Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Chemokine and receptor...
Mechanisms of CKRs signaling...
CKR relevant immune cell...
CKRs signaling reshapes tumor...
Therapeutic implications and...
Conclusions
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2026; 16(13):7324-7344. doi:10.7150/thno.134995 This issue Cite
Review
Biomechanical signaling of cytoskeleton and membrane reorganization in tumor immune chemotactic migration
Qijie Zhao1, Keliang Chen2, Minglu Zhou1, Jing Jin3,4 ![]()
1. Department of Pharmacy, West China Hospital, Sichuan University, Chengdu, 610041, People’s Republic of China.
2. Department of Biotherapy, West China Hospital, Sichuan University, Chengdu, 610041, People’s Republic of China.
3. Department of Radiation Oncology, Cancer center, West China Second University Hospital of Sichuan University, Chengdu, People’s Republic of China.
4. Key Laboratory of Birth Defects and Related Diseases of Women and Children (Sichuan University), Ministry of Education, Chengdu, People’s Republic of China.
Received 2026-3-24; Accepted 2026-5-14; Published 2026-6-4
Abstract

A prominent activity of the chemokine system is the regulation of immune cells trafficking in tumor microenvironment (TME). This elegant chemotaxis process is regulated by both chemokine receptors (CKRs) stimulation and intracellular factors, such as chemokine, actin cytoskeleton, cell adhesion molecules, membrane fluidity, metabolic reorganization that govern differences in tumor immune cells migration patterns. At the core of tumor immune cell migration and effector functions are three fundamental questions: how CKR signaling triggers morphological changes, how the interplay between actin/selectin and energy metabolism regulates force production, and how cytoskeletal signaling adapts to membrane fluidity. In this review, we discuss the potential influences of CKRs signaling of the actin/selectin cytoskeleton reorganization, membrane fluidity, force regulation, and metabolic reorganization in immune cell chemotaxis. Apparently, the activity of immune cells migration is not regulated solely by chemokine-receptors recognition, as biomechanical-metabolic signaling orchestrate is critical for migratory morphology regulation. By using current knowledge of general chemotaxis mechanisms as a framework, we hope to provide new insights into secondary changes mechanisms of cytoskeleton and morphology network in supporting the tumor immune cells migration. Unraveling the mechanisms underlying chemotaxis and coupled mechanical immune checkpoints holds immense potential for discovering novel therapeutic targets in cancer immunotherapy.
Keywords: Chemokine receptors (CKRs), Chemotaxis, Actin, Cytoskeleton, Metabolic reprograming
Introduction
The chemokine relevant immune contexture is widely recognized as an important determinant of outcomes in cancer patients[1]. Chemokine constitutes a large family of signaling proteins that interact with cell surface chemokine G protein-coupled receptors (GPCRs) [2]. The cross-talks between chemokine and their receptors (CKRs) regulate immune cells activation and trafficking in physiological and pathological conditions[3]. Tumor microenvironment (TME) is associated with chemokine signaling induced intricate intracellular network and metabolic changes, especially tumor and immune cells[4]. Specifically, among the TME, accumulating evidence shows that tumor-derived chemokine imposed several limitations to dampen T cell immunity, and immunosuppressive cells experienced hyperactivation[5]. Although the chemokine determined chemotaxis are crucial for migration of distinct type of immune cells[6], the physiological correlation between intracellular kinesin and structural proteins in immune cells remain unclear.
The immune cytoskeleton is composed of dynamic networks of filaments that drive cell shape, polarity, migration, and the assembly of intercellular contacts [7]. During the immune cells chemotactic migration, CKRs, integrins and the cytoskeleton are triggered in a hierarchical sequence[8]. Parameters of TME immune cell migration are deeply influenced by CKRs signaling, and alterations in cells speed and directional persistence were result in a flawed location strategy and defects in immune priming[6]. Precise spatiotemporal control of these processes is essential to launch an effective innate immune response[9, 10]. The chemokine-induced CKRs stimulation is coupled to G-protein and β-arrestin-mediated signaling cascades, which in turn, are coupled to ultrastructural changes and molecular processes that define immune cells phenotype and function[11]. Morphological fitness of immune cells, defined here as their propensity to remodel cell shape in response to motility and chemokine stimuli, therefore affects their efficiency in the TME[12]. Insight into the role of CKRs signaling in the direct extravasation of immune and immunosuppressive cells is important from the angle of molecules composing the complex cytoskeleton-reorganization machinery[13, 14]. While some intricate details of the immune cell dynamics mechanisms in TME remain unknown, there have amount knowledge available to aid in the progression of our understanding.
Another key facet of chemotaxis control relates to the high energetic cost of actin polymerization[9]. As immune cells like Tregs experience the metabolic framework of growing tumors, they activate distinct pathways necessary to accomplish their function[15]. Sustained chemokine-induced adaptations in immune cells require metabolic activity and endogenous signaling cascades, with changes in glycolysis and cholesterol metabolism emerging as prominent features[16, 17]. However, several lines of evidence indicate that the underlying situation is more complex. Understanding the immune cells chemotaxis relevant CKRs signaling and metabolic switch in the TME is crucial for improving immune cell-based therapies.
In this review, we highlight current knowledge of CKRs signaling alter the immune cells migration and organellar proteins possessing in the context of tumor, as well as signal transduction, cells polarization and metabolic adaptation. We further examine how these studies illuminate the ways in which CKR signaling–induced conformational changes enhance the regulation of intracellular molecules within the structural network of leading edge and the trailing edge (uropod), thereby improving morphological fitness and influencing immune cell migration and functions.
Chemokine and receptor recognition
CKRs, a family of small proteins (8–10 kDa) activated by chemokines, are crucial in driving immune microenvironment dysregulation and tumor progression[18]. Recognition between CKRs and chemokines exhibits diversity and promiscuity, involving multiple interaction interfaces that contribute to biased signaling and functional selectivity[19]. With the physical combination between chemokines and CKRs, cell exerts its structural and physiological functions change in TME (Figure 1).
Examples of immune CKRs activation structural model. This figure illustrates the structural and molecular recognition mechanisms of CKRs in TME immune cells. I. CKRs recognition and binding: The chemokine's N-terminus length and conformation dictate its insertion degrees in the CKR's transmembrane pocket, wherein the receptor's N-terminal domain dynamically interacted with chemokine by multiple sites. II. Stabilization and affinity reconstruction: Cellular cholesterol stabilizes activated-CKRs structural through hydrophobic interactions. Meanwhile, N-terminal tyrosine sulfation n CKRs could enhance the binding affinity by electrostatic interaction with GAGs. III. Immune cells activation and signaling transduction: After chemokine-CKRs recognition, the chemokine stimulated the helical structural rearrangement and forming a tight activation CKRs pocket. Additionally, G proteins and β-arrestin cooperation stimulates the structural rearrangement and downstream cAMP and kinase signaling cascades. cAMP: Cyclic adenosine monophosphate, CKRs: Chemokine receptors, GAGs: Glycosaminoglycans, CRS: N-terminal domain of the receptors, TME: Tumor Microenvironment.
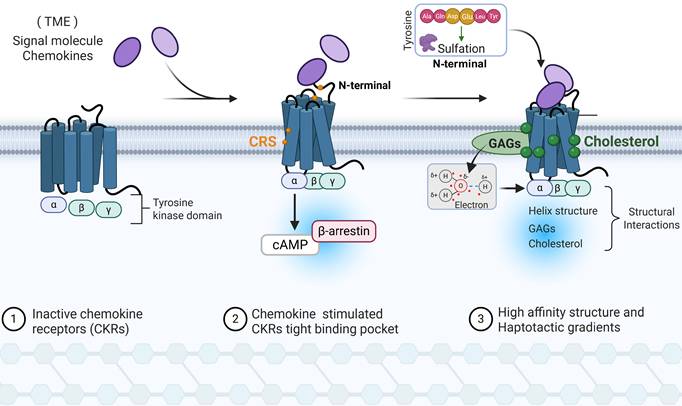
Chemokines N-terminal residues length determined its ability to penetrate the CKRs transmembrane helices binding pocket, with longer residues having deeper interior surfaces binding regions and selectively contributing to signaling biased agonism[20]. In compared with CCL5, CCL7 composed by longer residue and is more inclined to stimulate cAMP than β-arrestin signaling, one reason is binding to receptor residues closer to the intracellular side of the CCR1 conformations[20]. Qin et al. observed that CKRs N-terminus presented with conformation perpendicular to the membrane and extracellular half of helix shifted outward to forma top extra α-helical bending when interacting with chemokines[21]. A similar phenomenon was existed in CXCR2 when binding with CXCL8[22]. Meanwhile, different chemokine conformation like β-sheet interactions (CC motif) and no substantial protein-protein interface contacts (CXC motif), determine the recognition and interaction of CKRs type. Currently, multiple chemokine-CKRs complexes dynamic interaction sites have been proposed, termed as CRS1, CRS1.5, CRS2, etc.[23]. The signaling amplitude depends on the extent to which the receptor N terminus and CRS bind the chemokine, where different chemokine N-loop and distal flexible N-terminus epitope recognition acting as important initiators of signaling[24]. In particularly, human class A GPCRs subgroup of CKRs (CCR6, CCR7, CCR9, and CCR10) contain specific amino acids residue position, thereby only allow cognate chemokines with shorter N termini featuring and shallow binding mode[25]. After CKRs and chemokine interactions, CKRs presented with a tight ligand-binding pocket compared with the chemokine-free CKRs due to the inward shift of helix and extracellular loop[26].
In the chemokine and CKRs recognition, the CKRs extracellular N-terminal regions sulfated tyrosine residues increases chemokine binding affinity and potency in initial binding site [27]. Negatively charged residues (Asp and Glu) in the vicinity of tyrosine residues and these constitute the sequence motifs necessary for tyrosine sulfation. As a native pre-recognition complex modulation, tyrosine sulfation allowed CKRs to have more open and dynamic conformational states and facilitate ligand binding, as well as maintaining the long-range effects for the post-recognition [28]. As a feedback, tyrosine sulfation binding to CKRs-sulfopeptides can modulate the oligomerization state of chemokines and affect the ability of a chemokine to activate its receptor [27]. On the other hand, chemokines (basic) binding sulfated glycosaminoglycans (GAGs, acidic) selectively increase its concentration in the cell membrane with electrostatic and hydrogen bonding forces, wherein dimeric form interaction presented with higher affinity to CKRs[29]. The diminished affinity of chemokines for GAGs containing different heparin, heparan sulfate (HS) and chondroitin sulfate (CS) fine structures will directly influence the chemotactic and haptotactic gradients during the CKRs recognition. In addition, the activated CKRs structure is flourished with cholesterol, which binds at a specific transmembrane surface cavity and forms hydrophobic interactions with nearby residues[30]. The accumulated cholesterol could modulate CKRs-chemokine binding properties through orthosteric- and/or allosterical action. For instance, due to site-specific cholesterol consensus motif, cholesterol stabilizes the unique active conformation and maintains the tight helix structure of CKRs[31].
Mechanisms of CKRs signaling induced adhesive migration and morphogenetic processes
Cell activation and migration are complex processes involving CKRs signaling, spatial allostery, internal mechanical and biochemical mechanisms that coordinately generate forces governing cell structure and directionality[32]. In recently, targeting the intracellular actin cytoskeleton and depolymerization by ultrasmall diameter magnetic nanomotor has been applied in improving tumor immunotherapy resistance and TME[33]. In classical theory, actin produces force for diverse cell motile and morphogenetic processes, wherein actin filaments act as ‘tracks’ for myosin motor proteins and move concentrated towards ATP energy on the intracellular membrane side [13]. Actin-driven protrusions occur primarily at the front and myosin-driven contractile forces generate at the rear, causing cells to detach and move forward. Hence, intracellular force production and chemotaxis signaling are important for membrane deformation in many cellular processes (Figure 2).
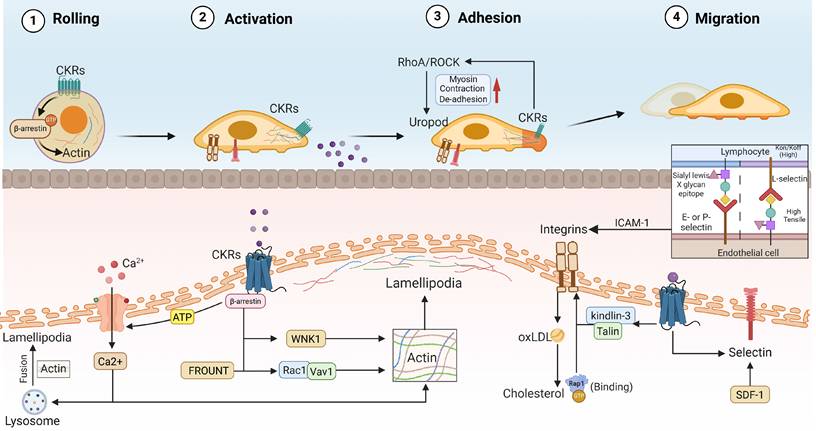
Formation of branched actin networks and CKRs signaling in immune cells migration. Immune cells morphological changes and polarization status modulated by transmembrane CKRs and cytoskeletal signaling in chemotaxis. I. Chemotactic rolling and adhesion: inside-out CKRs signaling like Talin and Kindlin-3 induced high-affinity conformations in integrins. Meanwhile, intercellular selectin recognition promoting the immune cell adhesion and rolling during the chemotaxis. II. Leading edge protrusion and actin polarization: Through the downstream β-arrestin, FROUNT, and Rac1/Vav1 signaling cascades, CKRs activation would promoting the actin polarization network and lamellipodia formation. III. Uropod contraction and de-adhesion: CKRs stimulates local intramembrane RhoA/ROCK signaling and non-muscle myosin rearrangement, wherein lysosome-modulated Ca²⁺ influx promote trailing edge contraction and detachment, thereby generating continuous forward force. CKRs: Chemokine receptors, oxLDL: Oxidized low-density lipoprotein, ICAM-1: Intercellular Adhesion Molecule 1.
Actins
Due to the β-arrestins scaffolding capacity in the actin assembly events is needed for gradient-sensing filopodia and lamellipodia formation, elongation of actin filaments extend lamellipodia ends and push the plasma membrane to form protrusion, therefore β-arrestins activation is important for cytoskeletal reorganization and initiation of cell migration[13, 34]. Actin polymerization and assembly within cellular protrusions enhance outward forces[35]. However, to date, only β-arrestin_1 has been observed to be associated with CXCL12-induced actin polymerization and directed cells (DCs) migration[36], which is closely associated with cognate CXCR4 nano-clustering and dynamics[37]. The Chemokine induced migration is a complex process involved in myriad proteins and intracellular pathways that coordinately to guide the polarized cell status and direction. Noteworthy, CKRs are usually concentrated at the leading front of motile cells, the membrane protrusion with correct receptor allows cell to positively sense chemoattractant gradients[38]. CXCL12 interaction promoted CXCR4 to form large nano-clusters with more than three receptors on the cell membrane, and this conformational intensification is important for receptor complex diffusion rates reduction, cell chemotactic sensitivity, actin cytoskeleton reorganization and integrin activation[38]. The CXCR4 receptor nanoclustering were temporarily confined in a region delimited by the F-actin cytoskeleton, which potentially increases the signal transmission stability. Omri et al. reported that actin polymerization drives actomyosin retrograde flow, generating “pushing” forces toward the membrane edge in immune cells, as well as contractile forces that “pull” the actins network to the leading front[39]. Additionally, upstream singling stimulation could accelerate actin dynamics and retrograde flow, consequently enhancing the actin polymerization and forces in immune cell migration[39]. In the case of T lymphocytes, pre-clustering of TCR oligomers with increased avidity has been reported to increase sensitivity to multivalent antigens by 100- to 3,000-fold[40]. Similarly, CCR5 and CCR7 transport to the plasma membrane also boosts the concept of CKRs local and polarized migration signaling[41]. The CCR7 activation triggered endomembranes Vav1/Rac1 signaling shows the ability to promote lead edge actin cytoskeleton reorganization and lamellipodia formation, subsequently promoting membrane protrusion and DCs migration[41]. Herein, CKRs activation induced local morphology change supporting the endomembrane actin signaling transduction and sustained directed migration. CCR7 signaling-induced ion influx kinase WNK1 was proved to be activated at the leading edge of T cells, thereby promoting osmosis- swelled the membrane at the leading edge and generating space where actin filaments can polymerize[12]. In addition, through binding with CCR2 or CCR5, intracellular protein FROUNT should amplify the chemokine-elicited Rac1-lamellipodium protrusion cascade[42]. FROUNT forms a complex with leading edge CCR2 microclusters and enhances the lamellipodium protrusion activity, which contribute to morphology asymmetric distribution and could be a mechanical immune checkpoint in immune disease. Hwang et al. demonstrated that exposure CXCL13 to CXCR5 would promote B cells three-dimensional lamellar-like pseudopods and F-actin-rich ridges in a short time[43]. On the other hand, CKRs-stimulated RhoA localizes to the cellular trailing edge, where RhoA/ROCK signaling promotes non-muscle myosin activation, uropod contraction and cell de-adhesion [44]. Local activation of RhoA not merely promoted actin and myosin recruitment, but also increased intracellular stress fibres flow-forced traction forces and actin flow[45]. CKRs are embedded in lipid membrane, at least partially, and its nano-clustering and dynamics might be influenced by membrane fluidity and actin polarization.
Ca2+
Chemokine activated CRKs have also be demonstrated to trigger Ca2+ flux and decrease cell adhesion, such as CXCR4, CCR5, etc. An increase in intracellular free Ca2+ is a sign of chemoattractant stimulation, and is linked to directional sensing, cytoskeleton structure, deformation tractive force and adhesions[45, 46]. Lorenzen et al. reported that activation of G protein subtype subunits through CCR5 leaded to Ca2+ flux enhancement, like Gq, Gi and Gβγ protein activation, wherein Gβγ subunit release after CCR5-Gq/Gi/o activation signaling bias potentially be responsible for eliciting Ca2+ flux[47]. With the CKRs concomitant ATP signaling, ATP releasing and degradation exerts a synergistic effect on Ca2+ flux through activating purinergic receptors and other Gi subunit[48]. As an important step in chemotaxis, Ca2+ flux is necessary for actin polymerization and cytoskeletal reorganization. Increased Ca2+ flux supports actin polymerization and ensures cells correct polarization and migration[49]. Due to the CKRs-induced Ca2+ flux, Ca2+-ATPases on the plasma membrane and endomembrane are responsible for Ca2+ homeostasis by pumping between extracellular space and intracellular stores[50]. Moreover, upon chemokine-stimulated Ca2+ flux, lysosomes fuse to the cell membrane in a localized area and contribute to local increases in ATP and lysosomal contents[48]. Especially, fusion of lysosomes with the plasma membrane at lamellipodia maintains cell shape remodeling and polarization through endomembranes replenishment and uropod disconnection[51]. Current research posits that lysosomes function as a signaling hub that integrates both extracellular and intracellular stimuli, thereby regulating cellular homeostasis[52]. Through post-fusion releasing with actomyosin cytoskeleton, lysosomes-Ca2+ signaling activated the actin-based motor myosin at the cell rear, ultimately facilitating the persistently intracellular force and immune cells migration[53].
Integrins
Chemokines-induced transmembrane adhesion protein (integrins) activation formed a critical link between the ECM and the cytoskeleton, as well as promoting the adhesion of immune cells to three-dimensional substratum[54]. As feedback, the activated integrins will drive excessive engagement and internalization of oxLDL, subsequently promoting the accumulation of cholesterol. Since chemokine-CKRs interaction caused a significant increase in the appearance of membrane actin-coupled protrusion (filopodia, lamellipodia and uropod), which are integrin-rich structures implicated in cell adhesion, and due to integrin-to-basement membrane for cells local/distal actin polymerization and forward movement[55]. Thus, in the transient cell-substrate interactions for movement, integrins and unspecific adhesion promoted forward force is highly dependent on actin polymerization and larger focal adhesions. Sheetz et al. has been indicated that integrins in the front of lamellipodia with enhanced cell cytoskeleton exhibit forward pull force to make itself moving [56]. Through integrative tension sensor (ITS) technique, Ying et al. observed that filopodium elongation is accompanied with myosin and integrins enrichment during cell adhesion movement, wherein integrins tension could generate in discrete foci (force nodes) along single filopodia with a spacing of ∼1μm[57]. In addition, filopodia integrins tension shows region-dependent, with weak tensions generated in the nodes at the filopodia tips and strong tensions generated in the nodes at the filopodia bases[57]. Increased adhesion maybe attributed to the result of inside-out integrins activation in immune cells, which involves the activation of the Rap1-GTPase and the integrin adaptor proteins Talin[58]. Mechanistically, surface membrane integrins undergo conformational switching to a high-affinity state via chemokine binding activation. It is widely accepted that integrins such as LFA1 are kept in low-affinity states with bent/closed conformations at quiescent condition, extended/closed conformers (intermediate affinity) to mediate rolling, and open/extended conformers (high affinity) by CKRs signaling inside-out stimulation to promote arrest and firm adhesion[59]. Activation of CKRs induced a high-affinity integrin structure to the endothelial cell interaction, which is potentially required for the firm binding of immune cells adhesion migration. The integrins high affinity conformation and adhesion ability require kindlin-3/Talin-1 binding to the β2-integrin cytoplasmic domain under the chemokines-CKRs recognized activation[59, 60]. Although all integrins can undergo activation, β2 integrins are exclusively expressed on the surface of leukocytes, neutrophils, lymphocytes, and monocytes, and show different degrees of activation under ultimate talin-1 or kindlin-3 coupling signals [61]. CKRs-activated integrins acted as a frictional interface with the environment, conveying tangential traction forces like a rake pulled.
Selectin
In tumor, the functional selectins expression was proved to be important for immune cells homing[62, 63]. Selectins are single-chain transmembrane glycoproteins that resemble C-type lectins, found on the surface of immune cells and endothelial cells, especially within the vascular network. Selectins have rapid association and dissociation rate constant (Kon/Koff) and a high tensile strength, supporting the observed transient interactions [64]. The selectin family which comprises 3 members like E-selectin, L-selectin and P-selectin bind to sialyl-Lewis x (sLeX) epitope carbohydrates[65], wherein the vessel wall chemoattractant signals for lymphocyte is controlled by the diversity of the selectin. The chemokine-triggered immune cells orientation movement could be arrested by selectin blocking, such as CXCR2+ neutrophils in inflammation [66]. Selectin- as well as chemokine-mediated inside-out signaling promoted efficient neutrophil recruitment [67]. In terms of this, selectin-mediated CD11a/CD18 neutrophil activation initiates slow rolling by transient interactions with intercellular adhesion molecule-1 (ICAM-1), which allows neutrophil to sample chemokines presented by the endothelial cell wall and further activation of β2 integrins[68]. For CKRs stimulus like chemokine and SDF-1α, SDF-1α-specific upregulation of selectin in endothelial cells has been observed [69]. Maryann et al. indicated that CXCR3-driven CD8+ T cells transfer is dependent on selectin and ICAM-1 activation, which is important for T cell trafficking across tumor vascular gateways [62]. Thus, CKRs-induced selectin facilitated the immune cells rolling to firm arrest within the TME, supporting strategic intervention of chemokine-dependent delivery of immune cells for cancer treatment. The cooperation of CKRs and selectin not only facilitates the early tumor seeding, but also promotes the immune lymphocytes trans-endothelial migration[70]. For example, the selectin-positive tumor endothelial cells exhibited immune-related pathways enrichment and enhanced CD8+ T cells infiltration, wherein CCL5 synergistically induced ACKR1+CD8+ T cells recruitment and activation[71]. In gastric adenocarcinoma, increased levels of selectin and CCR4 in CD4+/Foxp3+ Tregs contribute to Tregs recruitment and tumor progression[72]. Targeting both chemokines and selectins improved the tumor progression and tumor immune microenvironment rather than inhibiting selecting [73]. In rat sarcoma model, chemokine and selectin serum concentration were increased within tumor progress, whereas these parameters returned to baseline after tumor excision[74]. Collectively, investigating the cooperation among the CKRs signaling, actin cytoskeleton, cellular morphological changes and adhesion molecule, as well as underlying driving force, will provide insight into development of antitumor immunity dependent immune cell chemotaxis.
CKR relevant immune cell metabolic reprograming
OXPHS
In immune cells, processes such as cytoskeletal rearrangement, membrane trafficking, traction stress generation, migration, and proliferation all require a continuous energy supply. Mitochondria are usually located in the cytoplasm of cells, where they generate adenosine triphosphate (ATP) to empower cellular functions (Figure 3). Through real-time ATP biosensor and traction force microscopy, Curtis et al. observed positive correlation between cell traction forces (contractility) and energy consumption, indicating that enhanced cytoskeletal reorganization and integrins affinity require significant ATP utilization and glucose metabolic efficiency[75]. CXCL12-dependent leukocyte polarization and migration accompanied with mitochondrial redistribution[76]. Mitochondria respond to chemokine-stimulated and accumulated microtubule-organizing center. In gastric carcinomas, CCL21-CCR7 axis induced mitochondria pathway protects CD8+CCR7+ T lymphocytes from apoptosis[77]. The CKRs activation and mitochondrial metabolism energy supply in CD8+ T cells supported its high cytotoxicity and favor the interaction to tumor cells. In the cytoplasm, mitochondrial transcription factor A (TFAM) is essential for mitochondrial respiration and controls mitochondrial transcription, and packaging[78]. CKRs activation was proved to increase TFAM synthesis, thereby enhancing Th1 and Tregs responses[79]. The stable relationship between actin cytoskeleton and integrins adhesions may also result in a more efficient conversion of ATP to cellular traction forces, which is also critical for the regulation of intercellular adhesions and communications[75]. Due to TFAM is essential for mitochondrial respiration and ATP generation, TFAM-deficient Tregs presented with reduced proliferation and Foxp3 expression upon glucose deprivation in vitro TME[78]. Impaired mitochondrial ATP generation in Tregs contributes to an increase in CD8+ T cells, at least partially by reducing their glucose consumption and thereby alleviating glucose deprivation in the TME. Moreover, Eros et al. reported that decreased TFAM in immune cells will compromise the actin cytoskeleton and impair cellular motility in response to chemokine signaling, leading to spatial disorganization in the hypoxic TME[80]. The integrins induced P2X4 purinergic signaling and ATP generation is essential for effector immune cells adhesion and migration[81]. Li et al. reported that PEG-amine-QD nanoparticle exposure could stimulates purinergic signaling and early ATP release, driving integrin-dependent adhesion and crawling during immune cells recruitment[82]. Thus, targeting the ATP-dependent morphological dynamics interferes with the cellular interactions required for crawling and transmigration, thereby facilitating the clinical application of chemotaxis-targeted therapies.
Interplay between CKRs signaling and metabolic reprograming in immune cells. The chemotaxis and cytoskeleton rearrangement are highly energy-dependent manners. Activated-CKRs signaling promote the intracellular metabolic reprograming and energy requirements. I. OXPHOS: CKRs activation promotes the TFAM synthesis and mitochondrial activity, wherein the enhanced TCA cycle and ATP generation maintain energy requirement for stable morphological activities. II. Glycolysis: In part immune cells, CKR-driven PI3K/mTOR and HIF-1 signaling pathways upregulated the glycolytic enzymes like LDHA and PFKFB3, subsequently achieving rapid local ATP for actin rearrangement and extension effects. In addition, lactate accumulation in TME promoting the immunosuppressive effects and M2-like TAM chemokine secretion. III. Lipid and cholesterol metabolism: ApoE-mediated lipid rafts involved in stability of CKR transmembrane conformation, as well as activating mTOR/SREBP2 cascade to promote membrane fluidity and actin polarization. Meanwhile, cholesterol synthesis and mTOR/SREBP2 signaling were coordinately regulating the Tregs and M2-like TAMs accumulation. TFAM: Mitochondrial transcription factor A, TCA: Tricarboxylic Acid Cycle, TCR: T Cell Receptor, OXPHS: Oxidative Phosphorylation, LDHA: Lactate Dehydrogenase A, TAMs: Tumor associated macrophages, CKRs: Chemokine receptors, Tregs: Regulatory T cells, ApoE: Apolipoprotein E.
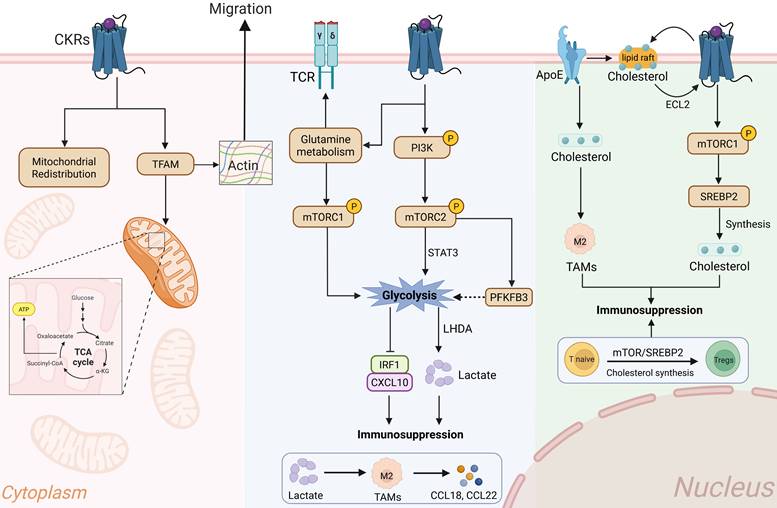
In line with this, CXCR4 blocking significantly decreased mitochondria number and oxidative phosphorylation, and induced mitochondria loss related non-apoptotic T cell death in T cell acute lymphoblastic leukemia (T-ALL) [83]. Mitochondrial inhibition potentially suppresses T cell activation through decreased myosin phosphorylation and ATP production, thereby weakening immune synapse adhesion strength and force transmission at the point of contact[84, 85]. Force-producing actin polymerization and actomyosin contraction as well as MMP delivery and membrane trafficking are energetically demanding processes [86]. Of note, the anti-tumor chemokine CXCL14 could inhibit mTOR phosphorylation signaling and tumor cells proliferation [87]. CXCL14-suppressed mTOR signaling and oxidative phosphorylation were also observed in leukemia-initiating stem cells and lymphocyte migration[88]. Targeting regulation mitochondrial respiration and metabolic reprograming may break the actin cytoskeletal reorganization dependent chemotaxis and warrants future consideration for certain cancer therapies.
Glycolysis
Glycolytic metabolism is a hallmark of tumor immune microenvironment. In tumor cells, chemokine induced tumor glycolysis like glycolytic enzymes GLUT1, HK2, and LDHA play a crucial role in both tumor progression and metastasis[89] (Figure 3). For tumor immunodepression, CCL20-CCR6 axis stimulated Tregs glycolysis and was deeply involved in its immunosuppressive effects[90]. The CCR6-induced Tregs activation closely associated with intracellular glycolysis, which simultaneously damaged the CD8+ T cells immune response and activity. The immune cells migration is thought to be primarily dependent on the glycolysis enzyme glucokinase, which is induced by the PI3K-mTORC2 signaling pathway. CCL20-CCR6 signaling induced the phosphorylation of Akt, mTOR, and STAT3 molecules in T cells and Tregs[91]. In compared with naïve T cells, the CKRs signaling more likely to enhance the glycolytic metabolism in Tregs. For Tregs, upregulation of glycolysis would promote actin-based cytoskeletal reorganization, thereby enhancing their migratory activity and immunosuppressive function[4]. The increased ATPase level and F-actin formation were significantly co-localized in Tregs. Similarly, CCR6 activation enhanced Tregs glycolysis and lactate production, and thereby achieving immunosuppressive activity toward CD8+ T cells[90]. Like other G protein–coupled receptors (GPCRs), CCR6 activation induces Tregs phospholipase C activation and release of intracellular Ca2+ levels [92]. Of note, CCR6 was found to be more frequently present in Tregs than in CD8+ T cells, which might preferentially promote Treg glycolysis, enabling them to outcompete CD8+ T cells for extracellular glucose. The CCL20-CCR6 axis inhibition not merely decreased compensatory glutamine metabolism, but also promoted the efficacy of anti-PD-1 therapy[90]. Glutamine is intimately involved in maintaining redox homeostasis, and acts as a metabolic hub to increase immune cells mTORC1-mediated glycolysis and enhancing TCR signaling[93]. Fluorescence lifetime microscopy revealed that activated T cells exhibit transient glycolytic activity, which promotes actin remodeling associated with outward immune synapse and receptor formation [94]. The enrichment of actin polymerization at the immunological synapse augments T cells outward forces[35]. The inhibitor of glycolysis, 2-deoxyglucose (2DG), leads to increased T cell activation potential in tumor[95]. Similarly, low levels of 2-DG would induce DCs metabolic stress and glycolytic reprogramming, subsequently enhancing CKRs expression and integrins-mediated immune cell adhesion[95]. In return, loss of adhesion force will lead to a significant downregulation of DCs intracellular metabolic rate and cellular ATP generation. During early stimulation, adhesion-dependent glycolytic activation in CKRS-stimulated DCs potentially enhances T cell activation, whereas glucose consumption appears to restrict DC responses and T cell activation at later time points. Intriguingly, in the depriving TME, higher lactate also facilitates M2-like polarization of tumor associated macrophages (TAMs) in a dose-dependent manner and exerts their tumor-supporting functions[96]. Under conditions of extracellular glucose deprivation, lactate-induced M2 macrophages exhibited an immunosuppressive and chemotactic profile [97]. Cellular energy metabolism and actin-based cell dynamics are tightly coupled processes. TAMs synergistically induced by lactate and chemokines exhibit immunosuppressive effects associated with CD8+ T cell dysfunction and Tregs accumulation. Spatial analysis revealed that TAMs localize to tumor regions enriched for hypoxia-responsive signatures [97]. Within these regions, the close physical proximity between TAMs and CD8+ T cells directly drives T cell dysfunction and exhaustion, though it remains incompletely understood whether metabolic competition plays a contributing driver. This phenotype was potentially fueled by glycolysis and characterized by actin cytoskeleton changes, cell protrusions, as well as adhesion and migration[98, 99]. To control these activities before and after polarization, macrophages continuously form actin-rich membrane protrusions and extend filopodia from their cell surface[99]. The lactate-derived TAMs had a significant higher mRNA level of CCL18, CCL22, and IL-10, which potentially forming a positive feedback loop[100]. Mechanistically, glycolytic reprogramming in macrophages could promote histone acetylation mediated chemokine transcription[101]. Intervention of 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 3 (PFKFB3) blocked an increased glycolytic flux under hypoxia, which is directly response to the PI3K/Akt/mTOR signaling cascade[102]. Inhibition of PFKFB3 reduced the cell-cell average junctional tension by perturbing glycolysis-driven actin dynamics and directly disrupting intercellular contact forces. Meanwhile, activated CXCR4/mTORC2 signaling axis contributes to downstream glycolysis regulator PFKFB3 and activates endothelial cells migration[103, 104]. During macrophages migration processes, ATP is rapidly consumed in the cytosol for the reorganization of cytoskeletal actin filaments and force expenditure[105]. Silencing CXCR4 can inhibit glycolytic metabolism, thereby directly influencing the macrophages polarization and migration[106]. The adaptive responses in glucose metabolism and glycolysis reprograming contribute to immunosuppressive cells actin cytoskeleton reorganization and chemotaxis activity.
In anti-tumor immunity, CXCL10-CXCR3 axis was observed to stimulate CD8+ T cells mTOR phosphorylation activation, thereby stimulating lactate dehydrogenase A (LDHA) mediated glycolysis and lactate generation[107]. Through F-actin polarization at the lamellipodia, chemokine-driven motility, and cellular adhesion, upregulated glycolysis promotes actin polymerization and density as an energy state integrator and sustains cell cytotoxic activity[108]. In cooperation with glycolysis, OXPHOS partially promoted F-actin content and cell elongation in the context of confined motility. The IRF1 and CXCL10 immunostimulatory molecules were usually reduced in highly glycolytic TME[109]. However, CXCR3-induced intracellular glycolysis promoted cells infiltration, ATP and tumor necrosis factor-α (TNF-α) secretion[107]. In this way, the CXCL10-CXCR3 and glycolytic genes additional signaling networks is important for CD8+ T cells survival, morphological fitness and cytotoxic activity in the surrounding glucose-deprived TME. In terms of this, raised fast ATP content and F/G-actin ratio indicated the CD8+ T cells immune synapses outward extension force and synaptic adhesion movement[108]. The mTOR metabolic signaling enhances glycolysis and plays a critical role in CD8+ memory T cells antitumor immunity[110]. Because memory T cells undergo extensive activation upon antigenic stimulation, they require a high glycolytic flux to sustain the bioenergetic demands and provide building blocks for cytoskeletal biomass[111]. In this process, the coordination between integrins and F-actin flow maintains high affinity within the immunological synapse, drives TCR clustering, and enhances ICAM-1 binding to facilitate the elongation of activated memory T cells[112, 113]. Of note, lactate-mediated inhibition of T cell motility was partially attributed to an interference with glycolysis activated upon engagement of the chemokine CXCR3-CXCL10 axis[9]. Chemotaxis of CD8+ T cells was suppressed by lactate and enhanced by glucose[114], which may be attributed to decreased CXCL10 levels in a highly glycolytic TME. Mao et al. engineered a multifunctional nanoparticle that targets the tumor mechanical microenvironment and AMP signaling, thereby reducing lactate levels by 50%, promoting extracellular matrix (ECM) and cytoskeletal reorganization, and enhancing effector T cell infiltration[115]. Glycolysis increases immune cells cytokine expression, and TCR or CKRs stimulated immune cells glycolysis was essential for their activation, biomechanics shape, membrane structure alteration and adhesive migration[116-118].
Cholesterol
Chemokine-dependent immune cell recruitment requires cholesterol, and higher lipid and cholesterol enhanced macrophages infiltration in TME[31, 119, 120] (Figure 3). It has been well known that cholesterol is an allosteric modulator of GPCRs and is important for active conformation of CKRs[31]. Cholesterol homeostasis is crucial for maintaining the deformability and fluidity of cell membranes, as well as the allosteric regulation of surface receptors. In the GPCR extracellular loop 2 (ECL2), cholesterol could induce site-specific conformational restraint and regulate CKRs signal transduction [121]. Immune cells chemotactic activity requires cytoskeleton-dependent membrane dynamics, wherein insoluble cholesterol sustain the transmembrane receptor protein and actin cytoskeleton, especially actin bundles formation, immune synapse, F-actin dynamic polarization and cell adhesion migration[122]. For example, successful CXCR4-mediated signaling and migration required the cholesterol-rich membrane microdomains known as lipid rafts[123]. Activated T cells presented with the movement of two-dimensional surface lipid rafts along actin filaments[124]. The CXCR4 lipid raft association was closely associated with rich cholesterol level[125]. In this instance, the diverse assembly states of CKR complexes may mediate the formation of specific raft domains and their colocalization with actin polymerization at the immune synapse. In addition, higher cholesterol and lipid rafts levels were accompanied with CKRs increasing in immune cells, rather than glycolysis or other canonical metabolic pathways[126]. Lipid raft integrity and cholesterol microdomains are critical for stabilizing T cells CKRs signaling and promoting F-actin polymerization at the periphery of the immune synapse[122]. Of note, the accumulation of actin polymerization at the immunological synapse augments immune cells adhesion force, thereby enhancing cytotoxicity against cancer cells[35]. In tumor cells, CXCL18 induced mTORC1 signaling was proved to increase de novo cholesterol synthesis by activating sterol regulatory element-binding protein-2 (SREBP2)[127]. Targeting mTORC1/SREBP2 directly enhanced metabolic-immunosuppressive role in tumor [128], which implied that CKRs induced mTOR signaling might plays an important role in cholesterol and lipid rafts regulation in Tregs and macrophage[101]. Of note, upregulated SREBP2 expression and the accompanying elevation in cholesterol promote enhanced Ca2+ signaling and coordinated F-actin polymerization in localized immune cells, thereby contributing to immune cells migratory force[129]. Intriguingly, through boost cholesterol synthesis, mTOR/SREBP2 signaling can induce metabolic reprogramming of naive CD4+ T cells and promote their conversion into Tregs and immunosuppressive function[130]. The cholesterol-dependent membrane lipid rafts not merely promote the actin cytoskeleton interactions and cell morpho-mechanical alterations, but also influence the differentiation status of immune cells[131, 132]. For the membrane lipid rafts regions cholesterol transport, apolipoprotein E (ApoE) was a necessary metabolic modulator in various tumor related immune cells, including macrophage and T cells[133]. The ApoE expression modulated the levels and distribution of cholesterol, directly involved in actin-dependent cells elongation and migration morphology[134, 135]. The ApoE inhibition directly decreased M2-like TAMs proportions and migration, which would be further promoted by CCR5 knockdown[136]. Moreover, lacking of CCR5 signaling also presented with defects in ApoE expressed immune cells trafficking[137]. Together, metabolic reprogramming and enhanced cytoskeletal reorganization play an integral role in signal transduction and force generation for immune cell chemotaxis and expansion within the TME.
CKRs signaling reshapes tumor immune cells metabolic-cytoskeleton coupling
T cell
Chemokine induced intracellular metabolic stress is associated with T lymphocyte migration and effect in the TME (Figure 4). The activated T cell adhesion and migration force were facilitated by ATP supplement and immune synapse-related actin cytoskeleton reorganization[81, 84]. The chemokine interaction with cognate CKRs simultaneously stimulates processes that increase intracellular nutrient and energy levels, thereby ensuring cell proliferation and biosynthetic processes. Kamnev et al. reported that the morphological fitness of T lymphocytes scales with the transcriptional activation of glycolysis, actin isoforms and membrane complex subunits[108]. In line with this, integrins induced effector T cells cytoskeletal reorganization and adhesion movement is ATP-dependent behavior[81]. Integrins activation signaling not merely increase the ATP generation, but also promote the Ca2+ to directly influence the membrane permeability. Both CD4+ and CD8+ T cells presented with increased cell size within 24 hours of activation, adapt the higher amounts of metabolites, and then followed by rapid cell division[138]. Meanwhile, T cell membrane domains and the actin cytoskeleton reorganized across increasing spatial scales, from highly ordered lipid rafts to filopodia and immunological synapses [139]. In T cells, CCL5-induced GLUT-1 activation is important in glucose transport and facilitates glucose uptake, which is essential for actin polymerization and directional cell migration[140]. Moreover, CCL5-induced CCR5 signaling activates the mTOR/4E-BP1 pathway to directly modulate mRNA translation, nutrient sensing and glycolysis[140], wherein activated mTORC1 in HIF-1/GLUT1 signaling is required for CD8+ T cells glucose metabolism and glycolysis in PI3K-Akt-independent manner[141]. Importantly, small changes in metabolic parameters are sufficient to lead to significant physiological cell migration changes, especially glycolysis-based rapid ATP production. Intervention of this process directly impaired the ATP generation and T cells migration, which was partially attributable to impaired adhesion and force transmission[94, 95, 140]. Mechanical forces spread across the T cells plasma membrane via a range of systems including integrins and adhesion proteins in conjunction with the actin cytoskeleton and mechanosensitive ion channels [139]. Interestingly, although vertebrate glucose transporters belong to the GLUT family, GLUT1 does not appear to be expressed on quiescent T lymphocytes [142]. With the membrane CXCR4 accumulation, GLUT1 expression was associated with increased glucose metabolism and higher migration ratio in immunopathy[143]. CXCR4 determined the fate of CD8-T lymphocytes immunological infiltration in type 1 diabetes[144]. In tumor, indigenous generation of particular chemokines force T cell activation and move towards the TME[145]. Lettau et al. reported that CXCR4-mediated HS1 phosphorylation is directly involved actin polymerization and adhesive migration, contributing to elongated cellular morphology and immunological synapse in T cells[146]. The TCR signaling transduction at the leading edge of T cells immunological synapse plays a crucial role in CXCR4-based chemotaxis[147]. The CXCR4 and F-actin were colocalized at the peripheral supramolecular activation cluster and redistribute by membrane kinetics in the T cells immune synapse[148]. It seems to support the concept that delivers adhesion receptors and proteins from the rear of the cell to the leading edge of the chemotaxis cell for extension and provides mechanical force to propel the cell forward.
Applying a mechanical load to T cells and Tregs chemotaxis requires actin network and metabolic reprograming cooperation. CKRs-induced glycolytic reprogramming provides crucial biomechanical and energetic support for specific T cells chemotaxis, morphological adaptation, and effector functions. I. Effector T Cells biomechanical-metabolic signaling: CKRs-activated PI3K/mTOR, PPARα and AMPK signaling cascades in T cells shifts metabolism from OXPHOS and fatty acid catabolism toward glycolysis, thereby promoting rapid ATP generation. The CKRs-dependent rapid ATP generation enhance the T cells lamellipodial actin polymerization and physical penetration within tumor. Meanwhile, upregulated GLUT1 and glycolytic enzymes HK1/PKM were synergized with this effect. II. Tregs immunosuppressive metabolic reprogramming: Tumor-driven chemokines would promote CKRs-PI3K/MTORC2 based high glycolytic level and support Tregs chemotaxis and actin polymerization. Meanwhile, CKRs-stimulated STAT3/Foxp3 cascade could reshapes Tregs glycolytic metabolism to adapt depriving TME, thereby promoting immune synapse formation and enhancing migration and immunosuppression. GCK: Glucokinase, OXPHS: Oxidative Phosphorylation, Tregs: Regulatory T cells, GLUT1: Glucose transporter 1, Foxp3: Forkhead box protein P3.
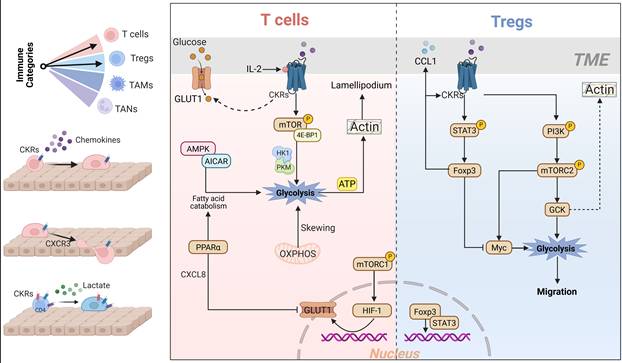
Following activation, effector CD8+ T cells metabolic skews from infantile mitochondrial oxidative phosphorylation (OXPHOS) to glycolysis[138], and is the major source of ATP to drive actin polymerization at the lamellipodium and related sustainment of motility[108]. Due to IL-2 ability in increasing CXCR3 and the migration of T cells in response to CXCL9 and CXCL10[149], IL-2 induced CD8+ T cells glycolysis fitted the proposed notion of CKRs-aligned ATP production and actin cytoskeleton re-organization[108]. During early stimulation, CD8+ T cells undergo drastic morphological changes, with the formation of lamellipodia and immune synapses driven by a rapid ATP supply. Surprisingly, CXCR3 seems to be prevailing modulator in CD8+ T cells trafficking across tumor vessels, while CCR5 and CCR2 did not contribute this chemotactic response[150]. At the membrane terminus, CXCR3 stimulation triggered the effectors of early integrin-dependent adhesion, actin polymerization and motility[151]. Chow et al. reported that CXCR3 signaling is essential for the anti-PD1-mediated CD8+ T cells residing within tumors[5]. The glycolysis fueled the ability of effector T cells lamellipodium thickness, adhesion and orientation during confined chemotaxis, as well as their cytotoxic activity to tumor[5]. Meanwhile, the increased glycolysis level would promote T cells actin-associated immunological synapse formation and outward adhesion force[35, 94]. Given the causal link between actin polarization and T cell glycolytic activity [94], F-actin polarization and its binding to glycolytic enzymes may at least partially explain the robust enhancement of cellular glycolysis during chemotactic morphological reshaping. Intriguingly, the glycolysis and mitochondrial OXPHOS might in different ways to influence T cells morphology and chemotaxis, actin branching at the lamellipodium extension required local glycolysis-derived ATP, while limited-OXPHOS might promote a separate actin pool involved in uropod contractility[108, 152]. In the tumor severe nutrient deprivation milieu, hypofunctional CKRs responses are characterized by a gradual decline metabolism and gradient chemotaxis in effector CD8+ T cells, thereby contributing to an immunosuppressive microenvironment and poor anti-tumor immune response[153]. In this regard, upon the CXCR3 engagement with CXCL10 within lactate environment, significant up-regulation of two rate-limiting enzymes like HK1 and pyruvate kinase M1/2 (PKM) have been observed in the CD4+ T cells glycolytic pathway, rather than CD8+ T cells[9]. Specifically, CXCR3 signaling increased the glycolytic metabolism was directly linked to actin cytoskeleton reorganization and migration force[9, 108]. Despite of this, in tumor or non-tumor systems, CXCR3 is important for localization of CD8+ T cells, and interfere with recombinant cognate chemokines contributes to CD8+ T cell infiltration and tumor regression[150]. The CXCR3+CD8+ T cells chemotaxis was presented with increased level of F-actin polymerization, which drives their high migratory velocity[154]. Noteworthy, orderly cytoskeleton assembly actin polymerization after effector CD8 T cells activation could promoted synaptic autocrine of CCL5 and cytotoxic granules exocytosis during the in mechanical migration within TME[155]. In such self-feedback loop, the synergistic regulation between actin and the T cell local synaptic membrane facilitates adhesion and homing to target cells, thereby enhancing chemotactic efficiency. Besides, CCL5 and its cognate CKRs decreased AMPK/AICAR axis mediated fatty acid catabolism extent in T cell chemotactic response and principally switch to glycolysis and migration [156]. Nguyen et al. observed that the co-localization of receptor CCR5 and actin at both the leading and trailing edges of T cells moving morphology, as well as the enrichment of cholesterol[157]. Generation of ATP from fatty acid catabolism in T-cell mitochondria is evoked by the stimulation of AMPK signaling through membrane receptor, intracellular Ca2+ or decreased intracellular ATP energy stress[158, 159]. Plausibly, in prostate cancer milieu, tumor-derived IL-8 will overactivated PPARα in CD8+ T cells, thereby decreasing glucose utilization by downregulating GLUT1 and HK2[160]. The agitation of cellular PPARα could reduce the use of glucose and promote the fatty acids utilization for metabolic needs [161]. Impeding of glycolysis and fast ATP generation could paralyze CD8+T cells and slack the cellular morphological adaptation[108, 160]. In this regard, due to the effector CD8+ T cells metabolic skewing from infantile mitochondrial OXPHOS to glycolysis[138], underscored the tight coordination between metabolic and actin polarization programs to sustain the T cells morphological adaptation forces, cytotoxic effects and chemotactic activity[108, 112].
Tregs
Tregs are a unique subset of CD4+ T cells that overcoming the hypoxic, acidic, and nutrient-deficient and maintaining immunosuppressive stress in tumor[162]. The high expression of CKRs likely drives the homing of Tregs to chemokine-rich microenvironments, where they suppress tumor antigen presentation and effector T cell responses through cell-cell interactions[163] (Figure 4). The Tregs immunological synapse formation and migratory interaction are accompanied by the dynamic reorganization of the actin cytoskeleton[4, 164]. Generally, Tregs maintain homeostatic frequencies of 5%–10% in blood and lymphoid tissue, while its accumulation exceeding 50% of total T cells in tumor[165]. How to balance the systemic Tregs depletion induced secondary autoimmunity remains the ‘Sword of Damocles’ for current Tregs-targeted anti-tumor immunotherapies. Intra-tumoral Tregs can be distinguished from peripheral Treg cells and other effector T cells by high expression of the CKRs like CXCR3, CCR4 and CCR8[163]. The CXCL10-CXCR4 mediates migration of Tregs and intracellular F-actin polymerization and Ca2+ influx, and underlies the basis of Tregs leading edge migration and immunosuppressive functions[166]. The ability to form a stable immune synapse is essential for the proper function of Tregs, TCR/CD3-driven F-actin reorganization would increase in a few minutes after Tregs stimulation[167]. The lack of actin mobility will impair the TCR signaling, as well as naïve CD4+ T cells differentiation into Tregs subsets[166]. With chemokine induction associated metabolic stress, unlike thymus natural Tregs, peripherally induced Tregs are more likely to selectively accumulate by chemotaxis and reforming metabolism to selectively buildup of in tumor mass[168, 169]. Intriguingly, through adhesion interactions at membrane surfaces and immune synapses, Tregs can transfer their ATP-derived cAMP into DCs, thereby diminishing DC immunogenicity [170]. However, with the CD74_deletion-mediated downregulation of Foxp3 expression and the inhibition of actin cytoskeleton organization, Tregs cell size and immune synapse formation are significantly reduced, thereby contributing to tumor rejection[171]. The cooperative engagement of CKRs and CD74 has been demonstrated to stimulate Ca2+ mobilization and F-actin polymerization, consequently promoting immune cells chemotaxis[172]. The successful establishment of CCL1-CCR8 interaction promotes Tregs Foxp3low to Foxp3high phenotypic characteristic in STAT3 phosphorylation dependent manners, as feedback, activated Foxp3high Tregs will further promote CCL1 secretion and epitopes CCR8 expression to achieve self-feeding mechanism[173]. The CCL1-induced Ca2+ flux and membrane autocrine loop potentiate the Tregs suppressive activities. Plausibly, as a master-regulator of Tregs, Fxop3 not merely a co-transcription factor for Tregs, and also modulates intracellular glucose metabolism [174]. In lactate rich and glucose deprivation TME, Fxop3-upregulation could repress Myc to restrict glycolysis and shifted cellular metabolism to OXPHOS, as well as compensating NAD+ consumption[175]. The glucose depletion and lactate production by tumors create a Tregs-favoring microenvironment, which may subsequently drive an immunosuppressive morphology and chemotactic interactions. Driven by tumor-derived cytokines and intracellular metabolic fueling, Tregs undergo actin cytoskeleton reorganization and exhibit enhanced mechanical deformability, which promotes their chemotaxis toward immune cells[4, 90, 176]. In this way, tumor can impair cytotoxic T cells through multiple extracellular desertification and hijack a physiologic mechanism of immune self-tolerance.
Meanwhile, Tregs also utilized glycolysis to fuel additional rapid energy demand by enhancing mTOR activity with outlier stimulation. The Tregs F-actin polarization and formation of leading-edge synapses were enriched with phospho-AKT, thereby facilitating the directed migration upon chemokine stimulation[177]. CCR4 induced glucose uptake in Tregs was parallelly accompanied with PI3K/mTORC2 cascade induced downstream enzyme glucokinase (GCK) activation, consequently enhancing glycolysis engaged for Tregs migration [178]. Synchronously, GCK could interact with actin to enhance cytoskeletal reorganization during CKRs-stimulated Tregs migration[179]. Glycolytic enzymes including GCK interact with actin to act as a glycolytic ATP feeder in Tregs, thus generating energy required for cytoskeletal rearrangements and migratory force. Unlike other hexokinases, GCK has low affinity manifestation to glucose, which might avoid glycolysis metabolites perturbations and maintain glycolytic flux of Tregs in tumor milieu, giving Tregs an advantage in harsh tumor milieu [180]. The co-localization of ATPase and GCK to the extracellular membrane of immune synapses was observed during actin elongation and morphological changes, whereas unstimulated Tregs displayed reduced F-actin and GCK expression[4]. In various cells, mTORC2 activation is positively linked with Myc expression, rather mTORC1, thereby regulating the glucose transport and glycolytic flux[181]. In terms of this, mTORC2 could regulate the glycolytic flux by fully activating AKT and controlling Myc and GCK expression, consequently modulating Tregs intracellular glycolysis rate-limiting steps [178]. Notably, inhibition of obligatory mTORC2 component Rictor could decrease cell chemotaxis and cytoarchitectural reorganization in an actin blunting-dependent manner[182], which in Tregs manifests as unpolarized cell morphology with limited size and poorly formed immune synapses[4]. Although single CKRs may not be sufficient for Tregs recruitment and function in tumors, specific CKRs activation and signaling transduction are required for Tregs cytoarchitectural reorganization and direct/indirect full immunosuppressive function in tumor[163].
Tumor associated macrophages (TAMs)
Macrophages are types of immune cells in defending against invading pathogens, and they participate in organ development, remodeling, healing and disease (Figure 5). Macrophages are also found in tumors with deleterious effects. In the tumor progression, macrophages density is positively correlated with microvessel counts and negatively correlated with patient prognosis[183]. Actin-rich structures in macrophages, such as basal podosomes and apical synapses, are essential for actin-dependent chemotaxis, phagocytosis, and cell-cell interactions within tumor foci[184]. Huang et al. observed that M2-like TAMs exhibit a tighter podosomes organization with compact actin polymerization fibers than M1 type[185]. Beyond directing macrophage migration, chemokine-CKR signaling drives macrophage polarization toward an M2-like TAM phenotype[186], which is potentially associated with actin reorganization, cell elongation and spindle-shaped morphology functions[187]. In the mouse model, CXCR2-dominated macrophages accumulation was markedly rescued under the CXCL1-inhibition, accompanying with selectively upregulated aerobic glycolysis and tumor adaptability[188]. In macrophages, glycolytic ATP synthesis localizes in filopodia and lammelipodia, where ATP is rapidly consumed during actin remodeling process and continuously membrane protrusions formation during the adhesion movement[99, 105]. Yuan et al. indicated that CCL5 skewed M0 macrophages toward an M2-like TAMs phenotype and migration, as well as promoted macrophages IL-10, IL-12 and TGF-β1 in tumor [189]. Of note, interleukin stimulation triggers the reorganization of actin-rich podosomes into rosette structures within M2 macrophages, enabling degradation of-and migration force through-dense extracellular matrices [190]. Similarly, CCL2 and CXCL12-induced M2-like TAMs polarization and recruitment also were observed in colorectal cancer[191]. There has been well documented for TAMs that could play a pro/M2- or an anti/M1-tumoral function. The CCL5/CCL2-producing tumor cells were able to chemoattract TAMs that co-expressed M1/M2 markers[192, 193]. For the intracellular structure, osteopontin (OPN) and actin expression levels are correlated with TAMs infiltration, wherein the OPN acts as the potent chemokine[194]. The integrin is highly expressed on TAMs and constitutes a major OPN receptor and M2 macrophages took on a more adhesive phenotype than M1-type[194, 195]. Chemokines exerted integrin activation has been approved in various cell types, including immune and tumor cells[196]. The CCR2 and integrin engagement leads to an interaction between activated Rac2 and Myosin 9, and promoting macrophages vascular endothelial growth factor A (VEGF-A) expression[197]. Meanwhile, Myosin 9 accumulated in the leading-edge lamellipodia extensions of macrophages (generated by Rac-induced actin polymerization) in a manner dependent on its motor activity, contributing to an elongated and enlarged morphology [198]. The Myosin 9 deficiency significantly weakened the CCL2-coupling in macrophage. Moreover, chemokine CCL18 stimulated PYK2 phosphorylation in TAMs potentially enhanced TAMs trans-endothelial migration and cancer-promoting functions[199]. The lack of PYK2 directly impairs the macrophages lamellipodia contractile activity and the forward immobilizing force during adhesion migration [200].
Molecular explanation for CKRs signaling regulated actin networks in TAMs and TANs cells migration and functions. CKRs activation promoting the TAMs and TANs migratory morphology and pro-tumor functions. I. TAMs polarization and migration: Activated CKRs promoting the PI3K/AKT2, Rac2, and WAVE2/Arp2/3 complexes signaling, consequently driving the actin nucleation and branched polymerization. Simultaneously, CKRs involved integrin signaling could enhance interaction between mechanosensor Piezo1 and PYK2 to regulate actin polarization and actomyosin formation. CKRs induced glycolysis-dependent M2-like TAMs polarization and the immunosuppressive cytokines secretion. II. TANs chemotactic morphology and migration: Chemokine-CKRs recognition could promote C-terminus LKIL motifs activation and LASP-1 phosphorylation to drive TANs actin polymerization. After CKRs activation, the VASP, α-tubulin, and local FAK signaling were synergistically to modulate TANs adhesion-contraction actin network and provide the adhesive migration force. Meanwhile, actin-myosin interaction driving the formation and of NETs and immunosuppressive function within TME. TAMs: Tumor associated macrophages, TANs: Tumor-associated neutrophil, NETs: Neutrophil extracellular traps, OPN: Osteopontin, VASP: Vasodilator-stimulated phosphoprotein, LASP-1: LIM and SH3 protein 1.
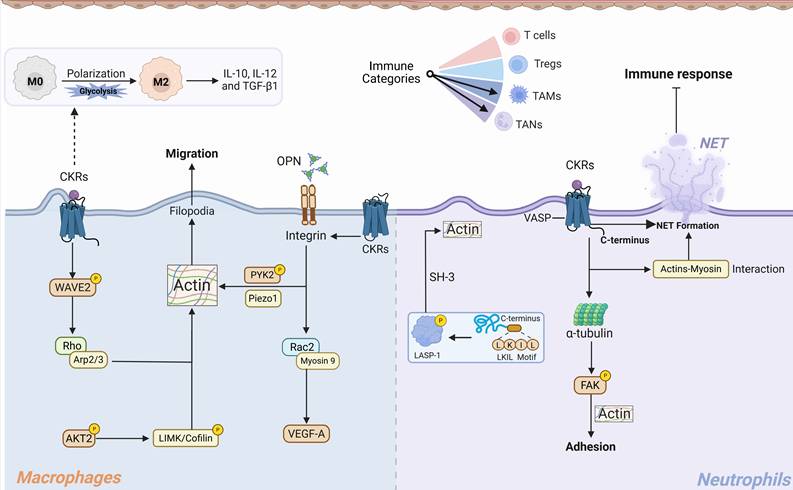
As a mechanical checkpoint, endogenous PYK2 could interact with stimulated CKRs to coordinate ordered F-actin polarization, which drives cellular force generation and significantly maintains TAMs stiffness and population[201]. The PYK2 senses mechanical signals via Piezo1 and integrins, triggering F-actin polymerization and translocating to the nucleus to regulate mechano-transduction and differentiation genes, thereby promoting monocyte-to-TAMs differentiation and recruitment[201].
The Arp2/3 complex is required for macrophage integrin functions and chemotaxis, forming networks involved in lamellipodia protrusion, phagocytosis, and cell adhesion[202]. In addition to an enlarged and elongated morphology after macrophage chemotactic activation, the Arp2/3-branched actin polymerization at leading edge protrusion facilitated the processive advancement force. The loss of Arp2/3 activation suppressed the actin nucleation and lamellipodia in favor of filopodia, thereby decreasing spread cell area and altering macrophages motility shape[202]. Recent experiments underscore the fundamental role of F-actin, not only in mediating protrusion but also in spatially organizing signaling at the phagocytic cup. The altered lamellipodia protrusion/retraction cycles and defects in migration persistence that deregulate the real chemotaxis drivers, a process wherein lamellipodia generates cytoskeletal struts that push against the membrane to drive leading edge advancement and boost cell speed[203]. The WAVE2 directly activated Arp2/3 has been observed in immune cells chemokine coreceptor CXCR4 or CCR5 dependent signaling, and leads to actin nucleation and filament branching[204]. The Rho–formin and Rho family–WASP–ARP2/3 pathways are involved in formation of filopodia and invadopodia (short actin-filled protrusions that degrade extracellular matrix)[204]. This WAVE2 phosphorylation event was involved in both CKRs stimulated Gαi-dependent and -independent pathways[204]. On the other hand, TAMs activation/polarization to distinct functional states is critically supported by metabolic shifts. The CKRs like CCR5 activation stimulated PI3K/AKT has been demonstrated to promote M2 macrophage polarization, thereby promoting tumor progression [205]. Upon activation of TAMs surface CKRs, a spectrum of phosphorylation cascades induces formation of actin filaments in the front and myosin contractile rings at the back and enhances cell adhesion and forward force[4, 98, 99, 102]. AKT was important in orchestrated actin cytoskeleton reorganization during cell migration, wherein AKT2 disruption will inhibit the downstream LIMK/Cofilin phosphorylation and directly contribute to the defects in TAMs actin polymerization and chemotaxis [206]. In summary, TAMs chemotaxis is a spatiotemporally orchestrated response in which a spectrum of phosphorylation cascades couples metabolic shifts with targeted actin reorganization, mechanisms essential for sustaining front-rear polarity and driving forward adhesive movement.
Tumor-associated neutrophil (TANs)
Neutrophils are the first immune cells to be recruited in response to infection or tissue injury to protect the host from harmful agents [207]. Chemotaxis-stimulated neutrophils presented with F-actin polymerization at the leading-edge synapsis and integrin conformational change during the adhesion migration[208]. Meanwhile, growing evidence suggests a crucial regulatory role for neutrophils in tumor establishment and progression[209]. Tumor-associated neutrophils (TANs) exhibit morphological and functional heterogeneity, their plasticity mediating bidirectional responses depending on the surrounding TME [210]. CXCR2 modulated tumor-associated neutrophil (TANs) infiltration has been demonstrated to be negatively associated with tumor prognosis[211]. The CXCR2 signaling was directly involved in F-actin and α-tubulin levels, as well as phagocytic ability [212]. Of note, ligand CXCL1 induced neutrophil adhesion and transmigration were highly relay on β2 integrin-dependent adhesion and actin polymerization, thereby forming larger size and more extracellular synapses[213]. Similarly, reduced extracellular CXCL8 decreases both β2 integrin levels and adhesion strength in TANs to the same extent, thereby reducing the anchoring of entrapped cells[214]. Chemokine stimulation-induced cytoskeletal reorganization promoted neutrophil spreading, wherein F-actin-rich lamellipodia are found at the leading edge [215]. Álvaro et al. reported that CXCR1/2 stimulation in TANs promoted neutrophil extracellular traps (NETs) formation, thereby inhibiting the immune response[216]. F-actin dynamics and myosin II function are essential for stimulus-induced NET formation, accompanying characteristic cell rounding and apparent contraction [215]. This may significantly contribute to the intriguing morphological phenomenon where TANs transition from a homogeneous to a more plastic and heterogeneous state following the chemotactic attraction by tumor cells. The CXCR2 signaling activation is essential for neutrophils mature and structural proteins integrity, and the lack of CXCR2 stimulation will contribute to F-actin polymerization and α-tubulin decrease[217]. The actin-tubulin cytoskeleton reorganization in neutrophils is essential for chemotaxis-induced phagocytosis and maturity [212]. CXCR2 expression by TANs is required for their homing to tumors where CXCR2 ligands are overexpressed [218]. During inflammation, CXCL8 inhibition was proved to abolish actin polymerization, thereby eliminating CXCR2 dependent neutrophils chemotaxis[219]. The CXCL8-induced cytoplasmic calcium concentration upregulation, local intracellular actin high-density polarization and cell rounding were important in neutrophils recruitment [219]. Meanwhile, N-terminal region of CXCR2 to CXCL8 epitope interaction was important for TANs chemotaxis and neutrophil extracellular trap (NET) formation[220, 221]. After stimulation, actins-myosin interactions, myosin that concentrated together with cortical actin along the periphery of the cell body forming the force to support NET expulsion[215, 222].
Furthermore, in tumor adaptation, vasodilator-stimulated phosphoprotein (VASP) has been reported as a promising biomarker [223]. As a processive actin polymerase, it promotes the formation of invasive membrane structures, leading to extracellular matrix remodeling. Neel et al. reported that VASP interacted with CXCR2 after CXCL8 stimulation in neutrophils, wherein free actin barbed ends will recruit VASP to the leading edge and phosphoprotein[224]. Similarly, in CXCR1- and CXCR2-expressing neutrophils, actin and microtubules were required for CXCL8-induced focal adhesion kinase (FAK) phosphorylation[225]. The phosphorylation of FAK was adhesion-dependent and was highly reliant on actin filaments and cytoskeleton reorganization to provide contractility at cell-substratum contact regions. Moreover, LIM and SH3 protein 1 (LASP-1), binds CXCR2 under both basal and ligand activated conditions, and co-localize at the leading edge of migrating cells and mediates cell chemotaxis [226]. The CXCR2 intracellular C-terminus LKIL motif (aa 327–330) interacts with the LASP1 and promotes LASP1 phosphorylation [226]. LASP-1 is an actin binding cytoskeletal protein, which potentially increases the neutrophils actin protein levels by inhibiting the depolymerization of actin[227, 228]. The LASP-1 contributes to cellular immunity and adhesion mechanisms, and predominantly binding to actin occurs via SH3 domain and the first nebulin repeat, and localized at the plasma membrane and in actin-rich subcellular protrusive structures [229]. These intricate coupling of chemokine signaling with dynamic cytoskeletal underlies remodeling the morphological plasticity of TANs, ultimately shaping their complex immune activities and adaptive roles within the TME.
Therapeutic implications and clinical translation
The developing landscape of tumor immunology highlights that immune cell chemotaxis is governed by an intricate "biomechanical-metabolic" axis, revealing novel mechano-checkpoint for clinical intervention. Recent studies reveal that a mechanosensitive immune checkpoint regulated by CKR signaling plays a direct role in immune-synapse cytoarchitecture and the efficacy of tumor immunotherapies[35, 37, 95]. Through distinguish the CKRs-dependent actin polymerization and integrin adhesion signaling may achieve the dual-pronged strategy that synchronously enhancing the effector CD8+ T cells physical penetrance and paralyzing immunosuppressive populations[5, 199, 201, 202]. Furthermore, due to actin-based chemotaxis is bioenergetically demanding, decoupling the metabolic-cytoskeletal connection presents another critical therapeutic strategy. Targeted intervention in either the mTORC2/GCK-mediated glycolysis essential for Treg immune synapses and migratory morphology[4, 178], or the PFKFB3-driven glycolytic flux required for TAM podosome formation and intercellular contact forces[102, 103], can selectively inhibit immune cell chemotaxis. Concurrently, alleviating TME deprivation or targeting metabolic supplementation can rescue the morphological fitness of exhausted CD8+ T cells[114, 115], re-establishing their structural capacity to execute cytotoxic functions.
CKR-directed biomechanical-metabolic therapies have great potential for translation into clinical practice. Advanced delivery systems, such as tumor-targeted magnetic nanomotors[33] or stimuli-responsive polymeric nanoparticles, offer a sophisticated solution for locally disrupting intracellular actin networks or purinergic signaling in immune cells within the TME[81, 82]. These nanotechnology-based interventions can effectively improve immune cells mechanical microenvironment and alter cytoskeletal affinities without broad off-target effects, which overcoming systemic toxicity and the redundancy of chemokine networks. Crucially, these mechano-modulators are highly compatible with combinatorial regimens. Pharmacologically activating specific CKRs and biomechanical signaling promotes initial effector T cell homing, which transforms immunologically 'cold' tumors into 'hot' ones and establishes a structural prerequisite that might synergistically amplifies the efficacy of anti-PD-1/PD-L1 therapy[5, 90]. To develop precision immunotherapy strategies, future research should focus on high-resolution dynamic imaging technologies and elucidate chemotaxis-induced cytoskeletal biomechanical regulation mechanisms, thereby strategically rewiring the chemotaxis, biomechanical, and metabolic forces that drive immune cells trafficking.
Conclusions
As evidence has been shown from the literature, chemokines are extensively studied and greatly contribute to the tumor immune reshapes. With the growing understanding of the biology and biogenesis of CKRs signaling in immune cells migration and actin cytoskeleton alteration, chemotaxis research has generated much excitement in the past decade. An important task is to link findings obtained with different experimental cues, taking into account CKRs conformations, signal transduction and cytoskeleton molecules parameters, as well as post-activation metabolic and polarization of TME immune cells, to reveal immune cells chemotaxis specific and activation signaling networks. Chemokines trigger receptor to activate context-specific signaling pathways through selectin, integrins, Ca2+, and actin modulation, and that in immune cells these functionally different cytoskeleton-binding proteins work in synergy with each other to promote morphology dynamics and force. In particularly, CKRs stimulation-induced immune cells migration appears to require dynamic interplay between actin polymerization and flux membrane protrusion, while metabolite from CKRs-induced metabolic reprogramming substantially contributes to the concentration of leading front actin filaments and flow-forced traction forces in cells. However, the underlying molecular mechanisms are still incompletely understood as the lacking of dynamic-resolution structures and signaling transduction in the stimulation of CKRs. How functionally distinct cytoskeleton molecules structures and cortex alteration of tumor immune cells response to CKRs signaling are only beginning to understand, as exemplified by the coordinate regulation of lamellipodia actin/integrins network and cell migration tractive force.
With critical roles in immune cells structural biochemical regulation, migration and immunosuppressive TME formation, the chemokine system embodies a key therapeutic target for improving tumor immune disorder. Here, we outlined the potential CKRs signaling cascades for immune cells migration that interfere with intracellular function through structural biomechanics and metabolic reprogramming signals. By modulating actin cytoskeleton reorganization, downstream integrin clustering, recruitment of metabolic reprograming for the morphological adaption, the CKRs help potentiate signaling in immune cells, thereby serving as trigger and modulators of immune cells migration required for functional responses in TME. Immune cells trafficking and effector functions within the TME are highly regulated process involving unique and complex combinations of chemokine stimulation, cellular mechanical force and intracellular cytoskeleton networks. Thus, we anticipate that future studies using advanced imaging methods, combined with biophysical approaches, will reveal interesting new insights into mechanical regulation of the chemokine-derived immune cells migration in the TME context and will uncover new links between biochemical and mechanical regulation of the cytoskeleton-reorganization. While efforts are still required to optimize these gaps, this anticipates great potential for the design of future tumor immunotherapy aimed at the chemokine system.
Acknowledgements
We apologize for the omission of any primary citations. We thank BioRender (https://BioRender.com) for the help with artwork.
Funding
This work was financially supported by National Natural Science Foundation of China (82504810 to ZQJ, and 82304395 to ZML); Qimingxing Research Fund for Young Talents (HXQMX0103); and Natural Science Foundation of Sichuan Province 2024NSFSC1739.
Author contributions
J.J. conceptualized and designed the review; Z.Q.J. and C.K.L. wrote the manuscript. Z.Q.J. and Z.M.L. conducted visualization; J.J. supervised the work. All authors reviewed and approved the manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Liu S, Tao Z, Lou J, Li R, Fu X, Xu J. et al. CD4(+) CCR8(+) Tregs in ovarian cancer: a potential effector Tregs for immune regulation. J Transl Med. 2023;21:803
2. Zlotnik A, Yoshie O. The chemokine superfamily revisited. Immunity. 2012;36:705-16
3. Hughes CE, Nibbs RJB. A guide to chemokines and their receptors. FEBS J. 2018;285:2944-71
4. Kishore M, Cheung KCP, Fu H, Bonacina F, Wang G, Coe D. et al. Regulatory T Cell Migration Is Dependent on Glucokinase-Mediated Glycolysis. Immunity. 2017;47:875-89.e10
5. Chow MT, Ozga AJ, Servis RL, Frederick DT, Lo JA, Fisher DE. et al. Intratumoral Activity of the CXCR3 Chemokine System Is Required for the Efficacy of Anti-PD-1 Therapy. Immunity. 2019;50:1498-512.e5
6. Di Pilato M, Kfuri-Rubens R, Pruessmann JN, Ozga AJ, Messemaker M, Cadilha BL. et al. CXCR6 positions cytotoxic T cells to receive critical survival signals in the tumor microenvironment. Cell. 2021;184:4512-30.e22
7. Pollard TD, Cooper JA. Actin, a central player in cell shape and movement. Science. 2009;326:1208-12
8. Guerra-Espinosa C, Jiménez-Fernández M, Sánchez-Madrid F, Serrador JM. ICAMs in Immunity, Intercellular Adhesion and Communication. Cells. 2024;13:339
9. Haas R, Smith J, Rocher-Ros V, Nadkarni S, Montero-Melendez T, D'Acquisto F. et al. Lactate Regulates Metabolic and Pro-inflammatory Circuits in Control of T Cell Migration and Effector Functions. PLoS Biol. 2015;13:e1002202
10. Bindea G, Mlecnik B, Tosolini M, Kirilovsky A, Waldner M, Obenauf AC. et al. Spatiotemporal dynamics of intratumoral immune cells reveal the immune landscape in human cancer. Immunity. 2013;39:782-95
11. Watts AO, Verkaar F, van der Lee MM, Timmerman CA, Kuijer M, van Offenbeek J. et al. β-Arrestin recruitment and G protein signaling by the atypical human chemokine decoy receptor CCX-CKR. J Biol Chem. 2013;288:7169-81
12. de Boer LL, Vanes L, Melgrati S, Biggs O'May J, Hayward D, Driscoll PC. et al. T cell migration requires ion and water influx to regulate actin polymerization. Nat Commun. 2023;14:7844
13. Lappalainen P, Kotila T, Jégou A, Romet-Lemonne G. Biochemical and mechanical regulation of actin dynamics. Nat Rev Mol Cell Biol. 2022;23:836-52
14. Hons M, Kopf A, Hauschild R, Leithner A, Gaertner F, Abe J. et al. Chemokines and integrins independently tune actin flow and substrate friction during intranodal migration of T cells. Nat Immunol. 2018;19:606-16
15. Kumar A, Das JK, Peng HY, Wang L, Ballard DJ, Ren Y. et al. Metabolic fitness of NAC1-deficient Tregs in the tumor microenvironment fuels tumor growth. JCI Insight. 2025;10:e186000
16. Jin J, Zhao Q, Wei Z, Chen K, Su Y, Hu X. et al. Glycolysis-cholesterol metabolic axis in immuno-oncology microenvironment: emerging role in immune cells and immunosuppressive signaling. Cell Biosci. 2023;13:189
17. Guo XJ, Zhu BB, Li J, Guo P, Niu YB, Shi JL. et al. Cholesterol metabolism in tumor immunity: Mechanisms and therapeutic opportunities for cancer. Biochem Pharmacol. 2025;234:116802
18. Bhat AA, Nisar S, Singh M, Ashraf B, Masoodi T, Prasad CP. et al. Cytokine- and chemokine-induced inflammatory colorectal tumor microenvironment: Emerging avenue for targeted therapy. Cancer Commun (Lond). 2022;42:689-715
19. Shao Z, Tan Y, Shen Q, Hou L, Yao B, Qin J. et al. Molecular insights into ligand recognition and activation of chemokine receptors CCR2 and CCR3. Cell Discov. 2022;8:44
20. Sanchez J, Lane JR, Canals M, Stone MJ. Influence of Chemokine N-Terminal Modification on Biased Agonism at the Chemokine Receptor CCR1. Int J Mol Sci. 2019;20:2417
21. Qin L, Kufareva I, Holden LG, Wang C, Zheng Y, Zhao C. et al. Structural biology. Crystal structure of the chemokine receptor CXCR4 in complex with a viral chemokine. Science. 2015;347:1117-22
22. Liu K, Wu L, Yuan S, Wu M, Xu Y, Sun Q. et al. Structural basis of CXC chemokine receptor 2 activation and signalling. Nature. 2020;585:135-40
23. Sonawani A, Kharche S, Dasgupta D, Sengupta D. Insights into the dynamic interactions at chemokine-receptor interfaces and mechanistic models of chemokine binding. J Struct Biol. 2022;214:107877
24. Stephens BS, Ngo T, Kufareva I, Handel TM. Functional anatomy of the full-length CXCR4-CXCL12 complex systematically dissected by quantitative model-guided mutagenesis. Sci Signal. 2020;13:eaay5024
25. Isaikina P, Tsai CJ, Dietz N, Pamula F, Grahl A, Goldie KN. et al. Structural basis of the activation of the CC chemokine receptor 5 by a chemokine agonist. Sci Adv. 2021;7:eabg8685
26. Zhang H, Chen K, Tan Q, Shao Q, Han S, Zhang C. et al. Structural basis for chemokine recognition and receptor activation of chemokine receptor CCR5. Nat Commun. 2021;12:4151
27. Ludeman JP, Stone MJ. The structural role of receptor tyrosine sulfation in chemokine recognition. Br J Pharmacol. 2014;171:1167-79
28. Sonawani A, Kharche S, Dasgupta D, Sengupta D. Allosteric modulation of the chemokine receptor-chemokine CXCR4-CXCL12 complex by tyrosine sulfation. Int J Biol Macromol. 2022;206:812-22
29. Rajarathnam K, Desai UR. Structural Insights Into How Proteoglycans Determine Chemokine-CXCR1/CXCR2 Interactions: Progress and Challenges. Front Immunol. 2020;11:660
30. Liu K, Shen L, Wu M, Liu ZJ, Hua T. Structural insights into the activation of chemokine receptor CXCR2. FEBS J. 2022;289:386-93
31. Lu M, Zhao W, Han S, Lin X, Xu T, Tan Q. et al. Activation of the human chemokine receptor CX3CR1 regulated by cholesterol. Sci Adv. 2022;8:eabn8048
32. SenGupta S, Parent CA, Bear JE. The principles of directed cell migration. Nat Rev Mol Cell Biol. 2021;22:529-47
33. Fan X, Chen H, Li Y, Feng Q, Tao F, Xu C. et al. Actin-Targeted Magnetic Nanomotors Mechanically Modulate the Tumor Mechanical Microenvironment for Cancer Treatment. ACS Nano. 2025;19:6454-67
34. Gera N, Swanson KD, Jin T. β-Arrestin 1-dependent regulation of Rap2 is required for fMLP-stimulated chemotaxis in neutrophil-like HL-60 cells. J Leukoc Biol. 2017;101:239-51
35. Lei K, Kurum A, Kaynak M, Bonati L, Han Y, Cencen V. et al. Cancer-cell stiffening via cholesterol depletion enhances adoptive T-cell immunotherapy. Nat Biomed Eng. 2021;5:1411-25
36. D'Agostino G, Artinger M, Locati M, Perez L, Legler DF, Bianchi ME. et al. β-Arrestin1 and β-Arrestin2 Are Required to Support the Activity of the CXCL12/HMGB1 Heterocomplex on CXCR4. Front Immunol. 2020;11:550824
37. García-Cuesta EM, Rodríguez-Frade JM, Gardeta SR, D'Agostino G, Martínez P, Soler Palacios B. et al. Altered CXCR4 dynamics at the cell membrane impairs directed cell migration in WHIM syndrome patients. Proc Natl Acad Sci U S A. 2022;119:e2119483119
38. Martínez-Muñoz L, Rodríguez-Frade JM, Barroso R, Sorzano CÓ S, Torreño-Pina JA, Santiago CA. et al. Separating Actin-Dependent Chemokine Receptor Nanoclustering from Dimerization Indicates a Role for Clustering in CXCR4 Signaling and Function. Mol Cell. 2018;70:106-19.e10
39. Matalon O, Ben-Shmuel A, Kivelevitz J, Sabag B, Fried S, Joseph N. et al. Actin retrograde flow controls natural killer cell response by regulating the conformation state of SHP-1. EMBO J. 2018;37:e96264
40. Kumar R, Ferez M, Swamy M, Arechaga I, Rejas MT, Valpuesta JM. et al. Increased sensitivity of antigen-experienced T cells through the enrichment of oligomeric T cell receptor complexes. Immunity. 2011;35:375-87
41. Laufer JM, Hauser MA, Kindinger I, Purvanov V, Pauli A, Legler DF. Chemokine Receptor CCR7 Triggers an Endomembrane Signaling Complex for Spatial Rac Activation. Cell Rep. 2019;29:995-1009.e6
42. Terashima Y, Onai N, Murai M, Enomoto M, Poonpiriya V, Hamada T. et al. Pivotal function for cytoplasmic protein FROUNT in CCR2-mediated monocyte chemotaxis. Nat Immunol. 2005;6:827-35
43. Hwang IY, Kim JS, Harrison KA, Park C, Shi CS, Kehrl JH. Chemokine-mediated F-actin dynamics, polarity, and migration in B lymphocytes depend on WNK1 signaling. Sci Signal. 2024;17:eade1119
44. Thelen M, Stein JV. How chemokines invite leukocytes to dance. Nat Immunol. 2008;9:953-9
45. Oakes PW, Wagner E, Brand CA, Probst D, Linke M, Schwarz US. et al. Optogenetic control of RhoA reveals zyxin-mediated elasticity of stress fibres. Nat Commun. 2017;8:15817
46. Colvin RA, Means TK, Diefenbach TJ, Moita LF, Friday RP, Sever S. et al. Synaptotagmin-mediated vesicle fusion regulates cell migration. Nat Immunol. 2010;11:495-502
47. Lorenzen E, Ceraudo E, Berchiche YA, Rico CA, Fürstenberg A, Sakmar TP. et al. G protein subtype-specific signaling bias in a series of CCR5 chemokine analogs. Sci Signal. 2018 11
48. Horioka M, Ceraudo E, Lorenzen E, Sakmar TP, Huber T. Purinergic Receptors Crosstalk with CCR5 to Amplify Ca(2+) Signaling. Cell Mol Neurobiol. 2021;41:1085-101
49. D'Agostino G, Saporito A, Cecchinato V, Silvestri Y, Borgeat A, Anselmi L. et al. Lidocaine inhibits cytoskeletal remodelling and human breast cancer cell migration. Br J Anaesth. 2018;121:962-8
50. Son K, Hussain A, Sehmi R, Janssen L. The Cycling of Intracellular Calcium Released in Response to Fluid Shear Stress Is Critical for Migration-Associated Actin Reorganization in Eosinophils. Cells. 2021;10:157
51. Sommer F, Torraca V, Xie Y, In 't Veld AE, Willemse J, Meijer AH. Disruption of Cxcr3 chemotactic signaling alters lysosomal function and renders macrophages more microbicidal. Cell Rep. 2021;35:109000
52. Tsujimoto K, Takamatsu H, Kumanogoh A. The Ragulator complex: delving its multifunctional impact on metabolism and beyond. Inflamm Regen. 2023;43:28
53. Vestre K, Persiconi I, Borg Distefano M, Mensali N, Guadagno NA, Bretou M. et al. Rab7b regulates dendritic cell migration by linking lysosomes to the actomyosin cytoskeleton. J Cell Sci. 2021;134:jcs259221
54. Wong HS, Jaumouillé V, Freeman SA, Doodnauth SA, Schlam D, Canton J. et al. Chemokine Signaling Enhances CD36 Responsiveness toward Oxidized Low-Density Lipoproteins and Accelerates Foam Cell Formation. Cell Rep. 2016;14:2859-71
55. Yamaguchi N, Zhang Z, Schneider T, Wang B, Panozzo D, Knaut H. Rear traction forces drive adherent tissue migration in vivo. Nat Cell Biol. 2022;24:194-204
56. Sheetz MP, Felsenfeld DP, Galbraith CG. Cell migration: regulation of force on extracellular-matrix-integrin complexes. Trends Cell Biol. 1998;8:51-4
57. Tu Y, Pal K, Austin J, Wang X. Filopodial adhesive force in discrete nodes revealed by integrin molecular tension imaging. Curr Biol. 2022;32:4386-96.e3
58. Kamioka Y, Ueda Y, Kondo N, Tokuhiro K, Ikeda Y, Bergmeier W. et al. Distinct bidirectional regulation of LFA1 and α4β7 by Rap1 and integrin adaptors in T cells under shear flow. Cell Rep. 2023;42:112580
59. Margraf A, Germena G, Drexler HCA, Rossaint J, Ludwig N, Prystaj B. et al. The integrin-linked kinase is required for chemokine-triggered high-affinity conformation of the neutrophil β2-integrin LFA-1. Blood. 2020;136:2200-5
60. Lefort CT, Rossaint J, Moser M, Petrich BG, Zarbock A, Monkley SJ. et al. Distinct roles for talin-1 and kindlin-3 in LFA-1 extension and affinity regulation. Blood. 2012;119:4275-82
61. Wen L, Moser M, Ley K. Molecular mechanisms of leukocyte β2 integrin activation. Blood. 2022;139:3480-92
62. Mikucki M, Skitzki J, Fisher D, Luster A, Evans S. Obligate role of CXCR3 chemokine receptor for trafficking of effector CD8 T cells in the tumor microenvironment. J IMMUNOL. 2012;188:46.14
63. Sanz-Ortega L, Andersson A, Carlsten M. Harnessing upregulated E-selectin while enhancing SDF-1α sensing redirects infused NK cells to the AML-perturbed bone marrow. Leukemia. 2024;38:579-89
64. Alon R, Hammer DA, Springer TA. Lifetime of the P-selectin-carbohydrate bond and its response to tensile force in hydrodynamic flow. Nature. 1995;374:539-42
65. Zhong K, Fan S, Yao S, Xu H, Bai S. A Atractylodes lancea polysaccharide inhibits metastasis of human osteosarcoma U-2 OS cells by blocking sialyl Lewis X (sLe(x) )/E-selectin binding. J Cell Mol Med. 2020;24:12789-98
66. Smith ML, Olson TS, Ley K. CXCR2- and E-selectin-induced neutrophil arrest during inflammation in vivo. J Exp Med. 2004;200:935-9
67. Cappenberg A, Kardell M, Zarbock A. Selectin-Mediated Signaling-Shedding Light on the Regulation of Integrin Activity in Neutrophils. Cells. 2022;11:1310
68. Bouti P, Webbers SDS, Fagerholm SC, Alon R, Moser M, Matlung HL. et al. β2 Integrin Signaling Cascade in Neutrophils: More Than a Single Function. Front Immunol. 2020;11:619925
69. Liu ZJ, Tian R, Li Y, An W, Zhuge Y, Livingstone AS. et al. Inhibition of tumor angiogenesis and melanoma growth by targeting vascular E-selectin. Ann Surg. 2011;254:450-6 discussion 6-7
70. Menzel L, Höpken UE, Rehm A. Angiogenesis in Lymph Nodes Is a Critical Regulator of Immune Response and Lymphoma Growth. Front Immunol. 2020;11:591741
71. Huang Q, Yang W, Wang F, Huang R, Wang Q, Li X. et al. SELP(+) TEC:CD8(+) T cell crosstalk associates with improved radiotherapy efficacy in cervical cancer. Mol Cancer. 2025;24:41
72. Enarsson K, Lundgren A, Kindlund B, Hermansson M, Roncador G, Banham AH. et al. Function and recruitment of mucosal regulatory T cells in human chronic Helicobacter pylori infection and gastric adenocarcinoma. Clin Immunol. 2006;121:358-68
73. Steele M, Fogler W, Magnani J, Hollingsworth M. A small molecule glycomimetic antagonist of E-selectin and CXCR4 (GMI-1359) delays pancreatic tumor metastasis and significantly alters the pancreatic tumor microenvironment. CANCER RES. 2016;76:902
74. Mishra R, Kovalska J, Janda J, Vannucci L, Rajmon R, Horak V. Tumor progression is associated with increasing CD11b+ cells and CCL2 in Lewis rat sarcoma. Anticancer Res. 2015;35:703-11
75. Schunk CT, Wang W, Sabo LN, Taufalele PV, Reinhart-King CA. Matrix stiffness increases energy efficiency of endothelial cells. Matrix Biol. 2024;133:77-85
76. Morlino G, Barreiro O, Baixauli F, Robles-Valero J, González-Granado JM, Villa-Bellosta R. et al. Miro-1 links mitochondria and microtubule Dynein motors to control lymphocyte migration and polarity. Mol Cell Biol. 2014;34:1412-26
77. Tang F, Chen JN, Zhang NN, Gong LP, Jiang Y, Feng ZY. et al. Expression of CCL21 by EBV-associated gastric carcinoma cells protects CD8(+)CCR7(+) T lymphocytes from apoptosis via the mitochondria-mediated pathway. Pathology. 2018;50:613-21
78. Fu Z, Ye J, Dean JW, Bostick JW, Weinberg SE, Xiong L. et al. Requirement of Mitochondrial Transcription Factor A in Tissue-Resident Regulatory T Cell Maintenance and Function. Cell Rep. 2019;28:159-71.e4
79. Tan S, Li S, Min Y, Gisterå A, Moruzzi N, Zhang J. et al. Platelet factor 4 enhances CD4(+) T effector memory cell responses via Akt-PGC1α-TFAM signaling-mediated mitochondrial biogenesis. J Thromb Haemost. 2020;18:2685-700
80. Yazicioglu YF, Marin E, Sandhu C, Galiani S, Raza IGA, Ali M. et al. Dynamic mitochondrial transcription and translation in B cells control germinal center entry and lymphomagenesis. Nat Immunol. 2023;24:991-1006
81. Hamoudi C, Lashgari A, Aoudjit F. Integrins α3 and α6 promote Th17 cell migration by activating the purinergic receptor P2X4. Purinergic Signal. 2026;22:8
82. Li C, Liu Q, Han L, Zhang H, Immler R, Rathkolb B. et al. The eATP/P2×7R Axis Drives Quantum Dot-Nanoparticle Induced Neutrophil Recruitment in the Pulmonary Microcirculation. Adv Sci (Weinh). 2024;11:e2404661
83. Xia J, Sun S, Jotte M, Uy G, Bohana-Kashtan O, Sorani E. et al. CXCR4 Blockade By BL-8040 in T Cell Acute Lymphoblastic Leukemia Decreases Mitochondrial Mass and Induces Non-Apoptotic Cell Death. BLOOD. 2019;134:2745
84. Krummel MF, Friedman RS, Jacobelli J. Modes and mechanisms of T cell motility: roles for confinement and Myosin-IIA. Curr Opin Cell Biol. 2014;30:9-16
85. Baixauli F, Martín-Cófreces NB, Morlino G, Carrasco YR, Calabia-Linares C, Veiga E. et al. The mitochondrial fission factor dynamin-related protein 1 modulates T-cell receptor signalling at the immune synapse. EMBO J. 2011;30:1238-50
86. Garde A, Sherwood DR. Fueling Cell Invasion through Extracellular Matrix. Trends Cell Biol. 2021;31:445-56
87. Li B, Li J, Wang L, Wei Y, Luo X, Guan J. et al. Yak-Derived CXCL14 Activates the Pro-Inflammatory Response of Macrophages and Inhibits the Proliferation and Migration of HepG2. Animals (Basel). 2023;13:3036
88. Dolinska M, Cai H, Månsson A, Shen J, Xiao P, Bouderlique T. et al. Characterization of the bone marrow niche in patients with chronic myeloid leukemia identifies CXCL14 as a new therapeutic option. Blood. 2023;142:73-89
89. Acevedo DS, Fang WB, Rao V, Penmetcha V, Leyva H, Acosta G. et al. Regulation of growth, invasion and metabolism of breast ductal carcinoma through CCL2/CCR2 signaling interactions with MET receptor tyrosine kinases. Neoplasia. 2022;28:100791
90. Pant A, Jain A, Chen Y, Patel K, Saleh L, Tzeng S. et al. The CCR6-CCL20 Axis Promotes Regulatory T-cell Glycolysis and Immunosuppression in Tumors. Cancer Immunol Res. 2024;12:1542-58
91. Kulkarni N, Meitei HT, Sonar SA, Sharma PK, Mujeeb VR, Srivastava S. et al. CCR6 signaling inhibits suppressor function of induced-Treg during gut inflammation. J Autoimmun. 2018;88:121-30
92. Gómez-Melero S, Caballero-Villarraso J. CCR6 as a Potential Target for Therapeutic Antibodies for the Treatment of Inflammatory Diseases. Antibodies (Basel). 2023;12:30
93. Toriyama K, Kuwahara M, Kondoh H, Mikawa T, Takemori N, Konishi A. et al. T cell-specific deletion of Pgam1 reveals a critical role for glycolysis in T cell responses. Commun Biol. 2020;3:394
94. Paillon N, Ung TPL, Dogniaux S, Stringari C, Hivroz C. Label-free single-cell live imaging reveals fast metabolic switch in T lymphocytes. Mol Biol Cell. 2024;35:ar11
95. Harjunpää H, Somermäki R, Saldo Rubio G, Fusciello M, Feola S, Faisal I. et al. Loss of β2-integrin function results in metabolic reprogramming of dendritic cells, leading to increased dendritic cell functionality and anti-tumor responses. Oncoimmunology. 2024;13:2369373
96. Sun K, Zhang X, Shi J, Huang J, Wang S, Li X. et al. Elevated protein lactylation promotes immunosuppressive microenvironment and therapeutic resistance in pancreatic ductal adenocarcinoma. J Clin Invest. 2025;135:e187024
97. Dong Y, Jiang W, Qiu Y, Xu Z, Cheng J, Ke P. et al. Lactate-driven IL8+ Tumor-Associated Macrophages Mediate Immunosuppression and Resistance to Immunotherapy in Clear Cell Renal Cell Carcinoma. Clin Cancer Res. 2026;32:1860-1873
98. N Md-B, Duncan-Moretti J, H Cd-C, Saldanha-Gama R, Paula-Neto HA, G GD. et al. Aerobic glycolysis is a metabolic requirement to maintain the M2-like polarization of tumor-associated macrophages. Biochim Biophys Acta Mol Cell Res. 2020;1867:118604
99. Venter G, Oerlemans FT, Wijers M, Willemse M, Fransen JA, Wieringa B. Glucose controls morphodynamics of LPS-stimulated macrophages. PLoS One. 2014;9:e96786
100. Ye H, Zhou Q, Zheng S, Li G, Lin Q, Wei L. et al. Tumor-associated macrophages promote progression and the Warburg effect via CCL18/NF-kB/VCAM-1 pathway in pancreatic ductal adenocarcinoma. Cell Death Dis. 2018;9:453
101. Covarrubias AJ, Aksoylar HI, Yu J, Snyder NW, Worth AJ, Iyer SS. et al. Akt-mTORC1 signaling regulates Acly to integrate metabolic input to control of macrophage activation. Elife. 2016;5:e11612
102. Fuhrmann DC, Brüne B. miR-193a-3p increases glycolysis under hypoxia by facilitating Akt phosphorylation and PFKFB3 activation in human macrophages. Cell Mol Life Sci. 2022;79:89
103. Braun M, Qorraj M, Büttner M, Klein FA, Saul D, Aigner M. et al. CXCL12 promotes glycolytic reprogramming in acute myeloid leukemia cells via the CXCR4/mTOR axis. Leukemia. 2016;30:1788-92
104. Ziegler M, Hughes C. The SDF-1/CXCR4 axis utilizes mTOR signal transduction to promote angiogenesis. FASEB J. 2014;28:supplement.278.8
105. Semba H, Takeda N, Isagawa T, Sugiura Y, Honda K, Wake M. et al. HIF-1α-PDK1 axis-induced active glycolysis plays an essential role in macrophage migratory capacity. Nat Commun. 2016;7:11635
106. Zhang Y, Zhang C, Feng R, Meng T, Peng W, Song J. et al. CXCR4 regulates macrophage M1 polarization by altering glycolysis to promote prostate fibrosis. Cell Commun Signal. 2024;22:456
107. Xin X, Li Z, Yan X, Liu T, Li Z, Chen Z. et al. Hepatocyte-specific Smad4 deficiency inhibits hepatocarcinogenesis by promoting CXCL10/CXCR3-dependent CD8(+)- T cell-mediated anti-tumor immunity. Theranostics. 2024;14:5853-5868
108. Kamnev A, Mehta T, Wielscher M, Chaves B, Lacouture C, Mautner AK. et al. Coordinated ARP2/3 and glycolytic activities regulate the morphological and functional fitness of human CD8(+) T cells. Cell Rep. 2024;43:113853
109. Cascone T, McKenzie JA, Mbofung RM, Punt S, Wang Z, Xu C. et al. Increased Tumor Glycolysis Characterizes Immune Resistance to Adoptive T Cell Therapy. Cell Metab. 2018;27:977-87.e4
110. Hu C, You W, Kong D, Huang Y, Lu J, Zhao M. et al. Tertiary Lymphoid Structure-Associated B Cells Enhance CXCL13(+)CD103(+)CD8(+) Tissue-Resident Memory T-Cell Response to Programmed Cell Death Protein 1 Blockade in Cancer Immunotherapy. Gastroenterology. 2024;166:1069-84
111. Zhang H, Liu J, Yang Z, Zeng L, Wei K, Zhu L. et al. TCR activation directly stimulates PYGB-dependent glycogenolysis to fuel the early recall response in CD8(+) memory T cells. Mol Cell. 2022;82:3077-88.e6
112. Comrie WA, Babich A, Burkhardt JK. F-actin flow drives affinity maturation and spatial organization of LFA-1 at the immunological synapse. J Cell Biol. 2015;208:475-91
113. Zeng YQ, Lu C, Dai Z. Editorial: Memory T Cells: Effectors, Regulators, and Implications for Transplant Tolerance. Front Immunol. 2016;7:7
114. Sasaki K, Nishina S, Yamauchi A, Fukuda K, Hara Y, Yamamura M. et al. Nanoparticle-Mediated Delivery of 2-Deoxy-D-Glucose Induces Antitumor Immunity and Cytotoxicity in Liver Tumors in Mice. Cell Mol Gastroenterol Hepatol. 2021;11:739-62
115. Mao Q, Cai H, Chen M, Xiao A, Jiao H, Jia F. et al. Reprogramming the Tumor Mechanical Microenvironment with Lactate Inhibition and Stimulator of Interferon Genes Activation to Potentiate Systematic Radio-Immunotherapy. ACS Nano. 2026;20:6808-22
116. Cho SH, Raybuck AL, Blagih J, Kemboi E, Haase VH, Jones RG. et al. Hypoxia-inducible factors in CD4(+) T cells promote metabolism, switch cytokine secretion, and T cell help in humoral immunity. Proc Natl Acad Sci U S A. 2019;116:8975-84
117. Zhou X, Fang D, Liu H, Ou X, Zhang C, Zhao Z. et al. PMN-MDSCs accumulation induced by CXCL1 promotes CD8(+) T cells exhaustion in gastric cancer. Cancer Lett. 2022;532:215598
118. Cho S, Raybuck A, Kemboi E, Haase V, Boothby M. et al. Hypoxia-Inducible Factors (HIF) in CD4 T cells promote metabolism, switch cytokine secretion, and T cell help in humoral immunity. Proc Natl Acad Sci U S A. 2019;116:8975-8984
119. Kamata T, Jin H, Giblett S, Patel B, Patel F, Foster C. et al. The cholesterol-binding protein NPC2 restrains recruitment of stromal macrophage-lineage cells to early-stage lung tumours. EMBO Mol Med. 2015;7:1119-37
120. Liu C, Yao Z, Wang J, Zhang W, Yang Y, Zhang Y. et al. Macrophage-derived CCL5 facilitates immune escape of colorectal cancer cells via the p65/STAT3-CSN5-PD-L1 pathway. Cell Death Differ. 2020;27:1765-81
121. van Aalst EJ, McDonald CJ, Wylie BJ. Cholesterol Biases the Conformational Landscape of the Chemokine Receptor CCR3: A MAS SSNMR-Filtered Molecular Dynamics Study. J Chem Inf Model. 2023;63:3068-85
122. Delaguillaumie A, Harriague J, Kohanna S, Bismuth G, Rubinstein E, Seigneuret M. et al. Tetraspanin CD82 controls the association of cholesterol-dependent microdomains with the actin cytoskeleton in T lymphocytes: relevance to co-stimulation. J Cell Sci. 2004;117:5269-82
123. Kucia M, Reca R, Miekus K, Wanzeck J, Wojakowski W, Janowska-Wieczorek A. et al. Trafficking of normal stem cells and metastasis of cancer stem cells involve similar mechanisms: pivotal role of the SDF-1-CXCR4 axis. Stem Cells. 2005;23:879-94
124. Yabuuchi S, Endo S, Baek K, Hoshino K, Tsujino Y, Vestergaard MC. et al. Raft-dependent endocytic movement and intracellular cluster formation during T cell activation triggered by concanavalin A. J Biosci Bioeng. 2017;124:685-93
125. Schabath H, Runz S, Joumaa S, Altevogt P. CD24 affects CXCR4 function in pre-B lymphocytes and breast carcinoma cells. J Cell Sci. 2006;119:314-25
126. van Os BW, Vos WG, Bosmans LA, van Tiel CM, Lith SC, den Toom MS. et al. Hyperlipidaemia elicits an atypical, T helper 1-like CD4(+) T-cell response: a key role for very low-density lipoprotein. Eur Heart J Open. 2023;3:oead013
127. Xiong X, Zhang S, Zhu W, Du J, Liao X, Hu S. et al. Androgen-ablative therapies inducing CXCL8 regulates mTORC1/SREBP2-dependent cholesterol biosynthesis to support progression of androgen receptor negative prostate cancer cells. Oncogene. 2024;43:3456-68
128. Tang W, Zhou J, Yang W, Feng Y, Mok M, Cheng A. Metabolic-immunoregulatory role of CCRK-mTOR signaling in NAFLD-associated hepatocellular carcinoma. CANCER RES. 2019;79:2804
129. Jiang Q, Tang YX, Zhou G. 7-Ketocholesterol promotes T cell migration through Ca(2+)-NFATc1 pathway-mediated F-actin polymerization and proinflammatory cytokine production in oral lichen planus. Front Immunol. 2026;17:1682589
130. Shi Y, Wu D, Wang Y, Shao Y, Zeng F, Zhou D. et al. Treg and neutrophil extracellular trap interaction contributes to the development of immunosuppression in sepsis. JCI Insight. 2024;9:e180132
131. Mitchell JS, Brown WS, Woodside DG, Vanderslice P, McIntyre BW. Clustering T-cell GM1 lipid rafts increases cellular resistance to shear on fibronectin through changes in integrin affinity and cytoskeletal dynamics. Immunol Cell Biol. 2009;87:324-36
132. Thomas S, Preda-Pais A, Casares S, Brumeanu TD. Analysis of lipid rafts in T cells. Mol Immunol. 2004;41:399-409
133. Dong L, Yang X, Wang Y, Jin Y, Zhou Q, Chen G. et al. Key Markers Involved in the Anticolon Cancer Response of CD8+ T Cells through the Regulation of Cholesterol Metabolism. J Oncol. 2021;2021:9398661
134. Lee S, Amici S, Tavori H, Zeng WM, Freeland S, Fazio S. et al. PMP22 is critical for actin-mediated cellular functions and for establishing lipid rafts. J Neurosci. 2014;34:16140-52
135. Bonacina F, Coe D, Wang G, Longhi MP, Baragetti A, Moregola A. et al. Myeloid apolipoprotein E controls dendritic cell antigen presentation and T cell activation. Nat Commun. 2018;9:3083
136. Huang W, Li W, Chen X, Xiang C, Luo K. APOE Drives Glioma Progression by Modulating CCL5/CCR5 Signaling in the Tumor Microenvironment and Inducing M2 Macrophage Polarization. Immunobiology. 2025;230:152895
137. Kuziel WA, Dawson TC, Quinones M, Garavito E, Chenaux G, Ahuja SS. et al. CCR5 deficiency is not protective in the early stages of atherogenesis in apoE knockout mice. Atherosclerosis. 2003;167:25-32
138. Wang R, Dillon CP, Shi LZ, Milasta S, Carter R, Finkelstein D. et al. The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation. Immunity. 2011;35:871-82
139. Garcia E, Ismail S. Spatiotemporal Regulation of Signaling: Focus on T Cell Activation and the Immunological Synapse. Int J Mol Sci. 2020 21
140. Chan O, Burke JD, Gao DF, Fish EN. The chemokine CCL5 regulates glucose uptake and AMP kinase signaling in activated T cells to facilitate chemotaxis. J Biol Chem. 2012;287:29406-16
141. Finlay DK, Rosenzweig E, Sinclair LV, Feijoo-Carnero C, Hukelmann JL, Rolf J. et al. PDK1 regulation of mTOR and hypoxia-inducible factor 1 integrate metabolism and migration of CD8+ T cells. J Exp Med. 2012;209:2441-53
142. Chakrabarti R, Jung CY, Lee TP, Liu H, Mookerjee BK. Changes in glucose transport and transporter isoforms during the activation of human peripheral blood lymphocytes by phytohemagglutinin. J Immunol. 1994;152:2660-8
143. Swainson L, Kinet S, Manel N, Battini JL, Sitbon M, Taylor N. Glucose transporter 1 expression identifies a population of cycling CD4+ CD8+ human thymocytes with high CXCR4-induced chemotaxis. Proc Natl Acad Sci U S A. 2005;102:12867-72
144. Zhong T, Li X, Lei K, Tang R, Zhou Z, Zhao B. et al. CXCL12-CXCR4 mediates CD57(+) CD8(+) T cell responses in the progression of type 1 diabetes. J Autoimmun. 2024;143:103171
145. Tokunaga R, Zhang W, Naseem M, Puccini A, Berger MD, Soni S. et al. CXCL9, CXCL10, CXCL11/CXCR3 axis for immune activation - A target for novel cancer therapy. Cancer Treat Rev. 2018;63:40-7
146. Lettau M, Kabelitz D, Janssen O. SDF1α-induced interaction of the adapter proteins Nck and HS1 facilitates actin polymerization and migration in T cells. Eur J Immunol. 2015;45:551-61
147. Allam AH, Charnley M, Pham K, Russell SM. Developing T cells form an immunological synapse for passage through the β-selection checkpoint. J Cell Biol. 2021 220
148. Pérez-Martínez M, Gordón-Alonso M, Cabrero JR, Barrero-Villar M, Rey M, Mittelbrunn M. et al. F-actin-binding protein drebrin regulates CXCR4 recruitment to the immune synapse. J Cell Sci. 2010;123:1160-70
149. Beider K, Nagler A, Wald O, Franitza S, Dagan-Berger M, Wald H. et al. Involvement of CXCR4 and IL-2 in the homing and retention of human NK and NK T cells to the bone marrow and spleen of NOD/SCID mice. Blood. 2003;102:1951-8
150. Mikucki ME, Fisher DT, Matsuzaki J, Skitzki JJ, Gaulin NB, Muhitch JB. et al. Non-redundant requirement for CXCR3 signalling during tumoricidal T-cell trafficking across tumour vascular checkpoints. Nat Commun. 2015;6:7458
151. Dagan-Berger M, Feniger-Barish R, Avniel S, Wald H, Galun E, Grabovsky V. et al. Role of CXCR3 carboxyl terminus and third intracellular loop in receptor-mediated migration, adhesion and internalization in response to CXCL11. Blood. 2006;107:3821-31
152. Campello S, Lacalle RA, Bettella M, Mañes S, Scorrano L, Viola A. Orchestration of lymphocyte chemotaxis by mitochondrial dynamics. J Exp Med. 2006;203:2879-86
153. Burkard T, Dreis C, Herrero San Juan M, Huhn M, Weigert A, Pfeilschifter JM. et al. Enhanced CXCR4 Expression of Human CD8(Low) T Lymphocytes Is Driven by S1P(4). Front Immunol. 2021;12:668884
154. Ito T, Hashizume H, Shimauchi T, Funakoshi A, Ito N, Fukamizu H. et al. CXCL10 produced from hair follicles induces Th1 and Tc1 cell infiltration in the acute phase of alopecia areata followed by sustained Tc1 accumulation in the chronic phase. J Dermatol Sci. 2013;69:140-7
155. Franciszkiewicz K, Boutet M, Gauthier L, Vergnon I, Peeters K, Duc O. et al. Synaptic release of CCL5 storage vesicles triggers CXCR4 surface expression promoting CTL migration in response to CXCL12. J Immunol. 2014;193:4952-61
156. Taub DD, Hesdorffer CS, Ferrucci L, Madara K, Schwartz JB, Goetzl EJ. Distinct energy requirements for human memory CD4 T-cell homeostatic functions. Faseb j. 2013;27:342-9
157. Kim HR, Na BR, Kwon MS, Ko YS, Han WC, Jun CD. Dynamic motile T cells highly respond to the T cell stimulation via PI3K-Akt and NF-κB pathways. PLoS One. 2013;8:e59793
158. Palma FR, Ratti BA, Paviani V, Coelho DR, Miguel R, Danes JM. et al. AMPK-deficiency forces metformin-challenged cancer cells to switch from carbohydrate metabolism to ketogenesis to support energy metabolism. Oncogene. 2021;40:5455-67
159. Lochner M, Berod L, Sparwasser T. Fatty acid metabolism in the regulation of T cell function. Trends Immunol. 2015;36:81-91
160. Xu F, Wang X, Huang Y, Zhang X, Sun W, Du Y. et al. Prostate cancer cell-derived exosomal IL-8 fosters immune evasion by disturbing glucolipid metabolism of CD8(+) T cell. Cell Rep. 2023;42:113424
161. Chekaoui A, Ertl HCJ. PPARα Agonist Fenofibrate Enhances Cancer Vaccine Efficacy. Cancer Res. 2021;81:4431-40
162. Wang Y, Huang T, Gu J, Lu L. Targeting the metabolism of tumor-infiltrating regulatory T cells. Trends Immunol. 2023;44:598-612
163. Moreno Ayala MA, Campbell TF, Zhang C, Dahan N, Bockman A, Prakash V. et al. CXCR3 expression in regulatory T cells drives interactions with type I dendritic cells in tumors to restrict CD8(+) T cell antitumor immunity. Immunity. 2023;56:1613-30.e5
164. Dohnke S, Moehser S, Surnov A, Kurth T, Jessberger R, Kretschmer K. et al. Role of Dynamic Actin Cytoskeleton Remodeling in Foxp3(+) Regulatory T Cell Development and Function: Implications for Osteoclastogenesis. Front Immunol. 2022;13:836646
165. Núñez NG, Tosello Boari J, Ramos RN, Richer W, Cagnard N, Anderfuhren CD. et al. Tumor invasion in draining lymph nodes is associated with Treg accumulation in breast cancer patients. Nat Commun. 2020;11:3272
166. Ito T, Ito N, Hashizume H, Takigawa M. Roxithromycin inhibits chemokine-induced chemotaxis of Th1 and Th2 cells but regulatory T cells. J Dermatol Sci. 2009;54:185-91
167. Janssen E, Kumari S, Tohme M, Ullas S, Barrera V, Tas JM. et al. DOCK8 enforces immunological tolerance by promoting IL-2 signaling and immune synapse formation in Tregs. JCI Insight. 2017;2:e94298
168. Lin YC, Mahalingam J, Chiang JM, Su PJ, Chu YY, Lai HY. et al. Activated but not resting regulatory T cells accumulated in tumor microenvironment and correlated with tumor progression in patients with colorectal cancer. Int J Cancer. 2013;132:1341-50
169. Chen W, Jin W, Hardegen N, Lei KJ, Li L, Marinos N. et al. Conversion of peripheral CD4+CD25- naive T cells to CD4+CD25+ regulatory T cells by TGF-beta induction of transcription factor Foxp3. J Exp Med. 2003;198:1875-86
170. Rueda CM, Jackson CM, Chougnet CA. Regulatory T-Cell-Mediated Suppression of Conventional T-Cells and Dendritic Cells by Different cAMP Intracellular Pathways. Front Immunol. 2016;7:216
171. Bonnin E, Rodrigo Riestra M, Marziali F, Mena Osuna R, Denizeau J, Maurin M. et al. CD74 supports accumulation and function of regulatory T cells in tumors. Nat Commun. 2024;15:3749
172. Klasen C, Ohl K, Sternkopf M, Shachar I, Schmitz C, Heussen N. et al. MIF promotes B cell chemotaxis through the receptors CXCR4 and CD74 and ZAP-70 signaling. J Immunol. 2014;192:5273-84
173. Barsheshet Y, Wildbaum G, Levy E, Vitenshtein A, Akinseye C, Griggs J. et al. CCR8(+)FOXp3(+) T(reg) cells as master drivers of immune regulation. Proc Natl Acad Sci U S A. 2017;114:6086-91
174. Sarkar T, Dhar S, Sa G. Tumor-infiltrating T-regulatory cells adapt to altered metabolism to promote tumor-immune escape. Curr Res Immunol. 2021;2:132-41
175. Angelin A, Gil-de-Gómez L, Dahiya S, Jiao J, Guo L, Levine MH. et al. Foxp3 Reprograms T Cell Metabolism to Function in Low-Glucose, High-Lactate Environments. Cell Metab. 2017;25:1282-93.e7
176. Ullrich KA, Derdau J, Baltes C, Battistella A, Rosso G, Uderhardt S. et al. IL-3 receptor signalling suppresses chronic intestinal inflammation by controlling mechanobiology and tissue egress of regulatory T cells. Gut. 2023;72:2081-94
177. Fan T, Huang X, Liu C, Liu R, Wang T, Ruan Q. Egress of murine regulatory T cells from the thymus requires TIPE2. Biochem Biophys Res Commun. 2018;500:376-83
178. Kishore M, Cheung KCP, Fu H, Bonacina F, Wang G, Coe D. et al. Regulatory T Cell Migration Is Dependent on Glucokinase-Mediated Glycolysis. Immunity. 2018;48:831-2
179. Yan Y, Huang L, Liu Y, Yi M, Chu Q, Jiao D. et al. Metabolic profiles of regulatory T cells and their adaptations to the tumor microenvironment: implications for antitumor immunity. J Hematol Oncol. 2022;15:104
180. Lenzen S. A fresh view of glycolysis and glucokinase regulation: history and current status. J Biol Chem. 2014;289:12189-94
181. Xu Z, Xu M, Liu P, Zhang S, Shang R, Qiao Y. et al. The mTORC2-Akt1 Cascade Is Crucial for c-Myc to Promote Hepatocarcinogenesis in Mice and Humans. Hepatology. 2019;70:1600-13
182. Kuehn HS, Jung MY, Beaven MA, Metcalfe DD, Gilfillan AM. Prostaglandin E2 activates and utilizes mTORC2 as a central signaling locus for the regulation of mast cell chemotaxis and mediator release. J Biol Chem. 2011;286:391-402
183. Chen JJ, Lin YC, Yao PL, Yuan A, Chen HY, Shun CT. et al. Tumor-associated macrophages: the double-edged sword in cancer progression. J Clin Oncol. 2005;23:953-64
184. Hanna SJ, McCoy-Simandle K, Leung E, Genna A, Condeelis J, Cox D. Tunneling nanotubes, a novel mode of tumor cell-macrophage communication in tumor cell invasion. J Cell Sci. 2019;132:jcs223321
185. Huang X, Li Z, Huang Y, Zhang Q, Cui Y, Shi X. et al. Vimentin intermediate filaments coordinate actin stress fibers and podosomes to determine the extracellular matrix degradation by macrophages. Dev Cell. 2025;60:1669-85.e6
186. Chiang Y, Tsai YC, Wang CC, Hsueh FJ, Huang CY, Chung SD. et al. Tumor-Derived C-C Motif Ligand 2 Induces the Recruitment and Polarization of Tumor-Associated Macrophages and Increases the Metastatic Potential of Bladder Cancer Cells in the Postirradiated Microenvironment. Int J Radiat Oncol Biol Phys. 2022;114:321-33
187. Shook PL, Wang-Heaton H, Casteel JL, Dalal S, Singh M, Yakubenko V. et al. Exogenous Ubiquitin Differentially Modulates the Phenotype and Function of M1 and M2 Macrophages. Cells. 2025;14:879
188. Wang S, Bai J, Zhang YL, Lin QY, Han X, Qu WK. et al. CXCL1-CXCR2 signalling mediates hypertensive retinopathy by inducing macrophage infiltration. Redox Biol. 2022;56:102438
189. Zhuang Y, Zhao X, Yuan B, Zeng Z, Chen Y. Blocking the CCL5-CCR5 Axis Using Maraviroc Promotes M1 Polarization of Macrophages Cocultured with Irradiated Hepatoma Cells. J Hepatocell Carcinoma. 2021;8:599-611
190. Cougoule C, Van Goethem E, Le Cabec V, Lafouresse F, Dupré L, Mehraj V. et al. Blood leukocytes and macrophages of various phenotypes have distinct abilities to form podosomes and to migrate in 3D environments. Eur J Cell Biol. 2012;91:938-49
191. Jiang H, Ge H, Shi Y, Yuan F, Yue H. CAFs secrete CXCL12 to accelerate the progression and cisplatin resistance of colorectal cancer through promoting M2 polarization of macrophages. Med Oncol. 2023;40:90
192. Manfroi B, De Grandis M, Moreaux J, Tabruyn S, Mayol JF, Quintero M. et al. The microenvironment of DLBCL is characterized by noncanonical macrophages recruited by tumor-derived CCL5. Blood Adv. 2021;5:4338-51
193. Möckel D, Bartneck M, Niemietz P, Wagner M, Ehling J, Rama E. et al. CCL2 chemokine inhibition primes the tumor vasculature for improved nanomedicine delivery and efficacy. J Control Release. 2024;365:358-68
194. Wei J, Marisetty A, Schrand B, Gabrusiewicz K, Hashimoto Y, Ott M. et al. Osteopontin mediates glioblastoma-associated macrophage infiltration and is a potential therapeutic target. J Clin Invest. 2019;129:137-49
195. Pham K, Huynh D, Le L, Delitto D, Yang L, Huang J. et al. E-cigarette promotes breast carcinoma progression and lung metastasis: Macrophage-tumor cells crosstalk and the role of CCL5 and VCAM-1. Cancer Lett. 2020;491:132-45
196. Drechsler M, de Jong R, Rossaint J, Viola JR, Leoni G, Wang JM. et al. Annexin A1 counteracts chemokine-induced arterial myeloid cell recruitment. Circ Res. 2015;116:827-35
197. Morrison AR, Yarovinsky TO, Young BD, Moraes F, Ross TD, Ceneri N. et al. Chemokine-coupled β2 integrin-induced macrophage Rac2-Myosin IIA interaction regulates VEGF-A mRNA stability and arteriogenesis. J Exp Med. 2014;211:1957-68
198. Hemkemeyer SA, Vollmer V, Schwarz V, Lohmann B, Honnert U, Taha M. et al. Local Myo9b RhoGAP activity regulates cell motility. J Biol Chem. 2021;296:100136
199. Huang C, Ou R, Chen X, Zhang Y, Li J, Liang Y. et al. Tumor cell-derived SPON2 promotes M2-polarized tumor-associated macrophage infiltration and cancer progression by activating PYK2 in CRC. J Exp Clin Cancer Res. 2021;40:304
200. Okigaki M, Davis C, Falasca M, Harroch S, Felsenfeld DP, Sheetz MP. et al. Pyk2 regulates multiple signaling events crucial for macrophage morphology and migration. Proc Natl Acad Sci U S A. 2003;100:10740-5
201. Xie W, Yu X, Yang Q, Ke N, Wang P, Kong H. et al. The Immunomechanical Checkpoint PYK2 Governs Monocyte-to-Macrophage Differentiation in Pancreatic Cancer. Cancer Discov. 2025;15:1740-65
202. Rotty JD, Brighton HE, Craig SL, Asokan SB, Cheng N, Ting JP. et al. Arp2/3 Complex Is Required for Macrophage Integrin Functions but Is Dispensable for FcR Phagocytosis and In vivo Motility. Dev Cell. 2017;42:498-513.e6
203. Davidson AJ, Millard TH, Evans IR, Wood W. Ena orchestrates remodelling within the actin cytoskeleton to drive robust Drosophila macrophage chemotaxis. J Cell Sci. 2019;132:jcs224618
204. Spear M, Guo J, Turner A, Yu D, Wang W, Meltzer B. et al. HIV-1 triggers WAVE2 phosphorylation in primary CD4 T cells and macrophages, mediating Arp2/3-dependent nuclear migration. J Biol Chem. 2014;289:6949-59
205. Yang H, He Y, Qu F, Zhu J, Deng L, Jiang F. et al. Maraviroc enhances Bortezomib sensitivity in multiple myeloma by inhibiting M2 macrophage polarization via PI3K/AKT/RhoA signaling pathway in macrophages. Cell Div. 2025;20:5
206. Zhang B, Ma Y, Guo H, Sun B, Niu R, Ying G. et al. Akt2 is required for macrophage chemotaxis. Eur J Immunol. 2009;39:894-901
207. Wang J. Neutrophils in tissue injury and repair. Cell Tissue Res. 2018;371:531-9
208. Bouti P, Klein B, Verkuijlen PJH, Schornagel K, van Alphen FPJ, Taris KH. et al. SKAP2 acts downstream of CD11b/CD18 and regulates neutrophil effector function. Front Immunol. 2024;15:1344761
209. Luyang H, Zeng F, Lei Y, He Q, Zhou Y, Xu J. Bidirectional role of neutrophils in tumor development. Mol Cancer. 2025;24:22
210. Que H, Fu Q, Lan T, Tian X, Wei X. Tumor-associated neutrophils and neutrophil-targeted cancer therapies. Biochim Biophys Acta Rev Cancer. 2022;1877:188762
211. Ji HZ, Liu B, Ren M, Li S, Zheng JF, Liu TY. et al. The CXCLs-CXCR2 axis modulates the cross-communication between tumor-associated neutrophils and tumor cells in cervical cancer. Expert Rev Clin Immunol. 2024;20:559-69
212. Delobel P, Ginter B, Rubio E, Balabanian K, Lazennec G. CXCR2 intrinsically drives the maturation and function of neutrophils in mice. Front Immunol. 2022;13:1005551
213. Wee JL, Schulze KE, Jones EL, Yeung L, Cheng Q, Pereira CF. et al. Tetraspanin CD37 Regulates β2 Integrin-Mediated Adhesion and Migration in Neutrophils. J Immunol. 2015;195:5770-9
214. Huh SJ, Liang S, Sharma A, Dong C, Robertson GP. Transiently entrapped circulating tumor cells interact with neutrophils to facilitate lung metastasis development. Cancer Res. 2010;70:6071-82
215. Sprenkeler EGG, Tool ATJ, Henriet SSV, van Bruggen R, Kuijpers TW. Formation of neutrophil extracellular traps requires actin cytoskeleton rearrangements. Blood. 2022;139:3166-80
216. Teijeira Á, Garasa S, Gato M, Alfaro C, Migueliz I, Cirella A. et al. CXCR1 and CXCR2 Chemokine Receptor Agonists Produced by Tumors Induce Neutrophil Extracellular Traps that Interfere with Immune Cytotoxicity. Immunity. 2020;52:856-71.e8
217. Mendonça MA, Souto FO, Micheli DC, Alves-Filho JC, Cunha FQ, Murta EF. et al. Mechanisms affecting neutrophil migration capacity in breast cancer patients before and after chemotherapy. Cancer Chemother Pharmacol. 2014;73:317-24
218. Bonavita O, Massara M, Bonecchi R. Chemokine regulation of neutrophil function in tumors. Cytokine Growth Factor Rev. 2016;30:81-6
219. Souto FO, Zarpelon AC, Staurengo-Ferrari L, Fattori V, Casagrande R, Fonseca MJ. et al. Quercetin reduces neutrophil recruitment induced by CXCL8, LTB4, and fMLP: inhibition of actin polymerization. J Nat Prod. 2011;74:113-8
220. Shi X, Wan Y, Wang N, Xiang J, Wang T, Yang X. et al. Selection of a picomolar antibody that targets CXCR2-mediated neutrophil activation and alleviates EAE symptoms. Nat Commun. 2021;12:2547
221. Gao J, Liu J, Lu J, Zhang X, Zhang W, Li Q. et al. SKAP1 Expression in Cancer Cells Enhances Colon Tumor Growth and Impairs Cytotoxic Immunity by Promoting Neutrophil Extracellular Trap Formation via the NFATc1/CXCL8 Axis. Adv Sci (Weinh). 2024;11:e2403430
222. Mannherz HG, Budde H, Jarkas M, Hassoun R, Malek-Chudzik N, Mazur AJ. et al. Reorganization of the actin cytoskeleton during the formation of neutrophil extracellular traps (NETs). Eur J Cell Biol. 2024;103:151407
223. Pitari GM, Cotzia P, Ali M, Birbe R, Rizzo W, Bombonati A. et al. Vasodilator-Stimulated Phosphoprotein Biomarkers Are Associated with Invasion and Metastasis in Colorectal Cancer. Biomark Cancer. 2018;10:1179299x18774551
224. Neel NF, Barzik M, Raman D, Sobolik-Delmaire T, Sai J, Ham AJ. et al. VASP is a CXCR2-interacting protein that regulates CXCR2-mediated polarization and chemotaxis. J Cell Sci. 2009;122:1882-94
225. Cohen-Hillel E, Yron I, Meshel T, Soria G, Attal H, Ben-Baruch A. CXCL8-induced FAK phosphorylation via CXCR1 and CXCR2: cytoskeleton- and integrin-related mechanisms converge with FAK regulatory pathways in a receptor-specific manner. Cytokine. 2006;33:1-16
226. Raman D, Sai J, Neel NF, Chew CS, Richmond A. LIM and SH3 protein-1 modulates CXCR2-mediated cell migration. PLoS One. 2010;5:e10050
227. Zhang S, Wang Z, Zhang Y, Dong X, Zhu Q, Yuan C. et al. LASP1 inhibits the formation of NETs and alleviates acute pancreatitis by stabilizing F-actin polymerization in neutrophils. Biochem Biophys Res Commun. 2025;744:151134
228. Raman D, Duvall-Noelle N, Richmond A. Chemokine-mediated nuclear translocation and novel nuclear role for LIM and SH3 Protein-1(LASP-1) in breast cancer. CANCER RES. 2012;72:P1-05 -1
229. Butt E, Howard CM, Raman D. LASP1 in Cellular Signaling and Gene Expression: More than Just a Cytoskeletal Regulator. Cells. 2022;11:3817
Author contact
![]() Corresponding author: Jing Jin M.D., Email: jinjing777edu.cn.
Corresponding author: Jing Jin M.D., Email: jinjing777edu.cn.