Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
1. Biology of efferocytosis and...
2. Current strategies for...
3. Nanomedicine-based strategies...
4. Therapeutic effects of...
5. Progress of...
6. Challenges and future...
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2026; 16(10):5537-5570. doi:10.7150/thno.128155 This issue Cite
Review
Advancements in nanomedicine for the therapeutic regulation of efferocytosis: opportunities and challenges
Zehuan Lin1, Xiyue Zhou1, Shuyun Liu1, Meihua Wan2, Jingping Liu1 ![]()
1. Department of General Surgery and NHC Key Laboratory of Transplant Engineering and Immunology, Frontiers Science Center for Disease-related Molecular Network, West China Hospital, Sichuan University, Chengdu 610041, China.
2. West China Center of Excellence for Pancreatitis, Institute of Integrated Traditional Chinese and Western Medicine, West China Hospital, Sichuan University, Chengdu 610041, China.
Received 2025-11-10; Accepted 2026-2-27; Published 2026-3-25
Abstract

Efferocytosis is a conserved event that plays an essential role in maintaining tissue homeostasis and immune balance. However, dysregulation of efferocytosis can induce disturbed apoptotic cells clearance and immune disorders, fueling an array of diseases, such as inflammatory diseases, autoimmunity, and cancers. The process of efferocytosis is dynamic and regulated by a complicated network composed of intracellular pathways and intercellular interactions, which pose significant challenges to current therapies. In recent years, nanomedicine-based strategies have shown potential in the precise regulation of efferocytosis for preventing or reversing pathology in diverse diseases. Although these results are encouraging, the opportunities and challenges in this field remain elusive and need to be comprehensively reviewed, which will be helpful for its improvement and future clinical translation. Here, we briefly introduce the key steps and signaling pathways of efferocytosis and its clinical relevance in diverse diseases. We highlight current advancements in nanomedicines for modulating efferocytosis, especially their design principles and therapeutic applications across a broad range of diseases, such as chronic inflammation, cardiovascular diseases, autoimmune diseases, neurodegenerative diseases and cancers. We discuss the ongoing clinical trials and discuss the challenges and limitations in this field, which may provide insights into developing precision nanomedicines to modulate efferocytosis for treating diverse forms of diseases.
Keywords: efferocytosis, nanomedicine, inflammation, metabolism, target drug delivery, macrophage
Introduction
Efferocytosis, a process by which phagocytes recognize and engulf apoptotic cells, is fundamental to maintain tissue homeostasis and the resolution of inflammation. Every day, billions of cells undergo apoptosis and must be cleared efficiently to prevent secondary necrosis and aberrant immune activation [1]. The process of efferocytosis is orchestrated by a cascade of molecular “find me” signals released by apoptotic cells, “eat me” signals displayed on their surface, and subsequent receptor recognition and digestion by phagocytes, which ensure that apoptotic debris is removed in an immunologically silent manner, along with the release of various immunoregulatory mediators (e.g., IL-10, TGF-β and lipid mediators) that promote inflammation resolution post-injury. Conversely, defective efferocytosis can result in the accumulation of apoptotic cells and cellular debris, fueling chronic inflammation and autoimmunity [2]. Interestingly, efferocytosis can play distinct roles in different conditions. For example, insufficient efferocytosis can exacerbate diseases such as atherosclerosis, autoimmune disorders and neurodegeneration, while tumor-associated macrophage (TAM)-mediated efferocytosis can contribute to the immunosuppressive tumor microenvironment [3-6]. Nevertheless, increasing evidence has indicated that precise modulation of efferocytosis may offer a promising therapeutic avenue for treating a variety of diseases.
Currently, some pharmacological approaches (e.g., small molecules, proteins) have been developed to manipulate efferocytosis, but their therapeutic potency is still limited for several reasons. For example, systemic blockade of the “don't-eat-me” signal CD47 with antibodies elicits macrophages to engulf tumor or plaque cells, but it can cause severe side effects such as anemia due to indiscriminate clearance of red blood cells [7]. Small-molecule modulators of efferocytic pathways (e.g., MerTK tyrosine kinase inhibitors) show promise in preclinical models, but their short in vivo half-time, poor bioavailability and lack of tissue or cell specificity often cause off-target effects and adverse reactions [8]. In recent years, nanomedicines have emerged as a robust means for the precise regulation of efferocytosis. For instance, various nanocarriers that can target efferocytic effector cells in distinct conditions (e.g., proinflammatory macrophages in plaques, TAMs in tumors) have been reported [9, 10], which can specifically modulate efferocytosis via either delivering diverse types of payloads (e.g., pathway inhibitors or agonists) [11] or mimicking the surface signals (e.g., the “eat me” signal phosphatidylserine) of apoptotic cells [12]. These advanced nanomedicines might have the potential to overcome the drawbacks of conventional treatments, enabling precise regulation of efferocytosis in diverse disease settings. Although the existing findings are encouraging, some critical problems, such as proper selection of targets, design and engineering strategies of nanomedicine, their mechanisms of action, and possible limitations, remain elusive and need to be comprehensively reviewed, which will be helpful for improvement and future clinical translation of these therapies.
In this review, we briefly introduce the key steps and signaling pathways of efferocytosis and its clinical relevance in diverse disease conditions. We highlight the advancements in nanomedicines for modulating efferocytosis, particularly in their design principles and therapeutic applications across a broad range of diseases, such as cancers, cardiovascular diseases, autoimmune and inflammatory disorders and neurodegenerative diseases. We also introduce the ongoing clinical trials and discuss the challenges and limitations in this field, which may provide insights into developing precision nanomedicines to modulate efferocytosis for treating diverse forms of diseases.
1. Biology of efferocytosis and its clinical relevance
Efferocytosis, a programmed process of apoptotic cells clearance by phagocytes, plays an essential role in regulating tissue homeostasis and immune reactions. In recent years, a growing body of studies has markedly expanded our understanding of this evolutionarily conserved process, revealing its central role in a broad spectrum of physiological and pathological contexts. These investigations have underscored the importance of efferocytosis in modulating innate immune responses, while disturbed efferocytosis may act as a key pathogenic driver in diverse diseases, such as chronic inflammatory disorders, autoimmune diseases, tumor immune evasion, and neurodegenerative conditions [2, 3, 5]. This section provides a concise overview of the key steps and pathways of efferocytosis and discusses the role of efferocytic dysregulation in the pathology of various diseases.
1.1. Key stages and signaling pathways of efferocytosis
Briefly, homeostatic efferocytosis comprises several key continual and coordinated steps, including recruitment, recognition, internalization, and degradation [13]. The process is initiated by the release of chemoattractants (“find-me” signals) from apoptotic cells, which serve to recruit phagocytes. Subsequent recognition is mediated by specific ligand-receptor interactions (“eat-me” signals). Cytoskeletal rearrangement of phagocytes facilitates the internalization of apoptotic cells, followed by phagosome-lysosome fusion and subsequent degradation of the cellular contents. These sequential events are tightly regulated by a network of molecular pathways that integrate external and intracellular signals to ensure efficient clearance, modulate immune cell trafficking, and promote anti-inflammatory responses.
In the “find me” stage, apoptotic cells release soluble chemoattractant signals, such as nucleotides (e.g., ATP, UTP), lipids (e.g., lysophosphatidylcholine, LPC), sphingosine-1-phosphate (S1P), and chemokines (e.g., CX3CL1), that serve as “find me” signals to create a gradient to recruit phagocytes (e.g., macrophages, dendritic cells) to the vicinity. These factors are likely actively released by apoptotic cells in a caspase-dependent manner before the cell membrane loses integrity and further bind to corresponding cognate receptors of phagocytes. For example, extracellular ATP/UTP, CX3CL1, and LPC can be detected by P2Y2 receptors, CX3CR1, and G2A receptors of phagocytes, respectively [14]. These chemoattractant-receptor interactions ensure that phagocytes are efficiently recruited to sites of cell death (Figure 1A).
Key stages and regulatory circuits of efferocytosis. (A) Find-me cues and recruitment: Apoptotic cells release ATP/UTP, LPC, and CX3CL1, which are sensed by P2Y2, G2A, and CX3CR1 on phagocytes, respectively, establishing chemotactic gradients that attract macrophages/dendritic cells. (B) Eat-me versus don't-eat-me recognition: Externalized phosphatidylserine (PtdSer) on apoptotic cells is bridged by MFG-E8 and Gas6 to αvβ3/β5 integrins and the TAM receptor (Tyro3, Axl, MerTK), or bound directly by TIM-4, initiating uptake; healthy cells are protected by the inhibitory CD47-SIRPα checkpoint. Downstream, the ELMO-DOCK complex activates Rac1 to drive actin remodeling, phagocytic cup formation, and internalization. (C) Digestion and resolution: Efferosomes mature and fuse with lysosomes (Rab5-to-Rab7 transition with LAP), leading to cargo degradation and release of IL-10 and TGF-β; LXR/PPAR programs upregulate ABCA1 for cholesterol efflux and maintain MerTK expression, supporting receptor availability for subsequent rounds. (D) Continual efferocytosis and metabolic coupling: Multiple rounds of uptake require metabolic rewiring—GLUT1-dependent glucose influx and a transient glycolytic burst with SLC16A1-mediated lactate export facilitate receptor recycling and proresolving signaling; mitochondrial FAO and the ETC sustain energy demands; apoptotic-cargo-derived arginine is converted via ARG1/ODC1 to putrescine, stabilizing Rac1 signaling and actin dynamics to maintain multiround efferocytosis. Abbreviations: AC: apoptotic cell; PtdSer: phosphatidylserine; TAM: Tyro3/Axl/MerTK family; LAP: LC3-associated phagocytosis; FAO: fatty-acid β-oxidation; ETC: electron transport chain.
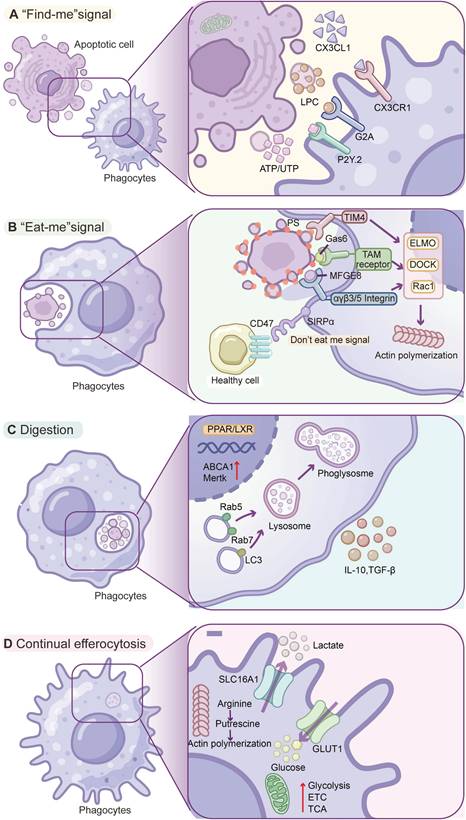
In the “eat me” stage, phagocytes recognize and bind apoptotic cells via specific surface cues. One of the most universal “eat me” signals is the externalization of phosphatidylserine (PtdSer) on the outer leaflet of the apoptotic cell membrane [15]. Phagocytes can bind PtdSer indirectly by bridging molecules (opsonins) or dedicated receptors. Key opsonins, including milk fat globule EGF-factor 8 (MFG-E8) and growth arrest-specific 6 (Gas6), bind PtdSer on apoptotic cells and simultaneously engage phagocyte receptors [13] MFG-E8 bridges PtdSer to integrins (αvβ3/β5) of phagocytes, and Gas6 (or Protein S) links PtdSer to the TAM family receptor tyrosine kinases (Tyro3, Axl, MerTK) of phagocytes [16, 17]. Some receptors on phagocytes can directly bind apoptotic cells. TIM family receptors (e.g., TIM-4) possess immunoglobulin domains that bind PtdSer [18] (Figure 1B); brain-specific angiogenesis inhibitor 1 (BAI1) can also directly bind PtdSer and initiate internalization via the ELMO/Dock/Rac signaling pathway [19].
Crucially, healthy living cells can avoid being mistakenly engulfed by displaying “don't eat me” signals [20] (Figure 1B). The prototypical don't-eat-me signal is CD47, a cell-surface protein ubiquitously expressed on host cells, which engages the receptor SIRPα on macrophages [21]. When SIRPα binds CD47, it delivers an inhibitory signal (via SHP-1/SHP-2 phosphatases) that halts the phagocytic machinery [21]. Recent studies have also identified CD24 on viable cells, which binds Siglec-10 on macrophages as a don't-eat-me signal, particularly exploited by cancer cells with high CD24 expression to suppress phagocytosis [21]. Through the balance of “eat me” signals versus “don't eat me” signals, phagocytes are able to discriminate target cells, thereby maintaining self-tolerance while clearing apoptotic cells.
Upon recognition and binding of apoptotic cells, phagocytes initiate engulfment by extending their plasma membrane around target cells. This process relies on phagocyte cytoskeletal rearrangement driven by actin polymerization [22]. In phagocytes, the transduction of signals from the engaged receptors converges on small GTPases (e.g., Rac1, Cdc42, and RhoA) that regulate actin, and activation of Rac1 and Cdc42 is required to drive the formation of phagocytic cups and then internalization of apoptotic cells [23] (Figure 1C). The TAM receptor MerTK or integrin engagement can trigger intracellular engulfment pathways. MerTK activation leads to recruitment of an ELMO1-Dock180 complex that acts as a guanine nucleotide exchange factor (GEF) to activate Rac1, inducing actin polymerization and membrane zippering around the corpse [24]. The dynamic cytoskeletal dance ensures that apoptotic cells can be fully enclosed in a membrane-bound phagosome (termed the efferosome when containing apoptotic cargo) within the macrophage.
Finally, in the digestion stage, after the phagocyte membrane encloses an apoptotic cell, the formed efferosome undergoes a series of maturation processes, including Rab5-to-Rab7 transition, phosphatidylinositol-3-phosphate (PI3P) acquisition, progressive acidification, and fusion with lysosomes for cargo degradation [1]. This process can be regulated by a noncanonical autophagy pathway, LC3-associated phagocytosis (LAP), in which autophagy-related proteins (e.g., LC3-II conjugation machinery and Beclin-1/Vps34 complexes) are recruited to single-membrane phagosomes rather than double-membrane autophagosomes [25] (Figure 1C). LAP leads to LC3 lipidation on the efferosome and thus promotes phagosome fusion and cargo digestion, which also actively suppresses proinflammatory signaling during apoptotic cells processing [23]. In brief, efferocytosis is a highly orchestrated process encompassing the release of find-me signals, specific recognition of apoptotic signals, engagement of phagocytic receptors, cytoskeleton-mediated engulfment, and LC3-assisted phagolysosomal digestion. This event culminates in the secretion of pro-decomposition mediators and the suppression of inflammation, thereby facilitating tissue repair and homeostasis.
1.2. Role of metabolic regulation in continual efferocytosis
An important feature of efferocytosis is the ability of phagocytes to ingest multiple apoptotic cells (ACs) over a period, referred to as continual efferocytosis. Continual efferocytosis can ensure efficient clearance when the number of ACs markedly exceeds that of macrophages. However, the massive materials (e.g., lipids, proteins, and nucleotides) from the engulfed ACs can induce metabolic overloading in phagocytes, and thus phagocytes must adapt metabolic rewiring to maintain continual efferocytosis. Upon engulfment of ACs, phagocytes rapidly upregulate glycolysis to meet the bioenergetic and redox demands of phagocytosis [26], and this transient “glycolytic burst” replenishes NAD⁺ (via conversion of pyruvate to lactate) and generates ATP to support actin-dependent engulfment (Figure 1D). Mitochondria have emerged as key regulators of continual efferocytosis. Mitochondrial oxidative metabolism plays a dominant role following the initial glycolytic burst, in which fatty acid β-oxidation supplies acetyl-CoA to the tricarboxylic acid (TCA) cycle and sustains electron transport chain (ETC) activity to fuel continual efferocytosis [27]. ACs-derived lipids can activate LXR and PPAR nuclear receptors, which also orchestrate a transcriptional program coupling metabolic shift with enhanced efferocytic capacity [28]. LXR/PPAR signaling upregulates genes for cholesterol efflux (e.g., ABCA1) to prevent lipid overload and drive the expression of MerTK and opsonins, thereby sustaining phagocytic receptor availability and function [28].
Other metabolic pathways are also involved in regulating continual efferocytosis. For example, arginine catabolism from the apoptotic cargo can be repurposed to sustain continual efferocytosis. ACs-derived arginine is metabolized by arginase 1 (ARG1) to ornithine and then by ornithine decarboxylase 1 (ODC1) to putrescine, and these polyamines can stabilize Dbl (MCF2) mRNA via HuR and thus activate Rac1 to drive actin remodeling and sustain continual efferocytosis [29]. Collectively, the current findings indicate that metabolic regulation of phagocytes is essential for sustaining, while disruption of this fine-tuned process can contribute to the onset and progression of diverse pathological conditions.
1.3. Pathological role of dysregulated efferocytosis in diverse diseases
Notably, defective or dysregulated efferocytosis has been recognized as a key pathological factor in a wide spectrum of diseases. When any step of the clearance process fails - whether due to overwhelming apoptotic burden, molecular interference with recognition signals, or an inhospitable tissue environment - the consequences can be deleterious. Accumulating apoptotic cells induce secondary necrosis and the release of excessive DAMPs that perpetuate inflammation and tissue damage [30]. Moreover, phagocytes with dysregulated efferocytosis can adopt aberrant phenotypes, such as overly immunosuppressive or foam-cell states, to promote disease development [5, 31]. In this section, we introduce the pathological role of dysregulated efferocytosis in diverse diseases.
1.4. Autoimmune diseases
Autoimmune diseases are serious chronic disorders that arise from loss of self-tolerance to self-antigens, leading to persistent inflammation and multiorgan tissue damage [32]. Defective efferocytosis has been associated with many autoimmune diseases, such as systemic lupus erythematosus (SLE). Patients with SLE exhibit large numbers of apoptotic bodies in tissues and circulation that are not properly cleared [33]. These remnants release nuclear autoantigens (DNA, histones, etc.) that drive autoantibody production and inflammatory cascades [34]. Multiple efferocytosis defects have been reported in lupus, such as macrophage-intrinsic dysfunction and the presence of autoantibodies against efferocytosis receptors [33]. Lupus patients often have antibodies against C1q and other opsonins involved in AC clearance. Genetic deficiencies in complement components or bridging molecules (C1q, MFG-E8, MerTK/Axl/Tyro3) are risk factors for lupus, underscoring the key role of efferocytosis in preventing autoimmunity [35]. Defects in efferocytosis during lupus chronically activate the immune system, breaking self-tolerance, inducing secondary necrosis and frustrating phagocytes in multiple tissues (e.g., kidneys).
In addition, a similar theme has been found in other autoimmune conditions, such as rheumatoid arthritis (RA). For instance, in the RA state, an imbalance of macrophage phenotypes in the joint leads to insufficient clearance of apoptotic neutrophils in the inflamed synovium, and the domination of pro-inflammatory macrophages and defective efferocytosis lead to the accumulation of cell debris and inflammatory cytokines in the local microenvironment, thereby perpetuating the degree of synovial inflammation and pannus formation [36]. These reports suggest that defective efferocytosis can break immune tolerance and fuel chronic inflammation across diverse forms of autoimmune diseases.
1.5. Cardiovascular diseases
Atherosclerosis is a chronic inflammatory disease of medium- and large-sized arteries that affects millions of people worldwide [37]. Although its mechanism is complicated, impaired clearance of ACs in atherosclerotic plaques has been proposed as a key driver of lesion progression [5]. During early atheromas, macrophages efficiently phagocytose apoptotic lipid-laden foam cells. However, as plaques enlarge, macrophages become dysfunctional due to oxidative stress, cholesterol overload, and inflammatory cytokines. Multiple mechanisms underlie efferocytosis defects in advanced plaques, such as ADAM17/10-mediated shedding of MerTK, impaired cholesterol efflux because of reduced ABCA1, and the upregulation of the “don't-eat-me” signal CD47 on atheroma cells [7]. The net result is defective efferocytosis: apoptotic foam cells accumulate in the plaque's core instead of being cleared. These uncleared corpses undergo secondary necrosis, forming a lipid-rich necrotic core that expands over time and destabilizes the plaque fibrous cap [5], potentially triggering acute thrombosis, heart attack or stroke [38, 39]. Human studies of advanced plaques have found abundant apoptotic debris and markers of impaired efferocytosis (e.g., elevated lesional CD47 and soluble MerTK reflecting MerTK receptor cleavage) [7, 40]. Thus, insufficient efferocytosis in plaques is considered a “linchpin” of atherosclerotic progression and instability. Conversely, enhancing efferocytosis reduced plaque size and promoted a more stable phenotype (with smaller necrotic cores and thicker fibrous caps) [41].
1.6. Neurodegenerative diseases
Neurodegenerative diseases, such as Alzheimer's disease (AD), are progressive, age-associated disorders characterized by neuronal loss and the accumulation of misfolded protein aggregates, leading to cognitive and functional decline [42]. In the central nervous system, microglia serve as the major resident phagocytes responsible for clearing apoptotic neurons, synapses, and protein aggregates [43]. However, microglial efferocytosis can become impaired or overwhelmed in neurodegenerative diseases. For instance, in AD brains, there is an accumulation of apoptotic or damaged neurons and excessive deposition of amyloid-β plaques, partly because microglia fail to effectively engulf and dispose of this debris [44]. This may be due to chronic microglial activation (skewing them to a pro-inflammatory state with reduced phagocytic capacity), genetic factors (many AD risk genes, such as TREM2, are linked to microglial phagocytosis function), or competitive binding of soluble factors that block the eat-me signals [45]. As a result, uncleared apoptotic cells and protein aggregates drive local inflammation and neurotoxicity in a vicious cycle to accelerate neurodegeneration. Moreover, in other neurodegenerative contexts (e.g., Parkinson's disease, ALS), evidence of defective clearance of dead neurons and myelin debris has been reported [46, 47]. Interestingly, if microglia are overactivated in certain contexts, they might also phagocytose stressed but viable neurons (a phenomenon observed in some models), indicating that proper calibration of efferocytosis is vital for maintaining neural homeostasis.
1.7. Inflammatory disorders
In the conditions of acute inflammation or tissue injury, timely efferocytosis is also critical for resolution. For example, in lung inflammation, such as acute respiratory distress syndrome (ARDS), massive neutrophil influx into the lungs followed by delayed neutrophil apoptosis and heightened formation of neutrophil extracellular traps (NETs), whereas impaired efferocytosis and NET clearance by phagocytes can exacerbate lung injury [48, 49]. Consistently, bronchoalveolar lavage fluid from ARDS patients reduces alveolar macrophage efferocytosis ex vivo, supporting a compartmentalized defect in corpse clearance within the lung [50]. These abnormalities favor the persistence of neutrophils and the propagation of hyperinflammation (cytokine storm-like state) that enhances alveolar-capillary damage [51]. Similar findings are also found in sepsis, where a dysregulated immune response can suppress macrophage function (sometimes called “immune paralysis”), leading to reduced clearance of microbes and apoptotic cells [52]. In the severe trauma or burn state, the inflammatory response can likewise overshoot, and if massive apoptotic immune cells and apoptotic cells cannot be cleared, systemic inflammatory response syndrome (SIRS) can result [53, 54]. Together, efficient tissue injury repair requires a switch from pro-inflammatory state to pro-resolving state, while defective efferocytosis tips the balance toward sustained inflammation and organ dysfunction.
1.8. Cancers
Disordered efferocytosis also plays a crucial role in the progression of multiple types of cancers because tumor cells often coopt efferocytic pathways as an immune evasion strategy [55]. Many types of cancer cells (e.g., acute myeloid leukemia, breast cancer, and ovarian cancer) overexpress CD47 (an “don't eat me” signal) and other antiphagocytic ligands (e.g., CD24) to directly thwart macrophage-mediated cell clearance [21]. Moreover, the tumor microenvironment (TME) is typically rich in apoptotic cells due to rapid cancer cell turnover or therapy-induced cell death, and tumor-associated macrophages (TAMs) avidly perform efferocytosis of these cells. This would seem beneficial, but in fact, TAM efferocytic activity drives them toward an M2-like, immunosuppressive phenotype [56]. Upon engulfing apoptotic tumor cells, TAMs secrete elevated immunosuppressive cytokines (e.g., IL-10 and TGF-β) and express immune checkpoint ligands, thereby establishing an immunosuppressive and tolerogenic milieu [57]. Indeed, elevated MerTK activity in TAMs has been associated with poor prognosis across multiple cancers, such as hepatocellular carcinoma and colorectal cancers [58], reflecting enhanced efferocytosis and subsequent suppression of cytotoxic T-cell responses. In essence, tumors exploit efferocytosis by evading phagocytosis when viable through “don't-eat-me” signals, yet leveraging TAM-mediated clearance of dead cells to dampen inflammation. Therefore, disordered efferocytosis in the TME has been considered an “immune checkpoint” that might be targeted to improve anti-tumor immunity [59].
In brief, dysregulated efferocytosis has been involved in diverse diseases. In some cases (atherosclerosis, autoimmune diseases, chronic inflammatory disorders), insufficient efferocytosis results in toxic or immunogenic material to accumulate that drives disease progression. In other cases (cancer), excess or contextually inappropriate efferocytosis by tumor-associated phagocytes contributes to an immunosuppressive microenvironment (Table 1). These insights set the stage for therapeutic interventions aiming to modulate efferocytosis - either boosting it to enhance clearance and resolution or tuning it down/changing its consequences to promote immunity.
Evidence of dysregulated efferocytosis in diverse diseases
| Diseases | Disregulated efferocytotic pathways | Pathological consequences | Model/Evidence level | Reference | |
|---|---|---|---|---|---|
| Autoimmune Diseases | Systemic Lupus Erythematosus (SLE)) | Defects in C1q/C3 opsonins; TAM receptors (MerTK, Axl, Tyro3); MFG-E8; SCARF1; TAM receptors. | Accumulation of apoptotic debris, release of nuclear autoantigens, autoantibody production, immune-complex deposition; lupus nephritis and multi-organ damage. | Human biopsy/serum; Mice, in vivo (lupus-prone models); macrophage & dendritic cell, in vitro | [33, 34, 60, 61] |
| Rheumatoid Arthritis (RA) | ADAM10/17; MerTK; MFG-E8; dysregulated macrophage phenotypes. | Persistent synovitis and pannus; defective apoptotic cell clearance in joints, chronic inflammation and cartilage/bone erosion. | Human synovium/serum; Mice, in vivo (CIA/K/BxN arthritis); macrophage & synoviocyte, in vitro | [36, 62] | |
| Inflammatory Bowel Disease (IBD) | RUBCN/LAP axis; MerTK; MFG-E8. | Impaired clearance of apoptotic cells in the intestinal milieu; barrier disruption, chronic colitis, dysbiosis-associated inflammation. | Human intestinal biopsy/serum; Mice, in vivo (DSS/TNBS/IL-10-/- colitis); macrophage & dendritic cell, in vitro | [63] | |
| Atherosclerosis | ADAM10/17; MerTK; LXR/PPAR-ABCA1/ABCG1 lipid-efflux axis; CD47-SIRPα. | Defective clearance of apoptotic foam cells, growth of lipid-rich necrotic core, fibrous cap thinning, plaque destabilization and thrombosis. | Human plaque; Mice, in vivo (ApoE-/-/Ldlr-/- models); foam cell, in vitro | [7, 38, 64] | |
| Neurodegeneration Diseases | Alzheimer's Disease (AD) | TREM2-TYROBP signaling; APOE; TAM receptors; LAP. | Impaired clearance of apoptotic neurons and amyloid-β/tau aggregates; chronic neuroinflammation and synapse loss. | Human postmortem/genetics; Mice, in vivo (APP/PS1, 5xFAD); microglia, in vitro | [43] |
| Parkinson's Disease (PD) | TREM2/TYROBP signaling; TAM receptors; altered debris handling. | Defective removal of α-synuclein aggregates and apoptotic dopaminergic neurons; persistent neuroinflammation; | Human postmortem/genetics; Mice, in vivo (MPTP-induced PD); microglia & astrocyte coculture, in vitro | [45-47] | |
| Inflammatory disorders | ARDS/ALI | Neutrophil extracellular traps (NETs) and DNASE1/1L3 degradation pathways; MerTK; MFG-E8; C5a-C5aR1; cytokine storm. | Delayed clearance of apoptotic cells and NETs, sustained hyperinflammation, alveolar injury, respiratory failure. | Human ICU cohorts/BALF; Mice, in vivo (LPS/acid/ventilation-induced ALI); neutrophil & macrophage, in vitro | [48] |
| Sepsis | NET overload and defective nucleases; (C5a-C5aR1); impaired MerTK; MFG-E8. | Inefficient corpse/NET clearance, immune paralysis, organ dysfunction. | Human ICU cohorts/blood; Mice, in vivo (CLP/LPS sepsis); neutrophil & macrophage, in vitro | [49] | |
| Acute organ injury | Overwhelming apoptotic burden with insufficient MerTK; MFG-E8; dysregulated resolution programs. | Accumulation of cell debris, persistent inflammation. | Human biopsy/serum; Mice, in vivo (ischemia-reperfusion/MI/AKI); macrophage, in vitro | [65-67] | |
| Cancers | CD47-SIRPα; CD24-Siglec-10; TAM receptors (MerTK/Axl); IL-10, TGF-β; tumor-associated macrophage (TAM) phenotypes | Immune evasion and T-cell suppression, M2-like TAM polarization, immunosuppressive tumor microenvironment, angiogenesis and metastasis. | Human tumor datasets/biopsies; early-phase clinical studies targeting CD47/SIRPα and MerTK/Axl; Mice, in vivo (syngeneic/xenograft and spontaneous tumor models); macrophage, in vitro. | [21, 59] | |
2. Current strategies for modulating efferocytosis
Given the critical role of efferocytosis in diverse diseases, researchers have begun exploring potential therapies for modulating efferocytosis. To date, some pharmacological approaches that aim to enhance efferocytosis (in contexts such as cardiovascular, autoimmune, or resolution of inflammation) or to inhibit efferocytosis checkpoints (in cancer) have been reported and achieve therapeutic effects to some extent. In this section, we outline the main strategies under investigation as well as their limitations that motivate new solutions (Table 2).
Conventional strategies for modulating efferocytosis
| Strategy Category | Target pathways | Mechanisms of action | Model/Evidence level | Limitations | Reference | ||
|---|---|---|---|---|---|---|---|
| Small molecule Drugs | LXRα/β (NR1H3/NR1H2) | Promote lipid efflux (ABCA1/ABCG1↑) Transactivate MerTK Reprogram macrophages toward a pro-resolving state | Clinical (Phase I); Mice, in vivo (atherosclerosis); macrophage & dendritic cell, in vitro | Systemic LXR agonists can cause hypertriglyceridemia/steatosis; pathway crosstalk with TLR can suppress LXR targets during infection; cell-type and context dependence. | [24, 68, 69] | ||
| PPARγ (e.g., pioglitazone) | Reprogram macrophages toward a pro-resolving state Cytoskeletal regulators (e.g. Rac1/Vav1 axis) Anti-inflammatory cytokines (IL-10) enhanced | Mice, in vivo (lung injury/MS models); macrophage & dendritic cell, in vitro | Metabolic side effects (weight gain, edema); variable efficacy across tissues; off-target nuclear receptor pleiotropy. | [70] | |||
| ALX/FPR2 (e.g., columbamine as a biased agonist) | Enhances LC3-associated efferocytosis (LAP) | Mice, in vivo (colitis models); macrophage & dendritic cell, in vitro | Selectivity and bias of agonists; receptor expression varies across tissues; translational evidence still emerging. | [71] | |||
| MerTK inhibitors (UNC2250, BMS-794833, etc.) | Pharmacologic MerTK inhibition generally suppresses efferocytosis Boost antitumor immunity by preventing clearance of apoptotic cells | Clinical (Phase I/II); Mice, in vivo (cancer models); macrophage & dendritic cell, in vitro | Impaired tissue resolution; potential toxicity if used outside oncology; off-target TAM inhibition (Axl/Tyro3). | [72-75] | |||
| Cytokines and Biologics | CD47-SIRPα axis (anti-CD47 mAbs; SIRPα-Fc fusion proteins) | Blocks the 'don't-eat-me' signal to enable macrophage engulfment of apoptotic/damaged cells and tumor cells | Clinical (Phase I/II/III) Mice, in vivo (cancer/atherosclerosis); macrophage & dendritic cell, in vitro | On-target anemia/thrombocytopenia; dosing/titration challenges (RBC sink); mixed Phase 3 outcomes and clinical holds in some indications. | [76, 77] | ||
| Recombinant MFG-E8 | Bridges phosphatidylserine on apoptotic cells to integrins (αvβ3/αvβ5) on phagocytes to augment recognition and internalization | Mice, in vivo (sepsis/ischemia/reperfusion); macrophage & dendritic cell, in vitro | Short half-life; context-dependent efficacy; manufacturing and delivery considerations. | [44, 78] | |||
| IL-10/IL-4/IL-13 | Pro-resolving cytokines that upregulate engulfment machinery (e.g. IL-10→Vav1→Rac1); Promote Macrophage alternative activation; Enhance clearance during resolution. | Clinical (selected biologics in development/approved, target-related); Mice, in vivo (atherosclerosis/ALI); macrophage & dendritic cell, in vitro | Systemic immunosuppression; dosing window for 'helpful vs harmful' responses; pleiotropy across tissues. | [28, 79, 80] | |||
| Gene Therapy | MERTK replacement in RPE (retinal dystrophy) | Gene replacement restores RPE efferocytosis of photoreceptor outer segments; Preserves photoreceptors in models of MERTK-associated retinopathy. | Mice, in vivo (RCS rat); macrophage & dendritic cell, in vitro | Vector delivery, durability and immunogenicity; indication-specific (ocular) rather than systemic. | [81] | ||
| Engineered efferocytic receptors (CHEF; e.g., TIM4-ELMO1 fusions) | Synthetic receptors couple PtdSer recognition to ELMO1-Rac1 signaling; Selectively boosting apoptotic-cell uptake. | Mice, in vivo (colitis/arthritis); macrophage & dendritic cell, in vitro | Cell engineering complexity; safety controls for over-phagocytosis; translational readiness. | [82] | |||
| CD47/SIRPα (e.g., CRISPR knockout) | Remove the CD47-SIRPα checkpoint to increase efferocytosis. | Mice, in vivo (cancer models) macrophage & tumor co-culture, in vitro | High editing efficiency needed; in vivo editing safety (on/off-target effects); delivery barriers; tumor heterogeneity | [83-85] | |||
| Macrophage ACSL1 inhibition; adoptive transfer of metabolically reprogrammed macrophages | Dampens NF-κB-driven inflammation; Shifts macrophages toward a pro-resolving state. | Mice, in vivo (atherosclerosis/diabetes); macrophage in vitro | Ex vivo manipulation and scalability; durability after transfer; disease specificity. | [86] | |||
2.1 Small molecule drugs
Small molecule modulators (mainly chemical compounds) provide a bidirectional lever to tune efferocytosis either enhancement in chronic inflammatory settings or inhibition in tumors to counter immunosuppression. Pharmacologically enhancing efferocytosis promotes apoptotic debris clearance and inflammation resolution in atherosclerosis. For example, activation of nuclear receptors, such as LXR via synthetic agonists (e.g., GW3965, T0901317), can transcriptionally upregulate efferocytic machinery MerTK in macrophages and augment efferocytosis to dampen inflammation [68, 87]. A PPARγ agonist (pioglitazone) was found to restore macrophage efferocytosis of apoptotic neutrophils and normalize the sterile inflammatory response in chronic granulomatous disease models in vivo [70]. In addition, the natural alkaloid columbamine was shown to promote LC3-associated phagocytosis (LAP), thereby enhancing macrophage efferocytosis and accelerating resolution in a murine colitis model [71].
In contrast, efferocytosis in the tumor microenvironment can reinforce immune suppression to promote cancer progression, and pharmacologic inhibitors of efferocytosis (e.g., MerTK) are being developed to boost an-titumor immunity [72]. In pancreatic ductal adenocarcinoma (PDAC) liver-metastasis models, an oral MerTK inhibitor (UNC2250, 10 mg kg⁻¹) was found to improve CD8⁺ T-cell function and reduce metastatic burden [73].
However, systemic administration of an efferocytotic inhibitor (e.g., MerTK) may increase the risks of autoimmunity and tissue injury, as evidenced by lupus-like disease in Mertk-deficient mice and exacerbated autoimmunity in TAM receptor knockout settings [33]. Moreover, the specificity of these small molecule inhibitors still has concerns, and some previously used MerTK inhibitors display polykinase activity, raising the risks of off-target effects and immunotoxicity. For example, BMS-794833 was originally developed against MET/VEGFR2 yet potently inhibits MerTK [74]. Additionally, broad stimulation of macrophages may induce systemic side effects, and LXR agonists frequently induce hepatic steatosis and hypertriglyceridemia, which has hindered clinical translation [88]. In addition, pharmacokinetics and delivery remain practical hurdles; for example, drug penetration into certain tissues (e.g., solid tumors) is often suboptimal, targeting lesion macrophages in atherosclerotic plaques is difficult [89, 90], and these limitations might be resolved using nanomedicine-based platforms.
2.2 Biologics and cytokines
Biologic agents, such as monoclonal antibodies and Fc-fusion proteins, may offer complementary ways to modulate efferocytosis by either removing antiphagocytic brakes or supplying pro-engulfment cues. One of the primary “don't-eat-me” pathways is CD47-SIRPα, which tumor cells exploit to evade clearance [21]. Multiple biological agents, such as anti-CD47 antibodies (e.g., magrolimab) and engineered SIRPα-Fc decoy receptors (e.g., TTI-621, ALX148/evorpacept), have been tested in clinical trials, as they may enhance macrophage recognition and engulfment of tumor cells [76, 77]. In addition, biologics can promote efferocytosis by providing opsonins or mimicking endogenous bridging molecules. Recombinant MFG-E8 protein binds phosphatidylserine on apoptotic cells and tethers them to phagocyte integrins (e.g., αvβ5), which can improve efferocytosis and reduce inflammation in models of sepsis and hemorrhagic shock [44, 66, 78]. Cytokine-based strategies may indirectly enhance efferocytosis by reprogramming macrophages. For example, IL-13 might drive a macrophage IL-10/Vav1/Rac1 circuit that increases phagosome formation and AC uptake during resolution [79].
However, biologics or cytokine-based treatments still face some challenges and biosafety concerns. For example, anti-CD47 antibodies can cause severe anemia because CD47 is highly expressed on red blood cells, SIRPα-directed decoys may have fewer anemia events, and infusion-type reactions predominate [91-93]. In addition, systemically administered antibodies or recombinant proteins also have suboptimal tissue penetration and cell-specific effects [94, 95]. Durable use of these drugs may elicit anti-drug antibodies (ADAs) that lower drug exposure and attenuate efficacy [96]. Systemic administration of cytokines (e.g., IL-4) has produced multisystem adverse events in early trials, possibly due to its overall immunosuppression, making it difficult to titrate the “just-right” dose that augments efferocytosis without off-target effects [97]. Together, while biologics offer receptor-targeting ability and have produced important clinical leads, their delivery issues and biosafety profiles also need to be improved.
2.3 Gene therapy
Gene therapy and gene-modulation strategies are emerging tools to tune efferocytosis by augmenting the phagocytic machinery in phagocytes or removing the inhibitory signals on target cells. Although most applications remain preclinical, proof-of-concept has been established that restoring or engineering efferocytic receptors can enhance AC clearance and dampen inflammation [82]. As a receptor-augmentation paradigm, gene replacement of phagocytic retinal pigment epithelium (RPE) restores outer-segment phagocytosis and improves retinal structure and function in rats, targeting the MERTK gene, which progressed to a phase I trial in 2016, suggesting that supplementing a phagocytic receptor can rescue defective clearance in vivo [81]. While the 2016 Phase I trial demonstrated acceptable ocular and systemic safety, with transient visual improvements in a subset of patients, subsequent-phase peer-reviewed clinical evidence and longer-term efficacy data remain limited, leaving the magnitude and durability of clinical benefit to be established [98].
Conversely, ex vivo CRISPR ablation of CD47 on tumor cells can increase macrophage phagocytosis and augment antitumor immunity in mice, and studies indicate that ~80% CD47 suppression might be required for phagocytosis to outpace tumor growth [83, 84]. In situ dual gene editing via CD47 knockdown plus IL-12 overexpression can reprogram tumor cells into immune-activating hubs, which suppress tumor growth in vivo by driving TAM re-education and boosting efferocytosis and antigen-presentation crosstalk [99]. Editing the phagocyte checkpoint SIRPα (e.g., CRISPR knockout or RNA silencing) renders macrophages “hyper-phagocytic,” improving engulfment of tumor targets and enhancing the performance of engineered macrophage therapies in preclinical studies [84, 85].
Although these results are promising, gene therapy to modulate efferocytosis remains early-stage and faces several challenges. For example, commonly used gene vectors, such as AAV, lack intrinsic tropism for macrophages in defined tissues, making off-target transduction likely unless one leverages cell-specific promoters, engineered capsids, or ligand-decorated carriers [100, 101]. These limitations might be partially resolved by using targeted nanocarriers; for example, it has been reported that collagen IV-targeted NPs can deliver payloads into atherosclerotic plaques, and pH-gated nanoplatforms selectively engage TAMs in acidic compartments [102, 103]. The possible biosafety and immune concerns of gene therapy also require caution. Viral vectors (e.g., AAV) can provoke innate and adaptive immunity, and durable transgene expression in vivo might induce unanticipated consequences. Furthermore, ethical and regulatory considerations for human gene editing, such as ex vivo CD47 editing or highly targeted in vivo editing, add complexity and cost. These hurdles may limit their clinical translation despite encouraging preclinical results. Thus, it is urgent to develop next-generation strategies to achieve more precise, safe, and efficacious modulation of efferocytosis.
3. Nanomedicine-based strategies for precision modulation of efferocytosis
Nanotherapeutics may offer a versatile platform to modulate efferocytosis with improved precision compared to conventional modalities. Nanocarriers can be functionalized to target phagocytic cells in specific tissues, maximizing the therapeutic index while minimizing systemic exposure [104]. Such targeted delivery not only improves pharmacokinetics and lesion retention but also enables combinatorial therapy. For example, NPs can co-deliver a “don't-eat-me” signal inhibitor alongside a pro-engulfment agent, simultaneously blocking immune evasion and triggering efferocytosis in tumors. Moreover, nanocarriers can be engineered to mimic apoptotic cells by decorating their surface with phosphatidylserine or entire cell membranes, thereby enhancing their binding to macrophages and intrinsically activating anti-inflammatory, pro-resolving programs during uptake [105, 106]. Stimuli-responsive nanoplatforms that selectively release drugs in specific pathological conditions (e.g., acidic pH, high ROS) can concentrate the therapeutic effect at disease sites while sparing healthy tissue [103]. In this section, we discuss cutting-edge nanotherapeutic strategies, such as boosting “find-me” or “eat-me” signals, targeting phagocytic receptors and blocking “don't-eat-me” pathways, and tactics for delivering interventions to specific phagocyte subsets (Figure 2, Table 3).
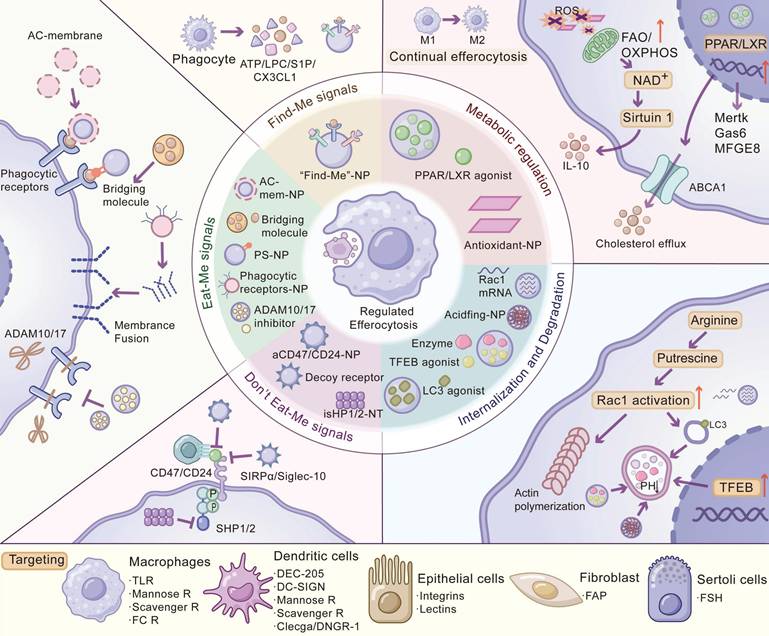
Nanotherapeutic interventions across efferocytosis and cell-specific targeting. The schematic centers on regulated efferocytosis and maps nanomedicine entry points across the process, with targeting cues summarized inline: nanoparticles that present or release ATP/UTP, LPC, S1P, or CX3CL1 (“find-me” NPs), recruit phagocytes to lesions; PS-decorated NPs and apoptotic-membrane-coated carriers mimic exposed PtdSer, while delivery of bridging molecules (Gas6, MFG-E8) or “receptor-NPs” upregulates/supplements MerTK and integrins, membrane fusion donates receptors, and ADAM10/17 inhibition prevents MerTK shedding to preserve uptake capacity; aCD47/aCD24-NPs multivalently block CD47-SIRPα or CD24-Siglec-10, decoy-receptor/peptide-modified NPs sequester inhibitory ligands, and macrophage-targeted SHP1/2 inhibition (iSHP1/2-NT) turns off intracellular brakes to restore phagocytosis; Rac1 mRNA or small-molecule boosters, LC3/LAP and TFEB agonists, acidifying materials, and lysosomal enzyme delivery enhance phagocytic-cup formation, efferosome maturation, lysosomal biogenesis, and efficient cargo digestion to prevent secondary necrosis; metabolic rewiring strategies—antioxidant nanozymes to temper excessive ROS, LXR/PPAR agonists to induce ABCA1-mediated cholesterol efflux and sustain MerTK, promotion of FAO/OXPHOS and the NAD⁺-sirtuin axis, and support of arginine→putrescine production to stabilize Rac1-actin dynamics—enable multi-round uptake and pro-resolving cytokines (e.g., IL-10); finally, receptor/ligand handles guide disease-site-specific delivery and combinations: macrophages (TLRs, mannose receptor, scavenger receptors, FcR), dendritic cells (DEC-205, DC-SIGN, mannose receptor, CLEC9A/DNGR-1), epithelial cells (integrins, lectins), fibroblasts (FAP), and Sertoli cells (FSH receptor). Abbreviations: PtdSer: phosphatidylserine; AC-mem: apoptotic cell membrane; LAP: LC3-associated phagocytosis; FAO: fatty acid β-oxidation; OXPHOS: oxidative phosphorylation.
Nanomedicine-based strategies for modulating efferocytosis
| Strategy Category | Applied nanomedicines | Target pathways | Formulations | Target cells | Biological outcomes | Reference |
|---|---|---|---|---|---|---|
| Regulation of “find-me” signals | PTX@NPpD-ATP | ATP/UTP, S1P, CX3CL1, LPC; dendritic-cell recruitment; antitumor immunity | Material: PLGA; Size: ~190 nm; Decorated: ATP Cargo: paclitaxel (PTX) | Dendritic cells; Macrophages | Enhance phagocyte recruitment to disease sites | [107] |
| S1P-PS-MMV@SPD | S1P (find-me); phosphatidylserine (eat-me) | Material: Macrophage-membrane nanovesicles (MMV); Size: ∼416.4 ± 1.9 nm; Decorated: S1P and PS Cargo: spermidine | Macrophages | Promote macrophage recruitment and uptake | [108] | |
| Regulation of “eat-me” signals | PSNP@DEM | Phosphatidylserine (PS); anti-inflammatory efferocytic signaling | Material:PLGA; Size:150-200 nm; Decorated: PS Cargo: dexamethasone | Alveolar/lung macrophages | Increase macrophage uptake; reduce inflammation; alleviate lung injury | [105] |
| Engineered neutrophil apoptotic bodies (eNAB) | Phosphatidylserine and other native eat-me signals | Material: Neutrophil apoptotic-body membrane cloaked mesoporous silica nanoparticles; Size: ~300 nm; Decorated: Neutrophil apoptotic-body membrane | Macrophages | Enable rapid binding/ingestion; reprogram toward pro-resolving phenotype | [109] | |
| mBM-MSC-EV | GAS6; MerTK signaling | Material: Extracellular vesicles derived from MSC; Size: 100-200 nm; Cargo: enrichment of GAS6 | Hepatic macrophages | Accelerate apoptotic-cell clearance; dampen inflammation | [67] | |
| Bispecific tumor-transforming nanoparticles (BiTNs) | Eat-me ligands; tumor-binding moieties | Material: PEG-PLGA; Size: ~100 nm; Decorated: conjugated with anti-HER2 and SLAMF7 | Macrophages; Tumor cells | Physically bridge targets to induce efferocytosis/phagocytosis | [110] | |
| Regulation of phagocytic receptors | MerTK-displaying hybrid-membrane nanovesicles (HMNVs) | MerTK receptor | Material: Hybrid membranes from MerTK-overexpressing macrophages; Size: 150-200 nm; Decorated: MerTK | Lesional macrophages | Donate functional MerTK; enhance local efferocytosis | [111] |
| PS-lipos-AuNC@LXR agonist | LXR; MerTK expression | Material: silver nanocubes and liposomes; Size: 100-150 nm; Decorated: PS Cargo: T0901317 | Macrophages | Upregulate MerTK; promote apoptotic-cell uptake and anti-inflammatory programs | [112] | |
| D&G@NPEOz | ADAM17 (MerTK shedding); LXR | Material: pH-responsive neutrophil membrane-based nanoplatform (NPEOz); Size: 50 nm; Cargo:GW280264X (an ADAM17 inhibitor) and desmosterol (an LXR agonist) | Brain phagocytes (e.g., microglia) | Prevent MerTK shedding; enhance efferocytosis (e.g., erythrophagocytosis) | [113] | |
| Efferocytosis-Inhibiting Nano-BMS | MerTK | Material: PLGA and mPEG-PLGA; Size: ~106 nm; Cargo: MerTK inhibitor (BMS777607) | Tumor-associated macrophages (TAMs) | Block efferocytosis to limit tumor immune evasion/growth | [114] | |
| Regulation of “don't eat me” signals | aCD47-DMSN | CD47-SIRPα axis; calreticulin ('eat-me') exposure | Material: mesoporous silica nanoparticle; Size: ~152 nm; Decorated: anti-CD47 antibodies; Cargo: doxorubicin | Tumor cells; Macrophages | Multivalent CD47 blockade; induce eat-me signaling; enhance phagocytosis | [115] |
| Single-walled carbon nanotubes (SWNTs) | SIRPα-SHP1 signaling | Material: PEG-functionalized SWNTs; Size: diameter of 5-6 nm; Cargo: inhibitor of SHP-1 | Plaque macrophages | Reactivate efferocytosis in plaques while minimizing anemia risk | [41] | |
| ZrMOF/C@P | CD47 checkpoint; STING pathway | Material: zirconium-based metal-organic framework and RAW264.7 cell membrane; Size: 190.6 nm; Decorated: Pep20, MMP2, and M2pep Cargo: STING agonists | Tumor cells; Macrophages | Block CD47; enhance phagocytosis with reduced hematologic toxicity | [116] | |
| Nano lysosome-targeting chimeras (LYTACs) | CD24-Siglec-10 axis | Material: LYTAC and N-acetylgalactosamine(GalNAc); Size: ~200 nm; Decorated: CD24 antibodies | Tumor cells; Macrophages | Disrupt CD24-Siglec-10; enhance macrophage phagocytosis | [117] | |
| Regulation of internalization and degradation | Silica nanoparticles | Rac1 GTPase; actin polymerization | Material: Silica | Macrophages | Boost Rac1 activity; improve phagocytic internalization | [118] |
| Engineered lysosome-acidifying nanoparticles | Lysosomal acidity; hydrolase activity | Material: poly(butylene tetrafluorosuccinate-co-succinate) polyesters; Size: ~100 nm | Macrophage lysosomes | Re-acidify lysosomes; recover degradative capacity | [119] | |
| Metabolic regulation for continual efferocytosis | MMR-Lobe | PPARγ; ABCA1/ABCG1; cholesterol efflux | Material: thiolated glycol chitosan; Size: 300-350 nm; Cargo: lobeglitazone | Macrophages | Promote cholesterol efflux; anti-inflammatory gene programs; sustain efferocytosis | [120] |
| AOzyme@ACM | ROS scavenging; mitochondrial integrity | Material: silica NPs core, Cu NPs and apoptotic cell membrane; Size: ~150 nm; Decorated: apoptotic cell membrane | Macrophages | Reduce oxidative stress; reactivate continual efferocytosis | [121] |
3.1 Strategies for regulating efferocytotic signaling pathways
3.1.1. Regulation of “find-me” signals
ACs actively release “find-me” cues, such as ATP/UTP, lysophosphatidylcholine (LPC), CX3CL1 (fractalkine), and sphingosine-1-phosphate (S1P), that establish chemotactic gradients to recruit phagocytes to sites of cell death. Based on this biology, nanoplatforms designed to augment or mimic find-me signaling and to enhance phagocyte recruitment have been reported. For example, NPs that present or release ATP in tumors increase intratumoral dendritic cell infiltration and potentiate antitumor immunity, which can be combined with checkpoint blockade therapy [107]. In parallel, engineered efferocytosis-mimicking nanovesicles that co-display S1P (find-me) and phosphatidylserine (eat-me) can enhance macrophage recruitment and AC uptake, and S1P-enriched extracellular vesicles have also been shown to drive macrophage chemotaxis [108, 122]. By strategies that aim to deliver or amplify “find-me” signals, nanoplatforms improve the homing ability of phagocytes to injured tissues. The chief advantage of this strategy is that it tackles the first limiting step in clearance: insufficient phagocyte presence.
However, limitations arise from the potential overstimulation leading to excessive inflammation if not precisely controlled. Another limitation is that simply recruiting phagocytes may not guarantee effective efferocytosis. For example, in atherosclerosis, simply recruiting more phagocytes to lesions might aid in clearing apoptotic cells, but it could also expand foam-cell burden and inflammation if not paired with restoration of lipid handling and efferocytic competence of those phagocytes [123]. Thus, find-me enhancement is most useful when phagocyte scarcity is a major bottleneck (e.g., “cold” tumors). Overall, this strategy excels in enhancing phagocyte localization and can potently amplify immune responses, but it should be deployed with mechanisms to contain their effects spatially and paired with complementary strategies to ensure recruited phagocytes can perform their clearance function.
3.1.2. Regulation of “eat-me” signals
The recognition (“eat-me”) phase of efferocytosis hinges on the exposure of phosphatidylserine (PS) on AC surfaces, which serves as a ubiquitous eat-me signal [124]. Externalized PS binds to phagocyte receptors directly or via opsonins and bridging molecules (e.g., Gas6, Protein S, MFG-E8), triggering engulfment signaling [13]. Nanotherapeutic strategies have increasingly focused on PS mimicry - decorating NP surfaces with PS or PS analogs to impersonate ACs and actively engage efferocytotic pathways. This approach exhibits two major advantages: (i) directing nanocarriers to phagocytes (macrophages, dendritic cells, etc.) and (ii) activating anti-inflammatory responses in those phagocytes akin to physiological efferocytosis [105, 106]. For example, PS-decorated PLGA NPs showed increased accumulation in lung macrophages and reduced inflammatory readouts in LPS-induced acute lung injury [105]. These findings align with earlier work showing that PS-presenting liposomes are preferentially taken up by macrophages and reprogram them toward a pro-resolving phenotype [65].
Besides PS coating, another approach is AC biomimetic NPs, including cell membrane-coated NPs and apoptotic body-like vesicles. By cloaking nanocarriers with the membrane of ACs, one can present a natural array of eat-me signals to phagocytes: engineered apoptotic vesicles derived from neutrophils (e.g., engineered neutrophil apoptotic bodies) can modulate skew macrophages toward a reparative, anti-inflammatory phenotype and reduce inflammatory cytokines in injury models, such as myocardial infarction, providing indirect evidence for improved efferocytosis [109]. By impersonating the body's own apoptotic cells, such biomimetics ensure that they are rapidly bound and ingested by phagocytes.
To potentiate recognition, strategies involving bridging molecule enhancement have been reported. Mesenchymal stem cell-derived extracellular vesicles (MSC-EVs) enriched in GAS6 accumulated in the injured liver and activated MerTK signaling in hepatic macrophages, as evidenced by increased p-MerTK by Western blot, which in turn accelerated the clearance of apoptotic hepatocytes, quantified by fluorescence-based efferocytosis assays in vitro: pHrodo-labeled apoptotic cells were co-incubated with BMDMs and quantified by fluorescence microscopy and dampened inflammation [67]; EVs with MFG-E8 expression also showed the ability to activate efferocytosis, with direct quantification of efferocytosis index via flow cytometry and confocal imaging [125]. This illustrates how supplementing the local environment with bridging factors can kick-start latent efferocytosis pathways. Furthermore, an interesting design is to use bispecific constructs, named bispecific nanobioconjugates, that carry one moiety binding tumor cells and an “eat-me” ligand to promote macrophage phagocytosis [110]. Such dual-targeted nanosystems can physically bring tumor cells and macrophages together to induce efferocytosis. This approach offers unique advantages in diseases characterized by defective recognition of apoptotic cells, such as atherosclerosis and autoimmune diseases. It should be noted that in neurodegenerative diseases, enhancing eat-me signals helps microglia recognize and clear dying neurons or protein aggregates. However, the overactivation of microglia could harm healthy neural connections; thus, deploying eat-me signal mimics must be done cautiously [126].
In addition, in the context of cancer, simply increasing efferocytosis via PS-coated nanoparticles could risk further polarizing TAMs toward an immunosuppressive state; encouragingly, innovative bispecific nanoconjugates offer a way to harness eat-me signals against live cancer cells. The challenges of this strategy include ensuring stable PtdSer exposure and potential saturation of phagocytic capacity, which could impair continual efferocytosis [27]. In summary, nanotherapies that amplify eat-me signaling offer a highly biomimetic and modular route for phagocyte targeting and pro-resolution immunoregulation, but they require careful calibration of signal strength and companion immunostimulatory payloads in settings where immune stimulation is desired, particularly cancer.
3.1.3. Regulation of phagocytic receptors
Regulation of (e.g., enhancing or inhibiting) phagocytic receptor activities has shown therapeutic promise. Phagocytic receptors such as MerTK are attractive checkpoints for therapeutic modulation. Nanotherapeutics provide multifaceted tools to enhance signaling through receptor delivery or upregulation [111] or inhibit or tune signaling at specific sites [127]. By targeting these receptors, one can directly influence the cell-intrinsic “appetite” of phagocytes for apoptotic prey, either sharpening it to clear dangerous debris or damping it to allow controlled antigen release.
MerTK (Mer tyrosine kinase) has garnered particular interest as a nodal point in efferocytosis signaling and a drug target. In diseases such as atherosclerosis, enhancing MerTK-driven clearance can help resolve necrotic plaques, whereas in cancer, inhibiting MerTK on TAMs can prevent them from clearing tumor cell corpses and help restore antitumor immune responses [72, 111]. Consequently, nanotherapeutics have been harnessed to modulate MerTK in both directions. A striking recent example is the targeted delivery of MerTK protein via a cell membrane-engineered NP. Hybrid nanovesicles coated with macrophage membranes overexpressing MerTK, these vesicles fused with lesional macrophages, effectively donating functional MerTK receptors to them, as confirmed by Western blot showing restored MerTK protein levels. Functionally, enhanced efferocytosis was directly quantified in vitro by flow cytometry (percentage of F4/80⁺ and CellTracker-labeled apoptotic-cell double-positive macrophages), complemented by confocal imaging-based assessment of macrophage uptake of fluorescently labeled apoptotic cells [111].
Also, the delivery of drugs that can enhance MerTK signals via nanocarriers is being explored. The nuclear receptor LXR drives MerTK expression and its ligands production [68], and delivery of an LXR agonist (T0901317) using PS-coated gold nanocarriers could increase macrophage MerTK expression and promote AC uptake [112]. Another strategy is preventing the loss of MerTK activity. In the inflammatory state, enzymes (e.g., ADAM17) can cleave the ectodomain of MerTK off the cell surface and thus inactivate its efferocytic function [40, 64]. To counteract this, neutrophil-mimicking nanoparticles (NPs) were constructed to deliver ADAM17 inhibitors for treating intracerebral hemorrhage, and such nanomedicine could inhibit ADAM17 and prevent local MerTK loss, as shown by Western blot (reduced ADAM17) and ELISA (decreased soluble MerTK), thereby boosting erythrophagocytosis (a form of efferocytosis) in the hemorrhagic brain with direct quantification in vitro by flow cytometry (fluorescent apoptotic RBCs co-incubated with BV2 cells) and visualization by confocal microscopy [113].
Conversely, strategies that aim to block phagocytic receptors can be beneficial in treating cancers. It has been found that TAMs suppress antitumor immunity through MerTK-dependent efferocytosis. Inhibiting MerTK signaling in the tumor microenvironment might convert immunosuppression into immunogenic responses [128]. Nanoformulations of MerTK inhibitors (e.g., UNC2025, BMS-794833) allow them to be delivered specifically to TAM-rich regions, minimizing off-target effects [114, 129]. Glycopolymeric NPs encapsulating a MerTK tyrosine kinase inhibitor (UNC2025) were shown to preferentially accumulate in TAMs and block efferocytosis to slow tumor growth [129]. As a TAM family receptor, Axl (together with MerTK and TYRO3), has also been implicated in efferocytosis of cancer cells and can be targeted [69]. TAM family receptors can be blocked using noncoding RNAs; for example, miR-34a suppresses AXL expression via targeting its 3′-UTR [130], and in a phase-1 trial, its liposomal mimic MRX34 achieved dose-dependent modulation of validated miR-34a targets (e.g., BCL2, CTNNB1, DNAJB1) with dexamethasone premedication, but it was halted for immune-mediated adverse events, underscoring the need for safer delivery [131]. Likewise, delivery of AXL siRNA by melittin-derived peptide NPs reduces metastatic burden with limited toxicity in different cancer models, including ovarian and uterine models [132]. Although these studies did not directly assess the impact of AXL blockade on efferocytosis, these promising findings may establish translatable precedents and motivate analogous designs that modulate AXL on tumor-associated phagocytes to influence efferocytosis in the tumor microenvironment. Besides, other phagocytic receptors, such as integrins, scavenger receptors, and immunoglobulin superfamily members like TIM-4, LILRB, and stabilin-2, are also vital for regulating efferocytosis, and whether they still have therapeutic potential is worthy of further investigation [24].
Rather than acting on signals emitted by apoptotic cells, this approach directly modulates receptors on phagocytes to reshape the execution of efferocytosis. Its comparative advantage is mechanistic precision: by upregulating or inhibiting a specific phagocytic receptor, one can augment or attenuate the phagocyte's intrinsic “appetite” for apoptotic targets in a controlled way. However, this strategy faces important limitations, including receptor-network complexity, potential compensatory pathways, and the challenges of targeting specific cell populations. First, the optimal receptor to target is disease dependent: in MASH mice, efferocytosis deficiency is mainly caused by TIM4 deficiency, while in atherosclerotic mice, efferocytosis deficiency in plaques is mediated by reduced MerTK [40, 133]. Second, because phagocytes express numerous redundant receptors (MerTK, Axl, TIM-4, integrins, scavenger receptors, etc.) for apoptotic cells [24], focusing on a single receptor may be insufficient in some contexts or may require coordinated modulation of complementary steps. Safety also warrants careful consideration: systemic enhancement of receptor-mediated clearance could theoretically increase inappropriate or excessive removal [134]. In this context, nanocarrier targeting offers a pragmatic mitigation strategy by confining receptor modulation to intended lesions and/or phagocyte subsets, thereby reducing systemic liabilities. Overall, nanocarrier-enabled phagocytic receptor modulation stands out for its directed mechanism and bidirectional tunability (up or down), but durable translational success will hinge on high-precision targeting and calibrated control of efferocytotic intensity to avoid unintended consequences at either extreme.
3.1.4. Regulation of “don't eat me” signals
To avoid being mistakenly engulfed, healthy cells highly express surface proteins (“don't-eat-me” signals) that actively inhibit phagocytosis. For example, CD47 is ubiquitously expressed and engages SIRPα receptors on macrophages to deliver a potent anti-phagocytic signal. Many pathological cells, notably cancer cells, can exploit this pathway by overexpressing CD47 to cloake themselves from immune clearance [7, 135]. Blocking CD47-SIRPα interactions by nanotherapies has emerged as a promising strategy to unleash phagocytes against targets, such as tumor cells or uncleared apoptotic debris. Anti-CD47-decorating NPs enable multivalent blockade of the CD47-SIRPα axis and co-delivery of chemotherapeutics (e.g., doxorubicin) to induce prophagocytic “eat-me” cues (e.g., calreticulin) and enhanced phagocytosis, as quantified in vitro by flow cytometry (percentage of CFSE⁺eFluor670⁺ double-positive BMDMs among total eFluor670⁺ BMDMs) [115]. Similarly, anti-CD47-loaded calcium-carbonate NPs (aCD47@CaCO3) were incorporated into a sprayable fibrin gel, and the degradation of CaCO3 NPs in the acidic tumor microenvironment led to the release of anti-CD47 and reduced tumor recurrence in melanoma post-surgical resection models [136]. Another interesting strategy is the “Trojan horse” NP. SHP1, a downstream phosphatase of the SIRPα signaling cascade, can transmit the “don't eat me” signal [137]. Macrophage-targeting single-walled carbon nanotubes (SWNTs) were used to deliver SHP1 inhibitors [41], which reactivated macrophage efferocytosis in plaques, as demonstrated by direct quantification in vitro (flow cytometry-based efferocytosis assay) and in vivo (reduced apoptotic cell accumulation in plaques via histological analysis) and without causing anemia in atherosclerotic mice and large animal (porcine) models [9, 41]. This exemplifies how nanotherapeutics can add a layer of specificity; unlike free anti-CD47 antibodies, nanotherapeutics can concentrate the anti-“don't-eat-me” effect only where it is needed (e.g., within TAMs or plaque macrophages).
Another approach is to use decoy receptors or peptides presented by NPs. A peptide derived from SIRPα's binding domain (Pep20) has been developed that binds CD47 without activating the inhibitory signal [138]. Surface modification of Pep20 on metal-organic framework NPs loaded with STING agonists has been shown to have antitumor potential by enhancing macrophage phagocytosis without causing anemia [116]. Functionalized NPs with fragments or membrane-presented forms of SIRPα may act as sponges, binding to CD47 on target cells and masking it. Representative samples include liposomes displaying the Fc-fused high-affinity SIRPα variant CV1 (Fc-CV1) on their surfaces [139]; microwave-responsive Prussian blue NPs cloaked with macrophage membranes overexpressing SIRPα (SIRPα-M@nanoPB) [140]; and EVs engineered to display SIRPα (EV-SIRPα) as nanocarriers [141].
Turning off inhibitory signals on phagocytes can convert them into more aggressive “hunters” of diseased cells. In addition to CD47, some other “don't eat me” signals are being targeted as well. CD24, which can bind Siglec-10 on TAMs, was recently identified as an antiphagocytic signal in multiple cancers, such as ovarian cancer [142] and hematological malignancies [143]. Recently, CD24-targeting therapies have gained traction, and multiple anti-CD24 antibodies have displayed a role in enhancing macrophage phagocytosis in preclinical reports and are going into early clinical trials [144, 145]. NP-based strategies can improve efficacy while reducing the side effects of antibody therapy and offer the potential for straightforward combinatorial treatment regimens through simple formulation adjustments. For instance, anti-CD24 antibodies conjugated to endocytic nanospheres have been developed to disrupt the CD24-Siglec10 axis, enhance macrophage phagocytosis and suppress tumor growth [117]. Ongoing research is extending this paradigm to solid tumors, often combining CD47/CD24 blockade with other therapies (e.g., cytotoxic drugs or immune checkpoint inhibitors) for synergistic effects [146].
Blocking inhibitory phagocytic axes such as the CD47-SIRPα pathway has become a prominent therapeutic strategy; antibodies and SIRPα-based decoy proteins targeting this axis have entered clinical trials, supporting its translational relevance as a druggable target [147]. The major limitation of this strategy is the potential for collateral damage to healthy cells that also express CD47 (notably red blood cells). Nanoparticle targeting offers a potential mitigation route by increasing local exposure in tumors or disease-relevant myeloid niches, thereby widening the therapeutic window and potentially reducing anemia risk. However, evidence for substantial mitigation of systemic toxicities remains largely preclinical and warrants rigorous clinical validation. Accordingly, in diseases requiring systemic therapy (e.g., hematologic malignancies), the application of this strategy demands careful optimization of its risk-benefit profile through dosing schemes, rational combinations, and proactive toxicity management [147]. Overall, nanotherapeutic blockade of anti-phagocytic signals is most compelling in cancer and selected localized chronic diseases where local delivery or cell-directed targeting can enhance specificity. Furthermore, patient stratification (e.g., selecting tumors with high CD47 expression) may be critical to maximize benefit-risk. In disease without clear “don't-eat-me” signal dependence, the anticipated therapeutic benefit is likely limited, while risks such as anemia or other hematologic toxicities remain substantial.
3.1.5. Regulation of internalization and degradation
After the recognition and binding stages, phagocytes can internalize the target cells and degrade them via phagolysosomes and lysosome routes [13]. This stage can also be inefficient when phagocytes are “overloaded” and/or their lysosomal function is impaired, resulting in abortive efferocytosis, such as incomplete digestion and secondary necrosis [148]. The small GTPase Rac1 governs actin polymerization during phagocytosis, and activation of Rac1 drives pseudopod extension and AC engulfment. Currently, nanotherapeutics are being developed to ensure that engulfment and processing proceed optimally. One emerging approach is to use macrophage-targeted NPs to modulate the RAC1-actin module that powers efferocytosis. For example, lipid nanoparticle (LNP)-mediated delivery of Rac1 mRNA to monocytes/macrophages could improve antifungal pathogen clearance in vivo, supporting the idea that boosting Rac1 can augment phagocyte internalization [118]. Interestingly, NP itself might affect the Rac1 pathway due to its intrinsic properties, and synthetic silica NPs were shown to induce strong Rac1 activation and actin polymerization in macrophages [149]. Efferocytotic macrophages can convert AC-derived arginine/ornithine to putrescine, which stabilizes Mcf2 mRNA and increases Rac1 activity to enable continual engulfment [29]. In this aspect, macrophage-targeted nanotherapy may in turn activate the Rac1 signaling pathway to restore efficient internalization during efferocytosis.
Nanomedicine-based approaches for enhancing lysosomal degradation capacity have also been reported. In advanced atherosclerotic lesions, repeated uptake of ACs and lipids can induce macrophage lysosomal stress, which impairs lysosomal acidity and degradative capacity [150]. Acidifying NPs, typically made of acidic or carboxyl-terminated polymers, can accumulate in endolysosomes and donate protons to re-acidify lysosomes and recover hydrolase activity [119]. A complementary strategy is to drive lysosomal biogenesis. TFEB is the principal regulator of lysosomal biogenesis, and it activates the coordinated lysosomal expression and regulation (CLEAR) network to increase lysosomal abundance and acidification [151]. TFEB agonists, such as trehalose, have been shown to induce lysosomal biogenesis and confer protection in disease models (e.g., atherosclerosis) [152]. Nanocarrier-based approaches can further enhance efficacy via targeted delivery of TFEB inducers that promote TFEB nuclear translocation, increasing their intralysosomal effective concentration and amplifying CLEAR-mediated degradation. Beyond small molecules, targeted lysosome enzyme supplementation might be feasible via delivery routes based on mannose- or mannose-6-phosphate, suggesting a potent path for macrophage-directed hydrolase augmentation [153].
The process of AC process and disposal is critical for sustaining efferocytosis. LC3-associated phagocytosis (LAP) is a non-canonical pathway in which LC3 is conjugated to single-membrane phagosomes, thereby accelerating fusion with lysosomes, and boosting LAP might couple phagosome maturation with efficient lysosomal clearance. Small molecule compounds (e.g., columbamine) can enhance LAP, and macrophage efferocytosis has been reported: researchers have performed direct in vivo quantification of systemic efferocytosis by measuring the CMFDA-positive apoptotic-cell-derived signal in the liver and spleen by flow cytometry and in tissues using TUNEL labeling of ACs together with macrophage staining to assess in situ efferocytosis [71]; nanoformulation of such agonists is a plausible strategy to enhance LAP signaling in phagocytes of lesion sites. Unlike canonical autophagy, LAP is mainly independent of the AMPK-mTORC1-ULK1 nutrient-sensing axis [25]. Although mTOR inhibitors or general autophagy inducers may enhance the overall lysosomal capacity [154], they might not be equated with LAP activation for efferocytosis intervention, given LAP's mTOR-independent control logic. Together, by fine-tuning the intracellular phase of efferocytosis, these nanomedicines may prevent secondary necrosis and pro-inflammatory leakage.
Augmenting the internalization and degradation steps of efferocytosis may be particularly beneficial in diseases where phagocytes are overwhelmed by apoptotic or necrotic debris and post-engulfment processing becomes rate-limiting, such as advanced atherosclerosis and neurodegenerative disorders [5, 155]. In cancer, however, the same strategy is highly context-dependent because efficient efferocytosis by tumor-associated macrophages often induces IL-10/TGF-β-linked immunosuppressive programs that can dampen antitumor immunity. Accordingly, for therapies aiming to enhance antitumor immunity, accelerating macrophage internalization/degradation is usually not favored in cancer. A key limitation of this strategy is sustainability and cellular capacity: accelerating internalization/degradation without concomitant support for metabolic and recycling programs may impose lipid, lysosomal and energetic burdens, leading to a transient increase in clearance followed by a functional decline in continual efferocytosis [156]. Thus, interventions that boost this phase may be more durable when paired with support for cholesterol homeostasis and mitochondrial adaptation.
3.1.6. Metabolic regulation for continual efferocytosis
Phagocytosis of ACs imposes significant metabolic demands on phagocytes, as a bolus of AC-derived materials, such as proteins, lipids, cholesterol, and nucleic acids, must be efficiently processed [27, 28]. When this metabolic adaptation is perturbed, macrophages lose capacity for subsequent rounds—internalizing one apoptotic cell but failing on the next—resulting in uncleared corpses and impaired resolution [29]. Strategies of metabolic regulation can sustain the ability of phagocytes to perform continual efferocytosis. One of the prominent targets is the LXR-PPAR pathway, since LXR upregulates ABCA1/G1 transporters for cholesterol efflux and preventing lipid overload [157]. Nanotherapeutics delivering cholesterol exporters or agonists of nuclear receptors (LXRα/β or PPARγ) have been utilized. By activating LXR/PPAR pathways, these treatments upregulate ApoE and ABCA1/G1 transporters to efflux excess cholesterol and induce an anti-inflammatory gene profile, effectively rejuvenating macrophages' ability to continue efferocytosis [120, 158, 159]. Designing LXR-agonist nanocarriers for macrophage receptor uptake yields selective macrophage delivery and circumvents hepatocyte uptake, thereby effectively blocking the effect of LXR agonists on hepatic adipogenesis [160], suggesting the superiority of cell-specific metabolic therapy.
Macrophages metabolize AC corpse-derived arginine via arginase-1 (Arg1) and ornithine decarboxylase (ODC) into polyamines (e.g., putrescine) [29], which can sustain the Rac1 signaling required for actin cytoskeletal rearrangements in continual efferocytosis [29]. Therapeutic enhancement of polyamine production or supplementation with exogenous polyamines has been shown to restore efferocytic capacity in macrophages facing heavy apoptotic loads [29]. To overcome the rapid turnover and short half-life of polyamines in vivo, NP-based formulations of polyamines or their precursors are being explored. Polymeric microparticles co-delivering the polyamine spermine and α-ketoglutarate, an intermediate of the tricarboxylic acid (TCA) cycle, have been found to reprogram macrophages toward a pro-resolving phenotype that is conducive to efferocytosis [156, 161]. By reprogramming cellular metabolism, such nanomedicines ensure that phagocytes have sufficient energy and biosynthetic capacity to perform continual efferocytosis.
Macrophages undergoing efferocytosis shift toward enhanced oxidative metabolism (increased fatty acid β-oxidation and OXPHOS) to supply ATP and pro-resolving molecules (e.g., IL-10) production [156, 162]. However, continual efferocytosis requires tight control of mitochondrial membrane potential and reactive oxygen species (ROS) [163]. Antioxidant nanozymes that scavenge excess ROS preserve mitochondrial integrity and sustain or reactivate efferocytosis in vivo [121]. For example, cerium oxide nanozymes cloaked with apoptotic neutrophil membranes reduced macrophage oxidative stress and reactivated continual efferocytosis, thereby accelerating inflammatory resolution and bone repair [164]. During efferocytosis, the NAD⁺-SIRT1 axis is required for robust IL-10 production [162, 165], and strategies that aim to bias macrophages toward an enhanced oxidative phenotype by activating PPAR or AMPK pathways may boost continual efferocytosis and inflammation resolution. Nanotherapeutics can be designed for targeted delivery of metabolic drugs, nutrient precursors, or instructive signaling molecules to boost mitochondrial fatty acid β-oxidation, polyamine biosynthesis, or cholesterol efflux.
The chief advantage of metabolic interventions is their potential to restore and sustain phagocyte “endurance”, thereby supporting long-term clearance of apoptotic cells and inflammatory resolution beyond a single engulfment event. Because these approaches frequently rely on transcriptional programs and organelle homeostasis (e.g., nuclear receptor-driven cholesterol efflux pathways), metabolic rewiring may take time and is therefore most pertinent to chronic settings featuring phagocyte overload or fatigue, such as atherosclerosis and chronic inflammatory/degenerative diseases. However, the ubiquity of metabolic pathways mandates tight specificity: LXR/PPAR signaling and redox balance operate across many cell types, so nontargeted modulation can dilute on-site efficacy and introduce systemic liabilities (for example, LXR agonist-linked hepatic lipogenesis and hypertriglyceridemia) [88]. Targeted delivery has therefore been proposed to avoid systemic metabolic side effects and expand the therapeutic window. In cancer, whether to promote macrophage cholesterol efflux is context dependent: in a metastatic ovarian cancer model, cholesterol efflux has been reported to drive tumor-promoting TAM programs [166]. Consistently, efferocytosis itself can reprogram macrophages toward immunosuppressive states.
Conversely, in other tumor settings, increased macrophage ABCA1 activity has been reported to reduce efferocytosis without influencing phagocytosis, illustrating that “ABCA1/LXR-linked” programs can also align with antitumor outcomes depending on the mechanism and cellular state [167]. Importantly, ABCA1 has been summarized as a facilitator of efferocytosis-related signaling in nontumor contexts, underscoring the need to choose directionality (promote versus restrain efflux programs) based on mechanism-of-action goals, tumor context, and TAM lineage/state. In summary, metabolic reprogramming strategies can prolong and potentiate the efferocytosis process by addressing the less visible but crucial constraint of phagocyte endurance; they may form a backbone for chronic inflammatory disease therapy and synergize with direct efferocytosis enhancers (find/eat-me cue modulation or receptor/engulfment agonism) to improve both uptake and processing.
3.2 Strategies for targeting diverse phagocyte types
In addition to the strategies that focus on which signals or pathways to modulate, an equally important consideration is which phagocytic cells are being targeted. Different phagocyte types and/or their tissue locations require different delivery strategies to achieve specific and efficient modulation. Nanotherapeutics can be engineered for targeting particular cell types by leveraging unique surface markers, tropism, or anatomical access routes [168-170]. In this section, we briefly discuss the strategies tailored to professional phagocytes (e.g., macrophages, DCs) and nonprofessional phagocytic cells (e.g., epithelial cells, fibroblasts, Sertoli cells).
3.2.1. Professional phagocytes
Macrophages
Macrophages are primary effectors of efferocytosis in most tissue types [171]. It is well documented that systemically administered NPs are mainly sequestered by the mononuclear phagocyte system, particularly Kupffer cells in the liver and red pulp macrophages in the spleen [172]. Instead, NPs with ligand decoration can actively target different macrophage subsets. M2-like macrophage subsets, such as TAMs, highly express the mannose receptor (CD206), and mannose-decorated NPs showed increased uptake in these macrophages [173]. Folate receptor-β (FRβ) is often upregulated on activated macrophages in inflamed tissues and can be targeted via folic acid modification. For example, [18F]fluoro-PEG-folate selectively images FRβ+ synovial macrophages in rheumatoid arthritis [174]. Antibodies or antibody fragments to macrophage scavenger receptors (MARCO, SR-A) or integrins (such as CD11b) have been attached to NP surfaces to preferentially target macrophages in plaques or sites of infection [175, 176].
The biomimetic/signal mimicry strategy involves wrapping NPs with signal molecules or membranes from cells that macrophages routinely interact with. For instance, PS-mimicking nanotherapeutics and apoptotic body biomimetics have enhanced uptake by macrophages [105, 177]. Coating PLGA NPs with bacterial outer membrane vesicles (OMVs) enables prolonged retention in the peritoneal cavity and efficient macrophage uptake after ip administration [178]. Additional strategies employ the concept of “hitchhiking” on circulating monocytes or macrophages. For example, loading of NPs into autologous monocytes ex vivo followed by reinfusion or injecting NPs endowed with cell-targeting specificity into the body, enabling them to attach to or be internalized by target cells, relies on these cells' natural homing to inflammation sites to ferry the therapy directly for targeting macrophages in injured tissues [179]. The route of administration may also affect the macrophage-targeting effect. Intravenous injection tends to favor uptake by liver and spleen macrophages [180]; inhalation effectively delivers NPs to alveolar macrophages in the lung [181].
Dendritic cells
Dendritic cells (DCs) are also important professional phagocytes that specialize in antigen processing and presentation [171]. Nanotherapeutics targeting DCs are especially useful for immunotherapies (vaccines, cancer immunotherapy) and for inducing tolerance in autoimmune disorders [182, 183]. Unlike macrophages, many DCs reside in or traffic through lymphoid organs and thus require delivery to lymph nodes or skin/mucosal surfaces. NP size influences the efficiency of DC targeting; after subcutaneous administration, small NPs (~20-200 nm) can drain via lymphatics to reach lymph node-resident DCs, while larger particles (~500-2000 nm) tend to remain at the injection site and be taken up by skin DCs that subsequently migrate to the node [184]. A wide array of DC surface receptors, such as C-type lectins (e.g., DEC-205, DC-SIGN, mannose receptor), Fc receptors, and scavenger receptors, have been exploited for broad DC-targeting delivery [185]. For example, PLGA NPs functionalized with an anti-DEC-205 antibody delivered melanoma antigens directly to DEC-205⁺ DCs, improving cross-presentation and boosting CD8⁺ T-cell responses compared with nontargeted formulations in preclinical models [186]. Mannan-coated antigen-loaded NPs have also been used for targeting DCs and have induced tolerogenic programs in autoimmune settings [187]. DC subsets might be targeted via specific surface receptors; for example, conventional dendritic cell 1 (cDC1) can be targeted via an XCL1-XCR1 axis [188]. CLEC9A-targeted nanovaccines exploit DNGR-1-mediated endocytosis in cDC1s to boost cross-presentation and antitumor efficacy [189].
In brief, DC-targeting nanotherapeutics require the comprehensive application of strategies, such as NP size and specific surface labeling (CLEC9A, DEC205, etc.). These interventions can boost the role of DCs in efferocytosis - whether to maintain self-tolerance (clearing apoptotic self-cells without inflammation) or to enhance immunity (efficiently processing tumor cell corpses into antigens for T cells). The result is a more controlled and effective linkage between innate clearance and adaptive immune activation.
3.2.2. Non-professional phagocytes
Epithelial cells
Epithelial cells in diverse organs can function as non-professional phagocytes, engulfing ACs to maintain local tissue homeostasis [1]. For instance, retinal pigment epithelial (RPE) cells in the eye can phagocytose shed photoreceptor outer segments, mammary epithelial cells (MECs) in the postpartum gland can phagocytose apoptotic cells during involution [190, 191], and lung airway epithelial cells engulf ACs via Rac1-dependent signaling [192]. Despite being less efficient than macrophages, these epithelial efferocytes also produce anti-inflammatory mediators that restrain inflammation [1]. Nanotherapeutic strategies are being designed to deliver payloads specifically to epithelial surfaces in the eye, mammary gland, respiratory tract, and intestine. One approach is ligand-directed targeting of epithelial receptors. Integrin-binding peptide (e.g., RGD)-decorated NPs showed enhanced binding and uptake by epithelial cells in target tissues [193]. In the intestine, NPs functionalized with lectins (such as Aleuria aurantia or tomato lectin) can bind to glycosylated apical proteins on M cells and enterocytes, boosting transcytosis across the epithelium [194]. Mucosal surfaces in the respiratory and ocular tract can be targeted via mucoadhesive lectins or antibodies to cell-specific surface proteins, which ensure that NPs can arrive at epithelial cells rather than being cleared [195]. Another strategy is topical or local delivery of complement receptor targeting. For example, aerosolized lipid-NPs can accumulate on airway epithelium, intravesical NPs have increased bladder epithelial retention, and oral/rectal nanoformulations enhance gastrointestinal epithelial access [196-198].
Fibroblasts
Fibroblasts, classically viewed as connective-tissue builders, nonetheless display certain non-professional phagocytic activity, including engulfment of ACs and matrix debris during tissue remodeling and repair [199]. Fibroblasts can internalize extracellular collagen via endocytic routes (e.g., uPARAP/Endo180) for lysosomal degradation, underscoring a capacity for matrix “phagocytosis-like” clearance [200]. In chronic fibrosis and the tumor stroma, activated fibroblasts (myofibroblasts or CAFs) are abundant and can profoundly shape immune infiltration and the local resolution milieu, indirectly influencing efferocytosis [201]. Fibroblast activation protein (FAP), which is highly expressed on activated fibroblasts in fibrotic and neoplastic lesions, enables selective delivery. FAP-targeted NPs have been shown to bind FAP+ fibroblasts in imaging and therapeutic contexts [202, 203]. Given the documented fibroblast uptake of apoptotic bodies and ECM, augmenting these clearance pathways with fibroblast-targeted NPs represents a forward-looking avenue to aid tissue cleanup when professional phagocytes are overwhelmed.
Sertoli cells
Sertoli cells are specialized non-professional phagocytes that continuously engulf apoptotic germ cells and residual bodies during spermatogenesis; this efferocytosis is indispensable for testicular homeostasis and fertility, in which TAM receptors (e.g., MerTK/AXL) play a central role [204]. However, sertoli cells are located behind the blood‒testis barrier (BTB), which limits systemic NP access and poses a major delivery challenge [205]. Nanomedicine may offer potent strategies to surmount the BTB for testicular drug delivery, and certain nanosystems (e.g., branched gold-copper nanocrystals) have been shown to penetrate into seminiferous tubules [206]. Nanomaterial design also influences BTB traversal, and optimizing size, shape and surface functionalization (e.g., FSH-peptide targeting of Sertoli cells) might enhance testis-specific delivery [207, 208]. Moreover, local intratesticular (intratubular) injection bypasses the BTB entirely, depositing nanocarriers directly into seminiferous tubules and leveraging Sertoli cell transcytosis to shuttle them across compartments [209]. By coupling ligand modification with local delivery, nanocarriers may deliver payloads that augment TAM signaling (e.g., MerTK ligands) and potentiate Sertoli efferocytosis, offering a rational strategy for male infertility or orchitis.
4. Therapeutic effects of efferocytosis-modulating nanomedicines
Inefficient clearance of apoptotic cells is a pathogenic feature in many conditions, from chronic inflammatory disorders where uncleared debris drives inflammation to tumors where phagocytic clearance paradoxically supports immune evasion. Nanotherapeutics designed to enhance or recalibrate efferocytosis are emerging as innovative interventions across these diverse pathologies. In this section, we discuss the therapeutic role of efferocytosis-modulating nanomedicines in diverse diseases, such as cardiovascular disease, autoimmune disease, neurodegenerative disease, inflammatory disorders, and cancers (Figure 3, Table S1).
Efferocytosis-modulating nanotherapeutics across disease settings: goals and actionable pathways. The radial layout summarizes therapeutic objectives and tractable pathways for efferocytosis-centered nanomedicine across five disease classes. Abbreviations: TAM: Tyro3/Axl/MerTK receptor family; LAP: LC3-associated phagocytosis.
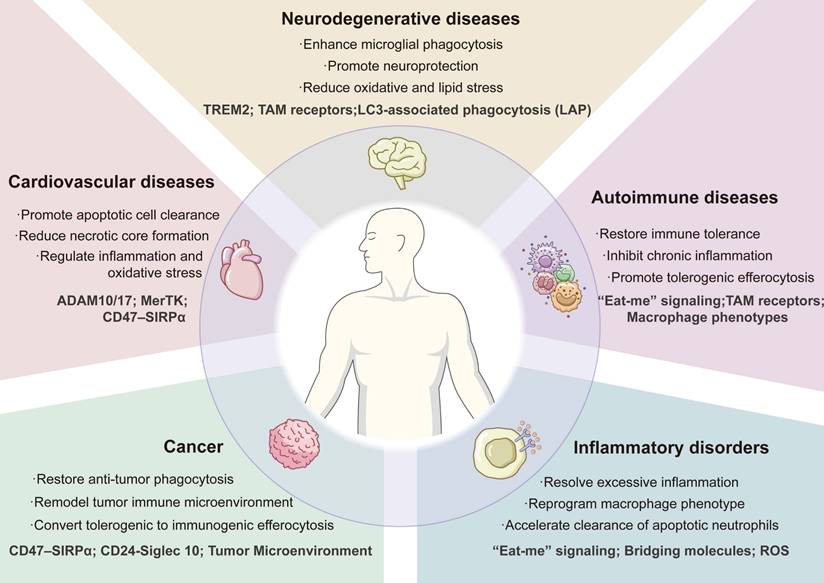
4.1 Cardiovascular diseases
Atherosclerotic plaques are characterized by accumulated apoptotic foam cells and a large necrotic core. In advanced lesions, macrophage phagocytic receptors such as MerTK are often cleaved or downregulated, whereas “don't-eat-me” signals (e.g., CD47) on dying foam cells are upregulated, collectively impairing AC clearance. Thus, efferocytosis-modulating nanomedicines for atherosclerosis focus on restoring phagocyte recognition and engulfment capacity, blocking inhibitory signals in plaques, and delivering pro-resolving cues to the inflammatory milieu. One strategy is to upregulate phagocytic receptors on lesional macrophages. For example, engineered hybrid-membrane nanovesicles (HMNVs) were constructed by fusing membranes from MerTK-overexpressing macrophages and transferrin receptor-overexpressing HEK293T cells, incorporating DOPE polymers and superparamagnetic iron oxide for magnetic navigation. In diabetic ApoE⁻/⁻ mice, HMNVs preferentially accumulate in the aorta to quell plaque inflammation under an external magnetic field and then fuse with recipient macrophage membranes and donate functional MerTK to restore efferocytosis [111]. This “receptor delivery” approach directly tackles the MerTK insufficiency known to exacerbate plaque necrosis. Macrophage-targeted delivery of liver X receptor (LXR) agonists into atherosclerotic plaque macrophages via polymeric PLGA-PEG nanoparticles (NP-LXR) increased cholesterol-efflux machinery (ABCA1/G1) and MerTK expression to damp inflammatory signaling [156]; sHDL NPs carrying LXR agonists (T0901317) enhanced atheroma targeting, boosted macrophage cholesterol efflux, and inhibited lesion progression in ApoE⁻/⁻ mice [210].
A complementary tactic addresses the prevalent “don't-eat-me” signal in plaques. Blockade of CD47 via antibodies can restore macrophage efferocytosis [7], but systemic anti-CD47 administration causes severe hematologic toxicity because of the high expression of CD47 on RBCs. To overcome this issue, macrophage-specific nanocarriers have been pursued; macrophage-specific single-walled carbon nanotubes (SWNTs) loaded with an SHP-1 inhibitor (downstream of CD47/SIRPα signaling) selectively target lesional phagocytes, reactivate efferocytosis, and reduce the necrotic core in ApoE⁻/⁻ mice without hematologic toxicity [41]. Moreover, this SHP-1-inhibitor-based nanomedicine can reduce AC accumulation and intraplaque inflammation without anemia in a porcine model of atherosclerosis, highlighting its translational potential [9]. Therefore, cell-specific checkpoint blockade via a nanoplatform can capture the pro-efferocytic benefit of CD47 inhibition while avoiding anemia and systemic immunosuppression.
In addition to the efferocytotic receptor and checkpoint, pro-resolving mediators or reprogramming cues can also be delivered by nanoplatforms to correct the inflammatory milieu that impairs macrophage efferocytosis. For example, ROS-responsive PEGylated black phosphorus nanosheets (BPNSs) decorated with the macrophage stabilin-2-targeting S2P peptide were used to deliver resolvin D1 (BPNSs@PEG-S2P/R), which can accumulate in lesional macrophages, release RvD1, and scavenge ROS, thereby reducing plaque burden in ApoE⁻/⁻ mice [211]. Nanotherapies that can modulate macrophage phenotype switching to enhance efferocytosis have also been reported. CpG-conjugated silver NPs (CpG-AgNPs) enhanced macrophage efferocytosis of CD47-positive apoptotic and foam cells via TLR9-mediated metabolic reprogramming (boosting fatty acid oxidation); the AgNP core also contributed to lesion control due to its intrinsic anti-inflammatory effect in ApoE⁻/⁻ mice [212]. Another biomimic nanoformulation composed of atorvastatin (AT) and metformin (Met)-loaded hyaluronic acid-coated and macrophage membrane-camouflaged NPs (HA-M@P@(AT+Met)) could target lesional macrophages and shift them toward M2-like phenotypes with enhanced efferocytosis via the ERK5-MerTK pathway, thereby reducing necrotic cores and stabilizing plaques in vivo [213]. This strategy that aims to deliver combination therapies in a targeted fashion underscores the combinatorial power of nanotherapeutics in tackling multiple facets of efferocytosis dysfunction simultaneously.
4.2. Autoimmune Disease
Autoimmune diseases are commonly characterized by a breakdown in self-tolerance and chronic inflammation, and defective efferocytosis has been recognized as a critical pathological factor. Nanomedicines that aim to enhance the clearance of apoptotic debris or modulate phagocytes toward tolerance are therefore of great interest in such diseases. Leveraging the natural “eat me” signal PS has emerged as an attractive strategy. By presenting PS on the nanocarrier surface, it engages phagocytes and induces anti-inflammatory, tolerogenic signaling during AC uptake [214]. An efferocytosis-informed nanoimitator (EINI) whose core complexes siIRF5 with a metformin-Zn²⁺ agent to modulate macrophage metabolism is cloaked by a ROS-responsive PS corona and is decorated with low-molecular-weight heparin (LMWH, antagonize P-selectin) for lesional targeting. In rheumatoid arthritis (RA) mice, EINI treatments could reprogram synovial inflammatory macrophages and mitigate autoimmune joint pathology [62]. Likewise, PS-liposome-coated gold nanocages carrying LXR agonists (T0901317) upregulated MerTK on macrophages and enhanced AC clearance while alleviating kidney damage in systemic lupus erythematosus (SLE) models [112]. Apoptotic-mimicking NPs have also been utilized for antigen-specific tolerization. PS-coated liposomes encapsulating insulin peptide were engulfed by macrophages, which then presented the antigens to lymphocytes in a tolerogenic context. This nanotherapy shifted macrophages toward an anti-inflammatory phenotype and increased Treg responses in non-obese diabetic mice [215]. Biomimetic NPs combining PS-mediated tolerogenic signaling with efferocytotic receptors or cellular metabolic modulators can restore defective clearance and quell autoimmune inflammation by resetting macrophage programs.
Naturally-derived nanomedicine, such as EVs with immunomodulatory effects, can be leveraged to re-educate macrophages and restore their efferocytotic capacity. For example, mesenchymal stem cell (MSC)-derived EVs have been used for treating lupus models, and intravenous infusion of MSC-EVs promoted an anti-inflammatory phenotype in recipient macrophages and enhanced their efferocytosis of ACs. As a result, lupus mice with MSC-EV therapy had reduced renal inflammation, increased clearance of nuclear debris, and corrected T-cell responses (reduced Th17 cells while expanded Treg cells) [216]. Furthermore, impaired extracellular nuclease activity (e.g., DNASE1L3 loss-of-function) permits the accumulation of apoptotic cell microparticle-associated chromatin DNA and promotes anti-dsDNA/anti-chromatin autoimmunity and lupus-like disease [217]. Accordingly, a promising therapeutic approach involves nanoparticle-mediated delivery of DNase to correct this enzymatic deficiency and enhance nucleic acid degradation. These reports illustrate a potent conceptual shift in treating autoimmune diseases: correction of intrinsic efferocytetic defects rather than broadly suppressing the immune system. The combinatorial modularity also showcases how nanotherapeutics can be tailored and layered to meet the complex immunological challenges of autoimmune diseases.
4.3. Neurodegenerative diseases
In neurodegenerative disorders, such as Alzheimer's and Parkinson's disease, inefficient clearance of apoptotic neurons and protein aggregates can amplify neuroinflammation and tissue injury. Microglia are the brain's resident macrophages and the principal efferocytes, making them a logical target for nanotherapeutics that aim to restore clearance functions while tempering inflammatory tone [43]. A synthetic efferocytic receptor (SER), fusing an amyloid-β-binding single-chain variable fragment to a TIM4 extracellular scaffold and an ELMO1 intracellular signaling module, was designed to reprogram microglial engulfment. These SER-encoded mRNAs were delivered by mannosylated lipid NPs (MERLINs). After intrathecal administration, MERLINs penetrate meningeal and ventricular spaces and are preferentially taken up by microglia, thereby enhancing Aβ clearance and attenuating neuroinflammation in Alzheimer's disease models [218]. Convergently, rejuvenating hypophagocytic, senescence-like microglia by silencing p16Ink4a with PLGA NPs delivered via the cisterna magna yields preferential microglial uptake, robust p16 knockdown, increased lysosomal activity and Aβ phagocytosis, with improved spatial learning and memory in Alzheimer's disease model mice [219]. These studies provide proof-of-concept that nanocarriers can deliver genetic instructions to reprogram microglial efferocytosis machinery in a disease-targeted manner.
Another therapeutic avenue is to use NPs that reprogramme how microglia handle toxic Aβ. For example, sugar-based amphiphilic macromolecule NPs (AM-NPs) that engage microglial scavenger receptors (SRA1, CD36, CD68) bind fibrillar Aβ, slow fibrillization and disrupt preformed fibers, dampen SR-mediated internalization, and enhance lysosomal degradation of Aβ, thereby reducing pro-inflammatory responses in microglia. This “eat smarter” paradigm illustrates how tuning substrate morphology and receptor usage can bias Aβ handling toward lysosomal clearance rather than pro-inflammatory signaling [220]. Also, TFEB-dependent lysosomal competence is critical for microglial activation states in tauopathy/AD, positioning lysosomal biogenesis and functioning as tractable targets for efferocytosis-oriented nanotherapeutics [221].
In addition, nanotherapeutics aiming to attenuate oxidative stress that impairs efferocytosis in microglia have also been reported. For example, Ceria (CeO2) nanoclusters might act as catalytic ROS scavengers for lowering microglial ROS and preserving endolysosomal handling of Aβ in cellular models [222]. While these results are promising, emerging solutions for overcoming the BBB barrier, such as RAGE-targeting systems, transferrin-receptor (TfR) “brain-shuttle” NPs/biologics, and nose-to-brain (intranasal) administration, may provide further therapeutic benefits [223-225]. More combinatorial approaches are likely essential because neurodegeneration intertwines protein aggregation, cell death, and chronic neuroinflammation, with microglial efferocytosis sitting at the nexus of injury resolution [3]. Safety concerns in the CNS require nanoplatforms with low immunogenicity and neuronal compatibility; accordingly, many efforts prioritize biocompatible carriers. Nanomedicines might help rebalance injury and repair in the brain, but rigorous work remains to secure precise human targeting and long-term safety.
4.4. Inflammatory disorders
Diverse forms of acute tissue injuries, such as acute lung injury (ALI), sepsis, acute kidney injury (AKI), myocardial infarction (MI), and intracerebral hemorrhage, are characterized by massive cell death, where timely efferocytosis is pivotal for returning inflamed tissues to homeostasis. To date, nanomedicines have been designed to promote resolution by boosting efferocytosis while tempering collateral inflammation [226]. PS-displaying or apoptotic membrane-coated nanocarriers preferentially engage lung macrophages and promote pro-resolving efferocytosis in ALI models. For example, an inhalable apoptotic membrane-coated antioxidant “nanozyme” (AOzyme@ACM) enhanced “eat-me” signaling and reduced alveolar inflammation in mouse ALI models [121]. Likewise, a “two-step” strategy, neutrophil-targeted apoptosis-restoring NPs followed by macrophage-targeted efferocytosis-restoring NPs, has also mitigated severe ARDS in preclinical models [227]. Sepsis involves systemic inflammation and often immunosuppression, while the MFGE8-containing exosomes derived from immature dendritic cells (IDC-exosomes) could augment macrophage efferocytosis and improve survival in rodent sepsis, suggesting that “bridge-molecule” augmentation may be a potent therapeutic target in this state [228]. Multiple types of acute organ injuries commonly couple oxidative stress and insufficient corpse clearance with reparative failure, which may be resolved by nanomedicines PS-presenting liposomes reprogrammed cardiac macrophages toward reparative phenotypes and improved postinfarct remodeling in MI models [65]. In AKI models, KIM-1 (a kidney injury protein)-targeted black phosphorus nanoplatform loaded with 4-octyl-itaconate (4-OI) accumulated in injured kidneys, which scavenged ROS and restored pathways linked to efferocytosis [229]. Neutrophil-like and pH-responsive pro-efferocytic NPs improved erythrophagocytosis and neurological recovery in mice with intracerebral hemorrhage [113].
Natural nanomedicines with efferocytosis-modulating effects have also been used. GAS6-enriched MSC-EVs enhanced efferocytosis in an ischemia/reperfusion-induced acute liver injury model [67]; some of them might be edging toward the clinic. Infusion of early apoptotic cells (Allocetra™-OTS) for sepsis/cytokine storm syndromes [230], while apoptotic extracellular vesicles (ApoEVs) represent a cell therapy-like modality with advantages in scalable manufacturing and controlled delivery compared with whole apoptotic cell infusion [231]. The multifunctional potential of nanoplatforms is compelling, as they can deliver payloads and actively engage macrophages to boost efferocytosis. Key challenges are safety and timing; mis-timed interventions can blunt necessary host responses, while well-timed therapies can facilitate resolution, underscoring the need for precise therapeutic windows. Nanomedicines with targeted delivery and controlled release effects are well suited to provide the required spatiotemporal control for such indications.
4.5. Cancers
Tumors coopt efferocytosis as an immune evasion mechanism. Many cancer cells overexpress “don't eat me” signals (notably CD47) to avoid phagocytosis, while the efferocytosis of apoptotic cancer cells by TAMs induces an immunosuppressive M2-like phenotype that dampens antitumor immunity. For cancer therapy, nanomedicines therefore pursue two complementary goals: enhancing the phagocytosis of tumor cells by TAMs and inhibiting the anti-inflammatory program initiated after the phagocytosis that causes immunosuppression. A prominent strategy is NP-mediated phagocytosis checkpoint blockade. For example, a degradable mesoporous silica NP co-delivering an anti-CD47 antibody and doxorubicin provided the two signals required for efficient engulfment—CD47 blockade to disable the inhibitory cue and doxorubicin-induced immunogenic cell death to expose calreticulin as an “eat-me” signal, thereby promoting macrophage phagocytosis and strengthening T-cell responses in murine tumor models [115]. Similarly, a mannose-decorated liposome that co-delivers the TLR7/8 agonist resiquimod (R848) and a high-affinity SIRPα decoy protein (CV1) drives uptake by M2-like TAMs, repolarizes them toward M1, blocks the CD47-SIRPα axis, and enhances phagocytosis; in the MC38 model, this nanoformulation markedly reduced tumor burden, increased cytotoxic T-cell infiltration, and—upon systemic administration—showed no detectable hematologic or histopathologic toxicity [232].
Beyond antibody formulations, researchers have cloaked NPs with genetically engineered cell membranes that display high-affinity SIRPα variants, enabling in situ binding to tumor cell CD47, local checkpoint blockade, and restoration of macrophage phagocytosis [233]. In addition to CD47, nanotherapeutics are being created to target alternative phagocytosis checkpoints such as CD24-Siglec-10, which certain cancers exploit to suppress macrophages [142]. A recent study conjugated an anti-CD24 monoclonal antibody to the surface of biodegradable nanospheres, which degraded cell membrane CD24, blocked CD24-Siglec-10 signaling, and augmented macrophage phagocytosis [117].
Another vital strategy is the suppression of phagocytic receptors on tumor-associated phagocytes to prevent the clearance of apoptotic tumor cells and their subsequent transition into an immunosuppressive state. The TAM receptor family (Tyro3, Axl, MerTK) is a prime target in this regard. A glycopolymer-based NP was developed to deliver a small-molecule MerTK inhibitor (UNC2025) to TAMs in the TME. These NPs accumulated in CD206+ TAMs and inhibited efferocytosis of apoptotic tumor cells, thereby preventing TAMs from entering an anti-inflammatory state [129]. In addition to blocking suppressive pathways, there is an interest in delivering “find-me” signals to tumors to recruit immune cells. The ATP-conjugated PLGA NPs could present ATP into tumors, thereby recruiting antigen-presenting cells while co-delivering paclitaxel to induce immunogenic cell death; combination with anti-PD-1 achieved complete regression in the CT26 colorectal tumor model and established immune memory [107], suggesting that efferocytosis-modulating NPs can synergize with other treatments, such as chemicals and anti-PD1 therapy. In the future, nanotherapeutics are proving to be the precise tools required to execute these nuanced interventions, holding promise for more effective and personalized cancer immunotherapy.
Collectively, current studies highlight unifying principles by which nanomedicines modulate efferocytosis for therapeutic gain: the ability to achieve precision targeting and controlled payload release, which minimizes off-target effects and enhances the therapeutic index. Common design elements include surface functionalization with ligands (e.g., phosphatidylserine mimics or macrophage-specific peptides) to facilitate phagocyte uptake, biomimetic properties that recapitulate apoptotic cell cues, and microenvironment-gated activation controls (e.g., pH- or ROS-sensitive release). While these shared mechanisms provide a foundational framework, disease-tailored designs remain essential because defective efferocytosis, tissue microenvironments and therapeutic goals differ substantially across indications. For instance, in atherosclerosis, nanomedicines aim to inhibit "don't-eat-me" signals (e.g., CD47) while boosting receptor activity (e.g., MerTK) and repairing lipid-handling programs needed for repeated rounds of efferocytosis; in autoimmune disease, nanomedicines often employ apoptotic mimicry to reimpose tolerogenic macrophage programs in a controlled manner. Cancer most clearly illustrates the need for mechanism-driven directionality: nanomedicines can enhance tumor cell phagocytosis via checkpoint blockade, yet they also inhibit TAM efferocytosis of apoptotic tumor cells and its downstream immunosuppressive program to restore antitumor immunity. Accordingly, whether to enhance or inhibit efferocytosis should be decided by the dominant pathogenic mechanism: enhancement is generally favored when the accumulation of apoptotic cells is a proximal driver of pathology (e.g., secondary necrosis, autoantigen exposure, and necrotic core formation), whereas inhibition may be rational when its downstream program is predicted to therapy resistance, most prominently in cancer. These considerations underscore that the translational challenge lies in precision—identifying the right compartment, phagocyte subset and disease stage for enhancement versus inhibition, while implementing spatiotemporal control to maximize benefits and minimize systemic liabilities.
5. Progress of efferocytosis-modulating therapies in clinical trials
Although the results from preclinical studies are promising, more important, their therapeutic potential requires further verification in clinical trials. Several translational strands have suggested that macrophage-engaging nanotherapies are approaching first-in-human evaluation, while closely related clinical advances, particularly localized or tumor-selective blockade of the CD47-SIRPα checkpoint, are already underway. For example, intratumoral delivery of the SIRPα-Fc decoy TTI-621 has shown activity in cutaneous T-cell lymphoma in a phase 1 setting (NCT02890368), and a protease-activated (masked) anti-CD47 antibody, SGN-CD47M, is being tested in a phase 1 trial in solid tumors (NCT03957096). In parallel, localized delivery of mRNAs encoding CD47 inhibitors has been reported preclinically as another route to confine target engagement [234, 235]. However, until now, the overall clinical results have remained preliminary (Table 4). For example, while intralesional TTI-621 demonstrated signs of activity in CTCL, definitive benefits have not been established, and the SGN-CD47M trial was terminated without reported efficacy (officially attributed to portfolio prioritization), indicating that these approaches require further assessment. These early experiences underscore a central design priority for next-generation efferocytosis-modulating nanomedicines: mitigating systemic toxicity through spatiotemporal control of target engagement and pharmacological activity. For instance, protease-sensitive nanosystems can carry “masked” payloads that remain inert during circulation and then release active drugs in enzyme-rich tumor sites, analogous to probody therapeutics, thereby sparing healthy cells [236, 237].
Clinical progress of efferocytosis-modulating approaches and representative clinically validated nanoparticle platforms
| Trial registration | Phase | Drug/Nanoparticle Type | Primary Endpoint | Key Results | Current Limitations |
|---|---|---|---|---|---|
| NCT02890368 | Phase 1 | TTI-621 (SIRPα-Fc decoy fusion protein) | Safety/tolerability (AEs); activity signals (lesion response metrics) | Intralesional administration showed antitumor activity in CTCL, with ≥50% reduction in CAILS score in 34% of patients; well tolerated; treatment-related AEs mostly grade 1-2. | Definitive benefits not established; terminated early; limited to specific indications like mycosis fungoides; requires further assessment for broader efficacy and systemic use. |
| NCT03957096 | Phase 1 | SGN-CD47M (protease-activated masked anti-CD47 antibody) | Safety/tolerability; PK; immunogenicity; preliminary antitumor activity | Terminated (sponsor decision/portfolio prioritization); no efficacy dataset established publicly. | Attributed to portfolio prioritization; no efficacy reported; potential systemic toxicity concerns; requires further evaluation for safety and activity in solid tumors. |
| NCT02719665 | Early-phase (pilot) | Specialized pro-resolving mediators (SPMs) enriched marine oil supplement | Changes in lipid mediators and leukocyte phenotype; safety. | Significant survival benefit in metastatic pancreatic cancer postgemcitabine; median OS 6.1 months for MM-398 + 5-FU/LV vs 4.2 months for 5-FU/LV alone; improved PFS and response rates. | Modest sample size; unblinded and short-term design; lack of clinically meaningful endpoints; preliminary evidence only; feasibility demonstrated but definitive benefits in inflammatory disorders not established. |
| NCT01494506 | Phase 3 | MM-398 (liposomal irinotecan) with or without 5-FU/LV | Overall survival (OS) | MM-398 with 5-FU/LV improved OS vs control (reported median OS benefit and favorable HR in final analyses). | Potential toxicities like diarrhea and neutropenia; limited to patients who failed prior gemcitabine; dose modifications may be needed. |
| NCT01960348 | Phase 3 | Patisiran (ALN-TTR02; lipid nanoparticle (LNP)-siRNA) | Change in modified Neuropathy Impairment Score +7 (mNIS+7) at 18 months. | Significant improvement in neuropathy scores; median change in mNIS+7: -6.0 points for patisiran vs +28.0 for placebo; overall survival and cardiac benefits observed; well-tolerated with infusion-related reactions. | Requires IV infusion; long-term follow-up in extension study; limited to hereditary transthyretin amyloidosis patients; no major limitations noted but ongoing monitoring for safety. |
| NCT04368728 | Phase 3 | BNT162b2 (LNP-mRNA vaccine) | Incidence of confirmed COVID-19 cases from 7 days after second dose; safety. | Vaccine efficacy of 95% against COVID-19; well-tolerated; immunogenicity confirmed in adolescents and adults. | requires ultracold storage; durability/boosting needs; variant immune escape; rare adverse events (e.g., myocarditis) require pharmacovigilance and risk-stratified use. |
| NCT04470427 | Phase 3 | mRNA-1273 (LNP-mRNA vaccine) | Number of participants with first occurrence of COVID-19 starting 14 days after second dose; safety. | Vaccine efficacy of 94.1% against COVID-19; well tolerated; serious adverse events low and comparable to placebo. | Similar class limitations: durability/boosting, variant drift, rare adverse events and ongoing postmarketing safety surveillance. |
Abbreviations: AEs: adverse events; CAILS: Composite Assessment of Index Lesion Severity; CTCL: cutaneous T-cell lymphoma; OS: overall survival; PK: pharmacokinetics; SPMs: specialized pro-resolving mediators; 5-FU/LV: 5-fluorouracil/leucovorin.
For the treatment of inflammatory disorders, specialized pro-resolving mediators (SPMs), including resolvins, protectins, maresins, and lipoxins, have advanced into early clinical evaluation (e.g., OMEGA-SPM-DOSE, NCT02719665), supporting the feasibility of resolution-directed interventions relevant to efferocytosis biology [238]. At the same time, prior regulatory successes, from liposomal doxorubicin (Doxil) to lipid-NP mRNA vaccines, demonstrate that NP platforms can achieve clinical and manufacturing acceptance when supported by robust CMC and safety packages [239, 240] (Table 4). Looking ahead, it is plausible that the first trials explicitly branded as “pro-efferocytic NPs” or “macrophage-checkpoint-inhibitor NPs” will emerge initially in oncology, with cardiovascular indications to follow contingent on safety, as suggested by large-animal studies in atherosclerosis that reduced vascular inflammation via pro-efferocytic nanotherapeutics [9].
The preclinical success across diverse disease models provides compelling evidence that targeted modulation of efferocytosis via nanotherapeutics can prevent and even reverse pathology. From the chronic inflammation of atherosclerosis to the fulminant cytokine storm of sepsis, efferocytosis-modualting nanomedicines are proving remarkably versatile in tilting the balance toward disease resolution. A recurrent theme in these examples is combinatorial design: integrating multiple mechanisms (targeting motifs, payloads and biomimetic surface cues) in one nanosystem to address the multifaceted nature of efferocytosis dysfunction. This modular combinability is a distinctive strength of nanotherapeutics and underpins strategies that co-deliver immune-modulating signals with disease-specific targeting. Equally important is transferability: the conceptual modules (PS coating, MerTK agonist, CD47 blocker, etc.) are, in many cases, interchangeable across diseases. For example, a PS-coated NP that works in lung injury might be adapted to target kidney inflammation, or a neutrophil-membrane vesicle used in stroke could be repurposed for myocardial infarction — the principles of efferocytosis are conserved, even if the context differs. This bodes well for accelerating development, as success in one domain can inform another. Additionally, the next steps will involve confirming these benefits in large animal models and early human trials.
6. Challenges and future perspectives
To date, advancements in this field are impressive, but some challenges and key problems still exist and need to be resolved. For example, targeted nanocarrier-mediated drug delivery helps confine phagocyte activity, but perfect specificity remains elusive. Liver Kupffer cells avidly sequester many formulations, and the amounts of drugs delivered to target tissues (e.g., solid tumors) are relatively small (~0.7% of the injected dose) [241, 242]. Moreover, marker overlap between target cells further complicates selectivity. For example, while CD206 (mannose receptor) is widely used to target M2-like TAMs, it is also expressed on Kupffer cells and some DCs or sinusoidal endothelial cells, and healthy tissue macrophages can be unintentionally engaged [243, 244]. A rational solution is “logic-gated” targeting that layers cell-type ligands with disease-site triggers (e.g., pH, ROS, enzymes) and controlled-release kinetics to achieve selective activation only where and when desired [245, 246].
Beyond targeting, achieving precise spatiotemporal control of efferocytosis modulation is critical because hyperactivated efferocytosis can suppress needed early inflammatory defenses against infection or tumors, whereas excessive or prolonged inhibition risks autoimmunity or non-resolving inflammation [247]. Thus, temporal and spatial control is required for nanomedicines, which might be achieved with stimuli-responsive or logic-gated nanomaterials that release payloads only in disease microenvironments. In some diseases (e.g., sepsis and lung infections), early enhancement of pro-resolving programs via efferocytosis can blunt antimicrobial defenses and worsen outcomes, whereas later delivery aids resolution and repair [52, 247]. This timing-dependent paradox might be resolved using a “two-phase” nanomedicine: a first phase to augment efferocytosis, followed by an adjuvant phase to ensure antigen presentation. Furthermore, many efferocytosis nodes are pleiotropic, and MerTK signaling and TGF-β not only enforce immune quiescence but can also drive profibrotic remodeling when chronically engaged [248]. Thus, anticipating and managing these pleiotropic outcomes will be part of the development process, engineering safety mechanisms such as limited exposure windows, using modules that self-destruct or release an inhibitor after a period, may all be part of future designs.
Immunogenicity and long-term safety remain major translational barriers for efferocytosis-targeting nanomedicines because engineered particles can be sensed as foreign and elicit innate and/or adaptive immune responses [249, 250]. Systemically administered nanoparticles may activate the complement cascade, which can reshape opsonization and phagocytic clearance and can also precipitate infusion reactions known as complement activation-related pseudoallergy (CARPA) [251]. Upon repeat dosing, carrier-directed antibodies can drive the accelerated blood clearance (ABC) phenomenon; a canonical example is PEGylated liposomes, where a priming dose can induce anti-PEG IgM that binds subsequent doses and promotes complement activation, thereby eroding the “stealth” benefit of PEG [252]. These complement-antibody interactions can reduce exposure and efficacy and may also increase acute safety liabilities, underscoring the need to prospectively evaluate and mitigate CARPA/infusion-reaction risk during clinical translation. Over longer time horizons, the intracellular fate of nanomaterials in professional phagocytes is pivotal: biopersistent particles can accumulate in lysosomes and disrupt autophagy-lysosome flux, contributing to “lysosomal nanotoxicity” phenotypes that may compromise phagocyte function [253]. Consequently, first-in-human development should prioritize immunocompatible, biodegradable materials, optimize dose and dosing frequency, and integrate repeat-dose and large-animal testing that is sensitive to anti-PEG/anti-carrier antibodies and complement-mediated reactions, thereby reducing ABC and CARPA risk.
Efferocytosis-modulating nanomedicine can also be combined with, rather than replace, current standards of care. In cancer, for example, preclinical studies have shown that inhibiting MerTK-mediated efferocytosis or blocking the CD47 checkpoint enhances responsiveness to PD-1/PD-L1 blockade [72]. In atherosclerosis, the clinically used drug statins may enhance the "eat-me" signals on macrophages [254], suggesting a potent synergistic interplay with nanotherapeutics that target efferocytosis. Thus, it is possible that efferocytosis-targeted nanomedicine will allow lower doses or shorter durations of some standard therapies (e.g., steroids) along with reduced side effects. Clinical trial designs will have to carefully incorporate these therapies with standard care, likely starting as an add-on in refractory cases, then moving to earlier lines if proven safe and beneficial. Regulatory considerations are also important, demonstrating that adding nanotherapeutics does not interfere with the pharmacokinetics or safety of standard drugs. Ultimately, the goal is to weave efferocytosis modulators into the fabric of existing treatment protocols, possibly as an induction therapy (induce remission in autoimmunity then maintain with conventional drugs) or as a maintenance add-on (keep giving periodically to prevent flares or plaque progression).
Achieving maximal clinical impact with these therapies will require robust biomarkers and imaging tools to monitor in vivo modulation of efferocytosis and inflammation resolution programs. To date, there is still no widely adopted, standardized clinical biomarker that has been rigorously validated to specifically track efferocytosis dynamics, reflecting the multi-step and context-dependent complexity of this process. Recent work has identified candidate indicators that could be leveraged as surrogate readouts, including soluble TAM receptors generated by proteolytic shedding (sTyro3, sAxl, and soluble Mer/sMerTK), which are measurable in blood or other biofluids and have been linked to dysregulated apoptotic cell clearance [255]. In atherosclerosis, myeloid expression of a cleavage-resistant MerTK variant lowers soluble Mer (sol-Mer) and improves lesional efferocytosis [40]. Similarly, in systemic lupus erythematosus, plasma sMer is increased during active disease and has been reported to decrease in parallel with improvements in disease activity (SLEDAI) after therapy [255]. These findings support soluble TAM receptors as candidate surrogate markers of efferocytosis dysregulation and therapeutic response, but clinical deployment will require standardized assays, longitudinal validation across cohorts and careful adjustment for inflammation-related confounding factors.
Beyond soluble mediators, transcriptomic efferocytosis-related gene signatures are being explored as candidate biomarkers. In oncology, studies integrating bulk RNA-seq with single-cell analyses and machine-learning feature selection have derived efferocytosis-related gene sets associated with prognosis and with differences in immune infiltration and predicted immunotherapy response [256]; in ulcerative colitis, multiomics integration identified efferocytosis-related hub genes whose baseline expression patterns correlated with clinical response to ustekinumab and showed moderate discriminative performance [257]. Overall, translating such panels into clinically actionable precision-medicine tools will require standardized assays, longitudinal sampling and validation against more direct measures of efferocytosis to establish both specificity and dynamic response ranges.
A parallel frontier in this field is the advancement of molecular imaging tools that can noninvasively quantify two crucial elements: the tissue burden of apoptotic cells and the biodistribution/lesional accumulation of nanomedicines at sites of disease. Radiolabeled Annexin A5/V (PS-binding) has been evaluated in clinical atherosclerosis studies [258]. Early clinical and validation studies suggest that Annexin-based imaging can highlight plaques with higher cell-death burden and features considered more “vulnerable”; however, the signal primarily reports cell-death burden and therefore provides only indirect evidence of impaired efferocytosis [259]. To more directly visualize efferocytosis, the pH-sensitive probe AnxA5-pHrodo developed by Stöhr and colleagues offers an elegant solution: it is dim at neutral pH but becomes brightly fluorescent once labeled apoptotic targets are internalized into acidic phagosomes/lysosomes, enabling flow cytometric and microscopic discrimination between surface-bound targets and truly engulfed cargo and thereby supporting quantitative assessment of efferocytic capacity [260].
Beyond imaging cell corpses, there is growing interest in noninvasively tracking the in vivo distribution and lesion-level deposition of therapeutic nanoparticles in real time. Theranostic nanomedicines can be equipped with imaging reporters, such as iron oxide/USPIO contrast for MRI, radiolabels for PET/SPECT, or fluorescent dyes for optical imaging, to quantify biodistribution and intralesional accumulation [261]. Clinically, in a pilot study of patients with advanced solid tumors treated with nanoliposomal irinotecan (nal-IRI), higher ferumoxytol-MRI-derived lesion metrics at 1-24 hours were significantly associated with subsequent lesion shrinkage, supporting ferumoxytol MRI as a noninvasive predictor of heterogeneous nanoparticle deposition [262]. Therefore, incorporating imaging readouts (e.g., MRI-traced nanoparticle deposition) into early-phase trials of efferocytosis-targeted nanomedicines could provide delivery/pharmacodynamic endpoints for patient-lesion stratification and early go/no-go decisions, thereby derisking larger outcome-focused studies.
One of the foremost translational challenges moving forward is to identify which patients will benefit most from efferocytosis-targeted therapies and to tailor interventions accordingly. Given the heterogeneity of efferocytosis across diseases and individuals, patient stratification based on molecular, cellular or disease-specific features is essential for maximizing efficacy. For example, patients with tumors highly overexpressing CD47 or having an abundance of CD47^+ tumor-derived extracellular vesicles in circulation may experience greater benefit from anti-CD47 agents. This is analogous to how HER2-positive breast cancer patients are selected for anti-HER2 therapy [263]. Likewise, in atherosclerosis, patients with high CD47 expression in plaques or with elevated soluble MerTK in blood might be prime candidates for an efferocytosis-enhancing nanotherapy. Stratification can also draw on gene signatures; as noted, at the transcriptomic level, efferocytosis-related gene panels have been proposed across indications and can be associated with prognosis and therapy response. If a tumor's RNA sequencing reveals an efferocytosis gene signature associated with immunosuppressive macrophages, the patient might benefit from an efferocytosis-inhibiting approach (for example, MerTK blockade).
Imaging-based companion diagnostics are particularly attractive for nanomedicines because delivery varies substantially across patients and lesions; clinical and translational studies have used ferumoxytol-MRI to estimate lesion uptake/permeability and to predict deposition and response to therapeutic nanoliposomal drugs [262]. This concept was highlighted by Cooley et al., who argued that pre-treatment imaging of nanoparticle delivery could serve as a stratification tool to improve trial success rates for nanomedicines [264]. In addition, novel PET tracers for apoptotic cells could identify patients with high apoptotic burden and low clearance; for example, an 18F-annexin PET scan might reveal which atherosclerosis patients have large areas of uncleared apoptotic cells in plaques, and these patients may be selected for pro-efferocytic therapy. Ultimately, a combination of biomarkers might be used to construct an “efferocytosis score” for patients, guiding enrollment in trials and individualized therapy plans. It should be noted that rigorous validation of these candidate biomarkers is needed; many have shown correlations in retrospective studies, but their predictive value in interventional settings remains to be proven.
Finally, there are significant scaled manufacturing and regulatory challenges that must be addressed to translate efferocytosis nanomedicines from bench to bedside. Nanomedicines typically present higher manufacturing complexity than small molecules or traditional biologics; achieving batch-to-batch consistency in sizes, surface attributes, and payloads under GMP is a well-recognized chemistry, manufacturing, and control (CMC) challenge (FDA-2017-D0759). Nanoplatforms often involve multiple components, each adding to cost and variability. This could affect commercial viability, especially if the target patient population is large (such as tens of millions with atherosclerosis). Future research needs to emphasize simplifying nanomedicine designs where possible or leveraging biomaterials that are easier to produce. Regulatory expectations are tightly coupled to manufacturing challenges: for complex nanoparticles, demonstrating robust CMC requires validated analytical methods to define and control critical quality attributes (CQAs)—including size distribution, morphology, surface properties/functionalization (e.g., ligand density, orientation and conformational state for actively targeted systems), and cargo loading/release, to support batch-to-batch consistency [265]. Fortunately, the field is actively advancing such methods, including single-particle tracking and advanced spectroscopy, to strengthen nanoparticle quality control. Additionally, modular manufacturing approaches offer promising solutions; for example, scalable "generic" liposomes or lipid nanoparticles (LNPs) can be produced first, followed by the addition of disease-specific ligands through post-insertion or late-stage conjugation steps to standardize the majority of the process [266]. Encouragingly, the mRNA-LNP experience during COVID-19 indicates that, with focused investment and demand, NP platforms can be scaled from lab to billions of doses. In the future, platformized and modular manufacturing processes will be pivotal to making efferocytosis-targeted nanotherapeutics commercially and clinically viable.
In summary, nanomedicine-based strategies for modulating efferocytosis have advanced considerably in recent years, and addressing the aforementioned challenges remains essential. In the future, interdisciplinary collaboration that integrates insights from immunology, materials science, and clinical medicine would foster innovative solutions. The field might be shifting from questions of feasibility (could this work?) to developments of nanoplatforms with advanced efficacy and biosafety (how do we make it work better and safer?). Ongoing research is poised to yield more refined targeting ligands and intelligent responsive nanoplatforms and to provide deeper insights into efferocytosis biology for therapeutic exploitation. Ultimately, modulating the body's natural ability to handle apoptotic cells will emerge as a mainstream therapeutic approach, enabled primarily by the precision and customizability of nanotherapeutics.
Abbreviations
ABCA1: ATP-binding cassette transporter A1; AC: apoptotic cell(s); AD: Alzheimer's disease; ADAM17: a disintegrin and metalloproteinase 17; ApoE: apolipoprotein E; ATP: adenosine triphosphate; AXL: AXL receptor tyrosine kinase; BTB: blood-testis barrier; CD24: cluster of differentiation 24; CD47: cluster of differentiation 47; CX3CL1: C-X3-C motif chemokine ligand 1 (fractalkine); IL-10: interleukin 10; LAP: LC3-associated phagocytosis; LPC: lysophosphatidylcholine; LXR: liver X receptor; MerTK: MER proto-oncogene tyrosine kinase; MFG-E8: milk fat globule-EGF factor 8 (lactadherin); NP: nanoparticle(s); PLGA: poly(lactic-co-glycolic acid); PPAR: peroxisome proliferator-activated receptor; PS: phosphatidylserine; PtdSer: phosphatidylserine; Rac1: Ras-related C3 botulinum toxin substrate 1; ROS: reactive oxygen species; SIRPα: signal regulatory protein alpha; TAM: Tyro3-Axl-Mer (receptor family); TFEB: transcription factor EB; TGF-β: transforming growth factor beta; TIM-4: T-cell immunoglobulin and mucin domain-containing protein 4; UTP: uridine triphosphate.
Supplementary Material
Supplementary table.
Acknowledgements
This study was partly supported by the National Natural Science Foundation of China (32271438 to JL, 82472129 to JL, 32071453 to JL, and 82570811 to SL), Sichuan Science and Technology Program (2024NSFSC0586 to JL, and 2024YFFK0199 to SL), and 1.3.5 Project for Disciplines of Excellence (ZYYC23001 to JL) of West China Hospital, Sichuan University. AI tools were not used for manuscript preparation, image generation, data collection, or data analysis.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Boada-Romero E, Martinez J, Heckmann BL, Green DR. The clearance of dead cells by efferocytosis. Nat Rev Mol Cell Biol. 2020;21:398-414
2. Kawano M, Nagata S. Efferocytosis and autoimmune disease. Int Immunol. 2018;30:551-8
3. Chen Y, Kou Y, Ni Y, Yang H, Xu C, Fan H. et al. Microglia efferocytosis: an emerging mechanism for the resolution of neuroinflammation in Alzheimer's disease. J Neuroinflammation. 2025;22:96
4. Werfel TA, Cook RS. Efferocytosis in the tumor microenvironment. Semin Immunopathol. 2018;40:545-54
5. Adkar SS, Leeper NJ. Efferocytosis in atherosclerosis. Nat Rev Cardiol. 2024;21:762-79
6. Cheng M, Chen S, Li K, Wang G, Xiong G, Ling R. et al. CD276-dependent efferocytosis by tumor-associated macrophages promotes immune evasion in bladder cancer. Nat Commun. 2024;15:2818
7. Kojima Y, Volkmer JP, McKenna K, Civelek M, Lusis AJ, Miller CL. et al. CD47-blocking antibodies restore phagocytosis and prevent atherosclerosis. Nature. 2016;536:86-90
8. Zhou X, Li D, Xia S, Ma X, Li R, Mu Y. et al. RNA-based modulation of macrophage-mediated efferocytosis potentiates antitumor immunity in colorectal cancer. J Control Release. 2024;366:128-41
9. Bamezai S, Zhang Y, Kumari M, Lotfi M, Alsaigh T, Luo L. et al. Pro-efferocytic nanotherapies reduce vascular inflammation without inducing anemia in a large animal model of atherosclerosis. Nat Commun. 2024;15:8034
10. Zheng J, Jiang J, Pu Y, Xu T, Sun J, Zhang Q. et al. Tumor-associated macrophages in nanomaterial-based anti-tumor therapy: as target spots or delivery platforms. Front Bioeng Biotechnol. 2023;11:1248421
11. Ramesh A, Kumar S, Nandi D, Kulkarni A. CSF1R- and SHP2-Inhibitor-Loaded Nanoparticles Enhance Cytotoxic Activity and Phagocytosis in Tumor-Associated Macrophages. Adv Mater. 2019;31:e1904364
12. Kraynak CA, Yan DJ, Suggs LJ. Modulating inflammatory macrophages with an apoptotic body-inspired nanoparticle. Acta Biomater. 2020;108:250-60
13. Moon B, Yang S, Moon H, Lee J, Park D. After cell death: the molecular machinery of efferocytosis. Exp Mol Med. 2023;55:1644-51
14. Park SY, Kim IS. Engulfment signals and the phagocytic machinery for apoptotic cell clearance. Exp Mol Med. 2017;49:e331
15. Nagata S. Apoptosis and Clearance of Apoptotic Cells. Annu Rev Immunol. 2018;36:489-517
16. Lam AL, Heit B. Having an Old Friend for Dinner: The Interplay between Apoptotic Cells and Efferocytes. Cells. 2021 10
17. Geng K, Kumar S, Kimani SG, Kholodovych V, Kasikara C, Mizuno K. et al. Requirement of Gamma-Carboxyglutamic Acid Modification and Phosphatidylserine Binding for the Activation of Tyro3, Axl, and Mertk Receptors by Growth Arrest-Specific 6. Front Immunol. 2017;8:1521
18. Kuroiwa M, Yamaguchi SI, Kato Y, Hori A, Toyoura S, Nakahara M. et al. Tim4, a macrophage receptor for apoptotic cells, binds polystyrene microplastics via aromatic-aromatic interactions. Sci Total Environ. 2023;875:162586
19. Park D, Tosello-Trampont AC, Elliott MR, Lu M, Haney LB, Ma Z. et al. BAI1 is an engulfment receptor for apoptotic cells upstream of the ELMO/Dock180/Rac module. Nature. 2007;450:430-4
20. Kelley SM, Ravichandran KS. Putting the brakes on phagocytosis: "don't-eat-me" signaling in physiology and disease. EMBO Rep. 2021;22:e52564
21. Liu Y, Wang Y, Yang Y, Weng L, Wu Q, Zhang J. et al. Emerging phagocytosis checkpoints in cancer immunotherapy. Signal Transduct Target Ther. 2023;8:104
22. Green DR, Oguin TH, Martinez J. The clearance of dying cells: table for two. Cell Death Differ. 2016;23:915-26
23. Elliott MR, Ravichandran KS. The Dynamics of Apoptotic Cell Clearance. Dev Cell. 2016;38:147-60
24. Mehrotra P, Ravichandran KS. Drugging the efferocytosis process: concepts and opportunities. Nat Rev Drug Discov. 2022;21:601-20
25. Heckmann BL, Green DR. Correction: LC3-associated phagocytosis at a glance (doi:10.1242/jcs.222984). J Cell Sci. 2019; 132
26. Morioka S, Perry JSA, Raymond MH, Medina CB, Zhu Y, Zhao L. et al. Efferocytosis induces a novel SLC program to promote glucose uptake and lactate release. Nature. 2018;563:714-8
27. Trzeciak A, Wang YT, Perry JSA. First we eat, then we do everything else: The dynamic metabolic regulation of efferocytosis. Cell Metab. 2021;33:2126-41
28. Doran AC, Yurdagul A Jr, Tabas I. Efferocytosis in health and disease. Nat Rev Immunol. 2020;20:254-67
29. Yurdagul A Jr, Subramanian M, Wang X, Crown SB, Ilkayeva OR, Darville L. et al. Macrophage Metabolism of Apoptotic Cell-Derived Arginine Promotes Continual Efferocytosis and Resolution of Injury. Cell Metab. 2020;31:518-33.e10
30. Poon IKH, Ravichandran KS. Targeting Efferocytosis in Inflammaging. Annu Rev Pharmacol Toxicol. 2024;64:339-57
31. Astuti Y, Raymant M, Quaranta V, Clarke K, Abudula M, Smith O. et al. Author Correction: Efferocytosis reprograms the tumor microenvironment to promote pancreatic cancer liver metastasis. Nat Cancer. 2024;5:808
32. Rosenblum MD, Remedios KA, Abbas AK. Mechanisms of human autoimmunity. J Clin Invest. 2015;125:2228-33
33. Shao WH, Cohen PL. Disturbances of apoptotic cell clearance in systemic lupus erythematosus. Arthritis Res Ther. 2011;13:202
34. Mistry P, Kaplan MJ. Cell death in the pathogenesis of systemic lupus erythematosus and lupus nephritis. Clin Immunol. 2017;185:59-73
35. Mahajan A, Herrmann M, Muñoz LE. Clearance Deficiency and Cell Death Pathways: A Model for the Pathogenesis of SLE. Front Immunol. 2016;7:35
36. Di Benedetto P, Ruscitti P, Vadasz Z, Toubi E, Giacomelli R. Macrophages with regulatory functions, a possible new therapeutic perspective in autoimmune diseases. Autoimmun Rev. 2019;18:102369
37. Engelen SE, Robinson AJB, Zurke YX, Monaco C. Therapeutic strategies targeting inflammation and immunity in atherosclerosis: how to proceed? Nat Rev Cardiol. 2022;19:522-42
38. Li M, Wang ZW, Fang LJ, Cheng SQ, Wang X, Liu NF. Programmed cell death in atherosclerosis and vascular calcification. Cell Death Dis. 2022;13:467
39. Libby P, Buring JE, Badimon L, Hansson GK, Deanfield J, Bittencourt MS. et al. Atherosclerosis. Nat Rev Dis Primers. 2019;5:56
40. Cai B, Thorp EB, Doran AC, Sansbury BE, Daemen MJ, Dorweiler B. et al. MerTK receptor cleavage promotes plaque necrosis and defective resolution in atherosclerosis. J Clin Invest. 2017;127:564-8
41. Flores AM, Hosseini-Nassab N, Jarr KU, Ye J, Zhu X, Wirka R. et al. Pro-efferocytic nanoparticles are specifically taken up by lesional macrophages and prevent atherosclerosis. Nat Nanotechnol. 2020;15:154-61
42. Knopman DS, Amieva H, Petersen RC, Chételat G, Holtzman DM, Hyman BT. et al. Alzheimer disease. Nat Rev Dis Primers. 2021;7:33
43. Butovsky O, Weiner HL. Microglial signatures and their role in health and disease. Nat Rev Neurosci. 2018;19:622-35
44. Zhang K, Zhang Y, Li Z, Chen J, Chang Y, Li Y. et al. Potentiating microglial efferocytosis by MFG-E8 improves survival and neurological outcome after successful cardiopulmonary resuscitation in mice. Brain Pathol. 2025;35:e13327
45. Subhramanyam CS, Wang C, Hu Q, Dheen ST. Microglia-mediated neuroinflammation in neurodegenerative diseases. Semin Cell Dev Biol. 2019;94:112-20
46. Konishi H, Koizumi S, Kiyama H. Phagocytic astrocytes: Emerging from the shadows of microglia. Glia. 2022;70:1009-26
47. Tremblay ME, Cookson MR, Civiero L. Glial phagocytic clearance in Parkinson's disease. Mol Neurodegener. 2019;14:16
48. Grégoire M, Uhel F, Lesouhaitier M, Gacouin A, Guirriec M, Mourcin F. et al. Impaired efferocytosis and neutrophil extracellular trap clearance by macrophages in ARDS. Eur Respir J. 2018 52
49. Liu X, Ou X, Zhang T, Li X, Qiao Q, Jia L. et al. In situ neutrophil apoptosis and macrophage efferocytosis mediated by Glycyrrhiza protein nanoparticles for acute inflammation therapy. J Control Release. 2024;369:215-30
50. Mahida RY, Scott A, Parekh D, Lugg ST, Belchamber KBR, Hardy RS. et al. Assessment of Alveolar Macrophage Dysfunction Using an in vitro Model of Acute Respiratory Distress Syndrome. Front Med (Lausanne). 2021;8:737859
51. Zhou X, Jin J, Lv T, Song Y. A Narrative Review: The Role of NETs in Acute Respiratory Distress Syndrome/Acute Lung Injury. Int J Mol Sci. 2024 25
52. Hotchkiss RS, Monneret G, Payen D. Sepsis-induced immunosuppression: from cellular dysfunctions to immunotherapy. Nat Rev Immunol. 2013;13:862-74
53. Baddam S, Burns B. Systemic Inflammatory Response Syndrome. StatPearls. Treasure Island (FL) ineligible companies. Disclosure: Bracken Burns declares no relevant financial relationships with ineligible companies.: StatPearls Publishing Copyright © 2025, StatPearls Publishing LLC. 2025
54. Huber-Lang M, Lambris JD, Ward PA. Innate immune responses to trauma. Nat Immunol. 2018;19:327-41
55. Qiu H, Shao Z, Wen X, Liu Z, Chen Z, Qu D. et al. Efferocytosis: An accomplice of cancer immune escape. Biomed Pharmacother. 2023;167:115540
56. Sica A, Schioppa T, Mantovani A, Allavena P. Tumour-associated macrophages are a distinct M2 polarised population promoting tumour progression: potential targets of anti-cancer therapy. Eur J Cancer. 2006;42:717-27
57. Lewis CE, Pollard JW. Distinct role of macrophages in different tumor microenvironments. Cancer Res. 2006;66:605-12
58. Wu G, Ma Z, Cheng Y, Hu W, Deng C, Jiang S. et al. Targeting Gas6/TAM in cancer cells and tumor microenvironment. Mol Cancer. 2018;17:20
59. Zhou Y, Yao Y, Deng Y, Shao A. Regulation of efferocytosis as a novel cancer therapy. Cell Commun Signal. 2020;18:71
60. Jorge AM, Lao T, Kim R, Licciardi S, El Khoury J, Luster AD. et al. SCARF1-Induced Efferocytosis Plays an Immunomodulatory Role in Humans, and Autoantibodies Targeting SCARF1 Are Produced in Patients with Systemic Lupus Erythematosus. J Immunol. 2022;208:955-67
61. Csorba K, Schirmbeck LA, Tuncer E, Ribi C, Roux-Lombard P, Chizzolini C. et al. Anti-C1q Antibodies as Occurring in Systemic Lupus Erythematosus Could Be Induced by an Epstein-Barr Virus-Derived Antigenic Site. Front Immunol. 2019;10:2619
62. Zhang S, Liu Y, Jing W, Chai Q, Tang C, Li Z. et al. Remodeling articular immune homeostasis with an efferocytosis-informed nanoimitator mitigates rheumatoid arthritis in mice. Nat Commun. 2023;14:817
63. Liu YQ, Li ZZ, Han YL, Wang QB. The role of efferocytosis in inflammatory bowel disease. Front Immunol. 2025;16:1524058
64. Garbin U, Baggio E, Stranieri C, Pasini A, Manfro S, Mozzini C. et al. Expansion of necrotic core and shedding of Mertk receptor in human carotid plaques: a role for oxidized polyunsaturated fatty acids? Cardiovasc Res. 2013;97:125-33
65. Harel-Adar T, Ben Mordechai T, Amsalem Y, Feinberg MS, Leor J, Cohen S. Modulation of cardiac macrophages by phosphatidylserine-presenting liposomes improves infarct repair. Proc Natl Acad Sci U S A. 2011;108:1827-32
66. Zhang F, Shah KG, Qi L, Wu R, Barrera R, Nicastro J. et al. Milk fat globule epidermal growth factor-factor 8 mitigates inflammation and tissue injury after hemorrhagic shock in experimental animals. J Trauma Acute Care Surg. 2012;72:861-9
67. Miao L, Yu C, Guan G, Luan X, Jin X, Pan M. et al. Extracellular vesicles containing GAS6 protect the liver from ischemia-reperfusion injury by enhancing macrophage efferocytosis via MerTK-ERK-COX2 signaling. Cell Death Discov. 2024;10:401
68. N AG, Bensinger SJ, Hong C, Beceiro S, Bradley MN, Zelcer N. et al. Apoptotic cells promote their own clearance and immune tolerance through activation of the nuclear receptor LXR. Immunity. 2009;31:245-58
69. Myers KV, Amend SR, Pienta KJ. Targeting Tyro3, Axl and MerTK (TAM receptors): implications for macrophages in the tumor microenvironment. Mol Cancer. 2019;18:94
70. Fernandez-Boyanapalli R, Frasch SC, Riches DW, Vandivier RW, Henson PM, Bratton DL. PPARγ activation normalizes resolution of acute sterile inflammation in murine chronic granulomatous disease. Blood. 2010;116:4512-22
71. Wu MY, Ge YJ, Wang EJ, Liao QW, Ren ZY, Yu Y. et al. Enhancement of efferocytosis through biased FPR2 signaling attenuates intestinal inflammation. EMBO Mol Med. 2023;15:e17815
72. Zhou Y, Fei M, Zhang G, Liang WC, Lin W, Wu Y. et al. Blockade of the Phagocytic Receptor MerTK on Tumor-Associated Macrophages Enhances P2X7R-Dependent STING Activation by Tumor-Derived cGAMP. Immunity. 2020;52:357-73.e9
73. Astuti Y, Raymant M, Quaranta V, Clarke K, Abudula M, Smith O. et al. Efferocytosis reprograms the tumor microenvironment to promote pancreatic cancer liver metastasis. Nat Cancer. 2024;5:774-90
74. Bae SH, Kim JH, Park TH, Lee K, Lee BI, Jang H. BMS794833 inhibits macrophage efferocytosis by directly binding to MERTK and inhibiting its activity. Exp Mol Med. 2022;54:1450-60
75. Nishida-Aoki N, Gujral TS. Polypharmacologic Reprogramming of Tumor-Associated Macrophages toward an Inflammatory Phenotype. Cancer Res. 2022;82:433-46
76. Petrova PS, Viller NN, Wong M, Pang X, Lin GH, Dodge K. et al. TTI-621 (SIRPαFc): A CD47-Blocking Innate Immune Checkpoint Inhibitor with Broad Antitumor Activity and Minimal Erythrocyte Binding. Clin Cancer Res. 2017;23:1068-79
77. Lakhani NJ, Chow LQM, Gainor JF, LoRusso P, Lee KW, Chung HC. et al. Evorpacept alone and in combination with pembrolizumab or trastuzumab in patients with advanced solid tumours (ASPEN-01): a first-in-human, open-label, multicentre, phase 1 dose-escalation and dose-expansion study. Lancet Oncol. 2021;22:1740-51
78. Hansen LW, Yang WL, Bolognese AC, Jacob A, Chen T, Prince JM. et al. Treatment with milk fat globule epidermal growth factor-factor 8 (MFG-E8) reduces inflammation and lung injury in neonatal sepsis. Surgery. 2017;162:349-57
79. Horst AK, Tiegs G, Diehl L. Contribution of Macrophage Efferocytosis to Liver Homeostasis and Disease. Front Immunol. 2019;10:2670
80. Bosurgi L, Cao YG, Cabeza-Cabrerizo M, Tucci A, Hughes LD, Kong Y. et al. Macrophage function in tissue repair and remodeling requires IL-4 or IL-13 with apoptotic cells. Science. 2017;356:1072-6
81. Ghazi NG, Abboud EB, Nowilaty SR, Alkuraya H, Alhommadi A, Cai H. et al. Treatment of retinitis pigmentosa due to MERTK mutations by ocular subretinal injection of adeno-associated virus gene vector: results of a phase I trial. Hum Genet. 2016;135:327-43
82. Morioka S, Kajioka D, Yamaoka Y, Ellison RM, Tufan T, Werkman IL. et al. Chimeric efferocytic receptors improve apoptotic cell clearance and alleviate inflammation. Cell. 2022;185:4887-903.e17
83. Martí-Díaz R, Sánchez-Del-Campo L, Montenegro MF, Hernández-Caselles T, Piñero-Madrona A, Cabezas-Herrera J. et al. Ex vivo engineering of phagocytic signals in breast cancer cells for a whole tumor cell-based vaccine. BMC Cancer. 2025;25:1029
84. Ray M, Lee YW, Hardie J, Mout R, Yeşilbag Tonga G, Farkas ME. et al. CRISPRed Macrophages for Cell-Based Cancer Immunotherapy. Bioconjug Chem. 2018;29:445-50
85. Zhang H, Huo Y, Zheng W, Li P, Li H, Zhang L. et al. Silencing of SIRPα enhances the antitumor efficacy of CAR-M in solid tumors. Cell Mol Immunol. 2024;21:1335-49
86. Wang T, Dong Y, Yao L, Lu F, Wen C, Wan Z. et al. Adoptive transfer of metabolically reprogrammed macrophages for atherosclerosis treatment in diabetic ApoE (-/-) mice. Bioact Mater. 2022;16:82-94
87. Esfahani NS, Wu Q, Kumar N, Ganesan LP, Lafuse WP, Rajaram MVS. Aging influences the cardiac macrophage phenotype and function during steady state and during inflammation. Aging Cell. 2021;20:e13438
88. Fessler MB. The challenges and promise of targeting the Liver X Receptors for treatment of inflammatory disease. Pharmacol Ther. 2018;181:1-12
89. Perera B, Wu Y, Nguyen NT, Ta HT. Advances in drug delivery to atherosclerosis: Investigating the efficiency of different nanomaterials employed for different type of drugs. Mater Today Bio. 2023;22:100767
90. Shen X, Pan D, Gong Q, Gu Z, Luo K. Enhancing drug penetration in solid tumors via nanomedicine: Evaluation models, strategies and perspectives. Bioact Mater. 2024;32:445-72
91. Oldenborg PA, Zheleznyak A, Fang YF, Lagenaur CF, Gresham HD, Lindberg FP. Role of CD47 as a marker of self on red blood cells. Science. 2000;288:2051-4
92. Son J, Hsieh RC, Lin HY, Krause KJ, Yuan Y, Biter AB. et al. Inhibition of the CD47-SIRPα axis for cancer therapy: A systematic review and meta-analysis of emerging clinical data. Front Immunol. 2022;13:1027235
93. Yang H, Xun Y, You H. The landscape overview of CD47-based immunotherapy for hematological malignancies. Biomark Res. 2023;11:15
94. Desai M, Kundu A, Hageman M, Lou H, Boisvert D. Monoclonal antibody and protein therapeutic formulations for subcutaneous delivery: high-concentration, low-volume vs. low-concentration, high-volume. MAbs. 2023;15:2285277
95. Tu L, Zou Z, Yang Y, Wang S, Xing B, Feng J. et al. Targeted drug delivery systems for atherosclerosis. J Nanobiotechnology. 2025;23:306
96. Velikova T, Sekulovski M, Peshevska-Sekulovska M. Immunogenicity and Loss of Effectiveness of Biologic Therapy for Inflammatory Bowel Disease Patients Due to Anti-Drug Antibody Development. Antibodies (Basel). 2024 13
97. Carlini V, Noonan DM, Abdalalem E, Goletti D, Sansone C, Calabrone L. et al. The multifaceted nature of IL-10: regulation, role in immunological homeostasis and its relevance to cancer, COVID-19 and post-COVID conditions. Front Immunol. 2023;14:1161067
98. Cheng SY, Punzo C. Update on Viral Gene Therapy Clinical Trials for Retinal Diseases. Hum Gene Ther. 2022;33:865-78
99. Lin M, Yang Z, Yang Y, Peng Y, Li J, Du Y. et al. CRISPR-based in situ engineering tumor cells to reprogram macrophages for effective cancer immunotherapy. Nano Today. 2022;42:101359
100. Stamataki M, Rissiek B, Magnus T, Körbelin J. Microglia targeting by adeno-associated viral vectors. Front Immunol. 2024;15:1425892
101. Wang JH, Gessler DJ, Zhan W, Gallagher TL, Gao G. Adeno-associated virus as a delivery vector for gene therapy of human diseases. Signal Transduct Target Ther. 2024;9:78
102. Fredman G, Kamaly N, Spolitu S, Milton J, Ghorpade D, Chiasson R. et al. Targeted nanoparticles containing the proresolving peptide Ac2-26 protect against advanced atherosclerosis in hypercholesterolemic mice. Sci Transl Med. 2015;7:275ra20
103. Tang M, Chen B, Xia H, Pan M, Zhao R, Zhou J. et al. pH-gated nanoparticles selectively regulate lysosomal function of tumour-associated macrophages for cancer immunotherapy. Nat Commun. 2023;14:5888
104. Sabit H, Pawlik TM, Radwan F, Abdel-Hakeem M, Abdel-Ghany S, Wadan AS. et al. Precision nanomedicine: navigating the tumor microenvironment for enhanced cancer immunotherapy and targeted drug delivery. Mol Cancer. 2025;24:160
105. Li Y, Li H, Hu Z, Zhang Y, Ding X, Huang X. et al. Phosphatidylserine-decorated delivery platform helps alleviate acute lung injury via potentiating macrophage targeting. J Lipid Res. 2025;66:100799
106. Zhang Q, Hu C, Feng J, Long H, Wang Y, Wang P. et al. Anti-inflammatory mechanisms of neutrophil membrane-coated nanoparticles without drug loading. J Control Release. 2024;369:12-24
107. Kwon S, Meng F, Tamam H, Gadalla HH, Wang J, Dong B. et al. Systemic Delivery of Paclitaxel by Find-Me Nanoparticles Activates Antitumor Immunity and Eliminates Tumors. ACS Nano. 2024;18:3681-98
108. Yuan S, Chai Y, Xu J, Wang Y, Jiang L, Lu N. et al. Engineering Efferocytosis-Mimicking Nanovesicles to Regulate Joint Anti-Inflammation and Peripheral Immunosuppression for Rheumatoid Arthritis Therapy. Adv Sci (Weinh). 2024;11:e2404198
109. Bao L, Dou G, Tian R, Lv Y, Ding F, Liu S. et al. Engineered neutrophil apoptotic bodies ameliorate myocardial infarction by promoting macrophage efferocytosis and inflammation resolution. Bioact Mater. 2022;9:183-97
110. Lu Y, Huntoon K, Lee D, Wang Y, Ha J, Qie Y. et al. Immunological conversion of solid tumours using a bispecific nanobioconjugate for cancer immunotherapy. Nat Nanotechnol. 2022;17:1332-41
111. Qiu S, Liu J, Chen J, Li Y, Bu T, Li Z. et al. Targeted delivery of MerTK protein via cell membrane engineered nanoparticle enhances efferocytosis and attenuates atherosclerosis in diabetic ApoE(-/-) Mice. J Nanobiotechnology. 2024;22:178
112. Xu N, Li J, Gao Y, Zhou N, Ma Q, Wu M. et al. Apoptotic cell-mimicking gold nanocages loaded with LXR agonist for attenuating the progression of murine systemic lupus erythematosus. Biomaterials. 2019;197:380-92
113. Fan L, Jin L, Tang T, Zheng Y, Chen Z, Lin H. et al. Neutrophil-like pH-responsive pro-efferocytic nanoparticles improve neurological recovery by promoting erythrophagocytosis after intracerebral hemorrhage. Theranostics. 2024;14:283-303
114. Wu Y, Wang C, Yan Y, Hao Y, Liu B, Dong Z. et al. Efferocytosis Nanoinhibitors to Promote Secondary Necrosis and Potentiate the Immunogenicity of Conventional Cancer Therapies for Improved Therapeutic Benefits. ACS Nano. 2023;17:18089-102
115. Luo JQ, Liu R, Chen FM, Zhang JY, Zheng SJ, Shao D. et al. Nanoparticle-Mediated CD47-SIRPα Blockade and Calreticulin Exposure for Improved Cancer Chemo-Immunotherapy. ACS Nano. 2023;17:8966-79
116. Hussain Z, Gou S, Liu X, Li M, Zhang H, Ren S. et al. Enhancing tumor immunotherapy with smart nanoparticles for reprogramming macrophages and blocking the CD47/Sirpα pathway. Mater Today Bio. 2025;32:101826
117. Wang K, Yu A, Liu K, Feng C, Hou Y, Chen J. et al. Nano-LYTACs for Degradation of Membrane Proteins and Inhibition of CD24/Siglec-10 Signaling Pathway. Adv Sci (Weinh). 2023;10:e2305364
118. Ma X, Tan X, Yu B, Sun W, Wang H, Hu H. et al. DOCK2 regulates antifungal immunity by regulating RAC GTPase activity. Cell Mol Immunol. 2022;19:602-18
119. Lo CH, O'Connor LM, Loi GWZ, Saipuljumri EN, Indajang J, Lopes KM. et al. Acidic Nanoparticles Restore Lysosomal Acidification and Rescue Metabolic Dysfunction in Pancreatic β-Cells under Lipotoxic Conditions. ACS Nano. 2024;18:15452-67
120. Choi JY, Ryu J, Kim HJ, Song JW, Jeon JH, Lee DH. et al. Therapeutic Effects of Targeted PPARɣ Activation on Inflamed High-Risk Plaques Assessed by Serial Optical Imaging In Vivo. Theranostics. 2018;8:45-60
121. Ji H, Zhang C, Xu F, Mao Q, Xia R, Chen M. et al. Inhaled Pro-Efferocytic Nanozymes Promote Resolution of Acute Lung Injury. Adv Sci (Weinh). 2022;9:e2201696
122. Liao CY, Song MJ, Gao Y, Mauer AS, Revzin A, Malhi H. Hepatocyte-Derived Lipotoxic Extracellular Vesicle Sphingosine 1-Phosphate Induces Macrophage Chemotaxis. Front Immunol. 2018;9:2980
123. Kim KW, Ivanov S, Williams JW. Monocyte Recruitment, Specification, and Function in Atherosclerosis. Cells. 2020 10
124. Li W. Eat-me signals: keys to molecular phagocyte biology and "appetite" control. J Cell Physiol. 2012;227:1291-7
125. Ma Z, Sun Y, Yu Y, Xiao W, Xiao Z, Zhong T. et al. Extracellular vesicles containing MFGE8 from colorectal cancer facilitate macrophage efferocytosis. Cell Commun Signal. 2024;22:295
126. Li Y, Chen S, Liu YJ. Microglial phagoptosis in development, health, and disease. Neurobiol Dis. 2026;218:107211
127. Zhou X, Liu X, Huang L. Macrophage-Mediated Tumor Cell Phagocytosis: Opportunity for Nanomedicine Intervention. Adv Funct Mater. 2021 31
128. Lahey KC, Gadiyar V, Hill A, Desind S, Wang Z, Davra V. et al. Mertk: An emerging target in cancer biology and immuno-oncology. Int Rev Cell Mol Biol. 2022;368:35-59
129. Shofolawe-Bakare O, Toragall VB, Hulugalla K, Mayatt R, Iammarino P, Bentley JP. et al. Glycopolymeric Nanoparticles Block Breast Cancer Growth by Inhibiting Efferocytosis in the Tumor Microenvironment. ACS Appl Nano Mater. 2024;7:28851-63
130. Cho CY, Huang JS, Shiah SG, Chung SY, Lay JD, Yang YY. et al. Negative feedback regulation of AXL by miR-34a modulates apoptosis in lung cancer cells. Rna. 2016;22:303-15
131. Hong DS, Kang YK, Borad M, Sachdev J, Ejadi S, Lim HY. et al. Phase 1 study of MRX34, a liposomal miR-34a mimic, in patients with advanced solid tumours. Br J Cancer. 2020;122:1630-7
132. Mills KA, Quinn JM, Roach ST, Palisoul M, Nguyen M, Noia H. et al. p5RHH nanoparticle-mediated delivery of AXL siRNA inhibits metastasis of ovarian and uterine cancer cells in mouse xenografts. Sci Rep. 2019;9:4762
133. Shi H, Wang X, Sloas C, Gerlach B, Yurdagul A Jr, Moore MP. et al. Impaired TIM4-mediated efferocytosis by liver macrophages contributes to fibrosis in metabolic dysfunction-associated steatohepatitis. Sci Transl Med. 2025;17:eadv2106
134. Brown GC, Neher JJ. Eaten alive!. Cell death by primary phagocytosis: 'phagoptosis'. Trends Biochem Sci. 2012;37:325-32
135. Chao MP, Majeti R, Weissman IL. Programmed cell removal: a new obstacle in the road to developing cancer. Nat Rev Cancer. 2011;12:58-67
136. Chen Q, Wang C, Zhang X, Chen G, Hu Q, Li H. et al. In situ sprayed bioresponsive immunotherapeutic gel for post-surgical cancer treatment. Nat Nanotechnol. 2019;14:89-97
137. Gresham HD, Dale BM, Potter JW, Chang PW, Vines CM, Lowell CA. et al. Negative regulation of phagocytosis in murine macrophages by the Src kinase family member, Fgr. J Exp Med. 2000;191:515-28
138. Wang H, Sun Y, Zhou X, Chen C, Jiao L, Li W. et al. CD47/SIRPα blocking peptide identification and synergistic effect with irradiation for cancer immunotherapy. J Immunother Cancer. 2020 8
139. Chang R, Chu X, Zhang J, Fu R, Feng C, Jia D. et al. Liposome-Based Co-Immunotherapy with TLR Agonist and CD47-SIRPα Checkpoint Blockade for Efficient Treatment of Colon Cancer. Molecules. 2023 28
140. Ji X, Qian X, Luo G, Yang W, Huang W, Lei Z. et al. Engineered macrophage nanoparticles enhance microwave ablation efficacy in osteosarcoma via targeting the CD47-SIRPα Axis: A novel Biomimetic immunotherapeutic approach. Bioact Mater. 2025;47:248-65
141. Kim YK, Hong Y, Bae YR, Goo J, Kim SA, Choi Y. et al. Advantage of extracellular vesicles in hindering the CD47 signal for cancer immunotherapy. J Control Release. 2022;351:727-38
142. Barkal AA, Brewer RE, Markovic M, Kowarsky M, Barkal SA, Zaro BW. et al. CD24 signalling through macrophage Siglec-10 is a target for cancer immunotherapy. Nature. 2019;572:392-6
143. Vavassori V, Ferrari S, Beretta S, Asperti C, Albano L, Annoni A. et al. Lipid nanoparticles allow efficient and harmless ex vivo gene editing of human hematopoietic cells. Blood. 2023;142:812-26
144. Feng M, Jiang W, Kim BYS, Zhang CC, Fu YX, Weissman IL. Phagocytosis checkpoints as new targets for cancer immunotherapy. Nat Rev Cancer. 2019;19:568-86
145. Li X, Tian W, Jiang Z, Song Y, Leng X, Yu J. Targeting CD24/Siglec-10 signal pathway for cancer immunotherapy: recent advances and future directions. Cancer Immunol Immunother. 2024;73:31
146. Gracia-Hernandez M, Suresh M, Villagra A. The advances in targeting CD47/SIRPα "do not eat me" axis and their ongoing challenges as an anticancer therapy. Oncotarget. 2024;15:462-5
147. Maute R, Xu J, Weissman IL. CD47-SIRPα-targeted therapeutics: status and prospects. Immunooncol Technol. 2022;13:100070
148. Aflaki E, Borger DK, Grey RJ, Kirby M, Anderson S, Lopez G. et al. Efferocytosis is impaired in Gaucher macrophages. Haematologica. 2017;102:656-65
149. Diesel B, Hoppstädter J, Hachenthal N, Zarbock R, Cavelius C, Wahl B. et al. Activation of Rac1 GTPase by nanoparticulate structures in human macrophages. Eur J Pharm Biopharm. 2013;84:315-24
150. Duewell P, Kono H, Rayner KJ, Sirois CM, Vladimer G, Bauernfeind FG. et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature. 2010;464:1357-61
151. Settembre C, Medina DL. TFEB and the CLEAR network. Methods Cell Biol. 2015;126:45-62
152. Sergin I, Evans TD, Zhang X, Bhattacharya S, Stokes CJ, Song E. et al. Exploiting macrophage autophagy-lysosomal biogenesis as a therapy for atherosclerosis. Nat Commun. 2017;8:15750
153. Seo J, Oh DB. Mannose-6-phosphate glycan for lysosomal targeting: various applications from enzyme replacement therapy to lysosome-targeting chimeras. Anim Cells Syst (Seoul). 2022;26:84-91
154. Napolitano G, Esposito A, Choi H, Matarese M, Benedetti V, Di Malta C. et al. mTOR-dependent phosphorylation controls TFEB nuclear export. Nat Commun. 2018;9:3312
155. Quick JD, Silva C, Wong JH, Lim KL, Reynolds R, Barron AM. et al. Lysosomal acidification dysfunction in microglia: an emerging pathogenic mechanism of neuroinflammation and neurodegeneration. J Neuroinflammation. 2023;20:185
156. Schilperoort M, Ngai D, Sukka SR, Avrampou K, Shi H, Tabas I. The role of efferocytosis-fueled macrophage metabolism in the resolution of inflammation. Immunol Rev. 2023;319:65-80
157. Chawla A, Boisvert WA, Lee CH, Laffitte BA, Barak Y, Joseph SB. et al. A PPAR gamma-LXR-ABCA1 pathway in macrophages is involved in cholesterol efflux and atherogenesis. Mol Cell. 2001;7:161-71
158. Smorodinsky NI, Cahalon L, Argov S, Witz IP, Shoenfeld Y. Naturally-occurring tumor-reactive autoantibodies: a monoclonal antibody from normal mice reacts with tumor cells and with DNA. Immunol Lett. 1988;18:43-9
159. Yang TM, Miao M, Yu WQ, Wang X, Xia FJ, Li YJ. et al. Targeting macrophages in atherosclerosis using nanocarriers loaded with liver X receptor agonists: A narrow review. Front Mol Biosci. 2023;10:1147699
160. Chen W, Schilperoort M, Cao Y, Shi J, Tabas I, Tao W. Macrophage-targeted nanomedicine for the diagnosis and treatment of atherosclerosis. Nat Rev Cardiol. 2022;19:228-49
161. Inamdar S, Tylek T, Thumsi A, Suresh AP, Jaggarapu M, Halim M. et al. Biomaterial mediated simultaneous delivery of spermine and alpha ketoglutarate modulate metabolism and innate immune cell phenotype in sepsis mouse models. Biomaterials. 2023;293:121973
162. Zhang S, Weinberg S, DeBerge M, Gainullina A, Schipma M, Kinchen JM. et al. Efferocytosis Fuels Requirements of Fatty Acid Oxidation and the Electron Transport Chain to Polarize Macrophages for Tissue Repair. Cell Metab. 2019;29:443-56.e5
163. Park D, Han CZ, Elliott MR, Kinchen JM, Trampont PC, Das S. et al. Continued clearance of apoptotic cells critically depends on the phagocyte Ucp2 protein. Nature. 2011;477:220-4
164. Wang H, Zhang Y, Zhang Y, Li C, Zhang M, Wang J. et al. Activating Macrophage Continual Efferocytosis via Microenvironment Biomimetic Short Fibers for Reversing Inflammation in Bone Repair. Adv Mater. 2024;36:e2402968
165. McCubbrey AL, Nelson JD, Stolberg VR, Blakely PK, McCloskey L, Janssen WJ. et al. MicroRNA-34a Negatively Regulates Efferocytosis by Tissue Macrophages in Part via SIRT1. J Immunol. 2016;196:1366-75
166. Goossens P, Rodriguez-Vita J, Etzerodt A, Masse M, Rastoin O, Gouirand V. et al. Membrane Cholesterol Efflux Drives Tumor-Associated Macrophage Reprogramming and Tumor Progression. Cell Metab. 2019;29:1376-89.e4
167. Bendre SV, Wang Y, Hajyousif B, K CR, Bhogale SG, Pradeep D. et al. Cholesterol efflux protein, ABCA1, supports anticancer functions of myeloid immune cells. Sci Adv. 2026;12:eadx5490
168. Fan D, Cao Y, Cao M, Wang Y, Cao Y, Gong T. Nanomedicine in cancer therapy. Signal Transduct Target Ther. 2023;8:293
169. Xu K, Duan S, Wang W, Ouyang Q, Qin F, Guo P. et al. Nose-to-brain delivery of nanotherapeutics: Transport mechanisms and applications. Wiley Interdiscip Rev Nanomed Nanobiotechnol. 2024;16:e1956
170. Lei W, Yang C, Wu Y, Ru G, He X, Tong X. et al. Nanocarriers surface engineered with cell membranes for cancer targeted chemotherapy. J Nanobiotechnology. 2022;20:45
171. Arandjelovic S, Ravichandran KS. Phagocytosis of apoptotic cells in homeostasis. Nat Immunol. 2015;16:907-17
172. Gustafson HH, Holt-Casper D, Grainger DW, Ghandehari H. Nanoparticle Uptake: The Phagocyte Problem. Nano Today. 2015;10:487-510
173. Dossou AS, Mantsch ME, Kapic A, Burnett WL, Sabnis N, Coffer JL. et al. Mannose-Coated Reconstituted Lipoprotein Nanoparticles for the Targeting of Tumor-Associated Macrophages: Optimization, Characterization, and In Vitro Evaluation of Effectiveness. Pharmaceutics. 2023 15
174. Verweij NJF, Yaqub M, Bruijnen STG, Pieplenbosch S, Ter Wee MM, Jansen G. et al. First in man study of [(18)F]fluoro-PEG-folate PET: a novel macrophage imaging technique to visualize rheumatoid arthritis. Sci Rep. 2020;10:1047
175. Pan D, Yan Y, Yang R, Xu YP, Chen F, Wang L. et al. PET imaging of prostate tumors with 18F-Al-NOTA-MATBBN. Contrast Media Mol Imaging. 2014;9:342-8
176. Wang Y, Zhang Y, Wang Z, Zhang J, Qiao RR, Xu M. et al. Optical/MRI dual-modality imaging of M1 macrophage polarization in atherosclerotic plaque with MARCO-targeted upconversion luminescence probe. Biomaterials. 2019;219:119378
177. Freeman MT, Parvaresh-Rizi A, Meenach SA. Enhanced Macrophage Uptake of Spray-Dried Phosphatidylserine-Loaded Microparticles for Pulmonary Drug Delivery Applications. J Drug Deliv Sci Technol. 2025 104
178. Wu N, Han Z, Lv W, Huang Y, Zhu J, Deng J. et al. Reprogramming peritoneal macrophages with outer membrane vesicle-coated PLGA nanoparticles for endometriosis prevention. Biomaterials. 2025;319:123198
179. Zhou Y, Wang X, Zhang D, Cui H, Tian X, Du W. et al. Precision-Guided Stealth Missiles in Biomedicine: Biological Carrier-Mediated Nanomedicine Hitchhiking Strategy. Adv Sci (Weinh). 2025;12:e2504672
180. He Y, Wang Y, Wang L, Jiang W, Wilhelm S. Understanding nanoparticle-liver interactions in nanomedicine. Expert Opin Drug Deliv. 2024;21:829-43
181. Cojocaru E, Petriș OR, Cojocaru C. Nanoparticle-Based Drug Delivery Systems in Inhaled Therapy: Improving Respiratory Medicine. Pharmaceuticals (Basel). 2024 17
182. Lin G, Wang J, Yang YG, Zhang Y, Sun T. Advances in dendritic cell targeting nano-delivery systems for induction of immune tolerance. Front Bioeng Biotechnol. 2023;11:1242126
183. Perez CR, De Palma M. Engineering dendritic cell vaccines to improve cancer immunotherapy. Nat Commun. 2019;10:5408
184. Manolova V, Flace A, Bauer M, Schwarz K, Saudan P, Bachmann MF. Nanoparticles target distinct dendritic cell populations according to their size. Eur J Immunol. 2008;38:1404-13
185. Apostolopoulos V, Thalhammer T, Tzakos AG, Stojanovska L. Targeting antigens to dendritic cell receptors for vaccine development. J Drug Deliv. 2013;2013:869718
186. Saluja SS, Hanlon DJ, Sharp FA, Hong E, Khalil D, Robinson E. et al. Targeting human dendritic cells via DEC-205 using PLGA nanoparticles leads to enhanced cross-presentation of a melanoma-associated antigen. Int J Nanomedicine. 2014;9:5231-46
187. Li S, Murakami D, Nagatoishi S, Liu Y, Tsumoto K, Katayama Y. et al. One-pot preparation of mannan-coated antigen nanoparticles using human serum albumin as a matrix for tolerance induction. J Colloid Interface Sci. 2023;649:955-65
188. Kroczek AL, Hartung E, Gurka S, Becker M, Reeg N, Mages HW. et al. Structure-Function Relationship of XCL1 Used for in vivo Targeting of Antigen Into XCR1(+) Dendritic Cells. Front Immunol. 2018;9:2806
189. Nguyen NT, Le XT, Lee WT, Lim YT, Oh KT, Lee ES. et al. STING-activating dendritic cell-targeted nanovaccines that evoke potent antigen cross-presentation for cancer immunotherapy. Bioact Mater. 2024;42:345-65
190. Finnemann SC, Bonilha VL, Marmorstein AD, Rodriguez-Boulan E. Phagocytosis of rod outer segments by retinal pigment epithelial cells requires alpha(v)beta5 integrin for binding but not for internalization. Proc Natl Acad Sci U S A. 1997;94:12932-7
191. Sandahl M, Hunter DM, Strunk KE, Earp HS, Cook RS. Epithelial cell-directed efferocytosis in the post-partum mammary gland is necessary for tissue homeostasis and future lactation. BMC Dev Biol. 2010;10:122
192. Juncadella IJ, Kadl A, Sharma AK, Shim YM, Hochreiter-Hufford A, Borish L. et al. Apoptotic cell clearance by bronchial epithelial cells critically influences airway inflammation. Nature. 2013;493:547-51
193. Liu J, Luo L, Xu F, Li G, Chen J, Teng L. et al. Cyclic RGD Peptide Targeting Coated Nano Drug Co-Delivery System for Therapeutic Use in Age-Related Macular Degeneration Disease. Molecules. 2020 25
194. Jepson MA, Clark MA, Hirst BH. M cell targeting by lectins: a strategy for mucosal vaccination and drug delivery. Adv Drug Deliv Rev. 2004;56:511-25
195. Prasher P, Sharma M, Singh SK, Gulati M, Jha NK, Gupta PK. et al. Targeting mucus barrier in respiratory diseases by chemically modified advanced delivery systems. Chem Biol Interact. 2022;365:110048
196. Rathi R, Sanshita, Kumar A, Vishvakarma V, Huanbutta K, Singh I. et al. Advancements in Rectal Drug Delivery Systems: Clinical Trials, and Patents Perspective. Pharmaceutics. 2022 14
197. Tam A, Kulkarni J, An K, Li L, Dorscheid DR, Singhera GK. et al. Lipid nanoparticle formulations for optimal RNA-based topical delivery to murine airways. Eur J Pharm Sci. 2022;176:106234
198. Marchenko IV, Trushina DB. Local Drug Delivery in Bladder Cancer: Advances of Nano/Micro/Macro-Scale Drug Delivery Systems. Pharmaceutics. 2023 15
199. Romana-Souza B, Chen L, Leonardo TR, Chen Z, DiPietro LA. Dermal fibroblast phagocytosis of apoptotic cells: A novel pathway for wound resolution. Faseb j. 2021;35:e21443
200. Engelholm LH, List K, Netzel-Arnett S, Cukierman E, Mitola DJ, Aaronson H. et al. uPARAP/Endo180 is essential for cellular uptake of collagen and promotes fibroblast collagen adhesion. J Cell Biol. 2003;160:1009-15
201. Yang D, Liu J, Qian H, Zhuang Q. Cancer-associated fibroblasts: from basic science to anticancer therapy. Exp Mol Med. 2023;55:1322-32
202. Xin L, Gao J, Zheng Z, Chen Y, Lv S, Zhao Z. et al. Fibroblast Activation Protein-α as a Target in the Bench-to-Bedside Diagnosis and Treatment of Tumors: A Narrative Review. Front Oncol. 2021;11:648187
203. Abbasi S, Khademi S, Montazerabadi A, Sahebkar A. FAP-Targeted Nanoparticle-based Imaging in Cancer: A Systematic Review. J Biomed Phys Eng. 2024;14:323-34
204. Chen Y, Wang H, Qi N, Wu H, Xiong W, Ma J. et al. Functions of TAM RTKs in regulating spermatogenesis and male fertility in mice. Reproduction. 2009;138:655-66
205. Klein JP, Mery L, Boudard D, Ravel C, Cottier M, Bitounis D. Impact of Nanoparticles on Male Fertility: What Do We Really Know? A Systematic Review. Int J Mol Sci. 2022 24
206. Lin Y, He R, Sun L, Yang Y, Li W, Sun F. Pentacle gold-copper alloy nanocrystals: a new system for entering male germ cells in vitro and in vivo. Sci Rep. 2016;6:39592
207. Fraser B, Wilkins A, Whiting S, Liang M, Rebourcet D, Nixon B. et al. Development of peptides for targeting cell ablation agents concurrently to the Sertoli and Leydig cell populations of the testes: An approach to non-surgical sterilization. PLoS One. 2024;19:e0292198
208. Fraser BA, Wilkins AL, De Iuliis GN, Rebourcet D, Nixon B, Aitken RJ. Development of a model for studying the developmental consequences of oxidative sperm DNA damage by targeting redox-cycling naphthoquinones to the Sertoli cell population. Free Radic Biol Med. 2023;206:50-62
209. Yu J, Xu J, Li H, Wu P, Zhu S, Huang X. et al. Gold nanoparticles retrogradely penetrate through testicular barriers via Sertoli-cells mediated endocytosis/exocytosis and induce immune response in mouse. Ecotoxicol Environ Saf. 2023;255:114827
210. Yuan W, Yu B, Yu M, Kuai R, Morin EE, Wang H. et al. Synthetic high-density lipoproteins delivering liver X receptor agonist prevent atherogenesis by enhancing reverse cholesterol transport. J Control Release. 2021;329:361-71
211. He Z, Chen W, Hu K, Luo Y, Zeng W, He X. et al. Resolvin D1 delivery to lesional macrophages using antioxidative black phosphorus nanosheets for atherosclerosis treatment. Nat Nanotechnol. 2024;19:1386-98
212. Tang C, Wang H, Guo L, Zou C, Hu J, Zhang H. et al. CpG-Conjugated Silver Nanoparticles as a Multifunctional Nanomedicine to Promote Macrophage Efferocytosis and Repolarization for Atherosclerosis Therapy. ACS Appl Mater Interfaces. 2023;15:52162-79
213. You P, Yang A, Sun Y, Zhang H, Zeng Y, Hao Y. et al. Pro-efferocytosis biomimetic nanocomplexes for targeted atherosclerosis therapy through promoting macrophage re-polarization and inhibiting senescence. Materials & Design. 2023;234:112316
214. Naeini MB, Bianconi V, Pirro M, Sahebkar A. The role of phosphatidylserine recognition receptors in multiple biological functions. Cell Mol Biol Lett. 2020;25:23
215. Garcia-Loza I, Perna-Barrull D, Aguilera E, Almenara-Fuentes L, Gomez-Muñoz L, Greco D. et al. Targeting macrophages with phosphatidylserine-rich liposomes as a potential antigen-specific immunotherapy for type 1 diabetes. J Autoimmun. 2024;145:103196
216. Zhang M, Johnson-Stephenson TK, Wang W, Wang Y, Li J, Li L. et al. Mesenchymal stem cell-derived exosome-educated macrophages alleviate systemic lupus erythematosus by promoting efferocytosis and recruitment of IL-17(+) regulatory T cell. Stem Cell Res Ther. 2022;13:484
217. Al-Mayouf SM, Sunker A, Abdwani R, Abrawi SA, Almurshedi F, Alhashmi N. et al. Loss-of-function variant in DNASE1L3 causes a familial form of systemic lupus erythematosus. Nat Genet. 2011;43:1186-8
218. Shao L, Zhang Y, Yang Z, Shi C, Yue X, Li C. et al. Synthetic efferocytic receptor microglia enhances anti-inflammatory clearance of amyloid-β for AD treatment in mice. Sci Adv. 2025;11:eads6613
219. Shin HJ, Kim IS, Choi SG, Lee K, Park H, Shin J. et al. Rejuvenating aged microglia by p16(ink4a)-siRNA-loaded nanoparticles increases amyloid-β clearance in animal models of Alzheimer's disease. Mol Neurodegener. 2024;19:25
220. Gebril HM, Aryasomayajula A, de Lima MRN, Uhrich KE, Moghe PV. Nanotechnology for microglial targeting and inhibition of neuroinflammation underlying Alzheimer's pathology. Transl Neurodegener. 2024;13:2
221. Wang B, Martini-Stoica H, Qi C, Lu TC, Wang S, Xiong W. et al. TFEB-vacuolar ATPase signaling regulates lysosomal function and microglial activation in tauopathy. Nat Neurosci. 2024;27:48-62
222. Liu HJ, Wang M, Wang Y, Shi S, Hu X, Zhang DB. et al. Ceria-nanocluster based therapy for Alzheimer's disease through the modulation of activated microglia and attenuation of amyloid-β deposition. Cell Biomater. 2025
223. Gandhi S, Shastri DH, Shah J, Nair AB, Jacob S. Nasal Delivery to the Brain: Harnessing Nanoparticles for Effective Drug Transport. Pharmaceutics. 2024 16
224. He X, Wang X, Yang L, Yang Z, Yu W, Wang Y. et al. Intelligent lesion blood-brain barrier targeting nano-missiles for Alzheimer's disease treatment by anti-neuroinflammation and neuroprotection. Acta Pharm Sin B. 2022;12:1987-99
225. Pizzo ME, Plowey ED, Khoury N, Kwan W, Abettan J, DeVos SL. et al. Transferrin receptor-targeted anti-amyloid antibody enhances brain delivery and mitigates ARIA. Science. 2025;389:eads3204
226. Yin C, Heit B. Cellular Responses to the Efferocytosis of Apoptotic Cells. Front Immunol. 2021;12:631714
227. Wang Y, Zhang LF, Zhang JJ, Yu SS, Li WL, Zhou TJ. et al. Spontaneous Inflammation Resolution Inspired Nanoparticles Promote Neutrophil Apoptosis and Macrophage Efferocytosis for Acute Respiratory Distress Syndrome Treatment. Adv Healthc Mater. 2025;14:e2402421
228. Miksa M, Wu R, Dong W, Komura H, Amin D, Ji Y. et al. Immature dendritic cell-derived exosomes rescue septic animals via milk fat globule epidermal growth factor-factor VIII [corrected]. J Immunol. 2009;183:5983-90
229. Wang Z, Xie G, Xie Y, Hu M, Hu Q, Yang M. et al. Kim-1 targeted black phosphorus nanoplatforms: Antioxidation and efferocytosis recovery for acute kidney injury treatment. Chemical Engineering Journal. 2024;496:154207
230. van Heerden PV, Abutbul A, Sviri S, Zlotnick E, Nama A, Zimro S. et al. Apoptotic Cells for Therapeutic Use in Cytokine Storm Associated With Sepsis- A Phase Ib Clinical Trial. Front Immunol. 2021;12:718191
231. Ng CY, Kee LT, Al-Masawa ME, Lee QH, Subramaniam T, Kok D. et al. Scalable Production of Extracellular Vesicles and Its Therapeutic Values: A Review. Int J Mol Sci. 2022 23
232. Jia D, Lu Y, Lv M, Wang F, Lu X, Zhu W. et al. Targeted co-delivery of resiquimod and a SIRPα variant by liposomes to activate macrophage immune responses for tumor immunotherapy. J Control Release. 2023;360:858-71
233. Yang Y, Liu Q, Wang M, Li L, Yu Y, Pan M. et al. Genetically programmable cell membrane-camouflaged nanoparticles for targeted combination therapy of colorectal cancer. Signal Transduct Target Ther. 2024;9:158
234. Levengood MR, Carosino CM, Zhang X, Lucas S, Ortiz DJ, Westendorf L. et al. Preclinical Development of SGN-CD47M: Protease-Activated Antibody Technology Enables Selective Tumor Targeting of the Innate Immune Checkpoint Receptor CD47. Mol Cancer Ther. 2025;24:471-84
235. Querfeld C, Thompson JA, Taylor MH, DeSimone JA, Zain JM, Shustov AR. et al. Intralesional TTI-621, a novel biologic targeting the innate immune checkpoint CD47, in patients with relapsed or refractory mycosis fungoides or Sézary syndrome: a multicentre, phase 1 study. Lancet Haematol. 2021;8:e808-e17
236. Autio KA, Boni V, Humphrey RW, Naing A. Probody Therapeutics: An Emerging Class of Therapies Designed to Enhance On-Target Effects with Reduced Off-Tumor Toxicity for Use in Immuno-Oncology. Clin Cancer Res. 2020;26:984-9
237. Li Y, Zhang C, Li G, Deng G, Zhang H, Sun Y. et al. Protease-triggered bioresponsive drug delivery for the targeted theranostics of malignancy. Acta Pharm Sin B. 2021;11:2220-42
238. Serhan CN, Levy BD. Resolvins in inflammation: emergence of the pro-resolving superfamily of mediators. J Clin Invest. 2018;128:2657-69
239. Barenholz Y. Doxil®-the first FDA-approved nano-drug: lessons learned. J Control Release. 2012;160:117-34
240. Patel R, Kaki M, Potluri VS, Kahar P, Khanna D. A comprehensive review of SARS-CoV-2 vaccines: Pfizer, Moderna & Johnson & Johnson. Hum Vaccin Immunother. 2022;18:2002083
241. Zhang YN, Poon W, Tavares AJ, McGilvray ID, Chan WCW. Nanoparticle-liver interactions: Cellular uptake and hepatobiliary elimination. J Control Release. 2016;240:332-48
242. Wilhelm S, Tavares AJ, Dai Q, Ohta S, Audet J, Dvorak HF. et al. Analysis of nanoparticle delivery to tumours. Nature Reviews Materials. 2016;1:16014
243. Cummings RD. The mannose receptor ligands and the macrophage glycome. Curr Opin Struct Biol. 2022;75:102394
244. Nielsen MC, Hvidbjerg Gantzel R, Clària J, Trebicka J, Møller HJ, Grønbæk H. Macrophage Activation Markers, CD163 and CD206, in Acute-on-Chronic Liver Failure. Cells. 2020 9
245. Tregubov AA, Nikitin PI, Nikitin MP. Advanced Smart Nanomaterials with Integrated Logic-Gating and Biocomputing: Dawn of Theranostic Nanorobots. Chem Rev. 2018;118:10294-348
246. Bai L, Wang XH, Song F, Wang XL, Wang YZ. "AND" logic gate regulated pH and reduction dual-responsive prodrug nanoparticles for efficient intracellular anticancer drug delivery. Chem Commun (Camb). 2015;51:93-6
247. Medeiros AI, Serezani CH, Lee SP, Peters-Golden M. Efferocytosis impairs pulmonary macrophage and lung antibacterial function via PGE2/EP2 signaling. J Exp Med. 2009;206:61-8
248. Kim KK, Dotson MR, Agarwal M, Yang J, Bradley PB, Subbotina N. et al. Efferocytosis of apoptotic alveolar epithelial cells is sufficient to initiate lung fibrosis. Cell Death Dis. 2018;9:1056
249. Szebeni J, Muggia F, Gabizon A, Barenholz Y. Activation of complement by therapeutic liposomes and other lipid excipient-based therapeutic products: prediction and prevention. Adv Drug Deliv Rev. 2011;63:1020-30
250. Reddy EC, Rand ML. Procoagulant Phosphatidylserine-Exposing Platelets in vitro and in vivo. Front Cardiovasc Med. 2020;7:15
251. La-Beck NM, Islam MR, Markiewski MM. Nanoparticle-Induced Complement Activation: Implications for Cancer Nanomedicine. Front Immunol. 2020;11:603039
252. Ishida T, Kiwada H. Accelerated blood clearance (ABC) phenomenon upon repeated injection of PEGylated liposomes. Int J Pharm. 2008;354:56-62
253. Uzhytchak M, Smolková B, Lunova M, Frtús A, Jirsa M, Dejneka A. et al. Lysosomal nanotoxicity: Impact of nanomedicines on lysosomal function. Adv Drug Deliv Rev. 2023;197:114828
254. Eberhardt N, Giannarelli C. Statins boost the macrophage eat-me signal to keep atherosclerosis at bay. Nat Cardiovasc Res. 2022;1:196-7
255. Wu J, Ekman C, Jönsen A, Sturfelt G, Bengtsson AA, Gottsäter A. et al. Increased plasma levels of the soluble Mer tyrosine kinase receptor in systemic lupus erythematosus relate to disease activity and nephritis. Arthritis Res Ther. 2011;13:R62
256. Wei L, Wen S, Dang T, Zeng T, Yang S, You Y. et al. Efferocytosis-related signatures identified via Single-cell analysis and machine learning predict TNBC outcomes and immunotherapy response. Sci Rep. 2025;15:38955
257. Wang JM, Xia WY, Liao YS, Yuan J. Multi-omics reveals efferocytosis-related hub genes as biomarkers for ustekinumab response in colitis. Front Immunol. 2025;16:1597528
258. Tahara N, Imaizumi T, Virmani R, Narula J. Clinical feasibility of molecular imaging of plaque inflammation in atherosclerosis. J Nucl Med. 2009;50:331-4
259. Tawakol A, Abohashem S, Zureigat H. Imaging Apoptosis in Atherosclerosis: From Cell Death, A Ray of Light. J Am Coll Cardiol. 2020;76:1875-7
260. Stöhr R, Deckers N, Schurgers L, Marx N, Reutelingsperger CP. AnnexinA5-pHrodo: a new molecular probe for measuring efferocytosis. Sci Rep. 2018;8:17731
261. Ehlerding EB, Grodzinski P, Cai W, Liu CH. Big Potential from Small Agents: Nanoparticles for Imaging-Based Companion Diagnostics. ACS Nano. 2018;12:2106-21
262. Ramanathan RK, Korn RL, Raghunand N, Sachdev JC, Newbold RG, Jameson G. et al. Correlation between Ferumoxytol Uptake in Tumor Lesions by MRI and Response to Nanoliposomal Irinotecan in Patients with Advanced Solid Tumors: A Pilot Study. Clin Cancer Res. 2017;23:3638-48
263. Wolff AC, Somerfield MR, Dowsett M, Hammond MEH, Hayes DF, McShane LM. et al. Human Epidermal Growth Factor Receptor 2 Testing in Breast Cancer: ASCO-College of American Pathologists Guideline Update. J Clin Oncol. 2023;41:3867-72
264. Cooley MB, Wegierak D, Exner AA. Using imaging modalities to predict nanoparticle distribution and treatment efficacy in solid tumors: The growing role of ultrasound. Wiley Interdiscip Rev Nanomed Nanobiotechnol. 2024;16:e1957
265. Desai N, Rana D, Patel M, Bajwa N, Prasad R, Vora LK. Nanoparticle Therapeutics in Clinical Perspective: Classification, Marketed Products, and Regulatory Landscape. Small. 2025;21:e2502315
266. Cruz-Samperio R, Hicks CL, Scott A, Gispert Contamina I, Elani Y, Richardson RJ. et al. Modular Bioorthogonal Lipid Nanoparticle Modification Platforms for Cardiac Homing. J Am Chem Soc. 2023;145:22659-70
Author contact
![]() Corresponding author: Jingping Liu (Email: liujingpingedu.cn); NHC Key Laboratory of Transplant Engineering and Immunology, West China Hospital, Sichuan University, No. 2222 Xinchuan Road, Chengdu 610041, China. Tel: +86-28-85164029, Fax: +86-28-85164030.
Corresponding author: Jingping Liu (Email: liujingpingedu.cn); NHC Key Laboratory of Transplant Engineering and Immunology, West China Hospital, Sichuan University, No. 2222 Xinchuan Road, Chengdu 610041, China. Tel: +86-28-85164029, Fax: +86-28-85164030.