, Zhang W. et al. m(6)A mRNA modification potentiates Th17 functions to inflame autoimmunity. Sci China Life Sci. 2023;66:2543-52
126. Zhou J, Zhang X, Hu J, Qu R, Yu Z, Xu H. et al. m(6)A demethylase ALKBH5 controls CD4(+) T cell pathogenicity and promotes autoimmunity. Sci Adv. 2021 7
127. Chen Z, Yan D, Guo S, Song Y, Zhang X, Gu W. et al. METTL3/miR-192-5p/SCD1 Axis Regulates Lipid Metabolism to Affect T Cell Differentiation in Asthma. Mediators Inflamm. 2025;2025:4955849
128. Fan L, Wu J, Wang H, Chen Q, He X, Wang Q. et al. METTL3-Mediated N6-Methyladenosine Methylation Modifies Foxp3 mRNA Levels and Affects the Treg Cells Proportion in Peripheral Blood of Patients with Asthma. Ann Clin Lab Sci. 2022;52:884-94
129. Rakshit S, Sunny JS, George M, Hanna LE, Leela KV, Sarkar K. T helper cell-mediated epitranscriptomic regulation via m6A RNA methylation bridges link between coronary artery disease and invasive ductal carcinoma. J Cancer Res Clin Oncol. 2022;148:3421-36
130. Li J, Feng H, Zhu J, Yang K, Zhang G, Gu Y. et al. Gastric cancer derived exosomal THBS1 enhanced Vγ9Vδ2 T-cell function through activating RIG-I-like receptor signaling pathway in a N6-methyladenosine methylation dependent manner. Cancer Lett. 2023;576:216410
131. Yu T, Nie FQ, Zhang Q, Yu SK, Zhang ML, Wang Q. et al. Effects of methionine deficiency on B7H3-DAP12-CAR-T cells in the treatment of lung squamous cell carcinoma. Cell Death Dis. 2024;15:12
132. Li R, Li S, Li H, Liu B, Wang Z, Lei H. et al. WTAP Accelerates Exhaustion of CD8(+) T Cells and Progression of Hepatocellular Carcinoma by Promoting m6A Modification and Translation of PD1 mRNA. Mediators Inflamm. 2025;2025:6217272
133. Diensthuber G, Novoa EM. Charting the epitranscriptomic landscape across RNA biotypes using native RNA nanopore sequencing. Mol Cell. 2025;85:276-89
134. Marinov GK, Williams BA, McCue K, Schroth GP, Gertz J, Myers RM. et al. From single-cell to cell-pool transcriptomes: stochasticity in gene expression and RNA splicing. Genome Res. 2014;24:496-510
135. Hess JF, Kohl TA, Kotrová M, Rönsch K, Paprotka T, Mohr V. et al. Library preparation for next generation sequencing: A review of automation strategies. Biotechnol Adv. 2020;41:107537
136. Papetti DM, Spolaor S, Nazari I, Tirelli A, Leonardi T, Caprioli C. et al. Barcode demultiplexing of nanopore sequencing raw signals by unsupervised machine learning. Front Bioinform. 2023;3:1067113
137. Yates KB, Tonnerre P, Martin GE, Gerdemann U, Al Abosy R, Comstock DE. et al. Epigenetic scars of CD8(+) T cell exhaustion persist after cure of chronic infection in humans. Nat Immunol. 2021;22:1020-9
138. Sen DR, Kaminski J, Barnitz RA, Kurachi M, Gerdemann U, Yates KB. et al. The epigenetic landscape of T cell exhaustion. Science. 2016;354:1165-9
139. Pai JA, Hellmann MD, Sauter JL, Mattar M, Rizvi H, Woo HJ. et al. Lineage tracing reveals clonal progenitors and long-term persistence of tumor-specific T cells during immune checkpoint blockade. Cancer Cell. 2023;41:776-90.e7
140. Qiu MZ, Wang C, Wu Z, Zhao Q, Zhao Z, Huang CY. et al. Dynamic single-cell mapping unveils Epstein-Barr virus-imprinted T-cell exhaustion and on-treatment response. Signal Transduct Target Ther. 2023;8:370
141. Peng T, Fang Q, Zhao Z, Chang Y, Zhao X, Li C. Multi-omics integration analysis identifies INPP4B as a T-cell-specific activation suppressor. Clin Transl Med. 2025;15:e70430
142. Ilyas M, Shah Q, Gul A, Ibrahim H, Fatima R, Babar MM. et al. Advances in CRISPR-Cas systems for epigenetics. Prog Mol Biol Transl Sci. 2024;208:185-209
143. Huang M, Ewadi A, Servatian N, Noormohamadi H, Aminov Z, Muzammil K. et al. Epigenetic mechanisms and next-gen editing platforms in hematology: From molecular basis to therapeutic frontiers. Crit Rev Oncol Hematol. 2025;215:104916
144. Wan YK, Hendra C, Pratanwanich PN, Göke J. Beyond sequencing: machine learning algorithms extract biology hidden in Nanopore signal data. Trends Genet. 2022;38:246-57
145. Xiao S, Duan S, Hong Y, Zhang J, Ma S, Caligiuri MA. et al. Loss of YTHDF2 enhances Th9 programming and CAR-Th9 cell antitumor efficacy. Nat Immunol. 2025;26:1501-15
146. Yin H, Zhang X, Yang P, Zhang X, Peng Y, Li D. et al. RNA m6A methylation orchestrates cancer growth and metastasis via macrophage reprogramming. Nat Commun. 2021;12:1394
Author contact
![]() Corresponding author: Li Cui, Email: licuiedu.cn.
Corresponding author: Li Cui, Email: licuiedu.cn.