Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
In vitro GBM models
Microfluidic GBM model for drug...
In vivo model for GBM treatment
Future perspectives and...
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2026; 16(10):5463-5500. doi:10.7150/thno.126324 This issue Cite
Review
Translational Models for Glioblastoma: Revolutionizing Drug Development and Personalized Medicine through Clinical Insights
Gaeun Lee1†, Yu Jin Kim2,3†, Sharon Jeeho Ham4, Hyeongjin Ahn1, Dayeong Choi1, Juhyeong Ha1, Jungseub Lee5, Hyung-Jin Lee6, Jaejoon Lim2,3 ![]() , Jungho Ahn1,4
, Jungho Ahn1,4 ![]()
1. Department of MetaBioHealth, Sungkyunkwan University, Suwon, Gyeonggi-do 16419, Republic of Korea.
2. Department of Neurosurgery, Bundang CHA Medical Center, CHA University, Yatap-dong 59, Seongnam 13496, Republic of Korea.
3. Department of Biomedical Science, College of Life Science, CHA University, Seongnam 13488, Republic of Korea.
4. Department of Biophysics, Institute of Quantum Biophysics, Sungkyunkwan University, Suwon-si, Gyeonggi-do, Republic of Korea.
5. Department of Mechanical and Aerospace Engineering, Seoul National University, Gwanak-gu, Seoul-si, Republic of Korea.
6. Department of Anatomy, College of Medicine, Chungbuk National University, Republic of Korea.
† These authors contributed equally to this work.
Received 2025-10-6; Accepted 2026-1-24; Published 2026-3-17
Abstract

Glioblastoma (GBM) remains one of the most aggressive and treatment resistant brain tumors and continues to present major challenges for effective therapeutic development. The failure of numerous late-stage clinical trials highlights the limited predictive value of conventional preclinical models. Although established cell lines, two-dimensional cultures, and animal models have been extensively employed, existing platforms fail to adequately recapitulate the complex tumor microenvironment, blood brain barrier function, and interpatient heterogeneity that drive therapeutic resistance in GBM.
To address this translational limitation, advanced experimental systems have been developed to more accurately reproduce key features of the human GBM microenvironment through the integration of microengineering approaches, biomaterials, and patient derived cells. This review focuses on recent advances in microfluidic GBM chip models and three-dimensional bioprinted GBM platforms, while also summarizing a broad range of in vitro and in vivo model systems extending from conventional 2D cultures and organoids to animal-based platforms.
Microfluidic and 3D bioprinting technologies enable controlled reconstruction of critical biophysical and biochemical features of the GBM microenvironment. These systems allow regulation of oxygen availability, drug exposure, and cellular interactions within engineered tumor constructs. As a result, patient specific GBM models with heterogeneous architectures can be generated with improved biological relevance and closer resemblance to in vivo tumor behavior. In parallel, in vivo systems remain indispensable for capturing systemic pharmacokinetics, host immune responses, and organ level toxicity. Among these, mouse models encompassing syngeneic, xenograft, and genetically engineered mouse models (GEMMs) continue to serve as the foundation of preclinical GBM research, while larger animal models have been explored to better approximate the physiological and immunological features of human disease.
Overall, this review discusses recent advances in GBM model development across microengineered in vitro platforms and in vivo animal systems and examines their potential to enhance translational accuracy, accelerate drug discovery, and support personalized therapeutic strategies. By emphasizing the technological scope and clinical relevance of these platforms, this work aims to provide practical guidance for the rational selection and integration of GBM model systems in the discovery and optimization of next generation therapeutics.
Introduction
Epidemiology/etiology
Glioblastoma (GBM) is the most aggressive form of glioma, a primary malignant brain tumor of the central nervous system (CNS), most commonly arising in the frontal and temporal lobes. It is characterized by rapid proliferation, extensive angiogenesis, and marked resistance to conventional therapies [1, 2].
GBM is the most prevalent and aggressive primary malignant brain tumor, accounting for approximately 15% of all brain tumors and nearly 50% of malignant brain tumors [3]. Globally, the incidence of GBM ranges from 3 to 5 cases per 100,000 individuals annually, with a slight male predominance (male-to-female ratio ~1.6:1) [4, 5]. The median age at diagnosis is 64 years, and the incidence peaks between 65 and 75 years of age, indicating that age-related genetic and epigenetic alterations may contribute to tumor development. Epidemiological data also suggest variations by ethnicity and geography, with higher rates among Caucasians and populations in urban or industrialized areas, possibly reflecting environmental exposures [6].
The etiology of GBM is multifactorial, involving genetic predisposition, environmental influences, and potential lifestyle factors. Genetic mutations are central to GBM pathogenesis, with somatic alterations in key genes such as TP53, EGFR, and PTEN frequently observed. These mutations disrupt critical pathways regulating cell growth, apoptosis, and DNA repair, driving the aggressive behavior of GBM. Mutations in isocitrate dehydrogenase (IDH) are more common in secondary GBM than in primary GBM and are associated with a better prognosis. Additionally, inherited genetic syndromes such as Li-Fraumeni syndrome, neurofibromatosis type 1, and Turcot syndrome further highlight the role of genetic predisposition in GBM etiology [7].
Environmental factors, particularly exposure to ionizing radiation, have been strongly linked to an elevated risk of GBM. Patients who have undergone therapeutic cranial irradiation for other cancers or those exposed to high levels of environmental radiation have an increased likelihood of developing gliomas later in life [8]. Occupational exposure to industrial chemicals, pesticides, and petrochemicals has also been suggested as a potential risk factor, though definitive evidence is still lacking. The role of lifestyle factors, such as smoking, alcohol consumption, and diet, in GBM risk remains unclear, but some studies suggest that poor dietary habits or chronic inflammation related to obesity may contribute to tumor development [9].
Recent studies have also explored the potential role of viral infections in GBM etiology. Human cytomegalovirus (HCMV) has been detected in a significant proportion of GBM tumors, raising questions about its involvement in tumor progression. HCMV may promote genetic instability and immune evasion, although the exact mechanisms and clinical significance remain under investigation [10].
Although extensive research has been conducted on its etiology and risk factors, GBM remains a highly lethal disease with limited prognosis. The median survival time following diagnosis is approximately 12-15 months, with a five-year survival rate of less than 5%. This poor clinical outlook emphasizes the critical need for a deeper understanding epidemiology of GBM and pathogenesis to guide the development of more effective treatment strategies [11].
Pathogenesis
GBM pathogenesis is characterized by a complex interplay of genetic, molecular, and cellular mechanisms that collectively drive its aggressive nature and resistance to therapy [12]. The tumor arises through the accumulation of genetic alterations, including mutations in key regulatory genes, TP53, EGFR, and PTEN, which disrupt essential cellular processes. TP53 mutations impair cell cycle control and DNA damage response, allowing genomic instability and tumor progression. EGFR amplification and mutations, such as the oncogenic EGFR variant III (EGFRvIII) variant, activate downstream signaling pathways, including PI3K/AKT and RAS/RAF/MEK, which promote cell proliferation, invasion, and resistance to apoptosis [13]. Loss of PTEN further enhances PI3K/AKT signaling, contributing to tumor cell survival and therapeutic resistance [2].
In addition to these genetic alterations, the molecular pathways involved in GBM pathogenesis include dysregulation of growth factor signaling and cell cycle checkpoints. Aberrant activation of the PI3K/AKT/mTOR pathway supports tumor growth and metabolic adaptation, while the RAS/RAF/MEK/ERK pathway enhances cell proliferation and migration [14]. Mutations in IDH1 and IDH2, often observed in secondary GBM, result in the production of the oncometabolite 2-hydroxyglutarate, which alters epigenetic regulation and facilitates tumorigenesis [15]. Furthermore, alterations in DNA repair mechanisms, p53 pathway defects and O6-methylguanine-DNA methyltransferase (MGMT) overexpression, contribute to therapeutic resistance by reducing the efficacy of alkylating agents [12].
GBM microenvironment
The GBM microenvironment is a highly complex and dynamic network comprising tumor cells, non-tumor stromal and immune cells, extracellular matrix (ECM), and soluble factors such as cytokines and metabolites [16]. The cellular components include tumor-associated macrophages (TAMs), microglia, astrocytes and endothelial cells, while the non-cellular components consist of ECM elements such as hyaluronic acid (HA), link proteins, tenascins, and proteoglycans like aggrecan, neurocan, versican and phosphacan [17]. This environment acts as a key regulator of GBM progression, supporting tumor invasion, proliferation, and therapeutic resistance [18].
Furthermore, a hypoxic environment is well known as a key factor in GBM treatment resistance by contributing to angiogenesis, invasion, and survival, and is directly correlated with poor patient prognosis. Changes in partial oxygen tension (pO2) are sensed by prolyl-4-hydroxylase 2 (PHD2), which regulates HIFs expression. Under hypoxic conditions, low partial oxygen tension suppresses PHD2, leading to the accumulation of HIF-1α. Dimerization of HIF-1α and HIF-1β induces upregulation of pro-angiogenic genes such as VEGF and fibroblast growth factor, thereby increasing angiogenesis, invasiveness, and tumor metastasis [19]. Simultaneously, GBM cells remodel the tumor microenvironment (TME) by secreting exosomes, cytokines, and other soluble factors that influence both surrounding and distant cells within TME. These secreted molecules form physical interactions with stromal and immune cells, reprogramming the behavior of surrounding cells and contributing to therapeutic resistance (Figure 1A). Through these multifaceted interactions, GBM cells exploit the TME to sustain their invasive nature and evade therapeutic interventions. One major obstacle in treating GBM is the blood-brain barrier (BBB), which acts as a highly selective and protective shield. The BBB allows only specific substances, primarily small and lipophilic molecules, to pass through. Effectively delivering anticancer agents to the brain to treat GBM is a significant challenge [16].
Schematic overview of GBM pathophysiology, TME, and therapeutic challenges.
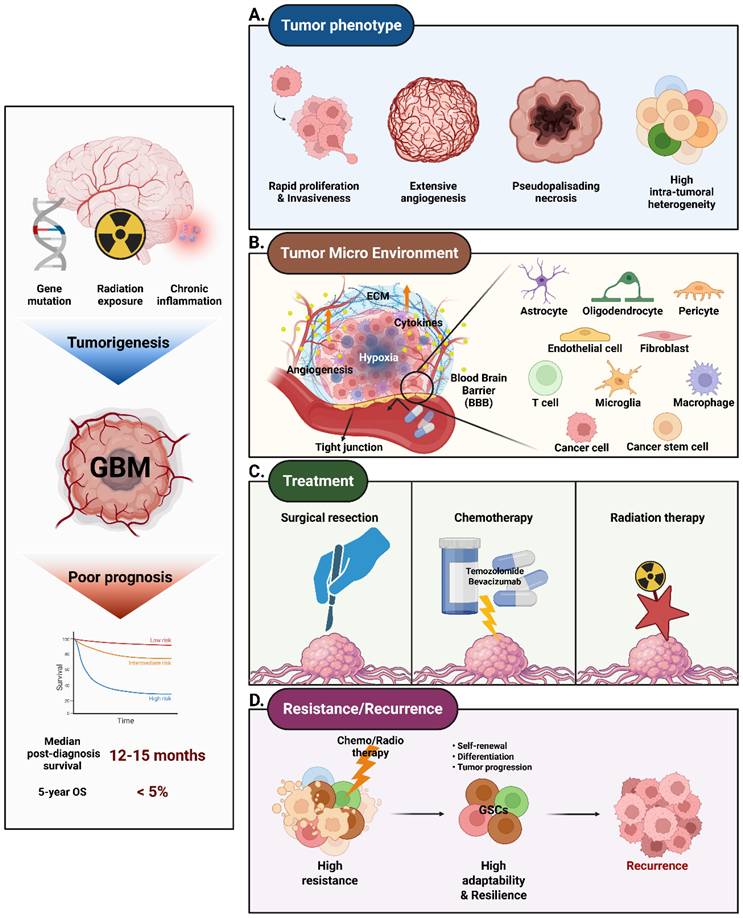
The immune cell population in the GBM microenvironment, including TAMs, microglia, and tumor-infiltrating lymphocytes (TILs), plays a crucial role in tumor progression [20]. TAMs and microglia, influenced by GBM-derived cytokines and growth factors, often acquire an immunosuppressive phenotype. These cells release anti-inflammatory molecules such as IL-10, transforming growth factor beta (TGF-β), and prostaglandins, promoting an environment conducive to tumor growth [21]. Furthermore, TAMs secrete pro-angiogenic factors like vascular endothelial growth factor (VEGF) and IL-8, enhancing the formation of abnormal vasculature that supports tumor survival and dissemination [22]. TAMs also contribute to immune evasion by expressing immune checkpoint molecules like PD-L1, which inhibit T cell activation [23].
Astrocytes and endothelial cells significantly contribute to the structural and functional remodeling of GBM TME (Figure 1B). Astrocytes establish gap junctions with tumor cells, facilitating the exchange of metabolic and ionic signals that support tumor proliferation and migration [24, 25]. They also secrete cytokines, including IL-6 and CXCL10, which promote tumor invasiveness. Additionally, GBM is characterized by its high vascularity. The tumor develops abnormally structured blood vessels that supply the necessary oxygen and nutrients but are also associated with increased permeability, leading to issues like brain edema. The endothelial cells are central to angiogenesis, forming abnormal and leaky blood vessels within the TME. These vessels provide nutrients and oxygen to the tumor but also create high interstitial pressure, hindering effective drug delivery. The resulting hypoxic environment further promotes tumor aggressiveness.
The ECM within GBM is uniquely adapted to the brain environment and plays an integral role in tumor progression and resistance. Key ECM components, such as HA, tenascins, and proteoglycans, regulate cell adhesion, migration, and invasion. Increased ECM stiffness mechanically promotes GBM proliferation and migration, which is directly correlated with decreased responsiveness to therapy in 3D models. ECM stiffness modulates YAP/TAZ signaling, increasing cytoskeletal tension and upregulating gene expression associated with chemotherapy resistance [26]. Specifically, the HA/collagen ratio influences integrin-mediated signaling and drug diffusion. Low-molecular-weight HA promotes tumor invasion, while high-molecular-weight HA can inhibit migration, but may sequester growth factors and limit drug permeability. This balance is often disrupted in GBM [27]. Additionally, tenascins and proteoglycans remodel the ECM, creating a structural and biochemical milieu that supports tumor progression and acts as a reservoir for growth factors that drive invasion and angiogenesis [28, 29].
GBM cells interact with the TME through direct and indirect mechanisms to enhance their adaptability and invasiveness. Direct interactions are mediated by gap junctions composed of connexin-43, enabling the exchange of ions, metabolites, and signaling molecules between tumor cells [25]. These interactions establish multicellular networks that facilitate coordinated invasion into healthy brain tissue. Indirect interactions involve the release of cytokines, such as VEGF and IL-6 and exosomes that transport bioactive molecules across the BBB.
These mechanisms act in a complex manner in the GBM microenvironment to promote angiogenesis and increase immune evasion, which contributes particularly to treatment resistance. [18]. Therefore, the need to develop therapeutic strategies targeting the GBM TME is emphasized to improve patient outcomes.
GBM recurrence
Recurrent GBM presents a major challenge in cancer treatment due to its aggressive behavior and high resistance to standard therapies [4]. Despite initial interventions such as surgery, radiation, and chemotherapy with temozolomide (TMZ), recurrence is almost inevitable (Figure 1D) [30]. This high recurrence rate is driven by several factors, including tumor heterogeneity, the presence of glioma stem-like cells (GSCs), and the TME [31]. GSCs are critical in driving GBM recurrence. These cells possess a heightened resistance to radiation and chemotherapy and contribute to tumor regrowth through their self-renewal capabilities [32]. GSCs are recognized as key regulators of tumor initiation and progression. They are closely associated with therapeutic resistance and recurrence by promoting cell survival, angiogenesis, invasion, and tumor dissemination. Studies have highlighted that GSCs promote not only tumor cell survival but also angiogenesis, invasion, and tumor spread, all of which are key factors in recurrence [31]. GSCs are characterized by their ability to self-renew and differentiate into various cell types found within the tumor, making them highly adaptable and resilient against treatment [33]. GSCs are also known to reside in specialized niches within the TME, such as perivascular regions, where they receive support from the surrounding blood vessels [34]. This close association with vasculature not only provides GSCs with necessary nutrients and oxygen but also contributes to their ability to promote angiogenesis through the secretion of pro-angiogenic factors like VEGF [35]. This process helps sustain tumor growth and creates a microenvironment that is conducive to recurrence [36]. Moreover, GSCs exhibit a high level of plasticity, allowing them to transition between different cellular states in response to therapeutic stress. This plasticity is a key factor in their ability to evade standard treatments, as GSCs can adapt to changing conditions and reinitiate tumor growth even after aggressive therapy [32]. GSCs are also highly efficient at repairing DNA damage, which further contributes to their resistance to radiation and chemotherapy [37]. The presence of GSCs within the tumor makes it difficult to achieve lasting remission, as these cells can repopulate the tumor and drive recurrence.
The majority of GBM recurrences occur within 2-3 cm of the original tumor site, largely due to the highly infiltrative nature of GBM cells [38, 39]. More than 90% of recurrences happen at or near the original tumor location, underscoring the challenge of achieving complete tumor removal even with aggressive surgery [40]. The TME, which consists of immune cells, blood vessels, and ECM components, plays a crucial role in supporting tumor cell survival and facilitating regrowth [41]. Radiation therapy, though initially effective, can alter the TME in a way that promotes a more aggressive tumor phenotype upon recurrence [42]. Changes in the ECM, increased inflammation, and hypoxia contribute to this enhanced aggressiveness by creating a supportive niche for GSCs, enhancing invasive potential, and reducing therapeutic efficacy [43].
Radiation-induced changes in the TME can lead to increased tumor cell invasion, altered cytokine signaling, and chronic inflammation, all of which help create an environment that supports tumor regrowth. These alterations not only promote a more favorable environment for the tumor but also reduce the efficacy of subsequent therapies [44]. Additionally, chemotherapeutic stress can induce GBM cells to transdifferentiate into endothelial cells, leading to vascular mimicry that further supports tumor growth and therapy resistance. This plasticity allows GBM cells to adapt under therapeutic pressure and form new tumor-derived blood vessels, ultimately contributing to recurrence [45].
Clinical presentation
The clinical presentation of GBM is generally related to the functional tumor location in the brain. Patients often present with focal neurological deficits such as motor weakness, sensory disturbances, and language dysfunction, depending on the specific brain regions involved by the tumor [46]. For instance, tumors located in the motor cortex commonly lead to weakness or paralysis, while those in the temporal lobe may cause speech difficulties or memory impairments [47]. Seizures are a presenting symptom in approximately 25% of patients with newly diagnosed GBM. These episodes tend to be focal but can generalize, and while anticonvulsants are commonly used, they are generally reserved for patients who have experienced these occurrences [48]. GBM may also present with non-specific symptoms such as headaches, which are often caused by increased intracranial pressure due to the mass effect of the tumor or obstructive hydrocephalus [49]. Cognitive disturbances, personality changes, and mood disorders are common when tumors affect the frontal lobes or other areas involved in executive function [50]. Additionally, symptoms such as confusion, memory loss, and fatigue may present in a significant number of patients, complicating early diagnosis since these symptoms are non-specific and can be attributed to various other neurological or systemic conditions [49]. The typical imaging characteristics of GBM include a heterogeneously enhancing lesion with a necrotic core, surrounded by a region of peritumoral edema on magnetic resonance imaging (MRI). These tumors are highly infiltrative, extending into the white matter, often involving structures including the corpus callosum [47]. This invasive nature contributes significantly to the heterogeneity of clinical presentations, as well as the challenge of achieving complete surgical resection, which in turn impacts prognosis. In some cases, GBM is presented as a thalamic tumor, which carries specific clinical features and challenges due to the deep location within the brain. Patients with thalamic GBM often experience symptoms like hydrocephalus, motor dysfunction, and cognitive changes. These tumors are considered largely unresectable due to their location, and the management is often limited to biopsy, followed by chemoradiation [51]. The clinical presentation of GBM is crucial in determining the management approach and expected outcomes.
Treatment of GBM
Understanding the epidemiology of GBM is crucial for developing strategies for early detection, prevention, and treatment (Figure 1C) [52]. This understanding also illuminates why GBM treatment is particularly challenging. Despite being the current standard of care, TMZ and radiotherapy exhibit limited efficacy in the treatment of GBM. GBM cells can develop resistance to TMZ, particularly in cases where high MGMT enzyme expression repairs the DNA damage caused by the drug. Although TMZ can cross the BBB, it may not reach sufficient concentrations in the tumor, thereby limiting its effectiveness [53]. Additionally, TMZ can cause side effects such as blood cell depletion and immunosuppression, which impact overall patient health. Although radiotherapy remains a mainstay in GBM treatment, it can inadvertently damage adjacent healthy brain tissue, resulting in cognitive impairments, neurological deficits, and long-term complications. These limitations underscore the urgent need for more effective treatment strategies.
Surgical resection
Surgical resection represents the foundational step in GBM treatment, aiming to achieve gross total resection (GTR) while preserving critical neurological function. However, complete removal is frequently limited by diffuse tumor infiltration into eloquent brain regions. The use of advanced technologies such as frameless stereotaxy, intraoperative MRI, and 5-aminolevulinic acid (5-ALA) fluorescence has significantly improved the accuracy of tumor localization and resection margins [54].
More recently, near-infrared II (NIR-II, 1000-1700 nm) fluorescence imaging has emerged as a promising adjunct, offering deeper tissue penetration, reduced scattering, and higher signal-to-background ratios compared to visible or NIR-I fluorescence, thereby enabling improved intraoperative visualization of tumor margins [55, 56]. In particular, NIR-II-guided imaging has shown potential for delineating infiltrative tumor boundaries beyond the contrast-enhancing regions, addressing a critical limitation of conventional fluorescence-guided surgery in GBM [57]. Furthermore, the integration of NIR-II imaging with targeted probes or nanomaterials may enable real-time, molecularly informed surgical guidance, improving resection precision in highly heterogeneous GBM tissues [58].
Preoperative mapping through functional MRI and diffusion tensor imaging (DTI) provides detailed visualization of brain networks, assisting in surgical planning and minimizing postoperative deficits [59], which provide detailed maps of both tumor boundaries and critical brain pathways. These modalities, combined with intraoperative imaging, allow for a tailored surgical approach that maximizes tumor removal while minimizing neurological deficits [60]. Despite these innovations, microscopic residual disease remains inevitable, making surgical intervention insufficient on its own.
Radiation therapy
Radiation therapy (RT) is a key component of the adjuvant treatment regimen, typically initiated after surgery. The standard approach involves external beam radiation therapy (EBRT) administered in 2 Gy fractions up to a total dose of 60 Gy over six weeks, in conjunction with concurrent TMZ [61]. This protocol has been shown to significantly improve overall survival compared to RT alone and is now widely adopted for newly diagnosed GBM. To enhance therapeutic precision and minimize collateral damage to healthy brain tissue, several advanced RT modalities have been developed. Intensity-modulated radiation therapy (IMRT) enables tailored dose distribution to match tumor contours, minimizing exposure to adjacent normal structures [62]. Stereotactic radiosurgery (SRS) delivers highly focused, high-dose radiation in a single or few sessions and is often used for recurrent or small lesions. Additionally, proton beam and carbon ion therapies offer improved dose conformity and reduced radiation exposure to non-cancerous brain regions, making them attractive alternatives to conventional photon-based RT [63]. Despite these technological advances, radioresistance and tumor recurrence remain significant challenges. Contributing factors include intratumoral heterogeneity, hypoxia-induced cellular adaptations, and activation of DNA repair pathways, all of which diminish RT efficacy [64].
Chemotherapy
Chemotherapy plays a central role in GBM treatment by complementing surgical resection and radiotherapy to control tumor progression and extend patient survival. Due to the highly infiltrative nature of GBM, complete surgical excision is rarely possible, making systemic therapy indispensable [65]. Among current options, TMZ, an alkylating cytotoxic agent and bevacizumab, a VEGF-targeting anti-angiogenic agent, are most widely used. Despite clinical benefits, treatment resistance remains a major limitation.
TMZ is administered as part of the Stupp regimen, which combines surgery, radiotherapy, and concurrent/adjuvant TMZ, significantly improving survival compared to radiotherapy alone. TMZ acts by methylating DNA, particularly at the O6 position of guanine, triggering mismatch repair cycles and apoptosis [66]. TMZ is typically administered as part of the Stupp protocol, which involves maximal safe surgical resection, followed by radiotherapy combined with daily TMZ for six weeks, and subsequent adjuvant TMZ cycles. This regimen has been shown to significantly improve median survival compared to radiotherapy alone, increasing the likelihood of patients surviving beyond two years [67]. However, the long-term efficacy of TMZ is often limited by acquired resistance mechanisms. One of the primary causes is the expression of MGMT, which repairs TMZ-induced DNA lesions and prevents apoptosis. High MGMT activity is associated with poor treatment response, whereas MGMT promoter methylation, which silences gene expression, is correlated with increased TMZ sensitivity and improved survival outcomes [68]. In addition to MGMT-related resistance, defects in the mismatch repair (MMR) pathway and enhanced base excision repair (BER) allow tumor cells to tolerate or correct TMZ-induced DNA damage, further diminishing its therapeutic efficacy [69]. GSCs characterized by their self-renewal capacity and resistance to conventional therapies, contribute significantly to treatment failure and tumor recurrence. These cells can survive cytotoxic stress and repopulate the tumor following treatment. To address TMZ resistance, several adjunct strategies are under investigation. These include MGMT inhibitors, such as O6-benzylguanine and lomeguatrib, although their clinical application is limited by systemic toxicity [70]. In addition, poly ADP-ribose polymerase (PARP) inhibitors have been investigated to block DNA repair pathways, thereby enhancing TMZ-induced cytotoxicity [71]. Targeting the TME, particularly angiogenesis, offers an additional therapeutic avenue. Bevacizumab, a monoclonal antibody against vascular endothelial growth factor (VEGF), reduces neovascularization and alleviates peritumoral edema, thereby improving progression-free survival and reducing corticosteroid dependence [72]. However, its impact on overall survival remains limited. To overcome VEGF inhibition, GBM cells activate alternative pro-angiogenic pathways, notably those mediated by PDGF and FGF. They also enhance their invasive capacity and undergo metabolic reprogramming to survive under hypoxic conditions [73]. These limitations have driven interest in multi-targeted kinase inhibitors that simultaneously block multiple angiogenic signals. Combination strategies integrating anti-angiogenic therapy with immune checkpoint blockade or metabolic pathway inhibitors are also under evaluation to overcome resistance and improve therapeutic outcomes [74].
In vitro GBM models
Conventional 2D models
In GBM research, various 2D cell models are extensively employed, each offering distinct advantages for investigating tumor biology, therapeutic responses, and genetic alterations [75]. These models include established cell lines, primary patient-derived cells, and advanced genetically engineered models, with co-culture systems emerging to better replicate the TME. Among the most widely used are immortalized patient-derived GBM cell lines, which offer cost-efficiency and ease of use in a range of experimental settings [76]. Prominent examples include U87MG, T98G, A172 and LN229 [75].
The U87MG cell line, a central model of GBM research for decades, is favored for its ease of culture and rapid proliferation [77]. It is commonly applied in drug screening and signal transduction studies. However, long-term culture has led to significant genetic drift from the original tumor, limiting its ability to fully represent GBM biology [78]. T98G, known for its resistance to both chemotherapy and radiotherapy, serves as a robust model for studying treatment resistance mechanisms [79]. It exhibits increased DNA repair capacity, including elevated DNA-PKcs activity, which contributes to its radioresistant phenotype [80]. A172 harbors mutations in the tumor suppressor gene TP53 and is primarily used to study cell cycle regulation, DNA repair, and tumor progression under p53-deficient conditions [81]. Similarly, LN229, another TP53-mutant line, is often employed in apoptosis-related research and in evaluating pro-apoptotic therapeutic agents [82]. Beyond established lines, primary patient-derived GBM cells offer greater clinical relevance. Isolated directly from tumor tissue, these cells retain patient-specific genetic heterogeneity, invasive capacity, and cancer stem-like traits when cultured under 2D conditions [83].
In particular, GSCs provide a more accurate model of GBM, reflecting key features such as self-renewal, therapy resistance, and tumorigenic potential. These cells are essential for studying disease progression, recurrence, and the development of therapies targeting tumor-initiating populations. GSC-based models—including neurospheres, organoids, and patient-derived xenografts—recapitulate the TME and are indispensable tools in preclinical research. However, their use presents challenges such as slow proliferation and potential loss of tumor characteristics during extended culture [84].
Microfluidic - 3D bioprinting fabrication
The development of GBM-on-a-chip platforms starts with the careful definition of components necessary to replicate the GBM TME. Key design considerations include the selection of pathophysiologically relevant cell types, the recreation of spatial tissue architecture, and the incorporation of physicochemical factors such as biochemical gradients, ECM stiffness, and shear stress. Beyond their structural role, dynamic flow and shear stress in GBM-on-a-chip platforms function as critical biological regulators of tumor behavior and drug transport. Interstitial flow within microfluidic GBM models has been shown to actively promote glioma cell migration and invasion by generating convective chemokine gradients that induce CXCR4/CXCL12-dependent autologous chemotaxis, resulting in enhanced directional motility within three-dimensional extracellular matrices [104]. In parallel, shear stress applied to vascularized or BBB-like GBM-on-a-chip systems modulates endothelial cell alignment, junctional integrity, and barrier permeability, thereby directly influencing therapeutic penetration across tumor-associated vasculature and BBB interfaces [105]. By enabling precise and reproducible control of these biomechanical and transport cues, microfluidic GBM-on-a-chip platforms provide a mechanistically grounded framework to interrogate glioma invasion dynamics and treatment delivery that cannot be captured in static in vitro cultures.
These criteria guide the engineering of microfluidic platforms through conventional techniques such as photolithography, replica molding, and soft lithography, with more recent advances incorporating high-resolution 3D bioprinting [106]. In soft lithography-based approaches, microchannel templates are first patterned on silicon wafers using photolithography. Polydimethylsiloxane (PDMS) is then poured onto these molds and cured to create flexible, transparent microstructures. Once bonded to glass or other polymeric substrates, these structures form closed microfluidic channels capable of simulating tissue-tissue interfaces, fluidic shear, and spatial compartmentalization characteristic of the brain tumor environment.
More recently, 3D bioprinting has emerged as a transformative tool in GBM chip development by enabling the layer-by-layer deposition of biofunctional materials and living cells with high spatial precision. This technique allows for the incorporation of diverse cell populations including GBM cells, endothelial cells, astrocytes, fibroblasts, and immune cells into ECM-mimetic hydrogels such as gelatin methacrylate (GelMA), alginate, and fibrin. By replicating the structural and biochemical complexity of native GBM tissues, bioprinting supports physiologically relevant mechanical environments essential for tumor progression and therapeutic evaluation [107, 108]. In another approach, a hybrid platform was fabricated by combining 3D bioprinted GBM and vascular compartments with a PDMS-based microfluidic channel network. Bioinks tailored to each cell type GelMA-alginate for tumor cells and GelMA-fibrin for endothelial cells enabled spatially organized tissue construction. This system not only permitted the application of dynamic shear stress via perfusion but also supported long-term monitoring of tumor spheroid behavior. When subjected to simulated microgravity conditions, the platform revealed that biomechanical unloading markedly suppressed spheroid formation and altered cellular morphology and signaling, highlighting the importance of physical forces in GBM biology and treatment resistance (Figure 3A) [102]. In comparison to other advanced in vitro GBM models, such as patient-derived organoids, microfluidic and 3D bioprinting-based platforms offer distinct advantages in controllability and standardization. While organoid models preserve patient-specific genetic and phenotypic features and recapitulate key histopathological traits, they provide limited control over spatiotemporal microenvironmental cues, as gradients of oxygen, nutrients, and therapeutics arise primarily through diffusion. In contrast, microfluidic systems enable reproducible regulation of biochemical and biomechanical parameters, including perfusion-driven transport, shear stress, and vascular barrier function, while 3D bioprinting allows deterministic control over tissue architecture and ECM composition. Together, these approaches complement organoid models by integrating patient-derived heterogeneity with programmable microenvironmental control [109].
The convergence of microfluidic engineering and 3D bioprinting has thus enabled the creation of advanced GBM-on-a-chip systems capable of recapitulating patient-specific tumor heterogeneity, vasculature, and drug response. These hybrid models provide a physiologically relevant framework for dissecting tumor-stroma interactions and conducting high-throughput therapeutic screening, offering a crucial bridge between oversimplified 2D cultures and complex in vivo systems (Figure 2).
Comparison of in vitro models for biomedical research.
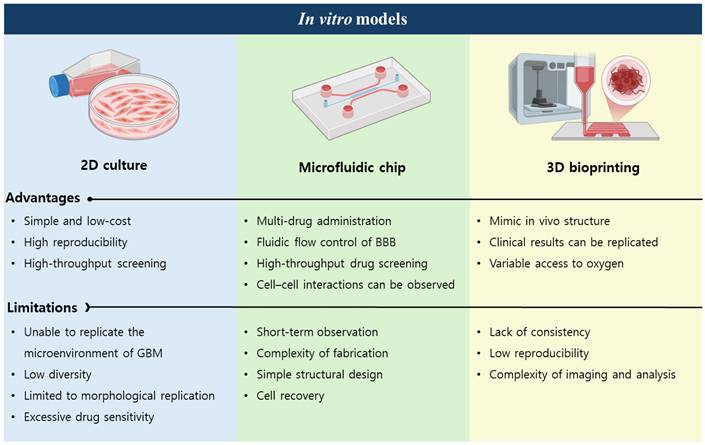
Introduction of GBM-on-a-chip
To develop biomimetic three-dimensional tumor models that accurately replicate the complexity of tumor pathophysiology, it is essential to incorporate the critical components of the TME. Among these, oxygen tension serves as a key regulator of cellular behavior, profoundly influencing processes such as proliferation, migration, and therapeutic resistance. This aspect is particularly significant in the case of GBM, the most common and aggressive form of primary malignant brain tumor, which presents a formidable therapeutic challenge due to its profoundly immunosuppressive microenvironment.
Conventional in vitro tumor models are typically cultured under normoxic conditions, approximating atmospheric oxygen levels, which do not faithfully reflect the dynamic and heterogeneous hypoxic conditions present within tumors in vivo. As a result, these models may produce findings that are not fully representative of the true biological behavior of tumors, potentially leading to inaccurate interpretations [41]. This limitation becomes especially critical when modeling metastasis. To accurately replicate the process of intravasation and dissemination observed in vivo, tumor cells migrating from the primary site must respond to a variety of microenvironmental cues, including fluctuating oxygen gradients.
In the context of GBM, the intricate interplay among hypoxia, immune evasion mechanisms, and the ECM creates a highly resilient TME that substantially diminishes the efficacy of conventional therapeutic modalities, including surgical resection, radiotherapy, and TMZ chemotherapy. Although emerging strategies such as immunotherapy and the integration of bioengineering technologies offer promising therapeutic avenues, their overall impact remains substantially constrained by the ability of the tumor to suppress and modulate immune responses. Microfluidic-based platforms present a promising solution to these challenges. These systems are capable of generating precisely controlled oxygen gradients with high spatial and temporal resolution, thereby enabling the creation of more physiologically relevant tumor models [113]. A microfluidic GBM model can recreate physiologically relevant oxygen gradients by employing gas-impermeable materials. This design enables the formation of a hypoxic core, a nutrient-deprived intermediate zone, and an oxygen-rich periphery through controlled oxygen and nutrient delivery via side channels (Figure 3B) [113]. By better simulating the oxygen dynamics of the native TME, microfluidic models provide valuable insights into how fluctuating oxygen levels and other microenvironmental factors influence critical tumor behaviors, including progression, invasion, and therapeutic resistance. This approach is especially effective for investigating complex malignancies like GBM, in which the TME plays a critical role in driving disease progression and shaping treatment response. TME is composed of diverse cellular constituents, including GSCs, endothelial cells, microglia, astrocytes, and neurons, as well as non-cellular components such as the ECM, dynamic hypoxia gradients, and various soluble signaling molecules (Figure 3C) [114]. As an example, a patient-specific GBM-on-a-chip platform replicates the complex and immunosuppressive microenvironment of GBM. This platform enables real-time analysis of subtype-dependent immunotherapeutic responses, including differences in T cell infiltration and macrophage polarization. Overall, the platform serves as a physiologically relevant tool to study dynamic tumor-immune interactions and supports the personalized screening of immunotherapeutic strategies [91]. Taken together, these elements collectively drive the aggressive phenotype of GBM and contribute to its notorious therapeutic resistance.
Microfluidic models of GBM. (A) Hybrid GBM-on-a-chip platform integrating 3D bioprinted tumor-vascular compartments with microfluidic perfusion. (i) Schematic of the microfluidic device featuring separate vascular and tissue compartments allowing independent control and perfusion. (ii) 3D bioprinting of GBM and endothelial cell compartments using GelMA-alginate and GelMA-fibrin bioinks, respectively. (iii) Cross-sectional illustration showing GBM spheroid formation within a 3D matrix under perfused conditions and shear stress. Reproduced from under [102] an open-access license, © 2021 Adv. Ther. (B) Graphical abstract illustrating the design of a GBM-on-a-chip platform that establishes hypoxic gradients to mimic the oxygen-deficient TME of GBM in the human brain. Live and dead cells are fluorescently labeled in green and red. Reproduced from under [110] an open-access license, © 2024 Cell Death Dis. (C) A schematic illustration of a GBM-on-a-chip platform representing the interaction between patient-derived GBM cells and primary human immune cells under immunosuppressive tumor microenvironmental conditions. Reproduced from under [91] an open-access license, © 2020 eLife. (D) Ex vivo culture of patient-derived GBM tissue within a microfluidic device. Schematic of a syringe pump-driven microfluidic chip enabling continuous tissue perfusion. Reproduced from [90] an open-access license, © 2019 Transl. Oncol. (E) A GBM-on-a-chip system incorporating a stiffness gradient on a fibronectin-conjugated polyacrylamide hydrogel substrate. (i) A schematic diagram illustrating the design of the GBM-on-a-chip platform with a stiffness gradient. (ii) Fluorescent imaging of U251 MG cells and HCT-116 cells, with live and dead cells labeled in green and red, respectively. Reproduced from under [111] an open-access license, © 2016 Sci. Rep. (F) Microfluidic platforms to demonstrate angiogenesis. Schematic illustration of a metastasis-on-a-chip model designed to recapitulate tumor-induced angiogenesis. Reproduced with permission from Ref. [112] © 2014 Biomicrofluidics.
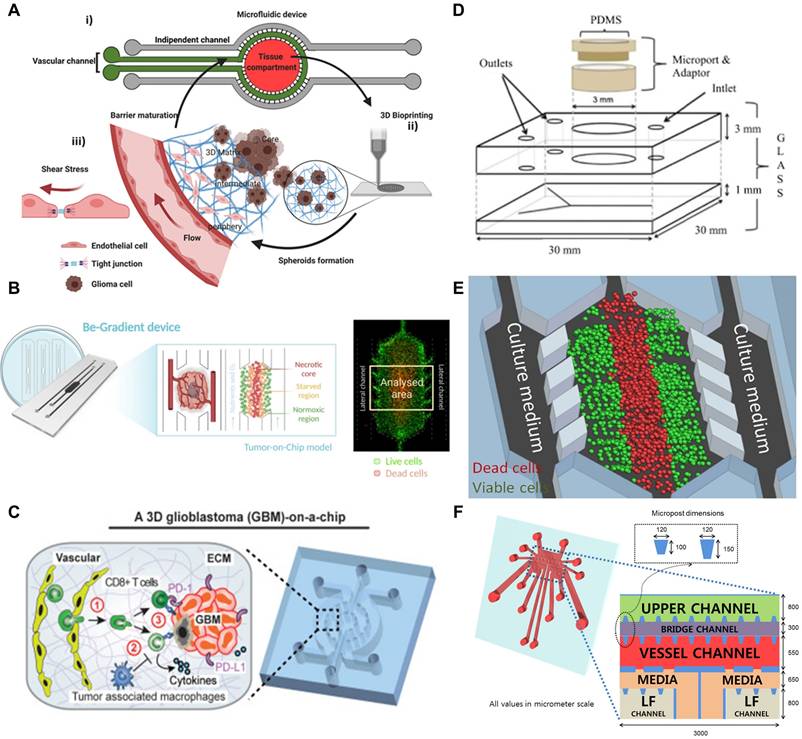
Therefore, to accurately model GBM in vitro, it is imperative to closely recapitulate both the cellular and non-cellular components of the TME. Microfluidic systems, with their capacity to precisely control environmental variables, represent an ideal platform for constructing such advanced and physiologically relevant GBM models.
Microfluidic GBM models
GBM remains a highly aggressive malignancy with poor prognosis, regardless of the treatment approach. To better study GBM biology under physiologically relevant conditions, microfluidic platforms capable of maintaining human GBM tissues ex vivo have been developed (Figure 3D) [90]. Although still in the early stages, these technologies offer promising opportunities for improving preclinical modeling. Larger patient-derived studies are expected to further validate their utility in predicting treatment responses and in developing more effective therapeutic strategies [90]. Among recent innovations, a microfluidic chip integrating a controllable stiffness gradient with orthogonal chemical stimulation has been introduced to investigate GBM cell behavior [115]. In this platform, a fibronectin-conjugated polyacrylamide hydrogel generates a longitudinal stiffness gradient, while chemical stimulation is delivered laterally via diffusion. Studies with U87MG cells demonstrated that increased stiffness promotes chemotaxis, and that the presence of an epidermal growth factor (EGF) gradient further accelerates migration. These results underscore the critical influence of biophysical and biochemical gradients in regulating GBM cell motility.
Efforts to recapitulate the perivascular niche (PVN) of GBM have led to the development of microvasculature-on-a-chip systems. These models recreate a microenvironment where brain tumor stem-like cells (BTSCs) preferentially localize near microvessels and exhibit distinct behavioral phenotypes either quiescent or invasive depending on their molecular subtype. Single-cell transcriptomic analyses revealed correlations between microvessel proximity and proneural or mesenchymal subtypes, effectively capturing patient-specific tumor heterogeneity [116]. Building on these approaches, a three-dimensional organotypic microfluidic platform has been constructed by integrating hydrogel-based biomaterials with engineered microvascular networks [117]. This system successfully maintained GSCs invasive morphology, proliferation, and stemness, mirroring in vivo observations. Moreover, the model identified the CXCL12-CXCR4 signaling axis as a critical mediator of GSCs invasion. Inhibition of this pathway with AMD3100 significantly reduced invasive behavior, highlighting the potential of this model for drug screening applications [118]. Microfluidic technology has enabled more realistic modeling of the GBM microenvironment.
Microfluidic platforms have also been instrumental in modeling the effects of hypoxia within the GBM microenvironment. One model precisely controlled oxygen and nutrient levels to replicate the formation of pseudopalisades, wherein dense clusters of tumor cells migrate away from hypoxic and nutrient-deprived zones caused by vascular occlusion [119, 120]. This system enabled real-time observation of hypoxia-driven migration, providing insights into tumor adaptation under metabolic stress [121].
Expanding upon this, another microfluidic system generated continuous gradients of oxygen and nutrients within a hydrogel matrix, establishing distinct tumor zones comprising normoxic, hypoxic, and necrotic regions. This model facilitated dynamic monitoring of processes such as proliferation, apoptosis, and reactive oxygen species (ROS) production, while also serving as a platform for evaluating the penetration and efficacy of therapeutics, particularly those targeting hypoxic tumor areas (Figure 3E) [111]. In addition, metastasis-on-a-chip systems have been developed to study tumor-endothelial interactions by forming perfusable microvessels that mimic natural vasculature (Figure 3F). These models enable investigation of key processes in cancer progression, including angiogenesis and tumor cell migration across endothelial barriers, and offer valuable tools for testing the efficacy of anti-angiogenic therapies [112]. Collectively, microfluidic GBM models provide a highly controlled environment for replicating key physiological and pathophysiological features of GBM. By enabling precise manipulation of microenvironmental conditions such as stiffness, chemical gradients, oxygen availability, and vascular interactions, these platforms are poised to significantly enhance the development of targeted therapies and personalized treatment strategies for GBM.
Building on these advances in microfluidic GBM modeling, patient-derived microengineered platforms have begun to extend these approaches toward clinically relevant translation. In the submitted manuscript, a microengineered GBM model was developed that integrates a 3D cellular network composed of normal human astrocytes and primary tumor cells isolated from surgically resected GBM tissue. Within a defined microfluidic environment, patient-derived GBM cells and astrocytes self-organize into interconnected 3D structures, enabling physiologically relevant investigation of patient-specific cellular interactions and therapeutic responses. Comparison of chip-based tumor responses with clinical outcomes following conventional chemotherapy revealed a strong correspondence between in vitro phenotypes and patient prognosis. Specifically, the chip derived from patient A exhibited a markedly disrupted barrier and minimal responsiveness to high-dose TMZ, consistent with the shortest progression-free survival and overall survival. In contrast, the patient B derived chip maintained robust barrier integrity and demonstrated pronounced TMZ sensitivity, aligning with the most favorable clinical outcome. Collectively, these observations indicate that patient-derived microfluidic GBM models can capture clinically meaningful, patient-specific therapeutic responses and highlight their potential as functional in vitro platforms for personalized therapy evaluation [122].
3D bioprinting GBM models
Bioprinting, a specialized form of 3D printing, enables the spatially controlled deposition of bioinks containing cells and biomaterials to engineer complex tissue-like constructs. In GBM research, bioprinting facilitates the development of in vitro models that better capture tumor heterogeneity, including interactions with the ECM, immune cells, and vasculature [118, 123]. Techniques such as extrusion-based bioprinting (EBB), digital light processing (DLP), and stereolithography (SLA) have been employed to construct physiologically relevant GBM models (Figure 4A).
3D Bioprinting approaches for modeling the GBM microenvironment. (A) 3D bioprinting and SEM characterization of U118 cell-laden hydrogel scaffolds, illustrating the formation and sprouting of GSC23 and U118 spheroids (white arrows) on day 15. Reproduced with permission from Ref. [123] © 2020 J. Biomed. Mater. Res. Part A (B) Schematic workflow of alginate-based extrusion bioprinting using transient Ca²⁺ crosslinking and representative images of multi-layered constructs consisting of a cancer cell core encapsulated by stromal cell layers. Reproduced with permission from Ref [124] © 2020 Adv. Biol. Regul. (C) Schematic of the fabrication process for cell-laden mini-brain constructs and corresponding images showing the spatial organization of GL261 GBM cells (red) and RAW264.7 macrophages within the bioprinted model (scale bar = 5 mm). Reproduced with permission from Ref. [125] © 2019 Adv. Mater. (D) A schematic representation of the digital light processing bioprinting method. Reproduced with permission from Ref [98]. (E) A schematic representation of stereolithography (SLA) bioprinting method. Reproduced from under [126] an open-access license, © 2024 Adv. Healthcare Mater. (F) Coaxially bioprinted GBM microenvironment showing the printing process and corresponding bright-field and fluorescence images of U118-RFP GBM cells in the outer shell and HUVEC-GFP endothelial cells embedded in the hydrogel core. Reproduced from under [95] an open-access license, © 2021 Front. Bioeng. Biotechnol. (G) A schematic illustration of GBM cell behavior within a GelMA and HAMA-based hydrogel encapsulating a dextran bead. Reproduced with permission from Ref [127]. © 2018 Colloids Surf. (H) Schematic illustration of a cross-sectional view of concentric-ring GBM-on-a-chip model integrating patient-derived GBM and endothelial cells. Reproduced with permission from Ref [103] © 2019 Nat. Biomed. Eng.
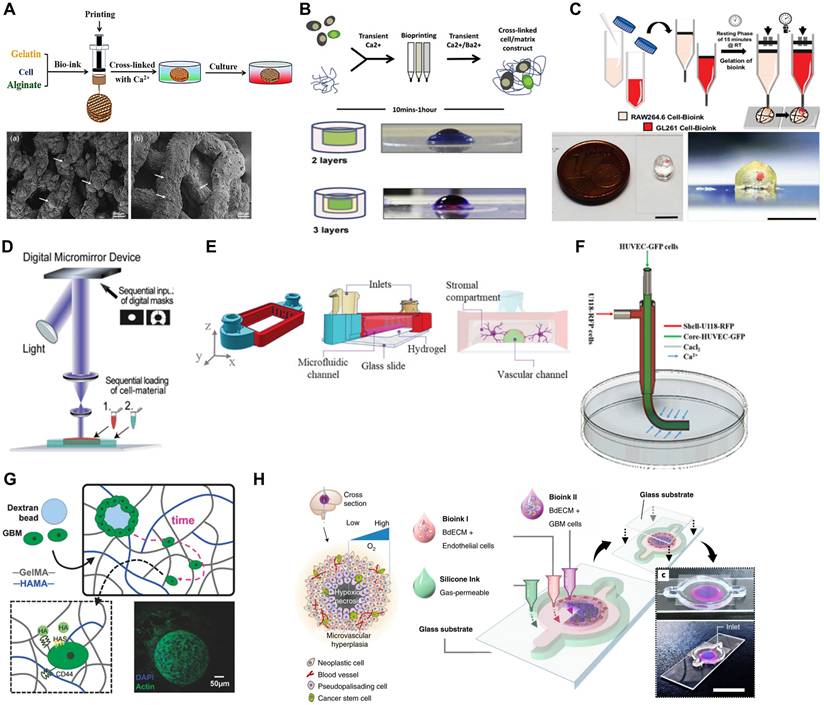
EBB remains the most frequently used technique for fabricating GBM constructs [128]. In this method, cell-laden hydrogels are extruded layer-by-layer to form 3D architectures [129, 130]. This technique supports high cell densities and is particularly suitable for co-printing GBM cells with stromal elements, such as endothelial cells and fibroblasts, within hydrogels [131, 132].
EBB has enabled the creation of models incorporating GSCs such as G144, G166, and G7 alongside endothelial cells, simulating tumor-vascular interactions and recapitulating chemoresistance mechanisms not observed in 2D cultures [124] (Figure 4B). The co-culture of GSCs with endothelial cells in a 3D bioprinted model allows researchers to observe how these cells interact, proliferate, and develop resistance to chemotherapeutic agents like TMZ. These processes are not typically observed in 2D culture systems. The 3D bioprinting model more accurately mimics the complex cell interactions and drug resistance mechanisms occurring in the TME [133]. The GAMs significantly contribute to GBM progression and invasiveness, yet their interaction with GBM cells remains poorly understood. A novel 3D-bioprinted mini-brain model, incorporating both GBM cells and macrophages, has been developed to study this crosstalk and test targeted therapies (Figure 4C). The model shows that GBM cells recruit and polarize macrophages into a GAM-specific phenotype, mirroring patient data. Additionally, GAMs enhance tumor growth and invasion, while therapeutic interventions targeting their interaction reduce tumor progression and improve chemotherapy sensitivity. This platform offers a valuable tool for advancing tumor biology research and evaluating new treatments [125]. In another study, researchers developed an advanced GBM model using a fibrin-based bioink incorporating patient-derived GBM cells, stromal cells, and perfusable vascular networks [101]. This model effectively mimics the TME, including cellular heterogeneity and vascular architecture, allowing detailed studies on tumor growth, invasion, and drug resistance. By incorporating patient-specific features and dynamic perfusion, the platform bridges the gap between traditional 2D cultures and in vivo models, offering a robust tool for personalized therapy screening and drug discovery.
DLP and SLA are other key technologies used in bioprinting GBM models [134]. These methods rely on light to cure bioinks layer by layer, allowing for the creation of highly detailed structures with region-specific properties. DLP and SLA are especially useful in generating GBM models with varying stiffnesses, which is critical for studying the mechanical influences on tumor growth and drug response (Figure 4D) [98, 135]. In GBM, the tumor core is often stiffer than the surrounding brain tissue, and this difference in stiffness affects cell behavior. DLP bioprinting has been used to create multi-stiffness GBM models that replicate this heterogeneity, enabling the study of how different stiffness environments influence glioma invasion, stemness, and resistance to therapies [136, 137].
For instance, a tri-regional GBM model was developed using DLP-based bioprinting, consisting of tumor, ECM and endothelial regions. By adjusting the stiffness of these regions, the model successfully replicated the biomechanical properties of the GBM microenvironment. This approach revealed that stiffer regions enriched in mesenchymal GBM subtypes exhibited greater invasiveness and drug resistance. The study highlighted the potential of 3D bioprinting to create physiologically relevant platforms for investigating the relationship between mechanical properties and tumor cell behavior [108].
Advanced 3D bioprinting method combined with SLA to create a microphysiological system for modeling GBM tumor environments. This system, GlioFlow3D, integrates human brain microvascular endothelial cells and glial cells into hydrogel-based channels within a rigid SLA-printed scaffold [126] (Figure 4E). The system is designed to replicate the vasculature and interstitial fluid flow dynamics seen in brain tissue. This model improves traditional methods by avoiding PDMS, which can absorb small molecules like chemotherapeutic agents such as TMZ, affecting the accuracy of drug testing. The 3D-printed system allows for real-time observation of cellular behavior and drug resistance, particularly highlighting the spatial differentiation of tumor cells near vascular structures, which exhibit higher resistance to chemotherapy. By simulating the TME, this method offers a robust platform for investigating complex biology of GBM and drug responses. These models have shown that GSCs near vascular interfaces often exhibit higher resistance to TMZ, mimicking the drug-resistant nature of perivascular GBM cells in vivo [126]. In another study, a self-organized TME array-on-a-chip was developed by integrating extrusion-based bioprinting with microfluidic technology [138]. This platform employs microstructured hydrogel pillars and vascular endothelial cells to form a perfusable vascular barrier around breast cancer spheroids, effectively replicating the TME. The design allows precise regulation of diffusion and shear forces, enabling tumor cell aggregation and self-assembly of vascular networks.
Building upon these advancements, recent studies have further refined bioprinted GBM models by integrating tumor-stromal interactions, vascular mimicry, and GSC enrichment strategies. By employing multi-cellular co-culture systems, researchers have sought to better replicate the TME and enable studies of heterogeneity, invasive behavior, and therapeutic resistance. One such approach involved the coaxial bioprinting of tubular glioma constructs, where glioma cells, U118 were co-cultured with HUVECs in a hydrogel-based fiber-like structure [95] (Figure 4F). This model successfully demonstrated that GBM cells actively secrete VEGF and bFGF, leading to the formation of vascular-like networks within the structure. Notably, U118 cells exhibited transdifferentiation into endothelial-like cells, reinforcing the hypothesis that GBM cells themselves contribute to vascular mimicry. However, the lack of fully perfusable vasculature within the bioprinted structure remained a limitation, as effective blood flow is essential for accurately replicating in vivo tumor conditions and drug diffusion properties. Similarly, expanding upon these findings, another study investigated the spatial organization of glioma and endothelial cells in a tumor-like construct using coaxial extrusion bioprinting [96]. The model integrated a shell-core hydrogel fiber system, with glioma cells in the shell and endothelial cells in the core, allowing researchers to examine tumor-endothelial cell interactions in a structured microenvironment. Results showed that glioma cells within this construct exhibited increased invasion rates and altered gene expression patterns associated with angiogenesis, reinforcing the concept that GBM cells play an active role in shaping their own vascular niche.
In another study, the role of glioma stem cells in tumor progression and therapy resistance was explored through 3D bioprinted glioma cell-laden scaffolds (Figure 4G) [94]. The model utilized a gelatin-alginate-fibrinogen (GAF) hydrogel scaffold to provide a supportive microenvironment for glioma cells. This environment enabled the maintenance of stemness markers, such as nestin and SOX2, and suppressed differentiation. Compared to traditional 2D cultures, cells within this 3D scaffold demonstrated enhanced epithelial-mesenchymal transition, a key process associated with tumor invasion and recurrence. Importantly, bioprinted glioma cells exhibited significantly higher resistance to TMZ, suggesting that 3D cultures better replicate the therapy-resistant nature of GSCs. Despite these promising findings, the gradual degradation of the hydrogel scaffold posed challenges for long-term experiments, highlighting the need for more stable bioinks that can support prolonged tumor studies. Recently, one notable implementation of this approach introduced a concentric-ring GBM-on-a-chip design, in which patient-derived GBM and endothelial cells were embedded within brain-derived decellularized ECM (BdECM) bioinks. The radial oxygen gradient established across the structure mimicked key tumor-stroma interactions and enabled the reproduction of hallmark pathological features, including hypoxia, pseudopalisading necrosis, and perivascular invasion. Furthermore, the model successfully predicted patient-specific responses to chemoradiotherapy, demonstrating its value as a platform for personalized treatment assessment (Figure 4H) [103].
Further efforts to improve GBM modeling have focused on the role of stromal cells in shaping the TME, particularly through bioprinted glioblastoma-mesenchymal stromal cell (MSC) co-cultures [99]. This model provided critical insights into how MSCs interact with GBM cells to alter the chemokine landscape, ultimately enhancing tumor invasiveness and immune evasion. Researchers observed that co-cultured glioma cells exhibited increased expression of CCL2, CCL5, and CXCL12, key chemokines involved in tumor progression, recruitment of immune cells, and resistance to therapy. This study emphasized the importance of tumor-stromal interactions in GBM research, yet the static nature of the bioprinted construct limited its ability to replicate dynamic cell migration and fluid exchange observed in vivo. Despite these advances, the study highlighted the need for improved methods to integrate functional blood vessels within the bioprinted structure, as tumor models lacking proper perfusion fail to capture key aspects of GBM metabolism and drug response.
The integration of patient-derived tissues into bioprinting-based GBM models requires careful ethical and biosafety evaluation to ensure responsible, safe, and reproducible research practices. Ethically, the use of human-derived materials demands fully informed and voluntary consent, including clear communication about tissue use, downstream applications, long-term storage, associated risks, and potential commercial implications [139]. Donor privacy must be strictly protected through de-identification procedures, and equitable representation of diverse patient populations is essential to avoid demographic bias [140]. All research involving patient-derived tissues must therefore be conducted under Institutional Review Board (IRB) approval. From a biosafety standpoint, human-derived samples must undergo screening for major pathogens and be handled within appropriate biosafety levels using certified biosafety cabinets, sterile techniques, and standardized operating procedures. Proper sterilization and disposal of biological waste, prevention of cross-contamination during storage, and verification of sample identity through routine cell line authentication are critical to maintaining safety and experimental reliability. For bioprinted constructs intended for translational or clinical applications, additional quality controls such as microbial contamination testing, analysis of residual biomaterials, and evaluation of cell viability and functional performance are required to ensure final product integrity and suitability for downstream use.
Biomaterials in bioprinted GBM models
The selection of biomaterials is crucial for developing bioprinted models that accurately recapitulate the mechanical and biochemical characteristics of the GBM microenvironment. Hydrogels such as hyaluronic acid (HA) and GelMA are widely utilized due to their brain ECM-mimicking properties and support for cell viability, proliferation, and migration [138, 141]. HA is particularly abundant in the brain ECM and plays a key role in facilitating tumor invasion by mediating cell-matrix interactions with glioma cells [199-201]. GelMA offers tunable mechanical properties, making it suitable for constructing scaffolds with varying stiffnesses, which is essential for modeling the biomechanical heterogeneity observed in GBM tissue [108, 142].
In recent studies, HA-based hydrogels have been used to encapsulate GSCs, enabling the formation of tumor organoids that closely resemble the in vivo characteristics of GBM. These organoids exhibit features such as cellular heterogeneity, stemness, and invasive behavior, providing a powerful tool for studying tumor evolution and drug resistance. The use of GelMA hydrogels in combination with DLP bioprinting has also allowed for the fabrication of multi-stiffness models that replicate the biomechanical heterogeneity of GBM, facilitating the study of stiffness-related tumor behavior [143, 144]. To further enhance the fidelity of GBM models, decellularized extracellular matrix (dECM) derived from brain tissue has emerged as a promising biomaterial [103, 145-147]. Brain dECM (BdECM) retains a wide range of native ECM components, including collagen IV, laminin, fibronectin, hyaluronic acid, and neurotrophic factors such as BDNF and NGF, which are largely absent in synthetic hydrogels [147]. These components provide essential biochemical and structural cues that regulate tumor-stroma interactions, cell migration, and matrix remodeling, facilitating the maintenance of glioma stemness and promoting tumor invasiveness. For example, region-specific dECM from cerebellum and cortex has been shown to differentially regulate astrocyte activation and metabolic rewiring under pathological conditions, suggesting that BdECM can recapitulate not only general but also localized brain microenvironments [145]. Moreover, dECM-based hydrogels support enhanced GBM cell viability, long-term culture, and making them valuable for drug screening applications [146]. Integration of dECM with biofabrication platforms, such as microfluidic devices or 3D bioprinting, further enables dynamic modeling of glioma progression and matrix-mediated therapy resistance [103]. Despite these advantages, the use of BdECM is still limited by batch variability, donor tissue availability, and the lack of standardized decellularization protocols. Nevertheless, it continues to gain recognition as a critical tool for developing more physiologically relevant GBM models.
Future in vitro models
As GBM research progresses, the development of in vitro platforms increasingly focuses on incorporating immune system components and systemic organ interactions to better reflect in vivo conditions. Two major directions include immune-related organ models and multi-organ systems.
Immune-related organ models
Integrating immune components into GBM models offers a promising avenue to study tumor-immune interactions and their impact on disease progression and treatment. Lymph nodes (LNs) and the spleen are particularly relevant due to their central roles in immune surveillance and systemic regulation.
LNs act as biological filters and hubs for immune cell activation, structured into distinct zones for B cells, T cells, and antigen-presenting cells [151, 152]. Recent advances in LN-on-a-chip technology replicate these microarchitectures using microfluidic platforms and 3D hydrogels, enabling controlled studies of immune cell dynamics, antigen presentation, and immunotoxicity (Figure 5A) [148]. Interestingly, the subcapsular sinus-on-a-chip further mimics fluid flow and adhesion cues found in native nodes, offering insights into immune cell trafficking and cancer metastasis (Figure 5B) [149]. Similarly, the spleen regulates immune responses and red blood cell clearance through its distinct red and white pulp regions [153]. The spleen-on-a-chip platform reproduces these filtration and circulatory dynamics using microstructured matrices to evaluate cell deformability, pathogen clearance, and drug effects in hematological disease contexts [154]. Despite the technical challenges of preserving immune cell viability and simulating dynamic responses, these immune organ-on-a-chip platforms are expected to greatly enhance the translational relevance of GBM models for immunotherapy testing.
Future in vitro models for GBM research. (A) Schematics of lymph node-on-a-chip systems. Microfluidic platform mimicking the human lymph node (LN) microenvironment by injecting different immune cell types. Reproduced from under [148] an open-access license, © 2020 Pharmaceutics. (B) Tumor- or inflammation-induced lymph node remodeling, characterized by dilation of afferent lymphatic vessels and the subcapsular sinus (SCS). Reproduced from under [149] an open-access license, © 2020 iScience. (C) Multi-organ-on-a-chip models incorporating GBM compartments. Schematic and photograph of a multi-interface organ-on-a-chip platform simulating oral drug delivery from the liver (HepG2) through the blood-brain barrier (BMECs and astrocytes) to brain tumors (U87MG), with collagen matrix interconnections. Reproduced with permission from Ref [150] © 2020 Biotechnol. Lett. (D) A biomimetic microfluidic system integrating intestinal (Caco-2), hepatic (HepG2), and GBM (U251) compartments for evaluating drug absorption, metabolism, and the synergistic anticancer effects of CPT-11 and TMZ on GBM cells. Reproduced with permission from Ref [88] © 2017 Analyst.
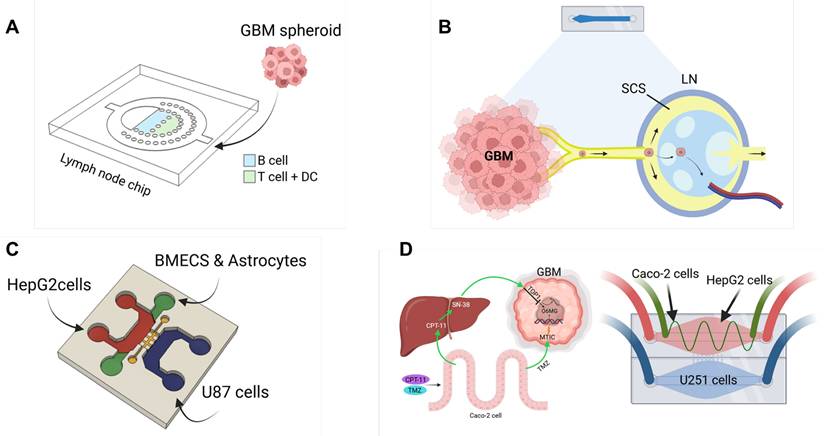
Advanced multi-organ systems
To address the complexity of GBM biology, advanced microfluidic platforms are being designed to link the brain tumor model with other functional organ modules. For instance, coupling GBM chips with BBB models enables the study of drug delivery and resistance [155]. Additionally, integrating organs such as the liver, which metabolizes drugs, or other immune organs, can help assess the systemic impacts of therapies (Figure 5C) [150]. One integrated microfluidic platform simulating intestine-liver-GBM interactions demonstrated that dual-drug combinations, such as irinotecan and TMZ, offer superior efficacy by accounting for metabolic and drug delivery (Figure 5D) [88]. Similarly, a liver-brain-GBM chip showed how hepatic metabolism and BBB permeability modulate drug responses to agents like paclitaxel, capecitabine, and TMZ [150]. These multi-organ platforms offer a system-level approach to studying GBM, bridging the gap between conventional in vitro assays and the complexity of human physiology. They are expected to enhance predictive accuracy for therapeutic screening and accelerate the development of personalized treatments [156].
Microfluidic GBM model for drug screening
Temozolomide (TMZ) as a single agent
TMZ is the primary chemotherapeutic agent used in the standard-of-care regimen for GBM, typically administered after surgical resection and radiotherapy [67]. TMZ exerts its cytotoxic effect by methylating guanine residues in DNA, leading to replication errors and cell death [161]. However, therapeutic resistance significantly limits its efficacy. This resistance primarily arises from the DNA repair enzyme MGMT, which reverses TMZ-induced DNA damage, along with the activation of alternative DNA repair mechanisms [162].
To address these challenges, advanced preclinical models have been developed to better evaluate TMZ efficacy. Microfluidic platforms allow real-time analysis of drug-induced cellular responses, such as changes in adhesion at the single-cell level, providing insights beyond traditional viability assays [163]. In one study, U87MG and U251 GBM cell lines were embedded within a microfluidic chip to assess the combined effects of TMZ and simvastatin. Drug efficacy was quantified via viability assays, immunofluorescence staining for apoptosis, and analysis of cell invasiveness, enabling simultaneous investigation of invasion, apoptosis, and autophagy pathways [157] (Figure 6A).
Microfluidic-based platforms for evaluating GBM drug response and combination therapies. (A) Schematic of GBM and the tumor-on-a-chip model featuring microfluidic chips with tumor and stromal compartments integrated with drug delivery channels. Reproduced from under [157] an open-access license, © 2020 Int. J. Mol. Sci. (B) Schematic of a 3D perfusion microfluidic chip for the evaluation of GBM cell invasion and drug response. Reproduced with permission from Ref [158]. © 2018 Biomed. Microdevices. (C) Gradient microfluidic chip featuring dual inlets and microwell arrays for TMZ and BEV screening via drug concentration gradients. Reproduced from under [159] an open-access license, © 2018 Sci. Rep. (D) DPEGDA-based brain cancer chip featuring gradient channels and 24 microwells for high-throughput drug combination analysis. Reproduced from under Ref [86] an open-access license, © 2016 Sci. Rep. (E) Schematic of a standardized microfluidic platform for modeling GBM spheroid-induced angiogenesis and drug screening. Reproduced with permission from Ref [160] © 2019 Lab Chip.
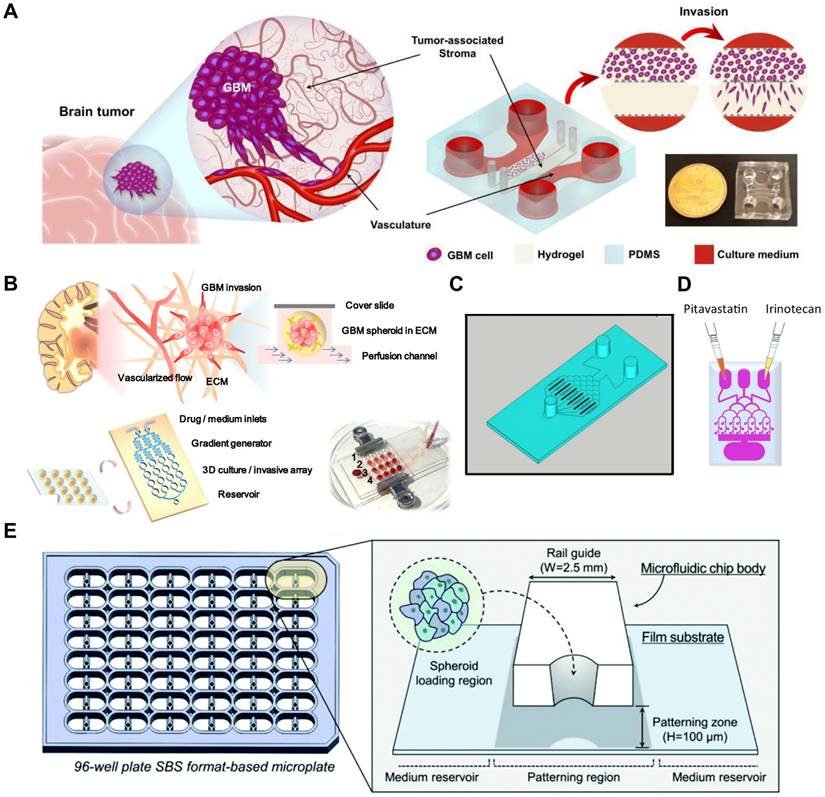
Other microfluidic systems replicate key aspects of the TME, including spatial tissue organization and continuous perfusion. These platforms support parallel manipulation of tumor spheroids and enable assessment of cell proliferation, viability, and invasiveness under dynamic drug exposure. Such models have demonstrated the effectiveness of agents like resveratrol and TMZ in reducing GBM invasiveness, underscoring their utility for personalized medicine and therapeutic screening (Figure 6B) [158]. Furthermore, a 3D bioprinted GBM vascular model was developed to better capture tumor heterogeneity and resistance patterns. This platform allows long-term culture and has shown that TMZ initially induces tumor regression, followed by recurrence, a phenomenon poorly modeled in 2D or suspended spheroid systems. By more accurately replicating in vivo-like tumor behavior, the bioprinted model provides a robust platform for optimizing TMZ-based therapies in a personalized context [100].
Combination therapy
Combination therapy has emerged as a promising strategy to overcome the limitations of monotherapy in GBM treatment. In particular, co-administration of TMZ with immune checkpoint inhibitors (ICIs) targeting PD-1 or CTLA-4 has demonstrated encouraging results in preclinical and early clinical studies. These regimens aim to restore anti-tumor immune surveillance while counteracting the immunosuppressive TME characteristic of GBM [164].
Another widely studied combination involves TMZ and bevacizumab, a monoclonal antibody targeting VEGF. Bevacizumab disrupts angiogenesis and enhances progression-free survival by impairing tumor vascular integrity. Microfluidic models have been instrumental in evaluating such combinations by enabling physiologically relevant drug exposure conditions. For example, a microfluidic platform incorporating 3D patient-derived GBM spheroids enabled precise control of drug diffusion and independent channel testing. This setup allowed detailed analysis of TMZ and bevacizumab interactions and their effects on tumor cell proliferation and viability (Figure 6C) [159].
Moreover, high-throughput drug screening is facilitated by microfluidic devices that allow parallel testing of multiple agents. In one study, a PEGDA-based hydrogel system supported the formation of uniform 3D GBM spheroids and was used to screen various drug combinations. The pairing of pitavastatin with irinotecan showed enhanced anti-tumor efficacy, demonstrating the potential of the platform in personalized medicine and rapid drug discovery (Figure 6D) [86].
In another example, a microfluidic chip incorporating pneumatic microstructures was used to trap U251MG GBM cells and generate uniform tumor arrays within a collagen matrix. This device enabled long-term culture for over a month and supported repeated drug testing cycles. Chemotherapeutic agents such as vincristine and bleomycin were evaluated using assays for viability and mitochondrial membrane potential, offering insights into drug-induced apoptosis [165]. These integrated systems collectively provide versatile platforms for investigating synergistic drug effects, understanding tumor heterogeneity, and developing optimized combination regimens tailored to individual patient responses.
Recently, the Sphero-IMPACT platform has been used to evaluate combination therapies in GBM, including anti-angiogenic drugs. In a co-culture model of U87MG spheroids and HUVECs within a fibrin matrix, endothelial invasion was observed and effectively inhibited by bevacizumab and sunitinib. This highlights the utility of platform in modeling tumor angiogenesis and screening vascular-targeted therapies in a reproducible, high-throughput format (Figure 6E) [160].
Immunotherapeutic investigations
Immunotherapy has emerged as a central focus in GBM treatment strategies due to the poor prognosis of disease and resistance to conventional therapies [166]. The GBM microenvironment shaped by the interplay of immune cells such as NK cells, TAMs, and T cells, plays a critical role in modulating immune responses and therapeutic efficacy [19]. However, the presence of the BBB significantly limits the delivery and efficacy of immunotherapeutic agents [167].
Microfluidic models have enabled the recreation of key features of the TME—such as immune cell infiltration, hypoxia, and cell-cell interactions—under physiologically relevant conditions. In one study, patient-derived GBM cells were used to develop an ex vivo microfluidic platform for personalized evaluation of responses to immune checkpoint inhibitors (ICIs), particularly anti-PD-1 therapies. This system revealed that mesenchymal GBM subtypes exhibited increased CD163⁺ TAMs and PD-1/PD-L1 signaling, contributing to immune evasion. Dual inhibition using nivolumab and a CSF-1R inhibitor (BLZ945) reduced TAM density and restored CD8⁺ T-cell activity, offering a tailored immunotherapeutic strategy [91]. Angiogenesis-focused microfluidic models have also elucidated immune-vascular crosstalk in GBM. A 3D angiogenesis-on-a-chip system demonstrated that TGF-β1 and αvβ3 integrin-mediated signaling drive macrophage polarization towards an M2 phenotype, promoting inflammation-induced angiogenesis. Targeting this pathway with combined αvβ3 and TGFβ-R1 inhibition suppressed neovascularization and modulated immunosuppressive signaling [168].
Advances in CAR-T cell therapies have further benefited from microfluidic technologies. BBB and blood-brain-tumor barrier (BBTB)-on-chip models derived from iPSCs have been used to test CAR-T constructs targeting EGFRvIII-overexpressing GBM cells. These platforms enabled analysis of CAR-T cell cytotoxicity, extravasation, and barrier integrity, demonstrating distinct behaviors of CAR-F263 and CAR-F269 variants [169, 170].
Additionally, antibody-drug conjugates (ADCs), which deliver cytotoxic payloads via tumor-specific monoclonal antibodies, have been evaluated using microfluidic models. Combining ADCs or CAR-T cells with ICIs on these platforms provides a unique opportunity to investigate resistance mechanisms and optimize combination regimens. Despite their utility, microfluidic models for GBM immunotherapy remain underdeveloped compared to those for other cancers. Challenges include replicating the full complexity of the immune microenvironment and the BBB. Limitations such as artificial materials, lack of immune diversity, and non-physiological cell ratios often reduce translational relevance. To advance GBM immunotherapy, there is a pressing need for standardized, reproducible microfluidic systems that faithfully recapitulate the immune and vascular microenvironments. Such platforms will be critical for evaluating new immunotherapeutic combinations and accelerating clinical translation.
While these examples highlight the potential of microfluidic platforms to inform immunotherapy design, current systems still face significant limitations in faithfully modeling immune-tumor dynamics [171]. Despite recent advances, incorporating immune cell populations such as TAMs and T cells into GBM-on-a-chip platforms remains a significant challenge. Primary immune cells offer physiological relevance but are limited by variability and poor long-term stability. On the other hand, immortalized cell lines provide consistency yet fail to capture the dynamic, context-dependent behavior of native immune cells. As a result, many platforms lack the cellular diversity and functional complexity necessary to reflect authentic immune-tumor interactions. Moreover, immune integration is often constrained by oversimplified co-culture systems, non-physiological ECM materials, and the absence of critical structural components such as the BBB and tumor vasculature [172]. These elements are essential for mediating immune infiltration, polarization, and therapeutic response but are frequently underrepresented or excluded altogether. Overcoming these limitations will be vital for enhancing the biological fidelity of GBM-on-chip systems and enabling their application in immunotherapy testing.
Phototherapy
Phototherapy is an advanced cancer treatment that utilizes light to specifically target and kill tumor cells. It operates through two main methods: photodynamic therapy (PDT) and photothermal therapy (PTT). PDT involves the use of a photosensitizer, a light-sensitive compound that produces ROS upon activation by specific wavelengths of light. These ROS cause localized damage to tumor cells, ultimately leading to cell death [173, 174]. PTT, in contrast, relies on photothermal agents to absorb light energy and convert it into heat. This localized heating raises the temperature in the TME, resulting in the destruction of cancer cells through protein denaturation and cell membrane disruption [175]. While PDT requires the combined action of light, oxygen, and a photosensitizer, PTT can often function independently of additional agents, though the inclusion of photothermal agents can significantly enhance its efficacy [176].
PDT has been effectively employed to treat various conditions, including skin disorders, ocular diseases, and cancers. Recent advancements have focused on improving its precision and therapeutic outcomes through nanotechnology. By integrating photosensitizers with nanomaterials, researchers have been able to enhance the generation of ROS and improve the targeted delivery of PDT agents. For example, microfluidic systems—miniaturized devices that allow precise control of experimental conditions—have been used to study the effects of PDT on GBM cells. One study utilized a microfluidic chip to explore how varying levels of photosensitizers, oxygen, and light influence therapeutic outcomes. Results indicated that increasing these factors significantly enhanced the cytotoxic effects of PDT. Another study demonstrated the potential of methylene blue (MB)-conjugated polyacrylamide nanoparticles (PAA NPs)-functionalized nanoparticles, optimized with a polyethylene glycol dimethacrylate (PEGDMA) cross-linker to enhance ROS production for PDT. The design minimized self-quenching, improved oxygen permeability, and was evaluated using advanced analytical tools to determine ROS composition and efficacy in a microfluidic platform. This approach identified near-optimal MB loading for effective light-dose-dependent killing of C6 GBM cells, highlighting the promise of combining nanotechnology with PDT [177].
Microfluidic technologies have become invaluable in phototherapy research, as they allow scientists to replicate complex tumor environments and evaluate therapeutic strategies in controlled settings. For instance, hydrogel-based microfluidic systems were utilized to develop a co-culture device for studying PTT and cancer cell migration, employing 10 w/v% GelMA hydrogels as semi-permeable barriers for culturing MCF7 breast carcinoma and U87MG GBM cells. This system demonstrated gold nanorod-mediated PTT effects on GBM and showed significant migration of U87MG cells toward VEGF-treated GelMA microchannels. Such hydrogel microfluidic devices provide a robust platform for evaluating PTT efficacy and analyzing migration behaviors of cancer cells with different motilities [178]. While PDT and PTT have advanced cancer phototherapy, their dependence on light restricts treatment of deep brain tumors. Recently, piezoelectric dynamic therapy (PZDT) has emerged as a non-optical, ultrasound-activated therapeutic strategy. Upon exposure to ultrasound, piezoelectric materials generate localized electric fields and induce charge separation, which in turn triggers a cascade of biochemical reactions, including enhanced ROS generation and disruption of cellular electrochemical homeostasis. Importantly, ultrasound exhibits superior tissue penetration compared to light, enabling PZDT to exert therapeutic effects in deep tumor regions that are largely inaccessible to PDT and PTT [179].
In vivo model for GBM treatment
The purpose of the preclinical process is to select final drug candidates to be applied to clinical trials through in vitro and in vivo studies and to determine safe doses for phase 1 clinical trials. Drug candidates screened through in vitro studies proceed to in vivo evaluations, where anticancer activity is verified and drug toxicity and pharmacokinetics studies are conducted [180]. In vivo models can replicate tumor behavior by reflecting actual diseases, which helps verify the efficacy and safety of drugs and discover new therapeutic strategies. Particularly, models that emulate key carcinogenic features, including the biological microenvironment, immune and inflammatory responses, and angiogenesis, provide new perspectives on tumor biology and facilitate a better understanding [181, 182]. This section provides a comprehensive review of the diverse in vivo animal models used in GBM research (Figure 7).
Comparative overview of mammalian animal models used in GBM research
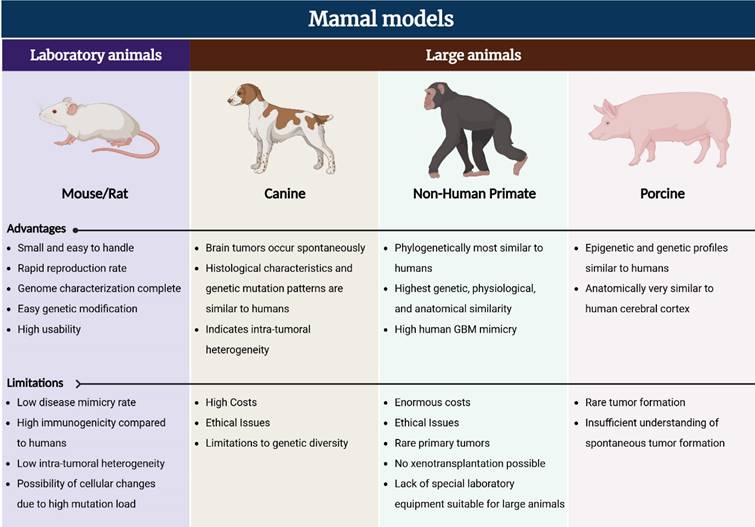
Mouse models
The mouse model is the most commonly used in vivo experimental model due to its relatively small size, rapid breeding essential for sustained experimental maintenance, and the thorough characterization of the mouse genome which provides experimental convenience [181]. Mice have been used in the development of several therapeutic strategies for human GBM and have provided valuable data, as they can mimic the complex genetic and physiological aspects of cancer and generate neoplastic tumors while maintaining tumor-host interactions. Particularly, mouse models are amenable to genetic manipulation in various ways, making them useful in assessing the effects of different mutations on tumorigenesis and treatment responses (Figure 8) [183, 184].
Classification of in vivo GBM mouse models based on tumor induction method.
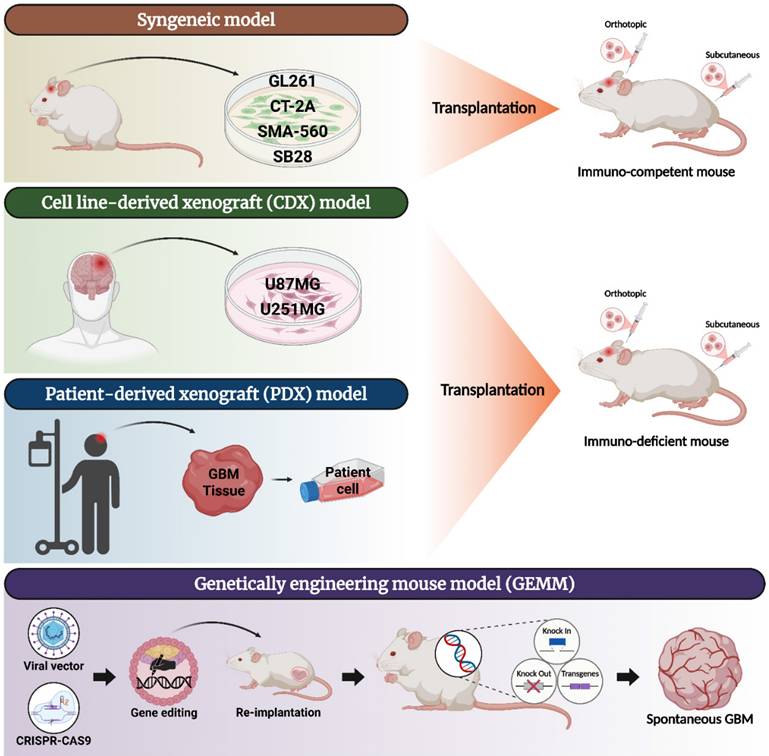
Syngeneic models
Syngeneic models are the oldest and most widely used preclinical models, being homogeneous in vivo models used between genetically identical individuals. This model is particularly useful for studying the interaction between the TME and the host immune system because it is immunocompetent, unlike other in vivo models that lack the immune capacity of experimental animals [185]. In the syngeneic GBM model, murine-derived brain tumor cell lines derived by various methods are used, such as GL261, CT-2A, SMA-560, and SB28.
GL261
GL261 is histologically very similar to human GBM, exhibiting features such as anaplastic and pleomorphic cells, active mitosis, and atypical nuclei, and as a model with a long history, tumor progression stages have been well studied, making it easy to interpret results and predict potential therapeutic responses [186, 187]. GL261 has been shown to be resistant to TMZ in vitro and expresses stem cell markers including mesenchymal properties such as CD133, CD44, nestin, and SOX2 in vivo [188, 189]. This allows it to be used to study the development of human GBM and to study glioma stem cells involved in chemotherapy and radiotherapy resistance.
The GL261 mouse model has been used in studies to discover novel therapeutics to develop standard treatments that do not significantly improve the overall survival rate of GBM patients [190, 191] and to develop administration methods to increase the delivery efficiency of TMZ [192]. It has also been actively utilized in immunotherapy development studies such as checkpoint-related immunotherapy and combination therapy with TMZ [193, 194], evaluation of the association between TMZ treatment and immune cell phenotypic changes in the TME [195], and alleviation of GBM-induced immunosuppression [196]. In addition, it has been widely used in studies to evaluate anti-GBM effects based on vaccines [197-199].
CT-2A
CT-2A, like GL261, recapitulates the characteristics of high-grade gliomas, including high cell density, pseudo-palisading necrosis, rapid abnormal mitosis, and microvascular proliferation, and exhibits proliferative and invasive properties similar to human GBM [200, 201]. The most notable feature of CT-2A is that given growth conditions for brain tumor stem cells, stemness can be enhanced, leading to significantly increased proliferation and invasiveness. It expresses stem cell markers such as CD133, Oct4, and nestin, which may be useful for studying glioma stem cells [201]. Additionally, the CT-2A glioma model is more resistant to checkpoint blockade therapy than the GL261 model [202, 203].
Due to their similar properties to GSCs, the CT-2A model is often utilized for tumor stem cell research in an immune-competent environment. The NS/CT-2A model, developed based on CT-2A cells cultured in neurospheres rather than monolayers, exhibited more abundant GSC markers, increased angiogenesis, and shorter mouse survival. It also showed an aggressive phenotype with decreased CD8+ T cells and increased regulatory T cells, which enhanced immunosuppressive properties, suggesting a clinically relevant platform, similar to the low immunogenicity of human GBM [204]. In addition, CT-2A has been actively utilized in various anti-GBM immunotherapy studies, including validating the combined effect of immunotoxin D2C7-IT and αCTLA-4/αPD-1/αPD-L1 therapy in a CT-2A mouse model overexpressing mutant EGFRvIII, a well-known glioma antigen [205], evaluating the efficacy of genetically engineered T cells targeting EGFRvIII in glioma stem cells [206], and evaluating the antitumor activity of mRNA-based multifunctional CAR T cells against malignant brain tumors [207].
SMA-560
SMA-560 histologically resembles dysplastic astrocytoma with highly cellular features showing focal necrosis and sporadic vascular proliferation. However, since nuclear atypia and strong expression of glial fibrillary acidic protein (GFAP) are also present, it is considered to morphologically reproduce human glioma [208, 209]. SMA-560 is resistant to TMZ both in vitro and in vivo and is slightly more sensitive to radiotherapy [210]. It also expresses the immunosuppressive molecule TGF-β, similar to human GBM, allowing the study of immunosuppressive effects on immunotherapy [211].
The SMA-560 model, characterized by TGF-β expression, has been used to investigate the correlation between the TGF-β pathway involved in angiogenesis, invasiveness, and immunosuppression and VEGF inhibition [212]. Additionally, this strategy has been applied to inhibit TGFβ using two antisense oligonucleotides targeting TGFβ1 and TGFβ2 [213]. In addition, there are studies that have evaluated tumor growth rates and toxicity by modifying SMA-560 cells. Genetic modification of tumor cells to secrete IL-2, IL-4, and TNFα enhanced antitumor immune responses and improved mouse survival [208]. Likewise, expression of a soluble CD70 ligand that stimulates CD27 signaling increased CD8⁺ T cell infiltration and extended survival [214]. It has also been used to study the reversal of the immunosuppressive GBM microenvironment via local delivery of IL-12 [215].
SB28
SB28 histologically shows hypervascularization and high cellularity, and infiltrates into the surrounding normal brain parenchyma [216]. Compared to other syngeneic mouse models such as GL261, SB28 is a more accurate representation of human GBM due to its lower immunogenicity, characterized by reduced tumor-infiltrating T cells, diminished MHC I expression, and significantly lower mutational load, making it an attractive model for the development of potential immunotherapeutic agents in preclinical trials [216-218].
The SB28 mouse model has been frequently used in immunotherapy and radiotherapy development studies. When radiation therapy was combined with a TGF-β inhibitor or anti-PD-L1 therapy, sensitivity was increased and survival was prolonged [219], and combination therapy with anti-CD40 monoclonal antibody and cyclooxygenase-2 (COX-2) inhibitor celecoxib was confirmed to increase anti-glioma activity by promoting type-1 immunity in both myeloid cells and T cells [218]. In addition, the effects of combination of mitogenic targeting drugs (spindle assembly checkpoint activators) and immune checkpoint blockade therapy on the TME and immunotherapy sensitivity were evaluated [220], and studies were also conducted on the association between immune evasion, a characteristic of GBM, and the intrinsic characteristics of the tumor and the special immune context of the brain using immune checkpoint inhibitors (ICIs) and the possibility of overcoming them [221, 222].
Although several syngeneic mouse models have been developed for the mechanism and successful preclinical studies of human GBM, they have limitations. The most significant limitation is that mice cannot guarantee that they accurately mimic real human diseases due to important genetic, immunological, molecular, and cellular differences from humans [223, 224]. In particular, most mouse-derived cell lines, such as GL261, CT-2A, and SMA-560, exhibit high immunogenicity compared to the low immunogenicity of human GBM [203, 208, 217, 225]. This property may not be suitable for studies for immunological evaluation because it increases sensitivity to immunotherapeutic agents. Unlike human GBM, which has a relatively low mutational load compared to other aggressive malignancies, GL261 has a high tumor mutational load with over 4000 non-synonymous exome mutations and a large number of predicted neoepitopes [217, 226]. It is therefore vulnerable to cellular changes that may occur during long-term cell culture in vitro [227]. In addition, CT-2A tends to have low invasion into the surrounding brain parenchyma, unlike human GBM, and SB28 is a homogenous cell line, in contrast to the high intra-tumoral heterogeneity of human GBM [225, 228]. As these significant differences from the properties and microenvironment of actual human brain tumors contribute to the failure of human clinical trials, development of advanced preclinical models that can most closely mimic human GBM is essential.
Xenograft models
Xenograft models have been developed to more closely reproduce human diseases and have long been used to study tumor development by subcutaneously or intracranially transplanting human-derived tumor cells into immunodeficient mice [229]. Since the biggest challenge in transplanting cells or tissues from other species is xenogenic immune rejection, immunodeficient mice are mainly used as hosts. Although immunodeficient mice are diverse, they are broadly classified into three types based on immunological differences: 1) athymic nude mice that do not produce T cells; 2) non-obese diabetic severe combined immunodeficiency mice (SCID and NOD-SCID) with depleted T and B cells; 3) NOD-SCID IL2R-γ null mice (NOG and NSG) with depleted T and B cells and extremely low NK cell activity [230]. Xenografts include cell line-derived xenografts (CDX) that transplant human GBM cell lines and patient-derived xenografts (PDX) that transplant patient-derived cells. Human GBM cell lines, including U87MG and U251MG, which are widely used in CDXs, are easy to handle and can mimic the histopathology of human GBM, and PDXs have improved ability to more closely reproduce actual disease, making xenograft models more likely to reproduce human GBM than syngeneic models [225].
Cell line-derived xenograft models
U87MG
The U87MG cell line is one of the most widely used human GBM cell lines and is thought to have genetic similarity to human GBM. It has mutations in hTERT, ATRX, and PTEN, no mutations in p53 and IDH1, and a methylated MGMT status, which are the same characteristics as human GBM [231, 232]. In addition, U87MG expresses CD133, forms neurospheres, and can be used in glioma stem cell research [233].
Since its establishment, the U87MG cell line has been widely used for a long time, and the xenograft mouse model has also been used in various research fields. Studies have been conducted to elucidate the effects and mechanisms of natural product extracts on glioma [234, 235], or to develop therapeutic strategies for human GBM by enhancing the delivery ability of anticancer drugs such as cisplatin or doxorubicin in combination with serum albumin [236, 237]. Additionally, it has been used in a wide range of studies, including comparative studies of bevacizumab doses for the treatment of gliomas [238], and molecular response studies using bevacizumab-resistance models [239].
U251MG
The U251MG cell line has a high proliferative capacity, most cells stain with Ki-67, and like U87MG, express CD133 and can form neurospheres [233, 240]. Genetically, it has mutations in hTERT, PTEN, and p53, but no IDH1 mutation and a methylated MGMT status [232]. It has the advantage of reproducing the histopathology of human GBM, such as invasive infiltration, cellular atypia and mitotic figures, and palisading necrosis [240, 241].
U251MG has also been used in studies to improve standard treatments, such as intracranial direct administration of bevacizumab [242] and combined effects of metformin and TMZ [243], as well as various functional studies to induce apoptosis of U251MG cells through modulation of proteins such as NF-E2-related factor 2 (Nrf2) [244] and hypoxia-induced BAX [245]. In addition, a comparative study on toxicity and anticancer efficacy between liposomal doxorubicin and free doxorubicin via convection-enhanced delivery (CED) was also conducted [246].
However, there are quite a few problems with xenograft models using cell lines. The first problem that arises is that they are limited in evaluating immunotherapy because they must be modeled in immunodeficient mice [225]. In addition, human GBM-derived cell lines consist of relatively homogeneous populations and thus lack the intratumor heterogeneity observed in patient tumors. Furthermore, long-term culture in serum-containing media can lead to genetic drift and transcriptomic alterations, limiting their ability to accurately recapitulate human GBM [247].
In particular, the U87MG model has many shortcomings. U87MG tumors transplanted into mice are well-defined as they are surrounded by reactive stellate cells without diffuse infiltration [2, 240, 241]. This is a significant difference from human GBM, which is characterized by diffuse infiltration. Additionally, necrotic features are rare, and there is little pseudo-palisading patterns or immune cell infiltrations [229]. Relative to human GBM, the model demonstrates increased vascularization, which may enhance therapeutic delivery. In contrast to clinical GBM, it remains responsive to both radiation and temozolomide treatment [248-250]. Another drawback of the U87MG cell line is the controversy over its authenticity. This issue was first raised when the laboratory that first isolated the U87MG cell line discovered that the U87MG cell line from the American Type Culture Collection (ATCC) had a different DNA profile from the original cells [77]. Therefore, although the U87MG cell line has been widely used and has contributed greatly to neuro-oncology research, concerns about authenticity and reproducibility have diminished its preference.
The U251MG cell line also has its drawbacks. Like U87MG, it is known to respond to both TMZ and radiation treatment [251-253]. The biggest problem is that in 1999, it was reported that the U373MG cell line was cross-contaminated with the U251MG cell line [241]. This is a similar issue to the authenticity of the U87MG cell line. Moreover, when the U251MG cell line was cultured for a long time, genetic changes accumulated, causing changes in various phenotypes, including cell morphology, proliferation rate, and cell surface marker expression [254]. Therefore, studies using the U87MG and U251MG cell lines may require careful approach and cell line validation.
Patient-derived xenograft models
The patient-derived xenograft (PDX) mouse model is one in which tumor tissue is obtained from patients, processed into single-cell suspensions or pieces, and injected immediately into the subcutaneous or intracranial microenvironment of mice [255]. PDXs are also human-derived cells, so immunodeficient mice must be used to prevent tumor rejection.
The main advantage of PDX models is that they can faithfully reproduce the characteristics of actual disease. Studies using GBM PDX models have shown that they reproduce the characteristics of human GBM, such as diffuse invasion, pseudo-palisading necrosis, and endovascular proliferation [36, 256]. Additionally, the characteristics and growth patterns of each GBM subtype were identified. PDX models using mesenchymal (MES) cells had a higher proliferation rate and increased invasiveness and vascularity compared to models using proneural cells [256-259]. This implies that the PDX model reflects the physiology of human GBM.
PDX models may have problems reproducing disease depending on the implantation location, so this must be carefully considered when designing. Orthotopic intracerebral implantation is preferred because it preserves the physiological environment of the BBB and cerebrospinal fluid, whereas subcutaneous implantation often fails to support normal tumor growth due to the markedly different microenvironment outside the brain [260]. A study investigating the genetic, epigenetic, and histopathological stability of orthotopic intracranial transplantation GBM PDX consistently reproduced primary tumor characteristics in contrast to subcutaneous transplantation, and long-term stability was demonstrated without evidence of mouse-specific tumor evolution [261]. Therefore, orthotopic PDX models can be useful for studies that measure and predict responses to novel therapies.
PDX models have been widely used in various studies due to their high reliability in reproducing human GBM. This model has been used to evaluate the anti-GBM effects and mechanisms of various potential agents, including diallyl trisulfide (DATS), a histone deacetylase inhibitor with reported anti-tumor activity [262], lisavanbulin alone or in combination with RT/TMZ [263], and combination therapy with a GSK-3 inhibitor and 1-[2-chloroethyl]-3-cyclohexyl-1-nitrosourea (CCNU) for chemo-resistant GBM [264]. Furthermore, studies such as the development and characterization of the GBM orthotopic PDX mouse model panel for drug efficacy evaluation have been conducted [265]. As such, the PDX model has been used in a wide variety of studies, helping to derive valuable results.
However, the PDX model also has disadvantages. First of all, as with CDX, because it uses immunodeficient mice, studies on the immune system and immune response to treatment cannot be conducted [225]. In addition, the establishment of patient-derived cells that can be used for xenografting is not easy and can take a long time, and the establishment success rate varies depending on the method and experience of each laboratory [266-268]. One of the problems encountered while culturing patient-derived cells is that GSC subpopulations within GBM can disappear after prolonged exposure to serum. GSCs differentiate in serum-containing conditions and can lose many primary tumor characteristics, which is a critical problem because GSCs are important for maintaining intra-tumoral heterogeneity, a hallmark feature of GBM [247]. Additionally, the use of patient-derived cells can be a double-edged sword. While PDX models can facilitate individualized studies because they use cells from a single individual patient, there can be significant variability between individual models, limiting standardization and reducing reproducibility of experimental results [184, 268].
Genetically engineered mouse models
As research on GBM continues, several genes such as EGFR, PTEN, and BRAF, which are commonly mutated, have been discovered, which are correlated with poor prognosis of patients [269-271]. Based on the association between these genetic changes and tumor progression, genetic research has become active and has led to the development of genetically engineered mouse models (GEMMs) [272]. GEMMs can be created by altering the genome and generating tumors through a variety of methods, including knockout and conditional knockout, replication-competent ALV LTR with a splice acceptor (RCAS), clustered regularly interspaced short palindromic repeats (CRISPR)-Cas9 technology, and other viral technologies [273]. Among these, CRISPR/Cas9 technology has significantly contributed to the creation of diverse GEMMs by enabling manipulation of single and multiple gene mutations [274].
GBM GEMMs have distinct advantages over other in vivo mouse models. Since these models induce tumors through genetic modification rather than direct injection of tumor cells into the brain, tumor development occurs in a manner similar to human GBM, allowing for a more accurate recapitulation. In addition, since there is no tumor cell injection process, the destruction of the BBB and the associated inflammatory phenomena can be avoided [227]. Additionally, unlike xenograft models, mice with an intact immune system can be used, making it possible to study the effects of immune cells or immunotherapy within the tumor environment [225]. Another advantage is that specific genes, such as P53 or PTEN, can be manipulated, allowing for in-depth studies of the role these genes play in tumorigenesis and treatment [275]. These clear advantages make it a useful in vivo model to investigate tumor-intrinsic and extrinsic factors in GBM from various perspectives.
GEMMs have been successfully used in various fields such as tumor microenvironmental impact analysis, tumor candidate gene and drug validation, therapeutic efficacy evaluation, and drug resistance mechanism analysis [276-278], and have been frequently used in GBM research, especially for evaluating therapies targeting genetic mutations [275, 279, 280]. In particular, GEMMs have been used to evaluate specific gene mutations and subsequent abnormal expression of related downstream signaling pathways in GBM. Overexpression of the common EGFR mutation (EGFRvIII) in GBM has been demonstrated to enhance high-grade gliomas and is associated with poor patient prognosis [271, 281], and additionally, the impact of PTEN inactivation on the development of high-grade gliomas was also evaluated [281]. A particularly clinically relevant finding is that IDH1 mutations are present at a low rate in primary GBM but at a high rate in secondary GBM and are also associated with increased survival [270]. Taken together, it has been demonstrated that EGFR amplification, PTEN loss, and IDH1 wild type are associated with primary GBM. Apart from this, GBM GEMMs were constructed by knocking out the tumor suppressor genes such as PTCH1, TP53, and NF1 [282], and it was reported that activation of the RAS pathway due to loss of TP53 and NF1 in CNS cells can cause malignant astrocytoma formation [283].
However, GEMMs also have disadvantages. First, tumor formation is often slow, which is time-consuming and expensive. In addition, unlike direct intracranial injection, GEMM tumors can occur in various locations within the brain, not at the desired location, which may limit the evaluation of some delivery methods that require precise location [284]. Another major disadvantage is that intra-tumoral heterogeneity may often be lacking compared to human GBM [227, 277].
Although the advantages of the mouse as an in vivo model are well established and have become the primary preclinical animal cancer model, its disadvantages are also evident, as described in this section. In addition to the disadvantages described for each mouse model, there are limitations due to significant differences between mice and humans. In particular, the mouse brain is lissencephalic, a key anatomical difference from the human brain, which results in a lack of brain gyration and cortical development [285, 286]. Besides, because the lifespan of mice is dozens of times shorter than that of humans, the risk of oncogenic mutations is lower due to fewer cell divisions. Significant differences between these preclinical animal models and humans lead to the failure of therapeutic clinical trials for GBM. Therefore, the need for animal cancer models that faithfully and reliably reproduce human cancer is steadily increasing.
Canine models
One of the conditions for an ideal preclinical model for human brain tumors is a tumor that arises spontaneously and grows within the parenchyma [287]. Canine models are mammals that frequently develop spontaneous brain tumors, and the characteristics of the tumors are similar to those in humans [288, 289]. A canine model in which brain tumors were induced by transplantation of tumor cells has also been reported, but it was used for purposes such as assessing the feasibility of CT-based estimation of tumor growth or training in tumor surgery rather than for studying anticancer therapy [288]. Research to explore anticancer therapies has largely been conducted using spontaneously occurring canine populations.
Unlike mouse models, canine GBMs exhibit endothelial proliferation, which is a major characteristic of human GBM and may be helpful in studying therapeutic strategies targeting endothelial cell proliferation [229, 289]. The detailed characteristics of canine gliomas include histological patterns including endothelial proliferation, GFAP expression, palisade necrosis, microinvasion, and hypercellular inflammation [229, 290]. It also showed similarities to human gliomas by showing neural progenitor markers such as vimentin and nestin and sphere-forming ability, and were recorded to have significantly higher expression of VEGF, VEGFR-1/2, EGFR-1, and PDGFα [291, 292]. A study was conducted to characterize the mutational process by comparing the molecular profiles between canine gliomas and human pediatric and adult gliomas. As a result, they showed alterations found in human gliomas such as TP53 and cell cycle pathways, receptor tyrosine kinases, and IDH1 R132, and canine gliomas showed high similarity to human pediatric gliomas based on the timing of mutations and DNA methylation patterns [293]. Another advantage of the canine model is that it can mimic the heterogeneity seen in human GBM patients. Canine gliomas occur across a wide range of age groups and breeds and show substantial heterogeneity, with different tumor types and grades, including astrocytomas, arising in distinct brain regions and exhibiting diverse immunological and genetic profiles [294, 295]. Therefore, the canine model may be a good preclinical model for GBM research.
The canine models have been proven and used in practice to be suitable models for studying human brain tumors, showing high similarity to humans [296, 297]. The similarity between the canine model and human gliomas has also been assessed from an immunological perspective. Glioma-associated microglia/GAMs are known to influence glioma progression and are polarized toward a tumor-supporting phenotype by TGF-β. In spontaneous canine gliomas, GAMs diffusely infiltrated canine astrocytomas, which correlated with an increased inflammatory cytokine environment, including TGF-β1. In addition, TGF-β1 was observed to promote tumor cell migration in vitro. This suggests that the GAM profile of high-grade canine astrocytomas is similar in many respects to that of adult high-grade gliomas [298]. Based on these similarities, it has been used in studies of anticancer drug delivery methods. The conjugation of doxorubicin to non-living bacteria-derived minicells and delivery to tumor cells via EGFR targeting was evaluated using a spontaneous canine brain tumor model [299]. The doxorubicin-loaded minicells were shown to be able to cross the BBB and selectively inhibit glioma cells while avoiding systemic toxicity, indicating strong potential for clinical application for the effective treatment of patients with brain cancer, leading to a phase 1 human clinical trial [300].
However, the high cost and ethical issues associated with using canines as animal models are major disadvantages and limitations [301]. Additionally, while spontaneous canine gliomas are useful for testing new treatments, they are not suitable for studying the effects of single mutations, for which genetic GEMMs are preferred [294]. Another limitation is the diversity of genetic composition. Crossbred canines with mixed backgrounds better mimic the diverse genetic background characteristics of humans, whereas inbred canines often lack genetic diversity [302].
Non-human primate models
Non-human primates (NHPs) are phylogenetically closest to humans, providing significantly higher genetic, physiological and anatomical similarities. Therefore, they can most closely reproduce actual human diseases and expected responses to treatment [303]. However, primary tumors in NHPs are rare, and transplantation using tumor cell lines from other animals is limited by immune rejection [304]. As a result, there are reports of cases that cause glioma in NHPs. It was demonstrated that treatment of Macaca mulatta (Rhesus monkeys) with fractionated whole-brain radiotherapy for 2 weeks resulted in the development of gliomas in most M. mulatta, which were histologically and genomically similar to human gliomas [305]. However, the period from radiotherapy to onset varied between 2.9 and 8.3 years, and this long latency may limit its application in clinical trials.
The NHP model may be an attractive preclinical model for successful clinical trials due to its advantage in best mimicking human physiology, but its practical use has been limited due to ethical concerns and prohibitive costs [306]. In particular, the lack of specialized experimental equipment required for genetic modification of large animal models makes it difficult to induce tumors in NHPs [307, 308]. Nevertheless, NHP models are being used to evaluate novel approaches for the treatment of gliomas. Carboplatin chemotherapy has shown limited clinical efficacy, which was thought to be due to its low tissue concentration when administered intravenously [309]. Therefore, ultrasound-induced BBB disruption was evaluated using a primate model to improve the delivery of intravenously administered carboplatin to brain tissue [310]. The results demonstrated that BBB opening using an implantable ultrasound transducer improves the distribution of carboplatin locally in the brain, leading to a phase 1 clinical trial.
The biggest limitations of the NHP model are ethical issues and enormous costs. There is a debate about whether studies using NHPs are necessary and justifiable, which has led to a decline in the use of NHPs in the United States and the European Union [311, 312].
Other animal models
Porcine models are another large animal model that can be used for cancer research. Minipigs of the Yucatan, Göttingen and Sinclair breeds grow up to 35-90 kg, which is similar to the weight of a human [313]. Previous studies have provided genome sequences of several laboratory pig breeds [314, 315], and comparative analyses of pig and human genomes have revealed similarities in epigenetic regulation and gene transcription profiles, which has had a major impact on the development of human genetic disease modeling [316, 317]. The pig brain is anatomically very similar to the human cortex, and can reproduce tumor infiltration, drug administration, and diffusion within the cortical region [313]. However, tumor formation is rare in pigs, and there is a lack of understanding of natural tumor formation in pigs due to the limited lifespan of livestock breeds prior to commercial exploitation [318]. Therefore, there is no evidence for spontaneous glioma formation in pigs, and pig models have been developed by inducing gliomas.
In 2013, the first pig model of glioma using human CDX was developed [319]. Landrace pigs, a domestic breed, were used, and U87MG cell lines were transplanted at 3 months of age. To overcome immune rejection, sufficient immunosuppression was performed using oral cyclosporine. Of the 15 pigs transplanted with U87MG, 14 showed macroscopic tumor formation and neurological symptoms within 30 days, and histopathological analysis showed increased cellularity, normal brain parenchyma infiltration, pseudo-palisading necrosis, and angiogenesis. Subsequently, in 2020, the first genetically engineered pig model of glioma induced by a lentiviral vector was developed [320]. Göttingen minipigs were injected with lentiviral vectors expressing PDGFB, constitutively active HRAS, and shRNA-p53. As a result, histopathological analysis revealed high-grade glioma features, including high cellularity, increased Ki-67 expression, astrocytic morphology, GFAP and oligodendrocyte transcription factor 2 (OLIG2) expression, and white and gray matter infiltration.
Future perspectives and conclusion
GBM remains one of the most formidable challenges in modern oncology due to its aggressive nature, high therapeutic resistance, and almost inevitable recurrence. Despite incremental advances in surgical techniques, radiation protocols, and chemotherapeutic agents, the median survival for GBM patients remains unacceptably short, underscoring a critical need for innovative strategies that can overcome the inherent complexities of this malignancy. Traditional 2D in vitro cell cultures, while valuable, often fail to adequately recapitulate the intricate cellular and molecular dynamics of the GBM TME, leading to a translational gap between preclinical findings and clinical outcomes. Consequently, promising therapeutic candidates that demonstrate efficacy in simplified model systems frequently fail to translate into meaningful clinical benefit for GBM patients. However, recent advancements in GBM microfluidic chip platforms and 3D bioprinting technologies offer a compelling path forward. These sophisticated modeling systems provide unprecedented opportunities to reconstruct the complex interplay of cellular and extracellular components that define the GBM TME, including vascularized networks, hypoxic gradients, dynamic cell-cell interactions, and the heterogeneous distribution of ECM components. By incorporating patient-derived GBM cells, immune cells, and stromal cells, these advanced models can more accurately mimic the intra-tumoral heterogeneity and personalized biology that are hallmarks of GBM. Moreover, these models enable researchers to investigate the dynamic interactions between GBM cells and the TME, elucidate the mechanisms of therapeutic resistance, and screen for novel therapeutic targets and drug combinations in a physiologically relevant context. Despite the rapid advancement of GBM-on-a-chip and 3D bioprinted platforms, several translational barriers must still be overcome before these models can meaningfully reduce failure rates in GBM clinical trials [321]. Key challenges include limited large-scale clinical validation linking model-predicted drug responses with patient outcomes, incomplete recapitulation of critical microenvironmental components such as functional vasculature, BBB interfaces, and immune interactions, and insufficient standardization across chip design, biomaterials, and analytical readouts [322].
In addition, integration into pharmaceutical development pipelines will require improved scalability, automation, and regulatory alignment to demonstrate added predictive value over existing preclinical models. In this context, advancing GBM-on-a-chip technologies toward high-throughput and automation-compatible formats will be essential for overcoming current translational bottlenecks. Transitioning from bespoke PDMS-based fabrication to scalable manufacturing strategies, such as injection molding of thermoplastics, offers a practical route to improving reproducibility, throughput, and compatibility with standardized pharmaceutical workflows. Together with plate-aligned microfluidic architectures that enable parallel drug and combination screening, these approaches are expected to facilitate regulatory alignment and accelerate the integration of GBM-on-a-chip platforms as predictive non-clinical testing tools [323].
Overview of GBM models using microfluidic and bioprinting platforms
| Model Type | Cell Source | Materials | Key Features | Advantages | Limitations | Applications | Ref |
|---|---|---|---|---|---|---|---|
| Microfluidic chip | GS5, GFP-HUVECs | PDMS | • Perivascular niche modeling | • Mimics the in vivo microenvironment • Dynamic tumor-EC interactions | • Incomplete mimicry • Short-term observation | • Microvasculature-on-a-Chip • Drug screening | [85] |
| U87MG | PEGDA hydrogel | • Integration of microwell arrays and microfluidic channels • 3D tumor spheroid formation | • Tunable drug release • Multi-drug administration • Massive parallel testing | • Static culture conditions • Diffusion-induced channel crosstalk | • High-throughput drug screening on 3D spheroid | [86] | |
| T98G, DRG neurons, Hippocampal neurons, Panc-1, A375, A549, MCF-7, SW620, PC-3 | PDMS | • Microgroove structure • Tumor neural microenvironment | • Physical separation with functional connection • Drug diffusion control • High-throughput drug screening | • Complexity of microfabrication | • Neuron-GBM-on-a-Chip • Drug screening | [87] | |
| U251, Caco-2, HepG2 | PDMS, mPES Hollow fiber, Matrigel | • Multi-organ co-culture • Gut-liver drug metabolism on GBM • Hollow fiber | • In vivo-like intestinal functionality • Dynamic drug delivery • Long-term culture capability | • Complexity of fabrication | • Gut-Liver-GBM on-a-chip • Prodrug metabolism studies, Drug screening | [88] | |
| GBM22, iPS-EC, Pericyte, Astrocyte | PDMS, fibrin | • BBB recapitulation • Vascularized GBM model | • In vivo-like BBB functionality • Quantitative 3D spatio-temporal analysis | • Absence of fluid flow • Absence of immune cells or neurons | • BBB-on-a-chip • Drug delivery across the BBB studies • Drug screening | [89] | |
| Patient-derived GBM | PDMS, ETFE | • Two bonded glass layers • Tissue chamber with microchannel network | • Ex vivo maintenance of human GBM tissue • Continuous tissue perfusion capability | • Short-term tissue maintenance • Limited correlation with patient outcomes • Complexity of fabrication | • Preclinical model • Personalized treatment • Drug screening | [90] | |
| Patient-derived GBM, hBMVECs, HMC3, U937 | PDMS, MMP-sensitive HA | • ex vivo GBM model • Recapitulation of the immunosuppressive microenvironment | • Predictability of clinical outcomes • High-throughput drug screening | • Use of allogeneic immune cells • Poor recapitulation of the BBB | • GBM-on-a-chip • Patient-specific screening for immunotherapy responses | [91] | |
| Patient-derived GBM, FNS, hCMEC/D3, HUVECs, HMVEC-L | PDMS, SU-8 negative photoresist, PDL, CollagenⅠ, Collagen Ⅳ, Laminin | • Perivascular niche modeling • Serum-free culture | • Maintenance of GSC stemness • High-throughput drug screening | • Insufficient recapitulation of the GBM microenvironment | • GSC-EC interaction studies • Drug screening | [92] | |
| Bioprinting | U87MG, SU3(GSC) | Gelatin, alginate, fibrin | • Cell-laden scaffold • Layering structure | • Precise control over cell density and spatial distribution • Improved cell-cell and cell-ECM interactions | • Complexity in long-term culture, Unknown reprogramming potential • Complexity of imaging and analysis | • GSCs studies • Glioma drug resistance studies • Drug screening | [93] |
| U118 | Gelatin, sodium alginate, fibrin | • Cell-laden scaffold • Layering structure • Epithelial-mesenchymal transition | • Enhanced GSC enrichment • Enhanced stemness and drug resistance | • Complexity of imaging and analysis | • GSCs studies • Epithelial-mesenchymal transition studies • Drug screening | [94] | |
| U118, HUVECs | Sodium alginate, Collagen | • Coaxial bioprinting • Cell-laden printing | • Multilayered tubular structures • Induction of angiogenesis by glioma cell | • Unclear necessity of tubular structure • Absence of fluid flow | • Tumor angiogenesis studies • Drug screening | [95] | |
| U118, GSC23 | Gelatin, alginate, fibrinogen | • Coaxial bioprinting • A separable core-shell design • Cell-laden printing | • Multilayered tubular structures • Facilitated analysis | • Loss of core cells near the shell • Decreased cell viability within the shell | • Drug resistance studies • Biomarker discovery • Drug screening | [96] | |
| GSC23, MSCs | Gelatin, alginate, fibrinogen | • Coaxial bioprinting • Self-assembled heterogeneous tumor fibers • Scaffold-free tissue constructs | • Cell viability and tissue organization • Promotion of ECM secretion | • Lumen collapse during long-term culture • Difficulty in removing dead cells | • Tumor studies • Drug screening | [97] | |
| GSCs(CW468, GSC23, 2907, 3264), THP-1 derived macrophage, Human astrocytes, NPCs | GM-HA, GelMA | • DLP-based 3D bioprinting system • Including M2 macrophages • Patient-derived ECM | • High-resolution spatial placement of cells and ECM • Reflection of pathological ECM characteristics | • Phototoxicity-induced cell damage • Limited scalability | • Therapeutic response profiling • Drug screening • Precision disease model | [98] | |
| GBM (DK-MG, U251MG), MSCs | CELLINK Laminink521, CELLINK Bioink biomaterial | • GBM-MSC interaction • Chemokine pattern analysis | • Reproducible platform • Manipulation of various variables • Recapitulation of chemokine signaling patterns | • Limitations of model standardization • Simplified model (GBM-MSC interactions only) | • Cell-microenvironment signaling validation • Drug screening | [99] | |
| GBM(isocitrate dehydrogenase-wild type), HUVECs | Gelatin, collagen I | • Perfusable vascular channel • 2GMFMT(mesoscopic fluorescence molecular tomography) | • Non-invasive • Real-time 3D imaging • Long-term Viability | • Limited Scalability for High-Throughput Testing • Limited single-cell imaging resolution | • Long-term monitoring of tumor behavior and drug response • Drug screening | [100] | |
| Patient-derived GBM, U-87MG, T98G, U373, 293T, Saos-2, MDA-MB-231, hAstro, hMG, human Pericyte, GL261 | Fibrin,Gelatin,Transglutaminase, Pluronic F127 | • Perfusable vascular channel • Multicellular 3D co-culture | • Brain-mimetic mechanical characteristics • Long-term Viability • Standardized fabrication protocol | • Tissue collapse during long-term culture • Limited reproducibility even with standardized protocols | • Long-term monitoring of tumor behavior and drug response • Personalized therapy screening | [101] | |
| Bioprinting / Microfluidic | A-172, HUVECs, hCMEC/D3 | PDMS, GelMA-Alginate, GelMA-Fibrin | • Microgravity condition • Cell-ECM mechanical interaction | • Gene expression analysis • Physiological flow-controlled perfusion • High-resolution imaging of junctional proteins | • Limited Long-term Viability • Incomplete mimicry | • Pre-clinical approach in brain tumor therapy • Drug screening • Drug delivery across the BBB studies | [102] |
| Patient-derived GBM, HUVECs | Brain dECM, Silicone ink | • Brain dECM • Oxygen gradient | • Highly biomimetic brain dECM • High heterogeneity recapitulation | • Complexity of fabrication • Inability to measure the oxygen gradient | • Patient-specific drug screening • Identification of mechanisms of therapeutic resistance | [103] |
Abbreviations: 293T, human embryonic kidney cells; A375, malignant melanoma; A549, non-small cell lung cancer; Caco-2, human colon cancer cell line; DRG, dorsal root ganglion; ETFE, ethylene tetrafluoroethylene; FNS, foetal neural stem; GelMA, gelatin methacryloyl; GL261, mouse glioblastoma cells; GSC, glioma stem cell; HA, hyaluronic acid; HepG2, human liver cancer cells; HMC3, human microglia cell line; hAstro, human astrocyte cells; hBMVECs, human brain microvascular endothelial cells; hCMEC/D3, human brain microvascular endothelial cell line; hMG, human microglia; hPericytes, human pericytes; HMVEC-L, human lung microvascular endothelial cells; HUVEC, human umbilical vein endothelial cells; MCF-7, breast cancer; MDA-MB-231, human breast cancer cells; MMP, matrix metalloproteinase; mPES, modified polyethersulfone; MSCs, mesenchymal stem cells; PDL, poly-D-lysine; PEGDA, photo-polymerizable polyethylene glycol diacrylate; PC-3, prostate cancer; Saos-2, human osteosarcoma cells; SW620, colorectal cancer; U118, human glioblastoma cells; U251, human glioblastoma cells; U373, human glioblastoma cells; U937, human monocytic cell line.
Comparative summary of in vivo GBM animal models: characteristics and research applications
| Animal models | Subtype | Method | Key Features | Pros | Cons | Applied Study Types | |
|---|---|---|---|---|---|---|---|
| Mouse | Syngeneic models | GL261 CT-2A SMA-560 SB28 | Syngeneic cell line implantation in immuno-competent host | • Histologically similar to hGBM • Recapitulates high-grade glioma • TMZ-resistant • Expresses stem markers • Checkpoint therapy resistant | • Historically well-validated model • Applicable to resistance studies, GSC research and immunotherapy studies | • High immunogenicity • High mutational load • Less invasive than hGBM • Lacks intra-tumoral heterogeneity | • Chemotherapy Immunotherapy • Combination therapy • GSC research • TME/Cytokine analysis • Drug delivery • Vaccines • CAR-T studies |
| Xenograft models | U87MG U251MG (CDX) | Xenogeneic cell line implantation in immuno-deficient host | • Genetic similarity to hGBM • Expresses CD133 • Forms tumorspheres Methylated MGMT status • High proliferation | • Historically well-validated model • Commonly used model • Applicable to GSC research • Reproducing the histopathology of hGBM | • Not suitable for immunotherapy studies • Lacks diffuse infiltration and necrosis • Phenotype drift by genetic alterations • Responsive to both radiation and TMZ • Authenticity concerns Cross-contamination issue | • Chemotherapy • Combination therapy • Natural compound studies • Resistance model studies • Apoptosis modulation • Drug delivery | |
| Patient- derived cell (PD) | Patient-derived cell implantation in immunosuppressed host | • Represents hGBM physiology • Requires site-specific consideration • Subtype-specific properties | • Closely mimics hGBM pathology • Orthotopic model reliably reflects primary tumor • Long-term stability | • Not suitable for immunotherapy studies • Time-consuming process • Inconsistent model engraftment rates • Loss of GSC characteristics during culture • Low reproducibility | • Combination therapy • Resistance model studies • Drug efficacy evaluation | ||
| GEMMs | Genetically induced glioma | Viral vector (RCAS-TCA vector) CRISPR/Cas9 | • Targeted gene modification (EGFR, PTEN, BRAF, TP53) • Tumor develops in a manner similar to hGBM | • High-fidelity GBM modeling • Preserves BBB integrity • Enables immunotherapy research • Enables in-depth gene function analysis in tumorigenesis and therapy | • Time- and cost-intensive due to delayed tumorigenesis • Inconsistent tumor sites restrict specific assessments • Lacks intra-tumoral heterogeneity | • Genetic pathway studies • TME analysis • Resistance mechanism studies | |
| Canine | Spontaneous/ transplanted glioma | Enrollment of spontaneous cases Syngeneic cell implantation | • Histologically similar to hGBM • Exhibits endothelial proliferation • Expresses neural progenitor markers • Forms tumorspheres | • High similarity to hGBM • Enables endothelial-targeted research • Mimics intra-tumoral heterogeneity | • High cost • Ethical concerns • Not suitable for mutation-specific studies • Lack of genetic diversity | • TME research • Drug delivery • Inflammatory cytokine studies | |
| NHP | Irradiation-induced glioma | Fractionated whole-brain radiotherapy | • Highest genetic, physiological, and anatomical similarity to humans • Closest phylogenetic proximity to humans | • Most accurately replicates human therapeutic responses | • Prohibitively high cost • Rare occurrence of primary tumors • Xenotransplantation limited by immune rejection | • BBB modulation • Chemotherapy • Drug delivery | |
| Porcine | Transplant/ Genetically induced glioma | Xenogeneic cell line implantation in immuno-deficient host Viral vector-based gene modification | • Comparable epigenetic/gene expression patterns to humans • Anatomically similar to the human cerebral cortex | • Capable of recapitulating tumor infiltration, drug delivery, and diffusion • Capable of overcoming immune rejection | • Rare tumor formation • Limited understanding of spontaneous tumor formation | • Human CDX model • Genetically modified model | |
To address these challenges and ultimately improve patient outcomes, future research efforts must prioritize the development and validation of advanced translational models that accurately reflect the complexities of GBM biology. These models should be used to: Identify novel therapeutic targets: By leveraging microfluidic and bioprinted models to screen for vulnerabilities within GBM cells and the TME, researchers can uncover new targets for drug development, including those that specifically target GSCs or modulate the immune response. Evaluate drug combinations: Due to the complex and multifactorial nature of GBM, combination therapies are expected to outperform single-agent treatments in terms of efficacy. Advanced models can facilitate the rapid and efficient evaluation of drug combinations, identifying synergistic interactions and optimizing treatment regimens. Personalize treatment strategies: The use of patient-derived cells in bioprinted models allows for the development of personalized treatment strategies that are tailored to the specific characteristics of each patient's tumor. This includes selecting the most effective drugs, optimizing drug dosages, and predicting treatment responses. Understand the mechanisms of resistance: By studying how GBM cells adapt and resist therapy within the TME, researchers can identify new strategies for overcoming resistance mechanisms and improving treatment outcomes. This includes investigating the role of epigenetic modifications, metabolic adaptations, and immune evasion. Together with these advanced in vitro models, the need for preclinical in vivo models with better GBM replication is emphasized to overcome the poor overall clinical predictability. As described in this review, in vivo models with better replication have been steadily developed for GBM mechanistic studies and effective therapeutic strategies, and this is currently in progress.
Artificial Intelligence (AI) and Machine Learning (ML) aid in optimizing complex 3D GBM models through two distinct mechanisms: constraint-driven design automation and high-dimensional phenotypic profiling. First, AI aids in optimizing chip design by automating the determination of complex geometric parameters. The design of microfluidic chips for GBM requires precise control over multiple variables, such as channel dimensions, fluidic resistance, and shear stress, to accurately mimic the vascular niche of tumor and interstitial pressure. Traditionally, this relies on iterative trial-and-error; however, recent methodologies in design automation utilize algorithms to automatically generate chip layouts that strictly satisfy these physiological constraints, reducing the barrier to adoption for non-experts [324, 325]. Furthermore, advanced optimization strategies, such as ML-driven cyclic optimization and Bayesian techniques, enable the systematic exploration of vast design spaces to predict optimal configurations for desired fluid behaviors [326, 327]. By automating these variables and predicting fluid dynamics such as plasma separation efficiency via Computational Fluid Dynamics (CFD) integration, AI significantly reduces the fabrication cycles required to produce physiologically relevant GBM microenvironments [328].
Second, ML aids in data interpretation by decoding the complex, non-linear relationships between cellular phenotypes and therapeutic responses. 3D GBM models generate massive datasets from high-content imaging and real-time biosensors that exceed human analytical capacity. Deep learning algorithms facilitate the interpretation of these data by performing morphological profiling, identifying subtle structural features in GBM spheroids, such as changes in nuclear texture or invasive edge morphology, that serve as early indicators of drug efficacy [329]. For instance, ML-based analysis has been shown to accurately classify drug mechanisms of action and predict concentration-dependent cytotoxicity in U87 GBM spheroids, outperforming traditional viability assays [330]. Additionally, AI-driven analytics can integrate these imaging features with multi-omics data and real-time sensing signals to predict patient-specific drug resistance pathways and monitor metabolic states [331, 332]. Collectively, these AI-driven capabilities transform GBM-on-a-chip from simple culture platforms into predictive precision medicine tools.
An emerging direction in cancer immunology and neuro-oncology is the establishment and utilization of PDXs in humanized mice with a human immune system [333]. As described previously, PDX has the advantage of being able to reproduce human GBM most closely with regard to histology and intra-tumoral heterogeneity. However, the main limitation is that immune-related studies cannot be performed because immunodeficient mice are used. The immune system plays an essential role in GBM treatment [334], and the inability to conduct studies such as immunotherapy that actively intervene in tumor killing is a significant limitation. Given the limitations of syngeneic and xenograft models or GEMMs in reproducing the histological and genetic characteristics, intra-tumoral heterogeneity, and immune system of human cancer, the need for humanized models is clear. Accordingly, a humanized cancer mouse model co-transplanted with human tumor and immune cells into NOD-SCID IL2R-γ null mice has been developed and is being used for immuno-oncology research with potential for clinical translation [335]. However, humanized mice are still limited in terms of modeling human immunity, especially in antibody response capacity [336]. Successful humanized immune reconstitution mouse models with continuous improvement are expected to increase the possibility of establishing a personalized approach to implanting patients' tumors to contribute to therapeutic decision-making.
CNS tumors are classified into several subtypes according to criteria selected by WHO [337]. In particular, the most important factor in classifying diffuse glioma as GBM is IDH status. IDH-wild type is classified as GBM, mutant type is classified as astrocytoma and oligodendroglioma, and IDH-mutant glioma is subdivided according to histologic grade and molecular profiles [337]. IDH mutant glioma is known to predict a better prognosis due to its longer overall survival compared to wild type [338].
However, IDH-mutant type is usually found in secondary tumors rather than primary tumors, suggesting that low-grade gliomas with IDH mutations often relapse to higher grades through malignant transformation [339]. Therefore, research on various types of gliomas, including GBM, is also considered important, and in August 2024, Vorasidenib, a treatment for IDH1/IDH2-mutant glioma developed in France, was approved by the FDA, which is the new drug for glioma in 25 years [340].
Research on IDH-mutant glioma is also actively underway, and mouse models suitable for the purpose have been established. In 2015, the GL261 cell line transfected with the IDH1R132H DNA vector was transplanted into C57BL/6N mice to create an mIDH1 murine model, and peptide immunization was evaluated [341]. Subsequently, in 2018, the role of IDH1R132H on glioma development was evaluated in mice using the RCAS/TVA retroviral vector system [342]. Additionally, a protocol to establish IDH1-mutant astrocytoma GEMM using CRISPR/Cas9 technology and adeno-associated virus (AAV) was proposed in 2023, but it has the limitation of an incubation period of more than 8 months, and troubleshooting is required [343]. In this way, research on various types of glioma as well as GBM and establishment of in vivo models are steadily progressing and have important implications.
In conclusion, overcoming the challenges of GBM requires a concerted effort to bridge the persistent translational gap between preclinical research and clinical application. This review emphasizes the need to move beyond technology-centric modeling toward integrated, clinically informed platforms that synergistically combine microfluidic and 3D bioprinted systems to more faithfully recapitulate GBM pathophysiology, therapeutic resistance, and patient-specific tumor heterogeneity. By embracing advanced modeling strategies that reconstruct the tumor microenvironment and enable personalized therapeutic evaluation, bioengineered GBM models can evolve from experimental research tools into clinically actionable systems for precision neuro-oncology. Ultimately, integrating these innovative platforms into clinical trials will be essential to accelerate the development of effective therapies and to transform the standard of care for patients suffering from this devastating disease.
Acknowledgements
This work was supported by grants from the National Research Foundation of Korea (RS-2024-00357185 and RS-2024-00350442 to Prof. Lim and Prof. Ahn, respectively).
Competing Interests
The authors declare no conflict of interest regarding the research, authorship, or publication of this article.
References
1. McKinnon C, Nandhabalan M, Murray SA, Plaha P. Glioblastoma: clinical presentation, diagnosis, and management. BMJ. 2021;374:n1560
2. Wen PY, Kesari S. Malignant gliomas in adults. New England Journal of Medicine. 2008;359(5):492-507
3. Ostrom QT, Gittleman H, Farah P, Ondracek A, Chen Y, Wolinsky Y. et al. CBTRUS statistical report: Primary brain and central nervous system tumors diagnosed in the United States in 2006-2010. Neuro Oncol. 2013 15 Suppl 2(Suppl 2):ii1-56
4. Louis DN, Perry A, Wesseling P, Brat DJ, Cree IA, Figarella-Branger D. et al. The 2021 WHO Classification of Tumors of the Central Nervous System: a summary. Neuro Oncol. 2021;23(8):1231-51
5. Ostrom QT, Patil N, Cioffi G, Waite K, Kruchko C, Barnholtz-Sloan JS. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2013-2017. Neuro Oncol. 2020;22(12 Suppl 2):iv1-iv96
6. Ohgaki H, Kleihues P. Epidemiology and etiology of gliomas. Acta Neuropathol. 2005;109(1):93-108
7. Sakthikumar S, Roy A, Haseeb L, Pettersson ME, Sundström E, Marinescu VD. et al. Whole-genome sequencing of glioblastoma reveals enrichment of non-coding constraint mutations in known and novel genes. Genome Biol. 2020;21(1):127
8. Braganza MZ, Kitahara CM, Berrington de González A, Inskip PD, Johnson KJ, Rajaraman P. Ionizing radiation and the risk of brain and central nervous system tumors: a systematic review. Neuro Oncol. 2012;14(11):1316-24
9. Charles LE, Burchfiel CM, Fekedulegn D, Kashon ML, Ross GW, Petrovitch H. et al. Occupational exposures and movement abnormalities among Japanese-American men: the Honolulu-Asia Aging Study. Neuroepidemiology. 2006;26(3):130-9
10. Söderberg-Nauclér C, Johnsen JI. Cytomegalovirus in human brain tumors: Role in pathogenesis and potential treatment options. World J Exp Med. 2015;5(1):1-10
11. Thakkar JP, Dolecek TA, Horbinski C, Ostrom QT, Lightner DD, Barnholtz-Sloan JS. et al. Epidemiologic and molecular prognostic review of glioblastoma. Cancer epidemiology, biomarkers & prevention. 2014;23(10):1985-96
12. Dean M, Fojo T, Bates S. Tumour stem cells and drug resistance. Nat Rev Cancer. 2005;5(4):275-84
13. Verhaak RG, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD. et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 2010;17(1):98-110
14. Mendelsohn J, Baselga J. Epidermal growth factor receptor targeting in cancer. Semin Oncol. 2006;33(4):369-85
15. Kaminska B, Czapski B, Guzik R, Król SK, Gielniewski B. Consequences of IDH1/2 mutations in gliomas and an assessment of inhibitors targeting mutated IDH proteins. Molecules. 2019;24(5):968
16. Baghban R, Roshangar L, Jahanban-Esfahlan R, Seidi K, Ebrahimi-Kalan A, Jaymand M. et al. Tumor microenvironment complexity and therapeutic implications at a glance. Cell Communication and Signaling. 2020;18:1-19
17. Reinhard J, Brösicke N, Theocharidis U, Faissner A. The extracellular matrix niche microenvironment of neural and cancer stem cells in the brain. Int J Biochem Cell Biol. 2016;81(Pt A):174-83
18. Quail DF, Joyce JA. The microenvironmental landscape of brain tumors. Cancer cell. 2017;31(3):326-41
19. Sharma P, Aaroe A, Liang J, Puduvalli VK. Tumor microenvironment in glioblastoma: Current and emerging concepts. Neurooncol Adv. 2023;5(1):vdad009
20. Martinez-Lage M, Lynch TM, Bi Y, Cocito C, Way GP, Pal S. et al. Immune landscapes associated with different glioblastoma molecular subtypes. Acta Neuropathol Commun. 2019;7(1):203
21. Cook J, Hagemann T. Tumour-associated macrophages and cancer. Current opinion in pharmacology. 2013;13(4):595-601
22. Turkowski K, Brandenburg S, Mueller A, Kremenetskaia I, Bungert AD, Blank A. et al. VEGF as a modulator of the innate immune response in glioblastoma. Glia. 2018;66(1):161-74
23. Ding XC, Wang LL, Zhang XD, Xu JL, Li PF, Liang H. et al. The relationship between expression of PD-L1 and HIF-1α in glioma cells under hypoxia. J Hematol Oncol. 2021;14(1):92
24. Guan X, Hasan MN, Maniar S, Jia W, Sun D. Reactive astrocytes in glioblastoma multiforme. Molecular Neurobiology. 2018;55:6927-38
25. Noronha C, Ribeiro AS, Taipa R, Castro DS, Reis J, Faria C. et al. Cadherin expression and EMT: A focus on gliomas. Biomedicines. 2021;9(10):1328
26. Cai X, Wang K-C, Meng Z. Mechanoregulation of YAP and TAZ in cellular homeostasis and disease progression. Frontiers in cell and developmental biology. 2021;9:673599
27. Zimmermann DR, Dours-Zimmermann MT. Extracellular matrix of the central nervous system: from neglect to challenge. Histochemistry and cell biology. 2008;130:635-53
28. Lundell A, Olin AI, Mörgelin M, Al-Karadaghi S, Aspberg A, Logan DT. Structural basis for interactions between tenascins and lectican C-type lectin domains: evidence for a crosslinking role for tenascins. Structure. 2004;12(8):1495-506
29. Rauch U. Brain matrix: structure, turnover and necessity. Biochemical Society Transactions. 2007;35(Pt 4):656-60
30. Vaz-Salgado MA, Villamayor M, Albarrán V, Alía V, Sotoca P, Chamorro J. et al. Recurrent Glioblastoma: A Review of the Treatment Options. Cancers (Basel). 2023 15(17)
31. Gimple RC, Bhargava S, Dixit D, Rich JN. Glioblastoma stem cells: lessons from the tumor hierarchy in a lethal cancer. Genes Dev. 2019;33(11-12):591-609
32. Cheng L, Bao S, Rich JN. Potential therapeutic implications of cancer stem cells in glioblastoma. Biochemical pharmacology. 2010;80(5):654-65
33. Bonnet D, Dick JE. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nature medicine. 1997;3(7):730-7
34. Tavazoie M, Van der Veken L, Silva-Vargas V, Louissaint M, Colonna L, Zaidi B. et al. A specialized vascular niche for adult neural stem cells. Cell stem cell. 2008;3(3):279-88
35. Yan H, Romero-Lopez M, Frieboes HB, Hughes CC, Lowengrub JS. Multiscale Modeling of Glioblastoma Suggests that the Partial Disruption of Vessel/Cancer Stem Cell Crosstalk Can Promote Tumor Regression Without Increasing Invasiveness. IEEE Trans Biomed Eng. 2017;64(3):538-48
36. Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB. et al. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature. 2006;444(7120):756-60
37. Wang Q-E. DNA damage responses in cancer stem cells: Implications for cancer therapeutic strategies. World journal of biological chemistry. 2015;6(3):57
38. Gaspar LE, Fisher BJ, Macdonald DR, Leber DV, Halperin EC, Schold Jr SC. et al. Supratentorial malignant glioma: patterns of recurrence and implications for external beam local treatment. International Journal of Radiation Oncology* Biology* Physics. 1992;24(1):55-7
39. Halperin EC, Burger PC, Bullard DE. The fallacy of the localized supratentorial malignant glioma. International journal of radiation oncology, biology, physics. 1998;2(15):505-9
40. Feldman L. Hypoxia within the glioblastoma tumor microenvironment: a master saboteur of novel treatments. Frontiers in Immunology. 2024;15:1384249
41. Anderson NM, Simon MC. The tumor microenvironment. Current Biology. 2020;30(16):R921-R5
42. Ngambenjawong C, Gustafson HH, Pun SH. Progress in tumor-associated macrophage (TAM)-targeted therapeutics. Advanced drug delivery reviews. 2017;114:206-21
43. Vescovi AL, Galli R, Reynolds BA. Brain tumour stem cells. Nat Rev Cancer. 2006;6(6):425-36
44. Barker HE, Paget JT, Khan AA, Harrington KJ. The tumour microenvironment after radiotherapy: mechanisms of resistance and recurrence. Nature Reviews Cancer. 2015;15(7):409-25
45. Lee G, Auffinger B, Guo D, Hasan T, Deheeger M, Tobias AL. et al. Dedifferentiation of glioma cells to glioma stem-like cells by therapeutic stress-induced HIF signaling in the recurrent GBM model. Molecular cancer therapeutics. 2016;15(12):3064-76
46. Posti J, Bori M, Kauko T, Sankinen M, Nordberg J, Rahi M. et al. Presenting symptoms of glioma in adults. Acta Neurologica Scandinavica. 2015;131(2):88-93
47. Esquenazi Y, Moussazadeh N, Link TW, Hovinga KE, Reiner AS, DiStefano NM. et al. Thalamic glioblastoma: clinical presentation, management strategies, and outcomes. Neurosurgery. 2018;83(1):76-85
48. Chaichana KL, Parker SL, Olivi A, Quiñones-Hinojosa A. Long-term seizure outcomes in adult patients undergoing primary resection of malignant brain astrocytomas. Journal of neurosurgery. 2009;111(2):282-92
49. Alexander BM, Cloughesy TF. Adult glioblastoma. Journal of Clinical Oncology. 2017;35(21):2402-9
50. Chieffo DPR, Lino F, Ferrarese D, Belella D, Della Pepa GM, Doglietto F. Brain tumor at diagnosis: from cognition and behavior to quality of life. Diagnostics. 2023;13(3):541
51. Grasso CS, Tang Y, Truffaux N, Berlow NE, Liu L, Debily M-A. et al. Functionally defined therapeutic targets in diffuse intrinsic pontine glioma. Nature medicine. 2015;21(6):555-9
52. Weller M, van den Bent M, Preusser M, Le Rhun E, Tonn JC, Minniti G. et al. EANO guidelines on the diagnosis and treatment of diffuse gliomas of adulthood. Nature reviews Clinical oncology. 2021;18(3):170-86
53. Papademetriou IT, Porter T. Promising approaches to circumvent the blood-brain barrier: progress, pitfalls and clinical prospects in brain cancer. Therapeutic delivery. 2015;6(8):989-1016
54. Zhao S, Wu J, Wang C, Liu H, Dong X, Shi C. et al. Intraoperative fluorescence-guided resection of high-grade malignant gliomas using 5-aminolevulinic acid-induced porphyrins: a systematic review and meta-analysis of prospective studies. PLoS One. 2013;8(5):e63682
55. Li Z, Du L, Du B, Ullah Z, Zhang Y, Tu Y. et al. Inorganic and hybrid nanomaterials for NIR-II fluorescence imaging-guided therapy of Glioblastoma and perspectives. Theranostics. 2025;15(12):5616-65
56. Roy S, Bag N, Bardhan S, Hasan I, Guo B. Recent progress in NIR-II fluorescence imaging-guided drug delivery for cancer theranostics. Adv Drug Deliv Rev. 2023;197:114821
57. Chen P, Liu Y, Huang H, Li M, Xie H, Roy S. et al. Genetically Engineered IL12/CSF1R-Macrophage Membrane-Liposome Hybrid Nanovesicles for NIR-II Fluorescence Imaging-Guided and Membrane-Targeted Mild Photothermal-Immunotherapy of Glioblastoma. Advanced Science. 2025:2500131.
58. Huang H, Li M, Gu J, Roy S, Jin J, Kuang T. et al. Bright NIR-II emissive cyanine dye-loaded lipoprotein-mimicking nanoparticles for fluorescence imaging-guided and targeted NIR-II photothermal therapy of subcutaneous glioblastoma. Journal of Nanobiotechnology. 2024;22(1):788
59. Dubey A, Kataria R, Sinha VD. Role of Diffusion Tensor Imaging in Brain Tumor Surgery. Asian J Neurosurg. 2018;13(2):302-6
60. Kircher MF, de la Zerda A, Jokerst JV, Zavaleta CL, Kempen PJ, Mittra E. et al. A brain tumor molecular imaging strategy using a new triple-modality MRI-photoacoustic-Raman nanoparticle. Nat Med. 2012;18(5):829-34
61. Chiu WT, Shen SC, Chow JM, Lin CW, Shia LT, Chen YC. Contribution of reactive oxygen species to migration/invasion of human glioblastoma cells U87 via ERK-dependent COX-2/PGE(2) activation. Neurobiol Dis. 2010;37(1):118-29
62. Burnet NG, Jena R, Burton KE, Tudor GS, Scaife JE, Harris F. et al. Clinical and practical considerations for the use of intensity-modulated radiotherapy and image guidance in neuro-oncology. Clin Oncol (R Coll Radiol). 2014;26(7):395-406
63. Alphandéry E. Glioblastoma Treatments: An Account of Recent Industrial Developments. Front Pharmacol. 2018;9:879
64. Wu Y, Song Y, Wang R, Wang T. Molecular mechanisms of tumor resistance to radiotherapy. Mol Cancer. 2023;22(1):96
65. Anjum K, Shagufta BI, Abbas SQ, Patel S, Khan I, Shah SAA. et al. Current status and future therapeutic perspectives of glioblastoma multiforme (GBM) therapy: A review. Biomedicine & Pharmacotherapy. 2017;92:681-9
66. Ortiz R, Perazzoli G, Cabeza L, Jiménez-Luna C, Luque R, Prados J. et al. Temozolomide: An Updated Overview of Resistance Mechanisms, Nanotechnology Advances and Clinical Applications. Curr Neuropharmacol. 2021;19(4):513-37
67. Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ. et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352(10):987-96
68. Fan CH, Liu WL, Cao H, Wen C, Chen L, Jiang G. O6-methylguanine DNA methyltransferase as a promising target for the treatment of temozolomide-resistant gliomas. Cell Death Dis. 2013;4(10):e876
69. Rivera AL, Pelloski CE, Gilbert MR, Colman H, De La Cruz C, Sulman EP. et al. MGMT promoter methylation is predictive of response to radiotherapy and prognostic in the absence of adjuvant alkylating chemotherapy for glioblastoma. Neuro Oncol. 2010;12(2):116-21
70. Middleton MR, Kelly J, Thatcher N, Donnelly DJ, McElhinney RS, McMurry TB. et al. O(6)-(4-bromothenyl)guanine improves the therapeutic index of temozolomide against A375M melanoma xenografts. Int J Cancer. 2000;85(2):248-52
71. M A, Xavier J, A SF, Bisht P, Murti K, Ravichandiran V. et al. Epigenetic basis for PARP mutagenesis in glioblastoma: A review. Eur J Pharmacol. 2023;938:175424
72. Tamura R, Tanaka T, Morimoto Y, Kuranari Y, Yamamoto Y, Takei J. et al. Alterations of the tumor microenvironment in glioblastoma following radiation and temozolomide with or without bevacizumab. Ann Transl Med. 2020;8(6):297
73. Casanovas O, Hicklin DJ, Bergers G, Hanahan D. Drug resistance by evasion of antiangiogenic targeting of VEGF signaling in late-stage pancreatic islet tumors. Cancer Cell. 2005;8(4):299-309
74. Gerstner ER, Batchelor TT. Antiangiogenic therapy for glioblastoma. Cancer J. 2012;18(1):45-50
75. Bigner D, Bigner S, Ponten J, Westermark B, Mahaley Jr M, Ruoslahti E. et al. Heterogeneity of genotypic and phenotypic characteristics of fifteen permanent cell lines derived from human gliomas. Journal of Neuropathology & Experimental Neurology. 1981;40(3):201-29
76. Kapałczyńska M, Kolenda T, Przybyła W, Zajączkowska M, Teresiak A, Filas V. et al. 2D and 3D cell cultures-a comparison of different types of cancer cell cultures. Archives of medical science. 2018;14(4):910-9
77. Allen M, Bjerke M, Edlund H, Nelander S, Westermark B. Origin of the U87MG glioma cell line: Good news and bad news. Sci Transl Med. 2016;8(354):354re3
78. Li A, Walling J, Kotliarov Y, Center A, Steed ME, Ahn SJ. et al. Genomic changes and gene expression profiles reveal that established glioma cell lines are poorly representative of primary human gliomas. Molecular Cancer Research. 2008;6(1):21-30
79. Roy K, Wang L, Makrigiorgos GM, Price BD. Methylation of the ATM promoter in glioma cells alters ionizing radiation sensitivity. Biochemical and biophysical research communications. 2006;344(3):821-6
80. Serafim RB, Da Silva P, Cardoso C, Di Cristofaro LFM, Netto RP, De Almeida R. et al. Expression profiling of glioblastoma cell lines reveals novel extracellular matrix-receptor genes correlated with the responsiveness of glioma patients to ionizing radiation. Frontiers in oncology. 2021;11:668090
81. Park C-M, Park M-J, Kwak H-J, Moon S-I, Yoo D-H, Lee H-C. et al. Induction of p53-mediated apoptosis and recovery of chemosensitivity through p53 transduction in human glioblastoma cells by cisplatin. International journal of oncology. 2006;28(1):119-25
82. Kusaczuk M, Krętowski R, Bartoszewicz M, Cechowska-Pasko M. Phenylbutyrate—A pan-HDAC inhibitor—Suppresses proliferation of glioblastoma LN-229 cell line. Tumor Biology. 2016;37:931-42
83. Ling FY, Reznik E, Korn JM, Lin F, Yang G, Malesky K. et al. Patient-derived glioblastoma cultures as a tool for small-molecule drug discovery. Oncotarget. 2020;11(4):443
84. Wang R, Chadalavada K, Wilshire J, Kowalik U, Hovinga KE, Geber A. et al. Glioblastoma stem-like cells give rise to tumour endothelium. Nature. 2010;468(7325):829-33
85. Xiao Y, Kim D, Dura B, Zhang K, Yan R, Li H. et al. Ex vivo Dynamics of Human Glioblastoma Cells in a Microvasculature-on-a-Chip System Correlates with Tumor Heterogeneity and Subtypes. Advanced Science. 2019;6(8):1801531
86. Fan Y, Nguyen DT, Akay Y, Xu F, Akay M. Engineering a brain cancer chip for high-throughput drug screening. Scientific reports. 2016;6(1):25062
87. Liu X, Zhang W, Zheng W, Jiang X. Micropatterned Coculture Platform for Screening Nerve-Related Anticancer Drugs. ACS Nano. 2021;15(1):637-49
88. Jie M, Mao S, Liu H, He Z, Li H-F, Lin J-M. Evaluation of drug combination for glioblastoma based on an intestine-liver metabolic model on microchip. Analyst. 2017;142(19):3629-38
89. Straehla JP, Hajal C, Safford HC, Offeddu GS, Boehnke N, Dacoba TG. et al. A predictive microfluidic model of human glioblastoma to assess trafficking of blood-brain barrier-penetrant nanoparticles. Proc Natl Acad Sci U S A. 2022;119(23):e2118697119
90. Olubajo F, Achawal S, Greenman J. Development of a Microfluidic Culture Paradigm for Ex vivo Maintenance of Human Glioblastoma Tissue: A New Glioblastoma Model? Translational Oncology. 2020;13(1):1-10
91. Cui X, Ma C, Vasudevaraja V, Serrano J, Tong J, Peng Y. et al. Dissecting the immunosuppressive tumor microenvironments in Glioblastoma-on-a-Chip for optimized PD-1 immunotherapy. Elife. 2020;9:e52253
92. Gerigk M, Bulstrode H, Shi HH, Tönisen F, Cerutti C, Morrison G. et al. On-chip perivascular niche supporting stemness of patient-derived glioma cells in a serum-free, flowable culture. Lab on a Chip. 2021;21(12):2343-58
93. Dai X, Ma C, Lan Q, Xu T. 3D bioprinted glioma stem cells for brain tumor model and applications of drug susceptibility. Biofabrication. 2016;8(4):045005
94. Wang X, Dai X, Zhang X, Ma C, Li X, Xu T. et al. 3D bioprinted glioma cell-laden scaffolds enriching glioma stem cells via epithelial-mesenchymal transition. Journal of biomedical materials research Part A. 2019;107(2):383-91
95. Wang X, Li X, Zhang Y, Long X, Zhang H, Xu T. et al. Coaxially Bioprinted Cell-Laden Tubular-Like Structure for Studying Glioma Angiogenesis. Front Bioeng Biotechnol. 2021;9:761861
96. Wang X, Li X, Dai X, Zhang X, Zhang J, Xu T. et al. Coaxial extrusion bioprinted shell-core hydrogel microfibers mimic glioma microenvironment and enhance the drug resistance of cancer cells. Colloids Surf B Biointerfaces. 2018;171:291-9
97. Dai X, Liu L, Ouyang J, Li X, Zhang X, Lan Q. et al. Coaxial 3D bioprinting of self-assembled multicellular heterogeneous tumor fibers. Scientific reports. 2017;7(1):1457
98. Tang M, Xie Q, Gimple RC, Zhong Z, Tam T, Tian J. et al. Three-dimensional bioprinted glioblastoma microenvironments model cellular dependencies and immune interactions. Cell research. 2020;30(10):833-53
99. Zielniok K, Rusinek K, Słysz A, Lachota M, Bączyńska E, Wiewiórska-Krata N. et al. 3D-Bioprinted Co-Cultures of Glioblastoma Multiforme and Mesenchymal Stromal Cells Indicate a Role for Perivascular Niche Cells in Shaping Glioma Chemokine Microenvironment. Cells. 2024 13(17)
100. Ozturk MS, Lee VK, Zou H, Friedel RH, Intes X, Dai G. High-resolution tomographic analysis of in vitro 3D glioblastoma tumor model under long-term drug treatment. Science advances. 2020;6(10):eaay7513
101. Neufeld L, Yeini E, Reisman N, Shtilerman Y, Ben-Shushan D, Pozzi S. et al. Microengineered perfusable 3D-bioprinted glioblastoma model for in vivo mimicry of tumor microenvironment. Science advances. 2021;7(34):eabi9119
102. Silvani G, Basirun C, Wu H, Mehner C, Poole K, Bradbury P. et al. A 3D-bioprinted vascularized glioblastoma-on-a-chip for studying the impact of simulated microgravity as a novel pre-clinical approach in brain tumor therapy. Advanced therapeutics. 2021;4(11):2100106
103. Yi H-G, Jeong YH, Kim Y, Choi Y-J, Moon HE, Park SH. et al. A bioprinted human-glioblastoma-on-a-chip for the identification of patient-specific responses to chemoradiotherapy. Nature biomedical engineering. 2019;3(7):509-19
104. Munson JM, Bellamkonda RV, Swartz MA. Interstitial flow in a 3D microenvironment increases glioma invasion by a CXCR4-dependent mechanism. Cancer research. 2013;73(5):1536-46
105. Buchanan CF, Verbridge SS, Vlachos PP, Rylander MN. Flow shear stress regulates endothelial barrier function and expression of angiogenic factors in a 3D microfluidic tumor vascular model. Cell adhesion & migration. 2014;8(5):517-24
106. Alves AH, Nucci MP, Mamani JB, Valle NM, Ribeiro EF, Rego GN. et al. The advances in glioblastoma on-a-chip for therapy approaches. Cancers. 2022;14(4):869
107. Parra-Cantu C, Li W, Quiñones-Hinojosa A, Zhang YS. 3D Bioprinting of Glioblastoma Model. Journal of 3D printing in medicine. 2020;4(2):113-25
108. Tang M, Tiwari SK, Agrawal K, Tan M, Dang J, Tam T. et al. Rapid 3D bioprinting of glioblastoma model mimicking native biophysical heterogeneity. Small. 2021;17(15):2006050
109. Li S, Liu X, Zhang L, Wang Q. Integrated applications of microfluidics, organoids, and 3D bioprinting in in vitro 3D biomimetic models. International Journal of Bioprinting. 2025;11(3):115-53
110. Bayona C, Alza L, Ranđelović T, Sallán MC, Visa A, Cantí C. et al. Tetralol derivative NNC-55-0396 targets hypoxic cells in the glioblastoma microenvironment: an organ-on-chip approach. Cell Death & Disease. 2024;15(2):127
111. Ayuso JM, Virumbrales-Muñoz M, Lacueva A, Lanuza PM, Checa-Chavarria E, Botella P. et al. Development and characterization of a microfluidic model of the tumour microenvironment. Scientific reports. 2016;6(1):36086
112. Lee H, Park W, Ryu H, Jeon NL. A microfluidic platform for quantitative analysis of cancer angiogenesis and intravasation. Biomicrofluidics. 2014 8(5)
113. Pombo Antunes AR, Scheyltjens I, Duerinck J, Neyns B, Movahedi K, Van Ginderachter JA. Understanding the glioblastoma immune microenvironment as basis for the development of new immunotherapeutic strategies. Elife. 2020;9:e52176
114. Uyar R. Glioblastoma microenvironment: The stromal interactions. Pathology-Research and Practice. 2022;232:153813
115. Dou J, Mao S, Li H, Lin J-M. Combination Stiffness Gradient with Chemical Stimulation Directs Glioma Cell Migration on a Microfluidic Chip. Analytical Chemistry. 2020;92(1):892-8
116. Charles N, Holland EC. The perivascular niche microenvironment in brain tumor progression. Cell Cycle. 2010Aug1;9(15):3012-3021
117. Chung M, Ahn J, Son K, Kim S, Jeon NL. Biomimetic model of tumor microenvironment on microfluidic platform. Advanced healthcare materials. 2017;6(15):1700196
118. Truong D, Fiorelli R, Barrientos ES, Melendez EL, Sanai N, Mehta S. et al. A three-dimensional (3D) organotypic microfluidic model for glioma stem cells-Vascular interactions. Biomaterials. 2019;198:63-77
119. Ayuso JM, Monge R, Martínez-González A, Virumbrales-Muñoz M, Llamazares GA, Berganzo J. et al. Glioblastoma on a microfluidic chip: Generating pseudopalisades and enhancing aggressiveness through blood vessel obstruction events. Neuro-oncology. 2017;19(4):503-13
120. Ahn J, Sei YJ, Jeon NL, Kim Y. Tumor Microenvironment on a Chip: The Progress and Future Perspective. Bioengineering. 2017;4(3):64
121. Ahn J, Kim D-H, Koo D-J, Lim J, Park T-E, Lee J. et al. 3D microengineered vascularized tumor spheroids for drug delivery and efficacy testing. Acta Biomaterialia. 2023;165:153-67
122. Ryoo M, Lee G, Jung J, Cho S, Ham SJ, Kang N. et al. Human blood-brain tumor barrier on a chip to investigate personalized treatment for glioblastoma patients. medRxiv. 2025:2025.12. 01.25341181.
123. Wang X, Li X, Ding J, Long X, Zhang H, Zhang X. et al. 3D bioprinted glioma microenvironment for glioma vascularization. Journal of Biomedical Materials Research Part A. 2021;109(6):915-25
124. Hermida MA, Kumar JD, Schwarz D, Laverty KG, Di Bartolo A, Ardron M. et al. Three dimensional in vitro models of cancer: Bioprinting multilineage glioblastoma models. Advances in biological regulation. 2020;75:100658
125. Heinrich MA, Bansal R, Lammers T, Zhang YS, Michel Schiffelers R, Prakash J. 3D-bioprinted mini-brain: a glioblastoma model to study cellular interactions and therapeutics. Advanced materials. 2019;31(14):1806590
126. Pun S, Prakash A, Demaree D, Krummel DP, Sciumè G, Sengupta S. et al. Rapid Biofabrication of an Advanced Microphysiological System Mimicking Phenotypical Heterogeneity and Drug Resistance in Glioblastoma. Advanced Healthcare Materials. 2024:2401876.
127. Chen JE, Pedron S, Harley BAC. The Combined Influence of Hydrogel Stiffness and Matrix-Bound Hyaluronic Acid Content on Glioblastoma Invasion. Macromol Biosci. 2017 17(8)
128. Ozbek II, Saybasili H, Ulgen KO. Applications of 3D Bioprinting Technology to Brain Cells and Brain Tumor Models: Special Emphasis to Glioblastoma. ACS Biomaterials Science & Engineering. 2024;10(5):2616-35
129. Murphy SV, Atala A. 3D bioprinting of tissues and organs. Nature biotechnology. 2014;32(8):773-85
130. Hölzl K, Lin S, Tytgat L, Van Vlierberghe S, Gu L, Ovsianikov A. Bioink properties before, during and after 3D bioprinting. Biofabrication. 2016;8(3):032002
131. Ma X, Liu J, Zhu W, Tang M, Lawrence N, Yu C. et al. 3D bioprinting of functional tissue models for personalized drug screening and in vitro disease modeling. Advanced drug delivery reviews. 2018;132:235-51
132. Pereira RF, Bártolo PJ. 3D bioprinting of photocrosslinkable hydrogel constructs. Journal of Applied Polymer Science. 2015 132(48)
133. Shukla P, Yeleswarapu S, Heinrich MA, Prakash J, Pati F. Mimicking tumor microenvironment by 3D bioprinting: 3D cancer modeling. Biofabrication. 2022;14(3):032002
134. Li H, Dai J, Wang Z, Zheng H, Li W, Wang M. et al. Digital light processing (DLP)-based (bio) printing strategies for tissue modeling and regeneration. Aggregate. 2023;4(2):e270
135. Şahin M, Öncü G, Yılmaz MA, Özkan D, Saybaşılı H. Transformation of SH-SY5Y cell line into neuron-like cells: Investigation of electrophysiological and biomechanical changes. Neuroscience Letters. 2021;745:135628
136. Bhattacharya S, Calar K, de la Puente P. Mimicking tumor hypoxia and tumor-immune interactions employing three-dimensional in vitro models. Journal of Experimental & Clinical Cancer Research. 2020;39(1):75
137. Wang P, Zhu W, Ma X, Tang M, You S, Lakshmipathy D. et al. A sequential 3D bioprinting and orthogonal bioconjugation approach for precision tissue engineering. 2020.
138. Lee G, Kim SJ, Park J-K. Fabrication of a self-assembled and vascularized tumor array via bioprinting on a microfluidic chip. Lab on a Chip. 2023;23(18):4079-91
139. World Medical Association Declaration of Helsinki. ethical principles for medical research involving human subjects. Jama. 2013;310(20):2191-4
140. Chosewood LC, Wilson DE. Biosafety in microbiological and biomedical laboratories: US Department of Health and Human Services, Public Health Service, Centers …; 2009
141. Yigci D, Sarabi MR, Ustun M, Atceken N, Sokullu E, Bagci-Onder T. et al. 3D bioprinted glioma models. Progress in Biomedical Engineering. 2022;4(4):042001
142. Chen JWE, Pedron S, Harley BA. The combined influence of hydrogel stiffness and matrix-bound hyaluronic acid content on glioblastoma invasion. Macromolecular bioscience. 2017;17(8):1700018
143. Elkin BS, Azeloglu EU, Costa KD, Morrison Iii B. Mechanical heterogeneity of the rat hippocampus measured by atomic force microscope indentation. Journal of neurotrauma. 2007;24(5):812-22
144. Wilson WR, Hay MP. Targeting hypoxia in cancer therapy. Nature Reviews Cancer. 2011;11(6):393-410
145. Yoon H, Kang JH, Cho SW, Park CG, Kim DW, Park TE. Brain-Decellularized ECM-Based 3D Myeloid Sarcoma Platform: Mimicking Adaptive Phenotypic Alterations in the Brain. Advanced Healthcare Materials. 2024 13(13)
146. Reginensi D, Ortiz DA, Denis B, Castillo S, Burillo A, Khoury N. et al. Region-specific brain decellularized extracellular matrix promotes cell recovery in an in vitro model of stroke. Scientific Reports. 2025 15(1)
147. Kim B-S, Kim J-U, Lee J, Ryu KM, Kim S-H, Hwang NS. Decellularized brain extracellular matrix based NGF-releasing cryogel for brain tissue engineering in traumatic brain injury. Journal of Controlled Release. 2024;368:140-56
148. Shanti A, Samara B, Abdullah A, Hallfors N, Accoto D, Sapudom J. et al. Multi-compartment 3D-cultured organ-on-a-chip: towards a biomimetic lymph node for drug development. Pharmaceutics. 2020;12(5):464
149. Birmingham KG, O'Melia MJ, Bordy S, Aguilar DR, El-Reyas B, Lesinski G. et al. Lymph node subcapsular sinus microenvironment-on-a-chip modeling shear flow relevant to lymphatic metastasis and immune cell homing. Iscience. 2020 23(11)
150. Li Z, Li D, Guo Y, Wang Y, Su W. Evaluation of hepatic drug-metabolism for glioblastoma using liver-brain chip. Biotechnology Letters. 2021;43:383-92
151. Buettner M, Bode U. Lymph node dissection-understanding the immunological function of lymph nodes. Clinical & Experimental Immunology. 2012;169(3):205-12
152. Willard-Mack CL. Normal structure, function, and histology of lymph nodes. Toxicologic pathology. 2006;34(5):409-24
153. Chaudhry SR, Luskin V, Panuganti KK. Anatomy, abdomen and pelvis, spleen. StatPearls [Internet]: StatPearls Publishing. 2023
154. Rigat-Brugarolas L, Elizalde-Torrent A, Bernabeu M, De Niz M, Martin-Jaular L, Fernandez-Becerra C. et al. A functional microengineered model of the human splenon-on-a-chip. Lab on a Chip. 2014;14(10):1715-24
155. Zhang F, Xu C-L, Liu C-M. Drug delivery strategies to enhance the permeability of the blood-brain barrier for treatment of glioma. Drug Design, Development and Therapy. 2015:2089-100
156. Zuchowska A, Skorupska S. Multi-organ-on-chip approach in cancer research. Organs-on-a-Chip. 2022;4:100014
157. Samiei E, Seyfoori A, Toyota B, Ghavami S, Akbari M. Investigating programmed cell death and tumor invasion in a three-dimensional (3D) microfluidic model of glioblastoma. International journal of molecular sciences. 2020;21(9):3162
158. Ma J, Li N, Wang Y, Wang L, Wei W, Shen L. et al. Engineered 3D tumour model for study of glioblastoma aggressiveness and drug evaluation on a detachably assembled microfluidic device. Biomedical Microdevices. 2018;20:1-16
159. Akay M, Hite J, Avci NG, Fan Y, Akay Y, Lu G. et al. Drug screening of human GBM spheroids in brain cancer chip. Scientific reports. 2018;8(1):15423
160. Ko J, Ahn J, Kim S, Lee Y, Lee J, Park D. et al. Tumor spheroid-on-a-chip: a standardized microfluidic culture platform for investigating tumor angiogenesis. Lab on a Chip. 2019;19(17):2822-33
161. Newlands ES, Blackledge GR, Slack JA, Rustin GJ, Smith DB, Stuart NS. et al. Phase I trial of temozolomide (CCRG 81045: M&B 39831: NSC 362856). Br J Cancer. 1992;65(2):287-91
162. Esteller M, Garcia-Foncillas J, Andion E, Goodman SN, Hidalgo OF, Vanaclocha V. et al. Inactivation of the DNA-repair gene MGMT and the clinical response of gliomas to alkylating agents. N Engl J Med. 2000;343(19):1350-4
163. Zhang Q, Mao S, Li W, Huang Q, Feng S, Hong Z. et al. Microfluidic adhesion analysis of single glioma cells for evaluating the effect of drugs. Science China Chemistry. 2020;63:865-70
164. Li X, Halldórsdóttir HR, Weller S, Colliander A, Bak M, Kempen P. et al. Enhancing Adoptive Cell Therapy by T Cell Loading of SHP2 Inhibitor Nanocrystals before Infusion. ACS Nano. 2022;16(7):10918-30
165. Liu W, Xu J, Li T, Zhao L, Ma C, Shen S. et al. Monitoring tumor response to anticancer drugs using stable three-dimensional culture in a recyclable microfluidic platform. Anal Chem. 2015;87(19):9752-60
166. Rocha Pinheiro SL, Lemos FFB, Marques HS, Silva Luz M, de Oliveira Silva LG, Faria Souza Mendes Dos Santos C. et al. Immunotherapy in glioblastoma treatment: Current state and future prospects. World J Clin Oncol. 2023;14(4):138-59
167. Rui Y, Green JJ. Overcoming delivery barriers in immunotherapy for glioblastoma. Drug Delivery and Translational Research. 2021;11(6):2302-16
168. Cui X, Morales RT, Qian W, Wang H, Gagner JP, Dolgalev I. et al. Hacking macrophage-associated immunosuppression for regulating glioblastoma angiogenesis. Biomaterials. 2018;161:164-78
169. Huang J, Li YB, Charlebois C, Nguyen T, Liu Z, Bloemberg D. et al. Application of blood brain barrier models in pre-clinical assessment of glioblastoma-targeting CAR-T based immunotherapies. Fluids Barriers CNS. 2022;19(1):38
170. Ijaz M, Tan Q, Yan Y, Zhang D, Chen Q, Zhang Y. et al. Overcoming barriers in glioblastoma: The potential of CAR T cell immunotherapy. Theranostics. 2025;15(14):7090
171. Maulana TI, Kromidas E, Wallstabe L, Cipriano M, Alb M, Zaupa C. et al. Immunocompetent cancer-on-chip models to assess immuno-oncology therapy. Advanced drug delivery reviews. 2021;173:281-305
172. Zhang C, Jin M, Zhao J, Chen J, Jin W. Organoid models of glioblastoma: advances, applications and challenges. American Journal of Cancer Research. 2020;10(8):2242
173. Finkel T. Signal transduction by reactive oxygen species. Journal of Cell Biology. 2011;194(1):7-15
174. Kim S, Ohulchanskyy TY, Bharali D, Chen Y, Pandey RK, Prasad PN. Organically Modified Silica Nanoparticles with Intraparticle Heavy-Atom Effect on the Encapsulated Photosensitizer for Enhanced Efficacy of Photodynamic Therapy. J Phys Chem C Nanomater Interfaces. 2009;113:12641-4
175. Li X, Lovell JF, Yoon J, Chen X. Clinical development and potential of photothermal and photodynamic therapies for cancer. Nat Rev Clin Oncol. 2020;17(11):657-74
176. Melamed JR, Edelstein RS, Day ES. Elucidating the fundamental mechanisms of cell death triggered by photothermal therapy. ACS Nano. 2015;9(1):6-11
177. Yoon HK, Lou X, Chen Y-C, Koo Lee Y-E, Yoon E, Kopelman R. Nanophotosensitizers engineered to generate a tunable mix of reactive oxygen species, for optimizing photodynamic therapy, using a microfluidic device. Chemistry of Materials. 2014;26(4):1592-600
178. Lee JM, Seo HI, Bae JH, Chung BG. Hydrogel microfluidic co-culture device for photothermal therapy and cancer migration. Electrophoresis. 2017;38(9-10):1318-24
179. Muhammad S, Maridevaru MC, Roy S, Chen D, Zeng W, Sun L. et al. Piezodynamic Therapy: Unleashing Mechanical Energy and Featuristic Next Generation Therapeutic Paradigms for Glioblastoma. ACS Nano. 2025;19(37):33008-58
180. Arrondeau J, Gan HK, Razak AR, Paoletti X, Le Tourneau C. Development of anti-cancer drugs. Discov Med. 2010;10(53):355-62
181. Slika H, Karimov Z, Alimonti P, Abou-Mrad T, De Fazio E, Alomari S. et al. Preclinical Models and Technologies in Glioblastoma Research: Evolution, Current State, and Future Avenues. Int J Mol Sci. 2023 24(22)
182. Sahu U, Barth RF, Otani Y, McCormack R, Kaur B. Rat and Mouse Brain Tumor Models for Experimental Neuro-Oncology Research. J Neuropathol Exp Neurol. 2022;81(5):312-29
183. Simeonova I, Huillard E. In vivo models of brain tumors: roles of genetically engineered mouse models in understanding tumor biology and use in preclinical studies. Cell Mol Life Sci. 2014;71(20):4007-26
184. Gómez-Oliva R, Domínguez-García S, Carrascal L, Abalos-Martínez J, Pardillo-Díaz R, Verástegui C. et al. Evolution of Experimental Models in the Study of Glioblastoma: Toward Finding Efficient Treatments. Frontiers in Oncology. 2021 10
185. Olson B, Li Y, Lin Y, Liu ET, Patnaik A. Mouse Models for Cancer Immunotherapy Research. Cancer Discov. 2018;8(11):1358-65
186. Newcomb EW, Zagzag D. The Murine GL261 Glioma Experimental Model to Assess Novel Brain Tumor Treatments. In: Meir EG, editor. CNS Cancer: Models, Markers, Prognostic Factors, Targets, and Therapeutic Approaches. Totowa, NJ: Humana Press. 2009 p. 227-41
187. Szatmári T, Lumniczky K, Désaknai S, Trajcevski S, Hídvégi EJ, Hamada H. et al. Detailed characterization of the mouse glioma 261 tumor model for experimental glioblastoma therapy. Cancer Sci. 2006;97(6):546-53
188. Azzalin A, Nato G, Parmigiani E, Garello F, Buffo A, Magrassi L. Inhibitors of GLUT/SLC2A Enhance the Action of BCNU and Temozolomide against High-Grade Gliomas. Neoplasia. 2017;19(4):364-73
189. Wu A, Oh S, Wiesner SM, Ericson K, Chen L, Hall WA. et al. Persistence of CD133+ cells in human and mouse glioma cell lines: detailed characterization of GL261 glioma cells with cancer stem cell-like properties. Stem Cells Dev. 2008;17(1):173-84
190. Lan CW, Chen HH, Sheu JJ. Deoxyelephantopin induces apoptosis and cell cycle arrest in GL261 glioblastoma cells. Naunyn Schmiedebergs Arch Pharmacol. 2024
191. Towner RA, Ihnat M, Saunders D, Bastian A, Smith N, Pavana RK. et al. A new anti-glioma therapy, AG119: pre-clinical assessment in a mouse GL261 glioma model. BMC Cancer. 2015;15:522
192. Enríquez Pérez J, Kopecky J, Visse E, Darabi A, Siesjö P. Convection-enhanced delivery of temozolomide and whole cell tumor immunizations in GL261 and KR158 experimental mouse gliomas. BMC Cancer. 2020;20(1):7
193. Wu S, Calero-Pérez P, Arús C, Candiota AP. Anti-PD-1 Immunotherapy in Preclinical GL261 Glioblastoma: Influence of Therapeutic Parameters and Non-Invasive Response Biomarker Assessment with MRSI-Based Approaches. Int J Mol Sci. 2020 21(22)
194. Dai B, Qi N, Li J, Zhang G. Temozolomide combined with PD-1 Antibody therapy for mouse orthotopic glioma model. Biochem Biophys Res Commun. 2018;501(4):871-6
195. Calero-Pérez P, Wu S, Arús C, Candiota AP. Immune System-Related Changes in Preclinical GL261 Glioblastoma under TMZ Treatment: Explaining MRSI-Based Nosological Imaging Findings with RT-PCR Analyses. Cancers (Basel). 2021 13(11)
196. Ueda R, Fujita M, Zhu X, Sasaki K, Kastenhuber ER, Kohanbash G. et al. Systemic inhibition of transforming growth factor-beta in glioma-bearing mice improves the therapeutic efficacy of glioma-associated antigen peptide vaccines. Clin Cancer Res. 2009;15(21):6551-9
197. Renner DN, Jin F, Litterman AJ, Balgeman AJ, Hanson LM, Gamez JD. et al. Effective Treatment of Established GL261 Murine Gliomas through Picornavirus Vaccination-Enhanced Tumor Antigen-Specific CD8+ T Cell Responses. PLoS One. 2015;10(5):e0125565
198. Kindy MS, Yu J, Zhu H, Smith MT, Gattoni-Celli S. A therapeutic cancer vaccine against GL261 murine glioma. J Transl Med. 2016;14:1
199. Ciesielski MJ, Apfel L, Barone TA, Castro CA, Weiss TC, Fenstermaker RA. Antitumor effects of a xenogeneic survivin bone marrow derived dendritic cell vaccine against murine GL261 gliomas. Cancer Immunol Immunother. 2006;55(12):1491-503
200. Seyfried TN, el-Abbadi M, Roy ML. Ganglioside distribution in murine neural tumors. Mol Chem Neuropathol. 1992;17(2):147-67
201. Binello E, Qadeer ZA, Kothari HP, Emdad L, Germano IM. Stemness of the CT-2A Immunocompetent Mouse Brain Tumor Model: Characterization In vitro. J Cancer. 2012;3:166-74
202. Liu CJ, Schaettler M, Blaha DT, Bowman-Kirigin JA, Kobayashi DK, Livingstone AJ. et al. Treatment of an aggressive orthotopic murine glioblastoma model with combination checkpoint blockade and a multivalent neoantigen vaccine. Neuro Oncol. 2020;22(9):1276-88
203. Nakashima H, Alayo QA, Penaloza-MacMaster P, Freeman GJ, Kuchroo VK, Reardon DA. et al. Modeling tumor immunity of mouse glioblastoma by exhausted CD8(+) T cells. Sci Rep. 2018;8(1):208
204. Riva M, Wouters R, Weerasekera A, Belderbos S, Nittner D, Thal DR. et al. CT-2A neurospheres-derived high-grade glioma in mice: a new model to address tumor stem cells and immunosuppression. Biol Open. 2019 8(9)
205. Chandramohan V, Bao X, Yu X, Parker S, McDowall C, Yu YR. et al. Improved efficacy against malignant brain tumors with EGFRwt/EGFRvIII targeting immunotoxin and checkpoint inhibitor combinations. J Immunother Cancer. 2019;7(1):142
206. Morgan RA, Johnson LA, Davis JL, Zheng Z, Woolard KD, Reap EA. et al. Recognition of glioma stem cells by genetically modified T cells targeting EGFRvIII and development of adoptive cell therapy for glioma. Hum Gene Ther. 2012;23(10):1043-53
207. Meister H, Look T, Roth P, Pascolo S, Sahin U, Lee S. et al. Multifunctional mRNA-Based CAR T Cells Display Promising Antitumor Activity Against Glioblastoma. Clin Cancer Res. 2022;28(21):4747-56
208. Sampson JH, Ashley DM, Archer GE, Fuchs HE, Dranoff G, Hale LP. et al. Characterization of a spontaneous murine astrocytoma and abrogation of its tumorigenicity by cytokine secretion. Neurosurgery. 1997;41(6):1365-72 discussion 72-3
209. Learn CA, Grossi PM, Schmittling RJ, Xie W, Mitchell DA, Karikari I. et al. Genetic analysis of intracranial tumors in a murine model of glioma demonstrate a shift in gene expression in response to host immunity. J Neuroimmunol. 2007;182(1-2):63-72
210. Weiss T, Schneider H, Silginer M, Steinle A, Pruschy M, Polić B. et al. NKG2D-Dependent Antitumor Effects of Chemotherapy and Radiotherapy against Glioblastoma. Clin Cancer Res. 2018;24(4):882-95
211. Tran TT, Uhl M, Ma JY, Janssen L, Sriram V, Aulwurm S. et al. Inhibiting TGF-beta signaling restores immune surveillance in the SMA-560 glioma model. Neuro Oncol. 2007;9(3):259-70
212. Mangani D, Weller M, Seyed Sadr E, Willscher E, Seystahl K, Reifenberger G. et al. Limited role for transforming growth factor-β pathway activation-mediated escape from VEGF inhibition in murine glioma models. Neuro Oncol. 2016;18(12):1610-21
213. Papachristodoulou A, Silginer M, Weller M, Schneider H, Hasenbach K, Janicot M. et al. Therapeutic Targeting of TGFβ Ligands in Glioblastoma Using Novel Antisense Oligonucleotides Reduces the Growth of Experimental Gliomas. Clin Cancer Res. 2019;25(23):7189-201
214. Miller J, Eisele G, Tabatabai G, Aulwurm S, von Kürthy G, Stitz L. et al. Soluble CD70: a novel immunotherapeutic agent for experimental glioblastoma. J Neurosurg. 2010;113(2):280-5
215. Vom Berg J, Vrohlings M, Haller S, Haimovici A, Kulig P, Sledzinska A. et al. Intratumoral IL-12 combined with CTLA-4 blockade elicits T cell-mediated glioma rejection. J Exp Med. 2013;210(13):2803-11
216. Genoud V, Marinari E, Nikolaev SI, Castle JC, Bukur V, Dietrich PY. et al. Responsiveness to anti-PD-1 and anti-CTLA-4 immune checkpoint blockade in SB28 and GL261 mouse glioma models. Oncoimmunology. 2018;7(12):e1501137
217. Johanns TM, Ward JP, Miller CA, Wilson C, Kobayashi DK, Bender D. et al. Endogenous Neoantigen-Specific CD8 T Cells Identified in Two Glioblastoma Models Using a Cancer Immunogenomics Approach. Cancer Immunol Res. 2016;4(12):1007-15
218. Kosaka A, Ohkuri T, Okada H. Combination of an agonistic anti-CD40 monoclonal antibody and the COX-2 inhibitor celecoxib induces anti-glioma effects by promotion of type-1 immunity in myeloid cells and T-cells. Cancer Immunol Immunother. 2014;63(8):847-57
219. Borrero-Garcia L, Reiners O, Gonzàlez-Juncà A, Okada H, Barcellos-Hoff M. RDNA-09. RADIATION PRIMES SB28 GLIOBLASTOMA FOR RESPONSE TO TGFβ AND PD-L1 NEUTRALIZING ANTIBODIES. Neuro-Oncology. 2019;21:vi208-vi
220. Genoud V, Espinoza FI, Marinari E, Rochemont V, Dietrich PY, McSheehy P. et al. Treating ICB-resistant glioma with anti-CD40 and mitotic spindle checkpoint controller BAL101553 (lisavanbulin). JCI Insight. 2021 6(18)
221. Simonds EF, Lu ED, Badillo O, Karimi S, Liu EV, Tamaki W. et al. Deep immune profiling reveals targetable mechanisms of immune evasion in immune checkpoint inhibitor-refractory glioblastoma. J Immunother Cancer. 2021 9(6)
222. Ijaz M, Ullah Z, Aslam B, Khurshid M, Chen P, Guo B. From promise to progress: the dynamic landscape of glioblastoma immunotherapy. Drug Discovery Today. 2024;29(11):104188
223. Schuh JC. Trials, tribulations, and trends in tumor modeling in mice. Toxicol Pathol. 2004;32(Suppl 1):53-66
224. Gawrylewski A. The trouble with animal models. Scientist (Philadelphia, Pa). 2007;21:45-51
225. Haddad AF, Young JS, Amara D, Berger MS, Raleigh DR, Aghi MK. et al. Mouse models of glioblastoma for the evaluation of novel therapeutic strategies. Neuro-Oncology Advances. 2021 3(1)
226. Samstein RM, Lee CH, Shoushtari AN, Hellmann MD, Shen R, Janjigian YY. et al. Tumor mutational load predicts survival after immunotherapy across multiple cancer types. Nat Genet. 2019;51(2):202-6
227. Lenting K, Verhaak R, Ter Laan M, Wesseling P, Leenders W. Glioma: experimental models and reality. Acta Neuropathol. 2017;133(2):263-82
228. Shelton LM, Mukherjee P, Huysentruyt LC, Urits I, Rosenberg JA, Seyfried TN. A novel pre-clinical in vivo mouse model for malignant brain tumor growth and invasion. J Neurooncol. 2010;99(2):165-76
229. Candolfi M, Curtin JF, Nichols WS, Muhammad AG, King GD, Pluhar GE. et al. Intracranial glioblastoma models in preclinical neuro-oncology: neuropathological characterization and tumor progression. J Neurooncol. 2007;85(2):133-48
230. Yoshida GJ. Applications of patient-derived tumor xenograft models and tumor organoids. J Hematol Oncol. 2020;13(1):4
231. Patil V, Pal J, Somasundaram K. Elucidating the cancer-specific genetic alteration spectrum of glioblastoma derived cell lines from whole exome and RNA sequencing. Oncotarget. 2015;6(41):43452-71
232. Sesen J, Dahan P, Scotland SJ, Saland E, Dang VT, Lemarié A. et al. Metformin inhibits growth of human glioblastoma cells and enhances therapeutic response. PLoS One. 2015;10(4):e0123721
233. Qiang L, Yang Y, Ma YJ, Chen FH, Zhang LB, Liu W. et al. Isolation and characterization of cancer stem like cells in human glioblastoma cell lines. Cancer Lett. 2009;279(1):13-21
234. Jeong JC, Kim JW, Kwon CH, Kim TH, Kim YK. Fructus ligustri lucidi extracts induce human glioma cell death through regulation of Akt/mTOR pathway in vitro and reduce glioma tumor growth in U87MG xenograft mouse model. Phytother Res. 2011;25(3):429-34
235. Hong JF, Song YF, Liu Z, Zheng ZC, Chen HJ, Wang SS. Anticancer activity of taraxerol acetate in human glioblastoma cells and a mouse xenograft model via induction of autophagy and apoptotic cell death, cell cycle arrest and inhibition of cell migration. Mol Med Rep. 2016;13(6):4541-8
236. Park CR, Kim HY, Song MG, Lee YS, Youn H, Chung JK. et al. Efficacy and Safety of Human Serum Albumin-Cisplatin Complex in U87MG Xenograft Mouse Models. Int J Mol Sci. 2020 21(21)
237. Marrero L, Wyczechowska D, Musto AE, Wilk A, Vashistha H, Zapata A. et al. Therapeutic efficacy of aldoxorubicin in an intracranial xenograft mouse model of human glioblastoma. Neoplasia. 2014;16(10):874-82
238. Pechman KR, Donohoe DL, Bedekar DP, Kurpad SN, Hoffmann RG, Schmainda KM. Characterization of bevacizumab dose response relationship in U87 brain tumors using magnetic resonance imaging measures of enhancing tumor volume and relative cerebral blood volume. J Neurooncol. 2011;105(2):233-9
239. Jahangiri A, Nguyen A, Chandra A, Sidorov MK, Yagnik G, Rick J. et al. Cross-activating c-Met/β1 integrin complex drives metastasis and invasive resistance in cancer. Proc Natl Acad Sci U S A. 2017;114(41):E8685-e94
240. Radaelli E, Ceruti R, Patton V, Russo M, Degrassi A, Croci V. et al. Immunohistopathological and neuroimaging characterization of murine orthotopic xenograft models of glioblastoma multiforme recapitulating the most salient features of human disease. Histol Histopathol. 2009;24(7):879-91
241. Jacobs VL, Valdes PA, Hickey WF, De Leo JA. Current review of in vivo GBM rodent models: emphasis on the CNS-1 tumour model. ASN Neuro. 2011;3(3):e00063
242. Liu YX, Liu WJ, Zhang HR, Zhang ZW. Delivery of bevacizumab by intracranial injection: assessment in glioma model. Onco Targets Ther. 2018;11:2673-83
243. Valtorta S, Lo Dico A, Raccagni I, Gaglio D, Belloli S, Politi LS. et al. Metformin and temozolomide, a synergic option to overcome resistance in glioblastoma multiforme models. Oncotarget. 2017;8(68):113090-104
244. Ji XJ, Chen SH, Zhu L, Pan H, Zhou Y, Li W. et al. Knockdown of NF-E2-related factor 2 inhibits the proliferation and growth of U251MG human glioma cells in a mouse xenograft model. Oncol Rep. 2013;30(1):157-64
245. Ozawa T, Hu JL, Hu LJ, Kong EL, Bollen AW, Lamborn KR. et al. Functionality of hypoxia-induced BAX expression in a human glioblastoma xenograft model. Cancer Gene Ther. 2005;12(5):449-55
246. Yamashita Y, Saito R, Krauze MT, Kawaguchi T, Noble C, Drummond DC. et al. Convection-enhanced delivery of liposomal doxorubicin in intracranial brain tumor xenografts. Targeted Oncology. 2006;1(2):79-85
247. Lee J, Kotliarova S, Kotliarov Y, Li A, Su Q, Donin NM. et al. Tumor stem cells derived from glioblastomas cultured in bFGF and EGF more closely mirror the phenotype and genotype of primary tumors than do serum-cultured cell lines. Cancer Cell. 2006;9(5):391-403
248. Akbarnejad Z, Eskandary H, Dini L, Vergallo C, Nematollahi-Mahani SN, Farsinejad A. et al. Cytotoxicity of temozolomide on human glioblastoma cells is enhanced by the concomitant exposure to an extremely low-frequency electromagnetic field (100Hz, 100G). Biomed Pharmacother. 2017;92:254-64
249. Cammarata FP, Torrisi F, Forte GI, Minafra L, Bravatà V, Pisciotta P. et al. Proton Therapy and Src Family Kinase Inhibitor Combined Treatments on U87 Human Glioblastoma Multiforme Cell Line. Int J Mol Sci. 2019 20(19)
250. Wachsberger PR, Burd R, Cardi C, Thakur M, Daskalakis C, Holash J. et al. VEGF trap in combination with radiotherapy improves tumor control in u87 glioblastoma. Int J Radiat Oncol Biol Phys. 2007;67(5):1526-37
251. Gong L, Gu J, Ge J, Wu X, Zhang C, Yang C. et al. Differential radiation response between normal astrocytes and glioma cells revealed by comparative transcriptome analysis. Onco Targets Ther. 2017;10:5755-64
252. Pan Q, Yang XJ, Wang HM, Dong XT, Wang W, Li Y. et al. Chemoresistance to temozolomide in human glioma cell line U251 is associated with increased activity of O6-methylguanine-DNA methyltransferase and can be overcome by metronomic temozolomide regimen. Cell Biochem Biophys. 2012;62(1):185-91
253. Shen H, Hau E, Joshi S, Dilda PJ, McDonald KL. Sensitization of Glioblastoma Cells to Irradiation by Modulating the Glucose Metabolism. Mol Cancer Ther. 2015;14(8):1794-804
254. Torsvik A, Stieber D, Enger P, Golebiewska A, Molven A, Svendsen A. et al. U-251 revisited: genetic drift and phenotypic consequences of long-term cultures of glioblastoma cells. Cancer Med. 2014;3(4):812-24
255. Tentler JJ, Tan AC, Weekes CD, Jimeno A, Leong S, Pitts TM. et al. Patient-derived tumour xenografts as models for oncology drug development. Nat Rev Clin Oncol. 2012;9(6):338-50
256. Teng J, da Hora CC, Kantar RS, Nakano I, Wakimoto H, Batchelor TT. et al. Dissecting inherent intratumor heterogeneity in patient-derived glioblastoma culture models. Neuro Oncol. 2017;19(6):820-32
257. da Hora CC, Schweiger MW, Wurdinger T, Tannous BA. Patient-Derived Glioma Models: From Patients to Dish to Animals. Cells. 2019 8(10)
258. Conroy S, Kruyt FAE, Wagemakers M, Bhat KPL, den Dunnen WFA. IL-8 associates with a pro-angiogenic and mesenchymal subtype in glioblastoma. Oncotarget. 2018;9(21):15721-31
259. Wang J, Miletic H, Sakariassen P, Huszthy PC, Jacobsen H, Brekkå N. et al. A reproducible brain tumour model established from human glioblastoma biopsies. BMC Cancer. 2009;9:465
260. Liu P, Griffiths S, Veljanoski D, Vaughn-Beaucaire P, Speirs V, Brüning-Richardson A. Preclinical models of glioblastoma: limitations of current models and the promise of new developments. Expert Rev Mol Med. 2021;23:e20
261. Golebiewska A, Hau AC, Oudin A, Stieber D, Yabo YA, Baus V. et al. Patient-derived organoids and orthotopic xenografts of primary and recurrent gliomas represent relevant patient avatars for precision oncology. Acta Neuropathol. 2020;140(6):919-49
262. Das A, Henderson F Jr, Lowe S, Wallace GCt, Vandergrift WA 3rd, Lindhorst SM. et al. Single agent efficacy of the HDAC inhibitor DATS in preclinical models of glioblastoma. Cancer Chemother Pharmacol. 2018;82(6):945-52
263. Burgenske DM, Talele S, Pokorny JL, Mladek AC, Bakken KK, Carlson BL. et al. Preclinical modeling in glioblastoma patient-derived xenograft (GBM PDX) xenografts to guide clinical development of lisavanbulin-a novel tumor checkpoint controller targeting microtubules. Neuro Oncol. 2022;24(3):384-95
264. Ugolkov A, Qiang W, Bondarenko G, Procissi D, Gaisina I, James CD. et al. Combination Treatment with the GSK-3 Inhibitor 9-ING-41 and CCNU Cures Orthotopic Chemoresistant Glioblastoma in Patient-Derived Xenograft Models. Transl Oncol. 2017;10(4):669-78
265. Nielsen CH, Alfsen MZ, Wick MJ, Rundle M, Gudbergsson JM, Jensen MM. et al. Abstract 89: Development and characterization of a panel of orthotopic glioblastoma multiforme (GBM) patient-derived xenograft (PDX) mouse models for drug efficacy evaluation. Cancer Research. 2019;79(13_Supplement):89 -
266. Jin KT, Du WL, Lan HR, Liu YY, Mao CS, Du JL. et al. Development of humanized mouse with patient-derived xenografts for cancer immunotherapy studies: A comprehensive review. Cancer Sci. 2021;112(7):2592-606
267. Xu Z, Kader M, Sen R, Placantonakis DG. Orthotopic Patient-Derived Glioblastoma Xenografts in Mice. Methods Mol Biol. 2018;1741:183-90
268. Patrizii M, Bartucci M, Pine SR, Sabaawy HE. Utility of Glioblastoma Patient-Derived Orthotopic Xenografts in Drug Discovery and Personalized Therapy. Front Oncol. 2018;8:23
269. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455(7216):1061-8.
270. Parsons DW, Jones S, Zhang X, Lin JC, Leary RJ, Angenendt P. et al. An integrated genomic analysis of human glioblastoma multiforme. Science. 2008;321(5897):1807-12
271. An Z, Aksoy O, Zheng T, Fan QW, Weiss WA. Epidermal growth factor receptor and EGFRvIII in glioblastoma: signaling pathways and targeted therapies. Oncogene. 2018;37(12):1561-75
272. Miyai M, Tomita H, Soeda A, Yano H, Iwama T, Hara A. Current trends in mouse models of glioblastoma. J Neurooncol. 2017;135(3):423-32
273. Huse JT, Holland EC. Genetically engineered mouse models of brain cancer and the promise of preclinical testing. Brain Pathol. 2009;19(1):132-43
274. Mao XY, Dai JX, Zhou HH, Liu ZQ, Jin WL. Brain tumor modeling using the CRISPR/Cas9 system: state of the art and view to the future. Oncotarget. 2016;7(22):33461-71
275. Lin F, de Gooijer MC, Roig EM, Buil LC, Christner SM, Beumer JH. et al. ABCB1, ABCG2, and PTEN determine the response of glioblastoma to temozolomide and ABT-888 therapy. Clin Cancer Res. 2014;20(10):2703-13
276. de Vries NA, Beijnen JH, van Tellingen O. High-grade glioma mouse models and their applicability for preclinical testing. Cancer Treat Rev. 2009;35(8):714-23
277. Hambardzumyan D, Parada LF, Holland EC, Charest A. Genetic modeling of gliomas in mice: new tools to tackle old problems. Glia. 2011;59(8):1155-68
278. Schmid RS, Vitucci M, Miller CR. Genetically engineered mouse models of diffuse gliomas. Brain Res Bull. 2012;88(1):72-9
279. McConville P, Hambardzumyan D, Moody JB, Leopold WR, Kreger AR, Woolliscroft MJ. et al. Magnetic resonance imaging determination of tumor grade and early response to temozolomide in a genetically engineered mouse model of glioma. Clin Cancer Res. 2007;13(10):2897-904
280. Lin F, de Gooijer MC, Hanekamp D, Chandrasekaran G, Buil LC, Thota N. et al. PI3K-mTOR Pathway Inhibition Exhibits Efficacy Against High-grade Glioma in Clinically Relevant Mouse Models. Clin Cancer Res. 2017;23(5):1286-98
281. Wei Q, Clarke L, Scheidenhelm DK, Qian B, Tong A, Sabha N. et al. High-grade glioma formation results from postnatal pten loss or mutant epidermal growth factor receptor expression in a transgenic mouse glioma model. Cancer Res. 2006;66(15):7429-37
282. Oldrini B, Curiel-García Á, Marques C, Matia V, Uluçkan Ö, Graña-Castro O. et al. Somatic genome editing with the RCAS-TVA-CRISPR-Cas9 system for precision tumor modeling. Nat Commun. 2018;9(1):1466
283. Maes W, Van Gool SW. Experimental immunotherapy for malignant glioma: lessons from two decades of research in the GL261 model. Cancer Immunol Immunother. 2011;60(2):153-60
284. Jahangiri A, Chin AT, Flanigan PM, Chen R, Bankiewicz K, Aghi MK. Convection-enhanced delivery in glioblastoma: a review of preclinical and clinical studies. J Neurosurg. 2017;126(1):191-200
285. Kerjan G, Gleeson JG. Genetic mechanisms underlying abnormal neuronal migration in classical lissencephaly. Trends Genet. 2007;23(12):623-30
286. Kelava I, Lewitus E, Huttner WB. The secondary loss of gyrencephaly as an example of evolutionary phenotypical reversal. Front Neuroanat. 2013;7:16
287. Bradford R, Darling JL, Thomas DG. The in-vitro chemosensitivity of three cell lines derived from the VM/DK spontaneous murine astrocytoma. J Neurol Neurosurg Psychiatry. 1986;49(12):1361-6
288. Salcman M, Scott EW, Schepp RS, Knipp HC, Broadwell RD. Transplantable canine glioma model for use in experimental neuro-oncology. Neurosurgery. 1982;11(3):372-81
289. Hicks J, Platt S, Kent M, Haley A. Canine brain tumours: a model for the human disease? Vet Comp Oncol. 2017;15(1):252-72
290. Herranz C, Fernández F, Martín-Ibáñez R, Blasco E, Crespo E, De la Fuente C. et al. Spontaneously Arising Canine Glioma as a Potential Model for Human Glioma. J Comp Pathol. 2016;154(2-3):169-79
291. Fernández F, Deviers A, Dally C, Mogicato G, Delverdier M, Cauzinille L. et al. Presence of neural progenitors in spontaneous canine gliomas: A histopathological and immunohistochemical study of 20 cases. Vet J. 2016;209:125-32
292. Dickinson PJ, Roberts BN, Higgins RJ, Leutenegger CM, Bollen AW, Kass PH. et al. Expression of receptor tyrosine kinases VEGFR-1 (FLT-1), VEGFR-2 (KDR), EGFR-1, PDGFRalpha and c-Met in canine primary brain tumours. Vet Comp Oncol. 2006;4(3):132-40
293. Amin SB, Anderson KJ, Boudreau CE, Martinez-Ledesma E, Kocakavuk E, Johnson KC. et al. Comparative Molecular Life History of Spontaneous Canine and Human Gliomas. Cancer Cell. 2020;37(2):243-57.e7
294. Bentley RT, Ahmed AU, Yanke AB, Cohen-Gadol AA, Dey M. Dogs are man's best friend: in sickness and in health. Neuro Oncol. 2017;19(3):312-22
295. Filley A, Henriquez M, Bhowmik T, Tewari BN, Rao X, Wan J. et al. Immunologic and gene expression profiles of spontaneous canine oligodendrogliomas. J Neurooncol. 2018;137(3):469-79
296. Paoloni M, Khanna C. Translation of new cancer treatments from pet dogs to humans. Nat Rev Cancer. 2008;8(2):147-56
297. Lindblad-Toh K, Wade CM, Mikkelsen TS, Karlsson EK, Jaffe DB, Kamal M. et al. Genome sequence, comparative analysis and haplotype structure of the domestic dog. Nature. 2005;438(7069):803-19
298. Toedebusch R, Grodzki AC, Dickinson PJ, Woolard K, Vinson N, Sturges B. et al. Glioma-associated microglia/macrophages augment tumorigenicity in canine astrocytoma, a naturally occurring model of human glioma. Neurooncol Adv. 2021;3(1):vdab062
299. MacDiarmid JA, Langova V, Bailey D, Pattison ST, Pattison SL, Christensen N. et al. Targeted Doxorubicin Delivery to Brain Tumors via Minicells: Proof of Principle Using Dogs with Spontaneously Occurring Tumors as a Model. PLoS One. 2016;11(4):e0151832
300. Jain KK. A Critical Overview of Targeted Therapies for Glioblastoma. Front Oncol. 2018;8:419
301. HICKS WH, BIRD CE, PERNIK MN, HAIDER AS, DOBARIYA A, ABDULLAH KG. et al. Large Animal Models of Glioma: Current Status and Future Prospects. Anticancer Research. 2021;41(11):5343-53
302. Schachtschneider KM, Schwind RM, Newson J, Kinachtchouk N, Rizko M, Mendoza-Elias N. et al. The Oncopig Cancer Model: An Innovative Large Animal Translational Oncology Platform. Front Oncol. 2017;7:190
303. Rogers J, Gibbs RA. Comparative primate genomics: emerging patterns of genome content and dynamics. Nat Rev Genet. 2014;15(5):347-59
304. Duncan BB, Dunbar CE, Ishii K. Applying a clinical lens to animal models of CAR-T cell therapies. Mol Ther Methods Clin Dev. 2022;27:17-31
305. Lonser RR, Walbridge S, Vortmeyer AO, Pack SD, Nguyen TT, Gogate N. et al. Induction of glioblastoma multiforme in nonhuman primates after therapeutic doses of fractionated whole-brain radiation therapy. J Neurosurg. 2002;97(6):1378-89
306. Carvalho C, Gaspar A, Knight A, Vicente L. Ethical and Scientific Pitfalls Concerning Laboratory Research with Non-Human Primates, and Possible Solutions. Animals (Basel). 2018 9(1)
307. Huszthy PC, Daphu I, Niclou SP, Stieber D, Nigro JM, Sakariassen P. et al. In vivo models of primary brain tumors: pitfalls and perspectives. Neuro Oncol. 2012;14(8):979-93
308. Lowenstine LJ, editor Neoplasms and Proliferative Disorders in Nonhuman Primates1986; New York, NY: Springer New York
309. Jacobs S, McCully CL, Murphy RF, Bacher J, Balis FM, Fox E. Extracellular fluid concentrations of cisplatin, carboplatin, and oxaliplatin in brain, muscle, and blood measured using microdialysis in nonhuman primates. Cancer Chemother Pharmacol. 2010;65(5):817-24
310. Goldwirt L, Canney M, Horodyckid C, Poupon J, Mourah S, Vignot A. et al. Enhanced brain distribution of carboplatin in a primate model after blood-brain barrier disruption using an implantable ultrasound device. Cancer Chemother Pharmacol. 2016;77(1):211-6
311. Lankau EW, Turner PV, Mullan RJ, Galland GG. Use of nonhuman primates in research in North America. J Am Assoc Lab Anim Sci. 2014;53(3):278-82
312. Commission E. Seventh report on the statistics on the number of animals used for experimental and other scientific purposes in the member states of the European Union. Accompanying document to the Report from the Commission to the Council and the European Parliament, Brussels, 512 2013, COM (2013) 859 final. 2013
313. Lind NM, Moustgaard A, Jelsing J, Vajta G, Cumming P, Hansen AK. The use of pigs in neuroscience: modeling brain disorders. Neurosci Biobehav Rev. 2007;31(5):728-51
314. Groenen MA. A decade of pig genome sequencing: a window on pig domestication and evolution. Genet Sel Evol. 2016;48:23
315. Schook LB, Collares TV, Hu W, Liang Y, Rodrigues FM, Rund LA. et al. A Genetic Porcine Model of Cancer. PLoS One. 2015;10(7):e0128864
316. Zhang C, Plastow G. Genomic Diversity in Pig (Sus scrofa) and its Comparison with Human and other Livestock. Curr Genomics. 2011;12(2):138-46
317. Schachtschneider KM, Madsen O, Park C, Rund LA, Groenen MA, Schook LB. Adult porcine genome-wide DNA methylation patterns support pigs as a biomedical model. BMC Genomics. 2015;16:743
318. Neff EP. Cancer modeling thinks big with the pig. Lab Anim (NY). 2019;48(3):75-8
319. Selek L, Seigneuret E, Nugue G, Wion D, Nissou MF, Salon C. et al. Imaging and histological characterization of a human brain xenograft in pig: the first induced glioma model in a large animal. J Neurosci Methods. 2014;221:159-65
320. Tora MS, Texakalidis P, Neill S, Wetzel J, Rindler RS, Hardcastle N. et al. Lentiviral Vector Induced Modeling of High-Grade Spinal Cord Glioma in Minipigs. Sci Rep. 2020;10(1):5291
321. Low LA, Tagle DA. Organs-on-chips: Progress, challenges, and future directions. Experimental Biology and Medicine. 2017;242(16):1573-8
322. Logun M, Zhao W, Mao L, Karumbaiah L. Microfluidics in malignant glioma research and precision medicine. Advanced biosystems. 2018;2(5):1700221
323. Ko J, Park D, Lee J, Jung S, Baek K, Sung KE. et al. Microfluidic high-throughput 3D cell culture. Nature Reviews Bioengineering. 2024;2(6):453-69
324. Emmerich M, Ebner P, Wille R, editors. Design automation for organs-on-chip. 2024 Design, Automation & Test in Europe Conference & Exhibition (DATE); 2024: IEEE.
325. McIntyre D, Lashkaripour A, Fordyce P, Densmore D. Machine learning for microfluidic design and control. Lab on a Chip. 2022;22(16):2925-37
326. Lv K, Zhang Y, Tang K, Huang W, Chen F, Chen M. et al. A Machine Learning-Driven Cyclic Optimizing Strategy for the Construction of Paper-Based Microfluidic Devices in the Early Diagnosis of Periodontitis. ACS sensors. 2025;10(9):7002-13
327. Kundacina I, Kundacina O, Miskovic D, Radonic V. Advancing microfluidic design with machine learning: a Bayesian optimization approach. Lab on a Chip. 2025;25(4):657-72
328. Lashkaripour A, Rodriguez C, Mehdipour N, Mardian R, McIntyre D, Ortiz L. et al. Machine learning enables design automation of microfluidic flow-focusing droplet generation. Nature communications. 2021;12(1):25
329. Riordon J, Sovilj D, Sanner S, Sinton D, Young EW. Deep learning with microfluidics for biotechnology. Trends in biotechnology. 2019;37(3):310-24
330. Goyal V, Blivis D, Titus SA, Itkin M, Zakharov A, Wilson K. et al. A Machine Learning Based Analysis Method for Small Molecule High Content Screening of 3D Cancer Spheroid Morphology. Molecular Pharmacology. 2025:100067.
331. Eş I, Ece E, Hacıosmanoğlu N, Güngen MA, Inci F. Artificial intelligence-assisted organ-on-chip systems. Smart Organ-on-Chip Devices: Elsevier. 2025 p. 61-9
332. Deng S, Li C, Cao J, Cui Z, Du J, Fu Z. et al. Organ-on-a-chip meets artificial intelligence in drug evaluation. Theranostics. 2023;13(13):4526
333. Buqué A, Galluzzi L. Modeling Tumor Immunology and Immunotherapy in Mice. Trends Cancer. 2018;4(9):599-601
334. Montoya ML, Kasahara N, Okada H. Introduction to immunotherapy for brain tumor patients: challenges and future perspectives. Neurooncol Pract. 2020;7(5):465-76
335. Ito M, Hiramatsu H, Kobayashi K, Suzue K, Kawahata M, Hioki K. et al. NOD/SCID/gamma(c)(null) mouse: an excellent recipient mouse model for engraftment of human cells. Blood. 2002;100(9):3175-82
336. Wang M, Yao LC, Cheng M, Cai D, Martinek J, Pan CX. et al. Humanized mice in studying efficacy and mechanisms of PD-1-targeted cancer immunotherapy. Faseb j. 2018;32(3):1537-49
337. Gritsch S, Batchelor TT, Gonzalez Castro LN. Diagnostic, therapeutic, and prognostic implications of the 2021 World Health Organization classification of tumors of the central nervous system. Cancer. 2022;128(1):47-58
338. Yan H, Parsons DW, Jin G, McLendon R, Rasheed BA, Yuan W. et al. IDH1 and IDH2 mutations in gliomas. N Engl J Med. 2009;360(8):765-73
339. Nobusawa S, Watanabe T, Kleihues P, Ohgaki H. IDH1 mutations as molecular signature and predictive factor of secondary glioblastomas. Clin Cancer Res. 2009;15(19):6002-7
340. Lamb Y. Vorasidenib: First Approval. Drugs. 2024 84
341. Pellegatta S, Valletta L, Corbetta C, Patanè M, Zucca I, Riccardi Sirtori F. et al. Effective immuno-targeting of the IDH1 mutation R132H in a murine model of intracranial glioma. Acta Neuropathol Commun. 2015;3:4
342. Philip B, Diana XY, Silvis MR, Shin CH, Robinson JP, Robinson GL. et al. Mutant IDH1 promotes glioma formation in vivo. Cell reports. 2018;23(5):1553-64
343. Shi DD, Lee JH, Wang AC, Khanal J, Gao W, Kaelin WG Jr. et al. Protocol to establish a genetically engineered mouse model of IDH1-mutant astrocytoma. STAR Protoc. 2023;4(2):102281
Author contact
![]() Corresponding authors: Jaejoon Lim, MD PhD, Department of Neurosurgery, Bundang CHA Medical Center, CHA University College of Medicine, Yatap-dong 59, Seongnam, 13496, Korea; Email: coolppengco.kr. Jungho Ahn, MD PhD, Department of Biophysics, Institute of Quantum Biophysics, Sungkyunkwan University, 2066, Seobu-ro, Jangan-gu, Suwon-si, Gyeonggi-do, Republic of Korea; Email: jhahn513edu.
Corresponding authors: Jaejoon Lim, MD PhD, Department of Neurosurgery, Bundang CHA Medical Center, CHA University College of Medicine, Yatap-dong 59, Seongnam, 13496, Korea; Email: coolppengco.kr. Jungho Ahn, MD PhD, Department of Biophysics, Institute of Quantum Biophysics, Sungkyunkwan University, 2066, Seobu-ro, Jangan-gu, Suwon-si, Gyeonggi-do, Republic of Korea; Email: jhahn513edu.