Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
1. Introduction
2. Structure and functions of...
3. PD-1/PD-L1 inhibitors
4. PD-L1 degraders
5. Co-inhibitors of PD-L1 and...
6. Conclusions and perspectives
Abbreviations
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2026; 16(11):6051-6080. doi:10.7150/thno.130935 This issue Cite
Review
Small molecules targeting the PD-1/PD-L1 axis for cancer immunotherapy
Jia-Yi Yin2#, Hui-Min Liu2#, Shao-long Li3#, Xin-Qian Ji2, Jun-Jie Wang2, Meng-Jie Fu2, Cong-Jun Liu4, Ning Wang5, Guo-Liang Lu6,7, Yan Li7,8, Hong-Min Liu2, Yi-Chao Zheng2, Xing-Jie Dai2 ![]() , Ying Liu1
, Ying Liu1 ![]()
1. Department of Pharmacy, Henan Province Engineering Research Center of Application & Translation of Precision Clinical Pharmacy, the First Affiliated Hospital of Zhengzhou University, Zhengzhou 450052, China.
2. Key Laboratory of Advanced Drug Preparation Technologies, Ministry of Education, China; State Key Laboratory of Metabolic Dysregulation & Prevention and Treatment of Esophageal Cancer; Key Laboratory of Henan Province for Drug Quality and Evaluation; Institute of Drug Discovery and Development; School of Pharmaceutical Sciences, Zhengzhou University, Zhengzhou 450001, China.
3. Department of Orthopaedics, Qilu Hospital, Shandong University Centre for Orthopaedics, Advanced Medical Research Institute, Cheeloo College of Medicine, Shandong University, Shandong, 250012, China.
4. School of Food and Health Engineering, Zhengzhou University of Technology, Zhengzhou 450000, China
5. School of Chinese Medicine, University of Hong Kong, 3, Sasson Road, Pokfulam, Hong Kong.
6. Auckland Cancer Society Research Centre, Faculty of Medical and Health Sciences, The University of Auckland, Private Bag 92019, Auckland 1142, New Zealand.
7. Maurice Wilkins Centre, The University of Auckland, Private Bag 92019, Auckland 1142, New Zealand.
8. Department of Biomedicine and Medical Diagnostics, School of Science, Auckland University of Technology. 34 St. Paul Street, Auckland 1010, New Zealand.
#These authors contributed equally to this work.
Received 2026-1-3; Accepted 2026-3-21; Published 2026-4-8
Abstract

PD-1/PD-L1 pathway, a key immune checkpoint, triggers T-cell exhaustion via binding and aiding tumor immune evasion. Although several anti-PD-1/PD-L1 monoclonal antibodies (mAbs) have been granted food and drug administration (FDA) approval, their high cost, poor oral bioavailability, and potential immunogenicity have led to a shift in research toward small molecules. This review summarizes the structure and function of PD-1/PD-L1 and, based on the PD-1/PD-L1 signaling process, focuses on three major classes of related compounds: small molecule inhibitors inducing PD-L1 dimerization or blocking PD-1/PD-L1 binding; PD-L1 degraders (e.g., Proteolysis-targeting chimeras (PROTACs) and Lysosome-targeting chimeras (LYTACs)) via the ubiquitin-proteasome or lysosomal pathway, overcoming membrane protein targeting; and dual-target inhibitors that enhance therapeutic efficacy by exerting synergistic effects. While small molecule drugs have advantages over monoclonal antibodies, including oral administration and reduced immunogenicity, they face drug resistance and toxicity challenges. This review aims to provide insights into the discovery of safe and effective antitumor immunotherapeutic agents.
Keywords: PD-1, PD-L1, inhibitors, degraders, dual-target inhibitors, immunotherapy, anticancer
1. Introduction
Cancer continues to be the second leading cause of mortality worldwide, underscoring the critical demand for safer and more potent therapeutic interventions [1]. Malignant cells evade immune surveillance through diverse immunosuppressive mechanisms [2]. Immune checkpoint inhibitors constitute a promising therapeutic paradigm to amplify antitumor immunity by targeting these regulatory molecules [3]. Since the first cytotoxic T-lymphocyte-associated antigen 4 (CTLA-4) monoclonal antibody, ipilimumab, was approved in 2011, tumor immunotherapy, particularly Immune Checkpoint Therapy (ICT), has undergone remarkable progress and rapid clinical translation [4]. To date, many ICT monoclonal antibody drugs have been authorized globally for the therapeutic intervention of more than 20 tumor types, encompassing over 100 indications [5]. The durable clinical responses achieved using ICT have provided unprecedented therapeutic benefits to patients [6]. However, as protein-based therapeutics, monoclonal antibodies are associated with inherent drawbacks, including strong immunogenicity, slow metabolic clearance, poor tissue penetration, high production costs. These factors collectively restrict the broader clinical applications of ICT [7].
PD-1 and PD-L1 represent the most extensively characterized immune checkpoints [8]. Upon ligand binding, PD-1 initiates inhibitory signal transduction via its two key cytoplasmic motifs, namely the immunoreceptor tyrosine-based switch motif (ITSM) and the immunoreceptor tyrosine-based inhibitory motif (ITIM). PD-L1, also known as CD274 or B7-H1, is a member of the B7 family and a type I transmembrane glycoprotein, serving as the primary ligand for the PD-1 [9]. PD-L1 is highly expressed in various malignant tumors, such as lung cancer, gastric cancer, liver cancer and so on. [10]. PD-1 engagement with PD-L1 induces T-cell exhaustion, which in turn permits tumor cells to elude immune surveillance [11]. However, immune checkpoint therapy targeting PD-1/PD-L1 also has the aforementioned limitations [12, 13]. These limitations have spurred the development of small molecule inhibitors, which offer several significant advantages over monoclonal antibodies, such as enhanced tumor penetration, minimal immunogenicity, reduced manufacturing costs, and the feasibility of oral administration (Table 1). Their favorable pharmacokinetic properties make them compelling candidates for augmenting the scope and efficacy of cancer immunotherapy [14]. Currently, most PD-1/PD-L1 inhibitors in clinical stages are in phase I, and many are based on a biphenyl core structure. The sluggish clinical progress has prompted researchers to shift their focus to PROTACs, LYTACs, dual-target inhibitors and other alternative modalities, aiming to provide novel insights for PD-1/PD-L1 drug development [15].
Comparison between PD-1/PD-L1 small molecule inhibitors and monoclonal antibodies
| Feature | PD-1/PD-L1 monoclonal antibodies | PD-1/PD-L1 small molecule inhibitors | Advantage of small molecules |
|---|---|---|---|
| Molecular size | Large (~150 kDa) | Small (<1 kDa) | Easier tissue penetration |
| Target binding | Extracellular PD-1/PD-L1 blockade | Target PD-1/PD-L1 interaction or intracellular regulators | Broader targeting possibilities |
| Route of administration | Intravenous infusion | Often oral or injectable | More convenient administration |
| Pharmacokinetics | Long half-life (weeks) | Shorter half-life (hours-days) | Better dose control and flexibility |
| Tumor penetration | Limited in solid tumors | Improved diffusion in tumor microenvironment (TME) | Better access in dense tumor tissue |
| Manufacturing | Complex biologic production | Chemical synthesis, scalable | Lower production cost |
| Storage and stability | Requires cold chain | More stable, easier storage | Improved accessibility worldwide |
| Immunogenicity | Potential immune-related antibody responses | Generally low immunogenicity | Reduced risk of anti-drug antibodies |
| Immune-related adverse events | Often prolonged due to long persistence | Potentially reversible (shorter duration) | Safer management of toxicity |
| Target spectrum | Highly specific for PD-1/PD-L1 | Can be dual-target or multi-pathway modulators | Expanded therapeutic scope |
| Intracellular accessibility | Cannot enter cells | Can modulate intracellular signaling molecules | Expanded druggable space |
| Combination potential | Limited by long exposure and toxicity overlap | Flexible combinational regimens | Improved adaptability |
| Cost | Very expensive | Potentially lower cost | Improved affordability |
Existing reviews on the PD-1/PD-L1 pathway have predominantly focus on monoclonal antibodies, with small molecule drugs often mentioned only peripherally or restricted to a single class of compounds [15-19]. In contrast, this review provides a comprehensive and systematic focus on small molecule drugs targeting the PD-1/PD-L1 axis. It delineates the structural characteristics and biological functions of this pathway, and traces the evolutionary progression of both small molecule inhibitors and degraders. A particular emphasis is placed on the recent advancements in dual-target inhibitors, which co-inhibit the PD-1/PD-L1 pathway alongside other key signaling cascades to enhance antitumor efficacy. By integrating discussions on mechanisms of action, structural classification, drug design strategies, and clinical challenges, this review aims to consolidate the core breakthroughs in the field and outline future directions for the development of next-generation PD-1/PD-L1-targeted small molecule therapeutics.
2. Structure and functions of PD-1/PD-L1
2.1. Structure of PD-1/PD-L1
PD-1 functions as a type I transmembrane glycoprotein (approximately 50-55 kDa) that is critical to regulating immune tolerance, especially in T cells [9]. It comprises an extracellular IgV-like domain, a transmembrane region, and an intracellular tail harboring critical signaling motifs [11]. The extracellular domain exhibits an α/β-sandwich immunoglobulin fold, consisting of nine β-strands linked by eight loops—notably, a flexible N-terminal loop is indispensable for ligand engagement. The structural integrity is maintained by a disulfide bond linking the cysteine residues at positions 54 and 123 [20]. The intracellular domain encompasses two key signaling motifs, namely an ITSM and ITIM, which are crucial for transducing inhibitory signals [21]. PD-1 shares a high degree of structural similarity with CD28 and CTLA-4, suggesting that they may utilize the same class of ligands, such as members of the B7 or MHC class families [22].
PD-L1, the most canonical ligand for PD-1, is a 290-amino-acid type I transmembrane protein of ~33 kDa belonging to the B7 family of immunoregulatory molecules. [9]. It plays a critical role in modulating T-cell activation and immune tolerance [23]. Its structure consists of an N-terminal signal peptide, an IgV-like domain containing N-linked glycosylation sites, an IgC-like domain conferring extracellular stability, a transmembrane domain, and a cytoplasmic tail featuring s-palmitoylation sites. N-linked glycosylation within these domains is essential for regulating PD-L1 expression and function activity [24, 25].
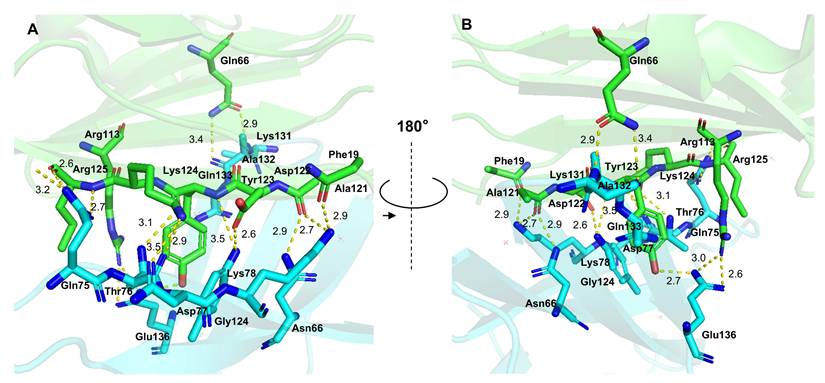
The PD-1/PD-L1 combination is primarily mediated by hydrophobic residues localized in the C, C', F, and G strands of both proteins, which form a central component critical for mediating immunosuppressive effects (Figure 1) [26]. Targeting and disrupting this protein-protein binding interface serve as a primary approach for small molecule inhibitors designed to block the PD-1/PD-L1 immune checkpoint [27].
Ribbon representation of human PD-1 (blue) and human PD-L1 (green) complex (PDB ID: 4ZQK). (A) Front view. (B) Back view. Yellow dashed lines indicate hydrogen bonds stabilizing the interaction. Sticks represent the interacting residues.
2.2. Functions of PD-1/PD-L1
PD-1 is mainly expressed on the surface of activated immune cells, including T cells, B cells, natural killer (NK) cells, macrophages, dendritic cells (DCs), and monocytes, especially on tumor-specific T cells and functionally exhausted T cells, PD-1 is highly expressed [28-30]. Upon PD-L1 association with PD-1, the tyrosine residues within the ITIM and ITSM motifs of PD-1 are phosphorylated by specific kinases, such as Lck or Src family kinases in T cells, or Lyn in B cells. This phosphorylation event promotes the association of Src homology 2 (SH2) domain-containing tyrosine phosphatase 2 (SHP2) with PD-1. Subsequently, SHP2 mediates the dephosphorylation of several critical downstream signaling molecules, including ZAP70, PI3K, and Ras, thereby suppressing signaling through the T cell receptor (TCR) and the co-stimulatory receptor CD28 [11]. The PD-1 signaling pathway further inhibits important T cell activation pathways such as PI3K/AKT and RAS-ERK1/2 [31]. The cumulative effect of this series of inhibitory signals is comprehensive, resulting in restricted T cell proliferation, reduced secretion of effector cytokines (such as IL-2 and IFN-γ), decreased cytotoxicity, and ultimately leading T cells to enter a state of functional impairment or “exhaustion” [32].
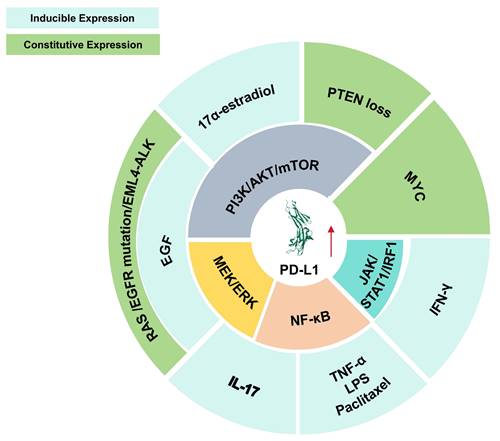
PD-L1 is widely expressed across various cancer types [33]. Its expression is induced by extrinsic stimuli (e.g., 17α-estradiol induces PD-L1 expression by activating the PI3K/AKT signaling pathway. Similarly, epidermal growth factor (EGF) upregulates PD-L1 by stimulating both the PI3K/AKT/mTOR and MEK/ERK pathways. Interleukin-17 (IL-17) enhances PD-L1 expression via the MEK/ERK and NF-κB signaling axes. Other stimuli, including tumor necrosis factor-alpha (TNF-α), lipopolysaccharide (LPS), and paclitaxel, also elevate PD-L1 levels by activating the NF-κB pathway. Additionally, interferon-gamma (IFN-γ) promotes PD-L1 expression through the JAK/STAT1/IRF1 signaling cascade.) or undergoes constitutive upregulation via oncogenic driver mutations and epigenetic alterations in cancer cells (e.g., phosphatase and tensin homolog (PTEN) functional impairment or loss activates the PI3K/AKT/mTOR signaling cascade, thereby enhancing PD-L1 expression. Genetic alterations in rat sarcoma virus (RAS) and epidermal growth factor receptor (EGFR), as well as EML4-ALK chromosomal translocations, concurrently activate both the PI3K/AKT/mTOR and MEK/ERK pathways, ultimately leading to PD-L1 upregulation. Furthermore, the MYC oncogene directly contributes to and promotes PD-L1 expression) (Figure 2) [34]. Besides binding to PD-1, PD-L1 has also been demonstrated to bind to B7-1 (CD80), inhibiting T cell costimulatory signaling by displacing CD80 from CD28 and further restricting T cell activation [35, 36]. In addition to indirectly promoting tumor growth and survival by inhibiting immunity through receptor binding, the intracellular domain of PD-L1 itself also facilitates tumor growth and metastasis via ligand-independent signaling mechanisms [37].
Mechanisms governing PD-L1 expression in cancer cells. PD-L1 expression in cancer cells is regulated through both inducible and constitutive mechanisms. Extrinsic stimuli, including EGF, 17α-estradiol, IFN-γ, TNF-α, IL-17, lipopolysaccharide, and paclitaxel, can enhance PD-L1 expression through multiple signaling cascades, such as PI3K/AKT/mTOR, MEK/ERK, NF-κB, and JAK/STAT1/IRF1. In parallel, constitutive PD-L1 upregulation may arise from oncogenic alterations and tumor-intrinsic events, including PTEN loss, RAS or EGFR activation, EML4-ALK rearrangement, and MYC-driven transcriptional regulation. Together, these regulatory inputs promote persistent PD-L1 expression in tumor cells and contribute to immune escape within the tumor microenvironment. The outer ring indicates upstream stimuli or genetic events, and the inner ring indicates intracellular pathways. Red arrows indicate upregulation.
Within the TME, tumor cells frequently exploit the PD-1/PD-L1 pathway to suppress T cell function and evade immune monitoring [38]. The interaction between PD-L1 and PD-1 induces phosphatase activation that attenuates TCR signaling, thereby impairing T cell activity and facilitating immune evasion [39, 40]. To establish this immunosuppressive state, tumors upregulate PD-L1 through diverse molecular strategies, ranging from gene amplification and epigenetic changes to the activation of oncogenic pathways and alterations at the transcription and post-translational levels [41]. Given its central role, disrupting the PD-1/PD-L1 interaction has emerged as a powerful therapeutic approach to reinvigorate T-cell immunity and treat a wide array of malignant tumors [42].
2.3. Downstream molecular mechanisms of PD-1/PD-L1-mediated immunosuppression
During T cell activation, several canonical signaling pathways are engaged. Classical TCR signaling is initiated upon antigen recognition, which triggers activation of the downstream tyrosine kinase ZAP70 through the CD3ζ chain. CD3ζ and ZAP70 are widely regarded as central “switches” in proximal TCR signaling [43, 44]. Activated ZAP70 subsequently phosphorylates the adaptor protein linker for activation of T cells (LAT), thereby initiating multiple critical downstream signaling cascades. (i) The NFAT signaling axis represents the most well-established pathway downstream of LAT. Phosphorylated LAT recruits phospholipase Cγ1 (PLCγ1), which catalyzes the hydrolysis of phosphatidylinositol 4,5-bisphosphate (PIP2) into diacylglycerol (DAG) and inositol trisphosphate (IP3) [45]. IP3 triggers intracellular Ca2+ release and activates calcineurin [46], which subsequently promotes the nuclear translocation of NFAT transcription factors [47]. (ii) In parallel, LAT recruit the adaptor protein Grb2, which associates with SOS to activate the Ras-Raf-MEK-ERK signaling cascade (the MAPK pathway). This cascade ultimately induces the formation of the AP-1 transcription factor complex [48]. (iii) Moreover, DAG generated from PLCγ1-medisted PIP2 hydrolysis activates protein kinase Cθ (PKCθ) [49], which stimulates the CARMA1-BCL10-MALT1 (CBM) complex, this leads to IKK activation and subsequent induction of the NF-κB signaling pathway [50]. (iv) Beyond these classical pathways, T cell activation also engages the PI3K/AKT/mTOR signaling axis. Although this pathway is typically associated with TCR signaling, it is prominently driven by CD28-mediated costimulation. Mechanistically, phosphorylated CD28 recruits PI3K, which catalyzes the conversion of PIP2 to PIP3, thereby activating AKT and subsequently mTORC1. Notably, this pathway represents one of the major targets of PD-1/PD-L1 immune checkpoint-mediated inhibition [51, 52].
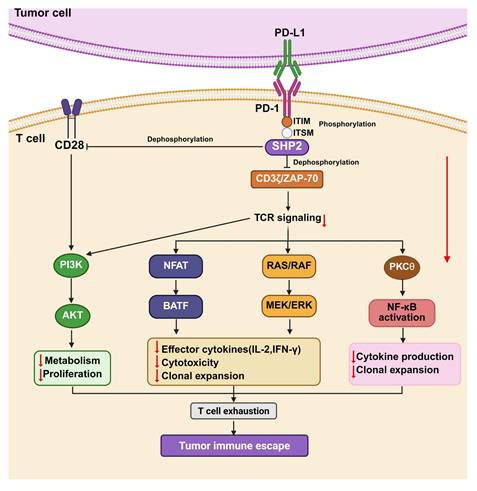
The canonical inhibitory function of the PD-1/PD-L1 pathway is primarily mediated by the recruitment and activation of SHP2 [53]. As described above, upon PD-L1 engagement, tyrosine residues within the ITIM and ITSM motifs of PD-1 are phosphorylated by specific kinases, leading to the recruitment of SHP2 (Figure 3). Once activated, SHP2 dephosphorylates multiple key downstream signaling molecules, including ZAP70, PI3K, and Ras, thereby suppressing signal transduction from both the TCR complex and the CD28 costimulatory receptor (Figure 3). Specifically, SHP2-mediated dephosphorylate of CD28 inhibits the PI3K/AKT/mTOR pathway [52, 54], thereby impairing T cell metabolic activation and proliferation. Concurrently, SHP2 promotes the dephosphorylation of ZAP70 and CD3ζ, reducing their interaction [55], and leading to diminished phosphorylation of LAT [56]. As LAT serves as a key signaling hub that orchestrates the NFAT, MAPK, and NF-κB pathways, its inactivation results in broad suppression of these downstream effector cascades, ultimately attenuating T cell activation and functional responses.
Downstream molecular mechanisms of PD-1/PD-L1-mediated immunosuppression. Upon PD-L1 binding, PD-1 undergoes cytoplasmic phosphorylation and recruits the phosphatase SHP2. Activated SHP2 dampens proximal TCR/CD28 signaling by dephosphorylating key signaling molecules, including CD3ζ, ZAP-70, and CD28, thereby weakening LAT-dependent signal propagation. As a consequence, multiple effector pathways, such as PI3K/AKT/mTOR, NFAT, Ras/RAF/MEK/ERK, and NF-κB, are coordinately suppressed. This signaling blockade limits T-cell activation, proliferation, cytokine production, and cytotoxic function, ultimately driving T-cell exhaustion and facilitating tumor immune evasion. Black arrows indicate activation, and T-shaped lines indicate inhibition. Red arrows denote changes in expression levels, with downward arrows indicating downregulation or inhibition.
The binding of PD-1 and PD-L1 activates multiple intracellular signal transduction cascades that suppress T cell function. Upon PD-1/PD-L1 ligation, the tyrosine phosphatase SHP2 is recruited to the dephosphorylated ITSM of PD-1, where it dephosphorylates proximal signaling molecules, such as ZAP70/CD3ζ, thereby attenuating TCR signaling [57]. These events lead to the downregulation of several signaling pathways, such as PI3KAKT/mTOR, NF-κB, and Ras-MAPK axes, ultimately inhibiting T cell activation, suppressing clonal proliferation, reducing proinflammatory cytokines, and promoting immune evasion. The functional consequences of PD-1/PD-L1 signaling extend beyond T cell modulation and vary across cancer types. In non-small cell lung carcinoma (NSCLC), the EML4-ALK fusion oncogene drives PD-L1 overexpression through activation of the MEK-ERK and PI3K-AKT pathways. Meanwhile, in diffuse large B-cell lymphoma (DLBCL), PD-1 engagement directly triggers AKT/mTOR signaling in malignant cells, a mechanism associated with poor clinical outcomes [58]. A comprehensive understanding of these signaling networks is essential for the rational design of effective therapeutic strategies. For instance, combining PD-1/PD-L1 immune checkpoint blockade with inhibitors targeting the MEK-ERK or PI3K-AKT pathways holds promise for synergistically enhancing antitumor immunity. Furthermore, targeting key nodes within the PD-1/PD-L1 signaling cascade—such as the phosphatase SHP2—represents an emerging and promising approach to augment the efficacy of cancer immunotherapies.
2.4. Targeted inhibition of the PD-1/PD-L1 axis is an effective strategy for cancer treatment
In the TME, PD-L1 expression is significantly upregulated, leading to engagement of the PD-1/PD-L1 axis and subsequent T cell dysfunction. This phenomenon has been documented across multiple cancer types, with distinct regulatory mechanisms and clinical implications. For example, in chronic lymphocytic leukemia (CLL), crosstalk between malignant cells and stromal components drives PD-L1 overexpression via the Notch-c-Myc-EZH2 signaling cascade, thereby enhancing resistance to autologous T lymphocyte-mediated killing [59]. In plasmablastic lymphoma (PBL), a subset of cases characterized by elevated PD-L1 expression in both cancer cells and the TME correlates with reduced overall survival [60]. In head and neck cancer, aneuploidy-associated chromosome 9p deletion contributes to immune evasion, while arm-level 9p loss accompanied by JAK2-PD-L1 co-deletion serve as a prognostic indicator of poor response to anti-PD-1 immunotherapy [61]. Collectively, these findings underscore the critical role of the PD-1/PD-L1 signaling pathway in tumor-mediated immune evasion and highlight the therapeutic potential of targeting this pathway to enhance antitumor immune responses.
Accumulating evidence demonstrates that targeting the PD-1/PD-L1 axis represents a promising strategy for remodeling the TME and enhancing antitumor immunity. In head and neck squamous cell carcinoma (HNSCC), unique spatial distribution characteristics of CD68⁺ and CD163⁺ leukocytes have been identified, with PD-L1-high (PD-L1hi) and PD-1-high (PD-1hi) cells preferentially accumulating around tumor foci. Notably, elevated peritumoral infiltration densities of PD-L1hiCD68hiCD163hi cells or PD-1hi T cells correlate with favorable patient survival, suggesting that PD-1/PD-L1 crosstalk between specific cellular subsets within the TME can modulate clinical outcomes [62]. In pancreatic ductal adenocarcinoma (PDAC), neoadjuvant radioimmunotherapy combining the GVAX vaccine, anti-PD-1 agents, and stereotactic body radiation (SBRT) reshapes the TME toward an antitumor immune profile. This combinatorial regimen increases infiltration of GZMB⁺CD8⁺ T cells, TH1, and TH17, while concurrently increasing the proportion of immunosuppressive M2-like tumor-associated macrophages (TAMs) [63]. In clear cell renal cell carcinoma (ccRCC), engineered chimeric antigen receptor T (CAR-T) cells targeting carbonic anhydrase IX (CAIX) and secreting anti-PD-L1 monoclonal antibodies restore robust antitumor immunity. This effect is achieved by enhancing tumor-killing cytotoxic activity, reducing immunosuppressive cell populations, and strengthening T follicular helper (Tfh)-B cell crosstalk within the TME [64]. Collectively, these findings underscore the pleiotropic role of PD-1/PD-L1 interactions in TME remodeling and reinforce that therapeutic targeting of this axis represents a well-established and effective therapeutic strategy for cancer.
3. PD-1/PD-L1 inhibitors
3.1. PD-L1 inhibitors
Based on their developmental status, PD-L1 small molecule inhibitors can be divided into those that have entered clinical trials and those still in preclinical research. The following section first introduces inhibitors that have reached clinical evaluation, followed by a summary of representative compounds at the preclinical stage.
3.1.1. Clinical-stage PD-L1 inhibitors
Gilead Sciences developed GS-4224 (evixapodlin) (1, Table 2), a C2-symmetric tetraaryl PD-L1 inhibitor [65]. Its symmetric structure enables simultaneous binding to two PD-L1 molecules, promoting PD-L1 dimerization and thereby blocking the PD-L1/PD-1 interaction (IC50 = 0.213 nM). In vitro, GS-4224 effectively reversed PD-L1- mediated T cell suppression, augmented immune-mediated tumor cell lysis, and demonstrated activity comparable to the PD-L1 antibody atezolizumab. In MC38 murine models harboring PD-L1 knock-in modification, GS-4224 achieved greater than 90% target occupancy and elicited robust antitumor efficacy. A phase I clinical trial (NCT04049617) reported favorable pharmacokinetics and tolerability, alongside dose-dependent T cell activation and cytokine induction, although the trial has now been terminated [65, 66].
Clinical small molecule PD-L1 inhibitors
| Compd. | Inventor | Structure | Biological data | Clinical Trail | Ref. |
|---|---|---|---|---|---|
| GS-4224 (1) | Gilead Sciences | 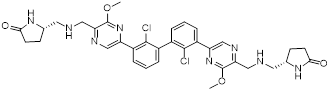 | IC50 = 0.213 nM | NCT04049617, phase I; (Terminated) | [65] |
| CCX559 (2) | ChemoCentryx, Inc. | - | IC50 = 0.47 nM | ACTRN12621001342808, phase I | [67, 68] |
| ASC61 (3) | GannexPharma Co., Ltd. | - | - | NCT05287399, phase I | [69] |
| MAX-10181 (4) | Maxi novel Pty, Ltd. | 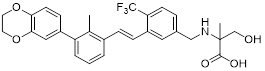 | IC50 = 18 nM | NCT05196360, phase I; (Unknown status) NCT04122339, phase I; (Unknown status) | [70] |
| BPI-371153 (5) | Betta Pharmaceuticals Co, Ltd. | - | IC50 = 4.2 nM | NCT05341557, phase I (Recruiting) | [72] |
| INCB086550 (6) | Incyte Corporation | 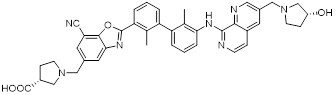 | IC50 = 3.1 nM | NCT03762447, phase I; (Completed)、 NCT04674748, phase I; (Terminated)、 NCT04629339, phase II; (Terminated)、 NCT05101369, phase I; (Completed) | [73, 74] |
| YPD-29B (7) | Tianjin Chasesun Pharmaceutical Co., Ltd. | 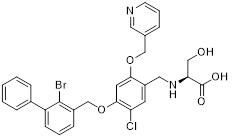 | IC50 < 0.1 pM | NCT04343859, phase I; (Unknown status) | [77] |
“-” means the data is unavailable (not public or non-existent).
ChemoCentryx Inc. synthesized CCX559 (2, Table 2), a substituted biphenyl-based small molecule that promotes PD-L1 dimerization and subsequent internalization, thereby effectively blocking PD-1 engagement (IC50 = 0.47 nM) [67]. In preclinical murine models, this agent demonstrated significant antitumor activity and induced reversible PD-L1 internalization. A phase I dose-escalation trial (ACTRN12621001342808) confirmed potent inhibition of PD-L1 binding to both PD-1 and CD80 in vitro. In MC38 tumor models, cell-surface PD-L1 expression was restored following treatment discontinuation, indicating the reversibility of its mechanism of action [68].
Wu et al. reported in a conference presentation that ASC61 (3, Table 2) was a PD-L1 inhibitor [69]. As a prodrug, this compound was metabolized in vivo to form its active metabolite, ASC61A, which induces PD-L1 dimerization and internalization, thereby effectively restoring T cell function. In CT-26-hPD-L1 tumor-bearing BALB/c mouse model, ASC61 exhibited the most potent antitumor activity among all tested compounds evaluated in the study.
MaxiNovel developed MAX-10181 (4, Table 2), an active PD-L1 inhibitor that exhibited potent activity and efficacy comparable to that of durvalumab (IC50 = 18 nM) [70]. A phase I trial (NCT04122339) conducted in Australia and China demonstrated good safety and tolerability, along with consistent pharmacokinetic profiles across diverse ethnic populations. Notably, among subjects with PD-L1 expression levels below 5%, disease control rates were comparable to those achieved with pembrolizumab. Furthermore, meaningful clinical activity responses were observed in individuals who were resistant or intolerant to prior PD-1/PD-L1 antibody therapy [71].
Wang et al. synthesized BPI-371153 (5, Table 2), a PD-L1 inhibitor that induced PD-L1 dimerization and enhanced its internalization (IC50 = 4.2 nM) [72]. This compound subsequently activated NFAT signaling and promoted IFN-γ release, leading to restored T cell function. In preclinical models, BPI-371153 significantly inhibited tumor growth and demonstrated favorable pharmacokinetic properties, including high oral bioavailability.
Koblish et al. discovered INCB086550 (6, Table 2), a biphenyl-based PD-L1 inhibitor [73]. In biochemical assays, this compound potently interfered with the PD-1/PD-L1 interaction while simultaneously promoting PD-L1 dimerization and subsequent internalization (IC50 = 3.1 nM). Mechanistic studies further revealed that treatment with INCB086550 induced transcriptional programs indicative of T cell activation in vivo [73]. This finding aligns with clinical observations from a phase I clinical trial (NCT03762447), in which INCB086550 demonstrated dose-dependent T cell activation [74, 75].
Chen et al. identified IMMH-010 as a candidate drug after in vitro and in vivo screening and structural modifications [76, 77]. IMMH-010 was identified as a prodrug that undergoes rapid in vivo metabolism to yield its bioactive form YPD-29B (7, Table 2). YPD-29B potently abrogated the PD-1/PD-L1 interaction (IC50 < 0.1 pM) and exhibited a markedly prolonged half-life within tumors relative to plasma, likely due to its high receptor-binding affinity, resulting in sustained intratumoral drug accumulation and improved therapeutic outcomes [78]. A phase I trial (NCT04343859) was completed, but its current status is unknown.
3.1.2. Preclinical-stage PD-L1 inhibitors
In addition to the compounds that have reached clinical evaluation, numerous PD-L1 small molecule inhibitors remain at the preclinical stage and display promising in vitro and in vivo activities.
Hermanowicz et al. described MM-129 (8, Table 3), a heterofused 1,2,4-triazine derivative [79]. This compound inhibited the expression of PD-L1 and induced G0/G1 phase cell cycle arrest, thereby reducing the proliferative viability of colorectal cancer cells and impairing DNA synthesis (IC50 = 3.1 μM in DLD-1 cell) [80]. When administered at 10 µmol/kg in murine models, MM-129 significantly impeded tumor progression without inducing nephrotoxicity or adverse hematological effects [81]. Furthermore, combination therapy with indoximod (IDO1 inhibitor) enhanced antitumor responses in colon cancer models, suggesting potential for combinatorial immunotherapy approaches [82].
Preclinical small molecule PD-L1 inhibitors
| Compd. | Inventor | Structure | Biological data | Ref. |
|---|---|---|---|---|
| MM-129 (8) | Medical University of Bialystok | 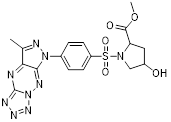 | IC50 = 3.1 μM (DLD-1 cell) | [79] |
| S4-1 (9) | China Pharmaceutical University | 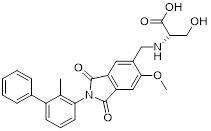 | IC50 = 6.1 nM | [83] |
| CB31 (10) | Chulalongkorn University | 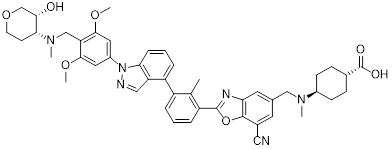 | IC50 = 0.2 nM | [84] |
| APBC (11) | Lanzhou University | 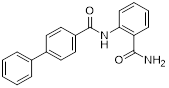 | IC50 = 27.82 μM | [85] |
| Pt-2 (12) | Sun Yat-sen University | 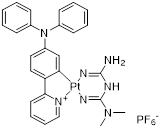 | IC50 = 490 nM | [86] |
| SA-49 (13) | Chinese Academy of Medical Sciences | 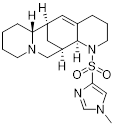 | - | [87] |
| BMS-202 (14) | BMS | 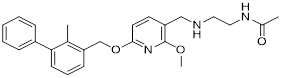 | IC50 = 18 nM | [88, 90] |
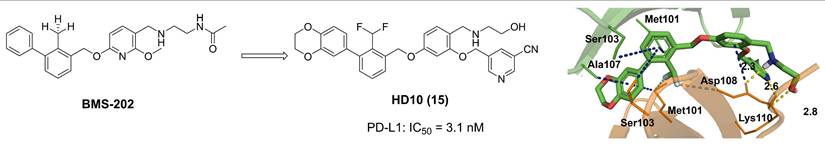
| HD10 (15) | Zhejiang University of Technology | 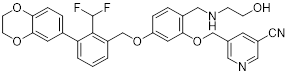 | IC50 = 3.1 nM | [93] |
| Compound 16 | China Pharmaceutical University | 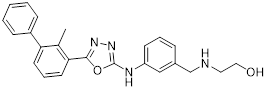 | IC50 = 27.8 nM | [94] |
| NPH 16 (17) | China Pharmaceutical University Southern Medical University | 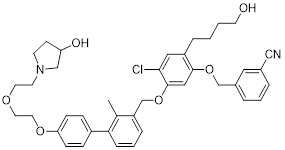 | IC50 = 24.4 nM | [95] |
| D6 (18) | Capital Medical University | 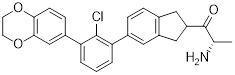 | IC50 = 4.8 nM | [96] |
| ARB-272572(19) | Arbutus Biopharma Inc, Xtal BioStructures Inc. | 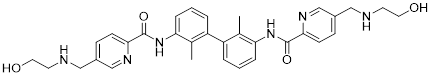 | IC50 = 400 pM | [97] |
| Anidulafungin (20) | College of Pharmacy, Jiangsu University | 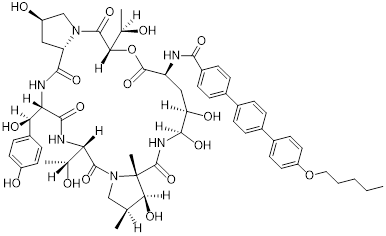 | - | [98] |
| CBPA (21) | Lanzhou University |  | IC50 = 57.67 μM (MC38 cell) IC50 = 77.45 μM (B16F10 cell) | [99] |
“-” means the data is unavailable (not public or non-existent).
Sun et al. designed PD-L1 inhibitors with biphenyl scaffolds, among which S4-1 (9, Table 3) exhibited the highest activity [83]. This compound interfered with PD-1/PD-L1 binding by inducing PD-L1 dimerization and cellular internalization, leading to its aberrant accumulation in the endoplasmic reticulum and potential proteolytic degradation (IC50 = 6.1 nM). In vitro, S4-1 markedly boosted T cell activation and strengthened the cytotoxic capacity of peripheral blood mononuclear cells (PBMCs) toward tumor cells. In murine models of lung and colorectal cancer, this compound exhibited robust antitumor efficacy. Notably, in a humanized colorectal cancer model, S4-1 achieved an 88.8% tumor growth inhibition rate, underscoring its promising therapeutic potential.
Prucksaritanont et al. developed novel tetra-aryl-scaffold PD-L1 inhibitors using ring fusion design and structural extension strategies, among which CB31 (10, Table 3) exhibited the strongest activity in blocking PD-1/PD-L1 binding (IC50 = 0.2 nM) [84]. This compound triggered the internalization of surface PD-L1, altered its glycosylation pattern leading to intracellular retention, and reduced the total PD-L1 protein levels (IC50 = 0.2 nM). It exhibited minimal off-target cytotoxicity, and enhanced TCR-dependent cytokine release as well as PBMC-mediated tumor cell killing.
Wang et al. developed APBC (11, Table 3), a PD-L1 inhibitor featuring a novel scaffold composed of a biphenyl ring and a carbamoylphenyl group [85]. This compound directly bound to the PD-L1 dimer, locked its conformation, and blocked the PD-1 binding site (IC50 = 27.82 μM). In MC38 mouse tumor models, APBC significantly increased the infiltration density of CD4⁺ and CD8⁺ T cells in tumor tissue, promoted cytokines production without inducing hepatotoxicity, and exhibited superior therapeutic efficacy and plasma stability.
Mao et al. developed Pt-2 (12, Table 3), a cyclometalated platinum-metformin conjugate that functions as a small molecule PD-L1 inhibitor [86]. The cyclometalated platinum moiety played multiple pivotal roles: it markedly enhanced the intracellular delivery efficiency of metformin, enabled its selective accumulation in lysosomes, endowed the conjugate with favorable photophysical properties for real-time cellular imaging, and provided potent chemotherapeutic activity against tumor cells. Notably, Pt-2 selectively accumulated in lysosomes, where it not only disrupts the PD-1/PD-L1 signaling pathway at the cell surface but also inhibited PD-L1 expression by the AMPK-TFEB pathway. Specifically, it activated AMPK to promote TFEB-mediated lysosomal biogenesis and function, thereby facilitating the lysosomal degradation of PD-L1 while concurrently suppressing its expression (IC50 = 490 nM).
Zhang et al. characterized SA-49 (13, Table 3) as a PD-L1 inhibitor within a library of novel aloperine analogs [87]. At the cellular level, SA-49 reduced both constitutive and IFN-γ-induced PD-L1 expression in NSCLC cells in a time- and concentration-dependent manner, exerted extremely low cytotoxicity on NSCLC cells, and enhanced the cytotoxicity of co-cultured T cells and NK cells against tumor cells. In vivo, SA-49 significantly inhibited the growth of Lewis tumor xenografts in C57BL/6 mice via intragastric administration, decreased the expression of PD-L1 in tumor tissues, increased the number of CD3+ T cells and reduced the number of FoxP3+ Treg cells, thus activating the TME. Meanwhile, it had no obvious effects on the body weight, major organs and serum biochemical indicators of mice, with no significant systemic toxicity observed.
Bristol-myers squibb (BMS) developed BMS-202 (14, Table 3) as a lead prototypical compound via structural optimization aligned with the hydrophobic pocket characteristics of the PD-L1 dimerization interface, complemented by comprehensive verification of its biological activity and underlying molecular mechanism [88-90]. BMS-202 disrupted PD-1/PD-L1 receptor-ligand complex formation by binding to a hydrophobic cleft generated upon PD-L1 dimerization (IC50 = 18 nM) [26]. Beyond immune checkpoint blockade, BMS-202 modulated extracellular matrix biology by suppressing type I collagen synthesis in fibroblasts and promoting branched-chain amino acid transaminase 1 (BCAT1) expression, thereby enhancing L-isoleucine catabolism and impairing the malignancy of glioblastoma multiforme. BMS-1166, an optimized derivative of BMS-202, introduced a 1,4-benzodioxane moiety at the terminus of the biphenyl ring and incorporated an m-cyanobenzyl alcohol group into the central benzene ring [91, 92]. This derivative induced PD-L1 dimerization, a mechanism consistent with that of BMS-202. However, BMS-1166 exhibited a significantly enhanced binding affinity, with its IC50 value decreased drastically to 1.4 nM.
Zhang et al. designed and developed compound HD10 (15, Table 3) as a novel PD-L1 inhibitor (IC50 = 3.1 nM) based on BMS-202 (Figure 4) [93]. HD10 bound PD-L1 with high affinity, and its co-crystal structure revealed critical interactions between its difluoromethylbiphenyl core and the PD-L1 dimer interface (PDB ID: 9ERY). At the cellular level, HD10 could effectively block the binding between hPD-1 293T cells and hPD-L1, promote the secretion of IFN-γ in PBMCs in a dose-dependent manner to restore T cell function without significant toxicity to PBMCs. It could significantly induce apoptosis in HCC827 and MDA-MB-231 cells with high PD-L1 expression, but had no obvious effect on A549 cells with low PD-L1 expression. In vivo, when administered orally at 50 mg/kg in the PD-1/PD-L1 humanized mouse model, the tumor growth inhibition rate (TGI) reached 57.31% without obvious toxicity. It could activate the immune system by increasing the proportion of tumor infiltrating CD3⁺ and CD8⁺ T cells, upregulating the expression of CXCR3/CXCL9/CXCL10 chemokines, and enhancing the levels of granzyme B and perforin. In addition, HD10 possesses favorable pharmacokinetic properties.
Discovery of HD10 and its co-crystal structure with PD-L1 (PDB ID: 9ERY). Yellow dashed lines depict hydrogen bonds. Gray dashed line indicates an electrostatic interaction. Blue dashed line highlights a π-sulfur interaction.
Wang et al. designed oxadiazole-based structures, among which compound 16 (Table 3) exhibited the strongest ability [94]. This compound bound to both human and murine PD-L1 and induced its dimerization (IC50 = 27.8 nM). It promoted time- and concentration-dependent internalization of cell-surface PD-L1, followed by degradation via a lysosome-dependent pathway, while upregulating the expression of lysosome-related genes. In vitro, it attenuated PD-1/PD-L1 binding and activated PBMCs-mediated antitumor immunity in a dose-dependent manner. In vivo, oral administration exhibited dose-dependent antitumor effects in both CT26 colon cancer and B16-F10 melanoma mouse models. At a dose of 160 mg/kg, it achieves a 75% tumor growth inhibition rate of CT26 tumors without obvious toxicity. Mechanistically, high-doses treatment reduced PD-L1 expression in tumor tissues, increased CD8⁺ cell infiltration, and elevated the expression of immune factors.
Xiao et al. designed PD-L1 inhibitors by introducing hydrophilic tail groups at the end of the biphenyl skeleton, among which NPH 16 (17, Table 3) exhibited the most potent activity (IC50 = 24.4 nM) [95]. It bound efficiently to the PD-L1 dimer through hydrogen bonds and hydrophobic interactions, and showed concentration-dependent binding ability to both human and murine PD-L1. In the HepG2/Jurkat coculture model, NPH 16 promoted tumor cell apoptosis in a dose-dependent manner. In vivo, it increased the infiltration of CD3⁺CD8⁺T cells and the level of IFN-γ in tumor tissues, while reduced PD-L1 expression, with no obvious organ toxicity. Meanwhile, it possessed excellent drug-like properties with a water solubility of 0.794 mg/mL and an oral bioavailability of 15.9%.
Lu et al. synthesized D6 (18, Table 3), a derivative based on the 5-phenylindoline [96]. This compound formed a stable binding with the hydrophobic pocket of the PD-L1 dimer, thereby blocking the PD-L1/PD-1 interaction (IC50 = 4.8 nM). In vitro, 100 nM D6 significantly promoted IFN-γ secretion from PBMCs and inhibited immune cells apoptosis. In vivo, D6 exhibited dose-dependent tumor growth inhibitory in the MC38 tumor-bearing mouse models. Mechanistically, it activated the TME by enhancing the infiltration and activation of CD8⁺ T cells, upregulating the levels of cytokines such as IFN-γ, and inhibiting tumor cell proliferation.
Moore et al. identified ARB-272572 (19, Table 3) as a potent PD-L1 inhibitor through a small molecule library [97]. Subsequent research demonstrated that ARB-272572 triggered PD-L1 dimerization and subsequent internalization into the cytosol, leading to reduced cell surface PD-L1 levels (IC50 = 400 pM). In a humanized PD-1/PD-L1 colorectal cancer mouse model, administration at 10 mg/kg for 7 days reduced average tumor volume by 60.4%, accompanied by decreased PD-L1 expression in tumors, increased peripheral blood CD3+/CD4+ T cells, and a reduction in regulatory T cells. In patients with chronic hepatitis B, ARB-272572 decreased the PD-L1 expression on the surface of dendritic cells and B cells, significantly enhanced the proliferation of HBV-specific T cells and IFNγ secretion, and elevated the frequency of HBsAg-specific B cells. These findings highlight the dual biological activity of ARB-272572 against both tumors and chronic viral infections.
Feng et al. conducted a virtual screening of 3,906 small molecules and approved drugs, including those used for immune regulation and antitumor traditional Chinese medicine, and identified anidulafungin (20, Table 3) inhibited PD-L1 expression [98]. Research found that anidulafungin directly bound to PD-L1, decreasing the PD-1/PD-L1 intermolecular binding energy from -63.9 kcal/mol to -36.8 kcal/mol, forcing PD-1 to deviate from its original binding site. In addition, it activated both systemic and local antitumor immunity, demonstrating markable antitumor efficacy in both in vitro and in vivo experimental models.
Wang et al. identified CBPA (21, Table 3) as a potent inhibitor of PD-1/PD-L1 signaling through screening [99]. CBPA bound to dimeric PD-L1 and occluded the PD-1 interaction surface on PD-L1. It restored T cell function and significantly promoted the secretion of proinflammatory cytokines such as IFN-γ and TNF-α by primary CD4+ T cells. In vivo, in the MC38 colon adenocarcinoma and B16F10 melanoma mouse models, intraperitoneal injection of CBPA at 10 mg/kg achieved tumor growth inhibition rates of 67.20% and 45.26% respectively, without obvious liver or kidney function damage or body weight loss. Mechanistically, CBPA significantly increased the infiltration of CD4+ and CD8+ T cells in the TME, promoted the secretion of perforin and granzyme B by intratumoral CD8+ T cells, and upregulated immune-related pathways such as antigen processing and presentation and T cell receptor signaling.
In the field of PD-1/PD-L1-targeted small molecule cancer immunotherapy, biphenyl-based and C2-symmetric tetraaryl compounds represent the most clinically translatable core chemotypes. These agents exert their effects either by inducing PD-L1 dimerization and internalization or by directly blocking the protein-protein interactions. Several such molecules have entered phase I clinical trials, demonstrating favorable pharmacokinetic profiles and promising antitumor activity. However, the field still faces multiple challenges associated with clinical failure, including pharmacokinetic issues (such as short half-lives and poor water solubility) [100], insufficient selectivity leading to off-target toxicity [101], immune-related adverse events (irAEs) [102], in vitro assay artifacts [103], clinical translation biases caused by species differences [104], and inadequate verification of target engagement [105]. When evaluating the strength of evidence in drug development, human target engagement date and clinical outcomes represent the most reliable validation, as their directly determine the clinical utility of a candidate drugs; In vivo studies serve as a critical intermediate link by recapitulating the complexity of the TME, thereby enabling assessment of antitumor activity, safety, and pharmacokinetic/pharmacodynamic (PK/PD) relationships; while cell assays are core screening tools for drug discovery, enabling rapid validation of targeted activity, preliminary safety, and structure-activity relationships (SAR), though they are limited by the gap between in vitro environments and the actual TME. Collectively, these three types of evidence form a complete chain for drug development, with their respective strengths and limitations complementing each other to support the advancement of PD-1/PD-L1-targeted small molecule therapeutics.
3.2. PD-1 inhibitors
In contrast to PD-L1 inhibitors, the development of small molecules targeting PD-1 itself has progressed more slowly. All currently reported compounds remain at the preclinical stage, and none has yet entered clinical trials.
Lu et al. discovered agents with potent inhibitory activity against the PD-1/PD-L1 interaction [106]. Among these, CH-4 exhibited the most potent ability to disrupt the binding between soluble PD-L1 (sPD-L1) and membrane-localized PD-1 in KG-1 cells. Subsequent structural optimization led to the identification of CH-4.7 (22, Figure 5), an analog of CH-4 that retains strong targeting ability while exhibiting minimal cytotoxicity. Specifically, CH-4.7 exhibited no cytotoxicity against Jurkat cells and KG-1 cells. It effectively blocked the binding of sPD-L1 to PD-1 on the surface of KG-1 cells without altering PD-1 protein expression, and its PD-1/PD-L1 inhibitory activity remains comparable to that of CH-4. Meanwhile, CH-4.7 reversed the inhibition of sPD-L1 on T cells cytokine secretion, significantly elevated effect of IL-2 and IFN-γ levels in PMA/PHA-activated Jurkat cells, maintained T cells activation function. Notably, its inhibitory efficacy at the cellular level surpasses that of PD-1 antibodies.
Structures of PD-1 inhibitors.
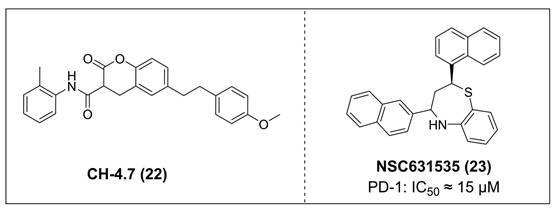
Patil et al. identified NSC631535 (IC50 ≈ 15 μM) (23, Figure 5) as a PD-1/PD-L1 inhibitor through screening of lead-like and large molecule databases [107]. Mechanistically, NSC631535 bound directly to PD-1, thereby inhibiting the PD-1/PD-L1 interaction. In vitro, at a concentration of 50 μM, NSC631535 exhibited an inhibitory rate of up to 65.5%. Dose-dependent experiments further revealed that its inhibitory activity is concentration-dependent.
Investigations into small molecule agents targeting the PD-1/PD-L1 axis have revealed that the research focus on PD-L1-targeted strategies [108]. These strategies include inhibitors that bind to the hydrophobic pocket of PD-L1 and induce its dimerization [109]. This research preference is largely attributable to the unique structural and biological properties of PD-L1. Structurally, its extracellular domain possesses well-defined dimerization interfaces and rigid hydrophobic pockets [109]. Biologically, PD-L1 expression is largely restricted to tumor tissues, making its blockade more likely to result in localized immune reactivation in the TME while minimizing systemic immune toxicity [110]. The core challenges in this area lie in the balance of efficacy and the optimization of druggability [111]. In contrast, PD-1 presents substantially greater challenges as a small-molecule target. Its extracellular binding region exhibits conformational flexibility and lacks stable hydrophobic pockets, rendering it less druggable [112, 113]. Moreover, as a core immune checkpoint receptor, PD-1 is expressed on various immune cells types and plays a critical role in maintaining systemic immune tolerance [114]. Blockade of PD-1 therefore leads to widespread immune activation, increasing the risk of irAEs. In addition, PD-1 can bind to PD-L1 and PD-L2. Selective inhibition of PD-L1 preserves the PD-1/PD-L2 axis, thereby helping to maintain immune homeostasis. Conversely, PD-1 inhibition disrupts both pathways, potentially leading to immune overactivation and an elevated risk of autoimmune reactions [115, 116]. These mechanistic and pharmacological distinctions have shaped the current landscape of drug development for the PD-1/PD-L1 pathway. Currently, antibody-based therapies dominate PD-1-targeted strategies, while small molecule drug development faces bottlenecks such as low binding affinity and insufficient selectivity [117]. As a result, PD-L1 has emerged as the mainstream target for small-molecule inhibitor development, while PD-1-targeted therapies remain primarily antibody-based, with small-molecule candidates lagging considerably behind in clinical progress [118].
4. PD-L1 degraders
4.1. PD-L1 PROTACs
PROTACs are heterobifunctional molecules that harness the ubiquitin-proteasome pathway to induce selective degradation of target proteins. These molecules consist of three key structural components: a ligand that interacts with target protein, an E3 ubiquitin ligase-recruiting ligand, and a chemical linker that connects these two functional moieties [119]. PROTACs exert their activity by promoting ubiquitination modification of the target protein, thereby flagging it for proteasomal recognition and degradation. This unique strategy not only enables effective modulate disease-associated proteins but also extends therapeutic intervention to protein subtypes traditionally considered “undruggable” or those that have developed resistance to conventional inhibitors. As such, PROTAC technology has opened new research avenues and expanded the therapeutic landscape in immuno-oncology and beyond [120, 121].
Chen et al. developed compound P22 (24, Figure 6) by conjugating the PD-L1 inhibitor BMS-1198 with the E3 ligase ligand pomalidomide via a rigid piperazine linker [122]. Compared to flexible or straight linkers, this rigid linker was more conducive to preserving the binding affinity between the molecule and PD-L1. In a Hep3B/OS-8/hPD-L1 and CD3+ T cell coculture model, P22 significantly promoted IFN-γ secretion in a dose-dependent manner, exhibiting efficacy superior to that of the clinical drug Keytruda. Furthermore, P22 moderately reduced PD-L1 protein level in MDA-MB-231 cells, and this degradation effect was dependent on the lysosomal pathway rather than the proteasomal pathway. For membrane proteins like PD-L1, PROTACs could induce protein endocytosis followed by lysosomal degradation. This phenomenon was a common pathway for PROTAC-mediated membrane protein degradation and not exclusive to LYTACs [123].
Structures of PD-L1 PROTACs.
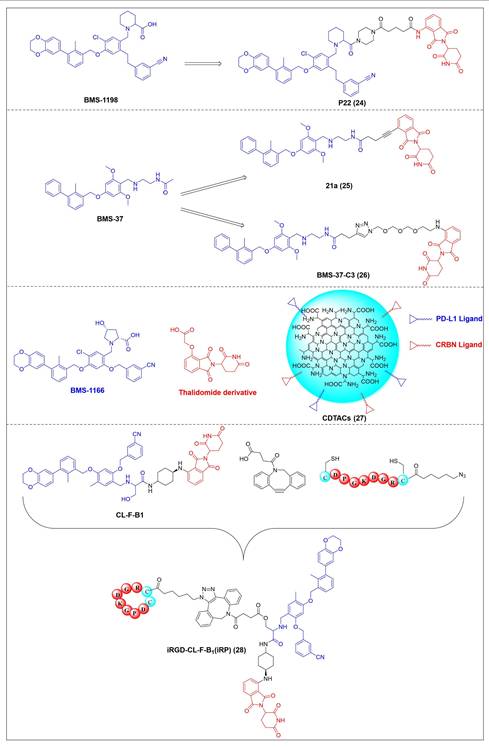
Wang et al. conjugated BMS-37 with thalidomide to generate a series of PROTACs, from which compound 21a (25, Figure 6) was identified as the most promising PD-L1 degrader [124]. In vitro, 21a degraded PD-L1 in a dose- and time-dependent manner via a proteasome-dependent pathway in various malignant cell lines including hematological, breast, colorectal and prostate cancer cell. Notably, it preferentially degraded cytoplasmic PD-L1 for degradation, thereby reducing its expression on the cell membrane. In vivo, 21a significantly decreased PD-L1 levels in MC-38 tumors, promoted CD8+ T cell infiltration, and upregulated genes associated with CD8+ T cell cytotoxicity. These effects translated into potent tumor growth inhibition without causing changes in mouse body weight.
Liu et al. synthesized PD-L1 degraders by conjugating BMS-37 with thalidomide via PEG-based linkers [125]. Among these, BMS-37-C3 (26, Figure 6) emerged as the most potent degrader. In A375 and B16-F10 melanoma cells, BMS-37-C3 degraded PD-L1 in a dose- and time-dependent manner. In the coculture model comprising A375 cells and T cells, BMS-37-C3 enhanced T cell-mediated tumor killing in a concentration-dependent fashion. Notably, its antitumor efficacy surpassed that of both the parent compound BMS-37 and the marketed monoclonal antibody atezolizumab.
Su et al. developed a class of carbon dot-based PROTACs (CDTACs) (27, Figure 6) by conjugating the PD-L1-binding probe BMS-1166 and the CRBN E3 ligase ligand thalidomide onto carbon dots (CDs) [126]. CDs are biocompatible, photostable, and fluorescently traceable nanomaterials that enable tumor accumulation via both the enhanced permeability and retention (EPR) effect and active PD-L1 targeting. In 2-12 h following intravenous injection, CDTACs accumulated in tumors through EPR and active targeting, with accumulation further enhanced by folic acid-modified dextran (FMD). Treatment with CDTACs combined with FMD increased intratumoral infiltration of CD8⁺ T cells, elevated levels of pro-inflammatory cytokines such as IFN-γ and TNF-α, and reduced immunosuppressive factors including IL-4 and IL-10. Notably, CDTACs efficiently degraded PD-L1, reshaped the TME, and demonstrated potent antitumor efficacy even in PD-L1 inhibitor-insensitive tumors such as B16-F10 melanoma.
Qi et al. connected BMS-1001 with pomalidomide through a rigid trans-1,4-diaminocyclohexyl linker to obtain CL-F-B1 [127]. In MC38 colon cancer cells, CL-F-B1 achieved 67.05% PD-L1 degradation at 5 μM after 24 h of incubation, with a DC50 of 2.32 μM. To address the challenge of targeted delivery, they further conjugated CL-F-B1 with the cyclic iRGD peptide via azide-alkyne cycloaddition (click chemistry) to generate the iRGD-CL-F-B₁ conjugate (iRP) (28, Figure 6). This conjugate could self-assemble into nanomicelles (iRP NPs), the iRGD peptide could mediate deep tumor penetration via αvβ3 integrins and neuropilin-1 (NRP-1). In vitro, iRP NPs exhibited 3.78-fold higher cellular uptake in MC38 cells efficiency compared to control PROTAC NPs. In vivo, iRP NPs exhibited high intratumoral accumulation in the MC38 tumor model. At a dose of 6 mg/kg, the formulation achieved an 80.88% TGI, promoted CD8+ T cell infiltration, and caused no damage to major organs.
4.2. PD-L1 LYTACs
LYTACs encompass three key structural components: a target protein-binding ligand, a ligand specific for the lysosomal targeting receptor (LTR), and a connecting linker [128]. Following binding to their respective targets, LYTACs assemble a ternary molecular complex with the LTR, which subsequently undergoes receptor-mediated endocytosis and lysosome-dependent degradation.
Banik et al. utilized the cation-independent mannose-6-phosphate receptor as LTRs by conjugating the PD-L1 antibody atezolizumab with a glycopeptide ligand that targets the cation-independent mannose-6-phosphate receptor (CI-M6PR), which resulted in the production of atz-LYTAC (29, Figure 7) [129]. The degradation activity of atz-LYTAC was associated with the cell surface expression level of CI-M6PR. In MDA-MB-231 cells, which exhibit low CI-M6PR expression, atz-LYTAC could reduce the cell surface PD-L1 levels by approximately 33%. In contrast, in HDLM-2 cells with high CI-M6PR expression, treatment with atz-LYTAC achieved approximately 70% PD-L1 degradation within 48 hours, which was significantly superior to that of the unconjugated parental antibody.
Structures of PD-L1 LYTACs.
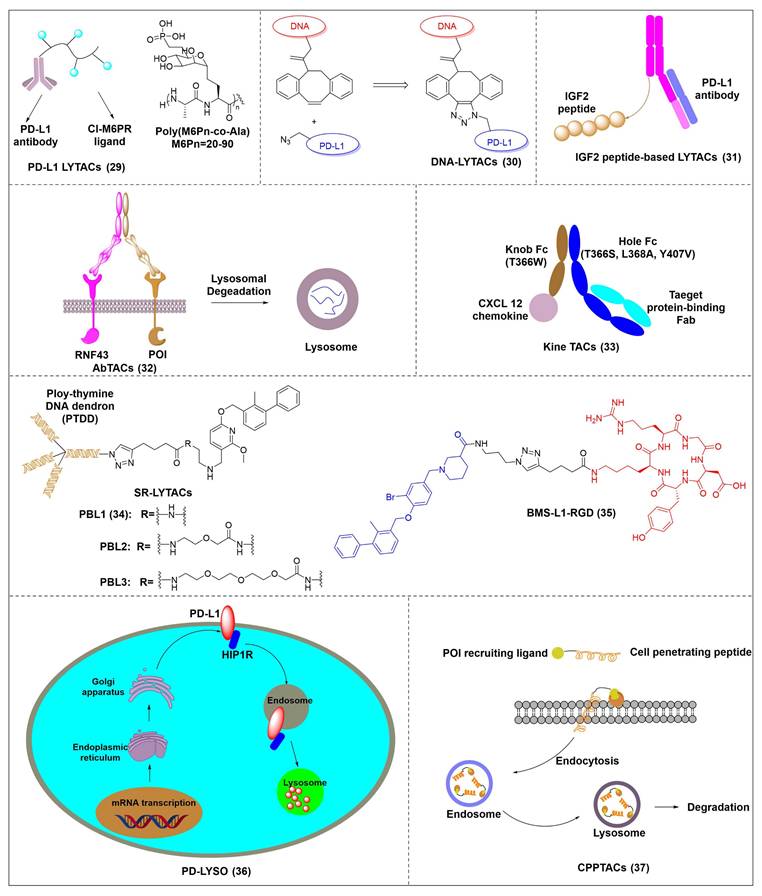
Liu et al. developed DNA aptamer-based covalent LYTACs (30, Figure 7) that targeting the CI-M6PR on one side and enabling bioorthogonal covalent conjugation-facilitated specific binding to PD-L1 on the other [130]. These covalent LYTAC not only abrogated the immune suppression of the PD-1/PD-L1 axis but also was the first found to inhibit STAT3 phosphorylation by degrading PD-L1, directly inducing immunogenic apoptosis of tumor cells, releasing damage-associated molecular patterns such as calreticulin, and promoting dendritic cell maturation and tumor-specific cytotoxic T cell responses.
Sitar et al. developed IGF2- derived peptides as ligands for CI-M6PR, which were subsequently fused to a PD-L1 antibody to generate peptide/protein-based LYTACs (31, Figure 7) [131]. The designed IGF2-derived polypeptides bound to IGF2R with high affinity and specificity and did not bind to IGF1R. Their corresponding LYTACs exhibited bispecific binding to both PD-L1 and IGF2R, induced the internalization and degradation of soluble and transmembrane PD-L1 in a time- and concentration-dependent manner, and significantly triggered the growth inhibition and lysis of tumor cells when co-incubated with PBMCs, with efficacy superior to that of traditional anti-PD-L1 antibodies. Meanwhile, this class of LYTACs showed no obvious cytotoxicity on their own.
Cotton et al. developed a novel cell-surface protein degradation technology called antibody-based PROTACs (AbTACs) (32, Figure 7) [132]. AbTACs induced internalization and lysosomal degradation of target proteins by connecting cell surface proteins with E3 ubiquitin ligase RNF43. The team developed a bispecific antibody named AC-1. Incubation of cells with 10 nM AC-1 for 12 hour initiated PD-L1 degradation, and maximal effect was achieved at 24 hours (Dmax = 63%, DC50 = 3.4 nM). In terms of broad-spectrum activity, treatment with 10 nM AC-1 for 24 hours achieved PD-L1 degradation in PD-L1-high-expressing cell lines of triple-negative breast cancer, non-small cell lung cancer and bladder cancer.
Wells et al. developed cytokine receptor-targeting chimeras (KineTACs) that exploits endogenous cytokine-mediated internalization of cognate receptors to deliver target proteins for lysosomal degradation [133]. Using a CXCL12-based KineTAC conjugated to a PD-L1 antibody, they successfully induced PD-L1 degradation in MDA-MB-231 cells. The degradation efficiency of this KineTAC correlated strongly with the binding affinity and dissociation rate of the antibody arm for PD-L1, while remaining unaffected by CXCR4 signaling or pH-dependent binding. The system exhibited high selectivity, downregulating PD-L1 without affecting other surface proteins, and retained its activity against murine PD-L1 in MC38 and CT26 cells.
Huang et al. reported the first DNA-based lysosome-targeted degradation strategy mediated by scavenger receptors (SRs), establishing SRs as novel LYTACs receptors [134]. By coupling polythymidine DNA dendrites with BMS-202, three compounds with different linker lengths (PBL1, PBL2, and PBL3) were synthesized. Among these, PBL1 (34, Figure 7) exhibited the most potent activity (DC50 = 110.4 nM) and effectively promoted PD-L1 degradation in cancer cells. Mechanistically, PBL1 was internalized by cells and targeted to lysosomes in a concentration- and time-dependent manner. In the immune co-culture model combined with PBMCs, PBL1 effectively enhanced the killing effect of immune cells on tumor cells. Collectively, PBL1 exhibited potent PD-L1 degradation activity, low off-target toxicity, and excellent anti tumor efficacy both in vitro and in vivo.
Zheng et al. designed cyclic Arg-Gly-Asp (cRGD)—a ligand for integrin αVβ3—and conjugated it to the BMS-8 via various linkers, generating three integrin-facilitated lysosomal degraders (IFLDs) for PD-L1 targeting [135]. Under the action of IFLDs, the target protein formed a ternary complex with the IFLD molecules and integrins, which was sequentially accompanied by endocytosis and lysosomal degradation. Among the constructs, BMS-L1-RGD (35, Figure 7) exhibited the most potent PD-L1 degradation activity in MDA-MB-231 cells. In the B16F10 tumor xenograft mouse model, BMS-L1-RGD significantly inhibited tumor growth and reduced tumor weight, markedly downregulated PD-L1 expression in tumor tissues and significantly elevated the level of tumor cell apoptosis. Furthermore, it decreased splenic metastasis, with no obvious body weight loss or other side effects were observed in the mice.
Xu et al. identified huntingtin-interacting protein 1-related (HIP1R) as an endogenous ligand that bound to the intracellular domain of PD-L1 and mediated its degradation via a lysosome-dependent pathway [136]. Based on this mechanism, the authors designed a chimeric peptide, designated PD-LYSO (36, Figure 7), which effectively induces lysosome-dependent degradation of PD-L1. By decreasing PD-L1 expression, PD-LYSO reduced the binding of tumor cells to PD-1, thereby enhancing T cell-mediated cytotoxicity.
He et al. developed a platform of CPP-mediated lysosome-targeting chimeras by exploiting the endosomal entrapment effect of cell-penetrating peptides (CPPs) and conjugating different CPPs to target small protein-binding molecules [137]. By linking BMS-8 with the CPP penetrating through a triazole linker, the authors generated a BMS-CPP conjugate (37, Figure 7). This conjugate could significantly degrade PD-L1 via lysosomal pathways (up to 75%-80%) without the “hook effect”, a common limitation of certain degradation platforms. Notably, BMS-CPP offered broad applicability—it was applicable to almost all cell types, exhibited high linker tolerance, possessed good tissue penetration, and had no immunogenicity.
This chapter focuses on small molecule PD-L1 degraders and provides a systematic overview of two core technologies: PROTACs and LYTACs. The former degrades PD-L1 via the ubiquitin-proteasome pathway, while the latter accomplishes this through receptor-dependent endocytosis and lysosomal degradation. Both types of degraders have developed innovative delivery systems such as carbon dots, iRGD-modified nanomicelles, and CPPs to enhance tumor targeting and tissue penetration. They have demonstrated significant tumor-suppressive effects in preclinical models and can exert efficacy against PD-L1 inhibitor-insensitive tumors. However, current research still has obvious limitations. First, all reported degraders remain in the preclinical stage, lacking support from human clinical trial data, and species differences may affect translational outcomes [138, 139]. Second, technical bottlenecks such as the difficulty in large-scale production of delivery systems, the impact of PROTACs linker design on degradation efficiency, and the immunogenicity risk of LYTACs have not been fully overcome [129, 140, 141]. Third, the structure-activity relationships (SAR) underlying degradation mechanisms remain insufficiently characterized [142]. Addressing these challenges will be critical for advancing PD-L1 degraders toward clinical application.
5. Co-inhibitors of PD-L1 and other targets
The clinical applicability of PD-1/PD-L1 monotherapies is often constrained by limited response rates, selectivity challenges, and acquired resistance [143]. Dual-target inhibitors offer a strategy to mitigate these limitations by exerting synergistic effects, enabling more durable and comprehensive tumor control, and potentially lowering toxicity compared to combination regimens. This approach represents a promising avenue for precision immuno-oncology [144]. Herein, we summarize the reported dual-targeted agents that combine PD-L1 inhibition with the modulation of key secondary targets, highlighting their therapeutic rationale and potential.
5.1. PD-L1/ HDAC inhibitors
Histone deacetylases (HDACs) are enzymes that catalyze the deacetylation of lysine residues on proteins. By modulating chromatin structure and non-histone protein functions, HDACs negatively regulate gene transcription at the epigenetic level and play a crucial role in cell proliferation, differentiation, apoptosis, and immune regulation [145]. HDAC can upregulate the expression of PD-L1 through epigenetic regulation and signaling pathway interactions in the TME promoting immune escape [146].
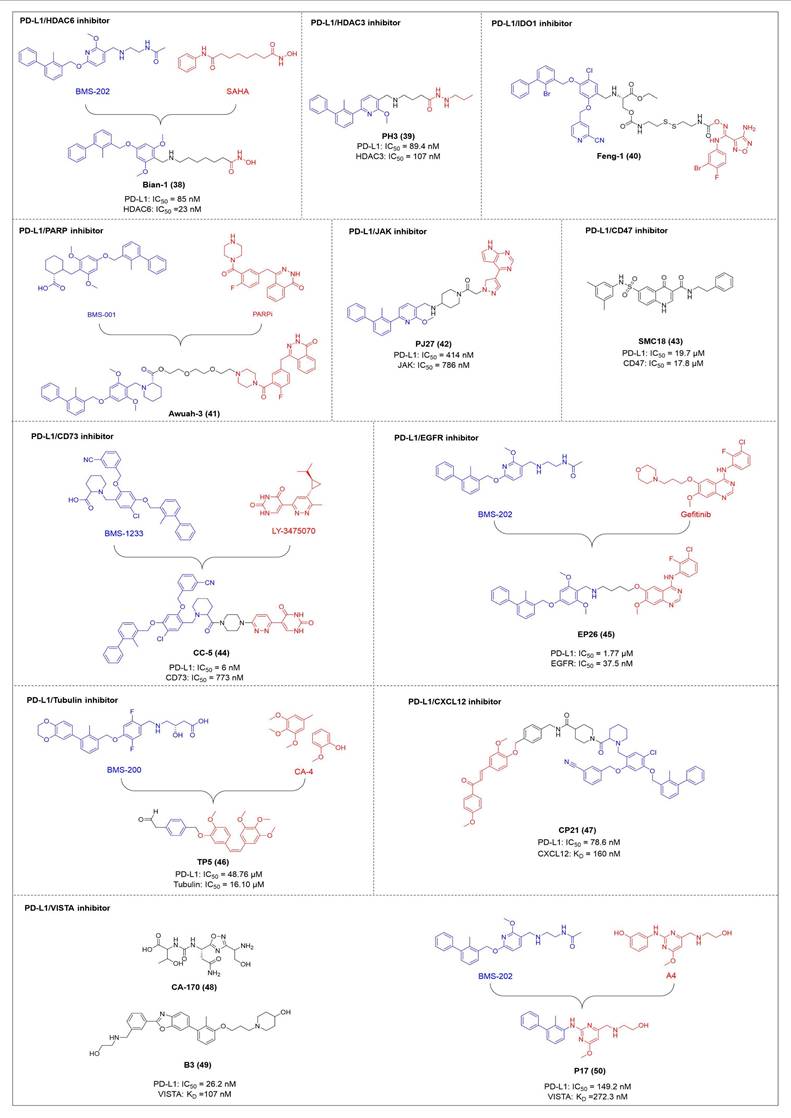
HDAC6 is a multifunctional cytoplasmic deacetylase involved in diverse pathological processes, including cancer and neurodegenerative diseases, through its regulation of microtubule stability and clearance of misfolded protein aggregates [147]. Notably, HDAC6 facilitated tumor immune evasion by deacetylating and stabilizing PD-L1 in tumor cells, thereby positioning it as a key therapeutic target that bridges epigenetic regulation and cancer immunotherapy [148]. Therefore, inhibiting HDAC6 exhibits a synergistic effect with anti-PD-L1 therapy [149]. Bian et al. synthesized a PD-L1/HDAC6 dual inhibitor, Bian-10 (PD-1/PD-L1: IC50 = 85 nM, HDAC6: IC50 = 23 nM) (38, Figure 8; Table 4) [150]. In vitro, it showed significant inhibitory activity on the proliferation of tumor cells such as B16 and CT26. In vivo, for the CT26 colon cancer cell xenograft model in nude mice, the tumor inhibition rates of the 50 mg/kg and 100 mg/kg dose groups reached 55% and 75%, respectively, which were superior to those of the positive control drugs BMS-202 (51%) and SAHA (53%).
Structures of co-inhibitors of PD-L1 and other targets.
Co-inhibitors of PD-L1 and other targets
| Compd. | Inventor | Target | Structure | Biological data | Ref. |
|---|---|---|---|---|---|
| Bian-10 (38) | China Pharmaceutical University | PD-L1/ HDAC6 | 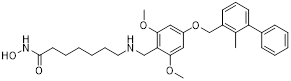 | IC50 = 85 nM (PD-L1); IC50 = 23 nM (HDAC6) | [150] |
| PH3 (39) | Southern Medical University | PD-L1/ HDAC3 | 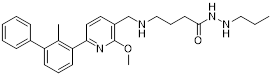 | IC50 = 89.4 nM (PD-L1); IC50 = 107 nM (HDAC3) | [153] |
| Feng-1 (40) | Chinese Academy of Medical Sciences | PD-L1/ IDO1 | 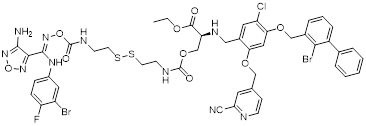 | - | [156] |
| Awuah-3 (41) | University of Kentucky | PD-L1/ PARP | 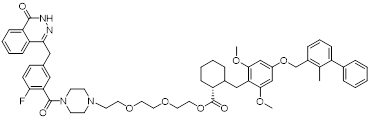 | - | [160] |
| PJ27 (42) | Henan University of Chinese Medicine | PD-L1/ JAK | 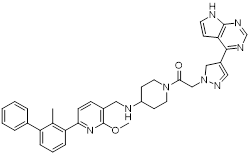 | IC50 = 414 nM (PD-L1); IC50 = 786 nM (JAK) | [164] |
| SMC18 (43) | Zhengzhou University | PD-L1/ CD47 | 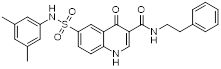 | IC50 = 19.7 μM (PD-L1); IC50 =17.8 μM (CD47) | [168] |
| CC-5 (44) | Wenzhou Medical University | PD-L1/ CD73 | 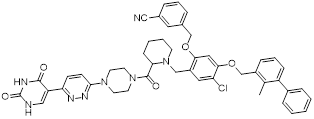 | IC50 = 6 nM (PD-L1); IC50 = 773 nM (CD73) | [172] |
| EP26 (45) | Southern Medical University | PD-L1/ EGFR | 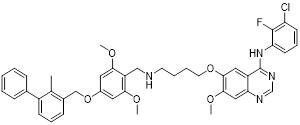 | IC50 = 1.7 μM (PD-L1); IC50 = 37.5 nM (EGFR) | [175] |
| TP5 (46) | Southern Medical University | PD-L1/ Tubulin | 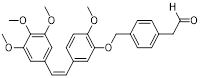 | IC50 = 48.76 μM (PD-L1); IC50 = 16.10 μM (Tubulin) | [178] |
| CP21 (47) | Hubei Polytechnic University | PD-L1/ CXCL12 | 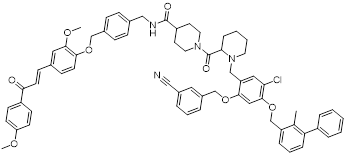 | IC50 = 78.6 nM (PD-L1); | [181] |
| CA-170 (48) | Curis | PD-L1/ VISTA | 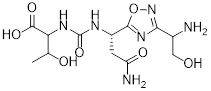 | - | [184, 186, 187] |
| B3 (49) | China Pharmaceutical University | PD-L1/ VISTA | 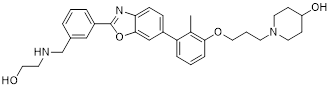 | IC50 = 26.2 nM (PD-L1); KD = 107 nM (VISTA) | [188] |
| P17 (50) | China Pharmaceutical University | PD-L1/ VISTA | 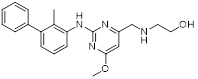 | IC50 = 149.2 nM (PD-L1); KD =272.3 nM (VISTA) | [189] |
“-” means the data is unavailable (not public or non-existent).
HDAC3, a class I HDAC family member, regulates the acetylation levels of histone and non-histone proteins, thereby influencing tumor progression and representing an important antitumor target [151]. Notably, combining HDAC3 inhibitors with PD-L1 antibodies has been shown to produce synergistic antitumor effects [152]. Wang et al. synthesized dual-target candidate drugs PH3 (39, Figure 8; Table 4) by incorporating the pharmacophores of HDAC3 inhibitors into the tails of PD-L1 inhibitors [153]. PH3 exerted potent inhibitory effects on both PD-1/PD-L1 (IC50 = 89.4 nM) and HDAC3 (IC50 = 107 nM), with remarkable selectivity over other HDAC isoforms. In vitro, PH3 inhibited the proliferation of various tumor cells, induced tumor cell apoptosis, and arrested the cell cycle at the G0/G1 phase. Additionally, in the coculture model, PH3 enhanced T cell-mediated tumor cell killing. In vivo, PH3 exhibited a dose-dependent antitumor effect in the B16-F10 melanoma mouse model, which were superior to those of NP19 monotherapy and the combination therapy of NP19+MS-275. No obvious toxicity was observed. Meanwhile, it increased the infiltration of CD3⁺CD8⁺ and CD3⁺CD4⁺ cells in the TME and activated antitumor immunity.
5.2. PD-L1/IDO1 inhibitors
Indoleamine 2,3-dioxygenase 1 (IDO1) catalyzes the depletion of tryptophan and the generation of kynurenine, thereby suppressing T-cell functional and promoting immune tolerance [154]. The concurrent PD-L1 and IDO1 inhibition can potentiate antitumor immunity [155]. Feng et al. designed PD-L1/IDO1 dual-target inhibitors incorporating a disulfide bonds linker. This disulfide bonds linker can only be cleaved in the TME/cells, enabling the targeted release of PD-L1/IDO1 inhibitors and thereby reducing off-target toxicity [156]. In vivo, Feng-1 (40, Figure 8; Table 4) exhibited significant antitumor activity in the mouse melanoma B16F10 subcutaneous xenograft model. In the mouse colon cancer CT26 model, Feng-1 achieved a TGI of 54.9%, outperforming both the PD-L1 inhibitor monotherapy (TGI = 40.6%) and the IDO1 inhibitor monotherapy (TGI = 32.1%). Regarding safety, Feng-1 treatment did not significantly affect body weight, spleen weight, or spleen index in mice, and induced a decreasing trend in regulatory T cell (CD4⁺CD25⁺Foxp3⁺) in the spleen.
5.3. PD-L1/PARP inhibitors
Poly (ADP-ribose) polymerases (PARPs) are a family of enzymes involved in DNA damage recognition and repair. They maintain genomic stability by catalyzing ADP-ribosylation (PARylation), thereby regulating the function of diverse proteins [157]. Research has shown that PARP inhibition can upregulate PD-L1 expression [158], and combined inhibition of PD-L1 and PARP has demonstrated synergistic antitumor effects in preclinical models [159]. Ofori et al. developed conjugates by linking BMS-001 to a PARP inhibitor pharmacophore [160]. Among them, Awuah-3 (41, Figure 8; Table 4) retained structural integrity upon incubation in PBS, DMEM medium and human liver microsomes. It exerted potent cytotoxicity across multiple cancer cell lines, including ovarian, lung and breast cancer cells, with activity 2- to 20-fold higher than that of the individual parent drugs. Notably, Awuah-3 showed exceptionally strong killing activity against the triple-negative breast cancer cell line MDA-MB-231, inducing early and late apoptosis in 60% of treated cells. In addition, Awuah-3 significantly downregulated the cell surface PD-L1 expression with an inhibitory effect superior to that of BMS-001. It also arrested the cell cycle at the G1 phase, modulated the progression of the S phase, and effectively restored the proliferative capacity of T cells. By synergistically blocking the PARP and PD-L1 dual signaling pathways, Awuah-3 exerted a robust antitumor effect.
5.4. PD-L1/JAK inhibitors
Janus kinases (JAK) are a non-receptor tyrosine kinase that regulates cytokine-mediated immune and inflammatory responses through the JAK-STAT signaling pathway, serving as therapeutic target for various autoimmune diseases and myeloproliferative disorders [161]. Activation of the JAK-STAT signaling pathway can upregulate PD-L1 expression on tumor cells through cytokine-mediated mechanisms (e.g., IFN-γ), thereby contributing to the formation of an immunosuppressive microenvironment and exhibiting functional interplay with the PD-1/PD-L1 immune checkpoint pathway [162, 163]. Wang et al. synthesized the PD-L1/JAK inhibitor PJ27 (PD-1/PD-L1: IC50 = 414 nM, JAK1: IC50 = 786 nM) (42, Figure 8; Table 4) [164]. In the LLC lung cancer mouse model, PJ27 exhibited a dose-dependent tumor growth inhibitory effect: at 50 mg/kg, its achieved a TGI rate of 56%, significantly outperforming the single-target PD-L1 inhibitor BMS-202 at the same dose. Meanwhile, PJ27 could effectively downregulate PD-L1 expression in tumor tissues and inhibited STAT3 phosphorylation. It also markedly promoted the infiltration of CD3⁺CD8⁺ cytotoxic T cells and CD3⁺CD4⁺ helper T cells into the TME, thereby enhancing antitumor immune response. In addition, no significant decrease in the body weight of mice was observed during the administration period, and there were no notable pathological damages to major organs such as the heart, liver, spleen, lungs, and kidneys.
5.5. PD-L1/CD47 inhibitors
CD47-mediated signaling inhibits macrophage-mediated phagocytosis [165, 166]. Collective inhibition of the PD-L1 and CD47 signaling pathways potentiated the tumor-suppressive effect in a synergistic manner [167, 168]. Jin et al. developed SMC18 (43, Figure 8; Table 4), a bispecific inhibitor targeting both the CD47/SIRPα (IC50 = 17.8 μM) and PD-1/PD-L1 (IC50 = 19.7 μM) signaling axes [168]. In vitro, 50 μM SMC18 showed no obvious cytotoxicity to tumor cells. It significantly inhibited the tyrosine phosphorylation of SIRPα and PD-1, effectively restore the phagocytic capacity of bone marrow-derived macrophages against MC38 and B16-OVA tumor cells, and meanwhile recover the IL-2 secretion Jurkat cells. In vivo, in the MC38 colorectal cancer-bearing mouse model, SMC18 as a single agent could significantly inhibit tumor growth, effectively promote the infiltration of CD8⁺ T cells and M1-type macrophages into tumor sites, and enhance the IFN-γ secretion capacity of CD8⁺ T cells in the TME. Moreover, SMC18 at these doses caused no obvious hematotoxicity or hepatotoxicity, and no pathological damage was observed in the major organs of mice. In addition, the combination of SMC18 with local radiotherapy exerted a significant synergistic antitumor effect, which further inhibited tumor growth without exerting adverse effects on the body weight of mice.
5.6. PD-L1/CD73 inhibitors
CD73 functions as an ecto-5'-nucleotidasethat converts extracellular AMP into adenosine, a potent immunosuppressive metabolite [169]. Combining CD73 suppression with PD-L1 inhibition can counteract adenosine-mediated immune suppression and enhance tumor control [170, 171]. Wang et al. covalently linked BMS-1233 (PD-L1 inhibitor) with LY-3475070 (CD73 inhibitor) to construct a series of bifunctional compounds, among which CC-5 (PD-L1: IC50 = 6 nM; CD73: IC50 = 773 nM) (44, Figure 8; Table 4) exhibited the best activity [172]. At the cellular level, CC-5 exhibited no significant toxicity to PBMCs and CT26 tumor cells, yet dose-dependently enhanced PBMCs-mediated killing activity of CT26 cells. Meanwhile, it effectively inhibited the PD-1/PD-L1 interaction, exerting immunomodulatory effects. In vivo, CC-5 showed remarkable tumor growth inhibitory activity in both the CT26 colorectal cancer and B16-F10 melanoma model. Additionally, CC-5 could significantly elevate the infiltration level of CD3⁺CD8⁺ cells in tumor tissues, effectively activating the TME and thereby exerting a synergistic antitumor effect.
5.7. PD-L1/EGFR inhibitors
EGFR is frequently overexpressed or mutated in epithelial malignancies [173]. EGFR signaling mediates PD-L1 overexpression in malignant cells [174]. Yang et al. integrated the biphenyl/pyridine core of BMS-202 with the quinazoline scaffold of gefitinib (EGFR inhibitor) to synthesize a series of bifunctional compounds, among which EP26 (45, Figure 8; Table 4) emerged as the most promising candidate [175]. EP26 exhibited inhibitory activity against both EGFR (IC50 = 37.5 nM) and PD-1/PD-L1 (IC50 = 1.77 μM). In vitro, Its antiproliferative effects across various glioblastoma (GBM) cell lines significantly surpassed that of control drugs such as Gefitinib. Mechanistically, EP26 induced G0/G1 phase cell cycle arrest, downregulated EGFR phosphorylation, and reactivated immunosuppressed T cells. Notably, EP26 possessed favorable pharmacokinetic properties with an oral bioavailability of 22.2% and blood-brain barrier penetration ability. In vivo, in GBM mouse model, EP26 administered at 100 mg/kg achieved a TGI rate of 92.0%, outperforming Gefitinib and NP19 (PD-L1 inhibitor), without obvious toxicity. Although EP26 increased the proportions of CD4⁺ and CD8⁺ cells in the TME, the increase was significantly lower than that of NP19. It exerted antitumor effects through the dual mechanisms of direct tumor inhibition and immune activation.
5.8. PD-L1/Tubulin inhibitors
Microtubules play essential role in cellular motility, division, and intracellular trafficking, while additionally impacting immune cell responses [176]. Concomitant inhibition of PD-L1 and microtubules exhibited synergistic antitumor efficacy [177]. Yang et al. adopted a hybridization strategy, integrated the key structural fragments of these two classes of inhibitors, and designed and synthesized 20 CA-4 analogs, among which TP5 (46, Figure 8; Table 4) exhibited the most potent activity [178]. In vitro, TP5 exhibited potent inhibitory effects on five cancer cell lines, including HepG2 and MC38, and paclitaxel-resistant A549/PTX cells, with an IC50 value of 0.8 μM in HepG2 cells and extremely low cytotoxicity to normal cells. TP5 inhibited tubulin polymerization (IC50 = 16.10 μM), disrupted the microtubule network, arrested the cell cycle at the G2/M phase, induced apoptosis, and suppressed tumor cell migration and colony formation. Additionally, TP5 moderately inhibited the PD-1/PD-L1 interaction (IC50 = 48.76 μM), showing similar binding affinities to both human and murine PD-L1. In vivo, intragastric administration of TP5 at 100 mg/kg achieved TGI rate of 57.9% in the humanized PD-1 melanoma mouse model, without significant hepatotoxicity, nephrotoxicity, cardiotoxicity, or myelosuppression. Meanwhile, TP5 upregulated the mRNA expression of CXCR3 and CXCL10, downregulated PD-L1 expression in tumor tissues, increased the proportion of tumor-infiltrating T cells, and activated the TME.
5.9. PD-L1/CXCL12 inhibitors
The chemokine CXCL12 (SDF-1) contributes to immunosuppression in the TME by recruiting PD-L1-expressing cells and promoting tumor cell survival and metastasis [179]. Dual inhibition of the PD-L1 and CXCL12 pathways enhances antitumor activity by mitigating these immunosuppressive mechanisms [180]. Chen et al. synthesized bifunctional compounds by conjugated the pharmacophores of PD-L1/CXCL12 inhibitors via a linker, among which CP21 (47, Figure 8; Table 4) exhibited the optimal activity [181]. CP21 embedded into the inner cavity of the PD-L1 dimer and bound to PD-L1 through hydrophobic interactions (IC50 = 78.6 nM), while it could bind to the hydrophobic cleft of CXCL12 (KD = 160 nM), thus exhibiting a stable binding mode. Pharmacokinetic studies showed that CP21 had a higher plasma exposure and a lower clearance rate upon intravenous administration, and also exhibited acceptable plasma exposure following oral dosing. In vivo, in B16-F10 melanoma and CT-26 colon cancer models, CP21 inhibited tumor growth in a dose-dependent manner with a better efficacy than monotherapy, and it also significantly increased the proportions of CD3⁺ and CD3⁺CD8⁺ T cells, as well as the CD8⁺/CD4⁺ ratio in tumor tissues, effectively activating the TME. Meanwhile, CP21 caused no obvious hepatic, renal or cardiac toxicity and induced no morphological abnormalities in tissues, exhibiting a good safety profile.
5.10. PD-L1/VISTA inhibitors
V-domain Ig suppressor of T-cell activation (VISTA), a PD-1 homolog and immune checkpoint, exhibited therapeutic synergy when co-blocked with PD-L1 [182, 183]. CA-170 (48, Figure 8; Table 4) was the first oral PD-L1/VISTA inhibitor to enter clinical trials (completed phase I and advanced to phase II) [184]. However, its binding mechanism was controversial. On the one hand, nuclear magnetic resonance (NMR) and homogeneous time-resolved fluorescence (HTRF) assays verify that it does not directly bind to PD-L1, as its core Ser-Asn-Thr (SNT) motif was derived from the BC loop of AUNP-12—a region distant from the PD-1/PD-L1 interface—thus failing to achieve specific binding. However, this did not negate the clinical potential of CA-170. It was speculated that its antitumor mechanism did not directly block the PD-1/PD-L1 interaction; instead, it might have acted on the downstream signaling molecules of the PD-1 pathway and regulated other T cell activation pathways [185]. On the other hand, cellular NMR experiments suggested that it bound to PD-L1 in cells without prevent the assembly of the PD-1/PD-L1 complex, but instead formed a functionally defective ternary complex [186]. Preclinical monotherapy or combination therapy with docetaxel and cyclophosphamide could effectively inhibit tumor growth or reduce metastasis and exhibited good safety. Adverse reactions were mainly mild to moderate, such as fatigue and nausea. The core advantages were convenient oral administration, dual-target synergy, and high production cost potential. However, there were still issues such as unclear binding mechanisms, narrow dose windows, and small sample sizes in some clinical data remain to be resolved [184, 187].
Wang et al. identified the benzo[d]oxazole derivative B3 (49, Figure 8; Table 4) as a dual inhibitor of PD-L1/VISTA [188]. This compound inhibited the PD-1/PD-L1 interaction (IC50 = 26.2 nM) and bound to VISTA (KD = 107 nM). Moreover, B3 induced the degradation of both PD-L1 and VISTA through the autophagy-lysosome pathway. In vitro, it restored T-cell function and enhanced the antitumor immunity of PBMCs. In vivo, treatment with B3 suppressed tumor growth in a dose-dependent manner in the CT26 mouse model, showing superior efficacy to positive drugs and their combination therapy. Additionally, it increased CD8⁺ T cell infiltration into tumors.
Sun et al. adopted a scaffold fusion strategy, in which the biphenyl structure of BMS-202 and the ethanolamine-pyrimidine ring structure of the VISTA inhibitor A4, were fused to synthesize a series of compounds. Among these, P17 (50, Figure 8; Table 4) was identified as the most promising candidate [189]. P17 induced PD-L1 dimerization (IC50 = 149.2 nM) and bound to VISTA (KD = 272.3 nM), thereby blocking its interaction with endogenous ligands. At the cellular level, P17 exhibited no significant cytotoxicity, concentration-dependently enhanced PBMCs antitumor activity, promoted IFN-γ secretion, and relieved VISTA-mediated T cell suppression. In vivo, its antitumor efficacy surpassed that of PD-L1 or VISTA single-target inhibitors. Additionally, P17 significantly increased the infiltration and activation of CD8+ T cells, downregulated the proportion of Tregs, and optimized TME. Moreover, it showed good safety with an oral bioavailability of 11.86%.
In the field of dual inhibitors of PD-L1 and other targets, a “pharmacophore hybridization” or “skeleton fusion” strategy was adopted for drug design (e.g., PD-L1/HDAC inhibitors, PD-L1/EGFR inhibitors, PD-L1/VISTA inhibitors). This approach leveraged well-defined immune synergistic mechanisms between targets (e.g., pathway complementarity and upstream-downstream regulation), solid preclinical data (e.g., tumor suppressive activity across multiple tumor models, favorable safety profiles, and partially optimized pharmacokinetic characteristics), and improved drug-like properties, with some agents (e.g., the PD-L1/VISTA inhibitor CA-170) having entered clinical validation. However, these bifunctional molecules face multiple core challenges in practical research and development. Achieving a balanced potency against both targets simultaneously proved difficult. Structurally, their increased complexity often led to suboptimal pharmacokinetic profiles, including low oral bioavailability and inadequate half-lives. The multi-domain design also raised the risk of off-target effects. Moreover, the majority of these inhibitors remained in the preclinical stage, lacking support from large-scale clinical data. Insufficiently in-depth research on synergistic mechanisms and drug resistance, as well as the lack of clear predictive biomarkers for efficacy, further limit their clinical application and development.
6. Conclusions and perspectives
Malignant tumor cells have demonstrated the ability to circumvent immune surveillance through a range of immunosuppressive mechanisms, and ICT has revolutionized the clinical landscape of cancer treatment [2, 3, 5]. The PD-1/PD-L1 axis is recognized as a central molecular mechanism underlying tumor immune escape [9]. Although the U.S. FDA has authorized multiple anti-PD-1/PD-L1 mAbs, these agents were limited by their macromolecular properties and efficacy bottlenecks. These challenges have steered the development of small molecule immuno-oncology agents toward three core strategies: inhibitors, degraders, and dual-target inhibitors. Although inhibitors and degraders share similar functions in inhibiting the PD-1/PD-L1 axis, they exert fundamentally distinct effects on immune homeostasis and present distinct profiles of irAEs. PD-L1 inhibition function by reversibly blocking the PD-1/PD-L1 interaction [190]. However, they are often insufficient to target the entire cellular PD-L1 pool. The residual PD-L1 retains partial physiological immune regulatory functions, causing mild and reversible disturbance to immune homeostasis with lower risk of mostly mild-to-moderate irAEs [191]. In contrast, PD-L1 degraders irreversibly eliminates the protein through the ubiquitin-proteasome or lysosomal pathway. This approach not only completely blocks the pro-tumor immunosuppressive function of PD-L1 but also abrogates its physiological role in maintaining immune tolerance, resulting to a more profound and sustained disturbance to immune homeostasis and a higher risk of irAEs [192]. However, the application of targeted delivery systems can enhance tumor-specific accumulation to reduce off-target effects, and the more thoroughly blockade of PD-L1-mediated pro-tumor signals (including ligand-independent ones) by degraders may offer more durable therapeutic efficacy [193]. From a biological standpoint, PD-L1 inhibition and degradation may result in different degrees of immune modulation, and careful consideration of immune homeostasis and safety will be important for future clinical translation.
Most PD-L1 inhibitors utilize a biphenyl core scaffold that adapts to the hydrophobic binding cavity of PD-L1 through rotation, inducing dimerization / endocytosis. For example, CCX559 [67, 68], S4-1 [83], INCB086550[73], BMS-202[88-90]. Some have adopted C2-symmetric or dual-site binding structures, such as GS-4224 [47] and CB31 [53], to bind two PD-L1 molecules. To overcome single-action limitations or optimize pharmacokinetics, heterocycles [94], metal ligands [86], and natural product scaffolds [79] have been incorporated, whereas terminal hydrophilic groups address the poor water solubility of pure aromatic structures [72, 95]. PD-L1 degraders employ a “ligand/carrier-linker-effector ligand” modular design: As heterobifunctional entities, PROTACs incorporate a PD-L1-binding ligand, a specific E3 ubiquitin ligase ligand, and a connecting linker segment—linker properties regulate the formation of the ternary complex [122, 124, 125], while LYTACs target lysosomes via LTR ligands with linkers maintaining dual-ligand activity; for example, CI-M6PR ligands [129-131], scavenger receptor ligands [134], cRGD [135], cell-penetrating peptides [137]. Dual-target inhibitors integrate PD-L1 active moieties (predominantly biphenyl scaffolds) with secondary target pharmacophores via “pharmacophore hybridization”. For example, hydroxamate for HDAC6/3 [150, 153], indolinone for IDO1 [156], benzimidazole for CD47 [168], furopyrimidine for CD73 [172], quinazoline for EGFR [175], with linkers of tailored length/rigidity/hydrophilic-lipophilic balance. Also adopted the “scaffold fusion” strategy to synergistically modulate the PD-1/PD-L1 pathway and another key target, balancing dual-target efficacy and drug-likeness [185, 188].
The research and development of PD-1/PD-L1 inhibitors has reached a key and sophisticated development stage [194]. Although small molecule inhibitors offer distinct advantages such as oral bioavailability, low immunogenicity, and strong tumor penetration, antibody-based therapies remains the mainstream clinical practice, with small molecule mostly serving as complementary options [18, 195]. Because small molecule inhibitors face multiple core bottlenecks in clinical translation. First, in terms of drug resistance, primary and adaptive resistance coexist with heterogeneous mechanisms, making preemptive intervention difficult. Second, insufficient target specificity predisposes these agents to off-target toxicity, which may disrupt normal immune homeostasis or induce organ damage [196]. Third, pharmacokinetics is affected by factors such as individual metabolic differences and short half-lives, which limits the practical advantages of oral administration [195]. Fourth, clinical validation mostly remains in the early stages, lacking large-scale phase II data for direct comparison with antibody therapies. For instance, published clinical data for INCB086550 have been suboptimal, and its phase II results remain undisclosed; moreover, predictive biomarkers for efficacy are not yet well defined. Fifth, tumor heterogeneity renders them difficult to adapt to different PD-L1 expression types and cancer types [197]. Finally, issues such as purity control and stability during production also constitute hidden challenges [198]. To address these limitations, future development of PD-1/PD-L1 inhibitors will mainly focus on three key strategic directions: optimization of combination regimens, epigenetic targeting, and development of novel drug modalities [199]. combination strategy focuses on “resistance and insufficient target binding”, epigenetic targeting directly addresses “abnormal target expression regulation and heterogeneity”, and novel drug modalities optimize around “safety, selectivity, and clinical translation efficiency”. In terms of drug combination strategies, compensatory activation of alternative targets and synergistic efficacy enhancement can be achieved through combination with other immunomodulatory targets and chemotherapy/radiotherapy/anti-angiogenic agents [8]. At the epigenetic targeting level, aimed at addressing abnormalities in PD-1/PD-L1 methylation, histone deacetylation, and chromatin remodeling, specific regulatory drugs can be developed [200]. With respect to advancing emerging drug modalities, next-generation small molecule inhibitors, PROTACs/LYTACs degraders, and bispecific antibodies will be leveraged to overcome the limitations of traditional antibodies, thereby improving targeting specificity and safety [17]. With the advancement of mechanistic research and technological innovations, PD-1/PD-L1 inhibitors are poised to propel tumor immunotherapy toward greater precision and efficiency.
Abbreviations
mAbs: monoclonal antibodies; FDA: food and drug administration; PROTACs: proteolysis-targeting chimeras; LYTACs: lysosome-targeting chimeras; CTLA-4: cytotoxic T-lymphocyte-associated antigen 4; ICT: immune checkpoint therapy; PD-1: programmed cell death protein 1; PD-L1: programmed death-ligand 1; ITSM: immunoreceptor tyrosine-based switch motif; ITIM: immunoreceptor tyrosine-based inhibitory motif; TME: tumor microenvironment; NK cells: natural killer cells; DCs: dendritic cells; SH2: Src homology 2; SHP2: Src homology 2 domain-containing tyrosine phosphatase 2; TCR: T cell receptor; EGF: epidermal growth factor; IL-17: interleukin-17; TNF-α: tumor necrosis factor-alpha; LPS: lipopolysaccharide; IFN-γ: interferon-gamma; PTEN: phosphatase and tensin homolog; RAS: rat sarcoma virus; EGFR: epidermal growth factor receptor; PLCγ1: phospholipase C gamma 1; PIP2: phosphatidylinositol 4,5-bisphosphate; DAG: diacylglycerol; IP3: inositol trisphosphate; PKCθ: protein kinase Cθ; CBM: CARMA1-BCL10-MALT1; NSCLC: non-small cell lung carcinoma; DLBCL: diffuse large B-cell lymphoma; CLL: Chronic lymphocytic leukemia; PBL: plasmablastic lymphoma; HNSCC: head and neck squamous cell carcinoma; PDAC: pancreatic ductal adenocarcinoma; SBRT: stereotactic body radiation; TAMs: tumor-associated macrophages; ccRCC: clear cell renal cell carcinoma; CAR-T: chimeric antigen receptor T cells; CAIX: carbonic anhydrase IX; Tfh: T follicular helper cells; PBMCs: peripheral blood mononuclear cells; BCAT1: branched-chain amino acid transaminase 1; TGI: tumor growth inhibition rate; irAEs: immune-related adverse events; PK/PD: pharmacokinetic/pharmacodynamic; SAR: structure-activity relationships; sPD-L1: soluble programmed death-ligand 1; AbTACs: antibody-based PROTACs; EPR: enhanced permeability and retention; FMD: folic acid-modified dextran; NRP-1: neuropilin-1; LTR: lysosomal targeting receptor; CI-M6PR: cation-independent mannose-6-phosphate receptor; SRs: Scavenger receptors; cRGD: cyclic Arg-Gly-Asp; IFLDs: integrin-facilitated lysosomal degraders; HIP1R: huntingtin-interacting protein 1-related; CPPs: cell-penetrating peptides; HDAC: histone deacetylase; IDO1: indoleamine 2,3-dioxygenase 1; PARP: poly (ADP-ribose) polymerase; JAK: Janus kinase; EGFR: epidermal growth factor receptor; VISTA: V-domain Ig suppressor of T-cell activation; NMR: nuclear magnetic resonance; HTRF: homogeneous time-resolved fluorescence; SNT: Ser-Asn-Thr.
Acknowledgements
Funding
This work received funding from the National Natural Science Foundation of China (82404425); Natural Science Foundation of Henan Province (252300421127); Fundamental Research of the Key Program in Higher Education by the Education Department of Henan Province (22ZX008); The Young Top Talent Program from Henan Association for Science and Technology; R&D of Key Project of Henan Province (241111312500).
Consent for publication
All participating authors have reviewed this paper and sanctioned its publication.
Author contributions
Jia-Yi Yin, Hui-Min Liu and Shao-Long Li contributed to the manuscript drafting; Jun-Jie Wang, Xin-Qian Ji, and Meng-Jie Fu were responsible for the collection of all relevant data; Cong-Jun Liu, Ning Wang, Guo-Liang Lu, Yan Li, Hong-Min Liu and Yi-Chao Zheng conducted critical review and revision of the manuscript; Xing-Jie Dai and Ying Liu were responsible for the study conception and review of the entire manuscript. All authors have read the final manuscript and approved its publication.
Competing interests
The authors have declared that no competing interest exists.
References
1. Kiri S, Ryba T. Cancer, metastasis, and the epigenome. Mol Cancer. 2024;23:154
2. Galassi C, Chan TA, Vitale I, Galluzzi L. The hallmarks of cancer immune evasion. Cancer Cell. 2024;42:1825-63
3. Boussiotis VA. Molecular and biochemical aspects of the PD-1 checkpoint pathway. N Engl J Med. 2016;375:1767-78
4. Kaushik I, Ramachandran S, Zabel C, Gaikwad S, Srivastava SK. The evolutionary legacy of immune checkpoint inhibitors. Semin Cancer Biol. 2022;86:491-8
5. Sharma P, Goswami S, Raychaudhuri D, Siddiqui BA, Singh P, Nagarajan A. et al. Immune checkpoint therapy-current perspectives and future directions. Cell. 2023;186:1652-69
6. Weiss SA, Kluger H. CheckMate-067: raising the bar for the next decade in oncology. J Clin Oncol. 2022;40:111-3
7. Wu Y, Chen W, Xu ZP, Gu W. PD-L1 distribution and perspective for cancer immunotherapy-blockade, knockdown, or inhibition. Front Immunol. 2019;10:2022
8. Yi M, Zheng X, Niu M, Zhu S, Ge H, Wu K. Combination strategies with PD-1/PD-L1 blockade: current advances and future directions. Mol Cancer. 2022;21:28
9. Chen RY, Zhu Y, Shen YY, Xu QY, Tang HY, Cui NX. et al. The role of PD-1 signaling in health and immune-related diseases. Front Immunol. 2023;14:1163633
10. Zhang J, Dang F, Ren J, Wei W. Biochemical aspects of PD-L1 regulation in cancer immunotherapy. Trends Biochem Sci. 2018;43:1014-32
11. Gao M, Shi J, Xiao X, Yao Y, Chen X, Wang B. et al. PD-1 regulation in immune homeostasis and immunotherapy. Cancer Lett. 2024;588:216726
12. Lemma EY, Letian A, Altorki NK, McGraw TE. Regulation of PD-L1 trafficking from synthesis to degradation. Cancer Immunol Res. 2023;11:866-74
13. Geng Q, Dong Y, Jin P, Xu J, Chen L, Du X. et al. Synthesis and preliminary evaluation of aminophenol derivatives as molecular glues blocking PD-1/PD-L1 interaction. J Mol Struct. 2023;1289:135900
14. Xu J, Kong Y, Zhu P, Du M, Liang X, Tong Y. et al. Progress in small molecule inhibitors targeting PD-L1. RSC Med Chem. 2024;15:1161-75
15. Fan Yang DL, Yue Wang, Bojie Wen, Yisheng Lai. Clinical progress of PD-1/PD-L1 small molecule inhibitors. HJMCe. 2024;12:77-86
16. Zhang F, Ramar S, Wang Y, Xu H, Zhang K, Awadasseid A. et al. Advances in cancer immunotherapy using small molecular inhibitors targeting the PD-1/PD-L1 interaction. Bioorg Med Chem. 2025;127:118238
17. Wang Z, Yuan L, Liao X, Guo X, Chen J. Reducing PD-L1 expression by degraders and downregulators as a novel strategy to target the PD-1/PD-L1 pathway. J Med Chem. 2024;67:6027-43
18. Javed SA, Najmi A, Ahsan W, Zoghebi K. Targeting PD-1/PD-L1 immune checkpoint inhibition for cancer immunotherapy: success and challenges. Front Immunol. 2024;15:1383456
19. Li X, Gao S, Shan C, Zhang Q, Tan Y, Yu X. et al. Advances in PD-1/PD-L1 pathway inhibitors in the treatment of thyroid cancer: mechanisms and clinical therapeutic perspectives. Front Immunol. 2025;16:1643421
20. Zak KM, Kitel R, Przetocka S, Golik P, Guzik K, Musielak B. et al. Structure of the complex of human programmed death 1, PD-1, and its ligand PD-L1. Structure. 2015;23:2341-48
21. Keir ME, Butte MJ, Freeman GJ, Sharpe AH. PD-1 and its ligands in tolerance and immunity. Annu Rev Immunol. 2008;26:677-704
22. Zhang X, Schwartz JC, Guo X, Bhatia S, Cao E, Lorenz M. et al. Structural and functional analysis of the costimulatory receptor programmed death-1. Immunity. 2004;20:337-47
23. Chen Y, Liu P, Gao F, Cheng H, Qi J, Gao GF. A dimeric structure of PD-L1: functional units or evolutionary relics? Protein Cell. 2010;1:153-60
24. Tan S, Zhang H, Chai Y, Song H, Tong Z, Wang Q. et al. An unexpected N-terminal loop in PD-1 dominates binding by nivolumab. Nat Commun. 2017;8:14369
25. Wang R, He S, Long J, Wang Y, Jiang X, Chen M. et al. Emerging therapeutic frontiers in cancer: insights into posttranslational modifications of PD-1/PD-L1 and regulatory pathways. Exp Hematol Oncol. 2024;13:46
26. Zak KM, Grudnik P, Guzik K, Zieba BJ, Musielak B, Dömling A. et al. Structural basis for small molecule targeting of the programmed death ligand 1 (PD-L1). Oncotarget. 2016;7:30323-35
27. Chen T, Li Q, Liu Z, Chen Y, Feng F, Sun H. Peptide-based and small synthetic molecule inhibitors on PD-1/PD-L1 pathway: A new choice for immunotherapy? Eur J Med Chem. 2019;161:378-98
28. Ortega MA, Boaru DL, De Leon-Oliva D, Fraile-Martinez O, García-Montero C, Rios L. et al. PD-1/PD-L1 axis: implications in immune regulation, cancer progression, and translational applications. J Mol Med (Berl). 2024;102:987-1000
29. Zabeti Touchaei A, Vahidi S. MicroRNAs as regulators of immune checkpoints in cancer immunotherapy: targeting PD-1/PD-L1 and CTLA-4 pathways. Cancer Cell Int. 2024;24:102
30. Wang Z, Chen T, Lin W, Zheng W, Chen J, Huang F. et al. Functional tumor specific CD8+ T cells in spleen express a high level of PD-1. Int Immunopharmacol. 2020;80:106242
31. Onuora S. Freeing PD-L1 alleviates autoimmunity. Nat Rev Rheumatol. 2022;18:185
32. Zhang J, Yu J, Liu M, Xie Z, Lei X, Yang X. et al. Small molecule modulators of tumor immune microenvironment. Bioorg Chem. 2024;145:107251
33. Molina-Arcas M, Downward J. Exploiting the therapeutic implications of KRAS inhibition on tumor immunity. Cancer Cell. 2024;42:338-57
34. Godiyal Y, Maheshwari D, Taniguchi H, Zinzuwadia SS, Morera-Díaz Y, Tewari D. et al. Role of PD-1/PD-L1 signaling axis in oncogenesis and its targeting by bioactive natural compounds for cancer immunotherapy. Mil Med Res. 2024;11:82
35. Zhao Y, Lee CK, Lin CH, Gassen RB, Xu X, Huang Z. et al. PD-L1:CD80 cis-heterodimer triggers the co-stimulatory receptor CD28 while repressing the inhibitory PD-1 and CTLA-4 pathways. Immunity. 2019;51:1059-73.e9
36. Beenen AC, Sauerer T, Schaft N, Dörrie J. Beyond cancer: regulation and function of PD-L1 in health and immune-related diseases. Int J Mol Sci. 2022;23:8599
37. Zheng H, Ning Y, Zhan Y, Liu S, Wen Q, Fan S. New insights into the important roles of tumor cell-intrinsic PD-1. Int J Biol Sci. 2021;17:2537-47
38. Filippone A, Lanza M, Mannino D, Raciti G, Colarossi C, Sciacca D. et al. PD-1/PD-L1 immune checkpoint as a potential target for preventing brain tumor progression. Cancer Immunol Immunother. 2022;71:2067-75
39. Liu Q, Guan Y, Li S. Programmed death receptor (PD-1)/PD-ligand (L)1 in urological cancers: the "all-around warrior" in immunotherapy. Mol Cancer. 2024;23:183
40. Cha JH, Chan LC, Li CW, Hsu JL, Hung MC. Mechanisms controlling PD-L1 expression in cancer. Mol Cell. 2019;76:359-70
41. Feng C, Zhang L, Chang X, Qin D, Zhang T. Regulation of post-translational modification of PD-L1 and advances in tumor immunotherapy. Front Immunol. 2023;14:1230135
42. Abaza A, Sid Idris F, Anis Shaikh H, Vahora I, Moparthi KP, Al Rushaidi MT. et al. Programmed cell death protein 1 (PD-1) and programmed cell death ligand 1 (PD-L1) immunotherapy: a promising breakthrough in cancer therapeutics. Cureus. 2023;15:e44582
43. Chan AC, Iwashima M, Turck CW, Weiss A. ZAP-70: a 70 kd protein-tyrosine kinase that associates with the TCR zeta chain. Cell. 1992;71:649-62
44. Courtney AH, Lo WL, Weiss A. TCR signaling: mechanisms of onitiation and propagation. Trends Biochem Sci. 2018;43:108-23
45. Lu W, Helou YA, Shrinivas K, Liou J, Au-Yeung BB, Weiss A. The phosphatidylinositol-transfer protein Nir3 promotes PI(4,5)P(2) replenishment in response to TCR signaling during T cell development and survival. Nat Immunol. 2023;24:136-47
46. Imboden JB, Stobo JD. Transmembrane signalling by the T cell antigen receptor. perturbation of the T3-antigen receptor complex generates inositol phosphates and releases calcium ions from intracellular stores. J Exp Med. 1985;161:446-56
47. Beals CR, Clipstone NA, Ho SN, Crabtree GR. Nuclear localization of NF-ATc by a calcineurin-dependent, cyclosporin-sensitive intramolecular interaction. Genes Dev. 1997;11:824-34
48. Finco TS, Kadlecek T, Zhang W, Samelson LE, Weiss A. LAT is required for TCR-mediated activation of plcgamma1 and the Ras pathway. Immunity. 1998;9:617-26
49. Idris I, Gray S, Donnelly R. Protein kinase C activation: isozyme-specific effects on metabolism and cardiovascular complications in diabetes. Diabetologia. 2001;44:659-73
50. Wegener E, Oeckinghaus A, Papadopoulou N, Lavitas L, Schmidt-Supprian M, Ferch U. et al. Essential role for IKB kinase beta in remodeling Carma1-Bcl10-Malt1 complexes upon T cell activation. Mol Cell. 2006;23:13-23
51. August A, Dupont B. CD28 of T lymphocytes associates with phosphatidylinositol 3-kinase. Int Immunol. 1994;6:769-74
52. Wang S, Liu C, Yang C, Jin Y, Cui Q, Wang D. et al. PI3K/AKT/mTOR and PD-1/CTLA-4/CD28 pathways as key targets of cancer immunotherapy (review). Oncol Lett. 2024;28:567
53. Chemnitz JM, Parry RV, Nichols KE, June CH, Riley JL. SHP-1 and SHP-2 associate with immunoreceptor tyrosine-based switch motif of programmed death 1 upon primary human T cell stimulation, but only receptor ligation prevents T cell activation. J Immunol. 2004;173:945-54
54. Hui E, Cheung J, Zhu J, Su X, Taylor MJ, Wallweber HA. et al. T cell costimulatory receptor CD28 is a primary target for PD-1-mediated inhibition. Science. 2017;355:1428-33
55. Sheppard KA, Fitz LJ, Lee JM, Benander C, George JA, Wooters J. et al. PD-1 inhibits T-cell receptor induced phosphorylation of the ZAP70/CD3zeta signalosome and downstream signaling to PKCtheta. FEBS Lett. 2004;574:37-41
56. Tocheva AS, Peled M, Strazza M, Adam KR, Lerrer S, Nayak S. et al. Quantitative phosphoproteomic analysis reveals involvement of PD-1 in multiple T cell functions. J Biol Chem. 2020;295:18036-50
57. Karyampudi L, Lamichhane P, Krempski J, Kalli KR, Behrens MD, Vargas DM. et al. PD-1 blunts the function of ovarian tumor-infiltrating dendritic cells by inactivating NF-κB. Cancer Res. 2016;76:239-50
58. Garcia-Lacarte M, Grijalba SC, Melchor J, Arnaiz-Leché A, Roa S. The PD-1/PD-L1 checkpoint in normal germinal centers and diffuse large B-cell lymphomas. Cancers (Basel). 2021;13:4683
59. Böttcher M, Bruns H, Völkl S, Lu J, Chartomatsidou E, Papakonstantinou N. et al. Control of PD-L1 expression in CLL-cells by stromal triggering of the Notch-c-Myc-EZH2 oncogenic signaling axis. J Immunother Cancer. 2021;9:e001889
60. Rosado FG, Coberly J, Gupta A, John G, Naina H, Koduru P. et al. PD-1/PD-L1 expressions in plasmablastic lymphoma with clinicopathological correlation. Ann Clin Lab Sci. 2021;51:174-81
61. William WN Jr, Zhao X, Bianchi JJ, Lin HY, Cheng P, Lee JJ. et al. Immune evasion in HPV(-) head and neck precancer-cancer transition is driven by an aneuploid switch involving chromosome 9p loss. Proc Natl Acad Sci U S A. 2021;118:e2022655118
62. Netzer C, von Arps-Aubert V, Mačinković I, von der Grün J, Küffer S, Ströbel P. et al. Association between spatial distribution of leukocyte subsets and clinical presentation of head and neck squamous cell carcinoma. Front Immunol. 2023;14:1240394
63. Wang J, Gai J, Zhang T, Niu N, Qi H, Thomas DL 2nd. et al. Neoadjuvant radioimmunotherapy in pancreatic cancer enhances effector T cell infiltration and shortens their distances to tumor cells. Sci Adv. 2024;10:eadk1827
64. Wang Y, Cho JW, Kastrunes G, Buck A, Razimbaud C, Culhane AC. et al. Immune-restoring CAR-T cells display antitumor activity and reverse immunosuppressive TME in a humanized ccRCC mouse model. iScience. 2024;27:108879
65. Odegard JM, Othman AA, Lin KW, Wang AY, Nazareno J, Yoon OK. et al. Oral PD-L1 inhibitor GS-4224 selectively engages PD-L1 high cells and elicits pharmacodynamic responses in patients with advanced solid tumors. J Immunother Cancer. 2024;12:e008547
66. Chai I, Kornyeyev D, Hsieh E, Magombedze G, Stapleton L, Hung M. et al. Effects of small molecule-induced dimerization on the programmed death ligand 1 protein life cycle. Scientific Reports. 2022;12:21286
67. Tapia G, Lundy J, Richardson GE, Zhao N, Ebsworth K, Yue H. et al. Preliminary data from an ongoing phase 1 dose-escalation study of CCX559, an orally administered small molecule PD-L1 inhibitor, in patients with advanced solid tumors. J Clin Oncol. 2022;40:2593
68. Sullivan KMC, Vilalta M, Ertl LS, Wang Y, Dunlap C, Ebsworth K. et al. CCX559 is a potent, orally-administered small molecule PD-L1 inhibitor that induces anti-tumor immunity. PLoS One. 2023;18:e0286724
69. Wu JJ, He H. Abstract 5529: in vivo efficacy evaluation of ASC61, an oral PD-L1 inhibitor, in two tumor mouse models. Cancer Research. 2022;82:5529 -
70. Chen R, Yuan D, Ma J. Advances of biphenyl small molecule inhibitors targeting PD-1/PD-L1 interaction in cancer immunotherapy. Future Med Chem. 2022;14:97-113
71. Slota A, Golebiowska-Mendroch K, Kocik-Krol J, Musielak B, Stec M, Weglarczyk K. et al. Characterization of clinically evaluated small molecule inhibitors of PD-L1 for immunotherapy. ACS Med Chem Lett. 2025;16:1359-64
72. Wang Y, Jing X, Chen H, Zhang H, Ma T, Zhang Y. et al. BPI-371153, an orally bioavailable small molecule PD-L1 inhibitor. Cancer Res. 2022;82:5444 -
73. Koblish HK, Wu L, Wang LS, Liu PCC, Wynn R, Rios-Doria J. et al. Characterization of INCB086550: a potent and novel small molecule PD-L1 inhibitor. Cancer Discov. 2022;12:1482-99
74. Piha-Paul S, Mitchell T, Sahebjam S, Mehnert J, Karasic T, O'Hayer K. et al. 419 Pharmacodynamic biomarkers demonstrate T-cell activation in patients treated with the oral PD-L1 inhibitor INCB086550 in a phase 1 clinical trial. J Immunother Cancer. 2020;8:419
75. Van Cutsem E, Prenen H, Delafontaine B, Spencer K, Mitchell T, Burris H. et al. 529 Phase 1 study of INCB086550, an oral PD-L1 inhibitor, in immune-checkpoint naive patients with advanced solid tumors. J Immunother Cancer. 2021;9:529
76. Wu Y, Yang Z, Cheng K, Bi H, Chen J. Acta Pharm Sin B. Acta Pharmaceutica Sinica B. 2022;12:4287-308
77. Jiang J, Zou X, Liu Y, Liu X, Dong K, Yao X. et al. Simultaneous determination of a novel PD-L1 inhibitor, IMMH-010, and its active metabolite, YPD-29B, in rat biological matrices by polarity-switching liquid chromatography-tandem mass spectrometry: application to ADME studies. Front Pharmacol. 2021;12:677120
78. Wang Y, Liu X, Zou X, Wang S, Luo L, Liu Y. et al. Metabolism and interspecies variation of IMMH-010, a programmed cell death ligand 1 inhibitor prodrug. Pharmaceutics. 2021;13:598
79. Hermanowicz JM, Pawlak K, Sieklucka B, Czarnomysy R, Kwiatkowska I, Kazberuk A. et al. MM-129 as a novel inhibitor targeting PI3K/AKT/mTOR and PD-L1 in colorectal cancer. Cancers (Basel). 2021;13:3203
80. Dong G, Jiang Y, Zhang F, Zhu F, Liu J, Xu Z. Recent updates on 1,2,3-, 1,2,4-, and 1,3,5-triazine hybrids (2017-present): the anticancer activity, structure-activity relationships, and mechanisms of action. Arch Pharm (Weinheim). 2023;356:e2200479
81. Hermanowicz JM, Kalaska B, Pawlak K, Sieklucka B, Miklosz J, Mojzych M. et al. Preclinical toxicity and safety of MM-129—first-in-class BTK/PD-L1 inhibitor as a potential candidate against colon cancer. Pharmaceutics. 2021;13:1222
82. Kwiatkowska I, Hermanowicz JM, Czarnomysy R, Surażyński A, Kowalczuk K, Kałafut J. et al. Assessment of an anticancer effect of the simultaneous administration of MM-129 and indoximod in the colorectal cancer model. Cancers. 2023;16:122
83. Sun C, Yin M, Cheng Y, Kuang Z, Liu X, Wang G. et al. Novel small molecule PD-L1 inhibitor induces PD-L1 internalization and optimizes the immune microenvironment. J Med Chem. 2023;66:2064-83
84. Ruengsatra T, Soponpong J, Nalinratana N, Jirapongwattana N, Dunkoksung W, Rattanangkool E. et al. Design, synthesis, and optimization of novel PD-L1 inhibitors and the identification of a highly potent and orally bioavailable PD-L1 inhibitor. Eur J Med Chem. 2024;277:116730
85. Wang F, Ye W, Wang S, He Y, Zhong H, Wang Y. et al. Discovery of a new inhibitor targeting PD-L1 for cancer immunotherapy. Neoplasia. 2021;23:281-93
86. Liu B, Liang BB, Cao WD, Su XX, Cao Q, Mao ZW. Platinum-metformin conjugates acting as promising PD-L1 inhibitors through the AMP-activated protein kinase mediated lysosomal degradation pathway. Angew Chem Int Ed Engl. 2024;63:e202410586
87. Zhang N, Dou Y, Liu L, Zhang X, Liu X, Zeng Q. et al. SA-49, a novel aloperine derivative, induces MITF-dependent lysosomal degradation of PD-L1. EBioMedicine. 2019;40:151-62
88. Ashizawa T, Iizuka A, Tanaka E, Kondou R, Miyata H, Maeda C. et al. Antitumor activity of the PD-1/PD-L1 binding inhibitor BMS-202 in the humanized MHC-double knockout NOG mouse. Biomed Res. 2019;40:243-50
89. Cai Y, Xiao M, Li X, Zhou S, Sun Y, Yu W. et al. BMS-202, a PD-1/PD-L1 inhibitor, decelerates the pro-fibrotic effects of fibroblasts derived from scar tissues via ERK and TGFβ1/Smad signaling pathways. Immun Inflamm Dis. 2022;10:e693
90. Yang X, Wang W, Ji T. Metabolic remodeling by the PD-L1 inhibitor BMS-202 significantly inhibits cell malignancy in human glioblastoma. Cell Death Dis. 2024;15:186
91. Chen FF, Li Z, Ma D, Yu Q. Small molecule PD-L1 inhibitor BMS1166 abrogates the function of PD-L1 by blocking its ER export. Oncoimmunology. 2020;9:1831153
92. Wherry EJ, Kurachi M. Molecular and cellular insights into T cell exhaustion. Nat Rev Immunol. 2015;15:486-99
93. Zhang F, Zhang H, Zhou S, Plewka J, Wang M, Sun S. et al. Design, synthesis, and evaluation of antitumor activity of 2-arylmethoxy-4-(2-fluoromethyl-biphenyl-3-ylmethoxy) benzylamine derivatives as PD-1/PD-L1 inhibitors. Eur J Med Chem. 2024;276:116683
94. Wang T, Cai S, Cheng Y, Zhang W, Wang M, Sun H. et al. Discovery of small molecule inhibitors of the PD-1/PD-L1 axis that promote PD-L1 internalization and degradation. J Med Chem. 2022;65:3879-93
95. Yang Z, Yang P, Xu J, Yang X, Zhou J, He H. et al. Discovery and crystallography study of novel resorcinol dibenzyl ether-based PD-1/PD-L1 inhibitors with improved drug-like and pharmacokinetic properties for cancer treatment. J Med Chem. 2025;68:12593-614
96. Lu T, Zhang J, Chen Q, Ni M, Zhang J, Wu Y. et al. Design, synthesis, evaluation, and SAR of 5-phenylisoindoline derivatives, a potent class of small molecule inhibitors targeting the programmed cell death-1/programmed cell death-ligand 1 (PD-1/PD-L1) interaction. J Med Chem. 2025;68:7291-312
97. Park JJ, Thi EP, Carpio VH, Bi Y, Cole AG, Dorsey BD. et al. Checkpoint inhibition through small molecule-induced internalization of programmed death-ligand 1. Nat Commun. 2021;12:1222
98. Feng C, Ge Y, Wang S, Li M, Chen Q, Dong H. et al. Discovery of small molecule PD-L1 inhibitors via virtual screening and their immune-mediated anti-tumor effects. Pharmaceuticals (Basel). 2025;18:1209
99. Wang F, Ye W, He Y, Zhong H, Zhu Y, Han J. et al. Identification of CBPA as a new inhibitor of PD-1/PD-L1 interaction. Int J Mol Sci. 2023;24:3971
100. Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev. 2001;46:3-26
101. Lin A, Giuliano CJ, Palladino A, John KM, Abramowicz C, Yuan ML. et al. Off-target toxicity is a common mechanism of action of cancer drugs undergoing clinical trials. Sci Transl Med. 2019;11:eaaw8412
102. Postow MA, Sidlow R, Hellmann MD. Immune-related adverse events associated with immune checkpoint blockade. N Engl J Med. 2018;378:158-68
103. Maddox CB, Rasmussen L, White EL. Adapting cell-based assays to the high throughput screening platform: problems encountered and lessons learned. JALA Charlottesv Va. 2008;13:168-73
104. Mak IW, Evaniew N, Ghert M. Lost in translation: animal models and clinical trials in cancer treatment. Am J Transl Res. 2014;6:114-8
105. Schenone M, Dančík V, Wagner BK, Clemons PA. Target identification and mechanism of action in chemical biology and drug discovery. Nat Chem Biol. 2013;9:232-40
106. Lu CH, Chung WM, Tsai CH, Cheng JC, Hsu KC, Tzeng HE. In vitro characterization of a small molecule PD-1 inhibitor that targets the PD-l/PD-L1 interaction. Sci Rep. 2022;12:303
107. DiFrancesco M, Hofer J, Aradhya A, Rufinus J, Stoddart J, Finocchiaro S. et al. Discovery of small molecule PD-1/PD-L1 antagonists through combined virtual screening and experimental validation. Comput Biol Chem. 2023;102:107804
108. Cheng B, Xiao Y, Xue M, Cao H, Chen J. Recent advances in the development of PD-L1 modulators: degraders, downregulators, and dovalent inhibitors. J Med Chem. 2020;63:15389-98
109. Xu X, Luo S, Zhao X, Tang B, Zhang E, Liu J. et al. Computational analysis of PD-L1 dimerization mechanism induced by small molecules and potential dynamical properties. Int J Biol Macromol. 2024;265:130921
110. Bocanegra A, Blanco E, Fernandez-Hinojal G, Arasanz H, Chocarro L, Zuazo M. et al. PD-L1 in systemic immunity: unraveling its contribution to PD-1/PD-L1 blockade immunotherapy. Int J Mol Sci. 2020;21:5918
111. Zou W, Luo X, Gao M, Yu C, Wan X, Yu S. et al. Optimization of cancer immunotherapy on the basis of programmed death ligand-1 distribution and function. Br J Pharmacol. 2024;181:257-72
112. Du J, Qin Y, Wu Y, Zhao W, Zhai W, Qi Y. et al. The design of high affinity human PD-1 mutants by using molecular dynamics simulations (MD). Cell Commun Signal. 2018;16:25
113. Mittal L, Srivastava M, Kumari A, Tonk RK, Awasthi A, Asthana S. Interplay among structural stability, plasticity, and energetics determined by conformational attuning of flexible loops in PD-1. J Chem Inf Model. 2021;61:358-84
114. Laba S, Mallett G, Amarnath S. The depths of PD-1 function within the tumor microenvironment beyond CD8(+) T cells. Semin Cancer Biol. 2022;86:1045-55
115. Hutchins B, Starling GC, McCoy MA, Herzyk D, Poulet FM, Dulos J. et al. Biophysical and immunological characterization and in vivo pharmacokinetics and toxicology in nonhuman primates of the anti-PD-1 antibody pembrolizumab. Mol Cancer Ther. 2020;19:1298-307
116. Sugiura D, Okazaki IM, Maeda TK, Maruhashi T, Shimizu K, Arakaki R. et al. PD-1 agonism by anti-CD80 inhibits T cell activation and alleviates autoimmunity. Nat Immunol. 2022;23:399-410
117. Wang T, Wu X, Guo C, Zhang K, Xu J, Li Z. et al. Development of inhibitors of the programmed cell death-1/programmed cell death-ligand 1 signaling pathway. J Med Chem. 2019;62:1715-30
118. Yang J, Hu L. Immunomodulators targeting the PD-1/PD-L1 protein-protein interaction: from antibodies to small molecules. Med Res Rev. 2019;39:265-301
119. Yang Z, Xu J, Yang X, Chen J. Targeted protein degradation with small molecules for cancer immunotherapy. Asian J Pharm Sci. 2025;20:101058
120. Li S, Chen T, Liu J, Zhang H, Li J, Wang Z. et al. PROTACs: novel tools for improving immunotherapy in cancer. Cancer Lett. 2023;560:216128
121. Dai XJ, Ji SK, Fu MJ, Liu GZ, Liu HM, Wang SP. et al. Degraders in epigenetic therapy: PROTACs and beyond. Theranostics. 2024;14:1464-99
122. Cheng B, Ren Y, Cao H, Chen J. Discovery of novel resorcinol diphenyl ether-based PROTAC-like molecules as dual inhibitors and degraders of PD-L1. Eur J Med Chem. 2020;199:112377
123. Nalawansha DA, Paiva SL, Rafizadeh DN, Pettersson M, Qin L, Crews CM. Targeted protein internalization and degradation by endosome targeting chimeras (ENDTACs). ACS Cent Sci. 2019;5:1079-84
124. Wang Y, Zhou Y, Cao S, Sun Y, Dong Z, Li C. et al. In vitro and in vivo degradation of programmed cell death ligand 1 (PD-L1) by a proteolysis targeting chimera (PROTAC). Bioorg Chem. 2021;111:104833
125. Liu Y, Zheng M, Ma Z, Zhou Y, Huo J, Zhang W. et al. Design, synthesis, and evaluation of PD-L1 degraders to enhance T cell killing activity against melanoma. Chin Chem Lett. 2023;34:107762
126. Su W, Tan M, Wang Z, Zhang J, Huang W, Song H. et al. Targeted degradation of PD-L1 and activation of the STING pathway by carbon-dot-based PROTACs for cancer immunotherapy. Angew Chem Int Ed Engl. 2023;62:e202218128
127. Qi Q, Zhang Z, ji X, Wang D. Rational design of PROTAC degraders and their spatiotemporal controlled delivery for enhanced tumor penetration and PD-L1 protein degradation. J Med Chem. 2025;68:22665-88
128. Chen X, Zhou Y, Zhao Y, Tang W. Targeted degradation of extracellular secreted and membrane proteins. Trends Pharmacol Sci. 2023;44:762-75
129. Banik SM, Pedram K, Wisnovsky S, Ahn G, Riley NM, Bertozzi CR. Lysosome-targeting chimaeras for degradation of extracellular proteins. Nature. 2020;584:291-7
130. Li Y, Liu X, Yu L, Huang X, Wang X, Han D. et al. Covalent LYTAC enabled by DNA aptamers for immune checkpoint degradation therapy. J Am Chem Soc. 2023;145:24506-21
131. Mikitiuk M, Barczyński J, Bielski P, Arciniega M, Tyrcha U, Hec A. et al. IGF2 peptide-based LYTACs for targeted degradation of extracellular and transmembrane proteins. Molecules. 2023;28:7519
132. Cotton AD, Nguyen DP, Gramespacher JA, Seiple IB, Wells JA. Development of antibody-based PROTACs for the degradation of the cell-surface immune checkpoint protein PD-L1. J Am Chem Soc. 2021;143:593-8
133. Pance K, Gramespacher JA, Byrnes JR, Salangsang F, Serrano JC, Cotton AD. et al. Modular cytokine receptor-targeting chimeras for targeted degradation of cell surface and extracellular proteins. Nat Biotechnol. 2023;41:273-81
134. Huang W, Yang C, Cheng S, Fu S, Chen X, Zhu Y. et al. A DNA-mediated lysosomal degradation strategy for targeted degradation of PD-L1 protein. J Med Chem. 2025;68:11829-40
135. Zheng J, He W, Li J, Feng X, Li Y, Cheng B. et al. Bifunctional compounds as molecular degraders for integrin-facilitated targeted protein degradation. J Am Chem Soc. 2022;144:21831-6
136. Wang H, Yao H, Li C, Shi H, Lan J, Li Z. et al. HIP1R targets PD-L1 to lysosomal degradation to alter T cell-mediated cytotoxicity. Nat Chem Biol. 2019;15:42-50
137. He W, Chen C, Zheng J, Li Y, Shi H, Zhou Y. et al. Targeted degradation of cell surface proteins through endocytosis triggered by cell-penetrating peptide-small molecule conjugates. Nat Commun. 2025;16:7575
138. Mullard A. Targeted protein degraders crowd into the clinic. Nat Rev Drug Discov. 2021;20:247-50
139. Sun X, Gao H, Yang Y, He M, Wu Y, Song Y. et al. PROTACs: great opportunities for academia and industry. Signal Transduct Target Ther. 2019;4:64
140. Wang C, Zhang Y, Xing D, Zhang R. PROTACs technology for targeting non-oncoproteins: advances and perspectives. Bioorg Chem. 2021;114:105109
141. He S, Gao F, Ma J, Ma H, Dong G, Sheng C. Aptamer-PROTAC conjugates (APCs) for tumor-specific targeting in breast cancer. Angew Chem Int Ed Engl. 2021;60:23299-305
142. Testa A, Hughes SJ, Lucas X, Wright JE, Ciulli A. Structure-based design of a macrocyclic PROTAC. Angew Chem Int Ed Engl. 2020;59:1727-34
143. Kamb A, Wee S, Lengauer C. Why is cancer drug discovery so difficult? Nat Rev Drug Discov. 2007;6:115-20
144. Bayat Mokhtari R, Homayouni TS, Baluch N, Morgatskaya E, Kumar S, Das B. et al. Combination therapy in combating cancer. Oncotarget. 2017;8:38022-43
145. de Ruijter AJ, van Gennip AH, Caron HN, Kemp S, van Kuilenburg AB. Histone deacetylases (HDACs): characterization of the classical HDAC family. Biochem J. 2003;370:737-49
146. Woods DM, Sodré AL, Villagra A, Sarnaik A, Sotomayor EM, Weber J. HDAC inhibition upregulates PD-1 ligands in melanoma and augments immunotherapy with PD-1 blockade. Cancer Immunol Res. 2015;3:1375-85
147. Simões-Pires C, Zwick V, Nurisso A, Schenker E, Carrupt PA, Cuendet M. HDAC6 as a target for neurodegenerative diseases: what makes it different from the other HDACs? Mol Neurodegener. 2013;8:7
148. M L, P PV, T K, M P, E S, J P. et al. Essential role of HDAC6 in the regulation of PD-L1 in melanoma. Mol Oncol. 2016;10:735-50
149. Fukumoto T, Fatkhutdinov N, Zundell JA, Tcyganov EN, Nacarelli T, Karakashev S. et al. HDAC6 inhibition synergizes with anti-PD-L1 therapy in ARID1A-inactivated ovarian cancer. Cancer Res. 2019;79:5482-9
150. Bian JL, Bi XL, Wang M, Li ZY, Xu X, Wang JB. et al. Dual PD-1/PD-L1&HDACs inhibitors, and their preparation methods and applications. CN113387840A.
151. Makgoba TB, Kapp E, Egieyeh S, Joubert J. HDAC3 inhibitors: a patent review of their broad-spectrum applications as therapeutic agents. Expert Opin Ther Pat. 2024;34:273-95
152. Li L, Hao S, Gao M, Liu J, Xu X, Huang J. et al. HDAC3 inhibition promotes antitumor immunity by enhancing CXCL10-mediated chemotaxis and recruiting of immune cells. Cancer Immunol Res. 2023;11:657-73
153. Wang Z, He H, Liao X, Yuan L, Sun S, Xu C. et al. Discovery of dual PD-L1/HDAC3 inhibitors for tumor immunotherapy. J Med Chem. 2025;68:8046-64
154. Anu RI, Shiu KK, Khan KH. The immunomodulatory role of IDO1-Kynurenine-NAD(+) pathway in switching cold tumor microenvironment in PDAC. Front Oncol. 2023;13:1142838
155. Abdulla M, Alexsson A, Sundström C, Ladenvall C, Mansouri L, Lindskog C. et al. PD-L1 and IDO1 are potential targets for treatment in patients with primary diffuse large B-cell lymphoma of the CNS. Acta Oncol. 2021;60:531-8
156. Feng ZQ CX, Zhou C. et al. Compounds contained disulfur bond, their preparation methods and pharmaceutical compo sitions and applications. CN115073442A.
157. Gibson BA, Kraus WL. New insights into the molecular and cellular functions of poly(ADP-ribose) and PARPs. Nat Rev Mol Cell Biol. 2012;13:411-24
158. Curtin NJ, Szabo C. Poly(ADP-ribose) polymerase inhibition: past, present and future. Nat Rev Drug Discov. 2020;19:711-36
159. Jiao S, Xia W, Yamaguchi H, Wei Y, Chen M-K, Hsu J-M. et al. PARP inhibitor upregulates PD-L1 expression and enhances cancer-associated immunosuppression. Clin Cancer Res. 2017;23:3711-20
160. Ofori S, Awuah SG. Small molecule poly(ADP-ribose) polymerase and PD-L1 inhibitor conjugates as dual-action anticancer agents. ACS Omega. 2019;4:12584-97
161. Xue C, Yao Q, Gu X, Shi Q, Yuan X, Chu Q. et al. Evolving cognition of the JAK-STAT signaling pathway: autoimmune disorders and cancer. Signal Transduct Target Ther. 2023;8:204
162. Mimura K, Teh JL, Okayama H, Shiraishi K, Kua LF, Koh V. et al. PD-L1 expression is mainly regulated by interferon gamma associated with JAK-STAT pathway in gastric cancer. Cancer Sci. 2018;109:43-53
163. Yi M, Niu M, Xu L, Luo S, Wu K. Regulation of PD-L1 expression in the tumor microenvironment. J Hematol Oncol. 2021;14:10
164. Wang Z, He H, Xu J, Liao X, Tan J, Xu C. et al. Discovery of the first dual PD-L1/JAK inhibitor with enhanced in vivo antitumor immunity. Eur J Med Chem. 2026;306:118605
165. Logtenberg MEW, Scheeren FA, Schumacher TN. The CD47-SIRPα immune checkpoint. Immunity. 2020;52:742-52
166. Jiang Z, Sun H, Yu J, Tian W, Song Y. Targeting CD47 for cancer immunotherapy. J Hematol Oncol. 2021;14:180
167. Wang Y, Ni H, Zhou S, He K, Gao Y, Wu W. et al. Tumor-selective blockade of CD47 signaling with a CD47/PD-L1 bispecific antibody for enhanced anti-tumor activity and limited toxicity. Cancer Immunol Immunother. 2021;70:365-76
168. Jin S, Wang H, Li Y, Yang J, Li B, Shi P. et al. Discovery of a novel small molecule as CD47/SIRPα and PD-1/PD-L1 dual inhibitor for cancer immunotherapy. Cell Commun Signal. 2024;22:173
169. Jin D, Fan J, Wang L, Thompson LF, Liu A, Daniel BJ. et al. CD73 on tumor cells impairs antitumor T-cell responses: a novel mechanism of tumor-induced immune suppression. Cancer Res. 2010;70:2245-55
170. Bach N, Winzer R, Tolosa E, Fiedler W, Brauneck F. The clinical significance of CD73 in cancer. Int J Mol Sci. 2023;24:11759
171. Roh M, Wainwright DA, Wu JD, Wan Y, Zhang B. Targeting CD73 to augment cancer immunotherapy. Curr Opin Pharmacol. 2020;53:66-76
172. Wang S, Kong Z, Shi Y, Shao C, Wang W, Su Z. et al. Discovery of small and bifunctional molecules targeting PD-L1/CD73 for cancer dual immunotherapy. J Med Chem. 2024;67:9447-64
173. Levantini E, Maroni G, Del Re M, Tenen DG. EGFR signaling pathway as therapeutic target in human cancers. Semin Cancer Biol. 2022;85:253-75
174. Rubio-Pérez L, Lázaro-Gorines R, Harwood SL, Compte M, Navarro R, Tapia-Galisteo A. et al. A PD-L1/EGFR bispecific antibody combines immune checkpoint blockade and direct anti-cancer action for an enhanced anti-tumor response. Oncoimmunology. 2023;12:2205336
175. Yang Z, Liu Z, Wan S, Xu J, Huang Y, He H. et al. Discovery of novel small molecule-based potential PD-L1/EGFR dual inhibitors with high druggability for glioblastoma immunotherapy. J Med Chem. 2024;67:7995-8019
176. McKenna ED, Sarbanes SL, Cummings SW, Roll-Mecak A. The tubulin code, from molecules to health and disease. Annu Rev Cell Dev Biol. 2023;39:331-61
177. Hayashi H, Nakagawa K. Combination therapy with PD-1 or PD-L1 inhibitors for cancer. Int J Clin Oncol. 2020;25:818-30
178. Yang X, Cheng B, Xiao Y, Xue M, Liu T, Cao H. et al. Discovery of novel CA-4 analogs as dual inhibitors of tubulin polymerization and PD-1/PD-L1 interaction for cancer treatment. Eur J Med Chem. 2021;213:113058
179. Miao L, Li J, Liu Q, Feng R, Das M, Lin CM. et al. Transient and local expression of chemokine and immune checkpoint traps to treat pancreatic cancer. ACS Nano. 2017;11:8690-706
180. Zeng Y, Li B, Liang Y, Reeves PM, Qu X, Ran C. et al. Dual blockade of CXCL12-CXCR4 and PD-1-PD-L1 pathways prolongs survival of ovarian tumor-bearing mice by prevention of immunosuppression in the tumor microenvironment. Faseb j. 2019;33:6596-608
181. Cheng B, Wang W, Liu T, Cao H, Pan W, Xiao Y. et al. Bifunctional small molecules targeting PD-L1/CXCL12 as dual immunotherapy for cancer treatment. Signal Transduct Target Ther. 2023;8:91
182. Wang L, Rubinstein R, Lines JL, Wasiuk A, Ahonen C, Guo Y. et al. VISTA, a novel mouse Ig superfamily ligand that negatively regulates T cell responses. J Exp Med. 2011;208:577-92
183. Zheng S, Zhang K, Zhang X, Xiao Y, Wang T, Jiang S. Development of inhibitors targeting the V-domain Ig suppressor of T cell activation signal pathway. J Med Chem. 2022;65:11900-12
184. Bang Y, Sosman J, Daud A, Meric-Bernstam F, Garcia-Corbacho J, Patel M. et al. Phase 1 study of CA-170, a first-in-class, orally available, small molecule immune checkpoint inhibitor (ICI) dually targeting VISTA and PD-L1, in patients with advanced solid tumors or lymphomas. J Immunother Cancer. 2018;6:114
185. Musielak B, Kocik J, Skalniak L, Magiera-Mularz K, Sala D, Czub M. et al. CA-170 - A potent small molecule PD-L1 inhibitor or not? Molecules. 2019;24:2804
186. Sasikumar PG, Sudarshan NS, Adurthi S, Ramachandra RK, Samiulla DS, Lakshminarasimhan A. et al. PD-1 derived CA-170 is an oral immune checkpoint inhibitor that exhibits preclinical anti-tumor efficacy. Commun Biol. 2021;4:699
187. Radhakrishnan VS, Bakhshi S, Prabhash K, Deshmukh C, Nag S, Lakshmaiah K. et al. Phase 2 trial of CA-170, a novel oral small molecule dual inhibitor of immune checkpoints VISTA and PD-1, in patients with advanced solid tumor and hodgkin lymphoma. Cancer Res. 2009;15:7412-20
188. Wang K, Cai S, Cheng Y, Qi Z, Ni X, Zhang K. et al. Discovery of benzo[d]oxazoles as novel dual small molecule inhibitors targeting PD-1/PD-L1 and VISTA pathway. J Med Chem. 2024;67:18526-48
189. Sun C, Cheng Y, Dong J, Hu L, Zhang Y, Shen H. et al. Novel PD-L1/VISTA dual inhibitor as potential immunotherapy agents. J Med Chem. 2025;68:156-73
190. Dai MY, Shi YY, Wang AJ, Liu XL, Liu M, Cai HB. High-potency PD-1/PD-L1 degradation induced by peptide-PROTAC in human cancer cells. Cell Death Dis. 2022;13:924
191. Yan T, Long M, Liu C, Zhang J, Wei X, Li F. et al. Immune-related adverse events with PD-1/PD-L1 inhibitors: insights from a real-world cohort of 2523 patients. Front Pharmacol. 2025;16:1519082
192. Ke ZB, Chen JY, Xue YT, Lin B, Huang Q, Huang XY. et al. Mechanical signal modulates prostate cancer immune escape by USP8-mediated ubiquitination-dependent degradation of PD-L1 and MHC-1. Cell Death Dis. 2025;16:413
193. Ding L, Chen X, Zhang W, Dai X, Guo H, Pan X. et al. Canagliflozin primes antitumor immunity by triggering PD-L1 degradation in endocytic recycling. J Clin Invest. 2023;3:133
194. Gao P, Li X, Duan Z, Wang Y, Li Y, Wang J. et al. Improvement of the anticancer efficacy of PD-1/PD-L1 blockade: advances in molecular mechanisms and therapeutic strategies. MedComm (2020). 2025;6:e70274
195. Xu Y, Du H, Guo W, Liu B, Yan W, Zhang C. et al. Discovery of highly potent small molecule PD-1/PD-L1 inhibitors with a novel scaffold for cancer immunotherapy. J Med Chem. 2024;67:4083-99
196. Liu L, Zhang H, Hou J, Zhang Y, Wang L, Wang S. et al. Discovery of novel PD-L1 small molecular inhibitors with potent in vivo anti-tumor immune activity. J Med Chem. 2024;67:4977-97
197. Fan P, Qi Z, Liu Z, Wang S, Wang Y, Kuai J. et al. High baseline levels of PD-L1 reduce the heterogeneity of immune checkpoint signature and sensitize anti-PD1 therapy in lung and colorectal cancers. Cell Death Dis. 2025;16:152
198. Mukherjee S, Rogers A, Creech G, Hang C, Ramirez A, Dummeldinger M. et al. Process development of a macrocyclic peptide inhibitor of PD-L1. J Org Chem. 2024;89:6651-63
199. Mandal K, Barik GK, Santra MK. Overcoming resistance to anti-PD-L1 immunotherapy: mechanisms, combination strategies, and future directions. Mol Cancer. 2025;24:246
200. Brat DJ. Expanding the bandwidth of checkpoint inhibitors for cancer using epigenetic regulators. J Clin Invest. 2025;135:e188611
Author contact
![]() Corresponding author: dxjedu.cn (Xing-Jie Dai) or 991536786com (Ying Liu).
Corresponding author: dxjedu.cn (Xing-Jie Dai) or 991536786com (Ying Liu).