Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
1. Introduction
2. Basic information on CCR2 and...
3. Regulatory network of...
4. The role of CCR2-dependent...
5. CCR2 and its ligands as...
6. Targeting CCR2 signaling as a...
7. Summary and outlook
Abbreviations
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2026; 16(11):5951-5991. doi:10.7150/thno.130426 This issue Cite
Review
Unraveling the pleiotropic effects of CCR2-dependent signal transduction in fibrosis development
Guozheng Sun1*, Jiaxing Wang2*, Jiayou Tang1*, Mingzhe Chen1,2, Zhe Zhang2, Huadong Zhao3, Zhenxiao Jin1, Xuezeng Xu1, Yang Yang2 ![]() , Jincheng Liu1
, Jincheng Liu1 ![]()
1. Department of Cardiovascular Surgery, Xijing Hospital, The Airforce Medical University, 127 Changle West Road, Xi'an 710032, China.
2. Xi'an Key Laboratory of Innovative Drug Research for Heart Failure, Faculty of Life Sciences and Medicine, Northwest University, 229 Taibai North Road, Xi'an 710069, China.
3. Department of General Surgery, Tangdu Hospital, The Airforce Medical University, 1 Xinsi Road, Xi'an 710038, China.
*These authors contributed equally to this work.
Received 2025-12-13; Accepted 2026-3-19; Published 2026-4-8
Abstract

Fibrosis is a pathological process characterized by the abnormal deposition of connective tissue across multiple organ systems. Given the high prevalence of fibrotic diseases and the limited availability of clinical treatment options, it has emerged as a major challenge in contemporary medicine. Chronic inflammation is widely recognized as a common pathological basis of various fibrotic disorders. In fibrosis progression, CCR2 acts as a critical signaling hub, initiating cascade reactions and contributing to the formation of a complex regulatory network. Studies have demonstrated that in most organ fibrotic processes, CCR2 primarily exerts pro-fibrotic effect by recruiting inflammatory monocytes, activating fibroblasts, and promoting extracellular matrix deposition. However, the function of CCR2 is not unidimensional. It may also play a regulatory role in promoting fibrosis regression under specific tissue and pathological contexts. CCR2 signaling exhibits dual regulatory properties at different stages of liver fibrosis. CCR2 promotes injury in the early phase, while participating in fibrosis reversal by mediating macrophage transition toward a reparative phenotype and facilitating extracellular matrix degradation. This stage-dependent behavior suggests that inappropriate timing of intervention may disrupt repair process, and the functional redundancy of the chemokine system may trigger compensatory adaptations. Together, these factors constitute the core translational challenges facing CCR2-targeted therapeutic strategies. This article systematically reviews the complex regulatory network and pivotal role of CCR2 signaling in fibrosis progression, summarizes the latest advances in the diagnosis and treatment of clinically relevant fibrotic diseases associated with this pathway, analyzes the specific challenges in translating CCR2-targeted therapies into clinical practice, and outlines future research directions.
Keywords: CCR2, fibrosis, regulatory network, diagnostic biomarkers, targeted therapy
1. Introduction
Fibrosis is generally considered to be the result of dysregulated tissue repair responses caused by chronic inflammation, with excessive accumulation of extracellular matrix (ECM) proteins as its primary characteristic [1]. Hypoxia, viral infections, allergies, and other factors can all lead to tissue damage [2, 3]. In response to tissue damage, fibroblasts from various sources initiate the healing response by remodeling the extracellular environment to restore tissue integrity. Typically, this profibrotic process shuts down once the tissue has healed [4]. However, persistent inflammatory injury can cause this repair response to become excessive or uncontrolled, leading to excessive ECM deposition, which in turn results in organ structural damage and functional decline [5]. Fibrosis occurs in almost all organs and tissues of the human body, including the liver, lungs, heart, and kidneys [6-9]. Among these, idiopathic pulmonary fibrosis (IPF) is a chronic progressive disease with variable disease course and high mortality rate [10]. Currently, clinical treatment options for IPF are limited, with no significantly effective therapeutic drugs available, and treatment can only partially slow the progression of IPF and reduce the risk of related complications. This also reflects the clinical treatment challenges faced in all fibrosis-related organ and tissue damage.
Although fibrosis in different organs or tissues exhibits specific clinical manifestations and hazards, most fibrotic diseases share a common pathogenic process. Persistent injury stimuli induce inflammatory cell infiltration and chemokine release, which in turn activate fibroblasts, ultimately leading to excessive ECM deposition [11, 12]. Within this pathological process, monocyte chemotactic protein-1 (CCL2/MCP-1)/C-C motif chemokine receptor 2 (CCR2) axis occupies a central regulatory position, playing a crucial role in the initiation and progression of fibrosis [13-15]. As a key regulator of monocyte/macrophage recruitment, the CCL2/CCR2 axis not only mediates the directed migration of inflammatory cells to sites of injury [16, 17]. More importantly, it directly participates in converting initial inflammatory signals into fibrotic effects. Myeloid-derived fibroblast precursors require CCL2/CCR2 signaling to migrate into tissues and differentiate into ECM-producing effector cells [13]. Furthermore, CCR2 knockout significantly reduces expression levels of fibrosis markers such as α-smooth muscle actin (α-SMA), fibronectin, and collagen I in the hearts of streptozotocin-induced diabetic cardiomyopathy mice, improving streptozotocin-induced cardiac dysfunction and fibrosis [18]. Consequently, CCR2-dependent signaling has emerged as a pivotal link between inflammatory initiation and fibrotic disease progression, making targeted modulation of the CCR2 pathway a promising therapeutic strategy for fibrotic disorders.
In this review, we first introduce the biological functions of CCR2 and CCR2 ligands, and focus on the complex regulatory networks and key roles of CCR2-dependent signaling in the fibrotic process. We then summarize the diagnostic and therapeutic potential of CCR2 signaling, thereby providing valuable insights for future research and clinical practice.
2. Basic information on CCR2 and its ligands
2.1 Brief introduction of CCR2
CCR2 is a functional chemokine receptor found in various organs and tissues, including the heart, liver, spleen, lungs, kidneys, brain, colon, bladder, skin, and bone marrow. Furthermore, CCR2 is widely expressed in various cell populations, such as monocytes, macrophages, endothelial cells (ECs), lymphocytes, dendritic cells (DCs), and T cells [19-22]. Studies have shown that interferon-γ (IFN-γ) acts in concert with bacterial lipopolysaccharide (LPS), tumor necrosis factor α (TNF-α), and interleukin-1β (IL-1β) in activating CCR2 expression [23]. Based on the carboxyl-terminal (C-terminal) tail, CCR2 is divided into two subtypes, CCR2A and CCR2B [24, 25]. CCR2B is primarily localized on the cell surface, while CCR2A is localized in the cytoplasm [24].
CCR2 belongs to the G protein-coupled receptors (GPCRs) family, with a spatial structure comprising a C-terminal domain, seven helical transmembrane domains connected by intracellular and extracellular hydrophilic loops, and an amino-terminal (N-terminal) domain [26]. The N-terminal of CCR2 determines the specificity of ligand binding. CCR2 can bind to multiple CC chemokines, exhibiting significant redundancy. This redundancy is crucial for maintaining the activity and stability of chemokines. CCR2 serves as a high-affinity receptor for multiple members of the MCP family. In humans, CCR2 has four ligands: CCL2 (MCP-1), CCL8 (MCP-2), CCL7 (MCP-3), and CCL13 (MCP-4) [27-29]. In mice, CCR2 has three ligands: CCL2 (JE/MCP-1), CCL7 (MCP-3), and CCL12 (MCP-5) [30]. Moreover, mouse CCL2 (mCCL2) and CCL12 are the closest homologues to human CCL2, so studying the roles of mouse CCL2 and CCL12 in diseases can reflect the functions of human CCL2 [31]. CCR2 also binds to cytokine-like 1 (Cytl1) and PC3-secreted microprotein (PSMP)/microseminoprotein (MSMP) [32-34]. Among these, Cytl1 has structural similarities with CCL2 and possesses chemotactic activity [32]. PSMP is a novel chemokine structurally distinct from CC chemokines, which can mediate monocyte recruitment and migration via activating the CCR2B/extracellular-signal-regulated kinase (ERK) pathway [32, 34].
The C-terminal residues of CCR2 bind to G proteins in CCR2 ligands, thereby activating downstream signaling pathways [26]. These signaling events recruit and activate proteins involved in cell transport, thus promoting cell migration along the chemokine gradients. Moreover, CCR2-mediated signaling is recognized as a key coordinator of numerous critical cellular activities, including inflammation, hematopoiesis, wound healing, tumor growth and metastasis, and fibrosis [17, 35-37]. Extensive research has shown that CCR2, which binds to CC chemokines, regulates the development of various diseases such as atherosclerosis and myocardial infarction (MI) by promoting bone marrow-derived monocytes mobilization into the bloodstream and migration to inflammatory sites [38, 39]. Receptor knockout experiments found that CCR2 knockout (CCR2 -/-) mice exhibit defects in macrophage recruitment, dendritic cell activation, and immune defense functions [40].
2.2 Brief introduction to CCR2 ligands
This section primarily introduces members of the MCP family that bind to CCR2 with high affinity, including the discovery, cellular expression, structure, and biological functions of these chemokines (Table 1).
A summary of MCP family members that bind to CCR2 with high affinity.
| CCR2 Ligands | Species specificity | Structural homology with human CCL2 | Target cells of chemotactic action | Biological function | Disease association |
|---|---|---|---|---|---|
| CCL2/MCP-1 | Both humans and mice | 68% | T cells, NKs, monocytes, macrophages, neutrophils, B cells, DCs, mast cells, endothelial cells, epithelial cells, microglia, fibroblasts, tumor cells | Inflammatory response, immune regulation, angiogenesis, tissue repair and regeneration, tumor growth and metastasis, fibrosis | Atherosclerosis, hypertension, cancer, diabetes, respiratory tract infection, osteoarthritis, RA, hepatic fibrosis, neurodegenerative diseases |
| CCL7/MCP-3 | Both humans and mice | 73% | T cells, NKs, monocytes, macrophages, DCs, eosinophils, neutrophils, basophils, endothelial cells, epithelial cells, fibroblasts, mast cells, astrocytes, stromal cells, tumor cells | Immune regulation, inflammatory responses, antiviral immunity, tissue regeneration | Cancer, allergic diseases, viral infection, cardiovascular disease, diabetes, AKI, ALI, osteoarthritis, neuropathic pain, pneumonia, renal tubulointerstitial fibrosis |
| CCL8/MCP-2 | Human only | 69% | T cells, NKs, DCs, monocytes, basophils, macrophages, fibroblasts, endometrial cells, mast cells | Inflammatory responses, Th2 immune response, skeletal muscle regeneration | Cancer, allergic diseases, AIDS, ARDS, graft-versus-host disease, IPF, preeclampsia, viral pneumonias |
| CCL12/MCP-5 | Mice only | 66% | Macrophages, T cells, astrocytes, endothelial cells, epithelial cells | Function similar to human CCL2 | Cancer, ALI, cardiovascular disease, ICH, IPF, osteoarthritis |
| CCL13/MCP-4 | Human only | 65% | T cells, NKs, DCs, monocytes, macrophages, eosinophils, basophils, mast cells, endothelial cells, epithelial cells, fibroblasts, chondrocytes, tumor cells | Function similar to human CCL7/8 | Cancer, allergic diseases, RA, cancer, SSc, Alzheimer's disease, cardiovascular disease |
Abbreviations: AIDS, acquired immune deficiency syndrome; AKI, acute kidney injury; ALI, acute lung injury; ARDS, acute respiratory distress syndrome; B cells, bursa dependent lymphocytes; CCL2/MCP-1, monocyte chemotactic protein-1; DCs, dendritic cells; ICH, intracerebral hemorrhage; IPF, Idiopathic pulmonary; fibrosis; NKs: natural killer cells; RA: Rheumatoid arthritis; SSc, Systemic sclerosis; T cells, thymus dependent lymphocytes.
CCL2
CCL2, also known as MCP-1, was first isolated from human glioma cells and human blood mononuclear leukocytes [27, 41]. Among proteins with similar sequences, the coding regions of human CCL2 and mCCL12 exhibit 68% identity [42]. CCL2 is a small molecule protein composed of 76 amino acid residues, secreted by various cells and most abundantly expressed in monocytes, macrophages, lymphocytes, and DCs [43-45]. Furthermore, CCL2 expression can be either persistent or inducible. Multiple mediators can induce CCL2 expression, including IL-1β, IL-6, TNF-α, transforming growth factor-β (TGF-β), IFN-γ, granulocyte-macrophage colony-stimulating factor (GM-CSF), and LPS [16, 46-50]. Research has demonstrated that CCL2 exhibits significant chemotactic activity toward monocytes, microglia, T cells, natural killer (NK) cells, and fibroblasts [17, 36, 51, 52]. CCL2 can also regulate disease progression by modulating the migration and infiltration of these immune cells. CCL2 initiates atherosclerosis by recruiting macrophages and monocytes and promoting these cells migration to the damaged vascular wall [53].
CCL7
CCL7, also known as MCP-3, was first discovered in the supernatant of human osteosarcoma cells MG-63 [28]. At the amino acid level, human CCL7 and CCL2 share 73% structural homology [28]. CCL7 is expressed in various cell types, including lymphocytes, DCs, NK cells, astrocytes, and stromal cells [54-57]. Under pathological conditions, CCL7 may also be expressed in tumor cells [58]. Furthermore, CCL7 is an effective chemotactic agent for various leukocytes, mediating the recruitment and migration of monocytes, macrophages, neutrophils, and eosinophils through interaction with multiple chemokine receptors [59-63]. Endothelial dysfunction and vascular lesions in diabetic mice can be effectively alleviated by inhibiting CCL7 [64].
CCL8
CCL8, also known as MCP-2, was initially identified in the supernatant of human osteosarcoma cells and exhibits 69% structural homology with CCL2 [28]. Various cell types produce CCL8, including monocytes, fibroblasts, endometrial cells, and mast cells. CCL8 plays a key role in inflammatory responses and allergic diseases by attracting multiple immune cells [65, 66].
CCL12
CCL12, also known as MCP-5, is present only in mice and not in humans, and was first identified in allergic pneumonia [67, 68]. The mCCL12 protein is 68% identical to human CCL2 protein [31]. The chemotactic effects of CCL12 are mediated by binding to the specific receptor CCR2 [68].
CCL13
Human CCL13, also known as MCP-4, was initially isolated from a human cardiac cDNA library using eotaxin as a probe [29]. At the amino acid level, human CCL13 is 65% homologous with human CCL2 [29]. Various tissues show high levels of CCL13 expression, and this expression increases significantly in tumor cell lines [69, 70]. Chondrocytes also secrete large amounts of CCL13, which exacerbates rheumatoid arthritis by promoting fibroblast-like synovial cells proliferation [71]. Moreover, CCL13 levels are significantly upregulated under the stimulation of pro-inflammatory cytokines [72, 73]. However, the Th2-type cytokine IL-4 inhibits CCL13 expression induced by TNF-α and IL-1β in peripheral blood mononuclear cells, but only minimally affects CCL13 expression in epithelial cells [74].
3. Regulatory network of CCR2-dependent signal transduction in fibrosis
Given that CCL2 is the most prominent member in terms of activity among CCR2 ligands, this section will first systematically elucidate the fundamental mechanisms of CCL2/CCR2 signal transduction [17]. Existing research indicates that under various internal and external stimuli, the expression of CCR2 and its ligands is finely regulated by multiple upstream factors at different levels. These regulatory factors modulate CCL2 expression through direct or indirect pathways, thereby profoundly influencing the balance between inflammatory responses and tissue repair during fibrosis [75, 76]. Accordingly, this section will provide a hierarchical and logically coherent systematic discussion, focusing on the upstream regulatory network, downstream effector pathways, and synergistic molecular interactions of CCR2 signaling in the progression of fibrosis.
3.1 CCL2/CCR2 signal transduction
The CCL2/CCR2 axis is the most widely studied mechanism for recruiting monocytes. Upon binding to CCL2, CCR2 undergoes a conformational change, which subsequently activates multiple intracellular G protein-mediated signaling pathways, including the phosphoinositide 3-kinase/ protein kinase B (PI3K/AKT) pathway, mitogen-activated protein kinase (MAPK) pathway, protein kinase C (PKC) pathway, and RAS/RAF/mitogen-activated protein kinase kinase (MEK)/ERK pathway [77-81]. These signaling pathways not only participate in cell recruitment and migration processes but also promote the production of various transcription factors and cytokines involved in cell proliferation, growth, and differentiation [82-84]. Furthermore, these pathways collectively coordinate biological processes such as cell survival, migration, apoptosis, angiogenesis, and inflammation [79-81, 85, 86]. And the CCL2/CCR2 axis is closely associated with the development of various diseases, such as atherosclerosis, stroke, pulmonary arterial hypertension, and cancer [53, 80, 87, 88]. Targeting the CCL2/CCR2 axis is considered a key strategy for treating these diseases.
3.2 Upstream regulatory factors of the CCR2 signal in the fibrosis process
The CCL2/CCR2 axis is a key signal driving tissue remodeling. We focus on exploring the upstream molecular regulatory network of CCL2. Studies have shown that CCL2 expression and activity are precisely regulated through multi-level, multi-pathway mechanisms, including transcriptional regulation and epigenetic modifications.
3.2.1 Transcriptional regulation
In terms of transcriptional regulation, we mainly discuss specific transcription activators and signal-sensing transcription activators (Figure 1). The specific transcription activator nuclear factor of activated T-cells (NFAT) regulates gene transcription by directly binding to target gene promoters or forming synergistic complexes with other transcription factors [89-91]. NFAT5 is a widely expressed transcription factor whose activity is regulated by extracellular tonicity [92]. Evidence suggests that NFAT5 stimulates CCL2 expression through two distinct mechanisms. First, NFAT5 activates CCL2 transcription by directly binding to TonE, a cis-acting element located upstream of the CCL2 transcription start site [93]. Secondly, high osmotic pressure activates NFAT5 in mesothelial cells, which interacts with the p65 subunit of nuclear factor-κB (NF-κB) to synergistically upregulate CCL2 expression, thereby promoting peritoneal fibrosis during continuous ambulatory peritoneal dialysis [75, 94]. NFATc3 expression is elevated in the lung tissue and lung macrophages of mice induced with bleomycin (BLM)-induced pulmonary fibrosis. NFATc3 promotes pulmonary fibrosis progression by regulating the expression of CCL2 and CXCL2 genes in macrophages [95].
Upstream regulatory network of CCR2 signaling in fibrosis. The specific transcription activator NFAT regulates gene transcription by directly binding to the CCL2 gene promoter. Signal-sensing transcription activators enter the nucleus after undergoing modifications such as phosphorylation in response to extracellular signals like hormones, growth factors, and stress, where they bind to specific DNA sequences to regulate CCL2 gene transcription. NF-κB and AP-1 serve as core transcription factors for the CCL2 gene, jointly binding adjacent sites on the CCL2 promoter to synergistically enhance CCL2 transcription. The JAK/STAT pathway and Notch pathway also exert significant transcriptional regulatory roles. These transcription factors often exhibit synergistic interactions and mutual regulation. AP-1, activator protein 1; CSF-1R, colony-stimulating factor 1 receptor; IFN-β, interferon-β; IKK, inhibitor of κB kinase; IL-1α, interleukin-1 α; IκB, inhibitor of κB; JAK, Janus kinase; NFAT5, activator nuclear factor of activated T-cells; NF-κB, p65 subunit of nuclear factor-κB; STAT, signal transducer and activator of transcription; TGF-β, transforming growth factor-β; TK, tyrosine kinase; TRAF, TNF receptor-associated factor; TSLP, thymic stromal lymphopoietin.
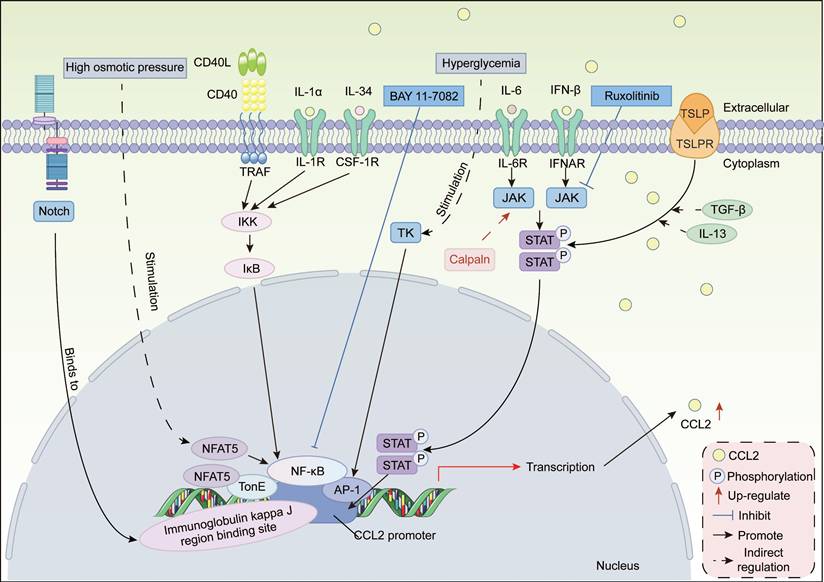
Signal-induced transcription activators sense extracellular signals such as hormones, growth factors, or stress, undergo modifications such as phosphorylation, and then enter the nucleus to bind to specific DNA sequences, thereby activating or inhibiting CCL2 gene transcription. Common types include NF-κB, activating protein-1 (AP-1), signal transducer and activator of transcription (STAT), and Notch.
NF-κB/AP-1
The CCL2 promoter region contains adjacent and evolutionarily conserved NF-κB and AP-1 binding sites, whose synergistic binding drives CCL2 transcriptional expression in pulmonary fibrosis [96, 97]. Point mutations or deletions in these binding sites significantly reduce CCL2 promoter activity, thereby impairing normal CCL2 transcription [97]. Notably, NF-κB and AP-1 serve as a common integrator pathway for multiple pro-fibrotic signals. CD40 enhances CCL2 secretion in activated human hepatic stellate cells (HSCs) by activating NF-κB [98]. Similarly, IL-34 enhances CCL2 expression by activating the NF-κB pathway, thereby promoting macrophage recruitment and polarization, exacerbating cardiac remodeling after myocardial ischemia-reperfusion (I/R) injury [99]. In an in vitro model of surgery-induced fibrosis in total knee arthroplasty, fibroblasts exacerbate joint fibrosis via the IL-1α/NF-κB/CCL2 signaling pathway [100]. Moreover, in peritoneal mesothelial cells, hyperglycemia stimulates CCL2 expression through the tyrosine kinase/AP-1 pathway [101]. The cross-organ conservation of this mechanism indicates that the NF-κB/AP-1 complex serves as a common pathway for various stromal cells, including fibroblasts and HSCs, to sense injury and initiate CCL2 expression. BAY 11-7082, an NF-κB inhibitor, reduces CCL2 expression by inhibiting NF-κB p65 activation in a rat myocardial I/R model, thereby decreasing infarct area and late-stage fibrosis [102]. This further confirms that CCL2 expression during fibrosis depends on transcriptional regulation by NF-κB and AP-1.
STAT family
Members of the STAT family, such as STAT1, STAT3, and STAT6, exhibit functional similarities across different fibrotic contexts. STAT family enhance CCL2 transcription efficiency by forming complexes with the CCL2 promoter or other transcription factors PU.1 and CEBPα [103]. In early intestinal inflammation, cells expressing Ly6Chigh enhance the expression of CCL2 and CCR2 genes via activating the JAK/STAT1 signaling pathway, which is associated with the pathogenesis, exacerbation, and chronicity of acute colitis [104]. In systemic sclerosis (SSc) mice, STAT6 deficiency leads to significant downregulation of CCL2 [105]. In hepatitis C virus (HCV)-induced liver fibrosis, the virus downregulates microRNA-449a (miRNA-449a) and miRNA-107 in the liver, thereby releasing inhibition of the IL-6/JAK1/STAT3 pathway. Activated STAT3 forms a transcriptional activation complex with PU.1 and CEBPα, which synergistically binds to the promoter and activates CCL2 expression [103]. Furthermore, in the context of high cholesterol and chronic myocardial ischemia, calpain increases collagen expression by enhancing JAK/STAT/CCL2 signaling, thereby promoting cardiac fibrosis [106]. Specifically, The JAK inhibitor Ruxolitinib can inhibit CCL2 transcription by blocking IFN-β-stimulated STAT1 phosphorylation in bone marrow-derived macrophages [107].
Notch pathway
The Notch signaling pathway is crucial for multicellular organisms, programmatically controlling cell fate and tissue differentiation during early development [108]. An evolutionarily conserved Notch/RBP-J binding site on the CCL2 promoter enables Notch signaling to directly activate CCL2 transcription [109]. In non-alcoholic steatohepatitis (NASH) mice, hepatocytes upregulate CCL2 via this site, further promoting MDMs infiltration of into the liver and advancing hepatic fibrosis [109]. Bone marrow-specific Notch activation promotes CCR2+ macrophage infiltration by upregulating CCL2 expression, ultimately exacerbating renal fibrosis. In addition, Brandt et al. utilized chimeric mice lacking Notch3 in hematopoietic cells and/or resident tissue cells to confirm that the development of renal fibrosis and inflammation following unilateral ureteral obstruction (UUO) is significantly associated with upregulation of CCL2 levels. And CCL2 upregulation is Notch3-dependent [110].
It is worth noting that the transcriptional regulatory network of CCL2 in a fibrotic context is extremely complex, with often synergistic and mutually regulatory interactions between different types of transcription factors. Compared to healthy individuals, in the late stages of oral submucous fibrosis, the expression of transcription factor genes cyclic AMP response element-binding protein (CREB), NF-κB, and NFAT5 is upregulated, synergistically promoting CCL2 expression [111]. Moreover, thymic stromal lymphopoietin (TSLP) is upregulated in the skin of SSc patients. And the TSLP-TSLPR-STAT3 signaling axis synergistically promotes CCL2 expression in fibroblasts by interacting with pro-fibrotic cytokines TGF-β and IL-13 [112]. Furthermore, STAT3 is essential for TSLP-induced CCL2 expression [112].
In summary, these transcription activators promote fibrosis progression by binding to specific sites on the CCL2 promoter to activate CCL2 gene expression. Future integration of multi-omics data and disease model information will comprehensively reveal the CCL2 transcriptional interaction network, providing innovative therapeutic strategies for fibrotic diseases.
3.2.2 Epigenetic modifications of CCL2
Some epigenetic mechanisms, such as histone modifications and miRNA regulation, influence CCL2 gene expression at the genetic information level [113] (Figure 2). Histone methylation is a type of chromatin modification. Different modification sites and degrees may affect gene transcriptional activation or silencing. ASH1-like histone lysine methyltransferase (ASH1L), a methyltransferase, is highly expressed in activated HSCs and hepatocellular carcinoma cells. Mechanistically, ASH1L significantly upregulates CCL2 transcriptional expression through directly binding to the CCL2 promoter region and catalyzing histone H3 lysine 27 trimethylation (H3K4me3) modification [114]. This epigenetic regulatory mechanism promotes the recruitment and polarization of M2-like macrophages, forming an immunosuppressive tumor microenvironment, ultimately accelerating liver fibrosis and hepatocellular carcinoma progression [114]. In contrast, in rat renal mesangial cells, TGF-β suppresses CCL2 expression by downregulating enhancer of Zeste homolog 2 (Ezh2) to inhibit H3K27me3 at the CCL2 gene promoter [115]. Increased CCL2 expression is associated with fibrosis and glomerular dysfunction in diabetic nephropathy (DN) [115].
A summary of the epigenetic mechanisms regulating CCL2 gene expression. Histone modifications, including methylation, acetylation, and deacetylation, regulate the transcriptional opening and closing of CCL2, thereby influencing CCR2 signaling-mediated cellular activities during fibrosis, thus ultimately altering the fibrotic process. miRNAs regulate post-transcriptional expression by binding to CCL2 and CCL12 to degrade mRNA or inhibit translation. ASH1L, ASH1-like histone lysine methyltransferase; BRD4, bromodomain-containing protein 4; Ezh2, enhancer of Zeste homolog 2; H3K4me3, Histone H3 lysine 27 trimethylation; HDAC1/2, histone deacetylases 1 and 2; HSCs, hepatic stellate cells; METTL3, methyltransferase-like 3; NASH, non-alcoholic steatohepatitis; NF-κB, p65 subunit of nuclear factor-κB; Sp1, specific protein 1; TGF-β, transforming growth factor-β; TNF-α, tumor necrosis factor α; 3'UTRs, 3' untranslated regions.
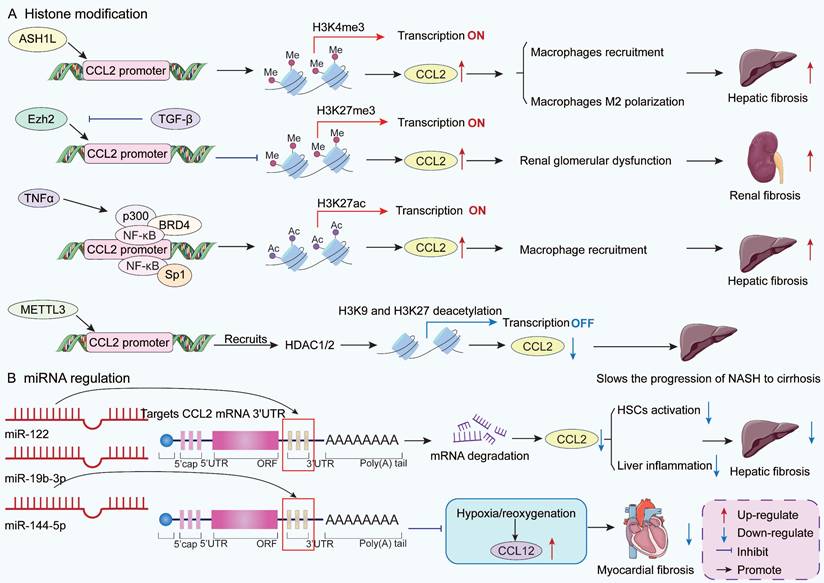
Histone acetylation is another important epigenetic mechanism. During liver injury, TNFα promotes histone acetyltransferase p300 interaction with NF-κB and bromodomain-containing 4 to form a complex in mouse liver sinusoidal ECs [116-118]. This complex acetylates H3K27 in the CCL2 enhancer and promoter regions, thereby opening the chromatin structure and enhancing CCL2 gene transcription [117]. Subsequently, CCL2 recruits CCR2+ monocyte-derived macrophages (MDMs) to the liver, promoting liver fibrosis and portal hypertension [117]. In addition, Specific Protein 1 (Sp1), a protein that binds to DNA and activates genes, promotes CCL2 transcriptional activation through regulating histone acetylation in the proximal promoter region of CCL2 gene with NF-κB interaction [119, 120]. Research has found that histone deacetylation negatively regulates CCL2 gene transcription [121]. Li et al. confirmed that methyltransferase-like 3 can directly bind to the CCL2 gene promoter and recruit histone deacetylases to induce histone H3K9 and H3K27 deacetylation in the CCL2 promoter region, thereby inhibiting CCL2 gene transcription in the liver [122]. This process protects the body from the progression of NASH. NASH is a critical step in the progression of non-alcoholic fatty liver disease (NAFLD) to cirrhosis [123].
miRNAs are a class of endogenous non-coding small single-stranded RNAs that regulate post-transcriptional expression of target genes by binding to their target genes to degrade mRNA or inhibit translation [124]. In patients infected with HCV, CCL2 expression is significantly upregulated and negatively correlated with the abundance of liver miR-12, suggesting that miR-122 may negatively regulate CCL2 [125, 126]. A dual luciferase gene reporter assay demonstrated that miR-122 downregulates CCL2 expression by binding to complementary sequences in the 3' untranslated regions (3'UTRs) of CCL2 mRNA, thereby alleviating liver inflammation [127]. An in vitro study found that miR-144-5p directly targets the 3'UTR of CCL12 to inhibit the upregulation of CCL12 and CCR2 levels in H9C2 cells induced by hypoxia/reoxygenation [128]. This process effectively reduces cell necrosis and fibrosis. Furthermore, hypoxia specifically inhibits miR-146b, thereby releasing TRAF6 inhibition and inducing CCL2 expression [129]. This pathway drives cardiac fibrosis and dysfunction and may lead to heart failure. Lan et al. compared the miRNA expression profiles between fibrotic and normal livers and found that miR-19b-3p levels were downregulated in activated HSCs. And miR-19b-3p expression was also downregulated in fibrotic human liver tissue. miR-19b-3p can directly bind to the 3'UTR region of CCR2 mRNA, leading to reduced CCL2 mRNA expression and thereby attenuating HSC activation [130].
In summary, CCL2 expression represents the complex outcome of multi-level epigenetic regulation integrating histone methylation/acetylation modifications and miRNA interference. These mechanisms collectively determine CCL2 expression levels within the tissue injury microenvironment, thereby acting as a key switch that drives the fibrosis process by regulating macrophage infiltration. Targeting these epigenetic regulatory nodes, such as ASH1L, p300, or specific miRNAs, hold promise for developing novel therapeutic strategies against diseases including liver fibrosis, diabetic nephropathy, and cardiac fibrosis.
Although progress has been made in studying regulatory factors such as transcription factors and epigenetic modifiers, the dynamic regulatory mechanisms of the CCR2/CCL2 axis in the context of fibrosis remain largely unknown. For example, the specificity of regulatory factors in different tissue microenvironments, the spatiotemporal expression patterns of modifiers, and their correlation with fibrosis stages remain unclear. A deeper understanding of these upstream regulatory networks could not only help reveal the molecular pathophysiological mechanisms of fibrosis but also provide new insights for developing therapies targeting the CCR2/CCL2 pathway to treat fibrotic diseases.
3.3 Downstream pathways and co-regulators of CCR2 in the fibrosis process
During the fibrosis process, CCR2 acts as a key regulatory factor, activating multiple downstream signaling pathways through binding with CCL2 and CCL7. The activation of these pathways further upregulates the expression of pro-fibrotic factors. Meanwhile, the CCL2/CCR2 axis synergistically interacts with certain pathways, collectively driving the onset and progression of fibrosis. Here, we briefly discuss important downstream pathways such as PI3K/AKT, TGF-β/Smad, and MAPK (Figure 3).
Downstream pathways and synergistic factors regulated by CCR2 during fibrosis. CCR2 activates multiple downstream signaling pathways such as PI3K/AKT, TGF-β/Smad, and MAPK by binding to CCL2, CCL7, and MSMP. The activation of these pathways accelerates the fibrosis process. Simultaneously, CCR2 signaling forms a feedback loop with the TGF-β pathway. The TGF-β pathway activated by CCR2 signaling further upregulates CCL2 and CCL7 expression, collectively driving the progression of fibrosis in multiple tissues and organs. AKT, protein kinase B; α-SMA, α-smooth muscle actin; ECM, extracellular matrix; ERK, extracellular signal-regulated kinase; HSCs, hepatic stellate cells; HIF-1α, hypoxia-inducible factor-1α; IL-6, interleukin-6; MEK, mitogen-activated protein kinase kinase; MPA, mycophenolic acid; MSMP, microseminoprotein; mTOR: mammalian target of rapamycin; NF-κB, p65 subunit of nuclear factor-κB; p38 MAPK, p38 mitogen-activated protein kinase; PI3K, phosphoinositide 3-kinase; STAT3, signal transducer and activator of transcription 3; TGF-β, transforming growth factor-β; VEGF-C, vascular endothelial growth factor C.
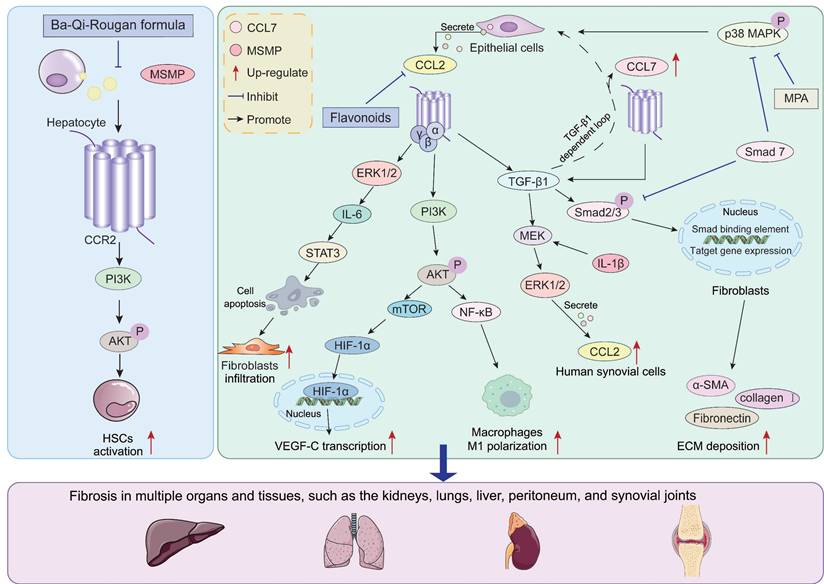
PI3K/AKT
The PI3K/AKT signaling pathway is a key regulator of cell growth, proliferation, and apoptosis [131, 132]. Multiple studies have confirmed that activation of the PI3K/AKT pathway is associated with fibroblast activation, epithelial cell damage, and macrophage polarization during fibrosis [133, 134]. Notably, the CCL2/CCR2 axis serves as a key upstream signal driving this process. After the CCR2 receptor is activated, its coupled GTP-binding protein βγ subunit (Gβγ) directly binds to the p110 catalytic subunit of PI3K, catalyzing the conversion of phosphatidylinositol 4,5-bisphosphate (PIP2) to phosphatidylinositol 3,4,5-trisphosphate (PIP3), thereby initiating the AKT phosphorylation cascade [135, 136].
Activation of the CCL2/CCR2/PI3K/AKT signaling axis exhibits pro-fibrotic functions across multiple organ fibrosis models. In obstructive nephropathy, the CCL2/CCR2 axis mediates hypoxia-inducible factor-1α (HIF-1α) expression by activating the PI3K/AKT/Mammalian target of rapamycin (mTOR) signaling pathway. Subsequently, HIF-1α drives vascular endothelial growth factor-C expression to regulate UUO-induced renal lymphangiogenesis [137]. CCR2-deficient mice exhibit suppressed lymphangiogenesis and reduced renal injury and fibrosis following UUO induction [137]. This pathway also serves as a key node regulating immune responses and inflammation, not only promoting M1 polarization of infiltrating macrophages but also mediating hepatic inflammatory responses in NAFLD models, thereby driving liver fibrosis progression [138]. Pure total flavonoids from citrus mitigate hepatic inflammation in NAFLD by inhibiting CCL2/CCR2/PI3K/AKT signaling, thereby slowing NAFLD progression to cirrhosis [138]. Furthermore, the CCR2/PI3K/AKT signaling axis directly participates in effector cell activation, such as mediating HSCs activation in liver fibrosis [139]. Ba-Qi-Rougan formula counteracts liver fibrosis by reducing MSMP expression and inhibiting MSMP-induced HSCs activation via the CCR2/PI3K/AKT pathway [139]. Consequently, targeting CCL2/CCR2 and its downstream PI3K/AKT signaling pathway has emerged as a promising strategy for intervening in fibrotic diseases.
TGF-β/Smad
The TGF-β/Smad signaling pathway is a core mechanism driving fibrosis. It primarily functions through TGF-β1 binding to the βRII receptor, activating the Smad2/3 complex to translocate into the nucleus, thereby upregulating collagen gene expression and promoting ECM deposition [140-142]. In-depth studies reveal a bidirectional feedback loop between this classical pathway and the CCL2/CCR2 axis, with this interaction synergistically amplifying pathological processes in multiple organ fibrosis models. On one hand, CCL2/CCR2 signaling serves as an upstream driver of TGF-β1 expression and functional enhancement. CCL2 not only directly induces TGF-β1 and its receptor TβRII expression in pulmonary fibroblasts but also stimulates collagen synthesis via autocrine or paracrine mechanisms, accelerating fibrosis progression [143, 144]. Functional knockout studies further validate this regulatory importance: in CCR2 knockout mice, not only were BLM-induced pulmonary TGF-β1 mRNA levels significantly lower than in wild-type (WT) mice, but fibroblast responsiveness to TGF-β1 stimulation was also impaired due to reduced TβRII and Smad3 expression, resulting in diminished myofibroblast generation [145]. Conversely, TGF-β signaling can also induce CCL2 expression in a vicious cycle. For instance, in renal proximal tubule cells, CCL2 expression is directly stimulated by TGF-β1 [146]. Furthermore, TGF-β and IL-1β synergistically activate the MEK/ERK1/2 pathway, significantly upregulating the expression of CCL2. This is a key mechanism for chronic synovial inflammation and fibrosis [147]. Overexpression of the inhibitory Smad7 not only blocks Smad2/3 phosphorylation but also suppresses TGF-β1-induced CCL2 upregulation by downregulating p38 MAPK activation, thereby alleviating peritoneal fibrosis [148, 149].
Notably, this bidirectional regulatory pattern may also apply to other CCR2 ligands. CCL7 expression is upregulated during fibrosis, and CCL7 promotes activation of the TGF-β/Smad3 signaling pathway, thereby increasing type I collagen secretion [150, 151]. Concurrently, CCL7 gene expression is stimulated by TGF-β [151]. This suggests potential synergistic effects between CCL7 and TGF-β within the fibrotic microenvironment, jointly promoting collagen biosynthesis in fibroblasts.
MAPK
The MAPK cascade is a highly conserved signaling pathway that transmits environmental stimuli into the cell nucleus to initiate intracellular responses [152]. At least three distinct MAPK families have been identified: p38 MAPK, c-Jun N-terminal kinase (JNK), and ERK.
p38 MAPK primarily functions as a hub for pro-inflammatory and pro-fibrotic signaling, and its activation is crucial for CCL2 production [153, 154]. In renal artery stenosis mice, blocking p38 MAPK directly suppressed TNF-α and TGF-β-induced CCL2 upregulation, thereby mitigating renal atrophy and fibrosis [155]. Similarly, in peritoneal mesothelial cells, p38 MAPK enhances fibroblast recruitment and infiltration by promoting CCL2 production, thereby driving peritoneal fibrosis [156]. Multiple intervention strategies have validated this mechanism. Shenkang injection alleviates diabetic nephropathy by inhibiting p38 MAPK/NF-κB signaling to reduce CCL2/CCR2 activation [157]. In rat renal fibroblasts, mycophenolic acid (MPA), an inhibitor of hypophosphimonosulfate dehydrogenase, effectively mitigates renal fibrosis by reducing TNF-α-induced CCL2 expression through downregulating p38 MAPK phosphorylation [158]. Collectively, these findings suggest that p38 MAPK activation constitutes a common pathway for CCL2 upregulation and subsequent inflammatory cascades.
In contrast, ERK1/2 primarily functions as a downstream effector activated by the CCL2/CCR2 axis, mediating cell survival and phenotypic maintenance. CCR2 rapidly activates ERK1/2 upon ligand stimulation [78]. Activated ERK1/2 promotes fibrosis through two distinct pathways. First, the CCL2/CCR2 axis suppresses fibroblast apoptosis via the ERK1/2/IL-6/STAT3 signaling pathway, contributing to pulmonary fibroblast survival and pulmonary fibrosis development [159]. Second, CCL2-enhanced macrophage inflammatory responses correlate with increased ERK1/2 phosphorylation and upregulation of miR-9 expression [160]. This differential upstream-downstream relationship suggests that p38 MAPK and ERK play functionally complementary roles in the CCL2/CCR2 signaling network: the former primarily drives CCL2 production and amplification, while the latter mediates CCL2-triggered cellular responses and maintenance.
In summary, during fibrosis progression, the CCL2/CCR2 axis collaborates with downstream cascades including PI3K/AKT, TGF-β/Smad, and MAPK to construct a complex molecular interaction network. The PI3K/AKT pathway, as a core regulator of metabolism and survival, directly activates inflammatory cascades via G proteins, promoting macrophage infiltration and fibroblast activation. The TGF-β/Smad pathway, as the primary executor of matrix deposition, forms a bidirectional positive feedback loop with CCL2/CCR2, directly coupling inflammatory signals to collagen synthesis. Members of the MAPK family assume differentiated yet complementary roles: p38 MAPK, as an upstream hub, is crucial for CCL2 production, forming an autocrine amplification loop, while ERK1/2 acts as a downstream effector mediating cell survival and inflammatory maintenance. The interplay of these three pathways ultimately transforms initial chemotactic signals into persistent tissue remodeling and irreversible fibrotic damage. Future research should further elucidate the spatiotemporal expression patterns of CCR2 within the fibrotic microenvironment and explore combined intervention strategies to achieve more precise anti-fibrotic therapies.
4. The role of CCR2-dependent signal transduction in the pathogenesis of fibrosis
4.1 Pulmonary fibrosis
Pulmonary fibrosis is a chronic, progressive lung disease with a poor prognosis [161]. Increasing evidence indicates that IPF is an epithelial cell-driven disease, where abnormally activated alveolar epithelial cells (AECs) produce mediators that promote fibroblast migration, proliferation, and differentiation into active myofibroblasts [162]. These processes result in loss of lung elasticity, reduced alveolar surface area for gas exchange, and respiratory dysfunction. Furthermore, the pathogenesis of IPF subtypes differs. Ligand-receptor analysis indicates a monocyte-macrophage chemotactic axis in the myeloid-rich IPF subtype, potentially involving CCL2-CCR2 signaling [163] (Figure 4).
The role of CCR2-dependent signaling in the pathogenesis of pulmonary fibrosis. Epithelial cell dysfunction is a key driver of pulmonary fibrosis, with CCR2 initiating damage to AECs through multiple pathways. Furthermore, bone marrow-derived inflammatory monocytes respond to CCL2 and CCL7 chemotactic signals, recruiting CCR2-dependently from the circulation to the lungs. Over time, these cells replace tissue-resident macrophages, differentiating into CCR2+ MDMs. CCR2+ MDMs highly express inflammatory genes and pro-fibrotic cytokines, driving inflammatory initiation and adverse remodeling. CCR2 signaling not only mediates macrophage polarization, enhancing MDMs infiltration to promote fibrotic progression, but also directly stimulates proliferation of resident pulmonary fibroblasts. Concurrently, bone marrow-derived fibroblasts require CCR2 signaling to be recruited into the alveolar interstitium in response to tissue fibrotic injury. In summary, CCR2 functions as both a regulator of AECs activity and a key factor governing macrophage infiltration and polarization, fibroblast recruitment, and fibroblast activation. AECs, alveolar epithelial cells; ADGRF5, adhesion G-protein coupled receptor F5; CCR2, C-C motif chemokine receptor 2; ECM, extracellular matrix; IL-10, interleukin 10; LRRK2, leucine-rich repeat kinase 2; MDMs, monocyte-derived macrophages; mTOR, mammalian target of rapamycin; NF-κB, p65 subunit of nuclear factor-κB; PAR1, protease-activated receptor-1; PGE2, prostaglandin E2; TGF-β1, transforming growth factor β1.
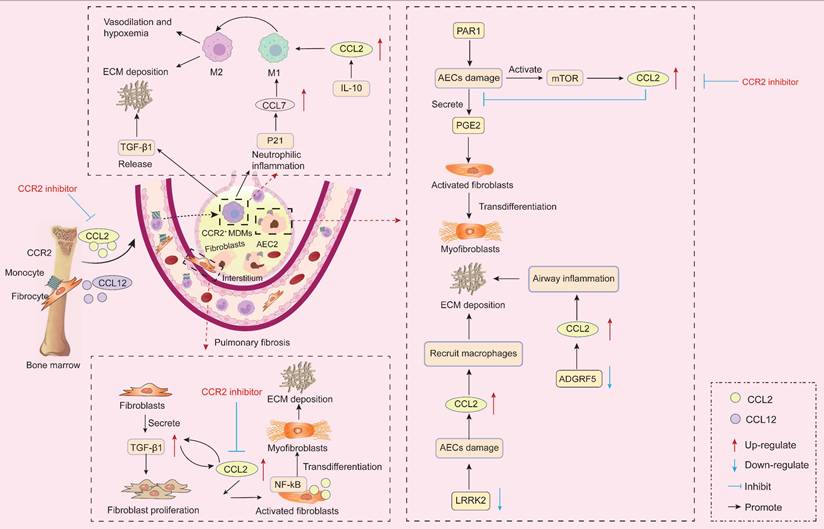
Epithelial cell dysfunction is a key driver of pulmonary fibrosis [164]. In the lung, AECs primarily maintain alveolar barrier integrity and represent one of the earliest response mechanisms to lung injury [165]. When AECs are injured, compromised epithelial barrier integrity can trigger abnormal fibroblast activation, increased ECM deposition, and structural lung damage. Adhesion G-protein coupled receptor F5 (ADGRF5) is a key regulator of pulmonary surfactant homeostasis in type II alveolar cells. Studies indicate ADGRF5 modulates CCL2 gene expression to maintain immune homeostasis [166]. Knockout of ADGRF5 induces airway inflammation mediated by type 2 immune responses and CCL2-induced inflammation [166]. In BLM-treated mice, leucine-rich repeat kinase 2 (LRRK2) expression was significantly reduced in type II AECs. Its deficiency caused severe functional impairment in these cells, manifested as impaired autophagy and accelerated cellular senescence. Furthermore, LRRK2-deficient type II AECs exhibited enhanced capacity to recruit pre-fibrotic macrophages via CCL2/CCR2 signaling, leading to progressive pulmonary fibrosis [167]. Furthermore, forkhead box F1 (FOXF1) is an endothelial transcription factor involved in pulmonary fibrosis. FOXF1 stimulates Rras transcription, thereby suppressing CCL2 expression. An in vitro experiment confirmed that FOXF1-deficient ECs promote pulmonary fibrosis by secreting CCL2 to stimulate macrophage migration and enhance pulmonary fibroblast activation [168]. Specifically, under steady-state conditions, AECs suppress fibrosis by inhibiting the conversion of fibroblasts to myofibroblasts through the secretion of the fibroblast inhibitor prostaglandin E2 (PGE2) [169, 170]. Studies reveal significantly elevated CCL2 expression in AECs from IPF patients [171]. A key pro-fibrotic mechanism of the CCL2/CCR2 interaction is the suppression of PGE2 production in AECs following lung injury, thereby promoting fibroblast-to-myofibroblast conversion and collagen deposition [169]. Furthermore, protease-activated receptor-1 activation on AECs may represent a crucial mechanism driving increased local CCL2 release in pulmonary fibrosis [172]. Notably, CCR2-/- mice are protected against experimental pulmonary fibrosis. This is because AECs from CCR2-/- mice produce more PGE2 than those from CCR2+/+ mice, thereby more effectively suppressing fibroblast proliferation [169]. Furthermore, a study revealed that injury in mouse and human primary AECs partially activates the mTOR pathway, leading to increased CCL2 and CCL12 production. These cytokines promote fibrosis through CCR2 activation [15]. Targeting the mTOR pathway to reduce CCL2 and CCL12 production in AECs may represent a viable anti-fibrotic strategy.
MDMs are also recognized as key mediators in the pathogenesis of pulmonary fibrosis [173, 174]. CCL2 mRNA and protein expression levels in lung epithelial cells from IPF patients are significantly elevated compared to healthy controls, sustaining macrophage recruitment and pulmonary infiltration under pathological conditions [175, 176]. Myeloid-derived inflammatory monocytes express CCR2 and recruit from the circulation in a CCR2-dependent manner in response to CCL2 and CCL7, replacing alveolar macrophages and interstitial macrophages over time to ultimately define CCR2+ MDMs [177, 178]. CCR2+ MDMs highly express inflammatory genes and pro-fibrotic cytokines, leading to inflammatory initiation and adverse remodeling [179, 180]. In cystic fibrosis mice with chronic inflammation, both inflammatory monocytes and CCR2+ MDMs increase in number alongside up-regulated CCL2 expression, while tissue-resident alveolar macrophages decrease [181, 182]. Moreover, abundant CCR2+MDMs exacerbate fibrosis by driving lung neutrophil-dominant inflammation and TGF-β-dependent pulmonary tissue remodeling [174]. Critically, in the cystic fibrosis context, pharmacological inhibition of CCR2 reduces pathological neutrophilic inflammation and TGF-β levels by attenuating MDMs recruitment [174]. Similarly, following chemotherapy, bone marrow-derived inflammatory monocytes respond to early fibrotic reactions and migrate to the lungs, where CCR2+ MDMs subsequently infiltrate lung tissue, thereby exacerbating radiation-induced pulmonary fibrosis [173]. Moreover, Groves et al. demonstrated through receptor knockout experiments that mice receiving CCR2-deficient bone marrow showed no pulmonary fibrosis 22 weeks after radiation exposure compared to controls [173]. Specifically, pulmonary hypertension (PH) is a fatal disease characterized by progressive pulmonary arteriolar fibrosis and remodeling. Recruitment of CCR2+ MDMs leads to pulmonary arteriolar fibrosis correlated with PH severity [183]. Specific CCR2 deficiency suppresses CCR2+ MDM infiltration, thereby reversing pulmonary arteriolar fibrosis and PH [183]. These findings suggest that targeted inhibition of CCR2 may represent a key therapeutic strategy for mitigating pulmonary fibrosis.
Additionally, single-cell RNA sequencing data from IPF patients indicate that the CCL2/CCR2 axis is critical for M1 polarization of macrophages [184]. M1 macrophages promote alveolar inflammation and activate myofibroblasts [184]. Following radiation or BLM exposure, P21 is upregulated in stressed lung epithelial cells, thereby promoting CCL7 production. CCL7 recruits macrophages by binding to CCR2 and enhances macrophage M1 polarization, ultimately exacerbating lung injury [185]. Notably, M2 macrophages also appear implicated in pulmonary fibrosis. Accumulation of M2 macrophages induced by granulocyte-macrophage colony stimulating factor (GM-CSF)/GM-CSFR and CCL2/CCR2 leads to pulmonary fibrosis, promoting vasodilation and hypoxemia, thereby developing hepatopulmonary syndrome [186]. Furthermore, IL-10 induces fibrosis through fibroblast recruitment and M2 macrophage activation, a process dependent on the CCL2/CCR2 axis [187]. Administration of anti-CCL2 neutralizing antibodies to IL-10-overexpressing mice attenuates pulmonary fibrosis, reducing pulmonary hydroxyproline content and total pulmonary collagen levels [187]. The CCL2-mediated M2 macrophage expansion pathway is also present in the lungs of congestive heart failure (CHF) patients, potentially exacerbating pulmonary fibrosis and worsening dyspnea [188].
In summary, CCR2 signaling mediates bone marrow monocyte recruitment to inflammatory tissues, enhances CCR2+ MDM infiltration, and ultimately promotes pulmonary fibrosis progression. Notably, Shichino et al. demonstrated that CCR2+ MDMs exert a protective effect in silica-induced mouse pulmonary fibrosis by inhibiting tissue remodeling-related gene expression, thereby preventing progression from nodular to diffuse fibrosis [189]. In distinct experimental models, Liang et al. reported that mouse lung-specific overexpression of CCL2 increased MDM infiltration in bronchoalveolar lavage fluid (BALF) and attenuated BLM-induced pulmonary fibrosis in a CCR2-dependent manner [190]. These findings suggest that CCR2+ MDMs may play variable roles in pulmonary fibrosis progression in a stage- and model-dependent manner. This variability likely correlates with differing activation states in macrophages resulting from variations in fibrotic stimuli and exposure duration [191, 192]. Currently, BLM, silica, and fluorescein isothiocyanate (FITC) are commonly used to establish fibrotic animal models. These agents target different pathways and possess distinct half-lives, and the proportions of MDMs and fibroblasts in the lungs of different models vary, potentially leading to differential activation of MDMs.
Additionally, Hadjicharalambous et al. documented differentially expressed lncRNAs in human lung fibroblasts following IL-1β activation, demonstrating that MIR3142HG positively regulates IL-8 and CCL2 release. They further established that reduced inflammatory responses in IPF fibroblasts correlate with diminished MIR3142HG expression [193]. Furthermore, immunohistochemical analysis of human lung tissue revealed that activated IPF fibroblasts exhibit enhanced contractility and produce abundant CCL2, with the NF-κB signaling pathway participating in CCL2 production and release by these fibroblasts [194]. The CCL2/CCR2 axis also upregulates endogenous TGF-β1 expression in pulmonary fibroblasts, enhancing their responsiveness to TGF-β1 and consequently increasing type I collagen levels [143, 145]. Moreover, fibroblasts from IPF patients exhibit excessive responsiveness to TGF-β1, IL-13, and CCL2, with these three factors reciprocally reinforcing the fibrotic response [143].
In summary, CCR2 signaling participates throughout the entire process of pulmonary fibrosis. CCR2 serves both as a regulator of AECs function and as a key factor modulating macrophage infiltration, fibroblast recruitment, and fibroblast activation. Beyond basic research, human genetics data also support the critical role of CCR2. Neehus et al. reported that homozygous mutations in the CCR2 gene directly cause human progressive polycystic lung disease, characterized by marked peribronchial and parenchymal lymphocytosis with peribronchiolar fibrosis, progressive obstructive airflow limitation, and recurrent secondary infections [195]. However, CCR2 signaling may also exert beneficial effects during pulmonary fibrosis progression by influencing specific immune cell subsets. Studies demonstrate increased proportions of the CCR2+CD4+ T cell subset in BALF from IPF patients, non-IPF pulmonary fibrosis patients, and experimental fibrotic mice [196]. This rare T cell subset possesses immunoregulatory functions, capable of suppressing T cell proliferation, alleviating pulmonary inflammation, and inhibiting IPF progression [196]. Future intervention strategies targeting CCR2 signaling must comprehensively consider its dual roles in promoting fibrosis and regulating immunity to achieve more precise treatment.
4.2 Cardiac fibrosis
Cardiovascular disease remains a leading cause of morbidity and mortality worldwide. The formation of scar tissue within the heart, known as myocardial fibrosis, represents a terminal feature in nearly all cardiac pathologies. Myocardial fibrosis is characterized by excessive deposition of ECM proteins, which lack the contractile capacity of cardiomyocytes. This leads to cardiac tissue stiffening, reduced compliance, and impaired function, ultimately progressing to heart failure [197, 198]. Cardiac diseases such as MI, hypertrophy induced by pressure or volume overload are all associated with progressive cardiac fibrosis [199, 200]. Extensive experimental evidence indicates CCL2 mediates cardiac fibrosis in models of ischemic, inflammatory, and stress-induced cardiomyopathies [201, 202] (Figure 5).
The role of CCR2-dependent signaling in the pathogenesis of cardiac fibrosis. During myocardial injury, microvascular endothelial cells release GMCSF to recruit monocytes that differentiate into CCR2+ MDMs. These initially exhibit an M1 proinflammatory phenotype and secrete CCL2, which subsequently induces their M2 transformation. The released TGF-β promotes fibroblast-to-myofibroblast conversion, driving fibrosis. At the level of cell-cell interactions, neutrophil S100A8/A9 upregulates CCL2 via the p38 MAPK/JNK/AP-1 pathway, exacerbating macrophage infiltration. Furthermore, mast cell-fibroblast crosstalk and the IFN-β/STAT1/CCL2 positive feedback loop between macrophages and fibroblasts amplify the fibrotic response and suppress cardiac reprogramming. Thus, CCR2 signaling plays a central role in cardiac fibrosis by regulating immune cell recruitment, phenotypic conversion, and multicellular interactions. AP-1, activating protein-1; CCR2, C-C motif chemokine receptor 2; ECM, extracellular matrix; GMCSF, granulocyte-macrophage colony-stimulating factor; IFN-β, interferon-β; JNK, c-Jun N-terminal kinase; MDMs, monocyte-derived macrophages; NF-κB, p65 subunit of nuclear factor-κB; p38 MAPK, p38 mitogen-activated protein kinase; SCF, stem cell factor; α-SMA, α-smooth muscle actin; TGF-β1, transforming growth factor β1; TNF-α, tumor necrosis factor α.
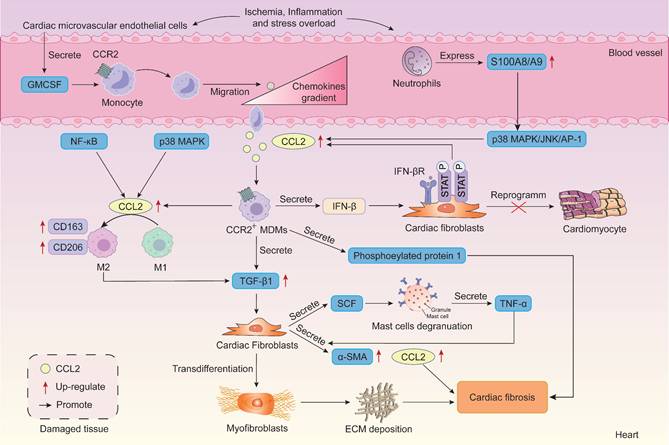
Studies reveal that recruited MDMs stimulate fibrosis in mouse hearts following aortic arch coarctation surgery. Conversely, resident macrophages suppress cardiac fibrosis. CCR2 expression on macrophages aids classification into resident (CCR2-) or circulating-derived (CCR2+) types [203]. Crucially, the fibrogenic effects of CCL2 primarily stem from the recruitment and activation of CCR2-expressing monocytes and macrophages, leading to production of the pro-fibrotic mediators TGF-β1 and type I collagen [204]. CCR2+ MDM infiltration is essential for adverse cardiac remodeling during stress overload [200]. Early interception of CCR2 signaling or selective depletion of proinflammatory Ly6ChighCCR2+ monocytes during stress overload attenuates late-stage pathological left ventricular remodeling, contractile dysfunction, and cardiac fibrosis [200]. In TAC-induced hypertrophic mice, TAC-stimulated neutrophils exhibit upregulation of S100A8/A9, which activates the p38 MAPK/JNK/AP-1 pathway to induce IL-1β and CCL2/CCL6 production. These chemokines promote CCR2+ macrophage infiltration into the injured heart [205]. Furthermore, CCR2+ macrophages mediate PAH-induced atrial fibrillation by secreting phosphoprotein 1 and exacerbate right atrial fibrosis [206].
CCR2 signaling also plays a crucial role in determining macrophage phenotype and ultimately fibrotic progression. During the inflammatory phase of early fibrosis, CCL2 exhibits pro-inflammatory effects similar to LPS, promoting M1 polarization of macrophages and driving the progression from valvular inflammation to valvular fibrosis [207]. Moreover, CCL2 is a key regulator of macrophage phenotype during MI healing, specifically promoting M1 polarization [208, 209]. Further studies indicate that the role of CCL2 in promoting M1 polarization is significantly attenuated when the p38 MAPK pathway and NF-κB pathway are inhibited [208]. In MI mice, cardiac CCL2 deficiency markedly reduced infarct size, collagen synthesis, and cardiac fibrosis, correlating with decreased total macrophage and M1 macrophage numbers in the infarct region [208]. Notably, during fibrosis progression, the CCL2/CCR2 axis also upregulates expression of M2 markers CD163 and CD206 on macrophages; these markers are highly expressed and correlate with fibrosis severity [49]. Specifically, following acute myocardial I/R injury, cardiac microvascular ECs release large amounts of GM-CSF to attract monocytes migrating to the heart. Under GM-CSF induction, monocytes differentiate into macrophages and switch to the pro-inflammatory M1 phenotype, releasing substantial amounts of inflammatory cytokines and CCL2 [47, 210]. Subsequently, CCL2 converts GM-CSF-induced M1 macrophages to the M2 phenotype. M2 macrophages release TGF-β to promote the transformation of fibroblasts into myofibroblasts, ultimately leading to cardiac fibrosis [47].
In summary, since macrophage functional phenotypes reflect responses to the local microenvironment and distinct temporal courses of inflammation, the role of CCR2 signaling in regulating macrophage polarization during fibrosis exhibits disease-stage specificity. In early stages, CCR2 signaling promotes macrophage skewing toward proinflammatory phenotypes, thereby driving the transition from chronic inflammation to fibrosis [207]. In later stages, CCR2 signaling promotes M2 polarization of macrophages to exacerbate fibrosis [49]. Therefore, when targeting CCR2 signaling for fibrotic disease treatment, we must fully consider the disease stage and the timing of targeted intervention. However, the traditional M1/M2 macrophage polarization model fails to meet precision medicine standards, hindering translational progress in clinical research. Recent advances in single-cell sequencing technology have facilitated deeper exploration of macrophage heterogeneity and plasticity. This suggests that future research should integrate single-cell transcriptomics and spatial analysis to track transcriptional changes in CCR2+ macrophages across different stages of fibrosis. This approach aims to identify novel subpopulation markers and regulatory pathways, thereby providing precise targets for intervention.
Cardiac fibrosis is mediated by cardiac fibroblasts activation, which differentiate into myofibroblasts under injury or stress. Studies reveal that activated CCR2 signaling induces the recruitment of bone marrow-derived fibroblast precursors to the heart, where these cells differentiate into fibroblasts in response to angiotensin II (Ang-II)-induced cardiac fibrosis [211, 212]. In vitro experiments demonstrate that CCL2, IL-6, and hypoxia directly promote the differentiation of cardiac fibroblasts into myofibroblasts [213]. CCL2 gene deficiency results in significantly reduced ability to recruit proinflammatory macrophages and decreased numbers of cardiac fibroblasts and myofibroblasts [214]. Specifically, Wen et al. intravenously administered nanoparticles containing the CCL2-binding peptide to AMI mice, neutralizing CCL2 to inhibit CCL2-induced myofibroblast differentiation. This resulted in reduced cardiac myofibroblast formation and decreased total collagen content [215]. Furthermore, Luo et al. emphasized that early MCs-fibroblast crosstalk and the stem cell factor (SCF)/MC/CCL2/monocyte/macrophage axis constitute key mechanisms driving myocardial fibrosis [216]. Specifically, fibroblasts trigger MC degranulation and TNF-α secretion by producing SCF. In turn, MC-secreted TNF-α stimulates fibroblasts to increase CCL2, α-SMA, and TGF-β expression, thereby exacerbating myocardial fibrosis [216].
Building upon these studies, we note recent landmark research published in Cell. Although this study did not directly track the presence of brain-derived fibroblasts in the heart, it revealed a novel brain-heart axis immune mechanism. Brain injury can drive fibrosis in distant organs, particularly the heart, by inducing innate immune memory in myeloid cells [217]. Specifically, Simats et al. found that monocytes/macrophages in the heart persistently exhibit pro-inflammatory alterations within three months after brain injury, ultimately leading to cardiac fibrosis and dysfunction [217]. Further mechanistic studies indicate that IL-1β is a key driver of this epigenetic remodeling. Blocking proinflammatory monocyte migration using CCR2 inhibitors significantly improves post-stroke cardiac dysfunction [217]. Although studies have demonstrated that signaling molecules such as CCL2 play important roles in regulating fibroblast fate, whether brain-derived fibroblasts or their precursor cells migrate to the heart via the brain-heart axis and promote fibrosis after brain injury requires further validation. Moreover, the identity, origin, and specific role of brain-derived fibroblasts in cardiac fibrosis remain controversial. Future studies should integrate lineage tracing and single-cell multi-omics technologies to systematically evaluate their contribution to cardiac pathological changes following brain injury.
The role of CCR2 signaling in MI is also a current research hotspot. During MI, elevated CCL2 levels recruit monocytes to participate in the inflammatory response, promoting the replacement of necrotic myocardium with granulation tissue [199]. This process contributes to the healing of infarcted myocardium. However, as CCL2 levels rise in the infarcted heart, CCL2 also appears to stimulate fibrous tissue deposition in the injured heart, leading to the development of cardiac interstitial fibrosis and inducing cardiac dysfunction [199]. Moreover, within the inflammatory microenvironment of MI, macrophages activate the IFN-β-IFNAR-p-STAT1 axis in cardiac fibroblasts (CFs) by secreting IFN-β, thereby stimulating CFs to secrete chemokines such as CCL2. Subsequently, CCL2 recruits more IFN-β-secreting macrophages, further amplifying its own expression, ultimately forming a self-reinforcing positive feedback loop between CFs and macrophages [218]. This positive feedback loop inhibits cardiac reprogramming (i.e. The process where CFs directly convert into cardiomyocytes in vivo to regenerate cardiac tissue), leading to adverse remodeling of the infarcted heart. Crucially, macrophages profoundly influence the post-MI repair process, thereby determining subsequent pathological remodeling. Following MI in adult rats, the reparative CCR2- cardiac resident macrophages (cRM) subpopulation is significantly depleted. The excessive recruitment of CCR2+ cRMs competitively inhibits the proliferation of reparative CCR2- cRMs, leading to myocardial fibrosis and scar formation [219]. Ding et al. targeted CCR2- cRMs for artificial intervention by developing a functional conductive cardiac patch capable of inducing CCR2- cRM renewal. This modulates the CCR2- cRM/CCR2+ cRM balance, fundamentally correcting the abnormal immune microenvironment and offering a novel approach to suppress cardiac fibrosis [219]. These findings indicate that CCL2 acts as both a key factor promoting healing and a primary culprit causing scar formation and adverse remodeling in infarcted myocardium during MI. However, some studies suggest that CCL2 overexpression may actually benefit cardiac repair post-MI. Morimoto et al. demonstrated in mice with cardiac-specific CCL2 overexpression that local cardiac CCL2 upregulation reduced infarct size and scarring, promoted myocardial IL-6 secretion and neovascularization, thereby preventing post-MI cardiac dysfunction [213]. CCL2/CCR2 also promotes cardiac repair in MI mice by activating the JNK/STAT3 pathway to induce cardiomyocyte proliferation, suppress myocardial apoptosis, and enhance post-MI angiogenesis [220].
In summary, CCR2 signaling exerts multifaceted effects in cardiac fibrosis, including promoting macrophage recruitment and activation, modulating macrophage phenotype, and mediating circulating fibroblast recruitment. Furthermore, extensive studies confirm that CCL2 is significantly induced in infarcted myocardium, aiding infarct healing while also inducing adverse remodeling of infarcted myocardium. However, the detrimental role of CCR2 signaling in post-MI cardiac remodeling and repair remains controversial. Future studies urgently require single-cell spatiotemporal analysis, cell-specific interventions, and more refined ligand-receptor functional characterization to elucidate the dual roles of CCR2 signaling across different repair phases and cell subpopulations, and to explore targeted cardiac intervention strategies.
4.3 Liver fibrosis
Liver fibrosis represents the healing response of the liver to various chronic insults, such as viral hepatitis, alcoholic liver disease, non-alcoholic steatohepatitis, and autoimmune disorders. It is characterized by the activation and transformation of HSCs into myofibroblasts, leading to excessive ECM deposition [221-224]. Persistent injury can cause severe liver dysfunction, progressing to cirrhosis or even hepatocellular carcinoma. Studies reveal upregulation of CCR2 expression in rodent and human fibrotic livers [225]. Extensive research indicates that CCR2 signaling exhibits dual roles in promoting fibrosis and facilitating regression across different stages of hepatic fibrosis (Figure 6). This contradictory function makes it a critical focus for understanding the dynamic regulation of liver fibrosis and developing targeted therapies.
The role of CCR2-dependent signaling in the pathogenesis of liver fibrosis. CCR2 signaling exhibits dual roles in promoting fibrosis and facilitating regression across different stages of liver fibrosis. A. In the early phase of liver injury, endothelial cell autophagy defects upregulate CCL2, enhancing HSCs migration via the ERK/AKT pathway while recruiting CCR2-expressing Ly6Chigh inflammatory monocytes to infiltrate the liver, directly stimulating HSCs activation. Activated HSCs further recruit macrophages via the CCL2/CCR2 pathway and induce their M2 polarization. M2 macrophages secrete IL-10 and TGF-β, which in turn sustain HSCs activation, forming a pro-fibrotic amplification loop. IL-33 promotes CCL2 upregulation and HSCs activation, while IL-19 signaling inhibits fibrosis by downregulating CCL2 expression. B. When liver injury ceases and enters the reparative phase, infiltrating macrophages switch to a reparative phenotype, secreting MMPs to degrade the ECM and promote fibrosis regression. This process partially dependent on CCR2 signaling. The increased proportion of CD11c+ dendritic cells during regression may synergistically promote fibrosis reversal, and CCR2 deficiency can impair this process. AKT, protein kinase B; CCR2, C-C motif chemokine receptor 2; ECM, extracellular matrix; ERK, extracellular-signal-regulated kinase; HSCs, hepatic stellate cells; IL-10, interleukin-10; MMPs, matrix metalloproteinases; PSMP, PC3-secreted microprotein; TGF-β1, transforming growth factor-β1.
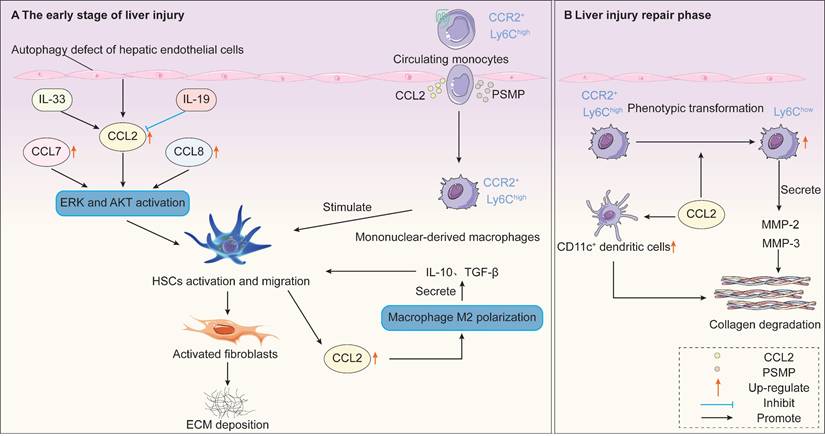
During early and persistent liver injury phases, CCR2 signaling drives fibrosis progression through multiple mechanisms. In the early stages of liver injury, CCR2 is primarily expressed on inflammatory monocytes. Inflammatory monocytes migrate from the bone marrow to the injured liver via CCR2-dependent recruitment. These monocyte-derived CCR2+ macrophages accumulate in the periportal regions of patients with NASH and advanced fibrosis [226]. Furthermore, these macrophages promote inflammation and angiogenesis while directly stimulating HSC activation [227]. In CCR2-deficient mice, impaired recruitment of monocyte subsets results in reduced HSC activation and attenuated liver fibrosis [228]. Furthermore, rats administered the human CCL2 mutant 7ND (i.e., with 7 amino acids deleted from the N-terminus of CCL2) via tail vein injection exhibited significantly reduced macrophage infiltration, suppressed HSCs activation, and inhibited liver fibrosis [229]. Furthermore, PSMP expression is markedly elevated in human cirrhotic tissues and in mice with experimental liver fibrosis [230]. Mechanistically, PSMP promotes inflammatory macrophage infiltration via CCR2 while directly activating HSCs, ultimately exacerbating liver fibrosis [230].
Substantial evidence indicates CCL2 may exert a direct pro-fibrotic effect by stimulating HSCs migration to injured liver tissue. CCL2 has indeed been demonstrated to activate HSCs in vitro and stimulate their migration in a dose-dependent manner [231]. Nicotinamide adenine dinucleotide phosphate (NADPH) oxidase is a key component in CCR2-mediated HSC activation and chemotaxis during liver fibrosis [232]. Following bile duct ligation (BDL), mRNA expression of liver CCR2, CCL2, CCL7, and CCL8 all increased [233]. In vitro experiments demonstrate that HSCs lacking CCR2 or p47phox (a key component of NADPH oxidase) exhibit impaired ERK and AKT activation, ROS production, and HSCs migration capacity upon stimulation with CCL2, CCL7, and CCL8 [233]. Among various fibrosis factors derived from bile duct epithelial cells (BECs), CCL2 produced by the innate immune system of the biliary tract is considered most critical in the development of liver fibrosis. CCL2 derived from BECs activates HSCs, thereby promoting periportal fibrosis [234]. Furthermore, activating the IL-19 signaling pathway downregulates CCL2 expression in Kupffer cells, thereby reducing HSCs activation and myofibroblast migration to alleviate CCL4-induced liver fibrosis [235]. Although HSCs are considered the primary source of type I collagen in fibrotic livers, bone marrow-derived fibroblasts are also implicated in the pathogenesis of liver fibrosis [236]. Persistent liver injury triggers the migration of circulating fibroblasts from the bone marrow to the liver, where they differentiate into myofibroblasts. This process is regulated by the CCR2 receptor [237].
When liver injury ceases or enters the repair phase, CCR2 signaling exhibits an opposite, fibrotic regression-promoting function. Studies reveal that CCR2 deficiency reduces inflammatory macrophage migration, leading to diminished HSC activation and ultimately resulting in attenuated liver fibrosis [238]. However, once chronic fibrotic injury resolves, the disease regression process is also delayed. This indicates that CCR2 also participates in fibrotic regression. During fibrosis regression, infiltrating macrophages can transition to a reparative phenotype, characterized by downregulated Ly6C expression and secretion of matrix metalloproteinases (MMPs) to degrade the ECM, thereby promoting fibrosis resolution [239, 240]. The recruitment and function of these reparative macrophages partially depend on CCR2 signaling. Findings from Mitchell et al. support this perspective. In CCR2-/- mice, fibrosis regression was significantly delayed after cessation of CCl₄ injury, accompanied by elevated tissue inhibitor of metalloproteinase 1 (TIMP-1) expression and reduced MMP-2 and MMP-13 expression [238]. This suggests that CCR2 deficiency impairs MMPs-mediated ECM degradation, potentially explaining the delayed fibrosis regression. Furthermore, Duffield et al. demonstrated that depletion of macrophage populations during injury or during the repair and recovery phases has markedly different effects on the overall fibrotic response [241]. Depletion of macrophages during the early injury phase reduces inflammatory responses, diminishes scar formation, and decreases myofibroblast numbers. In contrast, depletion of macrophages during the recovery phase leads to failure of ECM degradation and reduced repair efficiency [241]. Furthermore, other cell types may also participate in fibrosis regression. During regression, WT mice livers exhibit increased proportions of CD11c+ DCs, which may synergistically promote fibrosis reversal. In CCR2 ⁻/⁻ mice, alterations in this cell population further disrupt fibrosis regression kinetics [238].
Furthermore, we note the critical role of the CCR2 signaling pathway in the progression of diseases like viral hepatitis and NASH toward fibrosis. In patients with chronic HCV infection, CCL2 mRNA levels significantly increase in liver tissue as the disease advances [242]. Interaction between HCV core protein and gC1qR on macrophages may induce CCL2 secretion via the NF-κB signaling pathway [243]. Moreover, hepatic CCL2 mRNA levels correlate directly with histological changes and fibrosis severity [242]. In chronic HBV infection, the proportion of monocytes expressing CCR2 increases with disease progression. These cells promote natural killer T cell dysfunction, accelerating the progression from hepatitis to cirrhosis [244]. Moreover, in patients with chronic HBV-associated fibrosis, activated HSCs recruit macrophages via the CCL2/CCR2 pathway by upregulating CCL2, inducing their polarization toward the M2 phenotype. M2 macrophages not only exhibit marker expression positively correlated with fibrosis severity but also maintain HSCs activation by secreting cytokines like IL-10 and TGF-β, forming an amplification loop that exacerbates fibrosis [245]. Notably, CCL2 also plays a specific role in immune evasion during HBV infection. In advanced cirrhosis caused by chronic HBV infection, persistent liver inflammation may lead to increased spontaneous apoptosis of immune cells, resulting in decreased plasma CCL2 levels [246]. This reflects the terminal state of immune dysregulation.
In the pathogenesis of NASH, the CCL2/CCR2 axis plays a complex and seemingly contradictory central role, primarily manifested in recruiting and regulating monocytes/macrophages to influence hepatic inflammation, lipid metabolism, and fibrosis progression. In human NASH liver tissue, enhanced infiltration of CCR2-expressing CD11c+CD206+ immune cell subsets and increased hepatic CCL2 expression correlate with disease activity [247]. In NASH mice, inhibition of CCR2 reduced infiltration of liver CD11b+CD11c+F4/80+ monocytes (i.e., the functional homologs of human CD11c+CD206+ cells), thereby improving hepatic inflammation and fibrosis [247]. Furthermore, a choline-deficient amino acid-defined diet increased hepatic CCL2 expression and CCR2+ macrophage infiltration, accompanied by marked hepatic steatosis and fibrosis [248]. Additionally, autophagy defects in ECs exacerbated NASH fibrosis by upregulating inflammatory mediators, including CCL2, further underscoring the pivotal role of CCL2 in disease progression [249]. Inhibition of IL-33 signaling alleviates liver fibrosis by reducing α-SMA and CCL2 expression, thereby preventing NASH progression to hepatocellular carcinoma [250]. However, recent studies reveal that CCR2+ macrophage populations may possess important protective functions. Surprisingly, in CCR2 -/- mice fed a long-term high-fat diet, liver fibrosis significantly increased despite reduced overall macrophage infiltration [251]. One explanation is that in CCR2-deficient mice, CX3CR1/CCR2-expressing macrophages fail to appear in the liver, preventing the formation of macrophage aggregates. These macrophage aggregates may exert anti-fibrotic protective effects by clearing dead cells and toxic lipids [251]. CCR2 deficiency disrupts the formation of these protective aggregates, leading to accumulation of lipotoxicity and injury signals that instead promote fibrosis progression.
In summary, these findings suggest CCR2 may act as a double-edged sword in hepatic fibrosis. Under persistent injury conditions, CCR2 signaling drives inflammatory monocyte infiltration, amplifying inflammation, activating HSCs, and promoting ECM deposition. However, CCR2 also mediates reparative macrophage phenotype conversion, promotes ECM degradation, and resolves fibrotic scarring. Therefore, if CCR2 inhibitors are used in fibrotic patients, the timing of macrophage infiltration inhibition may be critical to avoid interfering with the recruitment and function of reparative macrophages. We also observed that while CCR2 inhibition has predominantly demonstrated a reduction in liver fibrosis, conflicting results may arise across different experimental models. Consequently, the therapeutic efficacy of CCR2 targeting may be highly dependent on the timing of intervention, the specific disease context, and the resulting balance of macrophage functions. This suggests that future therapeutic strategies must more precisely account for macrophage heterogeneity and their functional roles at different disease stages.
4.4 Renal fibrosis
Renal fibrosis represents a common and irreversible pathological pathway in the progression of various chronic kidney diseases to end-stage renal failure. In recent years, CCR2 has been recognized as a key mediator in renal fibrosis, with its expression increasing in multiple chronic kidney diseases such as diabetic nephropathy (DN), autoimmune nephropathy, and ischemic kidney injury [252-254].
The UUO model is a classic model of renal injury that simulates tubulointerstitial fibrosis associated with obstructive nephropathy. In UUO mouse kidneys, tubulointerstitial injury and progressive fibrosis correlate with elevated CCL2 expression and substantial accumulation of CCR2-expressing interstitial macrophages and T lymphocytes [255]. The CCL2/CCR2 axis facilitates monocyte recruitment, driving renal inflammation and fibrosis progression. Inflammatory CCR2+ macrophages entering the injured kidney during the early stages of tissue injury are a key cause of the subsequent fibrosis observed in UUO [256]. Inhibition of CCR2 significantly reduced macrophage infiltration and fibrosis in UUO mice [257]. Furthermore, studies revealed that overexpression of human Ang I in UUO mouse tubules reduced CCL2-activated macrophage migration and attenuated late-stage endothelial cell apoptosis, microvascular rarefaction, and fibrosis [258]. In contrast to Ang I, Ang II induces CCL2 expression in ECs, where CCL2 subsequently promotes macrophage infiltration and increases endothelial cell apoptosis [258]. This process negatively impacts survival rates in chronic kidney disease (CKD) patients. The CCL2/CCR2 axis also serves as a primary pathway for Twist1-mediated renal fibrosis in UUO mice. Increased Twist1 expression in macrophages significantly correlates with the severity of renal fibrosis in UUO mice [259]. Knocking out Twist1 partially inhibits CCL2-mediated macrophage chemotaxis and M2 macrophage polarization, thereby delaying the progression of renal fibrosis [259]. Furthermore, IL-15 has been demonstrated to reduce CCL2 expression in UUO mice, alleviating fibrosis and decreasing the likelihood of progression to CKD [260]. The role of CCR2 in renal fibrosis development is further evidenced by its recruitment of bone marrow-derived fibroblasts into the kidney, promoting the transformation of bone marrow-derived myofibroblasts and the expression of α-SMA and ECM proteins [261]. CCR2 deficiency disrupts fibroblast migration to the kidney in response to obstructive injury [262]. Specifically, Gonzalez et al. demonstrated that CCL7 acts as a direct pro-fibrotic cytokine for renal tubular cells, directly stimulating expression of TGF-β1 downstream mediators CTGF and type I collagen [263]. Furthermore, the CCL7/CCR2 axis exhibits dual roles in the progression of tubulointerstitial fibrosis, detrimental in early stages but beneficial in later stages. CCL7 promotes early inflammatory cell infiltration and ECM formation [263]. Regulatory T cells (Tregs), the most potent anti-inflammatory cells, contribute to terminating renal inflammation. In later stages, CCL7 maintains protective inflammation by limiting Treg recruitment, thereby attenuating tubulointerstitial fibrosis [263].
Diabetic nephropathy (DN) is a common microvascular complication in long-term diabetic patients, with fibrosis being one of its primary pathological changes. CCR2-mediated monocyte/macrophage recruitment also constitutes a major mechanism of DN-associated renal tissue injury [252]. CCR2 is further expressed in cells beyond monocytes, such as podocytes [264, 265]. Podocytes constitute a critical component of the glomerular filtration barrier and play a pivotal role in the pathophysiology of DN. Increased podocyte foot process width alongside reduced podocyte number and density directly correlates with elevated urinary albumin excretion rates and progressive renal injury in DN [266, 267]. In vitro studies revealed that CCL2 binding to CCR2 enhances podocyte chemotaxis with moderate effects on proliferation [268]. In type 1 diabetic mice, increased CCR2 expression in podocytes elevated renal fibrillin and type I collagen mRNA expression without enhancing renal macrophage infiltration [264]. Notably, the interaction between CCL2/CCR2 signaling and TGF-β1 may exacerbate podocyte apoptosis under diabetic conditions, leading to proteinuria [269]. Inhibition of CCR2 protects kidneys from diabetic injury in type 1 diabetic mice [269]. CCR2 expression is also increased in podocytes of DN patients [265]. In human podocytes, CCL2 binding to CCR2 significantly reduces renin levels via a Rho-dependent mechanism [265]. Renin downregulation is closely associated with the development of diabetic proteinuria [270]. These findings indicate a pathogenic role for the CCL2/CCR2 axis in DN progression, suggesting that targeting this pathway may represent a novel therapeutic strategy for improving DN outcomes.
Renal fibrosis is also a critical process in autoimmune nephropathies. Urinary CCL2 serves as a non-invasive biomarker for disease activity and treatment response in lupus nephritis patients [271]. In lupus nephritis-affected mice, the CCL2/CCR2 axis promotes glomerular macrophage infiltration and induces glomerular microvascular cell proliferation [271]. Furthermore, IL-22 binding to IL-22R on renal epithelial cells upregulates CCL2 via STAT3 signaling, thereby promoting macrophage infiltration and exacerbating lupus nephritis [272]. A prospective cohort study of 100 patients with autoimmune glomerulonephritis demonstrated that elevated levels of CCL2, sC5b-9, and TGF-β1 correlate with disease activity and poor outcomes [273]. Furthermore, CD68-positive interstitial cell infiltration accompanied by CCL2/CCR2 expression represents the most significant prognostic indicator for end-stage renal failure in idiopathic membranous nephropathy [274].
Renal fibrosis is also a key pathological feature in the progression from acute kidney injury (AKI) to CKD. Studies have demonstrated that sustained expression of GM-CSF by renal tubular cells in unilaterally I/R mice, sustained tubular cell expression of GM-CSF significantly increased CCL2 expression by macrophages during the transition from normal renal repair to adverse fibrosis. This led to increased infiltration of CCR2-positive immune cells, including macrophages, DCs, and cytotoxic T cells, into the renal interstitium, thereby promoting persistent tubular injury and interstitial fibrosis during the transition from AKI to CKD [254]. The gut microbiota-derived metabolite trimethylamine N-oxide (TMAO) also influences CCR2-mediated renal fibrosis. Choline-rich diets elevate TMAO levels by altering gut microbiota. TMAO promotes sustained high expression of CCR2 in injured kidneys, enhancing macrophage recruitment and infiltration, thereby exacerbating AKI following renal I/R injury and subsequent CKD progression [275]. In addition to its role in the aforementioned kidney diseases, Wilkening et al. analyzed increased expression of both CCR2 and CCL2 in human focal segmental glomerulosclerosis and demonstrated that infiltration of CCR2-expressing macrophages exacerbates renal injury and fibrotic remodeling [276]. Moreover, high sodium chloride intake upregulates CCL2 expression in 5/6 nephrectomized mice, where CCL2 subsequently recruits macrophages to the injured kidney, exacerbating renal fibrosis [277].
In summary, CCR2 signaling serves as a pivotal link connecting renal injury, inflammation, and fibrosis by recruiting and regulating distinct cellular subpopulations. The CCL2/CCR2 axis primarily mediates the infiltration of pro-inflammatory Ly6Chigh monocytes/macrophages into the kidney, thereby driving inflammatory responses, myofibroblast transdifferentiation, and ECM deposition. However, the biological effects of CCR2 signaling extend far beyond simple pro-fibrotic action. By interacting with CCL7, CCR2 signaling exhibits bidirectional regulatory effects across different stages of renal fibrosis. CCL7/CCR2 signaling drives injury in the early phase, while potentially promoting inflammation resolution and attenuating fibrosis in later stages by limiting Treg recruitment. It is crucial to emphasize that macrophages recruited by CCL2/CCR2 signaling during renal fibrosis are not a homogeneous population. Their function is highly dependent on specific subpopulations. Traditionally, macrophage subpopulations are distinguished by differential Ly6C expression. Ly6Chigh macrophages, derived from circulating monocytes, exhibit proinflammatory functions and have been identified as the primary injury-driven population in multiple organ fibroses via CCR2 signaling [200, 228, 278]. In contrast, Ly6Clow macrophages are typically regarded as tissue-resident macrophages primarily involved in immune regulation and tissue repair [279]. However, this paradigm is challenged in the specific context of renal fibrosis. Reports indicate that the Ly6Clow macrophage subpopulation exhibits a pro-fibrotic phenotype in I/R-induced renal injury mice [280]. Yang et al. demonstrated that while CCR2 deficiency suppresses inflammatory Ly6Chigh macrophages recruitment and improves AKI. Due to the compensatory replenishment of Ly6Clow macrophages in the kidneys during the chronic phase and their presentation of a pro-fibrotic phenotype, ischemia-induced renal fibrosis still worsens in CCR2-deficient mice [281]. This challenges the traditional reparative role of Ly6Clow macrophages, revealing the complexity and context-dependent nature of macrophage function in renal fibrosis progression. It also suggests that simply blocking CCR2 signaling globally may disrupt macrophage homeostasis and exacerbate fibrosis. Therefore, future therapeutic strategies require more precise targeting of specific signaling pathways or cell subpopulations at particular stages to achieve optimal therapeutic outcomes.
4.5 SSc
SSc is a rare autoimmune disease primarily characterized by fibrosis of the skin and internal organs, leading to diverse clinical symptoms and complications. Studies have shown that CCL2 levels in SSc patients are significantly higher than in controls, with a marked decrease following PGE1 treatment [282]. Importantly, the CCR2/CCL2 axis plays a crucial role in SSc by promoting the infiltration of inflammatory monocytes at lesion sites [283]. CCL2 mRNA is expressed on infiltrating monocytes and fibroblasts within SSc lesion skin [284]. Circulating CD14+ monocytes from some SSc patients show upregulation of the ECM component versican and CCL2. Interestingly, versican forms CCL2 chemotactic gradients that attract circulating monocytes and T cells to versican-rich site reservoirs, where these cells subsequently produce additional versican [285]. This positive feedback loop involving versican, CCL2, and monocyte influx significantly accelerates SSc progression [285]. Intraperitoneal injection of the CCL2 antagonist SKL-2841 reduces inflammatory monocyte and polymorphonuclear cell infiltration while markedly suppressing BLM-induced scleroderma fibrosis [286]. This suggests that blocking CCL2 may represent a novel strategy for controlling SSc progression.
One study reported that cultured SSc fibroblasts exhibited significantly elevated levels of spontaneously expressed CCL2 compared to fibroblasts from control skin [287]. Interestingly, exogenous administration of CCL2 stimulated autocrine induction of CCL2 mRNA. Thus, increased responsiveness of sclerotic fibroblasts to CCL2 may perpetuate the fibrotic response. Moreover, CCL2/CCR2 may stimulate a subset of quiescent fibroblasts to differentiate into metabolically active myofibroblasts via an autocrine mechanism in early stages of SSc [288]. Cross-species comparisons between human and mouse scleroderma reveal IL-13 and CCL2 as specific targets in SSc [289]. CCL2 expression is upregulated in IL-13-stimulated human skin fibroblasts, as well as in skin biopsy specimens from inflammatory SSc patients and sclerotic graft-versus-host disease mice [289]. In BLM-induced SSc mice, CCL2 promotes dermal fibrosis by directly upregulating ECM mRNA expression in fibroblasts and indirectly through cytokine-mediated effects released by immune cells recruited to the lesion site [290]. In SSc patients, CCL2 also induces infiltrating CD4+ T cells to differentiate into Th2 cells, which release large amounts of IL-4 and stimulate IL-4R-expressing fibroblasts to produce excessive ECM [291]. In CCL2 knockout mice, BLM treatment resulted in mild skin thickening and reduced collagen accumulation, indicating diminished responsiveness to fibrotic stimuli in the absence of CCL2 [292]. Tight skin 1 (Tsk-1) mice are a widely used SSc animal model. Immunohistochemistry revealed substantial CCL7 expression in the dermis of Tsk-1 mice [150]. Furthermore, in the reported experiment, the promoter of the mouse procollagen α2(I) gene was activated by CCL7 [150]. This suggests that CCL2 may also promote tissue fibrosis by activating ECM synthesis in fibroblasts, thereby contributing to collagen formation.
4.6 Colonic fibrosis
Colonic fibrosis represents the outcome of recurrent episodes of chronic colitis. In colitis-prone mice, both CD30L and CCR2 expression are increased in inflammatory monocytes. CD30L activates circulating monocytes via the CCL2/CCR2 axis and NF-κB pathway, enhancing inflammatory responses [293]. As an angiotensin receptor blocker, irbesartan reduces the accumulation of Ly6ChighCCR2+ monocytes and fibroblasts along with type I collagen expression in inflamed colons by inhibiting CCL2 production, ultimately suppressing intestinal fibrosis and tumor development [294]. The CCR2 ligand PSMP is expressed in patient colitis and colon tumor tissues and is significantly upregulated in dextran sulfate sodium (DSS)-induced colitis tissues in mice. PSMP chemotaxes Ly6Chigh monocytes from the circulation to inflamed colonic tissue in a CCR2-dependent manner, exacerbating colitis [295]. CCR2 also modulates ECM remodeling by upregulating the expression of TIMP-1 [296, 297]. During chronic colonic inflammation, CCR2+ monocytes migrate to the inflamed colon via the CCL2/CCR2 axis and differentiate into CCR2+ fibroblasts. Subsequently, CCR2+ fibroblasts infiltrate the colon and promote colonic fibrosis by inhibiting collagen degradation through TIMP-1 production [296].
4.7 Duchenne muscular dystrophy (DMD)
Muscle fiber necrosis and fibrosis are hallmark features of DMD, leading to fatal diaphragmatic weakness. The diaphragm of DMD (mdx) mice exhibits significantly increased expression of CCR2 and CCL2. CCR2 signaling mediates the recruitment of inflammatory Ly6Chigh monocytes and the accumulation of CD11bhigh MDMs, thereby promoting DMD progression [298]. In DMD mice, CCR2 deficiency reduces macrophage infiltration by blocking blood-borne inflammatory monocyte recruitment, accompanied by transient improvement in muscle damage and fibrosis [299]. This suggests CCR2 inhibition may offer a novel strategy for DMD management. However, this benefit is lost following expansion and pathogenic activation of muscle-resident macrophages, preventing sustained CCR2-mediated improvement in DMD. Mechanistically, these macrophages may induce diaphragmatic fibrosis in late-stage DMD mice by producing persistently elevated levels of fibroblast growth factor [300].
4.8 Others
Chronic pancreatitis (CP) is characterized by progressive, irreversible inflammation and fibrosis. In CP rats, PGE2 promotes disease progression by regulating TNF-α-induced CCL2 synthesis in pancreatic acinar cells, which in turn enhances macrophage infiltration [301]. Inhibiting cyclooxygenase COX-2 activity reduces PGE2 levels, thereby slowing the progression of pancreatitis and fibrosis [301]. Furthermore, the mutant CCL2 reduces serum CCL2 concentrations and impedes monocyte/macrophage recruitment, effectively suppressing di-n-butyl dichloride-induced experimental chronic pancreatitis and subsequent fibrosis in rats [302].
Oral submucosal fibrosis (OSMF) is a chronic disease with a high malignant transformation rate. Increased CCL2 expression in OSMF patients suggests CCL2 may promote OSMF by recruiting myofibroblasts [111, 303]. Experimental autoimmune orchitis (EAO) serves as an animal model for studying chronic testicular inflammation and fibrosis. During EAO, CCL2 expression increases, mediating leukocyte infiltration into the testicular parenchyma and recruiting macrophages, thereby promoting fibrosis in testicular inflammation [304]. Prostatic fibrosis leads to lower urinary tract dysfunction. Popovics et al. demonstrated in a benign prostatic hyperplasia mouse model that CCR2 deficiency impairs monocyte recruitment and macrophage infiltration, thereby improving urinary dysfunction and prostate fibrosis [305]. Furthermore, the CCL2/CCR2 axis directly participates in peritoneal mesothelial cell-associated epithelial-mesenchymal transition and ECM synthesis, inducing peritoneal dialysis (PD)-associated peritoneal fibrosis [306]. Elevated CCL8 levels in peritoneal effusions of PD patients correlate with increased risk of PD failure, which subsequently leads to peritoneal fibrosis and dysfunction [307].
In summary, the role of CCR2 signaling in organ-specific fibrosis exhibits remarkable complexity. Although CCR2-dependent signaling is widely recognized as a prominent pro-fibrotic pathway that promotes collagen deposition and tissue fibrosis by regulating the dynamic migration and functional activation of diverse immune cells, this effect is not universal. Taking liver fibrosis as an example, CCR2 signaling not only participates in fibrosis formation but also plays a crucial role in the regression phase through mechanisms such as mediating the transition of Ly6Chigh monocytes to a pro-reparative phenotype and promoting ECM degradation. This organ specificity and stage dependency represent the deep-rooted causes of current research controversies and translational challenges. Therefore, future studies should focus on developing stage-specific and organ-specific intervention strategies. On the temporal dimension, it is essential to identify the functional transition points of CCR2 signaling across different fibrosis stages and explore dynamic regulatory approaches characterized by early moderate interception and late protective enhancement. In the spatial dimension, the tissue origins and microenvironmental differences of CCR2+ cells in organs such as the lung, kidney, and heart should be elucidated to avoid simplistic extrapolation of findings from liver fibrosis. Furthermore, cell-specific delivery systems or ligand-selective antagonists should be employed to achieve fine-tuning rather than complete interception of CCR2 signaling.
5. CCR2 and its ligands as biomarkers of fibrosis
Fibrosis is a common pathological core of multiple organ chronic diseases, and dynamic monitoring is crucial for assessing disease progression. Research has found that the expression of CCR2 and CCR2 ligands in fibrotic diseases exhibits specificity, making them potential biomarkers for disease diagnosis and prognosis monitoring.
5.1 Pulmonary fibrosis
IPF is a progressive inflammatory lung disease that currently lacks effective molecular biomarkers reflecting disease activity or treatment response. Substantial evidence indicates significantly elevated CCL2 levels in both BALF and serum of IPF patients. High CCL2 levels are predictive of poor prognosis in IPF [308-310]. Serum CCL2 levels are associated with macrophage activation, and CCL2 upregulation increases the risk of death in IPF patients [311]. Moreover, compared with non-fibrotic children, children with pulmonary fibrosis have significantly elevated CCL2 levels and CCR2+ T cells in BALF, which are associated with the severity of interstitial lung disease (ILD) [312]. Although CCL2 levels were elevated in BALF and serum of ILD patients compared with healthy volunteers, only IPF patients had significantly higher CCL2 levels in BALF than in serum [308]. Therefore, simultaneous measurement of CCL2 levels in BALF and serum may help distinguish IPF from ILD.
Specifically, Brody et al. developed a radiotracer, ⁶⁴Cu-DOTA-ECL1i, utilizing positron emission tomography to track CCR2+ monocytes and macrophages [313]. They discovered that in lung tissue from IPF patients, CCR2+ monocytes and interstitial macrophages concentrated in perifibrotic regions [313]. Furthermore, increased ⁶⁴Cu-DOTA-ECL1i PET uptake correlated with CCR2+ cell infiltration. These findings support imaging CCR2+ cells within the fibrogenic niche in IPF, providing a molecular target for disease monitoring and personalized therapy [313]. Further studies revealed that in pulmonary fibrosis models, early CCR2-PET uptake signals not only reflect pulmonary inflammatory burden but also independently predict subsequent fibrosis severity and treatment response to anti-fibrotic drugs such as nintedanib [314]. To further elucidate the imaging capabilities of this novel PET tracer, Heo et al. evaluated its efficacy in imaging CCR2+ cells in the context of heart transplantation and MI. They observed strong and specific binding to regions containing CCR2+ cells, highlighting its potential for imaging human cardiac injury [315]. Additionally, transcriptomic analysis revealed that CCL8 expression in fibroblasts from IPF patients was higher than in controls, and CCL8 was involved in key pathways associated with IPF progression. This suggests that CCL8 may be a candidate biomarker for IPF diagnosis and survival prediction [316].
5.2 Liver fibrosis
Bai et al. analyzed expression dataset GSE84044 of hepatic fibrosis in the GEO database and identified 10 key genes in the protein-protein interaction network, including CCL2 [317]. Persistently elevated CCL2 levels are considered critical in triggering liver injury and subsequent fibrosis progression, and may serve as a predictive marker for cirrhosis progression [318]. Research has found that CCL2 expression is significantly upregulated in the tissues of patients with active cirrhosis [319]. Furthermore, liver CCL2 transcription levels are positively correlated with the degree of liver macrophage activation and patient MELD scores (an indicator of the severity of liver fibrosis [320, 321]. Although adult cirrhosis is often caused by alcohol abuse or hepatitis, the etiology in children remains unclear. A study conducted genetic sequencing on 14 children with syndromic cirrhosis and identified a recessive mutation in the FOCAD gene as the causative factor. Zebrafish lacking FOCAD exhibited liver damage accompanied by elevated CCL2 expression, suggesting that targeting the CCL2/CCR2 axis may represent a potential therapeutic approach for pediatric cirrhosis [322]. Furthermore, CCL2 levels were higher in patients with severe hepatitis recurrence compared to those with non-severe HCV recurrence [323]. Functional -2518 CCL2 promoter polymorphisms appear to influence liver CCL2 expression, making HCV patients more susceptible to severe hepatic inflammation and fibrosis [324]. CCL2 rs1024610 and the ATGC haplotype are plausible candidate markers for chronic HCV infection [325]. These data indicate that CCL2 possesses good predictive capability for identifying severe HCV infection.
5.3 Renal fibrosis
Diabetic nephropathy (DN) is a severe complication of diabetes. CCL2 is considered a marker for early-stage DN. With increasing serum and urinary CCL2 levels, the risk of developing new-onset microalbuminuria continues to rise, representing an early sign of DN [326]. Currently, DN is the leading cause of end-stage renal disease (ESRD). In DN patients, elevated plasma CCL2 levels are an independent risk factor for ESRD and have significant clinical value in assessing DN prognosis [327]. Studies have also found that elevated serum levels of IL-6, NF-κB, and CCL2 are closely associated with renal injury and poor prognosis in DN patients, and combined detection holds significant value for assessing patient status and prognosis [328].
Elevated early-stage urinary CCL2 levels have been demonstrated to correlate with tubulointerstitial fibrosis [329]. A meta-analysis revealed that increased urinary CCL2 levels in 596 chronic kidney disease patients predicted renal fibrosis on biopsy and were associated with accelerated eGFR decline [330]. Renal CCL2 mRNA expression and urinary CCL2 concentration correlate with the degree of obstruction in hydronephrosis and subsequent renal injury [331]. Furthermore, early urinary CCL2 is associated with the late development of interstitial fibrosis and tubular atrophy in renal allografts[332]. Elevated ratio of CCL2 to creatinine (CCL2: Cr) in early urine may aid in identifying patients with interstitial fibrosis and inflammation following kidney transplantation [333]. Furthermore, elevated serum CCL8 expression in patients with CKD is associated with advanced CKD staging, urine protein-to-creatinine ratio, and renal fibrosis [334].
5.4 SSc
SSc is an autoimmune disease characterized by vascular lesions and uncontrolled fibrosis of the skin and internal organs [335]. Delays in the diagnosis and treatment of SSc may lead to uncontrolled disease progression, underscoring the importance of identifying early diagnostic markers. Bioinformatics screening identified the tissue-specific gene CCL2 as an effective biomarker, providing new insights into the mechanisms of SSc [336]. Bioinformatics analysis revealed CCL2 as a common signature gene in both IPF and SSc [337]. CCL2 expression is elevated in the sclerotic skin of SSc patients, also increased in serum and BALF. Serum CCL2 levels are particularly high in patients with early diffuse cutaneous disease [338]. Another clinical study in SSc patients demonstrated that high levels of serum CCL2 are closely associated with increased skin fibrosis and increased risk of internal organ involvement in patients with SSc [339]. High circulating CCL2 levels can predict poorer survival outcomes in SSc patients [340]. A recent longitudinal analysis of SSc patients revealed a year-by-year decline in circulating CCL2 levels, accompanied by improvement in skin sclerosis [341]. Therefore, CCL2 can serve as a biomarker for assessing disease activity and severity of visceral involvement in SSc [342]. Furthermore, elevated serum CCL7 levels in SSc patients are positively correlated with the severity of skin fibrosis and pulmonary fibrosis [343]. Serum CCL13 is also specifically elevated in SSc patients [344]. However, data from Gambichler et al. indicate that CCL13 levels in SSc patients are not significantly different from those in healthy controls [345]. Therefore, further research data is needed to confirm whether CCL13 can be used as a biomarker for SSc.
5.5 Others
In patients with myeloproliferative neoplasms (MPN), the percentage of CCR2+ cells is significantly associated with the severity of myelofibrosis, and CCR2 expression on CD34+ cells correlates with high-risk classification of MF and the presence of circulating primitive cells [346]. Moreover, single nucleotide polymorphisms (SNPs) in CCL2 have been shown to influence the bone marrow microenvironment in MPN. The CCL2 rs1024611 SNP (i.e., CCL2 gene regulatory region -2518A/G substitution) alters its transcriptional activity, thereby upregulating CCL2 expression levels [347]. Analysis of MF patients indicates that homozygous status for the CCL2 -2518G allele is an independent prognostic factor for reduced overall survival [348]. Furthermore, the polymorphic allele (G) is more prevalent in patients who progressed to MF from polycythemia vera (PV) or essential thrombocythemia (ET), and its presence correlates with adverse clinical outcomes [347]. Collectively, these findings suggest that the CCL2 rs1024611 polymorphism may represent a candidate genetic susceptibility factor for MF and an independent risk factor for PV/ET-to-MF progression [349].
Beyond studies in MPN, the CCL2 -2518A/G polymorphism has demonstrated relevance in other diseases. Research in German SSc patients suggests the G variant may be associated with increased CCL2 gene transcriptional activity [350]. Overexpression of the CCL2 -2518 GG homozygote in patients suggests that carrying the CCL2 G allele is a risk factor for SSc, and this polymorphism may influence CCL2 expression in skin fibroblasts of SSc patients [350]. However, another study in European Caucasians failed to replicate this finding, reporting no association between this SNP and SSc susceptibility [351]. Additionally, the CCL2 -2518A/G polymorphism has been linked to acute pancreatitis, acute recurrent pancreatitis, and chronic pancreatitis [352]. Furthermore, serum CCL2 levels were significantly elevated in all patients with pancreatic inflammatory diseases, and the CCL2 -2518G allele was significantly overexpressed in patients with acute recurrent pancreatitis [352]. A genetic association study involving 79 Korean male patients with alcoholic chronic pancreatitis and 82 male controls further indicated that CCL2 polymorphisms may correlate with disease severity in alcoholic chronic pancreatitis [353].
In summary, these studies demonstrate that the specific expression levels of serum CCR2 ligands serve as a standard risk assessment and predictive tool for fibrotic diseases, enabling monitoring of disease progression and evaluation of prognosis. Moreover, following fibrotic injury, CCR2 PET imaging detects CCR2⁺ monocytes/macrophages localizing to periportal areas of fibrosis, with the number of CCR2⁺ cells reflecting disease activity and severity. Thus, the potential core value of CCR2 PET imaging lies in its ability to non-invasively localize and quantify CCR2⁺ monocytes/macrophages, thereby capturing spatiotemporal dynamics unattainable through conventional biopsy or blood tests. Therefore, CCR2-PET not only serves as a non-invasive biomarker for monitoring disease activity and progression but also holds promise as a tool for assessing drug efficacy in clinical trials, accelerating the development of anti-fibrotic drugs and the implementation of precision treatment strategies. Consequently, CCR2 and its ligands hold promise as potential biomarkers for fibrotic diseases, offering effective strategies for early diagnosis, dynamic monitoring, and stratified prevention and treatment.
However, translating these biomarkers from basic research to clinical application remains challenging. Circulating chemokine levels do not necessarily correlate with tissue levels, meaning plasma CCL2 concentrations may not accurately reflect the degree of CCL2 activation within the vascular wall. This limitation reduces the sensitivity of biomarkers. Combining CCR2 ligands with other clinical diagnostic parameters may enhance diagnostic effect. Furthermore, the metabolic networks within the body are highly complex, and disease manifestations exhibit significant heterogeneity. Selected biomarkers may show variations across different individuals, disease stages, or treatment interventions, thus limiting their clinical applicability. Therefore, future efforts should validate the feasibility of these biomarkers in larger, more representative clinical sample populations.
6. Targeting CCR2 signaling as a therapeutic strategy for fibrotic diseases
Extensive research has confirmed that CCR2 signaling is a key therapeutic target for treating various organ fibrotic diseases. The crucial role of CCR2 in the pathogenesis of fibrosis has driven the development of various treatment strategies, including small molecule antagonists, natural Chinese herbal preparations, gene therapy, and mesenchymal stem cell (MSC) transplantation (Table 2). These therapeutic strategies focus on inhibiting the activation of the CCL2/CCR2 axis and have demonstrated significant potential in preclinical studies and clinical trials. Among these, cenicriviroc (CVC) and NOX-E36 have entered the clinical trial phase (Table 3).
Preclinical studies of drugs targeting CCL2/CCR2 axis for the treatment of fibrotic diseases.
| Drug | Target | Experimental model | Method | Result | Refs |
|---|---|---|---|---|---|
| CVC | CCR2/CCR5 | Mice with liver fibrosis induced by CCl4 | 4 dosages of CVC (15 mg/kg body weight) per week, for 6 weeks, i.p. | Inflammatory FSCN1+ macrophages and HERC6+neutrophils ⬇, ECM accumulation ⬇, liver fibrosis ⬇ | [225] |
| Mice with intrabiliary injection of BV6 | 15 mg/kg/d, for 5 days, s.c. | Monocyte recruitment ⬇, macrophage accumulation ⬇, liver fibrosis ⬇ | [357] | ||
| Alcohol-fed mice | 15 mg/kg/d, for 6 weeks, s.c. | Alcohol-related liver mRNA⬇, TNF-α, IL-1β, IL-6, and CCL2 ⬇, proinflammatory Ly6Chigh MDMs ⬇ | [360] | ||
| Fibrotic steatohepatitis mice | 50mg/kg, two times a day, i.g. | Histological NASH activity and hepatic fibrosis ⬇, Ly6Chigh monocytes infiltration ⬇ | [359] | ||
| Diet-induced NASH mice | High-fat diet containing 0.015% CVC for 12 weeks | M1-like macrophages ⬇, M2-like macrophages ⬆, hepatic stellate cell activation ⬇, hepatic lipid accumulation and peroxidation ⬇, | [361] | ||
| Diet-induced NASH mice | 30 mg/kg/d for 4 weeks or 30 mg/kg/d for 14 weeks, i.p. | Ly6Chigh BM-derived macrophages accumulation ⬇, liver fibrosis ⬇ | [362] | ||
| Mdx mice (genetic homologue of DMD) | 20 mg/kg/d, for 4 weeks, i.p. | Total infiltrating macrophages ⬇, disease progression ⬇ | [363] | ||
| RS-102895 | CCR2 | Renal artery stenosis mice | 10 mg/kg/d, for 4 weeks, i.g. | Renal atrophy ⬇, iNOS+ and CD206+ macrophage accumulation ⬇ | [371] |
| RS-504393 | CCR2 | UUO mice | 2 mg/kg twice a day, i.g. | Macrophage infiltration and activation ⬇ | [257] |
| BLM-induced scleroderma mice | 4 mg/kg/d or 8 mg/kg/d, three days per week, for 4 weeks, i.d. | The number of mast cells and myofibroblasts in the skin ⬇, mRNA levels of TGF-β1 and collagen α1 ⬇, | [373] | ||
| Rheumatic heart disease rats | 5 mg/kg, once daily for 8 weeks, i.p. | Macrophage infiltration, inflammation, and fibrosis ⬇ | [207] | ||
| Bladder outlet obstruction rats | 5 mg/kg, for 6 weeks, i.g. | Macrophage infiltration in bladder tissue ⬇, bladder fibrosis ⬇ | [374] | ||
| NOX-E36 | CCR2 | Diabetic Apo E -/- mice | 20 mg/kg, three times a week for 4 weeks, c | Albuminuria ⬇, CCR2-expressing Ly6Chigh monocytes ⬇, restores glomerular endothelial glycocalyx and barrier function | [376] |
| Mice with type 2 diabetes | 50 mg/kg, three times per week, continued for 8 weeks, i.d. | Glomerular macrophages ⬇, diffuse glomerulosclerosis ⬇ | [377] | ||
| Mice with metabolic liver fibrosis | 20 mg/kg NOX-E36, s.c. | Intrahepatic levels of proinflammatory cytokines ⬇, early influx of Ly6Chigh MDMs ⬇, liver fibrosis ⬇ | [379] | ||
| INCB334 | CCR2 | Ang II-treated mice | 30 mg/kg/d, continued for 21 days, i.p. | Ly6Chigh monocyte and M2 macrophage accumulation ⬇, aortic collagen deposition, elastin loss, and BP ⬇ | [382] |
| SKL-2841 | CCL2 | BLM-induced scleroderma mice | 100 mg/kg SKL-2841, for 3 days or 3 weeks, i.p. | Infiltration of inflammatory mononuclear cells and polymorphonuclear cells ⬇ | [286] |
| OPL-CCL2-LPM | CCR2 | Rats of anti-thymocyte serum-induced mesangioproliferative glomerulonephritis | 50 or 100 microg/kg, from days 2, 4, 6, and 8, i.v. | CCR2+ MDMs ⬇, mesangial cell proliferation ⬇, ECM synthesis ⬇ | [383] |
| Anti-CCL2 NAb | CCL2 | Rats with suprarenal aortic constriction | 2 mg/kg/d, daily from 1 day before operation to day 28, i.v. | Macrophage accumulation ⬇, fibroblast proliferation ⬇, TGF-β ⬇ | [202] |
| Honokiol | CCL2 | UUO rats | 2.5 mg/kg, twice per day, for 14 days, i.g. | Tubulointerstitial fibrosis ⬇, expression of pro-fibrotic factors ⬇, accumulation of type I collagen and fibronectin ⬇ | [388] |
| Tianhuang formula | CCL2/MAPK/NF-κB | CCl4-induced liver fibrosis mice | 4.8 g/kg for 6 weeks, i.g. | Serum ALT, AST, and hepatic TG levels ⬇, expression of fibrosis and inflammation markers ⬇ | [389] |
| Quercetin | ERK1/2-C/EBPβ | Mice with experimental autoimmune myocarditis | 20mg/kg every other day, i.g. | CCL2 ⬇, Cardiac function ⬆, inflammation ⬇, fibrosis ⬇ | [390] |
| Dachaihu decoction | CCL2 | Mice with chronic pancreatitis | 14 g/kg/d, intragastrically | Macrophages infiltration ⬇, degree of fibrosis ⬇, IL-6 and fibronectin levels ⬇ | [387] |
| Fu-Gan-Wan | NF-κB/CCL2/CCR2 | CCl4-induced liver fibrosis mice | 9.75 g/kg or 19.5 g/kg, once a day for 4 weeks, i.g. | ALT and AST ⬇, collagen deposition ⬇, pro-fibrotic factors α-SMA, COL1α1, CTGF, TIMP-1, TGF-β1 ⬇ | [391] |
| Puerarin | CCL2, CCL7 | DSS-induced colitis mice | 25 and 50 mg/kg/d | Restores intestinal barrier integrity, proinflammatory cytokine production ⬇, colitis ⬇ | [392] |
| MI mice | 100 mg/kg/d for 28 days, i.p. | Monocytes/macrophages activation ⬇, TGF-β1 ⬇, cardiac fibrosis ⬇ | [394] | ||
| Arctigenin | ROS/ERK1/2/ NF-κB | UUO rats | 1 and 3mg/kg/d, for 11 consecutive days, by gastric gavage | CCL2 ⬇, macrophages infiltration ⬇, TNF-α ⬇, IL-1β ⬇, IFN-γ ⬇, TGF-β1 ⬇, oxidative stress and EMT of renal tubules ⬇ | [396] |
| Astragalus | TGF-β1 | Rats with peritoneal dialysis | 1000/2000/4000 mg/kg/d for 7 d, i.p. | CCL2 ⬇, monocytes/macrophages activation⬇, TGF-β1 ⬇ | [397] |
| AS-IV | NF-κB | LPS-induced H9C2 cells | AS-IV (5, 10, and 50 μM) groups | CCL2 ⬇, cell surface size ⬇, cardiac hypertrophy and fibrosis ⬇ | [399] |
| Streptozotocin-induced diabetic rats | 5 and 10 mg/kg/d for 8 weeks, p.o. | Serum levels of TNF-α, CCL2 and ICAM-1 ⬇, type I collagen production ⬇ | [400] | ||
| Curcumin | CCL2 | Pancreatic stellate cells | Curcumin at 1, 2.5, 5,10, 25 mM | Inhibits IL-1β and TNF-α-induced activation of AP-1 and MAPK, CCL2 ⬇, α-SMA level ⬇, type I collagen production ⬇ | [401] |
| CCl4-induced liver fibrosis mice | 200 mg/kg/d, once daily, for 4 weeks, i.g. | Ly6Chigh monocytes intrahepatic infiltration ⬇, kupffer cells activation ⬇, pro-inflammatory and pro-fibrogenic cytokines ⬇ | [402] | ||
| CCl4-induced liver fibrosis mice | 200 mg/kg body weight daily, for 6 weeks, i.g. | CCL2 ⬇, TNF-α and TGF-β1 ⬇, Gr1hi monocytes infiltration ⬇, inflammation and fibrosis ⬇ | [403] | ||
| Mice with NASH | 25 microg per mouse, every other day, i.p. | Intrahepatic gene expression of CCL2, CD11b, procollagen type I, α-SMA, and TIMP-1 ⬇, reactive oxygen species ⬇ | [404] |
Abbreviations: ALT, alanine aminotransferase; Ang II, angiotensin II; AP-1, activator protein 1; Apo E, apolipoprotein E; AST, aspartate aminotransferase; AS-IV, Astragaloside IV; BLM, bleomycin; BM, bone marrow; BP, blood pressure; CCl4, carbon tetrachloride; CCR2, C-C motif chemokine receptor 2; COL1α1, collagen type I α 1 chain; CTGF, connective tissue growth factor; CVC, cenicriviroc; DMD, duchenne muscular dystrophy; DSS, dextran sulfate sodium; ECM, extracellular matrix; EMT, epithelial-mesenchymal transition; ERK1/2, extracellular signal-regulated kinase 1/2; FSCN1, fascin actin-bundling protein 1; HERC6, HECT and RLD domain containing E3 ubiquitin protein ligase family member 6; ICAM-1, intercellular adhesion molecule 1; i.d., intradermal injection; IFN-γ, interferon-gamma; i.g., intragastric injection; IL-1β, interleukin-1 beta; iNOS, inducible nitric oxide synthase; i.p., intraperitoneal injection; i.v., intravenous injection; LPS, lipopolysaccharide; MAPK, mitogen-activated protein kinase; MDMs, monocyte-derived macrophages; MI, myocardial infarction; NASH, non-alcoholic steatohepatitis; NF-κB, p65 subunit of nuclear factor-κB; p.o., oral (by mouth); ROS, reactive oxygen species; s.c., subcutaneous injection; α-SMA, α-smooth muscle actin; TG, triglycerides; TGF-β, transforming growth factor β; TIMP-1, tissue inhibitor of metalloproteinases 1; TNF-α, tumor necrosis factor α; UUO, unilateral ureteral obstruction.
Clinical trials of drugs targeting the CCL2/CCR2 axis in the treatment of fibrotic diseases.
| Drug | Target | Experimental object | Research stage | Treatment outcomes and safety | Clinical limitations/reasons for failure | Trial identification | Refs |
|---|---|---|---|---|---|---|---|
| CVC | CCR2/CCR5 | Patients with NASH and stage 0-4 liver fibrosis | II | CVC 150 mg once daily was well tolerated, with a median treatment duration of 33.6 months | / | NCT03059446 | [364] |
| Patients with at least 6 months of PSC | II | Treatment with CVC 150 mg for 24 weeks was well tolerated and a modest decrease (median of 18%) in the endpoint of ALP | The main limitations are the small sample size and single-arm study design, which means that each participant was his or her own control | NCT02653625 | [355] | ||
| NASH patients with NAFLD activity score at screening (4 or ≥5) and fibrosis stage (≤2 or >2) | IIb | Antifibrotic benefit with good tolerability at year 1, achieves improvement in fibrosis by ≥1 stage and no worsening of NASH | Differences in responses among subgroups (e.g., region, sex, and T2DM) that may reflect the multifactorial nature of the disease or be associated with sample size of the subgroups | NCT02217475 | [365] | ||
| Patients with NASH and stage 2 or 3 liver fibrosis | III | Safe and well tolerated, primary endpoint—fibrosis regression (22.3% in the CVC group vs. 25.5% in the placebo group) and no worsening of NASH was not met | CVC inhibits Ly6Chigh macrophage infiltration but fails to eliminate pro-fibrotic TREM2⁺ scar-associated macrophages, thus failing to drive reversal of deposited collagen | NCT03028740 | [367] | ||
| NOX-E36 | CCL2 | Type 2 diabetic patients with albuminuria | IIa | Generally safe and well tolerated, the urinary albumin/creatinine ratio (ACR) from baseline to week 12 decreased by 29% (P < 0.05) | Baseline characteristics are not balanced between the groups due to small sample size, which contributes to a worse renal and metabolic outcome in the placebo group | NCT01547897 | [422] |
| Carlumab | CCL2 | Patients with progressive IPF disease within 12 months | II | The primary endpoint of percentage change in forced vital capacity did not demonstrate any therapeutic effect. SGRQ scores indicated a slight trend toward worsening during treatment. | The primary cause of failure is the excessive activation of compensatory mechanisms under CCL2 inhibition, which also involves pharmacodynamic limitations and patient population heterogeneity. | NCT00786201 | [385] |
Abbreviation: ALP, alkaline phosphatase; ACR, albumin/creatinine ratio; CVC, cenicriviroc; CCR2, C-C motif chemokine receptor 2; IPF, idiopathic pulmonary fibrosis; NAFLD, non-alcoholic fatty liver disease; NASH, non-alcoholic steatohepatitis; PSC, primary sclerosing cholangitis; T2DM, type 2 diabetes mellitus; TREM2, triggering receptor expressed on myeloid cells 2.
6.1 Small molecule antagonists
CVC
CVC is a novel oral dual-target CCR2/CCR5 antagonist with good safety and bioavailability [354, 355]. The anti-inflammatory and anti-fibrotic effects of CVC have been confirmed in many fibrotic animal models [356-358]. CVC has been demonstrated to improve hepatic inflammation and fibrosis by inhibiting inflammatory monocyte recruitment and reducing macrophage infiltration [225, 359]. In vitro studies revealed that CVC suppresses transcription of key pre-fibrotic genes in macrophages by inactivating the STAT1/NF-κB/ERK signaling pathway [225]. CVC also suppressed CCL2-induced increases in hepatic fatty acid synthase and adipose differentiation-related protein while enhancing acyl-coA oxidase 1 and proliferator-activated receptor gamma coactivator-α expression, suggesting a mechanism for mitigating hepatic steatosis [360]. CVC also reverses hepatic steatosis, insulin resistance, inflammation, and fibrosis in NASH mice by inhibiting macrophage M2 polarization [361, 362]. Furthermore, in acute sclerosing cholangitis mice treated with the apoptosis inhibitor BV6, CVC therapy alleviates liver damage and bile duct fibrosis [357]. CVC also reduces macrophage infiltration and disease severity in DMD mice [363].
Currently, clinical studies using CVC for the treatment of NASH-related liver fibrosis have made significant progress, and CVC is expected to become a commonly used drug for the clinical treatment of liver fibrosis. CVC has demonstrated good tolerability and safety in over 1,100 trial participants, including patients with cirrhosis and mild to severe liver dysfunction [355, 364]. In a Phase IIb clinical trial (NCT02217475), Friedman et al. found that one year of CVC treatment resulted in significant improvement in liver fibrosis, and no worsening of steatohepatitis in NASH patients [365]. One year later, their research team further reported that CVC exhibits sustained antifibrotic effects, with greater efficacy in patients with advanced fibrosis, particularly those with stage 3 fibrosis [365]. CVC is currently undergoing a Phase III clinical trial (NCT03028740) to evaluate and confirm the efficacy and safety of CVC treatment for adult NASH [366]. Results showed that while CVC is safe and well-tolerated in patients, its primary endpoint—fibrosis regression (22.3% in the CVC group vs. 25.5% in the placebo group) and no worsening of NASH (23.0% in the CVC group vs. 27.2% in the placebo group)—was not met [367]. Mechanistically, CVC selectively targets CCR2/CCR5 to block liver infiltration by Ly6Chigh proinflammatory MDMs, demonstrating clear efficacy in inhibiting the initiation of fibrotic signaling [359]. However, the failure of this Phase III trial highlights the challenges of targeting CCR2 during the fibrosis reversal phase.
As previously noted, the regression of liver fibrosis relies not only on suppressing inflammation but also requires the participation of reparative macrophages. Studies confirm that in the late stages of fibrosis, CCR2 also participates in recruiting reparative macrophages with pro-degradative functions. These cells promote ECM degradation by producing MMPs, serving as key drivers of active fibrosis reversal. Long-term CVC blockade, while suppressing pro-inflammatory monocyte infiltration, may also disrupt the recruitment of this reparative population, thereby undermining the intrinsic capacity for fibrosis regression. Further single-cell transcriptomics studies reveal that hepatic macrophages exhibit heterogeneity and adapt their phenotypes in response to microenvironmental signals within fatty liver, enabling compensatory interactions among distinct macrophage subpopulations [368]. Inhibiting the CCR2⁺ subset alone is insufficient to eliminate pro-fibrotic populations like TREM2⁺ scar-associated macrophages, making it difficult to drive active reversal of deposited collagen [369]. Moreover, Phase III trial data show that the CVC group even had a lower rate of fatty liver inflammation resolution than the placebo group, clearly demonstrating that anti-inflammatory strategies alone struggle to achieve histological reversal under sustained metabolic stress.
In summary, anti-fibrotic treatment requires precise timing control to avoid excessive blockade of the CCR2 receptor during the inflammatory suppression process, as this could impede the critical pathways for initiating tissue repair. Concurrently, future anti-fibrotic drug development should shift toward mechanism-complementary combination therapy strategies to achieve multidimensional interventions encompassing anti-inflammatory effects, metabolic regulation, and pro-regenerative repair. Recent studies reveal synergistic effects when combining CCR2 inhibitors with FGF21 agonists in alleviating steatohepatitis and fibrosis [370]. CVC inhibition blocks hepatic infiltration of inflammatory monocytes, while FGF21 agonists improve obesity-related metabolic dysfunction, confirming the therapeutic potential of integrating these approaches in NASH patients [370].
RS-504393
Research has shown that the small-molecule CCR2 antagonists RS-102895 and RS-504393 exhibit significant anti-inflammatory and anti-fibrotic effects [254]. The use of RS-102895 to block CCL2/CCR2 signaling can reduce renal fibrosis in UUO mice and renal vascular hypertension mice [257, 371]. Furthermore, treatment with RS504393 significantly reduced urinary albumin excretion and mesangial expansion while inhibiting the synthesis of pro-fibrotic and pro-inflammatory cytokines, thereby restoring renal function in type 2 diabetic mice [372]. In BLM-induced scleroderma mice, RS-504393 effectively reduces the levels of TGF-β1 and type I collagen in the skin [373]. Intraperitoneal injection of RS-504393 reduces valvular inflammation and fibrosis in rheumatic heart disease rats [207]. RS-504393 also blocks macrophage infiltration and bladder fibrosis in rats with bladder outlet obstruction [374]. However, RS-504393 remains in the preclinical development stage, and future clinical data are needed to assess its safety and efficacy.
NOX-E36
As a CCL2 antagonist, NOX-E36 specifically binds to and inhibits CCL2, demonstrating potent anti-fibrotic effects in DN animal models by restoring glomerular endothelial glycocalyx and barrier function [375, 376]. Furthermore, NOX-E36 protects DN mice from developing diffuse glomerulosclerosis [377]. NOX-E36 also accelerates the resolution of toxic and metabolic liver fibrosis in two experimental models by suppressing the early influx of Ly6Chigh monocytes, thereby shifting the hepatic macrophage balance toward the reparative Ly6Clow subpopulation [378, 379]. A Phase IIa study involving 75 patients with type 2 diabetes and proteinuria showed that NOX-E36 is safe, well-tolerated, and has renal protective effects.
Others
INCB334 is an efficient and orally bioavailable CCR2 antagonist with strong inhibitory activity against human CCR2 but moderate activity against mouse CCR2 [380, 381]. Inhibition of macrophage accumulation using INCB3344 prevents Ang II-induced vascular fibrosis and blood pressure elevation [382]. McIntosh et al. reported a novel CCR2-targeted drug, OPL-CCL2-LPM, comprised of the human CCL2 fused to a truncated form of the enzymatically active A1 domain of Shigella dysenteriae holotoxin [383]. Data from a rat model of anti-thymocyte serum-induced mesangioproliferative glomerulonephritis indicate that treatment with OPL-CCL2-LPM leads to CCR2+ MDMs depletion and reduces mesangial cell proliferation and ECM synthesis [383]. Therefore, treatment with LPM may slow down downstream fibrotic events occurring in many glomerulonephritis and nephrotic syndromes. Further clinical trials are needed to validate the safety, specificity, and efficacy of LPM.
In addition, long-term treatment with anti-CCL2 monoclonal neutralizing antibodies can inhibit macrophage accumulation, fibroblast proliferation, and TGF-β expression, thereby effectively alleviating myocardial fibrosis [202]. Moreover, this neutralizing antibody alleviated myocardial fibrosis without reducing myocardial hypertrophy, and improved diastolic dysfunction without affecting blood pressure or systolic function. Carlumab is a specific CCL2-inhibiting immunoglobulin G1κ monoclonal antibody [384]. However, in Phase II clinical trials, carlumab failed to demonstrate clinical benefit in IPF patients and even suggested a trend toward worsening prognosis in some dose groups [385]. The reason for this failure may be that compensatory mechanisms became overly activated under CCL2 inhibition. However, the failure of this therapy stems not from a single mechanism but from multiple factors, including pharmacokinetic limitations and patient population heterogeneity. From a pharmacodynamic perspective, carlumab may fail to achieve sustained and adequate CCL2 neutralization due to insufficient dosage, inadequate administration frequency, or antibody affinity limitations, resulting in incomplete target inhibition. Regarding patient heterogeneity, the tendency toward worsened prognosis in the medium-dose group (5 mg·kg⁻¹) may correlate with poorer baseline characteristics. Patients in this group were older, more obese, had lower baseline forced vital capacity and 6-minute walk distances, and exhibited higher rates of oral corticosteroid use [385]. This suggests that patients with higher disease severity or metabolic inflammation may exhibit poorer responses to this therapy, potentially even experiencing paradoxical worsening. In summary, although carlumab failed to demonstrate clinical benefit, the therapeutic value of targeting the CCL2/CCR2 axis remains intact. Developing next-generation interventions with optimized pharmacokinetic profiles or mechanisms that circumvent compensatory pathways retains research potential.
6.2 Natural Chinese herbal preparations
Many herbal medicine formulations have been shown to have good anti-fibrotic effects [386, 387]. Honokiol is a polyphenolic compound isolated from the bark of magnolia officinalis. This compound reduces renal interstitial fibrosis in UUO rats by inhibiting CCL2 expression [388]. Tianhuang formula improves liver fibrosis by inhibiting the CCL2/CCR2/MAPK/NF-κB signaling pathway [389]. Quercetin is a natural flavonoid compound with anti-inflammatory and cardioprotective effects. Quercetin downregulates CCL2 expression via the ERK1/2-C/EBPβ pathway, significantly improving cardiac inflammation and fibrosis in autoimmune myocarditis mice [390]. Dachaihu decoction has been shown to reduce CCL2 levels in the pancreas, thereby decreasing macrophage infiltration and fibrosis associated with chronic pancreatitis [387]. RNA sequencing combined with network pharmacology studies suggest that Fu-Gan-Wan exhibits potential in alleviating carbon tetrachloride-induced hepatic fibrosis, lipid peroxidation, and iron metabolism disorders in mice [391]. This effect is mediated by the NF-κB/CCL2/CCR2 and nuclear factor erythroid 2-related factor 2/heme oxygenase-1 (Nrf2/HMOX1) pathways [391]. Moreover, Puerarin exhibits strong anti-inflammatory effects by inhibiting the NF-κB signaling pathway and reducing CCL2 and CCL7 production in colon tissue [392, 393]. Puerarin also prevents myocardial fibrosis after MI by reducing CCL2 expression and inhibiting the TGF-β1 pathway [394]. Arctigenin downregulates CCL2 expression by inhibiting the ROS/ERK1/2/NF-κB pathway, ultimately reversing tubular EMT in the renal interstitial fibrosis process [395, 396].
Astragalus exhibits anti-inflammatory and anti-fibrotic effects in various diseases. Astragalus alleviates peritoneal fibrosis in PD rats by inhibiting CCL2 and TGF-β1 pathway [397]. Furthermore, the antifibrotic effects of astragalus saponin IV (AS-IV) have been confirmed in various animal models of cardiovascular diseases [398, 399]. AS-IV alleviates renal injury in streptozotocin-induced DN rats by inhibiting CCL2 expression mediated by NF-κB [400]. Activated pancreatic stellate cells (PSCs) play a key role in the pathogenesis of pancreatic fibrosis and inflammation. The polyphenolic compound curcumin inhibits the activation of AP-1 induced by IL-1β and TNF-α, thereby suppressing CCL2 production and ultimately inhibiting PSC activation [401]. Curcumin also reduces CCL2 secretion by inhibiting Kupffer cell activation, thereby reducing Ly6Chigh monocyte infiltration to prevent carbon tetrachloride-induced liver fibrosis [402, 403]. Curcumin can also effectively limit the progression of fibrosis in experimental fatty liver mice [404]. In brief, the evidence for the antifibrotic effects of these herbal formulations remains at the preclinical stage, and future clinical trials are needed to explore their toxicological characteristics, human safety thresholds, and clinical adverse reactions.
6.3 Gene therapy
Anti-CCL2 or CCR2 gene therapy can effectively reduce fibrosis, as demonstrated in various fibrotic animal models such as UUO mice and DMD mice [137, 257, 299]. Kitagawa et al. demonstrated that CCR2 gene knockout reduces UUO-induced renal interstitial fibrosis in mice [257]. Furuichi et al. also obtained similar results using a renal I/R model, with CCR2-deficient mice exhibiting resistance to renal injury and fibrosis [405]. Moreover, CCL2-deficient mice show reduced collagen fiber formation and are protected from BLM-induced skin fibrosis [292]. Tian et al. established a new strategy based on CCR2 small interfering RNA silencing (siCCR2) by loading multivalent siCCR2 with tetrahedron framework DNA nanostructure vehicle (tFNA-siCCR2) to alleviate liver fibrosis [406]. tFNA-siCCR2 can restore the immune cell landscape and establish an anti-fibrotic microenvironment by inhibiting the accumulation of macrophages and neutrophils in mouse livers [406]. Chen et al. synthesized FNA-siCCR2 targeting M1 macrophages to block macrophage accumulation in lung parenchyma, thereby improving chemoradiation-induced pulmonary fibrosis in mice [184].
Moreover, the truncated [1+9-76] CCL2 analogue (also known as 7ND) has been shown to be a weak inhibitor of CCL2/CCR2 signaling in mice [407]. In BLM-induced pulmonary fibrosis mouse lung tissue, 7ND mediates anti-fibrotic effects in mouse fibroblasts by reducing CCL2 and ECM protein levels [407]. Mesenchymal stem cells transduced with 7ND significantly mitigate BLM-induced lung injury [408]. 7ND also prevents liver fibrosis in rats by blocking macrophage infiltration and inhibiting HSCs activation [229]. Currently, the method of using mutant gene transfection to antagonize CCL2/CCR2 signaling has demonstrated good anti-fibrotic effects. Delivery of the N-terminal deletion mutant 7ND of the human CCL2 gene to skeletal muscle significantly improves renal fibrosis in UUO mice by reducing type I collagen deposition and TGF-β expression [229]. Furthermore, 7ND gene transfection therapy alleviates dimethylnitrosamine-induced liver fibrosis and interstitial fibrosis following experimental MI [409, 410]. 7ND gene transfection therapy can also effectively inhibit fibrosis of the infrapatellar fat pad in arthritis rats [411].
6.4 MSC transplantation
MSCs and derived exosomes (EVs) have emerged as promising alternative therapies for treating various organ fibroses due to their potent immunomodulatory capabilities [412, 413]. MSCs primarily reduce mature macrophages expressing high levels of CCR2 by inhibiting CCL2 production in fibroblasts and macrophages [413]. This finding supports MSC-based clinical treatment for SSc patients. Furthermore, MSC-derived EVs (MSC-EVs) reduce CCL2 production by inhibiting ERK1/2 phosphorylation [412]. This leads to decreased monocyte-macrophage recruitment to the lungs, effectively alleviating pulmonary fibrosis [412]. MSC-EVs also diminish CCL2 expression, macrophage infiltration, and production of fibrosis markers type I collagen and α-SMA in liver injury [414]. Furthermore, human amniotic fluid-derived stem cells therapy can reduce CCL2 expression in BALF from BLM-injured mice, thereby inhibiting collagen deposition and fibrosis progression [415].
In summary, these data support the clinical translation of targeting CCR2 signaling to alleviate fibrosis in patients. To maximize therapeutic efficacy, further clinical trials are needed to provide more beneficial databases to elucidate the efficacy of targeting CCR2.
6.5 Systematic comparison of different CCR2/CCL2 axis targeting strategies
Regarding oral bioavailability, small-molecule antagonists represent the only strategy with viable oral administration potential. INCB3344 demonstrated 47% oral bioavailability in mice [380]. In contrast, monoclonal antibodies, gene therapies, and MSCs therapies lack oral feasibility and require intravenous or local injection. Notably, oral bioavailability varies significantly among herbal formulations. Most active constituents, such as polysaccharides, flavonoids, and alkaloids, exhibit poor water solubility and generally low absorption rates.
Regarding target specificity and off-target risks, small-molecule antagonists exhibit high target specificity. INCB3344 demonstrates over 100-fold greater selectivity for CCR2 compared to highly homologous receptors such as CCR1 and CCR5. While off-target risks exist, they are generally manageable. Monoclonal antibodies exhibit exceptional target specificity, with anti-CCL2 antibodies demonstrating high antigenic epitope selectivity and low off-target risk. Natural herbal medicines exhibit the weakest targeting specificity. Their inhibition of the CCR2/CCL2 axis is an indirect effect, primarily achieved by modulating multiple signaling pathways including NF-κB, MAPK, and PI3K, with no evidence of direct binding to the target. Consequently, natural compound formulations carry a higher off-target risk. Gene therapy achieves highly specific gene silencing through siRNA sequence design. MSCs exert immunomodulatory effects via multi-targeted, non-specific mechanisms. Its off-target risk is moderate, as its distribution, survival, and differentiation within the body are difficult to precisely control. Allogeneic cells also carry the risk of immune rejection.
Clinical validation of small-molecule antagonists remains limited. CVC has demonstrated good safety and tolerability in patients with liver cirrhosis, with no adverse effects on body weight, liver/kidney weight, or liver function [364]. However, in a 24-week study evaluating CVC safety in patients with primary sclerosing cholangitis, 20 participants (83.3%) reported at least one treatment-emergent adverse event, including fatigue, rash, and dizziness, with most events classified as mild or moderate [355]. Although no serious adverse events were observed, data on the long-term hepatic and renal burden and drug interactions associated with oral administration of small-molecule antagonists require further experimental investigation. For monoclonal antibodies, the inability to achieve sustained, complete target coverage may paradoxically exacerbate inflammation through compensatory overexpression. Therefore, heightened vigilance is warranted regarding the risks of immunogenicity and compensatory activation with monoclonal antibodies. Traditional Chinese herbal formulations possess extensive historical usage experience but lack robust evidence-based medical support. Some herbs exhibit long-term hepatotoxicity and nephrotoxicity, with safety data insufficient for modern toxicological evaluation. Regarding gene therapy, although anti-CCL2 gene therapy has shown short-term efficacy in liver, kidney and joint fibrosis, its long-term safety has not been systematically evaluated. Mesenchymal stem cells have a low survival rate and poor phenotypic stability in inflammatory microenvironments, and there are potential risks such as tumorigenicity and abnormal differentiation caused by gene-modified cells. Therefore, the long-term safety of mesenchymal cell therapy also needs to be studied.
Small-molecule antagonists are relatively low-cost with well-established chemical synthesis processes. Monoclonal antibodies, however, incur higher costs due to complex production processes and stringent quality control requirements. Natural Chinese herbal medicines have lower costs, but establishing systems for extracting, purifying, and quality controlling active ingredients significantly increases expenses. Gene therapy is extremely costly, primarily due to high expenditures in viral vector production, purification, and quality control. MSCs therapy is also highly expensive, as the autologous cell preparation process is time-consuming and poses significant challenges in quality control.
Comparative analysis reveals that small-molecule antagonists offer a viable therapeutic strategy for fibrotic diseases due to their favorable oral bioavailability. However, their clinical translation faces significant barriers primarily due to target limitations. While monoclonal antibodies exhibit high specificity, the clinical failure of carlumab starkly highlights the systemic limitations of pure ligand-neutralization strategies when confronted with risks of compensatory activation. Traditional Chinese herbal medicines require identification of active components and key targets to serve as precise interventions for fibrosis treatment. Gene therapy and mesenchymal stem cell therapy offer advantages in locally inhibiting CCR2 signaling and mitigating compensatory risks, yet clinical translation remains challenged by delivery efficiency, long-term safety, and high costs.
7. Summary and outlook
As a key cytokine, CCR2 is considered a pivotal node in the progression of fibrotic diseases. CCR2-mediated fibrosis involves a series of cellular or molecular signaling pathways, forming a complex regulatory network with multiple levels and pathways. Extensive experimental data support that CCR2 signaling participates in nearly the entire process of fibrosis formation, being closely associated with inflammatory monocyte recruitment, macrophage polarization, fibroblast production and activation, and ECM deposition [208, 288, 290, 295]. Notably, CCR2 signaling may exert detrimental or beneficial effects at different stages of fibrosis [238, 263]. CCR2 not only promotes early fibrotic injury but also facilitates late-stage fibrosis resolution by enhancing macrophage MMPs secretion for ECM degradation and maintaining immunosuppressive T cell subsets, including Tregs and CCR2+CD4+ T cells [196, 238, 263]. In this context, blocking CCR2 alleviates early fibrotic injury but also delays fibrosis resolution [238-240]. This poses challenges for targeting CCR2 signaling in fibrotic disease therapies. We must not only inhibit the capacity of CCR2 signaling to amplify inflammation and fibrotic injury but also preserve matrix degradation and T cell immunosuppression.
Therefore, when targeting CCR2 signaling to treat fibrotic diseases, it is crucial to identify the stage of disease progression and select the optimal timing for targeted intervention. Prioritizing CCR2 antagonists during the fibrogenesis phase has demonstrated efficacy in multiple preclinical studies and clinical trials [365, 373, 379]. During the regression phase, combining MMP activators and immunomodulators enables precise regulation. Combined application of CVC and MMP1 has been demonstrated to reverse liver inflammation and fibrosis in vivo while reducing adverse reactions [416]. Furthermore, adoptive transfer of CD4⁺CD25⁺FoxP3⁺ Tregs alleviates BLM-induced pulmonary fibrosis in mice, accompanied by decreased CCL2 production and reduced circulating fibroblasts accumulation [417].
Currently, clinical trials targeting the CCL2/CCR2 axis have demonstrated promising anti-fibrotic effects, with most participants reporting significant relief of fibrotic symptoms [355, 365]. However, the failure of the CVC and carlumab clinical trials profoundly highlights the complex challenges encountered during the translation from basic research to clinical application. Based on the above analysis, we attribute the failures to four primary factors: (1) Limitations in target biology. The Phase III failure of CVC indicates that merely blocking CCR2⁺ monocyte infiltration is insufficient to reverse fibrosis driven by heterogeneous populations such as TREM2⁺ scar-associated macrophages [368, 369]. Moreover, excessive blockade of CCR2 signaling may also interfere with the recruitment of reparative macrophages during the late stages of liver fibrosis, thereby impairing the liver own capacity for fibrosis regression. (2) Redundancy and compensatory activation of signaling pathways. This is the primary reason anti-CCR2 therapies prove completely ineffective or even harmful in certain scenarios. The inherent redundancy of chemokine networks makes neutralizing a single ligand highly prone to triggering compensatory responses. For example, in FITC-induced pulmonary fibrosis mice, systemic CCL2 blockade failed to inhibit fibroblast recruitment, attributed to compensatory effects from other CCR2 ligands [418]. This aligns with the Phase II clinical trial of the CCL2 neutralizing antibody carlumab, which failed to improve IPF and demonstrated compensatory increases in serum free CCL2 levels [385]. Similarly, complete CCL12 knockout failed to protect BLM-injured mice from pulmonary fibrosis, whereas lung epithelial cell-specific CCL12 knockout did [15]. One explanation is that complete CCL12 deficiency leads to significant compensatory increases in other CCR2 ligands, such as CCL2 and CCL7, whereas CCL12-specific deletion attenuates this compensatory response while reducing the accumulation of pro-fibrotic macrophages [15]. A more specific case comes from Gurczynski et al. In the context of γ-herpesvirus infection following bone marrow transplantation, CCR2 deficiency not only failed to confer benefit but exacerbated pulmonary fibrosis [419]. The mechanism involves impaired recruitment of classical monocytes due to lost CCR2 signaling, but the body activates an alternative pathway. Massive infiltration of αGR1-resistant MHC II+ neutrophils that secrete IL-17, directly promoting fibroblast activation and ECM deposition [420]. (3) Limitations of disease models. The anti-fibrotic effects observed in preclinical experiments failed to replicate in humans, exposing the limitations of traditional rodent models in simulating human disease. (4) Patient Heterogeneity. As a highly heterogeneous progressive disease, IPF demonstrated in Phase II clinical trial of carlumab that patients with higher disease severity or metabolic inflammation may respond less favorably to the therapy. This outcome was attributed to the lack of detailed clinical stratification based on underlying conditions and metabolic inflammatory profiles.
The aforementioned clinical translation failures provide crucial insights for CCR2-targeting strategies. First, drug design must address redundancy. Simply neutralizing the CCL2 ligand may fail to achieve sustained, complete signal blockade, potentially exacerbating chemokine-driven processes through feedback activation. Therefore, to maximize therapeutic efficacy, we should consider multi-targeted combined inhibition of CCR2 signaling to circumvent compensatory mechanisms. Combined siRNA targeting Sparc, CCR2, and Smad3 demonstrated favorable anti-fibrotic effects in BLM-induced mice, with suppressed activation of both macrophages and fibroblasts [421]. Concurrently, future trials must prioritize precise patient stratification to mitigate impacts on therapeutic efficacy.
In summary, CCR2 signaling serves as a key driver of fibrosis, contributing to the progression of fibrosis in multiple organs and tissues. In the future, gaining a deeper understanding of the spatiotemporal regulatory mechanisms of CCR2 signaling will facilitate the development of more precise therapeutic strategies for fibrosis, enabling effective interventions that shift from inhibiting fibrosis progression to promoting its regression. Specifically, research should focus on the following key directions: (1) Precisely deciphering the functional heterogeneity of CCR2+ cells within the fibrotic microenvironment. It is recommended to combine high-throughput, high-dimensional techniques such as single-cell sequencing, spatial transcriptomics, and organoid models to systematically map the molecular characteristics and spatial distribution of CCR2+ cells across different stages of fibrosis. This will clarify the dynamic evolutionary relationship between fibrosis-promoting subpopulations (CCR2+Ly6Chigh macrophages) and reparative subpopulations. (2) Explore the potential application of CCR2/CCL2 as biomarkers for patient stratification and personalized therapy. Integrate clinical information with molecular phenotypes to establish a CCR2 axis-based activity-stratification system, providing evidence for guiding targeted drug administration. (3) Develop organ-specific targeted delivery systems to enhance tissue selectivity in CCR2 signaling intervention. Particularly in diseases like liver fibrosis and pulmonary fibrosis, prioritize designing organ-targeted nanocarriers or fusion proteins to achieve efficient local inhibition of fibrosis progression while minimizing systemic side effects such as immunosuppression.
Abbreviations
AECs: alveolar epithelial cells; ADGRF5: adhesion G-protein coupled receptor F5; AKT: protein kinase B; AP-1: activating protein-1; Ang-II: angiotensin II; AS-IV: astragalus saponin IV; α-SMA: α-smooth muscle actin; ASH1L: ASH1-like histone lysine methyltransferase; BALF: bronchoalveolar lavage fluid; CCL2/MCP-1: monocyte chemotactic protein-1; CCR2: C-C motif chemokine receptor 2; CCR2 -/-: CCR2 knockout; CFs: cardiac fibroblasts; CHF: congestive heart failure; CKD: chronic kidney disease; CREB: cyclic AMP response element-binding protein; cRM: cardiac resident macrophages; CVC: cenicriviroc; Cytl1: cytokine-like 1; DMD: Duchenne muscular dystrophy; DN: diabetic nephropathy; DSS: dextran sulfate sodium; EAO: Experimental autoimmune orchitis; ECs: endothelial cells; ECM: extracellular matrix; EMT: epithelial-mesenchymal transition; ERK: extracellular-signal-regulated kinase; ESRD: end-stage renal disease; Ezh2: enhancer of Zeste homolog 2; ET: essential thrombocythemia; FITC: fluorescein isothiocyanate; FOXF1: forkhead box F1; GM-CSF: granulocyte-macrophage colony-stimulating factor; GPCRs: G protein-coupled receptors; HCV: hepatitis C virus; HIF-1α: hypoxia-inducible factor-1α; H3K4me3: Histone H3 lysine 27 trimethylation; HSCs: hepatic stellate cells; IFN-γ: interferon-γ; IL-1β: interleukin-1β; ILD: interstitial lung disease; IPF: idiopathic pulmonary fibrosis; I/R: ischemia-reperfusion injury; JAK: janus kinase; JNK: c-Jun N-terminal kinase; LPS: lipopolysaccharide; LRRK2: leucine-rich repeat kinase 2; MAPK: mitogen-activated protein kinase; MDMs: monocyte-derived macrophages; MEK: mitogen-activated protein kinase kinase; MI: myocardial infarction; MMPs: matrix metalloproteinases; MPN: myeloproliferative neoplasms; MSMP: microseminoprotein; mTOR: mammalian target of rapamycin; NADPH: nicotinamide adenine dinucleotide phosphate; NAFLD: non-alcoholic fatty liver disease; NASH: non-alcoholic steatohepatitis; NETs: neutrophil extracellular traps; NFAT: activator nuclear factor of activated T-cells; NF-κB: p65 subunit of nuclear factor-κB; NK: natural killer; PGE2: prostaglandin E2; PH: pulmonary hypertension; PIP2: phosphatidylinositol bisphosphate 2; PI3K: phosphoinositide 3-kinase; PKC: protein kinase C; PSCs: pancreatic stellate cells; PSMP: PC3-secreted microprotein; PV: polycythemia vera; SCF: stem cell factor; scRNA-seq: single-cell RNA sequencing; SNPs: single nucleotide polymorphisms; Sp1: specific protein 1; SSc: systemic sclerosis; STAT: signal transducer and activator of transcription; TIMP-1: tissue inhibitor of metalloproteinase 1; TMAO: trimethylamine N-oxide; TNF-α: tumor necrosis factor α; Tregs: regulatory T cells; Tsk-1: tight skin 1; TSLP: thymic stromal lymphopoietin; 3'UTRs: 3' untranslated regions; UUO: unilateral ureteral obstruction; WT: wild-type.
Acknowledgements
Funding
This work was supported by the National Natural Science Foundation of China (82572480, 82370273, 82270526, and 82070264), The Talent Project of Air Force Military Medical University (2023JSYX23), Xijing Hospital's Promotion Plan (XJZT25CX49), The Innovation Capability Support Plan Project in Shaanxi Province (2024CX-GXPT-01), Natural Science Foundation of Shaanxi Province (2025SYS-SYSZD-087), Key Medical Research Projects in Xi'an City (24YXYJ0010), Research Plan Project of Shaanxi Institute of Basic Science (22JHQ053), Shaanxi Province Traditional Chinese Medicine Research and Innovation Talent Project (TZKN-CXRC-06), Qinchuangyuan Traditional Chinese Medicine Innovation Research and Development Transformation Project (2022-QCYZH-036).
Author contributions
G.Z.S., J.X.W., and J.Y.T. contributed equally to this work. Y.Y. and J.C.L. supervised the project and evaluated the results. G.Z.S., J.X.W., J.Y.T., M.Z.C., Z.Z., H.D.D., Z.X.J., and X.Z.X. collected and interpreted the data. Z.Z. provided some materials and technical help. All authors reviewed and approved the manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Wynn TA. Cellular and molecular mechanisms of fibrosis. J Pathol. 2008;214:199-210
2. Wang Q, Wang P, Qin Z, Yang X, Pan B, Nie F. et al. Altered glucose metabolism and cell function in keloid fibroblasts under hypoxia. Redox Biol. 2021;38:101815
3. Ackermann M, Verleden SE, Kuehnel M, Haverich A, Welte T, Laenger F. et al. Pulmonary Vascular Endothelialitis, Thrombosis, and Angiogenesis in Covid-19. N Engl J Med. 2020;383:120-8
4. Kisseleva T, Cong M, Paik Y, Scholten D, Jiang C, Benner C. et al. Myofibroblasts revert to an inactive phenotype during regression of liver fibrosis. Proc Natl Acad Sci U S A. 2012;109:9448-53
5. Travers JG, Kamal FA, Robbins J, Yutzey KE, Blaxall BC. Cardiac Fibrosis: The Fibroblast Awakens. Circ Res. 2016;118:1021-40
6. Zhu S, Chen X, Wang JN, Xu JJ, Wang A, Li JJ. et al. Circular RNA circUbe2k promotes hepatic fibrosis via sponging miR-149-5p/TGF-β2 axis. Faseb j. 2021;35:e21622
7. Hutchinson J, Fogarty A, Hubbard R, McKeever T. Global incidence and mortality of idiopathic pulmonary fibrosis: a systematic review. Eur Respir J. 2015;46:795-806
8. Czubryt MP, Hale TM. Cardiac fibrosis: Pathobiology and therapeutic targets. Cell Signal. 2021;85:110066
9. Li H, Dixon EE, Wu H, Humphreys BD. Comprehensive single-cell transcriptional profiling defines shared and unique epithelial injury responses during kidney fibrosis. Cell Metab. 2022;34:1977-98.e9
10. Richeldi L, Collard HR, Jones MG. Idiopathic pulmonary fibrosis. Lancet. 2017;389:1941-52
11. Savin IA, Zenkova MA, Sen'kova AV. Pulmonary Fibrosis as a Result of Acute Lung Inflammation: Molecular Mechanisms, Relevant In Vivo Models, Prognostic and Therapeutic Approaches. Int J Mol Sci. 2022 23
12. Hammerich L, Tacke F. Hepatic inflammatory responses in liver fibrosis. Nat Rev Gastroenterol Hepatol. 2023;20:633-46
13. Haudek SB, Xia Y, Huebener P, Lee JM, Carlson S, Crawford JR. et al. Bone marrow-derived fibroblast precursors mediate ischemic cardiomyopathy in mice. Proc Natl Acad Sci U S A. 2006;103:18284-9
14. Moore BB, Kolodsick JE, Thannickal VJ, Cooke K, Moore TA, Hogaboam C. et al. CCR2-mediated recruitment of fibrocytes to the alveolar space after fibrotic injury. Am J Pathol. 2005;166:675-84
15. Yang J, Agarwal M, Ling S, Teitz-Tennenbaum S, Zemans RL, Osterholzer JJ. et al. Diverse Injury Pathways Induce Alveolar Epithelial Cell CCL2/12, Which Promotes Lung Fibrosis. Am J Respir Cell Mol Biol. 2020;62:622-32
16. Wang XM, Hamza M, Wu TX, Dionne RA. Upregulation of IL-6, IL-8 and CCL2 gene expression after acute inflammation: Correlation to clinical pain. Pain. 2009;142:275-83
17. Raghu H, Lepus CM, Wang Q, Wong HH, Lingampalli N, Oliviero F. et al. CCL2/CCR2, but not CCL5/CCR5, mediates monocyte recruitment, inflammation and cartilage destruction in osteoarthritis. Ann Rheum Dis. 2017;76:914-22
18. Tan X, Hu L, Shu Z, Chen L, Li X, Du M. et al. Role of CCR2 in the Development of Streptozotocin-Treated Diabetic Cardiomyopathy. Diabetes. 2019;68:2063-73
19. Xu J, Ganguly A, Zhao J, Ivey M, Lopez R, Osterholzer JJ. et al. CCR2 Signaling Promotes Brain Infiltration of Inflammatory Monocytes and Contributes to Neuropathology during Cryptococcal Meningoencephalitis. mBio. 2021;12:e0107621
20. Kohno H, Koso H, Okano K, Sundermeier TR, Saito S, Watanabe S. et al. Expression pattern of Ccr2 and Cx3cr1 in inherited retinal degeneration. J Neuroinflammation. 2015;12:188
21. Christensen PJ, Du M, Moore B, Morris S, Toews GB, Paine R 3rd. Expression and functional implications of CCR2 expression on murine alveolar epithelial cells. Am J Physiol Lung Cell Mol Physiol. 2004;286:L68-72
22. Sebastiani G, Ventriglia G, Stabilini A, Socci C, Morsiani C, Laurenzi A. et al. Regulatory T-cells from pancreatic lymphnodes of patients with type-1 diabetes express increased levels of microRNA miR-125a-5p that limits CCR2 expression. Sci Rep. 2017;7:6897
23. Penton-Rol G, Polentarutti N, Luini W, Borsatti A, Mancinelli R, Sica A. et al. Selective inhibition of expression of the chemokine receptor CCR2 in human monocytes by IFN-gamma. J Immunol. 1998;160:3869-73
24. Park HK, Na YH, Nguyen HT, Nguyen LP, Hurh S, Seong JY. et al. Analysis of CCR2 splice variant expression patterns and functional properties. Cell Biosci. 2022;12:59
25. Charo IF, Myers SJ, Herman A, Franci C, Connolly AJ, Coughlin SR. Molecular cloning and functional expression of two monocyte chemoattractant protein 1 receptors reveals alternative splicing of the carboxyl-terminal tails. Proc Natl Acad Sci U S A. 1994;91:2752-6
26. Shao Z, Tan Y, Shen Q, Hou L, Yao B, Qin J. et al. Molecular insights into ligand recognition and activation of chemokine receptors CCR2 and CCR3. Cell Discov. 2022;8:44
27. Yoshimura T, Robinson EA, Tanaka S, Appella E, Kuratsu J, Leonard EJ. Purification and amino acid analysis of two human glioma-derived monocyte chemoattractants. J Exp Med. 1989;169:1449-59
28. Van Damme J, Proost P, Lenaerts JP, Opdenakker G. Structural and functional identification of two human, tumor-derived monocyte chemotactic proteins (MCP-2 and MCP-3) belonging to the chemokine family. J Exp Med. 1992;176:59-65
29. Garcia-Zepeda EA, Combadiere C, Rothenberg ME, Sarafi MN, Lavigne F, Hamid Q. et al. Human monocyte chemoattractant protein (MCP)-4 is a novel CC chemokine with activities on monocytes, eosinophils, and basophils induced in allergic and nonallergic inflammation that signals through the CC chemokine receptors (CCR)-2 and -3. J Immunol. 1996;157:5613-26
30. Shin H, Prasad V, Lupancu T, Malik S, Achuthan A, Biondo M. et al. The GM-CSF/CCL17 pathway in obesity-associated osteoarthritic pain and disease in mice. Osteoarthritis Cartilage. 2023;31:1327-41
31. Sarafi MN, Garcia-Zepeda EA, MacLean JA, Charo IF, Luster AD. Murine monocyte chemoattractant protein (MCP)-5: a novel CC chemokine that is a structural and functional homologue of human MCP-1. J Exp Med. 1997;185:99-109
32. Wang X, Li T, Wang W, Yuan W, Liu H, Cheng Y. et al. Cytokine-like 1 Chemoattracts Monocytes/Macrophages via CCR2. J Immunol. 2016;196:4090-9
33. Valtonen-André C, Bjartell A, Hellsten R, Lilja H, Härkönen P, Lundwall A. A highly conserved protein secreted by the prostate cancer cell line PC-3 is expressed in benign and malignant prostate tissue. Biol Chem. 2007;388:289-95
34. Pei X, Sun Q, Zhang Y, Wang P, Peng X, Guo C. et al. PC3-secreted microprotein is a novel chemoattractant protein and functions as a high-affinity ligand for CC chemokine receptor 2. J Immunol. 2014;192:1878-86
35. Hong KH, Ryu J, Han KH. Monocyte chemoattractant protein-1-induced angiogenesis is mediated by vascular endothelial growth factor-A. Blood. 2005;105:1405-7
36. Kudo-Saito C, Shirako H, Ohike M, Tsukamoto N, Kawakami Y. CCL2 is critical for immunosuppression to promote cancer metastasis. Clin Exp Metastasis. 2013;30:393-405
37. Frangogiannis NG, Dewald O, Xia Y, Ren G, Haudek S, Leucker T. et al. Critical role of monocyte chemoattractant protein-1/CC chemokine ligand 2 in the pathogenesis of ischemic cardiomyopathy. Circulation. 2007;115:584-92
38. Hüsing AM, Wulfmeyer VC, Gaedcke S, Fleig SV, Rong S, DeLuca D. et al. Myeloid CCR2 Promotes Atherosclerosis after AKI. J Am Soc Nephrol. 2022;33:1487-500
39. Thorp EB, Filipp M, Dima M, Tan C, Feinstein M, Popko B. et al. CCR2(+) monocytes promote white matter injury and cognitive dysfunction after myocardial infarction. Brain Behav Immun. 2024;119:818-35
40. Chiu BC, Freeman CM, Stolberg VR, Hu JS, Zeibecoglou K, Lu B. et al. Impaired lung dendritic cell activation in CCR2 knockout mice. Am J Pathol. 2004;165:1199-209
41. Yoshimura T, Robinson EA, Tanaka S, Appella E, Leonard EJ. Purification and amino acid analysis of two human monocyte chemoattractants produced by phytohemagglutinin-stimulated human blood mononuclear leukocytes. J Immunol. 1989;142:1956-62
42. Yoshimura T, Yuhki N, Moore SK, Appella E, Lerman MI, Leonard EJ. Human monocyte chemoattractant protein-1 (MCP-1). Full-length cDNA cloning, expression in mitogen-stimulated blood mononuclear leukocytes, and sequence similarity to mouse competence gene JE. FEBS Lett. 1989;244:487-93
43. Wang L, Zheng G, Wang P, Jia X. Unlocking the secrets of NPSLE: the role of dendritic cell-secreted CCL2 in blood-brain barrier disruption. Front Immunol. 2024;15:1343805
44. Popovic M, Laumonnier Y, Burysek L, Syrovets T, Simmet T. Thrombin-induced expression of endothelial CX3CL1 potentiates monocyte CCL2 production and transendothelial migration. J Leukoc Biol. 2008;84:215-23
45. Novoszel P, Holcmann M, Stulnig G, De Sa Fernandes C, Zyulina V, Borek I. et al. Psoriatic skin inflammation is promoted by c-Jun/AP-1-dependent CCL2 and IL-23 expression in dendritic cells. EMBO Mol Med. 2021;13:e12409
46. Messeha SS, Zarmouh NO, Antonie L, Soliman KFA. Sanguinarine Inhibition of TNF-α-Induced CCL2, IKBKE/NF-κB/ERK1/2 Signaling Pathway, and Cell Migration in Human Triple-Negative Breast Cancer Cells. Int J Mol Sci. 2022 23
47. Shen SC, Xu J, Cheng C, Xiang XJ, Hong BY, Zhang M. et al. Macrophages promote the transition from myocardial ischemia reperfusion injury to cardiac fibrosis in mice through GMCSF/CCL2/CCR2 and phenotype switching. Acta Pharmacol Sin. 2024;45:959-74
48. Zhang F, Zhou C, Wang X, Liu Y, Hou Y, Niu L. INHBA, transcriptionally activated by SPI1, facilitates gastric cancer progression by inducing macrophage recruitment and M2 polarization via activating the TGF-β signaling to increase CCL2. Pathol Res Pract. 2025;269:155920
49. Sierra-Filardi E, Nieto C, Domínguez-Soto A, Barroso R, Sánchez-Mateos P, Puig-Kroger A. et al. CCL2 shapes macrophage polarization by GM-CSF and M-CSF: identification of CCL2/CCR2-dependent gene expression profile. J Immunol. 2014;192:3858-67
50. Akhter N, Kochumon S, Hasan A, Wilson A, Nizam R, Al Madhoun A. et al. IFN-γ and LPS Induce Synergistic Expression of CCL2 in Monocytic Cells via H3K27 Acetylation. J Inflamm Res. 2022;15:4291-302
51. Lehmann MH, Masanetz S, Kramer S, Erfle V. HIV-1 Nef upregulates CCL2/MCP-1 expression in astrocytes in a myristoylation- and calmodulin-dependent manner. J Cell Sci. 2006;119:4520-30
52. Kruglov EA, Nathanson RA, Nguyen T, Dranoff JA. Secretion of MCP-1/CCL2 by bile duct epithelia induces myofibroblastic transdifferentiation of portal fibroblasts. Am J Physiol Gastrointest Liver Physiol. 2006;290:G765-71
53. Harrington JR. The role of MCP-1 in atherosclerosis. Stem Cells. 2000;18:65-6
54. Xue J, Zhang Y, Zhang J, Zhu Z, Lv Q, Su J. Astrocyte-derived CCL7 promotes microglia-mediated inflammation following traumatic brain injury. Int Immunopharmacol. 2021;99:107975
55. Inaba A, Tuong ZK, Riding AM, Mathews RJ, Martin JL, Saeb-Parsy K. et al. B Lymphocyte-Derived CCL7 Augments Neutrophil and Monocyte Recruitment, Exacerbating Acute Kidney Injury. J Immunol. 2020;205:1376-84
56. Qiu Y, Zeltzer S, Zhang Y, Wang F, Chen GH, Dayrit J. et al. Early induction of CCL7 downstream of TLR9 signaling promotes the development of robust immunity to cryptococcal infection. J Immunol. 2012;188:3940-8
57. Won Jun H, Kyung Lee H, Ho Na I, Jeong Lee S, Kim K, Park G. et al. The role of CCL2, CCL7, ICAM-1, and VCAM-1 in interaction of endothelial cells and natural killer cells. Int Immunopharmacol. 2022;113:109332
58. Vande Broek I, Asosingh K, Vanderkerken K, Straetmans N, Van Camp B, Van Riet I. Chemokine receptor CCR2 is expressed by human multiple myeloma cells and mediates migration to bone marrow stromal cell-produced monocyte chemotactic proteins MCP-1, -2 and -3. Br J Cancer. 2003;88:855-62
59. Zhou SL, Zhou ZJ, Hu ZQ, Huang XW, Wang Z, Chen EB. et al. Tumor-Associated Neutrophils Recruit Macrophages and T-Regulatory Cells to Promote Progression of Hepatocellular Carcinoma and Resistance to Sorafenib. Gastroenterology. 2016;150:1646-58.e17
60. Zouggari Y, Ait-Oufella H, Bonnin P, Simon T, Sage AP, Guérin C. et al. B lymphocytes trigger monocyte mobilization and impair heart function after acute myocardial infarction. Nat Med. 2013;19:1273-80
61. Wu Z, Bai X, Lu Z, Liu S, Jiang H. LINC01094/SPI1/CCL7 Axis Promotes Macrophage Accumulation in Lung Adenocarcinoma and Tumor Cell Dissemination. J Immunol Res. 2022;2022:6450721
62. Chen J, Shi S, Li X, Gao F, Zhu X, Feng R. et al. CCL7 promotes macrophage polarization and synovitis to exacerbate rheumatoid arthritis. iScience. 2025;28:112177
63. Ford J, Hughson A, Lim K, Bardina SV, Lu W, Charo IF. et al. CCL7 Is a Negative Regulator of Cutaneous Inflammation Following Leishmania major Infection. Front Immunol. 2018;9:3063
64. Chang TT, Li YZ, Mo HW, Chen C, Lin LY, Chang CC. et al. Inhibition of CCL7 improves endothelial dysfunction and vasculopathy in mouse models of diabetes mellitus. Sci Transl Med. 2024;16:eadn1507
65. Mei H, Dai X, Chen W, Luo R, Liu Y, Meng J. et al. Intervening CCL8-CCR8 Signaling Reduces Cutaneous Inflammation and Itch in Mice. J Invest Dermatol. 2025;145:1519-23.e4
66. Dulek DE, Newcomb DC, Goleniewska K, Cephus J, Zhou W, Reiss S. et al. Allergic airway inflammation decreases lung bacterial burden following acute Klebsiella pneumoniae infection in a neutrophil- and CCL8-dependent manner. Infect Immun. 2014;82:3723-39
67. Jia GQ, Gonzalo JA, Lloyd C, Kremer L, Lu L, Martinez AC. et al. Distinct expression and function of the novel mouse chemokine monocyte chemotactic protein-5 in lung allergic inflammation. J Exp Med. 1996;184:1939-51
68. Tsui P, Das A, Whitaker B, Tornetta M, Stowell N, Kesavan P. et al. Generation, characterization and biological activity of CCL2 (MCP-1/JE) and CCL12 (MCP-5) specific antibodies. Hum Antibodies. 2007;16:117-25
69. Liang H, Geng S, Wang Y, Fang Q, Xin Y, Li Y. Tumour-derived exosome SNHG17 induced by oestrogen contributes to ovarian cancer progression via the CCL13-CCR2-M2 macrophage axis. J Cell Mol Med. 2024;28:e18315
70. Du Y, Cai Y, Lv Y, Zhang L, Yang H, Liu Q. et al. Single-cell RNA sequencing unveils the communications between malignant T and myeloid cells contributing to tumor growth and immunosuppression in cutaneous T-cell lymphoma. Cancer Lett. 2022;551:215972
71. Iwamoto T, Okamoto H, Iikuni N, Takeuchi M, Toyama Y, Tomatsu T. et al. Monocyte chemoattractant protein-4 (MCP-4)/CCL13 is highly expressed in cartilage from patients with rheumatoid arthritis. Rheumatology (Oxford). 2006;45:421-4
72. Lamkhioued B, Garcia-Zepeda EA, Abi-Younes S, Nakamura H, Jedrzkiewicz S, Wagner L. et al. Monocyte chemoattractant protein (MCP)-4 expression in the airways of patients with asthma. Induction in epithelial cells and mononuclear cells by proinflammatory cytokines. Am J Respir Crit Care Med. 2000;162:723-32
73. Chakravorty SJ, Howie AJ, Girdlestone J, Gentle D, Savage CO. Potential role for monocyte chemotactic protein-4 (MCP-4) in monocyte/macrophage recruitment in acute renal inflammation. J Pathol. 2001;194:239-46
74. Nakamura H, Luster AD, Tateno H, Jedrzkiewicz S, Tamura G, Haley KJ. et al. IL-4 differentially regulates eotaxin and MCP-4 in lung epithelium and circulating mononuclear cells. Am J Physiol Lung Cell Mol Physiol. 2001;281:L1288-302
75. Küper C, Beck FX, Neuhofer W. NFAT5 contributes to osmolality-induced MCP-1 expression in mesothelial cells. Mediators Inflamm. 2012;2012:513015
76. Jiang Y, Wang Y, Ma P, An D, Zhao J, Liang S. et al. Myeloid-specific targeting of Notch ameliorates murine renal fibrosis via reduced infiltration and activation of bone marrow-derived macrophage. Protein Cell. 2019;10:196-210
77. Arefieva TI, Kukhtina NB, Antonova OA, Krasnikova TL. MCP-1-stimulated chemotaxis of monocytic and endothelial cells is dependent on activation of different signaling cascades. Cytokine. 2005;31:439-46
78. Jiménez-Sainz MC, Fast B, Mayor F Jr, Aragay AM. Signaling pathways for monocyte chemoattractant protein 1-mediated extracellular signal-regulated kinase activation. Mol Pharmacol. 2003;64:773-82
79. Yasui H, Kajiyama H, Tamauchi S, Suzuki S, Peng Y, Yoshikawa N. et al. CCL2 secreted from cancer-associated mesothelial cells promotes peritoneal metastasis of ovarian cancer cells through the P38-MAPK pathway. Clin Exp Metastasis. 2020;37:145-58
80. Chiu HY, Sun KH, Chen SY, Wang HH, Lee MY, Tsou YC. et al. Autocrine CCL2 promotes cell migration and invasion via PKC activation and tyrosine phosphorylation of paxillin in bladder cancer cells. Cytokine. 2012;59:423-32
81. Tang CH, Tsai CC. CCL2 increases MMP-9 expression and cell motility in human chondrosarcoma cells via the Ras/Raf/MEK/ERK/NF-κB signaling pathway. Biochem Pharmacol. 2012;83:335-44
82. Goto A, Komura S, Kato K, Maki R, Hirakawa A, Aoki H. et al. PI3K-Akt signalling regulates Scx-lineage tenocytes and Tppp3-lineage paratenon sheath cells in neonatal tendon regeneration. Nat Commun. 2025;16:3734
83. Haston S, Pozzi S, Carreno G, Manshaei S, Panousopoulos L, Gonzalez-Meljem JM. et al. MAPK pathway control of stem cell proliferation and differentiation in the embryonic pituitary provides insights into the pathogenesis of papillary craniopharyngioma. Development. 2017;144:2141-52
84. Yao M, Fang W, Smart C, Hu Q, Huang S, Alvarez N. et al. CCR2 Chemokine Receptors Enhance Growth and Cell-Cycle Progression of Breast Cancer Cells through SRC and PKC Activation. Mol Cancer Res. 2019;17:604-17
85. Roca H, Varsos Z, Pienta KJ. CCL2 protects prostate cancer PC3 cells from autophagic death via phosphatidylinositol 3-kinase/AKT-dependent survivin up-regulation. J Biol Chem. 2008;283:25057-73
86. Fang S, Tang H, Li MZ, Chu JJ, Yin ZS, Jia QY. Identification of the CCL2 PI3K/Akt axis involved in autophagy and apoptosis after spinal cord injury. Metab Brain Dis. 2023;38:1335-49
87. Amsellem V, Abid S, Poupel L, Parpaleix A, Rodero M, Gary-Bobo G. et al. Roles for the CX3CL1/CX3CR1 and CCL2/CCR2 Chemokine Systems in Hypoxic Pulmonary Hypertension. Am J Respir Cell Mol Biol. 2017;56:597-608
88. Ye Q, Jo J, Wang CY, Oh H, Zhan J, Choy TJ. et al. Astrocytic Slc4a4 regulates blood-brain barrier integrity in healthy and stroke brains via a CCL2-CCR2 pathway and NO dysregulation. Cell Rep. 2024;43:114193
89. Jiang Y, He R, Jiang Y, Liu D, Tao L, Yang M. et al. Transcription factor NFAT5 contributes to the glycolytic phenotype rewiring and pancreatic cancer progression via transcription of PGK1. Cell Death Dis. 2019;10:948
90. López-Rodríguez C, Aramburu J, Jin L, Rakeman AS, Michino M, Rao A. Bridging the NFAT and NF-kappaB families: NFAT5 dimerization regulates cytokine gene transcription in response to osmotic stress. Immunity. 2001;15:47-58
91. Fujiki T, Udono M, Kotake Y, Yamashita M, Shirahata S, Katakura Y. NFAT5 regulates transcription of the mouse telomerase reverse transcriptase gene. Exp Cell Res. 2010;316:3342-50
92. Hasler U. An example of functional interaction between NFAT5/TonEBP and nuclear factor-κB by hypertonic stress: aquaporin-2 transcription. Cell Cycle. 2011;10:364-5
93. Kojima R, Taniguchi H, Tsuzuki A, Nakamura K, Sakakura Y, Ito M. Hypertonicity-induced expression of monocyte chemoattractant protein-1 through a novel cis-acting element and MAPK signaling pathways. J Immunol. 2010;184:5253-62
94. Roth I, Leroy V, Kwon HM, Martin PY, Féraille E, Hasler U. Osmoprotective transcription factor NFAT5/TonEBP modulates nuclear factor-kappaB activity. Mol Biol Cell. 2010;21:3459-74
95. Nie Y, Zhai X, Li J, Sun A, Che H, Christman JW. et al. NFATc3 Promotes Pulmonary Inflammation and Fibrosis by Regulating Production of CCL2 and CXCL2 in Macrophages. Aging Dis. 2023;14:1441-57
96. Deng X, Xu M, Yuan C, Yin L, Chen X, Zhou X. et al. Transcriptional regulation of increased CCL2 expression in pulmonary fibrosis involves nuclear factor-κB and activator protein-1. Int J Biochem Cell Biol. 2013;45:1366-76
97. Martin T, Cardarelli PM, Parry GC, Felts KA, Cobb RR. Cytokine induction of monocyte chemoattractant protein-1 gene expression in human endothelial cells depends on the cooperative action of NF-kappa B and AP-1. Eur J Immunol. 1997;27:1091-7
98. Schwabe RF, Schnabl B, Kweon YO, Brenner DA. CD40 activates NF-kappa B and c-Jun N-terminal kinase and enhances chemokine secretion on activated human hepatic stellate cells. J Immunol. 2001;166:6812-9
99. Zhuang L, Zong X, Yang Q, Fan Q, Tao R. Interleukin-34-NF-κB signaling aggravates myocardial ischemic/reperfusion injury by facilitating macrophage recruitment and polarization. EBioMedicine. 2023;95:104744
100. Paish HL, Kalson NS, Smith GR, Del Carpio Pons A, Baldock TE, Smith N. et al. Fibroblasts Promote Inflammation and Pain via IL-1α Induction of the Monocyte Chemoattractant Chemokine (C-C Motif) Ligand 2. Am J Pathol. 2018;188:696-714
101. Lee SK, Kim BS, Yang WS, Kim SB, Park SK, Park JS. High glucose induces MCP-1 expression partly via tyrosine kinase-AP-1 pathway in peritoneal mesothelial cells. Kidney Int. 2001;60:55-64
102. Kim YS, Kim JS, Kwon JS, Jeong MH, Cho JG, Park JC. et al. BAY 11-7082, a nuclear factor-κB inhibitor, reduces inflammation and apoptosis in a rat cardiac ischemia-reperfusion injury model. Int Heart J. 2010;51:348-53
103. Sarma NJ, Tiriveedhi V, Crippin JS, Chapman WC, Mohanakumar T. Hepatitis C virus-induced changes in microRNA 107 (miRNA-107) and miRNA-449a modulate CCL2 by targeting the interleukin-6 receptor complex in hepatitis. J Virol. 2014;88:3733-43
104. Kii S, Kitamura H, Hashimoto S, Ikeo K, Ichikawa N, Yoshida T. et al. STAT1-mediated induction of Ly6c-expressing macrophages are involved in the pathogenesis of an acute colitis model. Inflamm Res. 2022;71:1079-94
105. Huang J, Puente H, Wareing NE, Wu M, Mayes MD, Karmouty-Quintana H. et al. STAT6 suppression prevents bleomycin-induced dermal fibrosis. Faseb j. 2023;37:e22761
106. Potz BA, Sabe AA, Sabe SA, Lawandy IJ, Abid MR, Clements RT. et al. Calpain inhibition decreases myocardial fibrosis in chronically ischemic hypercholesterolemic swine. J Thorac Cardiovasc Surg. 2022;163:e11-e27
107. Pattison MJ, MacKenzie KF, Elcombe SE, Arthur JS. IFNβ autocrine feedback is required to sustain TLR induced production of MCP-1 in macrophages. FEBS Lett. 2013;587:1496-503
108. Guo R, Han D, Song X, Gao Y, Li Z, Li X. et al. Context-dependent regulation of Notch signaling in glial development and tumorigenesis. Sci Adv. 2023;9:eadi2167
109. Kang J, Postigo-Fernandez J, Kim K, Zhu C, Yu J, Meroni M. et al. Notch-mediated hepatocyte MCP-1 secretion causes liver fibrosis. JCI Insight. 2023 8
110. Brandt S, Ballhause TM, Bernhardt A, Becker A, Salaru D, Le-Deffge HM. et al. Fibrosis and Immune Cell Infiltration Are Separate Events Regulated by Cell-Specific Receptor Notch3 Expression. J Am Soc Nephrol. 2020;31:2589-608
111. Yadahalli R, Sarode GS, Sarode SC, Khan ZA, Vyas N, Kharat AH. et al. CC group of chemokines and associated gene expression of transcription factors: Deciphering immuno-pathogenetic aspect of oral submucous fibrosis. Dis Mon. 2023;69:101351
112. Christmann RB, Mathes A, Affandi AJ, Padilla C, Nazari B, Bujor AM. et al. Thymic stromal lymphopoietin is up-regulated in the skin of patients with systemic sclerosis and induces profibrotic genes and intracellular signaling that overlap with those induced by interleukin-13 and transforming growth factor β. Arthritis Rheum. 2013;65:1335-46
113. Zheng Y, Wang Z, Wei S, Liu Z, Chen G. Epigenetic silencing of chemokine CCL2 represses macrophage infiltration to potentiate tumor development in small cell lung cancer. Cancer Lett. 2021;499:148-63
114. Du Y, Wu S, Xi S, Xu W, Sun L, Yan J. et al. ASH1L in Hepatoma Cells and Hepatic Stellate Cells Promotes Fibrosis-Associated Hepatocellular Carcinoma by Modulating Tumor-Associated Macrophages. Adv Sci (Weinh). 2024;11:e2404756
115. Jia Y, Reddy MA, Das S, Oh HJ, Abdollahi M, Yuan H. et al. Dysregulation of histone H3 lysine 27 trimethylation in transforming growth factor-β1-induced gene expression in mesangial cells and diabetic kidney. J Biol Chem. 2019;294:12695-707
116. Raisner R, Kharbanda S, Jin L, Jeng E, Chan E, Merchant M. et al. Enhancer Activity Requires CBP/P300 Bromodomain-Dependent Histone H3K27 Acetylation. Cell Rep. 2018;24:1722-9
117. Gao J, Wei B, Liu M, Hirsova P, Sehrawat TS, Cao S. et al. Endothelial p300 Promotes Portal Hypertension and Hepatic Fibrosis Through C-C Motif Chemokine Ligand 2-Mediated Angiocrine Signaling. Hepatology. 2021;73:2468-83
118. Brown JD, Lin CY, Duan Q, Griffin G, Federation A, Paranal RM. et al. NF-κB directs dynamic super enhancer formation in inflammation and atherogenesis. Mol Cell. 2014;56:219-31
119. Ping D, Boekhoudt G, Zhang F, Morris A, Philipsen S, Warren ST. et al. Sp1 binding is critical for promoter assembly and activation of the MCP-1 gene by tumor necrosis factor. J Biol Chem. 2000;275:1708-14
120. Boekhoudt GH, Guo Z, Beresford GW, Boss JM. Communication between NF-kappa B and Sp1 controls histone acetylation within the proximal promoter of the monocyte chemoattractant protein 1 gene. J Immunol. 2003;170:4139-47
121. Golec P, Bernatowicz PL, Tokajuk G, Kozłowski M, Kowal K. Histone deacetylases affect transcriptional regulation of CCL2 and CXCL8 expression by pulmonary fibroblasts in vitro. Adv Respir Med. 2017;85:307-12
122. Li X, Yuan B, Lu M, Wang Y, Ding N, Liu C. et al. The methyltransferase METTL3 negatively regulates nonalcoholic steatohepatitis (NASH) progression. Nat Commun. 2021;12:7213
123. Anstee QM, Reeves HL, Kotsiliti E, Govaere O, Heikenwalder M. From NASH to HCC: current concepts and future challenges. Nat Rev Gastroenterol Hepatol. 2019;16:411-28
124. Miranda KC, Huynh T, Tay Y, Ang YS, Tam WL, Thomson AM. et al. A pattern-based method for the identification of MicroRNA binding sites and their corresponding heteroduplexes. Cell. 2006;126:1203-17
125. Asselah T, Bièche I, Laurendeau I, Paradis V, Vidaud D, Degott C. et al. Liver gene expression signature of mild fibrosis in patients with chronic hepatitis C. Gastroenterology. 2005;129:2064-75
126. Nabih HK. The Significance of HCV Viral Load in the Incidence of HCC: a Correlation Between Mir-122 and CCL2. J Gastrointest Cancer. 2020;51:412-7
127. Tang Y, Jia W, Niu X, Wu L, Shen H, Wang L. et al. CCL2 is Upregulated by Decreased miR-122 Expression in Iron-Overload-Induced Hepatic Inflammation. Cell Physiol Biochem. 2017;44:870-83
128. Chen JY, Ruan HJ, Chen SY, Wang XQ, Wen JM, Wang ZX. MiR-144-5p/CCL12 Signaling Axis Modulates Ischemic Preconditioning-Mediated Cardio-protection by Reducing Cell Viability, Enhancing Cell Apoptosis, Fibrosis, and Pyroptosis. Appl Biochem Biotechnol. 2023;195:1999-2014
129. Chouvarine P, Legchenko E, Geldner J, Riehle C, Hansmann G. Hypoxia drives cardiac miRNAs and inflammation in the right and left ventricle. J Mol Med (Berl). 2019;97:1427-38
130. Lan T, Li C, Yang G, Sun Y, Zhuang L, Ou Y. et al. Sphingosine kinase 1 promotes liver fibrosis by preventing miR-19b-3p-mediated inhibition of CCR2. Hepatology. 2018;68:1070-86
131. Klampfleuthner FAM, Lotz B, Renkawitz T, Richter W, Diederichs S. Stage-Dependent Activity and Pro-Chondrogenic Function of PI3K/AKT during Cartilage Neogenesis from Mesenchymal Stromal Cells. Cells. 2022 11
132. Gündüz D, Troidl C, Tanislav C, Rohrbach S, Hamm C, Aslam M. Role of PI3K/Akt and MEK/ERK Signalling in cAMP/Epac-Mediated Endothelial Barrier Stabilisation. Front Physiol. 2019;10:1387
133. Wang Q, Yang X, Ma J, Xie X, Sun Y, Chang X. et al. PI3K/AKT pathway promotes keloid fibroblasts proliferation by enhancing glycolysis under hypoxia. Wound Repair Regen. 2023;31:139-55
134. Zhao H, Li C, Li L, Liu J, Gao Y, Mu K. et al. Baicalin alleviates bleomycin-induced pulmonary fibrosis and fibroblast proliferation in rats via the PI3K/AKT signaling pathway. Mol Med Rep. 2020;21:2321-34
135. Dong Y, Dong Y, Zhu C, Yang L, Wang H, Li J. et al. Targeting CCL2-CCR2 signaling pathway alleviates macrophage dysfunction in COPD via PI3K-AKT axis. Cell Commun Signal. 2024;22:364
136. Mizutani K, Roca H, Varsos Z, Pienta KJ. Possible mechanism of CCL2-induced Akt activation in prostate cancer cells. Anticancer Res. 2009;29:3109-13
137. Guo YC, Zhang M, Wang FX, Pei GC, Sun F, Zhang Y. et al. Macrophages Regulate Unilateral Ureteral Obstruction-Induced Renal Lymphangiogenesis through C-C Motif Chemokine Receptor 2-Dependent Phosphatidylinositol 3-Kinase-AKT-Mechanistic Target of Rapamycin Signaling and Hypoxia-Inducible Factor-1α/Vascular Endothelial Growth Factor-C Expression. Am J Pathol. 2017;187:1736-49
138. Wu L, Lu Z, He B, Yu J, Yan M, Jiang J. et al. Pure total flavonoids from citrus improve nonalcoholic steatohepatitis liver inflammatory responses by regulating the CCL2/CCR2-PI3K-Akt signal transduction pathway. Anat Rec (Hoboken). 2023;306:3169-77
139. Xue Y, Zhu W, Qiao F, Yang Y, Qiu J, Zou C. et al. Ba-Qi-Rougan formula alleviates hepatic fibrosis by suppressing hepatic stellate cell activation via the MSMP/CCR2/PI3K pathway. J Ethnopharmacol. 2024;329:118169
140. Feng L, Chen C, Xiong X, Wang X, Li X, Kuang Q. et al. PS-MPs promotes the progression of inflammation and fibrosis in diabetic nephropathy through NLRP3/Caspase-1 and TGF-β1/Smad2/3 signaling pathways. Ecotoxicol Environ Saf. 2024;273:116102
141. Fang J, Shu S, Dong H, Yue X, Piao J, Li S. et al. Histone deacetylase 6 controls cardiac fibrosis and remodelling through the modulation of TGF-β1/Smad2/3 signalling in post-infarction mice. J Cell Mol Med. 2024;28:e70063
142. Derynck R, Zhang YE. Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature. 2003;425:577-84
143. Murray LA, Argentieri RL, Farrell FX, Bracht M, Sheng H, Whitaker B. et al. Hyper-responsiveness of IPF/UIP fibroblasts: interplay between TGFbeta1, IL-13 and CCL2. Int J Biochem Cell Biol. 2008;40:2174-82
144. Gharaee-Kermani M, Denholm EM, Phan SH. Costimulation of fibroblast collagen and transforming growth factor beta1 gene expression by monocyte chemoattractant protein-1 via specific receptors. J Biol Chem. 1996;271:17779-84
145. Gharaee-Kermani M, McCullumsmith RE, Charo IF, Kunkel SL, Phan SH. CC-chemokine receptor 2 required for bleomycin-induced pulmonary fibrosis. Cytokine. 2003;24:266-76
146. Qi W, Chen X, Polhill TS, Sumual S, Twigg S, Gilbert RE. et al. TGF-beta1 induces IL-8 and MCP-1 through a connective tissue growth factor-independent pathway. Am J Physiol Renal Physiol. 2006;290:F703-9
147. Yoshimura H, Nakahama K, Safronova O, Tanaka N, Muneta T, Morita I. Transforming growth factor-beta stimulates IL-1beta-induced monocyte chemoattractant protein-1 expression in human synovial cells via the ERK/AP-1 pathway. Inflamm Res. 2006;55:543-9
148. Wang X, Li X, Ye L, Chen W, Yu X. Smad7 inhibits TGF-β1-induced MCP-1 upregulation through a MAPK/p38 pathway in rat peritoneal mesothelial cells. Int Urol Nephrol. 2013;45:899-907
149. Kavsak P, Rasmussen RK, Causing CG, Bonni S, Zhu H, Thomsen GH. et al. Smad7 binds to Smurf2 to form an E3 ubiquitin ligase that targets the TGF beta receptor for degradation. Mol Cell. 2000;6:1365-75
150. Ong VH, Evans LA, Shiwen X, Fisher IB, Rajkumar V, Abraham DJ. et al. Monocyte chemoattractant protein 3 as a mediator of fibrosis: Overexpression in systemic sclerosis and the type 1 tight-skin mouse. Arthritis Rheum. 2003;48:1979-91
151. Ong VH, Carulli MT, Xu S, Khan K, Lindahl G, Abraham DJ. et al. Cross-talk between MCP-3 and TGFbeta promotes fibroblast collagen biosynthesis. Exp Cell Res. 2009;315:151-61
152. Zhang T, Liu Y, Yang T, Zhang L, Xu S, Xue L. et al. Diverse signals converge at MAPK cascades in plant. Plant Physiol Biochem. 2006;44:274-83
153. Wada T, Furuichi K, Sakai N, Hisada Y, Kobayashi K, Mukaida N. et al. Involvement of p38 mitogen-activated protein kinase followed by chemokine expression in crescentic glomerulonephritis. Am J Kidney Dis. 2001;38:1169-77
154. Rovin BH, Wilmer WA, Danne M, Dickerson JA, Dixon CL, Lu L. The mitogen-activated protein kinase p38 is necesssary for interleukin 1beta-induced monocyte chemoattractant protein 1 expression by human mesangial cells. Cytokine. 1999;11:118-26
155. Wang D, Warner GM, Yin P, Knudsen BE, Cheng J, Butters KA. et al. Inhibition of p38 MAPK attenuates renal atrophy and fibrosis in a murine renal artery stenosis model. Am J Physiol Renal Physiol. 2013;304:F938-47
156. Kokubo S, Sakai N, Furuichi K, Toyama T, Kitajima S, Okumura T. et al. Activation of p38 mitogen-activated protein kinase promotes peritoneal fibrosis by regulating fibrocytes. Perit Dial Int. 2012;32:10-9
157. Zhou X, Wang S, Jin G, Zhou K, Cai Y, Zhao Z. Protective effects of Shenkang injection against diabetic kidney disease via p38 MAPK/NFκB/MCP-1/CCR2 pathway inhibition. Front Endocrinol (Lausanne). 2025;16:1596000
158. Chang HW, Wu VC, Wu KD, Huang HY, Hsieh BS, Chen YM. In rat renal fibroblasts, mycophenolic acid inhibits proliferation and production of the chemokine CCL2, stimulated by tumour necrosis factor-alpha. Br J Pharmacol. 2010;160:1611-20
159. Liu X, Das AM, Seideman J, Griswold D, Afuh CN, Kobayashi T. et al. The CC chemokine ligand 2 (CCL2) mediates fibroblast survival through IL-6. Am J Respir Cell Mol Biol. 2007;37:121-8
160. Carson WFt, Salter-Green SE, Scola MM, Joshi A, Gallagher KA, Kunkel SL. Enhancement of macrophage inflammatory responses by CCL2 is correlated with increased miR-9 expression and downregulation of the ERK1/2 phosphatase Dusp6. Cell Immunol. 2017;314:63-72
161. Buckley CD, Midwood KS. Tracing the origins of lung fibrosis. Nat Immunol. 2024;25:1517-9
162. Martinez FJ, Collard HR, Pardo A, Raghu G, Richeldi L, Selman M. et al. Idiopathic pulmonary fibrosis. Nat Rev Dis Primers. 2017;3:17074
163. Karman J, Wang J, Bodea C, Cao S, Levesque MC. Lung gene expression and single cell analyses reveal two subsets of idiopathic pulmonary fibrosis (IPF) patients associated with different pathogenic mechanisms. PLoS One. 2021;16:e0248889
164. Fließer E, Jandl K, Lins T, Birnhuber A, Valzano F, Kolb D. et al. Lung Fibrosis Is Linked to Increased Endothelial Cell Activation and Dysfunctional Vascular Barrier Integrity. Am J Respir Cell Mol Biol. 2024;71:318-31
165. Zheng C, Zhang L, Sun Y, Ma Y, Zhang Y. Alveolar epithelial cell dysfunction and epithelial-mesenchymal transition in pulmonary fibrosis pathogenesis. Front Mol Biosci. 2025;12:1564176
166. Kubo F, Ariestanti DM, Oki S, Fukuzawa T, Demizu R, Sato T. et al. Loss of the adhesion G-protein coupled receptor ADGRF5 in mice induces airway inflammation and the expression of CCL2 in lung endothelial cells. Respir Res. 2019;20:11
167. Tian Y, Lv J, Su Z, Wu T, Li X, Hu X. et al. LRRK2 plays essential roles in maintaining lung homeostasis and preventing the development of pulmonary fibrosis. Proc Natl Acad Sci U S A. 2021 118
168. Bian F, Lan YW, Zhao S, Deng Z, Shukla S, Acharya A. et al. Lung endothelial cells regulate pulmonary fibrosis through FOXF1/R-Ras signaling. Nat Commun. 2023;14:2560
169. Moore BB, Peters-Golden M, Christensen PJ, Lama V, Kuziel WA, Paine R 3rd. et al. Alveolar epithelial cell inhibition of fibroblast proliferation is regulated by MCP-1/CCR2 and mediated by PGE2. Am J Physiol Lung Cell Mol Physiol. 2003;284:L342-9
170. Kolodsick JE, Peters-Golden M, Larios J, Toews GB, Thannickal VJ, Moore BB. Prostaglandin E2 inhibits fibroblast to myofibroblast transition via E. prostanoid receptor 2 signaling and cyclic adenosine monophosphate elevation. Am J Respir Cell Mol Biol. 2003;29:537-44
171. Zhao T, Wu X, Zhao X, Yao K, Li X, Ni J. Identification and validation of chemokine system-related genes in idiopathic pulmonary fibrosis. Front Immunol. 2023;14:1159856
172. Mercer PF, Johns RH, Scotton CJ, Krupiczojc MA, Königshoff M, Howell DC. et al. Pulmonary epithelium is a prominent source of proteinase-activated receptor-1-inducible CCL2 in pulmonary fibrosis. Am J Respir Crit Care Med. 2009;179:414-25
173. Groves AM, Johnston CJ, Williams JP, Finkelstein JN. Role of Infiltrating Monocytes in the Development of Radiation-Induced Pulmonary Fibrosis. Radiat Res. 2018;189:300-11
174. Öz HH, Cheng EC, Di Pietro C, Tebaldi T, Biancon G, Zeiss C. et al. Recruited monocytes/macrophages drive pulmonary neutrophilic inflammation and irreversible lung tissue remodeling in cystic fibrosis. Cell Rep. 2022;41:111797
175. Antoniades HN, Neville-Golden J, Galanopoulos T, Kradin RL, Valente AJ, Graves DT. Expression of monocyte chemoattractant protein 1 mRNA in human idiopathic pulmonary fibrosis. Proc Natl Acad Sci U S A. 1992;89:5371-5
176. Car BD, Meloni F, Luisetti M, Semenzato G, Gialdroni-Grassi G, Walz A. Elevated IL-8 and MCP-1 in the bronchoalveolar lavage fluid of patients with idiopathic pulmonary fibrosis and pulmonary sarcoidosis. Am J Respir Crit Care Med. 1994;149:655-9
177. David BA, Kubes P. Exploring the complex role of chemokines and chemoattractants in vivo on leukocyte dynamics. Immunol Rev. 2019;289:9-30
178. Kuziel WA, Morgan SJ, Dawson TC, Griffin S, Smithies O, Ley K. et al. Severe reduction in leukocyte adhesion and monocyte extravasation in mice deficient in CC chemokine receptor 2. Proc Natl Acad Sci U S A. 1997;94:12053-8
179. Bajpai G, Bredemeyer A, Li W, Zaitsev K, Koenig AL, Lokshina I. et al. Tissue Resident CCR2- and CCR2+ Cardiac Macrophages Differentially Orchestrate Monocyte Recruitment and Fate Specification Following Myocardial Injury. Circ Res. 2019;124:263-78
180. Osterholzer JJ, Olszewski MA, Murdock BJ, Chen GH, Erb-Downward JR, Subbotina N. et al. Implicating exudate macrophages and Ly-6C(high) monocytes in CCR2-dependent lung fibrosis following gene-targeted alveolar injury. J Immunol. 2013;190:3447-57
181. Schupp JC, Khanal S, Gomez JL, Sauler M, Adams TS, Chupp GL. et al. Single-Cell Transcriptional Archetypes of Airway Inflammation in Cystic Fibrosis. Am J Respir Crit Care Med. 2020;202:1419-29
182. Brennan S, Sly PD, Gangell CL, Sturges N, Winfield K, Wikstrom M. et al. Alveolar macrophages and CC chemokines are increased in children with cystic fibrosis. Eur Respir J. 2009;34:655-61
183. Qiu F, Miao H, Hui H, Qiu L, Chen Y, Luo M. et al. MHCII(hi)LYVE1(lo)CCR2(hi) Interstitial Macrophages Promote Medial Fibrosis in Pulmonary Arterioles and Contribute to Pulmonary Hypertension. Circ Res. 2025;137:46-66
184. Li C, Feng X, Li S, He X, Luo Z, Cheng X. et al. Tetrahedral DNA loaded siCCR2 restrains M1 macrophage polarization to ameliorate pulmonary fibrosis in chemoradiation-induced murine model. Mol Ther. 2024;32:766-82
185. Liu X, Zeng L, Zhou Y, Zhao X, Zhu L, Zhang J. et al. P21 facilitates macrophage chemotaxis by promoting CCL7 in the lung epithelial cell lines treated with radiation and bleomycin. J Transl Med. 2023;21:314
186. Chen B, Yang Y, Yang C, Duan J, Chen L, Lu K. et al. M2 macrophage accumulation contributes to pulmonary fibrosis, vascular dilatation, and hypoxemia in rat hepatopulmonary syndrome. J Cell Physiol. 2021;236:7682-97
187. Sun L, Louie MC, Vannella KM, Wilke CA, LeVine AM, Moore BB. et al. New concepts of IL-10-induced lung fibrosis: fibrocyte recruitment and M2 activation in a CCL2/CCR2 axis. Am J Physiol Lung Cell Mol Physiol. 2011;300:L341-53
188. Puukila S, Lawrence MD, De Pasquale CG, Bersten AD, Bihari S, McEvoy-May J. et al. Monocyte chemotactic protein (MCP)-1 (CCL2) and its receptor (CCR2) are elevated in chronic heart failure facilitating lung monocyte infiltration and differentiation which may contribute to lung fibrosis. Cytokine. 2023;161:156060
189. Shichino S, Abe J, Ueha S, Otsuji M, Tsukui T, Kosugi-Kanaya M. et al. Reduced supply of monocyte-derived macrophages leads to a transition from nodular to diffuse lesions and tissue cell activation in silica-induced pulmonary fibrosis in mice. Am J Pathol. 2015;185:2923-38
190. Liang J, Jung Y, Tighe RM, Xie T, Liu N, Leonard M. et al. A macrophage subpopulation recruited by CC chemokine ligand-2 clears apoptotic cells in noninfectious lung injury. Am J Physiol Lung Cell Mol Physiol. 2012;302:L933-40
191. Murray PJ, Allen JE, Biswas SK, Fisher EA, Gilroy DW, Goerdt S. et al. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity. 2014;41:14-20
192. Xue J, Schmidt SV, Sander J, Draffehn A, Krebs W, Quester I. et al. Transcriptome-based network analysis reveals a spectrum model of human macrophage activation. Immunity. 2014;40:274-88
193. Hadjicharalambous MR, Roux BT, Feghali-Bostwick CA, Murray LA, Clarke DL, Lindsay MA. Long Non-coding RNAs Are Central Regulators of the IL-1β-Induced Inflammatory Response in Normal and Idiopathic Pulmonary Lung Fibroblasts. Front Immunol. 2018;9:2906
194. Walsh SM, Worrell JC, Fabre A, Hinz B, Kane R, Keane MP. Novel differences in gene expression and functional capabilities of myofibroblast populations in idiopathic pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol. 2018;315:L697-l710
195. Neehus AL, Carey B, Landekic M, Panikulam P, Deutsch G, Ogishi M. et al. Human inherited CCR2 deficiency underlies progressive polycystic lung disease. Cell. 2024;187:3460
196. Milger K, Yu Y, Brudy E, Irmler M, Skapenko A, Mayinger M. et al. Pulmonary CCR2(+)CD4(+) T cells are immune regulatory and attenuate lung fibrosis development. Thorax. 2017;72:1007-20
197. Kahan T. The importance of myocardial fibrosis in hypertensive heart disease. J Hypertens. 2012;30:685-7
198. Iles L, Pfluger H, Phrommintikul A, Cherayath J, Aksit P, Gupta SN. et al. Evaluation of diffuse myocardial fibrosis in heart failure with cardiac magnetic resonance contrast-enhanced T1 mapping. J Am Coll Cardiol. 2008;52:1574-80
199. Dewald O, Zymek P, Winkelmann K, Koerting A, Ren G, Abou-Khamis T. et al. CCL2/Monocyte Chemoattractant Protein-1 regulates inflammatory responses critical to healing myocardial infarcts. Circ Res. 2005;96:881-9
200. Patel B, Bansal SS, Ismahil MA, Hamid T, Rokosh G, Mack M. et al. CCR2(+) Monocyte-Derived Infiltrating Macrophages Are Required for Adverse Cardiac Remodeling During Pressure Overload. JACC Basic Transl Sci. 2018;3:230-44
201. Koyanagi M, Egashira K, Kitamoto S, Ni W, Shimokawa H, Takeya M. et al. Role of monocyte chemoattractant protein-1 in cardiovascular remodeling induced by chronic blockade of nitric oxide synthesis. Circulation. 2000;102:2243-8
202. Kuwahara F, Kai H, Tokuda K, Takeya M, Takeshita A, Egashira K. et al. Hypertensive myocardial fibrosis and diastolic dysfunction: another model of inflammation? Hypertension. 2004;43:739-45
203. Revelo XS, Parthiban P, Chen C, Barrow F, Fredrickson G, Wang H. et al. Cardiac Resident Macrophages Prevent Fibrosis and Stimulate Angiogenesis. Circ Res. 2021;129:1086-101
204. Sakai N, Wada T, Furuichi K, Shimizu K, Kokubo S, Hara A. et al. MCP-1/CCR2-dependent loop for fibrogenesis in human peripheral CD14-positive monocytes. J Leukoc Biol. 2006;79:555-63
205. Yu WJ, Jiang WX, Liu SJ, Li HH, Lin QY. Single-cell RNA sequencing reveals that myeloid S100A8/A9 is a novel regulator of the transition from adaptive hypertrophy to heart failure after pressure overload. Theranostics. 2025;15:8587-608
206. Li Z, Chen K, Wu W. Right atrial CCR2(+) macrophages mediate atrial fibrillation in rat with monocrotaline-induced pulmonary arterial hypertension. Int Immunopharmacol. 2026;172:115920
207. Bai L, Li Y, Xue Y, Lu Z, Meng Z, Lu C. et al. Inhibition of Macrophage Recruitment to Heart Valves Mediated by the C-C Chemokine Receptor Type 2 Attenuates Valvular Inflammation Induced by Group A Streptococcus in Lewis Rats. Front Biosci (Landmark Ed). 2024;29:303
208. Chen L, Pan D, Zhang Y, Zhang E, Ma L. C-C Motif Chemokine 2 Regulates Macrophage Polarization and Contributes to Myocardial Infarction Healing. J Interferon Cytokine Res. 2024;44:68-79
209. Cheng Y, Luo D, Zhao Y, Rong J. N-Propargyl caffeate amide (PACA) prevents cardiac fibrosis in experimental myocardial infarction by promoting pro-resolving macrophage polarization. Aging (Albany NY). 2020;12:5384-98
210. Inaba K, Inaba M, Romani N, Aya H, Deguchi M, Ikehara S. et al. Generation of large numbers of dendritic cells from mouse bone marrow cultures supplemented with granulocyte/macrophage colony-stimulating factor. J Exp Med. 1992;176:1693-702
211. Xu J, Lin SC, Chen J, Miao Y, Taffet GE, Entman ML. et al. CCR2 mediates the uptake of bone marrow-derived fibroblast precursors in angiotensin II-induced cardiac fibrosis. Am J Physiol Heart Circ Physiol. 2011;301:H538-47
212. Haudek SB, Cheng J, Du J, Wang Y, Hermosillo-Rodriguez J, Trial J. et al. Monocytic fibroblast precursors mediate fibrosis in angiotensin-II-induced cardiac hypertrophy. J Mol Cell Cardiol. 2010;49:499-507
213. Morimoto H, Takahashi M, Izawa A, Ise H, Hongo M, Kolattukudy PE. et al. Cardiac overexpression of monocyte chemoattractant protein-1 in transgenic mice prevents cardiac dysfunction and remodeling after myocardial infarction. Circ Res. 2006;99:891-9
214. Shen JZ, Morgan J, Tesch GH, Fuller PJ, Young MJ. CCL2-dependent macrophage recruitment is critical for mineralocorticoid receptor-mediated cardiac fibrosis, inflammation, and blood pressure responses in male mice. Endocrinology. 2014;155:1057-66
215. Wen J, Guan Y, Niu H, Dang Y, Guan J. Targeting cardiac resident CCR2+ macrophage-secreted MCP-1 to attenuate inflammation after myocardial infarction. Acta Biomater. 2024
216. Luo Y, Zhang H, Yu J, Wei L, Li M, Xu W. Stem cell factor/mast cell/CCL2/monocyte/macrophage axis promotes Coxsackievirus B3 myocarditis and cardiac fibrosis by increasing Ly6C(high) monocyte influx and fibrogenic mediators production. Immunology. 2022;167:590-605
217. Simats A, Zhang S, Messerer D, Chong F, Beşkardeş S, Chivukula AS. et al. Innate immune memory after brain injury drives inflammatory cardiac dysfunction. Cell. 2024;187:4637-55.e26
218. Correction to. Macrophages suppress cardiac reprogramming of fibroblasts in vivo via IFN-mediated intercellular self-stimulating circuit. Protein Cell. 2024;15:938
219. Ding C, Tang G, Sun Y, Fu X, Tian Y, Zhan J. et al. A functional cardiac patch promotes cardiac repair by modulating the CCR2(-) cardiac-resident macrophage niche and their cell crosstalk. Cell Rep Med. 2025;6:101932
220. Wang W, Chen XK, Zhou L, Wang F, He YJ, Lu BJ. et al. Correction: Chemokine CCL2 promotes cardiac regeneration and repair in myocardial infarction mice via activation of the JNK/STAT3 axis. Acta Pharmacol Sin. 2025;46:1494
221. Mederacke I, Hsu CC, Troeger JS, Huebener P, Mu X, Dapito DH. et al. Fate tracing reveals hepatic stellate cells as dominant contributors to liver fibrosis independent of its aetiology. Nat Commun. 2013;4:2823
222. Xie X, Lv H, Liu C, Su X, Yu Z, Song S. et al. Correction: HBeAg mediates inflammatory functions of macrophages by TLR2 contributing to hepatic fibrosis. BMC Med. 2022;20:180
223. Page A, Paoli PP, Hill SJ, Howarth R, Wu R, Kweon SM. et al. Alcohol directly stimulates epigenetic modifications in hepatic stellate cells. J Hepatol. 2015;62:388-97
224. Rosenthal SB, Liu X, Ganguly S, Dhar D, Pasillas MP, Ricciardelli E. et al. Heterogeneity of HSCs in a Mouse Model of NASH. Hepatology. 2021;74:667-85
225. Guo Y, Zhao C, Dai W, Wang B, Lai E, Xiao Y. et al. C-C motif chemokine receptor 2 inhibition reduces liver fibrosis by restoring the immune cell landscape. Int J Biol Sci. 2023;19:2572-87
226. Dambach DM, Watson LM, Gray KR, Durham SK, Laskin DL. Role of CCR2 in macrophage migration into the liver during acetaminophen-induced hepatotoxicity in the mouse. Hepatology. 2002;35:1093-103
227. Ehling J, Bartneck M, Wei X, Gremse F, Fech V, Möckel D. et al. CCL2-dependent infiltrating macrophages promote angiogenesis in progressive liver fibrosis. Gut. 2014;63:1960-71
228. Karlmark KR, Weiskirchen R, Zimmermann HW, Gassler N, Ginhoux F, Weber C. et al. Hepatic recruitment of the inflammatory Gr1+ monocyte subset upon liver injury promotes hepatic fibrosis. Hepatology. 2009;50:261-74
229. Imamura M, Ogawa T, Sasaguri Y, Chayama K, Ueno H. Suppression of macrophage infiltration inhibits activation of hepatic stellate cells and liver fibrogenesis in rats. Gastroenterology. 2005;128:138-46
230. She S, Wu X, Zheng D, Pei X, Ma J, Sun Y. et al. PSMP/MSMP promotes hepatic fibrosis through CCR2 and represents a novel therapeutic target. J Hepatol. 2020;72:506-18
231. Marra F, Romanelli RG, Giannini C, Failli P, Pastacaldi S, Arrighi MC. et al. Monocyte chemotactic protein-1 as a chemoattractant for human hepatic stellate cells. Hepatology. 1999;29:140-8
232. Bataller R, Schwabe RF, Choi YH, Yang L, Paik YH, Lindquist J. et al. NADPH oxidase signal transduces angiotensin II in hepatic stellate cells and is critical in hepatic fibrosis. J Clin Invest. 2003;112:1383-94
233. Seki E, de Minicis S, Inokuchi S, Taura K, Miyai K, van Rooijen N. et al. CCR2 promotes hepatic fibrosis in mice. Hepatology. 2009;50:185-97
234. Harada K, Chiba M, Okamura A, Hsu M, Sato Y, Igarashi S. et al. Monocyte chemoattractant protein-1 derived from biliary innate immunity contributes to hepatic fibrogenesis. J Clin Pathol. 2011;64:660-5
235. Ono N, Fujita T, Miki M, Nishiyama K, Izawa T, Aoyama T. et al. Interleukin-19 Gene-Deficient Mice Promote Liver Fibrosis via Enhanced TGF-β Signaling, and the Interleukin-19-CCL2 Axis Is Important in the Direction of Liver Fibrosis. Biomedicines. 2023 11
236. Kisseleva T, Uchinami H, Feirt N, Quintana-Bustamante O, Segovia JC, Schwabe RF. et al. Bone marrow-derived fibrocytes participate in pathogenesis of liver fibrosis. J Hepatol. 2006;45:429-38
237. Scholten D, Reichart D, Paik YH, Lindert J, Bhattacharya J, Glass CK. et al. Migration of fibrocytes in fibrogenic liver injury. Am J Pathol. 2011;179:189-98
238. Mitchell C, Couton D, Couty JP, Anson M, Crain AM, Bizet V. et al. Dual role of CCR2 in the constitution and the resolution of liver fibrosis in mice. Am J Pathol. 2009;174:1766-75
239. Ramachandran P, Iredale JP. Macrophages: central regulators of hepatic fibrogenesis and fibrosis resolution. J Hepatol. 2012;56:1417-9
240. Miura A, Hosono T, Seki T. Macrophage potentiates the recovery of liver zonation and metabolic function after acute liver injury. Sci Rep. 2021;11:9730
241. Duffield JS, Forbes SJ, Constandinou CM, Clay S, Partolina M, Vuthoori S. et al. Selective depletion of macrophages reveals distinct, opposing roles during liver injury and repair. J Clin Invest. 2005;115:56-65
242. Zhdanov KV, Gusev DA, Chirskiĭ VS, Sysoev KA, Iakubovskaia LA, Shakhmanov DM. et al. [Chronic HCV-infection and expression of mRNA of CC-chemokines and their receptors]. Zh Mikrobiol Epidemiol Immunobiol. 2008:73-8
243. Song X, Gao X, Wang Y, Raja R, Zhang Y, Yang S. et al. HCV Core Protein Induces Chemokine CCL2 and CXCL10 Expression Through NF-κB Signaling Pathway in Macrophages. Front Immunol. 2021;12:654998
244. Wu X, Zhao W, Miao Q, Shi S, Wei B, Luo L. et al. CCR2+TREM-1+ monocytes promote natural killer T cell dysfunction contributing towards HBV disease progression. Immunol Res. 2024;72:938-47
245. Xi S, Zheng X, Li X, Jiang Y, Wu Y, Gong J. et al. Activated Hepatic Stellate Cells Induce Infiltration and Formation of CD163(+) Macrophages via CCL2/CCR2 Pathway. Front Med (Lausanne). 2021;8:627927
246. Barathan M, Riazalhosseini B, Iyadorai T, Vellasamy KM, Vadivelu J, Chang LY. et al. Comparative expression of pro-inflammatory and apoptotic biosignatures in chronic HBV-infected patients with and without liver cirrhosis. Microb Pathog. 2021;161:105231
247. Parker R, Weston CJ, Miao Z, Corbett C, Armstrong MJ, Ertl L. et al. CC chemokine receptor 2 promotes recruitment of myeloid cells associated with insulin resistance in nonalcoholic fatty liver disease. Am J Physiol Gastrointest Liver Physiol. 2018;314:G483-g93
248. Miura K, Yang L, van Rooijen N, Ohnishi H, Seki E. Hepatic recruitment of macrophages promotes nonalcoholic steatohepatitis through CCR2. Am J Physiol Gastrointest Liver Physiol. 2012;302:G1310-21
249. Hammoutene A, Biquard L, Lasselin J, Kheloufi M, Tanguy M, Vion AC. et al. A defect in endothelial autophagy occurs in patients with non-alcoholic steatohepatitis and promotes inflammation and fibrosis. J Hepatol. 2020;72:528-38
250. Wakamatsu S, Jojima T, Hashiguchi M, Kishi H, Niitani T, Sakurai S. et al. Inhibition of IL-33 signaling ameliorate hepatic fibrosis with decreasing MCP-1 in a mouse model of diabetes and non-alcoholic steatohepatitis; comparison for luseogliflozin, an SGLT2 inhibitor. J Diabetes Complications. 2024;38:108650
251. Daemen S, Gainullina A, Kalugotla G, He L, Chan MM, Beals JW. et al. Dynamic Shifts in the Composition of Resident and Recruited Macrophages Influence Tissue Remodeling in NASH. Cell Rep. 2022;41:111660
252. Awad AS, Kinsey GR, Khutsishvili K, Gao T, Bolton WK, Okusa MD. Monocyte/macrophage chemokine receptor CCR2 mediates diabetic renal injury. Am J Physiol Renal Physiol. 2011;301:F1358-66
253. Zoshima T, Baba T, Tanabe Y, Ishida Y, Nakatani K, Nagata M. et al. CCR2- and CCR5-mediated macrophage infiltration contributes to glomerular endocapillary hypercellularity in antibody-induced lupus nephritis. Rheumatology (Oxford). 2022;61:3033-48
254. Xu L, Sharkey D, Cantley LG. Tubular GM-CSF Promotes Late MCP-1/CCR2-Mediated Fibrosis and Inflammation after Ischemia/Reperfusion Injury. J Am Soc Nephrol. 2019;30:1825-40
255. Vielhauer V, Anders HJ, Mack M, Cihak J, Strutz F, Stangassinger M. et al. Obstructive nephropathy in the mouse: progressive fibrosis correlates with tubulointerstitial chemokine expression and accumulation of CC chemokine receptor 2- and 5-positive leukocytes. J Am Soc Nephrol. 2001;12:1173-87
256. Braga TT, Correa-Costa M, Silva RC, Cruz MC, Hiyane MI, da Silva JS. et al. CCR2 contributes to the recruitment of monocytes and leads to kidney inflammation and fibrosis development. Inflammopharmacology. 2018;26:403-11
257. Kitagawa K, Wada T, Furuichi K, Hashimoto H, Ishiwata Y, Asano M. et al. Blockade of CCR2 ameliorates progressive fibrosis in kidney. Am J Pathol. 2004;165:237-46
258. Chang FC, Liu CH, Luo AJ, Tao-Min Huang T, Tsai MH, Chen YJ. et al. Angiopoietin-2 inhibition attenuates kidney fibrosis by hindering chemokine C-C motif ligand 2 expression and apoptosis of endothelial cells. Kidney Int. 2022;102:780-97
259. Wu Q, Sun S, Wei L, Liu M, Liu H, Liu T. et al. Twist1 regulates macrophage plasticity to promote renal fibrosis through galectin-3. Cell Mol Life Sci. 2022;79:137
260. Devocelle A, Lecru L, Ferlicot S, Bessede T, Candelier JJ, Giron-Michel J. et al. IL-15 Prevents Renal Fibrosis by Inhibiting Collagen Synthesis: A New Pathway in Chronic Kidney Disease? Int J Mol Sci. 2021 22
261. Xia Y, Entman ML, Wang Y. CCR2 regulates the uptake of bone marrow-derived fibroblasts in renal fibrosis. PLoS One. 2013;8:e77493
262. Reich B, Schmidbauer K, Rodriguez Gomez M, Johannes Hermann F, Göbel N, Brühl H. et al. Fibrocytes develop outside the kidney but contribute to renal fibrosis in a mouse model. Kidney Int. 2013;84:78-89
263. Gonzalez J, Mouttalib S, Delage C, Calise D, Maoret JJ, Pradère JP. et al. Dual effect of chemokine CCL7/MCP-3 in the development of renal tubulointerstitial fibrosis. Biochem Biophys Res Commun. 2013;438:257-63
264. You H, Gao T, Raup-Konsavage WM, Cooper TK, Bronson SK, Reeves WB. et al. Podocyte-specific chemokine (C-C motif) receptor 2 overexpression mediates diabetic renal injury in mice. Kidney Int. 2017;91:671-82
265. Tarabra E, Giunti S, Barutta F, Salvidio G, Burt D, Deferrari G. et al. Effect of the monocyte chemoattractant protein-1/CC chemokine receptor 2 system on nephrin expression in streptozotocin-treated mice and human cultured podocytes. Diabetes. 2009;58:2109-18
266. Steffes MW, Schmidt D, McCrery R, Basgen JM. Glomerular cell number in normal subjects and in type 1 diabetic patients. Kidney Int. 2001;59:2104-13
267. Pagtalunan ME, Miller PL, Jumping-Eagle S, Nelson RG, Myers BD, Rennke HG. et al. Podocyte loss and progressive glomerular injury in type II diabetes. J Clin Invest. 1997;99:342-8
268. Burt D, Salvidio G, Tarabra E, Barutta F, Pinach S, Dentelli P. et al. The monocyte chemoattractant protein-1/cognate CC chemokine receptor 2 system affects cell motility in cultured human podocytes. Am J Pathol. 2007;171:1789-99
269. Nam BY, Paeng J, Kim SH, Lee SH, Kim DH, Kang HY. et al. The MCP-1/CCR2 axis in podocytes is involved in apoptosis induced by diabetic conditions. Apoptosis. 2012;17:1-13
270. Doublier S, Salvidio G, Lupia E, Ruotsalainen V, Verzola D, Deferrari G. et al. Nephrin expression is reduced in human diabetic nephropathy: evidence for a distinct role for glycated albumin and angiotensin II. Diabetes. 2003;52:1023-30
271. Moloi MW, Rusch JA, Omar F, Ekrikpo U, Dandara C, Bello AK. et al. Urinary MCP-1 and TWEAK as non-invasive markers of disease activity and treatment response in patients with lupus nephritis in South Africa. Int Urol Nephrol. 2021;53:1865-73
272. Hu L, Hu J, Chen L, Zhang Y, Wang Q, Yang X. Interleukin-22 From Type 3 Innate Lymphoid Cells Aggravates Lupus Nephritis by Promoting Macrophage Infiltration in Lupus-Prone Mice. Front Immunol. 2021;12:584414
273. Khalili M, Bonnefoy A, Genest DS, Quadri J, Rioux JP, Troyanov S. Clinical Use of Complement, Inflammation, and Fibrosis Biomarkers in Autoimmune Glomerulonephritis. Kidney Int Rep. 2020;5:1690-9
274. Yoshimoto K, Wada T, Furuichi K, Sakai N, Iwata Y, Yokoyama H. CD68 and MCP-1/CCR2 expression of initial biopsies reflect the outcomes of membranous nephropathy. Nephron Clin Pract. 2004;98:c25-34
275. Ren Y, Wang Z, You L, Zhou J, Huang H, Chang S. et al. Gut-derived trimethylamine N-oxide promotes CCR2-mediated macrophage infiltration in acute kidney injury. Nephrol Dial Transplant. 2024;39:1876-89
276. Wilkening A, Krappe J, Mühe AM, Lindenmeyer MT, Eltrich N, Luckow B. et al. C-C chemokine receptor type 2 mediates glomerular injury and interstitial fibrosis in focal segmental glomerulosclerosis. Nephrol Dial Transplant. 2020;35:227-39
277. Sakata F, Ito Y, Mizuno M, Sawai A, Suzuki Y, Tomita T. et al. Sodium chloride promotes tissue inflammation via osmotic stimuli in subtotal-nephrectomized mice. Lab Invest. 2017;97:432-46
278. Lin SL, Castaño AP, Nowlin BT, Lupher ML Jr, Duffield JS. Bone marrow Ly6Chigh monocytes are selectively recruited to injured kidney and differentiate into functionally distinct populations. J Immunol. 2009;183:6733-43
279. Huen SC, Cantley LG. Macrophage-mediated injury and repair after ischemic kidney injury. Pediatr Nephrol. 2015;30:199-209
280. Clements M, Gershenovich M, Chaber C, Campos-Rivera J, Du P, Zhang M. et al. Differential Ly6C Expression after Renal Ischemia-Reperfusion Identifies Unique Macrophage Populations. J Am Soc Nephrol. 2016;27:159-70
281. Yang Q, Wang Y, Pei G, Deng X, Jiang H, Wu J. et al. Bone marrow-derived Ly6C(-) macrophages promote ischemia-induced chronic kidney disease. Cell Death Dis. 2019;10:291
282. Bandinelli F, Del Rosso A, Gabrielli A, Giacomelli R, Bartoli F, Guiducci S. et al. CCL2, CCL3 and CCL5 chemokines in systemic sclerosis: the correlation with SSc clinical features and the effect of prostaglandin E1 treatment. Clin Exp Rheumatol. 2012;30:S44-9
283. Haub J, Roehrig N, Uhrin P, Schabbauer G, Eulberg D, Melchior F. et al. Intervention of Inflammatory Monocyte Activity Limits Dermal Fibrosis. J Invest Dermatol. 2019;139:2144-53
284. Yamamoto T, Eckes B, Hartmann K, Krieg T. Expression of monocyte chemoattractant protein-1 in the lesional skin of systemic sclerosis. J Dermatol Sci. 2001;26:133-9
285. Masuda A, Yasuoka H, Satoh T, Okazaki Y, Yamaguchi Y, Kuwana M. Versican is upregulated in circulating monocytes in patients with systemic sclerosis and amplifies a CCL2-mediated pathogenic loop. Arthritis Res Ther. 2013;15:R74
286. Kimura M, Kawahito Y, Hamaguchi M, Nakamura T, Okamoto M, Matsumoto Y. et al. SKL-2841, a dual antagonist of MCP-1 and MIP-1 beta, prevents bleomycin-induced skin sclerosis in mice. Biomed Pharmacother. 2007;61:222-8
287. Yamamoto T, Eckes B, Krieg T. High expression and autoinduction of monocyte chemoattractant protein-1 in scleroderma fibroblasts. Eur J Immunol. 2001;31:2936-41
288. Carulli MT, Ong VH, Ponticos M, Shiwen X, Abraham DJ, Black CM. et al. Chemokine receptor CCR2 expression by systemic sclerosis fibroblasts: evidence for autocrine regulation of myofibroblast differentiation. Arthritis Rheum. 2005;52:3772-82
289. Greenblatt MB, Sargent JL, Farina G, Tsang K, Lafyatis R, Glimcher LH. et al. Interspecies comparison of human and murine scleroderma reveals IL-13 and CCL2 as disease subset-specific targets. Am J Pathol. 2012;180:1080-94
290. Yamamoto T, Nishioka K. Role of monocyte chemoattractant protein-1 and its receptor,CCR-2, in the pathogenesis of bleomycin-induced scleroderma. J Invest Dermatol. 2003;121:510-6
291. Distler JH, Jüngel A, Caretto D, Schulze-Horsel U, Kowal-Bielecka O, Gay RE. et al. Monocyte chemoattractant protein 1 released from glycosaminoglycans mediates its profibrotic effects in systemic sclerosis via the release of interleukin-4 from T cells. Arthritis Rheum. 2006;54:214-25
292. Ferreira AM, Takagawa S, Fresco R, Zhu X, Varga J, DiPietro LA. Diminished induction of skin fibrosis in mice with MCP-1 deficiency. J Invest Dermatol. 2006;126:1900-8
293. Mei C, Meng F, Wang X, Yan S, Zheng Q, Zhang X. et al. CD30L is involved in the regulation of the inflammatory response through inducing homing and differentiation of monocytes via CCL2/CCR2 axis and NF-κB pathway in mice with colitis. Int Immunopharmacol. 2022;110:108934
294. Hachiya K, Masuya M, Kuroda N, Yoneda M, Tsuboi J, Nagaharu K. et al. Irbesartan, an angiotensin II type 1 receptor blocker, inhibits colitis-associated tumourigenesis by blocking the MCP-1/CCR2 pathway. Sci Rep. 2021;11:19943
295. Pei X, Zheng D, She S, Ma J, Guo C, Mo X. et al. The PSMP-CCR2 interactions trigger monocyte/macrophage-dependent colitis. Sci Rep. 2017;7:5107
296. Kuroda N, Masuya M, Tawara I, Tsuboi J, Yoneda M, Nishikawa K. et al. Infiltrating CCR2(+) monocytes and their progenies, fibrocytes, contribute to colon fibrosis by inhibiting collagen degradation through the production of TIMP-1. Sci Rep. 2019;9:8568
297. Motomura Y, Khan WI, El-Sharkawy RT, Verma-Gandhu M, Verdu EF, Gauldie J. et al. Induction of a fibrogenic response in mouse colon by overexpression of monocyte chemoattractant protein 1. Gut. 2006;55:662-70
298. Mojumdar K, Liang F, Giordano C, Lemaire C, Danialou G, Okazaki T. et al. Inflammatory monocytes promote progression of Duchenne muscular dystrophy and can be therapeutically targeted via CCR2. EMBO Mol Med. 2014;6:1476-92
299. Wang Y, Wang X, Alabdullatif S, Homma ST, Alekseyev YO, Zhou L. Expansion and pathogenic activation of skeletal muscle-resident macrophages in mdx(5cv)/Ccr2(-/-) mice. Proc Natl Acad Sci U S A. 2025;122:e2410095122
300. Zhao W, Wang X, Ransohoff RM, Zhou L. CCR2 deficiency does not provide sustained improvement of muscular dystrophy in mdx5cv mice. Faseb j. 2017;31:35-46
301. Sun LK, Reding T, Bain M, Heikenwalder M, Bimmler D, Graf R. Prostaglandin E2 modulates TNF-alpha-induced MCP-1 synthesis in pancreatic acinar cells in a PKA-dependent manner. Am J Physiol Gastrointest Liver Physiol. 2007;293:G1196-204
302. Zhao HF, Ito T, Gibo J, Kawabe K, Oono T, Kaku T. et al. Anti-monocyte chemoattractant protein 1 gene therapy attenuates experimental chronic pancreatitis induced by dibutyltin dichloride in rats. Gut. 2005;54:1759-67
303. Sarode G, Sarode SC, Deshmukh R, Raktade P, Patil S. Myofibroblasts could be recruited in a chemokine (C-C motif) ligand 2-dependent manner in pathogenesis of oral submucous fibrosis. J Oral Pathol Med. 2017;46:443-7
304. Peng W, Kepsch A, Kracht TO, Hasan H, Wijayarathna R, Wahle E. et al. Activin A and CCR2 regulate macrophage function in testicular fibrosis caused by experimental autoimmune orchitis. Cell Mol Life Sci. 2022;79:602
305. Popovics P, Silver SV, Uchtmann KS, Arendt LM, Vezina CM, Ricke WA. CCR2(+) monocytes/macrophages drive steroid hormone imbalance-related prostatic fibrosis. Sci Rep. 2024;14:15736
306. Lee SH, Kang HY, Kim KS, Nam BY, Paeng J, Kim S. et al. The monocyte chemoattractant protein-1 (MCP-1)/CCR2 system is involved in peritoneal dialysis-related epithelial-mesenchymal transition of peritoneal mesothelial cells. Lab Invest. 2012;92:1698-711
307. Lee Y, Lee J, Park M, Seo A, Kim KH, Kim S. et al. Inflammatory chemokine (C-C motif) ligand 8 inhibition ameliorates peritoneal fibrosis. Faseb j. 2023;37:e22632
308. Suga M, Iyonaga K, Ichiyasu H, Saita N, Yamasaki H, Ando M. Clinical significance of MCP-1 levels in BALF and serum in patients with interstitial lung diseases. Eur Respir J. 1999;14:376-82
309. Shinoda H, Tasaka S, Fujishima S, Yamasawa W, Miyamoto K, Nakano Y. et al. Elevated CC chemokine level in bronchoalveolar lavage fluid is predictive of a poor outcome of idiopathic pulmonary fibrosis. Respiration. 2009;78:285-92
310. Yin YQ, Peng F, Situ HJ, Xie JL, Tan L, Wei J. et al. Construction of prediction model of inflammation related genes in idiopathic pulmonary fibrosis and its correlation with immune microenvironment. Front Immunol. 2022;13:1010345
311. Gui X, Qiu X, Tian Y, Xie M, Li H, Gao Y. et al. Prognostic value of IFN-γ, sCD163, CCL2 and CXCL10 involved in acute exacerbation of idiopathic pulmonary fibrosis. Int Immunopharmacol. 2019;70:208-15
312. Hartl D, Griese M, Nicolai T, Zissel G, Prell C, Reinhardt D. et al. A role for MCP-1/CCR2 in interstitial lung disease in children. Respir Res. 2005;6:93
313. Brody SL, Gunsten SP, Luehmann HP, Sultan DH, Hoelscher M, Heo GS. et al. Chemokine Receptor 2-targeted Molecular Imaging in Pulmonary Fibrosis. A Clinical Trial. Am J Respir Crit Care Med. 2021;203:78-89
314. Farooq H, Luehmann HP, Koenitzer JR, Heo GS, Sultan DH, Kulkarni DH. et al. Molecular imaging in experimental pulmonary fibrosis reveals that nintedanib unexpectedly modulates CCR2 immune cell infiltration. EBioMedicine. 2024;110:105431
315. Heo GS, Bajpai G, Li W, Luehmann HP, Sultan DH, Dun H. et al. Targeted PET Imaging of Chemokine Receptor 2-Positive Monocytes and Macrophages in the Injured Heart. J Nucl Med. 2021;62:111-4
316. Lee JU, Cheong HS, Shim EY, Bae DJ, Chang HS, Uh ST. et al. Gene profile of fibroblasts identify relation of CCL8 with idiopathic pulmonary fibrosis. Respir Res. 2017;18:3
317. Bai YM, Liang S, Zhou B. Revealing immune infiltrate characteristics and potential immune-related genes in hepatic fibrosis: based on bioinformatics, transcriptomics and q-PCR experiments. Front Immunol. 2023;14:1133543
318. Farci P, Wollenberg K, Diaz G, Engle RE, Lai ME, Klenerman P. et al. Profibrogenic chemokines and viral evolution predict rapid progression of hepatitis C to cirrhosis. Proc Natl Acad Sci U S A. 2012;109:14562-7
319. Marra F, DeFranco R, Grappone C, Milani S, Pastacaldi S, Pinzani M. et al. Increased expression of monocyte chemotactic protein-1 during active hepatic fibrogenesis: correlation with monocyte infiltration. Am J Pathol. 1998;152:423-30
320. Queck A, Bode H, Uschner FE, Brol MJ, Graf C, Schulz M. et al. Systemic MCP-1 Levels Derive Mainly From Injured Liver and Are Associated With Complications in Cirrhosis. Front Immunol. 2020;11:354
321. Tsuneyama K, Harada K, Yasoshima M, Hiramatsu K, Mackay CR, Mackay IR. et al. Monocyte chemotactic protein-1, -2, and -3 are distinctively expressed in portal tracts and granulomata in primary biliary cirrhosis: implications for pathogenesis. J Pathol. 2001;193:102-9
322. Moreno Traspas R, Teoh TS, Wong PM, Maier M, Chia CY, Lay K. et al. Loss of FOCAD, operating via the SKI messenger RNA surveillance pathway, causes a pediatric syndrome with liver cirrhosis. Nat Genet. 2022;54:1214-26
323. Micheloud D, Salcedo M, Bañares R, Rincón D, Lorente R, Muñoz-Fernández MA. et al. Serum levels of fibrosis biomarkers measured early after liver transplantation are associated with severe hepatitis C virus recurrence. Transpl Infect Dis. 2009;11:183-8
324. Mühlbauer M, Bosserhoff AK, Hartmann A, Thasler WE, Weiss TS, Herfarth H. et al. A novel MCP-1 gene polymorphism is associated with hepatic MCP-1 expression and severity of HCV-related liver disease. Gastroenterology. 2003;125:1085-93
325. Shao LN, Zhou SH, Wang N, Zhang ST, Liu M. Association between the Genetic Polymorphisms of CCL2, CCL5, CCL8, CCR2, and CCR5 with Chronic Hepatitis C Virus Infection in the Chinese Han Population. Immunol Invest. 2022;51:1182-97
326. Scurt FG, Menne J, Brandt S, Bernhardt A, Mertens PR, Haller H. et al. Monocyte chemoattractant protein-1 predicts the development of diabetic nephropathy. Diabetes Metab Res Rev. 2022;38:e3497
327. Schrauben SJ, Shou H, Zhang X, Anderson AH, Bonventre JV, Chen J. et al. Association of Multiple Plasma Biomarker Concentrations with Progression of Prevalent Diabetic Kidney Disease: Findings from the Chronic Renal Insufficiency Cohort (CRIC) Study. J Am Soc Nephrol. 2021;32:115-26
328. An Z, Qin J, Bo W, Li H, Jiang L, Li X. et al. Prognostic Value of Serum Interleukin-6, NF-κB plus MCP-1 Assay in Patients with Diabetic Nephropathy. Dis Markers. 2022;2022:4428484
329. Pérez-Arias AA, Méndez-Pérez RA, Cruz C, Zavala-Miranda MF, Romero-Diaz J, Márquez-Macedo SE. et al. The first-year course of urine MCP-1 and its association with response to treatment and long-term kidney prognosis in lupus nephritis. Clin Rheumatol. 2023;42:83-92
330. Mansour SG, Puthumana J, Coca SG, Gentry M, Parikh CR. Biomarkers for the detection of renal fibrosis and prediction of renal outcomes: a systematic review. BMC Nephrol. 2017;18:72
331. Stephan M, Conrad S, Eggert T, Heuer R, Fernandez S, Huland H. Urinary concentration and tissue messenger RNA expression of monocyte chemoattractant protein-1 as an indicator of the degree of hydronephrotic atrophy in partial ureteral obstruction. J Urol. 2002;167:1497-502
332. Ho J, Rush DN, Gibson IW, Karpinski M, Storsley L, Bestland J. et al. Early urinary CCL2 is associated with the later development of interstitial fibrosis and tubular atrophy in renal allografts. Transplantation. 2010;90:394-400
333. Ho J, Wiebe C, Gibson IW, Hombach-Klonisch S, Gao A, Rigatto C. et al. Elevated urinary CCL2: Cr at 6 months is associated with renal allograft interstitial fibrosis and inflammation at 24 months. Transplantation. 2014;98:39-46
334. Lee J, Lee Y, Kim KH, Kim DK, Joo KW, Shin SJ. et al. Chemokine (C-C Motif) Ligand 8 and Tubulo-Interstitial Injury in Chronic Kidney Disease. Cells. 2022 11
335. Carbone RG, Monselise A, Barisione E, Fontana V, Paredi P, Puppo F. Pulmonary hypertension in systemic sclerosis with usual interstitial pneumonia. Intern Emerg Med. 2023;18:1087-93
336. Jin J, Liu Y, Tang Q, Yan X, Jiang M, Zhao X. et al. Bioinformatics-integrated screening of systemic sclerosis-specific expressed markers to identify therapeutic targets. Front Immunol. 2023;14:1125183
337. Shan N, Shang Y, He Y, Wen Z, Ning S, Chen H. Common biomarkers of idiopathic pulmonary fibrosis and systemic sclerosis based on WGCNA and machine learning. Sci Rep. 2025;15:610
338. Carulli MT, Handler C, Coghlan JG, Black CM, Denton CP. Can CCL2 serum levels be used in risk stratification or to monitor treatment response in systemic sclerosis? Ann Rheum Dis. 2008;67:105-9
339. Scala E, Pallotta S, Frezzolini A, Abeni D, Barbieri C, Sampogna F. et al. Cytokine and chemokine levels in systemic sclerosis: relationship with cutaneous and internal organ involvement. Clin Exp Immunol. 2004;138:540-6
340. Wu M, Baron M, Pedroza C, Salazar GA, Ying J, Charles J. et al. CCL2 in the Circulation Predicts Long-Term Progression of Interstitial Lung Disease in Patients With Early Systemic Sclerosis: Data From Two Independent Cohorts. Arthritis Rheumatol. 2017;69:1871-8
341. Hasegawa M, Fujimoto M, Matsushita T, Hamaguchi Y, Takehara K, Sato S. Serum chemokine and cytokine levels as indicators of disease activity in patients with systemic sclerosis. Clin Rheumatol. 2011;30:231-7
342. Yalçinkaya Y, Çinar S, Artim-Esen B, Kamali S, Öcal L, Deniz G. et al. The relationship between vascular biomarkers and disease characteristics in systemic sclerosis: elevated MCP-1 is predominantly associated with fibrotic manifestations. Clin Exp Rheumatol. 2016;34(Suppl 100):110-4
343. Yanaba K, Komura K, Kodera M, Matsushita T, Hasegawa M, Takehara K. et al. Serum levels of monocyte chemotactic protein-3/CCL7 are raised in patients with systemic sclerosis: association with extent of skin sclerosis and severity of pulmonary fibrosis. Ann Rheum Dis. 2006;65:124-6
344. Yanaba K, Yoshizaki A, Muroi E, Hara T, Ogawa F, Shimizu K. et al. CCL13 is a promising diagnostic marker for systemic sclerosis. Br J Dermatol. 2010;162:332-6
345. Gambichler T, Yilmaz E, Höxtermann S, Kolios A, Moritz R, Bechara FG. et al. Serum CCL13 levels in patients with systemic sclerosis and controls. Br J Dermatol. 2011;165:216-8
346. Pozzi G, Carubbi C, Gobbi G, Tagliaferri S, Mirandola P, Vitale M. et al. Tracking fibrosis in myeloproliferative neoplasms by CCR2 expression on CD34(+) cells. Front Oncol. 2022;12:980379
347. Hodeib H, Abd El Hai D, Tawfik MA, Allam AA, Selim A, Elsawy AA. et al. CCL2 rs1024611Gene Polymorphism in Philadelphia-Negative Myeloproliferative Neoplasms. Genes (Basel). 2022 13
348. Masselli E, Carubbi C, Pozzi G, Percesepe A, Campanelli R, Villani L. et al. Impact of the rs1024611 Polymorphism of CCL2 on the Pathophysiology and Outcome of Primary Myelofibrosis. Cancers (Basel). 2021 13
349. Masselli E, Carubbi C, Cambò B, Pozzi G, Gobbi G, Mirandola P. et al. The -2518 A/G polymorphism of the monocyte chemoattractant protein-1 as a candidate genetic predisposition factor for secondary myelofibrosis and biomarker of disease severity. Leukemia. 2018;32:2266-70
350. Karrer S, Bosserhoff AK, Weiderer P, Distler O, Landthaler M, Szeimies RM. et al. The -2518 promotor polymorphism in the MCP-1 gene is associated with systemic sclerosis. J Invest Dermatol. 2005;124:92-8
351. Carulli MT, Spagnolo P, Fonseca C, Welsh KI, duBois RM, Black CM. et al. Single-nucleotide polymorphisms in CCL2 gene are not associated with susceptibility to systemic sclerosis. J Rheumatol. 2008;35:839-44
352. Cavestro GM, Zuppardo RA, Bertolini S, Sereni G, Frulloni L, Okolicsanyi S. et al. Connections between genetics and clinical data: Role of MCP-1, CFTR, and SPINK-1 in the setting of acute, acute recurrent, and chronic pancreatitis. Am J Gastroenterol. 2010;105:199-206
353. Lee SH, Ryu JK, Jeong JB, Lee KY, Woo SM, Park JK. et al. Polymorphisms of the MCP-1 and HSP70-2 genes in Korean patients with alcoholic chronic pancreatitis. Dig Dis Sci. 2008;53:1721-7
354. Madan U, Verma B, Awasthi A. Cenicriviroc, a CCR2/CCR5 antagonist, promotes the generation of type 1 regulatory T cells. Eur J Immunol. 2024;54:e2350847
355. Eksteen B, Bowlus CL, Montano-Loza AJ, Lefebvre E, Fischer L, Vig P. et al. Efficacy and Safety of Cenicriviroc in Patients With Primary Sclerosing Cholangitis: PERSEUS Study. Hepatol Commun. 2021;5:478-90
356. Lefebvre E, Moyle G, Reshef R, Richman LP, Thompson M, Hong F. et al. Antifibrotic Effects of the Dual CCR2/CCR5 Antagonist Cenicriviroc in Animal Models of Liver and Kidney Fibrosis. PLoS One. 2016;11:e0158156
357. Guicciardi ME, Trussoni CE, Krishnan A, Bronk SF, Lorenzo Pisarello MJ, O'Hara SP. et al. Macrophages contribute to the pathogenesis of sclerosing cholangitis in mice. J Hepatol. 2018;69:676-86
358. Yu D, Cai SY, Mennone A, Vig P, Boyer JL. Cenicriviroc, a cytokine receptor antagonist, potentiates all-trans retinoic acid in reducing liver injury in cholestatic rodents. Liver Int. 2018;38:1128-38
359. Krenkel O, Puengel T, Govaere O, Abdallah AT, Mossanen JC, Kohlhepp M. et al. Therapeutic inhibition of inflammatory monocyte recruitment reduces steatohepatitis and liver fibrosis. Hepatology. 2018;67:1270-83
360. Ambade A, Lowe P, Kodys K, Catalano D, Gyongyosi B, Cho Y. et al. Pharmacological Inhibition of CCR2/5 Signaling Prevents and Reverses Alcohol-Induced Liver Damage, Steatosis, and Inflammation in Mice. Hepatology. 2019;69:1105-21
361. Chen G, Yu Y, Zhu Y, Nagashimada M, Wang Y, Nagata N. et al. Cenicriviroc Suppresses and Reverses Steatohepatitis by Regulating Macrophage Infiltration and M2 Polarization in Mice. Endocrinology. 2024 165
362. Kruger AJ, Fuchs BC, Masia R, Holmes JA, Salloum S, Sojoodi M. et al. Prolonged cenicriviroc therapy reduces hepatic fibrosis despite steatohepatitis in a diet-induced mouse model of nonalcoholic steatohepatitis. Hepatol Commun. 2018;2:529-45
363. Liang F, Giordano C, Shang D, Li Q, Petrof BJ. The dual CCR2/CCR5 chemokine receptor antagonist Cenicriviroc reduces macrophage infiltration and disease severity in Duchenne muscular dystrophy (Dmdmdx-4Cv) mice. PLoS One. 2018;13:e0194421
364. Francque SM, Hodge A, Boursier J, Younes ZH, Rodriguez-Araujo G, Park GS. et al. Phase 2, open-label, rollover study of cenicriviroc for liver fibrosis associated with metabolic dysfunction-associated steatohepatitis. Hepatol Commun. 2024 8
365. Friedman SL, Ratziu V, Harrison SA, Abdelmalek MF, Aithal GP, Caballeria J. et al. A randomized, placebo-controlled trial of cenicriviroc for treatment of nonalcoholic steatohepatitis with fibrosis. Hepatology. 2018;67:1754-67
366. Anstee QM, Neuschwander-Tetri BA, Wong VW, Abdelmalek MF, Younossi ZM, Yuan J. et al. Cenicriviroc for the treatment of liver fibrosis in adults with nonalcoholic steatohepatitis: AURORA Phase 3 study design. Contemp Clin Trials. 2020;89:105922
367. Anstee QM, Neuschwander-Tetri BA, Wai-Sun Wong V, Abdelmalek MF, Rodriguez-Araujo G, Landgren H. et al. Cenicriviroc Lacked Efficacy to Treat Liver Fibrosis in Nonalcoholic Steatohepatitis: AURORA Phase III Randomized Study. Clin Gastroenterol Hepatol. 2024;22:124-34.e1
368. Guilliams M, Bonnardel J, Haest B, Vanderborght B, Wagner C, Remmerie A. et al. Spatial proteogenomics reveals distinct and evolutionarily conserved hepatic macrophage niches. Cell. 2022;185:379-96.e38
369. Ramachandran P, Dobie R, Wilson-Kanamori JR, Dora EF, Henderson BEP, Luu NT. et al. Resolving the fibrotic niche of human liver cirrhosis at single-cell level. Nature. 2019;575:512-8
370. Puengel T, Lefere S, Hundertmark J, Kohlhepp M, Penners C, Van de Velde F. et al. Combined Therapy with a CCR2/CCR5 Antagonist and FGF21 Analogue Synergizes in Ameliorating Steatohepatitis and Fibrosis. Int J Mol Sci. 2022 23
371. Kashyap S, Warner GM, Hartono SP, Boyilla R, Knudsen BE, Zubair AS. et al. Blockade of CCR2 reduces macrophage influx and development of chronic renal damage in murine renovascular hypertension. Am J Physiol Renal Physiol. 2016;310:F372-84
372. Kang YS, Lee MH, Song HK, Ko GJ, Kwon OS, Lim TK. et al. CCR2 antagonism improves insulin resistance, lipid metabolism, and diabetic nephropathy in type 2 diabetic mice. Kidney Int. 2010;78:883-94
373. Ishikawa M, Yamamoto T. Antifibrogenic effects of C-C chemokine receptor type 2 antagonist in a bleomycin-induced scleroderma model. Exp Dermatol. 2021;30:179-84
374. Wang W, Ai J, Liao B, Xiao K, Lin L, Chen H. et al. The roles of MCP-1/CCR2 mediated macrophage recruitment and polarization in bladder outlet obstruction (BOO) induced bladder remodeling. Int Immunopharmacol. 2021;99:107947
375. Oberthür D, Achenbach J, Gabdulkhakov A, Buchner K, Maasch C, Falke S. et al. Crystal structure of a mirror-image L-RNA aptamer (Spiegelmer) in complex with the natural L-protein target CCL2. Nat Commun. 2015;6:6923
376. Boels MGS, Koudijs A, Avramut MC, Sol W, Wang G, van Oeveren-Rietdijk AM. et al. Systemic Monocyte Chemotactic Protein-1 Inhibition Modifies Renal Macrophages and Restores Glomerular Endothelial Glycocalyx and Barrier Function in Diabetic Nephropathy. Am J Pathol. 2017;187:2430-40
377. Ninichuk V, Clauss S, Kulkarni O, Schmid H, Segerer S, Radomska E. et al. Late onset of Ccl2 blockade with the Spiegelmer mNOX-E36-3'PEG prevents glomerulosclerosis and improves glomerular filtration rate in db/db mice. Am J Pathol. 2008;172:628-37
378. Baeck C, Wehr A, Karlmark KR, Heymann F, Vucur M, Gassler N. et al. Pharmacological inhibition of the chemokine CCL2 (MCP-1) diminishes liver macrophage infiltration and steatohepatitis in chronic hepatic injury. Gut. 2012;61:416-26
379. Baeck C, Wei X, Bartneck M, Fech V, Heymann F, Gassler N. et al. Pharmacological inhibition of the chemokine C-C motif chemokine ligand 2 (monocyte chemoattractant protein 1) accelerates liver fibrosis regression by suppressing Ly-6C(+) macrophage infiltration in mice. Hepatology. 2014;59:1060-72
380. Xue CB, Wang A, Meloni D, Zhang K, Kong L, Feng H. et al. Discovery of INCB3344, a potent, selective and orally bioavailable antagonist of human and murine CCR2. Bioorg Med Chem Lett. 2010;20:7473-8
381. Brodmerkel CM, Huber R, Covington M, Diamond S, Hall L, Collins R. et al. Discovery and pharmacological characterization of a novel rodent-active CCR2 antagonist, INCB3344. J Immunol. 2005;175:5370-8
382. Moore JP, Vinh A, Tuck KL, Sakkal S, Krishnan SM, Chan CT. et al. M2 macrophage accumulation in the aortic wall during angiotensin II infusion in mice is associated with fibrosis, elastin loss, and elevated blood pressure. Am J Physiol Heart Circ Physiol. 2015;309:H906-17
383. McIntosh LM, Barnes JL, Barnes VL, McDonald JR. Selective CCR2-targeted macrophage depletion ameliorates experimental mesangioproliferative glomerulonephritis. Clin Exp Immunol. 2009;155:295-303
384. Brana I, Calles A, LoRusso PM, Yee LK, Puchalski TA, Seetharam S. et al. Carlumab, an anti-C-C chemokine ligand 2 monoclonal antibody, in combination with four chemotherapy regimens for the treatment of patients with solid tumors: an open-label, multicenter phase 1b study. Target Oncol. 2015;10:111-23
385. Raghu G, Martinez FJ, Brown KK, Costabel U, Cottin V, Wells AU. et al. CC-chemokine ligand 2 inhibition in idiopathic pulmonary fibrosis: a phase 2 trial of carlumab. Eur Respir J. 2015;46:1740-50
386. Yeqing C, Xinsheng F, Liping Z, Fangyuan H, Pengli W. Screening and evaluation of quality markers from Shuangshen Pingfei formula for idiopathic pulmonary fibrosis using network pharmacology and pharmacodynamic, phytochemical, and pharmacokinetic analyses. Phytomedicine. 2022;100:154040
387. Duan LF, Xu XF, Zhu LJ, Liu F, Zhang XQ, Wu N. et al. Dachaihu decoction ameliorates pancreatic fibrosis by inhibiting macrophage infiltration in chronic pancreatitis. World J Gastroenterol. 2017;23:7242-52
388. Chiang CK, Sheu ML, Lin YW, Wu CT, Yang CC, Chen MW. et al. Honokiol ameliorates renal fibrosis by inhibiting extracellular matrix and pro-inflammatory factors in vivo and in vitro. Br J Pharmacol. 2011;163:586-97
389. Lan T, Chen B, Hu X, Cao J, Chen S, Ding X. et al. Tianhuang formula ameliorates liver fibrosis by inhibiting CCL2-CCR2 axis and MAPK/NF-κB signaling pathway. J Ethnopharmacol. 2024;321:117516
390. Liu J, Li Z, Liu W, Jiang Z, Zhang X, Yuan Y. et al. Quercetin down-regulates MCP-1 expression in autoimmune myocarditis via ERK1/2-C/EBPβ pathway: An integrative approach using network pharmacology and experimental models. Int Immunopharmacol. 2025;154:114559
391. Shi H, Duan X, Dong J, Tao Y, Lei Y. RNA-seq combined network pharmacology reveals that Fu-Gan-Wan (FGW) inhibits liver fibrosis via NF-κB/CCL2/CCR2 and lipid peroxidation via Nrf2/HMOX1 signaling pathway. J Ethnopharmacol. 2024;326:117963
392. Xue L, Jin X, Ji T, Li R, Zhuge X, Xu F. et al. Luteolin ameliorates DSS-induced colitis in mice via suppressing macrophage activation and chemotaxis. Int Immunopharmacol. 2023;124:110996
393. Kwon EY, Choi MS. Luteolin Targets the Toll-Like Receptor Signaling Pathway in Prevention of Hepatic and Adipocyte Fibrosis and Insulin Resistance in Diet-Induced Obese Mice. Nutrients. 2018 10
394. Tao Z, Ge Y, Zhou N, Wang Y, Cheng W, Yang Z. Puerarin inhibits cardiac fibrosis via monocyte chemoattractant protein (MCP)-1 and the transforming growth factor-β1 (TGF-β1) pathway in myocardial infarction mice. Am J Transl Res. 2016;8:4425-33
395. Li A, Wang J, Zhu D, Zhang X, Pan R, Wang R. Arctigenin suppresses transforming growth factor-β1-induced expression of monocyte chemoattractant protein-1 and the subsequent epithelial-mesenchymal transition through reactive oxygen species-dependent ERK/NF-κB signaling pathway in renal tubular epithelial cells. Free Radic Res. 2015;49:1095-113
396. Li A, Zhang X, Shu M, Wu M, Wang J, Zhang J. et al. Arctigenin suppresses renal interstitial fibrosis in a rat model of obstructive nephropathy. Phytomedicine. 2017;30:28-41
397. Li Z, Zhang L, He W, Zhu C, Yang J, Sheng M. Astragalus membranaceus inhibits peritoneal fibrosis via monocyte chemoattractant protein (MCP)-1 and the transforming growth factor-β1 (TGF-β1) pathway in rats submitted to peritoneal dialysis. Int J Mol Sci. 2014;15:12959-71
398. Zhang S, Li S, Li X, Wan C, Cui L, Wang Y. Anti-fibrosis effect of astragaloside IV in animal models of cardiovascular diseases and its mechanisms: a systematic review. Pharm Biol. 2025;63:250-63
399. Chai CJ, Sun Y, Chi RF, Yang HY, Yang B, Li B. Astragaloside IV alleviates LPS-induced cardiomyocyte hypertrophy and collagen expression associated with CCL2-mediated activation of NF-κB signaling pathway. Biochem Biophys Res Commun. 2024;693:149367
400. Gui D, Huang J, Guo Y, Chen J, Chen Y, Xiao W. et al. Astragaloside IV ameliorates renal injury in streptozotocin-induced diabetic rats through inhibiting NF-κB-mediated inflammatory genes expression. Cytokine. 2013;61:970-7
401. Masamune A, Suzuki N, Kikuta K, Satoh M, Satoh K, Shimosegawa T. Curcumin blocks activation of pancreatic stellate cells. J Cell Biochem. 2006;97:1080-93
402. Zhao XA, Chen G, Liu Y, Chen Y, Wu H, Xiong Y. et al. Curcumin reduces Ly6C(hi) monocyte infiltration to protect against liver fibrosis by inhibiting Kupffer cells activation to reduce chemokines secretion. Biomed Pharmacother. 2018;106:868-78
403. Huang R, Liu Y, Xiong Y, Wu H, Wang G, Sun Z. et al. Curcumin protects against liver fibrosis by attenuating infiltration of Gr1hi monocytes through inhibition of monocyte chemoattractant protein-1. Discov Med. 2016;21:447-57
404. Vizzutti F, Provenzano A, Galastri S, Milani S, Delogu W, Novo E. et al. Curcumin limits the fibrogenic evolution of experimental steatohepatitis. Lab Invest. 2010;90:104-15
405. Furuichi K, Wada T, Iwata Y, Kitagawa K, Kobayashi K, Hashimoto H. et al. CCR2 signaling contributes to ischemia-reperfusion injury in kidney. J Am Soc Nephrol. 2003;14:2503-15
406. Tian T, Zhao C, Li S, Huang Z, Guo Y, Dai W. et al. Liver-Targeted Delivery of Small Interfering RNA of C-C Chemokine Receptor 2 with Tetrahedral Framework Nucleic Acid Attenuates Liver Cirrhosis. ACS Appl Mater Interfaces. 2023;15:10492-505
407. Kalderén C, Forsgren M, Karlström U, Stefansson K, Svensson R, Berglund MM. et al. A truncated analogue of CCL2 mediates anti-fibrotic effects on murine fibroblasts independently of CCR2. Biochem Pharmacol. 2012;83:644-52
408. Saito S, Nakayama T, Hashimoto N, Miyata Y, Egashira K, Nakao N. et al. Mesenchymal stem cells stably transduced with a dominant-negative inhibitor of CCL2 greatly attenuate bleomycin-induced lung damage. Am J Pathol. 2011;179:1088-94
409. Tsuruta S, Nakamuta M, Enjoji M, Kotoh K, Hiasa K, Egashira K. et al. Anti-monocyte chemoattractant protein-1 gene therapy prevents dimethylnitrosamine-induced hepatic fibrosis in rats. Int J Mol Med. 2004;14:837-42
410. Hayashidani S, Tsutsui H, Shiomi T, Ikeuchi M, Matsusaka H, Suematsu N. et al. Anti-monocyte chemoattractant protein-1 gene therapy attenuates left ventricular remodeling and failure after experimental myocardial infarction. Circulation. 2003;108:2134-40
411. Yoshimura H, Nakagawa Y, Muneta T, Koga H. A CCL2/MCP-1 antagonist attenuates fibrosis of the infrapatellar fat pad in a rat model of arthritis. BMC Musculoskelet Disord. 2024;25:674
412. Liang X, Li Y, Wu Y, Wu T, Huang D, Tang Z. et al. Human umbilical cord mesenchymal stem cell-derived microvesicles alleviate pulmonary fibrosis by inhibiting monocyte-macrophage migration through ERK1/2 signaling-mediated suppression of CCL2 expression. Stem Cell Res Ther. 2025;16:145
413. Gong P, Ding Y, Sun R, Jiang Z, Li W, Su X. et al. Mesenchymal stem cells alleviate systemic sclerosis by inhibiting the recruitment of pathogenic macrophages. Cell Death Discov. 2022;8:466
414. Mincheva G, Moreno-Manzano V, Felipo V, Llansola M. Extracellular vesicles from mesenchymal stem cells improve liver injury in rats with mild liver damage. Underlying mechanisms and role of TGFβ. Life Sci. 2025;364:123429
415. Sun D, Bu L, Liu C, Yin Z, Zhou X, Li X. et al. Therapeutic effects of human amniotic fluid-derived stem cells on renal interstitial fibrosis in a murine model of unilateral ureteral obstruction. PLoS One. 2013;8:e65042
416. Geervliet E, Karkdijk E, Bansal R. Inhibition of intrahepatic monocyte recruitment by Cenicriviroc and extracellular matrix degradation by MMP1 synergistically attenuate liver inflammation and fibrogenesis in vivo. Sci Rep. 2024;14:16897
417. Kamio K, Azuma A, Matsuda K, Usuki J, Inomata M, Morinaga A. et al. Resolution of bleomycin-induced murine pulmonary fibrosis via a splenic lymphocyte subpopulation. Respir Res. 2018;19:71
418. Moore BB, Murray L, Das A, Wilke CA, Herrygers AB, Toews GB. The role of CCL12 in the recruitment of fibrocytes and lung fibrosis. Am J Respir Cell Mol Biol. 2006;35:175-81
419. Gurczynski SJ, Procario MC, O'Dwyer DN, Wilke CA, Moore BB. Loss of CCR2 signaling alters leukocyte recruitment and exacerbates γ-herpesvirus-induced pneumonitis and fibrosis following bone marrow transplantation. Am J Physiol Lung Cell Mol Physiol. 2016;311:L611-27
420. Zhou X, Loomis-King H, Gurczynski SJ, Wilke CA, Konopka KE, Ptaschinski C. et al. Bone marrow transplantation alters lung antigen-presenting cells to promote TH17 response and the development of pneumonitis and fibrosis following gammaherpesvirus infection. Mucosal Immunol. 2016;9:610-20
421. Ding W, Pu W, Jiang S, Ma Y, Liu Q, Wu W. et al. Evaluation of the antifibrotic potency by knocking down SPARC, CCR2 and SMAD3. EBioMedicine. 2018;38:238-47
422. Menne J, Eulberg D, Beyer D, Baumann M, Saudek F, Valkusz Z. et al. C-C motif-ligand 2 inhibition with emapticap pegol (NOX-E36) in type 2 diabetic patients with albuminuria. Nephrol Dial Transplant. 2017;32:307-15
Author contact
![]() Corresponding authors: Yang Yang, MD., PhD., and Jincheng Liu, MD., PhD., Department of Cardiovascular Surgery, Xijing Hospital, The Airforce Medical University, 127 Changle West Road, Xi'an 710032, China, Telephone: +86 13379217366, Email address: yang200214yyedu.cn (Yang Yang), jinchengliufmmucom (Jincheng Liu).
Corresponding authors: Yang Yang, MD., PhD., and Jincheng Liu, MD., PhD., Department of Cardiovascular Surgery, Xijing Hospital, The Airforce Medical University, 127 Changle West Road, Xi'an 710032, China, Telephone: +86 13379217366, Email address: yang200214yyedu.cn (Yang Yang), jinchengliufmmucom (Jincheng Liu).